Данное изобретение относится к усовершенствованному способу проведения последнейстадии.получениягербицидаэтил-α-2-дихлор-5-[4-(дифторметил)-4,5-дигидро-3-метил-5-оксо-1Н-1,2,4триазол-1-ил] -4-фторбензолпропаноата реакцией диазотирования Меервейна и арилирования.
Гербицид, который имеет следующую структуру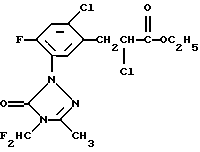
описан и заявлен в патенте США 5125958.
В обычной практике реакцию диазотирования Меервейна и арилирования проводят в две стадии. Сначала ариламин диазотируют в водном растворе, например, нитрита натрия, и затем раствор диазотированного амина добавляют к раствору соединения, которое должно быть арилировано.
В патенте США 5125958 последняя стадия получения данного гербицида включает диазотирование амина 4-дифторметил-4,5-дигидро-3-метил-5-оксо-1-(5-амино-2-фтор-4-хлорфенил)-1Н-1,2,4-триазола. Тем не менее, затем было обнаружено, что, когда пытались диазотировать этот амин в водном растворе, 2-фторзаместитель был потерян из-за гидролиза в соответствующий фенол. В соответствии с этим диазотирование, описанное в этом патенте, проводили двумя обычными стадиями, но в неводной среде, с трет-бутилнитритом в качестве источника нитрита. Диазотированный амин затем добавляли к этилакрилату (арилирование) для получения целевого гербицида. Несмотря на то, что эта процедура удовлетворительна для лабораторного получения в небольших количествах, она неудовлетворительна для крупномасштабного производства и коммерческого получения. Не только слишком ограничена подача трет-бутилнитрита, но получение в больших масштабах требует обращения с большими количествами диазосоединения - нежелательно опасной операции.
Неожиданно было обнаружено, что в подходящих условиях диазотирование и арилирование можно проводить одновременно водным нитритом натрия без потери 2-фторзаместителя и неожиданно с хорошими выходами.
Последовательность реакции диазотирование/арилирование представлена следующей схемой: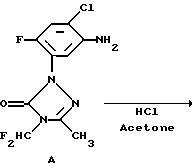
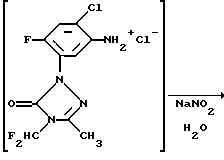
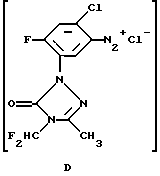
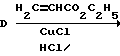
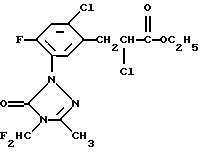
Как указано выше, в обычной практике сначала получают диазониевую соль и затем ее добавляют к соединению, которое должно быть арилировано. В способе по данному изобретению гидрохлоридную соль амина (А) диазотируют нитритом натрия в присутствии этилакрилата, материала, который должен быть арилирован. Диазонийхлорид (D), полученный диазотированием гидрохлорида амина нитритом натрия, реагирует с этилакрилатом в присутствии каталитического количества хлорида меди (I). Реакцию проводят при низкой температуре в ацетоне. Использование ацетона в качестве растворителя позволяет применять нитрит натрия в минимальном количестве воды. Ацетон облегчает также рециркуляцию используемого в качестве катализатора хлорида меди (I). Другим преимуществом данного способа является тот факт, что реакции диазотирования/арилирования, проводимые одновременно, минимизируют вероятность реакции продукта диазотирования с самим собой.
Способ начинается с получения раствора амина А в ацетоне, предпочтительно в атмосфере инертного газа, например азота. Приемлемое отношение ацетона к амину находится в диапазоне от 10 до 30 эквивалентов ацетона к одному эквиваленту амина, предпочтительно от 20 до 25 эквивалентов ацетона к одному эквиваленту амина. Раствор можно фильтровать для удаления любого нерастворимого материала, который может присутствовать. К раствору амина добавляют при перемешивании катализатор арилирования Меервейна, например, хлорид меди (I) или хлорид меди (II), предпочтительно хлорид меди (I), и опять-таки предпочтительно в атмосфере инертного газа (перемешивание продолжают на всем протяжении реакции).
В способе по настоящему изобретению имеет значение количество хлорида меди (I). Если его используют слишком мало, реакция идет более медленно и выход конечного продукта ниже. Слишком большое количество хлорида меди (I) в реакционной смеси приводит к дехлорированию конечного продукта. Следовательно, приемлемое отношение хлорида меди (I) к амину находится в диапазоне от 0,05 до 1,0 эквивалента хлорида меди (I) к одному эквиваленту амина, предпочтительно от 0,1 до 0,15 эквивалента к одному эквиваленту. Реакционную смесь затем охлаждают до температуры ниже 0oС, предпочтительно до около -10oС, и соль амина получают добавлением концентрированной хлористоводородной кислоты или безводного хлористого водорода, предпочтительно безводный хлористый водород вводят ниже поверхности реакционной смеси. Во время добавления реакционную смесь выдерживают при температуре от -20oС до 30oС, предпочтительно от -10oС до 10oС. Получение соли амина требует приблизительно от 30 до 120 мин, предпочтительно приблизительно от 45 до 90 мин. Имеет значение количество этилакрилата, которое затем добавляют в суспензию соли амина. Для получения оптимальных выходов по этому способу требуется большой избыток этилакрилата. Если избыток уменьшают, выход конечного продукта снижается. Было обнаружено, что приемлемое отношение этилакрилата к амину составляет от 5 до 20 эквивалентов этилакрилата к одному эквиваленту амина, предпочтительно от 10 до 15 эквивалентов к одному эквиваленту. Время, требуемое для добавления этилакрилата, относительно не важно. Однако температуру реакционной смеси поддерживают ниже 10oС в течение всего добавления и регулируют до некоторой степени добавлением этилакрилата. Температуру реакционной смеси затем доводят до около 0oС и к реакционной смеси добавляют водный раствор нитрита натрия. Эта стадия является критической стадией в способе настоящего изобретения. Реакции диазотирования/арилирования проводят одновременно.
Во время стадии диазотирования количество воды следует сохранять минимальным. Большое количество воды приводит к гидролизу диазо-промежуточного продукта, давая фенольный побочный продукт. Для минимизации образования побочных продуктов приемлемое отношение нитрита натрия к амину находится в диапазоне от 1,0 до 2,0 эквивалентов нитрита натрия к одному эквиваленту амина, предпочтительно от 1,4 до 1,7 эквивалентов к одному эквиваленту. Для поддержания минимального количества воды благоприятно использование для стадии диазотирования концентрированного водного раствора нитрита натрия. Насыщенный водный раствор нитрита натрия содержит около 40% (мас./мас.) нитрита натрия. Приемлемая концентрация водного раствора нитрита натрия, следовательно, составляет приблизительно от 20 до 40% (мас./мас.) нитрита натрия, предпочтительно от 35 до 40% нитрита натрия. Температуры реакционной смеси также важны в оптимизации реакции диазотирования/арилирования. Температуры выше 10oС повышают вероятность образования побочного продукта. Приемлемый диапазон температур реакционной смеси для оптимизации стадии способа составляет от -10 до 20oС, предпочтительно от 0 до 10oС. Скорость добавления водного раствора нитрита натрия также важна для оптимизации выхода. Высокая скорость добавления в способе по настоящему изобретению приводит к более низким выходам продукта. Оптимальная скорость добавления требует приблизительно от 1 до 6 ч, предпочтительно от 2 до 3 ч. Добавление водного раствора нитрита натрия проводят ниже поверхности перемешиваемой реакционной смеси. Реакционную смесь затем перемешивают в течение периода времени от 15 до 90 мин, предпочтительно от 20 до 40 мин, до завершения реакции. В этой точке получают выходы сырого продукта в диапазоне 80-88%. Чистоту продукта можно повысить промывкой с последующей перегонкой. Во время стадии промывки температуру поддерживают приблизительно от -10 до 20oС, предпочтительно от 5 до 10oС, и в предпочтительной последовательности промывки реакционную смесь промывают сначала разбавленной водной кислотой, например 5% хлористоводородной кислотой, затем разбавленным водным основанием, например 5% гидроксидом натрия, и в заключение раствором хлорида натрия. После завершения стадий промывки летучие материалы в реакционной смеси, т.е. ацетон/этилакрилат, можно удалить при температуре в реакционной емкости от 30 до 80oС, предпочтительно от 40 до 65oС, при пониженном давлении от 20 до 70 мм Hg, предпочтительно от 45 до 55 мм Hg. Удаление летучего материала считают законченным, когда концентрация этилакрилата ниже приблизительно 5%. Известно, что при определенных условиях этилакрилат полимеризуется. В качестве предупредительной меры к системе верхнего погона добавляют антиоксидант и ингибитор свободных радикалов. Однако нет никаких свидетельств тому, что в реакционном сосуде во время реакции, на стадиях промывки или во время удаления летучего материала имеет место полимеризация.
Для дальнейшей очистки продукта содержащий нелетучий материал продукт реакции можно перегонять в выпарном аппарате с коротким путем прохождения продукта или перегонном аппарате со стекающей пленкой жидкости. Чтобы повысить эффективность процесса перегонки, продукт реакции можно пропустить (стадия дегазации) через перегонный аппарат при температуре испарителя от 100 до 140oС и давлении от 10 до 20 мм Hg, предпочтительно от 120 до 130oС и давлении от 13 до 18 мм Hg для удаления любого оставшегося количества этилакрилата или других низкокипящих материалов. Дегазированный продукт реакции можно затем пропустить один раз через выпарной аппарат при температуре испарителя от 160 до 180oС и давлении от 1,0 до 5,0 мм Hg, предпочтительно от 170 до 180oС и давлении от 1,0 до 2,0 мм Hg. Нелетучий материал из первого прогона через выпарной аппарат можно затем пропустить во второй раз через выпарной аппарат при температуре испарителя от 160 до 200oС и давлении от 0,03 до 1,0 мм Hg, предпочтительно от 160 до 180oС и давлении от 0,03 до 0,5 мм Hg. Скорость пропускания через выпарной аппарат будет определяться размером выпарного аппарата. Понятно, что условия дегазации и перегонки будут изменяться в зависимости от используемого аппарата. Операция в предпочтительных условиях дает типичный выход в процентах перегнанного этил-α-2-дихлор-5-[4-(дифторметил)-4,5-дигидро-3-метил-5-оксо-1Н-1,2,4-триазол-1-ил] -4-фтор-бензолпропаноата 75-80% (относительно количества исходного амина) при чистоте около 91% (определено жидкостной хроматографией высокого давления).
Пример 1
Получение этил-α-2-дихлор-5-[4-(дифторметил)-4,5-дигидро-3-метил-5-оксо-1Н-1,2,4-триазол-1-ил]-4-фторбензолпропаноата, лабораторный масштаб
Колбу из полимера на 2 л с рубашкой снабжают термометром, трубкой для ввода азота, выходной трубкой, соединенной с угольной ловушкой (для минимизации утечки запаха этилакрилата) и устройством, состоящим из покрытой тефлоном иглы длиной 203,2 мм (8 inch) и большим диаметром, присоединенной к шприцу на 50 мл, который, в свою очередь, связан с поршнем шприца. В отдельном сосуде 140,4 г (0,46 моль-1,0 экв.) 4-дифторметил-4,5-дигидро-3-метил-5-оксо-1-(5-амино-2-фтор-4-хлорфенил)-1Н-1,2,4-триазола растворяют в 600 г (10,33 моль - 22,5 экв.) ацетона. Раствор охлаждают до 0oС и фильтруют в полимерную склянку. При перемешивании в раствор триазол/ацетон барботируют 56,2 г (1,54 моль - 3,4 экв.) безводного хлористого водорода в течение периода 15-20 мин. После завершения добавления прибавляют 6,1 г (0,06 моль - 0,13 экв.) хлорида меди(I). При продолжении перемешивания реакционную смесь охлаждают до 0oС в атмосфере азота и добавляют 640,4 г (6,33 моль - 13,8 экв. ) этилацетата. Реакционную смесь снова охлаждают до 0oС и добавляют раствор 48,1 г (0,69 моль - 1,5 экв.) нитрита натрия, растворенного в 88,1 г(4,9 моль - 10,7 экв.) воды. Раствор нитрит/вода добавляют к реакционной смеси со скоростью 0,48 мл/мин из шприца с выходным отверстием иглы ниже поверхности реакционной смеси. Полностью прибавление требует около 3,75 ч, после чего реакционную смесь перемешивают в течение дополнительных 45 мин при 0oС. Реакцию затем гасят 200 мл воды, после чего реакционную смесь перемешивают в течение около 15 мин. Органический слой отделяют и промывают по одной порции каждого из 200 мл водной 1н хлористоводородной кислоты, 200 мл воды и смеси 1:1 водного 5% гидроксида натрия и этилацетата, где каждый из объемов раствора гидроксида натрия и этилацетата равен объему органического слоя. Органический слой затем концентрируют при 70oС в вакууме, получая 193,3 г (анализ - 84,6%, выход сырого продукта - 86,4%) этил-α-2-дихлор-5-[4-(дифторметил)-4,5-дигидро-3-метил-5-оксо-1Н-1,2,4-триазол-1-ил]-4-фтор-бензолпропаноата. Концентрированный продукт пропускают через перегонный аппарат Pope со стекающей пленкой жидкости размером 50,8 мм (2 inch), 0,2322 м2 (0,25 ft2) при 160 и 0,8 мм Hg для удаления низкокипящих примесей. Нелетучий материал пропускают во второй раз через перегонный аппарат со стекающей пленкой жидкости при 220oС/1 мм Hg для отгонки продукта от высококипящих примесей, получая 134,9 г (анализ - 92%, выход перегнанного продукта - 65,6%) этил-α-2-дихлор-5-[4-(дифторметил)-4,5-дигидро-3-метил-5-оксо-1Н-1,2,4-триазол-1-ил]-4-фторбензолпропаноата.
Пример 2
Получение этил-α-2-дихлор-5-[4-(дифторметил)-4,5-дигидро-3-метил-5-оксо-1 Н-1,2,4-триазол-1-ил]-4-фторбензолпропаноата, масштаб пилотной установки
В соответствии со следующим способом получили несколько порций продукта. В реактор на 757 л (200 gallon) с рубашкой загружают посредством перепада давления ацетон, 207,472 кг (457,4 pound) (3578,8 г-моль (7,89 lb-moles), 22 экв.), затем 48,534 кг (107 pounds) (163,29 г-моль (0,36 lb-mole), 1,0 экв.) 98% 4-дифторметил-4,5-дигидро-3-метил-5оксо-1-(5-амино-2-фтор-4-хлорфенил)-1Н-1,2,4-триазола. Смесь перемешивают и необязательно фильтруют при комнатой температуре для удаления твердых примесей. Раствор перегружают в реактор на 757 л (200 gallon), перемешивают и продувают азотом. К нему затем добавляют 2,086 кг (4,6 pounds) (22,68 г-моль (0,05 lb-mol), 0,13 экв.) хлорида меди(I). После завершения добавления смесь охлаждают приблизительно до -10oС циркуляцией холодного солевого раствора через рубашку реактора. Реактор закрывают и продувку азотом прекращают. Затем безводный хлористый водород, 14,832 кг (32,7 pounds) (408,23 г-моль (0,9 lb-mole), 2,5 экв.) барботируют ниже поверхности реакционной смеси при скорости около 249,5 г (0,55 lb/мин, в то время как температуру реакционной смеси поддерживают между -10 и 10oС. Полное добавление требует около одного часа. После завершения добавления реактор открывают для сообщения с атмосферным давлением. Температуру реакционной смеси доводят приблизительно до 0oС и самотеком загружают 219,537 кг (484 pound) (2195,4 г-моль (4,84 lb-moles), 13,5 экв.) этилакрилата. После окончания добавления прибавляют 16,828 кг (37,1 pounds) (2195,4 г-моль (4,84 lb-moles, 1,5 экв.) нитрита натрия в 31,298 кг (69 pounds) (1737,2 г-моль (3,83 lb-moles), 10,6 экв.) воды со скоростью около 272,2 г (0,6 pound)/мин ниже поверхности реакционной смеси. Температуру реакционной смеси поддерживают между 0 и 5oС. Полное добавление требует около трех часов, после этого времени реакционную смесь перемешивают в течение 30 мин. В то время как температуру реакционной смеси поддерживают между 5 и 10oС, реакционную смесь промывают по очереди 66,678 кг (147 pounds) воды, 66,587 кг (146,8 pounds) водной 5% хлористоводородной кислоты, 69,399 кг (153 pounds) водного 5% гидроксида натрия и 68,038 кг (150 pounds) воды. Этилацетат и ацетон затем удаляют из реакционной смеси перегонкой при около 65oС/50 мм Hg. Для предотвращения полимеризации перегнанную смесь этилацетата и ацетона стабилизируют раствором, состоящим из 81,6 г (0,18 pound) 4-метоксифенола, 81,6 г (0,18 pound) фенотиазина и 25,401 кг (56 pounds) толуола, который циркулирует через холодильник и приемник дистиллята. Температуру продукта-остатка поддерживают приблизительно при 45oС, когда его затаривают в барабан для будущего использования. Выход сырого продукта, этил-α-2-дихлор-5[4-(дифторметил)-4,5-дигидро-3-метил-5-оксо-1Н-1,2,4-триазол-1-ил]-4-фторбензопропаноата составляет 69,036 кг (152,2 pounds). Ряд порций, полученных как описано выше, объединяют. Продукт объединения пропускают через перегонный аппарат со стекающей пленкой жидкости сначала при низком вакууме для удаления низкокипящего материала (дегазированного), затем дважды при высоком вакууме для получения перегнанного продукта. Для перегонки в перегонном аппарате со стекающей пленкой жидкости используют условия, приведенные в табл. 1.
Процентный выход перегнанного этил-α-2-дихлор-5-[4-(дифторметил)-4,5-дигидро)-3-метил-5-оксо-1Н-1,2,4-триазол-1-ил] -4-фторбензолпропаноата составляет 65% (на основе количества исходного амина), чистота 91% (как определено при помощи жидкостной хроматографии высокого давления).
Пример 3
Получение этил-α-2-дихлор-5-[4-(дифторметил)-4,5-дигидро-3-метил-5-оксо-1Н-1,2,4-триазол-1-ил]-4-фторбензолпропаноата, масштаб пилотной установки
В соответствии со следующей методикой получили несколько порций продукта. Реактор на 3785 л (1000 gallon) с рубашкой продувают азотом и в реактор загружают 231,331 кг (510,0 pounds) (775,7 г-моль (1,71 lb-moles), 1,0 экв. ) 98% 4-дифторметил-4,5-дигидро-3-метил-5-оксо-1-(5-амино-2-фтор-4-хлорфенил)-1Н-1,2,4-триазола. В него затем загружают 9,979 кг (22,0 pounds) (99,79 г-моль (0,22 lb-mole), 0,13 экв.) хлорида меди(I). Реактор герметизируют и снова продувают азотом. Затем в реактор при помощи нагнетательного поршневого насоса загружают ацетон 988,282 кг (2178,8 pounds) (17041,38 гмоль (37,57 lb-moles), 22,0 экв). После завершения добавления смесь перемешивают и охлаждают до температуры между -10 и 0oС путем циркуляции холодного солевого раствора через рубашку реактора. Затем безводный хлористый водород 70,669 кг (155,8 pounds) (1936,83 г-моль (4,27 lb-mole), 2,5 экв.) барботируют ниже поверхности реакционной смеси со скоростью около 1133,97 г (2,5 lb)/мин, в то время как температуру реакционной смеем поддерживают ниже 10oС. Добавление безводного хлористого водорода обычно заканчивается за время от одного до двух часов. После завершения подачи хлористого водорода реактор открывают для сообщения с атмосферным давлением. Температуру реакционной смеси затем доводят приблизительно до 0oС и в реактор при помощи нагнетательного поршневого насоса загружают 1045,6 кг (2305,2 pounds) (10477,9 г-моль, (23,1 lb-moles), 13,5 экв.) этилакрилата. После завершения добавления загружают ниже поверхности реакционной смеси 200,396 кг (441,8 pounds) водного 40 мас.% (1161,2 г-моль (2,56 lb-mole), 1,5 экв.) раствора нитрита натрия со скоростью около 816,5 г (1,8 lbs)/мин. Температуру реакционной смеси поддерживают между 0 и 5oС. Полное добавление требует около четырех часов, после этого времени реакционную смесь перемешивают в течение 30 мин. Реакцию считают законченной, когда количество непрореагировавшего амина (без растворителя), оставшегося в реакционной смеси, менее чем около 0,5% площади при определении газовой хроматографией. После завершения реакции реакционную смесь выдерживают при температуре от 5 до 10oС и промывают двумя порциями по 317,513 кг (700 pounds) водного 5% раствора хлористоводородной кислоты, двумя порциями по 330,667 кг (729 pounds) водного 5% раствора гидроксида натрия и одной порцией 324,317 кг (715 pounds) водного 10% раствора хлорида натрия. Этилацетат и ацетон затем удаляют из реакционной смеси перегонкой при температуре между 65 и 75oС и менее чем 10 мм Hg до тех пор, пока концентрация этилакрилата в растворе не станет менее чем приблизительно 5 %. Для предотвращения полимеризации дистиллят смеси этилакрилата и ацетона стабилизируют раствором, состоящим из 95,2 г (0,21 pound) 4-метоксифенола, 95,2 г (0,21 pound) фенотиазина и 12,700 кг (28 pounds) толуола, который циркулирует через холодильник и приемник дистиллята. Температуру продукта-остатка поддерживают приблизительно при 45oС, когда его затаривают в барабан для будущего использования. Выход сырого продукта, этил-α-2-дихлор-5-[4-(дифторметил)-4,5-дигидро-3-метил-5-оксо-1Н-1,2,4-триазол-1-ил]-4-фторбензолпропаноата этой реакции в этой точке обычно около 82%. Ряд порций, полученных как описано выше, объединяют. Продукт объединения пропускают через выпарной аппарат с коротким путем прохождения материала (SPE) сначала при низком вакууме для удаления низкокипящего материала (дегазация), затем дважды при высоком вакууме для получения перегнанного продукта. Для перегонки в SPE используют условия, приведенные в табл. 2.
Процентный выход перегнанного этил-α-2-дихлор-5-[4-(дифторметил)-4,5-дигидро-3-метил-5-оксо-1Н-1,2,4-триазол-1-ил]-4-фторбензолпропаноата составляет 80% (на основе количества исходного амина), чистота 91% (как определено жидкостной хроматографией высокого давления).

Описывается усовершенствованный способ получения известного гербицида - этил-α-2-дихлор-5-[4-(дифторметил)-4,5-дигидро-3-метил-5-оксо-1Н-1,2,4-триазол-1-ил] -4-фторбензолпропионата путем диазотирования 4-дифторметил-4,5-дигидро-3-метил-5-оксо-1-(5-амино-2-фтор-4-хлорфенил)-1Н-1,2,4-триазола и арилирования этилакрилата полученной диазониевой солью, причем диазотирование и арилирование осуществляют одновременно, при этом к производному 1,2,4-триазола в 10-30 эквивалентах ацетона последовательно добавляют 0,05-0,5 эквивалентов хлорида меди (I), хлористоводородную кислоту, этилакрилат и нитрит натрия. 2 з.п. ф-лы, 2 табл.
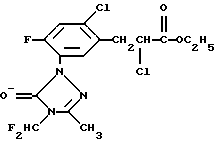
включающий диазотирование 4-дифторметил-4,5-дигидро-3-метил-5-оксо-1-(5-амино-2-фтор-4-хлорфенил)-1Н-1,2,4-триазола и арилирование этилакрилата полученной диазониевой солью, отличающийся тем, что диазотирование и арилирование осуществляют одновременно, при этом к перемешиваемому раствору 4-дифторметил-4,5-дигидро-3-метил-5-оксо-1-(5-амино-2-фтор-4-хлорфенил)-1Н-1,2,4-триазола в 10-30 эквивалентах ацетона добавляют от 0,05 до 0,5 эквивалентов хлорида меди (I), охлаждают раствор до температуры от -20 до 30oС и поддерживают температуру в этом диапазоне при перемешивании и добавлении хлористоводородной кислоты в количестве не более 3,4 эквивалентов на один мол. эквивалент 4-дифторметил-4,5-дигидро-3-метил-5-оксо-1-(5-амино-2-фтор-4-хлорфенил)-1Н-1,2,4-триазола, к полученной суспензии гидрохлоридной соли 4-дифторметил-4,5-дигидро-3-метил-5-оксо-1-(5-амино-2-фтор-4-хлорфенил)-1Н-1,2,4-триазола, при поддержании температуры ниже 10oС и перемешивании, добавляют от 5 до 20 эквивалентов этилакрилата, затем доводят температуру перемешиваемой реакционной смеси до около 0oС и добавляют от 1,0 до 2,0 эквивалентов нитрита натрия в течение 1-6 ч при поддержании температуры ниже приблизительно 20oС и продолжают перемешивание в течение 15-90 мин с последующим выделением целевого продукта.
| US 5125958, 1992 | |||
| US 4404019, 1988 | |||
| Гербицидная композиция (ее варианты) | 1981 |
|
SU1276244A3 |
| СПОСОБ ПОЛУЧЕНИЯ АРИЛТРИАЗОЛИНОНА | 1990 |
|
RU2060994C1 |
