Изобретение относится к серии новых производных 1-бифенилметилимидазола, обладающих гипотенсивной активностью, и поэтому предназначенных для лечения и профилактики гипертензии, включая заболевания сердца и системы кровообращения, а также к способам получения указанных соединений, к композициям, содержащим эти соединения, и к способам получения указанных композиций.
Известно, что ренин-ангиотензинная система обеспечивает один из наиболее важных механизмов, направленных на поддержание гомеостаза кровяного давления в живых организмах. При понижении кровяного давления или при снижении концентрации ионов натрия в жидкостях организма эта система активизируется. В результате этого активируются фермент ренин и ангиотензин-конвертирующий фермент (далее обозначаемый "АКФ") и воздействуют на ангиотензиноген, который сначала с помощью ренина разлагается с образованием ангиотензина I, обозначаемого далее "АI", а затем с помощью АКФ превращается в ангиотензин II, обозначаемый далее "А II". Так как А II способствует сильному сокращению кровеносных сосудов и ускоряет секрецию альдостерона, то активация указанной системы приводит к повышению кровяного давления. Ингибиторы или суппрессоры ренин-ангиотензинной системы, например ингибиторы ренина, АКФ-ингибиторы и АII-антагонисты, способствуют расширению кровеносных сосудов, вызывая тем самым снижение кровяного давления и улучшение функции кровообращения. Поэтому лечение сердечных заболеваний основано на использовании вышеуказанных агентов.
В настоящее время, лишь АКФ-ингибиторы находят клиническое применение, хотя и ведутся исследования по использованию в этих целях ингибиторов ренина и АII-антагонистов. Некоторые из них, например АII-антагонисты пептидного типа, такие, как саралазин, известны уже давно, тогда, как другие антагонисты, не пептидного типа, открыты совсем недавно (см. например, публикации Европатента N 28833, 28834, 245637, 253310, 323841, 324377, 380959, 399732, 399731 и 400835 и заявку на патент Японии Кока N 57-98270). Из указанных работ наиболее близким прототипом являются публикации Европатента N 253310 и N 324377.
В известной публикации раскрывается ряд производных 1-фенил-, 1-фенетил- или 1-бензилимидазола, обладающих способностью к ингибированию АII. Соединения, входящие в объем указанного прототипа, являются производными 1-бифенил-метилимидазола, которые, однако, отличаются от предлагаемых соединений природой заместителя в 4- или 5-положениях имидазола.
В публикации Европатента N 324377 также раскрывается ряд аналогичных соединений. Однако активности соединений указанного прототипа, а также соединения вышеназванного прототипа N 253310 являются недостаточными и для получения хороших клинических результатов необходимо использовать более сильные АII-антагонистические средства.
Изобретение относится к соединениям формулы
где4 R1 представляет собой алкильную группу, имеющую 1-6 атомов углерода;
R2 и R3 являются одинаковыми или различными и каждый представляет собой алкильную группу, имеющую 1-6 атомов углерода, или арильную группу, имеющую 6-10 атомов углерода;
R4 представляет собой атом водорода или алкильную группу, имеющую 1 6 атомов углерода;
R5 представляет собой карбокси-группу или группу формул -COOR5a или -CONR8R9, где R8 и R9 являются одинаковыми или различными и каждый из них представляет собой атом водорода, незамещенную алкильную группу, имеющую 1-6 атомов углерода, замещенную алкильную группу, имеющую 1-6 атомов углерода и замещенную карбокси-группу или алкоксикарбонильной группой, в которой алкильная часть имеет 1-6 атомов углерода, или R8 и R9 вместе представляют собой замещенную алкиленовую группу, которая имеет 2-6 атомов углерода и которая является замещенной, алкоксикарбонильной группой, в которой алкильная часть имеет 1-6 атомов углерода; R5a представляет собой алкильную группу, имеющую 1-6 атомов углерода, алканоилоксиалкильную группу, в которой каждая из алканоильной и алкильных частей имеет 1-6 атомов углерода, алкоксикарбонилоксиалкильную группу, в которой каждая из алкокси и алкильной частей имеет 1-6 атомов углерода, (5-метил-2-оксо-1,3-диоксолен-4-ил)метильную группу, или фталидильную группу,
R6 представляет собой атом водорода;
R7 представляет собой карбокси-группу или тетразол-5-ильную группу,
и его фармацевтически приемлемые соли и эфиры.
Изобретение относится также к фармацевтически приемлемым солям и сложным эфирам описанных выше соединений.
Изобретение также относится к фармацевтическим композициям, предназначенным для лечения и профилактики гипертензии и содержащим эффективное количество гипотензивного средства в сочетании с фармацевтически приемлемым носителем или разбавителем, где гипотензивное средство выбирают из группы, состоящей из соединений формулы I и их фармацевтически приемлемых солей и сложных эфиров.
Кроме того, изобретение относится к способам получения соединений формулы I и их фармацевтически приемлемых солей и сложных эфиров, которые подробно описаны ниже.
В предлагаемых соединениях, где R1, R2, R3, R4, R8, R9, являются алкильной группой, указанная алкильная группа представляет собой алкильную группу с 1-6 атомами углерода, которая может быть прямой или разветвленной группой, такой, как метил, этил, пропил, изопропил, бутил, изобутил, вторбутил, т-бутил, пентил, т-пентил, 2-метилбутил, 3-метилбутил, 1-этилпропил, 4-метилпентил, 3-метилпентил, 2-метилпентил, 1-метилпентил, 3,3-диметилбутил, 2,2-диметилбутил, 1,1-диметилбутил, 1,2-диметилбутил, 1,3-диметилбутил, 2,3-диметилбутил, 2-этилбутил, гексил и изогексил. R1 является предпочтительно прямой или разветвленной алкильной группой, содержащей 2-5 атомов углерода, и более предпочтительно прямой группой, а наиболее предпочтительно этилом, пропилом или бутилом. R2 и R3 могут быть одинаковыми или различными, представляют собой прямую или разветвленную алкильную группу, содержащую 1-4 атомов углерода, предпочтительно метильную, этильную, пропильную, изопропильную или т-бутильную группу, а более предпочтительно метильную или этильную группу, если R5 является карбокси-группой, или изопропильную или т-бутильную группу, если R5 является группой формулы -CONR8R9, R4 является предпочтительно прямой или разветвленной алкильной группой, имеющей 1-4 атомов углерода, а более предпочтительно метильной или этильной группой. Если R8 и R9 являются алкильным группами, то они могут быть одинаковыми или различными, и каждый из них предпочтительно является алкильной группой, содержащей 1-4 атомов углерода, более предпочтительно метильной, этильной, пропильной или бутильной группой, а наиболее предпочтительно метильной или этильной группой. В случае, если заместитель (b) является алкильной группой, то предпочтительно она является группой с 1-4 атомами углерода, а более предпочтительно метильной или этильной группой.
В случае, когда R2 и R3 представляют арильную группу, то этой группой является ароматическая карбоциклическая группа, которая имеет 6-14 (предпочтительно 6-10, а более предпочтительно 6 или 10) кольцевых атомов, R5 представляет собой карбоксильную группу или группу формулы -CONR8R9. В случае, если R5 является группой формулы -CONR8R9 и R8 и R9 являются алкильной группой, то эта группа может быть незамещенной алкильной группой, имеющей 1-6 атомов углерода, например такой, как группы, указанные выше, или замещенной алкильной группой, которая имеет 1-6 атомов углерода и которая является замещенной карбокси-группой или алкоксикарбонильной группой.
Если R8 и R9 вместе представляют замещенную алкиленовую группу, то эта группа содержит 2-6 атомов углерода и может быть прямой или разветвленной. В качестве заместителя, например, используется алкоксикарбонильная группа, алкильная часть которой содержит 1-6 атомов углерода.
Примерами таких заместителей являются метоксикарбонил, этоксикарбонил, пропоксикарбонил, бутоксикарбонил, изобутоксикарбонил, т-бутоксикарбонил, пентилоксикарбонил и гексилоксикарбонил, из которых предпочтительными являются карбокси, метоксикарбонильная и этоксикарбонильная группы.
Если R5 представляет карбоксильную группу, то соединение является карбоновой кислотой, и поэтому может образовывать сложные эфиры, в которых карбоксильная группа, представленная R5, замещается группой формулы -COOR5a, где R5a представляет собой сложноэфирный остаток (в случае карбоновой кислоты, R5a является атомом водорода). Оно может также образовывать соли. Природа образованного таким образом сложного эфира не имеет решающего значения, кроме того, что этот эфир должен удовлетворять фармацевтическим требованиям, то есть его токсичность не должна быть выше, а активность не должна быть ниже исходной кислоты. Однако, если данный сложный эфир используется в других целях, например в качестве промежуточного соединения для получения другого, возможно более активного соединения, то даже это ограничение можно не принимать во внимание и в данном случае приемлемым является любой, обычно используемый эфирный остаток, который может быть выбран на основании его функциональности и коммерческих преимуществ. Однако хорошо известно, что некоторые эфирные остатки придают соединениям определенные преимущества, например такие соединения легче и лучше абсорбируются in vivo, а поэтому при необходимости такие эфирные остатки могут быть использованы в изобретении.
Примерами указанных сложноэфирных остатков являются:
алкильные группы, имеющие 1-6 атомов углерода, такие, как группы, приведенные выше в качестве примеров для R1;
алканоилоксиалкильные группы, в которых каждая из алканоильных и алкильных частей имеет 1-6 атомов углерода и может быть любой из приведенных выше примеров для R1 и R4, соответственно, и предпочтительно алканоильная часть имеет 1-5 атомов углерода, а алкильная часть 1-4 атомов углерода, а более предпочтительно, если алканоильная часть имеет 2-5 атомов углерода и алкильная часть 1-2 атомов углерода, при этом примерами алканоилоксиалкильных групп являются формилоксиметил, ацетоксиметил, пропионилоксиметил, бутирилоксиметил, пивалоилоксиметил, валерилоксиметил, изовалерилоксиметил, гексаноилоксиметил, 1-(формилокси)этил, 1-(ацетокси)этил, 1-(пропионилокси)этил, 1-(бутирилокси)этил, 1-(пивалоилокси)-этил, 1-(бутиролокси)этил, 1-(пивалоилокси)этил, 1-(валерилокси)этил, 1-(изовалерилокси)этил, 1-(гексаноилокси)этил, 1-(формилокси)этил, 1-(ацетокси)этил, 2-(пропионилокси)этил, 2-(бутирилокси)этил, 2-(пивалоилокси)этил, 2-(валерилокси)-этил, 2-(изовалерилокси)этил, 2-(гексаноилокси)этил, 1-(формилокси)пропил, 1-(ацетокси)пропил, 1-(пропионилокси)пропил, 1-(бутирилокси)пропил, 1-(пивалоилокси) пропил, 1-(валерилокси)пропил, 1-(изовалерилокси)пропил, 1-(гексаноилокси)-пропил, 1-(ацетокси)бутил, 1-(пропиноилокси)бутил, 1-(бутирилокси)бутил, 1-(пивалоилокси)бутил, 1-(ацетокси)пентил, 1-(пропионилокси)фенил, 1-(бутирилокси)пентил, 1-(пивалоилокси)пентил и 1-(пивалоилокси)гексил, из которых предпочтительными являются формилоксиметил, ацетоксиметил, пропионилоксиметил, бутирилоксиметил, пивалоилоксиметил, 1-(формилокси)этил, 1-(ацетокси)этил, 1-(пропионилокси)этил, 1-(бутирилокси)этил и 1-(пивалоилокси)этил, а более предпочтительными являются ацетоксиметил, пропиноилоксиметил, бутирилоксиметил, пивалоилоксиметил, 1-(ацетокси)этил, 1-(пропионилокси)этил, 1-(бутирилокси)этил и 1-(пивалоилокси)этил, и наиболее предпочтительными являются пивалоилоксиметил и 1-пивалоилоксиэтил;
алкоксикарбонилоксиалкильные группы, в которых каждая из алкокси и алкильных частей имеет 1-6 атомов углерода, являются такими же, как они были определены выше для R1, соответственно, причем предпочтительные из них имеют 1-4 атомов углерода, а наиболее предпочтительно, если алкокси часть имеет 1-4 атомов углерода, а алкильная часть имеет 1-2 атомов углерода, примерами указанных алкоксикарбонилоксиалкильных являются метоксикарбонилоксиметил, этоксикарбонилоксиметил, пропоксикарбонилоксиметил, изопропоксикарбонилоксиметил, бутоксикарбонилоксиметил, изобутоксикарбонилоксиметил, пентилоксикарбонилоксиметил, гексилоксикарбонилоксиметил, 1-(метоксикарбонилокси)этил, 1-(этоксикарбонилокси)этил, 1-(пропоксикарбонилокси)этил, 1-(изопропоксикарбонилокси)этил, 1-(бутоксикарбонилокси) этил, 1-(изобутоксикарбонилокси)этил, 1-(пентилоксикарбонилокси)этил, 1-(гексилоксикарбонилокси)этил, 2-(метоксикарбонилокси)этил, 2-(этоксикарбонилокси)этил, 2-(пропоксикарбонилокси)этил, 2-(изопропоксикарбонилокси)этил, 2-(бутоксикарбонилокси)этил, 2-(изобутоксикарбонилокси)-этил, 2-(пентилоксикарбонилокси)этил, 2-(гексилоксикарбонилокси)-этил, 1-(метоксикарбонилокси)пропил, 1-(этоксикарбонилокси)-пропил, 1-(пропоксикарбонилокси)пропил, 2-(изопропоксикарбонилокси)пропил, 1-(бутоксикарбонилокси)пропил, 1-(изобутоксикарбонилокси)пропил, 1-(пентилоксикарбонилокси)пропил, 1-(гексилоксикарбонилокси)пропил, 1-(метоксикарбонилокси)бутил, 1-(этоксикарбонилокси)бутил, 1-(пропоксикарбонилокси)-бутил, 1-(изопропоксикарбонилокси)бутил, 1-(бутоксикарбонилокси)бутил, 1-(изобутоксикарбонилокси)бутил, 1-(метоксикарбонилокси)-пентил, 1-(этоксикарбонилокси)пентил, 1-(метоксикарбонилокси)-гексил и 1-(этоксикарбонилокси)гексил, предпочтительно метоксикарбонилоксиметил, этоксикарбонилоксиметил, пропоксикарбонилоксиметил, изопропоксикарбонилоксиметил, бутоксикарбонилоксиметил, изобутоксикарбонилоксиметил, 1-(метоксикарбонилокси)этил, 1-(этоксикарбонилокси)этил, 1-(пропоксикарбонилокси)этил, 1-(изопропоксикарбонилокси)этил, 1-(бутоксикарбонилокси)этил, 1-(изобутоксикарбонилокси)этил, 1-(метоксикарбонилокси)пропил, 1-(этоксикарбонилокси)пропил, 1-(пропоксикарбонилокси)пропил, 1-(изопропоксикарбонилокси)пропил, 1-(бутоксикарбонилокси)пропил, 1-(изобутоксикарбонилокси)пропил, 1-(метоксикарбонилокси)бутил, 1-(этоксикарбонилокси)бутил, 1-(пропоксикарбонилокси)бутил, 1-(изопропоксикарбонилокси)бутил, 1-(бутоксикарбонилокси)бутил, 1-(изобутоксикарбонилокси)бутил, более предпочтительно метоксикарбонилоксиметил, этоксикарбонилоксиметил, пропоксикарбонилоксиметил, изопропоксикарбонилоксиметил, бутоксикарбонилоксиметил, изобутоксикарбонилоксиметил, 1-(метоксикарбонилокси)этил, 1-(этоксикарбонилокси)этил, 1-(пропоксикарбонилокси)этил, 1-(изопропоксикарбонилокси)этил, 1-(бутоксикарбонилокси)этил и 1-(изобутоксикарбонилокси)этил, и наиболее предпочтительно метоксикарбонилоксиметил, этоксикарбонилоксиметил, изопропоксикарбонилоксиметил, 1-(метоксикарбонилокси) этил, 1-(этоксикарбонилокси)этил и 1-(изопропоксикарбонилокси)этил.
Предпочтительно, если бензольное кольцо, имеющее заместителей, представленных R6 и R7, находится в 3- или 4-положениях, а более предпочтительно в 4-положении бензильной группы, с которой оно связано, то есть предпочтительные соединения имеют формулу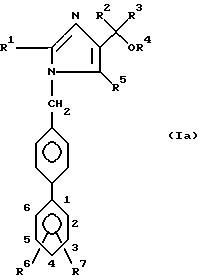
где R6 является атомом водорода;
R7 может быть карбоксильной группой или тетразол-5-ильной группой. Если R7 и является карбоксильной группой, то полученные в результате соединения могут образовывать соли или сложные эфиры. Указанные соли или сложные эфиры могут быть любой природы при условии, что они являются пригодными для терапевтического использования и фармацевтически приемлемыми. Если эти соли или сложные эфиры не предназначены для терапевтического использования, а предназначены для использования в качестве промежуточных соединений, для получения других, возможно более активных соединений, то даже указанным выше ограничением можно пренебречь. Примерами таких солей могут служить соли щелочных металлов, таких как натрий, калий или литий, соли щелочно-земельных металлов, таких как барий или кальций, соли других металлов, таких как магний или алюминий, соли органических оснований, таких как соли гванидина, триэтиламина или дихлоргексиламина, или соли с основными аминокислотами, такими как лизин или аргинин. Примерами сложноэфирных групп могут быть группы, представленные выше для R5a. Предпочтительно, если R7 находится во 2- или 3-положении фенильной группы, а более предпочтительно во 2-положении фенильной группы.
Предлагаемые соединения обязательно должны содержать, по крайней мере, один основной атом азота в имидазольном кольце, и поэтому они могут образовывать кислые аддитивные соли. Примерами таких кислых аддитивных солей являются аддитивные соли с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, серная кислота или фосфорная кислота, и аддитивные соли с органическими кислотами, такими как малеиновая кислота, фумаровая кислота, винная кислота или лимонная кислота.
Предпочтительными классами соединений изобретения являются:
соединения формулы I и их соли и сложные эфиры, в которых R5 представляет собой алкильную группу, имеющую 1-4 атомов углерода, алканоилоксиалкильную группу, в которой каждая алканоильная часть имеет 1-5 атомов углерода и алкильная части имеют 1-4 атомов углерода, алкоксикарбонилоксиалкильную группу, в которой каждая алкокси- и алкильная части имеют 1-4 атомов углерода, [5-метил-2-оксо-1,3-диоксолен-4-ил] метильную группы, фталидильную группу;
соединения, в которых R5a представляет собой C1-C4-алкильную группу, алканоилоксиалкильную группу, в которой алканоильная группа имеет 1-5 атомов углерода, а алкильная группа имеет 1-4 атомов углерода, алкоксикарбонилоксиалкильную группу, в которой алкокси-часть имеет 1-4 атомов углерода и алкильная часть имеет 1-2 атомов углерода, [5-метил-2-оксо-1,3-диоксолен-4-ил] метильную группу или фталидильную группу;
соединения, в которых R5a представляет собой пивалоилоксиметильную, этоксикарбонилоксиметильную, 1-(этоксикарбонилокси)этильную, изопропоксикарбонилоксиметильную, 1-(изопропоксикарбонилокси)этильную, (5-метил-2-оксо-1,3-диоксолен-4-ил)метильную или фталидильную группы;
соединения, в которых
R1 представляет собой алкильную группу, имеющую от 2 до 5 атомов углерода;
R2 и R3 являются одинаковыми или различными и каждый из них представляет собой алкильную группу, имеющую 1-4 атомов углерода, бензильную, нафтильную или фенильную группу;
R4 представляет собой атом водорода или алкильную группу, имеющую 1-4 атомов углерода;
R5 представляет собой группу формулы -COOR5a или группу формулы -CONR8R9, в которой R5a представляет собой алкильную группу, имеющую 1-4 атомов углерода, алканоилоксиалкильную группу, в которой алканоильная часть имеет 1-5 атомов углерода, а алкильная часть имеет 1-4 атомов углерода, алкоксикарбонилоксиалкильную группу, в которой алкокси-часть имеет 1-4 атомов углерода, а алкильная часть имеет 1-4 атомов углерода, [5-метил-2-оксо-1,3-диоксолен-4-ил]-метильную группу, в которой алкильная часть имеет 1-4 атомов углерода или фталидильную группу;
R8 и R9 являются одинаковыми или различными и каждая представляет собой атом водорода, алкильную группу, имеющую 1-4 атомов углерода, и замещенную алкильную группу, которая имеет 1-4 атомов углерода и которая является замещенной, карбокси-группой или алкоксикарбонильной группой, где алкильная часть имеет 2-5 атомов углерода; или
R8 и R9 вместе представляют собой замещенную алкиленовую группу, которая имеет 4 или 5 атомов углерода и которая является замещенной, по крайней мере, одним заместителем, выбранным из метоксикарбонильных групп и этоксикарбонильных групп;
бензольное кольцо, которое имеет заместители, представленные R6 и R7, находится в 3-4 положении бензильной группы, с которой он связано;
соединения, в которых R1 представляет собой алкильную группу, имеющую 2-5 атомов углерода; R2 и R3 являются одинаковыми или различными и каждая представляет собой алкильную группу, имеющую 1-4 атомов углерода, бензильную или фенильную группу;
R4 и представляет собой атом водорода, метильную или этильную группу;
R5 представляет собой группу формулы -COOR5a или группу формулы -COOR8R9, в которой R5a представляет собой алкильную группу, имеющую 1-4 атомов углерода, алканоилоксиалкильную группу, в которой алканоильная часть имеет 1-5 атомов углерода, а алкильная часть имеет метильную или этильную группу, циклоалканоилоксиалкильную группу, в которой циклоалканоильная часть имеет 6 или 7 атомов углерода, а алкильная часть является метильной или этильной группой, алкоксикарбонилоксиалкильную группу, в которой алкокси-часть имеет 1-4 атомов углерода, а алкильная часть является метильной или этильной группой, [5-метил-2-оксо-1,3-диоксолен-4-ил] -метильную группу или фталидильную группу;
R8 и R9 являются одинаковыми или различными и каждая представляет собой атом водорода, метильную группу, этильную группу или замещенную метильную или этильную группу, которая является замещенной, по крайней мере, одним заместителем, выбранным из карбокси-групп, метоксикарбонильных групп и этоксикарбонильных групп; или
R8 и R9 вместе представляют собой замещенную алкиленовую группу, которая имеет 4 или 5 атомов углерода и которая является замещенной, по крайней мере, одним заместителем, выбранным из метоксикарбонильных групп и этоксикарбонильных групп;
R6 представляет собой атом водорода;
R7 представляет собой карбокси-группу или тетразол-5-ильную группу во 2- или 3-положении бензольного кольца;
бензольное кольцо, которое имеет заместителей, представленных R6 и R7, находится в 4-положении бензильной группы, с которой оно связано;
соединения, в которых R1 представляет собой алкильную группу, имеющую 2-5 атомов углерода;
R2 и R3 являются одинаковыми или различными и каждый представляет собой атом водорода или алкильную группу, имеющую 1-4 атомов углерода;
R4 представляет собой атом водорода, метильную группу или этильную группу;
R5 представляет группу формулы -COOR5a или группу формулы -CONR8R9 в которой R5a представляет метильную, этильную, или бензильную группу, алканоилоксиметильную группу, в которой алканоильная часть имеет 1-5 атомов углерода, 1-(алканоилокси)этильную группу, в которой алканоильная часть имеет 1-5 атомов углерода, алкоксикарбонилоксиметильную группу, в которой алкокси-часть имеет 1-4 атомов углерода, 1-(алкоксикарбонилокси)этильную группу, в которой алкокси-часть имеет 1-4 атомов углерода, [5-метил-3-оксо-1,3-диоксолен-4-ил] метильную группу или фталидильную группу;
R8 и R9 являются одинаковыми или различными, и каждый представляет собой атом водорода, метильную группу или этильную группу, метоксикарбонилметильную группу, этоксикарбонилметильную группу или карбоксиметильную группу; или
R8 и R9 вместе представляют тетраметиленовую, пентаметиленовую, 1-карбокситетраметиленовую или 1-карбоксипентаметиленовую группу;
R6 представляет собой атом водорода;
R7 представляет собой карбокси-группу или тетразол-5-ильную группу во 2-положении бензольного кольца;
бензольное кольцо, которое имеет заместителей, представленных R6 и R7, находится в 4-положении бензильной группы, с которой оно связано;
соединения, в которых R1 представляет собой этильную, пропильную или бутильную группу;
R2 и R3 являются одинаковыми или различными и каждые представляют собой метильную группу;
R4 представляет атом водорода или метильную группу;
R5 представляет собой группу формулы COOR5a, в которой R5a представляет собой атом водорода, пивалоилоксиметильную группу, этоксикарбонилоксиметильную группу, 1-(этоксикарбонилокси) этильную группу, изопропоксикарбонилоксиметильную группу, 1-(изопропоксикарбонилокси)этильную группу, (5-метил-2-оксо-1,3- диоксолен-4-ил)метильную группу или фталидильную группу;
R6 представляет собой атом водорода;
R7 представляет собой карбокси-группу или тетразол-5-ильную группу во 2-положении бензольного кольца;
бензольное кольцо, которое имеет заместителей, представленных R6 и R7 находится в 4-положении бензильной группы с которой оно связано;
соединения, в которых R1 представляет собой этильную, пропильную или бутильную группу;
R2 и R3 представляют метильные группу;
R4 представляет собой атом водорода или метильную группу;
R5 представляет собой группу формулы -COOR5a в которой R5a представляет собой атом водорода, пивалоилоксиметильную группу, этоксикарбонилоксиметильную группу, 1-(этоксикарбонилокси)этильную группу, изопропоксикарбонилоксиметильную группу, 1-(изопропоксикарбонилокси)этильную группу, (5-метил-2-оксо-1,3-диоксолен-4-ил)метильную группу или фталидильную группу;
R6 представляет собой атом водорода;
R7 представляет собой карбокси-группу или тетразол-5-ильную во 2-положении бензольного кольца;
бензольное кольцо, которое имеет заместителей, представленных R6 и R7, находится в 4-положении бензильной группы, с которой оно связано.
Предлагаемые соединения могут содержать один или несколько асимметрических атомов углерода в своих молекулах, и поэтому могут образовывать оптические изомеры. Хотя все они могут быть представлены одной молекулярной формулой, однако изобретение включает в себя отдельные изомеры и их смеси, в том числе и рацематы. При использовании стереоспецифического синтеза или при использовании оптически активных соединений в качестве исходных материалов могут быть получены непосредственно отдельные изомеры, и с другой стороны, если имеется смесь изомеров, то отдельные изомеры могут быть получены с использованием стандартной техники разрешения.
Конкретные примеры соединений изобретения представлены в конце описания формулами I-1, I-2, I-3, I-4, I-5 и I-6.
Значения R1 R7 в этих формулах указаны выше.
Из них наиболее предпочтительными являются следующие соединения: 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил]-4-(1-гидрокси-1- метилэтил)имидазол-5-карбоновая кислота и ее фармацевтически приемлемые соли; пивалоилоксиметил 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил] -4-(1-гидрокси-1-метилэтил)имидазол-5-карбоксилат и его фармацевтически приемлемые соли; (5-метил-2-оксо-1,3-диоксолен-4-ил)метил 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил] -4-(1-гидрокси-1-метилэтил)имидазол-5-карбоксилат и его фармацевтически приемлемые соли; 1-[(2'-карбоксибифенил-4-ил)метил]-4-(1-гидрокси-1-метилэтил)- 2-пропилимидазол-5-карбоновая кислота и ее фармацевтически приемлемые соли; 1-[(2'-карбоксибифенил-4-ил)метил]-2-этил-4-(1-гидрокси-1- метилэтил)имидазол-5-карбоновая кислота и ее фармацевтически приемлемые соли; 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{ 4-[2-(тетразол-5-ил)фенил] фенил} метилимидазол-5-карбоновая кислота и ее фармацевтически приемлемые соли; 2-бутил-4-(1-гидрокси-1-метилэтил)-1-{ 4- [2-(тетразол-5-ил)фенил] фенил} метилимидазол-5-карбоновая кислота и ее фармацевтически приемлемые соли; пивалоилоксиметил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{ 4-[2-(тетразол-5-ил)фенил] фенил} метилимидазол-5-карбоксилат и его фармацевтически приемлемые соли; пивалоилоксиметил 2-бутил-4-(1-гидрокси-1-метилэтил)-1-{4-[2-(тетразол-5-ил)фенил]фенил} метилимидазол-5-карбоксилат и его фармацевтически приемлемые соли; 5-метил-2-оксо-1,3-диоксолен-4-ил)метил-4-(1-гидрокси-1-метилэтил)-2-пропил-1-{ 4-[2-(тетразол-5-ил)фенил] фенил} метилимидазол-5-карбоксилат и его фармацевтически приемлемые соли; (5-метил-2-оксо-1,3-диоксолен-4-ил)метил 2-бутил-4-(1-гидрокси-метилэтил)-1-{ 4-[2-(тетразол-5-ил)фенил] фенил} метилимидазол-5-карбоксилат и его фармацевтически приемлемые соли; этоксикарбонилоксиметил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{4-[2-(тетразол-5-ил)фенил]фенил} метилимидазол-5-карбоксилат и его фармацевтически приемлемые соли; изопропоксикарбонилоксиметил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{ 4-[2-(тетразол-5-ил)фенил] фенил} метилимидазол-5-карбоксилат и его фармацевтически приемлемые соли; 1-(этоксикарбонилокси)этил 4-(1-гидрокси-1-метил-этил)-2-пропил-1-{ 4- [2-(тетразол-5-ил)фенил] фенил} метилимидазол-5-карбоксилат и его фармацевтически приемлемые соли; 1-(изопропоксикарбонилокси)этил 4-(1-гидрокси-1-метилэтил)-2- пропил-1-{4-[2-(тетразол-5-ил)фенил] фенил} метилимидазол-5- карбоксилат и его фармацевтически приемлемые соли; пивалоилоксиметил 2-этил-4-(1-гидрокси-1-метилэтил)-1-{ 4-[2- (тетразол-5-ил)фенил] фенил} метилимидазол-5-карбоксилат и его фармацевтически приемлемые соли; (5-метил-2-оксо-1,3-диоксолен-4-ил)метил 2-этил-4-(1-гидрокси-1-метилэтил)-1-{ 4-[2-(тетразол-5-ил)фенил]фенил} метилимидазол-5-карбоксилат и его фармацевтически приемлемые соли; пивалоилоксиметил 1-[1(2'-карбоксибифенил-4-ил)-метил] -4-(1- гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилат и его фармацевтически приемлемые соли; (5-метил-2-оксо-1,3-диоксолен-4-ил)метил 1- [(2'-карбоксибифенил-4-ил)метил] -4-(1-гидрокси-1-метилэтил)-2-пропили- мидазол-5-карбоксилат и его фармацевтически приемлемые соли; фталидил 1-[(2'-карбоксибифенил-4-ил)метил]-4-(1-гидрокси-1- метилэтил)-2-пропилимидазол-5-карбоксилат и его фармацевтически приемлемые соли.
Соединения изобретения могут быть получены стандартными способами, обычно используемыми при получении соединений этого типа.
Например, в общих чертах, соединения могут быть получены реакцией соединения формулы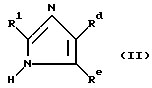
где R1 определен выше;
Rd является группой формулы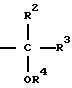
где R2, R3 и R4 определены выше,
или Rd представляет собой группу формулы -COORf, где Rf является карбокси-защитной группой, группу формулы -COR2 где R2 определен выше, или циано-группу;
Re представляет собой циано-группу, карбокси-группу или группу формулы -COORf, где Rf определен выше,
с соединением формулы
где R6 определен выше;
R7a является защищенной карбоксильной группой, циано-группой, защищенной тетразол-5-ил-группой, карбамоильной группой, или алкилкарбамоильной группой;
X является атомом галогена,
в результате чего получают соединение формулы
где Rd, Re, R1, R6 и R7a определены выше,
и в любом порядке, с последующим удалением защитных групп и при необходимости превращением указанной группы Rd в группу формулы
где R2, R3 и R4 определены выше,
и при необходимости с превращением указанной группы Re в группы R5, превращением указанной группы R7a в группу R7 или алкилированием или ацилированием гидроксильной группы в R4 с получением соединения формулы I, и необязательно, с последующим солеобразованием или этерификацией полученного продукта.
Предпочтительно, если Re является защищенной карбоксильной группой, когда R7a является защищенной карбоксильной группой, защищенной тетразолильной группой, и Re является циано-группой, когда R7a является защищенной карбоксильной группой или защищенной тетразолильной группой.
В частности, соединения изобретения могут быть получены в соответствии с приведенными в конце описания реакционными схемами.
Реакционная схема А. В этой реакционной схеме соединение формулы I получают с помощью реакции имидазол-5-карбоновой кислоты или ее сложного эфира формулы V с бифенилметилгалидом формулы III, с последующим, если необходимо, удалением защитных групп, превращением группы формулы -COOR5a в любую другую группу, представленную R5, превращением группы, представленной R7a, в любую другую группу, представленную R7, и/или алкилированием или ацилированием гидрокси-группы в R4, как показано на реакционной схеме А, где R1, R2, R3, R4, R5, R5a, R6, R7, R7a и X определены выше.
Если R7a является защищенной карбоксильной группой, то защитной группой может быть любой сложно-эфирный остаток из проиллюстрированных выше для R5a. Альтернативно, R7a может быть карбамоильной группой или замещенной карбамоильной группой формулы CONHR, где R представляет собой атом водорода или алкильную группу, имеющую 1-6 атомов углерода, например любую из групп, проиллюстрированных выше для R1. Примерами таких карбамоильных групп, которые могут быть представлены R7a, являются карбамоил, метилкарбамоил, этилкарбамоил, пропилкарбамоил, бутилкарбамоил, т-бутилкарбамоил, пентилкарбамоил, т-пентилкарбамоил и гексилкарбамоил, из которых предпочтительными являются карбамоил, т-бутилкарбамоил и т-пентилкарбамоил. Если R7a является защищенной тетразолильной группой, то защитной группой может быть любая защитная группа, которая обычно используется для блокирования тетразолильных групп в стандартных соединениях этого типа. Примерами подходящих защитных групп являются аралкильные группы, определенные и проиллюстрированные выше для R2, однако предпочтительными являются бензильная, дифенилметильная (бензгидрил) и трифенилметильная (тритил) группа, а наиболее предпочтительной является тритильная группа. X является атомом галогена, предпочтительно атомом хлора, брома или иода.
В стадии АI реакционной схемы A, соединение формулы Iа получают посредством реакции имидазол-5-карбоксилатного соединения формулы V с бифенилметиловым соединением формулы III. Обычно и предпочтительно реакция протекает в инертном растворителе и предпочтительно в присутствии основания.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. В принципе, нет конкретных ограничений на природу растворителя, при условии, если он не оказывает неблагоприятного воздействия на реакцию или реагенты и обладает способностью к растворению реагентов по крайней мере до определенной степени. Примерами подходящих растворителей являются углеводороды, предпочтительно ароматические углеводороды, такие, как толуол, простые эфиры, такие, как тетрагидрофуран или диоксан, спирты, такие, как метанол, этанол, или т-бутанол, амиды, такие, как N,N-диметилацетамид, N,N-диметилформамид, или N-метил-2-пирролидинон, кетоны, такие, как ацетон или метилэтилкетон, нитрилы, такие, как ацетонитрилы, и сульфоксиды, такие, как диметилсульфоксид. Из указанных растворителей предпочтительными являются амиды, кетоны, нитрилы и сульфоксиды.
Аналогично, природа основания, используемого в реакции, не является критической, и в этой реакции может быть использовало любое основание, способное реагировать с кислотой H-X. Примерами предпочтительных оснований, которые могут быть использованы в реакции, являются карбонаты щелочного металла, такие, как карбонат натрия или калия, гидриды щелочного металла, такие, как гидрид натрия, калия или лития, алкоксиды щелочного металла, такие, как метоксид натрия, этоксид натрия, т-бутоксид калия или метоксид лития, и бикарбонаты щелочных металлов, такие, как бикарбонат натрия или калия. Из указанных соединений предпочтительными являются карбонаты щелочных металлов, гидриды щелочных металлов или алкоксиды щелочных металлов.
Данная реакция может протекать в широком диапазоне температур и конкретное значение температуры не имеет решающего значения для данного изобретения. Но, в основном, подходящей температурой проведения реакции является от -10 до 100oC, а более предпочтительно является температура 0-80oC. Время, необходимое для проведения реакции также может варьироваться в широких пределах в зависимости от многих факторов, а именно от температуры реакции и природы реагентов и растворителя, участвующих в реакции. Однако, в предпочтительных условиях осуществления реакции, это время составляет от 30 мин до 24 ч, а более предпочтительно 1-16 ч.
После завершения реакции нужное соединение формулы Ia может быть восстановлено из реакционной смеси стандартными способами. Один из таких способов заключается в том, что удаляют растворитель путем дистилляции при пониженном давлении, смешивают полученный остаток с водой, экстрагируют остаток водонесмешиваемым растворителем, таким как этилацетат, осушают полученный экстракт, например безводным сульфатом натрия, и выделяют продукт из растворителя путем дистилляции. При необходимости полученный продукт может быть очищен стандартными способами, например, путем перекристаллизации, или с использованием хроматографической техники, например препаративной тонкослойной хроматографии или колоночной хроматографии.
Стадия A2 может включать в себя одну или (если это целесообразно) несколько следующих реакций:
I) удаление карбокси-защитных групп избирательно или неизбирательно из группы формулы -COOR5a и/или группы R7a для превращения ее (или их) в свободную карбоксильную группу, представленную R5 или R7;
II) этерификацию любой указанной свободной карбоксильной группы для получения группы, такой, как например, проиллюстрированная выше для R5;
III) превращение указанной свободной карбоксильной группы, представленной R5, в группу формулы -CONR8R9;
IV) удаление тетразолил-защитной группы;
V) превращение циано-группы, представленной R7a, в тетразолильную группу;
VI) превращение моноалкилкарбамоильной группы или карбамоильной группы, представленной R7a, сначала в циано-группу, а затем в тетразолильную группу;
VII) в случае, если R4 представляет собой три-замещенную силильную группу, аралкильную группу, алифатическую ацильную группу, алкоксиметильную группу, алкоксиалкоксиметильную группу, галогеналкоксиметильную группу, тетрагидропиранильную группу, тетрагидротиопиранильную группу, тетрагидротиенильную группу, тетрагидрофурильную группу или замещенную тетрагидропиранильную, тетрагидротиопиранильную, тетрагидротиенильную или тетрагидрофурильную группу, имеющую галогеновый или алкокси-заместитель, все из которых могут рассматриваться как гидрокси-защищенные группы, то осуществляют удаление защитной группы с получением соединения, в котором R4 является атомом водорода;
VIII) если R4 является гидроксильной группой, то осуществляют алкилирование или ацилирование этой группы.
I. Удаление карбокси-защитной группы.
Тип реакции, используемый для удаления карбокси-защитной группы зависит от природы удаляемой группы и хорошо известен специалистам в области органического синтеза.
Например, если карбокси-защитной группой является аралкильная группа, например бензильная или п-нитробензильная группа, то эта защитная группа может быть удалена путем каталитического восстановления в присутствии водорода при атмосферном давлении или сверхатмосферном давлении, например вплоть до 5 атм. Обычно и предпочтительно реакция протекает в инертном растворителе, предпочтительно в спирте, таком как метанол или этанол, или карбоновой кислоте, такой, как уксусная кислота, и в присутствии катализатора. В данной реакции может быть использован любой катализатор, обычно используемый для каталитической гидрогенизации или восстановления, предпочтительно палладированный уголь или окись платины.
Если карбокси-защитной группой является т-бутил или дифенилметил, то эта группа может быть удалена посредством реакции защищенного соединения с кислотой, предпочтительно минеральной кислотой, такой, как хлорводородной или серной кислотой, или органической кислотой, такой, как п-толуолсульфоновая кислота, в инертном растворителе (предпочтительно, в спирте, таком, как метанол или этанол, эфире, таком, как тетрагидрофуран или диоксан, воде или смеси воды и одного или нескольких вышеуказанных органических растворителей.
Если карбокси-защитной группой является силильная группа, то это может быть группа формулы SiRaRbRc, в которой Ra, Rb и Rc определены выше. В этом случае защитная группа может быть удалена посредством реакции защищенного соединения с кислотой, предпочтительно минеральной кислотой, такой, как уксусная кислота, трифторуксусная кислота, метансульфоновая кислота или п-толуолсульфоновая кислота, или солью фтора, такой, как, фторид тетрабутиламмония. Обычно и предпочтительно реакция протекает в инертном растворителе, предпочтительно в простом эфире, таком, как тетрагидрофуран или диоксан, спирте, таком, как метанол или этанол, таком, как N,N -диметилфориамид или N,N-диметилацетамид, воде или смеси воды и одного или нескольких растворителей, указанных выше.
Если карбокси-защитная группа является сложноэфирным остатком, то защитная группа может быть удалена путем гидролиза с использованием основания, предпочтительно гидроксида щелочного металла, такого, как гидроксид лития, гидроксид натрия или гидроксид калия, или карбоната щелочного металла, такого, как карбонат натрия или карбонат калия, в инертном растворителе, предпочтительно в спирте, таком, как метанол или этанол, в эфире, таком, как тетрагидрофуран или диоксан, в воде или в смеси воды и одного или нескольких органических растворителей, указанных выше. Если R4 является ацильной группой, то она может быть удалена в процессе реакции.
Реакция может быть осуществлена в широком диапазоне температур и точное значение реакционной температуры не играет решающей роли. В основном подходящей температурой реакции является температура 0-100oC, а более предпочтительной до 60oC. Время проведения реакции также может широко варьироваться в зависимости от многих факторов, а именно от температуры реакции и природы реагентов и растворителя. Однако при предпочтительных условиях проведения реакции, указанных выше, этот период составляет от 30 мин до 24 ч, а более предпочтительно 1-16 ч.
После завершения реакции, нужное соединение может быть выделено стандартными способами, выбор которых зависит от природы реакции. Например, если разблокирование осуществляют путем каталитического восстановления, то целевой продукт может быть восстановлен путем фильтрации катализатора и отгонки растворителя. Если разблокирование осуществляют с использованием кислоты, то целевой продукт может быть выделен путем сбора преципитата в реакционной системе путем фильтрации или путем концентрации реакционной смеси. Если разблокирование осуществляют путем щелочного гидролиза, то целевой продукт может быть выделен путем отгонки растворителя, а затем нейтрализации остатка водной кислотой, с последующим сбором полученного осадка в водном растворителе посредством фильтрации, альтернативно этот продукт может быть выделен путем нейтрализации водного слоя, полученного экстрагированием реакционной смеси водо-несмешивающимся органическим растворителем (таким, как этилацетат или диэтиловый эфир), затем экстрагирования нейтрализованного раствора водо-несмешивающимся органическим растворителем (таким, как этилацетат), с последующей отгонкой растворителя. Если необходимо, реакционный продукт может быть очищен стандартными методами, например, путем перекристаллизации или с использованием различной хроматографической техники, например препаративной тонкослойной хроматографии или колоночной хроматографии.
Каждая из защитных групп, представленных R5a и R7a, может быть избирательно элиминирована при соответствующем выборе защитных групп и специфических реакционных условий в целях ее удаления.
II. Этерификация.
Если полученное соединение содержит одну или несколько свободных карбоксильных групп, то эта группа или эти группы могут быть этерифицированы стандартными способами, известными в органической химии. Например, эта реакция может быть осуществлена посредством взаимодействия соответствующей карбоновой кислоты с соединением формулы R5b-Y, где R5b может быть любой из групп, определенных выше для R5a, а Y является атомом галогена, например атомом хлора, брома или иода, группой формулы -OSO3R5b, где R5b определен выше, или сульфонилокси-группой, такой, как метансульфонилокси или п-толуолсульфонилокси-группа. Реакцию проводят в присутствии основания, например органического амина, такого, как триэтиламин, пиридин или N-метилморфолин карбоната щелочного металла, такого, как карбонат натрия или калия, или бикарбоната щелочного металла, такого, как бикарбонат натрия или бикарбонат калия. Обычно и предпочтительно реакцию проводят в инертном растворителе, предпочтительно в амиде, таком, как N,N-диметилформамид или N,N-диметилацетамид, галогенированном углеводороде, предпочтительно галогенированном алифатическом углеводороде, таком, как метиленхлорид, кетоне, таком, как ацетон или метилэтилкетон, или эфире, таком, как тетрагидрофуран или диоксан. Если нужная сложноэфирная группа является алкильной группой, то реакцию осуществляют посредством взаимодействия карбоновой кислоты с соответствующим диалкилсульфатом.
Реакция может быть осуществлена в широком диапазоне температур и точное значение реакционной температуры не играет решающей роли. В основном подходящая температура реакции составляет от 0-120oC, а более предпочтительно 20-80oC. Время проведения реакции также может широко варьироваться в зависимости от многих факторов, в частности от температуры реакции и природы реагентов и растворителя. Однако при предпочтительных условиях проведения реакции, указанных выше, этот период составляет от 30 мин до 24 ч, а более предпочтительно 1-16 ч.
Если карбокси-защитной группой является C1-C6-алкильная группа, то реакция этерификации может быть проведена посредством взаимодействия карбоновой кислоты с C1-C6-спиртом, таким, как метанол, этанол, пропанол или гексанол, в присутствии кислотного катализатора, такого, как хлорводород или серная кислота, в инертном растворителе, например в одном из C1-C6-спиртов, который может быть использован как исходный материал, описанный выше, галогенированном углеводороде, таком, как метиленхлорид или в эфире, таком, как тетрагидрофуран или диоксан, при температуре 0-100oC в течение 1-24 ч или посредством взаимодействия соответствующей карбоновой кислоты с агентом галогенирования, например пентахлоридом фосфора, тионилхлоридом или оксалилхлоридом, в инертном растворителе, например галогенированном углеводороде, таким, как метиленхлорид, эфире, таком, как тетрагидрофуран или диоксан, или ароматическом углеводороде, таком, как бензол или толуол, приблизительно при комнатной температуре в течение периода времени от 30 мин до 5 ч с получением соответствующего ацилгалида, который затем подвергают реакции с соответствующим спиртом в инертном растворителе, например бензоле или метиленхлориде, в присутствии основания, например триэтиламина, а в случае т-бутилового сложного эфира, предпочтительно использовать т-бутоксид калия, приблизительно при комнатной температуре в течение периода времени от 30 мин до 10 ч. Целевое соединение может быть восстановлено стандартным способом, например способом, описанный выше для стадии A1.
III. Образование карбамоильных групп.
Конверсия карбоксильной группы, представленной R5, в группу формулы -CONR8R9, где R8 и R9 определены выше, может быть осуществлена стандартными способами, например посредством реакции соединения карбоновой кислоты, в котором R7-группа является защищенной, с соединением формулы
R8R9NH (VI),
где R8 и R9 определены выше.
Эта реакция предусматривает образование пептидной связи и хорошо известна в органической химии. Она может быть проведена в инертном растворителе, предпочтительно в галогенированном углеводороде, более предпочтительно в галогенированном алифатическом углеводороде, таком, как метиленхлорид или хлороформ, сложном эфире, таком, как этилацетат, простом эфире, таком, как тетрагидрофуран или диоксан, или амиде, таком, как N,N-диметилацетамид или N, N-диметилформамид, в присутствии конденсирующего агента.
Примерами конденсирующих агентов, которые могут быть использованы в указанной реакции, являются карбодиимиды, такие, как N,N-дициклогексилкарбодиимид или 1-(3-диметиламинопропил)-3- этилкарбодиимида гидрохлорид, фосфориловые соединения, такие, как дифенилфосфорилазид или диэтилфосфорилцианид, карбонилдиимидазол и трифенилфосфин - диэтилазодикарбоксилат. Из них наиболее предпочтительными являются карбодиимиды и дифенилфосфорилазид. Если используется фосфориловое соединение, то реакцию предпочтительно осуществляют в присутствии третичного амина, такого, как триэтиламин или N -метилморфолин.
Альтернативно в этой стадии реакция может быть осуществлена посредством взаимодействия карбоновой кислоты с (низший)алкилхлороформатом, таким, как этилхлороформатом или изобутилхлороформатом, в присутствии третичного амина, такого, как триэтиламин или N-метилформолин, с получением смешанного ангидрида кислоты или посредством взаимодействия карбоновой кислоты с N-гидроксисукцинимидом, N -гидроксибензотразолом или п-нитрофенолом или т. п. в присутствии карбодиимида, такого, как N,N-дициклогексилкарбодиимид, с получением соответствующего активного сложного эфира и с последующей реакцией смешанного ангидрида кислоты или активного сложного эфира с аминовым соединением формулы VI.
В качестве еще одной альтернативы реакция в этой стадии может быть осуществлена посредством взаимодействия карбоновой кислоты с галогенирующим агентом, таким, как пентахлорид фосфора, оксалилхлорид или тионилхлорид, в инертном растворителе, например галогенированном углеводороде, таком, как метиленхлорид, простом эфире, таком, как тетрагидрофуран или диоксан, или ароматическом углеводороде, таком, как бензол или толуол, с получением соответствующего ацилгалида, с последующей реакцией ацилгалида с аминовым соединением формулы VI.
Все указанные реакции могут быть проведены в широком диапазоне температур и точное значение температуры не играет решающей роли. В основном, подходящая температура реакции составляет от -20oC до 100oC, а более предпочтительно от -5oC до 50oC. Время проведения реакции также может широко варьироваться в зависимости от многих факторов, в частности от температуры реакции и природы реагентов и растворителя. Однако при предпочтительных условиях проведения реакции, указанных выше, этот период составляет от 30 мин до 24 ч, а более предпочтительно 1-16 ч.
После завершения реакции полученный продукт может быть выделен из реакционной смеси стандартными способами. Например, нерастворимый в реакционной системе материал отфильтровывают, к фильтрату добавляют водонесмешиваемый органический растворитель, такой, как этилацетат, и воду, слой органического растворителя отделяют и осушают в присутствии осушающего агента, такого, как безводный сульфат магния, а затем растворитель отгоняют с получением целевого продукта. При необходимости реакционный продукт может быть затем очищен стандартными способами, например путем перекристаллизации или с использованием различной хроматографической техники, например препаративной тонкослойной хроматографии или колоночной хроматографии.
IV. Удаление тетразолил-защитных групп.
Указанная реакция может быть осуществлена посредством взаимодействия защищенного соединения с кислотой. Обычно и предпочтительно указанную реакцию проводят в инертном растворителе.
Реакцию предпочтительно осуществляют в присутствии растворителя. В принципе нет конкретных ограничений на природу растворителя, при условии, если он не оказывает неблагоприятного воздействия на реакцию или реагенты, и обладает способностью к растворению реагентов по крайней мере до определенной степени. Примерами подходящих растворителей являются органическая кислота, такая как, уксусная кислота, простой эфир, такой, как тетрагидрофуран или диоксан, спирт, такой, как метанол, этанол или т-бутанол, или смесь любого из двух или более указанных растворителей. Из этих растворителей, наиболее предпочтительными являются вода, органическая кислота, спирт или их смесь.
В основном нет конкретных ограничений на кислоту, используемую в данной реакции, при условии, если она способна функционировать как кислота Бренстеда. Предпочтительными примерами таких кислот являются органические кислоты, такие, как уксусная кислота, муравьиная кислота, щавелевая кислота, метансульфоновая кислота, п-толуолсульфоновая кислота или трифтороуксусная кислота и неорганические кислоты, такие, как соляная кислота, бромистоводородная кислота, серная кислота или фосфорная кислота. Из них предпочтительными являются уксусная кислота, муравьиная кислота, трифторуксусная кислота или соляная кислота.
Реакция может протекать в широком диапазоне температур и точное значение температуры не играет решающей роли для данного изобретения. В основном подходящая температура реакции составляет от -10oC до 120oC, а более предпочтительно 0-100oC. Время, требуемое для проведения реакции, также может широко варьироваться и зависит от многих факторов, в частности от температуры реакции и природы реагентов и растворителя. Однако при предпочтительных условиях проведения реакции, указанных выше, этот период составляет 0,5-24 ч, а более предпочтительно 1-16 ч.
После завершения реакции полученный продукт может быть выделен из реакционной смеси стандартными способами. Например, после отгонки растворителя остаток растворяют в воде и водонесмешивающемся органическом растворителе. Органический слой, содержащий нужное соединение, отделяют и осушают безводным сульфатом магния. После отгонки растворителя получают нужное соединение. По необходимости реакционный продукт может быть очищен стандартными способами, например путем перекристаллизации или с использованием различной хроматографической техники, например препаративной тонкослойной хроматографии или колоночной хроматографии.
V. Конверсия циано-группы в тетразолильную группу.
В этой стадии циано-группу превращают в тетразолильную группу с помощью реакции циано-соединения с азидом щелочного металла.
Реакцию предпочтительно осуществляют в присутствии растворителя. В принципе нет конкретных ограничений на природу растворителя при условии, если он не оказывает неблагоприятного воздействия на реакцию или реагенты и обладает способностью к растворению реагентов по крайней мере до определенной степени. Примерами подходящих растворителей являются амиды, такие, как N, N-диметилфориамид или N,N -диметилацетамид, простые эфиры, такие, как диоксан или 1,2-диметоксиэтан, и сульфоксиды, такие, как диметилсульфоксид.
Примерами подходящих азидов щелочных металлов являются азид лития, азид натрия и азид калия, из которых предпочтительных является азид натрия. На количество используемого азида щелочного металла нет конкретных ограничений, однако предпочтительно использовать 1-5 экв. а более предпочтительно 1-3 экв. азида щелочного металла на 1 экв. циано-соединения.
Предпочтительно также, чтобы реакция протекала в присутствии галида аммония, например фторида аммония, хлорида аммония или бромида аммония, из которых предпочтительным является хлорид аммония. На количество используемого галида аммония нет конкретных ограничений, однако обычно используют 0,5-2 экв. а более предпочтительно 1-1,2 экв. галида аммония на 1 экв. циано-соединения.
Реакция может протекать в широком диапазоне температур и точное значение температуры не играет решающей роли для данного изобретения. В основном, подходящая температура реакции составляет 70-150oC, а более предпочтительно 80-120oC. Время, требуемое для проведения реакции, также может широко варьироваться в зависимости от многих факторов, в частности от температуры реакции и природы реагентов и растворителя. Однако при предпочтительных условиях проведения реакции, указанных выше, этот период составляет от 10 ч до 7 дней, а более предпочтительно 1-5 дней.
Альтернативно циано-группа может быть превращена в тетразолильную группу посредством взаимодействия циано-соединения с азидом триалкилолова или азидом триарилолова с последующей обработкой соединения олова кислотой, основанием или фторидом щелочного металла.
Реакцию циано-соединения с азидом триалкилолова или азидом триарилолова обычно и предпочтительно осуществляют в присутствии растворителя. В принципе нет конкретных ограничений на природу растворителя при условии, если он не оказывает неблагоприятного воздействия на реакцию или реагенты и обладает способностью растворять указанные реагенты по крайней мере до определенной степени. Примерами подходящих растворителей являются углеводороды, которыми могут быть алифатические или ароматические углеводороды, такие, как бензол, толуол, ксилол или гептан, галогенированные углеводороды, особенно галогенированные алифатические углеводороды, такие, как 1,2-дихлорэтан или хлороформ, простые эфиры, такие, как диоксан или 1,2-диметоксиэтан, амиды, такие, как N, N -диметилформамид или N,N -диметилацетамид, и сложные эфиры, такие, как этилацетат или бутилацетат.
Хотя на природу азида триалкилолова или азида триарилолова нет конкретных ограничений и в данной реакции может быть использовано любое соединение указанного типа, однако предпочтительным является азид триалкилолова, в котором каждая из алкильных групп, которые могут быть одинаковыми или различными, а лучше одинаковыми, имеет 1-4 атомов углерода, например, такой, как азид триметилолова, азид триэтилолова или азид трибутилолова, или азид триарилолова, в котором каждая из ариальных групп, которые могут быть одинаковыми или различными, а лучше одинаковыми, являются такими, как они были определены выше в отношении арильных групп, которые могут быть представлены R2, предпочтительно фенильными или замещенными фенильными группами, и примером указанного азида триарилолова является азид трифенилолова и азид тритолилолова. Количество используемого азида триалкилолова или азида триарилолова не является критическим, хотя предпочтительным является использование 1-3 экв. на 1 экв. циано-соединения, а более предпочтительным 1-2 экв. на 1 экв. циано-соединения.
Реакция циано-соединения с азидом триалкилолова или триарилолова может проходить в широком диапазоне температур и точное значение температуры не играет решающей роли для данного изобретения. В основном подходящая температура реакции составляет 60-150oC, а более предпочтительно 80-120oC. Время, требуемое для проведения реакции, также может широко варьироваться в зависимости от многих факторов, в частности от температуры реакции и природы реагентов и растворителя. Однако при предпочтительных условиях проведения реакции, указанных выше, этот период составляет от 8 ч до 7 дней, а более предпочтительно 1-5 дней.
Оловосодержащее соединение, полученное в результате вышеуказанной реакции, затем обрабатывают кислотой, основанием или фторидом щелочного металла в целях его превращения в тетразолиловое соединение. В реакции указанного типа могут быть использованы любая кислота, основание или фторид щелочного металла и примерами подходящих соединений являются кислоты, в частности минеральные кислоты, такие, как соляная кислота или серная кислота, основания, в частности неорганические основания, такие, как карбонаты щелочного металла и бикарбонаты щелочного металла, например карбонат натрия, карбонат калия, бикарбонат натрия или бикарбонат калия, или гидроксиды щелочных металлов, например гидроксид натрия или калия, фториды щелочного металла, такие, как фторид лития, фторид натрия или фторид калия.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. В принципе нет конкретных ограничений на природу растворителя при условии, что он не оказывает неблагоприятного воздействия на реакцию или реагенты и обладает способностью растворять указанные реагенты по крайней мере до определенной степени. Примерами подходящих растворителей являются растворители, перечисленные выше для реакции циано-соединения с азидом триалкилолова или триарилолова, и другие растворители, такие, как спирты, например метанол или этанол, вода или водные спирты. Реакция может проходить в широком диапазоне температур и точное значение температуры не играет существенной роли для настоящего изобретения. В основном подходящая для осуществления реакции температура составляет 0-100oC а предпочтительно комнатная температура. Время, требуемое для осуществления реакции, также может широко варьироваться в зависимости от многих факторов, в частности от температуры реакции и природы используемых реагентов и растворителя. Однако при предпочтительных условиях проведения реакции, указанных выше, этот период составляет от 30 мин до 3 дней, а более предпочтительно, 1-24 ч.
Еще один альтернативный способ превращения циано-группы в тетразолильную группу предусматривает реакцию циано-соединения с галидом триалкилолова или триарилолова в присутствии азида щелочного металла с последующей обработкой полученного соединения олова кислотой, основанием или фторида щелочного металла.
Реакция циано-соединения с галидом триалкилолова или триарилолова в присутствии азида щелочного металла обычно и предпочтительно протекает в присутствии растворителя. В принципе нет конкретных ограничений на природу используемого растворителя при условии, что он не оказывает неблагоприятного воздействия на реакцию или реагенты и обладает способностью растворять указанные реагенты по крайней мере до определенной степени. Примерами подходящих растворителей являются углеводороды, которые могут быть алифатическими или ароматическими углеводородами, такие, как бензол, толуол, ксилол или гептан, галогенированные углеводороды, в частности галогенированные алифатические углеводороды, такие, как 1,2-дихлорэтан или хлороформ, простые эфиры, такие, как диоксан или 1,2-диметоксиэтан, кетоны, такие, как ацетон или метилэтилкетон, амиды, такие, как N,N -диметилформамид или N,N -диметилацетамид, и сложные эфиры, такие, как этилацетат или бутилацетат.
Хотя нет конкретных ограничений на природу галида триалкилолова или триарилолова и в реакции указанного типа может быть использовано любое из таких соединений, однако предпочтительными соединениями являются галид триалкилолова, в котором каждая из алкильных групп, которые могут быть одинаковыми или различными, а предпочтительно одинаковыми, имеют 1-4 атомов углерода, например такие, как хлорид триметилолова, бромид тримеитлолова, хлорид триэтилолова или хлорид трибутилолова, или галид триарилолова, в котором каждая из арильных групп, которые могут быть одинаковыми или различными, а предпочтительно одинаковыми, являются такими, как они были определены выше в отношении арильных групп для R2, а предпочтительно фенильной или замещенной фенильной группой, при этом примерами вышеуказанных галида триалкилолова и галида триарилолова являются хлорид трифениоолова или хлорид тритолилолова. Количество галида триалкилолова или триарилолова, используемое в реакции, не является критическим, хотя предпочтительно использовать 1-3 экв. на 1 экв. циано-соединения, а более предпочтительно 1-2 экв. на циано-соединение.
Не существует также конкретных ограничений на азид щелочного металла, используемый в данной реакции. Примерами такого азида являются азид лития, азид натрия и азид калия, из которых предпочтительным является азид натрия. Количество используемого азида щелочного металла не является критическим, однако предпочтительно использовать 1-3 экв. на 1 экв. циано-соединения, а более предпочтительно 1-2 экв. на 1 экв. циано-соединения.
Реакция циано-соединения с галидом триалкилолова или триарилолова в присутствии азида щелочного металла может протекать в широком диапазоне температур и точное значение температуры не играет существенной роли для данного изобретения. В основном подходящая температура для осуществления реакции составляет 60-150oC, а предпочтительно 80-120oC. Время, требуемое для осуществления реакции, также может широко варьироваться в зависимости от многих факторов, в частности от температуры реакции и природы используемых реагентов и растворителя. Однако при предпочтительных условиях проведения реакции, указанных выше, этот период составляет от 8 ч до 7 дней, а более предпочтительно 1-5 дней.
Оловосодержащее соединение, полученное в результате вышеуказанной реакции, затем обрабатывают кислотой, основанием или фторидом щелочного металла в целях его превращения в третразолиловое соединение. Эта реакция в основном аналогична реакции оловосодержащего соединения, полученного посредством реакции циано-соединения с азидом триалкилолова или триарилолова с кислотой, основанием или фторидом щелочного металла, и может быть проведена с использованием таких же растворителей и реакционных условий.
VI. Конверсия алкилкарбомоильной группы или карбамоильной группы в циано-группу.
Для превращения алкилкарбамоильной группы в циано-группу алкилкарбамоильное соединение подвергают реакции с галогеновым соединением, являющимся галогенирующим агентом, предпочтительно хлорирующим агентом, например таким, как оксалилхдорид, оксихлорид фосфора или сульфонилхлорид. Количество используемого галогенового соединения не является критическим, хотя предпочтительно использовать 1-3 экв. а более предпочтительно 1-2 экв. на один экв. карбамоильного соединения.
Реакцию обычно и предпочтительно осуществляют в присутствии растворителя. В принципе нет конкретных ограничений на природу растворителя, при условии, что он не оказывает неблагоприятного воздействия на реакцию или реагенты и обладает способностью растворять указанные реагенты по крайней мере до определенной степени. Примерами подходящих растворителей являются углеводород, который может быть алифатическим, так и ароматическим углеводородом, таким, как бензол, толуол, ксилол или гептан, галогенированный углеводород, в частности галогенированный алифатический углеводород, такой, как метиленхлорид или хлороформ, простые эфиры, такие, как диоксан, тетрагидрофуран или диэтиловый эфир, и сложные эфиры, такие, как этилацетат или бутилацетат.
Реакция может протекать в широком диапазоне температур и точное значение температуры не играет решающей роли для данного изобретения. В основном, подходящая температура составляет от -10 до 100oC, а более предпочтительно 0-50oC. Время, требуемое для осуществления реакции, также может широко варьироваться в зависимости от многих факторов, в частности от температуры реакции и природы используемых реагентов и растворителя. Однако при предпочтительных условиях проведения реакции, указанных выше, этот период составляет от 10 мин до 16 ч, а более предпочтительно от 30 мин до 6 ч.
Для превращения карбамоильной группы в циано-группу карбамоильное соединение подвергают взаимодействию с дегидрирующим агентом, например уксусным ангидридом, трифторуксусным ангидридом, метансульфоновым ангидридом, трифторметансульфоновым ангидридом, оксалилхлоридом или сульфонилхлоридом, в присутствии органического амина, например триэтиламина, пиридин или N-метилморфолина.
Реакцию предпочтительно осуществляют в присутствии растворителя. В принципе нет конкретных ограничений на природу растворителя при условии, что он не оказывает неблагоприятного воздействия на реакцию или реагенты и обладает способностью растворять указанные реагенты по крайней мере до определенной степени. Примерами подходящих растворителей являются углеводороды, которые могут быть как алифатическими, так и ароматическими углеводородами, такими, как бензол, толуол, ксилол или гептан, галогенированными углеводородами, в частности галогенированными алифатическими углеводородами, такими, как метиленхлорид или хлороформ, простые эфиры, такие, как диоксан, тетрагидрофуран или диэтиловый эфир, и сложные эфиры, такие, как этилацетат или бутилацетат.
Реакция может протекать в широком диапазоне температур и точное значение температуры не играет существенной роли для данного изобретения. В основном подходящая температура составляет от -10 до 100oC, а более предпочтительно 0-50oC. Время, требуемое для осуществления реакции, также может широко варьироваться в зависимости от многих факторов, в частности от температуры реакции и природы используемых реагентов и растворителя. Однако при предпочтительных условиях проведения реакции, указанных выше, этот период составляет от 10 мин до 16 ч, а более предпочтительно от 30 мин до 6 ч.
Целевой продукт указанной реакции может быть выделен из реакционной смеси стандартными способами, например путем нейтрализации смеси слабым основанием, таким, как бикарбонат натрия с последующей обработкой продукта способом, аналогичным описанному для стадии A1 реакционной смеси A.
Полученное таким образом циано-соединение может быть затем превращено в соответствующее тетразолиловое соединение с использованием реакций, описанных выше.
VII. Удаление гидрокси-защитных групп.
Если R4 является три-замещенной силильной группой, аралкильной группой, ацильной группой, алкоксиметильной группой, тетрагидропиранильной группой, тетрагидротиопиранильной группой, тетрагидротиенильной группой, тетрагидрофурильной группой или замещенной тетрагидропиранильной, тетрагидротиенильной или тетрагидрофурильной группой, которые могут рассматриваться как гидрокси-защитные группы, то защитная группа может быть удалена с образованием соединения, в котором R4 является атомом водорода. Тип реакции, используемой для удаления защитной группы, зависит от природы защитной группы и для разблокирования соединений указанного типа могут быть использованы реакции, хорошо известные специалистам.
Если гидрокси-защитной группой является силильная группа, то она может быть удалена путем обработки защищенного соединения соединением, способным образовывать анион фтора, такой, как фторид тетрабутиламиония. Реакцию предпочтительно осуществляют в присутствии растворителя. В принципе нет конкретных ограничений на природу растворителя при условии, что он не оказывает неблагоприятного воздействия на реакцию или реагенты и обладает способностью растворять указанные реагенты по крайней мере до определенной степени. Примерами подходящих растворителей являются простые эфиры, такие, как тетрагидрофуран или диоксан.
Реакция может протекать в широком диапазоне температур и точное значение температуры не играет решающей роли. В основном подходящей температурой является комнатная температура. Время, требуемое для осуществления реакции, также может широко варьироваться в зависимости от многих факторов, в частности от температуры реакции и природы используемых реагентов и растворителя. Однако при предпочтительных условиях проведения реакции, указанных выше, этот период составляет 10-18 ч.
Если гидрокси-защитной группой является аралкильная группа, то разблокирование может быть осуществлено путем каталитического восстановления при температуре 0-80oC, а более предпочтительно 10-60oC, в растворителе в присутствии водорода и катализатора.
Реакцию предпочтительно осуществляют в присутствии растворителя. В принципе нет конкретных ограничений на природу растворителя при условии, что он не оказывает неблагоприятного воздействия на реакцию или реагенты и обладает способностью растворять указанные реагенты по крайней мере до определенной степени. Примерами подходящих растворителей являются спирты, такие, как метанол, этанол или изопропанол, простые эфиры, такие, как диэтиловый эфир, тетрагидрофуран или диоксан, ароматические углеводороды, такие, как толуол, бензол или ксилол, алифатические углеводороды, такие, как гексан, или циклогексан, сложные эфиры, такие, как этилацетат или пропилацетат, жирные кислоты, такие, как уксусная кислота, или смеси воды и одного или более из указанных органических растворителей.
В рассматриваемой реакции может быть использован любой катализатор, который обычно используют при каталитическом восстановлении. Примерами предпочтительных катализаторов являются палладированный уголь, скелетный никелевый катализатор гидрирования, окись платины, платиновая чернь, окись алюминия, покрытая родием, комплекс трифенилфосфина и хлорида родия и палладированный сульфат бария.
Используемое в реакции давление водорода не является критическим и может варьироваться в широких пределах, хотя реакцию предпочтительно осуществлять при давлении, которое превышает атмосферное давление в 1-3 раза.
Реакция может протекать в широком диапазоне температур и точное значение температуры не играет существенной роли для данного изобретения. В основном подходящая температура составляет 0-100oC, а более предпочтительно 10-50oC. Время, требуемое для осуществления реакции, также может широко варьироваться в зависимости от многих факторов, в частности от температуры реакции и природы используемых реагентов и растворителя. Однако при предпочтительных условиях проведения реакции, указанных выше, этот период составляет от 5 мин до 24 ч, а более предпочтительно от 30 мин до 16 ч.
Если гидрокси-защитной группой является алифатическая ацильная группа, ароматическая ацильная группа или алкокси-карбонильная группа, то она может быть удалена путем обработки защищенного соединения основанием.
В данном случае может быть использовано любое основание, если только оно не оказывает неблагоприятного воздействия на другие части соединения. Примерами предпочтительных оснований являются алкоксиды металлов, в частности алкоксиды щелочных металлов, такие, как метоксид натрия, карбонаты щелочных металлов, такие, как карбонат натрия или калия, гидроксиды щелочных металлов, такие, как гидроксид натрия или калия, и аммиак, предпочтительно в виде водного аммиака или в виде концентрированного раствора аммиака в метаноле.
Реакцию предпочтительно осуществляют в растворителе. В принципе нет конкретных ограничений на природу растворителя при условии, что он не оказывает неблагоприятного воздействия на реакцию или реагенты и обладает способностью растворять указанные реагенты по крайней мере до определенной степени. Примерами подходящих растворителей является вода, органические растворители, такие, как спирты, например метанол, этанол или пропанол, или простые эфиры, например тетрагидрофуран или диоксан, или смесь воды и одного или более указанных выше органических растворителей.
Реакция может протекать в широком диапазоне температур и точное значение температуры не играет решающей роли. В основном подходящая температура составляет 0-150oC, а более предпочтительно 0-60oC. Время, требуемое для осуществления реакции, также может широко варьироваться в зависимости от многих факторов, в частности от температуры реакции и природы используемых реагентов и растворителя. Однако при предпочтительных условиях проведения реакции, указанных выше, этот период составляет 1-20 ч, а предпочтительно 1-16 ч.
Если гидрокси-защитной группой является алкоксиметильная группа, алкоксиалкоксиметильная группа, галогеналкоксиметильная группа, тетрагидропиранильная группа, тетрагидротиопиранильная группа, тетрагидрофуранильная группа, тетрагидротиенильная группа или замещенная тетрагидропиранильная, тетрагидротиопиранильная, тотрагидрофуранильная или тетрагидротиенилная группа, имеющая по крайней мере один галогеновый или алкокси-заместитель, то она может быть удалена путем обработки защищенного соединения кислотой.
Конкретных ограничений на используемую кислоту в данной реакции не существует и в данном случае может быть использована любая кислота Бренстеда. Примерами предпочтительных кислот являются неорганические кислоты, в частности минеральные кислоты, такие, как соляная кислота или серная кислота, и органическая кислота, например карбоновые кислоты и сульфоновые кислоты, такие, как уксусная кислота или п-толуолсульфоновая кислота. В данной реакции могут быть также использованы сильные катионообменные смоляные кислоты, такие, как Dowex 50W (торговая марка).
Реакцию предпочтительно осуществляют в присутствии растворителя. В принципе нет конкретных ограничений на природу растворителя при условии, что он не оказывает неблагоприятного воздействия на реакцию или реагенты и обладает способностью растворять указанные реагенты по крайней мере до определенной степени. Примерами подходящих растворителей являются спирты, такие, как метанол или этанол, простые эфиры, такие, как тетрагидрофуран или диоксан, органические кислоты, такие, как муравьиная кислота или уксусная кислота, и смеси воды и одного или более из указанных растворителей.
Реакция может протекать в широком диапазоне температур и точное значение температуры не играет решающей роли. В основном подходящая температура составляет 0-50oC. Время, требуемое для осуществления реакции, также может широко варьироваться в зависимости от многих факторов, в частности от температуры реакции и природы используемых реагентов и растворителя. Однако при предпочтительных условиях проведения реакции, указанных выше, этот период составляет от 10 мин до 18 ч.
После завершения любой из вышеуказанных реакций целевое соединение данного изобретения может быть выделено из реакционной смеси стандартными способами, выбор которых зависит от природы реакции и реакционной среды. Один из таких способов заключается в том, что реакционную смесь нейтрализуют, удаляют любой нерастворимый материал, который может присутствовать в смеси, например, путем фильтрации, добавляют водонесмешиваемый органический растворитель, промывают водой и отгоняют растворитель. По необходимости полученный продукт может быть очищен стандартными способами, например путем перекристаллизации или с помощью различной хроматографической техники, такой, как препаративная тонкослойная хроматография или колоночная хроматография.
Иногда попутно в условиях, используемых для удаления гидрокси-защитной группы, одновременно может быть разблокирована защищенная карбокси-группа.
VIII. Алкилирование и ацилирование гидрокси-групп.
Алкилирование гидрокси-группы может быть осуществлено посредством реакции гидрокси-соединения с алкилгалидом, в котором алкильная группа имеет 1-6 атомов, предпочтительно метилиодидом, этилиодидом, этилбромидом, пропилиодидом, пропилбромидом или бутилиодидом или диалкилсульфатом, в котором алкильные группы имеют 1-6 атомов углерода и могут быть одинаковыми или различными, а предпочтительно одинаковыми, таким как, диметилсульфат или диэтилсульфат.
Реакцию предпочтительно осуществляют в присутствии растворителя. В принципе нет конкретных ограничений на природу растворителя при условии, что он не оказывает неблагоприятного воздействия на реакцию или реагенты и обладает способностью растворять указанные реагенты по крайней мере до определенной степени. Примерами подходящих растворителей являются амиды, такие, как N,N -диметилформамид, N,N-диметилацетамид, или N-метилпирролидинон, кетоны, такие, как ацетон или метилэтилкетон, или сульфоксиды, такие, как диметилсульфоксид.
Реакцию осуществляют в присутствии основания, природа которого не является критической, если только оно не оказывает неблагоприятного воздействия на реагенты или продукты. Примерами предпочтительных оснований являются гидриды щелочных металлов, такие, как гидрид натрия, гидрид калия или гидрид лития. Реакция может протекать в широком диапазоне температур и точное значение температуры не играет существенной роли для данного изобретения. В основном подходящая для осуществления реакции температура составляет 0-120oC, а предпочтительно 20-80oC. Время, требуемое для осуществления реакции, также может широко варьироваться в зависимости от многих факторов, в частности от температуры реакции и природы используемых реагентов и растворителя. Однако при предпочтительных условиях проведения реакции, указанных выше, этот период составляет от 30 мин до 24 ч, а более предпочтительно 1-16 ч. Ацилирование карбоксигруппы может быть также осуществлено стандартными способами, обычно используемыми в органической химии. Например, оно может быть осуществлено посредством реакции гидроксильного соединения с алканоилгалидом, содержащим 2-6 атомов углерода, таким, как ацетилхлорид, пропинилхлорид, бутурилбромид, валерилхлорид или гексаноилхлорид, с ангидридом карбоновой кислоты, в которой группа, происходящая от карбоновой кислоты, содержит 1-6, а предпочтительно 2-6, атомов углерода, например, таким, как смешанный ангидрид муравьиной и уксусной кислоты, ангидрид уксусной кислоты, ангидрид пропионовой кислоты, ангидрид валериановой кислоты или ангидрид гексановой кислоты, с алкоксикарбонилгалидом, в котором алкокси-группа содержит 1-6 атомов углерода, таким, как метоксикарбонилхлорид, метоксикарбонилбромид, этоксикарбонилхлорид, пропоксикарбонилхлорид, бутоксикарбонилхлорид или гексилоксикарбонилхлорид, с арилкарбонилгалидом, таким, как бензоилхлорид, бензоилбромид или нафтоилхлорид, с галогенили алкокси-алканоил-галидом, содержащим 2-6 атомов углерода, таким, как хлороацетилхлорид, дихлорацетилхлорид, трихлороацетилхлорид или метоксиацетилхлорид, или с алкенилхлоридом, содержащим 3-6 атомов углерода, таким, как акрилоилхлорид, метакрилоилхлорид, кротоноилхлорид, 3-метил-2-бутеноилхлорид или 2-метил-2-бутеноилхлорид.
Реакцию предпочтительно осуществляют в присутствии растворителя. В принципе нет конкретных ограничений на природу растворителя при условии, если он не оказывает неблагоприятного воздействия на реакцию или реагенты и обладает способностью растворять указанные реагенты по крайней мере до определенной степени. Примерами подходящих растворителей являются галогенированные углеводороды, в частности галогенированные алифатические углеводороды, такие, как метиленхлорид или хлороформ, сложные эфиры, такие, как этилацетат, и простые эфиры, такие, как тетрагидрофуран или диоксан. Реакцию осуществляют в присутствии основания, предпочтительно такого, как, органический третичный амин, например триэтиламин, пиридин, диэтилизопропиламин или 4-диметиламинопиридин. Реакция может протекать в широком диапазоне температур и точное значение температуры не играет существенной роли для данного изобретения. В основном подходящая для осуществления реакции температура составляет от -10oC до 120oC, а более предпочтительно 0-80oC. Время, требуемое для осуществления реакции, также может широко варьироваться в зависимости от многих факторов, в частности от температуры реакции и природы используемых реагентов и растворителя. Однако при предпочтительных условиях проведения реакции, указанных выше, этот период составляет от 30 мин до 24 ч, а более предпочтительно 1-16 ч.
После завершения любой из вышеуказанных реакций целевой продукт может быть выделен из реакционной смеси стандартными способами. Примеры таких способов выделения были уже указаны при описании выделения продукта в стадии A1.
Реакционная схема B.
Соединения формулы Iа, в которых R4 является атомом водорода, то есть, иначе говоря, соединения формулы Ib могут быть также получены в соответствии с представленной в конце описания реакционной схемой B, в формулах которой R1, R2, R3, R5a, R6, R7a и X определены значениями, указанными выше.
В стадии В1 соединение имидазол-5-карбоксилата формулы VII взаимодействует с бифенилметиловым соединением формулы III с образованием соединения формулы VIII. Эта реакция в основном аналогична реакции стадии А1 в реакционной схеме А и может быть осуществлена с использованием тех же реагентов и тех же реакционных условий.
В стадии В2 соединение формулы Ib получают с помощью реакции соединения формулы VIII с восстанавливающим агентом или с реактивом Гриньяра формулы R3a-Mg-X, где R3a представляет собой любую из групп, определенных выше для R3, и не является атомом водорода, а X определен выше.
Примерами восстанавливающих агентов, которые могут быть использованы в этой реакции, являются гидриды алкилалюминия, такие, как гидрид диизобутилалюминия, и борогидриды металлов, в частности щелочных металлов, такие, как борогидрид натрия или цианоборогидрид натрия. Из них предпочтительными являются гидрид алюминия и борогидрид натрия.
Реакция соединения формулы VIII с восстанавливающим агентом обычно и предпочтительно протекает в инертном растворителе. В принципе нет конкретных ограничений на природу растворителя при условии, если он не оказывает неблагоприятного воздействия на реакцию или реагенты и обладает способностью растворять указанные реагенты по крайней мере до определенной степени. Примерами подходящих растворителей являются углеводороды, особенно ароматические углеводороды, такие, как толуол или гексан, простые эфиры, такие, как тетрагидрофуран или диоксан, спирты, такие, как метанол или этанол, вода, и смеси воды с одним или несколькими из вышеуказанных органических растворителей. Предпочтительные растворители выбирают в зависимости от природы используемого восстанавливающего агента. Например, если восстанавливающим агентом является гидрид алкилалюминия, то предпочтительными растворителями являются углеводороды или простые эфиры, и альтернативно, если таким агентом является борогидрид щелочного металла, то предпочтительными растворителями являются спирты, вода или смеси воды со спиртом.
Реакция может протекать в широком диапазоне температур и точное значение температуры не играет существенной роли для данного изобретения. В основном подходящая для осуществления реакции температура составляет от -30 до 80oC, а более предпочтительно от -20 до 20oC, если восстанавливающим агентом является гидрид алкилалюминия, или от -30 до 80oC, а более предпочтительно 0-50oC, если указанным агентом является борогидрид щелочного металла. Время, требуемое для осуществления реакции, также может широко варьироваться в зависимости от многих факторов, в частности от температуры реакции и природы используемых реагентов и растворителя. Однако при предпочтительных условиях проведения реакции, указанных выше, этот период составляет от 30 мин до 24 ч, а более предпочтительно 1-16 ч.
Реакцию соединения формулы VIII с реагентом Гриньяра обычно и предпочтительно осуществляют в присутствии растворителя. В принципе нет конкретных ограничений на природу растворителя при условии, если он не оказывает неблагоприятного воздействия на реакцию или реагенты и обладает способностью растворять указанные реагенты по крайней мере до определенной степени. Примерами подходящих растворителей являются углеводороды, которые могут быть алифатическими или ароматическими, такие, как гексан или толуол, галогенированные углеводороды, в частности галогенированные алифатические углеводороды, такие, как метиленхлорид или 1,2-ди-хлорэтан, и простые эфиры, такие, как тетрагидрофуран или диэтиловый эфир, из которых предпочтительными являются простые эфиры и галогенированные углеводороды.
Реакция может протекать в широком диапазоне температур и точное значение температуры не играет существенной роли для данного изобретения. В основном подходящая для осуществления реакции температура составляет от -50oC до 100oC, а более предпочтительно от -10oC до 50oC. Время, необходимое для осуществления реакции, также может широко варьироваться в зависимости от многих факторов, в частности от температуры реакции и природы используемых реагентов и растворителя. Однако при предпочтительных условиях проведения реакции, указанных выше, этот период составляет от 30 мин до 24 ч, а более предпочтительно 1-16 ч.
После завершения любой из вышеуказанных реакций целевые соединения каждой реакции могут быть выделены из реакционной смеси стандартными способами. Например, реакционную смесь смешивают с водой или водным раствором хлорида аммония, перемешивают при комнатной температуре, а затем экстрагируют водонесмешивающимся растворителем, таким, как этилацетат. Экстракт промывают водой и осушают в присутствии осушающего агента, такого, как безводный сульфат магния, после чего растворитель отгоняют и, если необходимо, полученный продукт очищают стандартными способами, например путем перекристаллизации или с помощью различной хроматографической техники, такой, как препаративная тонкослойная хроматография или колоночная хроматография.
В представленной в конце описания реакционной схеме D сначала получают циано-соединение формулы XII, а затем это соединение превращают в соединение формулы I. В формулах схемы D R1, R2, R3, R4, R5, R6, R7, и X определены значениями, указанными выше, а R7b является защищенной карбоксигруппой, защищенной тетразолильной группой, каждая из которых может быть такой, как они были проиллюстрированы ранее для R7a.
В стадии D1 этой реакционной схемы, имидазол-5-карбонитриловое соединение формулы XI взаимодействует с бифенилметильным соединением формулы IIIa с получением соединения формулы XII. Эта реакция в основном аналогична реакции, описанной выше в стадии A1 реакционной схемы А, и может быть осуществлена с использованием тех же реагентов и тех же реакционных условий.
В стадии D2 полученное соединение формулы XII может быть подвергнуто любой одной или (в соответствующих случаях) несколькими из следующих реакций:
превращение циано-группы в 5-положении имидазольного кольца в карбоксильную группу;
удаление любой карбокси-защитной группы;
этерификация карбокси-группы в 5-положении имидазольного кольца или в бифенильной группе;
превращение карбоксильной группы в 5-положении имидазольного кольца в группу формулы -CONR8R9;
удаление тетразолил-защитной группы и осуществление алкилирования или ацилирования этой группы.
Превращение циано-группы в карбоксильную группу.
Указанную конверсию осуществляют путем гидролиза циано-группы в соединении формулы XII посредством карбамоильной группы. Эта реакция хорошо известна в химическом синтезе и может быть осуществлена с использованием реагентов, обычно применяемых в этих целях, например таких, как гидроксиды щелочных металлов, а именно гидроксида натрия, калия или лития.
Реакцию предпочтительно осуществляют в присутствии растворителя. В принципе нет конкретных ограничений на природу растворителя при условии, что он не оказывает неблагоприятного воздействия на реакцию или реагенты и обладает способностью растворять указанные реагенты по крайней мере до определенной степени. Примерами подходящих растворителей являются вода, спирты, такие, как метанол или этанол, простые эфиры, такие, как тетрагидрофуран или диоксан, или смесь любых двух или более из указанных растворителей, причем предпочтительный растворителем является водный растворитель.
Реакция может протекать в широком диапазоне температур и точное значение температуры не играет существенной роли для данного изобретения. В основном подходящая для осуществления реакции температура составляет 0-120oC, а более предпочтительно 20-100oC. Время, требуемое для осуществления реакции, также может широко варьироваться в зависимости от многих факторов, в частности от температуры реакции и природы используемых реагентов и растворителя. Однако при предпочтительных условиях проведения реакции, указанных выше, этот период составляет от 30 мин до 24 ч, а более предпочтительно 1-16 ч.
После завершения реакции целевой продукт может быть выделен из реакционной смеси стандартными способами. Например, один из стандартных способов выделения продукта включает в себя следующие процедуры: нейтрализацию реакционной смеси путем добавления минеральной кислоты, такой, как соляная кислота; при необходимости продукт формулы I осаждают, а затем выделяют фильтрацией, альтернативно, после нейтрализации реакционной смеси, растворитель отгоняют и полученный остаток очищают с помощью колоночной хроматографии, в результате чего получают целевой продукт, альтернативно, остаток смешивают с водой и с водонесмешиваемым растворителем, таким, как этилацетат, и полученную смесь экстрагируют органическим растворителем, после чего экстракт осушают в присутствии осушающего агента, такого, как безводный сульфат магния, и выделяют из растворителя с получением целевого продукта. По необходимости продукт может быть затем очищен стандартными способами, например путем перекристаллизации или с использованием различной хроматографической техники, такой, как препаративная тонкослойная хроматография или колоночная хроматография.
В этой реакции, где исходным материалом является соединение, в котором R4 является ацильной группой и/или R7b является сложноэфирной группой первичного или вторичного спирта, такого, как метанол, этанол или изопропанол, ацильная группа R4 и сложноэфирный остаток удаляют одновременно.
Удаление карбокси-защитных групп.
Эта реакция является аналогичной реакции I в стадии A2 реакционной схемы A и может быть осуществлена с использованием тех же реагентов и тех же реакционных условий.
Этерификация.
Эта реакция аналогична реакции II стадии A2 и может быть осуществлена с использованием тех же реагентов и тех же реакционных условий.
Конверсия карбоксильной группы в группу формулы -CONR8R9.
Эта реакция аналогична реакции III стадии A2 и может быть осуществлена с использованием тех же реагентов и тех же реакционных условий.
Удаление тетразолил-защитных групп.
Эта реакция аналогична реакции IV стадии A2 и может быть осуществлена с использованием тех же реагентов и тех же реакционных условий.
Удаление гидрокси-защитных групп.
Эта реакция аналогична реакции VIII стадии A2 и может быть осуществлена с использованием тех же реагентов и тех же реакционных условий.
Алкилирование и ацилирование гидроксильных групп.
Эта реакция аналогична реакции VIII стадии A2 и может быть осуществлена с использованием тех же реагентов и тех же реакционных условий.
В дополнение к схеме В получение некоторых исходных материалов, используемых в вышеуказанных реакционных схемах, показано в реакционных схемах G и H, представленных в конце описания. Значения R1, R2, R3 и R5a в формулах реакционных схем G и H определены выше, R10 является алкильной группой, содержащей 1-6 атомов углерода, например одной из групп, которые были проиллюстрированы выше для R1, а предпочтительно алкильной группой, содержащей 1-4 атомов углерода, а более предпочтительно метильной или этильной группой, R11 является атомом водорода или имидазолил-защитной группой, например аралкильной группой, такой, как тритильная группа, дифенилметилная группа, или бензильная группа, или C1-C4-алкоксиметильной группой, такой, как метоксиметильная группа, этоксиметильная группа, пропоксиметильная группа или бутоксиметильная группа, из которых предпочтительными являются тритильная, бензильная, метоксиметильная или этоксиметильная группы, а более предпочтительной является тритильная группа.
Реакционная схема G.
В реакционной схеме G получают соединение формулы V, где R4 является атомом водорода, то есть соединение формулы Va (которые являются исходным материалом в реакционных схемах А).
В стадии GI соединение формулы XVI получают путем реакции орто-эфирного соединения формулы XIX с диаминомалеонитрилом формулы XX. Реакцию предпочтительно осуществляют в присутствии растворителя. В принципе нет конкретных ограничений на природу растворителя при условии, что он не оказывает неблагоприятного воздействия на реакцию или реагенты и обладает способностью растворять указанные реагенты по крайней мере до определенной степени. Примерами подходящих растворителей являются ароматические углеводороды, такие, как бензол, толуол или ксилил, галогенированные углеводороды, в частности галогенированные алифатические углеводороды, такие, как 1,2-дихлорэтан или тетрахлорметан, простые эфиры, такие, как тетрагидрофуран или диоксал, и нитрилы, такие, как ацетонитрил.
Реакция может протекать в широком диапазоне температур и точное значение температуры не играет существенной роли для данного изобретения. В основном подходящая для осуществления реакции температура составляет 50-180oC, а предпочтительно 80-150oC. Время, необходимое для осуществления реакции также может широко варьироваться в зависимости от многих факторов, в частности от температуры реакции и природы используемых реагентов и растворителя. Однако при предпочтительных условиях проведения реакции, указанных выше, этот период составляет 1-24 ч, а более предпочтительно 2-10 ч.
Реакционный продукт формулы XVI может быть выделен путем сбора кристаллов, осажденных в реакционной системе, или путем отгонки растворителя. По необходимости продукт может быть затем очищен стандартными способами, например путем перекристаллизации или с помощью различной хроматографической техники, в частности препаративной тонкослойной хроматографии или колоночной хроматографии.
Стадия G2 включает в себя получение соединения имидазол-4,5-дикарбоновой кислоты формулы XVI, полученной в стадии G1 Эта реакция может быть осуществлена путем нагревания соединения формулы XVI при дефлегмации в присутствии водной минеральной кислоты, такой, как водный раствор соляной, серной или азотной кислоты, в течение 1-24 ч (предпочтительно 3-16 ч). Продукт формулы XXI может быть выделен путем сбора кристаллов, осажденных в реакционной смеси, при охлаждении путем фильтрации или отгонки растворителя.
Стадия G3 является необязательной стадией и включает в себя получение соединения сложного эфира двухосновной кислоты формулы XI путем блокирования карбокси-группы соединения имидазол-4,5-дикарбоновой кислоты формулы XXI, полученного в стадии G2. Эта реакция может быть осуществлена с помощью взаимодействия соединения XXI с соединением формулы R5b-Y, где R5b и Y являются такими, как они были определены выше.
Реакцию предпочтительно осуществляют в присутствии растворителя. В принципе нет конкретных ограничений на природу растворителя при условии, что он не оказывает неблагоприятного воздействия на реакцию или реагенты и обладает способностью растворять указанные реагенты по крайней мере до определенной степени. Примерами подходящих растворителей являются углеводороды, в частности ароматические углеводороды, такие, как бензол или толуол, галогенированные углеводороды, в частности галогенированные алифатические углеводороды, такие, как метиленхлорид или хлороформ, простые эфиры, такие, как тетрагидрофуран или диоксан, спирты, такие, как метанол, этанол или т-бутанол, амиды, такие, как N,N-диметилформамид, N,N-диметилацетамид или N-метил-2-пирролидон, кетоны, такие, как ацетаты или метилэтилкетон, нитрилы, такие, как ацетонитрил и сульфоксиды, такие, как диметилсульфоксид. Из них предпочтительными являются нитрилы, галогенированные углеводороды или амиды.
Предпочтительно также, чтобы реакция протекала в присутствии основания, природа которого не является критической, если только оно не оказывает неблагоприятного воздействия на другие части реагентов. Примерами предпочтительных оснований могут служить органические амины, такие, как триэтиламин, N, N-диизопропилэтиламин или N -метилфорфолин.
Реакция может протекать в широком диапазоне температур и точное значение температуры реакции не играет существенной роли. В основном подходящая для осуществления реакции температура, будет зависеть от природы исходных материалов, растворителя и основания. В данном случае предпочтительная температура составляет от -10oC до 100oC, а более предпочтительная 0-80oC. Время, необходимое для осуществления реакции, также может широко варьироваться в зависимости от многих факторов, в частности от температуры реакции и природы используемых реагентов и растворителя. Однако при предпочтительных условиях проведения реакции, указанных выше, этот период составляет 0,5-24 ч, а более предпочтительно 1-16 ч.
После завершения реакции целевое соединение может быть выделено из реакционной смеси стандартными способами. Например, после отгонки растворителя остаток смешивают с водой, смесь экстрагируют водонесмешиваемым органическим растворителем, таким, как этилацетат, экстракт осушают с использованием осушающего агента, такого, как безводный сульфат магния, а растворитель отгоняют. По необходимости продукт может быть затем очищен стандартными способами, например путем перекристаллизации или с помощью различной хроматографической техники, такой, как препаративная тонкослойная хроматография или колоночная хроматография.
Альтернативно соединение дикарбоновой кислоты формулы XXI может быть подвергнуто этерификации с получением сложного диэфира формулы IX. Выбор конкретной реакции в данном случае будет зависеть от природы сложноэфирного остатка R5b.
Например, если группа, представленная R5b, является C1C6-алкильной группой или аралкильной группой, такой, как бензильная группа, то соединение формулы IX может быть получено путем реакции соответствующей дикарбоновой кислоты с C1C6-спиртом, таким, как метанол, этанол, пропанол или гексанол, или с аралкиловым спиртом, таким, как бензиловый спирт, в присутствии кислотного катализатора, такого, как хлорводород или серная кислота, в инертном растворителе, например одном из C1-C6-спиртов, которые могут быть использованы в качестве исходных материалов, описанных выше, галогенированном углеводороде, таком, как метиленхлорид, или простом эфире, таком, как тетрагидрофуран или диоксан, при температуре 0-100oC, предпочтительно 20-80oC, в течение периода времени от 1 ч до 3 дней, а предпочтительно 16-24 ч, или путем обработки соответствующей дикарбоновой кислотой с галогенирующим агентом, например пентахлоридом фосфора, тионилхлоридом или оксалилхлоридом, в инертном растворителе, например галогенированном углеводороде, таком, как метилхлорид, простом эфире, таком, как тетрагидрофуран или диоксан, или ароматическом углеводороде, таком, как бензол или толуол, приблизительно при комнатной температуре в течение периода времени от 30 мин до 5 ч, предпочтительно 1-3 ч, с получением соответствующего ацилгалида, и с последующей реакцией ацилгалида с соответствующим спиртом (если получают т-бутиловый сложный эфир, то предпочтительно использовать т-бутоксид калия вместо спирта) в инертном растворителе, например в бензоле или метиленхлориде, в присутствии основания, например триэтиламина, примерно при комнатной температуре в течение перида времени от 30 мин до 10 ч.
Целевое соединение может быть выделено из реакционной смеси с использованием стандартной техники. Например, после отгонки растворителя, остаток растворяют в воде и водонесмешиваемом органическом растворителе, таком, как этилацетат, полученный в результате раствор нейтрализуют бикарбонатом натрия, затем органический слой отделяют и осушают в присутствии осушающего агента, такого, как безводный сульфат магния, затем растворитель отгоняют и получают целевой продукт. Этот продукт может быть (если это необходимо) очищен стандартными способами, например путем перекристаллизации, или с помощью различной хроматографической техники, такой, как препаративная тонкослойная хроматография или колоночная хроматография.
В стадии G4 соединение формулы Va получают с помощью реакции соединения сложного диэфира формулы IX с реагентом Гриньяра формулы R2aMgX или R3aMgX, где R2a,R3a и X определены выше.
Эта реакция в основном аналогична реакции, описанной выше для стадии B2 реакционной схемы В, и может быть осуществлена с использованием тех же реагентов и тех же реакционных условий.
Реакционная схема H.
В этих реакциях получают соединения формул XIIIa, XIa и VIIa, где каждый из R11 является атомом водорода, то есть соединения формул XIII, XI и VII и соединение формулы Va, которые являются исходными материалами, используемыми в реакционной схеме E, D, A и B, соответственно.
В стадии H1, которая является необязательной стадией, соединение формулы XVIa получают с помощью реакции динитрилового соединения XVI с соединением формулы R11a-X, где X определен выше, а R11a является любой из групп, определенных выше для R11 и не является атомом водорода, в присутствии основания.
Примерами подходящих соединений являются гидриды щелочных металлов, таких как гидрид лития или гидрид натрия, карбонаты щелочных металлов, такие, как карбонат натрия или калия, и алкоксиды щелочных металлов, такие, как метоксид натрия, этоксид натрия или т-бутоксид калия.
Реакцию предпочтительно осуществляют в присутствии растворителя. В принципе нет конкретных ограничений на природу растворителя при условии, если он не оказывает неблагоприятного воздействия на реакцию или реагенты и обладает способностью растворять указанные реагенты по крайней мере до определенной степени. Примерами подходящих растворителей являются галогенированные углеводороды, такие, как метиленхлорид или хлороформ, простые эфиры, такие, как тетрагидрофуран или диоксан, амиды, такие, как диметилформамид или диметилацетамид, и кетоны, такие, как ацетон или метилэтилкетон. Реакция может протекать в широком диапазоне температур и точное значение температуры не играет существенной роли для данного изобретения. В основном подходящая для осуществления реакции температура составляет 0-120oC, а более предпочтительно 20-80oC. Время, необходимое для осуществления реакции, также может широко варьироваться в зависимости от многих факторов, в частности от температуры реакции и природы используемых реагентов и растворителя. Однако при предпочтительных условиях проведения реакции, указанных выше, этот период составляет 1-24 ч, а более предпочтительно 3-8 ч.
После завершения реакции целевое соединение может быть выделено из реакционной смеси стандартными способами. Например, один из таких способов заключается в том, что к реакционной смеси добавляют воду, смесь экстрагируют водосмешиваемым органическим растворителем, таким, как этилацетат, экстракт промывают водой и осушают агентом, таким, как безводный сульфат магния, и отгоняют растворитель. По необходимости продукт может быть затем очищен стандартными способами, например путем перекристаллизации или с использованием различной хроматографической техники, такой, как препаративная тонкослойная хроматография или колоночная хроматография.
В стадии H2 соединение формулы XIIIa получают с помощью реакции динитрилового соединения формулы XVIa с реагентом Гриньяра формулы R2aMgX, в которой R2a и X определены выше, или с восстанавливающим агентом. Эта реакция в основном аналогична реакции, описанной выше в стадии B2 реакционной схемы B, и может быть осуществлена с использованием тех же реакционных условий.
Имидазолил-защитная группа соединения формулы XIIIa может быть (но необязательно) удалена путем обработки соединения формулы XIIIa стандартным способом в зависимости от природы защитной группы с получением соединения формулы XIII.
Например, если защитной группой является тритильная группа или алкоксиметильная группа, то она может быть удалена путем реакции защищенного соединения с кислотой.
Примерами подходящих кислот являются неорганические кислоты, такие, как соляная кислота или серная кислота, и органические кислоты, такие, как уксусная кислота, муравьиная кислота, метансульфоновая кислота или п-толуолсульфоновая кислота.
Реакцию предпочтительно осуществляют в присутствии растворителя. В принципе нет конкретных ограничений на природу растворителя при условии, если он не оказывает неблагоприятного воздействия на реакцию или реагенты и обладает способностью растворять указанные реагенты по крайней мере до определенной степени. Примерами подходящих растворителей являются простые эфиры, такие, как тетрагидрофуран или диоксан, спирты, такие, как метанол или этанол, кислоты, такие, как уксусная кислота, вода или смесь любых двух или более из вышеуказанных растворителей.
Реакция может быть осуществлена в широком диапазоне температур и точное значение температуры реакции не играет решающей роли для данного изобретения. В основном подходящая для осуществления реакции температура составляет 0-120oC, а более предпочтительно 10-100oC. Время, требуемое для осуществления реакции, также может широко варьироваться в зависимости от многих факторов, в частности от температуры реакции и природы используемых реагентов и растворителя. Однако при предпочтительных условиях проведения реакции, указанных выше, этот период составляет от 30 мин до 24 ч, а более предпочтительно 1-16 ч.
После завершения реакции целевое соединение может быть выделено из реакционной смеси стандартными способами. Например, один из таких способов заключается в том, что растворитель выпаривают и продукт очищают путем перекристаллизации или хроматографии или реакционную смесь нейтрализуют слабой кислотой, такой, как бикарбонат натрия, экстрагируют водонесмешиваемым органическим растворителем, таким, как этилацетат, и растворитель выпаривают. Полученный продукт, если это необходимо, может быть затем очищен путем перекристаллизации или путем хроматографии, например путем препаративной тонкослойной хроматографии или колоночной хроматографии.
Если имидазолил-защитной группой является аралкильная группа, такая, как бензильная или дифенилметильная группа, то она может быть удалена путем каталитической гидрогенизации. Эта реакция в основном аналогична реакции, описанной выше (I) стадии A2 реакционной схемы A, где карбокси-защитная группа является аралкильной группой, и может быть осуществлена с использованием тех же реагентов и тех же реакционных условий.
В стадии H3 полученное карбонильное соединение формулы XIIIa затем подвергают реакции с реагентом Гриньяра формулы R3aMgX, в которой R3a и X определены выше, или с восстанавливающим агентом с получением соединения формулы XIa. Эта реакция в основном аналогична реакции стадии B2 реакционной схемы B, описанной выше, и может быть осуществлена с использованием тех же реагентов и тех же реакционных условий.
По желанию имидазолил-защитная группа соединения формулы XIa может быть удалена с помощью в основном такой реакции, которая была описана как необязательная в стадии H2 реакционной схемы H, и может быть осуществлена с использованием тех же реагентов и тех же реакционных условий.
В стадии H4 соединение карбоновой кислоты формулы XXII получают путем гидролиза оставшейся циано-группы в 5-положении имидазольного кольца. Реакция может быть проведена с использованием гидроокиси щелочного металла, такой, как гидроокись натрия, калия или лития, в инертном растворителе, предпочтительно в воде, спирте, таком, как метанол или этанол, простом эфире, таком, как метанол или этанол, простом эфире, таком, как тетрагидрофуран или диоксан, или смеси любых двух или более из указанных растворителей. Реакция может протекать в широком диапазоне температур и точное знание температуры не играет существенной роли для данного изобретения. В основном подходящая для осуществления реакции температура составляет 0-120oC, а более предпочтительно 20-100oC. Время, необходимое для осуществления реакции, также может широко варьироваться в зависимости от многих факторов, в частности от температуры реакции и природы используемых реагентов и растворителя. Однако при предпочтительных условиях проведения реакции, указанных выше, этот период составляет 0,5-24 ч, а более предпочтительно 1-16 ч. После завершения реакции реакционный продукт может быть выделен стандартными способами. Например, реакционную смесь нейтрализуют путем добавления минеральной кислоты, такой, как соляная кислота, а если целевое соединение формулы XXII осаждается в реакционной среде, то оно может быть собрано путем фильтрации. Альтернативно целевое соединение может быть выделено следующим образом: после нейтрализации реакционной смеси растворитель отгоняют и остаток подвергают колоночной хроматографии, альтернативно, остаток смешивают с водой и водонесмешиваемым органическим растворителем и экстрагируют органический растворителем, после чего экстракт осушают в присутствии осушающего агента, такого, как безводный сульфат магния, растворитель отгоняют и получают целевой продукт. По необходимости этот продукт может быть затем очищен стандартными способами, например путем перекристаллизации, или с помощью различной хроматографической техники, такой, как препаративная тонкослойная хроматография или колоночной хроматографии.
В стадии H5, которая является необязательной стадией, соединение формулы Va получают путем этерификации соединения карбоновой кислоты формулы XXII и (не обязательно) с последующим разблокированием имидазолильной группы. Эта реакция этерификации в основном аналогична реакции II в стадии A2 реакционной схемы А, описанной выше, а реакция необязательного разблокирования в основном аналогична реакции, описанной для стадии H2 реакционной схемы H, и обе эти реакции могут быть осуществлены с использованием тех же реагентов и тех же реакционных условий.
В стадии H6 соединение формулы XXIII получают путем гидролиза соединения формулы XIIIa. Эта реакция в основном аналогична реакции, описанной выше для стадии H4 реакционной схемы H, и может быть осуществлена с использованием тех же реагентов и тех же реакционных условий.
В стадии H7 соединение формулы VIIa получают путем этерификации соединения формулы XXIII. Эта реакция в основном аналогична реакции, описанной выше для стадии H5 реакционной схемы H, и может быть осуществлена с использованием тех же реагентов и тех же реакционных условий.
По желанию имидазолил-защитная группа соединения формулы VIIa может быть удалена в основном с помощью реакции, аналогичной необязательной реакции, описанной выше для стадии H2 реакционной схемы H, которая может быть осуществлена с использованием тех же реагентов и тех же реакционных условий.
В стадии H8 соединение формулы Va получают с помощью реакции соединения формулы VIIa с реагентом Гриньяра и/или с восстанавливающим агентом, а затем необязательного разблокирования имидазолильной группы. Эта реакция в основном аналогична реакции, описанной выше для стадии B2 реакционной схемы B, а реакция необязательного разблокирования в основном аналогична реакции, описанной выше для стадии H2 реакционной схемы H, и эти обе реакции могут быть осуществлены с использованием тех же реагентов и реакционных условий.
Предлагаемые соединения могут образовывать соли. В принципе нет ограничений на природу солей при условии, что они являются фармацевтически приемлемыми и пригодными для терапевтического использования. Однако даже этим ограничением можно пренебречь, если эти соли предназначены лишь для использования в качестве промежуточных соединений для получения других, более активных соединений. Предлагаемые соединения могут образовывать соли с основанием. Примерами таких солей являются соли со щелочными металлами, такими, как натрий, калий или литий, соли со щелочноземельными металлами, такими, как барий или кальций, соли с другими металлами, такими, как магний или алюминий, соли органических оснований, таких, как дициклогексиламин, гуанидин или триэтиламин, и соли с основными аминокислотами, такими, как лизин или аргинин. Кроме того, предлагаемые соединения содержат основную группу в своей молекуле, и поэтому могут образовывать кислые аддитивные соли. Примерами таких кислых аддитивных солей являются соли с минеральными кислотами, в частности галогенводородными кислотами, такими, как фтористоводородная, бромистоводородная, иодистоводородная и хлористоводородная кислота, азотная кислота, карбоновая кислота, серная кислота или фосфорная кислота, соли с низшими алкилсульфоновыми кислотами, такими, как метансульфоновая кислота, трифторметансульфоновая кислота или этансульфоновая кислота, соли с органическими карбоновыми кислотами, такими, как уксусная кислота, муравьиная кислота, винная кислота, щавелевая кислота, малеиновая кислота, яблочная кислота, янтарная кислота или лимонная кислота, и соли с аминокислотами, такими, как глутаминовая кислота или аспарагиновая кислота. Предлагаемые соединения могут быть превращены в фармацевтически приемлемые соли путем обработки кислотой или основанием с использованием стандартных способов, хорошо известных специалистам.
Предлагаемые соединения являются по существу нетоксичными (для соединения примера 78 LD50 более 2000 мг/кг) и показывают хорошую ингибиторную активность против повышения кровяного давления, индуцированного ангиотензином II, и поэтому могут быть с успехом использованы для лечения или предупреждения заболеваний системы кровообращения в качестве гипотенсивного средства, или в качестве терапевтического лекарственного средства для лечения болезней сердца.
Биологическую активность предлагаемых соединений оценивали с помощью приведенных ниже экспериментов.
Оценка AII-рецептор-блокирующей активности путем ингибирования вазопрессорного ответа на введение ангиотензина III.
Биологическую активность каждого соединения оценивали путем определения дозы, требуемой для ингибирования вазопрессорного ответа на внутривенное введение ангиотензина II крысам при дозе 50% (LD50), Самцам крыс Wister-Imamichi (каждая весом 300-400 г) вводили внутрибрюшинную инъекцию 100 мг/кг тиобарбитал-натрия (Инактин (торговый знак)) и две канюли: одну канюлю вводили в бедренную артерию для измерения кровяного давления, а другую в бедренную вену для инъекции лекарственного средства. 50 нг/кг ангиотензина II вводили внутривенно с интервалом около 10 мин и наблюдали повышение кровяного давления (обычно около 50 мм рт. ст.). После получения стабильного вазопрессорного ответа на ангиотензин II вводили внутривенно испытываемое соединение. Через 2 мин опять вводили ангиотензин II и оценивали ингибиторную активность испытываемого соединения. Для расчета LD50 определяли процент ингибирования вазопрессорного ответа на ангиотензин II при наращивании концентрации вводимого испытываемого соединения. В данном эксперименте использовали ангиотензин II, растворенный в 0,5% альбумине бычьей сыворотки, а испытываемое соединение растворяли в 100% диметилсульфоксиде (ДМСО). Определенные таким образом ID50-величины представлены в таблице.
Помимо предлагаемых соединений, которые были идентифицированы в ряде примеров, иллюстрирующих их получение и представленных ниже, в данных экспериментах было использовано также соединение прототипа (обозначенное в таблице как соединение A), которое представляет собой 2-[4- (2-бутил-5-хлоро-4-хлорометилимидазол-1-ил-метил)фенил] бензойную кислоту, и которое описано в примере 118 публикации Европатента N 253310.
Предлагаемые соединения могут быть введены, например, перорально в виде таблеток, капсул, гранул, порошков, сиропов и т.п. или парентерально в виде инъекций, суппозиториев и т. п. Эти фармацевтические препараты могут быть получены стандартными способами с использованием адъювантов, хорошо известных специалистам, например таких, как наполнители, связующие, дезинтегрирующие агенты, замасливатели, стабилизаторы, модификаторы и т.п. Хотя доза активного ингредиента может варьироваться в зависимости от симптомов, возраста пациента, природы и тяжести заболевания, а также способа введения лекарственного средства, однако в случае перорального введения указанного средства взрослому человеку полная ежедневная доза предлагаемого соединения может составлять 1-1000 мг, а предпочтительно 5-300 мг и вводиться либо в виде однократной дозы, либо в виде разделенной дозы, например два или три раза в день, а в случае внутривенного введения эта доза может составлять 0,1-100 мг, а предпочтительно 0,5-30 мг и вводиться от одного до трех раз в день.
Приведенные ниже примеры иллюстрируют получение ряда предлагаемых соединений, а получения, следующие за этими примерами, иллюстрируют получение исходных материалов, используемых в указанных примерах.
Пример 1. Метил 1-[(2'-т- бутоксикарбонилбифенил-4-ил)метил] -2-бутил4-гидроксиметилимидазол-5-карбоксилат (соединение 1- 94).
1a) Диметил-1-[(2'-т-бутоксикарбонилбифенил-4-ил) метил]-2-бутилимидазол-4,5-динарбоксилат.
Раствор метоксида натрия, полученного из 0,69 г натрия и 40 мл метанола, добавляли к раствору 7,2 г диметил 2-бутил-имидазол-4,5-дикарбоксилата, полученного в соответствии с описанием, приведенным в получении 4, в 40 мл метанола и полученную смесь концентрировали путем выпаривания при пониженном давлении. Полученный остаток смешивали с бензолом и смесь концентрировали путем дистилляции при пониженном давлении. Затем эту операцию повторяли три раза и полученный в результате твердый остаток растворяли в 72 мл N,N - диметилацетамида. После этого к полученному раствору по капле добавляли раствор 10,41 г т-бутил-4-бромометилбифенил-2-карбоксилата в 100 мл N,N -диметилацетамида. Затем реакционную смесь размешивали влечение 1 ч при комнатной температуре и в течение 2 ч при 50-55oC. По истечение этого времени эту смесь смешивали с этилацетатом и водой и этилацетатный слой отделяли и осушали безводным сульфатом магния, после чего растворитель отгоняли при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью этилацетата и гексана (1:1), в результате получали 15,1 г целевого соединения в виде смолистого вещества.
Ядерный магнитный резонанс (ЯМР) (CDCl3) δ ppm: (0,90 3Н, т, J 7 Гц), 1,26 (9Н, т (триплет)), 1,1-2,0 (4Н, м, (мультиплет)), 2,70 (2Н, т. J 7 Гц), 3,81 (3Н, с, (синглет)), 4,90 (3Н, с.) 5,47 (2Н, с.) 6,95-7,85 (8Н, м).
1b). Метил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-4-гидроксиметилимидазол-5-карбоксилат.
42 мл гидрида диизобутилалюминия в виде 1,5 М раствора в толуоле по капле добавляли при температуре от -20oC до -15oC к раствору 16,0 г диметил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутилимидазол-4,5-дикарбоксилата, полученного как описано выше в стадии (а), в 200 мл тетрагидрофурана и полученную смесь оставляли на 16 ч при -5oC. По истечение этого времени, реакционную смесь смешивали с водным раствором хлорида аммония и этилацетата, а затем перемешивали в течение 1 ч. После этого осадок удаляли путем фильтрации. Затем этилацетатный слой отделяли, осушали безводным сульфатом магния и растворитель удаляли путем дистилляции при пониженном давлении. Затем остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя этилацетатом, в результате чего получали 12,0 г целевого соединения в виде кристаллов, т. пл. 99oC.
ЯМР (CDCl3) d ppm: 0,90 (3Н, т. J 7 Гц), 1,20 (9Н, с), 1,1-2,0 (4Н, м), 2,69 (2Н, т. J 7 Гц), 3,55 (1Н, шир.с), 3,78 (3Н, с), 4,84 (2Н, д. (дублет) J 5 Гц), 5,60 (2Н, с), 6,95-7,9 (8Н, м).
Пример 2. Этил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-2-бутил-4-гидроксиметилимидазол-5-карбоксилат (соединение 1-95).
2а). Диэтил-1-[(2'-т-бутоксикарбонилбифенил-4-ил)-метил]-2-бутилимидазол-4,5-дикарбоксилат.
Повторяли процедуру, описанную в примере 1а, но с использованием 8,0 г диэтил 2-бутил-имидазол-4,5-дикарбоксилата, полученного в соответствии с описанием в получении 3, и 10,41 г т-бутил 4-бромометилбифенил-2-карбоксилата, в результате чего получали 15,4 г целевого соединения в виде смолистого вещества.
ЯМР (CDCl3) d ppm: 0,90 (3Н, т. J 7 Гц), 1,1-2,0 (4Н, м), 1,24 (9Н, с), 1,26 (3Н, т. J 7 Гц), 1,39 (3Н, т. J 7 Гц), 4,40 (2Н, кв. J 7 Гц), 4,28 (2Н, кв. (квартет) J 7 Гц), 4,40 (2Н, кв. J 7 Гц), 5,50 (2Н, с), 7,0-7,9 (8Н, м).
2b). Этил-1[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-2-бутил -4-гидроксиметилимидазол-5-карбоксилат.
Повторяли процедуру, описанную в примере 1b, но с использованием 1,50 г диэтил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-2-бутилимидазол-4,5-дикарбоксилата, полученного в соответствии с описанием в стадии (а), и 3,9 мл гидрида диизобутилалюминия в виде 1,5 М раствора в толуоле, в результате чего получали 1,1 г целевого соединения в виде смолистого вещества.
ЯМР (CDCl3) d ppm: 0,90 (3Н, т. J 7 Гц), 1,24 (9Н, с), 1,30 (3Н, т. J 7 Гц), 1,1-2,0 (4Н, м), 2,68 (2Н, т. J 7 Гц), 3,60 (1Н, шир. с), 4,24 (2Н, кв. J 7 Гц), 4,84 (2Н, с.), 5,57 (2Н, с), 6,9-7,85 (8Н, м).
Пример 3. Метил 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил]-4 гидроксиметилимидазол-5-карбоксилат (соединение 1-5).
Раствор 0,36 г метил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-2-бутил-4-гидроксиметилимидазол-5-карбоксилата, полученного в соответствии с описанием в примере 1, в 4 мл 4 н. раствора хлорводорода в диоксане оставляли на 4 ч при комнатной температуре. По истечение этого времени реакционную смесь концентрировали путем выпаривания при пониженном давлении, а остаток перетирали с этилацетатом с образованием кристаллов, которые собирали путем фильтрации, в результате чего получали 0,35 г целевого соединения в виде его гидрохлорида, т. пл. 192-195oC (с разложением).
ЯМР (гексадейтерированный диметилсульфоксид) d ppm: 0,81 (3Н, т J 7 Гц), 1,22-1,35 (2Н, м), 1,43-1,56 (2Н, м), 3,00 (2Н, т. J 7 Гц), 3,82 (3Н, с), 4,81 (2Н, с), 5,77 (2Н, с), 7,18-7,75 (8Н, м).
Пример 4. 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-4-гидроксиметилимидазол-5-карбоновая кислота (соединение 1-96).
Раствор 2,01 г гидроксида-моногидрата лития в 96 мл воды добавляли к раствору 4,78 г метил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-2-бутил-4-гидроксиметилимидазол -5-карбоксилата, полученного в соответствии с описанием в примере 1, в 48 мл диоксана и полученную смесь размешивали при комнатной температуре в течение 18 ч. По истечение этого времени реакционную смесь выделяли из диоксана путем дистилляции при пониженном давлении и к водному остатку добавляли 49,6 мл 1 н. водной соляной кислоты. Осажденный кристаллы собирали путем фильтрации и последовательно промывали водой и диэтиловым эфиром, в результате чего получали 4,26 г целевого соединения, т. пл. 187oC (с разл.).
ЯМР (CDCl3) d ppm: 0,85 (3Н, т. J 7 Гц), 1,24 (9Н, с), 1,1-1,9 (4Н, м), 2,80 (2Н, т. J 7 Гц), 5,05 (2Н, с), 5,93 (2Н, с), 7,0-7,85 (8Н, м).
Пример 5. 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил]-4-гидроксиметил-имидазол-5 -карбоновая кислота.
Раствор 0,12 г 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-2-бутил-4-гидроксиметилимидазол-5-карбоновой кислоты, полученной в соответствии с описанием в примере 4, в 2 мл 4 н. раствора хлорводорода в диоксане оставляли при комнатной температуре на 5 ч, после чего растворитель удаляли путем дистилляции при пониженном давлении. Полученный остаток растирали в этилацетате и получали 0,11 г целевого соединения в виде его гидрохлорида, т. пл. 130-140oC (с размягчением).
ЯМР (ДМСО-d6) d ppm: 0,80 (3Н, т. J 7 Гц), 1,2-1,33 (2Н, м), 1,4-1,53 (2Н, м), 2,98 (2Н, т. J 7 Гц), 4,84 (2Н, с), 5,81 (2Н, с), 7,17-7,74 (8Н, м).
Пример 6. Пивалоилоксиметил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-4-гидроксиметилимидазол-5-карбоксилат (соединение 1-97).
350 мг карбоната калия добавляли к раствору 552 г 1-[(2'-т-бутоксикарбонлбифенил-4-ил)метил] -2-бутил-4-гидроксиметилимидазол-5-карбоновой кислоты, полученной в соответствии с описанием в примере 4, и 220 мг пивалоилоксиметилхлорида в 7 мл N,N-диметилацетамида, и полученную смесь размешивали при комнатной температуре в течение 5 ч. По истечение этого времени, реакционную смесь смешивали с этилацетатом и водой, этилацетатный слой отделяли и осушали безводным сульфатом магния, после чего растворитель отгоняли при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента этилацетат и получали 0,62 г целевого соединения в виде сиропа.
ЯМР (CDCl3) d ppm: 0,91 (3Н, т. J 7 Гц), 1,18 (9Н, с), 1,21 (9Н, с), 1,1-3,0 (4Н, м), 2,72 (2Н, т, J 7 Гц), 3,35 (1Н, шир.с), 4,85 (1Н, д. J 5 Гц), 5,61 (2Н, с), 5,90 (2Н, с), 6,95-7,9 (8Н, м).
Пример 7. Пивалоилоксиметил 2-бутил-1-[(2'-карбоксифенил-4-ил)метил]-4-гидроксиметилимидазол-5-карбоксилат (соединение 1-98).
Раствор 0,62 г пивалоилоксиметил 1-[(2'-т-бутоксикарбонлбифенил-4-ил)метил] -2-бутил-4-гидроксиметилимидазол-5-карбоксилата, полученного в соответствии с описанием в примере 6, в 10 мл 4 н. раствора хлорводорода в диоксане оставляли на 4 ч при комнатной температуре, после чего раствор концентрировали путем выпаривания при пониженном давлении. Сиропообразный остаток размешивали в диэтиловом эфире, после чего растворитель удаляли путем его сливания, а остаток осушали в вакууме, в результате чего получали 0,46 г гидрохлорида целевого соединения в виде порошка.
ЯМР (CDCl3) d ppm: 0,85 (3Н, т. J 7 Гц), 1,19 (9Н, с), 1,25-(1,45 (2Н, м), 1,65-1,80 (2Н, м), 2,99 (2Н, т J 7 Гц), 5,01 (2Н, с), 5,70 (2Н, с), 5,89 (2Н, с), 7,05-7,97 (8Н, м).
Пример 8. Метил-1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-2-бутил-4-метил-2-бутил-4-метоксиметил имидазол-5-карбоксилат (соединение 1-99).
0,057 г гидрида натрия (в виде 55 мас. дисперсии в минеральном масле) добавляли к раствору 0,478 г метил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-4-гидроксиметилимидазол-5-карбоксилата, полученного в соответствии с описанием в примере 1, в 5 мл N,N-диметилацетамиде и полученную смесь размешивали 30 мин при комнатной температуре. После этого, добавляли 0,125 мл иодометана, и реакционную смесь размешивали при 50oC в течение 3 ч. Затем эту смесь смешивали с этилацетатом и водой. Этилацетатный слой отделяли и осушали безводным сульфатом магния, а растворитель отгоняли при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью этилацетата и метиленхлорида (1: 1), в результате чего получали 0,30 г целевого соединения в виде смолистого вещества.
ЯМР (CDCl3) d ppm: 0,90 (3Н, т, J 7 Гц), 1,24 (9Н, с), 1,1-2,0 (4Н, м), 2,71 (2Н, т, J 7 Гц), 3,46 (3Н, с), 3,80 (4Н, с), 4,68 (2Н, с), 5,60 (2Н, с), 6,9-7,9 (8Н, м).
Пример 9. Метил 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил]-4-(метоксиметил) имидазол-5-карбоксилат (соединение 1-121).
Раствор 0,30 г метил 1-[(2'-т-бутоксикарбонилфенил-4-ил)-метил]-2-бутил-4-(метоксиметил)-имидазол -5-карбоксилата, полученного в соответствии с описанием в примере 8, в 3 мл 4 н раствора хлорводорода в диоксане оставляли на 5 ч при комнатной температуре, после чего растворитель отгоняли при пониженном давлении. Сиропообразный остаток перетирали в диэтиловом эфире и собирали путем фильтрации, в результате чего получали 0,26 г целевого соединения в виде его гидрохлорида, т. пл. 106-110oC (с размягчением).
ЯМР (ДМCО-d6) d ppm: 0,81 (3Н, т. J 7 Гц), 1,2-1,35 (2Н, м), 1,45-1,6 (2Н, м), 2,97 (2Н, т. J 7 Гц), 3,39 (3Н, с), 3,82 (3Н, с), 4,72 (2Н, с), 5,75 (2Н, с), 7,16-7,74 (8Н, м).
Пример 10. 1-бутил-1-[(2'-карбоксифенил-4-ил)метил]-4[(1-гидрокси-1-метил)этил]имидазол -5-карбоновая кислота (соединение 1-31).
10а) 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] 2-бутил-5-циано-4-[(1-гидрокси-1-метил) этил]-имидазол.
48 г гидрида натрия (в виде 55 мас. дисперсии в минеральном масле) добавляли при комнатной температуре и в атмосфере азота, размешивая при этом, к раствору 207 мг 2-бутил-4-циано-4-[(1-гидрокси-1-метил)этил] имидазола, полученного в соответствии с описанием в получении 7, в 10 мл N,N-диметилацетамида, и полученную смесь размешивали 30 мин, после чего добавляли 347 мг т-бутил-4-бромометилбифенил-2-карбоксилата. Затем реакционную смесь размешивали 2 ч при комнатной температуре, после чего ее выливали в смесь льда и хлорида натрия и экстрагировали этилацетатом. Экстракт осушали безводным сульфатом магния и концентрировали при пониженном давлении, в результате чего получали маслянистый сырой продукт. Продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью гексана и этилацетата (1:1), и получали 462 мг целевого соединения.
ЯМР (CDCl3) d ppm: 0,90 (3Н, т. J 7 Гц), 1,1-2,1 (4Н, м), 1,21 (9Н, с), 1,61 (6Н, с), 2,70 (2Н, т. J 7 Гц), 3,40 (1Н, с), 5,22 (2Н, с), 7,0-8,0 (8Н, м).
10b) 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил] -5-циано-4-[(1-гидрокси-1-метил) этил]имидазол.
Раствор 462 мг 1-[(2'-т-бутоксикарбонилбифенил-4-ил)-метил]-2-бутил-5-циано-4-[(1-гидрокси-1-метил)этил] имидазола, полученного в соответствии с описанием в стадии (а), в 10 мл 4 н. раствора хлорводорода в диоксане оставляли на ночь при комнатной температуре. После этого реакционную смесь концентрировали путем выпаривания при пониженном давлении и концентрат растворяли в метиленхлориде. Осадок собирали фильтрацией и осушали, в результате чего получали 457 мг гидрохлорида целевого соединения в виде бесцветного порошка, т. пл. 209-210oC.
ЯМР (ДМСО= d6) d ppm: 0,85 (3Н, т. J 7 Гц), 1,0-1,8 (4Н, м), 1,58 (6Н, с), 3,00 (2Н, т. J 7 Гц), 5,51 (2Н, с), 7,1-8.0 (8Н, м).
10 с) 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил] 4-[(1-гидрокси-1-метил)этил]имидазол -5-карбоновая кислота.
Раствор 314 мг 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил]-5-циано-4-[(1-гидрокси-1 -метил)этил] имидазола гидрохлорида, полученного в соответствии с описанием в стадии (b), в водном растворе 460 мг гидроксида натрия в 5 мл воды размешивали в масляной бане в течение 5 ч при 100oC. По истечение этого времени реакционную смесь охлаждали и ее pH доводили до значения 3-4 путем прибавления 1 н. водной соляной кислоты. Полученный бесцветный осадок собирали фильтрацией, промывали водой и осушали безводным сульфатом магния, в результате чего получали 244 мг, т.пл. 139-141oC.
ЯМР (ДМСО-d6) d ppm: 0,86 (3Н, т. J 7 Гц), 1,0-1,9 (4Н, м), 1,60 (6Н, с), 2,66 (2Н, т, J 7 Гц), 5,70 (2Н, с), 6,9-7,9 (8Н, м).
Пример 11. 2-бутил-1-[(2'-карбоксифенил-4-ил)метил] -4-(1-гидроксиэтил)-имидазол-5-карбоновая кислота (соединение 1-25).
11а) 4-ацетил-1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-5-цианоимидазол.
0,87 г карбоната калия и 2,4 г т-бутил-4'-бромометилбифенил-2-карбоксилата добавляли к раствору 1,2 г 4-ацетил-2-бутил-5-цианоимидазола, полученного в соответствии с описанием в получении 5, в 12 мл N,N-диметилацетамида, и полученную смесь размешивали 3 ч при комнатной температуре. После этого реакционную смесь разводили 100 мл этилацетата и промывали насыщенным водным раствором хлорида натрия. Водный слой еще раз экстрагировали 50 мл этилацетата, и объединенные экстракты промывали насыщенным водным раствором хлорида натрия. Растворитель удаляли путем дистилляции при пониженном давлении и полученный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью гексана и этилацетата (3:1), в результате чего получали 1,31 г целевого соединения.
ЯМР (CDCl3) d ppm: 0,93 (3Н, т. J 7 Гц), 1,1-2,1 (4Н, м), 1,23 (9Н, с), 2,58 (3Н, с), 2,75 (2Н, т, J 7 Гц), 5,32 (2Н, с), 7,0-8,0 (8Н, м).
11b) 4-ацетил-2-бутил-1-[(2'-карбоксибифенил-4-ил)метил] -5-цианоимидазол.
Раствор 1,3 г 4-ацетил-1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-2-бутил-5-цианоимидазола, полученный в соответствии с описанием в стадии (а), в 30 мл 4 н. раствора хлорводорода в диоксане оставляли на ночь при комнатной температуре, после чего раствор концентрировали при пониженном давлении. Концентрат очищали посредством колоночной хроматографии на силикагеле, элюируя смесью метиленхлорида и метанола (10:1), и получали бесцветное аморфное твердое вещество. Это вещество растирали в гексане, собирали фильтрацией и осушали, в результате чего получали 1,1 г целевого соединения, т. пл. выше 55oC (с размягчением).
ЯМР (CDCl3) d ppm: 0,84 (3Н, т. J 7 Гц), 1,0-2,0 (4Н, м), 2,54 (3Н, с), 2,66 (2Н, т. J 7 Гц), 5,17 (2Н, с), 6,8-7,0 (8Н, м).
11с) 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил]-5-циано-4-1-гидроксиэтил имидазол.
68 мг борогидрида натрия добавляли к раствору 718 г 4-ацетил-2-бутил-1-[(2-карбоксифенил-4-ил)метил] -5-циано-имидазола, полученного в соответствии с описанием в стадии (b), в смеси 20 мл изопропанола и 10 мл этанола и полученную смесь размешивали 3 ч при комнатной температуре. По истечение этого времени pH реакционной смеси доводили до значения 3 путем добавления 1 н. водной соляной кислоты, после чего растворитель отгоняли при пониженном давлении. Полученный остаток смешивали с метиленхлоридом и водой и метиленхлоридный слой отделяли. Водный слой экстрагировали три раза метиленхлоридом и объединенные экстракты осушали и концентрировали путем выпаривания при пониженном давлении. Полученный остаток растворял в 10 мл этилацетата и выдерживали при комнатной температуре. Затем осажденный твердый продукт собирали путем фильтрации и получали в результате 398 мг целевого соединения в виде бесцветного порошка, т. пл. 200-201oC.
ЯМР (ДМСО-d6) d ppm: 0688 (3Н, т. J 7 Гц), 1,0-2,0 (4Н, м), 1,54 (3Н, д. J 7 Гц), 2,68 (2Н, т. J 7 Гц), 4,91 (1Н, кв. J 7 Гц), 5,21 (2Н, с), 7,0-8,0 (8Н, м).
11d) 2-бутил-1-[(2'-карбоксибифенил-4-ил) метил]-4-(1-гидроксиэтил имидазол)-5-карбоновая кислота.
Смесь 300 мог 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил]-5-циано-4-(1-гидроксиэтил)имидазола, полученного в соответствии с описанием в стадии (c), и 3 мл 1 н. водного раствора гидроксида натрия размешивали в масляной бане, поддерживаемой 3 ч при 80oC. По истечение этого времени, реакционную смесь охлаждали, а затем немного подкисляли соляной кислотой, после этого смесь экстрагировали 4 раза метиленхлоридом (30 мл x 4). Комбинированные экстракты осушали и концентрировали досуха путем выпаривания при пониженном давлении, получая в результате аморфное твердое вещество. Это вещество очищали посредством колоночной хроматографии на силикагеле, элюируя смесью метиленхлорида и метанола (10: 1 и 3:1). Полученный из элюата твердый продукт растирали в диэтиловом эфире. Полученный порошок собирали путем фильтрации и осушали, в результате чего получали 72,3 мг целевого соединения в виде бесцветного порошка, т. пл. 168-170oC (с размягчением выше 140oC).
ЯМР (ДМСО-d6) d ppm: 0,84 (3Н, т. J 7 Гц), 1,0-2,0 (4Н, м), 1,52 (3Н, д. J 7 Гц), 2,3-2,8 (2Н, перекрыв. c пиком диметилсульфоксида) 4,93 (1Н, кв. J 7 Гц, 5,60 (2Н, шир.с.), 6,8-7,8 (8Н, м).
Пример 12. 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил]-4-(a--гидроксибензил)-имидазол-5-карбоновая кислота (соединение 1-80).
12a) 4-бензоил-1-[(2'-т-бутоксикарбонилобифенил-4-ил)метил]-2-бутил-5-цианоимидазол.
Повторяли процедуру, аналогичную описанной в примере 11a, но при этом использовали 1,27 г 4-бензоил-2-бутил-5-цианоимидазола, полученного в соответствии с описанием в получении 6, 1,74 г т-бутил-4'-бромометилбифенил-2-карбоксилата 0,69 г карбоната калия и 20 мл N,N-дилметилацетамида, после чего продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью гексана и этилацетата (2:1), и получали 2,1 г целевого соединения.
ЯМР (CDCl3) δ ppm: 0,93 (3Н, т. J 7 Гц), 1,0-2,1 (4Н, м), 1,23 (9Н, с), 2,79 (2Н, т. J 7 Гц), 5,38 (2Н, с), 7,1-8,0 (11Н, м), 7,3-7,7 (2Н, м).
12b) 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-5-циано-4-(a-гидроксибензил) имидазол.
50,5 мг борогидрида натрия добавляли к раствору 691 мг 4-бензиол-1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-5-цианоимидазола, полученного в соответствии с описанием в стадии (а), в 10 мл этанола и полученную смесь размешивали 1 ч при комнатной температуре. Затем реакционную смесь нейтрализовали водной соляной кислотой, после чего ее смешивали с этилацетатом и с насыщенным водным раствором хлорида натрия. Этилацетатный слой отделяли, осушали безводным сульфатом магния и концентрировали путем выпаривания при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью гексана и этилацетата (1:1, по объему) и получали 589 мг целевого соединения в виде бесцветного аморфного твердого вещества.
ЯМР (CDCl3) δ ppm: 0,89 (3Н, т. J 7 Гц), 1,0-2,0 (4Н, м), 2,68 (2Н, т, J 7 Гц), 5,18 (2Н, с), 5,89 (1Н, с), 7,0-8,0 (13Н, м).
12с) 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил]-5-циано-4-(a-гидроксибензил)имидазол.
Раствор 589 мг 1-[(2'-т-бутоксикарбонилбифенил)-4-ил)метил]-2-бутил-5-циано-4-(α-гидроксибензил)имидазола, полученного в соответствии с описанием в стадии (b), в 20 мл 4 н. раствора хлорводорода в диоксане оставляли на ночь при комнатной температуре, а затем концентрировали путем выпаривания при пониженном давлении. Остаток перетирали в гексане и собирали путем фильтрации, в результате чего получали 493 мг гидрохлорида целевого соединения в виде бесцветного порошка, т. пл. 95-97oC (с размягчением).
ЯМР (ДМСО-d6) δ ppm: 0,88 (3Н, т. J 7 Гц), 1,0-2,0 (4Н, м), 3,00- (2Н, т. J 7 Гц), 5,47 (2Н, с), 6,09 (1Н, с), 7,0-8,0 (13Н, м).
12d) 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил] -4-(альфа-гидроксибензил)имидазол-5-карбоновая кислота.
Смесь 450 мг 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил] -5-циано-4-(a-гидроксибензил)имидазола, полученного в соответствии с описанием в стадии (с), и 20 мл 1 н. водного раствора гидроокиси натрия размешивали в масляной бане, поддерживаемой при 100oC, в течение 7 ч. По истечение этого времени реакционную смесь охлаждали, а ее pH доводили до значения 3-4 путем добавления соляной кислоты. Полученный бесцветный осадок собирали фильтрацией, промывали водой и осушали, в результате чего получали 331 мг целевого соединения в виде бесцветного порошка, т. пл. 192-194oC.
ЯМР (ДМСО-d6) δ ppm: 0,80 (3Н, т. J 7 Гц), 1,0-2,0 (4Н, м), 2,69 (2Н, т. J 7 Гц), 5,69 (2Н, с), 6,32 (1Н, с), 6,9-7,9 (13Н, м).
Пример 13. Этил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-2-бутил-4-(1-гидрокси-1-метилэтил)имидазол-5-карбоксилат (соединение 1-118).
Повторяли процедуру, описанную в примере 1а, но с использованием 0,92 г этил 2-бутил-4-(1-гидрокси-1-метил-этил) имидазол-5-карбоксилата, полученного в соответствии с описанием в получении 8, и 1,28 г т-бутил-4'-бромометилбифенил-2-карбоксилата и получали в результате 1,23 г целевого соединения в виде кристаллов, т. пл. 92-93oC.
ЯМР (CDCl3) d ppm: 0,90 (3Н, т. J 7 Гц), 1,12 (3Н, т, J 7 Гц), 1,26 (9Н, с), 1,2-2,05 (4Н, м), 1,65 (4Н, с), 2,69 (2Н, т. J 7 Гц), 4,24 (2Н, кв. J 7 Гц), 5,52 (2Н, с), 5,73 (1Н, с), 6,88-7,9 (8Н, м).
Пример 14. Этил-2-бутил-1-[(2'-карбоксибифенил)-4-ил)метил]-4-(1-гидрокси-1-метилэтил)имидазол-5-карбоксилат (соединение 1-32).
Повторяли процедуру, описанную В примере 7, но с использованием 0,50 г этил-1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-4-(1-гидрокси-1-метилэтил) имидазол-5-карбоксилата, полученного в соответствии с описанием в примере 13, и 4 н. раствор хлорводорода в диоксане, в результате чего получали 0,45 г гидрохлорида целевого соединения в виде аморфного порошка, т. пл. выше 80oC (с размягчением).
ЯМР (ДМСО-d6) d ppm: 0,82 (3Н, т. J 7 Гц), 1,14 (3Н, т. J 7 Гц), 1,2-1,35 (2Н, м), 1,41-1,55 (2Н, м), 1,60 (6Н, с), 3,00 (2Н, т. J 7 Гц), 4,21 (2Н, кв. J 7 Гц), 5,63 (2Н, с), 7,14-7,75 (8Н, м).
Пример 15. Этил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилат (соединение 1-119).
Повторяли процедуру, описанную в примере 1а), но с использованием 0,845 г этил 4-(1-гидрокси-1-метилэтил-2-пропилимидазол-5-карбоксилата, полученного в соответствии с описанием в получении 9, и 1,22 г т-бутил-4-бромометилбифенил-2-карбоксилата и получали 1,31 г целевого соединения в виде смолистого вещества. Затем это соединение выдерживали при комнатной температуре в целях вызвать его кристаллизацию. Затем соединение перекристаллизовывали из смеси диизопропилового эфира и гексана, в результате чего получали чистое целевое соединение, т. пл. 90-91oC.
ЯМР (CDCl3) d ppm: 0,97 (3Н, т. J н Гц), 1,23 (3Н, т. J 7 Гц), 1,25 (4Н, с), 1,60 (6Н, с), 1,82 (2Н, секстет, J 7 Гц), 2,67 (2Н, т. J 7 Гц), 4,24 (2Н, кв. J 7 Гц), 5,51 5,72 (1Н, с), 6,87-7,85 (8Н, м).
Пример 16. Этил 1-[(2'-карбоксибифенил-4-ил)метил]-4-(1-гидрокси-1-метил-этил)-2-пропилимидазол-5-карбоксилат (соединение 1-50).
Повторяли процедуру, описанную в примере 7, но с использованием 0,80 г этил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилата, полученного в соответствии с описанием в примере 15, и 4 н. раствор хлорводорода в диоксане, в результате чего получали 0,67 г гидрохлорид целевого соединения в виде аморфного порошка.
ЯМР (ДМСО-d6) d ppm: 0,88 (3Н, т. J 7 Гц), 1,14 (3Н, т. J 7 Гц), 1,50-1,65 (2Н, м), 1,60 (6Н, с), 3,00 (2Н, т. J 7 Гц), 4,20 (2Н, кв. J 7 Гц), 5,63 (2Н, с), 7,13-7,75 (8Н, м).
Пример 17. 1-[(2'-карбоксибифенил-4-ил)метил]-4-(1-игдрокси-1-метилэтил)-2-пропилимидазол-5-карбоновая кислота (соединение 1-49).
Раствор 0,20 г этил 1-[(2'-карбоксибифенил-4-ил)метил]-4-(1-гидрокси-1-метилэтил)-2-пропилимидазол -5-карбоксилата гидрохлорида, полученного в соответствии с описанием в примере 16, в водном растворе 84 мг гидроксида-моногидрата лития в 5 мл воды размешивали 6 ч при комнатной температуре. По истечение этого времени к реакционной смеси добавляли по каплям 2 мл 1 н. водной соляной кислоты и полученный осадок собирали фильтрацией, в результате чего получали 0,17 г целевого соединения, т. пл. 176-179oC (с разложением).
ЯМР (ДМСО-d6) d ppm: 0,88 (3Н, т. J 7 Гц), 1,5-1,65 (2Н, м), 1,56 (6Н, с), 2,66 (2Н, т. J 7 Гц), 5,69 (2Н, с), 7,03-7,72 (8Н, м).
Этил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{ 4-[2(тетразол -5-ил)фенил] фенил}метилимидазол-5-карбоксидлат (соединение 2-7).
18а) Этил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{ 4-[2-(тритилтетразол -5-ил)фенил]фенил} метилимидазол-5-карбоксилат.
48 мг гидрида натрия (в виде 55 мас. дисперсии в минеральном масле) добавляли к раствору 0,26 г этил 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилата, полученного в соответствии с описанием в получении 9, в 5 мл N, N-диметилформамида и полученную смесь размешивали 30 мин при комнатной температуре. Затем добавляли раствор 0,72 г 4-[2-(тритилтетразол-5-ил)фенил] бензилбромида в 5 мл N,N-диметилформамида и реакционную смесь размешивали при комнатной температуре в течение 2 ч и при 60oC в течение 4 ч. По истечение этого времени смесь растворяли в этилацетате и раствор промывали три раза водой. После этого раствор осушали безводным сульфатом натрия, а затем растворитель отгоняли. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью гексана и этилацетата (1:1 по объему), в результате чего получали 0,62 г целевого соединения в виде аморфного твердого вещества. Это вещество кристаллизовали из диизопропилового эфира и получали в результате целевое соединение в виде кристаллов, т. пл. 167-168oC (с разложением).
ЯМР (CDCl3) d ppm: 0,88 (3Н, т. J 7 Гц), 1,08 (3Н, т, J 7 Гц), 1,5-1,8 (2Н, м), 1,64 (6Н, с), 2,52 (2Н, т. J 8 Гц), 4,12 (2Н, кв. J 7 Гц), 5,38 (2Н, с), 5,78 (1Н, с), 6,7-7,6 (22Н, м), 7,8-8,1 (1Н, м).
18b). Этил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{ 4-[2-(тетразол -5-ил)фенил]фенил} метилимидазол-5-карбоксилат.
Раствор 0,50 г этил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{ 4-[(2-(тритилтетразол -5-ил)фенил]фенил}метилимидазол-5-карбоксилата, полученного в соответствии с описанием в примере 18а, растворенного в 5 мл 4 н. раствора хлорводорода в диоксане, оставляли на ночь при комнатной температуре, после чего реакционную смесь концентрировали путем выпаривания при пониженном давлении. Полученный остаток растирали с диизопропиловым эфиром и получали 0,34 г гидрохлорида целевого соединения, т. пл. 100-103oC.
ЯМР (CDCl3) d ppm: 0,97 (3Н, т. J 7 Гц), 1,24 (3Н, т. J 7 Гц), 1,50-1,65 (2Н, м), 1,70 (6Н, с), 3,00 (2Н, т. J 8 Гц), 4,30 (2Н, кв. J 7 Гц), 5,70 (2Н, с), 6,9-7,8 (8Н, м).
Пример 19. 4-(1-гидрокси-1-метилэтил)-2-1-{4-[2-(тетразол-5-ил)фенил]-фенил}метилимидазол -5-карбоновая кислота (соединение 2-1).
3,65 мл 1 н. водного раствора гидроокиси натрий добавляли к раствору 0,31 г этил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{ 4-[(2-(тетразол -5-ил)фенил] фенил} метилимидазол-5-карбоксилата гидрохлорида, полученного в соответствии с описанием в примере 18b, в 6 мл метанола и полученную смесь оставляли на ночь при комнатной температуре. По истечение этого времени реакционную смесь концентрировали путем выпаривания при пониженном давлении для удаления метанола. Концентрат разбавляли водой и pH доводили до величины 3 путем добавления разбавленной соляной кислоты, после чего смесь экстрагировали этилацетатом. Органический экстракт осушали безводным сульфатом натрия, а затем концентрировали путем выпаривания при пониженном давлении. Полученный остаток растирали с диизопропиловым этилом и получали 0,15 г целевого соединения, т. пл. 166-169oC.
ЯМР (ДМСО-d6) d ppm: 0,85 (3Н, т. J 7,5 Гц), 1,54 (6Н, с), 1,4-1,6 (2Н, м), 2,58 (2Н, т. J 8 Гц), 5,64 (2Н, с), 6,94 (2Н, д. J 8,5 Гц), 7,06 (2Н, д. J 8,5 Гц), 7,5-7,7 (4Н, м).
Пример 20. Пивалоилоксиметил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{4-[2-(тетразол -5-ил)фенил] фенил}метилимидазол-5-карбоксилат (соединение 2-15).
20а). Пивалоилоксиметил 5-(1-гидрокси-1-метилэтил)-2-пропил-1-{ 4-[2-(тритилтетразолил-5-ил)фенил]фенил}метилимидазол-5-карбоксилат.
5,30 мл 1 н. водного раствора гидроксида натрия, а затем 5 мл тетрагидрофурана добавляли к раствору 0,76 г этил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{ 4-[(2-(тритилтетразол -5-ил)-фенил] фенил}метилимидазол-5-карбоксилата, полученного в соответствии с описанием в примере 18а, в 30 мл метанола, после чего полученную смесь размешивали при комнатной температуре 8 ч. Затем реакционную смесь концентрировали путем выпаривания при пониженном давлении с целью удаления метанола и тетрагидрофурана. К концентрату добавляли воду и pH смеси доводили до величины 4 путем добавления разбавленной соляной кислоты, охлажденной льдом. Затем смесь экстрагировали этилацетатом. Экстракт осушали безводным сульфатом магния и концентрировали путем выпаривания досуха. Остаток растворяли в 10 мл диметилацетамида и к полученному раствору добавляли 0,23 г карбоната калия и 0,13 мл пивалоилоксиметилхлорида. Затем смесь размешивали 4 ч при 50oC, после чего к ней добавляли 0,06 мл пивалоилоксиметилхлорида и полученную смесь размешивали еще 2 ч. После этого реакционную смесь разбавляли этилацетатом и три раза промывали водой. Органический слой отделяли, осушали безводным сульфатом натрия и концентрировали путем выпаривания при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью гексана и этилацетата (1:1 по объему), и получали 0,23 г целевого соединения в виде аморфного твердого продукта.
ЯМР (CDCl3) d ppm: 0,86 (3Н, т. J 7 Гц), 1,12 (9Н с), 1,62 (6Н, с), 1,4-169 (2Н, м), 2,51 (2Н, т, J 7 Гц), 5,37 (1Н, шир.с), 5,40 (2Н, с), 5,72 (2Н, с), 6,6-8,1 (23Н, м).
20b). Пивалоилоксиметил 4-(1-гидрокси-1-метилэтил)-2-пропил 1-{4-[2-(тетразол-5-ил)фенил] фенил}метилимидазол-5-карбоксилат.
5 мл 4 н. раствора хлорводорода в диоксане добавляли к 0,20 г пивалоилоксиметил-4-(1-гидрокси-1-метилэтил)-2-пропил-1-{ 4-[2-(тритилтетразол-5-ил)фенил] фенил} метилимидазол-5-карбоксилата, полученного в соответствии с описанием в примере 20а, и полученную смесь оставляли на ночь при комнатной температуре. По истечение этого времени реакционную смесь концентрировали досуха при пониженном давлении. Полученный остаток растирали с диизопропиловым эфиром для стимуляции кристаллизации и получали 0,13 г гидрохлорида целевого соединения в виде кристаллов, т. пл. 104-107oC.
ЯМР (ДМСО-d6) d ppm: 0,84 (3Н, т. J 7,5 Гц), 1,09 9Н, с), 1,35-1,50 (2Н, м), 1,56 (6Н, с), 2,88 (2Н, т J 8 Гц), 5,58 (2Н, с), 5,85 (2Н, с), 7,05 (2Н, д. J 8,5 Гц), 7,10 (2Н, д. J 8,5 Гц), 7,5-7,7 (4Н, м).
Пример 21. 2-бутил-4-(1-этил-1-гидроксипропил)-1-{ 4-[2-(тетразол-5-ил)фенил] фенил}метилимидазол-5-карбоновая кислота (соединение 2-40).
21а). Этил 2-бутил-4-(1-этил-1-гидроксипропил-)-1-{4-[2-(тритилтетразол -5-ил)фенил]фенил} метилимидазол-5-карбоксилат.
Повторяли процедуру, описанную в примере 18а, но с использованием 0,75 г этил 2-бутил-4-(1-этил-1-гидроксипропил)имидазол-5-карбоксилата, полученного в соответствии с описанием в получении 13, 0,12 г гидрида натрия (в виде 55 мас. дисперсии в минеральном масле) и 1,51 г 4-[2-(тритилтетразол-5-ил)фенил] бензилбромида и получали в результате 1,05 г целевого соединения в виде аморфного твердого продукта.
ЯМР (CDCl3) d ppm: 0,83 (6Н, триплет, J 7,5 Гц), 0,85 (3Н, триплет, J 6 Гц), 1,11 (3Н, т. J 7 Гц), 1,23-1,32 (2Н, м), 1,56-1,65 (2Н, м), 1,80-1,89 (2Н, м), 2,03-2,14 (2Н, м), 2,55 (2Н, т. J 8 Гц), 4,12 (2Н, кв. J 7,5 Гц), 5,37 (2Н, с), 5,64 (1Н, шир. с), 6,70 (2Н, д. J 8,5 Гц), 6,9-7,0 (6Н, м), 7,10 (2Н, д. J 8,5 Гц), 7,2-7,4 (10Н, м), 7,4-7,5 (2Н, м), 7,85-7,90 (1Н, м).
21b) 2-бутил-4-(1-этил-1-гидроксипропил)-1-{4-[(2-(тетразол-5-ил)фенил] фенил}метилимидазол-5-карбоновая кислота.
1,71 мл 1 н. водный соляной кислоты добавляли к раствору 0,65 г этил 2-бутил-4-(1-этил-1-гидроксипропил)-1-{ 4-[2-тритилтетразол-5-ил) фенил]фенил} метилимидазол-5-карбоксилата, полученного в соответствии с описанием в стадии (а), в 10 мл в метаноле и полученную смесь оставляли на ночь при комнатной температуре. После этого растворитель удаляли путем дистилляции при пониженном давлении, а концентрат снова растворяли в 10 мл метанола. Полученный раствор смешивали с 4,28 мл 1 н. водного раствора гидроксида натрия и оставляли на ночь при комнатной температуре. Затем реакционную смесь концентрировали путем выпаривания при пониженном давлении для удаления метанола, pH концентрата доводили до величины 3 путем добавления разбавленной водной соляной кислоты и осажденные кристаллы собирали путем фильтрации. Полученные таким образом кристаллы суспендировали в диизопропиловом эфире, а затем снова собирали путем фильтрации, в результате чего получали 0,35 г целевого соединения, т. пл. 181-183oC.
ЯМР (ДМСО-d6) d ppm: 0,74 (6Н, т. J 7,5 Гц), 0,79 (3Н, т. J 7,5 Гц), 1,1-1,3 (2Н, м), 1,40-1,55 (2Н, м), 1,67-1,80 (2Н, м), 1,90-2,05 (2Н, м), 2,59 (2Н, т. J 7,5) 5,67 (2Н, с), 6,88 (2Н, д. J 8,5 Гц), 7,5 (2Н, д. J 8,5 Гц), 7,5-7,7 (4Н, м).
Пример 22. 2-бутил-4-(1-гидрокси-1-метилэтил)-1-{ 4-[2-(тетразол -5-ил)фенил]фенил}метилимидазол-5-карбоновая кислота (соединение 2-2).
22а. Этил 2-бутил-4-(1-гидрокси-1-метилэтил)-1-{4-[2-(тритилтетразол-5-ил) фенил]фенил} метилимидазол-5-карбоксилат.
Повторяли процедуру, описанную в примере 18а, но с использованием 0,26 г этил-2-бутил-4-(1-гидрокси-1-метилэтил)имидазол-5-карбоксилата, полученного в соответствии с описанием в получении 8, 45,5 мг гидрида натрия (55 мас. дисперсия в минеральном масле) и 0,63 г 4-[2-(тритилтетразол-5-ил) фенил] бензилбромида, и получали 0,28 г целевого соединения в виде маслянистого продукта.
ЯМР (CDCl3) d ppm: 0,85 (3Н, т. J 7 Гц), 1,09 (3Н, т. J 7 Гц), 1,09 (3Н, т, J 7 Гц), 1,64 (6Н, с), 1,3-1,8 (4Н, м), 2,56 (2Н, т. J 8 Гц), 4,14 (2Н, кв. J 7 Гц), 5,38 (2Н, с), 5,78 (1Н, с), 6,6-7,6 (22Н, м), 7,7-8,1 (1Н, м).
22b). 2-бутил-4-(1-гидрокси-1-метилэтил)-1-{4-[2-(тетразол -5-ил)фенил] фенил}метилимидазол-5-карбоновая кислота.
Повторяли процедуру, описанную в примере 21 b, и путем обработки 0,28 г этил 2-бутил-4-(1-гидрокси-1-метилэтил)-1-{4-[2-(тритилтетразол -5-ил)фенил] фенил}метилимидазол-5-карбоксилата, полученного в соответствии с описанием в стадии (а), 0,42 мл 1 н. водной соляной кислоты, с последующей обработкой продукта 1,70 мл 1 н. водного раствора гидроксида натрия получали 78 г целевого соединения, т. пл. 138-141oC.
ЯМР (ДМСО-d6) d ppm: 0,81 (3Н, т. J 7,5 Гц), 1,15-1,35 (2Н, м), 1,4-1,6 (2Н, м), 1,53 (6Н, с), 2,58 (2Н, т. J 8,5 Гц), 5,64 (2Н, с), 6,94 (2Н, д. J 8,5 Гц), 7,06 (2Н, д, J 8,5 Гц), 7,15-7,70 (4Н, м).
Пример 23. 2-бутил-4-(1-гидрокси-1-метилпропил)-1-{4-[2-(тетразол -5-ил)фенил]фенил} метилимидазол-5-карбоновая кислота (соединение 2-38).
23а). 2-бутил-5-циано-4-(1-гидрокси-1-метилпропил)-1-{4-[2-тритилтетразол -5-ил)фенил]фенил}метилимидазол
Повторяли процедуру, описанную в примере 18а, но с использованием 465 мг 2-бутил-5-циано-4-(1-гидрокси-1-метилпропил)имидазола, полученного в соответствии с описанием в получении 19, 92 мг гидрида натрия (в виде 55 мас. дисперсии в минеральном масле), и 1,11 г 4-[2-(тритилтетразол-5-ил)фенил] бензилбромида и получали в результате 1,00 г целевого соединения в виде смолистого вещества.
ЯМР (CDCl3) d ppm: 0,86 (3Н, т. J 7,5 Гц), 0,87 (3Н, т. J 7 Гц), 1,21-1,34 (2Н, м), 1,54-1,66 (2Н, м), 1,60 (3Н, с), 1,82-1,97 (2Н, м), 2,51 (2Н, т. J 7,5 Гц), 3,22 (1Н, с), 5,04 (2Н, с), 6,87-7,52 (22 Н, м), 7,93-7,96 (1Н, м).
23b) 2-бутил-5-циано-4-(1-гидрокси-1-метилпропил)-1-{4-[2-(тетразол-5-ил)фенил] фенил}метилимидазол.
Смесь 1,00 г 2-бутил-5-циано-4-(1-гидрокси-1-метилпропил)-1-{ 4-[2-(тритилтетразол-5-ил)фенил] фенил}метилимидазола, полученного в соответствии с описанием, приведенным в стадии (а), и 25 мл 20% об./об. водной уксусной кислоты размешивали 2 ч при 60oC, после чего растворитель отгоняли при пониженном давлении. Остаточную воду и уксусную кислоту удаляли как толуоловый азеотроп путем дистилляции при пониженном давлении, а полученный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью метанола и метиленхлорида (1:9 и 1:4, по объему), в результате чего получали 0,65 г целевого соединения в виде стеклообразного вещества.
ЯМР (CDCl3) d ppm: 0,83 (3Н, т. J 7 Гц), 0,88 (3Н, т. J 7 Гц), 1,23-1,37 (2Н, м), 1,57 (3Н, с), 1,55-1,70 (2Н, м), 1,82-1,89 (2Н, м), 2,64 (2Н, т. J 7 Гц), 5,12 (2Н, с), 6,9-7,1 (4Н, м), 7,29-7,60 (3Н, м), 7,87 (1Н, д. J 7,5 Гц).
23с) 2-бутил-4-(1-гидрокси-1-метилпропил)-1-{4-[2-(тетразол -5-ил)фенил] фенил}метилимидазол-5-карбоновая кислота.
Смесь 360 мг 2-бутил-5-циано-4-(1-гидрокси-1-метилпропил)-1-{ 4-[2-(тетразол-5-ил)фенил] фенил} метилимидазола, полученного в соответствии с описанием в стадии (b), 266 мг гидроксида-моногидрата лития и 3,6 мл воды размешивали в масляной бане, поддерживаемой при 115oC, в течение 16 ч. По истечение этого времени реакционную смесь охлаждали и к смеси добавляли 6,4 мл 1 н. водной соляной кислоты, охлаждая при этом льдом. Осажденные кристаллы собирали фильтрацией и получали 302 мг целевого соединения, т. пл. 152-154oC.
ЯМР (ДМСО-d6) d ppm: 0,79 (3Н, т. J 7 Гц), 0,82 (3Н,т. J 7 Гц), 1,20-1,34 (2Н, м), 1,44-1,55 (2Н, м), 1,55 (3Н, с), 1,71-1,95 (2Н, м), 2,62 (2Н, т. J 7,5 Гц), 5,68 (2Н, АВ-квартет, d 0,10 ppm, J 17 Гц), 6,86-7,10 (4Н, м), 7,53-7,72 (4Н, м).
Пример 24. 4-(1-гидрокси-1-метилпропил)-2-пропил-1-{4-[2-(тетразол -5-ил)фенил]фенил} метилимидазол-5-карбоноваоя кислота (соединение 2-37).
24а) 5-циано-4-(1-гидрокси-1-метилпропил)-2-пропил-1-{4-[2-тритилтетразол -5-ил)фенил]фенил} метилимидазол.
Повторяли процедуру, описанную в примере 18а, но с использованием 380 мг 5-циано-4-(1-гидрокси-1-метилпропил)-2-пропилимидазола, полученного в соответствии с описанием в получении 20, 88 мг гидрида натрия (в виде 55 мас. дисперсии в минеральном масле) и 1,07 г 4-[2-(тритилтетразол-5-ил)фенил] бензилбромида, в результате чего получали 0,97 г целевого соединения в виде аморфного твердого вещества.
ЯМР (CDCl3) d ppm: 0,86 (3Н, т. J 8 Гц), 0,87 (3Н, т. J 7,5 Гц), 1,60 (3Н, с), 1,60-1,75 (2Н, м), 1,80-2,00 (2Н, м), 2,48 (2Н, т. J 8 Гц), 5,04 (2Н, с), 6,88 (2Н, д. J 8,5 Гц), 6,9-7,0 (4Н, м), 7,14 (2Н, д J 8,5 Гц), 7,2-7,4 (14Н, м), 7,45-7,55 (1Н, м).
24b) 5-циано-4-(1-гидрокси-1-метилпропил)-2-пропил-1-{4-[2-(тетразол -5-ил)фенил]фенил} метилимидазол.
Повторяли процедуру, описанную в примере 23b, и путем обработки 0,51 г 5-циано-4-(1-гидрокси-1-метилпропил)-2-пропил-19{ 4-[2-(тритилтетразол -5-ил)фенил]фенил}метилимидазола, полученного в соответствии с описанием в стадии (а), 75 об./об. водной уксусной кислотой получали 0,32 г целевого соединения в виде кристаллов, т. пл. 141-145oC.
ЯМР (CDCl3) d ppm: 0,84 (3Н, т. J 8 Гц), 0,90 (3Н, т. J 8,5 Гц), 1,52 (3Н, с), 1,5-1,7 (2Н, м), 1,75-1,90 (2Н, м), 2,65 (2Н, т. J 8 Гц), 5,27 (2Н, с), 7,03 (2Н, д. J 8,5 Гц), 7,14 (2Н, д. J 8,5 Гц), 7,45-7,63 (4Н, м).
24с) 4-(1-гидрокси-1-метилпропил)-2-пропил-1-{ 4-[2-(тетразол -5-ил)фенил]фенил}метилимидазол-5-карбоновая кислота.
Повторяли процедуру, описанную в примере 23с, и путем обработки 0,19 г 5-циано-4-(1-гидрокси-1-метилпропил)-2-пропил-1-{4-[2-(тетразол -5-ил)фенил] фенил} метилимидазола, полученного в соответствии с описанием в стадии (b), 0,15 г гидроксида-моногидрата лития получали 0,14 г целевого соединения в виде порошка, т. пл. 174-177oC.
ЯМР (CDCl3) Dd ppm: 0,88 (3Н, т. J 7,5 Гц), 0,94 (3Н, т. J 7,5 Гц), 1,50-1,65 (2Н, м), 1,63 (3Н, с), 1,85-2,05 (2Н, м), 2,76 (2Н, т. J 7,5 Гц), 5,80 (2Н, АВ-кварт. d 0,14 ppm, J 16,5 Гц), 7,01 (2Н, д. J 8,5 Гц), 7,11 (2Н, д. J 8,5 Гц), 7,48-7,75 (4Н, м).
Пример 25. Пивалоилоксиметил 1-[(2'-карбоксибифенил-4-ил)метил]-4-(1-гидрокси-1-метилэтил)-2-пропилимидазол -5-карбоксилат (соединение 3-1).
25а) Этил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-4-(1-гидрокси -1-метилэтил)-2-пропилимидазол-5-карбоксилат.
3,00 г т-бутоксида калия добавляли, охлаждая при этом льдом, к раствору 6 г этил 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилата, полученного в соответствии с описанием в получении 9, в 40 мл N,N-диметиалцетамида и полученную смесь размешивали 10 мин, после чего добавляли раствор 9.00 г т-бутил-4'-бромометилбифенил-2-карбоксилата в 40 мл N,N-диметилацетамида. Затем реакционную смесь размешивали 1 ч при комнатной температуре и 2 ч при 50oC, после чего смесь смешивали с водой и экстрагировали этилацетатом. Экстракт осушали безводным сульфатом магния, а растворитель отгоняли при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью гексана и этилацетата (1:1, по объему), и получали 11,6 г целевого соединения в виде твердого вещества с температурой размягчения 85oC.
ЯМР (CDCl3) d ppm: 0,97 (3Н, т. J 7 Гц), 1,23 (3Н, т. J 7 Гц), 1,25 (9Н, с), 1,60 (6Н, с), 1,82 (2Н, секстет, J 7 Гц), 2,67 (2Н, т. J 7 Гц), 4,24 (2Н, кв. J 7 Гц), 5,51 (2Н, с), 5,72 (1Н, с), 6,87-7,85 (8Н, м).
25b) 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-4-(1-гидрокси -1-метилэтил)-2-пропилимидазол-5-карбоновая кислота.
Раствор 4,8 г гидроксид-моногидрата лития в 100 мл воды добавляли к раствору 11,6 г этил-1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-4-(1-гидрокси -1-метилэтил)-2-пропилимидазол-5-карбоксилата, полученного в соответствии с описанием в стадии (а), в 60 мл диоксана и полученную смесь размешивали 16 ч при комнатной температуре. Диоксан отгоняли при пониженном давлении, а концентрат смешивали с ледяной водой и этилацетатом, после чего добавляли 114 мл 1 н. водной соляной кислоты. Слой этилацетата отделяли, осушали безводным сульфатом магния, а растворитель отгоняли путем дистилляции при пониженном давлении. Кристаллический остаток растирали в диизопропиловом эфире и собирали путем фильтрации, в результате чего получали 9,09 г целевого соединения, т. пл. 155-157oC.
ЯМР (CDCl3) d ppm: 0,85 (3Н, т. J 7,5 Гц), 1,23 (4Н, с), 1,53-1,65 (2Н, м), 1,65 (6Н, с), 2,91 (3Н, т. J 7,5 Гц), 5,90 (2Н, с), 7,09 (2Н, д. J 8 Гц), 7,21-7,48 (5Н, м), 7,75 (1Н, д. J 9 Гц).
25с) Пивалоилоксиметил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-4-(1-гидрокси -1-метилэтил)-2-пропил-имидазол-5-карбоксилат.
2,13 мл хлорметилпивалата и 3,99 г карбоната калия добавляли к раствору 6 г 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -4-(1-гидрокси -1-метилэтил)-2-пропилимидазол-5-карбоновой кислоты, полученной в соответствии с описанием в стадии (b), в 70 мл N,N-диметилацетамида и полученную смесь размешивали при комнатной температуре в течение 1 ч, и при 50oC в течение 2 ч. По истечение этого времени, реакционную смесь смешивали с этилацетатом и водой. Слой этилацетата отделяли и осушали безводным сульфатом магния, после чего растворитель удаляли путем перегонки при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле элюируя смесью этилацетата и гексана (1:1, по объему) и получали 6,80 г целевого соединения в виде кристаллов, т. пл. 106-107oC.
ЯМР (CDCl3) d ppm: 1,07 (3Н, т. J 7 Гц), 1,25 (9Н, с), 1,32 (9Н, с), 1,71 (6Н, с), 1,79-1,90 (2Н, м), 2,75 (2Н, т. J 8 Гц), 5,50 (1Н, с), 5,59 (2Н, с), 5,92 (2Н, с), 7,05 (2Н, д. J 8 Гц), 7,34-7,56 (4Н, м), 7,85 (1Н, д. J 7 Гц).
25d) Пивалоилоксиметил 1-[(2'-карбоксибифенил-4-ил)метил]-4-(1-гидрокси -1-метилэтил)-2-пропилимидазол-5-карбоксилат.
Смесь 6,6 г пивалоилоксиметил 1-[(2'-бутоксикарбонилбифенил-4-ил)метил] -4-(1-гидрокси -1-метилэтил-2-пропилимидазол-5-карбоксилата, полученного в соответствии с описанием в стадии (с), и 57 мл 4 н. раствора хлорводорода в диоксане размешивали 4 ч при комнатной температуре. После этого реакционную смесь концентрировали путем выпаривания при пониженном давлении и остаток растирали с этилацетатом в целях его кристаллизации, в результате чего получали 6,52 г целевого соединения в виде гидрохлорида, т. пл. 170-173oC.
ЯМР (ДМСО-d6) d ppm: 0,87 (3Н, т. J 7 Гц), 1,10 (9Н, с), 1,45-1,60 (2Н, м), 1,58 (6Н, с), 2,96 (2Н, т. J 7,5 Гц), 5,65 (2Н, с), 5,87 (2Н, с), 7,17 (2Н, д. J 8 Гц), 7,33 (2Н, д. J 8 Гц), 7,43-7,60 (3Н, м), 7,74 (1Н, д. J 8 Гц),
Пример 26. Изопропоксикарбонилоксиметил 1-[(2'-карбоксибифенил-4-ил)метил] -4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилат (соединение 3-13).
26а) Изопропоксикарбонилоксиметил 1-[(2'-т-бутоксикарбонил-бифенил-4-ил)метил]-4-(1-гидрокси-1-метилэтил)-2-пропил-имидазол-5-карбоксилат.
Повторяли процедуру, описанную в примере 25 (с), и целевое соединение получали в виде кристаллов (0,58 г, т. пл. 85-87oC) путем перемешивания смеси, содержащей 0,50 г 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоновой кислоты, полученной в соответствии с описанием в примере 25 (b), 0,19 г изопропоксикарбонилоксиметилхлорида и 0,33 г карбоната калия в 6 мл N,N-диметилацетамида, в течение 3 ч при комнатной температуре.
ЯМР (CDCl3) d ppm: 0,99 (3Н, т. J 7 Гц), 1,23 (9Н, с), 1,29 (6Н, д J 6 Гц), 1,63 (6Н, с), 1,70-1,85 (2Н, м), 2,68 (2Н, т. J 8 Гц), 4,89 (1Н, квинтет, J 6 Гц), 5,38 (1Н, с), 5,61 (2Н, с), 5,82 (2Н, с), 6,97 (2Н, д. J 8 Гц), 7,26-7,48 (5Н, м), 7,77 (1Н, д. J 8 Гц).
26b) Изопропоксикарбонилоксиметил 1-[(2'-карбоксибифенил-4-ил)метил]-4-(1-гидрокси-1-метилэтил)-2-пропилимидазол -5-карбоксилат.
Повторяли процедуру, описанную в примере 25b, и путем обработки 0,46 г изопропоксикарбонилоксиметил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилата, полученного в соответствии с описанием в стадии (а), 4 н. раствором хлорводорода в диоксане получали 0,36 г гидрохлорида целевого соединения в виде аморфного порошка, т. пл. 153-155oC.
ЯМР (CDCl3) d ppm: 0,98 (3Н, т. J 7 Гц), 1,29 (6Н, д. J 6 Гц), 1,50-1,65 (2Н, м), 1,75 (6Н, с), 3,13 (2Н, т. J 7 Гц), 4,90 (1Н, квинтет J 6 Гц), 5,55 (2Н, с), 5,82 (2Н, с), 7,02 (2Н, д. J 6,5 Гц), 7,21-7,57 (5Н, м), 7,96 (1Н, д. J 8 Гц).
Пример 27. Этоксикарбонилоксиметил 1-[(2'-карбоксибифенил-4-ил)метил]-4-(1-гидрокси-1-метилэтил)-2-пропилимидазол -5-карбоксилат (соединение 3-9).
27а) Этоксикарбонилоксиметил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -4-(1-гидрокси-1-метилэтил) -2-пропилимидазол-5-карбоксилат.
Следуя процедуре, описанной в примере 25с, получали 0,69 г целевого соединения в виде маслянистого вещества, исходя из 0,55 г 1-[(2'-т-бутоксикарбонилбефенил-4-ил)метил] -4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоновой кислоты, полученной в соответствии с описанием в примере 25b, 0,30 г этоксикарбонилоксиметилхлорида и 0,50 г карбоната калия.
ЯМР (CDCl3) d ppm: 0,99 (3Н, т. J 7 Гц), 1,23 (9Н, с), 1,29 (3Н, т. J 7 Гц), 1,64 (6Н, с), 1,74-1,85 (2Н, м), 2,69 (2Н, т. J 7,5 Гц), 4,21 (2Н, кв. J 7 Гц), 5,39 (1Н, с), 5,52 (2Н, с), 5,83 (2Н, с), 6,97 (2Н, g. J 8 Гц), 7,25-7,51 (5Н, м), 7,77 (1Н, д. J 6,5).
27b) Этоксикарбонилоксиметил 1-[(2'-карбоксифенил-4-ил)метил]-4-(1-гидрокси-1-метилэтил)-2-пропил-имидазол -5-карбоксилат.
Следуя процедуре, описанной в примере 25d, получали 0,48 г гидрохлорида целевого соединения в виде аморфного порошка (температура размягчения > 70oC) путем обработки 0,69 г этоксикарбонилоксиметил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -4-(1-гидрокси-1-метилэтил) -2-пропилимидазол-5-карбоксилата, полученного в соответствии с описанием в стадии (а), 4 н. раствором хлорводорода в диоксане.
ЯМР (ДМСО-d6) d ppm: 0,88 (3Н, т. J 7 Гц), 1,19 (3Н, т. J 7 Гц), 1,5-1,65 (2Н, м), 1,59 (6Н, с), 2,96 (2Н, т. J 7,5 Гц), 4,15 (2Н, кв. J 7 Гц), 5,64 (2Н, с), 5,84 (2Н, с), 7,18 (2Н, д. J 8 Гц), 7,32-7,61 (5Н, м), 7,74 (1Н, д. J 7 Гц).
Пример 28. 1-(изопропоксикарбонилокси) этил 1-[(2'-карбоксибифенил-4-ил)-метил] -4-(1-гидрокси-1-метилэтил) -2-пропилимидазол-5-карбо (соединение 3-14).
28а) 1-(изопропоксикарбонилокси) этил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-4-(1-гидрокси-1-метилэтил) -2-пропилимидазол-5-карбоксилат.
Следуя процедуре, описанной в примере 25с, получали 0,60 г целевого соединения в виде смолистого вещества путем размешивания в течение 16 ч при 60oC, 0,50 г 1-[(2'-т-бутоксикарбонилобифенил-4-ил)метил]-4-(1-гидрокси-1-метил-этил) -2-пропилимидазол-5-карбонвоой кислоты, полученной в соответствии с описанием в примере 25b, и 0,12 г 1-(изопропоксикарбонилокси) этилхлорида раствором 0,40 г карбоната калия в 6 мл N,N-диметилацетамида.
ЯМР (CDCl3) d ppm: 0,97 (3Н, т. J 7,5 Гц), 1,26 (4Н, с), 1,27 (6Н, дд дублет дублетов J 4,5 d 6 Гц), 1,42 (3Н, д. J 5,5 Гц), 1,64 (6Н, д. J 3 Гц), 1,75-1,80 (2Н, м), 2,65 (2Н, д. J 7,5 Гц), 4,86 (1Н, квин. J 6 Гц), 5,50 (2Н, с), 6,90 (1Н, кв. J 5,5 Гц), 6,97 (2Н, д. J 8,5 Гц), 7,26-7,50 (5Н, м), 7,78 (1Н, д J 8 Гц),
28b) 1-(изопропоксикарбонилокси) этил 1-[(2'-карбоксибифенил-4-ил)метил] -4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилат.
Следуя процедуре, описанной в примере 25d, получали 0,41 г гидрохлорида целевого соединения (в виде аморфного порошка с т. пл. 94-96oC) путем обработки 0,60 г 1-(изопропоксикарбонилокси) этил 1-[(2'-т-бутоксикарбонилбифенил)-2-ил метил]-4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилата, полученного в соответствии с описанием в стадии (а), 4 н. раствором хлорводорода в диоксане.
ЯМР (CDCl3) d ppm: 0,94 (3Н, т. J 7 Гц), 1,27 (6Н, дд. J 6,5&11 Гц), 1,47 (3Н, д. J 5,5 Гц), 1,50-1,65 (2Н, м), 1,76 (6Н, д. J 8,5 Гц), 3,08 (2Н шир. трипл. J 8 Гц), 4,86 (1Н, септет, J 7 Гц), 5,56 (2Н, с), 6,87 (1Н, кв. J 5,5 Гц), 7,04 (2Н, д. J 7,5 Гц), 7,27-7,65 (5Н, м), 7,97 (1Н, д. J 8 Гц).
Пример 29. (5-метил-2-оксо-1,3-диоксолен-4-ил)метил 1-[(2'-карбоксибифенил-4-ил)метил] -4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилат (соединение 3-25).
29а) (5-метил-2-оксо-1,3-диоксолен-4-ил)метил 1-[(2'-т-бутокси-карбонилбифенил-4-ил)метил] -4-(1-гидрокси-1-метилэтил) -2-пропилимидазол-5-карбоксилат.
Следуя процедуре, описанной в примере 25с, получали 0,65 г целевого соединения в виде смолистого вещества, исходя из 0,50 г 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -4-(1-гидрокси-1-метилэтил) -2-пропилмидазол-5-карбоновой кислоты, полученной в соответствии с описанием примера 25b, 0,27 г (5-метил-2-оксо-1,3-диоксолен-4-ил) метилбромида и 0,3 г карбоната калия в 6 мл N,N-диметилацетамида.
ЯМР (CDCl3) d ppm: 0,99 (3Н, т. J 6,5 Гц), 1,28 (9Н, с), 1,64 (6Н, с), 1,55-1,90 (2Н, м), 2,07 (4Н, с), 2,70 (2Н, т. J 7 Гц), 4,90 (2Н, с), 5,47 (2Н, с), 5,51 (1Н, с), 6,91 (2Н, д. J 8,5 Гц), 7,2-7,9 (6Н, м).
29b) (5-метил-2-оксо-1,3-диоксолен-4-ил)метил-1-[(2'-карбоксибифенил-4-ил)метил] -4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилат
Следуя процедуре, описанной в примере 25d, получали 0,54 г гидрохлорида целевого соединения в виде аморфного порошка (т. пл. 90-93oC) путем обработки 0,65 г (5-метил-2-оксо-1,3-диоксолен-4-ил)метил-1-[(2'-т-бутоксикарбонилбифенил -4-ил)метил]-4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилата, полученного в соответствии с описанием в стадии (а), 4 н. раствором хлорводорода в диоксане.
ЯМР (ДМСО-d6) d ppm: 0,88 (3Н, т. J 7,5 Гц), 1,5-167 (2Н, м), 1,59 (6Н, с), 2,11 (3Н, с), 3,00 (2Н, т J 7,5 Гц), 5,13 (2Н, с), 5,63 (2Н, с), 7,13 (2Н, д. J 8 Гц), 7,26-7,75 (6Н, м).
Пример 30. Пивалоилоксиметил 1-[(2'-карбоксибифенил-4-ил)метил]-4-(1-гидрокси-1-метилэтил)-2-пропилимидазол -5-карбоксилат (соединение 3-1).
30а) Пивалоилоксиметил 1-[(2'-т-бутоксикарбонилбефенил-3-ил)метил]-4-(1-гидрокси-1-метилэтил-2-пропилимидазол-5-карбоксилат.
Следуя процедуре, описанной в примере 25а, получали 0,81 г целевого соединения, исходя из 500 мг пивалоилоксиметил 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилата, полученного в соответствии с описанием в получении 22 ii, и 560 мг т-бутил 5'-бромометилбифенил-2-карбоксилата. Этот продукт имеет такую же точку плавления и такой же ЯМР-спектр, как и соединение, полученное в примере 25с.
30b) Пивалоилоксиметил 1-[(2-карбоксибифенил-4-ил)метил] -4-(1-гидрокси-1-метилэтил)-2-пропилимидазол -5-карбоксилат.
Следуя процедуре, описанной в примере 25d, получали 0,45 г гидрохлорида целевого соединения в виде кристаллов исходя из 0,5 г пивалоилоксиметил 1-[(2'-т-бутоксикарбонил-бифенил-4-ил)-метил] -4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилата, полученного в соответствии с описанием в стадии (а). Этот продукт имеет такую же точку плавления и такой же ЯМР-спектр, как и соединение, полученное в примере 25d.
Пример 31. Пивалоилоксиметил 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил] -4-(1-гидрокси-1-метилэтил)имидазол -5-карбоксилат (соединение 3-27).
31a) Метил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)-метил]-2-бутил-4-(1-гидрокси -1-метилэтил)имидазол-5-карбоксилат.
Следуя процедуре, описанной в примере 25a, получали 3,54 г целевого соединения в виде сиропа, исходя из 2,00 г метил-2-бутил-4-(1-гидрокси-1-метилэтил)имидазол-5-карбоксилата, полученного в соответствии с описанием в получении 21, и 3,03 г т-бутил-4'-бромометилбифенил-2-карбоксилата.
ЯМР (CDCl3) d ppm: 0,92 (3Н, т. J 7,5 Гц), 1,25 (9Н, с), 1,33-1,46 (2Н, м), 1,64 (6Н, с), 1,68-1,78 (2Н, м), 2,70 (2Н, т. J 8 Гц), 3,78 (3Н, с), 5,50 (2Н, с), 5,70 (1Н, с), 6,97 (2Н, д. J 8,5 Гц), 7,26-7,33 (3Н, м), 7,37-7,54 (2Н, м), 7,76-7,81 (1Н, м).
31b) 2-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-4-(1-гидрокси -1-метилэтил) имидазол-5-карбоновая кислота.
Следуя процедуре, описанной в примере 25b, получали 2,46 г целевого соединения в виде кристаллов (т. пл. 158-159oC) путем гидролиза 3,31 г метил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)-метил]-2-бутил-4-(1-гидрокси -1-метилэтил)имидазол-5-карбоксилата, полученного в соответствии с описание в стадии (a), с использованием 1,37 г гидроксида моногидрата лития.
ЯМР (CDCl3) d ppm: 0,84 (3Н, т. J 7,5 Гц), 1,23 (9Н, с), 1,25-1,38 (2Н м), 1,52-1,65 (2Н, м), 1,68 (6Н, с), 2,83 (2Н, т. J 6,5 Гц), 5,81 (2Н, с), 7,07 (2Н д. J 8,0 Гц), 7,22-7,28 (3Н м), 7,34-7,50 (2Н, м), 7,74-7,78 (1Н, м).
31с) Пивалоилоксиметил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-2-бутил-4-(1-гидрокси -1-метилэтил)имидазол-5-карбоксилат.
Следуя процедуре, описанной в примере 25с, получали 0,48 г целевого соединения в виде сиропа путем эстерификации 0,40 г 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-2-бутил-4-(1-гидрокси -1-метилэтил)имидазол-5-карбоновой кислоты, полученной в соответствии с описанием в стадии (b), с использованием хлорметилпивалата и карбоната калия.
ЯМР (CDCl3) d ppm: 0,92 (3Н, т. J 7,5 Гц), 1,17 (9Н, с), 1,24 (9Н, с), 1,32-1,47 (2Н, м), 1,63 (6Н, с), 1,66-1,79 (2Н, м), 2,69 (12Н, т. J 8 Гц), 5,41 (1Н, с), 5,51 (2Н, с), 5,83 (2Н, с), 6,97 (2Н, д, J 8 Гц), 7,25-7,28 (3Н, м), 7,38-7,51 (2Н, м), 7,75-7,79 (1Н, м).
31d) Пивалоилоксиметил 2-бутил-1-[(2'-карбоксифенил-4-ил)метил]-4-(1-гидрокси-1-метилэтил)имидазол -5-карбоксилат.
Следуя процедуре, описанной в примере 25d, получали 0,45 г гидрохлорида целевого соединения в виде аморфного твердого вещества (т. пл. 139 - 144oC, т. размягч. 127oC) путем обработки 0,48 г пивалоилоксиметил 1-[(2'-т-бутоксикарбонил-бифенил-4-ил)метил] -2-бутил-4-(1-гидрокси-1-метилэтил) имидазол-5-карбоксилата, полученного в соответствии с описанием в стадии (с), 4 н. раствором хлорводорода в диоксане.
ЯМР (ДМСО-d6) d ppm: 0,80 (3Н, т, J 7,5 Гц), 1,10 (9Н, с), 1,21-1,35 (2Н, м), 1,39-1,50 (2Н, м), 1,58 (6Н, с), 2,96 (2Н, т. J 7,5 Гц), 5,65 (2Н, с), 5,88 (2Н, с), 7,17 (2Н, д J 8,5 Гц), 7,32-7,34 (3Н, м), 7,43-7,49 (1Н, м), 7,55-7,61 (1Н, м), 7,73-7,75 (1Н, м).
Пример 32. Изопропоксикарбонилоксиметил 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил]-4-(1-гидрокси-1-метилэтил)имидазол-5-карбоксилат (соединение 3-39).
32а) Изопропоксикарбонилоксиметил 1-[(2'-т-бутоксикарлонилфенил-4-ил)метил]-2-бутил-4-(1-гидрокси -1-метилэтил)-имидазол-5-карбоксилат.
Следуя процедуре, описанной в примере 25с, получали 0,46 г целевого соединения в виде кристаллов (т. пл. 91-93oC), исходя из 0,40 г 1-[(2'-т-тубоксикарбонилбифенил-4-ил)метил] -2-бутил-4-(1-гидрокси -1-метилэтил)-имидазол-5-карбоновой кислоты, полученной в соответствии с описанием в примере 31b, 0,15 г изопропоксикарбонилоксиметилхлорида и 0,31 г карбоната калия.
ЯМР (CDCl3) d ppm: 0,92 (3Н, т. J 7,5 Гц), 1,23 (9Н, с), 1,29 (6Н, д. J 6 Гц), 1,35-1,45 (2Н, м), 1,63 (6Н, с), 1,65-1,80 (2Н, м), 2,71 (2Н, т. J 7,5 Гц), 4,90 (1Н, септет J 6 Гц), 5,39 (1Н, с), 5,51 (2.с), 5,82 (2Н, с), 6,98 (2Н, д. J 8 Гц), 7,25-7,30 (3Н, м), 7,35-7,52 (2Н, м), 7,75-7,80 (1Н, м).
32b) Изопропоксикарбонилоксиметил 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил]-4-(1-гидрокси-1-метилэтил)имидазол -5-карбоксилат.
Следуя процедуре, описанной в примере 25d, получали 0,39 г гидрохлорида целевого соединения в виде кристаллов (т. пл. 154-156oC) путем обработки 0,40 г изопропоксикарбонилоксиметил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-4-(1-гидрокси-1-метилэтил)имидазол-5-карбоксилата, полученного в стадии (а), 4 н. раствором хлорводорода в диоксане.
ЯМР (ДМСО-d6) d ppm: 0,81 (3Н, т. J 7,5 Гц), 1,21 (6Н, д. J 6,5 Гц), 1,23-1,36 (2Н, м), 1,38-1,52 (2Н, м), 1,59-(6Н, с), 2,98 (2Н, т. J 6,5 Гц), 4,79 (1Н, септет, J 6,5 Гц), 5,65 (2Н, с), 5,85 (2Н, с), 7,18 (2Н, д. J 8 Гц), 7,30-7,38 (3Н, м), 7,42-7,62 (2Н, м), 7,74 (1Н, д J 7,5 Гц).
Пример 33. (5-метил-2-оксо-1,3-диоксолен-4-ил)метил 2-бутил-1-[(2'карбоксибифенил-4-ил)метил] -4-(1-гидрокси-1-метилэтил-имидазол-5-карбоксилат (соединение 3-51).
33а) (5-метил-2-оксо-1,3-диоксолен-4-ил)метил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-2-бутил-4-(1-гидрокси -1-метилэтил)имидазол-5-карбоксилат.
Следуя процедуре, описанной в примере 25с, получали 0,43 г целевого соединения в виде кристаллов (т. пл. 156-157oC), исходя из 0,40 г 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-2-бутил-4-(1-гидрокси-1-метилэтил)имидазол-5-карбоновой кислоты, полученной в соответствии с описанием в примере 31b, 0,22 г (5-метил 2-оксо-1,2-диоксолен-4-ил) метилбромида, и 0,26 г карбоната калия в 5 мл N,N-диметилацетамида.
ЯМР (CDCl3) d ppm: 0,92 (3Н, т. J 7,5 Гц), 1,27 (9Н, с), 1,30-1,45 (2Н, м), 1,62 (6Н, с), 1,65-1,80 (3Н, м), 2,07-(3Н, с), 2,70 (2Н, т J 7,5 Гц), 4,89 (2Н, с), 5,46 (2Н, с), 5,55 (1Н, с), 6,91 (2Н, д. J 8,5 Гц), 7,26-7,50 (5Н, м), 7,76 (1Н, д. J 6,5 Гц).
33b) (5-метил-2-оксо-1,3-диоксолен-4-ил)метил-2-бутил-1-[(2'-карбоксибифенил -4-ил)метил]-4-(1-гидрокси-1-метил-этил)имидазол-5-карбоксилат.
Следуя процедуре, описанной в примере 25d, получали 0,26 г гидрохлорида целевого соединения в виде порошка (т. пл. более 70oC с размягчением), путем обработки 0,32 г (5-метил-2-оксо-1,3-диоксолен-4-ил)метил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -4-(1-гидрокси-1-метилэтил)имидазол -5-карбоксилата, полученного в соответствии с описанием в стадии (а), 4 н. раствором хлорводорода в диоксане.
ЯМР (ДМСО-d6) Dd ppm: 0,82 (3Н, т. J 7 Гц), 1,20-1,40 (2Н, м), 1,40-1,60 (2Н, м), 1,59 (6Н, с), 2,12 (3Н, с), 2,98 (2Н, т. J 7,5 Гц), 5,14 (2Н, с), 5,63 (2Н, с), 7,13 (2Н, д. J 7,5 Гц), 7,30-7,60 (5Н, м), 7,74 (1Н, д J 7,5 Гц),
Пример 34. Фталидил 1-[(2'-карбоксибифенил-4-ил)метил]-4-(1-гидрокси-1-метилэтил)-2-пропилимидазол -5-карбоксилат (соединение 3-26).
34а) Фталидил 1-[(2'-т-бутоксикарбонилбифенил-4-ил-метил]-4-(1-гидрокси-1-метилэтил) -2-пропилимизадол-5-карбоксилат.
Следуя процедуре, описанной в примере 25с, получали 0,62 г целевого соединения в виде кристаллов (т. пл.144oC), исходя из 0,50 г 1-[(2'-т-бутоксикарбонилбифенил)метил] -4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоновой кислоты, полученной в соответствии с описанием в примере 25b, 0,25 г 3-бромофталида и 0,3 г карбоната калия в 6 мл N,N-диметилацетамида.
ЯМР (CDCl3) d ppm: 0,97 (2Н, т. J 7,5 Гц), 1,25 (9Н, с), 1,62 (6Н, с), 1,75 (2Н, секстет, J 7,5 Гц), 2,66 (2Н, т. J 6,5 Гц), 5,38 (2Н, АВ-квартет, d0,10 ppm J 17 Гц), 5,42 (1Н, с), 6,69 (2Н, д. J 7,5 Гц), 7,15 (2Н, д. J 7,5 Гц), 7,28-7,89 (9Н, м).
34d) Фталидил 1-[(2'-карбоксибифенил-4-ил)метил]-4-(1-гидрокси-1-метилэтил)-2-пропилимидазол -5-карбоксилат.
В соответствии с процедурой, описанной в примере 25d, получали 0,37 г гидрохлорида целевого соединения в виде аморфного порошка (т. пл. 142-144oC) путем обработки 0,45 г фталидил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилата, полученного в стадии (а), 4 н. раствором хлорводорода в диоксане.
ЯМР (ДМСО-d6) d ppm: 0,92 (3Н, т. J 7,5 Гц), 1,50-1,70 (2Н, м), 1,59 (6Н, с), 3,00 (2Н, т, J 7,5 Гц), 5,65 (2Н, с), 7,01 (2Н, д. J 8 Гц), 7,27 (2Н, д. J 8 Гц), 7,36-7,98 (9Н, м).
Пример 35. Этил 4-гидроксиметил-2-пропил-1-{4-[2-(тетразол-5-ил)фенил] фенил}метил имидазол-5-карбоксилат (соединение 4-3).
35а) Диэтил 2-пропил-1-{4-[2-тритилтетразол-5-ил)фенил]фенил}метилимидазол-4,5-дикарбоксилат.
0,441 г т-бутоксид калия добавляли к раствору 1,00 г диэтил 2-пропилимидазол-4,5-дикарбоксилата, полученного в соответствии с описанием в получении 12, в 15 мл N,N-диметилацетамида и полученную смесь размешивают 30 мин при комнатной температуре. Раствор 2,19 г 4-[2-(тритилтетразол-4-ил)фенил] бензилбромида в 15 мл N,N-диметилацетамида по капле добавляли к реакционной смеси при комнатной температуре, после чего реакционную смесь размешивали 3 ч при комнатной температуре. По истечение этого времени смесь разводили водой, а затем экстрагировали этилацетатом. Экстракт осушали безводным сульфатом магния, а растворитель отгоняли путем перегонки. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью гексана и этилацетата (1: 1, по объему), и получали 2,24 г целевого соединения в виде аморфного твердого вещества.
ЯМР (CDCl3) d ppm: 0,88 (3Н, т. J 7,5 Гц), 1,20 (3Н, т. J 7,5 Гц), 1,39 (3Н, т. J 7,5 Гц), 1,59 (6Н, с), 1,61-1,72 (3Н, м), 2,55 (2Н, т. J 7,5 Гц), 4,20 (2Н, кв. J 7,5 Гц), 4,39 (2Н, кв. J 7,5 Гц), 5,30 (2Н, с), 6,78 (2Н, д. J 8 Гц), 6,92-7,52 (20Н, м), 7,90 (1Н, д. J 7,5 Гц).
35b) Этил 4-гидроксиметил-2-пропил-1-{4-[2-(тритилтетразол-5-ил)фенил] фенил}метилимидазол-5-карбоксилат.
10 мл 1,5 М раствора гидрида диизобутилалюминия в толуоле по капле добавляли при -20oC и в атмосфере азота к раствору 4,27 г диэтил 2-пропил-1-{ 4-[2-(тритилтетразол-5-ил)фенил] фенил} метилидимидазол-4,5-дикарбоксилата, полученного в стадии (а), в 50 мл тетрагидрофурана. Полученную смесь оставляли на 16 ч при 0oC, а затем смешивали с этилацетатом и с насыщенным водным раствором хлорида аммония и полученную смесь размешивали в течение 1 ч при комнатной температуре, осадок отфильтровывали, слой этилацетата отделяли и осушали безводным сульфатом магния, а растворитель отгоняли путем дистилляции при пониженном давлении. Кристаллический остаток промывали диизопропиловым эфиром и получали в результате 4,03 г целевого соединения, т. пл. 135-138oC.
ЯМР (CDCl3) d ppm: 0,94 (6Н, т. J 7,5 Гц), 1,29 (3Н, т J 7 Гц), 1,67-1,77 (2Н, м), 2,56 (2Н, т. J 7,5 Гц), 3,43 (1Н, шир. т. J 4 Гц), 4,25 (2Н, кв. J 7 Гц), 4,91 (2Н, д. J 4 Гц), 5,49 (2Н, с), 6,82 (2Н, д. J 7,5 Гц), 6,98-7,57 (20Н, м), 7,94 (1Н, д. J 7 Гц).
35с) Этил 4-гидроксиметил-2-пропил-1-{4-[(2(тетразол -5-ил)-фенил]фенил} метилимидазол-5-карбоксилат.
Раствор 0,28 г этил 4-гидроксиметил-2-пропил-{4-[2-(тритилтетразол-5-ил)фенил] фенил} метилимидазол-5-карбоксилата, полученного в стадии (b), в 4 мл 75% об./об. водной уксусной кислоты размешивали 2 ч при 60oC. Затем реакционную смесь концентрировали путем выпаривания при пониженном давлении, а остаток растворяли в толуоле. Полученный раствор снова концентрировали путем выпаривания при пониженном давлении для удаления по возможности большего количества воды и уксусной кислоты. После этого остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью метиленхлорида и метанола (9:1 и 4:1 по объему), и получали 0,20 г целевого соединения в виде аморфного твердого вещества.
ЯМР (CDCl3) d ppm: 0,80 (3Н, т. J 7,5 Гц), 1,20 (3Н, т. J 7,5 Гц), 1,45-1,65 (2Н, м), 2,44 (2Н, т. J 7,5 Гц), 4,20 (2Н, кв. J 7,5 Гц), 4,58 (2Н, с), 5,43 (2Н, с), 6,78 (2Н, д. J 7,5 Гц), 6,98 (2Н, д. J 7,5 Гц), 7,38-7,60 (3Н, м), 7,79 (1Н, д. J 7,5 Гц),
Примеры 36. 4-гидроксиметил-2-пропил-1-{4-[2-(тетразол-5-ил)фенил]фенил} метилимидазол -5-карбоновая кислота (соединение 4-1).
Смесь 0,20 г этил 4-гидроксиметил-2-пропил-1-{4-[2-(тетразол-5-ил)фенил] фенил} метилимидазол -5-карбоксилата, полученного в соответствии с описанием в примере 35с, и 0,10 г гидроксид-моногидрата лития в 3 мл воды размешивали 3 ч при комнатной температуре, после чего смесь оставляли при этой же температуре на 16 ч. Затем реакционную смесь смешивали с 2,38 мл 1 н. водной соляной кислоты, а полученный осадок собирали фильтрацией, в результате чего получали 150 мг целевого соединения, т. пл. 233oC (с разлож.).
ЯМР (ДМСО-d6) d ppm: 0688 (3Н, т. J 7,5 Гц), 1,59 2Н, секстет, J 7,5 Гц), 2,58 (2Н, т, J 7,5 Гц), 4,64 (2Н, с), 2,62 (2Н, с), 6,98 (2Н, д. J 8 Гц), 7,08 (2Н, д. J 8 Гц), 7,39-7,69 (3Н, м).
Пример 37. Пивалоилоксиметил 4-гидроксиметил-2-пропил-1-{ 4-[2-(тетразол-4-ил)фенил]фенил} метилимидазол -5-карбоксилат (соединение 4-4).
37а) 4-гидроксиметил-2-пропил-1-{4-[2-(тритилтетразол -5-ил)фенил]фенил} метилимидазол-5-карбоновая кислота.
Раствор 0,66 г моногидрата гидроокиси лития в 20 мл воды добавляли к раствору 1,22 г этил 4-гидроксиметил-2-пропил-1-{4-[2-(трититерразол-4-ил)фенил] фенил} метилимидазол-5-карбоксилата, полученного в соответствии с описанием в примере 35b, в 5 мл диоксана и полученную смесь размешивали 5 ч при 80oC. После этого реакционную смесь выделяли из диоксана путем дистилляции при пониженном давлении, а водный остаток смешивали со льдом и этилацетатом, затем добавляли 15,7 мл 1 н. водной соляной кислоты. Целевое соединение осаждалось и его собирали фильтрацией и промывали водой. Затем этилацетатный слой выделяли из фильтрата и осушали безводным сульфатом магния, а растворитель отгоняли путем дистилляции при пониженном давлении. Полученный остаток промывали диэтиловым эфиром и получали в результате целевое соединение в виде порошка. Две части целевого соединения объединяли (вместе они весили 0,98 г) и сразу, без дополнительной очистки или идентификации, использовали в последующей реакции этерификации.
37b) Пивалоилоксиметил 4-гидроксиметил-2-пропил-1-{4-[2-(тритилтетразол -5-ил)фенил]фенил} метилимидазол-5-карбоксилат.
0,30 г карбоната калия и 0,24 г пивалоилоксиметилхлорида добавляли к раствору 0,98 г 4-гидроксиметил-2-пропил-1-{4-[2-(тритилтетразол -5-ил)фенил] фенил} метилимидазол-5-карбоновой кислоты, полученной в стадии (а), в 10 мл N,N-диметилацетамида и полученную смесь размешивали при комнатной температуре в течение 6 ч. По истечение этого времени реакционную смесь смешивали с этилацетатом и водой. Этилацетатный слой отделяли и осушали безводным сульфатом магния, а растворитель отгоняли при пониженном давлении. Полученный остаток очищали с помощью хроматографии на силикагеле, элюируя смесью этилацетата и гексана (2:1, по объему) в результате чего получали 0,91 г целевого соединения в виде смолистого вещества.
ЯМР (CDCl3) d ppm: 0,89 (3Н, т. J 7,5 Гц), 1,18 (9Н, с), 1,70 (1Н, секстет, J 7,5 Гц), 2,52 (2Н, т. J 8 Гц), 3,35 (1Н, шир. с), 4,83 (2Н, с), 5,32 (2Н, с), 5,80 (2Н, с), 6,76 (2Н, д. J 8 Гц), 6,92-7,51 (20Н, м), 7,90 (1Н, д J 7,5 Гц).
37с). Пивалоилоксиметил 4-гидроксиметил-2-пропил-1-{4-[2-(тетразол-5-ил)фенил]фенил} метилимидазол -5-карбоксилат.
В соответствии с процедурой, описанной в примере 35с, 0,91 г пивалоилоксиметил 4-гидроксиметил-2-пропил-1-{4-[2-(тритилтетразол-5-ил)фенил]фенил} метилимидазола, полученного в стадии (b), детритилировали путем обработки 75 об./об. водной уксусной кислотой, в результате чего получали 0,42 г целевого соединения в виде порошка, т. пл. выше 60oC (с размягчен.).
ЯМР (CDCl3) d ppm: 0,94 (3Н, т. J 7,5 Гц), 1,14 (9Н, с), 1,72 (2Н, секстет, J 7,5 Гц), 2,61 (2Н, т. J 7,5 Гц), 2,90 (2Н, шир.с), 4,77 (2Н, с), 4,49 (2Н, с), 5,84 (2Н, с), 6,94 (2Н, д. J г Гц), 7,15 (2Н, д. J 8 Гц), 7,26-7,61 (3Н, м), 8,07 (1Н, д. J 7,5 Гц).
Пример 38. Метил 2-бутил-4-гидроксиметил-1-{4-[2-(тетразол-5-ил)фенил] фенил}метилимидазол -5-карбоксилат (соединение 4-47).
38а) Диметил 2-бутил-1-{4-[2-(тритилтетразол-5-ил) фенил] фенил}метилимидазол-4,5 дикарбоксилат.
Повторяли процедуру, описанную в примере 35а, но с использованием 0,50 г диметил-2-бутилимидазол-4,5-дикарбоксилата, полученного в соответствии с описанием, приведенным в получении 4, и 1,17 г 4[2-тритилтетразол-5-ил)фенил] бензилбромида, в результате чего получали 0,51 г целевого соединения в виде аморфного твердого вещества.
ЯМР (CDCl3) d ppm: 0,85 (3Н, т. J 7,5 Гц), 1,20- 1,80 (4Н, м), 2,59 (2Н, т. J 8,0 Гц), 3,73 (3Н, с), 3,92 (3Н, с), 5,30 (3Н,с), 6,6-7,6 (22Н, м), 7,8-8,0 (1Н, м).
38b) Метил 2-бутил-4-гидроксиметил-1-{4-[2-(тритилтетразол-5-ил)фенил] фенил}метилимидазол -5-карбсокилат.
В соответствии с процедурой, описанной в примере 35b, 0,51 г диметил 2-бутил-1-{4-[2-(тритилтетразол-5-ил)фенил]-фенил}метилимидазол -4,5-дикарбоксилата, полученного в стадии (а), восстанавливали с использованием 0,99 мл 1,5 М раствора гидрида диизобутилалюминия в толуоле, в результате чего получали 0,44 г целевого соединения в виде маслянистого вещества.
ЯМР (CDCl3) d ppm: 0,86 (3Н, т. J 7,5 Гц), 1,23-1,36 (2Н, м), 1,58-1,70 (2Н, м), 1,80-1,95 (1Н, м), 2,54 (2Н, т. J 8,0 Гц), 3,72 (3Н, с), 4,85 (2Н, д. J 6,0 Гц), 5,43 (2Н, с), 6,77 (2Н, д. J 8,5 Гц), 6,92-6,95 (4Н, м), 7,08 (2Н, д, J 8,5 Гц), 7,22-7,51 (14Н, м), 7,87-7,90 (1Н, м),
38с) Метил-2-бутил-4-гидроксиметил-1-{4-[2-(тетразол-5-ил)-фенил]фенил} метилимидазол-5-карбоксилат.
Раствор 0,44 г метил 2-бутил-4-гидроксиметил-1-{4-[2-(тритилтетразол-5-ил)фенил]фенил} метилимидазол -5-карбоксилата, полученного в стадии (b), в 10 мл метанола и 0,70 мл 1 н. водной соляной кислоты оставляли на ночь при комнатной температуре. После этого реакционную смесь концентрировали досуха путем дистилляции при пониженном давлении, а остаток растирали с диэтиловым эфиром, в результате чего получали 0,30 г гидрохлорида целевого соединения в виде твердого вещества.
ЯМР (ДМСО-d6) d ppm: 0,81 (3Н, т. J 7,5 Гц), 1,19-1,32 (2Н, м), 1,38-1,51 (2Н, м), 2,95 (2Н, т. J 7,5 Гц), 4,80 (2Н, с), 5,71 (2Н, с), 7,20-7,75 (8Н, м).
Пример 39. 2-бутил-4-гидроксиметил-1-{4-[2-(тетразол-5-ил) фенил]фенил} метилимидазол-5-карбоновоая кислота (соединение 4-46).
Повторяли процедуру, описанную в примере 36, но с использованием 0,30 г метил 2-бутил-4-гидроксиметил-1-{4-[2-(тетразол-5-ил)фенил]фенил} метилимидазол-5-карбоксилата, полученного в примере 38с, и 2,50 мл 1 н. водного раствора гидроксида натрия, в результате чего получали 95 мг целевого соединения в виде кристаллов, т. пл. 215-217oC.
ЯМР (ДМСО-d6) d ppm: 0,82 (3Н, т. J 7,5 Гц), 1,27 (2Н, м), 1,52 (2Н, м), 2,56 (2Н, т. J 7,5 Гц), 4,60 (2Н, с), 5,58 (2Н, с), 6,94 (2Н, д. J 8,5 Гц), 7,06 (2Н, д. J 8,5 Гц), 7,50 -7,70 (4Н, м).
Пример 40. Этил 4-(1-гидроксиэтил)-2-пропил-1-{ 4-[2-(тетразол -5-ил)фенил]фенил}метилимидазол-5-карбоксилат (соединение 4-30).
40а) Этил 4-формил-1-пропил-1-{ 4-[2-(тритилтетразол-4-ил)фенил]фенил} метилимидазол -5-карбоксилат.
6 г активированного диоксида марганца добавляли к раствору 2 г этил 4-гидроксиметил-2-пропил-1-{ 4-[2-(тритилтетразол-5-ил)фенил] фенил} метилимидазол -5-карбоксилата, полученного в примере 35b, в 40 мл метиленхлорида и полученную смесь размешивали 2,5 ч при комнатной температуре. После этого диоксид марганца отфильтровывали и фильтрат концентрировали путем выпаривания при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью этилацетата и гексана (1:1, по объему) в результате чего получали 1,45 г целевого соединения в виде кристаллов, т. пл. 177-179oC (с разложением).
ЯМР (CDCl3) d ppm: 0,88 (3Н, т. J 7,5 Гц), 1,29 (2Н, т. J 7 Гц), 1,74 (2Н, секстет, J 7,5 Гц), 2,57 (2Н, т. J 7,5 Гц), 4,29 (2Н, кв. J 7 Гц), 5,49 (2Н, с), 6,76 (2Н, д. J 8,5 Гц), 6,92-7,88 (20Н, м), 7,90 (1Н, д. J 7,5 Гц), 10,42 (1Н, с).
40b) Этил 4-(1-гидроксиэтил)-2-пропил-1-4-[2-(тритилтетразол-4-ил)фенил] фенил}метилимидазол-5-карбоксилат.
4,0 г 1 М раствора бромида метил марганца тетрагидрофуране добавляли по каплям при 10oC к раствору этил-4-формил-2-пропил-1-{4-[2-(тритилтетразол-4-ил)фенил]фенил}метилимидазол-5-карбоксилата, полученного в стадии (а), в 5 мл тетрагидрофурана и полученную смесь размешивали 3 ч при температуре от -10oC до 0oC. После этого реакционную смесь смешивали с этилацетатом и водным раствором хлорида аммония и полученную смесь размешивали 20 мин при комнатной температуре. Затем этилацетатный слой отделяли и осушали безводным сульфатом магния. Растворитель удаляли путем дистилляции при пониженном давлении, а остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесями этилацетата и метиленхлорида (1:4 и 1:2, по объему), в результате чего получали 1,23 г целевого соединения в виде вязкого масла.
ЯМР (CDCl3) d ppm: 0,78 (3Н, т, J 7,5 Гц), 1,22 (3Н, т, J 7 Гц), 1,54 (3Н, д. J 7 Гц), 1,68 (2Н, секстет, J 7,5 Гц), 2,50 (2Н, т. J 7,5 Гц), 3,82 (1Н, д. J 8 Гц), 4,18 (2Н, кв. J 7 Гц), 5,23 (1Н, квинтет J 7 Гц), 5,42 (2Н, с), 6,76 (2Н, д. J 8 Гц), 6,93-7,52 (20Н, м), 7,88 (1Н, д. J 7,5 Гц).
40с) Этил 4-(1-гидроксиэтил)-2-пропил-1-{4-[2-(тетразол-5-ил)фенил]фенил}метилимидазол -5-карбоксилат.
Следуя процедуре, описанной в примере 35с, 1,23 г этил 4-(1-гидроксиэтил)-2-пропил-1-{ 4-[2-(тритилтетразол-4-ил)фенил] фенил} метилимидазол-5-карбоксилата, полученного в стадии (b), обрабатывали 75% об./об. водной уксусной кислотой, в результате чего получили 0,82 г целевого соединения в виде аморфного твердого вещества.
ЯМР (CDCl3) d ppm: 0,85 (3Н, т. J 7,5 Гц), 1,24 (3Н, т. J 7 Гц), 1,42 (3Н, д. J 6 Гц), 1,59 (2Н, секстет J 7,5 Гц), 2,50 (2Н, т. J 7 Гц), 4,22 92Н, кв. J 7 Гц), 5,13-5,20 (1Н, м), 5,44 (2Н, АВ-квартет, d 0,12 ppm, J 16,5 Гц), 6,78 (2Н, д. J 8 Гц), 6,99 (2Н, д. J 8 Гц), 7,38-7,59 (3Н, м), 7,76 (1Н, д. J 7,5 Гц).
Пример 41. 4-(1-гидроксиэтил)-2-пропил-1-{4-[2-(тетразол-5-ил)фенил]фенил}метилимидазол -5-карбоновая кислота (соединение 4-29).
В соответствии с процедурой, описанной в примере 36, 0,82 этил 4-(1-гидроксиэтил)-2-пропил-1-{ 4-[2-(тетразол-5-ил)фенил]фенил}метилимидазол -5-карбоксилата, полученного в примере 40с, гидролизовали с использованием 0,43 г моногидрата гидроксида лития, в результате чего получали 0,50 г целевого соединения в виде порошка, т. пл. 198-201oC.
ЯМР (ДМСО-d6) d ppm: 0,86 (3Н, т. J 7,5 Гц), 1,38 (3Н, д. J 6,5 Гц), 1,55 (2Н, секстет J 7,5 Г3) 2,58 (2Н, т. J 8 Гц), 5,21 (1Н, кв. J 6,5 Гц), 5,61 (2Н, с), 6,95-7,08 (4Н, м), 7,51-7,70 (4Н, м).
Пример 42. Этил 4-(1-гидроксиэтил)-2-пропил-1-{ 4-[2-(тетразол-5-ил)фенил]фенил}метилимидазол -5-карбоксилат (соединение 4-30).
42а) Этил 4-(1-гидроксиэтил)-2-пропил-1-{ 4-[2-(тритилтетразол -4-ил)фенил]фенил}метилимидазол-5-карбоксилат.
Повторяли процедуру, описанную в примере 35а, но с использованием 114 мг этил 4-(1-гидроксиэтил-2-пропилимидазол-5-карбоксилата, полученного в соответствии с описанием, приведенным в получении 23 iii, 280 мг 4-[2-(тритилтетразол-4-ил)фенил] бензилбромида и 60 мг т-бутоксида калия, в результате чего получали 255 мг целевого соединения в виде вязкого масла. ЯМР-спектр этого соединения идентичен ЯМР-спектру соединения, полученного в примере 40b.
42b) Этил-4-(1-гидроксиэтил)-2-пропил-1-{4-[2-(тетразол -5-ил)фенил]фенил}метилимидазол-5-карбоксилат.
В соответствии с процедурой, описанной в пример 35с, 255 мг этил-4-(1-гидроксиэтил)-2-пропил-1-{ 4-[2-(тритилтетразол -4-ил)фенил]фенил}метилимидазол-5-карбоксилата, полученного в стадии (а), подвергали детритилированию путем обработки 75% об./об. водной уксусной кислотой, в результате чего получали 170 мг целевого соединения в виде аморфного твердого вещества. ЯМР-спектр этого соединения идентичен ЯМР-спектру соединения, полученного в примере 40с.
Пример 43. Этил-2-бутил-4-(1-гидроксиэтил)-1-{4-[2-(тетразол-5-ил)фенил] фенил}метилимидазол-5-карбоксилат (соединение 4-75).
43а) Этил 2-бутил-4-(1-гидроксиэтил-1-{4-[2-(тритилтетразол -5-ил)фенил] фенил}метилимидазол-5-карбоксилат.
Повторяли процедуру, описанную в примере 35а, но с использованием 400 мг этил 2-бутил-4-(1-гидроксиэтил)имидазол-5-карбоксилата, полученного в соответствии с описанием в получении 24 iii, 1,00 г 4_[2-(тритилтетразол-4-ил)фенил]-бензилбромида и 197 мг т-бутоксида калия, в результате чего получали 0,94 г целевого соединения в виде вязкого масла.
ЯМР (CDCl3) d ppm: 0,87 (3Н, т. J 7,5 Гц), 1,24 (3Н, т. J 7 Гц), 1,25-1,38 (2Н, м), 1,55 (3Н, д. J 6,5 Гц), 1,60-1,72 (2Н, м), 2,54 (2Н, т. J 8 Гц), 3,84 (1Н, д. J 6,5 Гц), 4,20 (4Н, кв. J 7 Гц), 5,25 (1Н, квинтет, J 6,5 Гц), 5,44 (2Н, с), 6,78 (2Н, д. J 8 Гц), 6,94-7,54 (20Н, м), 7,90 (1Н, д. J 7,5 Гц).
43 b) Этил 2-бутил-4-(1-гидроксиэтил)-1-{4-[2-(тетразол-5-ил)фенил]фенил}метилидимидазол -5-карбоксилат.
Следуя процедуре, описанной в примере 40с, 0,84 г этил 2-бутил-(4-1-гидроксиэтил)-1-{4-[2-(тритилтетразол -5-ил)фенил]фенил}метилимидазол-5-карбоксилата, полученного в соответствии с описанием в стадии (а), обрабатывали 75% об./об. водной уксусной кислотой, в результате чего получали 0,54 г целевого соединения в виде аморфного твердого вещества.
ЯМР (CDCl3) d ppm: 0,78 (3Н, т. J 7,5 Гц), 1,15-1,30 (2Н, м), 1,19 (3Н, т. J 7 Гц), 1,35 (3Н, д. J 6,5 Гц), 1,44-1,60 (2Н, м), 2,49 (2Н, т. J 8 Гц), 4,17 (2Н, кв, J 7 Гц), 5,09 (1Н, кв. J 6,5 Гц), 5,35 и 5,45 (каждый 1Н, АВ-квартет, J 16,5 Гц), 6,89 (2Н, д. J 8 Гц), 6,96 (2Н, д. J 8 Гц), 7,30-7,50 (3Н, м), 17,65 (1Н, д, J 7,5 Гц).
Пример 44. 2-бутил-4-(1-гидроксиэтил)-1-{4-[2-(тетразол-5-ил)фенил]фенил}метилимидазол-5-карбоновая кислота (соединение 4-74).
В соответствии с процедурой, описанной в примере 36, 0,54 г этил 2-бутил-4-(1-гидроксиэтил)-1-{ 4-[2-(тетразол-5-ил)фенил] фенил}метилимидазол -5-карбоксилата, полученного в примере 43b, гидролизовали, используя 245 мг моногидрата гидроксида лития, в результате чего получали 0,43 г целевого соединения в виде порошка, т. пл. 214-217oC.
ЯМР (ДМСО-d6) d ppm: 0,82 (3Н, т. J 7,5 Гц), 1,27 (2Н, секстет, J 7,5 Гц), 1,37 (3Н, д. J 6,5 Гц), 1,50 (2Н, квинтет, J 7,5 Гц), 2,58 (2Н, т. J 8 Гц), 5,20 (1Н, кв. J 6,5 Гц), 5,61 (2Н, с), 6,96 (2Н, д. J 8 Гц), 7,06 (2Н, д. J 8 Гц), 7,50-7,66 (4Н, м).
Пример 45. 2-бутил-1-[(2'-карбоксифенил-4-ил)метил] -4-(1-гидроксиэтил)-имидазол-5-карбоксамид (соединение 5-64).
45(а) 4-ацетил-1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутилимидазол-5-карбонитрил.
0,192 г гидрида натрия (в виде 55 мас. дисперсии в минеральном масле) добавляли к раствору 0,843 г 4-ацетил-2-бутилимидазол-5-карбонитрила, полученного в соответствии с описанием, приведенным в получении 24 i, в 17 мл N, N-диметилацетамида, и полученную смесь размешивали 20 мин при комнатной температуре. Затем добавляли 1,68 г т-бутил-4'-(бромометил)бифенил-2-карбоксилата и полученную смесь размешивали 2,5 ч при 55oC. После этого к смеси добавляли водный раствор хлорида натрия, а затем экстрагировали этилацетатом. Экстракт промывали водой и осушали безводным сульфатом магния, а растворитель удаляли путем дистилляции при пониженном давлении. Полученный маслянистый остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесями (4: 1 и 2:1, по объему) гексана и этилацетата, и получали 1,14 г целевого соединения в виде маслянистого вязкого вещества.
ЯМР (CDCl3) d ppm: 0,93 (3Н, т. J 7 Гц), 1,23 (9Н, с), 1,3-2,1 (4Н, м), 2,58 (3Н, с), 2,75 (2Н, т. J 7 Гц), 5,32 (2Н, с), 7,0-8,0 (8Н, м).
45b) 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-2-бутил-4-(1-гидроксиэтил)имидазол -5-карбонитрил.
0,098 г борогидрида натрия добавляли к раствору 1,18 г 4-ацетил-1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-имидазол-5-карбонитрила, полученного в стадии (а), в 30 мл этанола и полученную смесь размешивали 1 ч при комнатной температуре. Избыток борогидрида натрия удаляли путем добавления ацетона, после чего реакционную смесь концентрировали путем выпаривания при пониженном давлении. Остаток разбавляли водным раствором хлорида натрия и экстрагировали этилацетатом. Экстракт осушали и концентрировали путем выпаривания при пониженном давлении. Маслянистый осадок очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью этилацетата и гексана (3:2, по объему), и получали 1,18 г целевого соединения в виде вязкого масла.
ЯМР (CDCl3) d ppm: 0,92 (3Н, т. J 7,5 Гц), 1,25 (9Н, с), 1,3-1,5 (2Н, м), 1,60 (3Н, д. J 6,5 Гц), 1,6-1,8 (2Н, м), 2,6-268 (2Н, м), 5,00 (1Н, кв, J 6,5 Гц), 5,22 (2Н, с), 7,1-7,9 (8Н, м).
45с) 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-2-бутил-4-(1-гидроксиэтил) -имидазол-5-карбоксамид. 12 мл 1 н. водного раствора гидроксида натрия добавляли к раствору 0,52 г 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]2-бутил-4-(1-гидроксиэтил)имидазол -5-карбонитрила, полученного в стадии (b), в 3 мл этанола и полученную смесь нагревали с обратным холодильником в течение 3 ч. После этого реакционную смесь нейтрализовали путем добавления разбавленной водной соляной кислоты и экстрагировали этилацетатом. Экстракт промывали водой и осушали безводным сульфатом магния. Затем растворитель удаляли путем перегонки при пониженном давлении. Полученную смесь очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью этилацетата и гексана (4:1, по объему), а затем одним этилацетатом, в результате чего получали 0,14 г целевого соединения в виде аморфного твердого вещества.
ЯМР (CDCl3) d ppm: 0,90 (3Н, т. J 7,5 Гц), 1,23 (4Н, с), 1,2-1,5 (1Н, м), 1,6-1,8 (2Н, м), 1,66 (3Н, д. J 6,5 Гц), 2,63 (2.т. J 8 Гц), 5,11 (1Н, кв. J 6,5 Гц), 5,59 и 5,74 (каждый 1Н, АВ-квартет, J 16 Гц), 7,0-7,9 (8Н, м),
45d) 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил] -4-(1-гидроксиэтил)имидазол -5-карбоксамид.
Раствор 0,15 г 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-2-бутил-4-(1-гидроксиэтил)имидазол -5-карбоксамида, полученного в стадии (с), растворенный в 3 мл 4 н раствора хлорводорода в диоксане оставляли на ночь при комнатной температуре. Затем раствор концентрировали путем выпаривания при пониженном давлении. Остаток растирали в гексане и полученный таким образом порошок собирали путем фильтрации, в результате чего получали 0,105 г гидрохлорида целевого соединения в виде аморфного твердого вещества, т. пл. 212-214oC (с разл.).
ЯМР (CDCl3) d ppm: 0,94 (3Н, т. J 7,5 Гц), 1,3-1,6 (2Н, м), 1,59 (3Н, д. J 6,5 Гц), 1,6-2,0 (2Н, м), 3,0-3,4 (2Н, м), 5,16 (1Н, кв. J 6,5 Гц), 5,41 и 5,58 (каждый 1Н, АВ-кварт. J 15 Гц), 7,1-7,9 (8Н, м).
Пример 46. 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил]-4-(1-гидроксипропил)имидазол -5-карбоксамид (соединение 5-65).
46а) 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-4-пропионилимидазол-5-карбонитрил.
Повторяли процедуру, описанную в примере 45а, но с использованием 0,923 г 2-бутил-4-пропионилимидазол-2-карбонитрила, полученного в соответствии с описанием, приведенным в получении 25, 1,56 г т-бутил 4'-(бромометил)-бифенил-2-карбоксилата и 196 мг гидрида натрия (в виде 55 мас./мас. дисперсии в минеральном масле) в 20 мл N,N-диметилацетамида, в результате чего получали 1,84 г целевого соединения в виде маслянистого вязкого вещества.
ЯМР (CDCl3) d ppm: 0,91 (3Н, т. J 7 Гц), 1,0-2,1 (4Н, м), 1,25 (9Н, с), 2,72 (2Н, т. J 7 Гц), 3,02 (2Н, кв. J 7 Гц), 5,30 (2Н, с), 7,0-8,0 (8Н, м).
46b) 1-[(2'-т-бутоксикарбонилоксибифенил-4-ил)-метил]-2-бутил-4-(1-гидроксипропил)имидазол-5-карбонитрил.
Повторяли процедуру, описанную в примере 45b, но с использованием 451 мг 1-[(2'-т-бутоксикарбонилоксибифенил-4-ил)метил] -2-бутил-4-пропионил-имидазол-5-кирбонитрила, полученного в стадии (а), и 36 мг борогидрида натрия в 10 мл этанола, в результате чего получали 369 мг целевого соединения в виде вязкого масла.
ЯМР (CDCl3) d ppm: 0,91 (3Н, т. J 7 Гц), 0,99 (3Н, т. J 7 Гц), 1,0-2,3 (6Н, м), 1,25 (9Н, с), 2,70 (2Н, т. J 7 Гц), 3,16 (1Н, д. J 6,5 Гц), 4,74 (1Н, кв. J Гц), 5,21 (2Н, с), 7,0-8,0 (8Н, м). 46с) 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-2-бутил-4-(1-гидроксипропил)имидазол-5-карбоксамид.
20 мл 1 н. водного раствора гидроксида натрия добавляли к раствору 368 мг 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-4-(1-гидроксипропил)имидазол-5-карбонитрила, полученного в стадии (b), растворенного в 20 мл этанола, и полученную смесь нагревали с обратным холодильником в течение 6 ч. По истечение этого времени реакционную смесь обрабатывали в соответствии с процедурой, описанной в примере 45с, и получали 316 мг целевого соединения в виде аморфного твердого вещества.
ЯМР (CDCl3) d ppm: 0,89 (6Н, т. J 7 Гц), 1,0-2,3 (6Н, м), 1,24 (9Н, с), 2,61 (2Н, т. J 7 Гц), 4,76 (1Н, т. J 7 Гц), 5,52 и 5,83 (кажд. 1Н, АВ-кварт. J 17 Гц), 6,9-7,9 (8Н, м).
46d) 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил] -4-(1-гидроксиропил)имидазол -5-карбоксамид.
Повторяли процедуру, описанную в примере 45d, но с использованием 316 мг 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил -4-(1-гидросипропил)имидазол-5-карбоксамида, полученного в стадии (с), и 10 мл 4 н. раствора хлорводорода в диоксане, в результате чего получали 148 мг гидрохлорида целевого соединения в виде аморфного порошка, т. пл. выше 120oC (с размягчением).
ЯМР (ДМСО-d6) d ppm: 0,80 (3Н, т. J 7,5 Гц), 0,87 (3Н, т. J 7,5 Гц), 1,1-2,0 (6Н, м), 2,94 (2Н, т, J 7,5 Гц), 4,85 (1Н, т. J 7 Гц), 5,68 (2Н, с), 7,0-7,8 (8Н, м).
Пример 47. 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил]-4-(1-гидроксибутил) имидазол-5-карбоксамид (соединение 5-66).
47а) 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-2-бутил-4-бутирилимидазол -5-карбонитрил.
Повторяли процедуру, описанную в примере 45а, но с использованием 0,877 г 2-бутил-4-бутирилимидазол-5-карбонитрила, полученного в соответствии с описанием, приведенным в получении 26, 1,53 г т-бутил 4'-(бромометил)-бифенил-2-карбоксилата и 0,175 г гидрида натрия (в виде 55% мас./мас. дисперсии в минеральном масле) в 18 мл N,N-диметилацетамида, в результате чего получали 0,99 г целевого соединения в виде вязкого масла.
ЯМР (CDCl3) d ppm: 0,93 (3Н, т. J 7 Гц), 1,01 (3Н, т. J 7 Гц), 1,28 (9Н, с), 1,4-2,1 (6Н, м), 2,74 (2Н, т. J 7 Гц), 3,00 (2Н, т. J 7 Гц), 5,30 (2Н, с), 7,0-7,0 (8Н, м).
47b) 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-2-бутил-4-(1-гидроксибутил)имидазол-5-карбонитрил.
Повторяли процедуру, описанную в примере 45b, но с использованием 0,99 г 1-[(2'-т-бутоксикарбонилбифенил-4-ил)-метил] -2-бутил-4-бутирилимидазол-5-карбонитрила, полученного в стадии (а), и 0,077 г борогидрида натрия в 20 мл этанола, в результате чего получали 0,88 г целевого соединения в виде вязкого маслянистого вещества.
ЯМР (CDCl3) d ppm: 0,7-1,2 (6Н, м), 1,2-2,1 (8Н, м), 1,23 (9Н, с), 2,71 (2Н, т. J 7 Гц), 4,28 (1Н, д. J 6 Гц), 4,82 (1Н, кв. J 6 Гц), 5,28 (2Н, с), 7,0- 8,0 (8Н, м).
47с) 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-2-бутил-4-1-гидроксибутил имидазол-5-карбоксамид.
14 мл 1 н. водного раствора гидроксида натрия добавляли к раствору 0,86 г 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-4-(1-гидроксибутил)имидазол-5-карбонитрила, полученного в стадии (b), в 14 мл этанола и полученную смесь нагревали с обратным холодильником в течение 10 ч. По истечение этого времени реакционную смесь обрабатывали в соответствии с процедурой, описанной примере 45с, в результате чего получали 0,58 г целевого соединения в виде аморфного твердого вещества.
ЯМР (CDCl3) d ppm: 0,90 (3Н, т. J 7,5 Гц), 0,94 (3Н, т. J 7,5 Гц), 1,23 (9Н, с), 1,3-2,1 (8. т. J 8 Гц), 2,63 (2Н, т. J 8 Гц), 4,91 (1Н, т. J 7 Гц), 5,56 и 5,77 (каждый 1Н, АВ-квартет, J 16 Гц), 7,0-7,8 (8Н, м).
47d) 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил] -4-(1-гидроксибутил)имидазол -5-карбоксида.
Повторяли процедуру, описанную в примере 45d, но с использованием 0,58 г 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-4-(1-гидроксибутил)имидазол -5-карбоксамида, полученного в стадии (с), и 13 мл 4 н. раствора хлорводорода, в результате чего получали 0,55 г гидрохлорида целевого соединения в виде аморфного порошка, т. пл. выше 110oC (с размягчением).
ЯМР (ДМСО-d6) d ppm: 0,80 (3Н, т. J 7,5 Гц), 0,89 (3Н, т. J 7,5 Гц), 1,1-1,9 (8Н, м), 2,96 (2Н, т. J 7,5 Гц), 4,96 (1Н, т. J 7,5 Гц), 5,68 (2Н, с), 7,2-7,8 (8Н, м).
Пример 48. 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил]-4-(1-гидрокси)-2-метилпропил)имидазол-5-карбоксамид (соединение 5-67).
48а) 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-2-бутил-4-изобутирилимидазол-5-карбонитрил.
Повторяли процедуру, описанную в примере 45 (а), но с использованием 0,85 г 2-бутил-4-изобутирилимидазол-5-карбонитрила, полученного в соответствии с описанием приведенным в получении 27, 1,34 т-бутил-4'-(бромометил)бифенил-2-карбоксилата и 170 мг гидрида натрия (в виде 55% мас. /мас. дисперсии в минеральном масле) в 15 мл N,N-диметилацетамида, в результате чего получали 1,62 г целевого соединения в виде вязкого масла.
ЯМР (CDCl3) d ppm: 0,93 (3Н, т. J 7 Гц), 1,0-2,1 (4Н, м), 1,21 (6Н, д. J 7 Гц), 1,22 (9Н, с), 2,73 (2Н, т. J 7 Гц), 3,66 (1Н, септет J 7 Гц), 5,30 (2Н, с), 7,0-8,0 (8Н, м).
48b) 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-4-(1-гидрокси-2-метилпропил)имидазол-5-карбонитрил.
Повторяли процедуру, описанную в примере 45b, но с использованием 500 мг 1-[(2'-т-бутоксикарбонилбифенил-4-ил)-метил-2-бутил -4-изобутирилимидазол-5-карбонитрила, полученного в стадии (а), и 25 мг борогидрида натрия в 10 мл этанола, в результате чего получали 297 мг целевого соединения в виде маслянистого вязкого вещества.
ЯМР (CDCl3) d ppm: 0,7-1,2 (9Н, м), 1,0-2,5 (5Н, м), 1,27 (9Н, с), 2,70 (2Н, д. J 7 Гц), 3,01 (1Н, д. J 7 Гц), 4,54 (1Н, т. J 7 Гц), 5,23 (2Н, с), 7,0-7,0 (8Н, м).
48с) 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-4-(1-гидрокси)-2-метилпропил]имидазол-5-карбоксамид.
20 мл 1 н. водного раствора гидроксида натрия добавляли к раствору 297 мг 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]2-бутил-4-(1-гидрокси-2-метилпропил) имидазол-5-карбонитрила, полученного в стадии (b), в 20 мл этанола и полученную смесь нагревали с обратным холодильником в течение 8 ч. По истечение этого времени реакционную смесь обрабатывали в соответствии с процедурой, описанной в примере 45с, и получали 151 г целевого соединения в виде аморфного твердого вещества.
ЯМР (CDCl3) d ppm: 0,66 (3Н, д. J 7 Гц), 0,85 (3Н, т. J 7 Гц), 1601 (3Н, д. J 7 Гц), 1,0-2,4 (5Н, м), 1,22 (9Н, с), 2,59 (2Н, т. J 7 Гц), 4,40 (1Н, д. J 7 Гц), 5,53 и 5,83 (кажд. 1Н, АВ-квартет, J 17 Гц), 6,9-7,9 (8Н, м).
48d) 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил]4-(1-гидрокси-2-метилпропил)имидазол -5-карбоксамид.
Повторяли процедуру, описанную в примере 45d, но с использованием 151 мг 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-4-(1-гидрокси-2-метилпропил)-5-карлоксамида, полученного в стадии (с), и 5 мл 4 н. раствора хлорводорода в диоксане, в результате чего получали 119 мг гидрохлорида целевого соединения в виде аморфного порошка, т. пл. 131oC (с размягчением).
ЯМР (ДМСО-d6) d ppm: 0,73 (3Н, д. J 6,5 Гц), 0679 (3Н, т. J 7,5 Гц), 0,98 (3Н, д. J 6,5 Гц), 1,1-1,6 (4Н, м), 1,9-2,1 (1Н, м), 2,98 (2Н, т. J 7,5 Гц), 4,65 (1Н, д. J 8 Гц), 5,69 (2Н, с), 7,1-7,8 (8Н, м).
Пример 49. 1-[(2'-карбоксифенил-4-ил)метил]-4-(1-гидроксибутил)-2-пропоксиимидазол-5-карбоксамид (соединение 5-4).
49а) 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -4-бутирил-2-пропилимидазол -5-карбонитрил.
Повторяли процедуру, описанную в примере 45а, но с использованием 1,026 г 4-бутирил-2-пропилимидазол-5-карбонитрила, полученного в соответствии с описанием, приведенным в получении 28, 1,91 г т-бутил-4'-(бромометил)-бифенил-2-карбоксилата и 0,209 г гидрида натрия (в виде 55% мас./мас. дисперсии в минеральном масле) в 20 мл N,N-диметилацетамида, получали 1,70 г целевого соединения в виде вязкого маслянистого вещества.
ЯМР (CDCl3) d ppm: 1,00 (6Н, т. J 7,5 Гц), 1,25 (9Н, с), 1,7-1,9 (4Н, м), 2,70 (2Н, т. J 7,5 Гц), 2,99 (2Н,т, J 7,5 Гц), 5,31 (2Н, с), 7,1-7,9 (8Н, м).
49b) 1-[(2'-т-бутоксикарбонилбифенил-4-ил) метил]-4-(1-гидроксибутил)-2-пропилимидазол-5-карбонитрил.
Повторяли процедуру, описанную в примере 45b, но с использованием 1,13 г 1-[(2'-т-бутоксикарбонилбифенил-4-ил) метил]-4-бутирил-2-пропилимидазол-5-карбонитрила, полученного в стадии (а), и 0,091 г борогидрида натрия в 23 мл этанола, в результате чего получали 1,07 г целевого соединения в виде вязкого маслянистого вещества.
ЯМР (ДМСО-d6) d ppm: 0,87 (3Н, т. J 7,5 Гц), 0,90 (3Н, т. J 7,5 Гц), 1,17 (9Н, с), 1,2-1,4 (2Н, м), 1,5-1,7 (4Н, м), 2,67 (2Н, т. J 7,5 Гц), 4,58 (1Н, м), 5,34 (2Н, с), 5,41 (1Н, д. J 4,5 Гц), 7,1-7,7 (8Н, м).
49с) 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -4-(1-гидроксибутил)-2-пропилимидазол-5-карбоксамид.
16 мл 1 н. водного раствора гидроокиси натрия добавляли к раствору 1,07 г 1-[(2'-т-бутокси-карбонилбифенил-4-ил)метил]-4-(1-гидроксибутил)-2 -пропилимидазол-4-карбонитрила, полученного в стадии (b), в 16 мл этанола и полученную смесь обрабатывали в соответствии с описанием, приведенным в примере 45с, в результате чего получали 0,82 г целевого соединения в виде аморфного твердого вещества.
ЯМР (CDCl3) d ppm: 0,93 (3Н, т. J 7,5 Гц), 0,95 (3Н, т. J 7,5 Гц), 1,23 (9Н, с), 1,2-2,1 (6Н, м), 2,60 (2Н, т. J 8 Гц), 4,89 (1Н, т. J 7,5 Гц), 5,56 и 5,77 (каждый 1Н, АВ-квартет, J 16 Гц), 7,0-7,8 (8Н, м).
49d) 1-[(2'-карбоксибифенил-4-ил)метил] -4-(1-гидроксибутил)-2-пропилимидазол-5-карбоксамид.
Повторяли процедуру, описанную в примере 45d, но с использованием раствора 0,82 г 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-4-(1-гидроксибутил)-2-пропилимидазол-5-карбоксамида, полученного в стадии (с), в 17 мл 4 н. раствора хлорводорода в диоксане, в результате чего получали 0,78 г гидрохлорида целевого соединения в виде аморфного порошка, т. пл. 118-121oC (с размягчением).
ЯМР (CDCl3) d ppm: 0,90 (3Н, т. J 7,5 Гц), 0,93 (3Н, т. J 7,5 Гц), 1,1-1,5 (2Н, м), 167-2,1 (4Н, м), 2,9-3,1 (2Н, м), 5,00 (1Н, т. J 7,5 Гц), 5,4 и 5,56 (кажд. 1Н, АВ-квартет, J 1, 5 Гц), 7,1-7,9 (8Н, м).
Пример 50. 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил]-4-(1-гидрокси-1-метилэтил)имидазол-5-карбоксамид (соединение 5-69).
50а) 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил -4-(1-гидрокси-1-метилэтил) имидазол-5-карбоксамид.
10 мл 1 н. водного раствора гидроксида натрия добавляли к раствору 232 мг 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-4-(1-гидрокси-1-метилэтил)имидазол-5-карбонитрила, полученного в соответствии с описанием в примере 10а, в 10 мл этанола и полученную смесь нагревали с обратным холодильником в течение 3 ч. По истечение этого времени реакционную смесь обрабатывали способом, аналогичным описанному в примере 45с, в результате чего получали 185 мг целевого соединения в виде аморфного твердого вещества.
ЯМР (CDCl3) d ppm: 0,89 (3Н, т. J 7 Гц), 1,0-2,0 (4Н, м), 1,23 (9Н, с), 1,68 (6Н, с), 2,62 (2Н, т, J 7 Гц), 5,63 (2Н, с), 6,9-7,9 (8Н, м).
50b) 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил] -5-(1-гидрокси-1-метилэтил)имидазол -5-карбоксамид.
Повторяли процедуру, описанную в примере 45d, но с использованием 185 мг 1-[(2'-т-бутокси-карбонилбифенила-4-ил)метил]-2-бутил-4-(1-гидрокси -1-метилэтил)-имидазол-5-карбоксамида, полученного в стадии (а), и 10 мл 4 н. раствора хлорводорода в диоксане, в результате чего получали 88 мг гидрохлорида целевого соединения в виде аморфного твердого вещества, т. пл. 130-138oC (с размягчением).
ЯМР (ДМСО-d6) d ppm: 0,78 (3Н, т. J 7,0 Гц), 1,17-1,30 (2Н, м), 1,30-1,42 (2Н, м), 1,61 (6Н, с), 2,96 (2Н, т. J 7,5 Гц), 5,55 (2Н, с), 7,20-7,75 (8Н, м).
Пример 51. 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил]-4-(1-гидрокси-2-метил)-1-(1-метилэтил)пропил]имидазол-5-карбоксамид (соединение 5-333).
51а) 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-4-(1-гидрокси-2-метил -1-(1-метилэтил)пропил имидазол-5-карбонитрил.
51а) 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-4-(1-гидрокси-2-метил -2-(1-метилэтил)пропил]имидазол-5-карбонитрил.
Повторяли процедуру, описанную в примере 45а, но с использованием 282 мг 2-бутил-4-[(1-гидрокси-2-метил-1-(1-метилэтил)пропил] имидазол-5-карбонитрила, полученного в соответствии с процедурой, описанной в получении 30, 409 мг т-бутил-4'-(бромометил)бифенил-2-карбоксилата и 47 мг гидрида натрия (в виде 55% мас./мас. дисперсии в минеральном масле) в 5 мл N,N-диметилацетамида, в результате чего получали 513 мг целевого соединения в виде вязкого масла.
ЯМР (CDCl3) d ppm: 0,7-1,1 (15Н, м), 1,0-2,0 (4Н, м), 1,21 (9Н, с), 2,15-2,60 (2Н, м), 2,68 (2Н, т. J 7 Гц), 3,20 (2Н, с), 5,26 (2Н, с), 6,9-8.0 (8Н, м).
51b) 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-4-[1-гидрокси-2-метил -1-(1-метилэтил)-пропил]имидазол-5-карбоксамид.
10 мл 1 н. водного раствора гидроксида натрия добавляли к раствору 500 мг 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-4-[1-гидрокси-2-метил-1 -(1-метилэтил)пропил] имидазол-5-карбонитрила, полученного в стадии (а), в 10 мл этанола и полученную смесь нагревали с обратным холодильником в течение 20 ч. По истечение этого времени реакционную смесь обрабатывали в соответствии с описанием, приведенным в примере 45с, в результате чего получали 220 мг целевого соединения в виде аморфного вещества.
ЯМР (CDCl3) d ppm: 0,7-1,1 (15Н, м), 1,0-2,1 (4Н, м), 1,20 (9Н, с), 2,2-2,9 (4Н, м), 5,59 (2Н, с), 6,8-7,9 (8Н, м).
51с) 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил]-4-[1-гидрокси-2-метил-1-(1-метилэтил)пропил]имидазол-5-карбоксамид.
Повторяли процедуру, описанную в примере 45d, но с использованием 220 мг 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-4-[1-гидрокси-2-метил-1-(1-метилэтил)пропил] имидазол-5-карбоксамида, полученного в стадии (b), и 4,5 мл 4 н. раствора хлорводорода в диоксане, в результате чего получали 201 мг гидрохлорида целевого соединения в виде аморфного твердого вещества т. пл. 178-181oC.
ЯМР (ДМСО-d6) d ppm: 0,76 (3Н, т. J 7,5 Гц), 0,8-0,9 (12Н, м), 1,1-1,4 (4Н, м), 2,2-2,4 (2Н, м), 2,8-3,1 (2Н, м), 5,51 (2Н, с), 7,2-7,8 (8Н, м).
Пример 52. 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил]-4-гидроксиметилимидазол-5-карбоксамид (соединение 5-63).
52а) Сукцинимидо 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-2-бутил-4-гидроксиметилимидазол-5-карбоксилат.
206 мг N, N-дициклогексилкарбодиимида добавляли к суспензии 464 мг 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-4-гидроксиметилимидазол-5-карбоновой кислоты, полученной в соответствии с описанием в примере 4, и 140 мг N-гидроксисукцинимида в 10 мл тетрагидрофурана и полученную смесь размешивали 16 ч при комнатной температуре. После этого осажденный материал отфильтровывали, а фильтрат концентрировали путем выпаривания при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью метанола и метиленхлорида (1:15, по объему), и получали 0,52 г целевого соединения в виде кристаллов, т. пл. 107-109oC.
ЯМР (CDCl3) d ppm: 0,89 (3Н, т. J 7 Гц), 1,0-2,0 (4Н, м), 1,23 (9Н, с), 2,70 (2Н, т. J 7,5 Гц), 2,69 (4Н, с), 4,10 (1Н, шир.с), 4,96 (2Н, с), 5,56 (2Н, с), 7,00-7,90 (8Н, м).
52b) 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-2-бутил-4-гидроксиметилимидазол -5-карбоксамид.
0,5 мл концентрированного водного аммиака добавляли к раствору 0,60 г сукцинимидо 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-2-бутил-4-гидроксиметил-имидазол-5-карбоксилата, полученного в стадии (а), в 6 мл тетрагидрофурана и после этого сразу отделялось целевое соединение. Растворитель удаляли путем перегонки при пониженном давлении, а полученный остаток промывали диэтиловым эфиром и водой, в результате чего получали 0,38 г целевого соединения в виде порошка, т. пл. 222-224oC.
ЯМР (ДМСО-d6) d ppm: 0,85 (3Н, т. J 7 Гц), 1,19 (9Н, с), 1,0-1,9 (4Н, м), 2,57 (2Н, т. J 7,5 Гц), 4,52 (2Н, д. J 4,5 Гц), 5,63 (2Н, с), 5,83 (1Н, т. J 4,5 Гц( 6,95-7,8 (8Н, м).
52с) 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил] -4-гидрокси-метилимидазол-5-карбоксамид.
Раствор 0,28 г 1-[(2-т-бутоксикарбонилбифенил-4-ил)метил]-2-бутил-4-гидроксиметилимидазол -5-карбоксамида, полученного в стадии (b), в 3 мл 4 н. раствора хлорводорода в диоксане размешивали при комнатной температуре в течение 5 ч, а затем концентрировали путем выпаривания при пониженном давлении. Концентрат растирали со смесью этилацетата и диэтилового эфира и отвержденный материал собирали путем фильтрации, в результате чего получали 0,26 г гидрохлорида целевого соединения, которая размягчается при более чем 150oC, а разлагается при 235oC.
ЯМР (ДМСО-d6) d ppm: у0,80 (3Н, т. J 7,5 Гц), 1,20-1,31 (2Н, м), 1,43-1,54 (2Н, м), 2,96 (3Н, т. J 7,5 Гц), 4,68 (2Н, с), 5,71 (2Н, с), 7,21-7,75 (8Н, м).
Пример 53. N-метил-2-бутил-1-[(2'-карбоксибифенил-4-ил)метил] -4-гидроксиметилимидазол-5-карбоксамид (соединение 5-71).
53а) N-метил-1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-2-бутил -4-гидроксиметилимидазол-5-карбоксамид.
0,4 мл 40% об. раствора метиламина в воде добавляли при комнатной температуре к раствору 0,287 г сукцинимидо 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-4-гидрокси-метилимидазол-5-карбоксилата, полученного в соответствии с описанием в примере 52(а), в смеси 3 мл метиленхлорида и 2 мл метанола и полученную смесь оставляли на 16 ч при комнатной температуре. После этого раствор концентрировали путем выпаривания при пониженном давлении, а концентрат растворяли в этилацетате. Полученный раствор промывали водным раствором бисульфата калия и водным раствором бикарбоната натрия (последовательно), после чего раствор осушали безводным сульфатом магния. Затем растворитель удаляли при пониженном давлении, а остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя этилацетатом, в результате чего получали 176 мг целевого соединения в виде стеклообразного продукта.
ЯМР (CDCl3) d ppm: 0,85 (3Н, т. J 7 Гц), 1,23 (9Н, с), 1,0-2,0 (4Н, м), 2,54 (2Н, т. J 7,5 Гц), 2,91 (3Н, д. J 5 Гц), 4,70 (2Н, с), 5,62 (2Н, с), 6,9-7,85 (8Н, м), 8,38 (1Н, кв. J 5 Гц).
53b) N-метил-2-бутил-1-[(2'-карбонилбифенил-4-ил)метил] -4-гидроксиметилимидазол -5-карбоксамид.
Раствор N-метил-1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил -4-гидроксиметилимидазол-5-карбоксамида, полученного в стадии (а), в 2 мл 4 н. раствора хлорводорода в диоксане оставляли на 16 ч при комнатной температуре, а затем концентрировали при пониженном давлении. Полученный кристаллический осадок промывали смесью этилацетата и диэтилового эфира и получали 0,15 г гидрохлорида целевого соединения, т. пл. 205-208oC (с разлож.)
ЯМР (ДМСО-d6) d ppm: 0,81 (3Н, т. J 7,5 Гц), 1,25 (2Н, секстет, J 7,5 Гц), 1,49 (2Н, квинтет, J 7,5 Гц), 2,75 (3Н, д. J 4,5 Гц), 2,96 (2Н, т. J 8 Гц), 5,64 (2Н, с), 7,21-7,75 (8Н, м), 8,91 (1Н, кв. J 4,5 Гц).
Пример 54. N-этоксикарбонилметил-2-бутил-1-[(2'-карбоксифенил-4-ил)метил]-4-гидроксиметилимидазол-5-карбоксамид (соединение 5-126).
Повторяли процедуру, описанную в примере 53, но с использованием 0,307 г сукцинимидо 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-4-гидроксиметилимидазол-5-карбоксилата, полученного в соответствии с описанием в примере 52а, 89 мг этилглицинат-гидрохлорида и 0,089 мл триэтиламина, в результате чего получали 0,202 г гидрохлорида целевого соединения в виде аморфного порошка, т. пл. выше 80oC (с размягчением).
ЯМР (ДМСО-d6) Dd ppm: 0,80 (3Н, т. J 7,5 Гц), 1,18 (3Н, т. J 7 Гц), 1,20-1,33 (2Н, м), 1,47 (2Н, квинтет, J 7,5 Гц), 2,94 (2Н, т. J 8 Гц), 4,05 (2Н, д. J 6 Гц), 4,12 (2Н, кв. J 7 Гц), 4,72 (2Н, с), 5,63 (2Н, с), 7,24-7,75 (8Н, м), 9,37 (1Н, т. J 6 Гц).
Пример 55. N-карбоксиметил-2-бутил-1-[(2'-карбоксибифенил-4-ил)метил]-4-гидроксиметилимидазол-5-карбоксамид (соединение 5-125).
Повторяли процедуру, описанную в примере 53, но с использованием 0,32 г сукцинимидо 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-4-гидроксиметилимидазол -5-карбоксилата, полученного в примере 52а, 0,11 г т-бутилглицинат-гидрохлорида и 80 мг 4-диметиламинопиридина, в результате чего получали 0,21 г гидрохлорида целевого соединения в виде аморфного порошка, т. пл. > 110oC (с размягчением).
ЯМР (ДМСО-d6) d ppm: 0,81 (3Н, т. J 7,5 Гц), 1,25 (2Н, секстет J 7,5 Гц), 1,48 (2Н, квинтет, J 7,5 Гц), 2,95 (2Н, т. J 8,0 Гц), 3,98 (2Н, д. J 6 Гц), 4,71 (2Н, с), 5,64 (2Н, с), 7,26-7,75 (8Н, м), 9,22 (1Н, т. J 6 Гц).
Пример 56. N-[(S)-1-этоксикарбонилэтил-2-бутил-1-[(2'-карбоксибифенил-4-ил)метил]-4-гидроксиметилимидазол-5-карбоксамид (соединение 5-128).
Повторяли процедуру, описанную в примере 53, но с использованием 0,39 г сукцинимида 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-2-бутил-4-гидроксиметилимидазол -5-карбоксилата полученного в примере 52а, 0,13 г этил (S)-аланат-гидрохлорида и 0,21 мл триэтиламина, в результате чего получали 0,27 г гидрохлорида целевого соединения в виде аморфного твердого вещества.
ЯМР (ДМСО-d6) d ppm: 0682 (3Н, т. J 7,5 Гц), 1,17 (3Н, т. J 7 Гц), 1,20-1,35 (2Н, м), 1,34 (3Н, д. J 7 Гц), 1,43-1,58 (2Н, м), 2,98 (2Н, т. J 7,5 Гц), 4,10 (2Н, кв. J 7 Гц), 4,44 (1Н, квинтет, J 7 Гц), 4,70 (2Н, с), 5,63 (2Н, АВ-кв. d 0,10 ppm, J 16 Гц), 7,24-7,76 (8Н, м), 9,39 (1Н, д. J 7,5 Гц).
Пример 57. N-(2-этоксикарбонилэтил-2-бутил-1-[(2'-карбоксибифенил-метил] -4-гидроксиметилимидазол-5-карбоксамид (соединение 5-130).
Повторяли процедуру, описанную в примере 53, но с использованием 305 мг сукцинимидо 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-2-бутил-4-гидроксиметилмидазол-5-карбоксилата, полученного в соответствии с описанием в примере 52(а), 96 мг этил альфа-аланатгидрохлорида и 0,088 мл триэтиламина, в результате чего получали 0,20 г гидрохлорида целевого соединения в виде аморфного твердого вещества.
ЯМР (ДМСО-d6) d ppm: 0682 (3Н, т. J 7,5 Гц), 1,16 (3Н, т. J 7 Гц), 1,20-1,38 (2Н, м), 1,42-1,58 (2Н, м), 2,97 (2Н, т. J 7,5 Гц), 3,3-3,6 (4Н, м), 4,04 (2Н, кв. J 7 Гц), 4,60 (2Н, с),5,63 (2Н, с), 7,21-7,76 (8Н, м), 9,01 (1Н, шир. т.).
Пример 58. Метил(S)-N-{2-бутил-1-[(2'-карбоксибифенил-4-ил)метил]-4-гидроксиметилимидазол-5-карбонил}-пропинат (соединение 5-335).
Повторяли процедуру, описанную в примере 53, но с использованием 529 мг сукцинимидо 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-2-бутил-4-гидроксиметилимидазол-5-карбоксилата, полученного в соответствии с описанием в примере 52а, 180 мг метил (S)-пролинат-гидрохлорида и 0,2 мл триэтиламина, в результате чего получали 0,39 г гидрохлорида целевого соединения в виде аморфного порошка, т. пл. более 120oC (с размягчением).
ЯМР (ДМСО-d6) d ppm: 0,88 (3Н, т. J 7,5 Гц), 1,34 (2Н, секстет, J 7,5 Гц), 1,4-2,25 (6Н, м), 2,9-3,7 (2Н, м), 3,64 (3Н, с), 4,34 (1Н, т. J 7,5 Гц), 4,55 (2Н, с), 5,25 и 5,56 (кажд. 1Н, АВ-квартет, J 15,5 Гц), 7,26-7,77 (8Н, м).
Пример 59. 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил]-4-(1-гидрокси-2,2-диметилпропил) имидазол-5-карбоксамид (соединение 5-68).
59а) Метил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-4-формилимидазол-5-карбоксилат.
5,07 мл триэтиламина и 6,0 г комплекса триоксид серы/пиридина добавляли по очереди при температуре 10-15oC к раствору 3,0 г метил 1-[(2'-т-бутоксикарбонилбифенил-3-ил)-метил] -2-бутил-4-гидроксиметилимидазол -4-карбоксилата, полученного в соответствии с описанием в примере 1b, в 18 мл диметилсульфоксида и полученную смесь размешивали при той же температуре 45 мин. После этого реакционную смесь смешивали с водой и экстрагировали этилацетатом. Экстракт промывали водой, а затем водным раствором бикарбоната натрия, после чего экстракт осушали безводным сульфатом магния, а растворитель отгоняли путем дистилляции при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью гексана и этилацетата (1: 1, по объему), в результате чего получали 2,88 г целевого соединения в виде аморфного твердого вещества.
ЯМР (CDCl3) Dd ppm: 0,90 (3Н, т. J 7 Гц), 1,25 (9Н, с), 1,1-2,1 (4Н, м), 2,77-(2Н, т. J 8 Гц), 3,91 (3Н, с), 5,65 (2Н, с), 6,9-7,9 (8Н, м), 106 48 (1Н, с).
59b) Метил-1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-4-(1-гидрокси -2,2-диметилпропил)имидазол-5-карбоксилат.
2,77 мл 2 М раствора бромида т-бутилмагния в тетрагидрофуране добавляли при -55oC и в атмосфере азота к раствору 1,32 г метил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-4-формил имидазол-5-карбоксилата, полученного в стадии (а), в 26 мл тетрагидрофурана и полученную смесь размешивали при температуре -55oC (-50oC) в течение 30 мин. После этого реакционную смесь разбавляли 50 мл этилацетата и насыщенным водным раствором хлорида аммония. Органический слой отделяли и осушали безводным сульфатом магния, а растворитель удаляли путем дистилляции при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью гексана и этилацетата (2:1, по объему), в результате чего получали 0,87 г целевого соединения в виде аморфного твердого вещества.
ЯМР (CDCl3) d ppm: 0,90 (3Н, т. J 7,5 Гц), 0,93 (9Н, с), 1,0-2,0 (4Н, м), 1,19 (9Н, с), 2,68 (2Н, т. J 7,5 Гц), 3,41 (1Н, д. J 10 Гц), 3,74 (3Н, с), 4,92 (1Н, д. J 10 Гц), 5,59 (2Н, с), 6,9-7,9 (8Н, м).
59с) 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-4-(1-гидрокси-2,2-диметилпропил)имидазол-5-карбоновая кислота
В соответствии с процедурой, описанной в Примере 4 0,87 г метил-1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] 2-бутил-4-(1-гидрокси-2,2-диметилпропил)имидазол-5-карбоксилата, полученного в стадии (b), подвергали гидролизу с использованием 342 мг моногидрата гидроокиси лития, в результате чего получали 0,73 г целевого соединения в виде кристаллов, т. пл. 199-201oC (с разлож.).
ЯМР (ДМСО-d6) d ppm: 0,84 (3Н, т. J 7,5 Гц), 0689 (9Н, с), 1,16 (9Н, с), 1,22-1,4 (2Н, м), 1,58 (2Н, квинтет, J 7,5 Гц), 2,64 (2Н, т. J 7,5 Гц), 4,78 (1Н, с), 5,68 (2Н, АВ-квартет, d 0,14 ppm, J 17 Гц), 7,02 (2Н, д. J 8 Гц), 7,22-7,58 (5Н, м), 7,65 (1Н, д. J 7,5 Гц).
59d) Сукцинимидо 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-2-бутил-4-(1-гидрокси -2,2-диметилпропил)-имидазол-5-карбоксилат.
Повторяли процедуру, описанную в примере 52а, но с использованием 600 мг 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-4-(1-гидрокси-2,2-диметилпропил)имидазол-5-карбоновой кислоты, полученной в стадии (с), 172 мг N-гидроксисукцинимида и 250 мг N,N-дициклогексилкарбодиимида, в результате чего получали 663 мг целевого соединения в виде аморфного твердого вещества.
ЯМР (CDCl3) d ppm: 0,92 (3Н, т. J 7,5 Гц), 1,01 (9Н, с), 1,21 (9Н, с), 1,38 (2Н, секстет, J 7,5 Гц), 1,73 (2Н, квинтет J 7,5 Гц), 2,71 (2Н, т. J 7,5 Гц), 2,84 (4Н, с), 4,99 (1Н, д. J 7,5 Гц), 5,53 (2Н, с), 7,03 (2Н, д. J 8,5 Гц), 7,26-7,50 (5Н, м), 7,77 (1Н, д. J 8 Гц).
59е) 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-4-(1-гидрокси -2,2-диметилпропил)имидазол-5-карбоксамид.
Повторяли процедуру, описанную в примере 52b, но с использованием 0,66 г сукцинимидо 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-2-бутил-4-(1-гидрокси -2,2-диметил-пропил)имидазол-5-карбоксилата, полученного согласно описанию в стадии (d), в результате чего получали 0,33 г целевого соединения в виде аморфного твердого вещества.
ЯМР (CDCl3) d ppm: 0,89 3Н, т. J 7,5 Гц 0,96 (9Н, с), 1,22 (9Н, с), 1,34 (2Н, секстет, J 7,5 Гц), 1,64 (2Н, квинтет, J 7,5 Гц), 2,62 (2Н, т. J 7,5 Гц), 4,67 (1Н, д. J 5,5 Гц, 5,48 и 5,82 (кажд. 1Н, АВ-квартет, J 16 Гц), 7,02 (2Н, д. J 8,5 Гц), 7,23-7,50 (5Н, м), 7,76 (1Н, д. J 6,5 Гц).
59f) 2-бутил-1-[(2'-карбоксибифенил-4-ил)метил] -4-(1-гидрокси -2,2-диметилпропил) имидазол-5-карбоксамид.
Повторяли процедуру, описанную в примере 52с, но с использованием 326 мг 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -2-бутил-4-(1-гидрокси-2,2-диметилпропил)имидазол-5-карбоксамида, полученного в соответствии с описанием в стадии (е), в результате чего получали 228 мг гидрохлорида целевого соединения в виде порошкообразного твердого вещества, т. пл. 150-154oC (с размягчением).
ЯМР (ДМСО-d6) d ppm: 0,80 (3Н, т. J 7,5 Гц), 0,91 (9Н, с), 1,24 (2Н, секстет, J 7,5 Гц), 1,45 (2Н, квинтет, J 7,5 Гц), 2,99 (2Н, т. J 7,5 Гц), 4,78 (1Н, с), 5669 (2Н, с), 7,12 (2Н, д, J 8 Гц), 7,33-7,61 (5Н, м), 7,75 (1Н, д, J 8 Гц).
Пример 60. 1-[(2'-карбоксибифенил-4-ил)метил]-4-(1-гидрокси- 2,2-диметил-пропил)-2-пропилимидазол-5-карбоксамид (соединение 5-6).
60а) Диэтил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-2-пропилимидазол-4,5-дикарбоксилат.
Повторяли процедуру, описанную в примере 1а, но с использованием 9,0 г диэтил-2-пропил-имидазол-4,5-дикарбоксилата, полученного в соответствии с описанием, приведенным в получении 12, 12,3 г т-бутил 4-бромометилбифенил-2-карбоксилата и 4,1 г т-бутоксида калия в качестве основания, в результате чего получали 16,47 г целевого соединения в виде вязкого масла.
ЯМР (CDCl3) d ppm: 0,95 (3Н, т. J 7,5 Гц), 1,5-2,0 (2Н, м), 1,23 (9Н, с), 1,25 (3Н, т. J 7 Гц), 1,37 (3Н, т. J 7 Гц), 2,69 (2Н, т. J 7 Гц), 4,26 (2Н, кв. J 7 Гц), 4638 (2Н, кв. J 7 Гц), 5,48 (2Н, с), 7,0-7,9 (8Н, м).
60b) Этил - 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-4-гидроксиметил-2-пропилимидазол -5-карбоксилат.
В соответствии с процедурой, описанной в примере 1b, 16,47 г диэтил 1-[(2'-т-бутоксикарбонилоксибифенил-4-ил)метил] -2-пропилимидазол-4,5-дикарбоксилита, полученного в стадии (а), восстанавливали, используя 44,4 мл 1,5 М раствора гидрида диизобутилалюминия в тетрагидрофуране, в результате чего получали 10,83 г целевого соединения в виде кристаллов, т. пл. 108-110oC.
ЯМР (CDCl3) d ppm: 0,98 (3Н, т. J 7,5 Гц), 1,23 (9Н, с), 1,31 (3Н, т. J 7 Гц), 1,79 (2Н, секстет J 7,5 Гц), 2,67 (2Н, т. J 7,5 Гц), 4,27 (2Н, кв. J 7 Гц), 4,87 (2Н, с), 5,59 (2Н, с), 7,00 (2Н, д. J 8,5 Гц), 7,24-7,75 (5Н, м), 7,78 (1Н, д. J 7 Гц).
60с) Этил-1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -4-формил-2-пропилимидазол -5-карбоксилат
В соответствии с процедурой, описанной в примере 59а, 2,71 г этил 1-[(2'-т-бутоксикарбонилбифенил-4-ил) метил] -4-гидроксиметил-1-пропилимидазол-5-карбоксилата, полученного в стадии (b), подвергали окислению с использованием 4,6 мл триэтиламина и 5,5 г комплекса триоксида серы и пиридина в 17 мл диметилсульфоксида, в результате чего получали 2,57 г целевого соединения в виде кристаллов, т. пл. 117-199oC.
ЯМР (CDCl3) d ppm: 0,99 (3Н, т. J 7,5 Гц), 1,26 (9Н, с), 1,84 (1Н, секс. J 7,5 Гц), 2,73 (2Н, т. J 7,5 Гц), 4,40 (2Н, кв. J 7 Гц), 5,67 (2Н, с), 7,02 (2Н, д. J 8,5 Гц), 7,29-7,54 (5Н, м), 7,80 (1Н, д. J 8 Гц), 10,48 (1Н, с).
60d) Этил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -4-(1-гидрокси-2,2-диметилпропил)-2-пропилимидазол-5-карбоксилат.
В соответствии с процедурой, описанной в примере 59b, 1,14 г этил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -4-формил-2-пропилимидазол-5-карбоксилата, полученного в стадии (с), подвергали взаимодействию с 2,4 мл 2 М раствора т-бутил-магнийбромида в тетрагидрофуране, в результате чего получали 0,78 г целевого соединения в виде вязкого маслянистого вещества.
ЯМР (CDCl3) d ppm: 0,97 (3Н, т. J 7,5 Гц), 1,00 (9Н, с), 1,25 (9Н, с), 1,35 (3Н, т. J 7 Гц), 1,77 (2Н, секст. J 7,5 Гц), 2,68 (2Н, т. J 7,5 Гц), 3,46 (1Н, д. J 9 Гц), 4,29 (2Н, кв. J 7 Гц), 4,99 (1Н, д. 9 Гц), 5,62 (2Н, с), 7,00 (2Н, д. J 8 гц) 7,29-7,54 (5Н, м), 7,80 (1Н, д. J 7,5 Гц).
60е) 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -4-(1-гидрокси-2,2-диметилпропил) -2-пропилимидазол-5-карбоновая кислота.
В соответствии с процедурой, описанной в примере 4, 0,78 г этил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -4-(1-гидрокси-2,2-диметилпропил) -2-пропилимидазол-5-карбоксилата, полученного в стадии (d), подвергали гидролизу, используя 209 мг моногидрата гидроксида лития, в результате чего получали 0,62 г целевого соединения в виде кристаллов, т. пл. 207oC.
ЯМР (ДМСО-d6) d ppm: 0,88 (3Н, т. J 7,5 Гц), 0,89 (9Н, с), 1,15 (4Н, с), 1,63 (2Н, секстет, J 7,5 Гц), 2,63 (2Н, т. J 7,5 Гц), 4,79 (1Н, с), 5,63 и 5,76 (кажд. 1Н, АВ-квартет J 18,5 Гц), 7,02 (2Н, д. J 8 Гц), 7,22-7,67 (6Н, м).
60f) Сукцинимидо 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-4-(1-гидрокси-2,2-диметилпропил)-2-пропилимидазол-5-карбоксилат.
Повторяли процедуру, описанную в примере 52а, но с использованием 300 мг 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -4-(1-гидрокси-2,2-диметилпропил)-2-пропилимидазол-5-карбоновой кислоты, полученной в стадии (е), 110 мг N-гидроксисукцинимида и 130 мг N,N-дициклогексилкарбодиимида, в результате чего получали 321 мг целевого соединения в виде аморфного твердого вещества.
ЯМР (CDCl3) d ppm: 0,94 (3Н, т. J 7,5) 0698 (9Н, с), 1,18 (9Н, с), 1,75 (2Н, секстет, J 7,5 Гц), 2,64 (2Н, т. J 7,5 Гц), 3,12 (1Н, д. J 9,5 Гц), 4,98 91Н, д. J 9,5 Гц), 5,52 (2Н, с), 7,0-7,9 (8Н, м).
60g) 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -4-ил метил 4-(1-гидрокси-2,2-диметилпропил)-2-пропилимидазол-5-карбоксамид
Повторяли процедуру, описанную в примере 52b, но с использованием 0,13 г сукцинимидо 1-[(2'-т-бутоксикарбонил-бифенил-4-ил)метил] -4-(1-гидрокси-2,2-диметилпропил)-2-пропилимидазол-5-карбоксилата, полученного в стадии (f), в результате чего получали 0,12 г целевого соединения в виде стеклообразного вещества.
ЯМР (CDCl3) d ppm: 0,88 (3Н, т. J 7,5 Гц), 0,90 (9Н, с), 1,24 (9Н, с), 1,60 (2Н, секстет, J 7,5 Гц), 2,58 (2Н, т. J 7,5 Гц), 4,65 (1Н, д. J 6 Гц), 5,53 и 5,87 (кажд. 1Н, АВ-квартет, J 16 Гц), 7,02 (2Н, д. J 8 Гц), 7,23-7,48 (5Н, м), 7,78 (1Н д. J 6,5 Гц).
60h) 1-[(2'-карбоксибифенил-4-ил)метил] -4-(1-гидрокси-2,2-диметилпропил)-2-пропилимидазол-5-карбоксамид.
Повторяли процедуру, описанную в примере 52с, но с использованием 149 мг 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил] -4-(1-гидрокси-2,2-диметилпропил)-2-пропилимидазол-5-карбоксамида, полученного в стадии (g), в результате чего получали 96 мг гидрохлорида целевого соединения в виде порошка, т. пл. 160oC (с размягчен.).
ЯМР (ДМСО-d6) d ppm: 0,82 (3Н, т. J 7,5 Гц), 0,90 (9Н, с), 1,53 (2Н, секстет J 7,5 Гц), 2,97 (2Н, т. J 7,5 Гц), 4,79 (1Н, с), 5,69 (2Н, с), 7,19-7,75 (8Н, м).
Пример 61. (5-метил-2-оксо-1,3-диоксолен-4-ил)метил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{ 4-[2-(тетразол -5-ил)фенил]фенил}метилимидазол-5-карбоксилат (соединение 2-17).
61а) (5-метил-2-оксо-1,3-диоксолен-4-ил)метил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{4-[2-(тритилтетразол-5-ил)фенил]фенил}метилимидазол-5-карбоксилат.
Суспензию 0,97 г карбоната калия в 100 мл N,N-диметилацетамида нагревали при 60oC, а затем к нагретой суспензии, размешивая, по капле добавляли раствор 1,14 г (5-метил-2-оксо-1,3-диоксолен-4-ил)метил 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилата, полученного в соответствии с описанием, представленным в получении 31, и 2,35 г 4-[2-(тритилтетразол-5-ил)фенил] бензилбромида в 50 мл N,N-диметилацетамида. реакционную смесь размешивали 3,5 ч при 60oC, а затем разбавляли этилацетатом. Этилацетатный слой отделяли, промывали водой и осушали безводным сульфатом магния, а растворитель отгоняли путем дистилляции при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью гексана и этилацетата (1:1, по объему) в результате чего получали 1,4 г целевого соединения в виде аморфного твердого вещества. Этот продукт кристаллизовали из диизопропилового эфира и получали чистое целевое соединение, т. пл. 98-99oC (с разложением).
ЯМР (CDCl3) d ppm: 0,89 (3Н, т. J 7,5 Гц), 1,62 (6Н, с), 1,6-1,75 (2Н, м), 1,97 (3Н, с), 2,54 (2Н, т. J 8 Гц), 4,70 (2Н, с), 5,30 (2Н, с), 5,61 (1Н, с), 6,68 (2Н, д. J 7,5 Гц), 6,90-7,52 (20Н, м), 7,87(1Н, д. J 7,5 Гц).
61b) (5-метил-2-оксо-1,3-диоксолен-4-ил)метил-4-(1-гидрокси-1-метилэтил)-2-пропил-1-{ 4-[2-(тетразол 5-ил) фенил]фенил} метилимидазол-5-карбоксилат.
Смесь 1,4 г (5-метил-2-оксо-1,3-диоксолен-4-ил метил 4-(1-гидрокси-1метилэтил)-2-пропил-1-{ 4-[2-(тритилтетразол -4-ил)фенил]фенил}метелимидазол-5-карбоксилата, полученного в стадии (а), и 48 мл 75 об./об. водной уксусной кислоты размешивали в течение 1 ч при 60oC, после чего смесь концентрировали путем выпаривания при пониженном давлении. Остаток растворяли в толуоле, а полученный раствор концентрировали путем дистилляции при пониженном давлении, после чего эту операцию повторяли еще раз для удаления остатков воды и уксусной кислоты. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле элюируя смесями метанола и метиленхлорида (1: 9 и 1:4 по объему), в результате чего получали 0,73 г целевого соединения, т. пл. 170-172oC.
ЯМР (CDCl3) d ppm: 0,93 (3Н, т. J 7,5 Гц), 1,63 (6Н, с), 1,6-1,8 (2Н, м), 2,19 (3Н, с), 2,70 (2Н, т. J 7,5 Гц), 5,00 (2Н, с), 5,45 (2Н, с), 6,83 (2Н, д. J 8 Гц), 7,10 (2Н, д. J 8 Гц), 7,42-7,63 (3.м), 7,83 (1Н, дд. J 1 и 7,5 Гц),
Пример 62. Пивалоилоксиметил-4-(1-гидрокси-1-метилэтил)-2-пропил-1-{4-[2(тетразол -5-ил)фенил] фенил}метилимидазол-5-карбоксилат (соединение 2-15).
62а) Пивалоилоксиметил 4-(1-гидрокси-1-метилэтил) 2-пропил-1-{ 4-[2-(тритилтетразол-5-ил)фенил]фенил}метилимидазол -5-карбоксилат.
Повторяли процедуру, описанную в примере 61а, но с использованием 0,85 г пивалоилоксиметил 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилата, полученного в соответствии с описанием в получении 22 ii, 1,52 г 4_ [2-(тритилтетразол-5-ил)фенил] бензилбромида и 0,72 г карбоната калия, в результате чего получали 1,02 г целевого соединения в виде аморфного твердого вещества.
ЯМР-спектр этого соединения был идентичен спектру соединения, полученного в примере 20а.
62b) Пивалоилоксиметил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{ 4-[2-(тетразол -5-ил)фенил]фенил}метилимидазол-5-карбоксилат.
Пивалоилоксиметил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{ 4-[2-тритилтетразол-4-ил)фенил]фенил} метилимидазол-5-карбоксилат, полученный в соответствии с описанием в стадии (а), подвергали детритилированию согласно процедуре, описанной в примере 29b, в результате чего получали гидрохлорид целевого соединения с выходом 80%
Точка плавления и ЯМР-спектр этого соединения были идентичными т. пл. И ЯМР-спектру соединения, полученного согласно описанию в примере 20b.
Пример 63. Фталидил 4-(1-гидрокси-1-метилэтил)-2-пропил-1 -{ 4- [2-(тетразол-5-ил) фенил]фенил}метилимидазол-5-карбоксилат (соединение 2-65).
63а) Фталидил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{4-[2-(тритилтетразол -5-ил)фенил]фенил} метилимидазол-5-карбоксилат.
Повторяли процедуру, описанную в примере 61а, но с использованием 0,456 г фталидил 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилата, полученного согласно описанию в получении 32, 0,736 г 4-[2-(тритилтетразол-4-ил)фенил] бензилбромида и 0,366 г карбоната калия, в результате чего получали 0,196 г целевого соединения, т. пл. 118-120oC.
ЯМР (CDCl3) d ppm: 0,95 (3Н, т. J 7,5 Гц), 1,66 (6Н, с), 1,65-1,80 (2Н, м), 2,60 (2Н, т. J 7,5 Гц), 5,09 (2Н, с), 6,92-7,56 (2Н, м), 7,93 (1Н, дд. J 1 и 8 Гц).
63b) Фталидил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{4-[2-тетразол-5-ил)фенил]фенил} метилимидазол-5-карбоксилат.
В соответствии с процедурой, описанной в примере 61b, 0,196 г фталидил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{ 4-[2-(тритилтетразол-5-ил)фенил] фенил}метилимидазол-5-карбоксилата, полученного в стадии (а), и подвергали дитритилированию путем нагревания с 75% об./об. водной уксусной кислотой, в результате чего получали 0,110 г целевого соединения, т. пл.168-170oC.
ЯМР (CDCl3) d ppm: 0,92 (3Н, т. J 7,5 Гц), 1,57 (6Н, с), 1,60-1677 (6Н, с), 2,65 (2Н, т. J 7,5 Гц), 5,13 (2Н, с), 6,91-7,57 (12Н, м), 7,80 (1Н, д. J 7,5 Гц).
Пример 64. Изопропоксикарбонилоксиметил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{ 4-[2-(тетразол -5-ил)фенил] фенил} метилимидазол-5-карбоксилат (соединение 2-21).
64а) Изопропоксикарбонилоксиметил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{4-[2-(тритилтетразол-5-ил)фенил]фенил}метилимидазол-5-карбоксилат.
Повторяли процедуру, описанную в примере 61а, но с использованием 656 мг изопропоксикарбонилоксиметил-4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилата, полученного согласно описанию в получении 33, 1,20 г 4-[2-(тритилтетразол-5-ил)фенил]бензилбромида и 0,51 г карбоната калия, в результате чего получали 0,78 г целевого соединения в виде вязкого маслянистого продукта.
ЯМР (CDCl3) d ppm: 0,87 (3Н, т. J 7,5 Гц), 1,24 (6Н, д. J 6 Гц), 1,63 (6Н, с), 1,65-1,80 (2Н, м), 2,52 (2Н, т. J 7,5 Гц), 4,87 (1Н, квинтет, J 6 Гц), 5,35 (2Н, с), 5,42 (1Н, с), 5,66 (2Н, с), 6,74-7,87 (22Н, м), 7,87-7,96 (1Н, м).
64b) Изопропоксикарбонилоксиметил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{4-[2-(тетразол-5-ил)фенил]фенил} метилимидазол-5-карбоксилат.
В соответствии с процедурой, описанной в примере 61b, 0,78 г изопропоксикарбонилосиметил 4-(1-гидрокси-1-метил-этил)-2-пропил-1-{ 4-[2-(тритилтетразол-5-ил)фенил]фенил}метилимидазол-5-карбоксилата, полученного в стадии (а), подвергали детритилированию путем нагревания с 75% об./об. водной уксусной кислотой, в результате чего получали 0,48 г целевого получения в виде аморфного твердого вещества.
ЯМР (CDCl3) d ppm: 0,96 (3Н, т. J 7,5 Гц), 1,21 (6Н, д. J 6 Гц), 1,63 (6Н, с), 1,72 (2Н, секстет J 7,5 Гц), 2,60 (2Н, т. J 7,5 Гц), 4,72 (1Н, квинтет J 6,5 Гц), 5,33 (2Н, с), 5,76 (2Н, с), 6,77 (2Н, д. J 7,5 Гц), 6,92 (2Н, д. J 7,5 Гц), 7,37-7,60 (3Н, м.) 7,87 (1Н, д. J 7,5 Гц).
Пример 65. Этил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]2-этил-4-1-гидрокси-1-метилэтил имидазол-5-карбоксилат (соединение 1-130).
0,337 г т-бутоксида калия добавляли к раствору 0,68 г этил 2-этил-4-(1-гидрокси-1-метилэтил)имидазол-5-карбоксилита, полученного в соответствии с описанием в получении 37, в 7 мл N,N-диметилацетамида, и полученную смесь размешивали 10 мин при комнатной температуре. Затем к полученному раствору добавляли 1,04 г т-бутил-4'-бромометилбифенил-2-карбоксилата и реакционную смесь размешивали 4 ч при комнатной температуре. По истечение этого времени эту смесь смешивали с этилацетатом и водой. Этилацетатный слой отделяли, осушали безводным сульфатом магния и концентрировали путем выпаривания при пониженном давлении. Остаток очищали путем колоночной хроматографии на силикагеле, элюируя смесью этилацетата и гексана (1:1, по объему), в результате чего получали 1,32 г целевого соединения в виде смолы.
ЯМР (CDCl3) d ppm: 1,23 (9Н, с), 1,23 (3Н, т. J 7,5 Гц), 1,29 (3Н, т. J 7,5 Гц), 1,63 (6Н, с), 2,73 92Н, кв. J 7,5 Гц), 4,26 (2Н, кв. J 7,5 Гц), 5,54 (2Н, с), 6,73 (1Н, с), 6,98 (2Н, д. J 8,5 Гц), 7,5-7,9 (6Н, м).
Пример 66. Этил 1-[(2'-карбоксибифенил-4-ил)метил]-2-этил-4-(1-гидрокси-1-метилэтил) имидазол-5-карбоксилат (соединение 1-131).
Повторяли процедуру, описанную в примере 7, но с использованием 1,32 г этил 1-[(2'-т-бутоксикарбонилфенил-4-ил)метил] -2-этил-4-(1-гидрокси -1-метилэтил)имидазол-5-карбоксилата, полученного согласно описанию в примере 65, и 4 н. раствора хлорводорода в диоксане, в результате чего получали 0,94 г гидрохлорида целевого соединения в виде аморфного порошка.
ЯМР (ДМСО-d6) d ppm: 1,09 (3Н, т. J 7,5 Гц), 1,15 (3Н, т. J 7,5 Гц), 1,61 (6Н, с), 3,03 (2Н, кв. J 7,5 Гц), 4,22 (2Н, кв. J 7,5 Гц), 5,64 (2Н, с), 7,16 (2Н, д, J 8,5 Гц), 7,32-7,75 (6Н, м).
Пример 67. 1-[(2'-Карбоксибифенил-4-ил)метил]-2-этил-4-(1-гидрокси-1-метилэтил) имидазол-5-карбоновая кислота (соединение 1-132).
Повторяли процедуру, описанную в примере 17, но с использованием 0,40 г гидрохлорида этил 1-[(2'-карбоксифенил-4-ил)метил]-2-этил-4-(1-гидрокси-1-метилэтил) имидазол-5-карбоксилата, полученного в примере 66, и 0,18 г моногидрата гидроксида лития, в результате чего получали 0,25 г целевого соединения в виде аморфного порошка.
ЯМР (ДМСО-d6) d ppm: 1,17 (3Н, т, J 7,5 Гц), 1,64 (6Н, с), 2,85 (2Н, кв, J 7,5 Гц), 5,74 (2Н, с), 7,10 (2Н, д, J 8 Гц), 7,30-7,76 (6Н, м).
Пример 68. Этил 2-этил-4-(1-гидрокси-1-метилэтил)-1-{4-[2-(тетразол-5-ил -фенил]фенил} метилимидазол-5-карбоксилат (соединение 1-72).
65а) Этил 2-этил-4-(1-гидрокси-1-метилэтил)-1-{4-[2-(тритилтетразол -4-ил)фенил]фенил} метилимидазол-5-карбоксилат.
0,52 г т-бутоксид калия добавляли к раствору 1,00 г этил 2-этил-4-(1-гидрокси-1-метилэтил)имидазол-5-карбоксилата, полученного согласно описанию в получении 37, в 26 мл N,N-диметилацетамида, и полученную смесь размешивали 10 мин при комнатной температуре. К полученному раствору по капле добавляли раствор 2,71 г 4-[2-(тритилтетразол-5-ил)фенил]бензилбромида в 35 мл N,N-диметилацетамида, после чего реакционную смесь размешивали 4 ч при 50oC. После этого реакционную смесь обрабатывали способом, аналогичным описанному в примере 18а, в результате чего 2,01 г целевого соединения в виде кристаллов, т. пл. 150-152oC.
ЯМР (CDCl3) d ppm: 1,10 (3Н, т, J 7,5 Гц), 1,18 (3Н, т. J 7,5 Гц), 1,65 (6Н, с), 2,52 (2Н, кв. J 7,5 Гц), 4,14 (2Н, кв. J 7,5 Гц), 5,35 (2Н, с), 5,80 (1Н, с), 6,73 (2Н, д, J 8,5 Гц), 6,93-7,52 (20Н, м), 7,87 (1Н, д J 7,5 Гц).
68b) Этил 2-Этил-4-(1-гидрокси-1-метилэтил)-1-{4-[2-(тетразол-5 -ил)фенил] фенил}метилимидазол-5-карбоксилат.
Раствор 1,9 г этил 2-этил-4-(1-гидрокси-1-метилэтил)-1-{ 4-[2'-(тритилтетразол-5-ил)фенил] фенил}метилимидазола-5-карбоксилата, полученного в стадии (а), в 28 мл 75% об./об. водной уксусной кислоты размешивали 2 ч при 60oC. После этого реакционную смесь разбавляли 7 мл воды и охлаждали при комнатной температуре. Осажденный триэтиловый спирт удаляли путем фильтрации и фильтрат концентрировали путем выпаривания при пониженном давлении. Сиропообразный остаток кристаллизовали в диизопропиловом эфире, в результате чего получали 1,21 г целевого соединения, т. пл. 166-167o.
ЯМР (CDCl3) d ppm: 1,14 (3Н, т, J 7,5 Гц), 1,20 (3Н, т, J 7,5 Гц), 1,48 (6Н, с), 2,52 (2Н, кв. J 7,5 Гц), 4,19 (2Н, кв. J 7,5 Гц), 5,41 (2Н, с), 6,79 (2Н, д, J 8,5 Гц), 7,09 (2Н, д, J 8,5 Гц), 7,41-7,62 (3Н, м), 7,85 (1Н, д, J 7,5 Гц).
Пример 69. 2-Этил-4-(1-гидрокси-1-метилэтил)-1-{4-[(тетразол-5-ил)фенил] фенил}метилимидазол-5-карбоновая кислота.
Раствор 0,54 г моногидрата гидроксида лития в 10 мл воды добавляли к раствору этил 2-этил-4-(1-гидрокси-1-метил-этил-1-{4-[2-(тетразол-5-ил) фенил] фенил} метилимидазол-5-карбоксилата, полученного согласно описанию в примере 68b, в 10 мл диоксана и полученную смесь размешивали 4 ч при комнатной температуре. После этого диоксан удаляли путем выпаривания при пониженном давлении и к полученному водному остатку добавляли 12,6 мл 1 н. водной соляной кислоты. Осажденные кристаллы собирали путем фильтрации и получали в результате 0,93 г целевого соединения, т. пл. 179-181oC.
ЯМР (ДМСО-d6) d ppm: 1,09 (3Н, т, J 7,5 Гц), 1,55 (6Н, с), 2,63 (2Н, кв. J 7,5 Гц), 5,65 (2Н, с), 6,96 (2Н, д, J 8,5 Гц), 7,03 (2Н, д, J 8,5 Гц), 7,08-7,64 (4Н, м).
Пример 70. Этил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{4-[2-(тетразол -5-ил)фенил]фенил} метилимидазол-5-карбоксилат (соединение 2-7).
70а) Этил 1-(2'-цианобифенил-4-ил)метил-4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилат.
Повторяли процедуру, описанную в примере 68а, но с использованием 4,01 г этил 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилата, полученного согласно описанию в получении 9, 5,0 г 4'-бромометилбифенил-2-карбонитрила и 1,97 г т-бутоксида калия, в результате чего получали 6,86 г целевого соединения в виде кристаллов, т. пл. 92-93oC.
ЯМР (CDCl3) d ppm: 0,97 (3Н, т, J 7,5 Гц), 1,16(3Н, т, J 7 Гц), 1,65 (6Н, с), 1,74 (2Н, секстет, J 7,5 Гц), 2,67 (2Н, т, J 7,5 Гц), 4,24 (2Н, кв. J 7 Гц), 5,52 (2Н, с), 5,77 (1Н,с), 7,05 (2Н,д, J 8,5 Гц), 7,42-7,67 (5Н, м), 7,76 (1Н, д, J 8 Гц).
70b Этил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{ 4-[2-(тетразол -5-ил)фенил]фенил}метилимидазол-5-карбоксилат.
Раствор 2,00 г этил 1-(2'-цианобифенил-4-ил)метил-4-1-гидрокси-1-метилэтил-2-пропилимидазол -5-карбоксилата, полученного в стадии (а), и 2,00 г азида трибутилолова в 15 мл толуола размешивали 5 дней при 100oC. По истечение этого времени реакционную смесь концентрировали путем выпаривания при пониженном давлении, а остаток растворяли в 30 мл 4 н. раствора хлорводорода в диоксане. Раствор оставляли на 16 ч при комнатной температуре, после чего его концентрировали путем выпаривания при пониженном давлении. Остаток растирали в диизопропиловом эфире, в результате чего получали 2,00 г гидрохлорида целевого соединения.
ЯМР-спектр этого соединения был идентичен спектру соединения, полученного в примере 18b.
Пример 71. Этил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{4-[2-(тетразол -4-ил)фенил]фенил} метилимидазол-5-карбоксилат (соединение 2-7).
72а) Этил 1-{4-[2-(т-бутиламинокарбонил) фенил]фенил}метил-4-1-гидрокси-1-метилэтил-2-пропилимидазол-5-карбоксилат.
Повторяли процедуру, описанную в примере 68а, но с использованием 4,16 г этил 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилата, полученного согласно описанию в получении 9, 6,00 г N-т-бутил-4'-бромометилбифенил-2-карбоксамида, полученного согласно описанию в получении 38, и 2,14 г т-бутоксида калия, в результате чего получали 5,87 г целевого соединения в виде кристаллов, т. пл.145-146oC.
ЯМР (CDCl3) d ppm: 0,97 (3Н, т, J 7,5 Гц), 1,12 (9Н, с), 1,24 (3Н, т, J 7 Гц), 1,64 (6Н, с), 1,75 (2Н, секс. J 7,5 Гц), 2,66 (2Н, т, J 7,5 Гц), 4,25 (2Н, кв. J 7 Гц), 5,03 (1Н, с), 5,52 (2Н, с), 5,69 (1Н, с), 6,98 (2Н, д, J 8,5 Гц), 7,28-7,47 (5Н, м), 7,65 (1Н, д, J Гц).
71b) Этил 1-(2'-цианобифенил-4-ил)метил-4-(1-гидрокси-1-метилэтил)-2-пропилимидазол -5-карбоксилат.
0,345 мл оксалилхлорида по капле добавляли, охлаждая при этом льдом, к раствору 1,00 г этил 1-{4-[2-(т-бутиламинокарбонил)фенил]фенил}метил-4-(1-гидрокси-1-метилэтил) -2-пропилимидазол-5-карбоксилата, полученного в стадии (а), в 10 мл метиленхлорида, и полученную смесь размешивали 2 ч при той же температуре. После этого реакционную смесь разбавляли водным раствором бикарбоната натрия и этилацетата, затем слой этилацетата отделяли, осушали безводным сульфатом магния и концентрировали путем выпаривания при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью этилацетата и гексана (1:1, по объему), в результате чего получали 0,69 г целевого соединения в виде кристаллов.
Точка плавления и ЯМР-спектр этого соединения идентичны аналогичным показателям соединения, полученного в примере 70а.
71с) Этил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{ 4-[2-(тетразол -5-ил)фенил]фенил}метилимидазол-5-карбоксилат.
Повторяли процедуру, описанную в примере 70b, но с использованием этил 1-(2'-цианобифенил-4-ил)метил-4-(1-гидрокси-1-метилэтил)-2-проппилимидазол -5-карбоксилата, полученного в стадии (b), в результате чего получали целевое соединение с выходом 91%
ЯМР-спектр этого соединения идентичен спектру соединения, полученного в примере 18b.
Пример 72. Этил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{4-[2-(тетразол-5-ил)фенил]фенил} метилимидазол-5-карбоксилат (соединение 2-7).
72а) Этил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)-метил]-4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилат.
Повторяли процедуру, описанную в примере 68а, но с использованием 4,80 г этил 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилата, полученного согласно описанию в получении 9, 6,94 г т-бутил 4'-бромометилбифенил-2-карбоксилата и 2,28 г т-бутоксида калия, в результате чего получали 7,50 г целевого соединения в виде кристаллов, т. пл. 90-91oC.
ЯМР (CDCl3) d ppm: 0,97 (3Н, т, J 7 Гц), 1,23 (3Н, т. J 7 Гц), 1,25 (9Н, с), 1,60 (6Н, с),1,82 (1Н, секстет, J 7 Гц), 2,67 (2Н, т, J 7 Гц), 4,24 (2Н, кв. J 7 Гц), 5,51 (2Н, с), 5,72 (1Н, с), 6,87-7,85 (8Н,м).
72b) Этил 1-[(2'-карбоксибифенил-4-ил)метил] -4-(1-гидрокси-1-метилэтил)-2-пропилимидазол -5-карбоксилат.
Повторяли процедуру, описанную в примере 18b, но с использованием 0,80 г этил 1-[(2'-т-бутоксикарбонилбифенил-4-ил)метил]-4-(1-гидрокси-1-метилэтил) -2-пропилимидазол-5-карбокслита полученного в стадии (а), и 4 н. раствора хлорводорода в диоксане, в результате чего получали гидрохлорид целевого соединения в виде аморфного порошка.
ЯМР (ДМСО-d6) d ppm: 0,88 (3Н, т, J 7 Гц), 1,14 (3Н, т, J 7 Гц), 1,50-1,65 (2Н, м), 1,60 (6Н, с), 3,00 (2Н, т, J 7 Гц), 4,20 (2Н, кв. J 7 Гц), 5,63 (2Н, с), 7,13-7,75 (8Н, м),
72(с) Этил 1-[(2'-карбамоилбифенил-4-ил)метил]-4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилат.
3 мл оксалилхлорида по капле добавляли, охлаждая при этом льдом, к раствору 4,00 г гидрохлорида этил 1-[(2'-карбоксибифенил-4-ил)метил]-4-(1-гидрокси-1-метилэтил)-2-пропилимидазол -5-карбоксилата полученного в стадии (b), см. выше] в 40 мл метиленхлорида, и полученную смесь размешивали 2 ч при комнатной температуре. Затем реакционную смесь концентрировали путем выпаривания при пониженном давлении. После этого к остатку добавляли бензол и смесь снова концентрировали путем выпаривания при пониженном давлении в целях удаления оставшегося оксалилхлорида. Кристаллический остаток суспендировали в 100 мл этилацетата и смешивали с 15 мл концентрированного аммиака, охлаждая при этом льдом, после чего смесь размешивали 10 минут при комнатной температуре. Затем этилацетатный слой отделяли, промывали водой, осушали безводным сульфатом магния, и концентрировали путем выпаривания при пониженном давлении. Кристаллический остаток промывали диизопропиловым эфиром и получали 2,97 г целевого соединения с т.пл. 148-151oC.
ЯМР (CDCl3) d ppm: 0,96 (3Н, т, J 7,5 Гц), 1,19 (3Н, т, J 7 Гц), 1,64 (6Н, с), 1,73 (2Н, секстет, J 7,5 Гц), 2,65 (2Н, т J 7,5 Гц), 4,24 (1Н, кв, J 7 Гц), 5,36 (1Н, шир.с), 5,49 (2Н, с), 5,66 (1Н, шир.с), 5,76 (1Н, с), 6,99 (2Н, д, J 8 Гц), 7,32-7,53 (5Н, м), 7,71 (1Н, д, J 6 Гц).
72d) Этил 1-(2'-цианобефенил-4-ил)метил-4-(1-гидрокси-1-метилэтил)-2-пропилимидазол -5-карбоксилат.
264 мкл ангидрида трифторуксусной кислоты добавляли, охлаждая при этом в бане, содержащей смесь льда и хлорида натрия, к раствору 0,70 г этил 1-[(2'-карбамолибифенил-4-ил)-метил] -4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилата, полученного в стадии (с), и 0,43 мл триэтиламина в 7 мл метиленхлорида и полученную смесь размешивали 30 мин при той же температуре. После этого реакционную смесь разбавляли водным раствором бикарбоната натрия и этилацетата, этилацетатный слой отделяли, осушали безводным сульфатом магния и концентрировали путем выпаривания при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью этилацетата и гексана (1:1, по объему), в результате чего получали 0,60 г целевого соединения в виде кристаллов.
Температура плавления и ЯМР-спектр этого соединения идентичны аналогичным показателям соединения, полученного в примере 70а.
72e) Этил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{ 4-[2-(тетразол -5-ил)фенил]фенил}метилимидазол-5-карбоксилат.
Повторяли процедуру, описанную в примере 70b, но с использованием этил 1-(2'-цианобифенил-4-ил)метил-4-(1-гидрокси-1-метилэтил)-2-пропилимидазол -5-карбоксилата, полученного в стадии (d), в результате чего получали целевое соединение с выходом 90%
ЯМР-спектр этого соединения идентичен спектру соединения, полученному в примере 18b.
Пример 73. Пивалоилокси 4-(1-гидроксиэтил)-2-пропил-1-{4-[2-тетразол-5-ил)фенил]фенил} метилимидазол-5-карбоксилат (соединение 4-31).
73а) Пивалоилоксиметил 4-(1-гидроксиэтил)-2-пропил-1-{4-[2-(тритилтетразол -4-ил)фенил]фенил} метилимидазол-5-карбоксилат.
Раствор 196 мг моногидрата гидроокиси натрия в 15 мл воды добавляли к раствору 2,87 г этил 4-(1-гидроксиэтил0-2-пропил-1-{4-[2-(тритилтетразол -4-ил)фенил]фенил}метилимидазол-5-карбоксилата, полученного согласно описанию в примере 42а, в 30 мл диоксана и полученную смесь размешивали 16 ч при комнатной температуре. После этого к смеси добавляли небольшой кусочек сухого льда, а затем смесь концентрировали путем выпаривания при пониженном давлении досуха. Остаток растворяли в 40 мл N,N-диметилацетамида, и к раствору добавляли 0,45 г карбоната калия, а затем 1,1 мл пивалоилоксихлорида. Полученную смесь размешивали 3 ч при 50oC. После этого к реакционной смеси добавляли воду и этилацетат, этилацетатный слой отделяли, осушали безводным сульфатом магния и концентрировали путем выпаривания при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью этилацетата и гексана (1:1, по объему) и получали 2,41 г целевого соединения в виде аморфного порошка.
ЯМР (CDCl3) d ppm: 0,88 (3Н, т, J 7,5 Гц), 1,17 (9Н, с), 1,50 (3Н, д, J 6 Гц), 1,69 (2Н, секс. J 7,5 Гц), 2,51 (2Н, т, J 7,5 Гц), 3,62 (1Н, д, J 8 Гц), 5,17-5,291Н, м), 5,37 (1Н, д, J 16,5 Гц), 5,46 (1Н, д, J 16,5 Гц), 5,77 (1Н, д, J 5,5 Гц), 5,82 (1Н, д, J 5,5 Гц), 6,75 (2Н, д, J 8,5 Гц), 6,92-7,89 (20Р, м), 7,90 (1Н, д, J 7,5 Гц).
73b) Пивалоилоксиметил 9-(1-гидроксиэтил)-2-пропил-1-{4-[2-(тетразол-5-ил)фенил]фенил} метилимидазол -5-карбоксилат.
Повторяли процедуру, описанную в примере 35с, но с использованием 2,87 г пивалоилоксиметил 4-(1-гидроксиэтил)-2-пропил-1-{ 4-[2-(тритилтетразол -5-ил)фенил]фенил}метилимидазол-5-карбоксилата, полученного в стадии (а), и 75% об. /об. водной уксусной кислоты, в результате чего получали 1,21 г целевого соединения в виде порошка.
ЯМР (CDCl3) d ppm: 0,90 (3Н, т, J 7,5 Гц), 1,13 (9Н, с), 1,43 (3Н, д, J 6,5 Гц), 1,67 (2Н, секст. J 7,5 Гц), 2,55 93Н, т, J 7,5 Гц), 5,16 (1Н, кв. J 6,5 Гц), 5,40 (1Н, д, J 16,5 Гц), 5,51 (1Н, д, J 16,5 Гц), 5,80 (1Н, д, J 6 Гц), 5,85 (1Н, д, J 6 Гц), 6,86 (2Н, д, J 8 Гц), 7,08 (2Н, д, J 8 Гц), 7,40-7,61 (3Н, м), 7,92 (1Н, д, J 7,5 Гц),
Пример 74. 4-(1-Гидрокси-2,2-диметилпропил)-2-пропил-1-{4-[2-(тетразол -5-ил-фенил]фенил} метилимидазол-5-карбоксамид (соединение 5-37).
74а) 2-Пропил-4-пивалоил-1-{ 4-[2-(тритилтетразол-5-ил)фенил] фенил}метилимидазол -5-карбонитрил.
1,08 г т-бутоксида калия добавляли, охлаждая при этом льдом, к раствору 2,00 г 2-пропил-4-пивалоилимидазол-5-карбонитрила, полученного в соответствии с описанием, приведенным в получении 41, в 20 мл N,N-диметилацетамиде и полученную смесь размешивали 10 мин при той же температуре. Затем к раствору добавляли 6,10 г 4-[2-(тритилтетразол-5-ил)фенил]-бензилбромида и полученную смесь размешивали 4 ч при 50oC. После этого к смеси добавляли этилацетат и воду, а этилацетатный слой отделяли, осушали безводным сульфатом магния и концентрировали путем выпаривания при пониженном давлении. Сиропообразный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесями этилацетата и гексана (1: 3 и 1:2, по объему), в результате чего получали целевое соединение в виде кристаллов, т. пл. 107-100oC.
ЯМР (CDCl3) d ppm: 0,92 (3Н, т, J 7,5 Гц), 1,42 (9Н, с), 1,72 (2Н, секст. J 7,5 Гц), 2,50 (2Н, т, J 7,5 Гц), 5,09 (2Н, с), 6,92 (2Н, д, J 8 Гц), 7,13-7,53 (20Н, м), 7,95 (1Н, д, J 7 Гц).
74b) 4-(1-гидрокси-2,2-диметилпропил)-2-пропил-1-{4-[2-(тритилтетразол-5-ил)фенил] фенил}метилимидазол-5-карбонитрил.
Раствор 108 мг борогидрида натрия в 20 мл этанола добавляли к раствору 2,00 г 2-пропил-4-пивалоил-1-{4-[2-(тритилтетразол-5-ил)фенил] фенил}метилимидазол -5-карбонитрила, полученного в стадии (а), в 40 мл тетрагидрофурана и полученную смесь размешивали 2,5 ч при комнатной температуре. После этого реакционную смесь концентрировали путем выпаривания при пониженном давлении, а остаток растворяли в смеси этилацетата и воды. Этилацетатный слой отделяли, промывали водой, осушали безводным сульфатом магния и концентрировали путем выпаривания при пониженном давлении. Сиропообразный остаток кристаллизовали в смеси этилацетата и гексана (1:4, по объему) и получали 1,93 г целевого соединения в виде кристаллов, т. пл. 115-117oC.
ЯМР (CDCl3) d ppm: 0,87 (3Н, т, J 7,5 Гц), 0,99 (9Н, с), 1,64 (2Н, секст. J 7,5 Гц), 2,49 (2Н, т, J 7,5 Гц), 2,76 (1. д, J 7,5 Гц), 4,46 (1Н, д, J 7,5 Гц), 5,04 (2Н, с), 6,85-7,53 (22Н, м), 7,95 (1Н, д, J 7,5 Гц).
74с) 4-(1-Гидркоси-2,2-диметилпропил)-2-пропил-1-{ 4-[2-(тетразол-5-ил)фенил]фенил} метилимидазол-5-карбонитрил.
Суспензию 1,65 г 4-(1-гидрокси-2,2-диметилпропил)-2-пропил-1-{ 4-[2-(тритилтетразол -5-ил)]фенил}фенил метилимидазол-5-карбонитрила, полученного в соответствии с описанием в стадии (b), в 24 мл 75% об./об. водной уксусной кислоты размешивали 2 ч при 60oC. После этого 6 мл воды добавляли к реакционной смеси и охлаждали льдом. Осажденный тритиловый спирт удаляли путем фильтрации, а фильтрат концентрировали путем выпаривания при пониженном давлении, в результате чего получали 1,07 г целевого соединения в виде стеклообразного вещества.
ЯМР (CDCl3) d ppm: 0,87 (3Н, т, J 7,5 Гц), 0,92 (9Н, с), 1,63 (2Н, секс. J 7,5 Гц), 2,58 (2Н, т, J 7,5 Гц), 4,36 (1Н, с), 5,15 (2Н, с), 7,00 (2Н, д, J 8 Гц), 7,07 (2Н, д, J 8 Гц), 7,30-7,61 (3Н, м), 7,80 (1Н, д, J 7,5 Гц).
74d) 4-(1-Гидрокси-2,2-диметилпропил)-2-пропил-1-{ 4-[2-(тетразол -5-ил)фенил]фенил} метилимидазол-5-карбоксамид
Смесь 0,70 г 4-(1-гидрокси-2,2-идметилпропил)-2-пропил-1-{ 4-[2-(тетразол-5-ил)фенил]фенил}метилимидазол-5-карбонитрила, полученного согласно описанию в стадии (с), в 14 мл 1 н. водного гидроксида натрия и 7 мл этанола нагревали с обратным холодильником в течение 2 ч. После этого этанол удаляли из реакционной смеси путем выпаривания при пониженном давлении и к остатку добавляли этилацетат и 14 мл 1 н. водной соляной кислоты. Этилацетатный слой отделяли, осушали безводным сульфатом магния и концентрировали путем выпаривания при пониженном давлении, в результате чего получали 0,45 г целевого соединения в виде порошка, т. пл. 174-175oC.
ЯМР (ДМСО-d6) d ppm: 0,83 (3н, т, J 7,5 Гц), 0,88 (9Н, с), 1,44-1,63 (2Н, м), 2,46 (2Н, т, J 7,5 Гц), 4,45 (1Н, с), 5,39 (1Н, д, J 16 Гц), 5,77 (1Н, д, J 16 Гц), 6,20 (1Н, д, J 4,5 Гц), 6,91 (2Н, д, J 8,5 Гц), 7,04 (2Н, д, J 8,5 Гц), 7,47-7,63 (4Н, м).
Пример 75. 2-Бутил-4-(1-гидрокси-2,2-диметилпропил)-1-{4-[2-(тетразол-5-ил)фенил] фенил}метилимидазол-5-карбоксамид (соединение 5-99).
75а) 2-Бутил-4-пивалоил-1-{4-[2-(тритилтетразол-5-ил)фенил]фенил}метилимидазол -5-карбонитрил.
Повторяли процедуру, описанную в примере 74а, но с использованием 2,04 г 2-бутил-4-пивалоилимидазол-5-карбонитрила, полученного в соответствии с описание в получении 40, 5,6 г 4_[2-(тритилтетразол-5-ил)фенил]бензилбромида и 1,06 г т-бутоксида калия, в результате чего получали 5,43 г целевого соединения в виде кристаллов, т. пл. 103-103oC.
ЯМР (CDCl3) d ppm: 0,88 (3Н, т, J 7,5 Гц), 1,32 (2Н, секст. J 7,5 Гц), 1,41 (9Н, с), 1,66 (2Н, квин. J 7,5 Гц), 2,53 (2Н, т, J 7,5 Гц), 5,09 (2Н, с), 6,91-7,50 (22Н, м), 7,96 (1Н, д, J 7,5 Гц).
75b) 2-Бутил-4-(1-гидрокси-2,2-диметилпропил)-1-{4-[2-(тритилтетразол-5-ил)фенил]фенил} метилимидазол-5-карбонирил.
Повторяли процедуру, описанную в примере 74b, но с использованием 4,03 г 2-бутил-4-пивалоил-1-{ 4-[2-(тритилтетразол-5-ил)фенил] фенил} метилимидазол -5-карбонитрила, полученного согласно описанию в стадии (а), и 0,22 г борогидрида натрия, в результате чего получали 3,79 г целевого соединения в виде кристаллов, т. пл. 134-135oC.
ЯМР (CDCl3) d ppm: 0,85 (3Н, т, J 7,5 Гц), 0,99 (9Н, с), 1,27 (2Н, секст. J 7,5 Гц), 2,52-1,67 (2Н, м), 2,51 (2Н, т, J 7,5 Гц), 2,74 (1Н, д, J 7,5 Гц), 4,45 (1Н, д, J 7,5 Гц), 5,04 (2Н, с), 6,85-7,53 (22Н, м), 7,95 (1Н, д, J 7,5 Гц).
75с) 2-Бутил-4-(1-Гидркоси-2,2-диметилпропил)-1-{ 4-[2-(тетразол -5-ил)фенил]фенил}метилимидазол-5-карбонитрил.
Повторяли процедуру, описанную в примере 74с, но с использованием 1,00 г 2-бутил-4-(1-гидрокси-2,2-диметилпропил)-1-{4-[2-(тритилтетразол-5-ил)фенил] фенил} метилимидазол-5-карбонитрила, полученного согласно описанию в стадии (b), в 75% об./об. водной уксусной кислоты, в результате чего получали 0,65 г целевого соединения в виде стеклообразного вещества.
ЯМР (CDCl3) d ppm: 0,91 (3Н, т, J 7,5 Гц), 0,96 (9Н, с), 1,28-1,42 (2Н, м), 1,58-1,74 (2Н, м), 2,69 (2Н, т, J 7,5 Гц), 4,40 (1Н, с), 5,21 (2Н, с), 7,10-7,32 (4Н, м), 7,43-7,65 (3Н, м), 8,06 (1Н, д, J 8 Гц).
75d) 2-Бутил-4-(1-гидрокси-2,2-диметилпропил)-1-{ 4-[2-(тетразол -5-ил)фенил]фенил}метилимидазол-5-карбоксамид.
Повторяли процедуру, описанную в примере 74d, но с использованием 0,34 г 2-бутил-4-(1-гидрокси-2,2-диметилпропил)-1-{4-[2-(тетразол-5-ил)фенил]фенил} метилимидазол-5-карбонитрила, полученного согласно описанию в стадии (с), в 1 н. водном растворе гидроксида натрия, в результате чего получали 0,30 г целевого соединения в виде порошка, т. пл. 157-160oC.
ЯМР (ДМСО-d6) d ppm: 0,79 (3Н, т, J 7,5 Гц), 0,88 (9Н, с), 1,16-1,30 (2Н, м), 1,39-1,54 (2Н, м), 2,59 (2Н, т, J 7,5 Гц), 4,51 (1Н, с), 5,46 (1Н, д, J 16 Гц), 5,73 (1Н, д, J 16 Гц), 6,21 (1Н, д, J 4,5 Гц), 6,97 (2Н, д, J 8,5 Гц), 7,06 (2Н, д, J 8,5 Гц), 7,51-7,70 (4Н, м).
Пример 76. 4-(1-Гидрокси-2-метилпропил)-2-пропил-1-{4-[2-(тетразол -5-ил)-фенил]фенил} метилимидазол-5-карбоксамид (соединение 5-36).
76а) 4-Изобутирил-2-пропил-1-{4-[2-(тритилтетразол-5-ил)фенил]фенил}метилимидазол-5-карбонитрил.
Повторяли процедуру, описанную в примере 74а, но с использованием 0,97 г 4-изобутирил-2-пропилимидазол-5-карбонитрила, полученного согласно описанию в получении 39, 2,90 г 4-[2-(тритилтетразол-5-ил)фенил]бензилбромида и 0,56 г т-бутоксида калия, в результате чего получали 1,90 г целевого соединения в виде кристаллов, т. пл. 133-134oC.
ЯМР (CDCl3) d ppm: 0,91 (3Н, т, J 7,5 Гц), 1,22 (6Н, д, J 6,5 Гц), 1,69 (2Н, секстет, J 7,5 Гц), 2,54 (2Н, т, J 7,5 Гц), 3,64 (1Н, квин. J 6,5 Гц), 5,12 (2Н, с), 6,7-8,0 (23Н, м).
76b) 4-(1-Гидрокси-2-метилпропил)-2-пропил-1-{ 4-[2-(тритилтетразол-5-ил)фенил]фенил} метилимизазол-5-карбонитрил.
Повторяли процедуру, описанную в примере 75b, но с использованием 1,60 г 4-изобутирил-2-пропил-1-{ 4-[2-(тритилтетразол-5-ил)фенил] фенил} метилимидазол-5-карбонитрила, полученного согласно описанию в стадии (а), и 0,13 г борогидрина натрия, в результате чего получали 1,50 г целевого соединения в виде кристаллов, т. пл. 154-155oC.
ЯМР (CDCl3) d ppm: 0,87 (3Н, т. J 7,5 Гц), 0,94 (3Н, д, J 6,5 Гц), 1,00 (3Н, д, J 6,5 Гц), 1,66 (2Н, секст. J 7,5 Гц), 2,12 (1Н, секст. J 6,5 Гц), 2,50 (2Н, т, J 7,5 Гц), 4,54 (1Н, д, J 6 Гц), 5,04 (2Н, с), 6,85-6,95 (6Н, м), 7,14 (2Н, д, J 8,5); 7,23-7,53 (14Н, м), 7,94 (1Н, д, J 7,5 Гц).
76с) 9-(1-Гидрокси-2-метилпропил)-2-пропил-1-{ 4-[2-(тетразол -5-ил)фенил]фенил}метилимидазол-5-карбонитрил.
Повторяли процедуру, описанную в примере 74с, но с использованием 1,36 г 4-(1-гидрокси-2-метилпропил)-2-пропил-1-{ 4-[2-(тритилтетразол -5-ил)фенил] фенил} метилимидазол-5-карбонитрила, полученного в стадии (b), в 75% об./об. водной уксусной кислоты, в результате чего получали 0,87 г целевого соединения в виде стеклообразного вещества.
ЯМР (CDCl3) d ppm: 0,77 (3Н, д, J 6,5 Гц), 0,81 (3Н, д, J 7,5 Гц), 0,93 (3Н, д, J 6,5 Гц), 1,54 (2Н, секстет, J 7,5 Гц), 1,92-2,07 (1Н, м), 2,55 (2Н, т, J 7,5 Гц), 4,33 (1Н, д, J 7,5 Гц), 5,12 (2Н, с), 6,96-6,99 (4Н, м), 7,35-7,69 (3Н, м), 7,71 (1Н, д, J 7,5 Гц).
76d) 4-(1-Гидркоси-2-метилпропил)-2-пропил-1-{ 4-[2-(тетразол -5-ил)фенил]фенил}метилимидазол-5-карбоксамид.
Повторяли процедуру, описанную в примере 74d, но с использованием 0,90 г 4-(1-гидрокси-2-метилпропил)-2-пропил-1-{ 4-[2-(тетразол -5-ил)фенил]фенил} метилимидазол-5-карбонитрила, полученного в стадии (с), в 1 н. водного раствора гидроксида натрия, в результате чего получали 0,64 г целевого соединения в виде порошка, т. пл. 153-157oC.
ЯМР (ДМСО-d6) дельта, ppm: 0,69 (3Н, д, J 6,5 Гц), 0,81 (3Н, т, J 6,5 Гц), 0,99 (3Н, т, J 6,5 Гц), 1,49 (2Н, секст. J 7,5 Гц), 2,05 (1Н, квин. J 6,5 Гц), 2,68 (2Н, т, J 7,5 Гц), 4,45 (1Н, д, J 7,5 Гц), 5,55 (1Н, д, J 16,5 Гц), 5,70 (1Н, д, J 16,5 Гц), 7,02 (2Н, д, J 8,5 Гц), 7,08 (2Н, д, J 8,5 Гц), 7,51-7,71 (4Н, с).
Пример 77. 2-Бутил-4-(1-гидрокси-2-метилпропил)-1-{4-[2-(тетразол-5-ил)-фенил]фенил} метилимидазол-5-карбоксамид (соединение 5-98).
77а) 2-Бутил-4-изобутирил-1-{ 4-[2-(тритилтетразол-5-ил)фенил]фенил}метилимидазол-5-карбонитрил.
Повторяли процедуру, описанную в примере 74а, но с использованием 1,42 г 2-бутил-4-изобутирилимидазол-5-карбонитрила, полученного согласно описанию в получении 27, 4,49 г 4-[2-(тритилтетразол-5-ил)фенил]бензилбромида и 0,76 г т-бутоксида калия, в результате чего получали 3,04 г целевого соединения в виде кристаллов, т. пл. 115-116oC.
ЯМР (CDCl3)  ppm: 0,87 (3Н, т, J 7,5 Гц), 1,22 (6Н, д, J 6,г Гц), 1,31 (2Н, секстет, J 7,5 Гц), 1,63 (2Н, квин. J 7,5 Гц), 2,57 (2Н, т, J 7,5 Гц), 3,64 (1Н, септ. J 7,5 Гц), 5,11 (2Н, с), 6,90-7,52 (22Н, м), 7,96 (1Н, д, J 9 Гц).
ppm: 0,87 (3Н, т, J 7,5 Гц), 1,22 (6Н, д, J 6,г Гц), 1,31 (2Н, секстет, J 7,5 Гц), 1,63 (2Н, квин. J 7,5 Гц), 2,57 (2Н, т, J 7,5 Гц), 3,64 (1Н, септ. J 7,5 Гц), 5,11 (2Н, с), 6,90-7,52 (22Н, м), 7,96 (1Н, д, J 9 Гц).
77b) 2-Бутил-4-(1-гидрокси-2-метилпропил)-1-{ 4-[2-(тритилтетразол-5-ил)фенил]фенил} метилимидазол-5-карбонитрил.
Повторяли процедуру, описанную в примере 74b, но с использованием 2,00 г 2-бутил-4-изобутирил-1-{4-[2-(тритилтетразол-5-ил)фенил]фенил} метилимидазол -5-карбонитрила, полученного в стадии (а), и 0,22 г борогидрида натрия, в результате чего получали 1,68 г целевого соединения в виде кристаллов, т. пл. 127-128oC.
ЯМР (CDCl3) дельта, ppm: 0,85 (3Н, т, J 7,5 Гц), 0,93 (3Н, д, J 6,5 Гц), 1,00 (3Н, д, J 6,5 Гц), 1,26 (2Н, секстет, J 7,5 Гц), 1,59 (2Н, квин. J 7,5 Гц), 2,13 (1Н, секст. J 6,5 Гц), 2,52 (2Н, т, J 7,5 Гц), 4,53 (1Н, д, J 6 Гц), 5,04 (2Н, с), 6,85-7,52 (22Н, м), 7,95 (1Н, д, J 9 Гц).
77с) 2-Бутил-4-(1-Гидркоси-2-метилпропил)-1-{4-[2-(тетразол -5-ил)фенил] фенил}метилимидазол-5-карбонитрил.
Повторяли процедуру, описанную в примере 74с, но с использованием 1,29 г 2-бутил-4-(1-гидрокси-2-метилпропил)-1-{ 4-[2-(тритилтетразол-5-ил)фенил] фенил} метилимидазол-5-карбонитрила, полученного в стадии (b), в 75% об./об. водной уксусной кислоте, в результате чего получали 0,83 г целевого соединения в виде стеклообразного вещества.
ЯМР (CDCl3) дельта, ppm: 0,81 (3Н, д, J 6,5 Гц), 0,83 (3Н, т, J 7,5 Гц), 0,95 (3Н, д, J 6,5 Гц), 1,26 (2Н, секстет, J 7,5 Гц), 1,54 (2Н, квин. J 7,5 Гц), 1,97-2,09 (1Н, м), 2,59 (2Н, т, J 7,5 Гц), 4,37 (1Н, д, J 6,5 Гц), 5,14 (2Н, с), 6,98 (2Н, д, J 8,5 Гц), 7,05 (2Н, д, J 8,5 Гц), 7,32-7,60 (3Н, м), 7,77 (1Н, д, J 7,5 Гц).
77d) 2-Бутил-4-(1-гидрокси-2-метилпропил)-1-{4-[2-(тетразол -5-ил)фенил] фенил}метилимидазол-5-карбоксамид.
Повторяли процедуру, описанную в примере 74d, но с использованием 0,34 г 2-бутил-4-(1-гидрокси-2-метилпропил)-1-{ 4-[2-(тетразол -5-ил)фенил] фенил} метилимидазол-5-карбонитрила, полученного в стадии (с), в 1 н. водном растворе гидроксида натрия, в результате чего получали 0,24 г целевого соединения в виде порошка, т. пл. 155-157oC.
ЯМР (ДМСО-d6) дельта, ppm: 0,69 (3Н, д, J 6,5 Гц), 0,79 (3Н, т, J 7,5 Гц), 0,93 (3Н, д, J 6,5 Гц), 1,22 (2Н, секс. J 7,5 Гц), 1,45 (2Н, квин. J 7,5 Гц), 2,00-2,12 (1Н, м), 2,65 (2Н, т, J 7,5 Гц), 4,41 (1Н, д, J 8 Гц), 5,53 (1Н, д, J 16 Гц), 5,71 (1Н, д, J 16 Гц), 7,00 (2Н, д, J 8,5 Гц), 7,07 (2Н, д, J 8,5 Гц), 7,50-7,71 (4Н, м).
Пример 78. (5-Метил-2оксо-1,3-диоксолен-4-ил)метил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{ 4-[2-(тетразол-5-ил)фенил] фенил} метилимидазол-5-карбоксилат (соединение 2-17).
78а) (5-Метил-2оксо-1,3-диоксолен-4-ил)метил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{4-[2-(тритилтетразол -5-ил)фенил]фенил}метилимидазол.
Раствор 2,65 г моногидрата гидроокиси лития в 158 мл воды добавляли, охлаждая при этом льдом, к раствору 30 г этил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{ 4-[2-(тритилтетразол-5-ил)фенил] фенил} метилимидазол-5-карбоксилата, полученного согласно описанию в примере 18а, в 344 мл диоксана, и полученную смесь размешивали 20 ч при 5 10oC. По истечение этого времени к смеси добавляли небольшой кусочек сухого льда, после чего смесь концентрировали путем выпаривания при пониженном давлении до получения объема около 100 мл, затем к концентрату добавляли этилацетат и хлорид натрия, и полученную смесь размешивали. Этилацетатный слой отделяли осушали безводным сульфатом натрия и концентрировали путем выпаривания при пониженном давлении, в результате чего получали 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{ 4-[2-(тритилтетразол -5-ил)фенил] фенил}метилимидазол-5-карбоксилат лития в виде стеклообразного материала. Затем к раствору всего полученного карбоксилата лития в 160 мл N,N-диметилацетамида добавляли, охлаждая при этом льдом, 6,08 г карбоната калия, после чего к смеси по капле добавляли, охлаждая при этом льдом, раствор 11,2 г 4-хлорометил-5-метил-2-оксо-1,3-диоксолена (74 чистоты) в 26 мл N,N-диметилацетамида. Полученную смесь размешивали 3 ч при 50oC. По истечение этого времени к реакционной смеси добавляли воду и этилацетат и этилацетатный слой отделяли, осушали безводным сульфатом магния и концентрировали путем выпаривания при пониженном давлении. Остаток кристаллизовали в диизопропиловом эфире, в результате чего получали 29,3 г целевого соединения, т. пл. 98-100oC (с разл.).
ЯМР-спектр этого соединения идентичен спектру соединения, полученного в примере 61а.
78b) (5-Метил-2-оксо-1,3-диоксолен-4-ил)метил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{ 4-[2-(тетразол -5-ил)фенил] фенил}метилимидазол-5-карбоксилат.
75 мл воды добавляли к суспензии 29,3 г (5-метил-2-оксо-1,3-диоксолен-4-ил)метил 4-(1-гидрокси-1-метилэтил)-2-пропил-1{4-[2-(тритилтетразол -5-ил)фенил] фенил} метилимидазол-5-карбоксилата, полученного в стадии (а), в 225 мл уксусной кислоты, и полученную смесь размешивали 1,5 ч при 60oC. По истечение этого времени к смеси добавляли 75 мл воды и смесь охлаждали. Осажденный тритиловый спирт удаляли путем фильтрации и фильтрат концентрировали путем выпаривания при пониженном давлении. К остатку добавляли толуол и полученную смесь опять концентрировали путем выпаривания при пониженном давлении в целях удаления остаточной воды и уксусной кислоты. Остаток кристаллизовали в этилацетате, в результате чего получали 16,6 г целевого соединения в виде кристаллов, т. пл. 177-180oC (с разлож.).
ЯМР-спектр этого соединения идентичен спектру соединения, полученного в примере 61 (b).
Пример 79. (5-Метил-2-оксо-1,3-диоксолен-4-ил)метил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{ 4-[2-(тетразол -5-ил)фенил] фенил} метилмидазол-5-карбоксилат (соединение 2-17).
79а) Этил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{4-[2-(тритилтетразол-5-ил)фенил]фенил} метилимидазол-5-карбкосилат.
Раствор 1,00 г этил 1-(2'-цианобифенил-4-ил)метил-4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилата, полученного согласно описанию в примере 71b, и 1,00 г азида трибутилолова в 7,5 г толуола размешивали 5 дней при 100oC. Затем к смеси добавляли 2,5 г бикарбоната натрия и 20 мл воды и полученную смесь размешивали 8 ч при комнатной температуре. По истечение этого времени смесь разбавляли этилацетатом и подкисляли до pH 3 добавлением 3 н. водной соляной кислотой. Этилацетатный слой отделяли, осушали безводным сульфатом магния и концентрировали путем выпаривания при пониженном давлении, в результате чего получали этил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{ 4-[2-(тетразол -5-ил)фенил]фенил}метилимидазол-5-карбоксилат в виде сиропа. К раствору этого полученного сиропа в 15 мл пиридина добавляли 0,80 г тритилхлорида, и смесь размешивали 4 ч при 60oC. После этого реакционную смесь концентрировали путем выпаривания при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью этилацетата и гексана (1:1, по объему), и затем кристаллизовали из диизопропилового эфира, в результате чего получали 1,15 г целевого соединения в виде кристаллов.
ЯМР-спектр этого соединения идентичен спектру соединения, полученного в примере 18а.
79b) (5-метил-2-оксо-1,3-диоксолен-4-ил)метил 4-(1-гидрокси-1-метилэтил)-2-пропил-1-{4-[2-(тетразол -5-ил)фенил]фенил}метилимидазол-5-карбоксилат.
Повторяли процедуру, описанную в примере 78а и 78b, но с использованием этил 4-(1-гидрокси-1-метилэтил)-2-пропи-{ 4-[2-(тритилтетразол-5-ил)фенил] фенил} метилимидазол-5-карбоксилата, полученного в стадии (а), в результате чего получали целевое соединение с выходом 71%
ЯМР-спектр этого соединения идентичен спектру соединения, полученного согласно описанию в примере 61b.
Пример 80. Пивалоилоксиметил 2-этил-4-(1-гидрокси-1-метилэтил)-1-{4-[2-(тетразол-5-фенил]фенил}метилимидазол -5-карбоксилат (соединение 2-69).
80а) Пивалоилоксиметил 2-этил-4-(1-гидрокси-1-метилэтил)-1-{ 4-[2-(тритилтетразол-5-ил)фенил] фенил}метилимидазол-5-карбоксилат.
Повторяли процедуру, описанную в примере 78а, но с использованием 2,25 г этил 2-этил-4-(1-гидрокси-1-метилэтил)-1-{ 4-[2-(тритилтетразол-5-ил)фенил] фенил} метилимидазол-5-карбоксилата, полученного согласно описанию в примере 68а, а также 203 мг моногидрата гидроксида лития для гидролиза и 0,90 г пивалоилоксиметилхлорида для эстерификации, в результате чего получали 2,53 г целевого соединения в виде стеклообразного продукта, очищенного с помощью колоночной хроматографии на силикагеле с использованием в качестве элюента смесь этилацетата и гексана (1:1, по объему).
ЯМР (CDCl3) дельта, ppm: 1,14 (4Н, с), 1,19 (3Н, т, J 7,5 Гц), 1,64 (6Н, с), 2,50 (2Н, кв. J 7,5 Гц), 5,34 (2Н, с), 5,43 (1Н, с), 5,72 (2Н, с), 6,73 (2Н, д, J 8 Гц), 6,92-7,49 (20Н, м), 7,90 (1Н, д, J 8,5 Гц).
80b) Пивалоилоксиметил 2-этил-4-(1-гидрокси-1-метилэтил)-1-{ 4-[2-(тетразол -5-ил)фенил]фенил} метилимидазол-5-карбоксилат.
Повторяли процедуру, описанную в примере 78b, но с использованием 2,53 г пивалоилоксиметил 2-этил-4-(1-гидрокси-1-метилэтил)-1-{4-[2-(тритилтетразол-5-ил)фенил] фенил}метилимидазола-5-карбоксилата, полученного в стадии (а), и 28 мл 75% об./об. водной уксусной кислоты, в результате чего получали 1,70 г целевого соединения в виде стеклообразного продукта.
ЯМР (CDCl3) дельта, ppm: 1,09 (9Н, с), 1,24 (3Н, т, J 7,5 Гц), 1,59 (6Н, с), 2,64 (2Н, кв. J 7,5 Гц), 5,41 (2Н, с), 5,79 (2Н, с), 6,86 (2Н, д, J 8,6 Гц), 7,11 (2Н, д, J 8,5 Гц), 7,42-7,62 (4Н, м).
Пример 81. (5-Метил-2оксо-1,3-диоксолен-4-ил)метил 2-этил-4-(1-гидрокси-1-метилэтил)-1-{ 4-[2-(тетразол -5-ил)фенил] фенил}метилимидазол-5-карбоксилат (соединение 2-73).
81а) (5-Метил-2-оксо-1,3-диоксолен-4-ил)метил 2-этил-4-(1-гидрокси-1-метилэтил)-1-{ 4-[2-(тритилтетразол -5-ил)фенил] фенил}метилимидазол-5-карбоксилат.
Повторяли процедуру, описанную в примере 78а, но с использованием 2,25 г этил 2-этил-4-(1-гидрокси-1-метилэтил)-2-{4-[2-(тритилтетразол-5-ил) -фенил] фенил} метилимидазол-5-карбоксилата, полученного согласно описанию в примере 68а, а также 203 мг моногидрата гидроксида лития для гидролиза и 0,95 г 4-хлорометил-5-метил-2-оксо-1,3-диоксолена (74% чистоты) для этерификации, в результате чего получали 1,23 г целевого соединения в виде кристаллов, т. пл. 145oC.
ЯМР (CDCl3) дельта, ppm: 1,21 (3Н, т, J 7,5 Гц), 1,63 (6Н, с), 1,98 (3Н, с), 2,55 (2Н, кв. J 7,5 Гц), 4,783 (2Н, с), 5,30 (2Н, с), 5,59 (1Н, с), 6,69 (2Н, д, J 8 Гц), 6,90-7,53 (20Н, м), 7,87 (1Н, д, J 8 Гц).
81b) (5-Метил-2-оксо-1,3-диоксолен-4-ил)метил 2-этил-4-(1-гидрокси-1-метилэтил)-1-{4-[2-(тетразол -5-ил)фенил]фенил}метилимидазол-5-карбоксилат.
Повторяли процедуру, описанную в примере 78b, но с использованием 1,90 г (5-метил-2-оксо-1,3-диоксолен-4-ил)метил 2-этил-4-(1-гидрокси-1-метилэтил)-1-{4-[2-(тритилтетразол-5-ил)фенил] фенил}метилимидазол-5-карбоксилата, полученного согласно описанию в стадии (а), и 20 мл 75% об./об. водной уксусной кислоты, в результате чего получали 1,23 г целевого соединения в виде кристаллического порошка.
ЯМР (ДМСО-d6) дельта, ppm: 1,24 (3Н, т, J 7,5 Гц), 1,54 (6Н, с), 2,10 (3Н, с), 2,69 (2Н, квин. J 7,5 Гц), 4,99 (2Н, с), 5,44 (2Н, с), 6,86 (2Н, д, J 8,5 Гц), 7,08 (2Н, д, J 8,5 Гц), 7,50-7,65 (3Н, м),
Получение 1. 2-Бутилимидазол-4,5-дикарбонитрил.
Суспензию 51,4 г диаминомалеонитрила и 85,6 г триметилортовалерата в 300 мл ацетонитрила в масляной бане, поддерживаемой при 85oC, размешивали 6 ч. После этого реакционную смесь концентрировали путем выпаривания при пониженном давлении и концентрат очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью этилацетата и гексана (1:1, по объему), в результате чего получали 99 г 1-амино-2-N-(1-метоксипентилиден)аминосукционитрила. Это соединение полностью растворяли в 300 мл ксилола и полученный раствор размешивали в масляной бане, поддерживаемой при 150oC, в течение 8 ч, после чего реакционную смесь концентрировали до получения половины исходного объема и оставляли при комнатной температуре. Осажденные кристаллы собирали путем фильтрации и промывали небольшим количеством ксилола, в результате чего получали 55,2 г целевого соединения, т. пл. 109-111oC.
Получение 2. 2-Бутилимидазол-4,5-дикарбоновая кислота.
Раствор 100 г 2-бутилимидазол-4,5-дикарбонитрила, полученного согласно описанию в получении 1, в 1 л 6 н. водной соляной кислоты нагревали 7 ч с обратным холодильником, после чего реакционную смесь оставляли на ночь при комнатной температуре. Затем осажденные кристаллы собирали фильтрацией и промывали водой и небольшим количеством ацетона, в результате чего получали 84 г целевого соединения, т. пл. 261-263oC.
Получение 3. Диэтил 2-бутилимидазол-4,5-дикарбоксилат.
Сухой хлорводород барботировали через суспензию 40 г 2-бутилимидазол-4,5-дикарбоновой кислоты, полученной в соответствии с описанием в получении 2, в 600 мл этанола при комнатной температуре, размешивая при этом в течение 2 ч, и получали раствор. Этот раствор оставляли на 18 ч при комнатной температуре, после чего реакционную смесь концентрировали путем выпаривания при пониженном давлении. Концентрат смешивали с этилацетатом и с водным раствором бикарбоната натрия и нейтрализовали путем добавления бикарбоната натрия в виде порошка. Этилацетатный слой отделяли и осушали безводным сульфатом магния, а растворитель удаляли путем дистилляции при пониженном давлении. Полученный кристаллический остаток растирали со смесью диизопропилового эфира и гексана и собирали путем фильтрации, в результате чего получали 43 г целевого соединения, т. пл. 82-84oC.
Получение 4. Диметил 2-бутилимидазол-4,5-дикарбоксилат.
Повторяли процедуру, описанную в получении 3, используя 40 г 2-бутилимидазол-4,5-дикарбоновой кислоты, и вместо этанола использовали метанол, в результате чего получали 41,6 г целевого соединения в виде кристаллов, т. пл. 88oC.
Получение 5. 4-Ацетил-2-бутил-5-цианоимидазол.
5(i). 2-Бутил-1-тритилимидазол-4,5-дикарбонитрил.
1,25 г гидрида натрия (в виде 55 мас./мас. дисперсии в минеральном масле) добавляли, охлаждая при этом льдом, к раствору 5 г 2-бутилимидазол-4,5-дикарбонитрила, полученного в соответствии с описанием в получении 1, в 50 мл N,N-диметилформамида, и полученную смесь размешивали 15 мин. Затем добавляли 10 г тритилхлорида и реакционную смесь размешивали 6 ч при 50oC. После этого смесь смешивали с этилацетатом и водой и полученный продукт экстрагировали этилацетатом. Экстракт осушали безводным сульфатом магния, а растворитель удаляли путем дистилляции при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью этилацетата и гексана (1:5, по объему), в результате чего получали 9,83 г целевого соединения в виде сиропа, который оставляли для отверждения. Твердый продукт имеет т. пл.144-147oC (с разлож. И окрашив. при 94-98oC).
ЯМР (CDCl3) дельта, ppm: 0,60 (3Н, т, J 7 Гц), 0,5-1,4 (4Н, м), 2,03 (2Н, т, J 7 Гц), 7,0-7,6 (15Н, м).
5(ii). 4-Ацетил-2-бутил-циано-1-тритилимидазол.
11,1 мл 2 М раствора иодида метилмагния в диэтиловом эфире медленно по капле добавляли при комнатной температуре и в атмосфере азота, к раствору 4,5 г 2-бутил-1-тритилимидазол-4,5-дикарбонитрила, полученного в стадии i, в 45 мл тетрагидрофурана и полученную смесь размешивали 3 ч при комнатной температуре. По истечение этого времени к смеси, охлаждая льдом, по капле добавляли насыщенный водный раствор хлорида аммония. Тетрагидрофурановый слой отделяли, промывали насыщенным водным раствором хлорида натрия и концентрировали при пониженном давлении. Водный слой еще раз экстрагировали небольшим количеством этилацетатом. Экстракт промывали насыщенным водным раствором хлорида натрия, осушали и концентрировали путем выпаривания при пониженном давлении. Полученный экстракт объединяли с полученным выше концентратом и этот продукт очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью гексана и этилацетата (3:1, по объему). Полученный продукт кристаллизовали из смеси этилацетата и гексана, в результате чего получали 1,46 г целевого соединения, т. пл. 159-160oC (с разлож.).
ЯМР (CDCl3) дельта, ppm: 0,60 (3Н, т, J 7 Гц), 0,5-1,5 (4Н, м), 2,08 (2Н, т, J 7 Гц), 2,58 (3Н, с), 7,1-7,6 (15Н, м).
5δ. 4-Ацетил-2-бутил-5-цианоимидазол.
Суспензию 1,78 г 4-ацетил-2-бутил-5-циано-1-тритилимидазола, полученного в соответствии с описанием в стадии (ii) в 80% об./об. водной уксусной кислоте размешивали 1 ч при 60oC. Полученный таким образом раствор концентрировали досуха путем выпаривания при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью гексана и этилацетата (3:1, по объему), в результате чего получали 0,66 г целевого соединения в виде бесцветного твердого вещества с т. пл. 77-78oC.
ЯМР (CDCl3)  ppm: 0,93 (3Н, т, J 7 Гц), 1,0-2!6 (4Н, м), 2,72 (3Н, с), 2,89 (2Н, т, J 7 Гц).
ppm: 0,93 (3Н, т, J 7 Гц), 1,0-2!6 (4Н, м), 2,72 (3Н, с), 2,89 (2Н, т, J 7 Гц).
Получение 6. 4-Бензоил-2-бутил-5-цианоимидазол.
6(i). 4-Бензоил-2-бутил-5-циано-1-тритилимидазол.
В соответствии с процедурой, описанной в получении 5(ii), получали 10,3 г целевого соединения в виде аморфного твердого вещества посредством реакции раствора 10 г 2-бутил-1-тритил-имидазол-4,5-динитрила, полученного согласно описанию в получении 5δ, в 100 мл тетрагидрофурана с 25 мл 2 М раствора иодида фенилмагния в диэтиловом эфире.
ЯМР (CDCl3)  ppm: 0,67 93Н, т, J 7 Гц), 0,5-1,5 (4Н, м), 2,11 (2Н, т, J 7 Гц), 7,1-8,0 (20Н, м).
ppm: 0,67 93Н, т, J 7 Гц), 0,5-1,5 (4Н, м), 2,11 (2Н, т, J 7 Гц), 7,1-8,0 (20Н, м).
6( ). 4-Бензоил-2-бутил-5-цианоимидазол.
). 4-Бензоил-2-бутил-5-цианоимидазол.
Суспензию 10,3 г 4-бензоил-2-бутил-5-циано-1-тритилимидазола, полученного в стадии δ, в 80% об./об. водной уксусной кислоте размешивали 5 ч при 60oC. По истечение этого времени полученный таким образом раствор концентрировали путем выпаривания при пониженном давлении и концентрат очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью гексана и этилацетата (2:1, по объему). Полученный маслянистый продукт растворяли в тетрахлорметане и раствор оставляли при комнатной температуре для осаждения кристаллов, которые собирали путем фильтрации, в результате чего получали 4,46 г целевого соединения, т. пл. 121-122oC.
ЯМР (CDCl3)  ppm: 0,90 (3Н, т, J 7 Гц), 1,0-2,3 (4Н, м), 2,85 (2Н, т, J 7 Гц), 7,2-8,0 (5Н, м), 11,0-12,1 (1Н, шир. с).
ppm: 0,90 (3Н, т, J 7 Гц), 1,0-2,3 (4Н, м), 2,85 (2Н, т, J 7 Гц), 7,2-8,0 (5Н, м), 11,0-12,1 (1Н, шир. с).
Получение 7. 2-Бутил-5-циано-4-(1-гидрокси-1-метилэтил)имидазол.
7(i). 2-Бутил-5-циано-4-(1-гидрокси-1-метилэтил)-1-тритилимидазол.
1 мл 2 М раствора иодида метилмагния в тетрагидрофуране, размешивая при комнатной температуре, по капле добавляли к раствору 840 г 4-ацетил-2-бутил-5-циано-1-тритилимидазола, полученного в соответствии с описанием в получении 5δ, в 15 мл тетрагидрофурана и полученную смесь размешивали 1 ч при 40oC. Полученную смесь охлаждали, а затем к этой смеси по капле добавляли насыщенный водный раствор хлорида аммония. Слой тетрагидрофурана отделяли и концентрировали путем выпаривания при пониженном давлении. Концентрат очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью гексана и этилацетата (2:1, по объему), в результате чего получали 539 мг целевого соединения в виде бесцветного твердого вещества, т. пл. 151-152oC.
ЯМР (CDCl3) d ppm: 0,60 (3Н, т, J 7 Гц), 0,6-1,5 (4Н, м), 1,59 (6Н, с), 2,01 (2Н, т, 7 Гц), 3,78 (1Н, с), 7,0-7,6 (15Н, м).
7(ii). 2-Бутил-5-циано-4-(1-гидрокси-1-метилэтил)имидазол.
Смесь 1,3 г 2-бутил-5-циано-4-(1-гидрокси-1-метилэтил)-1-тритилимидазола, полученного в стадии (i), и 26 мл 75% об./об. водной уксусной кислоты размешивали 3 ч при 50oC, после чего растворитель удаляли путем дистилляции при пониженном давлении. Полученный остаток промывали тетрахлорметаном и очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью метиленхлорида и метанола (10:1, по объему), после чего продукт кристаллизовали в тетрахлорметане и получали в результате 0,6 г целевого соединения в виде бесцветных кристаллов, т. пл. 171-172oC.
ЯМР (CDCl3 + CD3OD) d ppm: 0,90 (3Н, т. J 7 Гц), 1,0-2,0 (4Н, м), 1,62 (6Н, с), 2,68 (2Н, т, J 7 Гц).
Получение 8. Этил 2-бутил-4-(1-гидрокси-2-метилэтил)имидазол-5-карбоксилат.
Раствор 5,36 г диэтил 2-бутилимидазол-4,5-дикарбоксилата, полученного согласно описанию в получении 3) в 100 мл тетрагидрофурана охлаждали до -30oC в атмосфере азота и к этому охлажденному раствору при -30 - (-20)oC по капле добавляли 32 и мл раствора бромида метилмагния (2,5 М в тетрагидрофуране). Затем реакционную смесь размешивали при 0oC в течение 1,5 ч и после этого смешивали с этилацетатом и водным раствором хлорида аммония. Этилацетатный слой отделяли и осушали безводным сульфатом магния, а растворитель отгоняли при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью метанола и метиленхлорида (1:20, по объему), в результате чего получали 5,01 г целевого соединения в виде маслянистого продукта.
ЯМР (CDCl3) d ppm: 0,90 (3Н, т, J 7 Гц), 1,32 (3Н, т, J 7 Гц), 1,2-2,0 (4Н, м), 1,64 (6Н, с), 2,70 (2Н, т, J 7 Гц), 4,33 (2Н, кв. J 7 Гц), 5,97 (1Н, шир. с), 10,2 (1Н, шир. с).
Получение 9. Этил 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилат.
В соответствии с процедурой, описанной в получении 8, получали 2,34 г целевого соединения в виде маслянистого продукта посредством реакции 3,01 г диэтил 2-пропилимидазол-4,5-дикарбоксилата, полученного согласно описанию в получении 12, с 16 мл 2,5 М раствора бромида метилмагния в тетрагидрофуране. Полученное соединение кристаллизовали, оставляя его при комнатной температуре, и получали продукт, т. пл. 69-71oC. После перекристаллизации этого продукта из диизопропилового эфира получали продукт, т. пл. 101-102oC.
ЯМР (CDCl3) d ppm: 0,96 (3Н, т, J 7 Гц), 1,35 (3Н, т, J 7 Гц), 1,64 (6Н, с), 1,81 (2Н, секстет, J 7 Гц), 2,68 (2Н, т, J 7 Гц), 4,35 (2Н, кв. J 7 Гц), 5,81 (1Н, с), 9,9 (1Н, шир.с).
Получение 10. 5-Пропилимидазол-4,5-дикарбонитрил.
Повторяли процедуру, описанную в получении 1, но с использованием 16,0 г диаминомалеонитрила и 24 г триметилортобутирата, в результате чего получали 18,7 г целевого соединения в виде кристаллов, т. пл. 141-144oC.
Получение 11. 2-Пропилимидазол-4,5-дикарбоновая кислота.
Повторяли процедуру, описанную в получении 2, но с использованием 18,2 г 2-пропилимидазол-4,5-дикарбонитрила, полученного в соответствии с описанием в получении 10, в результате чего получали 9,95 г целевого соединения в виде кристаллов, т. пл. 261-263oC.
Получение 12. Диэтил 2-пропилимидазол-4,5-дикарбоксилат.
Повторяли процедуру, описанную в получении 3, но с использованием 10,0 г 2-пропилимидазол-4,5-дикарбоновой кислоты, полученный в соответствии с описание в получении 11, в результате чего получали 9,55 г целевого соединения в виде кристаллов, т. пл. 81-83oC.
Получение 13. Этил 2-бутил-4-(1-этил-1-гидроксипропил)имидазол-5-карбоксилат.
В соответствии с процедурой, описанной в получении 8, получали 2,68 г целевого соединения с т. пл. 63-64oC в виде кристаллов, посредством реакции 2,68 г диэтил 2-бутилимидазол-4,5-дикарбоксилата, полученного в соответствии с описанием в получении 3, с 3,0 М раствором бромида этилмагния в диэтиловом эфире.
ЯМР (CDCl3) d ppm: 0,82 (6Н, т, J 7 Гц), 0,93 (3Н, т, J 7 Гц), 1,38 (3Н, т, J 7 Гц), 1,31-1,45 (2Н, м), 1,65-1,76 (2Н, м), 1,79-1,89 (2Н, м), 1,97-2,11 (2Н, м), 2,76 (2Н, т, J 7,5 Гц), 4,36 (2Н, кв. J 7 Гц), 5,70 (1Н, шир. с),
Получение 14. 2-Пропил-1-тритилимидазол-4,5-дикарбонитрил.
Повторяли процедуру, описанную в получении 5(i), но с использованием 7,8 г 2-пропилимидазол-4,5-дикарбонитрила, полученного согласно описанию в получении 10), 2,14 г гидрида натрия (в виде 55% мас./мас. дисперсии в минеральном масле) и 17,1 г тритилхлорида, в результате чего получали 14,6 г целевого соединения в виде кристаллов, т. пл. 107oC (с разложением и с окрашиванием в желтый цвет при 103oC).
ЯМР (CDCl3) d ppm: 0,52 (3Н, т, J 7 Гц), 1,07-1,21 (2Н, м), 2,03 (2Н, т, J 8 Гц), 7,19-7,48 (15Н, м).
Получение 15. 2-Бутил-5-циано-3-пропионил-1-тритилимидазол.
14 мл 3 М раствора бромида этилмагния в диэтиловом эфире при 10oC и в атмосфере азота по капле добавляли к раствору 8,33 г 2-бутил-1-тритилимидазол-4,5-дикарбонитрила, полученного согласно описанию в получении 5(i), в 83 мл тетрагидрофурана и полученную смесь размешивали 3 ч при комнатной температуре. После этого к реакционной смеси, охлаждая при этом льдом, добавляли смесь насыщенного водного раствора хлорида аммония и этилацетата. Слой этилацетата отделяли, промывали водой и осушали безводным сульфатом магния. Растворитель удаляли путем перегонки при пониженном давлении. Полученный таким образом кристаллический продукт промывали диизопропиловым эфиром и получали 4,56 г целевого соединения, т. пл. 140-143oC (с размягчением при 83oC).
ЯМР (CDCl3) d ppm: 0,61 (3Н, т. J 7 Гц), 0,84-1,14 (4Н, м), 1,18 (3Н, т. J 8 Гц), 2,08 (2Н, т, J 7 Гц), 3,03 (2Н, кв. J 7 Гц), 7,22-7,42 (15Н, м).
Получение 16. 5-Циано-4-пропионил-2-пропил-1-тритилимидазол.
Повторяли процедуру, описанную в получении 15, но с использованием 8,05 г 2-пропил-1-тритилимидазол-4,5-дикарбонитрила, полученного согласно описанию в получении 14, и 14 мл 3 М раствора бромида этилмагния в диэтиловом эфире, в результате чего получали 7,03 г целевого соединения в виде кристаллов, т. пл. 96oC (размягч. при 87oC).
ЯМР (CDCl3) d ppm: 0,52 (2Н, т, J 7 Гц), 1,05-1,3 (2Н, м), 1,18 (3Н, т, J 7 Гц), 2,05 (2Н, т, J 7 Гц), 3,03 (2Н, кв. J 7 Гц), 7,20-7,49 (15Н, м).
Получение 17. 2-Бутил-5-циано-4-(1-гидрокси-1-метилпропил)-1-тритилимидазол.
5 мл 1 М раствора бромида метилмагния в тетрагидрофуране при 10oC и в атмосфере азота по капле добавляли к раствору 2 г 2-бутил-5-циано-3-пропионил-1-тритилимидазола, полученного согласно описанию в получении 15, в 36 мл тетрагидрофурана и полученную смесь размешивали 1 ч при 20oC и еще 1 ч при 30oC. По истечение этого времени к реакционной смеси добавляли смесь насыщенного водного раствора хлорида аммония и этилацетата и полученную смесь хорошо взбалтывали. Этилацетатный слой отделяли и осушали безводным сульфатом магния. После этого осушающий агент удаляли путем фильтрации, а растворитель отгоняли при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью этилацетата и гексана 1:2 по объему, и получали 1,29 г целевого соединения в виде кристаллов, т. пл. 90-93oC.
ЯМР (CDCl3) d ppm: 0,60 (3Н, т, J 7 Гц), 0,80 (3Н, т, J 7 Гц), 0,80-1,00 (2Н, м), 1,00-1,13 (2Н, м), 1,58 (3Н, с), 1,75-2,05 (4Н, м), 3,98 (1Н, шир. с), 7,23-7,43 (15Н, м).
Получение 18. 5-Циано-4-(1-гидрокси-1-метилпропил)-2-пропил-1-тритилимидазол.
Повторяли процедуру, описанную в получении 17, но с использованием 5,00 г 5-циано-4-пропионил-2-пропил-1-тритилимидазола, полученного в соответствии с описанием в получении 16, и 12,5 мл 1 М раствора бромида метилмагния в тетрагидрофуране, в результате чего получали 3,32 г целевого соединения в виде кристаллического порошка, т. пл. выше 120oC (размягчение при 110oC).
ЯМР (CDCl3) d ppm: 0,50 (3Н, т, J 7 Гц), 0,80 (3Н, т, J 7 Гц), 1,07-1,12 (2Н, м), 1,58 (3Н, с), 1,74-2,00 (4Н, м), 3,90 (1Н, шир.с), 7,24-7,37 (15Н, м).
Получение 19. 2-Бутил-5-циано-4-(1-бутокси-1-метилпропил)имидазол.
Смесь 1,21 г 2-бутил-5-циано-4-(1-гидрокси-1-метилпропил)-1-тритилимидазола, полученного согласно описанию в получении 17, и 20 мл 75 об./об. водной уксусной кислоты размешивали в течение 1 ч при 50oC, после чего смесь охлаждали, а осажденные кристаллы тритилового спирта удаляли путем фильтрации. Фильтрат концентрировали путем выпаривания при пониженном давлении, оставшуюся воду и уксусную кислоту отгоняли как толуоловый азеотроп при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью метиленхлорида и метанола (5:1, по объему), и получали 0,47 г целевого соединения в виде кристаллического порошка, т. пл. 139-142oC.
ЯМР (ДМСО-d6) d ppm: 0,74 (3Н, т, J 7 Гц), 0,87 (3Н, т, J 7 Гц), 1,21-1,34 (2Н, м), 1,49 (3Н, с), 1,53-1,64 (2Н, м), 1,72 (2Н, кв. J 7,5 Гц), 2,56 (2Н, т, J 7 Гц), 5,45 (1Н, с).
Получение 20. 5-Циано-4-(1-гидрокси-1-метилпропил)-2-пропилимидазол.
Повторяли процедуру, описанную в получении 19, но с использованием 1,20 г 5-циано-4-(1-гидрокси-1-метилпропил)-2-пропил-1-тритилимидазола, полученного согласно описанию в получении 18, в результате чего получали 0,48 г целевого соединения в виде кристаллического порошка, т. пл. 157-159oC.
ЯМР (CDCl3) d ppm: 0,89 (3Н, т, J 7,5 Гц), 0,98 (3Н, т, J 7,5 Гц), 1,57 (3Н, с), 1,76 (2Н, кв. J 7,5 Гц), 1,83-2,08 (2Н, м), 2,00 (1Н, с), 2,67 (2Н, т, J 7,5 Гц).
Получение 21. Метил 2-бутил-4-(1-гидрокси-1-метилэтил)имидазол-5-карбоксилат.
Раствор 9,73 г диметил 2-бутилимидазол-4,5-дикарбоксилата, полученного согласно описанию в получении 4, в 100 мл тетрагидрофурана охлаждали до -30oC в атмосфере азота и к этому раствору при температуре -30oC - (-20oC) по капле добавляли 162 мл 1 М раствора бромида метилмагния в тетрагидрофуране. Полученную смесь размешивали 2,5 ч при 0oC, а затем к этой смеси добавляли этилацетат и водный раствор хлорида аммония. Этилацетатный слой отделяли и осушали безводным сульфатом магния, а растворитель отгоняли при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью метанола и метиленхлорида (1:20, по объему), в результате чего получали 7,15 г целевого соединения в виде маслянистого продукта. Это продукт кристаллизовали, выдерживая его при комнатной температуре, после чего получали продукт, т. пл. 60-65oC.
ЯМР (CDCl3)  ppm: 0,88 (3Н, т, J 7 Гц), 1,0-2,0 (4Н, м), 1,64 (6Н, с), 2,69 (2Н, т, J 7,5 Гц), 3,84 (3Н, с), 7,35 (2Н, шир.с).
ppm: 0,88 (3Н, т, J 7 Гц), 1,0-2,0 (4Н, м), 1,64 (6Н, с), 2,69 (2Н, т, J 7,5 Гц), 3,84 (3Н, с), 7,35 (2Н, шир.с).
Получение 22. Пивалоилоксиметил 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилат.
22(i). 4-(1-Гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоновая кислота.
Раствор 0,28 г моногидрата гидроксида лития в 5 мл воды добавляли к раствору 0,48 г этил 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилата, полученного согласно описанию в получении 9, в 5 мл метанола и полученную смесь размешивали 18 ч при комнатной температуре. По истечение этого времени pH реакционной смеси доводили до величины 2,3 путем добавления 6,67 мл 1 н. водной соляной кислоты и полученную смесь концентрировали путем фильтрации, в результате чего получали 0,20 г целевого соединения, т. пл. 232oC (с разложением).
ЯМР (ДМСО-d6) дельта, ppm: 0,87 93Н, т, J 7,5 Гц), 1,48 (6. с), 1,65 (2Н, секстет, J 7,5 Гц), 2,62 (2Н, т, J 7,5 Гц).
22δ. Пивалоилоксиметил 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилата.
1,76 мл N,N-диизопропилэтиламина добавляли к суспензии 1,14 г 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоновой кислоты полученной в стадии (i), в 12 мл N,N-диметилацетамида и полученную смесь размешивали при комнатной температуре в течение 10 мин, после чего к смеси добавляли 1,36 мл пивалоилоксиметилхлорида. Реакционную смесь размешивали 4 ч при 60oC, а затем ее смешивали с этилацетатом и водой. Этилацетатный слой отделяли и концентрировали путем выпаривания при пониженном давлении. Осажденные кристаллы растирали с диизопропиловым эфиром и собирали путем фильтрации, в результате чего получали 1,53 г целевого соединения, т. пл. 177oC.
ЯМР (CDCl3)  ppm: 0,99 (3Н, т, J 7,5 Гц), 1,22 (9Н, с), 1,62 (6Н, с), 1,76 (2Н, секст. J 7,5 Гц), 2,70 (2Н, т, J 7,5 Гц), 5,15 (1Н, шир.с), 5,95 (2Н, с).
ppm: 0,99 (3Н, т, J 7,5 Гц), 1,22 (9Н, с), 1,62 (6Н, с), 1,76 (2Н, секст. J 7,5 Гц), 2,70 (2Н, т, J 7,5 Гц), 5,15 (1Н, шир.с), 5,95 (2Н, с).
Получение 23. Этил 4-(1-гидроксиэтил)-2-пропилимидазол-5-карбоксилат.
23δ. 4-Ацетил-2-пропилимидазол-5-карбонитрил.
194 мл 1 М раствора бромида метилмагния в тетрагидрофуране по капле добавляли при температуре 10-15oC и в атмосфере азота к раствору 10 г 2-пропилимидазол-4,5-дикарбонитрила, полученного согласно описанию в получении 10, в 100 мл тетрагидрофурана и полученную смесь размешивали 30 мин при температуре 10 15oC. Затем реакционную смесь охлаждали и добавляли 200 мл этилацетата и 100 мл насыщенного водного раствора хлорида аммония. Эту смесь подкисляли путем добавления водного раствора бисульфата калия. Органический слой отделяли и осушали безводным сульфатом магния, а растворитель удаляли путем перегонки при пониженном давлении. Остаток подвергали колоночной хроматографии на силикагеле, элюируя смесью этилацетата и гексана (1:1, по объему), в результате чего получали 9,18 г целевого соединения в виде кристаллов, т. пл. 93-95oC.
ЯМР (CDCl3) d ppm: 0,99 (3Н, т, J 7,5 Гц), 1,83 (2Н, секс. J 7,5 Гц), 2,71 (3Н, с), 2,82 (2Н, т, J 8 Гц).
23ii. Этил 4-ацетил-2-пропилимидазол-4-карбоксилат.
Смесь 4,0 г 4-ацетил-2-пропилимидазол-5-карбонитрила, полученного в стадии (i), и 60 мл 6 н. водной соляной кислоты нагревали, размешивая при этом, с обратным холодильником в течение 8 ч. Затем реакционную смесь концентрировали путем выпаривания при пониженном давлении и концентрат растворяли в этаноле, после чего полученный раствор снова концентрировали таким же образом. Остаток растворяли в этаноле, а растворитель снова отгоняли. Эту последовательность растворений и концентраций осуществляли в целом пять раз, а затем остаток растворяли в 60 мл этанола. Через полученный раствор при комнатной температуре в течение 20 мин барботировали поток хлорводорода, после чего этот раствор оставляли на 16 ч при комнатной температуре. По истечение этого времени раствор концентрировали путем выпаривания при пониженном давлении. Концентрат растворяли в смеси этилацетата и водного раствора бикарбоната натрия и полученный раствор нейтрализовали путем добавления бикарбоната натрия. Этилацетатный слой отделяли и осушали безводным сульфатом магния. Растворитель удаляли путем перегонки при пониженном давлении, а остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью этилацетата и гексана (1:1, по объему), в результате чего получали 3,07 г целевого соединения в виде кристаллов при 76 78oC.
ЯМР (CDCl3)  ppm: 0,96 (3Н, т, J 7,5 Гц), 1,39 (3Н, т, J 7,0 Гц), 1,82 (2Н, секстет, J 7,5 Гц), 2,75 (3Н, с), 2,80 (2Н, т, J 7,5 Гц), 4,44 (2Н, кв. J 7 Гц).
ppm: 0,96 (3Н, т, J 7,5 Гц), 1,39 (3Н, т, J 7,0 Гц), 1,82 (2Н, секстет, J 7,5 Гц), 2,75 (3Н, с), 2,80 (2Н, т, J 7,5 Гц), 4,44 (2Н, кв. J 7 Гц).
23δ. Этил 4-(1-гидроксиэтил)-2-пропилимидазол-5-карбоксилат.
125 мл борогидрида натрия добавляли к раствору 1,50 г этил 4-ацетил-2-пропилимидазол-5-карбоксилата полученного в стадии (ii), в 15 мл этанола и полученную смесь размешивали 30 мин при комнатной температуре. Затем добавляли 2 л ацетона и смесь размешивали еще 10 мин. После этого смесь концентрировали путем выпаривания при пониженном давлении и концентрат растворяли в метаноле. Полученный раствор снова концентрировали путем выпаривания при пониженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесями метиленхлорида и метанола (1:20 и 1: 10, по объему), в результате чего получали 1,32 целевого соединения в виде кристаллов, т. пл. 151-153oC.
ЯМР (CDCl3 + ДМСО d6) d ppm: 0,95 (3Н, т, 7,5 Гц), 1,38 (3Н, т, J 7 Гц), 1,48 (3Н, д, J 6,5 Гц), 1,74 (2Н, секстет, J 7,5 Гц), 2,67 (2Н, т, J 8 Гц), 4,34 (2Н, кв. J 7 Гц), 5,28 (1Н, кв. J 6,5 Гц).
Получение 24. Этил 2-бутил-4-(1-гидроксиэтил) имидазол-5-карбоксилат.
24(i). 4-Ацетил-2-бутилимидазол-5-карбонитрил.
Повторяли процедуру, описанную в получении 23 (i), но с использованием 10 г 2-бутилимидазол-4,5-дикарбонитрила, полученного согласно описанию в получении 1, в результате чего получали 9,15 г целевого соединения в виде кристаллов, т. пл. 77-78oC.
ЯМР (CDCl3)  ppm: 0,93 (3Н, т, J 7 Гц), 1,0-2,1 (4Н, м), 2,72 (3Н, с), 2,89 (2Н, т, J 7 Гц).
ppm: 0,93 (3Н, т, J 7 Гц), 1,0-2,1 (4Н, м), 2,72 (3Н, с), 2,89 (2Н, т, J 7 Гц).
24 . Этил 4-ацетил-2-бутилимидазол-5-карбоксилат.
. Этил 4-ацетил-2-бутилимидазол-5-карбоксилат.
Повторяли процедуру, описанную в получении 23(ii), но с использованием 1,00 г 4-ацетил-2-бутилимидазол-5-карбонитрила, полученного в стадии δ, в результате чего получали 0,92 г целевого соединения в виде вязкого масла.
ЯМР (CDCl3)  ppm: 0,88 (3Н, т, J 7 Гц), 1,1-2,1 (4Н, м), 1,33 (3Н, т, J 7 Гц), 2,74 (3Н, с), 2,82 (2Н, т, J 7,5 Гц), 4,38 (2Н, кв. J 7 Гц).
ppm: 0,88 (3Н, т, J 7 Гц), 1,1-2,1 (4Н, м), 1,33 (3Н, т, J 7 Гц), 2,74 (3Н, с), 2,82 (2Н, т, J 7,5 Гц), 4,38 (2Н, кв. J 7 Гц).
24 . Этил 2-бутил-4-(1-гидроксиэтил)имидазол-5-карбоксила.
. Этил 2-бутил-4-(1-гидроксиэтил)имидазол-5-карбоксила.
Повторяли процедуру, описанную в получении 23ii, но с использованием 0,64 г этил 4-ацетил-2-бутилимидазол-5-карбоксилата, полученного в соответствии с описанием в стадии  , в результате чего получали 0,55 г целевого соединения в виде кристаллов, т. пл. 149oC.
, в результате чего получали 0,55 г целевого соединения в виде кристаллов, т. пл. 149oC.
ЯМР (CDCl3 + ДМСО-d6) δ ppm: 0,91 (3Н, т, 7,5 Гц), 1,37 (3Н, т, J 7 Гц), 1,3-1,42 (2Н, м), 1,50 (3Н, д, J 6,8 Гц), 1,69 (2Н, квин. J 7,5 Гц), 2,69 (2Н, т, J 8 Гц), 4,34 (2Н, кв. J 7 Гц), 5,26 (1Н, кв. J 6,5 Гц).
Получение 25. 2-Бутил-4-пропионилимидазол-5-карбонитрил.
Повторяли процедуру, описанную в получении 24  , но с использованием бромида этилмагния вместо бромида метилмагния, и получали в результате целевое соединение, т. пл. 84-85oC и выходом 51,9%
, но с использованием бромида этилмагния вместо бромида метилмагния, и получали в результате целевое соединение, т. пл. 84-85oC и выходом 51,9%
ЯМР (CDCl3) δ ppm: 0,95 (3Н, т, J 7 Гц), 1,0-2,2 (4Н, м), 1,28 (3Н, т, J 7,0 Гц), 2,88 (2Н, т, J 7 Гц), 3,15 (2Н, кв. J 7 Гц).
Получение 26. 2-Бутил-4-бутирилимидазол-5-карбонитрил.
Повторяли процедуру, описанную в получении 24  , но с использованием бромида пропилмагния вместо бромида метилмагния, в результате чего получали целевое соединение, т. пл. 91-92oC и выходом 57,2%
, но с использованием бромида пропилмагния вместо бромида метилмагния, в результате чего получали целевое соединение, т. пл. 91-92oC и выходом 57,2%
ЯМР (CDCl3) δ ppm: 1,02 (3Н, т, J 7,5 Гц), 1,11 (3Н, т, J 7,5 Гц), 1,3-1,6 (2Н, м), 1,7-2,0 (4Н, м), 2,88 (2Н, т, J 8 Гц), 3,13 (2Н, т, J 7,5 Гц).
Получение 27. 2-бутил-4-изобутирилимидазол-5-карбонитрил.
Повторяли процедуру, описанную в получении 24  , но с использованием бромида изопропилмагния вместо бромида метилмагния, в результате чего получали целевое соединение, т. пл. 88-89oC и выходом 36,2%
, но с использованием бромида изопропилмагния вместо бромида метилмагния, в результате чего получали целевое соединение, т. пл. 88-89oC и выходом 36,2%
ЯМР (CDCl3) δ ppm: 0,94 (3Н, т, J 7 Гц), 1,0-2,1 (4Н, м), 1,30 (6Н, д, J 7 Гц), 2,91 (2Н, т, J 7 Гц), 3,71 (1Н, септет, J 7 Гц).
Получение 28. 4-Бутирил-2-пропилимидазол-5-карбонитрил.
Повторяли процедуру, описанную в получении 24  , но с использованием 2-пропилимидазол-4,5-дикарбонитрила, полученного согласно описанию в получении 10, и бромида пропилмагния, в результате чего получали целевое соединение, т. пл 94-95oC и выходом 49,8%
, но с использованием 2-пропилимидазол-4,5-дикарбонитрила, полученного согласно описанию в получении 10, и бромида пропилмагния, в результате чего получали целевое соединение, т. пл 94-95oC и выходом 49,8%
ЯМР (CDCl3) δ ppm: 1,00 (3Н, т, J 7,5 Гц), 1,04 (3Н, т, J 7,5 Гц), 1,7-1,9 (4Н, м), 2,79 (2Н, т, J 7,5 Гц), 3,06 (2Н, т, J 7,5 Гц).
Получение 29. 2-Бутил-4-(1-гидрокси-1-метилэтил)имидазол-5-карбонитрил.
Повторяли процедуру, описанную в получении 23  , но с использованием 4-ацетил-2-бутилимидазол-5-карбонитрила, полученного согласно описанию в получении 24 (i), и раствора бромида метилмагния в тетрагидрофуране, в результате чего получали целевое соединение, т. пл. 171-172oC и с выходом 66,3%
, но с использованием 4-ацетил-2-бутилимидазол-5-карбонитрила, полученного согласно описанию в получении 24 (i), и раствора бромида метилмагния в тетрагидрофуране, в результате чего получали целевое соединение, т. пл. 171-172oC и с выходом 66,3%
ЯМР (CDCl3 + CD3OD) δ ppm: 0,91 (3Н, т, 7 Гц), 1,0-2,1 (4Н, м), 1,62 (6Н, с), 2,69 (2Н, т, J 7 Гц).
Получение 30. 2-Бутил-4-[1-гидрокси-2-метил-1-(1-метилэтил)пропил]имидазол-5-карбонитрил.
Повторяли процедуру, описанную в получении 23  , но с использованием 2-бутил-4-изобутирилимидазол-5-карбонитрила, полученного согласно описанию в получении 27, и раствор бромида изопропилмагния в тетрагидрофуране, в результате чего получали целевое соединение, т. пл. 63-65oC и выходом 87%
, но с использованием 2-бутил-4-изобутирилимидазол-5-карбонитрила, полученного согласно описанию в получении 27, и раствор бромида изопропилмагния в тетрагидрофуране, в результате чего получали целевое соединение, т. пл. 63-65oC и выходом 87%
ЯМР (CDCl3) δ ppm: 0,7-1,0 (3Н, м), 0,87 (6Н, д, J 7 Гц), 0,91 (6Н, д, J 7 Гц), 1,0-2,1 (4Н, м), 2,0-2,9 (2Н, м), 2,71 (2Н, т, J 7 Гц).
Получение 31. (5-Метил-2-оксо-1,3-диоксолен-4-ил)метил 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилат.
1,76 мл N,N-диизопропилэтиламина добавляли к суспензии 1,06 г 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоновой кислоты, полученной согласно описанию в получении 22  , в 10 мл N,N-диметилацетамида, и полученную смесь размешивали 10 мин при комнатной температуре, после чего к смеси добавляли 1,12 г (5-метил-2-оксо-1,3-диоксолен-4-ил)метилхлорида, и смесь размешивали 4 ч при 60oC. После этого реакционную смесь смешивали с этилацетатом и водой. Этилацетатный слой отделяли и концентрировали путем выпаривания при пониженном давлении и концентрат очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью метанола и метиленхлорида (1:15, по объему), в результате чего получали 1,14 г целевого соединения в виде вязкого маслянистого продукта.
, в 10 мл N,N-диметилацетамида, и полученную смесь размешивали 10 мин при комнатной температуре, после чего к смеси добавляли 1,12 г (5-метил-2-оксо-1,3-диоксолен-4-ил)метилхлорида, и смесь размешивали 4 ч при 60oC. После этого реакционную смесь смешивали с этилацетатом и водой. Этилацетатный слой отделяли и концентрировали путем выпаривания при пониженном давлении и концентрат очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью метанола и метиленхлорида (1:15, по объему), в результате чего получали 1,14 г целевого соединения в виде вязкого маслянистого продукта.
ЯМР (CDCl3) δ, ppm: 0,94 (3Н, т, J 7,5 Гц), 1,62 (6Н, с), 1,6-1,8 (2Н, м), 2,19 (3Н, с), 2,67 (2Н, т, J 8 Гц), 5,03 (2Н, с).
Получение 32. Фталидил 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилат.
Повторяли процедуру, описанную в Получении 31, но с использованием 1,06 г 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоновой кислоты, полученной согласно описанию в получении 22, и 1,15 г 3-бромофталида, в результате чего получали 1,63 г целевого соединения в виде аморфного твердого вещества.
ЯМР (CDCl3) d ppm: 0,92 (2Н, т, J 7,5 Гц), 1,64 (6Н, с), 1,6-1,75 (2Н, м), 2,63 (2Н, т, J 7,5 Гц), 7,63-7,79 (4Н, м), 7,91 (1Н, д, J 8,5 Гц).
Получение 33. Изопропоксикарбонилоксиметил 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоксилат.
Повторяли процедуру, описанную в получении 22 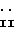 , но с использованием 1,06 г 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоновой кислоты, полученной в соответствии с описанием в получении 22
, но с использованием 1,06 г 4-(1-гидрокси-1-метилэтил)-2-пропилимидазол-5-карбоновой кислоты, полученной в соответствии с описанием в получении 22  , и 0,83 г изопропоксикарбонилоксиметилхлорида, в результате чего получали 1,22 г целевого соединения, т. пл. 144-146oC.
, и 0,83 г изопропоксикарбонилоксиметилхлорида, в результате чего получали 1,22 г целевого соединения, т. пл. 144-146oC.
ЯМР (CDCl3) δ ppm: 0,98 (3Н, т, J 7,5 Гц), 1,32 (6Н, д, J 6,5 Гц), 1,62 (6Н, с), 1,76 (2Н, секстет, J 7,5 Гц), 2,69 (2Н, т, J 7,5 Гц), 4,93 (1. квин. J 6,5 Гц), 5,95(2Н, с).
Получение 34. 2-Этилимидазол-4,5-дикарбонитрил.
Повторяли процедуру, описанную в получении 1, но с использованием 53,3 г диаминолалеонитрила и 91,3 г триетилортопропионата, в результате чего получали 59,5 г целевого соединения в виде кристаллов, т. пл. 179-181oC.
Получение 35. 2-Этилимидазол-4,5-дикарбоновая кислота.
Повторяли процедуру, описанную в получении 2, но с использованием 45,0 г 2-этилимидазол-4,5-дикарбонитрила полученного в соответствии с описанием в получении 34, в результате чего получали 31,2 г целевого соединения в виде кристаллов, т. пл. 265-268oC.
Получение 36. Диэтил 2-этилимидазол-4,5-дикарбоксилат.
Повторяли процедуру, описанную в получении 3, но с использованием 35,0 г 2-этилимидазол-4,5-дикарбоновой кислоты, полученной в соответствии с получением 35, в результате чего получали 38,7 г целевого соединения в виде кристаллов, т. пл. 83-84oC.
Получение 37. Этил 2-этил-4-(1-гидрокси-1-метилэтил)имидазол-5-карбоксилат.
Повторяли процедуру, описанную в получении 8, но с использованием 3,60 г диэтил 2-этилимидазол-4,5-дикарбоксилата, полученного в соответствии с описанием в получении 36, 60 мл 1 М раствора бромида метилмагния в тетрагидрофуране, в результате чего получали 2,05 г целевого соединения в виде кристаллов, т. пл. 181-184oC.
ЯМР (ДМСО-d6) d ppm: 1,22 (3Н, т, J 7 Гц), 1,33 (3Н, т, J 7,5 Гц), 1,50 (6. с), 2,65 (2Н, кв. J 7,5 Гц), 3,30 (1Н, шир.с), 4,31 (2Н, кв. J 7,5 Гц).
Получение 38. N-т-Бутил-4'-бромометилбифенил-2-карбоксамид.
38(i) N-т-Бутил-4'-метилбифенил-2-карбоксамид.
5,7 мл оксалилхлорида по капле добавляли, охлаждая при этом льдом, к раствору 6,91 г 4'-метилбифенил-2-карбоновой кислоты в 70 мл метиленхлорида, и полученную смесь размешивали 2 ч при комнатной температуре. Затем смесь концентрировали путем выпаривания при пониженном давлении и остаток растворяли в 70 мл тетрагидрофурана. После этого к раствору, охлаждая льдом, по капле добавляли раствор 7,5 мл т-бутиламина 4 50 мл тетрагидрофурана, и полученную смесь размешивали 10 мин при комнатной температуре. По истечение этого времени, реакционную смесь разбавляли водой и этилацетатом. Этилацетатный слой отделяли, промывали водным раствором бикарбоната натрия, а затем водным раствором хлорида натрия, осушали безводным сульфатом натрия и концентрировали путем выпаривания при пониженном давлении, в результате чего получали 7,48 г целевого соединения в виде кристаллов, т. пл. 105-106oC (после перекристаллизации из этилацетата и гексана).
ЯМР (CDCl3) d, ppm: 1,12 (9Н, с), 2,41 (3Н, с), 5,04 (1Н, шир.с), 7,2-7,5 (7Н, м), 7,71 (1Н, д, J 8 Гц).
38  . N-т-Бутил-4'-бромометилбифенил-2-карбоксамид.
. N-т-Бутил-4'-бромометилбифенил-2-карбоксамид.
4,39 г N-бромосукцинимида и 50 мг бензоилпероксида добавляли к раствору 6600 г N-т-бутил-4'-метилбифенил-2-карбоксамида, полученного согласно описанию в получении 38i, в 90 мл тетрахлорметана, и полученную смесь нагревали с обратным холодильником в течение 4 ч. После этого реакционную смесь охлаждали до комнатной температуры, промывали водой, осушали безводным сульфатом магния и концентрировали путем выпаривания при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью этилацетата и гексана (1:4, по объему), в результате чего получали 7,04 г целевого соединения в виде кристаллов, т. пл. 124-126oC.
ЯМР (CDCl3) δ ppm: 1,14 (9Н, с), 4,55 (2Н, с), 4,99 (1Н, шир. с), 7,30-7,32 (8Н, м).
Получение 39. 4-Изобутил-2-пропилимидазол-5-карбонитрил.
Повторяли процедуру, описанную в получении 23(i), но с использованием 8,24 г 2-пропилимидазол-4,5-дикарбонитрила, полученного согласно описанию в получении 10, и 103 мл 2 М раствора иодида изопропилмагния в диэтиловом эфире, в результате чего получали 45,0 г целевого соединения в виде кристаллов, т. пл. 90,5-91oC.
ЯМР (CDCl3) d ppm: 1,01 (3Н, т, J 7,5 Гц), 1,29 (6Н, д, J 6,5 Гц), 1,82 (2Н, секст. J 7,5 Гц), 2,81 (2Н, т, J 7,5 Гц), 3,66 (1Н, септет, J 6,5 Гц).
Получение 40. 2-Бутил-4-пивалоилимидазол-5-карбонитрил.
Раствор 10,4 г 2-бутилимидазол-4,5-дикарбонитрила, полученного согласно описанию в получении 1, в 150 мл метиленхлорида по капле добавляли, при 10-15oC и в атмосфере азота, к 100 мл 2 М раствора хлорида т-бутилмагния в диэтиловом эфире и полученную смесь размешивали 1 ч при той же температуре. Затем к реакционной смеси по капле добавляли 200 мл этилацетата и 100 мл водного раствора бисульфата калия и полученную смесь размешивали 20 мин при комнатной температуре. По истечение этого времени, нерастворившийся материал удаляли фильтрацией, а органический слой отделяли, осушали безводным сульфатом магния, и концентрировали путем выпаривания при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя смесью этилацетата и гексана (1,3, по объему), в результате чего получали 7,95 г целевого соединения в виде кристаллов, т. пл. 135-137oC.
ЯМР (CDCl3) d ppm: 0,95 (3Н, т, J 7,5 Гц), 1,42 (2Н, септет, J 7,5 Гц), 1,46 (9Н, с), 1,75 (2Н, квин. J 7,5 Гц), 2,79 (2Н, т, J 7,5 Гц).
Получение 41. 2-Пропил-2-пивалоилимидазоол-5-карбонитрил.
Повторяли процедуру, описанную в получении 40, но с использованием 3,2 г 2-пропилимидазол-4,5-дикарбонитрила, полученного согласно описанию в получении 10, и 33 мл 2 М раствора хлорида т-бутилмагния в диэтиловом эфире, в результате чего получали 2,35 г целевого соединения в виде кристаллов, т. пл. 176-178oC.
ЯМР (CDCl3) d ppm: 0,93 (3Н, т, J 7,5 Гц), 1,36 (9Н, с), 1,75 (2Н, секстет, J 7,5 Гц), 2,68 (2Н, т, J 7,5 Гц).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 1-БИФЕНИЛМЕТИЛИМИДАЗОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ГИПЕРТЕНЗИИ | 1992 |
|
RU2128173C1 |
| ПРОИЗВОДНЫЕ БИФЕНИЛА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2109736C1 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СЛОЖНЫЕ ЭФИРЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2086541C1 |
| ТРИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2072997C1 |
| СТЕРОИДНЫЕ СОЕДИНЕНИЯ, СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ | 1993 |
|
RU2097387C1 |
| α,ω ДИАРИЛАЛКАНЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2105752C1 |
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНКАРБОНОВОЙ КИСЛОТЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2091373C1 |
| ФУНГИЦИДНЫЕ ПРОИЗВОДНЫЕ ОКСЕТАНА И ИХ СОЛИ | 1992 |
|
RU2044736C1 |
| АЗАСТЕРОИДНЫЕ СОЕДИНЕНИЯ | 1991 |
|
RU2070204C1 |
| ПРОИЗВОДНЫЕ КАРБАПЕНЕМА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2097383C1 |

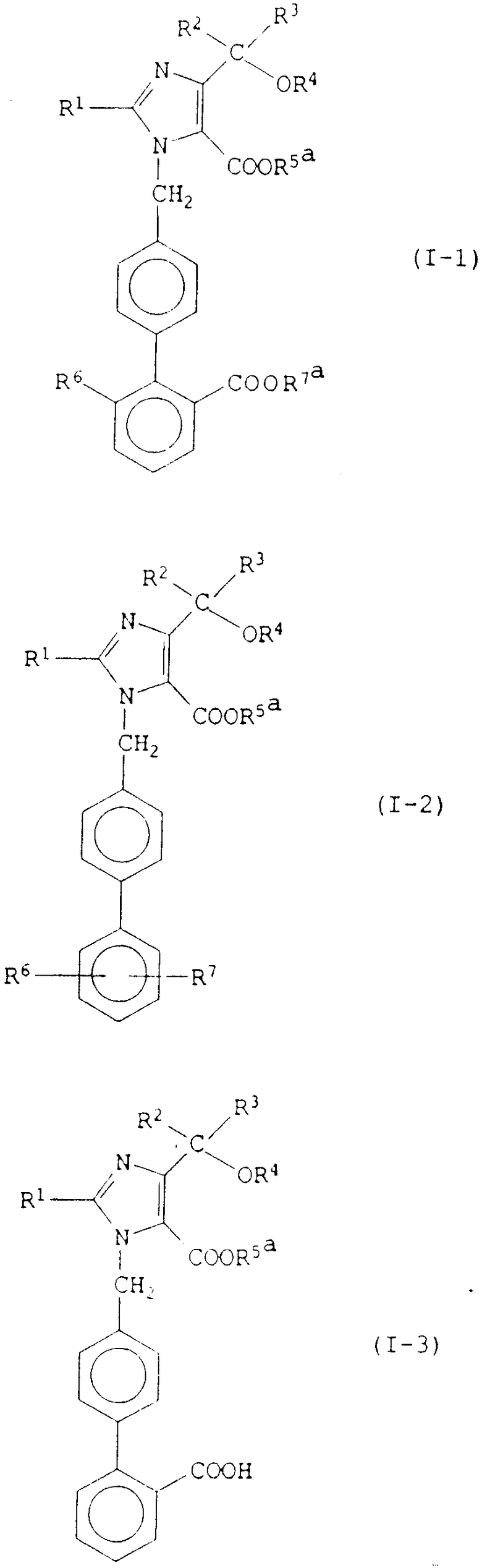
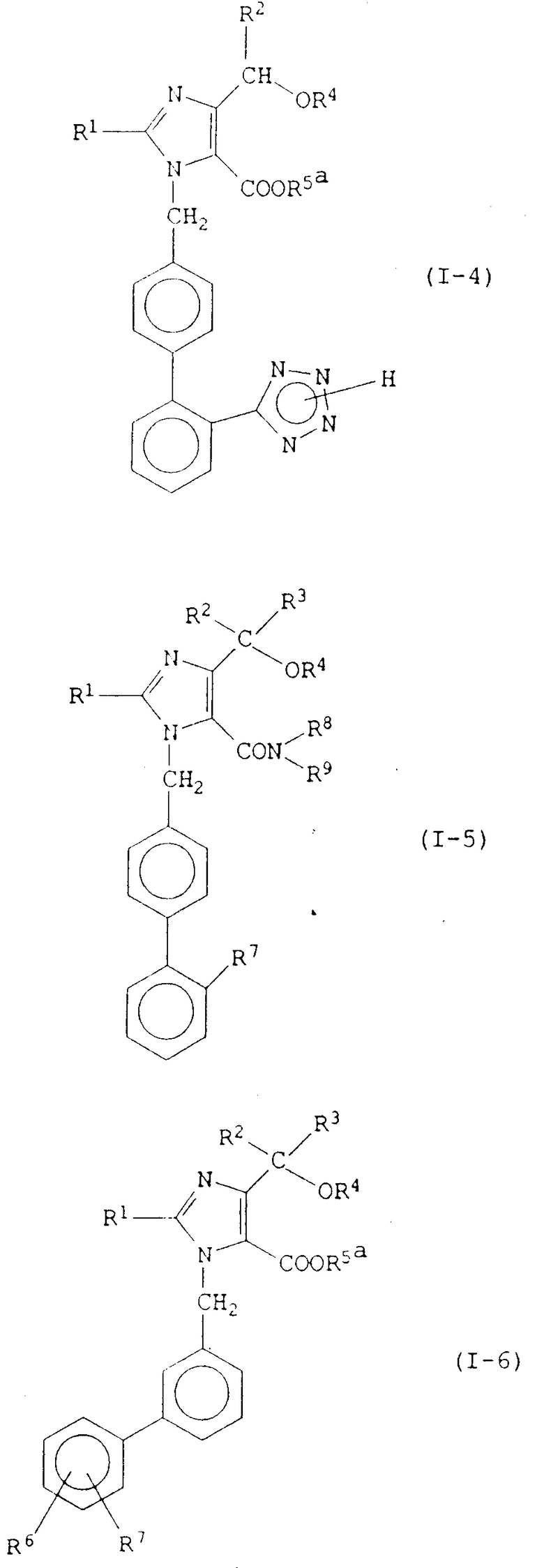
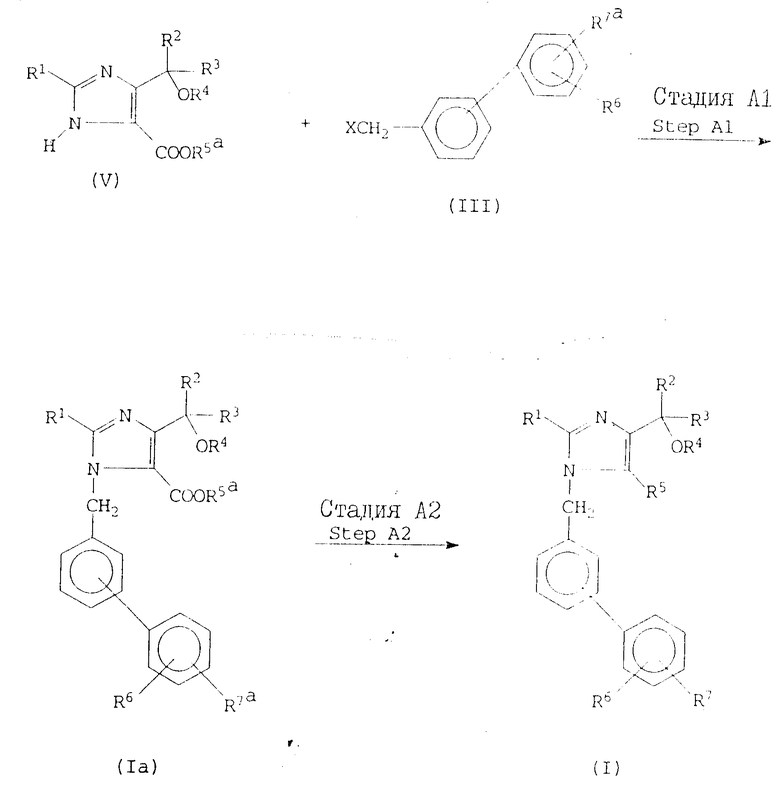
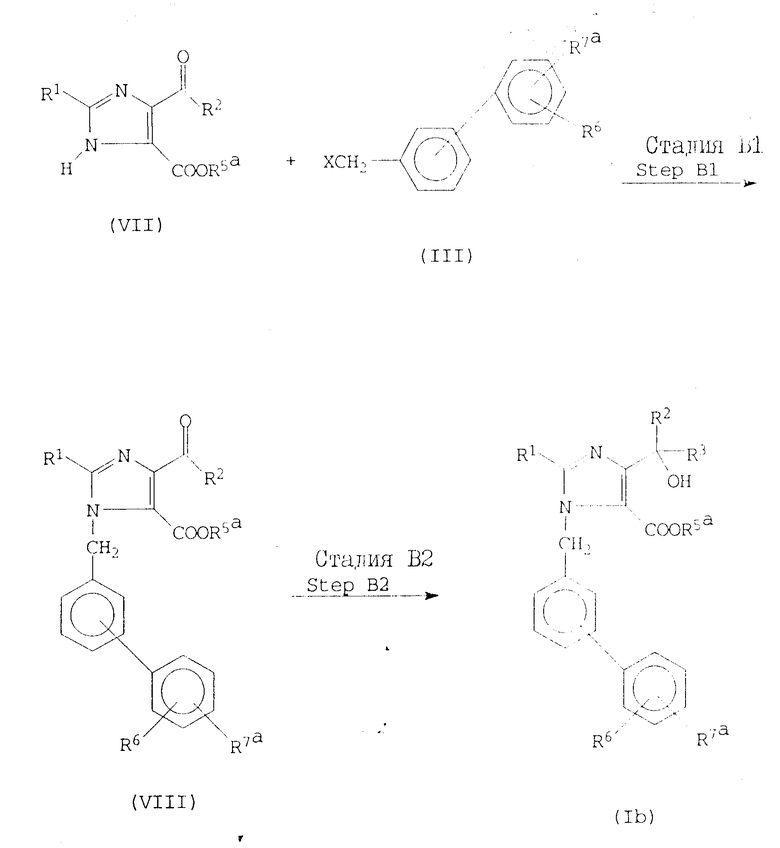
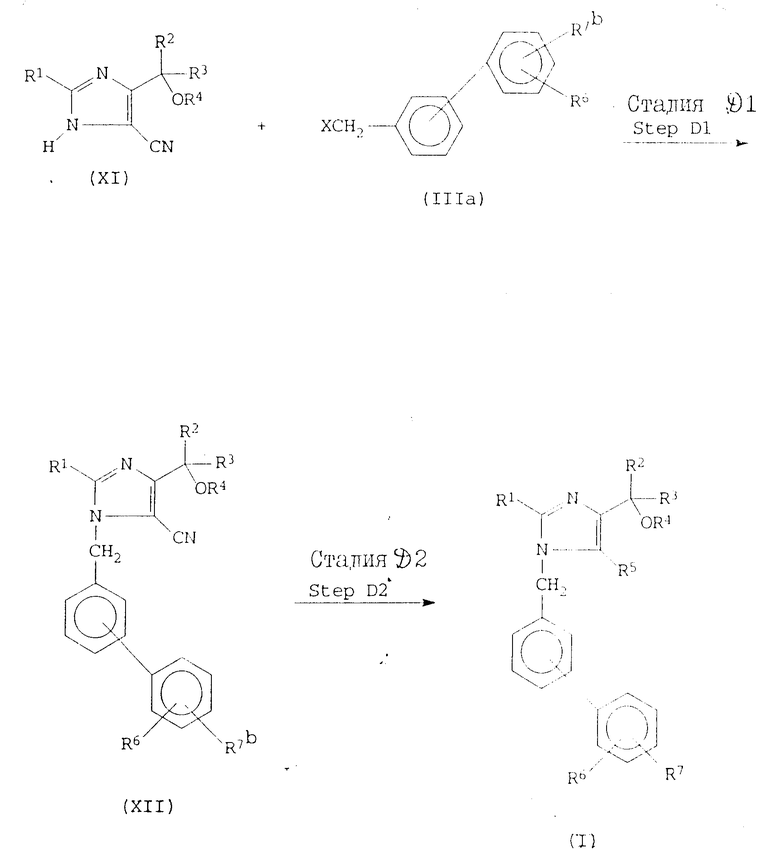

Изобретение относится к соединениям формулы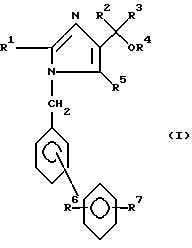
где R1 - алкил, или алкенил; R2 и R3 - водород, алкил, алкенил, циклоалкил, аралкил, арил, или арил, сплавленный с циклоалкилом; R4 - водород, алкил, алканоил, алкеноил, арилкарбонил, алкоксикарбонил, тетрагидропиранил, тетрагидротиопиранил, тетрагидротиенил, тетрагидрофурил, группу формулы -SiRaRbRc, Ra,Rb и Rc являются алкилом или арилом, алкоксиметилом, алкоксиалкокси метилом, галогеналкоксиметилом, аралкилом, арилом или алканоилоксиметоксикарбонилом; R5 - карбоксильная группа или группа формулы -CONR8R9, в которой R8 и R9 являются атомами водорода или алкилом, или R8 и R9 вместе образуют алкилен; R6 - водород, алкил, алкокси или галоген; R7 - карбоксильная группа или тетразол-5-ильная группа; или к их фармацевтически приемлемым солям и сложным эфирам. Указанные соединения и их фармацевтически приемлемые соли и сложные эфиры обладают гипотенсивной активностью и могут быть использованы для лечения и профилактики гипертензии. Предлагаемые соединения могут быть получены, inter alia, посредством реакции бифенилметилового соединения с имидазоловым соединением. 3 с. и 28 з. п. ф-лы, 1 табл.
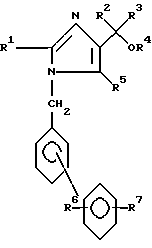
где R1 алкильная группа, имеющую 1 6 атомов углерода;
R2 и R3, одинаковые или различные, каждый алкильная группа, имеющая 1 6 атомов углерода, или арильная группа, имеющая 6 10 атомов углерода;
R4 водород или алкильная группа, имеющая 1 6 атомов углерода;
R5 карбоксигруппа или группа общей формулы COOR5а или -CONR8R9, где R8 и R9, одинаковые или различные, каждый водород, незамещенная алкильная группа, имеющая 1 6 атомов углерода, замещенная алкильная группа, имеющая 1 6 атомов углерода и замещенная карбоксигруппа или алкоксикарбонильная группа, в которой алкильная часть имеет 1 6 атомов углерода, или R8 и R9 вместе представляют собой замещенную алкиленовую группу, которая имеет 2 6 атомов углерода и является замещенной алкоксикарбонильной группой, в которой алкильная часть имеет 1 6 атомов углерода; R5a алкильная группа, имеющая 1 6 атомов углерода, алканоилоксиалкильная группа, в которой каждая из алканоилной и алкильных частей имеет 1 6 атомов углерода; алкоксикарбонилоксиалкильная группа, в которой каждая из алкокси- и алкильной частей имеет 1 6 атомов углерода, (5-метил-2-оксо-1,3-диоксолен-4-ил)метильная группа или фталидильная группа;
R6 водород;
R7 карбоксигруппа или тетразол-5-ильная группа
их фармацевтически приемлемые соли или эфиры.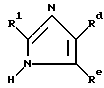
где R1 имеет указанные значения;
Rd является группой общей формулы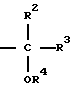
где R2 R4 определены в п.1,
Rd группа формулы -COORf, где Rf - карбоксизащитная группа, или Rd группа формулы -COR2, где R2 определен выше, цианогруппа;
Rl циано-, карбоксигруппа или группа формулы -CORf, где Rf определен выше,
с соединением общей формулы III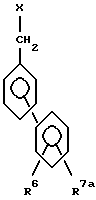
где R6 определен выше;
R7а защищенная карбоксильная группа, цианогруппа, защищенная тетразол-5-ильная группа, карбамоильная или алкилкарбамоильная группа;
X галоген;
с получением соединения общей формулы IV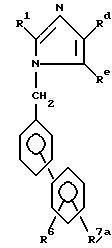
где Rd, Rl, R1, R6 и R7a определены выше,
и в любом порядке удаление защитных групп и, если необходимо, превращение указанной группы Rd в группу общей формулы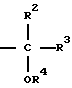
где R2, R3 и R4 определены выше,
и, если необходимо, превращение группы Rl в группу R5, превращение группы R7а в группу R7 или алкилирование или ацилирование гидроксильной группы в группу R4 с получением соединения формулы I,
и необязательно превращают в соль или сложный эфир полученного продукта.
Приоритет по признакам:
21.02.91 при R1 С1 С6-алкил; R2 и R3 одинаковые или различные, каждый алкильная группа, имеющая 1 6 атомов углерода, или арильная группа, имеющая 6 10 атомов углерода, R4 водород или алкильная группа, имеющая 1 6 атомов углерода; R5 карбоксигруппа или группа формулы -COOR5а, где R5а - алкильная группа, имеющая 1 6 атомов углерода, алканоилоксиалкильная группа, в которой каждая из алканоильной и алкильных частей имеет 1 6 атомов углерода, алкоксикарбонилоксиалкильная группа, в которой каждая из алкокси- и алкильной частей имеет 1 6 атомов углерода (5-метил-2-оксо-1,3-диоксолен-4-ил)метильной группы, R6 водород, R7 карбоксигруппа. 26.04.91 при R5а фталидильная группа, R7 тетразол-5-ил.
| EP, патент, 253310, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

