Целью изобретения является получение нового соединения и его терапевтически приемлемых солей, которые ингибируют экзогенно и эндогенно стимулированную секрецию желудочной кислоты и поэтому могут быть использованы в предотвращении лечении пептической язвы.
Предлагаемое изобретение также относится к использованию соединения, особенно его терапевтически приемлемых солей, для ингибирования секреции желудочной кислоты у млекопитающих, включая человека. В более общем смысле, предлагаемое соединение может быть использовано для предотвращения и лечения желудочно-кишечных воспалительных заболеваний и заболеваний, опосредованных желудочной кислотой, у млекопитающих, включая человека, таких как гастрит, желудочная язва, дуоденальная язва, рефлюкс-эзофагит и синдром Золлингера-Эллисона. Кроме того, предлагаемое соединение может быть использовано для лечения других желудочно-кишечных расстройств, когда желателен желудочный антисекреторный эффект, например, у пациентов с ульцерогенной аденомой поджелудочной железы и у пациентов с острым верхним желудочно-кишечным кровотечением. Оно также может быть использовано при интенсивной терапии, а также пре- и постоперационно для предотвращения кислотной аспирации и образования язвы. Предлагаемое соединение также может быть использовано для лечения или профилактики воспалительных состояний у млекопитающих, включая человека, особенно тех, которые имеют лизоцимальные ферменты. Особо следует отметить такие состояния, как ревматический артрит и подагра. Соединение также может оказаться полезным при лечении заболеваний, связанных с нарушениями костного обмена веществ, а также при лечении глаукомы.
Предлагаемое изобретение также касается фармацевтических композиций, содержащих соединение изобретения или его терапевтически приемлемые соли в качестве активного компонента.
В другом варианте предлагаемое изобретение относится к способам получения такого нового соединения и к использованию активного соединения для получения фармацевтических композиций, предназначенных для указанного выше медицинского применения.
Главной целью изобретения является получение соединения с высоким уровнем биологической доступности. Предлагаемое соединение также должно проявлять свойства высокой устойчивости при нейтральном рН и высокой потенции относительно ингибирования секреции желудочной кислоты. Биодоступность определяется как фракция, или процентное содержание вводимой дозы соединения, которое абсорбируется неизменяемым в общей крови. Потенцию в данной заявке определяют как значение ЭД50.
В многочисленных патентных документах раскрываются бензимидазолпроизводные, предназначенные для игибирования секреции желудочной кислоты. Среди них можно упомянуть патенты Великобритании NN 1500043 и 1525958, патенты США NN 4182766, 4255431 и 4599347, Европатент N 124495, патенты США NN 4555518, 4727150 и 4628098, Европатент N 208452 и реферат Дервент N 87-294449/42. В патенте США N 4539465 описываются бензимидазолпроизводные, предложенные для использования при лечении или предотвращении специальных желудочно-кишечных воспалительных заболеваний.
Соединения, описанные в известных технических решениях, являются эффективными ингибиторами секреции кислоты и поэтому пригодны в качестве противоязвенных соединений. С целью дальнейшего повышения полезности данного типа лекарственных веществ желательна более высокая биологическая ценность. Вместе с тем соединения должны иметь высокую потенцию в ингибировании секреции желудочной кислоты, а также высокую химическую устойчивость при нейтральном рН.
Установлено, что 2-[(2-пиридилметил)сульфинил]-1Н-бензимидазолы проявляют большую вариабильность в биологической доступности, а также в потенции и устойчивости, и очень трудно идентифицировать соединения, обладающие всеми тремя благоприятными свойствами. В известном уровне техники нет руководства относительно получения соединений с данной комбинацией свойств.
Обнаружено, что предлагаемое соединение имеет чрезвычайно высокую биологическую доступность, причем соединение весьма эффективно в качестве ингибитора секреции желудочной кислоты и проявляет высокую химическую устойчивость в растворе при нейтральном рН. Поэтому предлагаемое соединение может быть использовано в указанных выше ситуациях у млекопитающих, включая человека.
Предлагаемое соединение представляет собой 4-фтор-2-{[4-метокси-2-пиридинил)метил]сульфонил}-1Н-бензимидазол (соединение I), а также его физиологически приемлемые соли.
Предлагаемое соединение имеет асимметрический центр в атоме серы, т.е. существует как два оптических изомера (энантиомера). Оба чистых энантиомера, их рацемические смеси (50% каждого энантиомера) и неравные смеси находятся в пределах объема предлагаемогого изобретения. Также в пределах объема данного изобретения находятся два синтетических промежуточных соединения и способ их получения.
Предлагаемое соединение может быть получено следующим образом.
Осуществляют окисление 4-фтор-2-{[(4-метокси-2-пиридинил)метил]тио}-1Н-бензимида-зола (соединение II) с получением предлагаемого соединения. Данное окисление можно осуществлять с использованием такого окислителя, как азотная кислота, перекись водорода (не обязательно в присутствии ванадиевых соединений), перкислоты, эфиры перкислот, озон, четырехокись азота, иодозобензол, N-галосукцинимид, 1-хлор-бензотриазол, т-бутилгипохлорид, диазабицикло-[2,2,2] -октанбромокомплекс, метапериодат натрия, диоксид селена, диоксид марганца, хромовая кислота, церийаммония нитрат, бром, хлор и сульфурилхлорид. Окисление обычно имеет место в растворителе, таком как галоидзамещенные углеводороды, спирты, простые эфиры, кетоны.
Окисление также можно осуществлять ферментативно с использованием окисляющего фермента или микробиологически с использованием пригодного микроорганизма.
В зависимости от условий процесса и исходных материалов предлагаемое соединение получают либо в нейтральной, либо в солевой форме. Как нейтральное соединение, так и его соли включены в объем предлагаемого изобретения. Так, могут быть получены основные, нейтральные или смешанные соли, а также геми, мого-, сескви- или полигидраты.
Щелочные соли предлагаемого соединения приводятся в примерах их солей с Li+, Na+, K+, Mg2+, Ca+ и N+ R4, где R обозначает С1-С4-алкил. Особенно предпочтительны соли Na+, Ca+ и Mg2+. Наиболее предпочтительны соли Na+ и Mg2+. Такие соли могут быть получены путем взаимодействия соединения с основанием, способным высвобождать требуемый катион.
Примерами оснований, способных высвобождать такие катионы, являются перечисленные ниже вместе с реакционными условиями:
а) Соли, в которых катион представляет собой Li+, Na+ или К+, получают путем обработки предлагаемого соединения LiOH, NaOH или КОН в водной или неводной среде или путем обработки LiOR, LiNR2, LiNH2, NaOH, NaNH2, NaNR2, KOR, KNH2 или KNR2, где R обозначает алкильную группу, содержащую 1-4 атома углерода, в неводной среде.
б) Соли, в которых катион представляет собой Мg2+ или Са2+, получают путем обработки предлагаемого соединения MgOR2, Ca(OR)2 или СаН2, где R обозначает алкильную группу, содержащую 1-4 атома углерода, в неводном растворителе, таком как спирт (только для алкоголятов), например ROH, или в простом эфире, таком как тетрагидрофуран.
Полученные рацематы могут быть разделены на чистые энантиомеры. Это можно осуществить известными методами, например, из рацемических диастереомерных солей при помощи хроматографии или фракционированной кристаллизации.
Исходные вещества, описанные в примерах промежуточных соединений, могут быть получены с использованием способов, известных рer Se.
Для клинического применения предлагаемое соединение составляют в фармацевстические препараты для перорального, ректального, парентерального или других путей введения. Фармацевтический препарат содержит предлагаемое соединение обычно в комбинации с фармацевтически приемлемым носителем. Носитель может быть в форме твердого, полутвердого или жидкого разбавителя или в форме капсулы. Эти фармацевтические препараты составляют дополнительный предмет предлагаемого изобретения. Обычно количество активного соединения составляет от 0,1 до 95% по массе препарата, от 0,2 до 20% по массе в препаратах для парентерального применения и от 1 до 50% по массе в препаратах для перорального введения.
При получении фармацевтических препаратов, содержащих предлагаемое соединение в форме дозированых единиц для перорального введения, выбранное соединение можно смешивать с твердым, порошкообразным носителем, таким как лактоза, сахароза, сорбит, маннит, крахмал, амилопектин, производные целлюлозы, желатина или другой пригодный носитель, стабилизирующими веществами, такими как щелочные соединения, например карбонаты, гидроксилы и оксиды натрия, калия, кальция, магния и так далее, а также со смазочными веществами, такими как стеарат магния, стеарат кальция, стеарилфумарат натрия и полиэтиленгликолевые воски. Смесь затем перерабатывают в гранулы или отжимают в таблетки. Гранулы и таблетки могут покрываться энтеросолюбильной оболочкой, которая защищает активное соединение от разрушения, катализированного кислотой, пока лекарственная форма остается в желудке. Энтеросолюбильное покрытие выбирают из числа фармацевтически приемлемых веществ для нанесения энтеросолюбильного покрытия, например пчелиного воска, щеллака или анионных пленкообразующих полимеров, таких как целлюлоза-ацетатфталат, гидроксипропил-метилцеллюлозафталат, частично метил-эстерифицированные полимеры метакриловой кислоты и так далее, если предпочтительно, в сочетании с пригодным пластификатором. В покрытие могут добавляться различные красители для разграничения таблеток или гранул с различными активными соединениями или с различными количествами активного соединения, присутствующего в фармацевтическом препарате.
Мягкие желатиновые капсулы могут быть получены с содержанием смеси активного соединения предлагаемого изобретения, растительного масла, жира или другого пригодного наполнителя для мягких желатиновых капсул. Мягкие желатиновые капсулы также могут содержать гранулы активного вещества или гранулы с энтеросолюбильной оболочкой активного соединения. Твердые желатиновые капсулы могут также содержать активное соединение в комбинации с твердым порошкообразным носителем, таким как лактоза, сахароза, сорбит, маннит, картофельный крахмал, амилопектин, производные целлюлозы или желатина. Твердые желатиновые капсулы могут быть покрыты энтеросолюбильной оболочкой как описано выше.
Лекарственные формы для ректального введения моугт быть получены в форме суппозиториев, которые содержат активное вещество, смешанное с нейтральным жировым основанием, или в форме желатиновой ректальной капсулы, которая содержит активное вещество в смеси с растительным маслом, парафиновым маслом или другим пригодным наполнителем для желатиновых ректальных капсул, или в форме готовой к употреблению микроклизмы, или в форме сухого препарата микроклизмы, который переформулируют в пригодном растворителе непосредственно перед введением.
Жидкий препарат для перорального введения может быть получен в форме сиропов или суспензий, например растворов или суспензий, содержащих от 0,2 до 20% по массе активного ингредиента, а остальное составляет смесь этанола, воды, глицерина, пропиленгликоля и/или полиэтиленгликоля. Если необходимо, такие жидкие препараты могут содержать окрашивающие вещества, вкусовые вещества, сахарин и карбоксиметилцеллюлозы, а также другие загустители. Жидкие препараты для перорального введения также могут быть получены в форме сухого порошка, переформулируемого с пригодным растворителем перед использованием.
Растворы для парентерального введения могут быть получены как раствор предлагаемого соединения в фармацевтически приемлемом растворителе, предпочтительно при концентрации от 0,1 до 10 мас. Эти растворы также могут содержать стабилизаторы и/или буферы и могут изготавливаться в различных амппулах или пробирках для лекарственных форм в стандартной дозировке. Растворы для парентерального введения также могут быть получены в виде сухого препарата, переформулируемого с пригодным растворителем непосредственно перед использованием.
Обычная суточная доза активного вещества будет варьироваться в зависимости от различных факторов, таких как например, индицидуальное требование каждого пациента, путь введения и тип заболевания. Вообще, пероральные и парентеральные дозировки составляют от 5 до 500 мг в день активного вещества.
П р и м е р 1. Получение 4-фтор-2-{[(4-метокси-2-пиридинил)метил]сульфинил}-1Н-бензимидазола.
4-фтор-2-{ [(4-метокси-2-пиридинил)ме-тил] тио}-1Н-бензимидазол (1,31 г, 0,0045 моль) растворяют в метиленхлориде (60мл). Прибавляют NaHCO3 (0,76 г, 0,0090 моль), растворенный в воде (10 мл), и смесь охлаждают до 2оС. Затем при перемешивании прибавляют по каплям 84%-ную м-хлорпербензойную кислоту (1,64 г, 0,0045 моль). Перемешивание продолжают при 2оС в течение 15 мин. После разделения органический слой экстрагируют водным 0,20 М раствором NaOH (2 25 мл, 0,010 моль). Объединенные водные растворы нейтрализуют до рН 7-8 с применением 0,1 М НСl в присутствии метиленхлорида (100 мл). После разделения водный слой экстрагируют метиленхлоридом и объединенные органические растворы сушат в присутствии сульфата мегния. Раствор упаривают с получением указанного в заголовке соединения (1,06 г, 77%). Данные ЯМР-анализа конечного продукта приведены в табл.1.
П р и м е р 2. Получение 4-фтор-2-{[(4-метокси-2-пиридинил)- метил]сульфинил}-1Н-бензимидазола натриевой соли.
4-фтор-2-{[(4-метокси-2-пиридинил)метил]сульфинил}-1Н- бензимидазол (5,0 г, 16,3 ммоль), растворенный в дихлорметане (100 мл), и гидроксид натрия (0,64 г, 16,0 ммоль), растворенный в воде (100 мл), переносят в делительную воронку. Смесь встряхивают до равновесия, после чего разделяются фазы растворителей. Водный раствор промывают дихлорметаном (2х25 мл) и затем сушат вымораживанием. Остаток перекристаллизуют из этилацетата (простого диэтилового эфира). Выход: 4,7 г (89%) указанного в заголовке соединения. Данные ЯМР-анализа приведены в табл.1.
Получение промежуточных соединений.
П р и ме р 1.1: Получение 4-фтор-2-меркапто-1Н-бензимидазола.
1,2-диамино-3-фторбензол (1,6 г, 12,7 ммоль) и этилксантогенат калия (2,64 г, 16,5 ммоль) растворяют в этаноле (25 мл) и воде (6 мл). Смесь нагревают с обратным холодильником в течение 14 ч и затем концентрируют на роторном испарителе. Прибавляют воду (20 мл) и раствор подкисляют 2 М хлористоводородной кислотой. Осадок отфильтровывают и сушат. Таким образом получают указанное в заголовке соединение (1,23 г, 58%). Данные ЯМР-анализа приведены в табл.2.
П р и м е р 1.2: Получение 4-фтор-2-{[(4-метокси-2-пиридинилд)метил]тио} -1Н-бен-зимидазола, используемого в качестве исходного вещества.
К раствору 4-фтор-2-меркапто-1Н-бензимидазола (1,15 г, 0,068 моль) в метаноле (60 мл) прибавляют в указанном порядке NaOH (0,54 г, 0,014 моль), растворенную в воде (3 мл), и 4-метокси-2-хлорметилпиридин-хлоргидрат (1,32 г, 0,0068 моль), растворенный в метаноле (20 мл). Смесь нагревают с обратным холодильником в течение 1 ч, после чего раствор упаривают. Остаток распределяют между метиленхлоридом и водой. После разделения органический раствор сушат в присутствии сульфата магния и упаривают с получением масла, которое очищают на силикагеле (50 г) с использованием 1%-ного метанола в метиленхлориде в качестве элюента. Таким образом получают указанное в заготовке соединение (1,35 г, 69%). Данные ЯМР-анализа продукта приведены в табл.2.
Наилучшим способом осуществления предлагаемого изобретения на сегодня является использование натриевой соли соединения формулы I, т.е. соединения, описанного в примере 2.
В следующих вариантах технологии получения лекарственных препаратов приведены фармацевтические составы, содержащие предлагаемое соединение в качестве активного компонента.
Сироп.
Сироп, содержащий 1% (массы на объем) активного вещества, получают из следующих ингредиентов, г:
Соединение по примеру I 1,0 Сахар, порошок 30,0 Сахарин 0,6 Глицерин 5,0 Ароматизатор 0,05 Этанол 96%-ный 5,0
Дистиллированная вода до 100 мл
Сахар и сахарин растворяют в 60 г теплой воды. После охлаждения в сахарный раствор прибавляют активное соединение, а также глицерин и раствор ароматизаторов, растворенных в этаноле. Смесь разбавляют водой до конечного объема, равного 100 мл.
Таблетки с энтеросолюбильной оболочкой
Таблетку с энтеросолюбильной оболочкой, содержащую 50 мг активного соединения, получают из следующих компонентов, г: I. Соединение по примеру 1 в виде соли магния 500 Лактоза 700 Метилцеллюлоза 6
Сшитый поливинил- пирролидон 50 Стеарат магния 15 Карбонат натрия 6
Дистиллированная вода
Сколько
потребу-
ется
II. Целлюлоза- ацетатфталат 200 Цетиловый спирт 15 Изопропанол 2000 Метиленхлорид 2000
I. Соединение по примеру 1 в виде порошка смешивают с лактозой и гранулируют водным раствором метилцеллюлозы и карбоната натрия. Влажную массу пропускают через сито и гранулят сушат в печи. После сушки гранулят смешивают с поливинилпирролидоном и стератом магния. Сухую смесь отпрессовывают в таблеточные стержни (1000 таблеток), причем каждая таблетка содержит 50 мг активного вещества, с использованием таблетировочной машины с пуансонами диаметром 7 мм.
II. Раствор целлюлозы-ацетатфталата и цетилового спирта в смеси с изопропанола/метиленхлорида наносят на таблетки 1 в AcceIa CotaR, оборудовании для нанесения покрытий Manesty. Получают окончательную массу таблетки, равную 110 мг.
Раствор для внутривенного введения
Парентеральный состав для внутривенного применения, содержащий 4 мг активного соединения на мл, получают из следующих ингредиентов.
Соединение в соответствии с примером 2 4 г
Стерильная вода до конечного объема, равного 1000 мл
Активное соединение растворяют в воде до конечного объема, равного 1000 мл. Раствор фильтруют через 0,22 мкм фильтр и сразу же разливают в 10 мл стерильные ампулы. Ампулы герметизируют.
Капсулы
Капсулы, содержащие 30 мг активного соединения, получают из следующих ингредиентов, г:
Соединение в соответствии с примером 1 300 Лактоза 700
Микрокристаллическая целлюлоза 40
Низкозамещенная гидрокси- пропилцеллюлоза 62
Вторичный кислый фосфат натрия
Очищенная вода Сколько потребуется
Активное соединение смешивают с сухими ингредиентами и гранулируют раствором вторичного кислого фосфата натрия. Влажную массу пропускают через экструдер, образуют шарообразные частицы и сушат в сушильном аппарате с псевдоожиженным слоем.
500 г полученных гранул вначале покрывают раствором гидроксипропилметилцеллюлозы (30 г) в воде (750 г) с использованием машины для нанесения покрытий с псевдоожиженным слоем. После сушки гранулы покрывают вторым слоем, содержащим, г:
Гидроксипропил- метилцеллюлозафталат 70 Цетиловый спирт 4 Ацетон 200 Этанол 600
Гранулы с окончательным покрытием вводят в капсулы.
Суппозитории
Суппозитории получают с использованием сварочной методики, из следующих ингредиентов, г:
Соединение в соответствии с примером 1 4 Witepsol Н-15 180
Активное соединение гомогенно смешивают с Witepsol Н-15 при 41оС. Расплавленную массу вводят в предварительно изготовленные упаковки для суппозиториев до массы нетто 1,84 г. После охлаждения упаковки герметизируют термообработкой. Каждый суппозиторий содержит 40 мг активного соединения.
Биологические эффекты. Биологическая доступность. Выбор видов для тестирования.
Результаты испытаний на двух различных животных видах, крысы и собаки, варьируются в отношении измеренного уровня биологической доступности для одного и того же соединения. Заявитель полагает, что крыса является более релевантным видом для тестирования биологической доступности. Данное мнение основано на том, что метаболизм печени имеет более предоминантное воздействие на биологическую доступность, а метаболический характер печени человека относительно данного типа соединений совершенно схож с данным характером самца крысы (более, чем характер самки крысы, а также собаки). Кроме того, результаты испытания на биологическую доступность у самца крысы имеют тенденцию к более широкому распространению по сравнению с результатами испытаний на собаке, и поэтому модель самца крысы дает более четкие различия в биологической доступности между разными соединениями. Говоря иными словами, биологическая доступность, испытанная на самце крысы, как ожидают, дает лучшую оценку относительных различий у человека между разными испытуемыми соединениями по сравнению с результатами испытаний, полученными с использованием тех же соединений на собаке.
Оценка биологической доступности.
Биологическую доступность оценивают путем вычисления соотношения между площадью по кривой концентрации в плазме (АVC) после интрадуоденального (id) и внутривенного (iv) введения в отношении крысы или собаки. Используют низкие, терапевтически релевантные дозировки лекарственного средства. Данный метод является научно признанным как ценный для оценки биологической доступности (см. например, Роуланд М. и Тозер Т.Н. Клиническая фармакокинетика, 2-е издание, Ли энд Фэбигер, Лондон, 1989, с. 42). Данные, полученные как на крысе, так и на собаке, приведены в табл.3.
Модель грубого скрининга
Поскольку модель биологической доступности, описанная выше, является трудоемкой и требует много времени, а также большого количества анализов плазмы, используют модель грубого скрининга на основе относительных возможностей ингибировать кислотную секрецию (см. например, Гот А. Медиуинская фармакология. 7-е издание, Ц.В.Мосби Компани, Сент-Луис, 1974, с. 19). Таким образом вычисляют отношение (названное "Биологическая доступность" в табл.3) между значениями ЭД50 при внутривенном введении и ЭД50 при интрадуоденальном введении лекарственного препарата. Эти данные также приведены в табл.3.
Потенция
Потенцию для ингибирования кислотной секреции измеряют на крысе и собаке, внутривенно и интрадуоденально. Когда она релевантна потенции данного соединения у человека в отношении предлагаемого типа соединений, полагают, что потенция у человека соответствует уровню где-то между теми значениями, которые измерены на самце крысы и измерены на собаке. Данные потенции от двух животных видов приведены в табл.3.
Биологические исследования.
Ингибирование секреции желудочной кислоты у находящегося в сознании самца крысы.
Используют самцов крысы линии Sprague Dawley. Их снабжают канюлированными фистулами в желудке (полости) и верхней части двенадцатиперстной кишки для сбора желудочных секреций и введения испытуемых веществ соответственно. Перед испытаниями проводят 14-дневный восстановительный период после хирургического вмешательства.
Перед выделительными тестами животным не дают пищу (но не воду) в течение 20 ч. Желудок многократно промывают через желудочную канюлю и подкожно вводят 6 мл рингер-глюкозы. Кислотную секрецию стимулируют вливанием в течение 3,5 ч (1,2 мл/ч подкожно) пентагастрина и карбахола (20 и 110 нмоль/кг ч соответственно). В течение данного времени желудочные секреции собирают за 30-минутные фракции. Испытуемые вещества или наполнитель вводят внутривенно или интрадуоденально через 90 мин после начала стимуляции в объеме 1 мл/кг. Пробы желудочного сока титруют до рН 7,0 с помощью NаОН, 0,1 моль/л. Выход кислоты вычисляют как продукт объема титрованного раствора и концентрации. Дальнейшие вычисления основаны на групповых средних реакциях 4-5 крыс. Выход кислоты в течение периодов после введения испытуемых веществ или наполнителя выражают как фракционные реакции, устанавливая выход кислоты в течение 30-минутного периода перед введением на отметке 1,0. Процент ингибирования вычисляют из фракционных реакций, выведенных на основе испытуемого соединения и наполнителя. Значения ЭД50 получают из графического интерполирования логарифмических кривых зависимости доза эффект или вычисляют из экспериментов с однократной дозой, принимая в расчет аналогичный тангенс угла наклона для всех кривых зависимости доза эффект. Оценку биологической доступности производят путем вычисления отношения ЭД50 (iv) ЭД50 (id). Приведенные результаты основаны на секреции желудочной кислоты в течение второго часа после введения лекарственного средства/наполнителя.
Биологическая доступность у самца крысы
Используют взрослых самцов крыс линии Sprague Dawley. В один день перед проведением эксперимента всех крыс канюлируют в левую сонную артерию под анестезией. Крыс, используемых для внутривенных экспериментов, также канюлируют в яремную вену (см. Попович В. и Попович П. Журнал прикладной физиологии, 1960; 15, 727-728). Крыс, используемых для интрадуодельных экспериментов, также канюлируют в верхнюю часть двенадцадтиперстной кишки. Канюли выводят на заднюю часть шеи. Крыс помещают отдельно после проведения хирургического вмешательства и лишают пищи, но не воды, перед введением испытуемых веществ. Одинаковую дозу (4 мкмоль/кг) назначают внутривенно и интрадуоденально в виде болюса в течение приблизительно 1 млн (2 мл/кг).
Пробы крови (0,1-0,4 г) забирают многократно из сонной артерии с интервалами до 4 ч после введения дозировки. Пробы замораживают как можно быстрее и сохраняют до анализа испытуемого соединения.
Площадь под концентрацией крови в зависимости от кривой времени АVC определяют формулой линейных трапеций и экстраполируют до бесконечности путем деления последней определенной концентрации крови на постоянную скорости исключения в конечной фазе. Общую биологическую доступность (F,) после интрадуоденального введения вычисдляют по формуле
F,  × 100
× 100
Ингибирование секреции желудочной кислоты и биологическая доступность на собаках в сознании
Используют гончих собак любого пола. Их снабжают дуоденальными фистулами для введения испытуемых соединений или наполнителя, а также канюлированными вентрикулярными фистулами для сбора желудочных выделений.
Перед проведением выделительных исследований животных лишают пищи в течение приблизительно 18 ч, однако разрешают свободный доступ к воде. Секрецию желудочной кислоты стимулируют четырехчасовым вливанием дигидрохлорида гистамина (12 мл/ч) с дозировкой, продуцирующей около 80% индивидуального максимального секреторного ответа, и желудочный сок собирают в течение последовательных 30-минутных фракций. Испытуемое вещество или наполнитель назначают внутривенно или интрадуоденально через 1 ч после начала вливания гистамина с объемом 0,5 мл/кег массы тела. Кислотность проб желудочного сока определяют титрованием до рН 7,0, и вычисляют выход кислоты. Выход кислоты в течение периодов сбора после введения испытуемого вещества или наполнителя выражают как фракционные ответы, устанавливая выход кислоты во фракции перед введением на отметке 1,0. Процент инигибрования вычисляют из фракционных ответов на введение испытуемого соединения и наполнителя. Значения ЭД50 получают графическим интерполированием логарифмических кривых зависимости доза-эффект или вычисляют из экспериментов с однократной дозой, принимая в расчет аналогичный тангенс угла наклона для всех кривых зависимости доза эффект. Все результаты основаны на выходе кислоты через 2 ч после дозирования.
Пробу крови для анализа концентрации испытуемого соединения в плазме забирают с интервалами до 3 ч после дозирования. Плазму разделяют и замораживают в пределах 30 мин после сбора. АVС (площадь под кривой концентрация плазмы время), экстраполированную до бесконечности, вычисляют формулой линейных трапеций. Общую биологическую доступность (F,) после интрадуоденального введения вычисляют как 100 х (АUCid)/AUCiv).
Химическая устойчивость
Химическую устойчивость различных соединений предлагаемого изобретения определяют кинетически при низкой концнетрации при температуре 37оС в водном буферном растворе при различных значениях рН. Результаты в табл.3 показывают период полураспада (t1/2) при рН 7,0, т.е. временной период, после которого половина первоначального количества соединения остается неизменяемой.
Результаты биолоигческих тестов и испытаний на химическую устойчивость
Табл. 3 суммирует данные испытаний, имеющиеся для предлагаемого соединения и структурно родственного известного соединения, названного в табл. 3 Сравн. а именно 5-фторо-2-{[(4-изопропокси-2-пиридинил)метил]сульфинил}-1Н-бензимидазол, описанный в патенте США N 4727150. Как видно из табл.3, предлагаемое соединение имеет высокую биологическую доступность (F 96% у крысы), высокую потенцию (ЭД50iv 0,96 мкмоль/кг, ЭД50id 2,4 мкмоль/кг у крысы) и высокую химическую устойчивость (t 1/2 23 ч). Кроме того, принимая во внимание наиболее характерное свойство предлагаемого соединения, т.е. биологическую доступность, оно имеет намного более высокое значение (96% по сравнению с 31%), нежели известное соединение, и лучшие показатели в отношении других свойств (ЭД50iv 1,8 мкмоль/кг, ЭД50id 4,0 мкмоль/кг и t 1/2 14 ч) для известного соединения.


Использование: для ингибирования секреции желудочной кислоты. Сущность изобретения: 4-фтор-2-{[(4-метокси-2-пиридинил)метил]сульфинил}-1H-бензимидазол. (1) Б.Ф.С 14 H 12 F N3 02 S, выход 89% или его физиологически приемлемая соль, промежуточные 4-фтор-2-меркапто-1H-бензимидазол. Б.Ф. С7 H5 F N2 S и 4-фтор-2-{[(4-метокси-2-пиридинил)метил]тио}-1H-бензимидазол. (2) Б.Ф. C14 H12 F N3 O S. Реагент 1: соединение 2. Реагент 2: m хлорбензойная кислота. Условия реакции: с выделением в свободном виде или в виде соли. 7 з.п. ф-лы, 3 табл.
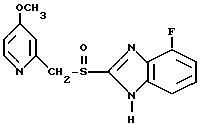
или его физиологически приемлемые соли.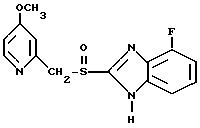
или его физиологически приемлемых солей, отличающийся тем, что окисляют 4-фтор- 2-{ [(4-метокси -2-пиридинил) метил]тио} -1Н-бензимидазол с выделением целевого продукта в свободном виде или в виде физиологически приемлемых солей.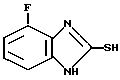
7. 4-Фтор-2-{ [(4-метокси-2- пиридинил)метил] тио} -1Н-бензимидазол формулы
8. Фармацевтическая композиция, обладающая ингибирующим секрецию желудочной кислоты действием, отличающаяся тем, что в качестве активного ингредиента содержит 0,1-95 мас. 4-фтор-2-{[ (4-метокси-2-пиридинил )метил]сульфинил} -1Н-бензимидазола формулы
в сочетании с фармацевтически приемлемым носителем.
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| 0 |
|
SU175464A1 | |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
