Изобретение направлено на новые соединения с высокой оптической чистотой, их использование в медицине, способ их приготовления и их использование в производстве фармацевтического препарата. Изобретение относится также к новым промежуточным продуктам для получения соединений данного изобретения.
Обоснование изобретения
Соединение 5-метокси-2-[[(4-метокси-3, 5-диметил-2- пиридинил)метил]сульфинил]-1Н-бензимидазол, имеющее общее название омепразол и его терапевтически приемлемые щелочные соли описаны в ЕР 5129 и ЕР 124 495 соответственно. Омепразол и его щелочные соли являются эффективными ингибиторами желудочной кислотной секреции и полезными в качестве противоязвенных средств. Соединения, являющиеся сульфоксидами, имеют центр асимметрии при атоме серы, то есть существуют в виде двух оптических изомеров (энантиомеров). Желательно получить соединения с улучшенными фармакокинетическими и метаболическими свойствами, которые будут давать улучшенную терапевтическую характеристику, такую как пониженная степень внутрииндивидуальной изменчивости. Настоящее изобретение дает такие соединения, которые являются новыми солями (-)-энантиомера омепразола.
Разделение энантиомеров омепразола на аналитическом уровне описано, например, в J. Chromatography, 532(1990), 305-19 и на препаративном уровне в DE 4035455. Последнее выполнено использованием диастереомерного простого эфира, который выделяется и после этого гидролизуется в кислом растворе. В кислых условиях, необходимых для гидролиза связанной группы, омепразол является довольно чувствительным и кислота должна быть быстро нейтрализована основанием во избежание разложения чувствительного к кислотам соединения. Применительно к вышеупомянутому случаю это осуществляется добавлением реакционной смеси, содержащей концентрированную серную кислоту, к концентрированному раствору NaOH. Это неблагоприятно, так как существует большой риск локального достижения значений pH в интервале 1-6, что может оказаться разрушительным для вещества. Кроме того, мгновенная нейтрализация будет создавать нагрев, которым будет трудно управлять при производстве в больших масштабах.
В дальнейшем аспекте данное изобретение дает новый метод получения новых соединений изобретения в крупных масштабах. Этот способ может также быть использован для крупномасштабного производства отдельных энантиомеров омепразола в нейтральной форме.
На известном уровне техники не существует никакого примера выделенной или идентифицированной соли оптически чистого омепразола, то есть отдельных энантиомеров омепразола или любой выделенной или идентифицированной соли любого оптически чистого аналога омепразола.
Подробное описание изобретения
Настоящее изобретение относится к новым Na+, Mg2+, Li+", K+ и Ca2+ солям (-)-энантиомера омепразола, то есть Na+, Mg2+, Li+, K+ и Ca2+ соли (-)-5-метокси-2-[[(4-метокси-3,5-диметил-2- пиридинил)метил]сульфинил]-1H-бензимидазола.
Особенно предпочтительными солями согласно изобретению являются Na+, Ca2 и Mg2+ соли, то есть натриевая соль (-)-5-метокси-2-[[(4-метокси-3,5-диметил-2-пиридинил) метил] -сульфинил]-1Н-бензимидазола, магниевая соль (-)-5-метокси-2-[[ (4-метокси-3,5-диметил-2-пиридинил)метил]сульфинил] -1Н-бензимидазола и кальциевая соль (-)-5-метокси-2-[[(4-метокси-3, 5-диметил-2-пиридинил)метил]сульфинил]-1H-бензимидазола.
Согласно изобретению, наиболее предпочтительными солями являются оптически чистые Na+ соли (-)-омепразола формулы (I)
и оптически чистые магниевые соли (-)-омепразола формулы (II)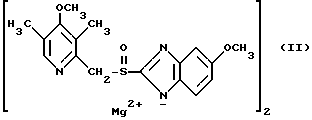
Выражение "оптически чистые Na+ соли омепразола" подразумевает Na-соль(-)-энантиомера омепразола, в значительной степени свободную от Na-соли (+)-энантиомера омепразола.
Отдельные энантиомеры омепразола до сих пор были получены только в виде сиропов, но не в виде кристаллических продуктов. Соли, определенные данным изобретением, легко могут быть получены посредством нового специфического метода, согласно одному из аспектов изобретения по получению отдельных энантиомеров омепразола. К тому же соли, однако, не в нейтральных формах, получаются в виде кристаллических продуктов. Поскольку оптически загрязненные соли энантиомеров омепразола возможно очистить кристаллизацией, они могут быть получены с очень высокой оптической чистотой, а именно с ≥ 99,8% энантиомерным избытком (e. e. ) даже из оптически загрязненного препарата. Кроме того, оптические чистые соли являются стабильными по отношению к рацемизации как в нейтральной pH, так и в щелочной pH, что было неожиданно, поскольку предполагалось, что известная депротонизация на углеродном атоме между пиридиновым циклом и хиральным атомом серы вызовет рацемизацию в щелочных условиях. Эта высокая стабильность по отношению к рацемизации делает возможным применение соли (-)-энантиомера омепразола в терапии.
Как упомянуто выше, специфический способ приготовления отдельных энантиомеров омепразола составляет дальнейший аспект изобретения, и этот метод может быть использован для получения отдельных энантиомеров омепразола в нейтральной форме, равно как и их солей.
Соединения согласно изобретению могут быть использованы для ингибирования желудочной кислотной секреции у млекопитающих животных и человека. В более общем значении соединения изобретения могут использоваться для лечения заболеваний желудка, связанных с нарушением кислотности, и желудочно-кишечных воспалительных заболеваний у млекопитающих и человека, таких как язва желудка, язва двенадцатиперстной кишки, рефлюкс-эзофагит и гастрит. Кроме того, соединения могут использоваться для лечения других желудочно-кишечных нарушений, при которых желателен желудочный антисекреторный эффект, например для пациентов, находящихся на NSAID терапии, пациентов с ульцероаденомой поджелудочной железы и пациентов с острым желудочно-кишечным кровотечением. Они могут также использоваться на пациентах в случаях интенсивной терапии, и до- и послеоперационно для предотвращения кислотной аспирации и стрессовой язвы. Соединения изобретения могут также использоваться для лечения или профилактики воспалительных состояний у млекопитающих, включая человека, особенно таких, при которых вовлекаются в патологический процесс лизоцимальные энзимы. Особенно могут быть упомянуты такие состояния как артрит и подагра. Соединения изобретения могут также быть полезными в лечении псориаза, а также в лечении Helicobacter инфекций.
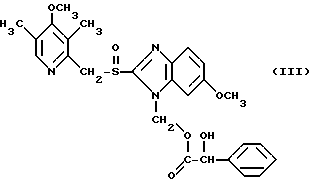
Кроме того, дальнейший аспект соединения составляет соединение III, которое является промежуточным продуктом, используемым в специфическом способе получения.
Получение
Оптически чистые соединения изобретения, то есть соли (-)-энантиомера приготавливают разделением двух стереоизомеров диастереоизомерной смеси следующего типа, 5-или 6-метокси-2-[[ (4-метокси-3,5-диметил-2-пиридинил)метил]сульфинил]-1- [ацилоксиметил]-1Н-бензимидазол формулы IV
где метокси заместитель в бензимидазольной составляющей находится в положении 5 или 6, и Acyl радикал является таким, как определено ниже, с последующим сольволизом выделенного диастереомера, содержащего (-)-энантиомер омепразола в щелочном растворе. Образовавшийся (-)-энантиомер омепразола затем выделяют нейтрализацией водных растворов солей (-)-энантиомеров омепразола нейтрализующим агентом, которым может быть кислота или сложный эфир такой как метилформиат.
Ацил (Acyl) составляющая в диастереомерном эфире может быть хиральной ацильной группой, такой, как манделоил, и центр асимметрии в хиральной ацильной группе может иметь как R, так и S конфигурацию.
Диастереомерные сложные эфиры могут быть разделены либо хроматографически либо фракционированной кристаллизацией.
Сольволиз обычно протекает в присутствии основания в протонном растворителе, таком как спирты или вода, но ацильная группа может также быть снята гидролизом с помощью основания в протонном растворителе, таком как диметилсульфоксид или диметилформамид. Реагирующей основой может быть OH- или R1O-, где R1 может представлять любую алкильную или арильную группу.
Для получения оптически чистых Na+ солей изобретения, то есть Na+ солей (-)-энантиомера омепразола, полученное соединение обрабатывают основанием, таким как NaOH, в водной или неводной среде, или с NaOR2, в котором R2 представляет алкильную группу, содержащую 1-4 атомов углерода или с NaNH2. Кроме того, щелочные соли, в которых катионы являются Li+ или К+, могут быть приготовлены с использованием литиевых или калиевых солей вышеупомянутых соединений. Для того чтобы получить кристаллическую форму Na+ соли, желательно добавление NaOH в неводную среду, такую как смесь 2-бутанона и толуола.
Для получения оптически чистой Mg2+ соли изобретения оптически чистую Na+ соль (-)-энантиомера обрабатывают водным раствором неорганической магниевой соли, такой как MgCl2, в результате чего Mg2+ соль выпадает в осадок. Оптически чистая Mg2+ соль может также быть приготовлена обработкой (-)- энантиомера омепразола основанием, таким как Mg(OR3)2, в котором R3 представляет алкильную группу, содержащую 1-4 атомов углерода, в неводном растворителе, таком как спирт (только для алкоголятов), например ROH, или в простом эфире, таком, как тетрагидрофуран. Аналогичным образом может быть приготовлена также щелочная соль, в которой катионом является Ca2+, с использованием водного раствора неорганической соли кальция, такой как CaCl2.
Примерами щелочных солей отдельных энантиомеров изобретения служат, как упомянуто выше, кроме соли натрия (соединение I) и соли магния (соединение II), служат также соли с Li+, К+ и Ca2+.
Для клинического применения (-)-энантиомеры, то есть оптически чистые соединения, изобретения составляются в виде фармацевтических форм для орального, ректального, парентерального или других способов введения. Фармацевтические составы содержат (-)-энантиомер изобретения обычно в комбинации с фармацевтически приемлемым носителем. Носитель может быть в форме твердого продукта, полутвердого продукта или жидкого растворителя, или капсулы. Эти фармацевтические препараты представляют дальнейшую цель изобретения. Обычно количество активного соединения составляет между 0,1-95 вес.% препарата между 0,2-20 вес. % в препаратах для парентерального применения и между 1-50 вес.% в препаратах для орального применения.
При приготовлении фармацевтических составов в форме стандартной дозы для орального применения оптически чистое соединение может быть смешано с твердым порошкообразным носителем, таким как лактоза, сахароза, сорбитол, маннитол, крахмал, амилопектин, производные целлюлозы, желатин или другие подходящие носители, стабилизирующими веществами, такими как щелочные соединения, например карбонаты, гидроокиси или окиси натрия, калия, кальция, магния и тому подобные, равно как со смазывающими веществами, такими как стеарат магния, стеарат кальция, натрий стеарил фумарат и полиэтиленгликолевые парафины. Затем смесь перерабатывается в гранулы или прессуется в таблетки. Гранулы и таблетки могут быть покрыты энтеросолюбильной оболочкой, которая защищает активное соединение от катализируемого кислотами разложения, пока дозированная форма остается в желудке. Энтеросолюбильное покрытие выбирается из фармацевтически приемлемых материалов для энтеросолюбильных оболочек, например воска, шеллака или анионных пленкообразующих полимеров и тому подобного, при желании с подходящим пластификатором. К оболочке могут добавляться различные красители для отличия таблеток или гранул с различными количествами данного активного соединения.
Мягкие желатиновые капсулы могут быть приготовлены в виде капсул, содержащих смесь активного соединения, растительного масла, жира или другого подходящего наполнителя для мягких желатиновых капсул. Мягкие желатиновые капсулы могут также быть покрыты энтеросолюбильной оболочкой, как описано выше.
Твердые желатиновые капсулы могут содержать гранулы или покрытые энтеросолюбильной оболочкой гранулы активного соединения. Твердые желатиновые капсулы могут также содержать активное соединение в комбинации с твердым порошкообразным носителем, таким как лактоза, сахароза, сорбитол, маннитол, картофельный крахмал, амилопектин, производные целлюлозы или желатин. Капсулы могут быть покрыты энтеросолюбильной оболочкой, как описано выше.
Стандартные дозы для ректального введения могут быть приготовлены в форме суппозиториев, содержащих в себе активное вещество, смешанное с нейтральным насыщенным основанием, или они могут быть приготовлены в форме желатиновой ректальной капсулы, которая содержит активное вещество в смеси с растительным маслом, парафиновым маслом или другим подходящим наполнителем для желатиновых ректальных капсул, или они могут быть приготовлены в форме готовой микроклизмы, или они могут быть приготовлены в форме состава для сухой микроклизмы для получения нужного состава в подходящем растворителе непосредственно перед введением.
Жидкий препарат для орального введения может быть приготовлен в форме сиропов или суспензий, например растворов или суспензий, содержащих от 0,2% до 20% по весу активного ингредиента и в остальном содержащих сахарозу или спирты сахаров и смесь этанола, воды, глицерина, пропиленгликоля и/или полиэтиленгликоля. При желании такие жидкие препараты могут содержать красители, корригенты, сахарин и карбокси- метилцеллюлозу или другие уплотняющие агенты. Жидкие препараты для орального введения могут также быть приготовлены в форме сухого порошка для получения нужного состава в подходящем растворителе непосредственно перед использованием.
Растворы для парентерального введения могут быть приготовлены как растворы оптически чистых соединений изобретения в фармацевтически приемлемых растворителях, предпочтительно в концентрации от 0,1 до 10% по весу. Эти растворы могут также содержать стабилизирующие агенты и/или буферы и могут выпускаться в ампулах или флаконах, содержащих стандартную дозу. Растворы для парентерального введения могут также быть приготовлены в виде сухих препаратов для получения импровизированного состава в подходящем растворителе перед употреблением.
Типичная суточная доза активного соединения будет зависеть от различных факторов, например таких, как индивидуальная потребность каждого пациента, путь введения к заболеванию. Вообще оральная и парентеральная дозы будут в интервале от 5 до 500 мг в сутки активного вещества.
Настоящее изобретение иллюстрируется следующими примерами. Направление оптического вращения энантиомеров омепразола будет меняться от (-) до (+), когда из несолевой формы получают натриевую соль, и наоборот, когда получают соль магния из натриевой соли.
Пример 1
Получение натриевой соли (+)-5-метокси-2-[[(4-метокси- 3,5-диметил-2-пиридинил)метил]сульфинил]-1H-бензимидазола
100 мг (0,3 ммоля) (-)-5-метокси-2-1[(4-метокси-3,5-диметил-2- пиридинил)метил]сульфинил]-1H-бензимидазола (загрязненного 3% (+)-изомера) растворили при перемешивании в 1 мл 2-бутанона. Добавили 60 мкл водного раствора 5,0 М гидроокиси натрия и 2 мл толуола. Образовавшаяся смесь была негомогенной. Для получения истинного раствора дополнительно добавили 2-бутанон (около 1 мл) и смесь перемешивали при температуре окружающей среды в течение ночи. Образовавшийся осадок отфильтровали и промыли диэтиловыми эфиром. Получили 51 мг (46%) названного в заглавии соединения в виде белых кристаллов т. пл. (разложен. ) 246-248oC. Оптическая чистота (e.e.), которая анализировалась методом хиральной колоночной хроматографии, составила ≥ 99,8%.
[α]
ЯМР-данные приведены ниже.
Пример 2
Получение натриевой соли (-)-5-метокси-2-[[(4-метокси-3,5-диметил- 2-пиридинил)метил]сульфинил]-1H-бенэимидазола
100 мг (0,3 ммоля) (+)-5-метокси-2- [[(4-метокси-3,5-диметил-2-пиридинил)метил]сульфонил]-1H- бензимидазола (загрязненного 3% (-)-изомера) растворили при перемешивании в 1 мл 2-бутанона. Добавили 60 мкл водного раствора 5,0 М гидроокиси натрия и 2 мл толуола. Образовавшаяся смесь была негомогенной. Для получения истинного раствора дополнительно добавили 2-бутанон (около 1 мл) и смесь перемешивали при температуре окружающей среды в течение ночи. Образовавшийся осадок отфильтровали и промыли диэтиловым эфиром. Получили 56 мг (51%) названного в заглавии соединения в виде белых кристаллов т. пл. (разложен. ) 247-249oC. Оптическая частота (e.e.), которая анализировалась методом хиральной колоночной хроматографии составила ≥ 99,8%.
[α]
ЯМР-данные приведены ниже.
Пример 3
Получение магниевой соли (+)-5-метокси-2-[[(4-метокси-3,5-диметил-2-пиридинил)метил] сульфинил]-1H-бензимидазола
2,9 мл 0,1 М раствора NaOH добавили к 0,10 г (0,29 ммоля) (+)-5-метокси-2-[[(4-метокси-3,5- диметил-2-пиридинил)-метил] сульфинил]-1H-бензимидазола. К этой смеси добавили 2 мл метиленхлорида и после смешивания в делительной воронке отделили водный раствор. Добавили по каплям раствор 14 мг (0,145 ммоля) MgCl2 в воде. Образовавшийся осадок отделили центрифугированием и выделили 52 мг (50%) продукта в виде аморфного порошка. Оптическая частота (е.е.) составила 98% и таким образом оказалась такой же, как у исходного материала. Оптическую чистоту определили методом хроматографии на аналитической хиральной колонке, [α]
Пример 4
Получение магниевой соли (+)-5-метокси-2-[[(4-метокси-3,5-диметил- 2-пиридинил)метил]сульфинил]-1H-бензимидазола
Натриевую соль (-)-5-метокси-2-[[(4-метокси-З, 5-диметил-2-пиридинил)метил]сульфинил]-1H-бензимидазола (0,500 г, 1,36 ммоля) растворили в воде (10 мл). К этой смеси добавили по каплям 10 мл водного раствора MgCl2•H2O (138 мг, 0,60 ммоля) и образовавшийся осадок отделяли центрифугированием. Получили 418 мг (86%) продукта в виде белого порошка. Оптическая чистота (е. е.) продукта составила 99,8% и таким образом оказалась такой же, как и у исходного продукта. Оптическую чистоту определили методом хроматографии на аналитической хиральной колонке.
[α]
Пример 5
Получение магниевой соли (-)-5-метокси-2-[[(4-метокси- З, 5-диметил-2-пиридинил)метил]сульфинил]-1H-бензимидазола
Натриевую соль (+)-5-метокси-2-[[(4-метокси-3,5-диметил-2- пиридинил)метил]сульфинил]-1H-бензимидазола (0,165 г, 0,5 ммоля) растворили в воде (3 мл). К этой смеси добавили по каплям 2 мл водного раствора MgCl2•H2O (46 мг, 0,23 ммоля) и образовавшийся осадок отделили центрифугированием. Получили 85 мг (51%) продукта в виде белого порошка. Оптическая чистота (е.е.) продукта составила 99,9%, что оказалось таким же или лучшим значением по сравнению с оптической чистотой исходного материала. Оптическую частоту определили хроматографически на аналитической хиральной колонке.
[α]
Пример 6
Увеличение оптической чистоты путем приготовления магниевой соли (-)-5-метокси-2-[[(4-метокси-3,5-диметил-2- пиридинил)-метил] сульфинил] -1Н-бензимидазола в неводном растворе с последующей кристаллизацией указанной соли
Магний (0,11 г, 4,5 ммоля) растворяют и вводят в реакцию с метанолом (50 мл) при 40oC с каталитическим количеством метиленхлорида. Реакцию проводят под азотом и заканчивают через пять часов. К раствору метоксида магния добавляют при комнатной температуре смесь двух энантиомеров (90% (-)-изомера и 10% (+)-изомера). 5-метокси-2-[[(4-метокси-3,5-диметил-2- пиридинил)метил] сульфинил] -1H-бензимидазола (2, 84 г, 8,2 ммоля). Смесь перемешивают в течение 12 часов, после чего добавляют небольшое количество воды (0,1 мл), чтобы осадить неорганические соли магния. После 30 минут перемешивания эти неорганические соли отфильтровывают и раствор концентрируют на ротавейпоре. Остаток теперь представляет собой концентрированный метанольный раствор энантиомерной смеси (т.е. целевого соединения, загрязненного (+)-изомером), с оптической чистотой (энантиомерный избыток, е.е.) 80%. Эту смесь разбавляют ацетоном (100 мл) и после перемешивания при комнатной температуре в течение 15 минут получают белый осадок. Дополнительное перемешивание в течение 15 минут и затем фильтрация дают 1,3 г (50%) целевого соединения в виде белых кристаллов. Хиральный анализ кристаллов и маточника осуществляют хроматографически на аналитической хиральной колонке. Найдено, что оптическая чистота кристаллов и маточного раствора составляет 98,4 е.е. и 64,4% е.е. соответственно. Таким образом, оптическая чистота (е.е.) увеличена с 80% до 98,4% просто путем кристаллизации соли магния из смеси ацетона и метанола. Продукт кристаллический, как показывает порошковая рентгеновская дифракция, а содержание магния составляет 3,44%, как показано атомной абсорбционной спектроскопией.
[α]
Пример 7
Увеличение оптической чистоты путем приготовления магниевой соли (+)-5-метокси-2-[[(4-метокси-3,5-диметил-2- пиридинил)-метил] сульфинил] -1Н-бензимидазола в неводном растворе с последующей кристаллизацией указанной соли
Магний (0,11 г, 4,5 ммоля) растворяют и вводят в реакцию с метанолом (50 мл) при 40o С с каталитическим количеством метиленхлорида. Реакцию проводят под азотом и оканчивают через пять часов. При комнатной температуре к раствору метоксида магния прибавляют смесь двух энантиомеров (90% (+)-изомера и 10% (-)-изомера) 5-метокси-2- [[(4-метокси-З,5-диметил-2-пиридинил)метил] сульфинил] -1H- бензимидазола (2, 84 г, 8,2 ммоля). Смесь перемешивают в течение 12 часов, после чего добавляют небольшое количество воды (0,1 мл), чтобы осадить неорганические соли магния. После 30 минут перемешивания неорганические соли отфильтровывают и раствор концентрируют на ротавейпоре. Остаток теперь представляет собой метанольный раствор энантиомерной смеси (т. е. целевого соединения, загрязненного (+)-изомером), с оптической чистотой (е. е.) 80%. Эту смесь разбавляют ацетоном (100 мл) и после перемешивания при комнатной температуре в течение одного часа получают белый осадок. Дополнительное перемешивание в течение 30 минут и затем фильтрация дают 0,35 г целевого соединения в виде белых кристаллов. Дополнительное перемешивание маточной жидкости в течение 24 часов при комнатной температуре дает еще 1,0 г (общий выход = 52%). Хиральный анализ кристаллов и второй маточной жидкости проводят хроматографически на аналитической хиральной колонке. Оптическая чистота первых кристаллов составляет 98,8% е.е. и 99,5% е.e. соответственно. Оптическая чистота маточной жидкости найдена равной 57% е.е. Таким образом, оптическая чистота (е.е.) увеличена с 80% до прибл. 99% просто путем кристаллизации магниевой соли из смеси ацетона и метанола. Первый осадок кристаллический, как показано порошковой рентгеновской дифракцией, и содержание магния составляет 3,49%, как показано атомной адсорбционной спектроскопией.
[α]
В следующих далее примерах описано получение промежуточных продуктов синтеза согласно изобретению.
Пример 8
Получение 6-метокси-2-[[(4-метокси-3,5-диметил-2-пиридинил) метил] -(R/S)-сульфинил]-1-[(R)-манделоилоксиметил]-1Н- бензимидазола
Раствор 3,4 гидроокиси натрия в 40 мл воды добавили к смеси 14,4 г (42 ммоля) кислого сульфата тетрабутиламмония и 6,4 г (42 ммоля) (R)-(-)-миндальной кислоты. Смесь экстрагировали 400 мл хлороформа. После разделения органический экстракт нагревали до кипения с обратным холодильником с 16,6 г (42 ммоля) рацемата 6- метокси-2-[[(4-метокси-3,5-диметил-2-пиридинил)метил] сульфинил] -1-[хлорометил] -1Н-бензимидазола. После упаривания растворителя остаток растворили в 100 мл дихлорметана и 700 мл этилацетата. Смесь промыли 3 х 200 мл воды и органический раствор высушили над MgSO4 и упарили. Сырой продукт очистили перекристаллизацией из 100 мл ацетонитрила, получив 8,1 г названного в заглавии соединения (38%) в виде диастереомерной смеси. ЯМР-данные приведены ниже.
Пример 9
Выделение более гидрофильного диастереомера 6-метокси-2-[[ (4-метокси-3,5-диметил-2-пиридинил)метил] -(R/S)-сульфинил]-1- [(R)-манделоилоксиметил] -1H-бензимидазола
Диастереомеры названного в заглавии соединения в Примере 8 разделили использованием обращенно-фазовой хроматографии (ВДЖХ). Приблизительно 300 мг диастереомерной смеси растворили в 10 мл горячего ацетонитрила, разбавленного 10 мл смеси водного 0,1 М ацетата аммония и ацетонитрила (70/30). Раствор вводили в колонку и элюировали смесью водного 0,1 М ацетата аммония и ацетонитрила (70/30). Более гидрофильный изомер было легче получить чистым, чем менее гидрофильный изомер. Разработана следующая методика для фракции, содержащей чистый изомер: экстракция дихлорметаном, промывка органического раствора 5% водным раствором бикарбоната натрия, высушивание над Na2SO4 и упаривание растворителя на роторном испарителе (в конце упаривания удаление ацетонитрила облегчалось дополнительным добавлением дихлорметана). Использованием в приведенной выше методике 1,2 г диастереомерной смеси получили более гидрофильный изомер, 410 мг, в чистом виде, представляющий бесцветный сироп.
ЯМР-данные приведены ниже.
Пример 10
Получение 6-метокси-2-[[(4-метокси-З, 5-диметил-2- пиридинил)метил] -(R/S)-сульфинил]-1-[(S)-манделоилоксиметил]- 1H-бензимидазола
Продукт получили из 0,1 г (202 ммоля) гидроокиси натрия в 100 мл воды, 34,4 г (101 ммоля) кислого сульфата тетрабутиламмония, 15,4 г (101 ммоля) (S)- (+)-миндальной кислоты и 39,9 г (101 ммоля) рацемата 6-метокси-2- [[4-метокси-З,5-диметил-2-пиридинил)метил]-сульфинил]-1- [хлорометил]-1H-бензимидазола использованием той же методики, что и в Примере 6. Перекристаллизация из 100 мл ацетонитрила дала 21,3 г, то есть 41% названного в заглавии соединения в виде диастереомерной смеси.
ЯМР-данные приведены ниже.
Пример 11
Выделение более гидрофильного диастереомера 6-метокси-2-[[(4- метокси-3,5-диметил-2-пиридинил) метил]-(R/S)-сульфинил]-1- [(S)-манделоилоксиметил] -1H-бензимидазола
Диастереомеры названного в заглавии соединения в Примере 10 разделили использованием обращенно-фазовой хроматографии (ВДЖХ) тем же способом, что и в Примере 9, но использованием диастереомерной смеси 6-метокси-2-[[4-метокси-3,5-диметил-2-пиридинил)метил] - (R/S)-сульфинил]-1-[(S)-манделоилоксиметил] -1H-бензимидазола вместо сложного эфира (R)-миндальной кислоты, использованного в Примере 9. Использованием 2,1 г диастереомерной смеси получили более гидрофильный изомер, 760 мг, в чистом виде, представляющий бесцветный сироп.
ЯМР-данные приведены ниже.
Пример 12
Получение (-)-5-метокси-2-[[(4-метокси-3,5-диметил-2- пиридинил)метил] -сульфинил]-1H-бензимидазола
0,23 г (0,45 ммоля) более гидрофильного диастереомера 6-метокси-2-[[4-метокси-3, 5-диметил-2-пиридинил)метил]-сульфинил]-1-[(R) -манделоилоксиметил] -1H-бензимидазола растворили в 15 мл метанола. Добавили раствор 36 мг (0,9 ммоля) гидроокиси натрия в 0,45 мл воды, и спустя 10 минут смесь упарили на роторном испарителе. Остаток распределили между 15 мл воды и 15 мл дихлорметана. Органический раствор экстрагировали 15 мл воды и к объединенным водным растворам добавили 85 мкл (1,4 ммоля) метилформиата. Через 15 минут смесь экстрагировали 3 х 10 мл дихлорметана. Органический раствор высушили над Na2SO4 и затем упарили. Получили 0,13 г (77%) названного в заглавии соединения в виде бесцветного сиропа. Оптическая чистота (е.е. ), которую анализировали хиральной колоночной хроматографией, составила 94%.
[α]
ЯМР-данные приведены ниже.
Пример 13
Получение (+)-5-метокси-2-[[(4-метокси-3,5-диметил-2- пиридинил)метил] -сульфинил]-1H-бензимидазола
0,76 г (1,5 ммоля) более гидрофильного диастереомера 6-метокси-2-[[(4-метокси-3, 5-диметил-2-пиридинил)метил] -сульфинил] -1-[(S)- манделоилоксиметил-1H-бензимидазола растворили в 50 мл метанола. Добавили раствор 0,12 мг (3,0 ммоля) гидроокиси натрия в 1,5 мл воды, и спустя 10 минут смесь упарили на роторном испарителе. Остаток распределили между 25 мл воды и 25 мл дихлорметана. Органический раствор экстрагировали 25 мл воды и к объединенным водным растворам добавили 200 мкл (3,2 ммоля) метилформиата. Через 15 минут смесь экстрагировали 3 х 25 мл дихлорметана. Органический раствор высушили над Na2SO4 и затем упарили. Получили 0,42 г (81%) названного в заглавии соединения в виде бесцветного сиропа. Оптическая чистота, (е.е.), которую анализировали хиральной колоночной хроматографией, составила 98%.
[α]
ЯМР-данные приведены в табл. 2 в конце описания.
Лучшим способом воплощения изобретения, известным в настоящее время, является использование натриевых солей, оптически чистых соединений изобретения, т.е. соединений, описанных в Примере 1 и Примере 2.
Фармацевтические препараты, включающие соединения изобретения в качестве активного ингредиента, иллюстрируются следующими составами.
Сироп
Сироп, содержащий 1% (вес. на объем) активного вещества, приготовлен из следующих ингредиентов:
Соединение настоящего изобретения - 1,0 г
Сахар, порошок - 30,0 г
Сахарин - 0,6 г
Глицерин - 5,0 г
Корригент - 0,05 г
Этанол 96% - 5,0 г
Дистиллированная вода q.s. (в нужном кол-ве) до общего объема - 100 мл
Сахар и сахарин растворили в 60 г теплой воды. После охлаждения добавили к раствору сахара активное соединение и также добавили глицерин и раствор корригентов, растворенных в этаноле. Смесь разбавили водой до конечного объема 100 мл.
Таблетки, покрытые энтеросолюбильной оболочкой
Таблетка, покрытая энтеросолюбильной оболочкой, содержащая 50 мг активного соединения, приготовлена из следующих ингредиентов:
I
Соединение изобретения в виде Mg соли - 500 г
Лактоза - 700 г
Метилцеллюлоза - 6 г
Сшитый поливинилпирролидон - 50 г
Стеарат магния - 15 г
Карбонат натрия - 6 г
Дистиллированная вода - (требуемое количество) q.s.
II
Фталат ацетат целлюлозы - 200 г
Цетиловый спирт - 15 г
Изопропанол - 2000 г
Метиленхлорид - 2000 г
I Соединение по изобретению, порошкообразное, смешали с лактозой и гранулировали с водным раствором метилцеллюлозы и карбоната натрия. Влажную массу пропустили под давлением через сито и гранулят высушили в печи. После высушивания гранулят смешали с поливинилпирролидоном и стеаратом магния. Сухую смесь спрессовали в таблеточные стержни (10 000 таблеток), каждая таблетка содержала 50 мг активного вещества, в машине для изготовления таблеток, используя перфораторы с диаметром 7 мм.
II Раствор фталата ацетат целлюлозы и цетилового спирта в изопропанол/метиленхлориде напыляли на таблетки I в Accela Cota ® , Manesty оборудовании для нанесения покрытия. Получили таблетки с конечным весом 110 мг.
Раствор для внутривенного введения
Парентеральный состав для внутривенного применения, содержащий 4 мг активного соединения на мл, приготовили из следующих ингредиентов:
Соединение изобретения - 4 г
Стерильная вода до конечного объема - 1000 мл
Активное соединение растворили в воде до конечного объема 1000 мл. Раствор профильтровали через 0,22 мкм фильтр и немедленно расфасовали в 10 мл стерильные ампулы. Ампулы запаяли.
Капсулы
Капсулы, содержащие 30 мг, приготовили из следующих ингредиентов:
Соединение изобретения - 300 г
Лактоза - 700 г
Микрокристаллическая целлюлоза - 40 г
Гидроксипропилцеллюлоза с низкой степенью замещения - 62 г
Динатриевая кислая (орто)фосфорнокислая соль - 2 г
Очищенная вода - q.s.
Активное соединение смешали с сухими ингредиентами и гранулировали с раствором динатриевой кислой (орто)-фосфорнокислой соли. Влажную массу пропустили под давлением через экструдер и превратили в шарики, высушили в сушилке во флюидизированном слое.
Раствор для нанесения покрытия:
Фталат гидроксипропилметилцеллюлозы - 70 г
Цетиловый спирт - 4 г
Ацетон - 200 г
Этанол - 600 г
Готовыми таблетками, покрытыми оболочкой, заполнены капсулы.
Суппозитории
Суппозитории приготовлены из следующих ингредиентов использованием способа сварки. Каждый суппозиторий содержал 40 мг активного соединения.
Соединение изобретения - 4 г
Витепсол Н-15 - 180 г
Активное соединение гомогенно смешивают с Витепсолом Н-15 при температуре 41oC. Расплавленной массой заполняют по объему предварительно изготовленные оболочки для суппозиториев до чистого веса 1,84 г. После охлаждения оболочки заплавляют путем нагрева. Каждый суппозиторий содержит 40 мг активного соединения.
Устойчивость к рацемизации при различных значениях pH
Стабильность оптически чистых соединений изобретения по отношению к рацемизации определяли при низких концентрациях в холодильнике в водных буферных растворах при pH 8; 9,3; 10 и 11,2. Стереохимическую стабильность определяли путем сравнения оптической чистоты (-)-изомера 5-метокси-2-[[4-метокси-3,5- диметил-2-пиридинил)метил]сульфинил]-1Н-бензимидазола в буферном растворе сразу после растворения и спустя несколько дней. Измерения осуществляли хроматографически на аналитической хиральной колонке. Удивительно высокая стереохимическая стабильность в щелочных средах для соединений изобретения подтверждается тем фактом, что даже спустя 21 день при pH 11,2 не наблюдается рацемизация испытуемого соединения. При pH 8; 9,3 и 10 более явно проявляется химическое разложение соединения, что значительно затрудняет выполнение измерений по рацемизации, однако в отсутствие этих значений pH заметная рацемизация наблюдалась после 16 дней.
В другом эксперименте по рацемизации с оптически чистыми соединениями изобретения, содержащем фосфатный буфер водный раствор (pH 11) натриевой соли (+)-изомера 5-метокси-2-[[(4-метокси-3, 5-диметил-2-пиридинил)метил] сульфинил] -1Н-бензимидазола (с= 10-5 М) нагревался в течение 26 часов при 37oC без какой-либо рацемизации за все время наблюдения.
Клиническое сравнение фармакокинетики натриевой соли (-)- энантиомера омепразола, натриевой соли (+)-энантиомера омепразола и натриевой соли омепразола
На фиг. 1 и 2 представлены данные двух исследований.
На фиг. 1 представлены средние уровни в плазме крови рацемического омепразола, (-)-энантиомера омепразола и (+)- энантиомера омепразола (здесь и далее обозначение (-)-омепразол и (+)-омепразол) в стационарном состоянии (7 день) у быстрых метаболизаторов после назначения 15 мг доз натриевой соли каждого соединения. Среднее значение AUC (-)-омепразола в стационарном состоянии было почти на 90% выше, чем таковое для рацемического омепразола, в то время как этот показатель для (+)-омепразола составил примерно одну треть от такового для рацемического омепразола.
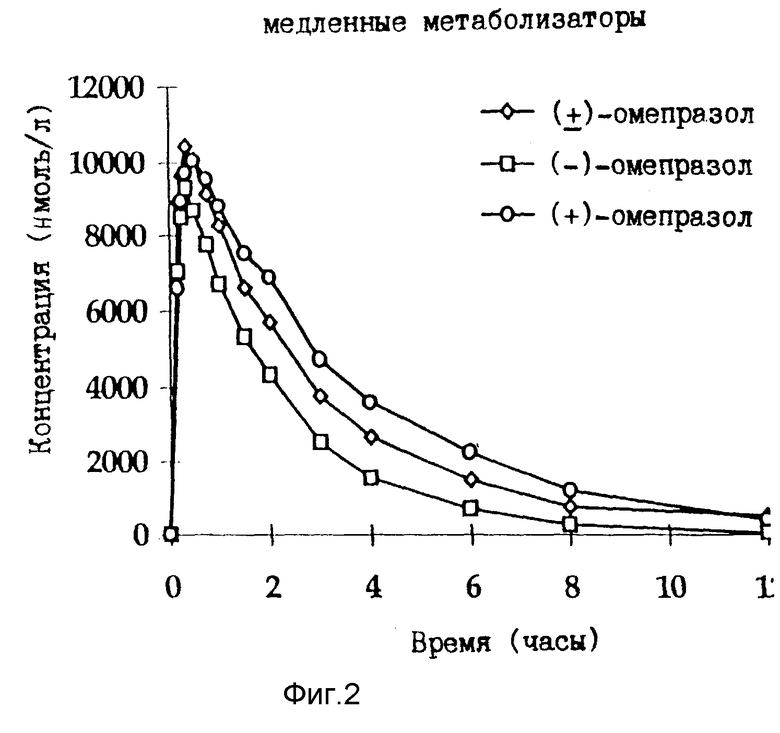
На фиг. 2 представлены средние уровни омепразола рацемата, (+)-омепразола и (-)-омепразола в плазме крови в стационарном состоянии (7 день) у медленных метаболизаторов после назначения 60 мг натриевых солей каждого соединения. У медленных метаболизаторов среднее значение AUC в стационарном состоянии (-)-омепразола было примерно на 30% ниже по сравнению с рацемическим омепразолом, в то время как AUC для (+)-омепразола было выше.
После корректировки в отношении различных доз было обнаружено, что AUC (-)-омепразола была примерно в 3 раза выше у медленных метаболизаторов по сравнению с быстрыми ("нормальными") метаболизаторами. С другой стороны, для (+)-омепразола различие в AUC между медленными и быстрыми метаболизаторами было более значительным (примерно в 30 раз). Рацемический омепразол, будучи смесью двух энантиомеров, проявляет примерно 10-кратное различие в AUC между медленными и быстрыми метаболизаторами.



Оптически чистые Na+, Mg2+, Li+, К+ или Са2+ соли (-)-5-метокси-2-[[(4-метокси-3,5-диметил-2-пиридинил) метил] сульфинил] -1H-бензимидазола обладают ингибирующей желудочную кислотную секрецию активностью. 4 c. и 13 з.п. ф-лы, 2 ил., 2 табл.
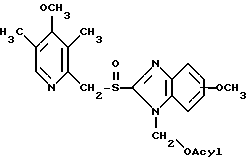
где ацил означает хиральную ацильную группу, имеющую R-или S-конфигурацию, разделяют для получения отдельных диастереоизомеров, после чего диастереомер, состоящий из ацилоксиметильного производного (-)-5-метокси-2-[[(4-метокси-3,5-диметил-2-пиридинил)метил] сульфинил] -1Н-бензимидазола, растворяют в щелочном растворе, где ацилоксиметильная группа гидролизуется с образованием (-)-5-метокси-2-[[(4-метокси-3,5-диметил-2-пиридинил)метил] сульфинил] -1Н-бензимидазола, который затем превращают в Na+, Мg2+, Li+ или Са2+ соль.
| DE 4035455 A1, 1992 | |||
| Котел для водяного отопления | 1926 |
|
SU5129A1 |
| Фазовая стоп-стартная система программного управления | 1958 |
|
SU124495A1 |
| Машковский М.Д | |||
| Лекарственные средства | |||
| -М.: Медицина, 1987, ч.2, с.30-34. | |||

