Это изобретение касается способа получения промежуточных соединений для β-лактамных антибиотиков. В частности, оно касается усовершенствованного способа получения сульфоксидов сложных эфиров 7-замещенного амино-3-экзометиленцефама.
Получение сложных эфиров сульфоксида 3-экзометиленцефама осуществляются известным двухстайдиынм способом, который включает превращение сложного эфира сульфоксида пенициллина в хлорсульфинилазетидинон с последующей циклизацией последнего до сложного эфира сульфоксида 3-экзометиленцефама. Сложный эфир сульфоксида пенициллина превращают в промежуточный хлорсульфинилазетидинон N-хлоргалогенизирующим агентом, как описано Kukolja в патент США N 4165315. Промежуточные соединения 4-хлорсульфинилазетидинона описаны и запатентованы Kukolja в патенте США N 4081440, Chou в патент США N 4075203 описывает получение сложного эфира сульфоксида 3-экзометиленцефама путем превращения сложного эфира сульфоксида пенициллина на первой стадии в 4-хлорсульфинилазетидион с помощью N-хлоргалогенизирующего агента в присутствии окиси алкилена и окиси кальция. Последний, Chou, в патент США N 4289695 описывает усовершенствованный способ получения сложных эфиров сульфоксидов 3-экзометиленцефама с использованием кислотного акцептора из сшитого поливинилпиридинового полимера на стадии I.
Kukolja в патенте США N 4052387 описывает циклизацию 4-хлорсульфинилазетидинонов катализатором Фриделя-Крафтса типа кислоты Льюиса, с помощью катализатора Фриделя-Крафтса типа протонной кислоты Bronsted a или с образующим обменный катион агентом. Позднее Chou в патенте США N 4190724 описывает и патентует усовершенствованный способ который включает проведение циклизации 4-хлорсульфинилазетидинона по Kukolia с помощью катализатора Фриделя-Крафтса в присутствии оксосоединений, таких как простые эфиры, кетоны или окиси фосфина. и соавторы в патенте США N 4950753, включенном в библиографическую справку, описывают дальнейшее усовершенствование способа Kukolja, которое включает проведение циклизации с катализатором Фриделя-Крафтса в присутствии как оксосоединений по методу Chou, так и ненасыщенных соединений, например, алкенов, таких как 1- или 2-гексан, циклоалкенов, таких как циклогексен, аллен или несопряженных циклоалкадиенов, таких как 1,4-циклогексадиен. Настоящее изобретение предусматривает также дальнейшее усовершенствование способа Kukolja, которое включает циклизацию по Фриделю-Крафтсу, проводимую в присутствии нитросоединения, например, нитрометана, нитроэтана или нитробензола.
На фиг. 1 представлен график, иллюстрирующий скорости реакции циклизации 4-хлорсульфинилазетидинона в различных условиях; на фиг. 2 - график, иллюстрирующий амплитуду сравнений скоростей реакций для фиг. 1.
В способе согласно изобретению хлорсульфинилазетидинон подвергают взаимодействию в инертном растворителе с катализатором Фриделя-Крафтса типа кислоты Льюиса, который образует комплекс с хлорсульфинилазетидиноном в присутствии нитросоединения. В предпочтительном осуществлении катализатором Фриделя-Крафтса является хлористое олово (4), а нитросоединением является нитрометан, и реакция протекает в присутствии оксоединения, например простого эфира, кетона или окиси фосфина и алкена, циклоалкена, аллена или циклодиена. Сложный эфир сульфоксида 3-экзометиленцефама получают с повышенным выходом, обычно в пределах 2-4% от выделенного проукта и повышенными скоростями реакций.
Способ согласно изобретению предусматривает сложный эфир сульфоксида 3-экзометиленцефама, представленный формулой I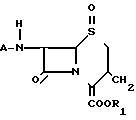 (1) где А представляет собой аминозащитную группу или группу формулы
(1) где А представляет собой аминозащитную группу или группу формулы
R- - R представляет собой остаток карбоновой кислоты RCOOH, и R1 представляет собой карбоксизащитную группу, полученный циклизацией хлорсульфинилазетидинона, представленного формулой 2
- R представляет собой остаток карбоновой кислоты RCOOH, и R1 представляет собой карбоксизащитную группу, полученный циклизацией хлорсульфинилазетидинона, представленного формулой 2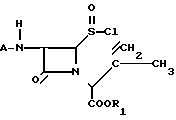
(2) с катализатором Фриделя-Крафтса типа кислоты Льюиса и нитросоединением в сущности безводных условиях в течение времени и при температуре, достаточных для получения соединения формулы 1.
Термин "остаток карбоновой кислоты" включает остатки боковых цепей в 7-м положении, известные в химии цефалоспорина и карбоцефалоспорина, и остатки боковых цепей в 6-м положении, известные в химии пенициллина. Обычно эти боковые цепи являются остатками карбоновых кислот с 1-20 углеродными атомами и представлены примерами, когда R является водородом, С1-С6-алкилом, С1-С6-алкилом, замещенным циано, карбокси, галогеном, амино, С1-С4-алкокси, С1-С4-алкилтио, трифторметилом или трифторметилтио; нафтилом, фенилом или замещенной фенильной группой формулы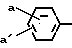 в которой а и а' независимо друг от друга являются водородом, галогеном, окси, С1-С4-алкокси, С1-С4-алканоилокси, С1-С4-алкилом, С1-С4-алкилтио, амино, С1-С4-алканоиламино, С1-С4-алксилсульфониламино, карбокси, карбамоилом, оксиметилом, аминометилом, карбоксиметилом, С1-С4-галоалкилом или С1-С4-пергалоидным алкилом; группой формулы
в которой а и а' независимо друг от друга являются водородом, галогеном, окси, С1-С4-алкокси, С1-С4-алканоилокси, С1-С4-алкилом, С1-С4-алкилтио, амино, С1-С4-алканоиламино, С1-С4-алксилсульфониламино, карбокси, карбамоилом, оксиметилом, аминометилом, карбоксиметилом, С1-С4-галоалкилом или С1-С4-пергалоидным алкилом; группой формулы 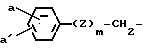 в которой а и а' имеют те же значения, которые определены для них выше. Z представляет собой атом кислорода или серы и m равно нулю или единице; арилметильной группой формулы
в которой а и а' имеют те же значения, которые определены для них выше. Z представляет собой атом кислорода или серы и m равно нулю или единице; арилметильной группой формулы
R3 -CH2- в которой R3 представляет собой нафтил, тиенил, фурил, бензотиенил, бензоаминотиазил, бензофурил, пиридил, 4-пиридилтио, пиримидил, пиридазинил, индолил, пиразолил, имидазолил, триазолил, тетразолил, оксазолил, тиазолил, оксадиазолил, триадиазолил и такие арильные группы, замещенные амино, окси, галогеном, С1-С4-алкилом, С1-С4-алкокси, фенилом, замещенным фенилом или С1-С4-алкилсульфониламино; замещенной метильной группой формулы R4- H- в которой R4 представляет собой циклогекс-1,4-диенил, фенил или замещенный фенил формулы
H- в которой R4 представляет собой циклогекс-1,4-диенил, фенил или замещенный фенил формулы 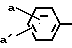 в которой а и а' имеют значения, определенные выше, или R4 является R3с его значениями, определенными выше, и Q представляет собой окси, C1-С4-алканоилокси, карбокси, сульфо, амино, сульфоамино или замещенную аминогруппу формулы -NH-
в которой а и а' имеют значения, определенные выше, или R4 является R3с его значениями, определенными выше, и Q представляет собой окси, C1-С4-алканоилокси, карбокси, сульфо, амино, сульфоамино или замещенную аминогруппу формулы -NH- -
-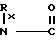 -Ry в которой Rx представляет собой водород или С1-С3-алкил, Ry представляет собой С1-С4-алкил, фурил, тиенил, фенил, галофенил, нитрофенил, стирил, галостирил или группу формулы -
-Ry в которой Rx представляет собой водород или С1-С3-алкил, Ry представляет собой С1-С4-алкил, фурил, тиенил, фенил, галофенил, нитрофенил, стирил, галостирил или группу формулы -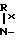 Rz в которой Rx имеет те же значения, как определено выше, и Rzпредставляет собой водород, С1-С3-алкилсульфонил, С1-С3-алкил или С1-С4-алканоил; или Q представляет собой замещенную аминогруппу формулы
Rz в которой Rx имеет те же значения, как определено выше, и Rzпредставляет собой водород, С1-С3-алкилсульфонил, С1-С3-алкил или С1-С4-алканоил; или Q представляет собой замещенную аминогруппу формулы 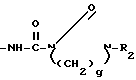 в которой Rz имеет те же значения, как определено выше, и q равно двум или трем; или Q представляет собой замещенную амино-группу формулы: -HNC(= O)-
в которой Rz имеет те же значения, как определено выше, и q равно двум или трем; или Q представляет собой замещенную амино-группу формулы: -HNC(= O)- -(C1-C4) или Q представляет собой бензамидогруппу формулы
-(C1-C4) или Q представляет собой бензамидогруппу формулы 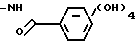 в которой х равен 1-3; или Q представляет собой пиридон или оксизамещенную пиридонилкарбониламиногруппу формулы
в которой х равен 1-3; или Q представляет собой пиридон или оксизамещенную пиридонилкарбониламиногруппу формулы 
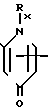 OH в которой Rx имеет значения, определенные выше; или Q представляет собой пиридилкарбониламиногруппу формулы
OH в которой Rx имеет значения, определенные выше; или Q представляет собой пиридилкарбониламиногруппу формулы 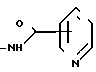 при этом такая группа необязательно замещена С1-С4-алкилом амино, карбокси, окси или галогеном; или Q представляет собой имидазолил или пиразолильную группу формулы
при этом такая группа необязательно замещена С1-С4-алкилом амино, карбокси, окси или галогеном; или Q представляет собой имидазолил или пиразолильную группу формулы 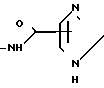 или
или 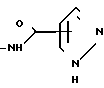 при этом такой имидазолил или пиразолил необязательно замещен С1-С4-алкилом, карбокси, имено или галогеном; или Q представляет собой бензопиридазин-4-оновую группу или ее таутомер, представленный формулой
при этом такой имидазолил или пиразолил необязательно замещен С1-С4-алкилом, карбокси, имено или галогеном; или Q представляет собой бензопиридазин-4-оновую группу или ее таутомер, представленный формулой 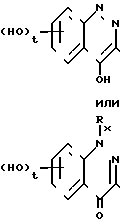
 где Rx имеет значения, как определено выше, и t равно от единицы до трех; или Q представляет собой бензпирановую группу формулы
где Rx имеет значения, как определено выше, и t равно от единицы до трех; или Q представляет собой бензпирановую группу формулы 
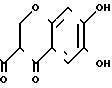 или R является группой формулы R5-
или R является группой формулы R5- - , R5-
- , R5-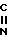 -
-

 R
R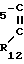 -
- где R5 является R3 или R4, как определено выше. R12 является водородом или галогеном и R6 представляет собой водород, С1-С4-алкил, С1-С4-алкил, замещенный галогеном, карбоксизамещенной алкильной или циклоалкильной группой, представленной формулой -
где R5 является R3 или R4, как определено выше. R12 является водородом или галогеном и R6 представляет собой водород, С1-С4-алкил, С1-С4-алкил, замещенный галогеном, карбоксизамещенной алкильной или циклоалкильной группой, представленной формулой -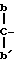 (CH
(CH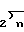 COR7 в которой b и b' независимо друг от друга являются водородом или С1-С3-алкилом, n равно нулю, единице, двум или трем; и b и b', взятые вместе с атомом углерода, с которым они связаны, образуют 3-6-членное карбоциклическое кольцо, и R7 представляет собой окси, С1-С4-амино, С1-С4-алкиламино или ди-(С1-С4-алкил)-амино; или R6 представляет собой С1-С4-алкил, замещенный фенилом или фенилом, замещенным одним или двумя одинаковыми или различными группами, выбранными среди С1-С4-алкила, окси, галогена, карбокси или замещенной карбоксигруппы; или R6представляет собой С1-С4-алкил, замещенный амино- или защищенной аминогруппой; или R6 представляет собой С1-С4-алкенил; или R6 является циклической лактамной группой формулы
COR7 в которой b и b' независимо друг от друга являются водородом или С1-С3-алкилом, n равно нулю, единице, двум или трем; и b и b', взятые вместе с атомом углерода, с которым они связаны, образуют 3-6-членное карбоциклическое кольцо, и R7 представляет собой окси, С1-С4-амино, С1-С4-алкиламино или ди-(С1-С4-алкил)-амино; или R6 представляет собой С1-С4-алкил, замещенный фенилом или фенилом, замещенным одним или двумя одинаковыми или различными группами, выбранными среди С1-С4-алкила, окси, галогена, карбокси или замещенной карбоксигруппы; или R6представляет собой С1-С4-алкил, замещенный амино- или защищенной аминогруппой; или R6 представляет собой С1-С4-алкенил; или R6 является циклической лактамной группой формулы 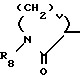 в которой V равно 2-4 и R8 является водородом или С1-С4-алкилом; или R6представляет собой арилметильную группу формулы:
в которой V равно 2-4 и R8 является водородом или С1-С4-алкилом; или R6представляет собой арилметильную группу формулы:
R3 -CH2- где R3 имеет те же значения, как определено выше.
Термин "карбоксизащитная группа", использованный в описании, относится к одной из групп производных сложного эфира карбоновой кислоты, обычно применяемых для блокирования или защиты группы карбоновой кислоты в процессе проведения реакций на других функциональных группах соединения. Примеры таких защитных групп карбоновой кислоты включают 4-нитробензил, 4-метилбензил, 3,4-диметтоксибензил, 2,4-диметоксибензил, 2,4,6-триметоксибензил, 2,4,6-триметилбензил, пентаметилбензил, 3,4-метилендиоксибензил, бензгидрил, 4,4'-диметоксибензгидрил, 2,2', 4,4'-тетраметоксибензгидрил, третбутил, трет-амил, трифенилметил, 4-метокситрифенилметил, 4,4'-диметокситрифенилметил: 4,4', 4''-триметокситрифенилметил, 2-фенилпроп-2-ил, триметилсилил, трет-бутилдиметилсилил, фенацил, 2,2,2-трихлорэтил, β-[ди-(н-бутил)-метилсилил] -этил, пара-толуолсульфонилэтил, 4-нитробензилсульфонилэтил, аллил, цинанамил, 1-(триметилсилилметил)-проп-1-ен-3-ил и тому подобные части молекулы. Вид применяемой карбоксизащитной группы не является критическим постольку, поскольку производная карбоновая кислота остается стабильной в условиях последующей (-их) реакции (-ий) на других положениях кольцевой системы, и может быть удалена в нужный момент без разрушения остальной молекулы. Предпочтительной защитной группой карбоновой кислоты является аллильная группа. Аналогичные карбоксизащитные группы, используемые в химии цефалоспоринов, пенициллинов и пептидов, могут быть также применены для защиты заместителей карбоксигрупп азетидинона. Дальнейшие примеры этих групп находим у Е. Haslam, "Protective Groups in Organic Chemistry", I. G. W. McOmie, Ed. , Plenum Press, New York, N. Y. , 1973, Chapter 5, and T. W. Greene, "Protective Groups in Organic Synthesis", John Wiley and Sons, New York, N. Y. , 1981, Chapter 5. Родственный термин "защищенная карбокси" означает, что карбоксигруппа замещена одной из перечисленных выше карбоксизащитных групп.
Термин "аминозащитная группа" относится к заместителям аминогруппы, обычно применяемым для блокировки или защиты аминофункциональности в процессе взаимодействия других функциональных групп на соединении. Примеры таких аминозащитных групп включают формильную группу, тритильную группу, фталимидогруппу. Упомянутый термин охватывает также аналогичные аминозащитные группы, используемые в химии цефалоспоринов, пеницилинов и пептидов. Дальнейшие примеры групп, относящихся к упомянутому выше термину описаны I. W. Barton, "Protective Groups in Organic Chemistry", I. G. W. NcOmie, Ed. , Plenum Press. New York, N. Y. , 1973, Chapter 2, and T. W. Greene, "Protective Groups in Organic Synthesis", John Wiley and Sons, New York, N. Y. , 1981, Chapter 7. Родственный термин "защищенная амино" означает, что аминогруппа замещена упомянутой аминозащитной группой.
В приведенном выше определении соединений, представленных формулой (1), С1-С6-алкил относится к алкильным группам с прямой и разветвленной цепью, таким как метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, н-пентил, н-гексил, 3-метилпентил и подобные алкильные группы; С1-С6-алкил, замещенный циано, относится к цианометилу, цианоэтилу, 4-цианобутилу и тому подобным; С1-С6-алкил, замещенный карбокси, относится к таким группам, как карбоксиметил, 2-карбоксиэтил 2-карбоксипропил, 4-карбоксибутил, 5-карбоксипентил и тому подобные; С1-С6-алкил, замещенный галогеном, относится к хлорметилу, бромметилу, 2-хлорэтилу, 1-бромэтилу, 4-хлорбутилу, 4-бромпентилу, 6-хлоргексилу, 4-фторбутилу, 3-фторпропилу, фторметилу, и тому подобным; С1-С6-алкил, замещенный аминогруппой, относится к таким группам, как 2-аминоэтил, аминометил, 3-аминопропил, и 4-аминобутил; С1-С6-алкил, замещенный С1-С4-алкоксигруппой, относится к метоксиметилу, 2-метоксиэтилу, 2-этоксиэтилу, этоксиметилу, 3-пропоксипропилу, 3-этоксибутилу, 4-трет-бутилоксибутилу, 3-метоксипентилу, 6-метоксигексилу и тому подобной группе. С1-С6-алкил, замещенный С1-С4-алкилтиогруппой, относится к таким группам как, например, метилтиометил, 2-метилтиоэтил, 3-этилтиопропил, 4-метилтиобутил, 5-этилтиогексил, 3-трет-бутилтиопропил и тому подобные группы; С1-С6-алкил, замещенный трифторметилом, является например, 2,2,2-трифторэтилом, 3,3,3-трифторпропилом, 4,4,4-трифторбутилом и подобные, и С1-С6-алкил, замещенный трифторметилтиогруппой, касается, например, трифторметилтиометила, 2-трифторметилтиоэтила, 2-трифторметилтиопропила, 4-трифторметилтиобутила, 5-трифторметилтиогексила и подобных С1-С6-алкильных замещенных групп.
Если в формуле 1 А является группой формулы
R-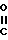 - и R представляет собой замещенную фенильную группу, в которой заместитель (-и) представлен (ы) символами а и а', то примерами таких групп являются галоидный фенил, такой как 4-хлорфенил, 3-бромфенил, 2-фторфенил, 2,4-дихлорфенил и 3,5-дихлорфенил; оксифенил, такой как 2-оксифенил, 3-оксифенил, 4-оксифенил, 2,4-диоксифенил, и 3,4-диоксифенил; алкоксифенил, такой как 2,6-диметоксифенил, 4-метоксифенил, 3-этоксифенил, 3,4-диметоксифенил, 4-трет-бутилоксифенил, 4-метокси-3-этоксифе- нил, и 4-н-пропоксифенил; алканоилоксифенил, такой как 2-ацетоксифенил, 4-пропионоксифенил, 4-формилоксифенил, 4-ацетоксифенил, 3-бутирилокисфенил и 3-ацетоксифенил; алкилфенил, такой как 4-метилфенил, 2-метилфенил, 2,4-диметилфенил, 3-трет-бутилфенил, 4-этилфенил, 4-этил-3-метилфенил, и 3,5-диметилфенил; алкилтиофенил, такой как 4-метилтиофенил, 3-н-бутилтиофенил, 2-этилтиофенил, 3,4-диметилтиофенил и 3-н-пропилтиофенил; аминофенил, такой как 2-аминофенил, 4-аминофенил, 3,5-диаминофенил, и 3-аминофенил; алканоиламино, такие как 2-ацетиламино, 4-ацетиламино, 3-пропиониламино и 4-бутириламино; алкилсульфониламино, такие как 3-метилсульфонил- амино, 4-метилсульфониламино, 3,5-(диметилсульфониламино)-фенил, 4-н-бутилсульфониламинофенил и 3-этилсульфониламино- фенил; карбоксифенил, такой как 2-, 3- или 4-карбоксифенил, 3,4-дикарбоксифенил и 2,4-дикарбоксифенил: карбамоилфенил, такой как 2-карбамоилфенил, 2,4-дикарбамоилфенил и 4-карбамоилфенил; оксиметилфенил, такой как 4-оксиметилфенил и 2-оксиметилфенил; аминометилфенил, такой как 2-аминометилфенил и 3-аминометилфенил; и карбоксифенил, такой как 2-карбоксиметилфенил, 4-карпбоксиметилфенил и 3,4-дикарбоксиметилфенил; и замещенные фенильные группы, несущие различные заместители, такие как 4-хлор-3-метилфенил, 4-фтор-3-оксифенил, 3,5-дихлор-4-оксифенил, 4-окси-3-хлорфенил, 4-окси-3 -метилфенил, 4-этил-3-оксифенил, 4-метокси-3-оксифенил, 4-трет-бутокси-2-оксифенил, 4-ацетиламино-3-метоксифенил, 3-амино-4-этилфенил, 2-аминометил-4- хлорфенил, 2-оксиметил-3-метоксифенил, 2-оксиметил-4-фторфенил, 2-ацетокси-аминофенил, 4-ацетокси-3-метоксифенил, 3-изопропилтио-4-хлорфенил, 2-метилтио- 4-оксиметилфенил, 4-карбокси-3+оксифенил, 4-этокси-3-оксифенил, 4-метилсульфониламино-2-карбоксифенил, 4-амино-3хлорфенил и 2-карбоксиметил-4-оксифенил.
- и R представляет собой замещенную фенильную группу, в которой заместитель (-и) представлен (ы) символами а и а', то примерами таких групп являются галоидный фенил, такой как 4-хлорфенил, 3-бромфенил, 2-фторфенил, 2,4-дихлорфенил и 3,5-дихлорфенил; оксифенил, такой как 2-оксифенил, 3-оксифенил, 4-оксифенил, 2,4-диоксифенил, и 3,4-диоксифенил; алкоксифенил, такой как 2,6-диметоксифенил, 4-метоксифенил, 3-этоксифенил, 3,4-диметоксифенил, 4-трет-бутилоксифенил, 4-метокси-3-этоксифе- нил, и 4-н-пропоксифенил; алканоилоксифенил, такой как 2-ацетоксифенил, 4-пропионоксифенил, 4-формилоксифенил, 4-ацетоксифенил, 3-бутирилокисфенил и 3-ацетоксифенил; алкилфенил, такой как 4-метилфенил, 2-метилфенил, 2,4-диметилфенил, 3-трет-бутилфенил, 4-этилфенил, 4-этил-3-метилфенил, и 3,5-диметилфенил; алкилтиофенил, такой как 4-метилтиофенил, 3-н-бутилтиофенил, 2-этилтиофенил, 3,4-диметилтиофенил и 3-н-пропилтиофенил; аминофенил, такой как 2-аминофенил, 4-аминофенил, 3,5-диаминофенил, и 3-аминофенил; алканоиламино, такие как 2-ацетиламино, 4-ацетиламино, 3-пропиониламино и 4-бутириламино; алкилсульфониламино, такие как 3-метилсульфонил- амино, 4-метилсульфониламино, 3,5-(диметилсульфониламино)-фенил, 4-н-бутилсульфониламинофенил и 3-этилсульфониламино- фенил; карбоксифенил, такой как 2-, 3- или 4-карбоксифенил, 3,4-дикарбоксифенил и 2,4-дикарбоксифенил: карбамоилфенил, такой как 2-карбамоилфенил, 2,4-дикарбамоилфенил и 4-карбамоилфенил; оксиметилфенил, такой как 4-оксиметилфенил и 2-оксиметилфенил; аминометилфенил, такой как 2-аминометилфенил и 3-аминометилфенил; и карбоксифенил, такой как 2-карбоксиметилфенил, 4-карпбоксиметилфенил и 3,4-дикарбоксиметилфенил; и замещенные фенильные группы, несущие различные заместители, такие как 4-хлор-3-метилфенил, 4-фтор-3-оксифенил, 3,5-дихлор-4-оксифенил, 4-окси-3-хлорфенил, 4-окси-3 -метилфенил, 4-этил-3-оксифенил, 4-метокси-3-оксифенил, 4-трет-бутокси-2-оксифенил, 4-ацетиламино-3-метоксифенил, 3-амино-4-этилфенил, 2-аминометил-4- хлорфенил, 2-оксиметил-3-метоксифенил, 2-оксиметил-4-фторфенил, 2-ацетокси-аминофенил, 4-ацетокси-3-метоксифенил, 3-изопропилтио-4-хлорфенил, 2-метилтио- 4-оксиметилфенил, 4-карбокси-3+оксифенил, 4-этокси-3-оксифенил, 4-метилсульфониламино-2-карбоксифенил, 4-амино-3хлорфенил и 2-карбоксиметил-4-оксифенил.
Если R представляет собой фенильную группу, и а' или а является С1-С4-галоалкилом или С1-С4-пергалоалкилом, то примеры таких заместителей включают хлорметил, иодометил, трихлорметил, 2-бром-2-метилпроцил, хлорпропил и фторметил.
Примеры, в которых R является группой, представленной формулой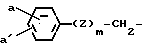 при m, равном нулю, включают фенилацетил, 4-оксифенилацетил, 4-хлорфенилацетил, 3,4-дихлорфенилацетил, 4-метоксифенилацетил, 3-этоксифенилацетил, 2-аминометилфенилацетил, 3-карбоксифенилацетил, 4-ацетоксифенилацетил, 3-аминофенилацетил и 4-ацетиламинофенилацетил, и при m, равном единице и Z, равном этому кислорода, включают: феноксиацетил, 4-хлорфеноксиацетил, 3-аминофеноксиацетил, 3-оксифеноксиацетил, 2-метоксифенонксиацетил, 2-метилтиофеноксиацетил, 4-ацетиламинофеноксиацетил, 3,4-диметилфеноксиацетил, и 3-оксиметилфеноксиацетил; и при m, равном единице, и Z, означающем атом серы, включат фенилтиоацетил, 4-хлорфенилтиоацетил, 3,4-дихлорфенилтиоацетил, 2-фторфенилтиоацетил, 3-оксифенилтиоацетил и 4-этоксифенилтиоацетил.
при m, равном нулю, включают фенилацетил, 4-оксифенилацетил, 4-хлорфенилацетил, 3,4-дихлорфенилацетил, 4-метоксифенилацетил, 3-этоксифенилацетил, 2-аминометилфенилацетил, 3-карбоксифенилацетил, 4-ацетоксифенилацетил, 3-аминофенилацетил и 4-ацетиламинофенилацетил, и при m, равном единице и Z, равном этому кислорода, включают: феноксиацетил, 4-хлорфеноксиацетил, 3-аминофеноксиацетил, 3-оксифеноксиацетил, 2-метоксифенонксиацетил, 2-метилтиофеноксиацетил, 4-ацетиламинофеноксиацетил, 3,4-диметилфеноксиацетил, и 3-оксиметилфеноксиацетил; и при m, равном единице, и Z, означающем атом серы, включат фенилтиоацетил, 4-хлорфенилтиоацетил, 3,4-дихлорфенилтиоацетил, 2-фторфенилтиоацетил, 3-оксифенилтиоацетил и 4-этоксифенилтиоацетил.
Примеры, где R представляет собой R3CH2-, где R3 означает арильную группу, включают 2-тиенилацетил, 3-тиенилацетил, 2-фурилацетил, 2-бензотиенилацетил, 2-бензофурилацетил, индол-2-илацетил, 1Н-тетразол-1-илацетил, оксазол-2-илацетил, оксазол-4-илацетил, тиазол-4-илацетил, 2-аминотиазол-4-илацетил, 1,3,4-оксадиазол-2-илацетил, 1,3,4-тиадиазол-2-илацетил, 5-этил-1,3,4-тиадиазол-2-илацеил и бенззоамино тиазоил и подобные арильные группы, необязательно замещенные амино, С1-С4-алкилсульфониламино, окси, гало, С1-С4-алкилом или С1-С4-алкоксигруппами.
Примеры, в которых R является замещенной метильной группой, представленной формулой R4-СH(Q)- и Q означает амино, карбокси, окси или сульфо, включают 2-карбокси-2-фенилацетил, 2-карбокси-2-(4-оксифенил)-ацетил, 2-амино-2-фенилацетил, 2-амино-2-(4-оксифенил)-ацетил, 2-амино-2й-(3-хлор-4-оксифенил)-ацетил, 2-амино-2-(циклогеск-1,4- диен-1-ил)-ацетил, 2-окси- 2-фенилацетил, 2-формилокси-2-фенилацетил, 2-сульфо-2-фенилацетил, 2-сульфо-2- (4-метилфенил)-ацетил, и 2-ацетокси-2-(3-оксифениил)-ацетил, 2-амино-2-(2-тиенил)-ацетил, 2- амино-2-(3-бензотиенил)-ацетил, 2-амино-2-(1Н-тетразол-1-ил)-ацетил, 2-окси-2-(1,3,4-тиадиахол-2-ил)-ацетил, 2-амино-2- (2аминотиазол-2-ил)-ацетил, 2-карбокси2-(2-тиенил)-ацетил, 2-карбокси-2-(бензотиен-2-ил)-ацетил и окси-2-(бензофур-2-ил); и если Q представляет особой замещенную аминогруппу, представленную формулой
-NH-C(O)-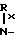 C(O)-Ry то примерами таких ацильных групп являются 2-(N-метил-N-бензоилкарбамоиламино)-2-фенилацетил, 2-(N-метил-N-циннамоилкарбамоиламино)-2-(2-фурил)-ацетил, 2-(N, N-диметилкарбамоилуреидо)-2-(4-хлорфенил)-ацетил, 2-[N-метил-N-(2-хлорциннамоил)-карбамоиламино] -2-(2-тиенил)- ацетил и 2-(N-этил-N-ацетилкарбамоиламино)-2-(4-оксифенил)- ацетил; и если Q является замещенной аминогруппой, представленной формулой:
C(O)-Ry то примерами таких ацильных групп являются 2-(N-метил-N-бензоилкарбамоиламино)-2-фенилацетил, 2-(N-метил-N-циннамоилкарбамоиламино)-2-(2-фурил)-ацетил, 2-(N, N-диметилкарбамоилуреидо)-2-(4-хлорфенил)-ацетил, 2-[N-метил-N-(2-хлорциннамоил)-карбамоиламино] -2-(2-тиенил)- ацетил и 2-(N-этил-N-ацетилкарбамоиламино)-2-(4-оксифенил)- ацетил; и если Q является замещенной аминогруппой, представленной формулой: 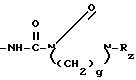 то примерами являются 2-[(3-метилимидазолидин-2-он-1-ил)-карбониламино] -2-фенилацетил, 2-[(3-ацетилимидазолидин-2-он-1-ил)-карбониламино] -2-фенилацетил, 2-(3-метилсульфонилимидазолидин-2-он-1ил)-2-(2-тиенил)-ацетил и 2-[(3-ацетилгексагидропиримидин-2-он-1-ил)-карбониламино] -2- фенилацетил; и если Q является оксизамещенной бензамидогруппой представленной формулой
то примерами являются 2-[(3-метилимидазолидин-2-он-1-ил)-карбониламино] -2-фенилацетил, 2-[(3-ацетилимидазолидин-2-он-1-ил)-карбониламино] -2-фенилацетил, 2-(3-метилсульфонилимидазолидин-2-он-1ил)-2-(2-тиенил)-ацетил и 2-[(3-ацетилгексагидропиримидин-2-он-1-ил)-карбониламино] -2- фенилацетил; и если Q является оксизамещенной бензамидогруппой представленной формулой 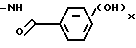 то примерами таких ацильных групп являются 2-(2,4-диоксибензамидо)-2-фенилацетил, 2-(4-оксибензамидо)-2-(4- оксифенил)-ацетил, 2-(3,4-диоксибензамидо)-2-(2-аминотиазол-4-ил)-ацетил, 2-(3,5-диоксибензамидо)2-(3-тиенил)-ацетил и 2-(2-оксибензамидо)- 2-(2-бензофурил)-ацетил.
то примерами таких ацильных групп являются 2-(2,4-диоксибензамидо)-2-фенилацетил, 2-(4-оксибензамидо)-2-(4- оксифенил)-ацетил, 2-(3,4-диоксибензамидо)-2-(2-аминотиазол-4-ил)-ацетил, 2-(3,5-диоксибензамидо)2-(3-тиенил)-ацетил и 2-(2-оксибензамидо)- 2-(2-бензофурил)-ацетил.
Если Q представляет собой оксизамещенную пиридинкарбониламиногруппу, то примеры включают, например, 2-оксипиридин-4-он-6-илкарбониламино и 3-оксипиридин-4-он-6- илкарбониламино. Если Q представляет собой пиридилкарбониламиногруппу, то примерами являются, например, пиридин-3-илкарбониламино, 4-аминопиридин-3-илкарбониламино, 5-хлорпиридин-2-илкарбониламино, 3-карбоксипиридин-4-ил- карбониламино и 4-аминопиридино-2-илкарбониламино. Если Q является имидазольной или пиразольной группой, как определено выше, то примеры включают, например, 2-аминоимидазол-4-илкарбониламино, 5-карбокси-2-метилимидазол-4-илкарбониламино, 5-карбоксипиразол- 3-илкарбониламино, 3-аминопиразол-4-илкарбониламино и 4-оксипиразол-5-илкарбониламино. Если Q является бензпиридазин-4-он-илкарбониламиногруппой, то примеры Q представлены формулами: 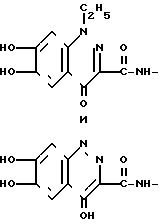
Примерами, когда R представляет собой кетогруппу или оксииминозамещенную группу, представленную формулами
R5- - и R5-
- и R5-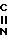 -
- являются кетогруппы 2-оксо-2-фенилацетил, 2-окси-2-(2-тиенил)-ацетил, 2-окси-2-(2-аминотиазол-4-ил)-ацетил и оксиминозаме- щенные группы 2-фенил-2-метоксиминоацетил, 2-(2-тиенил)-2-этоксиминоацетил, 2-(2-фурил)-2-метоксиминоацетил, 2-(2-бен- зотиенил)-2-карбоксиметоксииминоацетил, 2-(2-тиенил)-2- (2-карбоксиэтокси)-иминоацетил, 2-(2-амино-1,2,4-тиадиазол-4-ил)- 2-метоксииминоацетил, 2-(2-аминотиа- зол-4-ил)-2-метоксиимноацетил, 2-(2-хлортиазол-4-ил)-2-метоксииминоацетил, 2-(2-аминотиазол-4- ил)-2-(2-карбоксипропо- 2-ил)-оксииминоацетил, 2-(2-амино- тиазол-4-ил)-2-(2-карбамоилпроп-2-ил)-оксиимино- ацетил, 2-(5-амино-1,3,4-тиадиазол-2-ил)-2-метоксииминоацетил, 2-(2-аминотиазол-4-ил)-2-(пирролиджин-2-ое-ил)-оксиаминоацетил, 2-(2-аминотиазол-4-ил)-2-(1-метилпирролидин-2-он-3-ил)-оксииминоацетил, 2-фенил-2-(пирролидин-2-он-3-ил)-оксиимино-ацетил, 2-(2-аминооксазол-4-ил)-2-(1-этилпирролидин-2-ое-ил)- оксииминоацетил, 2-(2-аминотиазол-4-ил)-2-(1-этилпиперидин 2-ое- 3-ил)2-оксиминоацетил и 2-(2-фурил)-2-(пирролидин-2-он-3-ил)- оксииминоацетил.
являются кетогруппы 2-оксо-2-фенилацетил, 2-окси-2-(2-тиенил)-ацетил, 2-окси-2-(2-аминотиазол-4-ил)-ацетил и оксиминозаме- щенные группы 2-фенил-2-метоксиминоацетил, 2-(2-тиенил)-2-этоксиминоацетил, 2-(2-фурил)-2-метоксиминоацетил, 2-(2-бен- зотиенил)-2-карбоксиметоксииминоацетил, 2-(2-тиенил)-2- (2-карбоксиэтокси)-иминоацетил, 2-(2-амино-1,2,4-тиадиазол-4-ил)- 2-метоксииминоацетил, 2-(2-аминотиа- зол-4-ил)-2-метоксиимноацетил, 2-(2-хлортиазол-4-ил)-2-метоксииминоацетил, 2-(2-аминотиазол-4- ил)-2-(2-карбоксипропо- 2-ил)-оксииминоацетил, 2-(2-амино- тиазол-4-ил)-2-(2-карбамоилпроп-2-ил)-оксиимино- ацетил, 2-(5-амино-1,3,4-тиадиазол-2-ил)-2-метоксииминоацетил, 2-(2-аминотиазол-4-ил)-2-(пирролиджин-2-ое-ил)-оксиаминоацетил, 2-(2-аминотиазол-4-ил)-2-(1-метилпирролидин-2-он-3-ил)-оксииминоацетил, 2-фенил-2-(пирролидин-2-он-3-ил)-оксиимино-ацетил, 2-(2-аминооксазол-4-ил)-2-(1-этилпирролидин-2-ое-ил)- оксииминоацетил, 2-(2-аминотиазол-4-ил)-2-(1-этилпиперидин 2-ое- 3-ил)2-оксиминоацетил и 2-(2-фурил)-2-(пирролидин-2-он-3-ил)- оксииминоацетил.
Примеры, когда R представляет собой группу формулы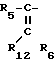 могут быть найдены у Hamashima, в патент США N 4634617, приведенном в библиографической справке. Примерными заместителями являются: для R12 - водород, для R5 - фенил, фурил, тиенил, оксазолил, изоксазолил, необязательно защищенный аминоизоксазолил, необязательно защищенный аминотиазолил, тиазолил и аминотиазолил, и для R6 - C1-С3-алкенил, в особенности метилен.
могут быть найдены у Hamashima, в патент США N 4634617, приведенном в библиографической справке. Примерными заместителями являются: для R12 - водород, для R5 - фенил, фурил, тиенил, оксазолил, изоксазолил, необязательно защищенный аминоизоксазолил, необязательно защищенный аминотиазолил, тиазолил и аминотиазолил, и для R6 - C1-С3-алкенил, в особенности метилен.
Если R6 является С1-С4-алкилом, замещенным фенилом, или замещенным фенилом, то такие группы представлены, например, бензилом, 4-оксибензилом, 4-хлорбензилом, 3-карбоксибензилом, 3-хлор-4-оксибензилом, 2-фенилэтилом, 1-фенилэтилом, 3-фенилпропилом, 4-окси-2-фенилпропилом, 3-фенилбутилом и тому подобными фенилалкильными группами.
Если R6 представляет собой С1-С4-алкил, замещенный амино, или защищенной аминогруппой, то примеры включают 2-аминоэтил, 3-аминопропил, 4-аминобутил, 2-аминопропил и такие группы, в которых аминогруппа защищена аминозащитной группой.
Если R6 представляет собой С2-С4-алкенильную группу, то примеры включают аллил, бутен-2, бутен-3, бутен-1 и тому подобные группы.
Катализаторы типа кислоты Льюиса характеризуются наличием вакантной орбитали, которая может принять подходящую электронную пару либо поделенную, например, на атоме кислорода, серы или галогенида, или на π-орбитали соединения типа основания Льюиса, чтобы образовать ковалентную связь. Примерами подходящих металлических катализаторов типа кислоты Льюиса являются хлористый алюминий, хлористое олово (4), бромистое олово (4), хлористый цинк, бромистый цинк, пятихлористый аммоний, четыреххлористый титан, хлористое железо (3), треххлористый галлий, четыреххлористый цирконий, бромистая ртуть (2), треххлористый хром и тому подобные галогениды металлов, проявляющие каталитическую активность типа Фриделя-Крафтса. Предпочтительными из этих катализаторов являются хлористое олово (4), четыреххлористый цирконий и четыреххлористый титан. Особенно предпочтительно хлористое олово (4). Применяют катализатор предпочтительно в количестве от 1,0 до 3,0 молей на моль хлористого тионила (2).
Нитросоединения включают С1-С6-нитроалканы и нитрозамещенные арилы и представлены нитрометаном, нитроэтаном, 1-нитропропаном, 2-нитропропаном, пара-нитротолуолом, альфанитротолуолом, и нитробензолом. Предпочтительны нитрометан, 1-нитропропан, нитроэтан и нитробензол. Особенно предпочтителен нитрометан. Применяемое нитросоединение используют предпочтительно в количестве от 1 до 4 молей на моль хлористого тионила (2) (выше передвен также как хлористый сульфинил).
4-Хлортионилазетидиноны формулы (2), использованные в способе согласно изобретению, являются известными соединениями и описаны Kukolja в патенте США N 4081440, приведенном в ссылке. Примерами исходных материалов, использованных в способе, являются трет-бутиловый эфир 3-метил-2-(4-хлорсульфинил-2-оксо-3-фенилацетиламино-1-азетидинил)- 3-бутеновой кислоты, трет-бутиловый эфир 3-метил-2-(4-хлорсульфинил-2-оксо-3-феноксиацетиламино-1- азетиднил)-2-бутеновой кислоты, дифенилметиловый эфир 3-метил-2- (4-хлорсульфинил-2-оксо-3-феноксиацетиламино-1-азетодинил)-3-бутеновой кислоты, пара-метоксибензиловый эфир 3-метил-2-(4-хлорсульфинил-2-оксо-3-фенилацетиламино-1- азетидинил)-3-бутеновой кислоты, пара-нитробензиловый эфир 3- метил-2-(4-хлорсульфинил-2-оксо-3-феноксиацетиламино-1- азетидинил)-3-бутеновой кислоты, дифенилметиловый эфир 3-метил-2- (4-хлорсульфинил-2-оксо-3-бензоиламино-1-азетидинил)3-бутеновой кислоты, пара-нитробензиловый эфир 3-метил-2-[4-хлортионил-2- оксо-3-(α-трет-бутилоксикарбониламинофенилацетиламино)-1- азетидинил] -3-бутеновой кислоты, бензиловый эфир 3-метил-2-(4- хлортионил-2-оксо-3-феноксиацетиламино-1-азетидинил)-3-бутеновой кислоты и бензгидриловый эфир 3-метил-2-(4-хлорсульфинил-2-оксо-3-ацетиламино-1-азетидинил)- 3-бутеновой кислоты. Предпочтительные азетидиноны представлены формулой (2), в которой А является группой формулы
R-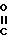 -
-
где R представляет собой бензил, феноксиметил или тиенилметил. Предпочтительной эфирной группой R1в формуле (2) является бензил или замещенный бензил, особенно пара-нитробензил.
Процесс проводят при температуре от -15 и до 60оС, предпочтительно в интервале от -10 до 0оС, в инертном органическом растворителе. Растворители, которые могут быть использованы, описаны Kukolja в патенте США N 4052387, который приведен в библиографической справке, и в котором описан процесс основной циклизации. Предпочтительными растворителями являются опротонные и включают ароматические углеводороды, такие как бензол, толуол, ксилол, хлорбензол и тому подобные, и галогенизировнные углеводороды, такие как хлороформ, хлористый метилен, четыреххлористый углерод, 1,2-дихлорэтан, 1,1,2-трихлорэтан и тому подобные. Особенно предпочтительными растворителями являются бензол и толуол.
Как отмечалось выше, процесс проводят в значительной мере в безводных условиях. Присутствие воды в следовых концентрациях допустимо, однако желательно поддерживать реакционную смесь в процессе возможно более сухой.
В предпочтительном осуществлении изобретения в процессе циклизации присутствует также оксосоединение. Оксо-соединения, используемые в настоящем процессе, описаны Chou в патенте США N 4190724, который включен в библиографическую справку, и выбраны из группы, вклю- чающей R2-О-R2, Z O , R2-
O , R2-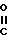 -R2, Z
-R2, Z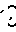 C= 0 и (R'2)3H -> O, где каждый R2 независимо друг от друга представляет собой С1-С4-алкил; каждый R2' независимо друг от друга является С1-С4-алкилом, С5-С6-циклоалкилом, фенилом или фенилом, замещенным С1-С4-алкилом, С1-С4-алкокси или галогеном; Z представляет собой (СН2)m-CH2-CH2-O-CH2-CH2- или -СН2-О-СН2СН2СН2-; m равно 4 или 5 и Z представляет собой группу формулы
C= 0 и (R'2)3H -> O, где каждый R2 независимо друг от друга представляет собой С1-С4-алкил; каждый R2' независимо друг от друга является С1-С4-алкилом, С5-С6-циклоалкилом, фенилом или фенилом, замещенным С1-С4-алкилом, С1-С4-алкокси или галогеном; Z представляет собой (СН2)m-CH2-CH2-O-CH2-CH2- или -СН2-О-СН2СН2СН2-; m равно 4 или 5 и Z представляет собой группу формулы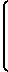 -
-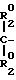
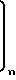 где каждый R2о является водородом или С1-С4-алкилом и n - целое число от 3 до 6. Предпочтительными оксо-соединениями являются диэтиловый эфир, ди-н-пропиловый эфир, ацетон и метилэтилкетон. Особенно предпочтителен диэтиловый эфир. Применяют оксосоединения в процессе предпочтительно в количестве, соответствующем от 0,75 до 2,0 молей на моль хлористого тионила формулы (2).
где каждый R2о является водородом или С1-С4-алкилом и n - целое число от 3 до 6. Предпочтительными оксо-соединениями являются диэтиловый эфир, ди-н-пропиловый эфир, ацетон и метилэтилкетон. Особенно предпочтителен диэтиловый эфир. Применяют оксосоединения в процессе предпочтительно в количестве, соответствующем от 0,75 до 2,0 молей на моль хлористого тионила формулы (2).
В другом предпочтительном осуществлении изобретения в процессе циклизации участвует ненасыщенное соединение. Ненасыщенное соединение, которое может быть использовано в процессе, может быть выбрано среди С2-С10-олефинов, С5-С10-циклоолефинов, несопряженных С5-С10-диолефинов, С3-С10-алленов и несопряженных С6-С10-циклодиолефинов. Примеры таких алкенов, алкадиенов, циклоалкенов, алленов и циклодиенов включают, например, алкены этилен, пропилен, 1-бутен, 2-бутен, 1-пентен, 2-пентен, 2-гексен, 1-гептен, 3-гептен, 1-октен, 2-нонен, 3-нонен, 1-децен, 5-децен и подобные алкены с концевой или неконцевой двойной связью; несопряженные алкадиены, такие как 1,4-пентадиен, 1,4-гексадиен, 3-метил-1,4-гексадиен, 1,5-гексадиен, 1,5-гептадиен, 1,6-гептадиен и подобные диены: несопряженные циклодиены, такие как 1,4-циклогексадиены, 1,4-циклогептадиен, и тому подобные: аллены, такие как аллен, метилаллен (1,2-бутадиен), диметилаллен (2,3-пентадиен) и тому подобные; циклоаклены, такие как 1-метилциклопент-2-ен, циклогексен, циклопентен, циклогептен, циклооктен и тому подобные. Алкены, алкадиены или аллены могут быть с прямой и разветвленной цепью и могут быть замещены инертной группой, предпочтительно на насыщенном углеродном атоме алкена. Например, ненасыщенное соединение может быть замещено алкилом, таким как метил, этил или изопропил; галогеном (предпочтительно в неаллильном положении); этерифицированной карбоксигруппой; ароматической группой, такой как фенил или толилом; нитро, циано и алкоксигруппой, такой как метокси или этокси и тому подобными апротонными заместителями, которые инертны в условиях процесса циклизации.
Алкены с неконцевыми двойными связями могут быть использованы либо в цис-, или в транс-форме. Предпочтительными ненасыщенными соединениями согласно изобретению являются алкены, например. 1-пентен, 2-пентен, 1-гексен, 2-гексен, 1-гептен, 1-октен и 1-децен и циклоалкены, такие как циклопентен и циклогексен. Особенно предпочтителен 1-гексен.
Ненасыщенные соединения, применяемые в процессе, предпочтительно присутствуют в количестве, соответствующем от одного моля до двух молей на моль хлористого тионила формулы (2). Особенно предпочтительно количество в пределах от одного до полутора моля ненасыщенного соединения на моль хлористого тионила формулы (2). Лучших результатов достигают с 1 молем ненасыщенного соединения, особенно 1-гексена, на моль соединения формулы (2).
В особенно предпочтительном осуществлении изобретения соединение формулы (1) получают взаимодействием 4-(хлортионилазетидин-2-она формулы (2) с 1,5-3,0 молями хлористого олова на моль азетидинона, 0,75-2,0 молями этилового эфира на моль азетидинона, с 1-2 молями 1-гексена на моль азетидинона и 1-4 молями нитрометана на моль азетидинона, в инертном растворителе, в значительной мере безводных условиях, в интервале температур от -10 до 0оС. Лучший выход получен при использовании 2,5 молярных эквивалентов нитрометана и 1,0 молярном эквиваленте 1-гексана с этиловым эфиром и хлористым оловом (4).
Способ согласно изобретению обычно осуществляют следующим образом.
4-Хлорсульфинилазетидином формулы (2) растворяют в безводном органическом растворителе. Раствор охлаждают до температуры примерно 0-15оС. В раствор, охлажденный до -10оС, добавляют нитросоединение и перемешивают. Катализатор, и, если желают, оксосоединение и ненасыщенное соединение в растворителе, охлажденном до -10оС, добавляют к раствору хлористого тионила. Раствор перемешивают в атмосфере азота и подвергают взаимодействию при комнатной температуре. Комплекс выделяют из реакционной смеси, например, фильтрацией или центрифугированием, промывают инертным растворителем и разлагают низшим спиртом, таким как метиловый спирт. Сложный эфир сульфоксида 3-экзометиленцефама, выпавший в осадок, отфильтровывают, промывают и сушат для последующего использования.
Как уже упоминалось выше, использование перечисленных выше нитросоединений в известном процессе циклизации приводит к повышенным скоростям реакции и выходам сложного эфира сульфоксида 3-экзометиленцефама. Выходы сложного эфира сульфоксида, обычно получаемые на 2-4% выше, чем выход контрольных препаратов. Такие увеличенные выходы и повышенные скорости реакций имеют существенное экономическое значение при производстве больших количеств сложного эфира сульфоксида 3-экзометиленцефама. Сложный эфир сульфоксида 3+экзометиленцефама формулы (1) используют в качестве промежуточного соединения при получении цефалоспориновых антибиотиков, например, цефаклора, т. е. 7 β-фенилглициламино-3-хлор-3-цефем-4-карбоновой кислоты известными методами. Промежуточное соединение формулы (1) может быть также использовано для получения цефалексина, т. е. 7 β-фенилглицинамино-3-метил-3-цефем-4-карбоновой кислоты. Следовательно, повышенные выходы, реализованные в способе согласно изобретению, приводят к увеличенному выходу этих ценных соединений антибиотиков.
Механизм действия нитросоединений для достижения увеличенных выходов соединения формулы (1) при повышенных скоростях реакций до настоящего времени не определен. Существует возможность того, что нитросоединения представляют большую степень свободы для нерастворимого комплекса металла и хлористого тионила участвовать в реакциях по замыканию кольца (циклизации) в противовес к образованию побочных продуктов. Однако, до настоящего времени не определено, влияет ли нитросоединение на выбор желательной реакции, чтобы ингибировать конкурирующие реакции, как описано, или действует иным образом.
Следующая экспериментальная часть предусматривает дальнейшее описание изобретения, не ограничивая его объема.
Экспериментальная часть
Общее. Нитросоединения (нитрометан, нитроэтан, 1-нитропропан и нитробензол) сушили глиноземом В (активность 1). Толуол сушили, используя молекулярное сито типа 4  . Все реакции проводили в атмосфере сухого азота. Анализ продуктов 3-метилцефама (формулы 1) проводили с использованием высокопроизводительной жидкости хроматографии (колонка диаметром 4,5 мм, длиной 5,0 см, наполнение октил, 3 мк длина волны 254 нм, скорость тока 1,7 мл/мин, подвижная фаза - 32% тетрагидрофурана, 0,5% фосфорной кислоты в воде). Спектр ЯМР на ядре 1Н получали в СДCl3 и регистрировали на спектрометре ОЕ-300 (300 МГц). Сокращения приняты следующие: с-синглет, д-дублет, т-триплет, кв-квартет, м-мультиплет.
. Все реакции проводили в атмосфере сухого азота. Анализ продуктов 3-метилцефама (формулы 1) проводили с использованием высокопроизводительной жидкости хроматографии (колонка диаметром 4,5 мм, длиной 5,0 см, наполнение октил, 3 мк длина волны 254 нм, скорость тока 1,7 мл/мин, подвижная фаза - 32% тетрагидрофурана, 0,5% фосфорной кислоты в воде). Спектр ЯМР на ядре 1Н получали в СДCl3 и регистрировали на спектрометре ОЕ-300 (300 МГц). Сокращения приняты следующие: с-синглет, д-дублет, т-триплет, кв-квартет, м-мультиплет.
Получение 1.
3-Метил-2-(2-хлортионил-4-оксо-3-имидо-1-азетидинил)-3- бутеновая кислота (далее называемая хлористым тионилом),
Получение соединения, указанного в названии примера, проводили следующим образом. Трехгорлую колбу с круглым днищем емкостью 1,0 литр снабжали механической мешалкой, ловушкой Дина-Старка, холодильником с подводом азота и термометром. Затем реакционный сосуд загружали 29,48 г влажного полимера (сополимер дивинилбензола и 4-винилпиридина, содержание воды 44,9% ) и 796 мл толуола. Эту взвесь нагревали с обратным холодильником (при 110-112оС) в атмосфере азота до тех пор, пока с помощью аппарата Дина-Старка собирали 160 мл азеотропной смеси с толуолом. Суспензию охлаждали до 60оС на ледяной бане перед добавлением 49,28 г (чистота 97,8% , 96,1 ммоля) (1В)-6-[(феноксиацетил)-амино] -пенициллоновой кислоты, 1-оксида сложного (4-нитрофенил)-метилового эфира и 21,86 г, 12,1 ммоля, 1,26 эквивалента) N-хлорфталимида в реакционную колбу. Толуол (20 мл) использовали для промывки реагентов в колбе. Смесь нагревали с обратным холодильником два часа (отсчет начинали при 109оС) и собирали дополнительную азеотропную смесь с толуолом (59 мл). Затем реакционную смесь немедленно охлаждали до 15оС, фильтровали и твердый осадок полимера и фталимида на фильтре промывали 104 мл толуола и за ненадобностью выбрасывали. Желтый раствор хлористого тионила использовали либо сразу, или хранили при 0-5оС до предстоящего использования в атмосфере аргона.
Спектр ЯМР на 1Н: (CДCl3, 300 МГц, млн. доли) δ: 1,92 (с, 3Н, СН3), 4,50-4,56 (АВ, 2Н, J = 15,1 Гц, боковая цепь СН2), 4,99 (с, 1Н, олефиновый СН2), 5,06 (с, 1Н, -СНСООпара-итробензол), 5,22 (д, 1Н, J = 1,6 Гц, олефиновый СН2), 5,25-5,33 (АВ, 2Н, J = 12,9 Гц, пара-нитробензол СН2), 5,53 (д, 1Н, J = 4,6 Гц, Н азетидинона), 6,27 (дд, 1Н, J = 4,6 Гц и 10,8 Гц, Н азетидинона), 6,90 (дд, 2Н, J = 7,4 Гц и 8,5 Гц, боковая цепь АгН), 7,01 (т, 1Н, J = 7,4 Гц боковая цепь АгН), 7,30 (дд, 2Н, J = 7,4 Гц и 8,5 Гц, боковая цепь АгН), 7,50 (АА ВВ, 2Н, J = 9,9 Гц, пара-нитробензол АгН), 7,98 (д, 1Н, J = 10,8 Гц, N-Н), 8,23 (AA' ВВ', 2Н, J = 8,9 Гц, пара-нитробензол АгН).
В следующих примерах получали 1-оксид (4-нитрофенил)-метилового эфира 3-метилен-7-[(феноксиацетил)-аСН2аминоцефам-4- карбоновой кислоты формулы (1), В таблицах IA, IB и IC сведены результаты примеров.
П р и м е р 1. Трехгорлую колбу с круглым дном, емкостью 500 мл снабжали механической мешалкой, вводом азота и термометром. В реакционный сосуд загружали 220 мл раствора хлористого тионила (25,5 ммоля в расчете на 1-оксид (4-нитроенил)-метилового эфира (1В)-6-([(феноксиацетил)-амино] -пенициллановой кислоты. В отдельную 50 мл колбу добавляли 10 мл толуола и диэтиловый эфир (2,5 мл, 1,75 г, 23,9 ммоля) и раствор охлаждали до 0оС и до -5оС. Хлористое олово (4) (5,1 мл, (11,36 г), 43,6 ммоля)) добавляли к раствору толуола и диэтилового эфира, и полученную суспензию немедленно охлаждали до 0оС в бане со смесью сухого льда и ацетона. Толуоловый раствор хлористого тионила охлаждали до 15оС, и суспензию хлористого олова (4), диэтилового эфира и толуола добавляли и хлористому тионилу а 5-10 с, используя 5 мл промывку толуолом. Полученную суспензию нагревали экзотермически до 21-23оС и перемешивали в течение 18 ч в атмосфере азота.
Суспензию фильтровали, промывали 25 мл толуола и возвращали в реакционный сосуд с 70 мл метилового спирта. Полученную суспензию перемешивали и оставляли для кристаллизации при 21-23оС на 15 мин. Суспензию затем охлаждали до 0-5оС и перемешивали еще 3 ч 45 мин. Через два часа этой обработки метиловым спиртом добавляли 15 мл деионизированной воды.
Затем суспензию фильтровали и продукт промывали 30 мл метилового спирта. Светлоокрашенное твердое вещество сушили в вакуумной печи при 45-60оС в течение времени от 15 до 16 ч. Анализ выхода и чистоты продукта формулы (1), 1-оксида (4-нитрофенил)-метилового эфира 3-метилен-7-[(феноксиацетил)-амино] -цефам-4-карбо-новой кислоты, проведенный повторной жидкостной хроматографией высокого давления, скорректировал выход выделенного продукта, равный 68,1% (9,21 г, расчетное количество - 12,74 г), и чистота продукта определена равной 94,2% .
Спектр ЯМР на 1Н (СДCl3, 300 МГц, млн доли) δ: 3,57-3,74 (АВ, 2Н, J = 14,1 Гц, СН2), 4,55 (с, 2Н, боковая цепь СН2), 4,89 (Д, 1Н, J = 4,80 Гц, Н азетидинона), 5,28 (с, 2Н, СН2 пара-нитробензола), 5,31 (с, 1Н, -СНСОО + + пара-нитробензол), 5,48 (д, 1Н, J = 1,50 Гц, СН2-экзо), 6,03 (дд, 1Н, J = 4,80 Гц и 10,70 Гц, Н ацетидинона), 6,93 (дд, J = 8,30 Гц и 7,40 Гц, АгН боковой цепи), 7.0 (т, 1Н, J = Гц, АнГ боковой цепи), 7,29 (дд, 2Н, J = 8,33 Гц и 7,40 Гц, АгН боковой цепи), 7,49 (АА ВВ, 2Н, J = 8,90 Гц, АгН пара-нитробензола), 8,11 (д, 1Н, J = 10,70 Гц, N-H), 8,25 (АА ВВ, 2Н, J = 8,90 Гц, АгН пара-нитробензола).
П р и м е р 2. Повторяли процесс по при меру 1 (выше), за исключением того, что толуоловый раствор хлористого тионила охлаждали до -50оС перед добавлением суспензии хлористого олова (4), диэтилового эфира и толуола при 0оС. Во время перемешивания метилового спирта не добавляли воду. Уточненный анализ выделенного продукта формулы (1) показал выход 66,3% (8,45 г, теоретическое - 12,33 г) и чистоту, определенную равной 66,7% методом повторной жидкостной хроматографии высокого давления.
П р и м е р 3. Повторяли процесс, описанный в примере 1 выше, за исключением того, что толуоловый раствор хлористого тионила охлаждали до -10оС перед добавлением к нему суспензии хлористого олова (4), диэтилового эфира и толуола, охлажденной до 0оС. В процессе обработки метиловым спиртом воду не добавляли. Уточненный анализ выделенного продукта формулы (1) показал выход 66,3% (8,45 г, теоретический - 12,33 г) и чистоту 96,7% , определенные повторной жидкостной хроматографией высокого давления.
П р и м е р 4. Повторяли процесс, описанный в примере 1 выше, за исключением того, что в реакции использовали диэтиловый эфир. Во время перемешивания с метиловым спиртом воду не добавляли. Уточненный анализ выделенного продукта формулы (1) показал выход 50,5% (10,15 г при теоретическом выходе 19,58 г) и чистоту 97,3% , определенные методом повторной высокопроизводительной жидкостной хроматографии.
П р и м е р 5. Трехгорлую колбу с круглым дном, емкостью 500 мл, снабжали механической мешалкой, подводом азота и термометром. В реакционный сосуд загружали 192 мл толуолового раствора хлористого тионила (25,5 ммоля в расчете на 1-оксид (4-нитрофенил)-метилового эфира (1В)-6[(яеноксиацетил)-амино] - пенициллановой кислоты. В отдельную колбу на 50 мл загружали 10 мл толуола. Этот толуол охлаждали до (0)-(-5)оС до добавления хлористого олова (4) (5,1 мл, (11,36 г), 43,6 ммоля). Полученный раствор немедленно охлаждали до -10оС в бане со смесью ацетон-сухой лед. К раствору хлористого тионила добавляли нитрометан (3,5 мл, (3,94 г), 65,6 ммоля), и полученный раствор охлаждали до -10оС в баке со смесью ацетон-сухой лед. К охлажденному до -10оС раствору хлористого тионила в течение 5-10 с добавляли раствор хлористого олова (4) в толуоле, используя 5 мл толуоловую промывку. Полученную суспензию экзотермически нагревали до 21-23оС и перемешивали 4 ч в атмосфере азота.
Суспензию фильтровали, промывали 25 мл толуола и возвращали в реакционный сосуд с 70 мл метилового спирта. Полученную суспензию перемешивали и позволяли кристаллизоваться при 21-23оС в течение 15 мин. Затем суспензию охлаждали до 0-5оС и перемешивали еще 3 ч 45 мин. Через два часа этой обработки метиловым спиртом добавляли 15 мл деионизированной воды.
Суспензию фильтровали и продукт промывали 30 мл метилового спирта. Светлоокрашенное твердое вещество сушили в вакуумной печи при 45-60оС в течение 15-16 ч. Уточненный анализ выделенного продукта формулы (1) показал выход 50,9 процента (7,96 г, расчетное значение - 12,74 г) и чистоту 97,4% , определенную повторной жидкостной хроматографией высокого давления.
П р и м е р 6. Повторяли процесс по примеру 5 выше, за исключением того что 1-гексен (3,2 мл, 2,17 г, 25,8 ммоля) также добавляли к толуоловому раствору хлористого тионила перед добавлением смеси хлористого олова (4) и толуола. Уточненный анализ выделенного соединения формулы (1) показал выход 65,2% (8,47 г, расчетное значение - 12,74 г) и чистоту 98,0% , определенную повторной высокопроизводительной жидкостной хроматографией.
П р и м е р 7. Трехгорлую 500 мл колбу с круглым дном оборудовали механической мешалкой, подводом азота и термометром. В реакционный сосуд загружали 179 мл толуолового раствора хлористого тионила (24,7 ммолей в расчете на 1-оксид (4-нитрофнил)-метилового эфира (1В)-6-[(феноксиацетил)-амино] -пенициллановой кислоты. В отдельную 50 мл колбу загружали 10 мл толуола и диэтиловый спирт (2,4 мл, 1,70 г, 22,9 ммоля) и раствор охлаждали до 0 - минус 5оС. К раствору толуола и диэтилового эфира добавляли хлористое олова (4) (4,9 мл, 10,91 г, 41,9 ммоля), полученную суспензию немедленно охлаждали и выдерживали при 0оС в ванне на смеси ацетон-сухой лед. К раствору хлористого тионила добавляли 1-гексен (3,1 мл (2,10 г), 25,0 ммолей), и полученный раствор охлаждали до 10оС. К хлористому тионилу добавляли суспензию хлористого олова (4), диэтилового спирта и толуола при 0оС в течение 5-10 с, используя 5 мл толуоловую промывку. Полученную суспензию экзотермически нагревали до 21-23оС и перемешивали 4 ч в атмосфере азота.
Суспензию фильтровали, промывали 25 мл толуола и возвращали в реакционный сосуд с 80 мл метилового спирта. Полученную суспензию перемешивали и позволяли кристаллизовать при 21-23оС в течение 15 мин. Затем суспензию охлаждали до 0-5оС и перемешивали еще 3 ч 45 мин. При перемешивании с метиловым спиртом воду не добавляли.
Суспензию фильтровали и продукт промывали 30 мл метилового спирта. Светлоокрашенное твердое вещество сушили в вакуумной печи при 45-60оС в течение 15-16 ч. Уточненные результаты анализа выделенного продукта формулы (1) показали выход 70,0% (8,9 г при расчетном значении 12,34 г) и чистоту 97,1% , определенную методом высокоэффективной жидкостной хроматографии, проводимой повторно.
П р и м е р 8. Процесс по примеру 5 повторяли за исключением того, что использовали диэтиловый эфир (2,5 мл, 1,76 г, 23,9 ммоля), как описано в примере 1. Анализ выделенного продукта формулы (1) показал, что выход равен 71,1% (9,60 г, расчетное значение - 12,74 г) и чистота - 94,3% , определенная повторной жидкостной высокоэффективной хроматографией.
П р и м е р 9. 500 мл трехгорлую колбу с круглым дном оборудовали механической мешалкой, подводом азота и термометром. В реакционный сосуд загружали 179 мл толуолового раствора хлористого тионила (24,7 ммоля в расчете на 1-оксид (4-нитрофенил)-метиловый эфир (1В)-6-[(феноксиацетил)-амино)-пенициллановой кислоты. В отдельную колбу, емкостью 50 мл, загружали 10 мл толуола и диэтиловый эфир (2,4 мл (1,70 г), 22,9 ммоля) и раствор охлаждали до 0 - минус 5оС. К раствору толуола и диэтилового эфира добавляли хлористое олово (4) (4,9 мл, 10,91 г, 41,0 ммоля) и полученную суспензию немедленно охлаждали и выдерживали при 0оС в ванне с ацетоном и сухим льдом. К раствору хлористого тионила добавляли 1-гексен (3,1 мл, (2,10 г), 25,0 ммолей) и нитрометан (3,3 мл, (3,72 г), 60,9 ммоля), и полученный раствор охлаждали до 0оС в ванне с ацетоном и сухим льдом. К охлажденному до 0оС хлористому тионилу в течение 2-10 с добавляли охлажденную до 0оС суспензию хлористого олова (4), диэтилового эфира и толуола, используя 5 мл промывку толуолом. Полученную суспензию нагревали экзотермически до 21-23оС и перемешивали в атмосфере азота четыре часа.
Суспензию фильтровали, промывали 25 мл толуола и возвращали в реакционный сосуд с 80 мл метилового спирта. Полученную суспензию перемешивали и оставляли на 15 мин для кристаллизации. Затем суспензию охлаждали до 0-5оС и перемешивали еще 3 ч 45 мин. В процессе обработки метиловым спиртом воду не добавляли.
Суспензию фильтровали и продукт промывали 30 мл метилового спирта. Светлоокрашенное твердое вещество сушили в вакуумной печи при 45-60оС в течение 15-16 ч. Уточненный анализ выделенного продукта формулы (1) показал выход 72,2% (9,16 г, расчетное значение - 12,34 г) и чистоту 97,3% , определенную повторной жидкостной хроматографией высокого давления.
П р и м е р 10. Повторяли процесс по примру 5 (выше), за исключением того, что использовали диэтиловый эфир (2,5 мл, (1,76 г), 23,9 ммоля), как описано в примере 1, и нитробензол (6,6 мл (8,21 г), 64,1 ммоля) использовали вместо нитрометана. Суспензию хлористого тионила, хлористого олова (4), диэтилового эфира, толуола и нитробензола перемешивали только 90 мин при 21-23оС вместо 4 ч. Уточненный анализ выделенного продукта формулы (1) показал выход 70,5% (9,50 г при теоретическом значении 12,74 г) и чистoту 94,6% , определенную анализом жидкостной хроматографии высокого давления, проведенном повторно.
П р и м е р 11. Повторяли процесс по примеру 5 выше, за исключением того, что использовали диэтиловый эфир (2,5 мл, (1,76 г), 23,9 ммоля), как описано в примерено 1, и вместо нитрометана использовали 1-нитропропан (6,7 мл, (5,68 г), 63,8 ммоля). Суспензию хлористого тионила, хлористого олова (4), диэтилового эфира, толуола и 1-нитропропана перемешивали только 90 мин при 21-23оС вместо 4 ч. Уточненный анализ выделенного продукта формулы (1) показал выход 69,7% (9,13 г, расчетное значение 12,74 г) и чистоту 97,2% , определенную методом высокопроизводительной жидкостной хроматографии, проведенным повторно.
П р и м е р 12. Повторяли процесс по примеру 5, за исключением того, что использовали диэтиловый эфир (2,5 мл, (1,76 г), 23,9 ммоля как описано в примере 1, и нитроэтан (4,6 мл, (4,80 г), 64,0 ммоля) использовали вместо нитрометана. Суспензию хлористого тионила, хлористого олова (4), диэтилового эфира, толуола и нитроэтана также перемешивали только 90 мин при 21-23оС вместо 4 ч. Уточненный анализ выделенного продукта формулы 1й) показал выход 69,5% (9,18 г, теоретическое значение 12,74 г) и чистоту 96,4% , определенную методом повторной жидкостной хроматографии высокого давления.
П р и м е р 13. 500 мл трехгорлую колбу с круглым дном снабжали механической мешалкой, подводом азота и термометром. В реакционный сосуд загружали 220 мл толуолового раствора хлористого тионила (25,5 ммоля в расчете на 1-оксид (4-нитрофенил)-метилового эфира (1В)-6-[(феноксиацетил)-амино] - пенициллановой кислоты. Этот раствор хлористого тионила охлаждали до 15оС перед добавлением хлористого титана (4) (5,6 мл, (9,69 г), 51,1 ммоля) в неразбавленном виде с помощью градуированной пипетки. Полученную суспензию немедленно нагревали и выдерживали при 60оС в атмосфере азота в течение всего времени реакции 4 ч.
Суспензию фильтровали, промывали 25 мл толуола и возвращали в реакционный сосуд с 70 мл метилового спирта. Полученную суспензию перемешивали и оставляли на 15 мин для кристаллизации при 21-23оС. Затем суспензию охлаждали до 0-5оС и перемешивали еще 3 ч 45 мин. При перемешивании с метиловым спиртом воду не добавляли.
Суспензию фильтровали и продукт промывали 30 мл метилового спирта. Светлоокрашенное твердое вещество сушили в вакуумной печи при 45-60оС в течение 15-16 ч. Уточненный анализ выделенного продукта формулы (1) показал выход 65,4% (8,82 г при расчетном значении 12,74 г) и чистоту 94,4% , определенную повторной жидкостной хроматографией высокого давления.
П р и м е р 14. Повторяли процесс по примеру 13 выше, за исключением того, что к толуоловому раствору хлористого тионила до его охлаждения до 15оС добавляли нитрометан (2,8 мл, 3,16 г, 51,7 ммоля) и затем добавляли хлористый титан (4). Уточненный анализ выделенного продукта формулы (1) показал выход 68,8% (9,45 г при расчетном значении 12,74 г) и чистоту 92,7% , определенную повторным анализом высокопроизводительной жидкостной хроматографии.
П р и м е р 15. Повторяли процесс по примеру 13, за исключением того, что к толуоловому раствору хлористого тионила добавляли до его охлаждения до 15оС нитрометан (3,5 мл, (3,94 г), 64,6 ммоля) и 1-гексен (3,2 мл )2,17 г), 25,8 ммоля) и добавляли хлористый титан (4). Суспензию хлористого тионила, хлористого титана (4), нитрометана и 1-гексена также перемешивали 3 ч при 60оС вместо 4 ч. Уточненный анализ выделенного продукта формулы (1) показал выход 68,2 (8,99 г при теоретическом значении 12,74 г) и чистоту 96,7, % определенную поворотным анализом методом высокопроизводительной жидкостной хроматографии.
П р и м е р 16. 500 мл трехгорлую колбу с круглым дном снабжали механической мешалкой, подводом для азота и термометром. В реакционный сосуд загружали 216 мл толуолового раствора хлористого тионила (25,5 ммоля в расчете на 1-оксид (4-нитрофенил)-метилового эфира (1В)-6-[(феноксиацетил)-амино] -пициллановой кислоты. Этот раствор хлористого тионила охлаждали до 5оС. В перчаточном боксе, заполненном азотом, взвешивали в 50 мл колбе с закругленным дном хлористый цирконий (4) (20,3 г, (99,6% чистоты), 86,8 ммоля)). 50 мл колбу закупоривали и 25 мл толуола добавляли через пробку с помощью шприца. Суспензию хлористого титана (4) и толуола добавляли к охлажденному до 5оС толуоловому раствору хлористого тионила, используя дополнительную 25 мл промывку толуолом. Полученную суспензию экзотермически нагревали до 21-23оС и перемешивали 48 ч в атмосфере азота. Анализ высокопроизводительной жидкостной хроматографией показал, что реакция не завершилась, так что суспензию нагревали и выдерживали при 60оС 3 ч.
Суспензию фильтровали и возвращали в реакционный сосуд с 70 мл метилового спирта. Никакого продукта не кристаллизовалось в результате метаноловой обработки и не был выделен продукт формулы (1).
П р и м е р 17. 500 мл трехгорлую колбу с круглым дном оборудовали механической мешалкой, подводом для азота и термометром. В реакционный сосуд загружали 136 мл толуолового раствора хлористого тионила (19,3 ммоля в расчете на 1-оксид (4-нитрофенил)-метилового эфира (1В)-6-[(феноксиацетил)-амино] - пенициллановой кислоты. Раствор хлористого тионила охлаждали до 15оС. В колбе с круглым дном, емкостью 50 мл, взвешивали хлористый цирконий (4) (7,7 г, (99,6% чистоты), 32,9 ммоля) в перчаточном боксе, заполненном азотом. 50 мл колбу закупоривали и в нее с помощью шприца вводили 25 мл толуола и диэтиловый эфир (1,9 мл, (1,34 г), 18,1 ммоля). Суспензию хлористого циркония (4), диэтилового эфира и толуола охлаждали до 0оС в ванне с ацетоном и сухим льдом, прежде чем ее добавляли к толуоловому раствору хлористого тионила. Толуол (5 мл) использовали для промывки реагентов. Полученную суспензию оставляли для экзотермического нагревания до 21-23оС и перемешивали 16 ч в атмосфере азота. Анализ высокопроизводительной жидкостной хроматографией показал, что реакция не завершилась, так что суспензию нагревали и выдерживали при 60оС 1 ч.
Суспензию фильтровали и возвращали в реакционный сосуд с 55 мл метилового спирта. Полученную суспензию перемешивали и оставляли для кристаллизации при 21-23оС на 15 мин. К суспензии добавляли деионизированную воду (10 мл) и полученную суспензию охлаждали затем до 0-5оС и перемешивали еще один час.
Суспензию фильтровали и продукт промывали 20 мл метилового спирта. Светлоокрашенный продукт в виде твердого вещества сушили в вакуумной печи при 45-60оС в течение 15-16 ч. Уточненный анализ выделенного продукта формулы (1) показал выход 32,1% (3,36 г, при расчетном значении 9,64 г) и чистоту 92,0% , определенную повторной высокопроизводительной жидкостнoй хроматографией.
П р и м е р 18. 500 мл трехгорлую колбу с круглым дном оборудовали механической мешалкой, подводом для азота и термометром. В реакционный сосуд загружали 216 мл толуолового раствора хлористого тионила (25,5 ммоля в расчете на 1-оксид (4-нитрофенил)-метилового эфира (1В)+6-[(феноксиацетил)-амино] - пенициллановой кислоты. К раствору хлористого тионила добавляли нитрометан (80 мл, (90,16 г), 1,48 моля) и полученный раствор охлаждали до 15оС. В 50 мл колбе с круглым дном взвешивали хлористый цирконий (10,1 г, (99,6% чистоты), 43,2 ммоля) в перчаточном боксе в атмосфере азота. 50 мл колбу закупоривали и с помощью шприца в нее вводили толуол (25 мл). Суспензию хлористого циркония (4) и толуола охлаждали до 0оС в ванне ацетона с сухим льдом прежде чем ее добавлять к охлажденному до 15оС раствору хлористого тионила и нитрометана в толуоле. Раствор выделял тепло на начальной стадии, но выпадение в осадок твердого вещества началось через 6 мин. Реакционную смесь экзотермически нагревали до 21й-23оС и перемешивали 105 мин. В процессе реакции добавляли еще нитрометан (35 мл, (39,45 г), 646,2 ммоля) для поддержания гомогенности реакционной смеси. Реакционный раствор переносили в колбу с круглым дном (для вакуумного испарения), используя дополнительный нитрометан (35 мл, (39,45 г), 646,2 ммоля) для облегчения переноса.
Реакционный раствор упаривали до масла и добавляли 70 мл метилового спирта. Полученную суспензию охлаждали и перемешивали при 0-5оС в течение 100 мин. Добавляли деионизированную воду (15 мл) добавляли через 50 мин обработки метиловым спиртом.
Суспензию фильтровали и продукт промывали 30 мл метилового спирта. Светлоокрашенное твердое вещество сушили в вакуумной печи при 45-60оС в течение 15-16 ч. Уточненный анализ выделенного продукта формулы (1) показал выход 66,2% (8,76 г при расчетном значении 12,74 г) и чистоту 96,4% , определенную повторной жидкостной хроматографией высокого давления.
Данные приведены в таблице.
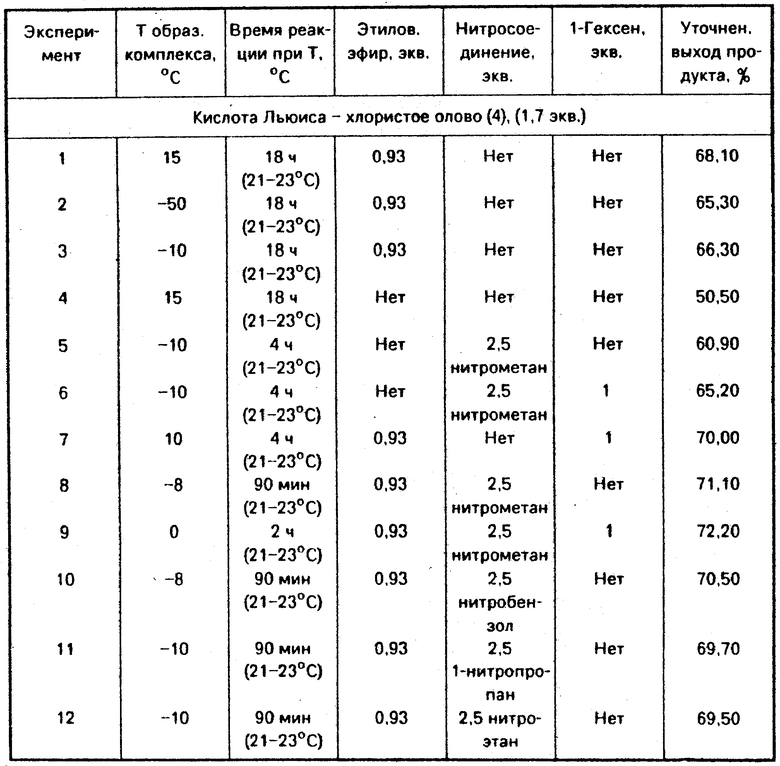
На фиг. 1 представлены изотермические реакции нитрометана, модифицированного хлористым оловом (4), способствующим закрытию кольца. Приводим следующие определения символов, использованных на фиг. 1 и 2
Реакции проводили в соответствии с экспериментом 1, без нитрометана и с добавлением нитрометана (2,5 эквивалента= = при установленной температуре хлористого тионила. Фигура 1 показывает, что реакции в присутствии нитрометана приводят к более скорому закрытию кольца в сравнении с реакциями без нитрометана. Далее, выход экзометилена с нитрометаном выше, чем без него.
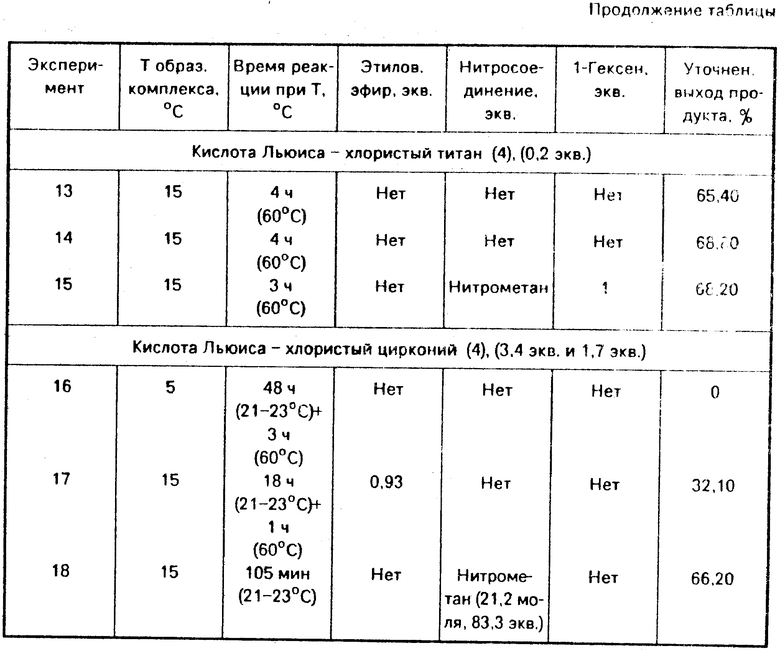
На фиг. 2 представлено математическое сравнение скоростей реакций и показано, что в первые 30 мин реакции скорость закрытия кольца (% образования экзометилена) повышена до 85, 191 и 41% при 15, 0 и -15оС соответственно. Математически А1 представлено уравнением Y = 3,6467Х и А2 представлено равенством Y = 1,9667Х. В1 математически представлено Y = 68000Х, тогда как В2 представлено равенством Y = 0,23333Х. С1представлено Y = 0,10333Х и С2 представлено равенством Y = 7,3333Х10-2. (56) Патент США N 4165315, кл. С 07 D 205/08, 1980.
Патент США N 4081440, кл. С 07 D 205/08, 1978.
Патент США N 4075203, кл. С 07 D 501/02, 1978.
Патент США N 4289695, кл. С 07 D 205/08, 1981.
Патент США N 4052387, кл. С 07 D 501/02, 1977.
Патент США N 4190724, кл. С 07 D 501/02, 1980.
Патент США N 4950753, кл. С 07 D 501/02, 1990.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ В КОЛЬЦЕ 2-АМИНО-1,2,3,4-ТЕТРАГИДРОНАФТАЛИНЫ ИЛИ 3-АМИНОХРОМАНЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ КИСЛОТНО-АДДИТИВНЫЕ СОЛИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2057751C1 |
| 6-ГЕТЕРОЦИКЛИЧЕСКИЗАМЕЩЕННЫЕ 4-АМИНО-1,2,2А,3,4,5-ГЕКСАГИДРОБЕНЗ-(CD)-ИНДОЛЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2062775C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ТИАЗОЛИДИНОНА ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМОЙ СОЛИ | 1990 |
|
RU2036915C1 |
| 4-АМИНО-6-ЗАМЕЩЕННЫЕ ТЕТРАГИДРОБЕНЗ[C,D]ИНДОЛЫ | 1992 |
|
RU2073672C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТЕТРАГИДРОБЕНЗ (C,D)ИНДОЛОВЫХ АГОНИСТОВ СЕРОТОНИНА | 1990 |
|
RU2007393C1 |
| СПОСОБ ПОЛУЧЕНИЯ 5-ЗАМЕЩЕННЫХ ПИРРОЛО (2,3-α)ПИРИМИДИНОВ | 1993 |
|
RU2127274C1 |
| ЗАМЕЩЕННЫЕ В КОЛЬЦЕ 2-АМИНО-1,2,3,4-ТЕТРАГИДРОНАФТАЛИНЫ ИЛИ 3-АМИНОХРОМАНЫ | 1994 |
|
RU2105756C1 |
| ПРОИЗВОДНЫЕ ТРИПЕПТИДОВ В ВИДЕ R- ИЛИ RS-ФОРМЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ НЕТОКСИЧНЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2077538C1 |
| ПРОИЗВОДНЫЕ 1-КАРБА-(1-ДЕТИА)-ЦЕФЕМА | 1993 |
|
RU2107067C1 |
| ЗАМЕЩЕННЫЕ ФЕНИЛФЕНОЛЬНЫЕ СОЕДИНЕНИЯ, ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2095340C1 |

Сущность изобретения: хлорсульфинилазетидин ф-лы (1), где А - аминозащитныя группа и R - карбоксизащитная группа, подвергают циклизации в сухом инертном органическом растворителе в присутствии катализатора типа кислоты Льюиса при температуре и времени достаточных для получения ф-лы (2). Процесс проводят в присутсвии нитросоединения, выбранного из групп, содержащей нитро C1-C6 -алканы и нитробензол. 34 з. п. ф-лы, 2 ил. Структура ф-л 1 и 2: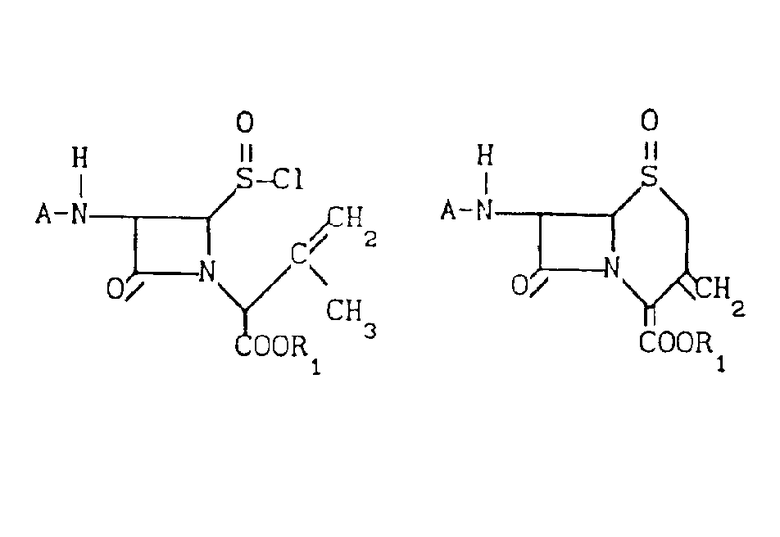
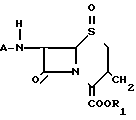
где А - аминозащитная группа;
R1 - карбоксизащитная группа,
путем циклизации хлорсульфинилазетидинона общей формулы II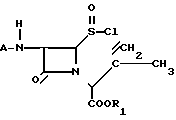
где А и R1 имеют указанные значения,
в сухом инертном органическом растворителе в присутствии катализатора типа кислоты Льюиса при температуре и времени, достаточных для получения соединения формулы I, с последующим выделением целевого продукта, отличающийся тем, что циклизацию проводят в присутствии нитросоединения, выбранного из группы, содержащей нитро(C1 - C6)-алканы и нитробензол.
R2-O-R2; Z O ; R2-
O ; R2- -R2
-R2
Z C= 0; (R3)3-P→ 0,
C= 0; (R3)3-P→ 0,
где каждый R2 независимо друг от друга является C1 - C4-алкилом;
каждый R3 независимо друг от друга является C1 - C4-алкилом, C5- C6-циклоалкилом, фенилом или фенилом, замещенным C1 - C4-алкилом, C1 - C4-алкоксигруппой или галогеном;
Z -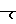 CH
CH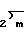 ; -CH2CH2-O-CH2CH2-
; -CH2CH2-O-CH2CH2-
или
-CH2-O-(CH2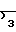
m = 4 или 5;
Z' - группа общей формулы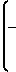
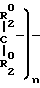
где R4 - водород или C1 - C4-алкил;
n = 3 - 6.
R-C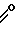
где R - бензил, феноксиметил или 2-тиенил;
R1 - бензил или замещенный бензил.
R2 - O - R2 или
R2-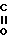 -R2
-R2
14. Способ по п. 13, отличающийся тем, что оксосоединение представляет собой диэтиловый эфир или ацетон.
R-C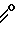
где R - феноксиметил,
группа R1 - паранитробензил, оксосоединение - диэтиловый эфир, ненасыщенное соединение - C5 - C8-алкен.
R-C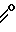
где R - бензил, феноксиметил или 2-тиенил,
R1 - бензил или замещенный бензил.
R-C
где R - бензил, феноксиметил или 2-тиенил,
R1 - бензил или замещенный бензил.
