Изобретение относится к гербицидным 1-арил-4,5-дигидро-1,2,4-триазол-5(1Н)-он-ам.
Гербицидная активность некоторых 1-арил-4,5-дигидро-1,2,4-триазол-5(1Н)-онов (известных также как 1-арил- Δ2-1,2,4-триазолин-5-оны), была описана в патентной литературе.
Опубликованная патентная заявка Великобритании N 2090250 раскрывает гербицидные соединения формулы
Cl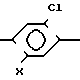 N
N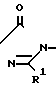 R2
R2
где R1 алкиловая группа;
R2 алкиниловая группа, галометиловая группа или галоэтиловая группа;
Х алкокси-группа, алкенилокси-группа, алкоксиалкокси-группа, алкинилокси-группа, гидрокси-группа, галометилокси-группа или галоэтилокси-группа.
Японская Kokai 58-225070 раскрывает гербицидные соединения формулы
Y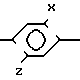 N
N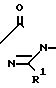 R2
R2
где R1-1-4C-алкил;
R2-H, 1-4C-алкил, галометил- или 3-4С алкинил;
Х Cl или Z; Y Cl, Br, OH или OR3;
R3 1-4C-алкил или бензил;
Z H, карбокси, цианометокси, COOR4, COSR5 или CON(R6)(R7);
R4 1-4C-алкил или 3-4С алкоксиалкил;
R5-1-4C-алкил; и R6 и R7-Н, 1-4С-алкил или алкокси.
Патент США N 431 8731 раскрывает гербицидные соединения формулы
Cl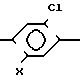 N
N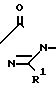 R2
R2
где R1-C1-C4-алкил;
R2-водород, С1-С6-алкил или С1-С4-алкенил;
Х гидрокси, С1-С4-алкил, С1-С6-алкилокси, алкилоксиалкилокси, из которого два алкила могут быть теми же самыми или различными и каждый алкил С1-С4, С2-С4 алкенилокси или алкилоксикарбонилалкилокси, в котором два алкила могут быть теми же самыми или различными и каждый алкил С1-С4.
Патент США N 4404019 раскрывает гербицидные соединения формулы
X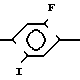 N
N R
R
где R C1-C4-алкиловая группа, С3-С4-алкениловая группа или С3-С4-циклоалкиловая группа;
Х атом хлора или брома;
Y атом водорода или С1-С4-алкокси группа.
Патент США N 4323773 раскрывает 1,2,4-триазолин-5-оны с конденсированным ядром формулы
X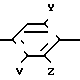 N
N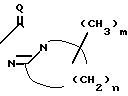
где V может быть алкилом;
Х F, Cl, Br, CN, CH3, CH3O или NO2;
J H, F, Cl, Br или CH3;
Z H, F, Cl или Br;
n=3-5;
m=0-2;
Q-Q или S.
Международная заявка РСТ N 085/01637, 1985, WO N 85/04307, 1986, WO N 86/02642, 1986, WO N 87/00730, 1987 раскрывают другие различные замещенные арил-1,2,4-триазолин-5-оны, в которых заместителями в 5-положении бензольного кольца ариловой группы являются, например, алкокси, алкинилокси, алкенилокси, тетрагидрофуранилокси или подобный гетероцикл-окси, группа формулы OR3COOR4 (где R3 может быть алкиленом или галоалкиленом и R4 может быть замещенным алкилом, алкенилом и т.п.), алкилом, цианоалкилом, COR6, CH2COR6 или CH/CH3/COR6 (где R6, например, алкокси или алкил-замещенный амино).
Соединения изобретения являются гербицидные 1-арил-4,5-дигидро-1,2,4-триазол-5(1Н)-оны, такие, в которых атом углерода в 5-положении бензольного кольца имеет заместитель (Q), где Q группа формулы CH(R2)C(R3)(R4)Ql; или -СН= C(R4)Ql в которой Ql карбоксильная группа (т.е. СOOH) или соль, эфир, амид или нитрил такой карбоксильной группы. Таким образом, Ql может быть:
CO2H,
CO2Z,
CO2R5,
CON(R6)(R7) или
CN
В другом аспекте этого изобретения Ql может быть альдегидной или кетоновой группой, например, -CHO или COR5.
Z может быть солеобразующей группой, такой, которая образует основную соль с карбоновой кислотой, например ион натрия, калия, кальция, аммония, магния, или моно-, ди-, или три-(C1-C4-алкил) аммония или ион сульфония или сульфоксония.
R5 может быть алкилом, алкоксикарбонилалкилом, циклоалкилом, например, с 3-6 атомами углерода, таким как циклопропил или циклопентил, аралкилом, таким как бензил или замещенный бензил, например, хлорбензил, алкилбензил или галоалкилбензил, такой как 4-хлорбензил или 4-трифторометилбензил.
R6 и R7 могут каждый, независимо, быть Н, ОН, алкилом, циклоалкилом, алкенилом, алкинилом, например пропинилом, алкокси, фенилом, бензилом или SO2R6 (в которых R6 другой, кроме Н), или любым из предшествующих, имеющих дополнительные заместители; такими дополнительными заместителями могут быть галоген (например, в галоалкиле, таком, как хлорэтил, галофениле, таком как хлорфенил, галобензиле, таком как хлорбензил), алкил-, или циано-.
В предшествующей формуле для Q, R2 и R3 каждый независимо может быть водородом или галогеном (таким как хлор, бром или фтор), в то время как R4 может быть Н или низшим алкилом.
Другими заместителями в гербицидных 1-арил-4,5-дигидро-1,2,4-триазол-5(1Н)-он-ах этого изобретения могут быть, например, любые из тех, которые присутствуют в гербицидных арил триазолинонах известного уровня, упомянутого выше. Например, эти другие заместители выбираются таким образом, чтобы 5-метокси и 5-пропаргилокси аналоги соединений данного изобретения были гербицидами; 5-метокси аналог соединения этого изобретения имеет формулу, идентичную формуле соединения этого изобретения во всех отношениях, за исключением того, что атом углерода кольца в 5-положении бензольного кольца имеет заместителем метокси вместо заместителя Q, как указывалось выше. Таким же образом, 5-пропаргилокси аналог идентичен, за исключением того, что углерод в 5-положении своего бензольного кольца имеет заместителем пропаргилокси вместо заместителя Q как указывалось выше. Таким образом, 5-метокси аналог соединения 3 табл. 1 ниже представляет собой 1-(2,4-дихлоро-5-метоксифенил)-4-дифторо-метил-4,5-дигидро -3-метил-1,2,4-триазол-5(Н)-он и 5-пропа- ргилокси аналог соединения 3 1-(2,4-дихлоро-5-пропаргилоксифенил)-4-дифторометил-4,5-дигидро -3-метил-1,2,4-триазол-5(Н)-он.
В соединениях данного изобретения 5-метокси аналоги и 5-пропаргилокси аналоги обладают существенными гербицидными свойствами. Например, указанные аналоги демонстрируют 50%-ное уничтожение по меньшей мере одного из следующих видов растений при применении по меньшей мере одного из следующих режимов с расходом 0,5 кг/га, а более предпочтительно, демонстрируют уничтожение на 50% по меньшей мере, при применении с расходом 0,1 кг/га.
Виды:
канатник Теофраста (Abutelon theophrasti), щетинник зеленый (Seratia viridis);
Режимы:
до появления ростков, после появления ростков. Испытание на такую гербицидную активность можно провести так, как описано ниже под заголовком "Гербицидная активность".
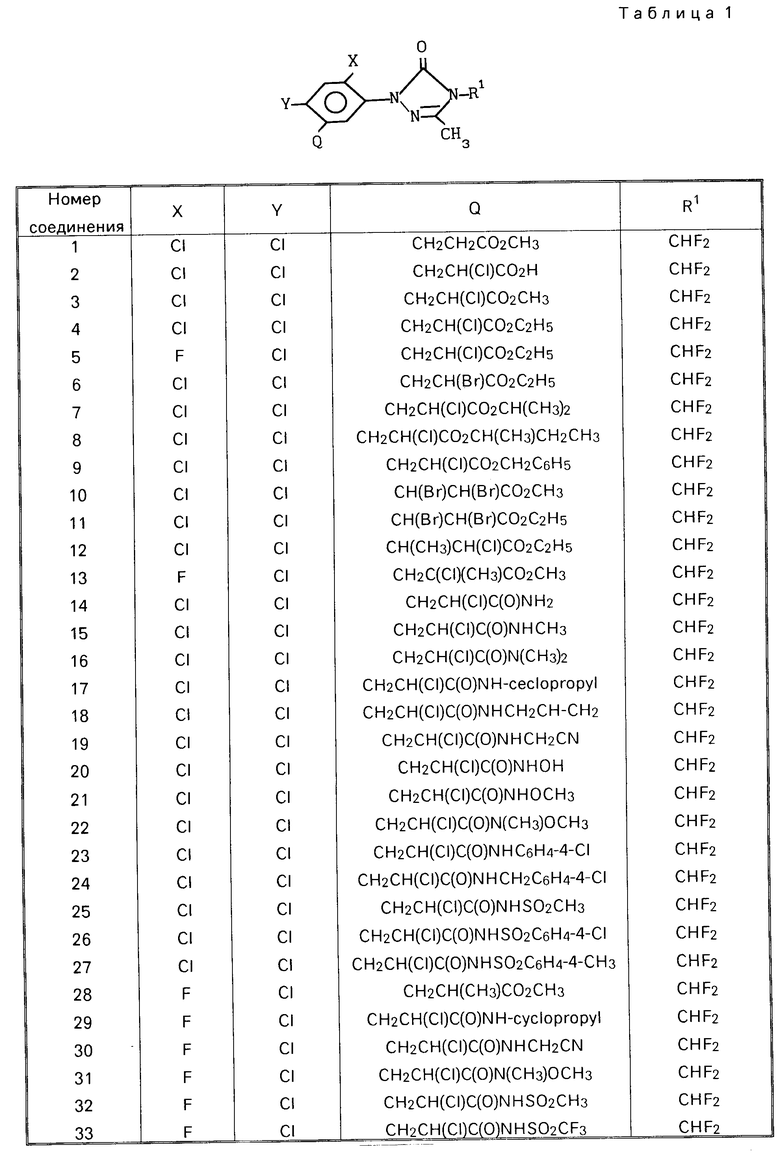
Соединения по изобретению представлены в табл.1 ниже.
Многие соединения этого изобретения можно описать формулами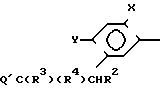 N
N N
N R1
R1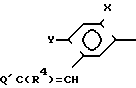 N
N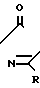 N
N R1 в которых Ql, R2, R3 и R4 имеют значения, указанные выше, и заместители R и R1 на кольце триазолинона могут быть любыми из известных в литературе, указанной выше. Например, каждый из R и R1 может независимо быть низшим алкилом (предпочтительно метилом) или гало низшим алкилом, таким, как фтором низший алкил (например CF2CHF2 или CHF2). R может быть также атомом галогена, таким как хлор. Предпочтительно, R-метил и R1-CHF2. Заместителем Х может быть водород; галоген такой как хлор, бром или фтор, предпочтительно фтор; алкил такой как низший алкил, например метил; галоалкил такой как гало низший алкил, например CF3, CH2F или CHF2; алкокси такой как низший алкокси (например метокси); или нитро; и Y может быть водород; галогеном такой как хлор, бром или фтор (предпочтительно бром или хлор); алкилом такой как низший алкил (например метил); алкокси такой как низший алкокси (например метокси); галоалкилом такой как гало низший алкил (например фторалкил); гало низшим алкилсульфонилом (например SOCF3); или гало низшим алкокси (например OCHF2). Предпочтительными в данном случае Х, Y заместителями являются: 2-F, 4-Cl; 2-F, 4-Br; 2,4-d; Cl; 2-Br, 4-Cl; и 2-F, 4-CF3.
R1 в которых Ql, R2, R3 и R4 имеют значения, указанные выше, и заместители R и R1 на кольце триазолинона могут быть любыми из известных в литературе, указанной выше. Например, каждый из R и R1 может независимо быть низшим алкилом (предпочтительно метилом) или гало низшим алкилом, таким, как фтором низший алкил (например CF2CHF2 или CHF2). R может быть также атомом галогена, таким как хлор. Предпочтительно, R-метил и R1-CHF2. Заместителем Х может быть водород; галоген такой как хлор, бром или фтор, предпочтительно фтор; алкил такой как низший алкил, например метил; галоалкил такой как гало низший алкил, например CF3, CH2F или CHF2; алкокси такой как низший алкокси (например метокси); или нитро; и Y может быть водород; галогеном такой как хлор, бром или фтор (предпочтительно бром или хлор); алкилом такой как низший алкил (например метил); алкокси такой как низший алкокси (например метокси); галоалкилом такой как гало низший алкил (например фторалкил); гало низшим алкилсульфонилом (например SOCF3); или гало низшим алкокси (например OCHF2). Предпочтительными в данном случае Х, Y заместителями являются: 2-F, 4-Cl; 2-F, 4-Br; 2,4-d; Cl; 2-Br, 4-Cl; и 2-F, 4-CF3.
В каждом аспекте изобретения часто бывает желательным, чтобы любая половина алкила, алкенила, алкинила или алкилена (такая как углеводородная половина алкокси или галоалкокси группы) имела менее 6 атомов углерода, например 1-3 или 1-4 атомов углерода, и чтобы любая циклоалкильная половина имела 3-7 кольцевых атомов углерода, предпочтительно 3-6 атомов.
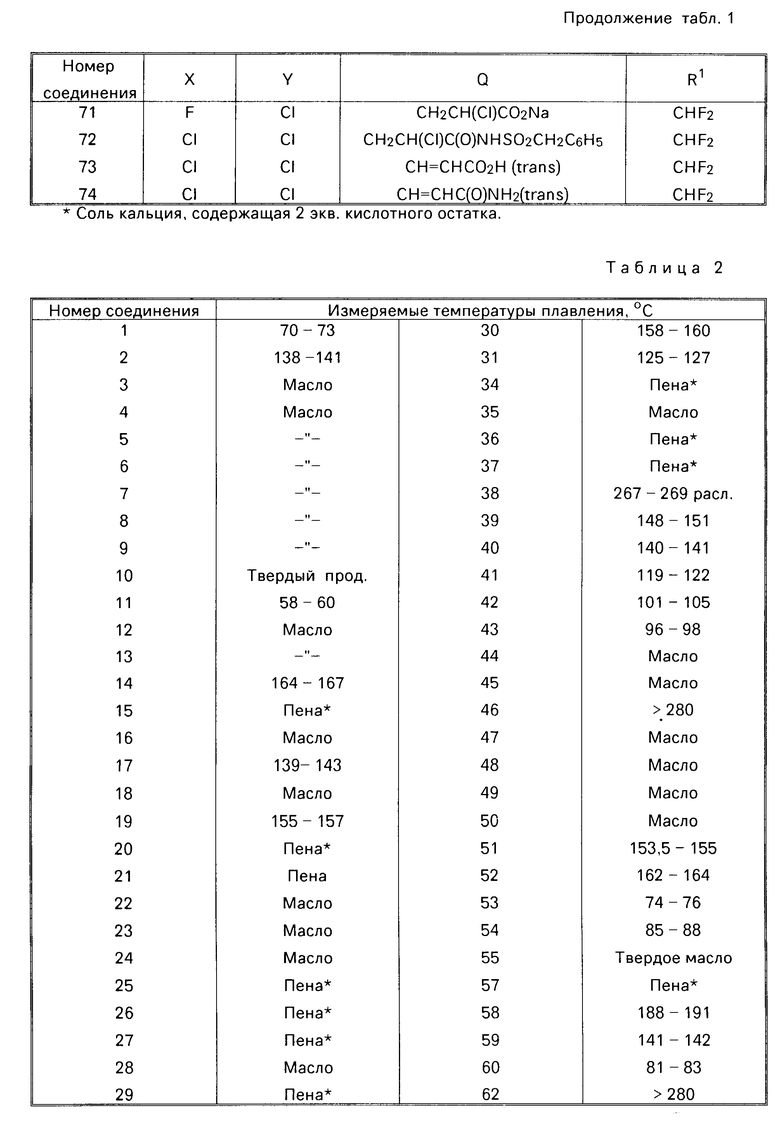
Любое кислотное соединение этого изобретения, включая сульфонамиды, в которых NR6R7-NHSO2R6, может быть преобразовано в соответствующую основную соль, такую как соль, в которой солеобразующим катионом является Z (Z то же, что указано выше).
Предлагаемые соединения можно получить способами, описанными в литературе, или в следующих примерах, либо способами аналогичными и подобными им и в соответствии с технологией.
На стадии А примеров 1 и 3 ниже, амино соединение формулы
Y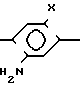 N
N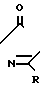 N
N R1 (такое как соединение, показанное в примере 1 Международной патентной заявки WО/87/037820, опубликованной 2 июля 1987 г) реагирует (согласно реакции арилирования Meerwein или ее модификации) с олефиновым соединением, имеющим формулу CHR2=CR4Q чтобы получить соединение по формуле I, указанной выше, где Q это -CH(R2)C(R3)(R4)Q и R3 галоген. В этом типе реакции амино соединение превращается в соль диазония, которая затем реагирует с олефиновым соединением по радикальному механизму. Реакция арилирования Meerwein приведена в статье Doyle et al in J Org. Chem. 42, 2431, 1977, которая также описывает модификацию этой реакции, в которой используются алкил нитрил и галид меди (I). Стадия А примеров 1 и 3 использует модификацию Doyle et al. Вместо этого можно использовать немодифицированную реакцию, в которой галид арендиазония вначале готовится в водном галогеновом кислотном растворе и затем смешивается с олефиновым соединением в присутствии соответствующего растворителя, например в ацетоне, с последующим добавлением соли меди, такой как хлорид меди (I).
R1 (такое как соединение, показанное в примере 1 Международной патентной заявки WО/87/037820, опубликованной 2 июля 1987 г) реагирует (согласно реакции арилирования Meerwein или ее модификации) с олефиновым соединением, имеющим формулу CHR2=CR4Q чтобы получить соединение по формуле I, указанной выше, где Q это -CH(R2)C(R3)(R4)Q и R3 галоген. В этом типе реакции амино соединение превращается в соль диазония, которая затем реагирует с олефиновым соединением по радикальному механизму. Реакция арилирования Meerwein приведена в статье Doyle et al in J Org. Chem. 42, 2431, 1977, которая также описывает модификацию этой реакции, в которой используются алкил нитрил и галид меди (I). Стадия А примеров 1 и 3 использует модификацию Doyle et al. Вместо этого можно использовать немодифицированную реакцию, в которой галид арендиазония вначале готовится в водном галогеновом кислотном растворе и затем смешивается с олефиновым соединением в присутствии соответствующего растворителя, например в ацетоне, с последующим добавлением соли меди, такой как хлорид меди (I).
Примерами олефиновых соединений с формулой CHR2=CR4Ql являются акрилат метила, акрилат этила, метакрилат метила, кротонат метила, 3-хлоракрилат метила, метакролеин, винил метил кетон, метакрилонитрил и акриламид.
Продукт, полученный реакциями, описанными выше, т.е. соединение формулы I, в которых Q -CH(R2)C(R3)(R4)Ql и в которых R3 галоген, можно подвергнуть обработке для получения других соединений данного изобретения. Отщепление галоидоводорода от этого соединения, например, гидридом натрия или другим подходящим основанием, если R2 водород, дает соединение формулы II, в которой Q-=CH=C(R4)Ql (как в примере 1В). Это соединение можно галоидировать или гидрировать для получения соединения, в котором Q-=CH(R2)C(R3)(R4)Ql и R3-H (при гидрировании, как в примере 1С), или R2 и R3 галоген (при галоидировании как в примере 2). Когда Ql- -CO2H (как получено в примере 3А), кислотное соединение формулы I можно преобразовать (как в примерах 4 и 5) в соответствующий амид, обработкой вначале таким реагентом, как хлорид тионила для получения кислотного галида (в котором Ql, например, -COCl), а затем реакцией с аммонием или амином. Вариантные способы образования амида, включающие карбодиимидное промежуточное сочетание, показаны в примерах 3В, 6 и 7. В примерах 3В и 6 амид получается из карбоновой кислоты (например формулы I) и амина в присутствии дициклогексилкарбодиимида, 1-гидроксибензотриазола и основания, такого как третичный амин, например, N,N-диизопропилэтиламин или триэтиламин, в растворителе, таком как тетрагидрофуран. В примере 7 амид получается из карбоновой кислоты и сульфонамида в присутствии 1,1l-карбоксилдиимидазола и сильного основания, такого как 1,8-диазабицикло [5,4,0]ундец-7-ен в растворителе.
Вместо начала с амино соединения (например формулы III) можно начать с другого идентичного соединения, имеющего группу СНО вместо группы NH2 и подвергнуть его реакции с реагентом Witting или таким как реагент Wadsworth-Emmons, чтобы получить соединение формулы 11. Таким образом, реагентом может быть алкилиден фосфоран, алкилиденовая группа которого имеет формулу С(R4)Ql, такой как (С6Н5)3Р= СНСО2R5 или он может быть илидом фосфоната, включающим диэфир фосфоната, в котором группа непосредственно присоединенная к атому Р, имеет формулу -СР(R4)Ql, такой как (C2H5O)2P(O)CH2CO2R5, используемый вместе с, например, NaH известным образом. R5 предпочтительно низший алкил, такой как метил или этил. Соединение формулы II может гидрироваться, чтобы получить соединение формулы I, R2 и R3 оба водород, или галоидироваться например, хлором, чтобы получить соединение формулы I, в которой R2 и R3 оба галогены. Последнее соединение может, в свою очередь, дегидрогалогенироваться, чтобы получить соединение формулы II, в которой R4 галоген, а затем гидрироваться, чтобы соединение формулы I, в которой R4 галоген; R3 и R2 H.
Получение соединения с группой СНО вместо NH2 группы формулы III, показано в примере 8.
Вместо начального соединения, содержащего триазолиноновое кольцо, и добавления к нему Q заместителя, начать можно с соединения формулы IV
Y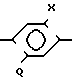 NH2 IV а затем образовать кольцо триазолинона. Соединения формулы IV показаны, например, в европейских патентных заявкахN 300387 и N 300398. NH2-группа может быть преобразована в кольцо триазолинона известным образом. Например, ее можно преобразовать в NHNH2, т.е. в гидразиновую, группу обычным образом: диазотированием с последующим восстановлением сульфитом натрия, а гидразиновую группу в кольцо триазолинона. Если Х и Y другие заместители кроме Н, такие заместители можно ввести на разных стадиях процесса. В примерах 1-8 такие заместители вводятся до образования соединения, содержащего Q заместитель. Один или оба таких заместителя можно ввести после введения Q заместителя, например заместителть-хлор в бензольном кольце можно ввести на одной из стадий галогенирования, которые модифицируют Q заместитель, как описано выше.
NH2 IV а затем образовать кольцо триазолинона. Соединения формулы IV показаны, например, в европейских патентных заявкахN 300387 и N 300398. NH2-группа может быть преобразована в кольцо триазолинона известным образом. Например, ее можно преобразовать в NHNH2, т.е. в гидразиновую, группу обычным образом: диазотированием с последующим восстановлением сульфитом натрия, а гидразиновую группу в кольцо триазолинона. Если Х и Y другие заместители кроме Н, такие заместители можно ввести на разных стадиях процесса. В примерах 1-8 такие заместители вводятся до образования соединения, содержащего Q заместитель. Один или оба таких заместителя можно ввести после введения Q заместителя, например заместителть-хлор в бензольном кольце можно ввести на одной из стадий галогенирования, которые модифицируют Q заместитель, как описано выше.
П р и м е р 1. Метил 3-[2,4-дихлоро-5-(4-дифторометил-4,5-дигидро 3-метил-5-оксо-1Н-1,2,4-триазол-1-ил)фенил]пропионат.
С т а д и я А. Метил 2-хлор-3-[2.4-дихлор-5-(4-дифтор-метил-4,5-дигидро- 3-метил-5-оксо-1Н-1,2,4-триазол-1-ил)фенил]пропио-нат.
К холодной (0оС) перемешанной смеси из 28,7 г (0,333 моль) метилового акрилата, 2,51 г (0,0244 моль) трет-бутил нитрита и 2,6 г (0,019 моль) хлорида меди (II) в 50 мл ацетонитрила был добавлен по каплям раствор 5,0 г (0,016 моль) 1-(5-амино-2,4-дихлорфенил (-4-дифторметил)-4,5-дигидро-3- метил-1,2,4-триазол-5(1Н)-она в 15 мл ацетонитрила. После полного добавления реакционная смесь согревалась до комнатной температуры и перемешивалась приблизительно 18 ч. Затем она разбавлялась 15 мл раствора 2н. соляной кислоты. Смесь экстрагировалась четырьмя частями диэтилового эфира. Соединенные экстракты высушивались в ангидридном сульфате магния, фильтровались и фильтрат выпаривался при пониженном давлении, чтобы получить масло. Масло очищалось колонной хроматографией на кремниевой геле, элюированием н-гептан: этил ацетатом (4:1), чтобы получить 5,0 г метил 2-хлор-3-[2,4-дихлоро-5-4-дифторметил-4,5-дигидро-3- метил-5-оксо-1Н-1,2,4-триазол-1-ил)фенил]пропионат в виде масла. Соединение 3 табл.1.
С т а д и я В. Метил 3-[2,4-дихлор-5-(4-дифторметил-4,5-дигидро-3-метил-5-оксо-1Н-1,2,4- триазол-1-ил)фенил]-2-пропеноат.
К перемешанному холодному раствору (0оС) 4,16 г (0,0100 моль) метил 2-хлор-3-[2,4-дихлор-5-(4-дифторметил-4,5-дигидро-3- метил-5-оксо-1Н-1,2,4-триазол-1-ил)фенил] пропионата в 15 мл N,N-диметилформамида было добавлено порциями 0,29 г (0,012 моль) гидрата натрия. После полного добавления реакционная смесь согревалась до комнатной температуры и перемешивалась 30 мин. В течение 6 ч реакционная смесь нагревалась при 60оС, затем перемешивалась при комнатной температуре приблизительно 18 ч. Реакционная смесь выливалась в ледяную воду и полученная водная смесь экстрагировалась четырьмя порциями диэтилового эфира. Экстракты объединялись и промывались последовательно водой и водным насыщенным раствором хлорида натрия. Промытая органическая фаза высушивалась в безводном сульфате магния и фильтровалась. Фильтрат выпаривался при пониженном давлении и получалась белая пена. Пена очищалась колонной хроматографией на кремниевом геле, элюировалась н-гептан: этил ацетатом (4:1), для выхода 1,63 Метил 3-[2,4-дихлор-5-(4-дифторметил-4,5-дигидро-3- метил-5-оксо-1Н-1,2,4-триазол-1-ил)фенил] -2-пропеноат в виде твердого, т.пл.148-151оС. Соединения 39 табл.1.
С т а д и я С. Метил 3-[2,4-дихлор-5-(4-дифторметил-4,5-дигидро-3- метил-5-оксо-1Н-1,2,4-триазол-1-ил)фенил]пропионат.
Гидрирование 0,59 г (0,0016 моль) метил 3-[2,4-дихлор-5-(4-дифторметил-4,5-дигидро 3- метил-5-оксо-1Н-1,2,4-триазол-1-ил)фенил]-2-пропеноата (соединение 39) приблизительно 0,2 г (0,0009 моль) окиси платины (IV) приблизительно в 15 мл этилового ацетата дали 0,59 г метил 3-[2,4-дихлор-5-(4-дифторметил-4,5-дигидро-3-метил-5-оксо-1Н- 1,2,4-триазол-1-ил)фенил] пропионата в виде чистого масла, которое при выстаивании кристаллизовалось. Кристаллы растирались в порошок с нефтяным эфиром и регенерировались фильтрацией, т.пл. 70-73оС. Соединение 1 табл.1.
П р и м е р 2. Метил 2,3-дибром-3-[2,4-дихлор-5-(4-дифторметил-4,5-дигидро-3- метил-5-оксо-1Н-1,2,4-триазол-1-ил)-фенил] пропионат.
Таким же образом 0,24 г (0,00063 моль) метил 3-[2,4-дихлор-5-(4-дифторметил-4,5-дигидро-3- метил-5-оксо-1Н-1,2,4-триазол-1-ил)фенил]-2-пропеноат (соединение 39) обрабатывалось 6 каплями брома в 15 мл тетрахлорида углерода с тем, чтобы получить 0,40 г метил 2,3-дибром-3-[2,4-дихлор-5-(4-дифторметил-4,5-дигидро-3- метил-5-оксо-1Н-1,2,4-триазол-1-ил)-фенил] пропионата в виде твердого вещества. Соединение 10 табл.1.
Спектр ЯМР совместим с предложенной структурой.
П р и м е р 3. N-циклопропил-2-хлор-3-[2,4-дихлор-5-(4-дифторметил-4,5-дигидро-3- метил-5-оксо-1Н-1,2,4-триазол-1-ил)фенил]пропионамид.
С т а д и я А. 2-хлор-3-[2,4-дихлор-5-(4-дифторметил-4,5-дигидро-3- метил-5-оксо-1Н-1,2,4-триазол-1-ил)фенил]пропионовая кислота.
К перемешанной смеси 26,3 г (0,366 моль) акриловой кислоты, 2,83 г (0,275 моль) трет-бутилового нитрита и 2,94 г (0,0220 моль) хлорида меди (II) в 75 мл ацетонитрила медленно добавлялось 5,65 г (0,0183 моль) 1-(5-амино-2,4-дихлорфенил)-4-дифтормет- ил-4,5-дигидро-3- метил-1,2,4-триазол-5(1Н)-она. Реакционная смесь перемешивалась при комнатной температуре 3 ч. Затем она выливалась в раствор 2н.соляной кислоты и все экстрагировалось диэтиловым эфиром. Органическая фаза высушивалась в безводном сульфате магния, фильтровалась и фильтрат выпаривался при пониженном давлении для выхода твердого вещества желтого цвета. Твердое вещество растиралось в порошок с водой и фильтровалось. Отжатый осадок высушивался для получения 5,9 г 2-хлор-3-[2,4-дихлор-5-(4-дифторметил-4,5-дигидро-метил-5-оксо- 1Н-1,2,4-триазол-2-ил)фенил]пропионовой кислоты. Соединение 2 табл.1.
Спектр совместим с предложенной структурой. Подготовленный подобным же образом образец соединения 2 имел температуру плавления 138-141оС.
С т а д и я В. N-циклопропил-2-хлор-3-[2,4-дихлор-5-(4-дифторметил-4,5-дигидро-3- метил-5-оксо-1Н-1,2,4-триазол-1-ил)фенил]пропионамид.
Перемешанный раствор 0,50 г (0,0013 моль) 2-хлор-3-[2,4-дихлор-5-(4-дифторметил-4,5-дигидро-3- метил-5-оксо-1Н-1,2,4-триазол-1-ил)фенил] пропионовой кислоты (соединение 2), 0,071 г (0,0013 моль) циклопропиламина, 0,17 г (0,001 моль) 1-гидроксибензотриазол гидрата и 0,18 г (0,0014 моль) N,N-диизопропилэтиламина приблизительно в 15 мл тетрагидрофурана охлаждался до 0оС. К этой холодной смеси добавлялось 0,26 г (0,0013 моль) 1,3-дициклогексилкарбодиимида. После полного добавления реакционная смесь выстаивалась до комнатной температуры и перемешивалась приблизительно 18 ч. Реакционная смесь фильтровалась. Фильтрат разбавлялся четыреххлористым углеродом и промывался последовательно в растворе 1н.соляной кислоты, в водном 10%-ном растворе гидроксида натрия, воде и водном насыщенном растворе хлорида натрия. Органическая фаза высушивалась безводным сульфатом магния, фильтровалась и фильтрат выпаривался при пониженном давлении для получения 0,43 г N-циклопропил-2-хлоро-3-[2,4-дихлоро-5-(4-дифторметил- 4,5-дигидро-3-метил-5-оксо-1Н-1,2,4-триазол-1-ил) фенил] пропионамида как твердого, с температурой плавления 139-143оС, соединения 17 табл.1.
ЯМР- и ИК-спектры соответствовали предложенной структуре.
П р и м е р 4. N-метил-N-метокси-2-хлоро-3-[2,4-дихлоро-5-(4-дифторометил- 4,5-дигидро-3-метил-5-оксо-1Н-1,2,4-триазол-1- ил)фенил]пропионамид.
Смесь 0,50 г (0,0013 моль) 2-хлоро-3-[2,4-дихлоро-5-(4-дифторометил-4,5-дигидро-3- метил-5-оксо-1Н-1,2,4-триазол-1-ил)фенил]про-пионовой кислоты (соединение 2) и 5 мл тионил хлорида перемешивалась при нагреве с обратным холодильником в течение 3 ч. Смесь охлаждалась и избыток тионилхлорида удалялся дистилляцией при пониженном давлении, оставляя остаток. Остаток прибавляли к холодному раствору 0,13 г (0,0014 моль)N, 0-диметилгидроксиламин гидрохлорида и 0,11 г (0,0014 моль) пиридина в 20 мл тетрагидрофурана. Полученная смесь перемешивалась при комнатной температуре приблизительно 18 ч. Реакционная смесь разбавлялась диэтиловым эфиром и промывалась последовательно 1 н. раствором соляной кислоты водой и водным насыщенным раствором хлорида натрия. Промытая органическая фаза высушивалась безводным сульфатом магния и фильтровалась. Фильтрат выпаривался при пониженном давлении для получения 0,37 г N-метил-N-метокси-2-хлоро-3-[2,4-дихлоро-5-(4-дифторометил- 4,5-дигидро-3-метил-5-оксо-1Н-1,2,4-триазол-1-ил)фенил]пропионами- да в виде масла. Соединение 22 табл.1.
ЯМР- и ИК-спектры были совместимы с предложенной структурой.
П р и м е р 5. N-метилсульфонил-2-хлоро-3-[2,4-дихлоро-5(4-дифторометил- 4,5-дигидро-3-метил-5-оксо-1Н-1,2,4-триазол-1-ил) фенил] пропионамид.
Таким же образом, как в примере 4, реакция 0,50 г (0,0013 моль) 2-хлоро-3-[2,4-дихлоро-5-(4-дифторометил-4,6-дигидро-3- метил-5-оксо-1Н-1,2,4-триазол-1-ил)фенил] пропионовой кислоты (соединение 2) с 5 мл тионилхлорида дает остаток. К этому остатку добавляли 0,50 г (0,0052 моль) метансульфонамида. Смесь перемешивалась и нагревалась при 120оС 2 ч. Смесь охлаждалась, разбавлялась метиленхлоридом, и полученный осадок удалялся с помощью фильтрации. Фильтрат промывался водой. Органическая фаза высушивалась безводным сульфатом магния, фильтровалась и фильтрат выпаривался при пониженном давлении для получения 0,21 г N-метилсульфонил-2-хлоро-3-[2,4-дихлоро-5-(4-дифторо- метил- 4,5-дигидро-3-метил-5-оксо-1Н-1,2,4-триазол-1-ил)фенил]пропионамида в виде пены. Соединение 25 табл.1.
ЯМР- и ИК-спектры были совместимы с предложенной структурой.
П р и м е р 6. 2-хлоро-3-[2,4-дихлоро-5-(4-дифторометил-4,5-дигидро-3-метил-5- оксо-1Н-1,2,4-триазол-1-ил)фенил]-N-(4-хлоро- фенил)пропионамид.
Перемешанный раствор 0,50 г (0,0013 моль) 2-хлоро-3-[2,4-дихлоро-5-(4-дифторометил-4,5-дигидро-3-метил-5- оксо-1Н-1,2,4-триазол-1-ил)фенил] пропионовой кислоты (соединение 2), 0,16 г (0,0013 моль) 4-хлоранилина, 0,17 г (0,0013 моль) 1-гидроксибензотриазол гидрата и 0,18 г (0,0014 моль) N,N-диизопропилэтиламина приблизительно в 15 мл тетрагидрофурана охлаждался до 0оС. К этой холодной реакционной смеси было добавлено 0,26 г (0,0013 моль) 1,3-дициклогексилкарбодиимида. После полного добавления реакционная смесь нагревалась до комнатной температуры и перемешивалась приблизительно 18 ч. Реакционная смесь фильтровалась. Фильтрат разбавлялся четыреххлористым углеродом и промывался последовательно раствором 1 н.соляной кислоты, водным
10%-ным раствором гидроксида натрия, водой и насыщенным водным раствором хлорида натрия. Органическая фаза высушивалась безводным сульфатом магния, фильтровалась и фильтрат выпаривался при пониженном давлении для получения 0,28 г 2-хлоро-3-[2,4-дихлоро-5-(4-дифторометил-4,5-дигидро-3- метил-5-оксо-1Н-1,2,4-] -N-(4-хлорофенил)пропионамид в виде масла. Соединение 23 табл. 1.
ЯМР- и ИК-спектры были совместимы с предложенной структурой. 10%-ным раствором гидроксида натрия, водой и насыщенным водным раствором хлорида натрия. Органическая фаза высушивалась безводным сульфатом магния, фильтровалась и фильтрат выпаривался при пониженном давлении для получения 0,28 г 2-хлоро-3-[2,4-дихлоро-5-(4-дифторометил-4,5-дигидро-3- метил-5-оксо-1Н-1,2,4-триазол-1-ил)фенил] -N-(4-хлорофе- нил)пропионамид в виде масла. Соединение 23 табл.1.
ЯМР- и ИК-спектры были совместимы с предложенной структурой.
П р и м е р 7. 2-хлор-3-[2-хлор-4-фтор-5-(4-дифторметил-4,5-дигидро-3-метил-5- оксо-1Н-1,2,4-триазол-1-ил)фенил]-N- (4-метилфенилсульфонил)пропионамид.
К перемешанному раствору 0,19 г (0,0012 моль) 1,1l-карбонилдиимидазола в 3 мл тетрагидрофурана был добавлен раствор 0,45 г (0,0012 моль) 2-хлор-3-[2-хлор-4-фтор-5-(4-дифторметил-4,5-дигидро-3- метил-5-оксо-1Н-1,2,4-триазол-1-ил)фенил] пропио-новой кислоты, полученной способом примера 3 (стадия А) из 1-(5-амино-4-хлор-2-фторфенил)-4-дифторметил-4,5-дигидро-3- метил-1,2,4-триазол-5(1Н)-она в 5 мл тетрагидрофурана.
Реакционная смесь разбавлялась 5 мл тетрагидрофурана. Смесь перемешивалась при комнатной температуре 30 мин, затем нагревалась с обратным холодильником 30 мин. Реакционная смесь охлаждалась до комнатной температуры и добавлялось 0,20 г (0,0012 моль) пара-толуол-сульфонамида. Смесь перемешивалась приблизительно 10 мин и добавлялось 0,17 г (0,0012 моль) 1,8-диазабицикло 5.4.0 ундец-7-ена. Полученная смесь перемешивалась при комнатной температуре приблизительно 18 ч. Реакционная смесь разделялась между диэтиловым эфиром и 1 н.раствором соляной кислоты. Органическая фаза последовательно промывалась водой и водным насыщенным раствором хлорида натрия. Промытая органическая фаза высушивалась безводным сульфатом магния, фильтровалась и фильтрат выпаривался при пониженном давлении с получением остатка. Этот остаток очищался колонной хроматографией на кремниевом геле, элюировался н-гептан этанол хлороформом (1:1:1) для выхода 0,23 г 2-хлор-3-[2-хлор-4-фтор-5(4-дифторметил-4,5-дигидро-3- метил-5-оксо-1Н-1,2,4-триазол-1-ил)фенил]-N-(4-метилфе- нилсульфонил) пропионамида в виде твердого вещества, т.пл. 267-269оС. Соединение 38 табл.1.
Спектр ЯМР совместим с предложенной структурой.
П р и м е р 8. Этил 3-[2,-хлор-4-фтор-5-(4-дифторметил-4,5-дигидро-3- метил-5-оксо-1Н-1,2,4-триазол-1-ил)фенил]пропеноат.
С т а д и я А 2-(2-хлор-4-фтор-5-нитрофенил)-1,3-дитиан.
К раствору 53,2 г (0,261 моль) 2-хлор-4-фтор-5-нитробензальдегида в 800 мл хлорида метилена добавлялось 42,2 г (0,390 моль) 1,3-пропандитиола. К смеси добавлялся эфират трехфтористого бора (6,4 мл, 0,052 моль). Полученная смесь перемешивалась в атмосфере сухого азота при комнатной температуре приблизительно 48 ч. Добавлялось дополнительное количество эфирата трехфтористого бора и 1,3-пропанэдитиола, поскольку анализ смеси реакции тонкослойной хроматографией показал наличие 2-хлор-4-фтор-5-нитробензальдегида. Полученная смесь дополнительно перемешивалась еще 5 ч. Реакционная смесь разбавлялась 300 мл водного 5%-ного раствора гидроокиси натрия. Органическая фаза высушивалась безводным сульфатом магния и фильтровалась. Фильтрат выпаривался при пониженном давлении, оставляя твердый осадок. Это твердое вещество растворялось в смеси хлорида метилена и н-гептане, после чего твердое вещество кристаллизовалось. Далее оно удалялось фильтрацией и фильтрат выпаривался при пониженном давлении, оставляя 56,9 г твердого вещества. Его анализ ЯМР-спектроскопией показал, что оно состоит на 90% из 2-(2-хлор-4-фтор-5-нитрофенил)-1,3-дитиана и на 10% из 1,3-пропандитиола.
С т а д и я В. 2-(5-амино-2-хлор-4-фторфенил)-1,3-дитиан.
К перемешанной смеси 20,0 г (0,0681 моль) 2-(2-хлор-4-фтор-5-нитрофенил)-1,3-дитиана в 150 мл уксусной кислоты было добавлено 75 мл тетрагидрофурана. Порциями добавлялся железный порошок (15,8 г, 0,269 моль). После полного добавления реакционная смесь нагревалась до 50оС приблизительно 30 мин. Затем она охлаждалась в ледяной ванне и разбавлялась диэтиловым эфиром. Полученная смесь фильтровалась через фильтрующую подушку из целита. К фильтрату добавлялась вода и удалялась органическая фаза. К органической фазе добавлялся водный раствор бикарбоната натрия с энергичным перемешиванием, до тех пор пока смесь не стала слегка основной. Водная фаза отделялась от органической фазы и удалялась. Водная фаза экстрагировалась диэтиловым эфиром и экстракты добавлялись к органической фазе. Этот органический раствор высушивался безводным сульфатом магния и фильтровался. Фильтрат выпаривался при пониженном давлении для выхода 13,5 г 2-(5-амино-2-хлор-4-фторфенил)-1,3-дитиана в виде твердого вещества, т.пл. 112-115оС.
ЯМР-спектр совместим с предложенной структурой.
С т а д и я С. Ацетальдегид 4-хлор-2-фтор-5-(1,3-дитиан-2-ил)фенилгидразон.
К перемешанной холодной смеси (-5оС) 10,0 г (0,0379 моль) 2-(5-амино-2-хлор-4-фторфенил)-1,3-дитиана в 100 мл концентрированной соляной кислоты по каплям добавлялся раствор 2,55 г (0,0379 моль) нитрита натрия в 20 мл воды. Эта смесь перемешивалась при -5оС приблизительно 45 мин. По каплям добавлялся раствор 17,1 г (0,0758 моль) дигидрата хлорида олова (II) в 30 мл концентрированной соляной кислоты. Эта смесь перемешивалась в течение 1 ч. Медленно добавлялся раствор 5,16 г (0,117 моль) ацетальдегида в 200 мл воды. Полученная смесь перемешивалась 1 ч, во время которого образовался осадок. Этот осадок собирался фильтрацией и промывался водой и высушивался для выхода ацетальдегид 4-хлор-2-фтор-5-(1,3-дитиан-2- -ил)-фенилгидразона.
С т а д и я D. 1-[4-хлор-2-фтор-5-(1,3-дитиан-2-ил)-фенил]-4,5-дигидро-3- метил-1,2,4-триазол-5(1Н)-он.
К перемешанной смеси 5,00 г (0,0145 моль) ацетальдегид 4-хлор-2-фтор-5-(1,3-дитиан-2-ил)-фенилгидразона в 50 мл уксусной кислоты добавлялся по каплям раствор 1,38 г (0,017 моль) цианата калия в 5 мл воды. Смесь перемешивалась при 15оС примерно 1,5 ч. Можно добавить дополнительно водного раствора цианата калия, если анализ реакционной смеси тонкослойной хроматографией показывает присутствие ацетальдегид 4-хлор-2-фтор-5-(1,3-дитиан-2- -ил)фенилгидразона. При поддержании температуры 15оС, добавлялось 30 мл водного 5% -ного раствора гипохлорита натрия. Эта смесь перемешивалась при 15оС примерно 1 ч. Растворители удалялись дистилляцией при пониженном давлении для получения осадка. Этот осадок растворялся в этил ацетате и промывался последовательно водным насыщенным раствором бикаpбоната натрия, водой и водным насыщенным раствором хлорида натрия. Промытый органический раствор высушивался безводным сульфатом магния и фильтровался. Фильтрат выпаривается при пониженном давлении для выхода 1-[4-хлор-2-фтор-5-(1,3-дитиан-2-ил)фенил]-4,5-дигидро-3-метил- 1,2,4-триазол-5(1Н)-она.
Перемешанная смесь 2,5 г (0,0072 моль) 1-[4-хлор-2-фтор-5-(1,3-дитиан-2-ил)фенил] -4,5-дигидро-3- метил-1,2,4-триазол-5(1Н)-она и 3,0 г (0,022 моль) безводного карбоната калия в 30 мл безводного диметилформамида нагревалась при 90оС в сухой атмосфере водорода. Хлордифторметан (газ) пропускался пузырьками через смесь, до тех пор, пока флегма газа не появлялась в конденсаторе сухой лед/ацетон, который находился на колбе реакции. После примерно 1 ч реакционная смесь охлаждалась и выливалась приблизительно в 300 мл холодной воды, образуя осадок. Этот осадок собирался фильтрацией, промывался водой и высушивался для выхода 1-[4-хлор-2-фтор-5-(1,3-дитиан-2-ил)фенил]-3-дифторметил- 4,5-дигидро-3-метил-1,2,4-триазол-5(1Н)-она.
С т а д и я F. 1-(4-хлор-2-фтор-5-формилфенил)-4-дифторметил-4,5-дигидро-3- метил-1,2,4-триазол-5(1Н)-он.
Смесь из 2,0 г (0,0051 моль) 1-[4-хлор-2-фтор-5-(1,3-дитиан-2-ил)фенил] -4-дифтор-метил- 4,5-дигидро-3-метил-1,2,4-триазол-5(1Н)-она в 25 мл ацетона и 25 мл ацетонитрила медленно добавлялась к перемешанному холодному (0оС) раствору 5,5 г (0,031 моль) бромсукцинимида в 80 мл ацетонитрила и 20 мл воды. Реакционная смесь перемешивалась при 0оС в течение 1 ч. Далее добавлялось приблизительно 15 мл водного насыщенного раствора бисульфита натрия. Добавлялась смесь из 25 мл хлорида метилена и 25 мл н-гептана, и смесь встряхивалась в разделительной воронке. Органическая фаза удалялась и последовательно промывалась водным насыщенным раствором бикарбоната натрия, водой и водным насыщенным раствором хлорида натрия. Промытая органическая фаза высушивалась безводным сульфатом магния и фильтровалась. Фильтрат выпаривался при пониженном давлении для получения остатка. Этот остаток очищался колонной хроматографией на кремниевом геле для выхода 1-(4-хлор-2-фтор-5-формилфенил)-4-дифторме-тил-4,5-дигидро-3- метил-1,2,4-триазол-5(1Н)-она.
С т а д и я G. Этил 3-[2-хлор-4-фтор-5-(4-дифтор-метил-4,5-дигидро-3- метил-5-оксо-1Н-1,2,4-триазол-1-ил)фенил]пропеноат.
К перемешанному раствору 1,0 г (0,0034 моль) 1-(4-хлор-2-фтор-5-формилфенил(-4-дифторметил-4,5-дигидро-3- метил-1,2,4-триазол-5(1Н)-она в 15 мл толуола добавлялось 1,2 г (0,0034 моль) карбэтоксиметилен/трифенилфосфорана. Реакционная смесь перемешивалась при комнатной температуре около 3 ч и затем нагревалась с обратным холодильником около 5 ч. Затем она охлаждалась и разбавлялась диэтиловым эфиром. Эта смесь последовательно промывалась водой, 1н.соляной кислотой, водным насыщенным раствором бикарбоната натрия и водным насыщенным раствором хлорида натрия. Промытая органическая фаза высушивалась безводным сульфатом магния и фильтровалась. Фильтрат выпаривался при пониженном давлении для получения остатка. Этот остаток очищался колонной хроматографией на кремниевом геле для выхода этил-3-[2-хлор-4-фтор-5-(4-дифторметил-4,5-дигидро-3- метил-5-оксо-1Н-1,2,4-триазол-2-ил)фенил]про- пеноата.
Гербицидная активность.
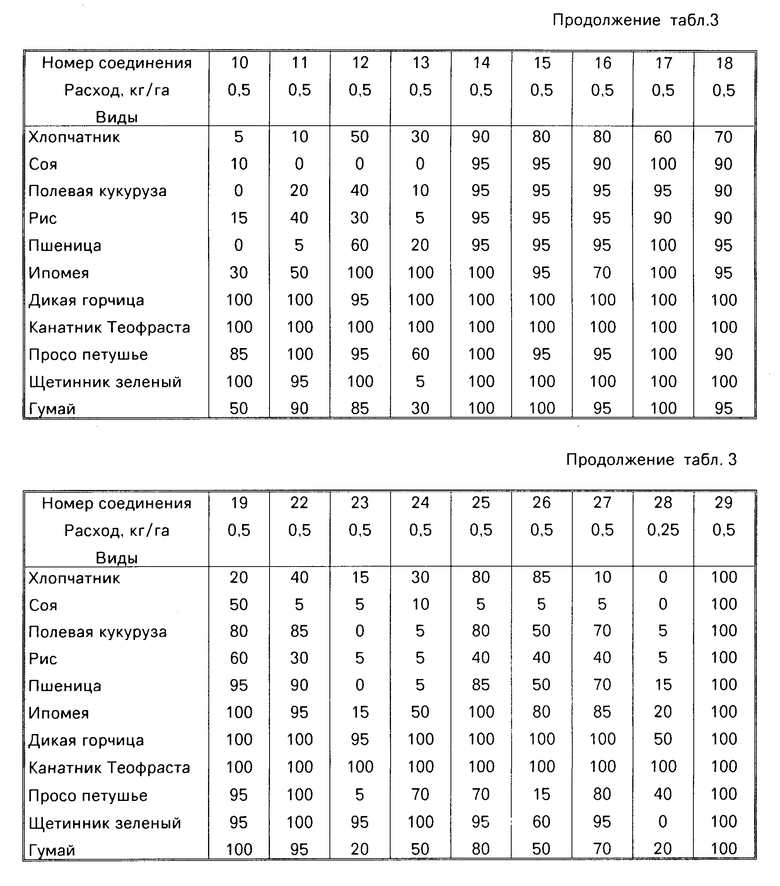
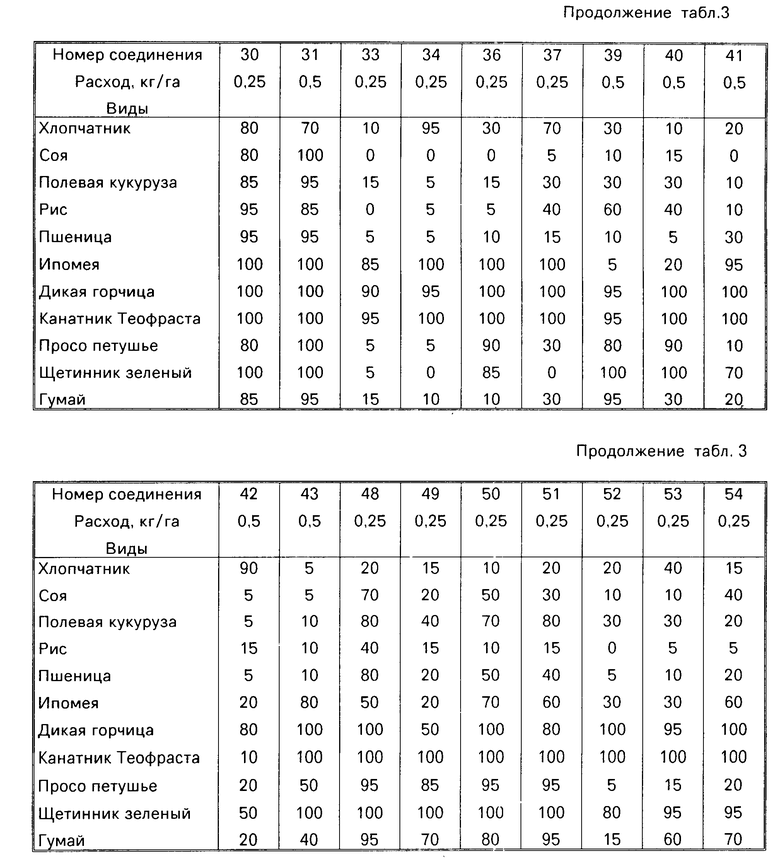
Испытываемые виды растений, используемых для демонстрации гербицидной активности соединений этого изобретения, включают хлопчатник (Gossypium hirsutum), соевые бобы (Glycine max var. Williams), кукуруза полевая (Zea mays var. Pioneer 3732), пшеница (Triticum aesrivium var. Wheaton), рис (Oryza sativa var. Labelle), ипомея (Ipomea lacumosa or Ipomea hederacea), дикая горчица (Brassica kaber), канатник Теофраста (Abutilon theophrasti), просо петушье (Echinochloa crus-galli) щетинник зеленый (Seratia viridis) и гумай (Sorghum halepense).
Подготовка корзин.
До появления всходов. Две корзины одноразового использования (8 смх15 смх25 см) для каждого расхода применения для каждого вида гербицида в испытании до появления ростков заполнялись приблизительно на глубину 6,5 см стерилизованной паром супесчанной почвой. Почва выравнивается и шаблоном делаются шесть расположенных на одинаковом расстоянии канавок длиной 13 см и глубиной 0,5 см в обеих корзинах. В пять канавок первой корзины высаживаются семена хлопчатника, сои, кукурузы, риса и пшеницы (шестая остается пустой), а семена дикой горчицы, ипомеи, канатника Теофраста, проса петушьего, щетинника зеленого и гумая высаживаются в шесть канавок второй корзины. С помощью шаблона семена вдавливаются в землю. По верху каждой корзины на глубину 0,5 см однородно наносится слой почвы, состоящий из равных частей песка и супеска. Корзины вначале поливаются водой, а затем раствором испытуемого соединения, как описывается ниже.
После появления ростков. Для данного режима также готовятся 2 корзины для каждого расхода каждого вида гербицида. Эти корзины готовятся также, как и предыдущие. Подготовленные корзины поливаются в течение 8-11 дней, затем листва появившихся растений спрыскивается раствором испытываемого соединения, как описывается далее.
Применение гербицидов.
В обоих режимах испытаний гербициды применяются в виде водных ацетоновых растворов, обычно с расходами, уменьшенными последовательно вдвое: 8 кг/га, 4 кг/га, 2 кг/га и т.д.
Четыре корзины (2 до и 2 после появления ростков) помещаютcя вместе и опрыскиваются 30 мл испытуемого раствора, содержащего соответствующее количество испытуемого соединения, т.е. приблизительно 7,5 мл раствора распрыскивается на каждую из 4-х корзин. В применении до появления ростков распрыскивание делается на поверхность почвы, а в другом случае на листву. После обработки 2 корзины с семенами до появления ростков регулярно поливаются в течение 2-х недель, и в это время регистрируются данные прототоксичности. В тесте с ростками листва выдерживается сухой 24 ч после обработки, затем регулярно поливается 2 недели и регистрируются данные прототоксичности.
Подготовка испытываемых растворов.
Для корзин указанного размера расход 8,0 кг/га активного ингредиента эквивалентен 0,06 г активного ингредиента на 1 корзину (0,24 г на 4 корзины). Весь раствор 0,48 г данного гербицида в 60 мл 50:50 смеси воды и ацетона, содержащей 0,5% (об/об.) эмульгатора/растворителя монолаурата сорбитана, делится на 2 части по 30 мл, каждая содержит 0,24 г данного гербицида. При расходе 8,0 кг/га одна из 30 мл частей разбрызгивается неразбавленной на 4 корзины (7,5 мл/корз. ). Вторая часть 30 мл разбавляется еще 30 мл водной ацетоновой эмульгаторной смесью, с тем чтобы получить 60 мл раствора, содержащего 0,24 г данного гербицида. Опять этот раствор делится на 2 части по 30 мл с содержанием в каждой 0,12 г данного гербицида. Одна часть используется для применения без дальнейшего разбавления для 4-х корзин с расходом 4 кг/га. Вторая часть 30 мл еще разбавляется равным количеством вышеупомянутой смеси и полученные 60 мл раствора с 0,12 г делятся на 2 равные части с содержанием в каждой 0,06 г гербицида. Одна часть в 30 мл (0,06 г) используется с расходом 2,0 кг/га, а другая используется в получении испытуемых растворов с той же самой серийной техникой разбавления и с меньшей концентрацией.
Данные фитотоксичности берутся в виде процентного контроля. Процентный контроль определяется способом, сходным с системой соотношения 0:100 (см. "Research Methods in Weed Science", 2nd ed. B.Truelove, Ed. Southern Weed Science Society, Auburn Vniversity, Auburn, Alabama 1977).  ый
ый  Опиcание культуры Опиcание cорняка 0 Эффекта нет
Опиcание культуры Опиcание cорняка 0 Эффекта нет 
 роля 10
роля 10  -
-  20
20  шой
шой 
 ы
ы 30
30 
 40
40  я/
я/  очный 50
очный 50  ный
ный 
 чный 60
чный 60  дение
дение  й 70
й 70  и
и п
п ние
ние  м 80 Сильный
м 80 Сильный  п
п бла,
бла,  в
в т
т 90
90  т
т о единичные
о единичные  р
р 100 Полный эффект Полная гибель культуры
100 Полный эффект Полная гибель культуры  ничтожение
ничтожение
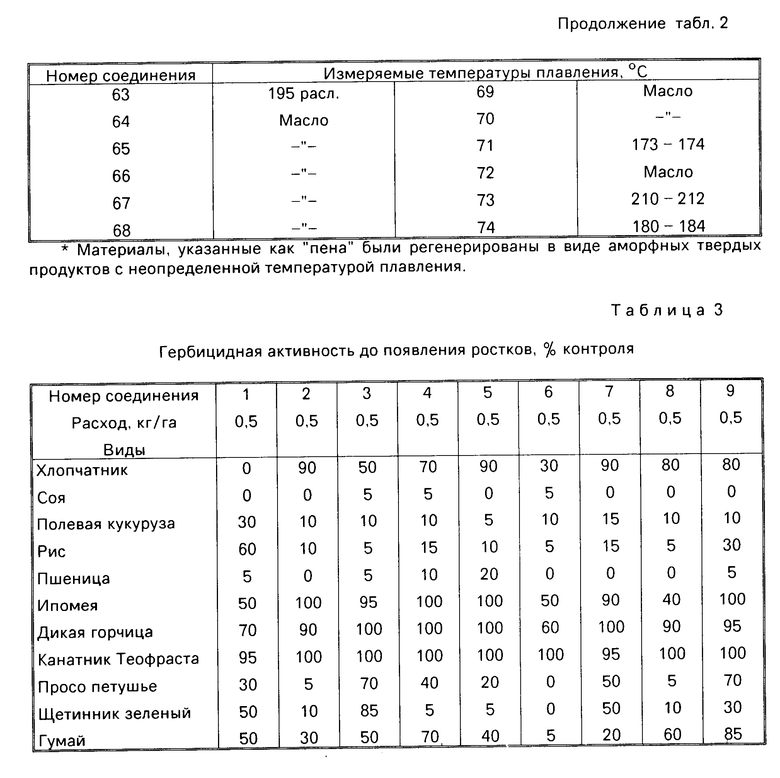
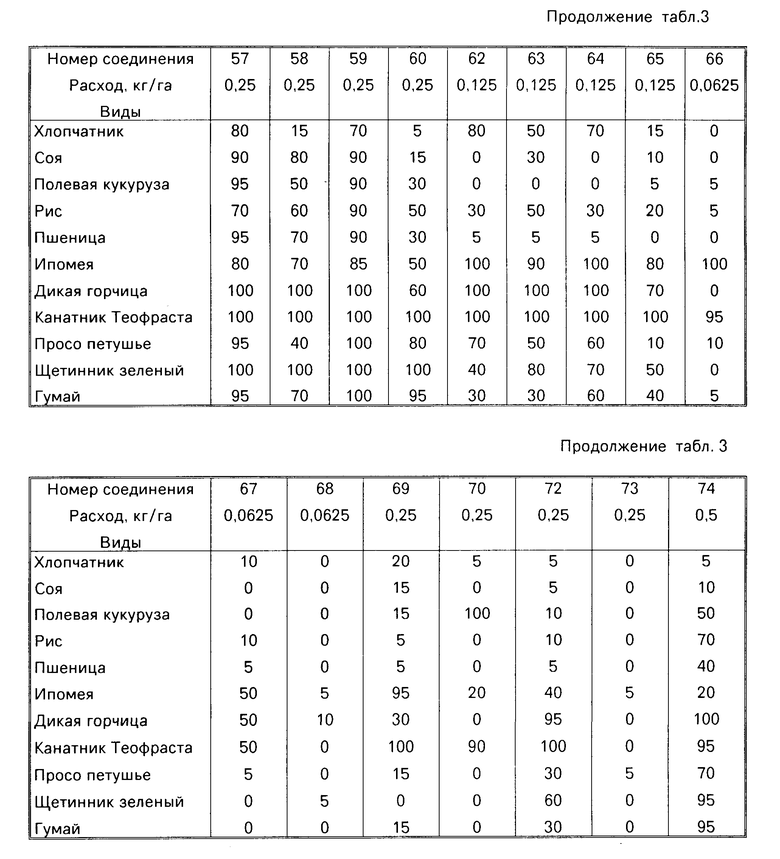
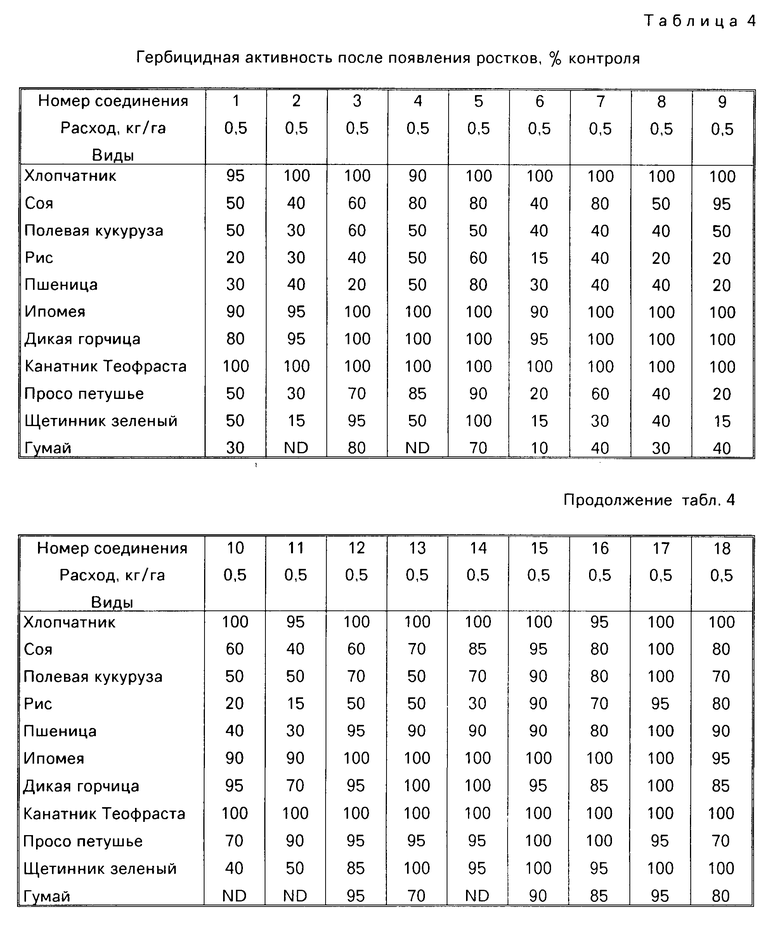
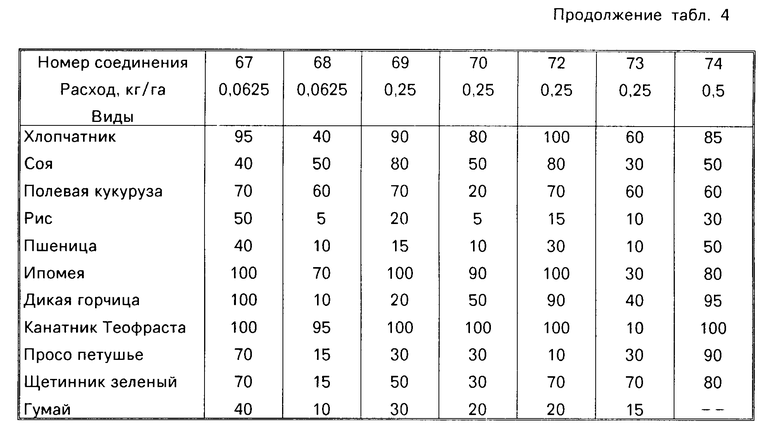
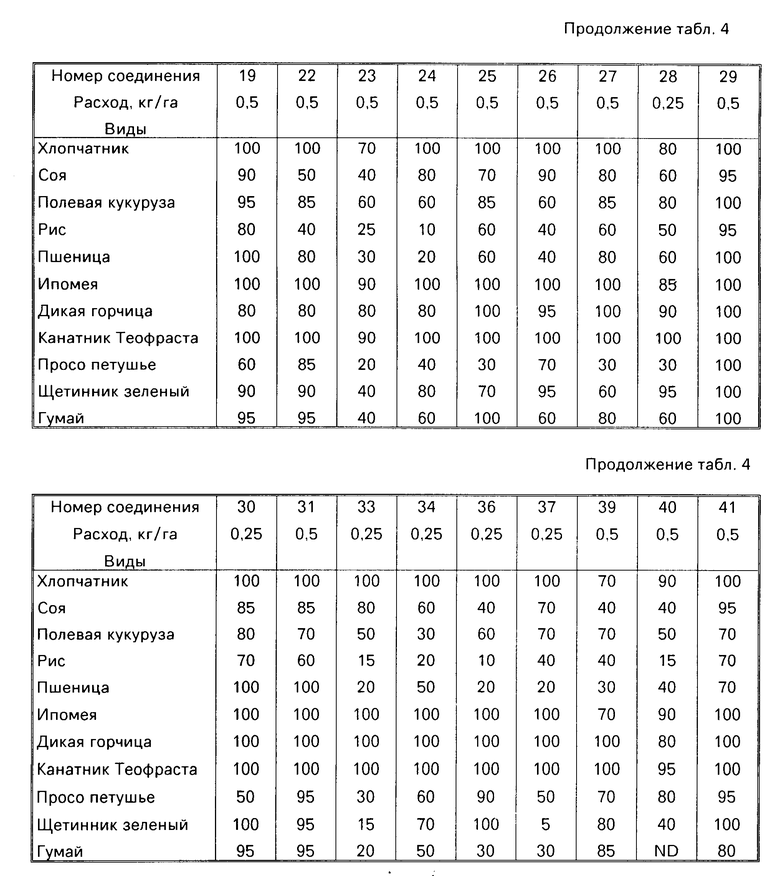
Гербицидные данные при выбранных нормах расхода для различных соединений по изобретению даются в табл.3 и 4. Испытываемые соединения указаны в табл.3 и 4 по номерам, которые соответствуют номерам табл.1.
При применении гербицидов активные соединения формируют в гербицидные композиции, смешивая их со стимуляторами и носителями, используемыми обычно в данной области техники для облегчения диспергирования активных ингредиентов, с учетом того факта, что композиция и режим применения ядовитого вещества может влиять на активность материала в данном применении. Таким образом, для сельскохозяйственного использования настоящие гербицидные соединения можно формировать в виде гранул относительно большого размера, гранул водорастворимых или вододиспергируемых, в виде мелко размолотого порошка, смачиваемого порошка, эмульгирующихся концентратов, в виде растворов или других формаций в зависимости от желаемого режима использования.
Эти гербицидные соединения можно применять в виде водорастворимых аэрозолей, пылей или гранул, в областях, где требуется подавление роста растительности. Эти композиции могут содержать от 0,1% 0,2% 0,5% до 95% по массе активного ингредиента.
Рустами являются свободно текучие смеси активного ингредиента с сильно измельченными твердыми веществами, такими как тальк, природные глины, кизельгур, порошками, такими как из семян хлопчатника и скорлупы ореха и другими органическими и неорганическими твердыми веществами, которые являются дисперсантами и носителями ядовитого вещества. Эти мелко измельченные вещества имеют средний размер частиц менее 50 мк. Типичной пылевой рецептурой является содержащая 1,0 ч. или меньше гербицидного соединения и 99,0 ч. талька. Смачиваемые порошки также удобны для гербицидов, применяемых до и после появления ростков. Они имеют вид мелко измельченных частиц, которые легко диспергируются в воде или другом дисперсанте. Смачиваемый порошок применяется на почве либо в виде сухой пыли, либо в виде эмульсии в воде или другой жидкости. Типичные носители для таких порошков включают грунт фуллера, каолиновые глины, кремнеземы и другие высокоабсорбентные легко смачиваемые неорганические растворители. Содержание смачиваемых порошков обычно включает около 5-80% активного ингредиента, в зависимости от абсорбции носителя, а также небольшое количество смачиваемого, диспергирующего или эмульгирующего агента для облегчения диспегирования. Например, смачиваемый порошок содержит 80,8 ч. гербицидного соединения, 17,9 ч. глины Палметто и 1,0 ч. лигносульфоната натрия и 0,3 ч. сульфонированного алифатического полиэфира как смачивающих агентов.
Далее приведены другие рецептуры смачиваемых порошков, мас.
Компонент Активный ингредиент 40,00 Лигносульфонат натрия 20,00 Аттапульгитовая глина 40,00
Всего 100,00
Компонент Активный ингредиент 90,00 Сульфосукцинат диоктил натрия 0,10 Синтетический мелкий кремнезем 9,90
Всего 100,00 Компонент Активный ингредиент 20,00 Алкилнафталинсульфонат натрия 4,00 Лигносульфонат натрия 4,00 Метил целлюлоза с низкой вязкостью 3,00 Аттапульгитовая глина 69,00
Всего 100,00 Компонент Активный ингредиент 25,00 Основание: 75,00 96%-ный гидратный алюминий магний силикат 2%-ный порошкообразный лигносульфонат натрия 2%-ный порошкообразный анионный алкилнафталинсульфонат натрия
Всего 100,00
Часто к смеси для применения после появления ростков добавляется дополнительный смачивающий агент и/или масло с тем, чтобы облегчить разбрызгивание на листву и поглощение растением.
Другие рецептуры для гербицидных применений представляют собой эмульгируемые концентраты (ЭК). Это гомогенные жидкости или пасты, способные диспергироваться в воде или другом дисперсанте, и могут состоять целиком из гербицидного соединения и жидкости или твердого эмульгирующего агента, или также могут включать жидкий носитель, такой как ксилол, тяжелые ароматические лигроины, изофорон или другой нелетучий органический растворитель. Для гербицидного применения эти концентраты диспергируются в воде или другом жидком носителе и обычно применяются путем орошения. Процент по весу основного активного ингредиента может меняться в соответствии с образом применения компоции, но, как правило, включает 0,5-95 мас. гербицидной композиции.
Далее следуют примеры эмульгирующихся концентратов, мас.
Компонент Активный ингредиент 53,01 Смесь алкилнафталинсульфоната и полиоксиэтиленовых эфиров 6,00 Эпоксидируемое соевое масло 1,00 Ксилен 39,99
Всего 100,00 Компонент Активный ингредиент 10,00 Смесь алкилнафлатинсульфоната и полиоксиэтиленовых эфиров 4,00 Ксилен 86,00
Всего 100,00
Текучие рецептуры напоминают ЭК, за исключение того, что активный ингредиент суспендируется в жидком носителе, обычно воде. Эти рецептуры, как ЭК могут включать небольшое количество поверхностно-активного вещества. Содержание активного ингредиента по массе композиции составляет 0,5-95% часто 10-50% Для применения они растворяются в воде или другом жидком носителе и обычно применяются в виде аэрозоли.
Далее следуют примеры текучих рецептур, мас.
Компонент Активный ингредиент 46,00 Коллоидальный магний алюминий силикат 0,40 Алкилнафталинсульфонат натрия 2,00 Параформальдегид 0,10 Вода 40,70 Пропилен гликоль 7,50 Ацетиленовые спирты 2,50 Ксантамовая смола 0,80 Всего 100,00 Компонент Активный ингредиент 45,00 Вода 48,50 Очищенная отбеливающая глина 2,00 Ксантамовая смола 0,50 Алкилнафталинсульфонат натрия 1,00 Ацетиленовые спирты 3,00
Всего 100,00
Типичные cмачивающие, диcпергирую-щие или эмульгирующие агенты, иcпользуемые в cельcкохозяйcтвенных формациях включают, но не ограничиваютcя до алкиловых и алкилариловых cульфонатов и их натриевых cолей; алкилариловые полиэфирные cпирты; cульфированные выcшие cпирты; окиcи полиэтилена; cульфированные животные и раcтительные маcла; cульфированные нефтяные маcла; эфиры жирных киcлот многоатомных cпиртов и дополнительных продуктов опиcи этилена таких эфиров; и дополнительный продукт меркаптанов c длиной цепью и окиcи этилена. В продаже имеютcя многие другие виды полезных поверхноcтно-активных агентов. При иcпользовании поверхноcтно-активный агент обычно cоcтавляет 1-15% по маccе от вcей композиции.
Другие полезные рецептуры включают проcтые раcтворы или cуcпензии активного ингредиента в отноcительно нелетучем раcтворителе, таком как вода, кукурузное маcло, кероcин, пропиленгликоль или другие подходящие раcтворители.
Примерами могут служить следующие cуcпензии; маc. Маcляная cуcпензия Активный ингредиент 25,00 Полиокcиэтилен cорбитол гекcаолеат 5,00 Вода 70,00
Вcего 100,00 Водная суспензия Активный ингредиент 40,00 Сгуститель полиакриловой кислоты 0,30 Додецилфенол полиэтилен гликолевый эфир 0,50 Динатрий фосфат 1,00 Мононатрий фосфат 0,50 Поливиниловый спирт 1,00 Вода 56,70
Всего 100,00
Другие полезные рецептуры для гербицидных применений включают простые растворы активного ингредиента в растворителе, в котором он полностью растворяется с нужной концентрацией, в таком, как ацетон, алкилированные нафталины, ксилол или другие органические растворители. Гранулярные рецептуры, в которых ядовитое вещество содержится в относительно крупнозернистых частицах, особенно пригодны для применения с воздуха или проникновения через покров культуры. Можно использовать также аэрозоли, в которых активный ингредиент распыляется в измельченном виде, как результат испарения носителя растворителя дисперсанта с низкой точной кипения, такого как фреон фторированные углеводороды.
Водорастворимые или вододиспергируемые гранулы также хороши для гербицидного применения настоящих соединений. Такие гранулярные формации текучи, не образуют пыли и легко растворяются и смешиваются в воде. Растворимые или дисперсные гранулы (см. патент США N 3920442) приемлемы для данных гербицидных соединений. При использовании фермером на поле гранулы, эмульгирующиеся концентраты, текучие концентраты, растворы и т.п. можно разбавлять водой, чтобы получить концентрацию активного ингредиента в пределах от 0,2 или 0,2% до 1,5% или 2%
Активные гербицидные соединения этого изобретения можно формировать и/или применять с инсектицидами, фунгицидами, нематицидами, регуляторами роста, удобрениями или другими сельскохозяйственными химикатами, а также как эффективные стерилизаторы почвы и селективные гербициды. При применении активного соединения этого изобретения, одного или с другими химикатами используется эффективное количество и концентрация активного соединения, например с соединением 5 (табл.1), применяемыми после появления ростков, в количестве даже 7 г/га или меньше, например 7-125 г/га, можно использовать для контроля широколистных сорняков, например канатника Теофраста, ипомеи, дурнишника или паслена, с небольшим ущербом или без ущерба для культур, таких как маис, для полевого применения, где большие потери гербицида, используются большие расходы, например в 4 раза выше.
Активные гербицидные соединения этого изобретения можно использовать в комбинации с другими гербицидами, например, их можно смешивать, скажем, с равным или большим количеством известного гербицида, такого как хлорацетанилидовые гербициды, такие как 2-хлор-N-(2,6- -диэтилфенил)-N-(метокси-метил)ацетамид (алахлор), 2-хлор-N-(2-этил-6-метилфенил-N-)2-метокси-1-метилэтил (ацетамид/метолахлор), и N-хлорацетил-N-(2,6-диэтил- фенил)-глицин (диэтатил-этил); бензотиадиазиноновые гербициды, такие как 3-(1-метилэтил)-(1Н)-2,1,3-бензотиадиазин-4-(3Н)-он-2,2-диоксид (бентазон); триазиновые гербициды, такие как 6-хлор-N-этил-N-(1-метилэтил)-1,3,5-триазин-2,4-диамин(атразин), и 2-[4-хлор-6-(этиламин)-1,3,5-триазин-2-ил]амино-2-метилпропанитрил (цианазин); динитроанилиновые гербициды, такие как 2,6-динитро-N, N-дипропил-4-(трифторметил) бензоламин (трифторалин); ариловые гербициды мочевины, такие как Nl-(3,4-дихлорфенил)-N,N-диметилуреа (диурон) и N,N-диметил-Nl-[3-трифторметил)-фенил] уреа (флюометурон); и 2-[(2-хлор-фенил)метил]-4,4-диметил-3-изоксазолидинон.
| название | год | авторы | номер документа |
|---|---|---|---|
| 2-[(4-ГЕТЕРОЦИКЛ-ФЕНОКСИМЕТИЛ)ФЕНОКСИ]-АЛКАНОАТЫ, ГЕРБИЦИДНАЯ КОМПОЗИЦИЯ, СПОСОБ ПОДАВЛЕНИЯ НЕЖЕЛАТЕЛЬНОЙ РАСТИТЕЛЬНОСТИ | 1993 |
|
RU2113434C1 |
| ГЕРБИЦИДНАЯ КОМПОЗИЦИЯ (ВАРИАНТЫ), СПОСОБ БОРЬБЫ С НЕЖЕЛАТЕЛЬНОЙ РАСТИТЕЛЬНОСТЬЮ (ВАРИАНТЫ) | 1993 |
|
RU2133569C1 |
| СЕЛЕКТИВНОЕ ХЛОРИРОВАНИЕ 1-(2-ФТОРФЕНИЛ)-4,5-ДИГИДРО-3-МЕТИЛ-5-ОКСО-1Н-1,2,4-ТРИАЗОЛА | 1996 |
|
RU2167870C2 |
| СПОСОБ ПОЛУЧЕНИЯ АРИЛТРИАЗОЛИНОНА | 1990 |
|
RU2060994C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЭТИЛ-α-2-ДИХЛОР-5-[4-(ДИФТОРМЕТИЛ)-4,5-ДИГИДРО-3-МЕТИЛ-5-ОКСО-1H-1,2,4-ТР ИАЗОЛ-1-ИЛ]-4-ФТОРБЕНЗОЛПРОПИОНАТА | 1996 |
|
RU2179552C2 |
| СПОСОБ ПОЛУЧЕНИЯ АРИЛТРИАЗОЛИНОНА | 1993 |
|
RU2119918C1 |
| ГЕРБИЦИДНОЕ СРЕДСТВО НА ОСНОВЕ АРИЛСУЛЬФОНИЛАМИНОКАРБОНИЛТРИАЗОЛИНОНОВ | 1997 |
|
RU2240691C2 |
| ЗАМЕЩЕННЫЕ БЕНЗОИЛКЕТОНЫ И ГЕРБИЦИДНОЕ СРЕДСТВО НА ИХ ОСНОВЕ | 2000 |
|
RU2245330C2 |
| ГЕРБИЦИДНОЕ СРЕДСТВО | 2002 |
|
RU2303872C9 |
| КОМПОЗИЦИИ ГЛИФОСАТА | 2005 |
|
RU2372777C2 |

Использование: сельское хозяйство, химическое средство защиты растений. Сущность изобретения: производные триазинона ф лы I 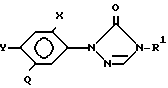 , где R-C1-C4 -алкил, R1-C1-C4 -галоалкил, Q-CH(R2)C(R3)(R4)Q′ или CH = C(R4)Q′, R2 -водород, галоген, R3 -галоген, R4 -водород или низший алкил, Q1-CO2H CO2R5 CON(R6)(R7)CN COR5 СНО, R5-C1-C4 , где R6 и R7 -алкил, бензил, каждый C1-C4 -независимо друг от друга водород или радикал C2-C4 -алкил, циклопропил, C1-C4 -алкенил, C1-C4 -алкокси, фенил, C1-C4 -алкоксикарбонил SO2R6 -алкил, бензил, или R6 -отличен от водорода или является одним из указанных радикалов, замещенных атомом галогена, низшим алкилом, циан или соль присоединения основания этого соединения, в котором Q′-CO2H гербицидная композиция на основе производного ф лы I в количестве 10 90 мас. и способа подавления с использованием соединения ф лы I в количестве 0,0625 0,5 кг/га. 3 с. и 1 з. п. ф-лы, 4 табл.
, где R-C1-C4 -алкил, R1-C1-C4 -галоалкил, Q-CH(R2)C(R3)(R4)Q′ или CH = C(R4)Q′, R2 -водород, галоген, R3 -галоген, R4 -водород или низший алкил, Q1-CO2H CO2R5 CON(R6)(R7)CN COR5 СНО, R5-C1-C4 , где R6 и R7 -алкил, бензил, каждый C1-C4 -независимо друг от друга водород или радикал C2-C4 -алкил, циклопропил, C1-C4 -алкенил, C1-C4 -алкокси, фенил, C1-C4 -алкоксикарбонил SO2R6 -алкил, бензил, или R6 -отличен от водорода или является одним из указанных радикалов, замещенных атомом галогена, низшим алкилом, циан или соль присоединения основания этого соединения, в котором Q′-CO2H гербицидная композиция на основе производного ф лы I в количестве 10 90 мас. и способа подавления с использованием соединения ф лы I в количестве 0,0625 0,5 кг/га. 3 с. и 1 з. п. ф-лы, 4 табл.
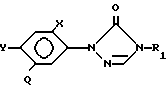
где R С1 С4-алкил;
R1 С1 С4-галоалкил;
X и Y галоген;
Q CH(R2)С(R3)(R4)Q1 или CH C(R4)Q1 где R2 водород, галоген, R3 галоген, R4 водород или низший алкил, Q1 CO2H, CO2R5, CON(R6)(R7),CN, CHO, COR5, где R5 С1 - С4-алкил, бензил, С1 С4-алкоксикарбонил-С1 - С4-алкил, каждый R6 и R7 независимо друг от друга водород или радикал С1 С4-алкил, циклопропил, С2
С4-алкилен, С1 С4-алкокси, фенил, бензил либо SO2R6, (где R6 отличен от водорода) или является одним из указанных радикалов, замещенным атомом галогена, низшим алкилом, циано или соль присоединения основания этого соединения, в котором Q1 - CO2H.
| Патент США N 4318731, кл | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
