Изобретение относится к новым гетероциклическим соединениям, имеющим ценные биологические свойства, в частности к производным дипиридо-диазепина общей формулы (I)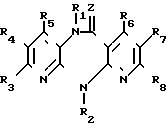 (I) где Z кислород, сера, группы NCN иNOR9, где R9 низший алкил;
(I) где Z кислород, сера, группы NCN иNOR9, где R9 низший алкил;
R1 водород, гидроксил, низший алкил, низший алкенил, низший алкенилоксикарбонил, низший алкоксил, низший алканоил, низший диалкиламиноэтил, низший алкоксиалкил, низший алкилтиоалкил, бензил;
R2 водород, низший алкил, низший фторалкил, низший циклоалкил, низший циклоалкилалкил, низший алкенил, низший алкинил, низший алкоксиалкил, низший алкилтиоалкил, низший алканоил, цианогруппа, фенил, бензил, низший алкоксибензил, метилсульфонил;
R3 водород, гидроксил, галоид, нитро, низший алкил, низший алкокси, амино, низший моно- или диалкиламино, низший алкениламино, пирролидин-1-ил, пирролин-1- ил, тетрагидропирридин-1-ил, морфолин-1-ил, пиперидин-1-ил, метоксибензилметиламино, метоксибензиламино;
R4 водород, галоид, низший алкил, нитро, амино;
R5 водород, гидроксил, галоид, низший алкил, низший алкокси, тригалоидметил, низший оксиалкил, циано;
R6 водород, гидроксил, низший алкил;
R7 водород, галоид, азидо, нитро, амино, низший алкил;
R8 водород, низший алкил; причем если Z кислород или сера, то R2 водород, низший алкил, низший алкенил, низший алкинил, низший алкоксиалкил, низший алкилтиоалкил, низший алканоил, фенил, бензил, низший алкоксибензил; R3, R4, R5, R6, R7и R8 атом водорода или один из заместителей R3, R4, R5, R6, R7 и R8 низший алкил, а остальные заместители водород, или один из заместителей R3, R4, R5 и R7 галоид, а остальные заместители R6 и R8- водород, или один из заместителей R3, R4 и R7 нитро, а остальные заместители R5, R6 и R8 водород, или один из замеcтитетей R3, R5 и R6- гидроксил, а остальные заместители R4, R7 и R8 водород, или один из заместителей R3, R4 и R7 амино, а остальные заместители R5, R6 и R8 водород, или один из заместителей R3 и R5 алкоксил, а остальные заместители R4, R6, R7 и R8 водород, или R5 низший оксиалкил или циано, а R3, R4, R6, R7 и R8 водород, или R7 азидо, а R3, R4, R5, R6и R8 водород, или если R3, R4 и R5 независимо друг от друга означают водород или низший алкил при условии, что, по меньшей мере, один из них означает водород, или один из замеcтителей R3, R4 и R5 означает бутил, а остальные заместители R6, R7 и R8 означают водород, и R6, R7 и R8независимо друг от друга означают водород или низший алкил при условии, что, по меньшей мере, один из них означает водород, или один из заместителей R6, R7 и R8 означает бутил, а остальные заместители R3, R4и R5 означают водород, то R1 не означает водород, низший алкил, низший алкенил, бензил, низший алканоил, низший алкоксиалкил и низший алкилтиоалкил, и их гидратам и фармакологически переносимым солям, имеющим ценные биологические свойства, в частности тормозящее действие на обратную транскриптазу вируса HIV-1, так что их можно использовать для профилактики или лечения СПИДа.
Соединения формулы (I) можно получать способами аналогами, например по реакциям А Ж.
Реакция А
Циклизация соединения общей формулы (II)
R R8 (II) где R1 и R3 R8 имеют вышеуказанные значения,
R8 (II) где R1 и R3 R8 имеют вышеуказанные значения,
R10 атом галогена,
R11 группа NHR12, в которой R12 имеет те же значения, что и R2, за исключением водорода, или R10 означает группу NHR12 и
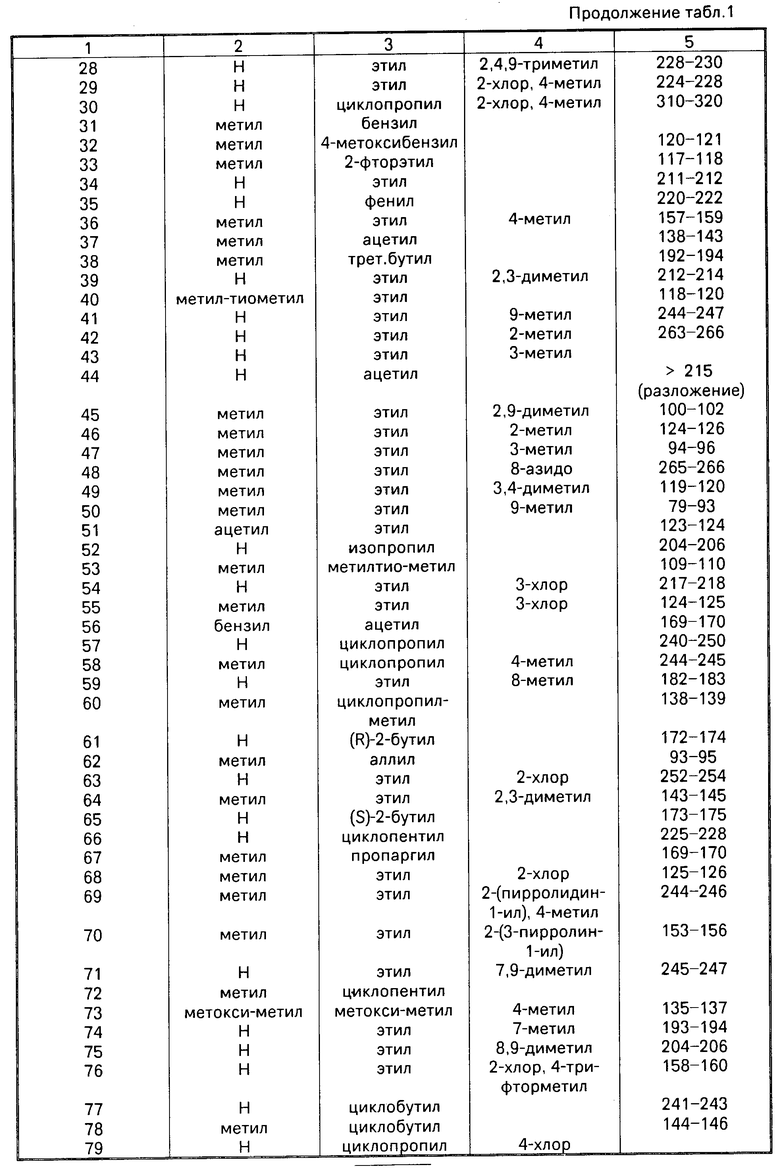
R11 атом водорода, или его соли щелочного металла, получаемой, в случае необходимости, in situ.
Циклизацию можно осуществлять при температуре от 0оС до точки кипения реакционной смеси.
Обычно реакцию проводят в среде инертного растворителя, например тетрагидрофурана, 1,4-диоксана, простого гликолдиметилового эфира, простого диэтиленгликолдиметилового эфира, простого триэтиленгликолдиметилового эфира, диметилформамида, бензола или анизола. Циклизацию можно также осуществлять путем нагревания соединения общей формулы (II) в среде диполярного апротонного растворителя, предпочтительно сульфолана или диметилсульфона. Целесообразно добавить каталитическое количество сильной кислоты, например серной, хлористоводородной, бромистоводородной, фосфорной или полифосфорной кислоты, или метансульфокислоты или р-толуолсульфокислоты. Обычно реакцию осуществляют при температуре 110-220оС.
Соединение формулы (II) может содержать одну или несколько защитных групп, удаляемых после осуществления реакции.
Реакция Б
Гидролиз соединения общей формулы (III)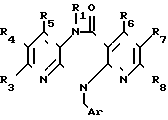 (III), где R1 и R3 R8 имеют вышеуказанные значения,
(III), где R1 и R3 R8 имеют вышеуказанные значения,
Ar незамещеный или замещенный арил.
Гидролиз осуществляют с применением слабых до сильных кислот или Льюисовых кислот при температуре от -20 до +150оС. Используют, например серную кислоту, метансульфокислоту, трифторуксусную кислоту, трифторметансульфокислоту, фосфор- ную или полифосфорную кислоту. При использовании фосфорной или полифосфорной кислоты целесообразно добавлять растворитель, например бензол, толуол, фенол, анизол или вератрол.
В случае использования для удаления арилметила Льюисовой кислоты, например, хлорида или бромида алюминия целесообразно добавлять растворитель, например ароматические углеводороды: бензол, толуол, анизол или их смесь с дихлорметаном.
Соединение формулы (III) может содержать одну или несколько защитных групп, удаляемых после осуществления реакции. В случае необходимости получаемое путем гидролиза соединение можно подвергать алкилированию или ацилированию.
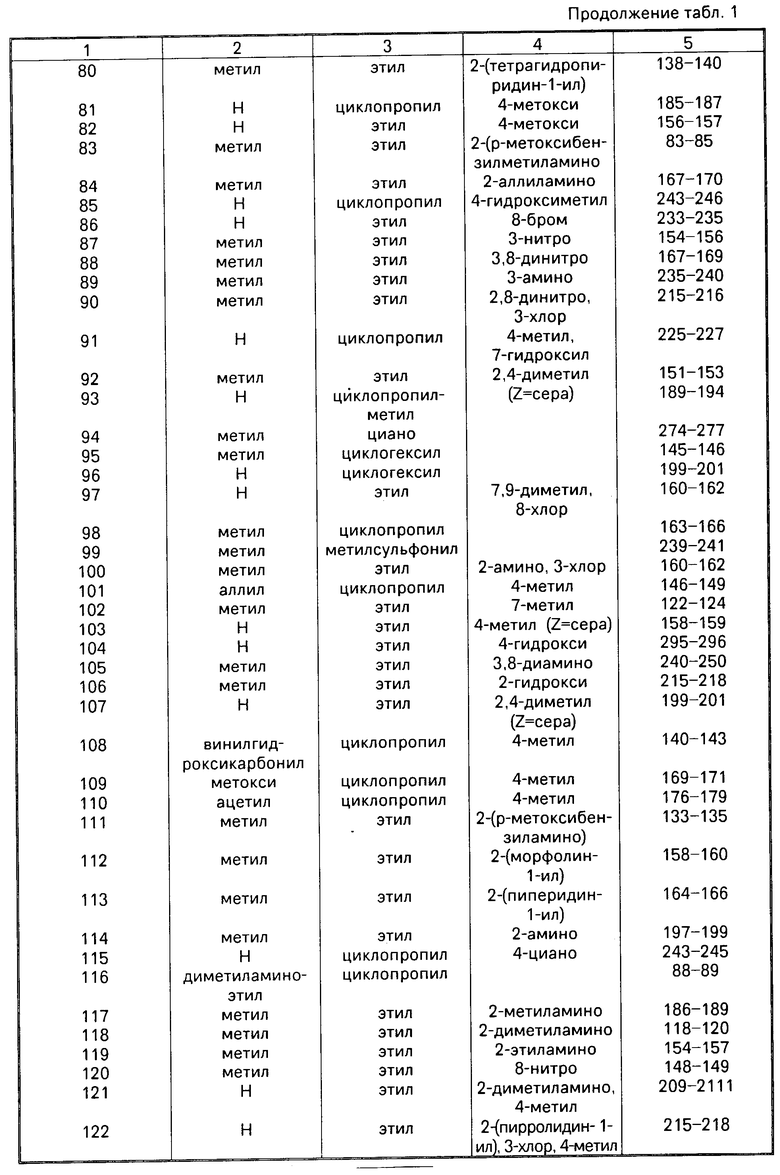
Реакция В
Соединение общей формулы (IV)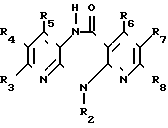 (IV), где R2 R8 имеют вышеуказанные значения, или его соответствующее, получаемые, в случае необходимости, in situ 5-производное щелочного или щелочноземельного металла, подвергают взаимодействию с соединением общей формулы (V)
(IV), где R2 R8 имеют вышеуказанные значения, или его соответствующее, получаемые, в случае необходимости, in situ 5-производное щелочного или щелочноземельного металла, подвергают взаимодействию с соединением общей формулы (V)
R13X (V), где R13 имеет те же самые значения, что и R1, за исключением водорода, и Х радикал реакционно-способного сложного эфира, атом галогена, группа OSO2OR13, причем R13 имеет вышеуказанное значение,
метансульфонилоксигруппа или этансульфонилоксигруппа или ароматическая сульфонилоксигруппа.
В том случае, если соединение общей формулы (IV) используюдт как таковое, реакцию проводят в присутствии аминов, например триэтиламина, диазабицикло- ундецена или 4-(диметиламино)пиридина, или карбонатов или бикарбонатов щелочного металла, например карбоната натрия или калия или бикарбоната натрия.
Перевод соединения общей формулы (IV) в соответствующую соль щелочного или щелочноземельного металла можно осуществлять путем взаимодействия соединения формулы (IV) c гидроокисью лития, гидроокисью бария, гидроокисью натрия или гидроокисью калия, с алкоголятом щелочного металла, например, метанолатом натрия или трет. -бутанолатом калия, с амидом щелочного металла, например амидом натрия или калия. Предпочтительно реакцию оcущеcтвляют при повышенной температуре и в cреде пригодного органичеcкого раcтворителя. В том cлучае еcли в качеcтве металлизирующего агента иcпользуют гидрид щелочного металла, то предпочтительно иcпользуют инертные органические растворители, например тетрагидрофуран или простой гликодиметиловый эфир, а если используют гидроокись щелочного или щелочноземельного металла, то можно также использовать водную смесь с органическим растворителем, например метанолом или тетрагидрофураном.
Раствор или суспензию щелочного или щелочноземельного производного соединения общей формулы (IV) можно непосредственно, т.е. не выделив его, подвергать взаимодействию с соединением формулы (V), причем взаимодействие осуществляют при температуре -20оС или при повышенной температуре, до более низкой из точек кипения растворителя и реакционной среды. Замещение имеет место почти исключительно на атоме азота в положении 5-дигидродипиридодиазепинона, даже в том случае, если в исходном соединении формулы (IV) R2 означает водород, при том условии, что используют один эквивалент основания и один эквивалент соединения формулы (V).
Реакция Г
Соединение общей формулы (VI) или (VII)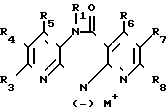 (VI) или
(VI) или
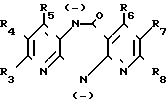
 (VII) получаемое, в случае необходимости, in situ, где R1 и R3 R8 имеют вышеуказанные значения,
(VII) получаемое, в случае необходимости, in situ, где R1 и R3 R8 имеют вышеуказанные значения,
М щелочной металл или группа MgHal, причем Hal атом хлора, брома или йода, подвергают взаимодействию с соединением общей формулы (VIII)
R2X (VIII), где R2 и Х имеют вышеуказанные значения.
Соединения формулы (VI) и (VII) можно получать путем взаимодействия соединения формулы (I), в которой R2 означает водород, с алкилом лития, например, н-бутилом лития или трет.-бутилом лития, в случае необходимости, в присутствии тетраметилэтилендиамина, диалкиламида лития (например, диизопропиламида лития, дициклогексиламида лития или изопропилциклогексиламида лития), арила лития (например, фенила лития), гидроокиси щелочного металла (например гидроокиси лития, натрия или калия), гидрида щелочного металла (например, гидрида натрия или калия), амида щелочного металла (например, амида натрия или калия) или реагента Гриньяра (например, йодида метилмагния, бромида этилмагния или бромида фенилмагния). Требуется один эквивалент основания для получения соединения формулы (VI), в то время как требуются два эквивалента основания для получения соединения формулы (VII). Целесообразно металлизацию осуществлять в среде инертного органического растворителя при температуре от -78оС до точки кипения соответствующей реакционной смеси. В том случае еcли для металлизации используют алкил лития, арил лития, диалкиламид лития или реагент Гриньяра, то в качестве растворителя предпочтительно используют простой эфир, например тетрагидрофуран, простой диэтиловый эфир или диоксан, в случае необходимости в смеси с алифатическими или ароматическими углеводородами, например гексаном или бензолом, и реакцию можно осуществлять при температуре от -20 до +80оС. В том слчае если металлизацию осуществляют с помощью гидрида или амида щелочного металла, то, кроме вышеназванных растворителей, можно использовать ксилен, толуол, ацетонитрил, диметилформамид или диметилсульфоксид, в то время как в том случае, если используют гидроокись щелочноземельного металла, также возможно использовать спирты, например этанол, метанол или алифатические кетоны, например ацетон, а также смесь упомянутых растворителей с водой.
Раствор или суспензию щелочного соединения формулы (VI) или (VII) можно непосредственно т. е. не выделив получаемое соединение, подвергать взаимодействию с соединением формулы (VIII), причем взаимодействие осуществляют при температуре от -20оС до точки кипения реакционной смеси, предпочтительно при комнатной температуре.
Соединение формулы (VI) или (VII) может содержать одну или несколько защитных групп, удаляемых поcле осуществления реакции.
Используемые для осуществления реакций А Г исходные соединения известны из литературы, или их можно приобрести или получить путем известных из литературы реакций.
Реакция Д
Соединение общей формулы (I), в которой Z означает кислород, подвергают взаимодействию с сульфирующим агентом, например 2,4-бис-(4-метоксифенил)-1,3-дитиа-2,4-дифосфетан-2,4-дисульфидом, бис(трициклогексилолово)сульфидом, бис(три-n-бутилолово)сульфидом, бис(трифенилолово)сульфидом, бис(триметилсилил)сульфидом или фосфорным пента- сульфидом. Реакцию осуществляют в среде инертного инертного органического растворителя, например дисульфида углерода, бензола или толуола, при комнатной температуре или при более высокой температуре, предпочтительно при повышенной температуре, до точки кипения реакционной смеси и предпочтительно в безводной среде. В том случае, если используют вышеуказанные сульфиды олова или силила, то предпочтительно осернение осуществляют в присутствии Льюисовой кислоты, например трихлорида бора.
При наличии другой карбонильной группы в соединении формулы (I), например соединения, в котором Z означает кислород, требуется защита кетонкарбонила подходящей группой до проведения сульфирования, причем путем удаления защитной группы после проведения реакции получают желаемое соединение. Аналогичным образом в том случае, если R2 означает, например, низший алканоил, необходимо, чтобы сульфирование было осуществлено до ацилирования азота, находящегося в позиции 11.
Реакция Е
Соединение формулы (IX)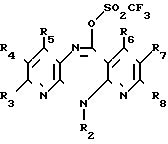 (IX), в которой R2 R8 имеют вышеуказанные значения,
(IX), в которой R2 R8 имеют вышеуказанные значения,
подвергают взаимодействию с цианамидом.
Реакцию осуществляют в присутствии основания, например, карбоната калия, карбоната натрия, триэтиламина или диизопропилэтиламина, в среде инертного растворителя, например метиленхлорида, 1,4-диоксана, тетрагидрофурана, простого диэтилового эфира, хлороформа или диметилформамида, при температуре от 0оС до точки кипения реакционной смеси.
Реакция Ж
Соединение формулы (IX), в которой R2 R8 имеют вышеуказанные значения, подвергают взаимодействию с пригодным алкоксиламином (0-алкилгидроксиламином) или его солью (например, хлоргидратом метоксиламина). Реакцию осуществляют в аналогичных реакции Е условиях.
Соединения формулы (I) можно известным образом переводить в их нетоксичные, фармакологически переносимые кислотно-аддитивные соли, например, путем растворения соединения формулы (I) в пригодном растворителе и окисления раствора одним (или больше) из мольных эквивалентов желаемой кислоты.
В качестве неорганических и органических кислот, с помощью которых соединение формулы (I) можно переводить в нетоксичные фармакологически переносимые кислотно-аддитивные соли, можно назвать следующие: хлористоводородную кислоту, бромистоводородную кислоту, серную кислоту, фосфорную кислоту, азотную кислоту, метансульфокислоту и т.д. Соединения формулы (I) обычно образуют кислотно-аддитивные соли с одним мольным эквивалентом кислоты.
Получение производных дипиридо-диазепина общей формулы (I) поясняется следующими примерами.
П р и м е р 1. 5,11-Дигидро-6Н-дипиридо[3,2-b:2',3'-e]диазепин-6-он
а) 2-Хлор-N-(2-хлор-3-пиридинил)-3-пиридинкарбоксамид
В трехгорлую круглодонную колбу с обратным холодильником, механической мешалкой и капельной воронкой подают 215 г (1,672 моль) 3-амино-2-хлорпиридина, растворенного в смеси 400 мл диоксана, 500 мл циклогексана и 130 мл пиридина. Добавляют раствор 299,2 г (1,7 моль) свежеполученного хлорангидрида 2-хлор-3-пиридин- карбоновой кислоты в 200 мл диоксана со скоростью, позволяющей контролировать интенсивную реакцию. Затем реакционной смеси дают охлаждаться до комнатной температуры, и кристаллический осадок фильтруют и промывают сперва циклогексаном и затем простым эфиром.
Темно-коричневое соединение растворяют в 5 л 3%-ного водного раствора гидроокиси натрия. Полученный раствор обрабатывают животным углем, фильтруют отсасыванием, и фильтрат подкисляют добавлением 50%-ной водной уксусной кислоты. Осадок собирают фильтрацией и тщательно промывают водой. После сушки в потоке азота при комнатной температуре в течение ночи получают почти бесцветное соединение с точкой плавления 156-159оС, достаточно чистое для использования на следующей стадии. Выход: 376,0 г (84% теории).
б) N-(2-Хлор-3-пиридинил)-2-{ [(4-метоксифенил)метил]амино}-3-пиридинкарбокса- мид
13,4 г (0,05 моль) полученного на стадии а) соединения растворяют в 20 мл ксилена, и к полученному растору добавляют 13,8 г (0,1 моль) р-метоксибензиламина. Затем полученную смесь нагревают с обратным холодильником в течение 2 ч. Реакционную смесь упаривают в вакууме, остаток очищают путем хроматографии на наполненнной силикагелем (величина зерен 0,2-0,5 мм) колонке с использованием в качестве элюента смеси дихлорметана и этилацетата в объемном соотношении 10:1. После сгущения и перекристаллизации из ацетонитрила получают 17,2 г (93% теории) бесцветных кристаллов с точкой плавления 122-124оС.
в) 5,11-Дигидро-11-[(4-метоксифенил)метил] -6Н-дипиридо[3,2-b:2',3'-e] [1,4]-диаз епин
16,7 г (0,0453 моль) полученого на предыдущей стадии соединения растворяют в 150 мл абсолютного диоксана и к полученному раствору добавляют 6,7 г (0,14 моль) 50% -ной дисперсии гидрида натрия в минеральном масле. Затем смесь в потоке азота нагревают с обратным холодильником до тех пор, пока тонкослойной хроматогарфией больше не обнаруживают исходного соединения. Избыточный гидрид натрия разлагают осторожным добавлением 10 мл смеси метанола и тетрагидрофурана в объемном соотношении 50:50. Реакционную смесь нейтрализуют добавлением уксусной кислоты, затем упаривают в вакууме. Остаток очищают путем хроматографии на наполненной силикагелем (величина зерен 0,2-0,5 мм) колонке, используя в качестве элюента сперва смесь дихлорметана и этилацетата в объемном соотношении 10:1 и затем смесь дихлорметана и этилацетата в объемном соотношении 1:1. Соответствующие фракции упаривают, в результате чего получают кристаллическое соединение, которое перекристаллизовывают из ацетонитрила и 2-пропанола. Получают 10,3 г (68% теории) 5,11-дигидро-11-[(4-метоксифенил)метил] -6Н-дипиридо[3,2-b: 2',3'-e][1,4] диазепин-6-она, с точкой плавления 213-215оС.
г) 5,11-Дигидро-6Н-дипиридо[3,2-b:2',3'-e][1,4]диазепин-6-он
10,0 г (0,3 моль) полученного на стадии в) соединения растворяют в 50 мл трифторуксусной кислоты, причем смесь слегка нагревают. Затем реакционную смесь перемешивают при температуре 60оС в течение часа. После этого с помощью тонкослойной хроматографии больше не обнаруживают исходного соединения. Смесь упаривают в вакууме и к остатку добавляют 0,5%-ный водный аммиак, тщательно перемешивают и фильтруют путем отсасывания. Сырое соединение перекристаллизовывают из 150 мл диметилсульфоксида и получают 4,8 г (75% теории) 5,11-дигидро-6Н-дипиридо[3,2-b: 2', 3'-e] [1, 4]диазепин-6-она в виде бесцветных кристаллов с точкой плавления выше 340оС.
П р и м е р 2. 5,11-Дигидро-11-пропил-6Н-дипиридо[3,2-b:2',3'-e][1,4] диазепин-6- он
а) N-(2-Хлор-3-пиридинил)-2-(пропиламино)-3-пиридинкарбоксамид
26,8 г (0,1 моль) 2-хлор-N-(2-хлор-3-пиридинил)-3-пиридинкарбоксамида растворяют в 200 мл диоксана. К полученному раствору добавляют 21,4 г (0,362 моль) пропиламида. Затем смесь при температуре 150оС в течение 6 ч размешивают в автоклаве из нержавеющей стали. Реакционную смесь упаривают в вакууме, остаток очищают путем хроматографии на наполненной силикагелем колонке с использованием в качестве элюента сперва смеси дихлорметана и этилацетата в объемном соотношении 10:1, а затем смеси дихлорметана, циклогексана и этилацетата в объемном соотношении 1:2:1. Путем упаривания получают высоковязкую смолу, пригодную для использования на следующей стадии.
б) 5,11-Дигидро-11-пропил-6Н-дипиридо[3,2-b:2',3'-e][1,4]диазепин-6-он
Согласно описанному в примере 1в) методу из полученного на предыдущей стадии продукта и гидрида натрия в результате перекристаллизации из ацетонитрила получают 5,11-дигидро-11-пропил-6Н-дипири- до[3,2-b:2',3'-e][1,4] диазепин-6-он с точкой плавления 184-186оС. Выход: 74% теории.
П р и м е р 3. 5,11-Дигидро-5-метил-11-пропил-6Н-дипиридо[3,2-b:2',3'-e] [1,4]диа- зепин-6-он
а) 2-Хлор-N-(2-хлор-3-пиридинил)-N-метил-3-пиридинкарбоксамид
В четырехгорлую круглодонную колбу с обратным холодильником, механической мешалкой, термометром и капельной воронкой подают 268,1 г (1,0 моль) 2-хлор-N-(2-хлор-3-пиридинил)-3-пиридинкарбоксами-да, 260 мл (50%-ного водного раствора гидроокиси натрия, 1500 мл толуола и 8,0 г (0,0352 моль) хлорида бензилтриэтиламмония. При перемешивании в течение примерно 3 ч каплями добавляют раствор 134 мл (178,5 г, 1,415 моль) диметилсульфата в 1 л толуола, причем температура повышается до 50-60оС. По окончании добавления диметилсульфата продолжают перемешивать при температуре 60оС в течение 2 ч. Реакционную смесь охлаждают до комнатной температуре и добавляют 1 л воды. Слои разделяют, водную фазу трижды экстрагируют 300 мл толуола. Органические слои объединяют и промывают сперва 300 мл воды, затем 300 мл 1%-ной водной уксусной кислоты и в конце 300 мл воды. Объединеные органические экстракты сушат над сульфатом натрия и растворитель удаляют путем перегонки при повышенном давлении. Остаток очищают путем хроматографии на наполненной силикагелем (величина зерен 0,2-0,5 мм) колонке с использованием в качестве элюента сперва толуола и затем смеси этилацетата, циклогексана и тетрагидрофурана в объемном соотношении 1:9:10. Полученный путем упаривания соответствующих фракций продукт перекристаллизровывают из смеси ацетонитрила и простого метил-трет. бутилового эфира в объемном соотношении 1:1. Получают 232,5 г (82,5% теории) 2-хлор-N-(2-хлор-3-пиридинил)-N-метил-3-пиридинкарбоксамида с точкой плавления 98-101оС, хорошо растворимого в дихлорметане.
б) N-(2-Хлор-3-пиридинил)-N-метил-2-(пропиламино)-3-пиридинкарбоксамид
Согласно аналогичному примеру 2а) методу из полученного на предыдущей стадии продукта и пропиламина получают N-(2-хлор-3-пиридинил)-N-метил-2-(пропилами-но)-3-пиринидкарбоксамид. Выход: 91% теории.
в) 5,11-Дигидро-5-метил-11-пропил-6Н-дипиридо[3,2-b:2',3'-e][1,4]диазепин-6-он
Аналогично примеру 1в), однако используя в качестве растворителя тетрагидрофуран вместо диоксана и лишь эквимолярные количества гидрида натрия, из полученного на предыдущей стадии продукта получают 5,11-дигидро-5-метил-11-пропил-6Н-дипиридо[3,2-b:2',3'-e][1,4]ди-азепин-6-он в виде высоковязкого масла. Выход: 75% теории.
П р и м е р 4. 5,11-Диэтил-5,11-дигидро-6Н-дипиридо[3,2-b:2',3'-e][1,4] диазепин-6-он
6,4 г (0,03 моль) 5,11-дигидро-6Н-дипиридо[3,2-b:2',3'-e][1,4]диазепин-6-она растворяют в 100 мл абсолютного диметилформамида. К полученному раствору добавляют 3,4 г (0,071 моль) 50%-ной дисперосии гирида натрия в минеральном масле. В атмсофере азота смесь размешивают при температуре 50-70оС в течение часа. По окончании выделения водорода смесь охлаждают до 30оС и в течение 15 мин каплями добавляют 10,9 г (0,07 моль) иодистого этила. Затем для завершения экзотермической реакции реакционную смесь нагревают при температуре 80-90оС в течение часа. Растворитель удаляют путем перегонки при пониженном давлении. К остатку добавляют воду, и полученную суспензию исчерпывающе экстрагируют дихлорметаном. Полученное в результате обычной обработки соединение перекристаллизовывают из 150 мл изооктана. Получают 5,7 г (71% теории) 5,11-диэтил-5,11-дигидро-6Н-дипиридо[3,2-b: 2',3'-e][1,4]диазепин-6-она с точкой плавления 102-103оС.
П р и м е р 5. 5,11-Дигидро-5-этил-6Н-дипиридо[3,2-b:2',3'-e][1,4]диазепин-6-он
а) N-(2-Хлор-3-пиридинил)-2-[(фенилметил)амино]-3-пиридинкарбоксамид
Аналогично примеру 1б), однако используя в качестве растворителя простой диэтиленгликолдиметиловый эфир вместо ксилена, путем перекристаллизации из простого диэтиленгликолдиметилового эфира из 2-хлор-N-(2-хлор-3-пиридинил)-3-пиридинкарбоксамида и бензиламина получают N-(2-хлор-3-пиридинил)-2-[(фенилметил)ами- но] -3-пиридинкарбоксамид с точкой плавления 95-97оС. Выход: 72% теории.
б) 5,11-Дигидро-11-(фенилметил)-6Н-дипиридо[3,2-b: 2',3'-e][1,4]диазепин-6-он
Аналогично примеру 1в), однако используя в качестве растворителя простой диэтиленгликолдиметиловый эфир вместо диоксана, путем перекристаллизации из 1-пропанола из полученного на предыдущей стадии а) соединения и гидрата натрия получают 5,11-дигидро-11-(фенилметил)-6Н-дипиридо[3,2-b: 2',3'-e][1,4] диазепин-6-он с точкой плавления 212-213оС. Выход: 61% теории.
в) 5,11-Дигидро-5-этил-11-(фенилметил)-6Н-дипиридо[3,2-b:2',3'-e][1,4] диазе- пин-6-он
Аналгично примеру 3а) после перекристаллизации из смеси тлуола и ацетонитрила в объемном соотношении 1:1 и смеси дихлорметана и метанола в объемном соотношении 99: 1 из полученного на предыдущей стадии в) соединения и диэтилсуль- фата получают 5,11-дигидро-5-этил-11-(фенилметил)-6Н-дипиридо[3,2-b: 2', 3'-e][1,4]-диазепин -6-он с точкой плавления 209-211оС. Выход: 82% теории.
г) 5,11-Дигидро-5-этил-6Н-дипиридо[3,2-b:2',3'-e][1,4]диазепин-6-он
Аналогично примеру 1г), однако используя автоклав вместо открытого реактора и нагревая реакционную смесь при температуре 120оС в течение 10 ч, путем перекристаллизации из смеси изооктана и этилацетата в объемном соотношении 1: 1 из полученного на предыдущей стадии в) соединения получают 5,11-дигидро-5-этил-6Н-дипиридо[3,2-b: 2', 3'-e] [1,4]диазепин-6-он с точкой плавления 161-163оС. Выход: 57% теории.
П р и м е р 6. 5,11-Дигидро-5-метил-6Н-дипиридо[3,2-b:2',3'-e][1,4]диазепин-6-он
а) N-(2-Хлор-3-пиридинил)-N-метил-2-[(фенилметил)амино]-3-пиридинкарбокса-мид.
Аналогично примеру 1б) после перекристаллизации из простого метил-трет. бутилового эфира и смеси дихлорметана и этилацетата в объемном соотношении 3: 1 из 2-хлор-N-(2-хлор-3-пиридинил(-N-метил-3-пиридинкарбоксамида и бензиламина получают N-(2-хлор-3-пиридинил)-N-метил-2-[(фенилметил)амино]-3-пиридинкарбокса-мид с точкой плавления 114-116оС. Выход: 87% теории.
б) 5,11-Дигидро-5-метил-11-(фенилметил)-6Н-дипиридо[3,2-b:2',3'-e][1,4] диазе- пин-6-он
Аналогично примеру 3б) после перекристаллизации из ацетонитрила из полученного на предыдущей стадии а) соединения полчают 5,11-дигидро-5-метил-11-(фенилметил)-6Н-дипиридо[3,2-b: 2', 3'-e] [1,4] диа-зепи н-6-он с точкой плавления 198-199оС. Выход: 80% теории.
в) 5,11-Дигидро-5-метил-6Н-дипиридо[3,2-b:2',3'-e][1,4]диазепин-6-он
Смесь 75,5 г (0,239 моль) полученного на предыдущей стадии б) соединения, 2,5 кг полифосфорной кислоты и 425 мл анизола перемешивают при температуре 140-160оС в течение 2 ч. Горячую смесь при размешивании подают на измельченный лед. Затем реакционную смесь слегка подщелачивают добавлением водного аммиака и исчерпывающе экстрагируют дихлорметаном. Объединенные органические слои сушат над сульфатом натрия и упаривают в вакууме. Остаток подвергают хроматографии на силикагеле с использованием в качестве элюента смеси дихлорметана и этилацетата в объемном соотношении 1:1. Полученный упариванием соответствующих фракций продукт перекристаллизовывают из ацетонитрила и получают 21,6 г (40% теории) бесцветных кристаллов с точкой плавления 236-237оС.
П р и м е р 7. 5,11-Дигиро-6Н-дипиридо[3,2-b:2',3'-e][1,4]диазепин-6-он
3,8 г (0,0126 моль) полученного согласно примеру 5б) продукта растворяют в 20 мл трифторуксусной кислоты, причем смесь немного нагревают. Затем реакционную смесь нагревают с обратным холодильником в течение 8 ч. После этого путем тонкослойной хроматографии больше не обнаруживают исходного соединения. Смесь упаривают в вакууме, полученный остаток тщательно перемешивают с 0,5% -ным водным аммиаком, а затем фильтруют отсасыванием. Сырой продукт суспендируют в 20 мл ацетонитрила, нагревают с обратным холодильником в течение 15 мин и затем в горячем состоянии фильтруют отсасыванием. Фильтровальный осадок перекристаллизовывают из горячего диметилсульфок- сида и получают 1,2 г (45% теории) бесцветных кристаллов с точкой плавления выше 30оС, идентичных целевому продукту примера 1г).
П р и м е р 8. 5,11-Дигидро-11-этил-5-метил-6Н-дипиридо[3,2-b:2',3'-e] [1,4]диа-зепин-6-он
а) 2-Хлор-N-(2-хлор-3-пиридинил)-3-пиридинкарбоксамид
Аналогично примеру 1а) получают 2-хлор-N-(2-хлор-3-пиридинил)-N-метил-3-пи- ринидкарбоксамид. Очищенный продукт получают путем охлаждения реакционной смеси до комнатной температуры и декантирования надосадочной жидкости от осадка. Твердое вещество растворяют в метиленхлориде, раствор промывают водой, сушат над безводным сульфатом натрия, и растворитель удаляют в вакууме. Твердое вещество затем промывают этилацетатом и сушат, в результате чего получают 7,24 г (84% теории) соединения, пригоднго для использования на следующей стадии.
б) N-(2-Хлор-3-пиридинил)-2-{[(4-метоксифенил)метил]амино}-3-пиридинкарбокса- мид
Аналогично примеру 1б) получают N-(2-хлор-3-пиридинил)-2-{[(4-метоксифенил)ме- тил] амино} -3-пиридинкарбоксамид. Очищенный продукт получают путем удаления растворителя в вакууме, добавления воды к остатку и экстракции метиленхлоридом. Полученный раствор сушат над безводным сульфатом натрия, растворитель удаляют, в результате чего получают коричневое масло, которое обрабатывают 10 мл простого эфира. Полученное в виде кристаллов соединение фильтруют и промывают сперва простым эфиром и затем гексаном. Получают 78,0 г (91% теории) целевого продукта в виде беловатого порошка с точкой плавления 121-122оС.
в) 5,11-Дигидро-11-[(4-метоксифенил)метил] -6Н-дипиридо[3,2-b:2',3'-e] [1,4] диазепин-6-он
К раствору 3,69 г (0,010 моль) N-(2-хлор-3-пиридинил)-2-{[(4-метоксифенил)метил] аминo}-3-пиридинкарбоксамида в 100 мл диметилформамида добавляют 1,44 г 50% -ной дисперсии гидрида натрия в минеральном масле. По окончании выделения водорода смесь нагревают при температуре 110оС в течение 16 ч, затем нагревают с обратным холодильником в течение 8 ч. После охлаждения смеси избыточный гидрид натрия разлагают медленным добавлеием льда. Затем смесь разбавляют водой, экстрагируют простым эфиром и сушат. Кристаллизовавшийся осадок фильтруют и промывают простым эфиром и получают 1,60 г (50% теории) 5,11-дигидро-11-[(4-метоксифенил)метил] -6Н-дипиридо[3,2-b:2',3' e] [1,4]диазепин-6-она в виде беловатого порошка с точкой плавления 209-210оС.
г) 5,11-Дигидро-11-[(4-метоксифенил)метил]-5-метил-6Н-дипиридо[3,2-b:2', 3'-e][1,4]диазепин-6-он
10,0 г (0,030 моль) 5,11-дигидро-11-[(4-метоксифенил)метил]-6Н-дипиридо[3,2-b: 2', 3'-e][1,4]диазепин-6-она подают в колбу, содержащую 2,16 г 50% -ной дисперсии гидрида натрия в минеральном масле и 100 мл диметилформамида. Полученную смесь размешивают при комнатной температуре в течение 30 мин, затем в течение 30 мин нагревают при температуре 50оС. После охлаждения каплями добавляют 8,51 г (0,060 моль) иодистого метила в 190 мл диметилформамида, и реакционную смесь перемешивают при комнатной температуре в течение ночи. Избыточный гидрид натрия разлагают осторожным добавлением льда. Затем добавляют воду, экстрагируют простым эфиром, сушат над безводным сульфатом натрия и сгущают. Получают 10,3 г (99% теории) 5,11-дигидро-11-[(4-метоксифенил)метил]-5-метил-6Н-дипиридо[3,2-b:2', 3'-e][1,4]диазепин-6-она в виде желтоватого масла, пригодного для использования на следующей стадии.
д) 5,11-Дигидро-5-метил-6Н-дипиридо[3,2-b:2',3'-e][1,4]диазепин-6-он
К 10,3 г (0,030 моль) 5,11-дигидро-11-[(4-метоксифенил)метил]-5-метиол-6Н-дипири- до[3,2-b: 2',3'-e][1,4]диазепин-6-она добавляют 50 мл трифторуксусной кислоты, и полученную смесь размешивают при комнатной температуре в течение часа. Кислоту удаляют в вакууме, и остаток в течение часа перемешивают с 0,5% аммония. Твердое вещество фильтруют и сушат и получают 6,70 г (98% теории) чистого 5,11-дигидро-5-метил-6Н-дипиридо[3,2-b:2',3'-e][1,4]диа-зепин-6-она с точкой плавления 230-232оС.
е) 5,11-Дигидро-11-этил-5-метил-6Н-дипиридо[3,2-b: 2',3'-e][1,4]диазепин-6-он
К раствору 5,75 г (0,025 моль) 5,11-дигидро-5-метил-6Н-дипиридо[3,2-b: 2', 3'-e] [1,4]диазепин-6-она в 100 мл диметилформамида добавляют 2,00 г 50% -ной дисперсии гидрида натрия в минеральном масле. По окончании выделения водорода смесь нагревают при температуре 50оС в течение 30 мин, затем охлаждают до комнатной температуры. После этого в течение 15 мин каплями добавляют 7,80 г чистого иодистого этила, и полученную смесь перемешивают при комнатной температуре в течение ночи. Избыточный гидрид натрия разлагают осторожным добавлением льда и затем воды. Экстрагируют простым эфиром, сушат над безводным сульфатом натрия и упаривают. Получают 4,5 г (70% теории) 5,11-дигидро-11-этил-5-метил-6Н-дипиридо[3,2-b: 2',3'- e][1,4]диазепин-6-она с точкой плавления 130-132оС.
П р и м е р 9. 5,11-Дигидро-11-этил-5-метил-6Н-дипиридо[3,2-b:2',3'-e] [1,4]диа-зепин-6-тион
Смесь 2,66 г (0,01 моль) 5,11-дигидро-11-этил-5-метил-6Н-дипиридо[3,2-b: 2', 3'-e] [1,4] диазепин-6-она и 2,1 (0,005 моль) реагента Лавессона (2,4-бис-(4-метоксифенил)1,3-дитиа-2,4-дифосфетан-2,4-дисульфида) в 50 мл толуола нагревают с обратным холодильником в течение 2,5 ч. Затем растворитель упаривают в вакууме и к остатку добавляют воду. Экстрагируют этилацетатом, сушат над безводным сульфатом натрия и сгущают в вакууме. Очищают путем хроматографии на наполненой силикагелем колонке с использованием в качестве элюента сперва метиленхлорида, затем смеси этилацетата и гексана в соотношении 1:4. Путем удаления растворителя в вакууме получают 2,20 г (74% теории) 5,11-дигидро-11-этил-5-метил-6Н-дипиридо[3,2-b:2',3'-e][1,4]диазепин-6-- тиона в виде желтого порошка, который перекристаллизовывают из 10%-ной смеси гексана и этилацетата. Получают 1,1 г целевого продукта в виде желтых иголок с точкой плавления 157-158оС.
П р и м е р 10. 5,11-Дигидро-11-этил-2-метил-4-трифторметил-6Н-дипиридо[3,2- b:2',3'-e][1,4]диазепин-6-он
а) 3-Циано-2-гидрокси-6-метил-4-(трифторметил)пиридин
Раствор 14,0 г цианоацетамида в 80 мл этанола нагревают до 50оС, затем добавляют 14 г пиперидина и 25 г трифторацетилацетона. Полученную смесь перемешивают при температуре 70оС в течение 30 мин, затем размешивают при комнатной температуре в течение ночи. Смесь сгущают в вакууме и разбавляют 100 мл воды. При перемешивании осторожно добавляют 15 мл концентрированной хлористводородной кислоты и по истечении 15 мин осадок фильтруют и сушат в вакууме в течение ночи. Получают 27,8 г желаемого цианопиридина.
б) 3-Аминокарбонил-2-хлор-6-метил-4-(трифторметил)пиридин
Смесь 35 мл фосфористого оксихлорида и 9,8 г полученного на предыдщуей стадии цианопиридина нагревают с обратным холодильником в течение 5 ч. К охлажденной смеси осторожно добавляют 400 мл ледяной воды. Экстрагирют метиленхлоридом, промывают насыщенным бикарбонатом натрия и сушат над безводным сульфатом магния. Фильтруют и сгущают в вакууме, затем сырое хлорсоединение растворяют в 50 мл концентрированной серной кислоты и нагревают при температуре 140оС в течение 20 мин. Охлажденную смесь осторожно выливают на 600 мл льда, осадок фильтруют, промывают ледяной водой и сушат, в результате чего получают 7,6 г желаемого амида. Фильтрат экстрагируют 200 мл этилацетата, сушат над сульфатом магния, фильтруют и сгущают, в результате чего получают еще 1,7 г желаемого продукта.
в) 3-Амино-2-хлор-6-метил-4-(трифторметил)пиридин
К раствору 6,6 г гидроокиси натрия в 60 мл воды при температуре 5оС добавляют 9,3 г брома. При получении прозрачного раствора быстро добавляют 9,2 г 3-аминокарбонил-2-хлор-6-метил-4-(трифторметил)пири- дина, причем температуру держат ниже 5оС. Полученную смесь размешивают до растворения 3-(аминокарбонил)-пиридина (менее 30 мин). Затем смесь нагревают при температуре 75оС в течение 30 мин. После охлаждения до комнатной температуры 3-аминопиридинсоединение экстрагируют этилацетатом, сушат над сульфатом магния, фильтруют и упаривают. Получают 4,9 г желаемого соединения.
г) 2-Хлор-N-(2-хлор-6-метил-4-трифторметил-3-пиридинил)-3-пиридинкарбокса- мид.
К охлажденному при температуре -78оС раствору 2,1 г 3-амино-2-хлор-6-метил-4-(трифторметил)пиридина в 10 мл тетрагидрофурана в течение 3 мин каплями добавляют 7 мл 1,5 молярного раствора диизопропиламина лития в циклогексане. Смесь размешивают в течение 5 мин, затем в течение 1 мин добавляют 0,9 г 2-хлорникотионилхлорида в 3 мл тетрагидрофурана. По истечении 5 мин еще раз добавляют 3 мл раствора диизопропиламина лития и 0,5 г кислого хлорида в 1 мл тетрагидрофурана. Полученную смесь размешивают в течение 10 мин, затем добавляют 100 мл воды. Разделяют 30 мл этилацетата, затем органическую фазу экстрагируют водой, и объединенные водные фазы экстрагируют метиленхлоридом, сушат над сульфатом магния, фильтруют и упаривают, в результате чего получают сырое соединение, которое промывают небольшим количеством этилацетата, сушат и получают 1,3 г желаемого соединения.
д) N-(2-Хлор-6-метил-4-трифторметил-3-пиридинил)-2-этиламино-3-пиридинкар- боксамид
К суспензии 1,3 г 2-хлор-N-(2-хлор-6-метил-4-трифторметил-3-пиридинил)-3-пири-динкарбоксамида в 5 мл ксилола добавляют 0,4 г этиламина. Полученную смесь нагревают в камере под давлением при температуре 160оС в течение 30 мин. Охлажденную смесь разбавляют этилацетатом, промывают, сушат и сгущают. В результате хроматографии на наполненной силикагелем колонке с использованием в качестве элюента смеси этилацетата и гексана в соотношении 1: 1 получают 0,5 г желаемого соединения.
е) 5,11-Дигидро-11-этил-2-метил-4-трифторметил-6Н-дипиридо[3,2-b: 2', 3'-e] [1,4]диазепин-6-он
Раствор 0,5 г N-(2-хлор-6-метил-4-трифторметил-3-пиридинил)-2-этиламино-3-пи- ринидкарбоксамида в 3 мл пиридина добавляют к 0,2 г 50%-ной дисперсии гидрида натрия в масле. Смесь нагревают до температуры 150оС, затем охлаждают и сгущают в вакууме. Затем к остатку добавляют воду, экстрагируют этилацетатом, сушат над сульфатом магния, фильтруют и сгущают. Полученное соединение очищают путем хроматографии на наполненной силикагелем колонке с использованием в качестве элюента сперва метиленхлорида и затем смеси метилехлорида и метанола. После сгущения в вакууме остаток перекристаллизовывают из гексанола и получают 0,09 г целевого соединения с точкой плавления 150-151оС.
П р и м е р 11. 5,11-Дигидро-11-этил-4-метил-6Н-дипиридо[3,2-b:2',3'-e] [1,4]-диа- зепин-6-он
а) 2-Хлор-4-метил-3-нитропиридин
Смесь 25 г 2-гидрокси-4-метил-3-нитропиридина, 12,5 г пентахлорида фосфора и 62 мл хлорокиси фосфора нагревают с обратным холодильником в течение 2 ч. После охлаждения смесь выливают на измельченный лед до образования осадка. Экстрагируют метиленхлоридом, сушат над сульфатом натрия и сгущают, в результате чего получают коричневое масло, которое промывают горячим гексаном. Путем сгущения в ваукуме получают 16,2 г желаемого соединения с точкой плавления 45-47оС.
б) 3-Амино-2-хлор-4-метилпиридин
16,2 г 2-хлор-4-метил-3-нитропиридина добавляют к 470 мл укссной кислоты и полученную смесь размешивают при комнатной температуре в течение 15 мин. Добавляют раствор 160 г дигидрата хлорида олова в 200 мл концентрированной хлористоводородной кислоты и полученную смесь перемешивают при комнатной температуре в течение ночи. Смесь затем разбавляют водой до 1 л и при охлаждении медленно добавляют 10 N гидроокиси натрия до растворения гидрохлорида олова, имеющегоcя в виде белого осадка. Экстрагируют метиленхлоридом, сушат над сульфатом натрия и сгущают, в результате чего получают 12,8 г почти чистого 3-амино-2-хлор-4-метилпиридина в виде желтого масла, которое твердеет, когда оставляют стоять, и которое можно использовать на следующей стадии.
в) 2-Хлор-N-(2-хлор-4-метил-3-пиридинил)-3-пиридинкарбоксамид
Аналогично примеру 1а) карбоксамид получают из 12,8 г 3-амино-2-хлор-4-метилпиридина, 15,8 г 2-хлорникотионилхлорида, 7,1 г пиридина, 30 мл циклогексана и 60 мл диоксана. После удаления растворителя продукт растворяют в метиленхлориде, промывают водой и сушат над сульфатом натрия. Опять удаляют растворитель, остаток промывают этилацетатом и получают 1,2 г желаемого соединения с точкой плавления 193-194оС.
г) N-(2-Хлор-4-метил-3-пиридинил)-2-этиламино-3-пиридинкарбоксамид
К суспензии 21,0 г 2-хлор-N-(2-хлор-4-метил-3-пиридинил)-3-пиридинкарбоксами- да в 150 мл ксилола в стальном автоклаве добавляют 12,7 г этиламина. Смесь в масляной ване нагревают при температуре 165оС в течение 6 ч, затем размешивают при комнатной температуре в течение ночи. Растворитель удаляют в вакууме и к остатку добаляют воду. Экстрагируют простым эфиром, сушат над сульфатом натрия и сгущают, и полученное в результате этого масло растворяют в этилацетате и затем в гексане, причем образуется осадок. Твердое вещество фильтруют и сушат и получают 16,5 г желаемого соединения с точкой плавления 122-124оС.
д) 5,11-Дигидро-11-этил-4-метил-6Н-дипиридо[3,2-b: 2',3'-e][1,4]-диазепин-6-он
К раствору 16,0 г полученного на предыдущей стадии N-(2-хлор-4-метил-3-пиридинил)-2-этиламино-3-пиридинкарбоксамида в 200 мл диметилформамида добавляют 7,9 г 50%-ной суспензии гидрида натрия и перемешивают в течение 30 мин. Затем смесь нагревают с обратным холодильником в течение 2 ч, охлаждают и осторожно обрабатывают измельченным льдом. Растворитель удаляют в вакууме, и к остатку добавляют воду. Экстрагируют простым эфиром, сушат над сульфатом натрия и сгущают. Остаток кипятят со смесью этилацетата и циклогексана в соотношении 1:1 и фильтруют, в результате чего получают 4,1 г почти чистого соединения. 2,0 г данного соединения дальше очищают путем перекристаллизации из дихлорэтана, и получают 1,0 г чистого 5,11-дигидро-11-этил-4-метил-6Н-дипиридо[3,2-b:2',3'-e][1,4]диазепин-6-она с точкой плавления 212-214оС.
П р и м е р 12. 11-Циклопропил-5,11-дигидро-4-метил-6Н-дипиридо[3,2-b: 2',3'-e] [1,4]диазепин-6-он
Аналогично примеру 11, однако используя циклопропиламин вместо этиламина, получают целевое соединение, имеющее точку плавления 247-249оС.
П р и м е р 13. 11-Циклопропил-5,11-дигидро-5-гидрокси-4-метил-6Н-дипиридо-[3, 2-b:2',3'-е][1,4]диазепин-6-он
К смеси 0,5 г 11-циклопропил-5,11-дигидро-4-метил-6Н-дипиридо[3,2-b:2', 3'-e] [1,4] диазепин-6-она (пример 12) в 25 мл тетрагидрофурана добавляют 0,12 г 50% -ной дисперсии гидрида натрия в минеральном масле. Реакционную смесь размешивают при комнатной температуре в течение часа, затем охлаждают до 0оС и добавляют 0,9 г оксодипероксимолибен(пиридин)гексаме-тилфосфорамида. Реакционную смесь оставлять стоять до достижения комнатной температуры и перемешивают в течение ночи. К смеси добавляют воду и растворители удаляют в вакууме. Экстрагируют теплым этилацетатом, сгущают в вакууме и очищают путем хроматографии на наполненой силикагелем колонке, используя в качестве элюента этилацетат. Получают 0,05 г чистого 11-циклопропил-5,11-дигидро-5-гидрокси-4-метил-6Н-дипиридо[3,2-b: 2', 3'-e] [1, 4] диазепин-6-она с точкой плавления 239-241оС. Выход: 9,5% теории.
П р и м е р 14. 5,11-Дигиро-11-этил-2-метокси-5-метил-6Н-дипиридо[3,2-b: 2',3'-e][1,4]-диазеп ин-6-он
а) 3-Амино-2-бром-6-метоксипиридин
К раствору 2,5 г 5-амино-2-метоксипиридина в 15 мл уксусной кислоты добавляют 1,6 г ацетата натрия. К получаемому раствору каплями добавляют 3,0 г брома. Смесь перемешивают в течение 20 мин, затем ее добавляют к раствору 10 г гидроокиси натрия в 100 мл воды. Экстрагируют 50 мл этилацетата, сушат над безводным сульфатом магния и сгущают в вакууме. Полученный продукт очищают путем хроматографии на наполненной силикагелем колонке, используя в качестев элюента смесь этилацетата и гексана в соотношении 4:1. Получают 2,7 г желаемого соединения, пригодного для использования на следующей стадии.
б) N-(2-Бром-6-метокси-3-пиридинил)-2-хлор-3-пиридинкарбоксамид
К раствору 2,7 г 3-амино-2-бром-6-метоксипиридина в 20 мл метиленхлорида и 1 мл пиридина добавляют 2,2 г 2-хлорнико- тионилхлорида, и полученную смесь размешивают в течение 20 мин. Затем смесь разбавляют 100 мл метиленхлорида, промывают 100 мл воды, сушат над безводным сульфатом магния и сгущают. Полутвердый остаток насыщают гексаном, фильтруют и сушат, в результате чего получают 4,1 г целевого продукта, пригодного для использования на следующей стадии.
в) N-(2-Бром-6-метокси-3-пиридинил)-2-хлор-N-метил-3-пиридинкарбоксамид
К 10 мл диметилсульфоксида добавляють 0,3 г 50%-ной дисперсии гидрида натрия в минеральном масле и нагревают до 50оС. После охлаждения до комнатной температуры добавляют 2,0 г N-(2-бром-6-метокси-3-пиридинил)-2-хлор-3-пиридинкар-боксамида, и полученный раствор перемешивают в течение 10 мин. Добавляют 0,4 мл иодистого метила и размешивают в течение 30 мин. К реакционной смеси добавляют 10 мл воды и затем 100 мл этилацетата. Органическую фазу четыре раза промывают 100 мл воды, сушат над безводным сульфатом магния, сгущают и очищают путем хроматографии на наполненной силикагелем колонке, используя в качестве элюента сперва метиленхлорид и затем смесь метиленхлорида и этанола в соотношении 98:2. Получают 1,9 г желаемого соединения, пригодного для использования на следующей стадии.
г) N-(2-Бром-6-метокси-3-пиридинил)-2-этиламино-N-метил-3-пиридинкарбоксамид
К раствору 1,9 г N-(2-бром-6-метокси-3-пиридинил)-2-хлор-N-метил-3-пиридинкар- боксамида в 5 мл ксилола добавляют 0,7 г этиламина. Смесь подают в автоклав, закрывают и нагревают при температуре 150оС в течение 4 ч. Затем раствор разбавляют этилацетатом, промывают водой, сушат над безводным сульфатом магния, сгущаюти и очищают путем хроматографии на наполненой силикагелем колонке (элюент смесь этилацетата и гексана в соотношении 1:4). Получают 1,5 г желаемого соединения, пригодного для использования на следующей стадии.
д) 5,11-Дигидро-11-этил-2-метокси-5-метил-6Н-дипиридо[3,2-b: 2', 3'-e] [1,4]диа-зе пин-
К раствору 1,4 г N-(2-бром-6-метокси-3-пиридинил)-2-этиламино-N-метил-3-пири-динкарбоксамида в 20 мл ксилола добавляют 0,9 г 50%-ной дисперсии гидрида натрия в минеральном масле, и полученную смесь нагревают с обратным холодильником в течение 2 ч. После охлаждения к смеси добавляют метанол, разбавляют этилацетатом и промывают водой. Органическую фазу сушат над безводным сульфатом магния, сгущают и очищают путем хроматографии на наполненной силикагелем колонке, используя в качестве элюента смесь этилацетата и гексана в соотношении 1:4. Получают довольно чистое соединение, которое дважды перекристаллизовывают из смеси этилацетата и гексана и получают 0,52 г чистого 5,11-дигидро-11-этил-2-метокси-5-метил-6Н- дипиридо[3,2-b:2',3'-e] [1,4]диазепин-6-она с точкой плавления 116-118оС.
П р и м е р 15. 5,11-Дигидро-11-этил-2-(N-пирролидино)-6Н-дипиридо[3,2-b:2',3'-e] [1,4]диазепин-6-он
а) 5,11-Дигидро-11-этил-2-окси-5-метил-6Н-дипиридо[3,2-b:2',3'-e][1,4] диазепин- 6-он
К раствору 0,3 г 5,11-дигидро-11-этил-2-метокси-5-метил-6Н-дипиридо[3,2-b:2,'3'-e][1,4]диазеп ин-6-она в 2 мл уксусной кислоты добавляют 2 мл 48% -ной бромистоводородной кислоты, и полученную смесь быстро нагревают и кипятят с обратным холодильником в течение 5 мин. К реакционной смеси добавляют 10 мл 10%-ной гидроокиси натрия, и продукт экстрагируют этилацетатом, сушат над безводным сульфатом магния и сгущают. Получают твердое вещество, которое перекристаллизовывают из этилацетата, в результате чего получают 0,08 г желаемого соединения с точкой плавления 215-218оС.
б) 5,11-Дигидро-11-этил-5-метил-2-трифторметансульфонилгидрокси-6Н-дипи- ридо[3,2-b:2',3'-e][1,4]диазепин-6-он
К раствору 0,2 г 5,11-дигидро-11-этил-2-гидрокси-5-метил-6Н-дипиридо[3,2-b: 2', 3'- e] [1,4] диазепин-6-она в 4 мл метиленхлорида в атмосфере азота добавляют 0,2 мл диизопропилэтиламина и затем 0,2 мл ангидрида трифторметансульфоновой кислоты. Полученную смесь перемешивают в течение часа, затем разбавляют 20 мл метиленхлорида и промывают водой. Органическую фазу сушат над безводным сульфатом магния и сгущают. В результате хроматографии на наполненной силикагелем колонке с использованием в качестве элюента смеси этилацетата и гексана в соотношении 1:3 получают довольно чистое соединение, пригодное для использования на следующей стадии.
в) 5,11-Дигидро-11-этил-5-метил-2-(N-пирролидино)-6Н-дипиридо-[3,2-b:2', 3'-e]-[ 1,4]диазепин-6-он
0,25 г 5,11-дигидро-11-этил-5-метил-2-трифторметансульфонилгидрокси-6Н-дипи- ридо[3,2-b:2',3'-e][1,4]диазепин-6-она растворяют в 1 мл пиридина и нагревают с обратным холодильником в течение 30 мин. Охлажденный раствор разбавляют этилацетатом, промывают водой, и органическую фазу сушат над безводным сульфатом магния и сгущают. Полученный маслянистый остаток выкристаллизовывают из смеси этилацетата и гексана и получают 0,11 г 5,11-дигидро-11-этил-5-метил-2-N-(пирролиди-но)-6Н-дипиридо[3,2-b: 2', 3'-e][14]диазе- пин-6-она с точкой плавления 185-188оС.
П р и м е р 16. 5,11-Дигидро-11-этил-2-метокси-4-метил-6Н-дипиридо[3,2-b:2',3'-e][1,4]диазеп ин-6-он
а) 2-Метокси-4-метил-5-нитропиридин
К раствору 19,0 г 2-хлор-4-метил-5-нитропиридина в 100 мл метанола добавляют 26,1 г метилата натрия, и полученную смесь нагревают с обратным холодильником в течение 12 ч. После охлаждения смесь выливают в 1 л воды и экстрагируют этилацетатом и промывают водой. Органическую фазу сушат над безводным сульфатом магния и сгущают, и полученный остаток растворяют в горячем простом эфире и фильтруют. Путем перекристаллизации из простого эфира получают 10,2 г желаемого соединения, пригодного для использования на следующей стадии.
б) 5-Амино-2-метокси-4-метилпиридин
К раствору 5,1 г 2-метокси-4-метил-5-нитропиридина в 40 мл уксусной кислоты медленно добавляют 41 г дигидрата хлорида олова и 40 мл концентрированной хлористоводородной кислоты, поддерживая температуру ниже 35оС. Полученную смесь перемешивают при комнатной температуре в течение 2 ч, затем оставляют стоять в холодильнике в течение ночи. Твердое вещество собирают, твердое вещество и надосадочную жидкость отдельно подщелачивают добавлением 20% -ного раствора гидроокиси натрия. Экстрагиурют хлороформом, объединяют, сушат над безводным сульфатом магния и сгущают. В результате получают 3,9 г желаемого соединения в виде твердого вещества, пригодного для использования на следующей стадии.
в) 3-Амино-2-бром-6-метокси-4-метилпиридин
К смеси 3,9 г 5-амино-2-метокси-4-метилпиридина в 25 мл уксусной кислоты и 4,0 г ацетата натрия добавляют 4,8 г брома. Полученную смесь перемешивают в течение 20 мин, затем ее добавляют к раствору 15 г гидрокси натрия в 200 мл воды. Экстрагирют хлороформом, сушат над безводным сульфатом магния, сгущают и очищают путем хроматографии на наполненной силикагелем колонке, используя в качестве элюента смесь метиленхлорида и этилацетата в соотношении от 19: 1 до 4:1 в результате чего получают 4,5 г соединения, пригодного для использования на следющей стадии.
г) N-(2-Бром-6-метокси-4-метил-3-пиридилин)-2-хлор-3-пиридинкарбоксамид
К раствору 4,5 г 3-амино-2-бром-6-метокси-4-метилпиридина в метиленхлориде добавляют 3,5 г 2-хлорникотионилхлорида. Полученную смесь перемешивают при комнатной температуре в течение ночи и растирают с простым диизопропиловым эфиром. Твердый осадок фильтруют, получают 6,0 г желаемого соединения, пригодного для использования на следующей стадии.
д) N-(2-Бром-6-метокси-4-метил-3-пиридинил)-2-этиламино-3-пиридинкарбокс- амид
Смесь 2,1 г N-(2-бром-6-метокси-4-метил-3-пиридинил)-2-хлор-3-пиридинкарбо-ксамида, 10 мл диоксана и 0,5 г этиламина нагревают в автоклаве при температуре 140оС в течение 5 ч. Охлажденную смесь разбавляют этилацетатом, промывают водой, и органическую фазу сушат над безводным сульфатом магния и сгущают. Полученное соединение очищают путем хроматографии на наполненной силикагелем колонке с использованием в качестве элюента смеси метиленхлорида и этилацетата в соотношении 99:1 и выкристаллизовывают из простого диизопропилового эфира. Получают 0,95 г желаемого соединения.
е) 5,11-Дигидро-11-этил-2-метокси-4-метил-6Н-дипиридо[3,2-b: 2', 3'-e] [1,4]диа-зе пин-
К раствору 0,54 г N-(2-бром-6-метокси-4-метил-3-пиридинил)-2-этиламино-3-пири- динкарбоксамида в 4 мл пиридина добавляют 0,14 г 50%-ной дисперсии гидрида натрия в минеральном масле. Полученную смесь нагревают с обратным холодильнигком в течение 1,5 ч. Охлажденную смесь разбавляют этилацетатом, промывают водой, и органическую фазу сушат над безводным сульфатом магния и сгущают. Остаток промывают простым диизопропиловым эфиром и горячим этилацетатом и затем выкристаллизовывают из этанола, в результате чего получают 0,2 г 5,11-дигиро-11- этил-2-метокси-4-метил-6Н-дипиридо[3,2-b:2', 3'-e][1,4]диазепин-6-она с точкой плавления 249-251оС.
Аналогично описанным выше методом получают приведенные в табл. 1 соединения примеров 17-122.
П р и м е р 123. Гемигидрат 8-амино-5,11-дигидро-11-этил-5-метил-6Н-дипири-до[3,2-b:2',3'-e][1,4]диазепи н-6-она
а) 2-Этиламино-3-нитропиридин
Размешанную смесь 8,60 г (0,054 моль) 2-хлор-3-нитропиридина, 5,37 г (0,12 моль) этиламина и 10 мл ксилола нагревают в автоклаве при температуре 100оС в течение 3 ч. После охлаждения растворитель удаляют в вакууме и к остатку добавляют воду. Экстрагирют метиленхлоридом, сушат над сульфатом натрия и сгущают в вакууме. Получают 10,0 г желаемого соединения в виде желтого масла, пригодного для использования на следующей стадии.
б) 3-Амино-2-этиламинопиридин
Аналогично примеру 11б) из 9,1 г 2-этиламино-3-нитропиридина получают 6,5 г вышеуказаного соединения.
в) 2-Хлор-N-(2-этиламино-3-пиридинил)-5-нитро-3-пиридинкарбоксамид
Раствор 2,21 г 2-хлор-5-нитроникотионилхлорида (полученного тем, что нитруют 2-оксиникотиновую кислоту с последующим переводом в 2-хлор-5-нитроникотиновую кислоту, которую затем обрабатывают тионилхлоридом) в 10 мл тетрагидрофурана в течение 15 мин медленно добавляют к охлаждаемой и размешиваемой смесим 1,34 г 3-амино-2-этиламинопиридина, 1,29 г диизопропилэтиламина и 40 мл тетрагидрофурана. Полученную смесь перемешивают при комнатной температуре в течение ночи, затем сгущают в вакууме. Путем обработки метиленхлоридом получают 2,30 г желаемого соединения в виде осадка, пригодного для использования на следующей стадии. Т.пл. 185-186оС.
г) 5,11-Дигидро-11-этил-8-нитро-6Н-дипиридо[3,2-b:2',3'-e][1,4]диазепин-6-он
Раствор 1,80 г 2-хлор-N-(2-этиламино-3-пиридинил)-5-нитро-3-пиридинкарбоксами- да в 25 мл ксилола нагревают с обратным холодильником в течение 4 ч. После сгущения в вакууме остаток очищают путем хроматографии на наполненной силикагелем колонке, используя в качестве элюента 50%-ную смесь этилацетата и гексана. Получают 0,93 г желаемого соединения.
д) 5,11-Дигидро-11-этил-5-метил-8-нитро-6Н-дипиридо[3,2-b:2',3'-e][1,4] диазепин 6-он
0,72 г вышеуказанного соединения, имеющего точку плавления 148-149оС, получают аналогично примеру 8г) из 0,93 г 5,11-дигидро-11-этил-8-нитро-6Н-дипиридо[3,2- b:2',3'-e][1,4]диазепин-6-она.
е) Гемигидрат 8-амино-5,11-дигидро-11-этил-5-метил-6Н-дипиридо[3,2-b:2', 3'-e] [1,4]диазепин-6-она
Аналогично примеру 11б) из 0,23 г 5,11-дигидро-11-этил-5-метил-8-нитро-6Н-дипи- ридо[3,2-b:2',3'-e][1,4]диазепин-6-она после перекристаллизации из смеси 1,2-дихлорэтана и гексана получают 0,060 г целевого соединения в виде желто-коричневого порошка, имеющего точку плавления 193-194оС.
П р и м е р 124. 6-Цианоимино-5,11-дигиро-11-этил-2,5-диметил-6Н-дипиридо- [3,2- b:2',3'-e][1,4]диазепин
Смесь 0,25 г (0,63 ммоль) 5,11-дигидро-11-этил-6-метансульфонилгидрокси-2,4-ди- метил-6Н-дипиридо[3,2-b: 2', 3'-e] [1,4]диа-зепина, 0,034 г (0,8 ммоль) цианамида, 5 мл 1,4-диоксана и 0,11 г (0,8 ммоль) карбоната калия нагревают при комнатной температуре в течение 10 дней. Затем смесь сгущают в вакууме, остаток разделяют между этилацетатом и водой. Органическую фазу сушат, фильтруют и сгущают в качестве элюента 10%-ной смеси этилацетата и метиленхлорида. Получают 0,025 г целевого соединения с точкой плавления 230-233оС.
П р и м е р 125. 5,11-Дигидро-11-этил-6-метоксиимино-2,4-диметил-6Н-дипиридо- [3,2-b:2',3'-e][1,4]диазепин
а) 5,11-Дигидро-11-этил-6-метансульфонилгидрокси-2,4-диметил-6Н-дипиридо- [3,2-b:2',3'-e][1,4]диазепин
К раствору 0,314 г (1,2 ммоль) 5,11-дигидро-11-этил-2,4-диметил-6Н-дипиридо [3,2-b:2',3'-e][1,4]диазепин-6-она в 15 мл метиленхлорида, содержащему 0,25 мл (14 ммоль) диизопропилэтиламина, добавляют 0,24 мл (14 ммоль) ангидрида трифторметансульфоновой кислоты. Полученную смесь в атмосфере аргона нагревают с обратным холодильником в течение 3 ч. Затем добавляют этилацетат (примерно 200 мл) и раствор трижды промывают водой и четыре раза соляным раствором. Сушат над сульфатом магния, затем раствор сгущают в вакууме, и остаток сушат в высоком вакууме в течение 2 ч. Остаток растворяют в 20 мл метиленхлорида, добавляют 0,23 г (14 моль) цианида тетраэтиламмония. Полученный раствор перемешивают при комнатной температуре в течение ночи, затем реакционную смесь сгущают в вакууме. Остаток растворяют в 100 мл этилацетата, и раствор промывают водой и соляным раствором. Раствор затем сушат над сульфатом магния и сгущают в вакууме. Остаток подвергают хроматографии на силикагеле с использованием в качестве элюента 5%-ной смеси этилацетата и гексана. Полученное твердое вещество выкристаллизовывают из гептана и получают 0,033 г желаемого соединения в виде красных кристаллов с точкой плавления 154-155оС.
б) 5,11-Дигиро-11-этил-6-метоксиимино-2,4-диметил-6Н-дипиридо-[3,2-b:2', 3'-e] [1,4]диазепин
Раствор 0,3 г (0,75 ммоль) 5,11-дигидро-11-этил-6-метансульфонилгидрокси-2,4-ди- метил-6Н-дипиридо[3,2-b: 2',3'-e][1,4]диа-зепина, 0,15 г (1,8 ммоль) гидрохлорида метоксиламина и 0,3 г (2 ммоль) диизопропилэтиламина в метиленхлориде размешивают при комнатной температуре в течение 4 дней. Органическую фазу промывают водой, сушат и фильтруют. Затем раствор сгущают в вакууме, и остаток подвергают хроматографии на силикагеле с использованием в качестве элюента 20%-ной смеси этилацетата и гексана. Получают 0,7 г целевого продукта с точкой плавления 164-166оС.
П р и м е р 126. 5,11-Дигидро-6Н-11-циклопропил-4-метил-дипиридо[3,2-b: 2',3'- e][1,4]диазепин-6-тион
Смесь 5,0 г (18,77 ммоль) 5,11-дигидро-6Н-11-циклопропил-4-метил-дипиридо[3,2- b: 2',3'-e][1,4]диазепин-6-она и 3,8 г (9,40 ммоль) димерного р-метоксифенилтиенофосфин-сульфида (агент Лавессона) в 100 мл толуола нагревают с обратным холодильником в течение 2,5 ч. Раствор охлаждают до комнатной температуры и оставляют стоять в течение ночи. Толуол удаляют путем перегонки, и остаток подвергают хроматографии на наполненной силикагелем колонке (элюент смесь метиленхлорида и этилацетата в соотношении 6 1), в результате чего получают желтое масло, которое твердеет, когда его оставляют стоять. После перекристаллизации из смеси простого этилового эфира и простого петролейного эфира получают ярко-желтое твердое вещество, которое сушат в высоком вакууме при температуре 80оС в течение 12 ч. Выход: 1,7 г (32,0% теории), т. пл. 189-194оС.
Как уже указывалось, соединения формулы (I) обладают тормозящим действием в отношении обратной транскриптазы вируса HIV-1. Из опытов, проведенных с данными соединениями, известно, что они тормозят активность обратной транскриптазы HIV, зависящей от РНК ДНК-полимеразы.
Опыт с применением обратной транскриптазы
Теория опыта.
Среди энзимов, курирующих вирус иммунодефицита человека НIV-1, находится и обратная транскриптаза (1), названная так потому, что она транскрибирует копию ДНК с матрицей РНК. Ее активность можно определять количественно в рамках опыта (2) с применением энзима без клеток, основанного на наблюдении способности обратной транскриптазы использовать синтетическую матрицу [поли r(C), снабженный олиго d(G)] для транскриптазы радиомаркированной, поддающейся осаждению кислотой однонитьевой ДНК, причем 3Н-dGTР используется в качестве субстрата.
Материалы:
а) Выделение энзима
Обратная транскриптаза из штамма LAV вируса иммунодефицита человека НIV-1 (1) выделяется из штамма бактерии JM109 (3), экспримирующей клон рBRTprtl + ДНК, контролироуемый промотором lac в экспрессионном векторе рIB121 (4). Культивированную в среде 2ХYT (при 37оС и 225 об/мин) (5) в течение ночи культуру, в которую в целях положительной селекции подают 100 мкг/мл ампициллина (5), инокулируют при разбавлении 1:40 в среде М9, содержащей 10 мкг/мл тиамина, 0,5% казаминокислот и 50 мкг/мл ампициллина. Культуру инкубируют при температуре 37оС при 225 об/мин до достижения оптической плотности 0,3-0,4 при 540. В этот момент добавляют репрессорный ингибитор изопропил- β-D-тиогалактопираносид в количестве 0,5 ммоль и продолжают инкубировать в теченеие 2 ч, после чего сгущают и остаток повторно суспендируют в 50 ммоль трис-буфера, содержащего 0,6 ммоль этиледиаминотетрауксусной кислоты и 0,375 моль хлорида натрия и добавлением лизозима в количестве 1 мл на мл переваривают в течение 30 мин на льду. Клетки растворяют подачей в 0,2% поверхностно-активного вещества NP-40, а затем подают в 1 М хлорида натрия.
После удаления нерастворимых нежелательных компонентов путем центрифугирования осаждают белок путем добавления 3 объемов насыщенного водного сульфата аммония, после чего сгущают и остаток повторно суспендируют в буфере обратной транскриптазы (50 ммоль трис, рН 7,5, 1 ммоль этилендиаминотетрауксусной кислоты, 5 моль ДТТ (1,4-димеркапто-2,3-бутандиола), 0,1% поверхностно-активного вещества NP-40, 0,1 моль хлорида натрия и 50% глицерина), и хранят при температуре -70оС до использования.
б) Состав исходной реакционной cмеcи Компоненты Содержание
компонентов 1 М Трис (рН 7,4) 100 ммоль 1 М ДТТ 40 ммоль 1 М хлорида натрия 120 ммоль 1% -ное поверхностно- активное вещество NP-40 0,1% 1 М хлорида магния 4 ммоль [поли r(C)/олиго d(G)] (5:1) 2 мкг/мл 3Н-dGTP (8 мкМ) 0,6 мкмоль
Осуществление опыта.
Аликвоты реакционной смеси хранят при температуре -20оС. Смесь является стабильной, и каждый раз для использования ее размораживают. В известном опыте (6) с применением энзима используют систему микротитрующих плит с 96 углублениями. 50 ммоль трис (рН 7,4), носитель (растворитель для разбавления исследуемых соединений), или исследуемые соединения в носителе подают в микротитрующие плиты с 96 углублениями (10 мкл на углубление, 3 углубления на исследуемое соединение). Размораживают обратную транскриптазу HIV, разбавляют ее в 50 ммоль трис (рН 7,4), так что 15 мкл разбавленного энзима содеражт 0,001 единицу (1 единица то количество энзима, которое трансформирует 1 мкмоль субстрата в минуту при температуре 25оС), и подают 15 мкл в каждое углубление. Добавляют 20 мкл 0,12-0,5 М этилендиаминотетрауксусной кислоты к первым трем углублениям микротитрующей плиты. Этилендиаминотетрауксусная кислота образует хелатный комплекс с магниевыми ионами и предотвращает обратную транскрипцию. 25 мкл указанной реакционной смеси добавляют во все углубления и дают инкубироваться при комнатной температуре в течение часа. Опыт заканчивают путем осаждения ДНК в каждом углублении в результате 50 мкл 10%-ной трифторуксусной кислотой с 1% пирофосфатом натрия. Микротитрующую плиту инкубируют при температуре 4оС в течение 15 мин, и осадок фиксируют на стекловолокнистой бумаге 30 (фирмы Schleicher Schuell) с помощью полуавтоматического аппарата Skatron. Затем фильтры промывают еще 5%-ной трихлоруксусной кислотой с 1%-ным пирофосфатом натрия, промывают 70%-ным водным этанолом, сушат и подают в трубки для сцинтилляции (6). В каждую трубку подают 2 мл сцинтиллирующего средства; подсчет осуществляют в счетчике бета-частиц Beckman.
Процент торможения вычисляли по следующему уравнению:

 → срм число отсчетов в минуту
→ срм число отсчетов в минуту
Результаты опыта сведены в табл. 2.
В литературе не описаны соединения аналогичной структуры, которые обладают таким тормозящим действием на обратную транскриптазу вируса HIV-1, что позволяет применять их для профилактики или лечения СПИДа и болезней, связанных с инфекцией вирусом HIV.
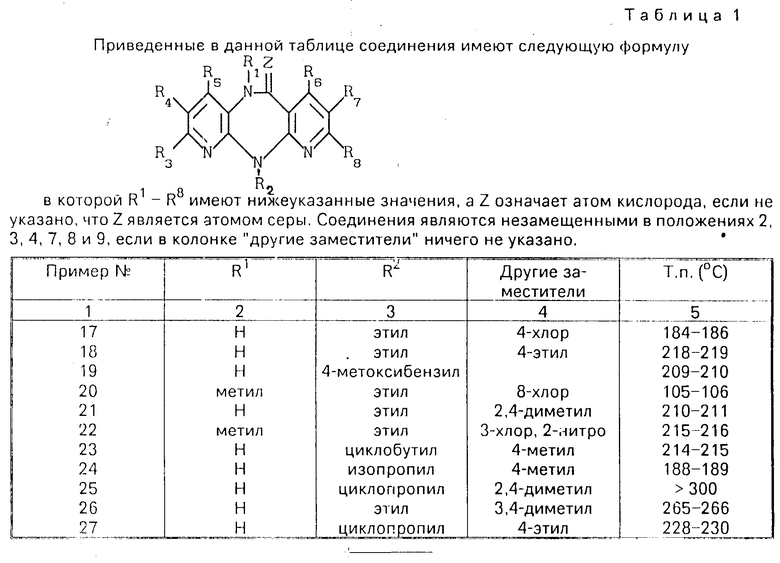


Использование: в фармакологии, в частности в качестве биологически активных веществ, тормозящих действие на обратную транскриптазу вируса HIV-1. Сущность изобретения: продукт производные дипиридо-диазепина и их гидраты ф-лы I (см.чертеж), где Z кислород, сера, группа NCN или -NOR9 при R9 -алкил; R1 водород, гидроксил, алкил, алкенил, алкенилоксикарбонил, алкокси, алканоил, диалкиламиноэтил, алкоксиалкил, алкилтиоалкил, бензил; R2 водород, алкил, фторалкил, циклоалкил, циклоалкилалкил, алкенил, алкинил, алкоксиалкил, алкилтиоалкил, алканоил, циано, фенил, бензил, алкоксибензил, метилсульфонил; R3 водород, гидроксил, галоид, цитро, алкил, алкокси, амино, моно- или диалкиламино, алкениламино, пирролидин-1-ил, пирролин-1-ил, тетрагидропиридин-1-ил, морфолин-1-ил, пиперидин-1-ил, метоксибензилметиламино, метоксибензиламино; R4 водород, галоид, алкил, нитро, амино; R5 водород, гидроксил, галоид, алкил, алкокси, тригалоидметил, оксиалкил, циано; R6 водород, гидроксил, алкил; R7 водород, галоид, азидо, нитро, амино, алкил; R8 водород, алкил, причем во всех упомянутых алкилах он является низшим. 1 з.п. ф-лы, 1 ил. 2 табл.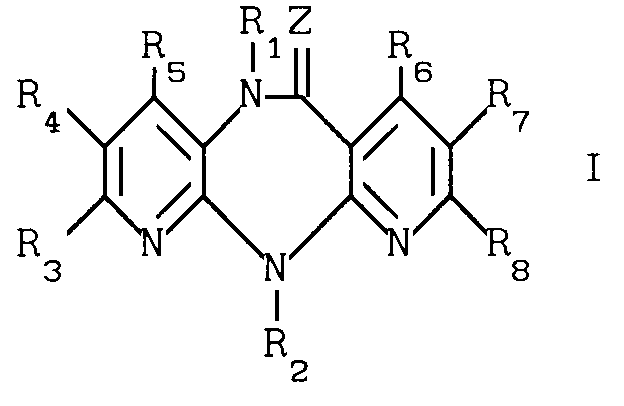
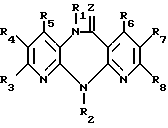
где Z кислород, сера, группы NCN и -NOR9, где R9 низший алкил;
R1 водород, гидроксил, низший алкил, низший алкенил, низший алкенилоксикарбонил, низший алкоксил, низший алканоил, низший диалкиламиноэтил, низший алкоксиалкил, низший алкилтиоалкил, бензил;
R2 водород, низший алкил, низший фторалкил, низший циклоалкил, низший циклоалкилалкил, низший алкенил, низший алкинил, низший алкоксиалкил, низший алкилтиоалкил, низший алканоил, цианогруппа, фенил, бензил, низший алкоксибензил, метилсульфонил;
R3 водород, гидроксил, галоид, нитро, низший алкил, низший алкокси, амино, низший моно- или диалкиламино, низший алкениламино, пирролидин-1-ил, пирролин-1-ил, тетрагидропиридин-1-ил, морфолин-1-ил, пиперидин-1-ил, метоксибензилметиламино, метоксибензиламино;
R4 водород, галоид, низший алкил, нитро, амино;
R5 водород, гидроксил, галоид, низший алкил, низший алкокси, тригалоидметил, низший оксиалкил, циано;
R6 водород, гидроксил, низший алкил;
R7 водород, галоид, азидо, нитро, амино, низший алкил;
R8 водород, низший алкил;
причем, если Z кислород или сера, R2 водород, низший алкил, низший алкенил, низший алкинил, низший алкоксиалкил, низший алкилтиоалкил, низший алканоил, фенил, бензил, низший алкоксибензил, R3, R4, R5, R6, R7 и R8 водород, или один из заместителей R3, R4, R5, R6, R7 и R8 низший алкил, а остальные заместители водород, или один из заместителей R3, R4, R5, R7 галоид, а остальные заместители R6 и R8 - водород, или один из заместителей R3, R4 и R7 означает нитро, а остальные заместители R5, R6 и R8 водород, или один из заместителей R3, R5 и R6 гидроксил, а остальные заместители R4, R7 и R8 водород, или один из заместителей R3, R4 и R7 амино, а остальные заместители R5, R6 и R8 водород, или один из заместителей R3 и R5 - алкоксил, а остальные заместители R4, R6, R7 и R8 - водород, или R5 низший оксиалкил или циано, а R3, R4, R6, R7, и R8 водород, или R7 азидо, а R3, R4, R5, R6 и R8 водород, или если R3, R4 и R5 независимо друг от друга водород или низший алкил при условии, что по меньшей мере один из них означает водород, или один из заместителей R3, R4 и R5 бутил, а остальные заместители R6, R7 и R8 водород, или, наоборот, R6, R7 и R8 независимо друг от друга водород или низший алкил при условии, что по меньшей мере один из них означает водород, или один из заместителей R6, R7 и R8 бутил, а остальные заместители R3, R4 и R5 водород, то R1 не может быть водород, низший алкил, низший алкенил, бензил, низший алканоил, низший алкоксиалкил, низший алкилтиоалкил и низший алкоксил,
и их гидраты, обладающие биологической активностью.
Приоритет по признакам:
17.11.89 при Z кислород, сера, группы NCN и -NOR9, где R9 низший алкил, R1 водород, низший алкил, низший алкенил, низший алканоил, бензоил, R2 низший алкил, низший фторалкил, низший циклоалкил, низший алкенил, низший алкинил, низший алкоксиалкил, низший алкилтиоалкил, фенил, бензил, низший алкоксибензил, R3 водород, галоид, нитро, низший алкил, низший алкокси, амино, низший моно- или диалкиламино, низший алкениламино, R4 водород, галоид, низший алкил, нитро, амино, R5 водород, галоид, низший алкил, низший алкоксил, тригалоидметил, низший оксиалкил, циано, R6 водород, низший алкил, R7 водород, амино, низший алкил, R8 водород, низший алкил;
06.09.90 при R1 гидроксил, низший алкенилоксикарбонил, низший алкоксил, низший диалкиламиноэтил, низший алкоксиалкил, низший алкилтиоалкил, бензил, R2 низший алканоил, R3 гидроксил, пирролидин-1-ил, пирролин-1-ил, тетрагидропирридин-1-ил, морфолин-1-ил, пиперидин-1-ил, метоксибензилметиламино, метоксибензиламино, R5 гидроксил, R6 - гидроксил, R7 галоид, азидо, нитро; 19.10.90 при R2 - цианогруппа;
16.11.90 при R2 низший циклоалкилалкил, метилсульфонил, если Z кислород или сера, R2 водород, низший алкил, низший алкенил, низший алкинил, низший алкоксиалкил, низший алкилтиоалкил, низший алканоил, фенил, бензил, низший алкоксибензил, R3, R4, R5, R6, R7 и R8 водород, или один из заместителей R3, R4, R5, R6, R7 и R8 низший алкил, а остальные заместители - водород, или один из заместителей R3, R4, R5 и R7 - галоид, а остальные заместители R6 и R8 водород, или один из заместителей R3, R4 и R7 нитро, а остальные заместители R5, R6 и R8 водород, или один из заместителей R3, R5 и R6 гидроксил, а остальные заместители R4, R7 и R8 водород, или один из заместителей R3, R4 и R7 амино, а остальные заместители R5, R6 и R8 водород, или один из заместителей R3 и R5 алкоксил, а остальные заместители R4, R6, R7 и R8 водород, или R5 низший оксиалкил или циано, а R3, R4, R6, R7 и R8 - водород, или R7 азидо, а R3, R4, R5, R6 и R8 водород, или если R3, R4 и R5 независимо друг от друга водород или низший алкил при условии, что по меньшей мере один из них означает водород, или один из заместителей R3, R4 и R5 - бутил, а остальные заместители R6, R7 и R8 водород, или, наоборот, R6, R7 и R8 независимо друг от друга водород или низший алкил при условии, что по меньшей мере один из них означает водород, или один из заместителей R6, R7 и R8 бутил, а также остальные заместители R3, R4 и R5 водород, то R1 не может быть водород, низший алкил, низший алкенил, бензил, низший алканоил, низший алкоксиалкил, низший алкилтиоалкил и низший алкоксил.
| БЛОК ДЛЯ ПОДВЕСКИ АНТЕННЫ | 0 |
|
SU344543A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
