Изобретение относится к получению 1-арил-1,2,4-триазолин-5-онов в среде трет-бутанола.
Патент США 4980480 описывает процесс получения 1-арил-1,2,4-триазолин-5-онов с помощью обработки арилтриазолидинона галогеноватистой кислотой, ее солью или галогеном. Триазолидинон может быть получен реакцией соответствующего арилгидразина с альдегидом с образованием соответствующего гидразона, с последующей обработкой последнего цианатом щелочного металла и получением триазолидинона. Предпочтительна среда, в которой триазолидинон по крайней мере частично растворим. Уксусная кислота упоминается как наиболее подходящая среда.
В соответствии с одним аспектом изобретения заявителя 1-арил-1,2,4-триазолидин-5-он обрабатывают в среде трет-бутанола галогеноватистой кислотой, ее солью или галогеном до образования соответствующего триазолинона. Реакция протекает быстро при относительно низких температурах с получением высокого выхода за короткое время реакции.
По другому аспекту изобретения триазолидинон может быть получен с высоким выходом в среде трет-бутанола с помощью реакции арилгидразона с цианатом щелочного металла. Арилгидразон, в свою очередь, может быть получен реакцией арилгидразина с альдегидом в трет-бутаноле.
Среда трет-бутанола может представлять собой безводный трет-бутанол или предпочтительно смесь трет-бутанола и воды.
При использовании среды трет-бутанола триазолинон может быть получен с высоким выходом. Среди особых преимуществ, триазолинон может быть получен с высоким выходом и высокой степенью чистоты при больших объемах получения, например с 2 фунт-молями (907 молей) арилгидразина в качестве исходного материала при использовании реактора объемом 500 галлонов (1893 л). Начиная с арилгидразина или арилгидразона, стадии процесса могут проводиться последовательно в одном реакторе с помощью последовательного добавления реагентов без необходимости выделения или очистки промежуточных продуктов. Без дальнейшей очистки, кроме как промывки водой, триазолиноновый продукт может быть выделен и прямо использован в последующих реакциях, таких, как дифторметилирование по 11-4 азоту триазолинонового кольца или хлорирование бензольного кольца, при получении продукта как в промышленном, так и в лабораторном масштабе. В ранее существующих способах в качестве среды выбиралась смесь уксусная кислота/вода (патент США 4980480). При разработке методик получения в крупных масштабах при использовании среды уксусная кислота/вода обнаружено, что результаты - переменны. Выход триазолинона и его чистота были значительно ниже, чем было получено в лабораторных условиях, а образование повышенного количества побочных продуктов реакции обычно требовало, чтобы триазолинон очищали перед использованием его в последующих реакциях.
Выделение триазолинона или триазолидинона может быть легко выполнено при отгонке азеотропа трет-бутанол/вода 88/12 (вес/вес) (т.кип. 80oC), который может быть возвращен для использования в последующем получении. Тем самым продукт может быть извлечен без помощи экстракции органическим растворителем или без интенсивного удаления растворителя в больших количествах, причем высококипящих растворителей, таких, как вода (т.кип. 100oC) или уксусная кислота (т. кип. 118oC). Способ этого изобретения, поэтому, будет требовать более низких затрат энергии, обеспечивать более чистый и менее дорогой продукт и будет пригодным для использования при получении в больших объемах.
В большинстве случаев использование спиртсодержащих сред для настоящего способа не желательно. В условиях, которые используются для окисления триазолидонона спирты могут окисляться до гидроперекисей. Гидроперекиси большинства спиртов, которые обычно могут использоваться в качестве реакционной среды, могут затем разлагаться до альдегидов и кетонов. При рециркулировании трет-бутанольной среды такие альдегиды или кетоны могут конкурировать в образовании гидразона, тем самым загрязняя реакционную систему нежелательными продуктами. Однако гидроперекись трет-бутанола, не имеющая альфа-водородов, может разлагаться, регенерируя трет-бутанол, тем самым снижая потенциальное загрязнение. Дополнительно гидроперекиси спиртов, содержащие альфа-водороды, могут проявлять нестабильность и потенциально могут разлагаться со взрывом. Трет-бутанол, образующий более стабильные гидроперекиси, снижает эту проблему.
Соединения, получаемые по настоящему способу являются промежуточными продуктами при получении гербицидов, таких, которые приведены в патенте США 4818275, который включен сюда в качестве ссылки, например такого, как соединение номер 1 таблицы 1 из этого патента. Различные исходные материалы, последовательности реакций и промежуточные продукты, включенные для получения гербицидов патента 4818275, проиллюстрированы в патенте США 4980480, описание которого включено сюда в качестве ссылки.
Один аспект этого изобретения может быть описан следующей схемой: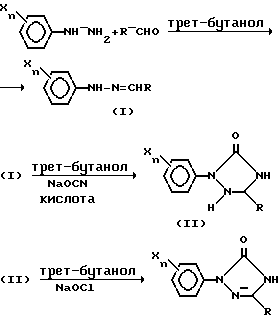
где
R - низший алкил;
X - независимо друг от друга галоген, низший алкил, низший алкоксил, нитро, гидрокси, -NHSO2R1, -N(SO2R')2 или N(R')SO2R',
где
R' - низший алкил и n целое число от 0 до 3
Предпочтительно фенильное кольцо - незамещенное (т.е. n = 0) и R - метил.
В каждом аспекте изобретения предпочтительно, чтобы замещенные или незамещенные алкильные радикалы имели менее семи атомов углерода.
Используемый здесь термин "низший" для "алкил", "алкокси" и других подобных радикалов означает прямую или разветвленную углеводородную цепочку из 1-3 атомов углерода.
Термин "гала" или "галоген" означает фтор, хлор или бром.
Термин "арил" означает фенил или замещенный фенил.
Термин "трет-бутанольная среда" означает трет-бутанол или смеси трет-бутанол/вода, в которых реагенты и продукты этого способа могут растворяться или диспергироваться. Термин "88/12" "95/5" и "70/30", применяющиеся с выражениями трет-бутанол/вода, трет-бутанольная среда, азеотроп трет-бутанола и с подобными им, означает весовое соотношение смеси трет-бутанол/вода. Таким образом, азетроп 88/12 означает, что он содержит 88 весовых частей трет-бутанола и 12 весовых частей воды.
По одному аспекту настоящего изобретения алифатический альдегид реагирует в трет-бутанольной среде с арилгидразином до образования соответствующего арилгидразинона. Реакция может проводиться при температуре от -10oC до 60oC. Наиболее предпочтителен интервал температур от приблизительно 5oC до 30oC. Реагенты могут использоваться в эквимолярных концентрациях или вплоть до 20%-ного молярного избытка альдегида. Наиболее предпочтительным является 1-10%-ный молярный избыток альдегида. Количество используемой трет-бутанольной среды может составлять от 50 до 2000 г среды на моль арилгидразина. Количество трет-бутанольной среды, используемое в любой конкретной реакционной системе, будет изменяться в зависимости от конкретных реагентов и условий реакции. Реагенты могут быть растворены в трет-бутанольной среде по отдельности и затем альдегид добавлен к арилгидразину. Предпочтительный способ использует приблизительно 100 г трет-бутанольной среды на моль альдегида и приблизительно 350 г трет-бутанольной среды на моль фенилгидразина.
Согласно второму аспекту настоящего изобретения арилгидразон реагирует с цианатом щелочного металла и источником протонов в среде трет-бутанола до образования арилтриазолидинона. Эту реакцию проводят при температуре от приблизительно -10oC до 60oC в течение периода от одного часа до 24 часов. Наиболее предпочтительное проведение реакции за период приблизительно от 2 до 5 часов при температуре от приблизительно 0oC до 35oC. В этой реакции может использоваться 20% молярный избыток цианата. Предпочтительно используют 5-10% молярный избыток цианата. Подходящими цианатами являются цианаты натрия, калия, кальция или стабильные цианат-доставляющие соли. Наиболее предпочтителен цианат натрия. В качестве источника протонов применяют любую слабую органическую кислоту, например уксусную кислоту, пропионовую кислоту или масляную кислоту. Наиболее предпочтительна уксусная кислота. В реакции могут использовать 20% эквивалентный избыток кислоты (основанный на цианате натрия и его примеси карбоната натрия). Это количество органической кислоты может быть уменьшено, если арилгидразин, используемый при получении арилгидразона, был по качеству плохим или старым или имели неожиданно плохой выход триазолинона. Органическая кислота в количестве, требуемом для этой реакции, прибавляется вместо этой реакции к трет-бутанольной среде при получении арилгидразона. Количество органической кислоты, внесенное при получении гидразона может быть вплоть до 50% (моль/моль) используемого исходного гидразина. Предпочтительно органическая кислота составляет приблизительно 10% - 16% (моль/моль) исходного гидразина. Если добавление кислоты отделяется от реакции, то обе реакции и образование гидразона, и образование триазолидинона выполняются при температурах от приблизительно 30oC до 40oC.
Так как присутствие уксусной кислоты обуславливает увеличение числа побочных реакций и полимеризацию, предпочтительно, чтобы используемый избыток уксусной кислоты был не более 5% молярных. Трет-бутанольная среда должна присутствовать в количестве, достаточном для растворения гидразона. Предпочтительной концентрацией трет-бутанольной среды является приблизительно 400 г на каждый моль гидразона.
Согласно третьему аспекту изобретения арилтриазолидинон окисляют в среде трет-бутанола до получения требуемого арилтриазолинона. Реакцию могут проводить при температуре от приблизительно 0oC до 60oC. Предпочтительно реакцию проводят в течение периода 2-4 часа при температуре от приблизительно 10oC до 40oC. Окисление могут выполнять при использовании галогена, галогеноватистой кислоты или соли галогеноватистой кислоты. В качестве окислителя выбирают гипохлорит натрия. Могут быть использованы водные растворы гипохлорита натрия концентрации от приблизительно 5% до 25% (вес/вес). Наиболее предпочтительны растворы концентрации от приблизительно 10% до 15%. Молярный избыток гипохлорита натрия может составлять вплоть до 40%. Наиболее предпочтителен молярный избыток вплоть до приблизительно 10%.
По окончании реакции трет-бутанол/вода отгоняются от реакционной смеси в виде азеотропа. Этот азеотроп может в дальнейшем повторно использоваться в последующих процессах. Триазолинон высаживается из оставшейся воды и собирается фильтрацией. Если необходимо, удаление остаточной воды может быть проведено прямым нагреванием при атмосферном давлении или в вакууме или при добавлении гептана и последующем удалении азеотропа гептан/вода нагреванием при атмосферном давлении или в вакууме.
Трет-бутанольная среда, используемая в этом изобретении, может быть безводной или может содержать воду. Количество трет-бутанольной среды и соотношение трет-бутанольной среды и соотношение трет-бутанол/вода должны быть такими, чтобы реагенты были по крайней мере частично растворены. Количество используемой трет-бутанольной среды и соотношение трет-бутанол/вода могут выгодно изменяться в зависимости от ряда критериев, включающих выход продуктов, снижение загрязнений, специфические реагенты, тип установки получения и стоимость материалов. Предпочтительно вышеприведенные реакции выполняются в реакционной среде, которая содержит от приблизительно 50 до 1000 г трет-бутанола на каждый моль первоначального реагента, гидразина, гидразона или триазолидинона. Вес воды, первоначально содержащийся в реакционной среде, меньше, чем вес трет-бутанола в реакционной среде. Предпочтительно трет-бутанольная среда, вводимая в реакционную систему, имеет соотношение трет-бутанол/вода между 95/5 и 70/30. Предпочтительно в систему вводится трет-бутанольная среда 88/12, являющаяся азеотропом, возвращенным из погона предшествующего получения. Показано, что в начале или в течение процесса рецикловый азеотроп трет-бутанол/вода может быть пополнен дополнительным количеством воды, трет-бутанола или смеси трет-бутанол/вода для создания нужной среды, для улучшения проведения процесса, для лучшего растворения реагентов или для других целей. Тем самым состав среды может изменяться и может содержать рецикловый азеотроп, так же как и другие компоненты.
Вышеприведенный процесс проводят преимущественно при атмосферном давлении, хотя это не является существенной характеристикой при условии, что температура реакции подходящим образом изменяется с давлением.
В приведенных выше стадиях процесса реакционная среда предпочтительно подвергается по крайней мере слабому перемешиванию.
Изобретение может быть использовано на практике для периодических процессов с большей по объему загрузкой при получении более чем 100 фунт-молей (45,359 молей) продукта, применяя подобное количество исходного материала. Предпочтительно получение арилтриазолинона из арилгидразона или арилгидразина проводят в одном реакторе при последовательном добавлении индивидуальных реагентов без выделения промежуточных продуктов. Однако фенилгидразон или триазолидинон можетт быть выделен отдельно и затем использован в качестве исходного материала для последующих стадий процесса. Предпочтительно используют загрузку процесса, дающую от приблизительно 1 (454 молей) до 20 фунт-молей (9072 молей) фенилтриазолинона и использующую подобное молярное количество фенилгидразина в качестве исходного материала. Экономичнее производство в большем объеме, если могут быть получены высокие выходы. Экспериментальная установка получения триазолинона с реактором на 500 галлонов (1893 л) использующая трет-бутанольную среду, обеспечивает выход 88,6% (пример 6). Это сравнивают с процессом получения в больших объемах (реакторы на 50 (189,3 л) и 500 галлонов (1883 л)) при использовании в качестве среды уксусной кислоты, для которого максимальный выход составлял 64% (пример 8).
Изобретение может быть реализовано на практике при использовании коммерчески доступных исходных материалов или тех, которые могут быть получены по методикам, описанным в литературе, или их модификаций, которые известны. Следующие далее примеры иллюстрируют данное изобретение.
Пример 1.
Получение 4,5-дигидро-3-метил-1-фенил-1,2,4-триазол-5 (1H)-она.
К раствору 54,4 г (0,500 молей) фенилгидразина в 150 г трет-бутанол/воды (88/12), охлажденному до 0 - 5oC, при перемешивании прибавляют по каплям в течение 20 минут раствор 54,0 г (0,525 молей) ацетальдегида в 54 г трет-бутанол/воды (88/12). При добавлении температура реакционной смеси повышается до 8oC. После окончания добавления реакционную массу перемешивают 5 минут, вносят в одну порцию суспензию 39,8 г (91,5% чистоты, 0,560 молей) цианата натрия в 90 г воды. При добавлении температура реакционной смеси повышается до 12oC. Дополнительные 27 г воды, используемые для смывки оставшейся суспензии цианата натрия из его контейнера, вносят в реакционную смесь. После окончания добавления реакционную смесь охлаждают до 0 - 5oC и прибавляют по каплям в течение 15 минут 39,3 г (0.655 молей - 5% эквивалентный избыток, приведенный на эквиваленты цианата натрия и его примеси карбоната натрия) уксусной кислоты. После окончания добавления реакционную смесь перемешивают приблизительно 2 часа, позволяя температуре реакции повыситься до приблизительно 20oC.
Во время этого двухчасового перемешивания готовят 12,4% (вес/вес) водный раствор гипохлорита натрия. Эту операцию проводят с помощью пропускания 41,8 г (0,590 молей) хлора (газ) через трубку, соединенную с трубкой воронки и выведенную к нижней поверхности перемешиваемого раствора 48,3 г (1,210 молей) гидроокиси натрия в 261,9 г смеси лед/вода.
После окончания двухчасового перемешивания реакционную смесь охлаждают до приблизительно 10oC и прибавляют по каплям в течение 40 минут 321 г (0,537 молей) 12,4% раствор гипохлорита натрия. После окончания добавления реакционную смесь дополнительно перемешивают 40 минут, а затем оставляют на 18 часов, в течение которых температура смеси поднимается до комнатной. Затем из реакционной смеси отгоняют трет-бутанольную среду с помощью медленного нагревания перемешиваемой реакционной смеси до приблизительно 100oC. После окончания отгонки, затвердевший осадок отфильтровывают, промывают 500 мл воды. Твердый осадок сушат и получают 82,5 г 4,5-дигидро-3-метил-1-фенил-1,2,4-триазол-5(1H)-она (93,1% выход). Газохроматографический анализ продукта показывает его 98,9% чистоту.
Пример 2.
Получение 1-(4-хлорфенил)-4,5-дигидро-3-метил-1,2,4-триазол-5 (1H)-она.
К раствору 71,3 г (0,500 молей) 4-хлорфенилгидразина в 175 мл трет-бутанол/воды (88/12) при охлаждении до 0 - 5oC и при перемешивании прибавляют по каплям в течение 20 минут раствор 23,4 г (0,525 молей) ацетальдегида в 54 г трет-бутанол/воды (88/12). После окончания добавления реакционную смесь перемешивают пять минут и вносят в одну порцию суспензию 39,8 г (91,5% чистоты - 0,560 молей - 12% молярный избыток) цианата натрия в 90 г воды. Дополнительное количество воды используется для смывки остатков суспензии цианата натрия из его контейнера и переносится в реакционную смесь, которую охлаждают до 0 - 5oC и прибавляют по каплям в течение 15 минут 39,3 г (0,655 молей - 5% эквивалентный избыток по эквиваленту цианата натрия и его примеси карбоната натрия) уксусной кислоты. После окончания добавления реакционную смесь перемешивают приблизительно 2 часа, в течение которых предусматривают повышение температуры до 20oC. Спустя эти 2 часа реакционную смесь охлаждают до 10oC и прибавляют по каплям в течение 40 минут 321 г (0,537 молей) 12,4% водного раствора гипохлорида натрия. После окончания добавления реакционную смесь перемешивают дополнительно 40 минут, затем отгоняют трет-бутанол/водную среду при медленном нагревании смеси до приблизительно 100oC. После окончания отгонки остаток промывают водой, сушат и получают 1-(4-хлорфенил)-4,5-дигидро-3-метил-1,2,4-триазол-5 (1H)-он.
Пример 3.
Получение 1-(4-хлор-2-фторфенил)-4,5-дигидро-3-метил-1,2,4-триазол-5 (1H)-она.
К раствору 80,3 г (0,500 молей) 4-хлор-2-фторфенил-гидразина в 180 мл трет-бутанол/воды (88/12) при охлаждении до 0 - 5oC при перемешивании прибавляют по каплям в течение 20 минут раствор 23,4 г (0,525 молей) ацетальдегида в 54 г трет-бутанол/воды (88/12). После окончания прибавления реакционную смесь перемешивают 5 минут и вносят в одну порцию 39,8 г (91,5% чистоты - 0,560 молей - 12% молярный избыток) цианата натрия в 90 г воды. Дополнительное количество воды, используемое для смывки остатков суспензии натрия из его контейнера, переносится в реакционную смесь, которую охлаждают до 0 - 5oC и прибавляют по каплям в течение 15 минут 39,3 г (0,655 молей - 5% эквивалентный избыток по эквиваленту цианата натрия и его примеси карбоната натрия) уксусной кислоты. После окончания добавления реакционную массу перемешивают приблизительно 2 часа, в течение которых допускается повышение температуры до 20oC. Спустя эти 2 часа смесь охлаждают до 10oC и прибавляют по каплям в течение 40 минут 321 г (0,537 молей) 12,4% водного раствора гипохлорита натрия. После окончания добавления реакционную смесь дополнительно перемешивают 40 минут, затем от реакционной массы отгоняют трет-бутанол/водную среду при медленном нагревании смеси до приблизительно 100oC. После окончания отгонки остаток промывают водой, сушат и получают 1-(4-хлор-2-фторфенил)-4,5-дигидро-3-метил-1,2,4-триазол-5 (1H)-он.
Пример 4.
Получение 1-(2,4-дихлор-5-метилсульфониламинофенил)-4,5-дигидро-3-метил-1,2,4- триазол-5 (1H)-она.
К раствору 117,3 г (0,500 молей) 2,4-дихлор-5-(метилсульфониламино)фенилгидразина в 200 мл трет-бутанол/воды (88/12) при охлаждении до 0 - 5oC при перемешивании прибавляют по каплям в течение 20 минут раствор 23,4 г (0,525 молей - 5% избыток) ацетальдегида в 54 г трет-бутанол/воды, перемешивают 5 минут и вносят в одну порцию суспензию 39,8 г (91,5% чистоты - 0,560 молей - 12% молярный избыток) цианата натрия в 90 г воды. Дополнительное количество воды, используемое для смыва остатков суспензии цианата натрия из его контейнера, переносится в реакционную массу, которую охлаждают до 0 - 5oC и прибавляют по каплям в течение 15 минут 39,3 г (0,655 молей - 5% эквивалентный избыток по эквиваленту цианата/натрия и его примеси карбоната натрия) уксусной кислоты. После окончания добавления реакционную смесь перемешивают приблизительно 2 часа, в течение которых допускается повышение температуры до 20oC. Спустя 2 часа, реакционную смесь охлаждают до 10oC и прибавляют по каплям в течение 40 минут 321 г 12,4% водного раствора гипохлорита натрия. После окончания добавления смесь перемешивают дополнительно 40 минут, отгоняют от реакционной массы трет-бутанол/водную среду при медленном нагревании смеси до приблизительно 100oC. После окончания отгонки остаток промывают водой, сушат и получают 1-(2,4-дихлор-5-метил сульфониламинофенил)-4,5-дигидро-3-метил-1,2,4-триазол-5-(1H)-он.
Пример 5.
Получение 4,5-дигидро-3-метил-1-фенил-1,2,4-триазол-5 (1H)-она.
В раствор на 50 галлонов (189,3 л) загружают 61,1 фунтов (27,7 кг) трет-бутанол/воды (88/12), охлаждают до 5oC, вносят 22,0 фунтов (9,98 г) (0,203 фунт-молей) (92,1 молей) фенилгидразина, затем, поддерживая температуру реакционной смеси при 5oC, прибавляют в течение 90 минут раствор 9,3 фунтов (4,22 кг) (0,211 фунт-молей) (95,7 молей) ацетальдегида в 20 фунтах (9,07 кг) трет-бутанол/воды (88/12). После окончания добавления вносят в течение 5 минут смесь 15,6 фунтов (7,08 кг) (0,240 фунт-молей) (109 молей) 85% чистоты цианата натрия в 44,5 фунтах (20,2 кг) воды. При добавлении температура реакции повышается приблизительно на 5 - 10oC. Реакционную смесь снова охлаждают до 5oC, перемешивают 30 минут и вносят в течение 30 - 45 минут, поддерживая температуру приблизительно 10oC, 14,8 фунтов (6,71 кг) (0,247 фунт-молей) (112 молей) уксусной кислоты. После окончания добавления реакционную смесь перемешивают 3 часа при приблизительно 10oC до окончания реакции. Границу реакции и концентрацию промежуточного фенилтриазолидинона определяют газохроматографически. После окончания реакции раствор 12,6 фунтов (5,72 кг) хлористого натрия в 35,8 фунтах (16,2 кг) воды перемешивают с реакционной смесью. Водную фазу отделяют при температуре реакционной смеси приблизительно 15oC. После чего, поддерживая температуру смеси при 20oC, прибавляют в течение 3 часов 120,5 фунтов (54,7 кг) водного 11,1% (вес/вес) гипохлорита натрия (0,180 фунт-молей) (81,6 молей). После окончания добавления реакционную смесь перемешивают 1 час при 20oC, затем вносят 67,8 фунтов (30,8 кг) воды, отгоняют 73,4 фунтов (33,3 кг) трет-бутанол/воды (88/12) при температуре отгона 80oC. Остаток охлаждают до 0oC, твердый продукт отфильтровывают, сушат при 80oC/ мм H в течение 24 часов и получают 32,2 фунта (14,6 кг) (выход 91%) 4,5-дигидро-3-метил-1-фенил-1,2,4-триазол-5 (1H)-он.
Пример 6.
Получение 4,5-дигидро-3-метил-1-фенил-1,2,4-триазол-5 (1H)-она.
В реактор объемом 500 галлонов (1893 л) загружают 71,5 фунтов (32,4 кг) воды и 534 фунта (242,2 кг) трет-бутанол/водной среды (88/12), перемешивают, охлаждают до 5oC, прибавляют 220 фунтов (99,8 кг) (2,03 фунт-молей) (921 молей) фенилгидразина, затем, поддерживая температуру реакции приблизительно 0 - 9oC, прибавляют в течение 3 часов 20 минут раствор 93 фунтов (42,2 кг) (2,11 фунт-молей) (957 молей) ацетальдегида в 207 фунтах (93,9 кг) трет-бутанол/воды (88/12). Спустя это время в течение 10 минут вносят смесь 155 фунтов (70,3 кг) (2,38 фунт-молей) (1080 молей) цианата натрия 80%-ной чистоты в 991 фунтах (450 кг) воды. Добавление вызывает повышение температуры до 14oC. Реакционную смесь охлаждают до приблизительно 8oC при перемешивании в течение 50 минут. Затем, поддерживая температуру реакции приблизительно 6 - 12oC, вносят в течение 95 минут 148 фунтов (67,1 кг) (2,47 фунт-молей) (1120 молей) уксусной кислоты. После окончания добавления реакционную смесь перемешивают 3 часа при температуре приблизительно 10oC до окончания реакции. Течение реакции контролируется с помощью тонкослойной хроматографии. Спустя это время к реакционной смеси добавляют раствор 125,8 фунтов (57,1 кг) хлористого натрия в 358 фунтах (162,4 кг) воды, перемешивают в течение 30 минут, выдерживают 1 час для отделения водной фазы, которую спустя это время отделяют от реакционной смеси. Смесь охлаждают до приблизительно 7oC и прибавляют в течение 8 часов 1234,3 фунтов (559,9 кг) 11,2% (вес/вес) водного гипохлорита натрия (1,86 фунт-молей) (843,7 молей), во время добавления температура реакционной смеси поднимается до приблизительно 13oC. После добавления реакционную смесь перемешивают 1 час и добавляют 678 фунтов (307,5 кг) воды. Смесь нагревают до приблизительно 102oC, удаляют отгонкой азеотроп трет-бутанол/вода (88/12) при температуре отгона 80oC. Остаток перемешивают с 50 фунтами (22,7 кг) воды, охлаждают до приблизительно 0oC. Выпавший твердый продукт отделяют центрифугированием и получают 342 фунта (155,1 кг) сырого продукта, который по анализу высушенной пробы содержит 318,5 фунтов (144,5 кг) (88,6% выход) 4,5-дигидро-3-метил-1-фенил-1,2,4-триазол-5 (1H)-она (98,9% чистоты).
Пример 7.
Получение 3-метил-1-фенил-1,2,4-триазолидин-5-она.
В реактор объемов 500 галлонов (1893 л) загружают 61,1 фунт (27,7 кг) трет-бутанола (88/12 азеотроп с водой), перемешивают, охлаждают до 5oC, прибавляют 22,0 фунта (9,98 кг) (0,203 фунт-молей) (92,1 молей) ацетальдегид фенилгидразона. Затем, поддерживая температуру реакционной смеси 0 - 5oC, прибавляют в течение 5 минут 15,6 фунтов (7,08 кг) (0,204 фунт-молей) (109 молей) цианата натрия 85%-ной чистоты в 44,5 фунтах (20,2 кг) воды, перемешивают 30 минут при 5oC. Затем, поддерживая температуру реакционной смеси приблизительно 10oC, прибавляют в течение 30 - 45 минут 14,8 фунтов (6,71 кг) (0,247 фунт-молей) (12 молей) уксусной кислоты. После окончания добавления реакционную смесь перемешивают 3 часа при температуре приблизительно 10oC до окончания реакции. Граница реакции и концентрация промежуточного фенилтриазолидинона определяется газохроматографически. Спустя это время раствор 12,6 фунтов (5,71 кг) хлористого натрия в 35,8 фунтах (16,2 кг) воды перемешивают вместе с реакционной массой. Водную фазу отделяют при температуре реакционной смеси приблизительно 15oC. Затем к реакционной смеси прибавляют 67,8 фунтов (30,8 кг), отгоняют 73,4 фунтов (33,3 кг) трет-бутанол/воды (88/12) при верхней температуре 80oC. Остаток охлаждают до 0oC, твердый продукт отфильтровывают, сушат при 80oC/5 мм H в течение 24 часов и получают 3-метил-1-фенил-1,2,4-триазолидин-5-он.
Пример 8.
Получение 4,5-дигидро-3-метил-1-фенил-1,2,4-триазол-5 (1H)-она при использовании в качестве растворителя уксусной кислоты.
В реактор объемом 50 галлонов (189,3 л) помещают раствор 12 фунтов (5,44 кг) (0,111 фунт-молей) (50,3 молей) фенилгидразина в 115 фунтах (52,2 кг) ледяной уксусной кислоты при охлаждении до приблизительно 5 - 10oC, при перемешивании прибавляют раствор 5,2 фунтов (2,36 кг) (0,087 фунт-молей) (39,5 молей) ацетальдегида в 6,1 фунтах (2,77 кг) уксусной кислоты со скоростью, позволяющей поддерживать температуру реакции приблизительно 12 - 15oC. Полное добавление требует приблизительно около 5 минут. Немедленно после окончания добавления прибавляют холодный раствор (приблизительно 15oC) 7,6 фунтов (3,45 кг) (0,117 фунт-молей) (53,1 молей) цианата натрия в 72 фунтах (32,7 кг) воды со скоростью, которая требуется для поддержания температуры реакции приблизительно 8 -12oC. Общее добавление требует приблизительно 30 минут. После окончания добавления реакционную смесь перемешивают 20 минут, затем нагревают до 20oC и вносят со скоростью, обеспечивающей температуру реакции 25 - 40oC, раствор 13,3 фунтов (6,03 кг) (0,179 фунт-молей) гипохлорита натрия в 119,7 фунтах (54,3 кг) воды. Полное добавление требует приблизительно 1 час. После окончания добавления реакционную смесь перемешивают 1 час, затем отгоняют в вакууме (50 мм H) приблизительно 220 фунтов (99,8 кг) (примерно 72%) растворителя, уксусная кислота/вода, вносят 82 фунта (37,2 кг) воды, образовавшуюся суспензию охлаждают до 25oC. Конечный продукт выделяют центрифугированием, промывают на центрифуге водой, сушат при 70oC в вакууме и получают 14,8 фунтов (6,71 кг) 84,4% чистоты 4,5-дигидро-3-метил-1-фенил-1,2,4-триазол-5- (1H)-он (64,2% выход).
Изобретение относится к получению 1-арилтриазолинона формулы I, где R - низший алкил; Х - галоген, низший алкил, нитро, гидрокси, -NHSO2R', -Ni(SO2R')2, -N(R')SO2R'; R' - низший алкил; n - целое число от 0 до 3, обработкой арилтриазолидинона формулы II галогеноватистой кислотой или ее солью в среде трет-бутанол - вода при весовом соотношении (95-70):(5-30) соответственно. Среди особых преимуществ такого способа получения - высокая степень чистоты при высоком выходе продукта. 8 з.п. ф-лы. 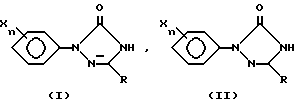
где R является низшим алкилом;
X представляет собой независимо друг от друга галоген, низший алкил, нитро, гидрокси, -NHSO2R', -N(SO2R')2, -N(R')SO2R',
где R' является низшим алкилом и n равно целому числу от 0 до 3,
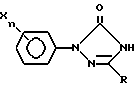
включающий обработку арилтриазолидинона общей формулы II
где R, X и n имеют указанные выше значения,
галогеноватистой кислотой или ее солью, отличающийся тем, что процесс осуществляют в среде трет.-бутанол - вода при весовом соотношении (95 - 70) : (5 - 30) соответственно.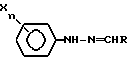
в которой X, n и R имеют значения, определенные в п. 1,
с цианатом щелочного металла в присутствии органической кислоты в среде трет.-бутанол - вода.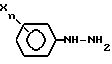
где X и n имеют значения, определенные в п. 1,
с альдегидом общей формулы V
R - C(O)H,
где R имеет значения, определенные в п. 1,
в среде трет.-бутанол - вода.
| US 4980480, 1990 | |||
| US 4818275, 1989 | |||
| Торфодобывающая машина с вращающимся измельчающим орудием | 1922 |
|
SU87A1 |
| Огнетушитель | 0 |
|
SU91A1 |
| US 4818275, 1989 | |||
| Приспособление для построения разверток поверхностей | 1972 |
|
SU441004A1 |
| EP 0431373, 1991. | |||
