Изобретение относится к ряду новых оксетановых производных, молекулярная структура которых характеризуется наличием четырехчленного кольца, содержащего атом кислорода (т.е. оксетанового кольца), и которые обладают сельскохозяйственной и фармацевтической противогрибковой или фунгицидной активностью. Данное изобретение представляет также способ получения этих соединений, а также способы и композиции, содержащие их, для защиты животных, включая людей, и растений от поражения грибками.
Хотя уже известны многие различные типы триазольных производных, имеющих сельскохозяйственную противогрибковую или фунгицидную активность, почти все из них не имеют оксетанового фрагмента, который является характерной чертой соединений предлагаемого изобретения.
В европейском патенте N 106515 раскрывается использование некоторых производных 2-(триазолилметил)оксетан-4-она в качестве промежуточных продуктов в процессе получения противогрибковых агентов (которые не являются производными оксетана), но эти промежуточные соединения в структурном отношении отличаются от соединений предлагаемого изобретения, и было найдено, что сами по себе они не обладают какой-либо противогрибковой активностью.
Кроме того, в европейском патенте N 318214 описывается применение ограниченного ряда 2-(триазолилметил)-оксетановых производных в качестве противо- грибковых агентов, хотя данное предшествующее описание касается главным образом соответствующих тетрагидрофураниловых соединений, а 2-(триазолилметил)-оксетановые производные, конкретно раскрытые в нем, отличаются от соединений предлагаемого изобретения характером заместителя в 4-положении оксетанового кольца.
В настоящее время мы обнаружили ряд новых соединений триазола, имеющих оксетановый скелет, которые отличаются в структурном отношении триазольных производных и которые обладают превосходными фармацевтическими и сельскохозяйственными противогрибковыми или фунгицидными активностями.
Целью изобретения является предоставление в качестве новых веществ ряда новых производных оксетана.
Следующий целью изобретения является предоставление способа получения этих соединений.
Еще одной целью изобретения является предоставление композиций, включающих эти соединения, и подходящих для использования в качестве или фармацевтических, или сельскохозяйственных противогрибковых или фунгицидных агентов, и предоставление способов использования этих соединений и композиций для фармацевтических и сельскохозяйственных целей.
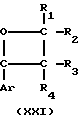
Новые производные оксетана изобретения могут быть представлены формулой
H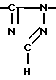 CR8R
CR8R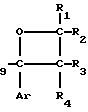 (I) в которой R1 и R2 независимо выбраны из группы, состоящей из атомов водорода и алкильных групп, имеющих от 1 до 6 атомов углерода, или R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют циклоалкильную группу, имеющую от 3 до 6 атомов углерода; R3 и R4независимо выбраны из группы, состоящей из атомов водорода, алкильных групп, имеющих от 1 до 6 атомов углерода, и фенильных групп, или R3 и R4вместе с атомом углерода, к которому они присоединены, образуют циклоалкильную группу, имеющую от 3 до 6 атомов углерода; или R1 и R3 и атомы углерода, к которым они присоединены, вместе образуют циклоалкильную группу, имеющую 5 или 6 кольцевых атомов и сконденсированную с оксетановым кольцом; R2 выбран из группы, состоящей из атомов водорода и алкильных групп, имеющих от 1 до 6 атомов углерода; и R4 выбран из группы, состоящей из атомов углерода, алкильных групп, имеющих от 1 до 6 атомов углерода, и фенильных групп; Ar представляет собой фенильную группу, замещенную группами R5, R6 и R7, где R5, R6 и R7независимо выбраны из группы, состоящей из атомов водорода, атомов галогена, алкильных групп, имеющих от 1 до 6 атомов углерода, алкоксигрупп, имеющих от 1 до 6 атомов углерода, галоидированных алкильных групп, имеющих от 1 до 6 атомов углерода, и галоидированных алкоксигрупп, имеющих от 1 до 6 атомов углерода; и R8 и R9 независимо выбраны из группы, состоящей из атомов водорода и алкильных групп, имеющих от 1 до 4 атомов углерода; и их солями.
(I) в которой R1 и R2 независимо выбраны из группы, состоящей из атомов водорода и алкильных групп, имеющих от 1 до 6 атомов углерода, или R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют циклоалкильную группу, имеющую от 3 до 6 атомов углерода; R3 и R4независимо выбраны из группы, состоящей из атомов водорода, алкильных групп, имеющих от 1 до 6 атомов углерода, и фенильных групп, или R3 и R4вместе с атомом углерода, к которому они присоединены, образуют циклоалкильную группу, имеющую от 3 до 6 атомов углерода; или R1 и R3 и атомы углерода, к которым они присоединены, вместе образуют циклоалкильную группу, имеющую 5 или 6 кольцевых атомов и сконденсированную с оксетановым кольцом; R2 выбран из группы, состоящей из атомов водорода и алкильных групп, имеющих от 1 до 6 атомов углерода; и R4 выбран из группы, состоящей из атомов углерода, алкильных групп, имеющих от 1 до 6 атомов углерода, и фенильных групп; Ar представляет собой фенильную группу, замещенную группами R5, R6 и R7, где R5, R6 и R7независимо выбраны из группы, состоящей из атомов водорода, атомов галогена, алкильных групп, имеющих от 1 до 6 атомов углерода, алкоксигрупп, имеющих от 1 до 6 атомов углерода, галоидированных алкильных групп, имеющих от 1 до 6 атомов углерода, и галоидированных алкоксигрупп, имеющих от 1 до 6 атомов углерода; и R8 и R9 независимо выбраны из группы, состоящей из атомов водорода и алкильных групп, имеющих от 1 до 4 атомов углерода; и их солями.
Изобретение также представляет фармацевтическую композицию для профилактики или лечения грибковых заражений, которая включает фунгидно или фунгистатически эффективное количество противогрибкового агента, в которой антигрибковый агент выбран из группы, состоящей из соединений формулы I и фармацевтически приемлемых их солей, определенных выше.
Далее изобретение представляет способ профилактики или лечения грибковых заражений, который включает применение или назначение фунгицидно или фунгистатически эффективного количества противогрибкового агента животному, например, млекопитающему, которым может быть человек, и в котором противогрибковый агент выбран из группы, состоящей из соединений формулы I и их фармацевтически приемлемых солей, определенных выше.
Изобретение представляет также сельскохозяйственную композицию для защиты растений и веществ для воспроизводства растений от поражения грибками, которая включает фунгицидно или фунгистатически эффективное количество соединения формулы I или его соли, определенных выше, в смеси с сельскохозяйственным носителем или разбавителем.
Изобретение представляет также способ защиты растений и веществ для воспроизводства растений от поражения грибками, который предусматривает применение к указанным растениям или веществам для их воспроизводства, или к месту распространения их, фунгицидно или фунгистатически эффективного количества соединения формулы I или его соли, определенных здесь выше.
Изобретение также представляет несколько новых способов получения соединений предлагаемого изобретения, которые описываются более подробно ниже.
В соединениях изобретения, в которых R1, R2, R3, R4, R5, R6 или R7представляет алкильную группу, данная группа может быть алкильной группой с прямой или разветвленной цепью, имеющей от 1 до 6 атомов углерода. Примеры таких групп включают метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, изопентил, трет-пентил, неовентил, гексил, изогексил, 2-метилбутил, 4-метилпентил, 3-метилпентил, 2-метилпентил, 3,3-диметилбутил, 2,2-диметилбутил, 1,1-диме- тилбутил, 1,2-диметилбутил, 1,3-диметилбутил и 2,3-диметилбутил группы, из которых предпочтительны указанные алкильные группы, содержащие от 1 до 4 атомов углерода.
В тех случаях, когда R1 и R2 или R3 и R4 вместе с атомом углерода, к которому они присоединены, образуют циклоалкильную группу, имеющую от 3 до 6 атомов углерода, образуется спиродвойная кольцевая система с оксетановым кольцом. Такие циклоалкильные группы включают циклопропильную, циклобутильную, циклопентильную и циклогексильную группы, из которых циклопентильная и циклогексильная группы являются предпочтительными, и наиболее предпочтительной является циклогексильная группа. Предпочтительно только один из R1 и R2 или R3 и R4 образуют такую спиросистему, и более предпочтительно такой парой является R3 и R4.
Когда R1 и R3 вместе с атомами углерода, к которым они присоединены, образуют циклоалкильную группу, последний является циклопентильная или циклогексильная группа, сконденсированная с оксетановым кольцом, и предпочтительно циклогексильная группа. Остальные группы, R2 и R4 могут быть атомами водорода или алкильными группами, или в случае R4 фенильной группой.
Из групп и атомов, которые могут быть представлены символами R1, R2, R3 и R4, мы особенно предпочитаем, чтобы они были одинаковыми или различными, и каждый из них представлял атом водорода или алкильную группу, содержащую от 1 до 4 атомов углерода. Более предпочтительно один из R1 и R2 должен представлять атом водорода, а другой алкильную группу, содержащую от 1 до 4 атомов углерода, и один из R3 и R4 должен представлять атом водорода или алкильную группу, содержащую от 1 до 4 атомов углерода, а другой должен представлять алкильную группу, содержащую от 1 до 4 атомов углерода. Еще более предпочтительно: один из R1 и R2 представляет атом водорода, а другой метильную или этильную группу, и один из R3 и R4 представляет собой атом водорода или метильную группу, а другой представляет метильную группу. Наиболее предпочтительно: R1 и R4 оба представляют метильную группу, а R2 и R3оба представляют атом водорода; или R1 представляет этильную группу, R4представляет метильную группу и R2 и R3 оба представляют атомы водорода; или R1 и R2 оба представляют атомы водорода и R3 и R4 оба представляют метильные группы; или R1, R3 и R4 все представляют собой метильные группы и R2 представляет собой атом водорода.
Ar представляет собой фенильную группу, имеющую от 1 до 5 заместителей, представленных символами R5, R6 и R7, т.е. он может быть замещенной или незамещенной фенильной группой, которая имеет 1,2 или 3 заместителя. Когда имеется два или три из указанных заместителей, они могут быть одинаковыми или различными.
Когда R5, R6 или R7 представляет атом галогена, им может быть атом фтора, хлора, брома или йода, предпочтительно им является атом фтора или атом хлора.
Когда R5, R6 или R7 представляет собой алкильную группу, ею может быть группа, определенная выше.
Когда R5, R6 или R7 представляет алкокси группу, ею может быть алкоксигруппа с прямой или разветвленной цепью, имеющая от 1 до 6 атомов углерода. Примеры таких групп включают метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси, трет-бутокси, пентилокси, изопентилокси, трет-пентилокси, неопентилокси, гексилокси, изогексилокси, 2-метилбутокси, 4-метилпентилокси, 3-метилпентилокси, 2-метилпентилокси, 3,3-диметилбутокси, 2,2-диметилбутокси, 1,1-диметилбутокси, 1,1-диметилбутокси, 1,3-диметилбутокси и 2,3-диметилбутокси группы. Из них мы предпочитаем алкокси-группы, содержащие от 1 до 4 атомов углерода.
Когда R5, R6 или R7 представляет собой галоидированную алкильную группу, она имеет от 1 до 6 атомов углерода и может быть алкильной группой с прямой или разветвленной цепью, имеющей один или более, предпочтительно от 1 до 5 (или менее, если имеется меньше способных к замещению положений), более предпочтительно от 1 до 3, галоидных заместителей. Примеры их включают любую из алкильных групп, примеры которых приведены выше, но более предпочтительно группы, имеющие от 1 до 4, и наиболее предпочтительно 1 или 2 атома углерода, в которых один или более атомов водорода замещены атомом галогена (например, атомом фтора, хлора, брома или йода). Конкретные примеры включают трифторметил, трихлорметил, дифторметил, дихлорметил, дибромметил, фторметил, хлорметил, бромметил, иодметил, 2,2,2-трихлорэтил, 2,2,2-трифторэтил, 2-бромэтил, 2-хлорэтил, 2-фторэтил и 2,2-дибромэтил группы, из которых мы предпочитаем трифторметил, трихлорметил, дифторметил, 2-бромэтил, 2-хлорэтил и 2-фторэтильная группы, особенно трифторметильную группу.
Когда R5, R6 или R7 представляет собой галоидированную алкоксигруппу, она имеет от 1 до 6 атомов углерода и может быть алкоксигруппой с прямой или разветвленной цепью, имеющей один или более, предпочтительно от 1 до 5 (или меньше, если имеется меньшее число замещаемых положений), более предпочтительно от 1 до 3 галоидных заместителей. Примеры включают любую из алкоксигрупп, примеры которых приведены выше, в которой один или более атомов водорода замещены атомами галогена (например, атомом фтора, хлора, брома или йода). Конкретные примеры включают трифторметокси трихлорметокси, дифторметокси, дихлорметокси, дибромметокси, фторметокси, хлорметокси, бромметокси, иодметокси, бромдифторметокси, хлордифторметокси, 2,2,2-трихлорметокси, 2,2,2-трифторэтокси, 2-бромэтокси, 2-хлорэтокси, 2-фторэтокси и 2,2-дибромэтокси группы, из которых мы предпочитаем трифторметокси группу.
Когда фенильная группа Ar имеет единственный заместитель, он предпочтительно находится в 2-(орто) или 4-(пара-) положении, более предпочтительно 4-положении. Когда имеется два таких заместителя, они находятся предпочтительно в 2,4- или 2,6-положениях, более предпочтительно 2,4-положениях. Когда имеется три таких заместителя, они предпочтительно находятся в 2,4,5-или 2,4,6-положениях, более предпочтительно в 2,4,6-положениях. Мы предпочитаем, чтобы было один и два таких заместителя.
Из групп и атомов, которые могут быть представлены символами R5, R6и R7, мы особенно предпочитаем, чтобы они независимо были выбраны из группы, состоящей из атомов водорода, атомов галогена и галоидированных алкильных групп, имеющих от 1 до 4 атомов углерода, более предпочтительно из группы, состоящей из атомов водорода и атомов галогена, в этих случаях ими являются предпочтительно атомы водорода, хлора, фтора или брома. В одном предпочтительном классе таких соединений один из R5, R6 и R7 представляет собой атом водорода, а другие два являются одинаковыми или различными и каждый представляет атом галогена или галоидированную алкильную группу, имеющую от 1 до 4 атомов углерода; более предпочтительно один и R5, R6 и R7 представляет атом водорода, а два других являются одинаковыми или различными, и каждый представляет атом галогена; еще более предпочтительно, один из R5, R6 и R7представляет атом водорода, а другие два являются одинаковыми или различными и каждый представляет собой атом хлора, фтора или брома.
Более предпочтительными группами, представленными символом Ar, являются о-хлорфенильная, п-хлорфенильная, п-фторфенильная, п-бромфенильная, 2,4-дифторфенильная, 2,6-дифторфенильная, 2,4-дихлорфенильная, 2-хлор-4-фторфенильная, 4-хлор-2-фторфенильная, 6-хлор-2-фторфенильная, 4-трифторметилфенильная и 4-трифторметоксифенильная группы, из которых п-хлорфенильная, п-фторфенильная, 2,4-дифторфенильная, 2-хлор-4-фторфенильная и 4-хлор-2-фторфенильная группы являются наиболее предпочтительными.
Когда R8 или R9 представляет алкильную группу, это может быть алкильная группа с прямой или разветвленной цепью, имеющая от 1 до 4 атомов углерода. Примеры таких групп включают метильную, этильную, пропильную, изопропильную, бутильную, изобутильную, втор-бутильную и трет-бутильную группы, из которых мы предпочитаем алкильную группу, содержащую 1 или 2 углеродных атома. Более предпочтительно один из R8 и R9 представляет атом водорода, а другой представляет атом водорода или алкильную группу, имеющую от 1 до 4 атомов углерода, и еще более предпочтительно оба из R8 и R9 представляют атомы водорода.
Соединения изобретения включают несколько основных атомов азота и могут, следовательно, образовывать кислотно-аддитивные соли. Нет какого-либо конкретного ограничения в отношении характера этих солей, при условии, что когда они предназначаются для терапевтического использования, они являются фармацевтически приемлемыми, и когда они предназначены для сельскохозяйственного использования, они являются приемлемыми в сельскохозяйственном отношении. Когда они предназначаются для нетеравпевтического или несельскохозяйственного применения, например, в качестве промежуточных продуктов при получении других, и особенно более активных, соединений даже и данное ограничение не является необходимым. Примеры таких кислотно-аддитивных солей включают: соли с минеральными кислотами, особенно галоидоводородной кислотой (такой, как соляная, фтористоводородная, бромистоводородная кислота или иодистоводородная кислота), или с другой минеральной кислотой (такой, как серная кислота, азотная, надхлорная кислота или фосфорная кислота); соли с органической карбоновой кислотой, такой как щавелевая, малеиновая, янтарная кислота или лимонная кислота; и соли сульфоновой кислоты, например, с алкансульфоновой или галоидалкансульфоновой кислотой, такой как метансульфокислота, трифторметансульфокислота или этансульфокислота, или с арилсульфоновой кислотой, такой как бензолсульфо- или п-толуолсульфокислота. Предпочитаются нитраты и оксалаты.
Соединения изобретения обязательно содержат несколько асимметричных атомов углерода в их молекулах, каждый из которых может существовать в R-конфигурации или t-конфигурации, и таким образом могут образовывать стереоизомеры. Хотя все они представлены здесь единственной молекулярной формулой, изобретение включает как индивидуальные, разделенные изомеры, так и смеси, включающие их рацематы. Когда применяются приемы стереоспецифического синтеза, могут получаться непосредственно индивидуальные изомеры; с другой стороны, если получается смесь изомеров, индивидуальные изомеры могут быть получены с помощью общепринятых приемов расщепления.
Стереохимия 4-положения оксетанового кольца, по-видимому, не оказывает влияния на активность соединений изобретения. Однако стереохимия 3-положения похоже является важной. Когда соединение имеет два заместителя, один в 3-положении, и один в 4-положении оксетанового кольца, например, 3,4-диметилоксетановое соединение, предпочтительной конфигурацией является (2R, 3S) конфигурация или диастереомерная (2S, 3R) конфигурация, или рацемат (1:1 смесь) двух диастереомеров, которые могут быть представлены как или (2R*, 3S*) или (2S*, 3R*), более предпочтительно (2R, 3S, 4R) конфигурация или диастереомерная (2S, 3R, 4S) конфигурация, или рацемат двух диастереомеров, который может быть представлен, или как (2R*, 3S*, 4R*), или (2S*, 3R*, 4S*).
Примеры конкретных соединений изобретения даны в формулах I-1, I-2 и I-3, в которых заместители имеют значения, определенные в соответствующей из табл. 1-3 соответственно, т. е. табл.1 относится к формуле I-1, табл. 2 к формуле I-2 и табл.3 к формуле I-3.
H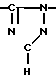 CH
CH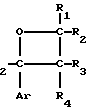 (I-1)
(I-1)
H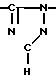 CH
CH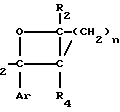 (I-2)
(I-2)
H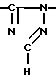 CH
CH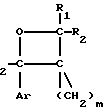 (I-3)
(I-3)
Из соединений проиллюстрированных выше соединения 1-7, 1-21, 1-114, 1-118, 1-145, 1-146, 1-246, 1-277, 1-137, 1-355, 1-383, 1-393, 1-409, 1-447, 1-470, 1-483, 1-486, 1-487, 1-493, 1-497 и 1-508 являются предпочтительными и соединения 1-7, 1-114, 1-145, 1-146, 1-246, 1-277, 1-317, 1-383, 1-447, 1-483 и 1-486 являются более предпочтительными.
Наиболее предпочтительными соединениями являются соединения 1-7, 2-(2,4-ди-фторфенил)-3,4-диметил-2-[(1 -1,2,4-триазол-1-ил)метил] оксетан, особенно (2
-1,2,4-триазол-1-ил)метил] оксетан, особенно (2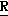 *, 3
*, 3 *, 4
*, 4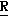 *)-2-(2,4-дифторфенил)-3,4-диметил-2- [(1Н-1,2,4-триазол-1-ил) метил]оксетан изомер;
*)-2-(2,4-дифторфенил)-3,4-диметил-2- [(1Н-1,2,4-триазол-1-ил) метил]оксетан изомер;
1-383, 2-(4-хлорфенил)-3,4-диметил-2-[(1Н-1,2,4-триазол-1-ил)метил]оксетан, особенно (2 *, 3
*, 3 *, 4
*, 4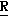 *)-2-(4-хлорфенил)-3,4-диметил-2-[(1
*)-2-(4-хлорфенил)-3,4-диметил-2-[(1 -1,2,4-триазол-1-ил)метил]оксетан изо- мер;
-1,2,4-триазол-1-ил)метил]оксетан изо- мер;
1-483, 4-этил-2-(4-фторфенил)-3-метил-2-[(1 -1,2,4-триазол-1-ил)метил] оксетан, особенно (2
-1,2,4-триазол-1-ил)метил] оксетан, особенно (2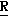 *, 3
*, 3 *, 4
*, 4 *)-4-этил-2-(4-фторфенил)-3-метил-2-[(1
*)-4-этил-2-(4-фторфенил)-3-метил-2-[(1 -1,2,4-триазол-1-ил)ме- тил] оксетан изомер;
-1,2,4-триазол-1-ил)ме- тил] оксетан изомер;
1-486, 2-(4-фторфенил)-3,4-диметил-2-[(1 -1,2,4-триазол-1-ил)метил]оксетан, особенно (2
-1,2,4-триазол-1-ил)метил]оксетан, особенно (2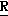 *, 3
*, 3 *, 4
*, 4 *)-2-(4-фторфенил)-3,4-диметил-2-[(1
*)-2-(4-фторфенил)-3,4-диметил-2-[(1 -1,2,4-триазол-1-ил)метил]оксетан изо- мер.
-1,2,4-триазол-1-ил)метил]оксетан изо- мер.
Также являются предпочтительными соли, особенно нитраты и оксалаты, вышеуказанных соединений.
Соединения изобретения могут приготавливаться различными методами, некоторые из них являются хорошо известными в области техники приготовления соединений этого типа. Например, в общем смысле слова, соединения могут приготавливаться любым из следующих способов А, В, С и D, которые являются новыми методами и сами по себе образуют часть изобретения.
С п о с о б А.
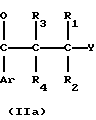
В данном процессе соединение формулы
H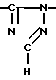 CR8R
CR8R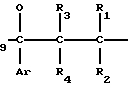 Y (II) в которой R1, R2, R3, R4, R8, R9 и Ar имеют значения, определенные выше; R10 представляет атом водорода или гидроксизащищающую группу; и Y представляет нуклеофильную отщепляемую группу или атом, обрабатывают основанием, чтобы вызвать циклизацию и получить соединение формулы I, которое может, если это желательно, быть подвергнуто реакции образования соли.
Y (II) в которой R1, R2, R3, R4, R8, R9 и Ar имеют значения, определенные выше; R10 представляет атом водорода или гидроксизащищающую группу; и Y представляет нуклеофильную отщепляемую группу или атом, обрабатывают основанием, чтобы вызвать циклизацию и получить соединение формулы I, которое может, если это желательно, быть подвергнуто реакции образования соли.
С п о с о б В.
Альтернативный способ получения соединений изобретения включает взаимодействие соединения формулы
Y-CR8R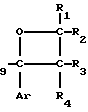 (III) в которой R1, R2, R3, R4, R8, R9, Ar и Y имеют вышеуказанные значения, с соединением формулы
(III) в которой R1, R2, R3, R4, R8, R9, Ar и Y имеют вышеуказанные значения, с соединением формулы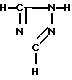 (IV) т. е. 1
(IV) т. е. 1 -1,2,4-триазолом, в присутствии основания для получения соединения формулы I, которое может, если это желательно, быть затем подвергнуто реакции образования соли.
-1,2,4-триазолом, в присутствии основания для получения соединения формулы I, которое может, если это желательно, быть затем подвергнуто реакции образования соли.
С п о с о б С.
Еще один способ получения соединений изобретения включает взаимодействие соединения формулы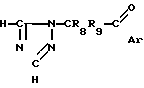 (V) в которой R8, R9 и Ar имеют вышеуказанные значения, с соединением формулы
(V) в которой R8, R9 и Ar имеют вышеуказанные значения, с соединением формулы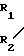 C
C C
C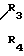 (VI) в которой R1, R2, R3 и R4 имеют вышеуказанные значения, или с его функциональным эквивалентом для получения соединения формулы (I), которое может, если это желательно, быть затем подвергнуто реакции образования соли.
(VI) в которой R1, R2, R3 и R4 имеют вышеуказанные значения, или с его функциональным эквивалентом для получения соединения формулы (I), которое может, если это желательно, быть затем подвергнуто реакции образования соли.
С п о с о б D.
Следующий способ включает расширение кольца соответствующего эпоксисоединения формулы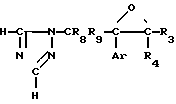 (VII) в которой R3, R4, R8, R9 и Ar имеют вышеуказанные значения, и затем, если это желательно, получившееся соединение подвергают реакции образования соли.
(VII) в которой R3, R4, R8, R9 и Ar имеют вышеуказанные значения, и затем, если это желательно, получившееся соединение подвергают реакции образования соли.
Далее более подробно описаны данные реакции.
С п о с о б А.
В способе А соединение формулы II циклизуется путем обработки основанием.
В соединении формулы II гидроксизащищающей группой, представленной символом R10, может быть любая из групп, обычно используемых в реакциях данного типа, и, поскольку она не остается в конечном продукте, ее природа не является существенной, и она может быть выбрана из широкого ряда таких защищающих групп, которые известны единственно на основе ее функциональности в реакции.
Примеры таких групп включают:
алифатические ацильные группы предпочтительно: алканоильные группы, имеющие от 1 до 25 атомов углерода, более предпочтительно от 1 до 20 атомов углерода, и наиболее предпочтительно от 1 до 6 атомов углерода (такие, как формил, ацетил, пропионил, бутирил, изобутирил, пивалоил, валерил, изовалерил, гексаноил, гептаноил, октаноил, лауроил, миристоил, тридеканоильная, пальмитоильная и стеароильная группы); галоидированные алканоильные группы, имеющие от 2 до 6 атомов углерода, особенно галоидированные ацетильные группы (такие, как хлорацетильная, дихлорацетильная, трихлорацетильная и трифторацетильная группы); низшие алкоксиалканоильные группы, в которых алкоксичасть предпочтительно имеет от 1 до 3 атомов углерода, и алканоильная часть имеет от 2 до 6 атомов углерода и представляет предпочтительно ацетильную группу (такую, как метоксиацетильная группа); и ненасыщенные аналоги таких групп, особенно алкеноильную или алкиноильную группы, имеющие от 3 до 6 атомов углерода (такие, как акрилоильная, метакроильная, пропиолоильная, кротоноильная, изокротоноильная и ( )-2-метил-2-бутеноильная группы);
)-2-метил-2-бутеноильная группы);
ароматические ацильные группы, предпочтительно арилкарбонильные группы, в которых арильная часть имеет от 6 до 14, более предпочтительно от 6 до 10 и наиболее предпочтительно 6 или 10 кольцевых атомов углерода и представляет карбоциклическую группу, которая не замещена или имеет от 1 до 5, предпочтительно от 1 до 3 заместителей, предпочтительно: незамещенные группы (такие, как бензоильная, α-нафтоильная и β-нафтоильная группы); галоидированные арилкарбонильные группы (такие как 2-бромбензоильная и 4-хлорбензоильная группы); низшие алкилзамещенные арилкарбонильные группы, в которых алкильный или каждый алкильный заместитель предпочтительно имеет от 1 до 4 атомов углерода (такие, как 2,4,6-триметилбензоильная и 4-толуолильная группы); низшие алкоксизамещенные арилкарбонильные группы, в которых алкокси или каждый алкоксизаместитель предпочтительно имеет от 1 до 4 атомов углерода (такие, как 4-анизоильная группа); нитрозамещенную арилкарбонильную группу (такую, как 4-нитробензоильная или 2-нитробензоильная группы); низшие алкоксикарбонилзамещенные арилкарбонильные группы, в которых алкоксикарбонил или каждый алкоксикарбонильный заместитель предпочтительно имеет от 2 до 5 атомов углерода [такие, как 2-(метоксикарбонил)-бензоильная группа] и арилзамещенные арилкарбонильные группы, в которых арильный заместитель такой же, как указано выше, за исключением того, что, если он является замещенным дополнительной арильной группой, то тогда арильная группа сама по себе замещена арильной группой (такие, как 4-фенилбензоильная группа);
тризамещенные силильные группы, в которых все три или два, или один из заместителей являются алкильными группами, имеющими от 1 до 4 атомов углерода, и ни один, один или два заместителя представляют собой арильные группы, как указано выше, но предпочтительно фенильные или замещенные фенильные группы, предпочтительно: три (низший алкил) силильные группы (такие, как триметилсилил, триэтилсилил, изопропилметилсилил, трет-бутилдиметилсилил, метилдиизопропилсилил, метилди-трет-бутилсилил и триизопропилсилильные группы); и три(низший алкил)силильные группы, в которых одна или две алкильные группы заменены арильными группами (такие, как фенилметилсилил, дифенилбутилсилил, дифенил-трет-бутилсилил, дифенилизопропилсилил и фенилдиизопропилсилильные группы);
алкоксиальные группы, в которых алкокси и алкильная части каждая имеет от 1 до 4 атомов углерода, особенно алкоксиметильные группы, и такие группы, которые имеют по крайней мере один, предпочтительно от 1 до 5, более предпочтительно от 1 до 3 и наиболее предпочтительно 1, заместителей, предпочтительно: низшие алкоксиметильные группы и другие алкоксиалкильные группы (такие, как метоксиметил, 1,1-диметил-1-метоксиметил, этоксиметил, пропоксиметил, изопропоксиметил, бутоксиметил, и трет-бутоксиметильная группы); низшие алкококсизамещенные низшие алкоксиметильные группы (такие, как 2-метоксиэтоксиметильная группа); галоидированные низкие алкоксиметильные группы (такие, как 2,2,2-трихлорэтоксиметильная и бис(2-хлорэтокси)метильная группы] и низшие алкоксизамещенные этильные группы (такие, как 1-этоксиэтильная и 1-изопропоксиэтильная группы);
другие замещенные этильные группы, предпочтительно: галоидированные этильные группы (такие, как 2,2,2-трихлорэтильная группа); и арилселенилзамещенные группы, в которых арильная часть является такой, как указано выше [такую, как 2-(фенилселенил)этильная группа] аралкильные группы, предпочтительно алкильные группы, имеющие от 1 до 4, более предпочтительно от 1 до 3 и наиболее предпочтительно от 1 до 2 углеродных атомов, которые являются замещенными 1-3 арильными группами, как указаны и проиллюстрированы выше, которые могут быть незамещенными (такие, как бензил, α-нафтилметил, β-нафтилметил, дифенилметил, трифенилметил, α-нафтилдифенилметил и 9-антрилметильная группа) или замещенными в арильной части низшей алкильной группой, низшей алкоксигруппой, нитрогруппой, атомом галогена, цианогруппой или алкилендиокси группой, имеющей от 1 до 3 атомов углерода, в которых алкильная или алкокси-группа могут быть такими, как определены и проиллюстрированы выше, и алкилендиоксигруппа представляет собой предпочтительно метилендиоксигруппу (такие, как 4-метилбензил, 2,4,6-триметилбензил, 3,4,5-триметилбензил, 4-метоксибензил, 4-метоксифенилдифенилметил, 2-нитробензил, 4-нитробензил, 4-хлорбензил, 4-бромбензил, 4-цианобензил, 4-цианобензилдифенилметил, бис-(2-нитрофенил)метил и пиперонильная группы), алкоксикарбонильные группы, особенно, такие группы, которые имеют от 2 до 7, более предпочтительно от 2 до 5 углеродных атомов и которые могут быть незамещенными (такие, как метоксикарбонил, этоксикарбонил, трет-бутоксикарбонил и изобутоксикарбонильная группы) или замещенными атомом галогена или тризамещенной силильной группой, например, три(низший алкилсилил)группой (такие, как 2,2,2-трихлорэтоксикарбонил и 2-триметилсилилэтоксикарбонильная группы);
алкенилоксикарбонильные группы, в которых алкенильная часть имеет от 2 до 6, предпочтительно от 2 до 4 атомов углерода (такие, как винилоксикарбонил и аллилоксикарбонильная группы), и аралкилоксикарбонильные группы, в которых аралкильная часть является такой же, как указана и проиллюстрирована выше, и в которых арильное кольцо, если оно является замещенным, предпочтительно имеет один или два низших алкокси- или нитрозаместителя (такие, как бензилоксикарбонил, 4-метоксибензилоксикарбонил, 3,4-диметоксибензилоксикарбонильная группы). Из них мы предпочитаем аралкильные и три(низший алкил)силильные группы, наиболее предпочтительно бензильную и триметилсилильную группы. Примеры нуклеофильных удаляемых групп и атомов, которые могут быть представлены символом Y, включают: атомы галогена, такие, как хлор, бром и иод;
низшие алкансульфонилокси группы, у которых алкильная часть имеет от 1 до 6, предпочтительно от 1 до 4 атомов углерода (такие, как метансульфонилокси и этансульфонилокси группы);
галоидированные низшие алкансульфонилоксигруппы, в которых алкильная часть имеет от 1 до 6, предпочтительно от 1 до 4 атомов углерода и является замещенной по крайней мере одним атомом галогена и может быть пергалоидированной (такие, как трихлорметансульфонилокси и пентафторэтансульфонилокси группы); и
арилсульфонилокси группы, в которых арильная часть является такой же, как указана и проиллюстрирована выше (такие, как бензолсульфонилокси и п-толуолсульфонилокси группы).
Из них мы предпочитаем низшие алкансульфонилоксигруппы и атомы галогена, и более предпочтительно метансульфонилоксигруппу и атом хлора.
Реакция в данном методе включает циклизацию соединения формулы II путем обработки его основанием, предпочтительно более чем одним эквивалентом основания, и обычно и предпочтительно в растворителе, для замыкания кольца.
Когда R10 представляет собой гидроксизащитную группу, которая может удаляться в присутствии основных условий, реакция на этой стадии может выполняться с использованием исходного материала формулы II, имеющего такую гидроксизащитную группу. В противном случае, необходимо использовать соединение, свободное от такой группы, в данном случае в зависимости от того, как было получено соединение формулы II, может быть необходимым сначала удалить гидроксизащитную группу.
Реакция обычно и предпочтительно проводится в присутствии растворителя, характер которого не имеет большого значения, при условии, что он не оказывает вредного влияния на реакцию и при условии что он растворяет исходное вещество по крайней мере до некоторой степени. Примеры подходящих растворителей включают: алифатические углеводороды, такие как гексан, гептан, лигроин и петролейный эфир; ароматические углеводороды, такие как бензол, толуол и ксилол; галоидированные углеводороды, включающие алифатические и ароматические галоидированные углеводороды, такие как метиленхлорид, хлороформ, четыреххлористый углерод, дихлорэтаны, хлорбензол и дихлорбензолы; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диэтиленгликольдиметиловый эфир; спирты, такие как метанол, этанол, пропанол, изопропанол, бутанол, изобутанол, трет-бутанол, изоамиловый спирт, диэтилегликоль, глицерин, октанол, циклогексанол, и этиленгликольмонометиловый эфир (например, продаваемый под торговым наименованием "Метилцеллозольв");
нитросоединения, такие как нитроэтан и нитробензол; нитрилы, такие как ацетонитрил и изобутиронитрил; амиды, такие как формамид, диметилформамид, диметилацетамид, гексаметилфосфорный триамид и 1,3-диметил-2-имидазолидинон; и сульфоксиды, такие как диметилсульфоксид и сульфолан.
Аналогичным образом нет никаких-либо особенных ограничений в отношении характера основания, используемого для реакции, и обычно любое основание, используемое в реакциях данного типа в равной степени может использоваться здесь. Примеры предпочтительных оснований включают неорганические основания, такие как карбонаты щелочного металла (например, карбонат натрия или карбонат калия); кислые карбонаты щелочных металлов (например, кислый карбонат натрия или бикарбонат калия); гидриды щелочных металлов (например, гидрид лития, гидрид натрия или гидрид калия); гидроокиси щелочных металлов (например, гидроокись натрия, гидроокись калия или гидроокись бария); цианиды щелочных металлов (например, цианид натрия или цианид калия); алкоголяты щелочных металов (например, метилат натрия, этилат натрия или трет-бутилат калия); азиды щелочных металлов (например, азид лития или азид натрия); и меркаптиды щелочных металлов (например, метилмеркаптид натрия или этилмеркаптид натрия). Другие предпочтительные основания включают: органические основания, особенно третичные амины, такие, как триэтиламин, трибутиламин, диизопропилэтиламин, N-метилморфолин, пиридин, 2-(N,N-диметиламино)пиридин, N, N-диметиланилин, N,N-диэтиланилин, 1,5-диазобицикло(4.3.0)нон-5-ен, 1,4-диазабицикло(2.2.2)- октан(ДАВСО) и 1,8-диазабицикло(5.4.0)- ундец-7-ен; и органические основания металлов, такие как бутиллитий и литий диизопропиламид.
Для того, чтобы промотировать реакцию более эффективно, она может осуществляться в присутствии одной или более четвертичных солей аммония, таких как хлористый бензилтриэтиламмоний или хлористый тетрабутиламмоний, или кроун эфиров, таких, как дибензо-18-кроун-6.
Реакция происходит в широком интервале температур, и выбираемая конкретно температура реакции не является критической для изобретения. В основном мы находим удобным проводить реакцию при температуре в интервале от -78оС до точки кипения применяемого растворителя, предпочтительно от -20 до 100оС. Время, требуемое для реакции, может аналогично широко варьировать в зависимости от многих факторов, а именно от температуры реакции и природы реагента или применяемого растворителя. Однако, в большинстве случаев, период времени от 10 мин до 24 ч является обычно достаточным.
После завершения данной реакции желаемый продукт может быть выделен из реакционной смеси общепринятыми методами. Пример такого технологического приема включает: добавление не смешиваемого с водой органического растворителя к реакционной смеси; промывание органической фазы водой; сушку промытой органической фазы; и, наконец, отгонку органического растворителя для получения желаемого продукта. Если необходимо, получающееся в результате соединение может в дальнейшем очищаться с помощью обычных технологических приемов, таких как перекристаллизация, переосаждение или различные приемы хроматографии, а именно хроматография на колонке или препаративная тонкослойная хроматография.
С п о с о б В.
В данном способе соединение формулы I получают путем взаимодействия соединения формулы III с 1 -1,2,4-триазолом, в присутствии основания.
-1,2,4-триазолом, в присутствии основания.
Реакция представляет собой реакцию замещения такого же вида, как и реакция в способе А, и может проводиться с использованием аналогичных реагентов и условий реакции. Предпочтительно реакция дополнительно осуществлется в присутствии неорганической соли, особенно галогенида щелочного металла, такого как йодистый натрий или бромистый литий.
С п о с о б С.
В данном способе, соединение формулы I получают путем взаимодействия соединения формулы V с этиленовоненасыщенным соединением формулы VI, причем предпочтительно используют фотохимическую реакцию, и обычно и предпочтительно в присутствии растворителя.
Реакция предпочтительно проводится с помощью облучения реакционной смеси светом, длина волны которого зависит от природы олефина формулы VI и кетона формулы V, но предпочтительно составляет от 280 до 350 нм. Предпочтительно устранять свет, имеющий длину волны, меньшую чем 280 нм, например, путем использования подходящего фильтра или проведения реакции в растворителе, который препятствует прохождению света с данной длиной волны, например, бензоле или их смеси. Кроме того, нет особых ограничений в отношении природы применяемого растворителя, при условии, что он не оказывает вредного влияния на реакцию или участвующие в реакции реагенты. Примеры подходящих растворителей включают: углеводороды, особенно ароматические углеводороды, такие как бензол, толуол или ксилол; спирты, такие как метанол, этанол или изопропанол; нитрилы, такие как ацетонитрил или бензонитрил; простые эфиры, такие как диэтиловый эфир или тетрагидрофуран, и низшие алифатические и циклоалифатические углеводороды, такие как пентан, гексан или циклогексан. Можно использовать один единственный из этих растворителей или смесь любых двух или более растворителей.
Реакция может проводиться в широком интервале температур, и конкретная температура реакции не является критичной для изобретения. В основном мы находим удобным проводить реакцию при температуре от -20 до +80оС, более предпочтительно от 10 до 40оС. Время, требуемое для реакции, может также широко варьировать в зависимости от многих факторов, а именно температуры реакции и природы реагентов. Однако при условии, что реакция проводится в предпочтительных условиях, описанных выше, период от 1 ч до 4 дней является обычно достаточным.
С п о с о б D.
Реакция расширения кольца в данном способе может быть достигнута путем взаимодействия соединения формулы VII с реагентом типа известного для расширения кольца соединений данного вида, например Corley реагентом, таким как метилид диметилсульфоксония; или галогенидом триметилсульфоксония, таким как хлористый триметилсульфоксоний или йодистый триметилсульфоксоний; или с помощью реакции переноса нуклеофильного метилена, например, в присутствии натрий-диметил-N-(пара-толуолсульфонил)сульфоксимина (J.Am.Chem.Soc. 1973, с. 4287 и далее). Реагент расширения кольца обычно применяется в количестве 2 эквивалента или более на эквивалент соединения формулы VII, более предпочтительно от 2 до 3 эквивалентов. Реакция предпочтительно проводится в присутствии основания, предпочтительно основного соединения щелочного металла, например гидрида щелочного металла, такого как гидрид натрия; гидроокиси щелочного металла, такой как гидроокись натрия или гидроокись калия; или алкоголята щелочного металла, такого как трет-бутилат калия.
Не существует особых органичений в отношении природы применяемого растворителя при условии, что он не оказывает вредного влияния на реакцию или на участвующие в ней реагенты. Примеры подходящих растворителей включают сульфоксиды, такие как диметил сульфоксид, и простые эфиры, такие как тетрагидрофуран.
Реакция может проходить в широком интервале температур, и конкретная температура реакции не является критической для изобретения. В основном мы находим удобным проводить реакцию при температуре от -20 до +130оС, более предпочтительно от 0оС до температуры окружающей среды.
В любом из способов, описанных выше, когда синтезируется рацемат, он может оптически разделяться с помощью любого из известных методов, например, с помощью образования кристаллической соли с оптически активной кислотой, такой как 1-камфорсульфоновая кислота, и затем выделения одного активного соединения из рацемата.
После завершения любой из реакций, описанных выше, желаемое соединение может быть выделено из реакционной смеси общепринятыми методами. Пример такого технического приема включает добавление к реакционной смеси не смешиваемого с водой органического растворителя, промывание органической фазы водой, сушку промытой органической фазы, и, наконец, отгонку органического растворителя для получения желаемого продукта. Если необходимо, получающееся в результате соединение может в дальнейшем очищаться с использованием общепринятых технических приемов очистки, например перекристаллизации, переосаждения, или различных приемов хроматографии, а именно хроматографии на колонке или препаративной тонкослойной хроматографии.
Получение исходных материалов.
Исходный материал, используемый в способе А, соединение формулы II, где R2 атом водорода, который является соединением формулы IIа, может быть получен, как показано в следующей реакционной схеме А:
Реакционная схема А: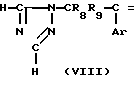

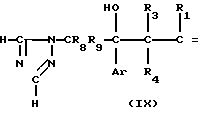

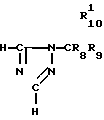
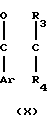
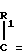


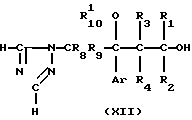
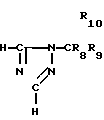
В указанной выше формуле R1, R2, R3, R4, R8, R9, R10, Y и Ar имеют значения, определенные выше; и R10 представляет гидроксизащищающую группу, которой может быть любая из гидроксизащищающих групп, определенных и приведенных в качестве примеров выше в отношении R10.
В стадии А1 данной реакционной схемы кетоновое соединение формулы VIII подвергается взаимодействию с этиленовоненасыщенным соединением Гриньяра формулы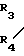 C
C 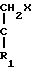 (XIII) в которой R1, R3 и R4 имеют значения, определенные выше, и Х представляет MgZ, где Z атом галогена, например, хлор, фтор, бром или йод), давая соединение формулы IX.
(XIII) в которой R1, R3 и R4 имеют значения, определенные выше, и Х представляет MgZ, где Z атом галогена, например, хлор, фтор, бром или йод), давая соединение формулы IX.
Эта реакция обычно и предпочтительно проводится в присутствии растворителя. Не существует особых ограничений в отношении природы используемого растворителя, при условии, что он не оказывает вредного влияния на реакцию и что он может растворять исходный материал по крайней мере в некоторой степени. Примеры предпочтительных растворителей включают простые эфиры, такие как диэтиловый эфир диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диэтиленгликольдиметиловый эфир.
Реакция может проводиться в широком интервале температур, и конкретная температура реакции не является критической для изобретения. В основном, мы находим удобным проводить реакцию при темпераутре от -20 до +30оС. Время, требуемое для реакции, может также широко варьировать в зависимости от многих факторов, а именно от темпераутры реакции и природы реагентов и используемого растворителя. Однако при условии, что реакция проводится в предпочтительных условиях, указанных выше, период от 30 мин до 3 ч является обычно достаточным.
В некоторых случаях реакция более предпочтительно может проводиться в присутствии кислоты Льюиса, такой как хлористый цинк, хлористое олово, хлористый титан, эфират трехфтористого бора или хлористый диэтилалюминий.
Исходный материал, используемой в данной стадии, может быеть легко получен согласно способу, описанному в Японской патентной предварительной публикации N Sho 63-46075 или Японской Патентной Публикации N Sho 63-5390.
В стадии А2 реакционной схемы гидроксигруппа в соединении формулы IX защищается подходящей защищающей группой. Нет особых ограничений в отношении природы вводимой защищающей группы, при условии, что она защищает гидроксигруппу и предотвращает ее участие в реакциях последующих стадий. Любая защищающая группа, известная для реакций данного типа, может в равной степени быть использована здесь, и подробности о таких группах и способах, применяемых для их введения, могут быть найдены в Green et al, "Proter tive Groups in Orgavic Synthesis, гл. 2, опубл. Wiley-Interscience (1981).
Примеры таких защищающих групп даны выше, и способ, выбираемый для включения такой защищающей группы будет, конечно, зависеть от природы вводимой группы и является хорошо известным. Они могут быть найдены в работе Protective Groups in Organic Synthesis, на которую дана ссылка выше.
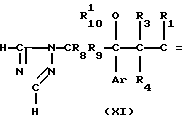
В стадии А3, карбонильное соединение формулы XI получается с помощью окислительного расщепления винильного соединения формулы X, полученного, как описано в стадии А2.
Данная реакция обычно и предпочтительно проводится в присутствии растворителя. Нет особых ограничений в отношении природы применяемого растворителя, при условии, что он не оказывает вредного влияния на реакции и что он может растворять исходный материал по крайней мере в некоторой степени. Примеры предпочтительных растворителей включают галоидированные углеводороды, особенно галоидированные алифатическиие углеводороды, такие как метиленхлорид, или хлороформ, сложные эфиры, такие как этилацетат или пропилацетат; кетоны, такие как ацетон; или простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан или диметоксиэтан.
Нет также особых ограничений в отношении характера реагента, используемого для окисления, при условии, что он может окислительно расщеплять двойную связь, давая карбонильную группу, и здесь в равной степени может использоваться любой окисляющий агент, известный для реакций данного типа. Реакция может предпочтительно осуществляться с помощью пропускания озона через растворитель (такой как галоидированый углводород или сложный эфир) при подходящей темпераутре, например при темпераутре от -78оС до комнатной температуры, а затем путем обработки продукта сульфидом, таким как диметилсульфид. Альтернативно, реакция может осуществляться при подходящей темпераутре, например порядка от 0 до 50оС, с использованием от 2 до 4 эквивалентов щелочно-металлической соли метапериодной кислоты, такой как метапериодат натрия, в подходящем растворителе, смеси воды и эфира или кетона, и в присутствии каталитического количества окиси осмия, такой как тетраокись осмия. Время, требуемое для реакции, может широко изменяться в зависимости, главным образом, от температуры реакции и от характера используемого исходного материала и растворителя, и обычно реакция заканчивается в пределах периода от 3 до 10 ч.
На стадии А4 спирт формулы XII получается с помощью восстановления карбонильного соединения формулы XI или с помощью использования соединения Гриньяра.
Данная реакция восстановления обычно и предпочтительно проводится в присутствии растворителя. Нет никаких особых ограничений относительно природы используемого растворителя, при условии, что он не оказывает вредного влияния на реакцию и что он может растворять по крайней мере до некоторой степени исходный материал. Примеры предпочтительных растворителей включают простые эфиры, такие как тетрагидрофуран, диоксан или диметоксиэтан; и спирты, такие как метанол, этанол или пропанол.
Восстановление может осуществляться в соответствии с обычными методами и предпочтительно с помощью использования обычных восстанавливающих агентов, таких как гидриды металлов, предпочтительно, боргидридов щелочного металла (например, боргидрида натрия).
Реакция может протекать в широком интервале температур, и точная температура реакции не является критической для данного изобретения. Обычно мы считаем удобным осуществлять реакцию при температуре от -30оС до комнатной температуры. Время, требуемое для реакции, может также широко варьировать в зависимости от многих факторов, а именно, в зависимсоти от температуры реакции и характера используемых реагентов и растворителей. Однако при условии, что реакция проводится в предпочтительных условиях, описанных выше, обычно достаточным является период реакции от 5 мин до 3 ч.
Реакция с использованием реактива Гриньяра может проводиться, например, с помощью реактива Гриньяра формулы R12-X, в которой R12представляет собой алкильную группу, имеющую от 1 до 4 атомов углерода (например, метильную, этильную, пропильную, изопропильную, бутильную, изобутильную, втор-бутильную или трет-бутильную группу), и Х представляет собой MgZ, где Z представляет атом галогена. Данная реакция обычно и предпочтиельно проводится в растворителе и может осуществляться в соответствии с общепринятой методикой.
Нет особых ограничений относительно характера применяемого растворителя, при условии, что он не оказывает вредного влияния на реакцию или на вовлеченные в реакцию реагенты и что он может растворять реагенты по крайней мере до некоторой степени. Примеры подходящих растворителей включают простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан или диэтиленгликоль-диметиловый эфир.
Реакция может протекать в широком интервале температур, и точная темпераутра реакции не является критической для изобретения. Обычно мы считаем удобным проводить реакцию при температуре от -20 до +30оС. Время, требуемое для реакции, также может варьировать в зависимости от многих факторов, а именно, от температуры реакции и характера реагентов. Однако при условии, что реакция проводится при описанных выше предпочтительных условиях, обычно достаточным будет период от 30 мин до 3 ч.
На стадии А5 соединение формулы IIa получается с помощью превращения свободной гидроксигруппы спиртового соединения формулы XII в нуклеофильную удаляемую группу Y. Данная реакция обычно и предпочтительно проводится в растворителе, в присутствии или в отсутствии основания. В то же самое время, если необходимо, гидроксизащищающая группа R10 также может удаляться.
Характер реакции, конечно, зависит от характера нуклеофильной удаляемой группы, которую необходимо ввести.
Например, в случае галоидирования, реакция может осуществляться с помощью подвержения взаимодействию соединения формулы XII с обычным галоидирующим агентом типа, хорошо известного для реакции галоидирования данного вида. Примеры предпочтительных галоидирующих агентов включают тионилгалогениды, такие как тионилхлорид, тионилбромид или тионилиодид; сульфонилгалогениды, такие, как сульфонилхлорид или сульфонилбромид; тригалогениды фосфора, такие как треххлористый фосфор, или трехбромистый фосфор; пентагалогениды фосфора, такие как пятихлористый фосфор, пятибромистый фосфор или пятииодистый фосфор; и оксигалогениды фосфора, такие как хлорокись фосфора или бромокись фосфора. Из них мы предпочитаем оксигалогениды фосфора и тионилгалогениды.
В случае сульфонилирования реакция может осуществляться с помощью введения соединения формулы XII в реакцию с соединением формулы R11SO2-O-SO2 R11 (где R11 представляет низшую алкильную группу, предпочтительно имеющую от 1 до 4 атомов углерода (такую как метильная или этильная группа), галоидированную низшую алкильную группу, предпочтительно имеющую от 1 до 4 атомов углерода (такую, как трифторметильная или пентафторэтильная группа), или арильную группу (такую как бензол- или п-толуолгруппа) или с соединением формулы R11SO2-Y (где R11 и Y имеют значения, определенные выше, и Y представляет предпочтительно галоген, например, атом хлора).
Данная реакция обычно и предпочтительно проводится в присутствии растворителя. Нет особых ограничений относительно природы используемого растворителя, при условии, что он не оказывает вредного влияния на реакцию и что он по крайней мере до некоторой степени может растворять исходный материал. Примеры предпочтительных растворителей включают ароматические углеводороды, такие как бензол, толуол или ксилол; галоидированные углеводороды, особенно галоидированные алифатические углеводороды, такие как метиленхлорид или хлороформ; сложные эфиры, такие как этилацетат или пропилацетат; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан или диметоксиэтан и амиды, особенно амиды жирных кислот, такие как диметилформамид, диметилацетамид или гексаметилфосфорный триамид.
Аналогичным образом нет каких-либо особых ограничений в отношении характера используемого основания при условии, что он может использоваться как основание в обычных реакциях данного вида. Примеры предпочтительных оснований включают неорганические основания, особенно основные соединения щелочного металла, такие как гидриды щелочных металлов (например, гидрид лития, гидрид натрия или гидрид калия); органические основания, особенно третичные амины, такие как триэтиламин, диизопропилэтиламин, N-метилморфолин, пиридин, 4-(N, N-диметиламино)пиридин, N, N-диметиланилин, 1,5-диазабицикло(4.3.0)-нон-5-ен, 1,4-диазабицикло(2.2.2)октан, 1,8-диазабицикло(5.4.0)ундец-7-ен (ДВИ) и органические основания в виде соединений металлов, особенно щелочных металлов, такие как бутиллитий или литийдиизопропиламид.
Реакция может проходить в широком интервале температур, и точная темпераутра реакции не является существенной для изобретения. Обычно мы считаем удобным осуществлять реакцию при температуре от -20 до 50оС и более предпочтительно от -15оС до комнатной температуры. Время, требуемое для реакции, также может широко изменяться в зависимости от многих факторов, а именно от температуры реакции и от характера применяемых реагентов и растворителей. Однако, если реакция проводится в предпочтительных условиях, описанных выше, обычно достаточным является период от 5 мин до 10 ч.
При желании гидроксизащищающая группа может удаляться или в этот момент времени, или непосредственно перед стадией А5, для получения соединения формулы IIa, в которой R10 представляет собой атом водорода.
Хотя условия реакции для осуществления снятия защиты гидроксизащитной группой изменяются в зависимости от характера защищающей группы, реакция может осуществляться в соответствии с методами, хорошо известными в технике, например, следующими.
Когда в качестве гидроксизащитной группы используется силильная группа, она может обычно удаляться с помощью обработки соединения соединением, способным образовывать анион фтора, таким как тетрабутиламмонийфторид. Данная реакция обычно и предпочтительно проводится в присутствии растворителя. Нет особых ограничений в отношении характера используемого для реакции растворителя, при условии, что он не оказывает вредного воздействия на реакцию и может растворять исходное вещество по крайней мере до некоторой степени. Предпочтительные растворители включают простые эфиры, такие как тетрагидрофуран или диоксан.
Реакция может протекать в широком интервале температур, но точная температура реакции не является существенной для изобретения. Обычно мы находим удобным осуществлять реакцию примерно при комнатной температуре. Время, требуемое для реакции, также может широко варьировать в зависимости от многих факторов, а именно от температуры реакции и от характера реагентов. Однако при условии, что реакция проводится в предпочтительных условиях, описанных выше, обычно достаточным является период от 10 до 18 ч.
Когда в качестве гидроксизащищающей группы применяется аралкилоксикарбонильная группа или аралкильная группа, она обычно может удаляться с помощью контактирования защищенного соединения с восстанавливающим агентом. Например, реакция может осуществляться с помощью введения защищенного соединения в реакцию каталитического восстановления при комнатной температуре с использованием такого катализатора, как палладий на активированном угле, платина на никеле Ренея, предпочтительно в присутствии растворителя. Нет особых ограничений в отношении характера используемого для реакции растворителя, при условии, что он не оказывает вредного влияния на реакцию, и что он может по крайней мере до некоторой степени растворять исходный материал. Примеры предпочтительных растворителей включают спирты, такие как метанол или этанол; простые эфиры, такие как тетрагидрофуран или диоксан; и жирные кислоты, такие как уксусная кислота; или смесь одного или более этих органических растворителей и воды.
Реакция может происходить в широком интервале температур, но точная температура реакции не является критической для изобретения. Обычно мы считаем удобным осуществлять реакцию при температуре от 0оС до комнатной температуры. Время, требуемое для реакции, также может широко меняться в зависимости от многих факторов, а именно от температуры реакции и характера реагентов. Однако при условии, что реакция проводится в предпочтительных условиях, описанных выше, обычно достаточным является период от 5 мин до 12 ч.
Альтернативно аралкильная и аралкилоксикарбонильная защитная группа может удаляться с помощью обработки защищенного соединения металлическим литием или натрием в жидком аммиаке или спирте, таком как метанол или этанол, при температуре от -78 до -20оС.
Альтернативно аралкильная или аралкилоксикарбонильная группа может удаляться с помощью реакции защищенного соединения с сочетанием хлористого алюминия и иодистого натрия или алкилсилилгалогенида, такого как триметилсилилиодид. Реакция обычно и предпочтительно проводится в присутствии растворителя. Нет никаких особых ограничений, связанных с характером растворителя, используемого для реакции, при условии, что он не оказывает вредного влияния на реакцию и по крайней мере до некоторой степени может растворять исходный материал. Примеры предпочтительных растворителей включают нитрилы, такие как ацетонитрил, и галоидированные углеводороды, особенно галоидированные алифатические углеводороды, такие как метиленхлорид или хлороформ, и смеси любых двух или более из них.
Реакция может иметь место в широком интервале температур, но точная температура реакции не является критической для изобретения. Обычно мы находим удобным осуществлять реакцию при температуре от 0 до 50оС.
В данном процессе, когда субстрат, соединение формулы XII, содержит один или более атомов серы, предпочтительным реагентом является сочетание хлористого алюминия и иодистого натрия.
Когда гидроксизащитной группой является алифатическая ацильная группа, ароматическая ацильная группа или алкоксикарбонильная группа, она может удаляться с помощью обработки защищенного соединения основанием в присутствии растворителя. Нет особых ограничений в отношении характера используемого основания при условии, что, когда удаляется защитная группа, не испытывают пагубного влияния другие части соединения. Примеры предпочтительных оснований включают алкоголяты металлов, такие как карбонат натрия или карбонат калия, гидроокиси щелочных металлов, такие как гидроокись натрия или гидроокись калия, или смесь концентрированного водного аммиака и метанола. В отношении характера используемого растворителя нет каких-либо особых ограничений, и обычный растворитель, обычно используемый для обычных реакций гидролиза, может использоваться здесь в равной степени. Примеры предпочтительных растворителей включают воду, органические растворители, такие как спирты (например, метанол, этанол или пропанол) и простые эфиры (например, тетрагидрофуран или диоксан), или смесь любых двух или более из этих растворителей, особенно смесь воды и одного или более органических растворителей.
Реакция может протекать в широком интервале температур, но точная температура реакции не является критической для изобретения. Обычно, для того, чтобы подавить побочные реакции, мы находим удобным осуществлять реакцию при температуре от 0 до 150оС. Время, требуемое для реакции, также может широко меняться в зависимости от многих факторов, а именно от температуры реакции и от характера реагентов, растворителя и используемого основания. Однако при том условии, что реакция проводится в предпочтительных условиях, описанных выше, обычно достаточным является период от 1 до 10 ч.
Когда гидроксизащитной группой является алкоксиметильная группа или замещенная этильная группа, она обычно может удаляться с помощью обработки защищенного соединения кислотой, предпочтительно в присутствии растворителя. Подходящими используемыми кислотами предпочтительно являются соляная кислота, смесь уксусной и серной кислот, п-толуолсульфокислота; альтернативно может также использоваться сильно кислая катионообменная смола, такая как Дауэкс (торговая марка) 50. Нет особых ограничений относительно характера используемого растворителя при условии, что он не оказывает пагубного влияния на реакцию и может растворять исходный материал, по крайней мере, до некоторой степени. Предпочтительными растворителями являются спирты, такие как метанол или этанол, простые эфиры, такие как тетрагидрофуран или диоксан, смеси любых двух или более из них, и смеси любого одного или более из них и воды.
Реакция может происходить в широком интервале температур, но точная температура реакции не является критической для изобретения. Обычно мы считаем удобным осуществлять реакцию при температуре от 0 до 50оС. Время, требуемое для реакции, может также широко варьировать в зависимости от многих факторов, а именно, от температуры реакции и от характера применяемых реагентов и кислоты. Однако при том условии, что реакция проводится в предпочтительных условиях, описанных выше, обычно достаточным является период от 10 мин до 18 ч.
Когда гидроксизащищающей группой является алкенилоксикарбонильная группа, она может обычно удаляться с помощью обработки защищенного соединения основанием в условиях реакции, аналогичных условиям, применяемым, когда гидроксизащитной группой является алифатическая ацильная группа, ароматическая ацильная группа или алкоксикарбонильная группа.
Далее, когда гидроксизащитной группой является аллилоксикарбонильная группа, реакция снятия защиты может осуществляться просто с использованием сочетания палладия и трифенилфосфина или тетракарбонила никеля, который обладает тем преимуществом, что уменьшает до предела побочные реакции.
После завершения каждой из реакций, описанных выше, желаемые соединения могут выделяться из реакционной смеси в соответствии с обычными способами. Один из примеров таких приемов включает добавление не смешиваемого с водой органического растворителя к реакционной смеси, промывку органического основания водой, сушку промытой органической фазы, и отгонку органического растворителя с получением желаемого продукта. Если необходимо, получающиеся в результате соединения могут далее очищаться с помощью хорошо известных приемов, например с помощью перекристаллизации, переосаждения, или различных приемов хроматографии, а именно препаративной тонкослойной хроматографии или хроматографии на колонке.
Альтернативный способ получения соединения формулы IIa иллюстрируется с помощью следующей схемы реакции В:
Схема реакции В
Triz-CR8R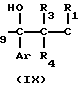 = CH2
= CH2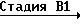
Triz-CR8R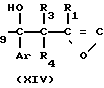 H2
H2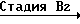 Triz-CR8R
Triz-CR8R OH
OH
Стадия В3 _→ (IIa)
В приведенных выше формулах R1, R3, R4, R8, R9, R12 и Ar имеют значения, определенные выше, и Triz обозначает 1 -1,2,4-триазол-1-ильную группу, которая имеет формулу
-1,2,4-триазол-1-ильную группу, которая имеет формулу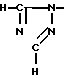
Альтернативно, соединение формулы XII, в котором R12 представляет атом водорода, иными словами соединение формулы XIIc, может получаться из соединения формулы XV, смотри схему реакции А, как показано на следующей схеме реакции С:
Реакционная схема С
Tr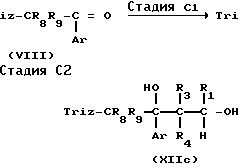 z-CR8R
z-CR8R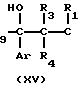 = O
= O
В указанной выше реакционной схеме, R1, R3, R4, R8, R9, Ar и Triz имеют значения, определенные выше.
Соединение формулы VIII, которое является исходным материалом в реакционной схеме А, и таким образом в реакционных схемах, указанных выше, может получаться, как показано в реакционной схеме Е:
Реакционная схема Е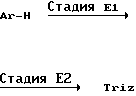
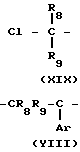 C
C
В указанной выше реакционной схеме R8, R9 и Ar имеют значения, определенные выше.
Соединение формулы III, в которой R8 и R9 оба представляют атомы водорода, которое представляет соединение формулы IIIа, используется в качестве исходного материала в способе В, может получаться, как показано в следующей реакционной схеме F:
Реакционная схема F
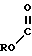
HO 
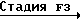 Y
Y 
В указанной выше реакционной схеме R1, R2, R3, R4, R и Ar имеют значения, определенные выше.
В стадии F1 указанной выше реакционной схемы производное глоксиловой кислоты формулы XX подвергается фотохимической реакции с соединением формулы VI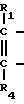
 (VI) в которой R1, R2, R3 и R4 имеют значения, определенные выше.
(VI) в которой R1, R2, R3 и R4 имеют значения, определенные выше.
Соединение формулы III, в которой R8 и R9 могут представлять или атомы водорода, или атомы углерода, имеющие от 1 до 4 атомов углерода, и Y представляет атом хлора, может быть получено, как показано на реакционной схеме G:
Схема реакции G
Cl
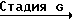
Данная реакция может проводиться с помощью обычных приемов, например, как описано авторами T.Sato и K.Tamura в журнале Tetrahedron Letters, т. 25, с. 1821-1824, 1984. Соединение формулы XIX, которое является исходным материалом, может быть получено, как показано в стадии Е1 схемы реакции Е.
Соединение формулы XII (смотри схему реакции А), в которой R10представляет атом водорода, а R2 метильную группу, т.е. соединение формулы XIIe, может быть также получено, как показано на схеме реакции Н.
Схема реакции Н
Triz = CH2
= CH2 Triz
Triz CR8R
CR8R OH
OH
В приведенных выше формулах R1, R3, R4, R8, R9, Ar и Triz имеют значения, определенные выше.
После завершения каждой из описанных выше реакций желаемые соединения могут выделяться из реакционной смеси с помощью обычных средств. Пример такого приема включает добавление к реакционной смеси не смешиваемого с водой органического растворителя, промывку органической фазы водой, сушку промытой органической фазы, и отгонку органического растворителя с получением нужного продукта. Если необходимо, получающиеся соединения могут далее очищаться с помощью обычных приемов очистки, например с помощью перекристаллизации, переосаждения или различных приемов хроматографии, а именно хроматографии на колонке или препаративной тонкослойной хроматографии.
Соединения предлагаемого изобретения обладают разнообразными видами ценной биологической активности, как показано с помощью следующих экспериментов, что делает их полезными в качестве как сельскохозяйственных, так и фармацевтических противогрибковых агентов.
Получение соединений предлагаемого изобретения иллюстрируется следующими примерами, в то время как получение некоторых исходных веществ, используемых в этих примерах, иллюстрируется следующими ниже препаративными примерами.
П р и м е р 1. (2R*,3S*,4R*)-2-(2,4-Ди-фторфенил)-3,4-диметил-2-[(1Н-1,2,4-три- азол-1-ил)метил]оксетан, а также его оксалат и нитрат.
309 мг водного раствора метилмеркаптана натрия, имеющего концентрацию около 15% добавляли к раствору 389 мг (2R*,3S*,4S*)-2-(2,4-дифторфенил)[-4-(метан- сульфонилокси)-3-метил-1Н- (1Н-1,2,4-триазол-1-ил)-2-пентанола] полученного в соответствии с препаративным примером 2(с), из 4,5-эпоксисоединения, имеющего низкую полярность стереоизомеров, полученных в соответствии с препаративным примером 2(а), представляющим собой один из изомеров оксетана в положении С4/ в 10 мл метанола. Полученную смесь перемешивали в течение ночи при комнатной температуре и затем переливали в смесь воды со льдом, после чего проводили экстракцию этилацетатом. Экстракт промывали насыщенным водным раствором хлористого натрия и сушили над безводным сульфатом натрия, после чего растворитель удаляли дистилляцией при пониженном давлении. Полученный в результате масляный остаток (256 мг) подвергали колонной хроматографии через силикагель, используя в качестве элюента смесь гексана и этилацетата в объемном соотношении 1:1, в результате чего получали целевое соединение в виде масла.
Масс-спектр (m/z): 279 (М+), 246, 231, 224, 213, 197, 182, 167, 149, 141, 127, 113, 101, 83.
ЯМР-спектр (СDСl3), δ ч./млн. 0,83 (3Н, дублет дублетов, J 7,2 Гц); 1,18 (3Н, дублет, J 7 Гц); 3,18 (1Н, квинтет, J 7 Гц); 4,50 (1Н, дублет, J 14,5 Гц); 4,58 (1Н, квинтет, J 7 Гц); 4,94 (1Н, дублет, J 14,5 Гц); 6,8-6,9 (2Н, мультиплет); 7,4-7,5 (1Н, мультиплет); 7,87 (1Н, синглет); 8,24 (1Н, синглет).
ИК-спектр (жидкая пленка), λмакс. см-1: 3100, 1610, 1600.
33 мг оксалиновой кислоты добавляли к раствору 100 мг оксетана, синтезированного в соответствии с описанным выше, растворенного в этилацетате, после чего с целью осаждения кристаллов добавляли гексан. Полученные кристаллы, имеющие температуру плавления 145-150оС, собирали фильтрацией с получением 92 мг оксалата целевого соединения.
ЯМР-спектр (гексадейтерированный диметилсульфоксид), δ ч./млн. 0,74 (3Н, дублет дублетов, J 7 Гц и 2 Гц); 1,10 (3Н, дублет, J 7 Гц); 2,7-4,5 (2Н, широкий синглет); 3,18 (1Н, квинтет, J 7 Гц); 4,57 (1Н, дублет, J 14,5 Гц); 4,66 (1Н, квинтет, J 7 Гц); 4,93 (1Н, дублет, J 14,5 Гц); 7,05 (16, дублет триплетов, J 8,5 Гц, 2,5 Гц); 7,2-7,4 (2Н, мультиплет); 8,80 (1Н, синглет); 8,40 (1Н, синглет).
ИК-спектр (Нуйол-торговое наименование), λмакс. см-1: 3500, 3130, 28800-2300, 1740, 1610, 1600.
По 1 мл азотной кислоты (плотность 1,38) и диэтилового эфира добавляли к раствору 100 мг оксетана, синтезированного в соответствии с описанным выше, в хлористом метилене. Выпавшие кристаллы собирали фильтрацией с получением 115 мг нитрата целевого соединения, т.пл. 160-177оС.
ЯМР-спектр (гексадейтерированный диметилсульфоксил), δ ч./млн. 0,75 (3Н, дублет дублетов, J 7 и 2 Гц); 1,1 (3Н, дублет, J 7 Гц); 3,19 (1Н, квинтет, J 7 Гц); 4,59 (1Н, дублет, J 14,5 Гц); 4,70 (1Н, квинтет, J 7 Гц); 4,93 (1Н, дублет, J 14,5 Гц); 7,05 (1Н, дублет триплетов, J 8,5 и 3 Гц); 7,2-7,3 (1Н, мультиплет); 7,89 (1Н, синглет); 8,53 (1Н, синглет).
П р и м е р 2. (2R*,3S*,4S*)-2-(2,4-Дифторфенил)-3,4-диметил-2-[(1Н-1,2,4- триазол-1-ил)метил]оксетан.
Это вещество представляет собой стереоизомер соединения примера 1 в положении С4.
Следуя методике, аналогичной описанной в примере 1, но используя 221 мг (2R*, 3S*,4R*)-2-(2,4-дифторфенил)-4-(метан- сульфонилокси)-3-метил- 1-(1Н-1,2,4-триазол-1-ил)-2-пентанол (стереоизомер исходного соединения примера 1 в положении С4, полученный обработкой стереоизомера из препаративного примера 2(а) в соответствии с описанным в препаративных примерах 2(в) и 2(с)), получали 43 мг целевого соединения в виде масла.
Масс-спектр (М/Z): 280 (М+ + 1), 279, 270, 256, 234, 224, 197, 179, 165, 151, 142, 127, 113, 101, 82.
ЯМР-спектр (СДСl3), δ ч./млн: 0,86 (3Н, дублет дублетов, J 7 Гц и 2,5 Гц); 1,11 (3Н, дублет, J 7 Гц); 2,73 (1Н, квинтет, J 7 Гц); 4,23 (1Н, квинтет, J 7 Гц); 4,42 (1Н, дублет, J 14,5 Гц); 4,81 (1Н, дублет, J 14,5 Гц); 6,8-7,0 (2Н, мультиплет); 7,5-7,7 (1Н, мультиплет); 7,93 (1Н, синглет), 8,28 (1Н, синглет).
ИК-спектр (жидкая пленка), λмакс. см-1: 1610, 1600. Перекристаллизацией такого масла из этилацетата получали чистый образец с температурой плавления 105-107оС.
П р и м е р 3. (2R*,3R*,4R*)-2-(2,4-Ди-фторфенил)-3,4-диметил-2-[(1Н-1,2,4-три- азол- 1-ил)метил]оксетан.
Это вещество представляет собой стереоизомер соединения примера 1 в положении С3.
Следуя методике, аналогичной описанной в примере 1, но используя 255 мг (2R*, 3R*,4S*)-2-(2,4-дифторфенил)-4-(ме- тансульфонилокси)-3-метил- 1-(1Н-1,2,4-триазол-1-ил)-2-пентанол (который получали мезилированием в соответствии с описанным в препаративном примере 2(с) главного стереоизомерного компонента продукта, полученного в соответствии с препаративным примером 4 и представляющего собой один изомер в положении С4), получали 135 мг целевого соединения, имеющего температуру плавления 100-110оС.
Масс-спектр (m/z): 279 (М+), 235, 224, 215, 197, 182, 166, 153, 141, 133, 127, 113, 101, 83.
ЯМР-спектр (СДСl3), δ ч./млн: 1,37 (3Н, дублет, J 6 Гц); 1,39 (3Н, дублет, J 7 Гц); 2,80 (1Н, квинтет, J 7 Гц); 4,63 (1Н, квинтет, J 7 Гц); 4,67 (1Н, дублет, J 14 Гц); 4,94 (1Н, дублет, J 14 Гц); 6,6-6,8 (2Н, мультиплет); 7,0-7,2 (1Н, мультиплет); 7,77 (1Н, синглет); 8,02 (1Н, синглет).
ИК-спектр (Нуйол), λмакс см-1: 1610, 1490.
П р и м е р 4. (2R*,3S*,4R*)-2-(2-хлорфенил)-3,4-диметил-2-[(1Н-1,2,4-триазол-1- ил)метил]оксетан.
11 мг 60% (мас.) дисперсии гидрида натрия в минеральном масле добавляли к раствору 51 мг (2R*,3S*,4S*)-2-(2-хлорфенил)-4-(метансульфонилокси)-3-метил-1- (1Н-1,2,4-триазол-1-ил)-2-пентанола (который синтезировали по методике, аналогичной описанной в препаративном примере 2(b) и 2(с), являющегося производным 4,5-эпоксисоединения с низкой полярностью стереоизомеров в положении С4, полученных по методике, аналогичной описанной в препаративном примере 2(а) в 2 мл диметилформамида; затем смесь перемешивали в течение 2 ч при 60оС. По истечении этого времени смесь переливали в смесь воды со льдом и экстрагировали этилацетатом. Экстракт промывали водой и сушили над безводным сульфатом натрия и растворитель удаляли дистилляцией при пониженном давлении. Полученный в результате масляный остаток очищали методом препаративной тонкослойной хроматографии на силикагеле с использованием в качестве проявляющего растворителя смеси гексана с этилацетатом в объемном соотношении 1:2, с получением 10 мг целевого соединения в виде масла.
Масс-спектр (m/z): 280 (М+ + 2), 278 (М+), 259, 242, 222, 197, 195, 141, 139, 129, 111, 101, 89, 75.
ЯМР-спектр (СДСl3), δ ч./млн. 0,95 (3Н, дублет, J 7 Гц); 1,18 (3Н, дублет, J 7 Гц); 3,20 (1Н, квинтет, J 7 Гц); 4,60 (1Н, квинтет, J 7 Гц); 4,80 (1Н, дублет, J 14,5 Гц); 5,19 (1Н, дублет, J 14,5 Гц); 7,2-7,6 (4Н, мультиплет); 7,84 (1Н, синглет); 8,12 (1Н, синглет).
П р и м е р 5. (2R*,3S*,4S*)-2-(2-хлорфенил)-3,4-диметил)-1-[(1Н-1,2,4-триазол-1- ил)метил]оксетан.
Это соединение представляет собой стереоизомер соединения примера 4 в положении Е4.
Следуя методике, аналогичной описанной в примере 4, но используя 50,7 мг (2R*, 3S*, 4R*)-2-(2-хлорфенил)-4-(метансуль- фонилокси)-3-метил-1- (1Н-1,2,4-триазол-1-ил)-2-пентанола (который синтезировали по методике, аналогичной описанной в препаративных примерах 2(b) и 2(с) и который представляет собой производное 4,5-эпоксисоединения высокой полярности стереоизомеров в положении С4, полученного по методике, аналогичной описанной в препаративном примере 2(а)), получали 10 мг целевого соединения в виде масла.
Масс-спектр (m/z): 280 (М+ + 2), 278 (М+), 266, 253, 242, 222, 195, 141, 139, 129, 125, 111, 101, 89.
ЯМР-спектр (СДСl3), δ ч./млн. 0,99 (3Н, дублет, J 7 Гц); 1,12 (3Н, дублет, J 7 Гц); 2,80 (1Н, квинтет, J 7 Гц); 4,19 (1Н, квинтет, J 7 Гц); 4,66 (1Н, дублет, J 14,5 Гц); 5,17 (1Н, дублет, J 14,5 Гц); 7,2-7,7 (4Н, мультиплет); 7,90 (1Н, синглет); 8,20 (1Н, синглет).
П р и м е р 6. (2R*,3S*,4R*)-2-(2-хлор-4-фторфенил)-3,4-диметил-2-[(1Н-1,2,4- триазол-1-ил)метил]оксетан.
Следуя методике, аналогичной описанной в примере 4, но используя 133 мг (2R*, 3S*, 4S*)-2-(2-хлор-4-фторфенил)-4-(ме- тансульфонилокси)-3- метил-1-(1Н-1,2,4-триазол-1-ил)-2-пентанола (который синтезировали способом, аналогичным методике, описанной в препаративном примере 2 и который является производным 4,5-эпоксисоединения с низкой полярностью стереоизомеров в положении С4, полученного по методике, аналогичной описанной в препаративном примере 2(а)), получали 86 мг целевого соединения в виде масла.
Масс-спектр (m/z): 297 (М+ + 2), 295 (М+), 280, 278, 260, 251, 240, 216, 215, 214, 213, 182, 160, 159, 158, 157, 147, 129, 123, 107, 94, 82.
ЯМР-спектр (СДСl3), δ ч./млн: 0,94 (3Н, дублет, J 7 Гц); 1,18 (36, дублет, J 7 Гц); 3,18 (1Н, квинтет, J 7 Гц); 4,62 (1Н, квинтет, J 7 Гц); 4,80 (1Н, дублет, J 14,5 Гц); 5,14 (1Н, дублет, J 14,5 Гц); 6,9-7,0 (1Н, мультиплет); 7,12 (1Н, дублет дублетов, J 8,5 и 2,5 Гц); 7,50 (1Н, дублет дублетов, J 8,5 Гц и 6,0 Гц); 7,83 (1Н, синглет); 8,17 (1Н, синглет).
П р и м е р 7. (2R*,3S*,4S*)-2-(2-Хлор-4-фторфенил)-3,4-диметил-2-[(1Н-1,2,4- триазол-1-ил)метил]оксетан.
Это соединение представляет собой стереоизомер соединения примера 6 в положении С4.
Следуя методике, аналогичной описанной в примере 4, но используя 32 мг (2R*, 3S*, 4R*)-2-(2-хлор-4-фторфенил)-4-(ме- тансульфонилокси)-3- метил-1-(1Н-1,2,4-триазол-1-ил)-2-пентанола (который синтезировали способом, аналогичным методике, описанной в препаративном примере 2(b) и 2(с) и который является производным 4,5-эпоксисоединения повышенной полярности стереоизомеров в положении С4, полученного по методике, аналогичной описанной в препаративном примере 2(а), получали 14 мг целевого соединения в виде масла.
Масс-спектр (m/z): 296 (М+): 279, 260, 213, 211, 204, 182, 171, 159, 157, 143, 129, 123, 107, 94, 82.
ЯМР-спектр (СДСl3), δ ч./млн: 0,98 (3Н, дублет, J 7 Гц); 1,15 (3Н, дублет, J 7 Гц); 2,79 (1Н, квинтет, J 7 Гц); 4,18 (1Н, квинтет, J 7 Гц); 4,67 (1Н, дублет, J 14,5 Гц); 5,10 (1Н, дублет, J 14,5 Гц); 6,9-7,1 (1Н, мультиплет); 7,17 (1Н, дублет дублетов, J 8,5 Гц и 2,5 Гц); 7,68 (1Н, дублет дублетов, J 8,5 и 6,5 Гц); 7,90 (1Н, синглет); 8,22 (1Н, синглет).
П р и м е р 8. (2R*,3R*)-2-(2-Хлор-4-фторфенил)-3,4-диметил-2-[(1Н-1,2,4-триазол- 1-ил)метил]оксетан.
Это вещество представляет собой стереоизомер соединения примера 6 в положении С3.
Следуя методике, аналогичной описанной в примере 4, но используя 139 мг (2R*, 3R*)-2-(2-хлор-4-фторфенил)-4-(метан-сульфонилокси)-3-метил- 1-(1Н-1,2,4-триазол-1-ил)-2-пентанола (который получали мезилированием основного стереоизомерного компонента, изомерного в положении С4, синтезированного способом, аналогичным описанному в препаративном примере 4), получали 91 мг целевого соединения в виде кристаллов с температурой плавления 145-147оС.
Масс-спектр (m/z): 297 (М+ + 2), 295, 260, 240, 213, 211, 198, 178, 159, 158, 149, 129, 123, 109, 98, 83.
ЯМР-спектр (СДСl3), δ ч./млн. 1,32 (дублет, J 7 Гц); 1,5 (3Н, дублет, J 7 Гц); 2,81 (1Н, квинтет, J 7 Гц); 4,59 (1Н, квинтет, J 7 Гц); 4,91 (2Н, синглет); 6,7-6,8 (1Н, мультиплет); 7,09 (1Н, дублет дублетов, J 8,5 и 2,5 Гц); 7,20 (1Н, дублет дублетов, J 8,5 и 6, Гц); 6,69 (1Н, синглет); 7,94 (1Н, синглет).
П р и м е р 9. (2R*,3S*,4S*)-2-(4-Хлор-2-фторфенил)-3,4-диметил-2-[(1Н-1,2,4- триазол-1-ил)метил]оксетан.
Следуя методике, аналогичной описанной в примере 4, но используя 260 мг (2R*, 3S*, 4R*)-2-(2-фтор-4-хлорфенил)-4-(ме- тансульфонилокси)-3- метил-1-(1Н-1,2,4-триазол-1-ил)-2-пентанола (который синтезировали по методу, аналогичному описанному в препаративных примерах 2(b) и 2(с) и который является производным 4,5-эпоксисоединения с повышенной полярностью стереоизомеров в положении С4, полученного по методу, аналогичному описанному в препаративном примере 2(а), получали 150 мг целевого соединения в виде масла.
Масс-спектр (m/z): 297 (М+ + 2), 295 (М+), 280, 250, 240, 215, 214, 206, 179, 159, 158, 143, 129, 123, 109, 99, 82, 94.
ЯМР-спектр (СДСl3), δ ч./млн. 0,84 (3Н, дублет дублетов, J 7 и 2,5 Гц); 1,10 (3Н, дублет, J 7 Гц); 2,74 (1Н, квинтет, J 7 Гц); 4,21 (1Н, квинтет, J 7 Гц); 4,40 (1Н, дублет, J 14,5 Гц); 4,80 (1Н, дублет, J 14,5 Гц); 7,14 (1Н, дублет дублетов, J 10,5 и 2,0 Гц); 7,20 (1Н, дублет дублетов, J 8 и 2,0 Гц); 7,56 (1Н, триплет, J 8 Гц); 7,91 (1Н, синглет); 8,22 (1Н, синглет).
ИК-спектр (жидкая пленка), λмакс. см-1: 3120, 1610, 1570.
П р и м е р 10. (2R*,3S*,4R*)-2-(2-фтор-4-хлорфенил)-3,4-диметил-2-[(1Н-1,2,4- триазол-1-ил)метил]оксетан.
Это вещество представляет собой соединение примера 9 в положении С4.
Следуя методике, аналогичной описанной в примере 4, но используя 390 мг (2R*, 3S*, 4S*)-2-(2-фтор-4-хлорфенил)-4-(метансульфонилокси)-3- метил-1-(1Н-1,2,4-триазол-1-ил)-2-пентанола (который синтезировали по способу, аналогичному описанному в препаративных примерах 2(b) и 2(с) и который является производным 4,5-эпоксисоединения с пониженной полярностью стереоизомеров в положении С4, полученного по способу, аналогичному описанному в препаративном примере 2(а)), получали 220 мг целевого соединения в виде масла.
Масс-спектр (m/z): 297 (М+ + 2), 295 (М+), 247, 240, 215, 214, 182, 159, 158, 157, 147, 129, 122, 107, 94, 82.
ЯМР-спектр (СДСl3), δ ч./млн. 0,83 (3Н, дублет дублетов, J 7 и 2, Гц); 1,17 (3Н, дублет, J 7 Гц); 3,19 (1Н, квинтет, J 7 Гц); 4,49 (1Н, дублет, J 14,5 Гц); 4,57 (1Н, квинтет, J 7 Гц); 4,90 (1Н, дублет, J 14,5 Гц); 7,0-7,1 (1Н, мультиплет); 7,15 (1Н, дублет дублетов, J 8,5 и 2,0 Гц); 7,46 (1Н, триплет, J 8,5 Гц); 7,86 (1Н, синглет); 8,21 (1Н, синглет).
ИК-спектр (жидкая пленка), λмакс. см-1: 3110, 1610, 1570.
П р и м е р 11. (2R*,3S*,4R*)-2-(2,6-ди-фторфенил)-3,4-диметил-2-[(1Н-1,2,4- триазол-1-ил)метил]оксетан.
Следуя методике, аналогичной описанной в примере 4, но используя 120 мг (2R*, 3S*, 4S*)-2-(2,6-дифторфенил)-4-(метан-сульфонилокси)-3-метил- 1-(1Н-1,2,4-триазол-1-ил)-2-пентанола (который синтезировали по способу, аналогичному описанному в препаративных примерах 2(b) и 2(с) и который является производным 4,5-эпоксисоединения с пониженной полярностью стерео- изомеров в положении С4, полученного по способу, аналогичному описанному в препаративном примере 2(а)), получали 63 мг целевого соединения в виде масла.
Масс-спектр (m/z): 297 (М+), 224, 197, 179, 166, 151, 141, 127, 113, 101, 82.
ЯМР-спектр (СДСl3), δ ч./млн. 1,00 (3Н, дублет триплетов, J 7 и 1 Гц); 1,24 (3Н, дублет, J 7 Гц), 3,20 (1Н, квинтет, J 7 Гц); 4,57 (1Н, квинтет, J 7 Гц); 4,60 (дублет, J 14,5 Гц); 4,92 (1Н, дублет, J 14,5 Гц); 6,86 (2Н, триплет, J 9 Гц); 7,2-7,3 (1Н, мультиплет); 7,87 (1Н, синглет); 8,31 (1Н, синглет).
П р и м е р 12. (2R*,3S*,4S*)-2-(2,6-ди-фторфенил)-3,4-диметил-2-[(1Н-1,2,4- триазол-1-ил)метил]оксетан.
Это соединение представляет собой стереоизомер соединения примера 11 в положении С4.
Следуя методике, аналогичной описанной в примере 4, но используя 121 мг (2R*, 3S*,4R*)-2-(2,6-дифторфенил)-4-(метан- сульфонилокси)-3-метил- 1-(1Н-1,2,4-триазол-1-ил)-2-пентанола (который синтезировали по способу, аналогичному описанному в препаративных примерах 2(b) и 2(с) и который является производным 4,5-эпоксисоединения с повышенной полярностью стереоизомеров в положении С4, полученного по способу, аналогичному описанному в препаративном примере 2(а)), получали 54 мг целевого соединения в виде масла.
Масс-спектр (m/z): 279 (М+), 261, 235, 224, 206, 197, 179, 166, 153, 141, 133, 123, 113, 95, 82.
ЯМР-спектр (СДСl3), δ ч./млн: 0,99 (3Н, дублет триплетов, J 7 и 2 Гц); 1,10 (3Н, дублет, J 7 Гц); 2,82 (1Н, квинтет, J 7 Гц); 4,38 (1Н, квинтет, J 7 Гц); 4,55 (1Н, дублет, J 14,5 Гц); 4,83 (1Н, дублет, J 14,5 Гц); 6,8-7,0 (2Н, мультиплет); 7,2-7,4 (1Н, мультиплет); 7,92 (1Н, синглет); 8,33 (1Н, синглет).
П р и м е р 13. (2R*,3S*)-2-(2,4-дифторфенил)-3-метил-2-[(1Н-1,2,4-триазол-1-ил)ме- тил]оксетан.
Следуя методике, аналогичной описанной в примере 1, но используя 100 мг (2R*, 3S*)-2-(2,4-дифторфенил)-4-(метансульфонилокси)-3-метил-1- (1Н-1,2,4-триазол-1-ил)-2-бутанола (который синтезировали методом, аналогичным описанному в препаративном примере 7), получали 54 мг целевого соединения в виде масла.
Масс-спектр (m/z): 265 (М+), 247, 232, 224, 184, 165, 153, 141, 133, 127, 113, 101, 94, 82.
ЯМР-спектр (СДСl3), δ ч./млн. 0,93 (3Н, дублет дублетов, J 7 Гц и 2 Гц); 3,15 (1Н, секстет, J 7 Гц); 3,99 (1Н, триплет, J 7 Гц); 4,37 (1Н, дублет дублетов, J 7 и 6 Гц); 4,48 (1Н, дублет, J 7 Гц); 4,90 (1Н, дублет, J 14,5 Гц); 6,8-7,0 (2Н, мультиплет); 7,2-7,4 (1Н, мультиплет); 7,90 (1Н, синглет); 8,30 (1Н, синглет).
П р и м е р 14. 2-(2,4-Дифторфенил)-4-метил-2-[(1Н-1,2,4-триазол-1-ил)-метил]оксе- тан.
Следуя методике, аналогичной описанной в примере 4, но используя 590 мг 2-(2,4-дифторфенил-4-(метансульфонилокси)-4-ме- тил-(1Н-1,2,4- триазол-1-ил)-2-бутанола (который синтезировали из хлористого аллилмагния аналогичным методом по способу, описанному в отдельной Японской патентной публикации N Sho 59-517 и в препаративных примерах 2, 3 и 4), получали один из стереоизомеров в положении С4 в качестве основного продукта в виде масла.
Масс-спектр (m/z): 265 (М+), 250, 224, 202, 184, 183, 142, 141, 133, 127, 119, 113, 99, 83.
ЯМР-спектр (СДСl3), δ ч./млн. 1,28 (3Н, дублет, J 7 Гц); 2,35 (1Н, дублет квартетов, J 8 и 6 Гц); 3,10 (1Н, дублет дублетов, J 8 и 7 Гц); 4,34 (1Н, дублет, J 14,5 Гц); 4,3-4,5 (1Н, мультиплет); 4,72 (1Н, дублет, J 14,5 Гц); 6,8-6,9 (2Н, мультиплет); 7,4-7,5 (1Н, мультиплет); 7,90 (1Н, синглет); 8,20 (1Н, синглет).
ИК-спектр (жидкая пленка), λмакс. см-1: 1610, 1600, 1500.
П р и м е р 15. (2R*,3S*)-2-(2,4-Дифторфенил)-3-метил-2-[(1Н-1,2,4-триазол-1-ил)- метил]оксетан и его нитрат.
Следуя методике, аналогичной описанной в примере 1, но используя 72 мг (2R*, 3S*)-2-(2,4-дихлорфенил)-4-(метансуль- фонилокси)-3-метил-1- (1Н-1,2,4-триазол-1-ил)-2-бутанола (который синтезировали способом, аналогичным описанному в препаративном примере 7), получали 25 мг целевого соединения в виде масла.
ЯМР-спектр (СДСl3), δ ч. /млн. 1,05 (3Н, дублет, J 7 Гц); 3,15 (1Н, мультиплет); 3,90 (1Н, дублет дублетов, J 6 и 5,5 Гц); 4,38 (1Н, дублет дублетов, J 7 и 6 Гц); 4,67 (1Н, дублет, J 14,5 Гц); 5,16 (1Н, дублет, J 14,5 Гц); 7,1-7,6 (3Н, мультиплет); 7,88 (1Н, синглет); 8,20 (1Н, синглет).
ИК-спектр (СНСl3), λмакс. см-1: 1590, 1275, 1137, 975.
Около 0,1 мл эфирного раствора, содержащего 5% азотной кислоты, добавляли к раствору 17 мг названного оксетана в 0,4 мл диэтилового эфира при охлаждении системы льдом. Затем смесь обрабатывали традиционными способами с получением 17 мг нитрата целевого соединения в виде кристаллов с температурой плавления 162-165оС.
П р и м е р 16. (2R*,3S*)-2-(2,4-дихлорфенил)-3,4-диметил-2-[(1Н-1,2,4-триазол-1- ил)метил]оксетан и его нитраты.
0,5 мл водного раствора, содержащего 15 мас./об. метилмеркаптана натрия, добавляли к раствору 54 мг смеси в соотношении 1:1 С4стереоизомеров мезилата (который получали из смеси в соотношении 1:1 С4стереоизомеров (2R*,3S*)-2-(2,4-дихлорфенил)-3-метил-1-(1Н-1,2,4-триазол-1- ил)- 2,4-пентандиола (который синтезировали способом, аналогичным описанному в препаративном примере 4) в 1 мл диметилформамида. Затем полученную смесь перемешивали в течение 2 ч при 50-60оС и после этого обрабатывали согласно методике, аналогичной описанной в примере 1. Полученный сырой продукт очищали методом препаративной тонкослойной хроматографии через силикагель с использованием смеси (в соотношении 3:1) этилацетата и гексана в качестве проявляющего агента, с получением 8 мг стереоизомера А (пониженная полярность) и 10 мг стереоизомера В (повышенная полярность), причем оба вещества были в виде масла.
Стереоизомер А (2R*,3S*,4S*):
ЯМР-спектр (СДСl3), δ ч./млн. 0,98 (3Н, дублет, J 7 Гц); 1,03 (3Н, дублет, J 7 Гц); 2,80 (1Н, квинтет, J 7 Гц); 4,19 (1Н, квинтет, J 7 Гц); 4,62 (1Н, дублет, J 15 Гц); 5,12 (1Н, дублет, J 15 Гц); 7,31 (1Н, дублет дублетов, J 9 и 2 Гц); 7,46 (1Н, дублет, J 2 Гц); 7,71 (1Н, дублет, J 9 Гц); 7,90 (1Н, синглет); 8,20 (1Н, синглет).
Стереоизомер В (2R*,3S*,4R*):
ЯМР-спектр (СДСl3), δ ч./млн. 0,92 (3Н, дублет, J 7 Гц); 1,17 (3Н, дублет, J 7 Гц); 3,17 (1Н, квинтет, J 7 Гц); 4,59 (1Н, квинтет, J 7 Гц); 4,72 (1Н, дублет, J 15 Гц); 5,15 (1Н, дублет, J 15 Гц); 7,20 (1Н, дублет дублетов, J 9 и 1,5 Гц); 7,38 (1Н, дублет, J 1,5 Гц); 7,50 (1Н, дублет, J 9 Гц); 7,81 (1Н, синглет); 8,13 (1Н, синглет).
Каждый из таких стереоизомеров превращали в его нитрат традиционными способами, путем добавления эфирного раствора, содержащего 5% азотной кислоты к раствору соответствующего оксетанового соединения при охлаждении системы льдом, с получением желаемых нитратов в виде кристаллов.
Нитрат стереоизомера А имел температуру плавления 150-165оС.
Нитрат стереоизомера В имел температуру плавления 160-164оС.
П р и м е р 17. 2-(2,4-Дифторфенил)-2-[(1Н-1,2,4-триазол-1-ил)метил]-3,3,4- триметилоксетан.
Следуя методике, аналогичной описанной в препаративном примере 2(а), два стереоизомера 4,5-эпоксисоединения синтезировали из 2-(2,4-дифторфенил)-3,3-диметил-1-(1Н-1,2,4-триазол-1-ил)-4- пентен-2-ола. Затем стереоизомер с пониженной полярностью превращали в 2-(2,4-дифторфенил)-3,3-диметил-1-(1Н-1,2,4-триазол-1-ил)-2,4- пентандиол в соответствии с методикой, описанной в препаративном примере 4. Затем полученное вещество мезилировали по методике, описанной в препаративном примере 2(с) с получением 2-(2,4-дифторфенил)-3,3-диметил-4-(метан-сульфонилокси)-1-(1Н- 1,2,4-триазол-1-ил)-2-пентанола.
10 мг гидрида натрия добавляли к раствору 77 мг 2-(2,4-дифторфенил-3,3-диметил-4-(метансульфонилокси)-1-(1Н-1,2,4- триазол-1-ил)-2-пентанола в 1,5 мл тетрагидрофурана при охлаждении системы льдом и перемешивании, после чего добавляли 1 мл метанола. Через 10 мин реакционную смесь разбавляли водой и экстрагировали этилацетатом. Полученный в результате сырой продукт очищали методом колонной хроматографии на силикагеле с использованием смеси этилацетата и гексана в соотношении 3:2 в качестве элюента. Полученное в результате соединение перекристаллизовывали из циклогексана с получением 39 мг чистого образца целевого соединения с температурой плавления 112-113оС.
ЯМР-спектр (СДСl3), δ ч./млн. 0,86 (3Н, синглет); 1,24 (3Н, дублет, J 6 Гц); 1,36 (3Н, синглет); 4,72 (1Н, дублет, J 15 Гц); 4,82 (1Н, квартет, J 6 Гц); 5,02 (1Н, дублет, J 15 Гц); 6,5-7,4 (3Н, мультиплет); 7,61 (1Н, синглет); 7,94 (1Н, синглет).
П р и м е р 18. 2-(2,4-Дифторфенил)-2-[(1Н-1,2,4-триазол-1-ил)-метил]-3,3,4- триметилоксетан.
Это вещество представляет собой стереоизомер соединения примера 17 в положении С4.
Следуя методике, аналогичной описанной в препаративном примере 2(а), два стереоизомера 4,5-эпоксисоединения синтезировали из 2-(2,4-дифторфенил)-3,3-диметил-1-(1Н-1,2,4-триазол-1-ил)-4- пентен-2-ола. Затем стереоизомер с повышенной полярностью превращали в 2-(2,4-дифторфенил)-3,3-диметил-1-(1Н-1,2,4-три- азол-1-ил)-2,4- пентандиол в соответствии с методикой, описанной в препаративном примере 4. Затем это вещество мезилировали по методике, описанной в препаративном примере 2(с) с получением 2-(2,4-ди- фторфенил)-3,3-диметил-4-(метансульфонилокси)-1-(1Н- 1,2,4-триазол-1-ил)-2-пентанола.
Следуя методике, аналогичной описанной в примере 17, но используя 4-мезилат, полученный в соответствии с описанным выше, получали целевое соединение и перекристаллизовывали его из смеси бензола с гексаном с получением чистого образца с температурой плавления 93-94оС.
ЯМР-спектр (СДСl3), δ ч./млн. 0,93 (3Н, дублет, J 2 Гц); 1,34 (3Н, синглет); 1,44 (3Н, дублет, J 6,5 Гц); 4,50 (1Н, квартет, J 6,5 Гц); 4,81 (2Н, синглет); 6,6-7,1 (2Н, мультиплет); 7,23 (1Н, триплет дублетов, J 9 и 6 Гц); 7,60 (1Н, синглет); 7,96 (1Н, синглет).
П р и м е р 19. (2R*,3S*,4R*)-2-(4-Три- фторметоксифенил)-3,4-диметил-2-[(1Н-1,2,4- триазол-1-ил)метил]оксетан.
Следуя методике, аналогичной описанной в примере 4, но используя 250 мг (2R*, 3S*,4S*)-2-(4-трифторметоксифенил)-4-(метансульфонилокси)-3- метил-1-(1Н-1,2,4-триазол-1-ил)-2-пентанола (который синтезировали по методике, аналогичной описанной в препаративном примере 2(b) и 2(с) и который является производным 4,5-эпоксисоединения с пониженной полярностью стереоизомеров в положении С4, полученного по методике, аналогичной описанной в препаративном примере 2(а)), получали 82 мг целевого соединения, в виде масла.
Масс-спектр (m/z): 328 (М+ + 1), 308, 272, 245, 214, 197, 190, 189, 175, 161, 141, 129, 115, 95, 82.
ЯМР-спектр (СДСl3), δ ч. /млн. 0,70 (3Н, дублет, J 7,5 Гц); 1,16 (3Н, дублет, J 6,5 Гц); 3,18 (1Н, квинтет, J 7,5 Гц); 4,35 (1Н, дублет, J 14,5 Гц); 4,39 (1Н, дублет квартетов, J 7,5 и 6,5 Гц); 4,70 (1Н, дублет, J 14,5 Гц); 7,2-7,4 (4Н, мультиплет); 7,93 (1Н, синглет); 8,23 (1Н, синглет).
ИК-спектр (жидкая пленка), λмакс. см-1: 1610, 1590, 1500, 1260, 1020.
П р и м е р 20. (2R*,3S*,4R*)-2-(4-Хлорфенил)-3,4-диметил-2-[(1Н-1,2,4-триазол- 1-ил)метил]оксетан.
Следуя методике, аналогичной описанной в примере 4, но используя 230 мг (2R*, 3S*, 4S*)-2-(4-хлорфенил)-4-(метансуль- фонилокси)-3-метил-1- (1Н-1,2,4-триазол-1-ил)-2-пентанола (который синтезировали по методике, аналогичной описанной в препаративном примере 2(b) и 2(с), и который является производным 4,5-эпоксисоединения с пониженной полярностью стереоизомеров в положении С4, полученного по методике, аналогичной описанной в препаративном примере 2(а)), получали 112 мг целевого соединения с температурой плавления 111-118оС.
Масс-спектр (m/z): 278 (М+ + 1), 256, 222, 197, 164, 149, 141, 139, 129, 111, 104.
ЯМР-спектр (СДСl3), δ ч./млн. 0,70 (3Н, дублет, J 7,25 Гц); 1,17 (3Н, дублет, J6,45 Гц); 3,16 (1Н, квинтет, J 7,25 Гц); 4,41 (1Н, дублет, J 14,5 Гц); 4,4-4,53 (1Н, мультиплет); 4,76 (1Н, дублет, J 14,5 Гц); 7,25 (2Н, дублет, J 8,46 Гц); 7,36 (2Н, дублет, J 8,46 Гц); 7,98 (1Н, синглет); 8,42 (1Н, синглет).
П р и м е р 21. 2-(4-Хлорфенил)-3,3-диметил-2-[(1Н-1,2,4-триазол-1-ил)метил]оксетан.
160 мг гидрида натрия (в виде 60 мас./мас. дисперсии в минеральном масле) добавляли к раствору 750 мг 2-(4-хлорфенил)-3,3-диметил-4-метансульфонилокси-1-(1Н-1,2,4- триазол-1-ил)-2-бутанола в 12 мл диметилформамида и полученную в результате смесь перемешивали при комнатной температуре в течение 3 ч. К концу указанного времени реакционную смесь переливали в смесь воды со льдом и экстрагировали этилацетатом. Экстракт промывали водой, водным раствором хлористого аммония, сушили над безводным сульфатом магния и концентрировали выпариванием при пониженном давлении. Полученный в результате остаток очищали методом препаративной тонкослойной хроматографии с использованием смеси этилацетата с гексаном в объемном соотношении 1:4 в качестве проявляющего растворителя, с получением 170 мг (выход 31%) целевого соединения, температура плавления 90-102оС.
ЯМР-спектр (СДСl3, 60 МГц), δ ч./млн. 0,90 (3Н, синглет); 1,38 (3Н, синглет), 4,11 (1Н, дублет, J 6 Гц); 4,50 (1Н, дублет, J 6 Гц); 4,60 (1Н, дублет, J 14 Гц); 5,00 (1Н, дублет, J 14 Гц); 6,85-7,30 (4Н, мультиплет); 7,56 (1Н, синглет); 7,81 (1Н, синглет).
Масс-спектр (m/z): 277 (М+), 222, 195, 137.
П р и м е р ы 22-32. С использованием методики, аналогичной описанной в примере 21, синтезировали следующие вещества:
П р и м е р 22. 2-(2,4-Дифторфенил)-2-[(1Н-1,2,4-триазол-1-ил)метил]-оксетан, в виде масла, с выходом 56%
ЯМР-спектр (СДСl3), δ ч./млн. 2,66-2,77 (1Н, мультиплет), 2,93-3,04 (1Н, мультиплет); 4,15-4,23 (1Н, мультиплет); 4,34 (1Н, дублет, J 14,9 Гц); 4,43-4,52 (1Н, мультиплет); 4,73 (1Н, дублет, J 14,9 Гц); 6,81-6,93 (2Н, мультиплет); 7,39-7,48 (1Н, мультиплет); 7,94 (1Н, синглет); 8,27 (1Н, синглет).
Масс-спектр (m/z): 251 (М+), 170, 141.
П р и м е р 23. 2-(2,4-Дифторфенил)-3-метил-2-[(1Н-1,2,4-триазол-1-ил)метил]оксе- тан, в виде масла, с выходом 73%
ЯМР-спектр (СДСl3), δ ч./млн. 0,93 (3Н, дублет дублетов, J 7 и 2 Гц); 3,15 (1Н, секстет, J 7 Гц); 3,99 (1Н, триплет, J 7 Гц); 4,37 (1Н, дублет дублетов, J 7 и 6 Гц); 4,48 (1Н, дублет, J 14,5 Гц); 4,90 (1Н, дублет, J 14,5 Гц); 6,8-7,0 (2Н, мультиплет); 7,2-7,4 (1Н, мультиплет); 7,90 (1Н, синглет); 8,30 (1Н, синглет).
Масс-спектр (m/z): 265 (М+), 247, 232, 224, 184, 183.
П р и м е р 24. 2-(2,4-Дифторфенил)-3-этил-2-[(1Н-1,2,4-триазол-1-ил)метил]оксе-тан, в виде масла, с выходом 39%
ЯМР-спектр (СДСl3), δ ч./млн. 0,70 (3Н, триплет, J 7,5 Гц); 1,03-1,15 (1Н, мультиплет); 1,46-1,56 (1Н, мультиплет); 2,87-2,98 (1Н, мультиплет); 4,07 (1Н, мультиплет); 4,34 (1Н, мультиплет); 4,48 (1Н, дублет, J 14,5 Гц); 4,91 (1Н, дублет, J 14,5 Гц); 6,80-6,94 (2Н, мультиплет); 7,44-7,53 (1Н, мультиплет); 7,88 (1Н, синглет); 8,22 (1Н, синглет).
Масс-спектр (m/z): 279 (М+), 224, 197, 141, 127.
П р и м е р 25. 2-(2,4-Дифторфенил)-3,3-диметил-2-[(1Н-1,2,4-триазол-1-ил)метил] оксетан, температура плавления 78оС, выход 72%
ЯМР-спектр (СДСl3), δ ч./млн. 1,04 (3Н, синглет); 1,43 (3Н, синглет); 4,18 (1Н, дублет, J 5,6 Гц); 4,62 (1Н, дублет, J 5,6 Гц); 4,79 (1Н, дублет дублетов, J 14,1 и 1,6 Гц); 5,01 (1Н, дублет, J 14,1 Гц); 6,68-6,83 (2Н, мультиплет); 7,1 (1Н, двойной дублет дублетов, J 6,5, 6,5 и 2,0 Гц); 7,62 (1Н, синглет); 7,95 (1Н, синглет).
Масс-спектр (m/z): 279 (М+), 261, 226, 197, 179, 167, 149.
П р и м е р 26. 2-(2,4-Дифторфенил)-3-фенил-2-[(1Н-1,2,4-триазол-1-ил)метил] оксетан получали в виде масла, выход 50%
ЯМР-спектр (СДСl3), δ ч./млн. 4,28 (1Н, триплет, J 7,5 Гц); 4,53-4,62 (2Н, мультиплет); 5,00 (2Н, дублет, J 14,9 Гц); 6,37-6,45 (1Н, мультиплет); 6,83-6,90 (1Н, мультиплет); 7,00-7,15 (5Н, мультиплет); 7,47-7,56 (1Н, мультиплет); 7,98 (1Н, синглет); 8,36 (1Н, синглет).
П р и м е р 27. 2-(4-Хлорфенил)-3-этил-2-[(1Н-1,2,4-триазол-1-ил)метил] оксетан получали в виде масла с выходом 40%
ЯМР-спектр (СДСl3), δ ч./млн. 0,63 (3Н, триплет, J 7,3 Гц); 0,91-1,01 (1Н, мультиплет); 1,16-1,32 (1Н, мультиплет); 2,84-2,93 (1Н, мультиплет); 4,05-4,09 (1Н, мультиплет); 4,22-4,27 (1Н, мультиплет); 4,38 (1Н, дублет, J 14,7 Гц); 4,74 (1Н, дублет, J 14,7 Гц); 7,25-7,39 (4Н, мультиплет); 7,97 (1Н, синглет); 8,32 (1Н, синглет).
П р и м е р 28. 3,3-Диметил-2-(4-метилфенил)-2-[(1Н-1,2,4-триазол-1-ил)метил]ок-сетан, температура плавления 76-77оС, выход 83%
ЯМР-спектр (СДСl3), δ ч./млн. 0,93 (3Н, синглет); 1,41 (3Н, синглет); 2,29 (3Н, синглет); 4,19 (1Н, дублет, J 5,6 Гц); 4,56 (1Н, дублет, J 5,6 Гц); 4,63 (1Н, дублет, J 14,1 Гц); 5,07 (1Н, дублет, J 14,1 Гц); 6,99-7,02 (2Н, мультиплет); 7,07-7,10 (2Н, мультиплет); 7,69 (1Н, синглет); 7,80 (1Н, синглет).
Масс-спектр (m/z): 257 (М+), 202, 175.
П р и м е р 29. 2-(4-Изопропилфенил)-3,3-диметил-2-[(1Н-1,2,4-триазол-1-ил)ме- тил] оксетан получали в виде масла, выход 67%
ЯМР-спектр (СДСl3), δ ч./млн. 0,92 (3Н, синглет); 1,21 (6Н, дублет, J 7,3 Гц); 1,41 (3Н, синглет); 2,80-2,90 (1Н, мультиплет); 4,19 (1Н, дублет, J 5,6 Гц); 4,55 (1Н, дублет, J 5,6 Гц); 4,62 (1Н, дублет, J 14,3 Гц); 6,97-7,15 (4Н, мультиплет); 7,69 (1Н, синглет); 7,77 (1Н, синглет).
Масс-спектр (m/z): 285 (М+), 230, 203.
П р и м е р 30. 2-(4-Метоксифенил)-3,3-диметил-2-[(1Н-1,2,4-триазол-1-ил)метил] оксетан получали в виде масла, выход 29%
ЯМР-спектр (СДСl3), δ ч./млн. 0,92 (3Н, синглет); 1,41 (3Н, синглет); 3,77 (3Н, синглет); 4,56 (1Н, дублет, J 5,8 Гц); 4,59 (1Н, дублет, J 5,8 Гц); 4,62 (1Н, дублет, J 14,3 Гц); 5,06 (1Н, дублет, J 14,3 Гц); 6,80-6,88 (2Н, мультиплет); 6,96-7,24 (2Н, мультиплет); 7,69 (1Н, синглет); 7,81 (1Н, синглет).
Масс-спектр (m/z): 273 (М+), 218, 191.
П р и м е р 31. 3-Этил-2-(4-фторфенил)-2-[(1Н-1,2,4-триазол-1-ил)метил] оксетан с температурой плавления 43-47оС получали с выходом 33%
ЯМР-спектр (СДСl3), δ ч./млн. 0,66 (3Н, триплет, J 7,25 Гц); 0,87-1,01 (1Н, мультиплет); 1,20-1,32 (1Н, мультиплет); 2,82-2,92 (1Н, мультиплет); 4,08 (1Н, триплет, J 6,2 Гц); 4,27 (1Н, триплет, J 6,2 Гц); 4,39 (1Н, дублет, J 14,9 Гц); 4,76 (1Н, дублет, J 14,9 Гц); 7,09 (2Н, триплет, J 8,9 Гц); 7,30-7,34 (2Н, дублет дублетов, J 8,9 и 2 Гц); 7,98 (1Н, синглет); 8,37 (1Н, синглет).
Масс-спектр (m/z): 262 (М+), 246, 206, 179.
П р и м е р 32. 2-(2,4-Дихлорфенил)-3,3-диметил-2-[(1Н-1,2,4-триазол-1-ил)метил] оксетан с температурой плавления 90-100оС получали с выходом 60%
ЯМР-спектр (СДСl3), δ ч./млн. 1,12 (3Н, синглет); 1,50 (3Н, синглет); 4,13 (1Н, дублет, J 5,8 Гц); 4,62 (1Н, дублет, J 5,8 Гц); 5,02 (1Н, дублет, J 14,3 Гц); 5,27 (1Н, дублет, J 14,3 Гц); 7,08 (1Н, дублет дублетов, J 1,95 и 8,45 Гц); 7,17 (1Н, дублет, J 8,45 Гц); 7,35 (1Н, дублет, J 1,95 Гц); 7,66 (1Н, синглет); 8,05 (1Н, синглет).
Масс-спектр (m/z): 315 (М+4)+, 313/(М+2)+/ 311 (М+), 276, 256, 231, 229, 199, 173.
П р и м е р 33. (2R*,3S*,4S*)-2-(2,4-Ди-фторфенил)-4-этил-3-метил-2-[(1Н-1,2,4-три- азол-1-ил)метил]оксетан.
960 мг (0,24 ммоль) гидрида натрия (в виде 60% мас./мас. дисперсии в минеральном масле) добавляли при охлаждении системы льдом к раствору 77,9 мг (0,2 ммоль) 2-(2-дифторфенил-4-метансульфонилокси-3-метил-1-(1Н-1,2,4-триазол- 1-ил)-2-гексанола в 4,9 мл диметилформамида. Затем полученную в результате смесь перемешивали в течение 3 ч при комнатной температуре. К истечению этого времени реакционную смесь переливали в 20 мл смеси воды со льдом и экстрагировали 40 мл этилацетата. Экстракт промывали насыщенным водным раствором хлористого натрия, сушили над безводным сульфатом натрия и концентрировали выпариванием при пониженном давлении. Остаток очищали методом препаративной тонкослойной хроматографии с использованием смеси гексана с этилацетатом в объемном соотношении 2:1 в качестве проявляющего растворителя, с получением 13,0 мг (выход 22,2%) целевого соединения в виде масла.
ЯМР-спектр (СДСl3), δ ч./млн. 0,82 (3Н, триплет, J 7,5 Гц); 0,87 (3Н, дублет дублетов, J 2,42 и 6,9 Гц); 1,18-1,34 (1Н, мультиплет); 1,36-1,50 (1Н, мультиплет); 2,76 (1Н, квинтет, J 6,9 Гц); 3,98 (1Н, квартет, J 6,91 Гц); 4,39 (1Н, дублет, J 14,5 Гц); 4,79 (1Н, дублет, J 14,5 Гц); 6,82-6,97 (2Н, мультиплет); 7,56-7,65 (1Н, мультиплет); 7,88 (1Н, синглет); 8,18 (1Н, синглет).
Масс-спектр (m/z): 294/(М+ 1)+/ 224, 211.
П р и м е р 34-35. С использованием методики, аналогичной описанной в примере 3, синтезировали следующие соединения.
П р и м е р 34. (2R*,3S*,4S*)-4-этил-2-(4-фторфенил)-3-метил-2-[1Н-1,2,4-триазол- 1-ил)метил]оксетан получали в виде масла с выходом 7,5%
ЯМР-спектр (СДСl3), δ ч/млн. 0,73 (3Н, дублет, J 7,1 Гц); 0,82 (3Н, триплет, J 7,6 Гц); 1,11-1,44 (2Н, мультиплет);2,68 (1Н, квинтет, J 17,1 Гц); 4,01 (1Н, квартет, J 7,1 Гц); 4,29 (1Н, дублет, J 14,9 Гц); 4,59 (1Н, дублет, J 14,9 Гц); 7,08-7,15 (2Н, мультиплет); 7,31-7,38 (2Н, мультиплет); 7,97 (1Н, синглет); 8,26 (1Н, синглет).
Масс-спектр (m/z): 276 /(М+ + 1)+/, 193.
П р и м е р 35. (2R*,3S*,4S*)-2-(4-хлорфенил)-4-этил-3-метил-2-[(1Н-1,2,4-триазол- 1-ил)метил]оксетан получали в виде масла с выходом 8,0%
ЯМР-спектр (СДСl3), δ ч. /млн. 0,69 (3Н, дублет, J 7,0 Гц); 0,79 (3Н, триплет, J 7,5 Гц); 1,11-1,29 (1Н, мультиплет); 1,31-1,44 (1Н, мультиплет); 2,69 (1Н, квинтет, J 7,0 Гц); 4,29 (1Н, дублет, J 14,9 Гц); 4,59 (1Н, дублет, J 14,9 Гц); 7,31 (2Н, дублет, J 8,7 Гц); 7,40 (2Н, дублет, J 8,7 Гц); 7,97 (1Н, синглет); 8,26 (1Н, синглет).
Масс-спектр (m/z): 292 /(М + 1)+/, 209.
П р и м е р 36. 3-трет-бутил-2-(4-фторфенил)-2-[(1,2,4-триазол-1-ил)метил]оксетан.
2,02 г (20 ммоль) триэтиламина и 2,29 г (20 ммоль) хлористого метансульфонила добавляли при охлаждении системы льдом, к раствору 1,54 г (5 ммоль) 2-(4-фторфенил)-3,5-диметил-1-(1Н-1,2,4-триазол-1-ил)-2,4- гександиола в 75 мл хлористого метилена и полученную в результате смесь перемешивали в течение 3 ч при той же температуре и затем при комнатной температуре в течение 1 ч. К концу указанного времени реакционную смесь перемешивали в 80 мл смеси льда с водой и экстрагировали 150 хлористого метилена. Экстракт промывали водой и сушили над безводным сульфатом натрия и концентрировали выпариванием при пониженном давлении. Остаток очищали методом колонной хроматографии на силикагеле с использованием смеси гексана и этилацетата в объемном соотношении 6:5 в качестве элюента, с получением 113 мг (выход 7,8%) целевого соединения с температурой плавления 128оС.
ЯМР-спектр (СДСl3), δ ч/млн. 1,06 (9Н, синглет); 2,28 (1Н, дублет дублетов, J 12,1 и 14,30 Гц); 2,64 (1Н, дублет дублетов, J 1,21 и 14,3 Гц); 4,92 (1Н, дублет, J 14,5 Гц); 5,21 (1Н, дублет, J 14,5 Гц); 4,97 (1Н, дублет дублетов, J 1,21 и 12,1 Гц); 7,01-7,11 (4Н, мультиплет); 7,77 (1Н, синглет); 7,80 (1Н, синглет).
Масс-спектр (m/z): 289 (М+), 207.
П р и м е р 37. 3-трет-бутил-2-(4-хлорфенил)-2-[(1Н-1,2,4-триазол-1-ил)метил]оксалат.
Следуя методике, аналогичной описанной в примере 36, целевое соединение получали в виде масла, выход 3,4%
ЯМР-спектр (СДСl3), δ ч./млн. 1, 10 (9Н, синглет); 2,27 (1Н, дублет дублетов, J 11,7 и 13,9 Гц); 2,62 (1Н, широкий дублет, J 13,9 Гц); 4,96 (1Н, дублет, J 12,5 Гц); 5,19 (1Н, дублет, J 12,5 Гц); 7,16 (2Н, дублет, J 8,3 Гц); 7,30 (2Н, дублет, J 8,3 Гц); 7,81 (1Н, широкий синглет); 7,86 (1Н, широкий синглет).
Масс-спектр (m/z): 305 (М+), 223.
П р и м е р 38. 2-(4-Хлорфенил)-4-изопропил-2-[(1Н-1,2,4-триазол-1-ил)-метил] оксетан.
Следуя методике, аналогичной описанной в примере 36, но используя 700 мг (2,26 ммоль) 2-(4-хлорфенил)-5-метил-1-(1Н-1,2,4-триазол-1-ил)-2,4-гександиола, получали два стереоизомера в положении 4 целевого соединения.
Выход соединения (А), имеющего более низкую полярность, составил 53,5 мг (8,1% ) и выход соединения (В) с более высокой полярностью составил 28,9 мг (4,4%).
Соединение (А): в виде масляного вещества.
ЯМР-спектр (СДСl3), δ ч. /млн. 1,06 (3Н, дублет, J 6,8 Гц); 1,07 (3Н, дублет, J 6,8 Гц); 1,96 (1Н, септет, J 6,8 Гц); 2,27 (1Н, дублет дублетов, J 12,1 и 14,1 Гц); 2,65 (1Н, дублет дублетов, J 1,8 и 14,1 Гц); 4,99 (1Н, дублет, J 14,5 Гц); 5,16 (1Н, дублет, J 14,5 Гц); 4,99-5,06 (1Н, мультиплет); 7,04 (2Н, дублет, J 8,7 Гц); 7,29 (2Н, дублет, J 8,7 Гц); 7,82 (1Н, синглет); 7,88 (1Н, синглет).
Масс-спектр (m/z): 291 (М+), 209.
Соединение (В):
ЯМР-спектр (СДСl3), δ ч./млн. 1,00 (3Н, дублет, J 7,3 Гц); 1,02 (дублет, J 7,3 Гц); 1,88-2,00 (1Н, мультиплет); 2,77 (1Н, дублет дублетов, J 3,2 и 14,5 Гц); 2,94 (дублет дублетов, J 12,1 и 14,5 Гц); 3,94 (1Н, дублет дублетов, J 3,2; 6,4 и 12,1 Гц); 4,45 (1Н, дублет, J 14,3 Гц); 4,70 (1Н, дублет, J 14,3 Гц); 7,14 (2Н, дублет, J 8,7 Гц); 7,32 (2Н, дублет, J 8,7 Гц); 7,69 (1Н, синглет); 7,92 (1Н, синглет).
Масс-спектр (m/z 291 (М+), 209.
П р и м е р 39. 3,3,4-Триметил-2-фенил-(1Н-1,2,4-триазол-1-ил)метилоксетан.
4,0 мл (37,6 ммоль) 2-метил-2-бутена добавляли к раствору 1,1 г (5,88 ммоль) 2-(1Н-1,2,4-триазол-1-ил)ацетофенона в 15 мл смеси ацетонитрила и бензола в соотношении (об. ) 2: 1. Затем смесь облучали с использованием ртутной лампы среднего давления мощностью 450 Вт (Ганновеа Ко. Инк.) в течение 15 ч. По истечении этого времени реакционную смесь концентрировали выпариванием при пониженном давлении и остаток подвергали очистке на колонке с силикагелем с использованием смеси гексана с этилацетатом в объемном соотношении 1: 1 в качестве элюента, с получением 409,7 мг (выход 27%) целевого соединения с температурой плавления 102оС.
ЯМР-спектр (СДСl3), δ ч./млн. 0,76 (3Н, синглет), 1,26 (3Н, дублет, J 6,4 Гц); 1,36 (3Н, синглет); 4,64 (1Н, дублет, J 14,1 Гц); 4,82 (1Н, квартет, J 5,4 Гц); 5,10 (1Н, дублет, J 14,1 Гц); 7,1-7,3 (5Н, широкий мультиплет); 7,65 (1Н, синглет); 7,76 (1Н, синглет).
Масс-спектр (m/z): 258 (М + 1)+: 188, 175, 152, 144, 129.
П р и м е р ы 40-44. С использованием методики, аналогичной описанной в примере 39, синтезировали следующие соединения.
П р и м е р 40. 2-(4-Изопропилфенил)-2-(1Н-1,2,4-триазол-1-ил)метил-3,3,4- триметилоксетан с температурой плавления 53-60оС получали с выходом 4,4%
ЯМР-спектр (СДСl3), δ ч./млн. 0,76 (3Н, синглет); 1,20 (6Н, дублет, J 6,85 Гц); 1,26 (3Н, дублет, J 6,45 Гц); 1,35 (3Н, синглет); 2,84 (1Н, септет, J 6,85 Гц); 4,62 (1Н, дублет, J 14,1 Гц); 4,81 (1Н, квартет, J 6,45 Гц); 5,10 (1Н, дублет, J 14,1 Гц); 6,96-7,12 (4Н, широкий мультиплет); 7,69 (1Н, синглет); 7,74 (1Н, синглет).
Масс-спектр (m/z): 299 (М+), 230, 217.
П р и м е р 41. 2-(4-Метоксифенил)-2-(1Н-1,2,4-триазол-1-ил)метил-3,3,4- триметилоксетан в виде масла получали с выходом 2,5%
ЯМР-спектр (СДСl3), δ ч./млн. 0,75 (3Н, синглет); 1,26 (3Н, дублет, J 6,44 Гц); 1,34 (3Н, синглет); 3,76 (3Н, синглет); 4,63 (1Н, J 14,1 Гц); 4,81 (1Н, квартет, J 6,44 Гц); 5,09 (1Н, дублет, J 14,1 Гц); 6,80 (2Н, дублет, J 8,87 Гц); 6,94-7,03 (2Н, широкий); 7,69 (1Н, синглет); 7,83 (1Н, синглет).
Масс-спектр (m/z): 287 (М+), 218, 205, 135.
П р и м е р 42. 2-(2,4-Дихлорфенил)-2-(1Н-1,2,4-триазол-1-ил)метил-3,3,4- триметилоксетан с температурой плавления 85-104оС получали с выходом 3,2%
ЯМР-спектр (СДСl3), δ ч./млн. 0,92 (3Н, синглет); 1,24 (3Н, дублет, J 6,45 Гц); 1,44 (3Н, синглет); 4,81 (1Н, квартет, J 6,45 Гц); 5,00 (1Н, дублет, J 14,3 Гц); 5,26 (1Н, дублет, J 14,3 Гц); 7,06 (1Н, дублет дублетов, J 2,02 и 8,86 Гц); 7,16 (1Н, дублет, J 8,86 Гц); 7,32 (1Н, дублет, J 2,02 Гц); 7,63 (1Н, синглет); 7,94 (1Н, синглет).
Масс-спектр (m/z): 325 (М+), 312, 243, 175.
П р и м е р 43. 2-(4-Фторфенил)-2-(1Н-1,2,4-триазол-1-ил)метил-3,3,4- триметилоксетан с температурой плавления 108оС получали с выходом 15%
ЯМР-спектр (СДСl3), δ ч./млн. 0,74 (3Н, синглет); 1,26 (3Н, дублет, J 6,45 Гц); 1,36 (3Н, синглет); 4,65 (1Н, дублет, J 14,1 Гц); 4,83 (1Н, квартет, J 6,45 Гц); 5,08 (1Н, дублет, J 14,1 Гц); 6,92-7,07 (4Н, широкий мультиплет); 7,67 (1Н, синглет); 7,90 (1Н, синглет).
Масс-спектр (m/z): 276 (М+), 206, 147.
П р и м е р 44. 2-(4-Хлорфенил)-2-(1Н-1,2,4-триазол-1-ил)метил-3,3,4-триметилок- сетан с температурой плавления 111-114оС получали с выходом 2%
ЯМР-спектр (СДСl3), δ ч./млн. 0,72 (3Н, синглет); 1,24 (3Н, дублет, J 6,35 Гц); 1,33 (3Н, синглет); 4,62 (1Н, дублет, J 14,2 Гц); 4,80 (1Н, квартет, J 6,35 Гц); 5,06 (1Н, дублет, J 14,2 Гц); 6,8-7,24 (4Н, широкий мультиплет); 7,65 (1Н, синглет); 7,91 (1Н, синглет).
Масс-спектр (m/z): 292 (М+), 279, 222, 209.
П р и м е р 45. 2-(2,4-Дифторфенил)-3,3-диметил-2-[(1Н-1,2,4-триазол-1-ил)метил] оксетан.
2-Метилпропен барботировали через 5 мл бензола при 0оС до 25% увеличения объема системы. Затем к раствору добавляли 6 мл ацетонитрила и 695,2 мг (3,115 ммоль) 2',2'-дифтор-2-(1Н-1,2,4-триазол-1-ил)ацето- фенона и полученную в результате смесь облучали с ртутно-дуговой лампы среднего давления мощностью 450 Вт (Ганновеа Ко. Инк) при 15оС в течение 15 ч. По истечении этого времени реакционную смесь концентрировали и выпаривали при пониженном давлении и затем остаток подвергали хроматографии на колонке с силикагелем с получением 450 мг смеси целевого соединения с непрореагировавшим исходным материалом. Эту смесь растворяли в 5 мл метанола и к полученному в результате раствору добавляли 200 мг (5,27 ммоль) борогидрида натрия, после чего смесь перемешивали в течение 3 ч при той же температуре. К концу этого времени реакционную смесь разбавляли водой и экстрагировали этилацетатом. Затем органический слой сушили над безводным сульфатом магния и концентрировали выпариванием при пониженном давлении с получением светло-желтого остатка. Этот остаток подвергали хроматографированию на колонке с силикагелем с использованием в качестве элюента смеси гексана с этилацетатом в объемном соотношении 1: 1, с получением 197 мг (0,706 ммоль) выход 23% целевого соединения с температурой плавления 76-78оС.
Полученный продукт был во всех отношениях идентичен соединению, полученному в примере 25.
П р и м е р 46. 2-(4-Фторфенил)-3,3-диметил-2-[(1Н-1,2,4-триазол-1-ил)метил] оксетан.
Этот пример иллюстрирует синтез целевого соединения путем фотореакции.
Следуя методике, аналогичной описанной в примере 39, но используя 2-метилпропен и 4'-фтор-2-(1Н-1,2,4-триазол-1- ил)ацетофенон, с выходом 2% получали целевое соединение с температурой плавления 110оС.
ЯМР-спектр (СДСl3), δ ч./млн. 0,92 (3Н, синглет); 1,43 (3Н, синглет); 4,20 (1Н, дублет, J 5,6 Гц); 4,58 (1Н, дублет, J 5,6 Гц); 4,64 (1Н, дублет, J 14,1 Гц); 5,06 (1Н, дублет, J 14,1 Гц); 6,97 (2Н, триплет, J 8,86 Гц); 7,00-7,09 (2Н, широкий мультиплет); 7,68 (1Н, синглет); 7,99 (1Н, синглет).
Масс-спектр (m/z): 261 (М+), 179, 123.
П р и м е р 47. 8-(2,4-Дифторфенил)-7-окса-8-(1Н-1,2,4-триазол-1-ил)метилбицик- ло (4.2.0)октан.
Следуя методике, аналогичной описанной в примере 39, но используя циклогексан и 2',4'-дифтор-2-(1Н-1,2,4-триазол-1-ил)ацетофенон, получали смесь стереоизомеров целевого соединения в положении 8 в соотношении 1:1, в виде смолы с выходом 31%
ЯМР-спектр (СДСl3), δ ч. /млн. 1,0-2,2 (широкий мультиплет); 2,64 (широкий дублет триплетов, J 4,1 Гц); 3,03 (квартет, J7,15 Гц); 4,52 (дублет, J 14,5 Гц); 4,60 (мультиплет); 4,70 (дублет, J 14,5 Гц); 4,89 (дублет, J 14,5 Гц); 5,11 (дублет, J 14,5 Гц); 6,65-6,96 (мультиплет); 7,31 (двойной дублет дублетов, J 1,61, 8,05 и 8,05 Гц); 8,17 (синглет); 7,95 (синглет); 7,81 (синглет).
П р и м е р 48. 2-(2,4-Дифторфенил-3,3,4,4-тетраметил-2-[(1Н-1,2,4-триазол-1- ил) метил]оксетан.
Следуя методике, аналогичной описанной в примере 39, но используя 2,3-диметил-2-бутен и 2',4'-дифтор-2-(1Н-1,2,4- триазол-1-ил)ацетофенон, получали 85 мг (выход 15%) целевого соединения с температурой плавления 100-101оС.
ЯМР-спектр (СДСl3), δ ч./млн. 0,93 (3Н, дублет, J 1,61 Гц); 1,27 (3Н, синглет); 1,42 (3Н, синглет); 1,58 (3Н, синглет); 4,48 (1Н, дублет дублетов, J 1,61; 14,10 Гц); 4,90 (1Н, дублет, J 14,10 Гц); 6,65-6,82 (2Н, мультиплет); 7,20 (1Н, двойной дублет дублетов, J 2,01; 9,01 и 9,01 Гц); 7,60 (1Н, синглет); 7,91 (1Н, синглет).
Масс-спектр (m/z): 307 (М+), 294, 279, 224, 205, 167, 149, 141.
П р и м е р 49. (2R*,4R*)-2-(4-фторфенил)-3,4-диметил-2-[(1Н-1,2,4-триазол-1-ил)- метил]оксетан.
Следуя методике, аналогичной описанной в примере 39, но используя цис-2-бутен и 950 мг (5,68 ммоль) 4'-фтор-2-(1Н-1,2,4-триазол-1-ил)ацетофенона, получали 80 мг (выход 5,3%) смеси стереоизомеров целевого соединения в положении 3 в соотношении 1:1, в виде масла.
ЯМР-спектр (СДСl3), δ ч./млн. 0,69 (дублет, J 7,25 Гц); 1,16 (дублет, J 6,45 Гц); 1,37 (дублет, J 6,45 Гц); 2,79 (квинтет, J 7,25 Гц); 3,15 (квинтет, J 7,25 Гц); 4,39 (дублет, J 14,50 Гц); 4,50 (мультиплет); 4,60 (мультиплет); 4,59 (дублет, J 14,1 Гц); 4,73 (дублет, J 14,5 Гц); 4,92 (дублет, J 14,1 Гц); 6,91-7,15 (мультиплет); 7,21-7,32 (мультиплет); 7,76 (синглет); 7,94 (синглет); 7,08 (синглет); 8,27 (синглет).
Масс-спектр (m/z): 261 (М+), 206, 179, 148, 133, 123.
П р и м е р 50. 2-(4-Метилфенил)-2-[(1Н-1,2,4-триазол-1-ил)метил]оксетан.
0,98 г (25,5 ммоль) гидрида натрия (в виде 60 мас./мас. дисперсии в минеральном масле) добавляли к 20 мл диметилсульфоксида и полученную в результате смесь перемешивали в течение 1 ч при 80оС. К концу этого времени добавляли 5,62 г (25,5 ммоль) иодистого триметилсульфоксония при охлаждении системы льдом, после чего смеси давали нагреваться до комнатной температуры. Затем смесь перемешивали в течение 30 мин и добавляли раствор 2,2 г (11,6 ммоль) 4'-метил-2-(1Н-1,2,4-триазол-1-ил)ацетофенона в 5 мл диметилсульфоксида, полученную смесь перемешивали в течение 6 ч при 50оС. После охлаждения реакционной смеси ее переливали в 20 мл смеси льда с водой и экстрагировали 200 мл этилацетата. Затем экстракт дважды промывали 100 мл порциями насыщенного водного раствора хлористого натрия, после чего ее сушили над безводным сульфатом натрия и концентрировали выпариванием при пониженном давлении с получением 2,7 г маслянистого соединения. Это масло подвергали колонной хроматографии на силикагеле смеси гексана с этилацетатом в объемном соотношении 1:1 в качестве элюента, с получением 2,3 г (выход 86,3%) целевого соединения в виде масла.
ЯМР-спектр (СДСl3), δ ч./млн. 2,37 (3Н, синглет); 2,6-2,7 (1Н, мультиплет); 2,91-2,98 (1Н, мультиплет); 4,02-4,10 (1Н, мультиплет); 4,29 (1Н, дублет, J 14,7 Гц); 4,52 (1Н, дублет, J 14,7 Гц); 4,38-4,46 (1Н, мультиплет); 7,22 (4Н, синглет); 7,98 (1Н, синглет); 8,22 (1Н, синглет).
Масс-спектр (m/z): 229 (М+), 214, 198, 184, 172, 159, 148, 119.
ИК-спектр (жидкая пленка), λмакс. см-1: 3140, 2980, 2950, 1505, 1271.
П р и м е р ы 51-57. С использованием методики, аналогичной описанной в примере 50, синтезировали следующие соединения.
П р и м е ер 51. 2-(4-Хлорфенил)-2-[(1Н-1,2,4-триазол-1-ил)метил]оксетан с температурой плавления 74-75оС получали с выходом 61%
ЯМР-спектр (СДСl3), δ ч./млн. 2,62-2,75 (1Н, мультиплет); 2,95-3,05 (1Н, мультиплет); 4,06-4,14 (1Н, мультиплет); 4,30 (1Н, дублет, J 14,5 Гц); 4,39-4,49 (1Н, мультиплет); 4,53 (1Н, дублет, J 14,5 Гц); 7,27 (2Н, дублет, J 8,46 Гц); 7,38 (2Н, дублет, J 8,46 Гц); 7,98 (1Н, синглет); 8,24 (1Н, синглет).
Масс-спектр (m/z): 249 (М+), 169, 139.
П р и м е р 52. 2-(2,4-Дифторфенил)-2-[(1Н-1,2,4-триазол-1-ил)метил] оксетан в виде масла получали с выходом 45%
ЯМР-спектр (СДСl3), δ ч./млн. 2,66-2,77 (1Н, мультиплет); 2,93-3,04 (1Н, мультиплет); 4,15-4,23 (1Н, мультиплет); 4,34 (1Н, дублет, J 14,9 Гц); 4,43-4,52 (1Н, мультиплет); 4,73 (1Н, дублет, J 14,9 Гц); 6,81-6,93 (2Н, мультиплет); 7,39-7,48 (1Н, мультиплет); 7,94 (1Н, синглет); 8,27 (1Н, синглет).
Масс-спектр (m/z): 251 (М+), 170, 141.
П р и м е р 53. 2-(4-Изопропилфенил)-2-[(1Н-1,2,4-триазол-1-ил)метил] оксетан в виде масла получали с выходом 87%
ЯМР-спектр (СДСl3), δ ч. /млн. 1,27 (6Н, дублет, J 7,3 Гц); 2,17-2,72 (1Н, мультиплет); 2,90-3,01 (2Н, мультиплет); 4,02-4,07 (1Н, мультиплет); 4,29 (1Н, дублет, J 14,7 Гц); 4,38-4,47 (1Н, мультиплет); 4,52 (1Н, дублет, J 14,7 Гц); 7,27 (4Н, синглет); 7,99 (1Н, синглет); 8,25 (1Н, синглет).
Масс-спектр (m/z): 257 (М+), 175, 147.
П р и м е р 54. 2-(4-Трифторметилфенил)-2-[(1Н-1,2,4-триазол-1-ил)метил] оксетан в виде масла получали с выходом 42%
ЯМР-спектр (СДСl3), δ ч./млн. 2,66-2,72 (1Н, мультиплет); 3,00-3,11 (1Н, мультиплет); 4,07-4,15 (1Н, мультиплет); 4,32 (1Н, дублет, J 14,9 Гц); 4,40-4,48 (1Н, мультиплет); 4,57 (1Н, дублет, J 14,9 Гц); 7,47 (2Н, дублет, J 8,1 Гц); 7,68 (2Н, дублет, J 8,1 Гц); 7,99 (1Н, синглет); 8,27 (1Н, синглет).
Масс-спектр (m/z): 283 (М+), 201, 173.
П р и м е р 55. 2-(4-Фторфенил)-2-[(1Н-1,2,4-триазол-1-ил)метил]оксетан с температурой плавления 65-66оС получали с выходом 55%
ЯМР-спектр (СДСl3), δ ч./млн. 2,61-2,81 (1Н, мультиплет); 2,94-3,04 (1Н, мультиплет); 4,06-4,16 (1Н, мультиплет); 4,30 (1Н, дублет, J 14,7 Гц); 4,40-4,48 (1Н, мультиплет); 4,53 (1Н, дублет, J 14,7 Гц); 7,06-7,12 (2Н, мультиплет); 7,27-7,33 (2Н, мультиплет); 7,99 (1Н, синглет); 8,25 (1Н, синглет).
Масс-спектр (m/z): 233 (М+), 151, 123.
П р и м е р 56. 2-(4-Метоксифенил)-2-[(1Н-1,2,4-триазол-1-ил)метил]оксетан с температурой плавления 66-67оС получали с выходом 46%
ЯМР-спектр (СДСl3), δ ч./млн. 2,61-2,71 (1Н, мультиплет); 2,90-3,02 (1Н, мультиплет); 3,82 (3Н, синглет); 4,04-4,14 (1Н, мультиплет); 4,30 (1Н, дублет, J 14,7 Гц); 4,44-4,47 (1Н, мультиплет); 4,51 (1Н, дублет, J 14,7 Гц); 6,93 (2Н, дублет, J 8,86 Гц); 7,25 (2Н, дублет, J 8,86 Гц); 7,98 (1Н, синглет); 8,21 (1Н, синглет).
Масс-спектр (m/z): 245 (М+), 163, 135.
П р и м е р 57. 2-(4-Хлорметилфенил)-2-[(1Н-1,2,4-триазол-1-ил)метил] оксетан в виде масла получали с выходом 59%
ЯМР-спектр (СДСl3), δ ч./млн. 2,66-2,77 (1Н, мультиплет); 2,94-3,04 (1Н, мультиплет); 4,08-4,23 (1Н, мультиплет); 4,34 (1Н, дублет, J 14,7 Гц); 4,39-4,51 (1Н, мультиплет); 4,73 (1Н, дублет, J 14,7 Гц); 7,08-7,18 (2Н, мультиплет); 7,37-7,43 (1Н, мультиплет); 7,94 (1Н, синглет); 8,27 (1Н, синглет).
Масс-спектр (m/z): 245 (М+), 163, 135.
П р и м е р 58. 2-(4-Метилфенил)-2-[1-(1,2,4-триазол-1-ил)этил]оксетан.
Следуя методике, аналогичной описанной в примере 50, но используя 4'-метил-2-(1Н-1,2,4-триазол-1-ил)пролиофенон получали в виде масла смесь стереоизомеров (с различной конфигурацией триазольной группы) целевого соединения в примерном соотношении 2:1 (выход 49,3%).
ЯМР-спектр (СДСl3), δ ч./млн. 1,33 (дублет, J 7,25 Гц); 2,37 (синглет); 2,44-2,53 (мультиплет); 2,70-2,80 (мультиплет); 3,98-4,06 (мультиплет); 4,37-4,47 (мультиплет); 4,74 (квартет, J 7,25 Гц); 7,20 (синглет); 7,99 (синглет); 8,34 (синглет).
Масс-спектр (m/z): 243 (М+), 228, 213, 147, 119.
П р и м е р 59. 2-(2,4-Дифторфенил)-2-[(1Н-1,2,4-триазол-1-ил)метил]оксетан.
101 мг (4,2 ммоль) гидрида натрия (в виде 60% мас./мас. дисперсии в минеральном масле) добавляли к 10 мл диметилсульфоксида и полученную в результате смесь перемешивали в течение 30 мин при 80оС. После истечения этого времени смесь охлаждали, добавляли 928 мг (4,2 ммоль) иодистого триметилсульфоксония и температуре реакции давали повышаться до комнатного значения. Затем смесь перемешивали в течение 30 мин, после чего добавляли 501 мг (2,11 ммоль) 2-(2,4-дифторфенил)-2-[(1Н-1,2,4-триазол-1-ил)метил]оксирана и полученную смесь перемешивали при 50оС еще в течение 16 ч. По истечении указанного времени реакционную смесь охлаждали, переливали в 50 мл смеси воды со льдом и экстрагировали 100 мл этилацетата. Экстракт промывали насыщенным водным раствором хлористого натрия, сушили над безводным сульфатом натрия и концентрировали выпариванием при пониженном давлении. Остаток подвергали хроматографии на колонке с силикагелем, проводя элюирование смесью гексана и этилацетатом в соотношении 2:3 (по объему) с получением 238 мг (выход 45%) целевого соединения в виде масла.
Полученный продукт был полностью идентичен соединению, полученному в соответствии с методикой примера 52.
П р и м е р 60. 5-Окса-4-фенил-[(1,2,4-триазол-1-ил)метил]спиро(2.3)гексан.
33,2 мг (0,496 ммоль) гидрида натрия (в виде 60% мас./мас. дисперсии в минеральном масле) добавляли, при охлаждении системы льдом, к раствору 69,8 мг (0,207 ммоль) 1-[1-(метансульфонилоксиметил)циклопро-пан-1-ил]-1-фенил-2-(1Н- 1,2,4-триазол-1-ил)этанола в диметилформамиде и полученную в результате смесь перемешивали в течение 2 ч при комнатной температуре. По истечении этого времени реакционную смесь переливали в ледяную воду и экстрагировали этилацетатом. Экстракт промывали насыщенным водным раствором хлористого натрия, сушили над безводным сульфатом магния и концентрировали выпариванием при пониженном давлении. Полученный в результате остаток подвергали колонной хроматографии на силикагеле, элюировали смесью гексана с этилацетатом в объемном соотношении 1:1, с получением 5 мг (выход 9,9%) целевого соединения в виде масла.
ЯМР-спектр (СДСl3), δ ч./млн. 0,3-0,9 (4Н, мультиплет); 4,37 (1Н, дублет, J 5,46 Гц); 4,44 (1Н, дублет, J 14,8 Гц); 4,54 (1Н, дублет, J 5,46 Гц); 4,85 (1Н, дублет, J 14,80 Гц); 7,19-7,41 (5Н, мультиплет); 7,88 (1Н, синглет); 8,32 (1Н, синглет).
Масс-спектр (m/z): 242 (М+), 188, 172, 159.
П р и м е р 61. (2R*,3S*,4S*)-2-(4-Хлорфенил)-3,4-диметил-2-[(1Н-1,2,4-триазол-1- ил)метил]оксетан.
28 мг (1,003 ммоль) гидрида натрия (в виде 60% мас./мас. дисперсии в минеральном масле) добавляли при охлаждении льдом к раствору 200 мг (0,59 ммоль) (2R*, 3S*,4S*)-2-(4-хлорфенил)-3-метил-4-ме- тансульфонилокси-1-(1Н- 1,2,4-триазол-1-ил)пентанола в 5 мл диметилформамида и полученную в результате смесь перемешивали в течение 1,5 ч. По истечении этого времени реакционную смесь обрабатывали способом, аналогичным описанному в примере 60 с получением 32,3 мг (выход 19,7%) целевого соединения в виде масла.
ЯМР-спектр (СДСl3), δ ч./млн. 0,73 (3Н, дублет, J 7,25 Гц); 1,06 (3Н, дублет, J 6,04 Гц); 2,66 (1Н, квинтет, J 7,25 Гц); 4,26 (1Н, мультиплет); 4,31 (1Н, дублет, J 14,9 Гц); 7,30 (2Н, дублет, J 8,66 Гц); 7,40 (2Н, дублет, J 8,66 Гц); 8,01 (1Н, синглет); 8,38 (1Н, синглет).
Масс-спектр (m/z): 278 (М + 1)+, 222, 195, 139.
П р и м е р ы 62-66. С использованием методики, аналогичной описанной в примере 61, синтезировали следующие соединения.
П р и м е р 62. (2R*,3R*,4S*)-2-(4-Хлорфенил)-3,4-диметил-2-[(1Н-1,2,4-триазол-1- ил)метилоксетан с температурой плавления 75-118оС получали с выходом 8,0%
ЯМР-спектр (СДСl3), δ ч./млн. 1,39 (3Н, дублет, J 6,04 Гц); 1,42 (3Н, дублет, J 4,88 Гц); 3,14 (1Н, квинтет, J 7,66 Гц); 4,60 (1Н, дублет, J 14,5Гц); 4,74 (1Н, дублет, J 14,5 Гц); 4,92 (1Н, квинтет, J 6,54 Гц); 7,10 (2Н, дублет, J 8,46 Гц); 7,26 (2Н, дублет, J 8,46 Гц); 7,83 (1Н, синглет); 8,13 (1Н, синглет).
Масс-спектр (m/z): 277 (М+), 222, 195, 139.
П р и м е р 63. (2R*,3R*,4R*)-2-(4-Хлорфенил)-3,4-диметил-2-[(1Н-1,2,4-триазол-1- ил)метил] оксетан с температурой плавления 113-121оС получали с выходом 70%
ЯМР-спектр (СДСl3), δ ч./млн. 1,37 (3Н, дублет, J 5,64 Гц); 1,38 (3Н, дублет, J 7,65 Гц); 2,77 (1Н, квинтет, J 7,25 Гц); 4,51-4,64 (1Н, мультиплет); 4,59 (1Н, дублет, J 14,3 Гц); 4,93 (1Н, дублет, J 14,3 Гц); 7,08 (2Н, дублет, J 8,46 Гц); 7,25 (2Н, дублет, J 8,46 Гц); 7,78 (1Н, синглет); 8,05 (1Н, синглет).
Масс-спектр (m/z): 278 /(М + 1)/, 222, 195, 139.
П р и м е р 64. (2R*,3S*,4R*)-2-(4-Бромфенил)-3,4-диметил-2-[(1Н-1,2,4-триазол-1- ил)метил]оксетан в виде масла получали с выходом 62,3%
ЯМР-спектр (СДСl3), δ ч./млн. 0,70 (3Н, дублет, J 7,66 Гц); 1,16 (3Н, дублет, J 6,45 Гц); 3,16 (1Н, квинтет, J 7,66 Гц); 4,38 (1Н, дублет, J 14,5 Гц); 4,40-4,54 (1Н, мультиплет); 4,72 (1Н, дублет, J 14,5 Гц); 7,19 (2Н, дублет, J 8,66 Гц); 7,52 (2Н, дублет, J 8,66 Гц); 7,95 (1Н, синглет); 8,30 (1Н, синглет).
Масс-спектр (m/z): 322 /(М + 1)+/, 303, 266, 241, 185.
П р и м е р 65. (2R*,3S*,4R*)-2-(4-Три-фторметилфенил)-3,4-диметил-2-[(1Н-1,2,4- триазол-1-ил)метил]оксетан получали в виде масла с выходом 45%
ЯМР-спектр (СДСl3), δ ч. /млн. 0,71 (3Н, дублет, J 7,7 Гц); 1,17 (3Н, дублет, J 6,4 Гц); 3,23 (1Н, квинтет, J 7,7 Гц); 4,40 (1Н, дублет, J 14,5 Гц); 4,42-4,48 (1Н, мультиплет); 4,75 (1Н, дублет, J 14,5 Гц); 7,44 (2Н, дублет, J 8,1 Гц); 7,65 (2Н, дублет, J 8,1 Гц); 7,95 (1Н, синглет); 8,30 (1Н, синглет).
Масс-спектр (m/z): 312 /(М + 1)+/, 229, 173.
П р и м е р 66. (2R*,3R*)-2-(4-хлор-2-фторметилфенил)-3,4-диметил-2-[(1Н-1,2,4- триазол-1-ил)метил] оксетан с температурой плавления 114-116оС получали с выходом 93%
ЯМР-спектр (СДСl3), δ ч./млн. 1,30-1,41 (6Н, мультиплет); 2,72-2,84 (1Н, мультиплет); 4,54-4,8 (1Н, мультиплет); 4,65 (1Н, дублет, J 13,31 Гц); 4,95 (1Н, дублет, J 13,31 Гц); 6,98-7,38 (3Н, мультиплет); 7,72 (1Н, синглет); 8,07 (1Н, синглет).
Масс-спектр (m/z): 296 /(М + 1)+/, 256, 240, 215, 159.
П р и м е р 67. (2R*,3S*,4R*)-2-(4-Хлорфенил)-3,4-диметил-2-[(1Н-1,2,4-триазол-1- ил)метил]оксетан.
38 мг (0,950 ммоль) гидрида натрия (в виде 60% мас./мас. дисперсии в минеральном масле) добавляли при 0оС к суспензии 107 мг (0,351 ммоль) 2-(4-хлорфенил)-3,4-диметил-2-метансульфонилоксиметилоксета- на, 51,5 мг (0,746 ммоль) 1Н-1,2,4-триазола и 45,6 мг (0,304 ммоль) иодистого натрия в 10 мл диметилимидазолидинона и полученную смесь перемешивали при комнатной температуре в течение 30 мин и после этого при 90оС в течение 12 ч. После этого к полученной смеси добавляли водный раствор тиосульфата натрия и проводили экстракцию смесью этилацетата и гексана в объемном соотношении 1: 1. Экстракт промывали водой, сушили над безводным сульфатом магния и концентрировали выпариванием при пониженном давлении. Полученный остаток подвергали хроматографии на колонке с силикагелем, элюировали смесь гексана и этилацетата в объемном соотношении 1:1 с получением 76,3 мг (выход 78%) целевого соединения.
Полученное таким образом соединение было идентично соединению, полученному в примере 20.
П р и м е р 68. (2R*,3S*,4R*)-2-(4-Хлорфенил)-3,4-диметил-2-[(1Н-1,2,4-триазол-1- ил)метил]оксетан нитрат.
0,19 мл концентрированного водного раствора азотной кислоты (около 61%) добавляли к раствору 300 мг (1,08 ммоль) (2R*,3S*,4R*)-2-(4-хлорфенил)-3,4-диметил- 2-[(1Н-1,2,4-триазол-1- ил)метил]оксетана (полученного в соответствии с методикой примера 20) в смеси 5 мл диэтилового эфира и 1 мл метанола. Полученную в результате смесь отделяли от растворителя дистилляцией при пониженном давлении и полученный в результате остаток смешивали со смесью диэтилового эфира и гексана. Осажденные кристаллы собирали фильтрацией с образованием 351 мг (выход 95,4%) целевого соединения с температурой плавления 129-142оС.
Элементный анализ для C14Н17СlN4O4.
Вычислено, C 49,35; H 5,03; Cl 10,40; N 16,44.
Найдено, C 49,28; H 5,15; Cl 10,32; N 16,64.
П р и м е р 69. (2R*,3S*,4R*)-2-(4-Хлорфенил)-3,4-диметил-2-[(1Н-1,2,4-триазол-1- ил)метил]оксетан оксалата.
Раствор 97,2 мл оксалиновой кислоты в этилацетате прикапывали к раствору 300 мг (1,08 ммоль) (2R*, 3S*,4R*)-2-(4-хлорфенил)-3,4-диметил-2-[(1Н-1,2,4-триазол-1- ил)метил]оксетана (полученного согласно примеру 20) в 5 мл этилацетата и осажденные кристаллы собирали фильтрацией. После сушки получали 229 мг (выход 57,7% ) целевого соединения, с температурой плавления 135-144оС.
ИК-спектр (КBr), λмакс. см-1: 3424, 3119, 2908, 2516, 1967, 1731, 1610.
ЯМР-спектр (гексадейтерированный диметилсульфоксид), ч./млн. 0,61 (3Н, дублет, J 7,25 Гц); 1,09 (3Н, дублет, J 6,45 Гц); 3,18 (1Н, квинтет, J 7,65 Гц); 3,37 (широкий синглет); 4,51 (1Н, триплет, J 7,25 Гц); 4,60 (1Н, дублет, J 14,5 Гц); 4,89 (1Н, дублет, J 14,5 Гц); 7,34 (2Н, дублет, J 8,86 Гц); 7,41 (1Н, дублет, J 8,86 Гц); 7,88 (1Н, синглет); 8,34 (1Н, синглет).
П р и м е р 70. 2-(2,4-Дихлорфенил)-3,3,4-триметил-2-[(1Н-1,2,4-триазол-1-ил)ме- тил] оксетан.
84 мг (2,1 ммоль) гидрида натрия (в виде 60% мас./мас. суспензии в минеральном масле) добавляли к раствору 400 мг (0,95 ммоль) 2-(2,4-дихлорфенил)-3,3-диметил-4-метилсульфонилокси-1-[(1Н-1,2,4- триазол-1-ил]-2-пентанола в 5 мл N,N-диметилформамида, и затем смесь перемешивали при 90оС в течение 2 ч. После этого реакционную смесь отстаивали до охлаждения и затем влили в воду со льдом и экстрагировали этилацетатом. Экстракт промыли водой и сушили над безводным сульфатом натрия, после чего растворитель удаляли дистилляцией при пониженном давлении. Остаток промывали гексаном с получением 252,4 мг неочищенного продукта, который затем очищали хроматографией на колонке с силикагелем, элюируя смесью 1:1 по объему гексана и этилацетата, с получением 220,7 мг (выход 71,4% ) целевого соединения, с температурой плавления 118-125оС. Это соединение является стереоизомером, изомерным в положении С4 соединения, полученного в примере 42.
Масс-спектр (m/z): 328, 325, 290, 256, 243, 173.
Масс-спектр (СДСl3), δ ч./млн. 1,06 (3Н, синглет); 1,42 (3Н, синглет); 1,49 (3Н, дублет, J 6,3 Гц); 4,36 (1Н, квардруплет, J 6,3 Гц); 4,83 (1Н, дублет, J 14,2 Гц); 5,36 (1Н, дублет, J 14,2 Гц); 7,10 (1Н, двойной дублет, J 2,2 и 8,4 Гц); 7,28 (1Н, дублет, J 8,4 Гц); 7,35 (1Н, дублет, J 2,2 Гц); 7,63 (1Н, синглет); 7,99 (1Н, синглет).
П р и м е р 71. (2R*,3S*,4R*)-2-(4-Хлорфенил)-3,4-диметил-[(1Н-1,2,4-триазол-1-ил)- метил]оксетан.
25 мг (0,38 ммоль) 1 -1,2,4-триазола и 25 мг (0,19 ммоль) карбоната калия добавляли к раствору 245 мг (1 ммоль) 2-хлорметил-2-(4-хлорфенил)-3,4-диметилоксетана (смесь 3:1 изомеров 4
-1,2,4-триазола и 25 мг (0,19 ммоль) карбоната калия добавляли к раствору 245 мг (1 ммоль) 2-хлорметил-2-(4-хлорфенил)-3,4-диметилоксетана (смесь 3:1 изомеров 4 * и 4
* и 4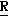 *) и затем смесь перемешивали при 130оС в течение 5 ч. После этого реакционную смесь отстаивали до охлаждения и затем влили в воду со льдом и экстрагировали этилацетатом. Экстракт промывали насыщенным водным раствором хлористого аммония и сушили над безводным сульфатом натрия, после чего растворитель удаляли дистилляцией при пониженном давлении с получением 260 мг остатка. Затем остаток очищали хроматографией на колонке с силикагелем с получением 56,2 мг (выход 81%) целевого соединения с температурой плавления 111-118оС.
*) и затем смесь перемешивали при 130оС в течение 5 ч. После этого реакционную смесь отстаивали до охлаждения и затем влили в воду со льдом и экстрагировали этилацетатом. Экстракт промывали насыщенным водным раствором хлористого аммония и сушили над безводным сульфатом натрия, после чего растворитель удаляли дистилляцией при пониженном давлении с получением 260 мг остатка. Затем остаток очищали хроматографией на колонке с силикагелем с получением 56,2 мг (выход 81%) целевого соединения с температурой плавления 111-118оС.
Масс-спектр (m/z): 278 /(М+ 1)+/, 222, 195, 141.
П р и м е р ы 72-75. По методике примера 4 были получены следующие соединения.
П р и м е р 72. (2R*,3S*,4S*)-2-(2,4-Ди-фторфенил)-4-изопропил-3-метил-2-[(1Н-1,2,4- триазол-1-ил)метил]оксетан кислый оксалат, с температурой плавления 140-145оС, выход 5%
ЯМР-спектр (СДСl3), δ ч. /млн. 0,69 (3Н, дублет, J 7 Гц); 0,88 (3Н, дублет дублетов, J 2,5 и 7 Гц); 0,90 (3Н, дублет, J 7 Гц); 1,25 (1Н, мультиплет); 2,79 (1Н, мультиплет); 3,61 (1Н, дублет дублетов, J 7,5 и 9,5 Гц); 4,40 (1Н, дублет, J 14,5 Гц); 4,78 (1Н, дублет, J 14,5 Гц); 6,8-7,0 (2Н, мультиплет); 7,60 (1Н, триплет дублетов, J 7 и 9 Гц); 7,87 (1Н, синглет); 8,19 (1Н, синглет).
П р и м е р 73. (2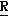 *, 3
*, 3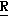 *, 4
*, 4 *)-2-(2,4-дифторфенил)-4-изопропил-3-метил-2-[(1Н- 1,2,4-триазол-1-ил)метил] оксетан кислый оксалат с температурой плавления 151-154оС, выход 5%
*)-2-(2,4-дифторфенил)-4-изопропил-3-метил-2-[(1Н- 1,2,4-триазол-1-ил)метил] оксетан кислый оксалат с температурой плавления 151-154оС, выход 5%
ЯМР-спектр (СДСl3), δ ч./млн. 0,81 (3Н, дублет, J 6,5 Гц); 1,02 (3Н, дублет, J 6,5 Гц); 1,47 (3Н, дублет, J 6,5 Гц); 2,29 (1Н, мультиплет); 3,19 (1Н, мультиплет); 4,17 (1Н, дублет дублетов, J 6,5 и 10,5 Гц); 4,65 (1Н, дублет, J 13 Гц); 4,42 (1Н, дублет, J 13 Гц); 6,7-7,2 (3Н, мультиплет); 7,52 (1Н, синглет); 8,13 (1Н, синглет).
П р и м е р 74. (2R*,3R*,4R*)-2-(2,4-Дифторфенил)-4-изопропил-3-метил-2-[(1Н- 1,2,4-триазол-1-ил)метил]оксетан в виде масла с выходом 35%
ЯМР-спектр (СДСl3), δ ч. /млн. 0,77 (3Н, дублет, J 7 Гц); 0,93 (3Н, дублет, J 7 Гц); 1,40 (3Н, дублет дублетов, J 1 и 7,5 Гц); 1,76 (1Н, мультиплет); 2,84 (1Н, мультиплет); 4,09 (1Н, дублет дублетов, J 7 и 8 Гц); 4,67 (1Н, дублет дублетов, J 1,5 и 14 Гц); 4,90 (1Н, дублет, J 14 Гц); 6,6-6,8 (2Н, мультиплет); 7,07 (1Н, триплет дублетов, J 6,5 и 8,5 Гц); 7,66 (1Н, синглет); 8,03 (1Н, синглет).
П р и м е р 75. (2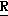 *,3
*,3 *,4
*,4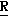 *)-3,4-диметил-2-фенил-2-[(1Н-1,2,4-триазол-1-ил)ме- тил] оксетан в виде масла, выход 27%
*)-3,4-диметил-2-фенил-2-[(1Н-1,2,4-триазол-1-ил)ме- тил] оксетан в виде масла, выход 27%
ЯМР-спектр (СДСl3), δ ч. /млн. 0,61 (3Н, дублет, J 7,4 Гц); 1,08 (3Н, дублет, J6,5 Гц); 3,09 (1Н, мультиплет); 4,28 (1Н, дублет, J 14,7 Гц); 4,32 (1Н, мультиплет); 4,67 (1Н, дублет, J 14,7 Гц); 7,21-7,34 (5Н, мультиплет); 7,85 (1Н, синглет); 8,15 (1Н, синглет).
Масс-спектр (m/z): 244 (М+1)+, 227, 188, 161.
П р и м е р 76. (2R*,3S*,4R*)-2-(4-фторфенил)-3,4-диметил-2-[(1Н-1,2,4-триазол-1- ил)метил]оксетан.
Следуя методике, аналогичной той, которая описана в примере 71, получали 110 мг (выход 38,3%) целевого соединения в виде масла.
Спектр ядерно-магнитного резонанса (СДСl3), δ млн.дол.
0,70 (3Н, дублет, J 7,65 Гц);
1,17 (3Н, дублет, J 6,44 Гц);
3,15 (1Н, квинтет, J 7,56 Гц);
4,38 (1Н, дублет, J 14,51 Гц);
4,39-4,52 (1Н, мультиплет);
4,73 (1Н, дублет, J 14,51 Гц);
7,08 (2Н, триплет, J 8,66 Гц);
7,22-7,32 (2Н, мультиплет);
7,95 (1Н, синглет);
8,30 (1Н, синглет).
Масс-спектр (m/z): 262 (М+1)+, 244, 218, 206, 179.
П р е п а р а т и в н ы й п р и м е р 1. (3S*,4R*)-4-(2,4-Дифторфенил)-3-метил-5- (1Н-1,2,4-триазол-1-ил)- 1,4-пентандиол и (2R*,3S*)-2-(2,4-дифторфенил)-3-метил-[1- (1Н- 1,2,4-триазол-1-ил]-2,4-пентандиол.
2,2 мл (4,4 ммоль) 2М раствора борандиметилсульфидного комплекса в тетрагидрофуране добавили в условиях охлаждения льдом и при перемешивании к раствору 200 мг (0,72 ммоль) (2R*,3S*)-2-(2,4-дифторфенил)-3-метил-1-(1Н-1,2,4-триазол-1-ил)- 4-пентен-2-ола (который был синтезирован по методике, описанной в Японской Патентной предварительной публикации N Sho 60-36468) в 5 мл тетрагидрофурана. Смеси позволяли нагреваться до комнатной температуры, после чего ее перемешивали при этой температуре в течение 15 мин и затем при 50оС в течение 20 мин. После этого смесь снова охладили водой со льдом и к смеси добавили 1 мл 15 вес./объем-ного водного раствора гидроокиси натрия и 1 мл 30 вес./объем-ного водного раствора перекиси натрия. Затем реакционную смесь перемешивали при комнатной температуре в течение 30 мин и затем при 50-60оС в течение 2 ч, после чего смесь разбавили этилацетатом и затем промыли насыщенным водным раствором хлорида натрия. После сушки смесь освободили от растворителя дистилляцией при пониженном давлении. Получающийся маслообразный остаток подвергали хроматографии на колонке с силикагелем с использованием смеси этилацетата и гексана в диапазоне от 5:1 до 10:1 по объему в качестве элюента, с получением 30 мг второго указанного целевого соединения в виде масла. ЯМР-спектры показали структуру соединения, которое представляет собой смесь 1:1 изомеров А и В в С4-положении.
Изомер А (основные пики).
ЯМР-спектр (СДСl3), δ ч./млн. 0,80 (3Н, дублет дублетов, J 7 и 3,5 Гц); 1,18 (3Н, дублет, J 6 Гц); 4,90 (2Н, синглет); 5,97 (1Н, синглет); 7,79 (1Н, синглет); 8,05 (1Н, синглет).
Изомер В (основные пики).
ЯМР-спектр (СДСl3), δ ч./млн. 0,76 (3Н, дублет дублетов, J 7 и 1,5 Гц); 1,27 (3Н, дублет, J 6 Гц); 5,47 (1Н, синглет); 7,70 (1Н, синглет); 7,93 (1Н, синглет).
Затем колонку элюировали последовательно этилацетатом и 1 об. метанола в этилацетате с получением 139 мг первого указанного целевого соединения, которое перекристаллизовывали из смеси этилацетата и гексана с получением чистого образца с температурой плавления 121- 122оС.
ЯМР-спектр (СДСl3), δ ч./млн. 0,78 (3Н, дублет дублетов, J 7 и 1 Гц); 1,6-2,0 (2Н, мультиплет); 2,4 (1Н, мультиплет); 3,3-4,0 (3Н, мультиплет); 4,62 (1Н, дублет, J 14 Гц); 4,93 (1Н, дублет дублетов, J 14 и 1 Гц); 5,52 (1Н, широкий пик); 6,5-7,0 (2Н, мультиплет); 7,43 (1Н, триплет дублетов, J 9 и 7 Гц); 7,73 (1Н, синглет); 7,97 (1Н, синглет).
П р е п а р а т и в н ы й п р и м е р 2. (2R*,3S*, 4S*)-2-(2,4-Дифторфенил)-4-метансульфо-нилокси)-3-метил- 1-(1Н-1,2,4-триазол-1-ил)-2-пентанол.
2(а) (2R*, 3S*,4S*)-2-(2,4-дифторфенил)-4,5-эпокси-3-метил-1-(1Н-1,2,4- триазол-1-ил)-2-пентанол
1,360 г (6,19 ммоль) 3-хлорпероксибензойной кислоты (чистота 80%) добавляли при 0оС к раствору 960 мг (3,44 ммоль) (2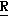 *,3
*,3 *)-2-(2,4-дифторфенил)-3-метил-1- (1
*)-2-(2,4-дифторфенил)-3-метил-1- (1 -1,2,4-триазол-1-ил)- 4-пентен-2-ол (который был синтезирован по методике, описанной в Японской Патентной предварительной заявке N Sho 60-36468) в 30 мл метиленхлорида. Через 5 мин после добавления реакционную смесь перемешивали и перемешивание продолжали при комнатной температуре в течение ночи. Затем реакционную смесь разбавили этилацетатом, после чего промыли последовательно водным раствором сульфита натрия, водным раствором бикарбоната натрия и насыщенным водным раствором хлорида натрия. После сушки смесь освобождали от растворителя дистилляцией при пониженном давлении. Получающийся остаток подвергали колоночной хроматографии на силикагеле с использованием 1:2 по объему смеси этилацетата и гексана в качестве элюента с получением 320 мг стереоизомера А целевого соединения (имеющего пониженную полярностью с температурой плавления 160-180оС и 206 мг целевого стереоизомера В требуемого соединения (имеющего повышенную полярность) в виде масла.
-1,2,4-триазол-1-ил)- 4-пентен-2-ол (который был синтезирован по методике, описанной в Японской Патентной предварительной заявке N Sho 60-36468) в 30 мл метиленхлорида. Через 5 мин после добавления реакционную смесь перемешивали и перемешивание продолжали при комнатной температуре в течение ночи. Затем реакционную смесь разбавили этилацетатом, после чего промыли последовательно водным раствором сульфита натрия, водным раствором бикарбоната натрия и насыщенным водным раствором хлорида натрия. После сушки смесь освобождали от растворителя дистилляцией при пониженном давлении. Получающийся остаток подвергали колоночной хроматографии на силикагеле с использованием 1:2 по объему смеси этилацетата и гексана в качестве элюента с получением 320 мг стереоизомера А целевого соединения (имеющего пониженную полярностью с температурой плавления 160-180оС и 206 мг целевого стереоизомера В требуемого соединения (имеющего повышенную полярность) в виде масла.
Стереоизомер А (2R*,3S*,4S*):
ЯМР-спектр (СДСl3), δ ч. /млн. 0,82 (3Н, дублет, J 7,3 Гц); 1,74 (1Н, квинтет, J 7,3 Гц); 2,54 (1Н, дублет дублетов, J 4,0 и 2,8 Гц); 2,89 (1Н, триплет, J 4,4 Гц); 3,3-3,4 (1Н, мультиплет); 4,90 (2Н, АВ-дублет, J 14,5 Гц); 5,06 (1Н, широкий синглет); 6,6-6,8 (2Н, мультиплет); 7,3-7,5 (1Н, мультиплет); 7,79 (1Н, синглет); 7,80 (1Н, синглет).
Стереоизомер В: (2R*,3S*,4R*):
ЯМР-спектр (СДСl3), δ ч. /млн. 0,94 (3Н, дублет, J 6,9 Гц); 1,86 (1Н, квинтет, J 6,9 Гц); 2,74 (1Н, дублет дублетов, J 4,8 и 2,8 Гц); 2,95 (1Н, триплет, J 4,8 Гц); 3,2-3,3 (1Н, мультиплет); 4,80 (2Н, АВ-дублет, J 13,7 Гц); 4,92 (1Н, синглет); 7,6-7,8 (2Н, мультиплет); 7,3-7,5 (1Н, мультиплет); 7,78 (1Н, синглет); 7,86 (1Н, синглет).
2(b) (2R*, 3S*, 4S*)-2-(2,4-дифторфенил)-3-метил-1-(1Н-1,2,4-триазол-1- ил)-2,4-пентандиол.
84 мг (2,16 ммоль) литийалюминийгидрида добавили в атмосфере азота к раствору 320 мг (1,08 ммоль) (2R*,3S*,4S*)-2-(2,4-дифторфенил)-4,5-эпокси-3-метил-1-(1Н-1,2,4- триазол-1-ил)-2-пентанола (стереоизомер А), полученного, как описано в стадии (а) выше, в 20 мл диэтилового эфира в условиях охлаждения льдом и при перемешивании. Через 10 мин реакционную смесь кипятили с обратным холодильником и это продолжали в течение 1 ч. В конце этого времени смесь охлаждали и медленно добавили 2 мл воды; затем смесь перемешивали в течение 10 мин. Нерастворенные вещества реакционной смеси удаляли фильтрованием с помощью целитного фильтра и затем остаток промыли этилацетатом. Объединенные фильтрат и промывные воды сушили и удаляли растворитель дистилляцией при пониженном давлении. Получающийся маслообразный остаток подвергали хроматографии на колонке с силикагелем с использованием 5:5:1 по объему смеси этилацетата, хлороформа и гексана в качестве элюента с получением 240 мг целевого соединения.
ЯМР-спектр (СДСl3), δ ч. /млн. 0,80 (3Н, дублет дублетов, J 6,9 и 3,0 Гц); 2,00 (1Н, квартет дублетов, J 6,9 и 1,6 Гц); 3,86 (1Н, квартет дублетов, J 6,9 и 1,6 Гц); 4,62 (1Н, дублет, J 14 Гц); 4,93 (2Н, синглет); 6,7-6,9 (2Н, мультиплет); 7,50 (1Н, триплет дублетов, J 8,9 и 6,5 Гц); 7,82 (1Н, синглет); 8,13 (1Н, синглет).
2(с) (2R*, 3S*, 4S*)-2-(2,4-дифторфенил)-4-метансульфонилокси)-3-метил- 1-(1Н-1,2,4-триазол-1-ил)-2-пентанол
140 мг (1,22 ммоль) метансульфонилхлорида добавили при 0оС к раствору 213 мг (0,72 ммоль) (2R*,3S*,4S*)-2-(2,4-дифторфенил)-3-метил-1-(1Н-1,2,4-триазол-1- ил)-2,4- (0,72 ммоль) (2R*, 3S*, 4S*)-2-(2,4-дифторфенил)-3-метил-1-(1 -1,2,4-триазол-1-ил)-2,4- пентандиол (полученного как описано в стадии (b) выше) в 4 мл пиридина, и смесь перемешивали в течение 2,5 ч. После этого пиридин удаляли дистилляцией при пониженном давлении. Получающийся остаток смешивали с разбавленным водным раствором бикарбоната натрия и экстрагировали этилацетатом. Экстракт сушили над безводным сульфатом натрия и удаляли растворитель выпариванием при пониженном давлении с получением 270 мг целевого соединения в виде масла.
-1,2,4-триазол-1-ил)-2,4- пентандиол (полученного как описано в стадии (b) выше) в 4 мл пиридина, и смесь перемешивали в течение 2,5 ч. После этого пиридин удаляли дистилляцией при пониженном давлении. Получающийся остаток смешивали с разбавленным водным раствором бикарбоната натрия и экстрагировали этилацетатом. Экстракт сушили над безводным сульфатом натрия и удаляли растворитель выпариванием при пониженном давлении с получением 270 мг целевого соединения в виде масла.
ЯМР-спектр (СДСl3), δ ч. /млн. 0,79 (3Н, дублет дублетов, J 6,5 и 0,8 Гц); 1,49 (3Н, дублет, J 6,5 Гц); 2,6-2,8 (1Н, мультиплет); 3,08 (3Н, синглет); 4,85 (2Н, АВ-дублет, J 13,9 Гц); 5,3-5,4 (1Н, мультиплет); 6,6-6,8 (2Н, мультиплет); 7,2-7,4 (1Н, мультиплет); 7,76 (1Н, синглет); 7,81 (1Н, синглет).
П р е п а р а т и в н ы й п р и м е р 3. (2R*,3R*)-2-(2,4-дифторфенил)-4,5-эпокси-3- метил-1-(1Н-1,2,4-триазол-1-ил)-2-пентанол.
635 мг (3,13 ммоль) 3-хлорпероксибензойной кислоты (чистота 85%) добавили к раствору 514 мг (1,84 ммоль) (2R*,3R*)-2-(2,4-дифторфенил)-3-метил-1-(1 -1,2,4-триазол- 1-ил)- 4-пентен-2-ола (который был приготовлен по методике, описанной в Японской Патентной предварительной публикации N Sho 60-36468) в 15 мл метиленхлорида. Через 5 мин температуре реакционной смеси позволяли увеличиться до комнатной температуры и смесь перемешивали 2 ч. После этого реакционную смесь разбавили этилацетатом и промыли последовательно водным раствором сульфита натрия, водным раствором бикарбоната натрия и насыщенным водным раствором хлорида натрия. Затем органический слой сушили над безводным сульфатом натрия и растворитель удаляли дистилляцией при пониженном давлении. Получающийся остаток подвергали хроматографии на колонке с силикагелем с использованием 2:1 по объему смеси этилацетата и гексана в качестве элюента, с получением 472 мг целевого соединения в виде твердого вещества.
-1,2,4-триазол- 1-ил)- 4-пентен-2-ола (который был приготовлен по методике, описанной в Японской Патентной предварительной публикации N Sho 60-36468) в 15 мл метиленхлорида. Через 5 мин температуре реакционной смеси позволяли увеличиться до комнатной температуры и смесь перемешивали 2 ч. После этого реакционную смесь разбавили этилацетатом и промыли последовательно водным раствором сульфита натрия, водным раствором бикарбоната натрия и насыщенным водным раствором хлорида натрия. Затем органический слой сушили над безводным сульфатом натрия и растворитель удаляли дистилляцией при пониженном давлении. Получающийся остаток подвергали хроматографии на колонке с силикагелем с использованием 2:1 по объему смеси этилацетата и гексана в качестве элюента, с получением 472 мг целевого соединения в виде твердого вещества.
Продукт был определен как смесь 3:1 двух стереоизомеров по отношению к атому углерода в положении С4.
Перекристаллизация из смеси этилацетата и гексана привела к получению основного изомера с температурой плавления 106-109оС.
ЯМР-спектр (СДСl3), δ ч./млн. 1,30 (3Н, дублет, J 6 Гц); 1,90 (1Н, широкий квинтет); 2,00 (1Н, дублет дублетов, J 4 и 3 Гц); 2,35 (1Н, триплет, J 4 Гц); 2,85 (1Н, мультиплет); 4,53 (1Н, дублет, J 14 Гц); 4,89 (1Н, дублет дублетов, J 14 и 1,5 Гц); 4,9 (1Н, широкий пик); 6,5-7,0 (2Н, мультиплет); 7,3-7,7 (1Н, мультиплет); 7,79 (1Н, синглет); 7,91 (1Н, синглет).
П р е п а р а т и в н ы й п р и м е р 4. (2R*,3R*, 4S*)-2-(2,4-дифторфенил)-3-метил-1-(1Н-1,2,4-триазол-1- ил)-2,4-пентандиол.
53 мг (1,40 ммоль) литийалюминий гидрида добавили в атмосфере азота к раствору 207 мг (0,70 ммоль) (2R*,3R*)-2-(2,4-дифторфенил)-4,5-эпокси-3-метил-1-(1Н-1,2,4- триазол-1-ил)-2-пентанола (примерно 3:1 смесь изомеров в положении С4, полученная, как описано в препаративном примере 3) в 4 мл диэтилового эфира при охлаждении льдом и перемешивали. Через 10 мин реакционную смесь нагревали и кипятили с обратным холодильником и это продолжали в течение 1 ч. После этого смесь охладили и медленно добавили 1 мл воды, после чего смесь перемешивали 10 мин. Нерастворимые вещества удалили фильтрованием с помощью целитного фильтра, после чего остаток промыли этилацетатом. Объединенные фильтрат и промывные жидкости сушили над безводным сульфатом натрия и растворитель удаляли дистилляцией при пониженном давлении. Получающийся маслообразный остаток подвергали хроматографии на колонке с силикагелем с использованием 5:5:1 по объему смеси этилацетата, хлороформа и гексана в качестве элюента, с получением 160 мг целевого соединения.
Этот продукт был определен как смесь 3:1 двух стереоизомеров по отношению к углеродному атому в положении С4.
Перекристаллизация из смеси бензола и гексана привела к получению основного изомера с температурой плавления 145-146оС.
ЯМР-спектр (СДСl3), δ ч./млн. 1,05 (3Н, дублет, J 6,5 Гц); 1,25 (3Н, дублет, J 6,5 Гц); 2,20 (1Н, квартет дублетов, J 6,5 и 1 Гц); 3,03 (1Н, широкий синглет); 3,74 (1Н, широкий квартет, J 6,5 Гц); 4,51 (1Н, дублет, J 14 Гц); 4,77 (1Н, дублет дублетов, J 14,1 Гц); 5,33 (1Н, синглет); 6,5-7,0 (2Н, мультиплет); 7,1-7,6 (1Н, мультиплет); 7,65 (1Н, синглет); 7,89 (1Н, синглет).
П р е п а р а т и в н ы й п р и м е р 5. (2R*,3R*)-3-(2,4-дихлорфенил)-3-гидрокси-2- метил-(1Н- 1,2,4- триазол-1-ил)-бутаналь и (2S*,3R*)-3-(2,4-дихлорфенил)-3-гидрокси-2- метил-4-(1Н-1,2,4- триазол-1-ил)бутаналь.
290 мг (1,32 ммоль) метилпериодата натрия и 1 мг тетраокиси осмия добавили к раствору 139 мг (0,45 ммоль) смеси 1:1 (2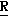 *,3
*,3 *)-4-(2,4-дихлорфенил)-3-метил-1- (1
*)-4-(2,4-дихлорфенил)-3-метил-1- (1 -1,2,4-триазол-1-ил)-4- пентен-2-ола и его (2
-1,2,4-триазол-1-ил)-4- пентен-2-ола и его (2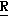 *,3
*,3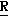 *)-изомера (который был синтезирован по методике, описанной в Японской патентной предварительной публикации N Sho 60-36468) в 2,8 мл объемной смеси 5:2 тетрагидрофурана и воды, и смесь перемешивали в течение ночи при комнатной температуре. После этого реакционную смесь разбавили этилацетатом и промыли насыщенным водным раствором хлорида натрия. Органический слой сушили над безводным сульфатом натрия и растворитель удаляли дистилляцией при пониженном давлении. Получающийся маслообразный остаток подвергали хроматографии на колонке с 4 г силикагеля с использованием смеси 4:5 по объему этилацетата и гексана в качестве элюента с получением 27 мг (2
*)-изомера (который был синтезирован по методике, описанной в Японской патентной предварительной публикации N Sho 60-36468) в 2,8 мл объемной смеси 5:2 тетрагидрофурана и воды, и смесь перемешивали в течение ночи при комнатной температуре. После этого реакционную смесь разбавили этилацетатом и промыли насыщенным водным раствором хлорида натрия. Органический слой сушили над безводным сульфатом натрия и растворитель удаляли дистилляцией при пониженном давлении. Получающийся маслообразный остаток подвергали хроматографии на колонке с 4 г силикагеля с использованием смеси 4:5 по объему этилацетата и гексана в качестве элюента с получением 27 мг (2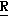 *,3
*,3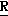 *)-изомера, 25 мг смеси (2
*)-изомера, 25 мг смеси (2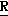 *, 3
*, 3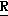 *)-изомера и (2
*)-изомера и (2 *,3
*,3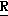 *)-изомера и 31 мг (2
*)-изомера и 31 мг (2 *,3
*,3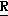 *)-изомера в указанной последовательности.
*)-изомера в указанной последовательности.
Перекристаллизация (2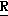 *,3
*,3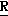 *)-изомера из бензола привела к получению чистого образца с температурой плавления 150-151оС.
*)-изомера из бензола привела к получению чистого образца с температурой плавления 150-151оС.
Перекристаллизация (2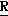 *, 3
*, 3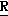 *)-изомера из смеси бензола и этилацетата привела к получению чистого образца с температурой плавления 155-157оС.
*)-изомера из смеси бензола и этилацетата привела к получению чистого образца с температурой плавления 155-157оС.
(2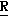 *,3
*,3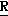 *)-изомер:
*)-изомер:
ИК-спектр поглощения (СHСl3), νмакс. см-1: 3400, 1715.
ЯМР-спектр (СДСl3) δ ч./млн. 0,96 (3Н, дублет, J 7 Гц); 3,47 (1Н, квартет дублетов, J 7,3 Гц); 4,64 (1Н, дублет, J 14 Гц); 5,30 (1Н, широкий пик); 5,42 (1Н, дублет, J 14 Гц); 7,11 (1Н, дублет дублетов, J 8,2 Гц); 7,31 (1Н, дублет, J 2 Гц); 7,52 (1Н, дублет, J 8 Гц); 7,7 (1Н, синглет); 7,85 (1Н, синглет); 9,88 (1Н, дублет, J 3 Гц).
(2S*,3R*)-изомер:
ЯМР-спектр (СДСl3) δ ч./млн. 1,40 (3Н, дублет, J 7 Гц); 3,52 (1Н, квартет дублетов, J 7, 1,5 Гц); 4,50 (1Н, дублет, J 14 Гц); 5,42 (1Н, дублет, J 14 Гц); 5,5 (1Н, широкий пик); 7,11 (1Н, дублет дублетов, J 8,2 Гц); 7,35 (1Н, дублет, J 2 Гц); 7,54 (1Н, дублет, J 8 Гц); 7,77 (1Н, синглет); 7,86 (1Н, синглет); 9,39 (1Н, дублет, J 1,5 Гц).
П р е п а р а т и в н ы й п р и м е р 6. (2S*,3R*)-3-(2,4-дихлорфенил)-2-метил-4- (1Н-1,2,4-триазол-1-ил)-1,3-бутандиол и (2R*,3R*)-3-(2,4-дихлорфенил)-2-метил-4- (1Н-1,2,4-триазол-1-ил)- 1,3-бутандиол.
15 мг боргидрида натрия добавили в условиях охлаждения льдом и при перемешивании к раствору 85 мг смеси 1:1 (2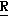 *, 3R*)-3-(2,4-дихлорфенил)-3-гидрокси-2-ме- тил-4-(1
*, 3R*)-3-(2,4-дихлорфенил)-3-гидрокси-2-ме- тил-4-(1 -1,2,4- триазол-1-ил)бутаналя и его (2
-1,2,4- триазол-1-ил)бутаналя и его (2 *,3
*,3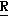 *)-изомера (полученных, как описано в препаративном примере 5) в 1,5 мл метанола. Через 10 мин реакционную смесь разбавили этилацетатом, после чего смесь промыли насыщенным водным раствором хлорида натрия. Органический слой дистиллировали для удаления растворителя при пониженном давлении с получением неочищенного продукта. Этот продукт подвергали хроматографии на колонке с 3 г силикагеля с использованием этилацетата в качестве элюента с получением 31 мг (2
*)-изомера (полученных, как описано в препаративном примере 5) в 1,5 мл метанола. Через 10 мин реакционную смесь разбавили этилацетатом, после чего смесь промыли насыщенным водным раствором хлорида натрия. Органический слой дистиллировали для удаления растворителя при пониженном давлении с получением неочищенного продукта. Этот продукт подвергали хроматографии на колонке с 3 г силикагеля с использованием этилацетата в качестве элюента с получением 31 мг (2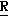 *,3
*,3 *)-изомера целевого соединения.
*)-изомера целевого соединения.
Его перекристаллизация из смеси бензола и гексана привела к получению чистого образца с температурой плавления 120-122оС.
Затем колонку элюировали этилацетатом, содержащим 7 об. метанола с получением 33 мг (2 *,3
*,3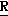 *)-изомера целевого соединения.
*)-изомера целевого соединения.
Его перекристаллизация из смеси бензола и гексана привела к получению чистого образца с температурой плавления 176-177оС.
(2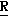 *,3
*,3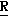 *)-изомер:
*)-изомер:
ЯМР-спектр (СДСl3) δ ч. /млн. 1,39 (3Н, дублет, J 7 Гц); 2,85 (1Н, мультиплет); 3,40 (1Н, синглет); 3,45 (1Н, синглет); 4,50 (1Н, дублет, J 14 Гц); 5,31 (1Н, дублет, J 14 Гц); 7,05 (1Н, дублет дублетов, J 8 и 2 Гц); 7,25 (1Н, дублет, J 2 Гц); 7,52 (1Н, дублет, J 8 Гц); 7,66 (1Н, синглет); 7,91 (1Н, синглет).
(2 *,3
*,3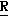 *)-изомер:
*)-изомер:
ИК-спектр (Нуйол) νмакс см-1: 3400, 3140.
ЯМР-спектр (СДСl3: С3О) 1:1 по объему) δ ч./млн. 0,77 (3Н, дублет, J 7 Гц); 2,9 (1Н, мультиплет); 3,6-4,3 (2Н, мультиплет); 4,74 (1Н, дублет, J 14,5 Гц); 5,44 (1Н, дублет, J 14,5 Гц); 7,04 (1Н, дублет дублетов, J9,2 Гц); 7,30 (1Н, дублет, J 2 Гц); 7,48 (1Н, дублет, J 9 Гц); 7,67 (1Н, синглет); 8,07 (1Н, синглет).
П р е п а р а т и в н ы й п р и м е р 7. (2R*,3S*)-2-(2,4-дихлорфенил)-4-метансуль- фонилокси)-3-метил-1-(1Н- 1,2,4-триазол-1-ил)-2-бутанол.
61 мг (0,61 ммоль) триэтиламина и 64 мг (0,56 ммоль) метансульфонилхлорида добавили при 0оС при перемешивании к раствоpу 74 мг (0,231 ммоль) (2 *, 3
*, 3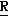 *)-3- (2,4-дихлорфенил)-2-метил-4-(1
*)-3- (2,4-дихлорфенил)-2-метил-4-(1 -1,2,4-триа- зол-1-ил)- 1,3-бутандиола в 2 мл метиленхлорида. Через 15 мин реакционную смесь смешали с разбавленным водным раствором бикарбоната натрия и экстрагировали этилацетатом. Экстракт промыли насыщенным водным раствором хлорида натрия и растворитель удалили выпариванием при пониженном давлении, с получением 92 мг неочищенного продукта.
-1,2,4-триа- зол-1-ил)- 1,3-бутандиола в 2 мл метиленхлорида. Через 15 мин реакционную смесь смешали с разбавленным водным раствором бикарбоната натрия и экстрагировали этилацетатом. Экстракт промыли насыщенным водным раствором хлорида натрия и растворитель удалили выпариванием при пониженном давлении, с получением 92 мг неочищенного продукта.
ЯМР-спектр (СДСl3), δ ч./млн. 0,70 (3Н, дублет, J 7 Гц); 3,07 (3Н, синглет); 2,9-3,4 (1Н, мультиплет); 4,22 (1Н, дублет дублетов, J 10 и 5 Гц); 4,59 (1Н, дублет, J 14,5 Гц); 4,71 (1Н, дублет дублетов; J 10 и 7 Гц); 5,18 (1Н, широкий пик); 5,51 (1Н, дублет, J 14,5 Гц); 7,04 (1Н, дублет дублетов, J 8 и 2 Гц); 7,27 (1Н, дублет, J 2 Гц); 7,43 (1Н, дублет, J 8 Гц); 7,73 (1Н, синглет); 7,81 (1Н, синглет).
П р е п а р а т и в н ы й п р и м е р 8. (2R*,4R*)-2-(4-Хлорфенил)-2-этоксикарбо- нил-3,4-диметилоксетан.
2-Бутен барботировали в 200 мл бензола при 0оС до тех пор, пока раствор бензола не увеличился до 1,25 раза от его первоначального объема. Затем к смеси добавили 24,3 г (114,09 ммоль) этил-4-хлорфенилглиоксалата и полученную смесь освещали ртутной лампой среднего давления мощностью 450 Вт (Hahnovea Co. Inc.) при 15оС в течение 3 ч. После этого реакционную смесь концентрировали выпариванием при пониженном давлении и полученный остаток подвергали хроматографии на колонке с силикагелем, элюируя смесью 10:1 по объему гексана и этилацетата с получением 23,4 г (выход 76%) целевого соединения с температурой кипения 141- 142оС/2,7 мм рт.ст.
ЯМР-спектр (СДСl3) δ ч. /млн. 0,76 (дублет, J 7,66 Гц); 1,23-1,31 (мультиплет); 1,34 (дублет, J 6,04 Гц); 2,83 (квинтет, J 7,25 Гц); 3,55 (квинтет, J 7,25 Гц); 4,16-4,32 (мультиплет), 4,56 (квинтет, J 6,45 Гц); 5,06 (квинтет, J 7 Гц); 7,267,45 (мультиплет).
Масс-спектр (m/z): 268 (М+), 224, 213, 195, 178, 167.
П р е п а р а т и в н ы й п р и м е р 9. (2R*,3S*, 4R*)-2-(4-Хлорфенил)-3,4-диметил-2-гидро- ксиметилоксетан и (2R*, 3R*, 4R*)-2-(-4-хлорфенил)-3,4-диметил-2-гидро- ксиметилоксе- тан.
3,2 мл (3,2 ммоль) 1М раствора литийалюминийгидрида в тетрагидрофуране добавили по каплям при 0оС к раствору 920 мг (3,42 ммоль) смеси 1:1 (2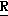 *, 3
*, 3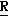 *, 4
*, 4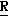 *)-2-(4-хлорфенил)-3,4-диметил-2-этоксикарбонил- оксетана и его (2
*)-2-(4-хлорфенил)-3,4-диметил-2-этоксикарбонил- оксетана и его (2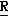 *, 3
*, 3 *,4
*,4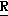 *)-изомера (полу-ченных, как описано в препаративном примере 8) в 7 мл тетрагидрофурана, и полученную смесь перемешивали при той же температуре в течение 30 мин. После этого к реакционной смеси последовательно добавили насыщенный водный раствор хлористого аммония и 1 нормальный водный раствор соляной кислоты. Неочищенные продукты, полученные экстракцией реакционной смеси этилацетатом, подвергали хроматографии на колонке с силикагелем, элюируя смесью 5: 1 по объему гексана и этилацетата, с получением 223,4 мг изомера А целевого соединения, имеющего (2
*)-изомера (полу-ченных, как описано в препаративном примере 8) в 7 мл тетрагидрофурана, и полученную смесь перемешивали при той же температуре в течение 30 мин. После этого к реакционной смеси последовательно добавили насыщенный водный раствор хлористого аммония и 1 нормальный водный раствор соляной кислоты. Неочищенные продукты, полученные экстракцией реакционной смеси этилацетатом, подвергали хроматографии на колонке с силикагелем, элюируя смесью 5: 1 по объему гексана и этилацетата, с получением 223,4 мг изомера А целевого соединения, имеющего (2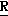 *,3
*,3 *,4
*,4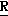 *) конфигурацию и 349,5 мг изомера В целевого соединения, имеющего (2
*) конфигурацию и 349,5 мг изомера В целевого соединения, имеющего (2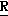 *,3
*,3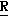 *,4
*,4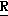 *) конфигурацию.
*) конфигурацию.
Изомер А:
ЯМР-спектр (СДСl3), δ ч./млн. 0,70 (3Н, дублет, J 7,44 Гц); 1,24 (3Н, дублет, J 6,44 Гц); 3,33 (1Н, дублет, J 7,66 Гц); 3,63 (1Н, дублет, J 12,08 Гц); 3,87 (1Н, дублет, J 12,08 Гц); 4,96 (1Н, дублет, квартетов, J 6,44 и 7,66 Гц); 7,25 (2Н, дублет, J 8,46 Гц); 7,35 (2Н, дублет, J 8,46).
Масс-спектр (m/z): 223, 195, 181, 167, 153, 139.
Изомер В:
ЯМР-спектр (СДСl3), δ ч./млн. 1,36 (3Н, дублет, J 6,04 Гц); 1,36 (3Н, дублет, J 7,25 Гц); 2,18 (1Н, широкий дублет дублетов, J 5,53 и 7,25 Гц); 2,65 (1Н, квинтет, J 7,25 Гц); 3,71 (1Н, дублет дублетов, J 5,53 и 12,09 Гц); 4,02 (1Н, дублет дублетов, J 7,25 и 12,09 Гц); 4,56 (1Н, дублет квартетов, J 6,04 и 7,25 Гц); 7,24 (2Н, дублет, J 8,46 Гц); 7,33 (2Н, дублет, J 8,46 Гц).
Масс-спектр (m/z): 195, 167, 139, 129, 125, 115.
П р е п а р а т и в н ы й п р и м е р 10. 2-(4-Хлорфенил)-3,4-диметил-2-метансуль- фонилоксиметилоксетан.
0,13 мл (1,68 ммоль) метансульфонил хлорида, после чего 0,24 мл (1,708 ммоль) триэтиламина добавили при 0оС к раствору 123 мг (0,543 ммоль) 2-(4-хлорфенил)-3,4-диметил-2-гидроксиметилоксетана в 10 мл метиленхлорида, и полученную смесь перемешивали в течение 4 ч, позволяя при этом температуре реакции увеличиться до комнатной температуры. После этого к смеси добавили водный раствор бикарбоната натрия и затем экстрагировали метиленхлоридом. Экстракт сушили над безводным сульфатом магния и концентрировали выпариванием при пониженном давлении. Полученный осадок подвергали хроматографии на колонке с силикагелем, элюируя смесью 10:1 по объему гексана и этилацетата с получением 107 мг (выход 65%) целевого соединения.
ЯМР-спектр (СДСl3), δ ч./млн. 0,72 (3Н, дублет, J 7,25 Гц); 1,21 (3Н, дублет, J6,44 Гц); 3,02 (3Н, синглет); 3,25 (1Н, квинтет, J 7,25 Гц); 4,33 (1Н, дублет, J 11,68 Гц); 4,60 (1Н, дублет, J 11,68 Гц); 5,04 (1Н, дублет квартетов, J 6,44 и 7,25 Гц); 7,28 (1Н, дублет, J 8,86 Гц); 7,37 (2Н, дублет, J 8,86 Гц).
П р е п а р а т и в н ы й п р и м е р 11. (2R*,4R*)-2-хлорметил-2-(4-хлорфенил)-3,4-диметилоксетан.
2-Бутен барботировали в 180 мл бензола при 0оС до тех пор, пока раствор бензола не увеличивался по объему до 1,25 раза его первоначальной величины. Затем к раствору добавили 15 г (79,34 ммоль) 4-хлорфенацил хлорида, после чего смесь освещали ртутной лампой среднего давления мощностью 450 Вт (Hahnovea Co. Inc.) в течение 15 ч. После этого реакционную смесь концентрировали выпариванием при пониженном давлении. Затем ее дистиллировали при пониженном давлении с получением 15,24 мг (78,4%) целевого соединения с температурой кипения 132-133оС/3,2 мм рт.ст. Исходя из ЯМР-спектров это соединение является смесью 1:3 α- и β-изомеров по отношению к С3-положению.
Масс-спектр (m/z): 246 ((М+2)+), 244 (М+), 195, 190, 155, 151, 141, 139.
ЯМР-спектр (СДСl3) δ ч./млн. 0,72 (дублет, J 7,00 Гц); 1,24 (дублет, J 6,40 Гц); 1,33 (дублет, J 7,00 Гц); 1,34 (дублет, J 6,40 Гц); 2,74 (дублет квартетов, J 7,00 и 7,00 Гц); 3,24 (дублет квартетов, J 7,54 и 7,54 Гц); 3,82 (дублет, J 11,62 Гц); 3,90 (дублет, J 11,62 Гц); 3,91 (дублет, J 11,62 Гц); 4,00 (дублет, J 11,62 Гц); 4,55 (дублет квартетов, 6,40 и 7,00 Гц); 5,05 (дублет квартетов, J 6,40 и 7,00 Гц); 7,23-7,40 (мультиплет).
П р е п а р а т и в н ы й п р и м е р 12. (2R*,3R*)-2-(4-Хлорфенил)-3-метил-2-(триметил-силилокси)-1-(1Н- 1,2,4-триазол-1-ил)-4-пентен.
2,47 мл (19,4 ммоль) триметилсилилхлорида и 1,7 г (24,3 ммоль) имидазола добавили к раствору 900 мг (3,24 ммоль) (2R*, 3R*)-3-метил-2-(4-хлорфенил)-(1Н-1,2,4-три- азол-1-ил)-2- пентанола в 20 мл 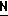 ,
,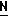 -диметилформамида, и затем смесь перемешивали при 50оС в течение 4 ч. После этого реакционную смесь влили в воду и экстрагировали этилацетатом. Экстракт промывали водой и сушили над безводным сульфатом натрия, после чего растворитель удаляли дистилляцией при пониженном давлении с получением 1,3 г целевого соединения в форме неочищенного масла.
-диметилформамида, и затем смесь перемешивали при 50оС в течение 4 ч. После этого реакционную смесь влили в воду и экстрагировали этилацетатом. Экстракт промывали водой и сушили над безводным сульфатом натрия, после чего растворитель удаляли дистилляцией при пониженном давлении с получением 1,3 г целевого соединения в форме неочищенного масла.
ЯМР-спектр (СДСl3), δ ч./млн. 0,17 (9Н, синглет), 0,88 (3Н, дублет, J 6,7 Гц); 2,7-2,85 (1Н, мультиплет); 4,47 (1Н, дублет, J 14,7 Гц); 4,82 (1Н, дублет, J 14,7 Гц); 5,05-5,2 (2Н, мультиплет), 5,5-5,7 (1Н, мультиплет); 7,2-7,3 (4Н, мультиплет), 7,54 (1Н, синглет), 7,78 (1Н, синглет).
П р е п а р а т и в н ы й п р и м е р 13. (2R*,3R*)-2-(4-Хлорфенил)-3-метил-2-триметилси- лилокси-1-(1Н-1,2,4-триазол-1-ил)бутаналь.
Озон вдували в раствор 190 мг (0,54 ммоль) (2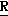 *,3
*,3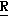 *)-3-метил-2-(хлорфенил)-2-(триметилсилилокси)-1-(1Н- 1,2,4-триазол-1-ил)-4-пентена в 10 мл метанола в условиях охлаждения сухим льдом в ацетоне. Завершение реакции было подтверждено тонкослойной хроматографией, после чего к реакционной смеси был добавлен порошок иодида калия в подходящих количествах. Затем к смеси добавили водный раствор карбоната натрия, который затем экстрагировали этилацетатом. Экстракт промывали последовательно 10 вес./об.-ным водным раствором тиосульфата натрия и водой и сушили над безводным сульфатом натрия. Затем растворитель удаляли дистиллированием при пониженном давлении с получением 140 мг (74%) целевого соединения в виде масла.
*)-3-метил-2-(хлорфенил)-2-(триметилсилилокси)-1-(1Н- 1,2,4-триазол-1-ил)-4-пентена в 10 мл метанола в условиях охлаждения сухим льдом в ацетоне. Завершение реакции было подтверждено тонкослойной хроматографией, после чего к реакционной смеси был добавлен порошок иодида калия в подходящих количествах. Затем к смеси добавили водный раствор карбоната натрия, который затем экстрагировали этилацетатом. Экстракт промывали последовательно 10 вес./об.-ным водным раствором тиосульфата натрия и водой и сушили над безводным сульфатом натрия. Затем растворитель удаляли дистиллированием при пониженном давлении с получением 140 мг (74%) целевого соединения в виде масла.
ЯМР-спектр (СДСl3) δ ч./млн. 0,17 (96, синглет); 1,28 (3Н, дублет, J 6,9 Гц); 3,04 (1Н, квартет, J 6,9 Гц); 4,48 (1Н, дублет, J 15 Гц); 4,67 (1Н, дублет, J 15 Гц); 7,04 (2Н, дублет, J 8,6 Гц); 7,27 (2Н, дублет, J 8,6 Гц); 7,55 (1Н, синглет); 7,88 (1Н, синглет); 9,325 (1Н, дублет, J 1,26 Гц).
П р е п а р а т и в н ы й п р и м е р 14. (2R*,3R*)-2-(4-хлорфенил)-3-метил-2-триме- тилсилилокси-1-(1Н- 1,2,4-триазол-1-ил)-4-пентанол.
0,45 мл (0,38 ммоль) диэтилалюминий хлорида (в виде 0,84 молярного раствора в гексане) добавили к раствору 111,1 мг (0,316 ммоль) (2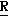 *,3
*,3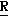 *)-3-метил-2-(4-хлорфенил)-2-(триметилсилилокси)-1-(1
*)-3-метил-2-(4-хлорфенил)-2-(триметилсилилокси)-1-(1 -1,2,4-триа- зол-1-ил)бутаналь в 10 мл тетрагидро- фурана, и затем смесь охладили до температуры -78оС с помощью сухого льда в ацетоне. К смеси добавили 1,39 мг (1,14 ммоль) метилмагнийбромида (в виде 0,82 молярного раствора в диэтиловом эфире) и далее перемешивали в течение 3 ч при той же температуре. После этого реакционную смесь влили в воду и экстрагировали этилацетатом. Экстракт промывали последовательно водой и водным раствором хлорида натрия. Затем смесь сушили над безводным сульфатом магния и растворитель удаляли дистилляцией при пониженном давлении с получением 85,4 мг (73%) целевого соединения в виде масла. Исходя из ЯМР-спектра продукта соединение представляло собой смесь 1:5 С4-изомеров.
-1,2,4-триа- зол-1-ил)бутаналь в 10 мл тетрагидро- фурана, и затем смесь охладили до температуры -78оС с помощью сухого льда в ацетоне. К смеси добавили 1,39 мг (1,14 ммоль) метилмагнийбромида (в виде 0,82 молярного раствора в диэтиловом эфире) и далее перемешивали в течение 3 ч при той же температуре. После этого реакционную смесь влили в воду и экстрагировали этилацетатом. Экстракт промывали последовательно водой и водным раствором хлорида натрия. Затем смесь сушили над безводным сульфатом магния и растворитель удаляли дистилляцией при пониженном давлении с получением 85,4 мг (73%) целевого соединения в виде масла. Исходя из ЯМР-спектра продукта соединение представляло собой смесь 1:5 С4-изомеров.
ЯМР-спектр (СДСl3), δ ч. /млн. 0,087 (синглет); 0,284 (синглет); 0,77 (дублет, J 6 Гц); 0,94 (дублет, J 6,5 Гц); 1,08 (дублет, J 6 Гц); 1,28 (дублет, J 6,9 Гц); 2,0-2,1 (мультиплет); 3,5-3,65 (мультиплет); 4,14 (дублет, J 13,8 Гц); 4,69 (дублет, J 13,8 Гц); 4,7-4,85 (мультиплет); 6,74 (дублет, J 8,67); 7,2-7,35 (мультиплет); 7,88 (синглет).
П р е п а р а т и в н ы й п р и м е р 15. (2R*,3R*)-2-(4-хлорфенил)-3-метил-1-(1Н-1,2, 4-триазол-1-ил)-2,4- пентандиол.
0,35 мл (0,35 ммоль) тетрабутиламмоний фторида (в виде 1 молярного раствора в тетрагидрофуране) добавили к раствору 85,4 мг (0,233 ммоль) (2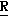 *, 3
*, 3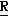 *)-3-метил-2-(4-хлорфенил)-2-(триметилсилилокси)-1-(1
*)-3-метил-2-(4-хлорфенил)-2-(триметилсилилокси)-1-(1 -1, 2,4- триазол-1-ил)-4-пентанола (полученного в препаративном примере 7) в 2,5 мл тетрагидрофурана, и затем смесь перемешивали при комнатной температуре в течение 30 мин. После этого реакционную смесь влили в воду и экстрагировали этилацетатом. Экстракт промыли водой и сушили над безводным сульфатом магния. Затем растворитель удаляли дистилляцией при пониженном давлении с получением 64 мг (93%) целевого соединения. Исходя из ЯМР-спектров это соединение представляло собой смесь 5:1 (2
-1, 2,4- триазол-1-ил)-4-пентанола (полученного в препаративном примере 7) в 2,5 мл тетрагидрофурана, и затем смесь перемешивали при комнатной температуре в течение 30 мин. После этого реакционную смесь влили в воду и экстрагировали этилацетатом. Экстракт промыли водой и сушили над безводным сульфатом магния. Затем растворитель удаляли дистилляцией при пониженном давлении с получением 64 мг (93%) целевого соединения. Исходя из ЯМР-спектров это соединение представляло собой смесь 5:1 (2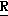 *,3
*,3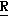 *,4
*,4 *)-изомера и (2
*)-изомера и (2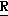 *,3
*,3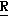 *,4
*,4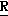 *)-изомера.
*)-изомера.
ЯМР-спектр (СДСl3), δ ч./млн, 0,81 (дублет, J 7 Гц); 1,01 (дублет, J 7 Гц); 1,12 (дублет, J 7 Гц); 1,25 (дублет, J 7 Гц); 1,99 (квартет, J 7 Гц); 3,68 (квартет, J 7 Гц); 4,44 (АВ-дублет, J 15 Гц); 4,75 (АВ-дублет, J 15 Гц); 7,1-7,4 (мультиплет); 7,68 (синглет); 7,74 (синглет); 8,00 (синглет).
П р е п а р а т и в н ы й п р и м е р 16. Этил 3-(4-хлорфенил)-2,2-диметил-3-гидрокси-4-(1Н-1,2,4-тризол-1-ил) бутаноат.
30 мл (30 ммоль) диэтилалюминийхлорида (в виде 1,0 молярного раствора в гексане) добавили к суспензии 2,0 г (30 ммоль) цинка и 0,14 г (1 ммоль) бромида меди в 30 мл тетрагидрофурана. Затем по каплям к смеси добавили раствор 1,33 г (6 ммоль) 4-хлор-2-(1 -1,2,4-триазол-1-ил)ацетофено- на и 6,7 г (30 ммоль) этил 2-бромизобутирата в 30 мл тетрагидрофурана, после чего смесь перемешивали при комнатной температуре в течение 18 ч. После этого к реакционной смеси добавили воду и 1 нормальную водную соляную кислоту с получением осадка, который затем отфильтровывали с помощью целитного фильтра и промывали этилацетатом. Фильтрат и промывные растворы объединяли и последовательно промывали 1 нормальной водной соляной кислотой, водным раствором хлорида аммония и насыщенным водным раствором хлорида натрия. Затем раствор сушили и концентрировали выпариванием при пониженном давлении с отделением осадка, который очищали хроматографией на колонке с силикагелем, элюируя смесью 1:4 по объему гексана и этилацетата с получением 1,89 г (93%) целевого соединения в виде масла.
-1,2,4-триазол-1-ил)ацетофено- на и 6,7 г (30 ммоль) этил 2-бромизобутирата в 30 мл тетрагидрофурана, после чего смесь перемешивали при комнатной температуре в течение 18 ч. После этого к реакционной смеси добавили воду и 1 нормальную водную соляную кислоту с получением осадка, который затем отфильтровывали с помощью целитного фильтра и промывали этилацетатом. Фильтрат и промывные растворы объединяли и последовательно промывали 1 нормальной водной соляной кислотой, водным раствором хлорида аммония и насыщенным водным раствором хлорида натрия. Затем раствор сушили и концентрировали выпариванием при пониженном давлении с отделением осадка, который очищали хроматографией на колонке с силикагелем, элюируя смесью 1:4 по объему гексана и этилацетата с получением 1,89 г (93%) целевого соединения в виде масла.
Масс-спектр (m/z): 337 (М+), 292, 252, 222, 139.
ЯМР-спектр (СДСl3), δ ч./млн. 1,21 (3Н, синглет); 1,21 (3Н, триплет, J 7 Гц); 1,23 (3Н, синглет); 4,10 (2Н, квартет, J 7 Гц); 4,63 (1Н, дублет, J 14 Гц); 5,13 (1Н, дублет, J 14 Гц); 5,37 (1Н, широкий синглет); 7,05-7,40 (4Н, мультиплет); 7,65 (1Н, синглет); 7,92 (1Н, синглет).
П р е п а р а т и в н ы е п р и м е р ы 17-32.
По методике препаративного примера 16 были получены следующие соединения.
П р е п а р а т и в н ы й п р и м е р 17. Октил 3-(2,4-дифторфенил)-3-гидрокси-4-(1 -1,2,4-триазол-1-ил)бутаноат, в виде масла, выход 42%
-1,2,4-триазол-1-ил)бутаноат, в виде масла, выход 42%
ЯМР-спектр (СДСl3), δ ч. /млн. 0,5-1,70 (15Н, мультиплет); 2,65 (1Н, дублет, J 15 Гц); 3,21 (1Н, дублет, J 15 Гц); 3,95 (2Н, триплет, J 6 Гц); 4,36 (1Н, дублет, J 13 Гц); 4,68 (1Н, дублет, J 13 Гц); 5,10 (1Н, широкий синглет); 6,55-7,00 (2Н, мультиплет); 7,10-7,80 (1Н, мультиплет); 7,81 (1Н, синглет); 8,15 (1Н, синглет).
Масс-спектр (m/z): 395 (М+), 313, 201, 183.
П р е п а р а т и в н ы й п р и м е р 18. Октил-3-(2,4-дифторфенил)-3-гидрокси-2- метил-4-(1Н-1,2,4-триазол-1- ил)бутаноат, в виде масла, выход 42%
ЯМР-спектр (СДСl3), δ ч. /млн. 0,70-1,95 (15Н, мультиплет); 3,28 (1Н, квартет, J 7 Гц); 4,115 (2Н, триплет, 6 Гц); 4,46 (1Н, дублет, J 13 Гц); 4,85 (1Н, дублет, J 13 Гц); 5,18 (1Н, синглет); 6,50-6,95 (2Н, мультиплет); 7,10-7,65 (1Н, мультиплет); 8,17 (1Н, синглет).
Масс-спектр (m/z): 409 (М+), 327, 224, 215, 197.
П р е п а р а т и в н ы й п р и м е р 19. Этил-3-(2,4-дифторфенил)-3-гидрокси-4- (1 -1,2,4-триазол-1-ил)бутаноат, в виде масла, выход 62%
-1,2,4-триазол-1-ил)бутаноат, в виде масла, выход 62%
ЯМР-спектр (СДСl3), δ ч. /млн. 1,10 (3Н, триплет, J 7 Гц); 2,65 (1Н, дублет, J 16 Гц); 3,20 (1Н, дублет, J 16 Гц); 3,99 (2Н, квартет, J 7 Гц); 4,39 (1Н, дублет, J 14 Гц); 4,68 (1Н, дублет, J 14 Гц); 5,25 (1Н, широкий синглет); 6,50-7,00 (2Н, мультилпет); 7,15-7,75 (1Н, мультиплет); 7,76 (1Н, синглет); 8,12 (1Н, синглет).
Масс-спектр (m/z): 312 (М+), 229, 182, 141.
П р е п а р а т и в н ы й п р и м е р 20. Этил-3-(2,4-дифторфенил)-3-гидрокси-2-ме- тил-4-(1 -1,2,4-триазол-1-ил) бутаноат, в виде масла, выход 58%
-1,2,4-триазол-1-ил) бутаноат, в виде масла, выход 58%
ЯМР-спектр (СДСl3), δ ч. /млн. 0,90 (3Н, дублет, J 7 Гц); 1,23 (3Н, триплет, J 7 Гц); 3,19 (1Н, квартет, J 7 Гц); 4,13 (2Н, квартет, J 7 Гц); 4,41 (1Н, дублет, J 13 Гц); 4,77 (1Н, дублет, J 13 Гц); 5,10 (1Н, широкий синглет); 6,45-7,00 (2Н, мультиплет); 7,20-7,65 (1Н, мультиплет); 7,66 (1Н, синглет); 7,95 (1Н, синглет).
Масс-спектр (m/z): 325 (М+), 243, 224, 197, 182, 141.
П р е п а р а т и в н ы й п р и м е р 21. Этил-3-(2,4-дифторфенил)-2-этил-3-гидрокси- 4-(1 -1,2,4-триазол-1-ил)бутаноат, с температурой плавления 100-105оС, выход 57%
-1,2,4-триазол-1-ил)бутаноат, с температурой плавления 100-105оС, выход 57%
ЯМР-спектр (СДСl3), δ ч. /млн. 0,78 (3Н, триплет, J 7 Гц); 1,32 (3Н, триплет, J 7 Гц); 1,00-2,10 (2Н, мультиплет); 3,07 (1Н, дублет дублетов, J 4 и 10 Гц); 4,22 (2Н, квартет, J 7 Гц); 4,30 (1Н, дублет, J 14 Гц); 4,84 (1Н, дублет, J 14 Гц); 5,10 (1Н, широкий синглет); 6,47-6,92 (2Н, мультиплет); 7,10-7,60 (1Н, мультиплет); 7,61 (1Н, синглет); 7,92 (1Н, синглет).
Масс-спектр (m/z): 339 (М+), 297, 257, 224, 211, 182, 141.
П р е п а р а т и в н ы й п р и м е р 22. Этил-3-(2,4-дифторфенил)-3-гидрокси-2,2- диметил-4-(1 -1,2,4- триазол-1-ил)бутаноат, с температурой плавления 90оС, выход 70%
-1,2,4- триазол-1-ил)бутаноат, с температурой плавления 90оС, выход 70%
ЯМР-спектр (СДСl3), δ ч. /млн. 1,20 (3Н, дублет, J 2,0 Гц); 1,25 (3Н, триплет, J 7,3 Гц); 1,30 (3Н, синглет); 4,17 (2Н, дублет квартетов, J 1,2 и 7,3 Гц); 4,65 (1Н, дублет дублетов, J 1,8 и 14,1 Гц); 5,36 (1Н, дублет дублетов, J 2,4 и 14,1 Гц); 5,42 (1Н, синглет); 6,59-6,82 (2Н, мультиплет); 7,55-7,64 (1Н, мультиплет); 7,73 (1Н, синглет); 8,02 (1Н, дублет, J 1,6 Гц).
Масс-спектр (m/z): 339 (М+), 294, 257, 224, 141.
П р е п а р а т и в н ы й п р и м е р 23. Этил-3-(2,4-дифторфенил)-3-гидрокси-2-фе- нил-4-(1Н-1,2,4-триазол- 1-ил)бутаноат в виде масла, выход 9%
ЯМР-спектр (СДСl3), δ ч. /млн. 1,20 (3Н, триплет, J 7 Гц); 4,10 (2Н, квартет, J 7 Гц); 4,38 (1Н, синглет); 4,78 (2Н, синглет); 6,00 (1Н, мультиплет); 6,20-7,30 (3Н, мультиплет); 7,08 (5Н, широкий синглет); 7,70 (1Н, синглет); 8,05 (1Н, синглет).
Масс-спектр (m/z): 387 (М+), 342, 305, 231, 224, 141.
П р е п а р а т и в н ы й п р и м е р 24. Этил-3-(4-хлорфенил)-3-гидрокси-2-метил-4-(1 -1,2,4-триазол-1-ил)бутаноат, с температурой плавления 75-90оС, выход 57%
-1,2,4-триазол-1-ил)бутаноат, с температурой плавления 75-90оС, выход 57%
ЯМР-спектр (СДСl3), δ ч. /млн. 1,03 (3Н, триплет, J 7 Гц); 1,30 (3Н, триплет, J 7 Гц); 2,98 (1Н, квартет, J 7 Гц); 4,16 (2Н, квартет, J 7 Гц); 4,35 (1Н, дублет, J 13 Гц); 4,65 (1Н, дублет, J 13 Гц); 5,00 (1Н, широкий синглет); 7,00-7,30 (4Н, мультиплет); 7,70 (1Н, синглет); 7,81 (1Н, синглет);
Масс-спектр (m/z): 323 (М+), 276, 241, 222.
П р е п а р а т и в н ы й п р и м е р 25. Этил-3-(4-хлорфенил)-2-этил-3-гидрокси-4-(1 -1,2,4-триазол-1-ил) бутаноат, в виде масла, выход 91%
-1,2,4-триазол-1-ил) бутаноат, в виде масла, выход 91%
ЯМР-спектр (СДСl3) δ ч. /млн. 0,82 (3Н, триплет, J 7,6 Гц); 1,22-1,39 (2Н, мультиплет); 1,31 (3Н, триплет, J 7,1 Гц); 1,63-1,77 (1Н, мультиплет); 4,18-4,29 (2Н, мультиплет); 4,39 (1Н, дублет, J 14,1 Гц); 4,64 (1Н, дублет, J 14,1 Гц); 7,20-7,28 (4Н, мультиплет); 7,75 (1Н, синглет); 7,93 (1Н, синглет).
Масс-спектр (m/z): 337 (М+), 255, 222, 139.
П р е п а р а т и в н ы й п р и м е р 26. Этил-3-(4-фторфенил)-2-этил-3-гидрокси-4-(1 -1,2,4-триазол-1-ил)бутаноат, в виде масла, выход 35%
-1,2,4-триазол-1-ил)бутаноат, в виде масла, выход 35%
ЯМР-спектр (СДСl3) δ ч. /млн. 0,82 (3Н, триплет, J 7,5 Гц); 1,32 (3Н, триплет, J 7,1 Гц); 0,94-1,77 (3Н, мультиплет); 2,80 (1Н, квартет, J 3,6 Гц); 4,16-4,33 (2Н, мультиплет); 4,41 (1Н, дублет, J 14,1 Гц); 4,56 (1Н, дублет, J 14,1 Гц); 6,89-7,02 (2Н, мультиплет); 7,22-7,29 (2Н, мультиплет). 7,77 (1Н, синглет), 8,02 (1Н, синглет).
Масс-спектр (m/z): 321 (М+), 303, 276, 239.
П р е п а р а т и в н ы й п р и м е р 27. Этил-3-(4-фторфенил)-3-гидрокси-2,2-диме- тил-4-(1 -1,2,4-триазол-1- ил)бутаноат, в виде масла, выход 87%
-1,2,4-триазол-1- ил)бутаноат, в виде масла, выход 87%
ЯМР-спектр (СДСl3), δ ч./млн. 1,20-1,28 (9Н, мультиплет); 4,07-4,19 (2Н, мультиплет); 4,71 (1Н, дублет, J 14,1 Гц); 5,14 (1Н, дублет, J 14,1 Гц); 6,89-7,38 (4Н, мультиплет); 7,75 (1Н, синглет); 8,04 (1Н, синглет).
Масс-спектр (m/z): 321 (М+), 276, 239, 206.
П р е п а р а т и в н ы й п р и м е р 28. Этил-2-этил-3-гидрокси-(4-метилфенил)-4- (1 -1,2,4-триазол-1-ил)бутаноат, в виде масла, выход 75%
-1,2,4-триазол-1-ил)бутаноат, в виде масла, выход 75%
ЯМР-спектр (СДСl3), δ ч./млн. 0,81 (3Н, триплет, J 7,3 Гц); 0,94-1,03 (1Н, мультиплет); 1,31 (3Н, триплет, J 7,3 Гц); 1,23-1,41 (1Н, мультиплет); 1,63-1,75 (1Н, мультиплет); 4,19-4,27 (2Н, мультиплет); 4,39 (1Н, дублет, J 14,1 Гц); 4,62 (1Н, дублет, J 14,1 Гц); 7,05-7,63 (4Н, мультиплет); 7,76 (1Н, синглет); 7,83 (1Н, синглет).
Масс-спектр (m/z): 317 (М+), 235, 202.
П р е п а р а т и в н ы й п р и м е р 29. Этил-3-гидрокси-2,2-диметил-3-(4-метилфе- нил)-4-(1 -1,2,4-триазол-1-ил)бутаноат, в виде масла, выход 82%
-1,2,4-триазол-1-ил)бутаноат, в виде масла, выход 82%
ЯМР-спектр (СДСl3), δ ч. /млн. 1,20-1,28 (9Н, мультиплет); 2,27 (3Н, синглет); 4,09-4,19 (2Н, мультиплет); 4,71 (1Н, дублет, J14,1 Гц); 5,12 (1Н, дублет, J 14,1 Гц); 7,03 (2Н, дублет, J 8,1 Гц); 7,76 (1Н, синглет); 7,99 (1Н, синглет).
Масс-спектр (m/z): 318 (М++1), 272, 235, 202.
Препаративный пример 30. Этил-3-гидрокси-3-(4-изопропилфенил)-2,2-диметил-4- (1 -1,2,4-триазол-1-ил)бутаноат, в виде масла, выход 85%
-1,2,4-триазол-1-ил)бутаноат, в виде масла, выход 85%
ЯМР-спектр (СДСl3), δ ч. /млн. 1,17-1,23 (15Н, мультиплет); 2,75-2,88 (1Н, мультиплет); 4,05-4,16 (2Н, мультиплет); 4,73 (1Н, дублет, J 14,5 Гц); 5,09 (1Н, дублет, J 14,5 Гц); 5,18-5,24 (1Н, широкий синглет); 7,07-7,09 (2Н, мультиплет); 7,25-7,28 (2Н, мультиплет); 7,74 (1Н, синглет); 7,96 (1Н, синглет).
Масс-спектр (m/z): 345 (М+), 263, 230.
П р е п а р а т и в н ы й п р и м е р 31. Этил-3-гидрокси-3-(4-метоксифенил)-2,2-ди- метил-4- (1 -1,2,4- триазол-1-ил)бутаноат, в виде масла, выход 95%
-1,2,4- триазол-1-ил)бутаноат, в виде масла, выход 95%
ЯМР-спектр (СДСl3), δ ч. /млн. 1,21-1,26 (9Н, мультиплет); 3,75 (3Н, синглет); 4,07-4,21 (2Н, мультиплет); 4,70 (1Н, дублет, J 14,5 Гц); 5,10 (1Н, дублет, J 14,5 Гц); 5,08-5,13 (1Н, широкий синглет); 6,73-6,79 (2Н, мультиплет); 7,25-7,30 (2Н, мультиплет); 7,74 (1Н, синглет); 7,97 (1Н, синглет).
Масс-спектр (m/z: 334 (М++1), 251, 218.
П р е п а р а т и в н ы й п р и м е р 32. Этил-2-этил-3-гидрокси-3-фенил-4-(1 -1,2,4- триазол-1-ил)бутаноат, с температурой плавления 72-79оС, выход 44%
-1,2,4- триазол-1-ил)бутаноат, с температурой плавления 72-79оС, выход 44%
ЯМР-спектр (СДСl3), δ ч./млн. 0,81 (3Н, триплет, J 7,5 Гц); 1,33 (3Н, триплет, J 7,1 Гц); 1,32-1,44 (1Н, мультиплет); 1,94-2,04 (1Н, мультиплет); 2,86 (1Н, квартет, J 3,6 Гц); 4,18-4,32 (2Н, мультилпет); 4,42 (1Н, дублет, H 14,1 Гц); 4,66 (1Н, дублет, J 14,1 Гц); 7,16-7,33 (5Н, мультиплет); 7,75 (1Н, синглет); 7,84 (1Н, синглет).
Масс-спектр (m/z): 303 (М+), 258, 221, 188.
П р е п а р а т и в н ы й п р и м е р 33. 3-(4-Хлорфенил)-2,2-диметил-4-(1Н-1,2,4- триазол-1-ил)-1,3-бутандиол.
1,9 г (50,2 ммоль) боргидрида натрия добавили к раствору 1,75 г (5,2 ммоль) этил-3-(4-хлорфенил)-2,2-диметил-3-гидрокси-4-(1 -1,2,4-триазол-1-ил)
-1,2,4-триазол-1-ил)
бутаноатав 25 мл метанола, и затем смесь кипятили с обратным холодильником в течение 3 ч. После этого реакционной смеси позволили охладиться и затем смесь влили в воду со льдом и экстрагировали этилацетатом. Экстракт промыли водным раствором аммонийхлорида, сушили и концентрировали выпариванием при пониженном давлении с получением 1,5 г (98%) целевого соединения с температурой плавления 120-135оС.
Масс-спектр (m/z): 295 (М+), 222, 139, 83.
ЯМР-спектр (СДСl3), δ ч./млн. 0,82 (3Н, синглет); 0,90 (3Н, синглет); 3,23-3,30 (3Н, мультиплет); 4,56 (1Н, дублет, J 14,7 Гц); 5,27 (1Н, дублет, J 14,7 Гц); 5,25 (1Н, синглет); 7,24-7,48 (4Н, мультиплет); 7,70 (1Н, синглет); 8,25 (1Н, синглет).
П р е п а р а т и в н ы е п р и м е р ы 34-44. По методике препаративного примера 33 были получены следующие соединения.
П р е п а р а т и в н ы й п р и м е р 34. 3-(2,4-Дифторфенил)-4-(1 -1,2,4-триазол-1- ил)-1,3-бу- тандиол с температурой плавления 89-102оС, выход 89%
-1,2,4-триазол-1- ил)-1,3-бу- тандиол с температурой плавления 89-102оС, выход 89%
ЯМР-спектр (СДСl3), δ ч./млн. 1,80-2,60 (2Н, мультиплет); 3,35-3,90 (2Н, мультиплет); 4,50 (4Н, широкий синглет); 6,50-6,95 (2Н, мультиплет); 7,15-7,85 (1Н, мультиплет); 7,65 (1Н, синглет); 8,00 (1Н, синглет).
Масс-спектр (m/z): 270 (М+), 253, 224, 187, 141.
П р е п а р а т и в н ы й п р и м е р 35. 3-(2,4-Дифторфенил)-2-метил-4-(1 -1,2,4- триазол-1- ил)-1,3-бутандиол, с температурой плавления 93-113оС, выход 89%
-1,2,4- триазол-1- ил)-1,3-бутандиол, с температурой плавления 93-113оС, выход 89%
ЯМР-спектр (СДСl3),δ ч./млн. 0,84 (3Н, дублет дублетов, J 1,2 и 7,3 Гц); 2,17-2,38 (1Н, мультиплет); 2,50 (1Н, широкий синглет); 3,82 (1Н, дублет дублетов, J 5,2 и 11,3 Гц); 4,77 (1Н, дублет, J 14,1 Гц); 4,96 (1Н, дублет дублетов, J 1,6 и 14,1 Гц); 5,30 (1Н, широкий синглет); 6,68-6,79 (2Н, мультиплет); 7,35-7,46 (1Н, мультиплет); 7,75 (1Н, синглет); 7,92 (1Н, синглет).
П р е п а р а т и в н ы й п р и м е р 36. 3-(2,4-Дифторфенил)-2-этил-4-(1 -1,2,4-три- азол-1-ил)-1,3-бутандиол, в виде аморфного порошка, выход 94%
-1,2,4-три- азол-1-ил)-1,3-бутандиол, в виде аморфного порошка, выход 94%
ЯМР-спектр (СДСl3), δ ч./млн. 0,83 (3Н, триплет, J 7,5 Гц); 0,90-1,53 (3Н, мультиплет); 3,96 (1Н, дублет дублетов, J 4,4 или 11,7 Гц); 4,11 (1Н, дублет дублетов, J 2,0 и 11,7 Гц); 4,78 (1Н, дублет, J 14,1 Гц); 4,95 (1Н, дублет дублетов, J 1,2 и 14,1 Гц); 5,20 (1Н, широкий синглет); 6,88-6,80 (2Н, мультиплет); 7,31-7,40 (1Н, мультиплет); 7,74 (1Н, синглет); 7,90 (1Н, синглет).
П р е п а р а т и в н ы й п р и м е р 37. 3-(2,4-Дифторфенил)-2,2-диметил-4-(1 -1,2, 4-триазол- 1-ил)-1,3- бутандиол, температура плавления 94-104оС, выход 92%
-1,2, 4-триазол- 1-ил)-1,3- бутандиол, температура плавления 94-104оС, выход 92%
ЯМР-спектр (СДСl3), δ ч. /млн. 0,93 (3Н, дублет, J 2,0 Гц); 1,13 (3Н, дублет, J 2,0 Гц); 3,49 (1Н, дублет, J 11,5 Гц); 3,55 (1Н, дублет, J 11,5 Гц); 4,60 (1Н, дублет дублетов, J 2,0 и 13,9 Гц); 5,30 (1Н, дублет дублетов, J 2,8 и 13,9 Гц); 5,82 (1Н, широкий синглет); 6,60-6,85 (2Н, мультиплет); 7,60-7,57 (1Н, синглет); 8,02 (1Н, синглет).
Масс-спектр (m/z): 298 (М++1), 224, 182, 141.
П р е п а р а т и в н ы й п р и м е р 38. 3-(2,4-Дифторфенил)-2-фенил-4-(1 -1,2,4- триазол-1- ил)-1,3-бутандиол, в виде масла, выход 95%
-1,2,4- триазол-1- ил)-1,3-бутандиол, в виде масла, выход 95%
ЯМР-спектр (СДСl3) δ ч./млн. 2,70-4,60 (5Н, мультиплет); 4,88 (1Н, широкий синглет); 5,25 (1Н, дублет дублетов, J 4,8 и 7,2 Гц); 6,50-7,60 (3Н, мультиплет); 7,20 (5Н, широкий синглет); 7,70 (1Н, синглет); 7,95 (1Н, синглет).
П р е п а р а т и в н ы й п р и м е р 39. 3-(4-Изопропилфенил)-2,2-диметил-4-(1 -1, 2,4-три- азол-1-ил)-1,3- бутандиол, в виде масла, выход 98%
-1, 2,4-три- азол-1-ил)-1,3- бутандиол, в виде масла, выход 98%
ЯМР-спектр (СДСl3), δ ч. /млн. 1,10-1,23 (12Н, мультиплет); 2,04-2,90 (1Н, мультиплет); 3,44 (1Н, дублет, J 13,1); 3,48 (1Н, дублет, J 14,1 Гц); 7,06-7,30 (4Н, мультиплет); 7,72 (1Н, синглет); 7,96 (1Н, синглет).
Масс-спектр (m/z): 304 (М+), 230, 221.
П р е п а р а т и в н ы й п р и м е р 40. 3-(4-Метоксифенил)-2,2-диметил-4-(1 -1,2, 4-триазол-1-ил)-1,3- бутандиол, в виде масла, выход 61%
-1,2, 4-триазол-1-ил)-1,3- бутандиол, в виде масла, выход 61%
ЯМР-спектр (СДСl3) δ ч. /млн. 0,81 (3Н, синглет); 1,21 (3Н, синглет); 3,54 (1Н, дублет, J 11,5 Гц); 3,66 (1Н, дублет, J 11,5 Гц); 3,78 (3Н, синглет); 4,60 (1Н, дублет, 14,3 Гц); 4,98 (1Н, дублет, J 14,3 Гц); 6,72-6,82 (1Н, мультиплет); 7,15-7,20 (2Н, мультиплет); 7,72 (1Н, синглет); 8,05 (1Н, синглет).
П р е п а р а т и в н ы й п р и м е р 41. 2,2-Диметил-2-этил-3-фенил-3-(1 -1,2,4- триазол-1- ил)-1,3-бутандиол, в виде масла, выход 24%
-1,2,4- триазол-1- ил)-1,3-бутандиол, в виде масла, выход 24%
ЯМР-спектр (СДСl3) δ ч. /млн. 0,84 (3Н, триплет, J 7,5 Гц); 1,22-1,40 (2Н, мультиплет); 1,73-1,87 (1Н, мультиплет); 3,80 (1Н, дублет дублетов, J 4,4 и 5,6 Гц); 4,04 (1Н, дублет дублетов, J 14,1 Гц); 4,80 (1Н, дублет, J 14,1 Гц); 7,19-7,38 (5Н, мультиплет); 7,98 (1Н, синглет); 8,09 (1Н, синглет).
Масс-спектр (m/z): 261 (М+), 240, 207, 188.
П р е п а р а т и в н ы й п р и м е р 42. 3-(4-Хлорфенил)-2-метил-4-(1 -1,2,4-три- азол-1-ил)-1, 3-бутандиол, в виде масла, выход 34%
-1,2,4-три- азол-1-ил)-1, 3-бутандиол, в виде масла, выход 34%
ЯМР-спектр (СДСl3) δ ч. /млн. 0,85 (3Н, триплет, J 7,1 Гц); 2,08 (1Н, дублет триплетов, J 3,3 и 7,0 Гц); 2,30-3,20 (2Н, широкий пик); 3,64 (1Н, дублет дублетов, J 6,8 и 11,1 Гц); 3,85 (1Н, дублет дублетов, J 3,3 и 11,1 Гц); 4,56 (1Н, дублет, J 14,2 Гц); 4,79 (1Н, дублет, J 14,2 Гц); 7,21-7,31 (4Н, мультиплет); 7,84 (1Н, синглет); 8,09 (1Н, синглет).
П р е п а р а т и в н ы й п р и м е р 43. 3-(4-Хлорфенил)-2-этил-4-(1 -1,2,4-триазол- 1-ил)-1,3-бутандиол в виде масла, выход 26%
-1,2,4-триазол- 1-ил)-1,3-бутандиол в виде масла, выход 26%
ЯМР-спектр (СДСl3) δ ч. /млн. 0,82 (3Н, триплет, J 7,6 Гц); 1,23-1,39 (2Н, мультиплет); 1,62-1,74 (1Н, мультиплет); 4,19-4,30 (2Н, мультиплет); 4,41 (1Н, дублет, J 14,4 Гц); 7,22-7,28 (4Н, мультиплет); 7,78 (1Н, синглет); 8,05 (1Н, синглет).
Масс-спектр (m/z): 295 (М+), 222, 213.
П р е п а р а т и в н ы й п р и м е р 44. 2-Этил-3-(4-фторфенил)-4-(1 -1,2,4-триазол- 1-ил)-1,3- бутандиол, темпераутра плавления 65-75оС, выход 58%
-1,2,4-триазол- 1-ил)-1,3- бутандиол, темпераутра плавления 65-75оС, выход 58%
ЯМР-спектр (СДСl3) δ ч. /млн. 0,85 (3Н, триплет, J 7,5 Гц); 1,30-1,37 (2Н, мультиплет); 1,70-1,85 (1Н, мультиплет); 3,51-3,69 (2Н, мультиплет); 3,77-3,84 (1Н, мультиплет); 4,02-4,07 (1Н, мультиплет); 4,57 (1Н, дублет, J 14,1 Гц); 4,77 (1Н, дублет, J 14,1 Гц); 6,92-7,90 (4Н, мультиплет); 8,04 (1Н, синглет); 8,26 (1Н, синглет).
Масс-спектр (m/z): 279 (М+), 206, 197.
ЭКСПЕРИМЕНТ 1. Лечебная активность против ржавчины риса.
Сеяницы риса (разновидность: "Sachikaze") на стадии 4-5 листа инокулировали грибком Puricularia oryzal путем опрыскивания их суспензией спор грибка и выдерживания сеянцев во влажной камере (относительная влажность: 100%) при 20-22оС. Спустя 24 ч сеянцы риса опрыскивали водной суспензией испытумого соединения в концентрации 10 ч./млн. в количестве 30 мл на три горшка. Сеянцы риса затем хранили во влажной камере в течение дополнительных 6 дней. В качестве контроля несколько растений были подвержены действию грибка, но их не обрабатывали каким-либо противогрибковым агентом.
Показатель активности определялся на основе ряда повреждений, образованных на верхних двух листьях каждого растения. Результаты показаны в табл. 4. В данной и последующих таблицах соединения предлагаемого изобретения идентифицируются номером одного из последующих примеров, в которых описано их получение.
Показатель активности оценивался на основе степени заболевания, которая определялась с помощью проверки невооруженным глазом, и показатель дается с помощью следующих обозначений (то же самое применимо к последующим экспериментам):
5 отсутствие болезни
4 степень заболевания была 10% или меньше от степени необработанного растения
3 степень заболевания 10-30% от заболевания необработанного растения
2 степень заболевания 30-50% от степени заболевания необработанного растения
1 степень заболевания 50-70% от степени заболевания необработанного растения
0 степень заболевания была 70% или больше по сравнению с необработанным растением и почти такая же, как степень заболевания необработанного растения.
ЭКСПЕРИМЕНТ 2. Активность предотвращения ризоктониоза растений риса.
Саженцы риса (разновидность Hihonbare) в стадии 4-5 листа опрыскивали водной суспензией испытуемого соединения в концентрации 100 ч./млн. (30 мл. /3 горшка). Саженцы затем хзранились в течение 24 ч при комнатной температуре, после чего их инокулировали Rhizostinia solani путем помещения 4-5 семян овса, на которых предварительно культивировался грибок вокруг основания каждого саженца. Саженцы затем хранили во влажной камере (относительная влажность 100% ) в течение 5 дней при 25-27оС. Показатель активности дан на основе высоты повреждений, образовавшихся на саженцах риса.
Результаты показаны в табл. 5.
ЭКСПЕРИМЕНТ 3. Лечебная активность против ризоктониоза растений риса.
Саженцы риса (роазновидность Hohanbare) на стадии 4-5 листа инокулировали Rhizostinia solani путем помещения 4-5 зерен овса, на которых предварительно культивировался грибок, вокруг основания каждого саженца риса, и хранились во влажной камере (относительная влажность 100%) при 25-27оС. Спустя 24 ч саженцы риса опрыскивали водной суспензией испытуемых соединений в концентрации 10 ч./млн. (30 мл/3 горшка), и их продолжали выдерживать во влажной камере еще в течение 5 дней. Показатель активности дан на основе высоты повреждений, образовавшихся на саженцах риса.
Результаты показаны в табл. 6.
ЭКСПЕРИМЕНТ 4. Профилактическая активность против ризоктониоза растений риса.
Саженцы риса (разновидность Hihon bare) на стадии 3-4 листа, выращенные в горшках, заливались водой на глубину 1 см. Затем по отношению к воде в горшках применялось испытуемое соединение в количестве, соответствующем 100 г на 10 акров. После того как саженцы выдерживали в теплице в течение 7 дн, их инокулировали Rhizostinia solani путем помещения 4-5 зерен овса, предварительно зараженных грибком, вокруг основания каждого саженца. Саженцы затем вдыерживались во влажной камере (относительная влажность 100%) в течение 5 дн при 25-27оС. Показатель активности приводился на основании высоты повреждений, образовавшихся на саженцах риса.
Результаты показаны в табл. 7.
ЭКСПЕРИМЕНТ 5. Активность против гибберилеза растений риса при пропитке семян.
10 г семян риса (разновидность Tangin bozu), которые предварительно были инфицированы грибком Jibberella fuji Huroi путем опрыскивания их суспензией спор грибка, погружали в 20 мл водной суспензии испытуемого соединения в концентрации 100 ч./млн. на 3 дня. В конце данного периода времени обработанные семена плотно высевали в почву в горшках, и горшки помещали в теплицу на 3 недели при 20-30оС. Показатель активности приводился на основании числа поврежденных сеянцев.
Результаты показаны в табл. 8.
ЭКСПЕРИМЕНТ 6. Лечебная активность против листовой ржавчины пшеницы.
Сеянцы пшеницы (разновидность Норин N 61) на стадии 1,5 листа инокулировали грибком Puccinia recondita путем разбрызгивания спор грибка на сеянцы. Сеянцы хранили во влажной камере (относительная влажность 100%) в течение 24 ч при 20-22оС, после чего их переносили в теплицу с температурой 15-20оС. Через 2 дня сеянцы опрыскивали водной суспензией испытуемого соединения в концентрации 3 ч./млн. (30 мл/3 горшка). Сеянцы затем непрерывно выдерживали в теплице в течение 10 дней. Показатель активности приводился на основании пораженной болезнью площади первого листа в конце данного времени.
Результаты приведены в табл. 9.
ЭКСПЕРИМЕНТ 7. Лечебная активность против настоящей мучнистой росы ячменя.
Сеянцы ячменя (разновидность Секисинрики) на стадии первого листа инкулировали кондициями erysiphe granunis f.sp. hordei путем распыления спор грибка на сеянцы, которые затем выдерживали в теплице при 15-20оС. Спустя один день сеянцы опрыскивали водной суспензией испытуемого соединения в концентрации 3 ч./млн. (30 мл/3 горшка), и их продолжали хранить в теплице при данной температуре еще в течение 10 дней. Показатель активности приводился на основании пораженной болезнью площади первого листа в конце данного периода времени.
Результаты показаны в табл. 10.
ЭКСПЕРИМЕНТ 8. Профилактическая активность по предотвращению ложной мучнистой росы огурцов.
Саженцы огурцов (разновидность Сагамиханпаку) на стадии 3-4 листа опрыскивали водной суспензией испытуемого соединения в концентрации 300 ч. /млн. (30 мл/3 горшка). Саженцы затем хранили в течение 24 ч при комнатной температуре, после чего их инокулировали кондициями Sphaerotheca fuliginaa путем обрызгивания саженцев спорами грибка. После того как саженцы были подвергнуты вдерживанию в теплице при 20-30оС в течение 7 дней, оценивали показатель активности на основании пораженной болезнью площади третьего и четвертого листьев.
Результаты показаны в табл. 11.
ЭКСПЕРИМЕНТ 9. Профилактическая активность предотвращения парши яблони.
Саженцы яблони в стадии 3-4 листа опрыскивали водной суспензией испытуемого соединения с концентрацией 300 ч./млн. (30 мл/3 горшка). Саженцы затем хранили в течение 24 ч при комнатной темпераутре, после чего их инокулировали грибком Venturia inaegualis путем опрыскивания их суспензией спор грибка. После инокулирования саженцы выдерживали во влажной камере (относительная влажность 100%) в течение 3 дней при 20-22оС, а затем переносили в теплицу при 20-22оС на 10 дней. Показатель активности приводился на основании пораженной болезнью площади третьего и четвертого листьев в конце данного периода времени.
Результаты приведены в табл. 12.
ЭКСПЕРИМЕНТ 10. Противогрибоквая активность.
Грибковый диск диаметром около 4 мм инокулировали на среде агара (2% по весу солодового экстракта, 1% глюкозы, 0,3% пентона и 2% агара) в чашке Петри диаметром 9 см, путем помещения диска в центр среды. Образцы бумажных дисков приготавливали путем пропитки диска (диаметром 8 мм, толщиной 0,7 мм) 30 мкл ацетонового раствора, содержащего 300 ч./млн. испытуемого соединения, и подвергали его сухой стерилизации. Образцы диска помещали в круг на расстоянии примерно 1 см от края пятна выросшего грибка спустя три дня после инокуляции. Образцы затем выдерживали при 25оС в течение 5 дней, после чего противогрибковая активность определялась с помощью визуального наблюдения заражения образцов грибком.
Испытуемыми грибками были: Aspergillus niger; Gliocladium virens и Fusarium moliniforme.
Результаты показаны в табл. 13.
ЭКСПЕРИМЕНТ 11. Активность по предохранию древесины.
Процедуру, описанную в эксперименте 10, повторяли, за исключением того, что испытуемыми грибками были Coriоlus versicolor и Tyromyces palustris, которые ранее описаны в I/S А-9302.
Результаты показаны в табл. 14.
Соединения формулы (1) и их соли полезны как фунгициды и фунгистатические агенты для сельскохозяйственного, садоводческого и аналогичных видов использования. Для такого применения они могут преобразовываться или формулироваться в общепринятые препаративные формы для такого использования, и они могут применяться по отношению к растениям, частям растений, воспроизводительным частям растений или к местам распространения или к среде, содержащей такие растения, части растений или части для их воспроизводства, как это хорошо известно для обычных фунгицидов и других сельскохозяйственных химикатов.
В частности, было найдено, что они являются высокоэффективными противогрибковыми агентами, не причиняя вреда растениям, по отношению к которым они применяются.
Так, например, ризоктониоз, важная болезнь, поражающая рисовые культуры, может предотвращаться при использовании соединения в форме препарата для опрыскивания или распыления, или для применения в условиях погружения. Когда соединения используются для обработки почвы или семян, они проявляют особенную эффективность по предотвращению выпревания или увядания таких культур, как свекла, хлопок или огурцы, являющиеся результатом заражения грибком Rhizostonia и почвенных инфекционных болезней, таких как склероциальная гниль баклажанов или огурцов и др. или войлочная болезнь (белая ножка) картофеля.
В количестах, используемых на практике, такие культуры, как рис, томаты, картофель, хлопок, баклажаны, огурцы или фасоль, не повреждаются соединениями настоящего изобретения.
Кроме того, данные соединения могут также применяться по отношению к садам, неполевым угодьям, лесам и аналогичным.
Отражая активность соединения настоящего изобретения, изобретение далее представляет композиции, которые содержат одно или более соединений изобретения вместе с носителем и необязательно другими вспомогательными агентами, если необходимо. Эти соединения могут формулироваться в виде препаратов типа, обычно применяемого для сельскохозяйственного или садоводческого использования в виде дустов, грубых дустов, микрогранул, тонкодисперсных гранул, смачиваемых порошков, эмульгируемых концентратов, водных или масляных суспензий и аэрозолей. Конечно нет необходимости использовать совершенно чистую форму соединения изобретения в композиции, и, конечно, очистка может проводиться на любой стадии или может быть отложена, и получающееся неочищенное вещество может использоваться в качестве активного ингредиента композиции.
Применяемым носителем в таких композициях могут быть природные или синтетические и органические или неорганические вещества; они обычно применяются для того, чтобы способствовать достижению активным ингредиентом обрабатываемого субстрата, и для того, чтобы облегчить хранения,транспортировку или обращение с активным соединением. Он может быть твердым, жидким или газообразным.
Подходящие твердые носители включают неорганические вещества, такие как глины (примерами которых являются бентонит, каолинит, монтмориллонит, и аттапульгит), тальк, слюда, агальматолит, пирофиллит, пемза, вермикулит, гипс, карбонат кальция, доломит, диатомовая земля, карбонат магния, аппатит, цеолит, кремниевый ангидрид и синтетический силикат кальция; растительные органические вещества, такие, как щелуха орехов (например, различные виды орехов, в скорлупе и других) соевая мука, табачный порошок, порошок ореховой скорлупы, пшеничная мука, древесная мука, крахмал и кристаллическая целлюлоза; синтетические или природные высокомолекулярные полимеры, особенно смолы, такие как кумароновая смола, нефтяные смолы, алкидные смолы, поливинилхлорид, полиалкиленгликоли, кетоновые смолы, сложноэфирные смолы, ксантоновая смола, смола копала и дамарровая смола; воски, такие как карнаубский воск и пчелиный воск; или мочевина.
Примеры подходящих жидких носителей включают парафиновые или нафтеновые углеводороды, такие как керосин, минеральное масло, машинное масло и вазелин или белое масло; ароматические углеводороды, такие как бензол, толуол, ксилол, растворитель нафта, этилбензол, кумол, и метилнафталин; галоидированные углеводороды, особенно хлорированные углеводороды, такие как четыреххлористый углерод, хлороформ, трихлорэтилен, монохлорбензол и о-хлортолуол; простые эфиры, такие, как диоксан и тетрагидрофуран; кетоны, такие как ацетон, метилэтилкетон, диизобутилкетон, циклогексанон, ацетофенон и изофорон; сложные эфиры, такие как этилацетат, амилацетат, этиленгликольацетат, диэтиленгликольацетат, дибутилмалеат и диэтилсукцинат; спирты, такие как метанол, этанол, изопропанол, гексанол, этиленгликоль, диэтиленгликоль, циклогексанол, и бензиловый спирт; эфироспирты, такие как этиленгликоль-моноэтиловый эфир, этиленгликоль-монофениловый эфир, диэтиленгликоль-моноэтиловый эфир и диэтилен- гликоль-монобутиловый эфир; другие полярные растворители, такие как диметилформамид и диметилсульфоксид; и вода.
Подходящие газообразные носители включают воздух, азот, двуокись углерода и фторуглеродные пропелленты, такие как вещества, продаваемые под товарным знаком "Фреон", они могут смешиваться известным образом, давая пропеллент.
Композиции изобретения могут содержать один или более поверхностных агентов и/или полимеров для улучшения свойств композиций и для того, чтобы способствовать их диспергированию, эмульгированию, распределению, проникновению связи, или для регулирования дезинтеграции, улучшения текучести или придания коррозионной стойкости композиции, или для стабилизации активного соединения. Могут применяться любые из общепринятых классов поверхностно-активных агентов (неионные, анионные, катионные или амфотерные), но предпочитается применять неионные и/или анионные поверхностно-активные агенты, благодаря чему может улучшаться смачивание, адгезия и абсорбция и достигаться желательные эффекты.
Примеры подходящих неионных поверхностно-активных агентов включают аддукты полимеризации окиси этилена с высшими спиртами, такими как лауриловый спирт, стеариловый спирт и олеиловый спирт; аддукты полимеризации окиси этилена с алкилфенолами, такими как изооктилфенол или нонилфенол; аддукты полимеризации окиси этилена с алкилнафтолами, такими как бутилнафтол или октилнафтол; и аддукты полимеризации окиси этилена с высшими жирными кислотами, такими как пальмитиновая кислота, стеариновая или олеиновая кислота; аддукты полимеризации окиси этилена с моно- или диалкилфосфорными кислотами, такими как стеарилфосфорная кислота или дилаурилфосфорная кислота; аддукты полимеризации окиси этилена с аминами, такими как додециламин; амиды или этоксилированные амиды высших жирных кислот, такие как стеарамид, сложные эфиры высших жирных кислот и многоатомных спиртов, такие как сорбитан, и аддукты полимеризации окиси этилена с ними; сложные эфиры высших жирных кислот и глицеринборатов или этоксилированных глицеринборатов; глицериды и сахароэфиры жирных кислот; и аддукты полимеризации окиси этилена с окисью пропилена.
Примеры подходящих анионных поверхностно-активных агентов влкючают соли высших жирных кислот, т.е. мыла, например, олеат натрия; соли, например натриевые и кальциевые соли сульфоновых кислот и сами эти кислоты, например, лигнинсульфоновой кислоты и арилсульфонатные соли, такие как изопропилнафталинсульфонат натрия, метиленбионафталинсульфонат натрия, лигнинсульфонат натрия или додецилбензолсульфонат натрия, или алкилсульфонатные соли, особенно диалкилсульфосукцинаты натрия, такие как диоктилсульфосукцинат натрия или 2-этилгексенсульфонат натрия; соли, например, натриевые, аммониевые и аминовые соли, сульфатов полиоксиэтиленалкиларилового эфира или сульфатов полиоксиэтиленалкилового эфира или свободных кислот; или соли фосфатов полиоксиэтиленалкил арилового эфира или полиоксиэтиленалкилфосфатов; алкилсульфатные соли, такие как лаурилсульфат натрия или олеилсульфатная соль амина.
Примеры подходящих катионных поверхностно-активных веществ включают высшие алифатические амины и конденсаты окиси этилена; четвертичные аммониевые соли, например, хлориды; N-алкиламинацетаты; и N-алкиламиноксиды.
Примеры амфотерных поверхностно-активных веществ включают бетаины и поверхностно-активные вещества аминокислотного типа.
Кроме того, композиции настоящего изобретения могут использоваться в сочетании с соединениями с высоким молекулярным весом или другими агентами готовых препаративных форм, например: защитными коллоидами, такими как казеин, желатин, аравийская камель, альбумин, клей, альгинат натрия, карбоксиметилцеллюлоза, метилцеллюлоза, гидроксиэтилцеллюлоза или поливиниловый спирт; с диспергирующими агентами, такими как полифосфат натрия; неорганическими диспергирующими агентами, такими как бентонит или веегам; стабилизаторами; связующими агентами; и антифризными агентами. Для более широкого применения и экономии труда композиция изобретения может, если необходимо, сочетаться с одними или более другими сельскохозяйственными химикатами, например фунгицидами, инсектицидами, гербицидами, регуляторами роста растений и удобрениями.
Вышеупомянутые носители и разнообразные вспомогательные агенты могут использоваться по одному или в любом нужном сочетании в зависимости от типа препарата, вида применения и других факторов. Аналогичные факторы также важны при определении концентрации активного ингредиента в препаративной форме.
Например, дусты могут содерожать от 0,1 до 25% по весу активного соединения, причем остаток является твердым носителем. Смачиваемые порошки могут содержать, например, от 1 до 90% предпочтительно от 25 до 80% по весу соединения, а остальное является твердым диспергирующим и смачивающим агентом, если требуется, вместе с защитным коллоидным агентом, тиксотропным агентом и пеногасящим или противопенящим агентом.
Гранулы могут содержать от 1 до 35% по весу активного соединения, причем большую часть остатка составляет твердый носитель. Активное соединение гомогенно смешивается с твердым носителем, или приклеивается к поверхности, или адсорбируется на поверхности носителя; диаметр каждой гранулы составляет предпочтительно от 0,2 до 1,5 мм.
Эмульсируемые концентраты могут обычно содержать, например, от 5 до 50% по весу активного соединения и от 5 до 20% по весу эмульгирующего агента, а остаток составляет жидкий носитель, если требуется, вместе с ингибитором коррозии.
Масляные препараты могут содержать от 0,5 до 5% по весу активного соединения, а остаток составляет жидкий носитель, такой как керосин.
Аэрозоли могут содержать от 0,1 до 5% по весу активного соединения и необязательно ароматизирующий агент, а остаток составляет масляный и/или водный носитель, и пропеллент, такой как сжиженный нефтяной газ, фторуглерод или двуокись углерода.
Композиции изобретения могут применяться, например, по отношению к заливным или другим полям, перед или после появления болезни у растений, или по отношению к растениям, уже зараженным вредными грибками; подходящей обычно является концентрация активного ингредиента от 10 до 500 ч./млн. особенно для применения по отношению к листьям и стеблям растений и к почве, посредством чего может достигаться эффективное подавление болезни.
Композиция изобретения может смешиваться с другим фунгицидом для достижения более широкого спектра фунгицидного действия, и, в некоторых случаях, можно ожидать достижения синергического эффекта. Подходящие другие фунгициды включают: фунгициды карбаматного типа, такие как 3,3-этиленбис/тетрагидро-4,6-диметил-2Н-1,3,5-тиадиазин-2-тион, этиленбисдитиокарбамат цинка или марганца, бис(диметилдитиокарбамоил)дисульфид, пропиленбисдитиокарбамат цинка, метил 1-(бутилкарбамоил)-2-бензимидазолкарба- мат, 1,2-бис(3-метоксикарбонил-2-тиоуреидо)бензол и бисдиметилдитиокарбамоилцинк-этиленбисдитиокарбамат;
функцигиды дикарбоксимидного типа, такие как N-трихлорметилтио-4-циклогексен-1,2-дикарбоксимид и N-тетрахлорэтилтио-4-циколгексен-1,2-дикарбоксимид;
фунгициды оксазинового типа, такие, как 5,6-дигидро-2-метил-1,4-оксазин-3-карбоксанидид-4,4-диоксид;
фунгициды нафтахинонового типа, такие как 2,3-дихлор-1,4-нафтохинон;
и другие фунгициды, такие как 3-гидрокси-5-метилизоксазол, 5-этокси-3-трихлорметил-1,2,4-тиадиазол, 2,4-дихлор-6-(о-хло- ранилино)-1,3,5-триазин, 2,3-дициано-1,4-дитиоантрахинон, 8-хинолят меди, полиоксин, валидамицин, тетрахлоризофталонитрил, 2-(1-метилпропил)-4,6-динитрофенол, ββ -диметилакрилат, гидроокись трифенилолова, фитомицин, динитрометилгептилфенилкротонат, 5-бутил-2-диметиламино-6-метилпиримидин-4-ол, 6-(3,5-дихлор-4-метил- фенил)-3-(2Н)пиридазинон, 6-(3-бромфенил)-3-(2Н)-пиридазинон, N-(2,6-диметилфенил)-N-метоксиацетанилинметиловый сложный эфир и бис(8-гуанидинооктил)амидацетат.
Композиции изобретения могут смешиваться с инсектицидами.
Подходящие инсектициды включают: фосфорсодержащие инсектициды, такие как 0,0-диэтил 0-(2-изопропил-4-метил-6-пиримидинил)фосфоротиоат, 0,0-диэтил S-[2-(этилтио)этил] фосфродитиоат, 0,0-диметил 0-(3-метил-4-нитрофенил)тиофосфат, 0,0-диметил S-(N-метилкарбамоилметил)фосфородитиоат, 0,0-диметил S-(N-метил-N- формилкарбамоилметил)фосфородитиоат, 0,0-диметил S-[2-(этилтио)этил] фосфонодитиоат, 0,0-диметил-1-гидрокси-2,2,2-трихлорэтилфосфонат, 0,0-диэтил-0-(5- фенил-3-изоксазолил)фосфоротиоат, 0,0-диметил-0-(3-метил-4-метилмеркаптофенил)-тиофосфат, 0-этил-0-п-цианофенил фенилфосфонотиоат, 0,0-диметил-S-(1,2-дикарбоэтоксиэтил)фосфородитиоат, 2-хлор-1- (2,4,5-трихлорфенил)винилдиметил-фосфат, 2-хлор-1-(2,4-дихлорфенил)винилдиметил-фосфат, 0,0-диметил-0-п-цианофенил-фосфоротиоат, 2,2-дихлорвинилдиметилфосфат, этилмеркаптофенилацетат, 0,0-диме- тилфосфородитиоат, S-[(6-хлор-2-оксо-3-бензооксазолинил)-метил] -0,0-диэтилфос-фородитиоат, 4-метилтиофенил-дипропилфосфат, 2-хлор-1-(2,4-дихлорфенил)винилдиэтилфосфат, 0,0-диэтил-0-(3-оксо-2- фенил-2Н-пиридазин-6-ил)фосфоротиоат, 0,0-диметил S-(1-метил-2-этилсульфинил)этилфосфоротиоат, 0,0-диметил S-фталимидометилфосфородитиоат, диметил- метилкарбомоилэтилтиоэтил-тиофосфоро- тиоат, 0,0-диэтил S-(N-этоксикарбонил-N-метилкарбамоилметил)фосфородитиоат, 0,0-диметил-S-[2-метокси-1,3,4-диадиазол-5(4Н)-онил-(4)-метил] дитиофосфат, 2-метокси-4Н-1,3,2-бензодиоксафосфорин-2- сульфид, 0,0-диэтил-0-(3,5,6-трихлор-2-пиридид)фосфоротиоат, 0, S-диметил-N-ацетилфосфороамилотиоат, 0-2,4-дихлорфенил 0-этил-S-пропилфосфородитиоат, 0,0-диэтил S-(2-хлор-1-фталимидоэтил)фосфородитиоат и 0-6-этокси-2-этилпиримидин-4-ил 0,0-диметилфосфоротиоат;
инсектициды карбаматного типа, такие как 1-нафтил N-метилкарбамат, S-метил-N-[метилкарбамоилокси] тиоацетоимидат, 2-втор-бутил-фенил-N-метилкарбамат, 2-изопропоксифенил-N-метилкарбамат, 1,3-бис-(карбамоилтио)-2-(N,N-диметиламино)- пропан-гидрохлорид, и 2-диметиламино-5,6-диметилпиримидин-4-ил-диметилкарба- мат;
и другие инсектициды, такие как никотинсульфат, милбемицид D, 6-метил-2,3-хиноксалиндитиоциклический, S,S-дитиокар- бонат, 2,4-динитро-6-втор-бутилфенилдиметилакрилат, 1,1-бис-(п-хлорфенил)-2,2, 2-трихлорэтанол, азоксибензол, ди(п-хлорфенил)-циклопропил-карбинол, изопропил-4,4'-дихлорбензилат, этил 4,4'-дихлорбензилат, этил 0-бензоил(3-хлор-2,6-диметоксибензогидроксимат, изопропил 4,4'-дибромбензилат, гидроокись трициклогексилолова, гексакис(β, β-диметилфенэтил)-дистан- оксан, 2-(4-трет-бутилфенокси)циклогексил-пропинилсульфид, 3-метил-1,5-бис- (2,4-ксилил)-1,3,5-триазапента-1,4-диен, 2,4, 5,4'-тетрахлордифенилсульфон, гексахлоргексагидрометанобензодиоксатиепиноксид, кислый оксалат 5-диметиламино-1,2,3-тритиана и машинное масло.
Однако природа или характер любого такого дополнительного инсектицида не является, критическим, т.е. определяющим. Дополнительно, при желании, соединения настоящего изобретения могут смешиваться с другими общепринятыми сельскохозяйственными или садоводческими материалами, такими как акарициды, нематоциды, гербициды, регуляторы роста растений, навоз или вещества, кондиционирующие почву, для получения композиций, имеющих более широкий спектр применения и/или для снижения трудовых затрат.
Количество используемого соединения настоящего изобретения варьирует в зависимости от погодных условий, типа применения, времени применения, способа применения, характера окружающей среды, характера болезни, типа растения и различных других известных факторов, но предпочтительно соединение может применяться в количестве от 0,1 до 100 г эффективного ингредиента на ар, предпочтительно от 5 до 40 г. Эмульгируемые концентраты, смачиваемые порошки, суспензионные концентраты и аналогичные предпочтительно применяются с помощью разбавления заданного количества, например, с 1-10 л воды на ар, а гранулы обычно применяются без разбавления. При желании к воде, используемой для разбавления, могут добавляться другие добавки, такие как агенты, распределения или спредеры, например поверхностно-активные агенты, полиоксиэтилен-смоляная кислота, лигнинсульфонаты, соли абиетиновой кислоты, динафтилметандисульфонат, парафин.
Соединения настоящего изобретения могут также использоваться в качестве фармацевтических веществ для лечения грибковых заражений, например, кожи, в этих случаях они обычно назначаются для тонического или местного применения, или кишечных инфекционных болезней, в случае которых они могут назначаться орально или парэнтерально. Считают, что они представляют особую ценность при лечении острого микоза, такого как кандидомикоз или кандидиаз.
Когда соединение настоящего изобретения применяется для фармацевтических целей, оно может назначаться в форме любой общепринятой фармацевтической готовой препаративной формы, характер которой, как известно, зависит от способа назначения и характера состояния, подвергаемого лечению. Так, соединения изобретения могут формулироваться в виде обычных дозированных форм, обычно в смеси с фармацевтическим разбавителем или носителем. Для орального назначения соединения могут преобразовываться, например, в форму таблеток, капсул, гранул, порошков или сиропов. Для парэнтерального назначения они могут формулироваться в виде инъекционных препаратов в подходящей жидкости или в виде медицинских свечей. Для топического назначения они могут формулироваться в виде мазей, кремов, порошков, жидкостей или аэрозолей. Эти фармацевтические препараты могут производиться с помощью общепринятых средств с использованием вспомогательных агентов, обычно известных в данной области, таких как эксципиенты, разбавители, диспергирующие средства, связующие, дезинтеграторы, смазочные агенты, стабилизаторы, корригенты и аналогичные.
Дозировка и частота применения могут варьировать в зависимости от симптомов, возраста и веса тела пациента, а также от способа назначения, и обычно соединения изобретения могут назначаться орально в суточной дозе от 50 до 2000 мг для взрослого, предпочтительно в дозе от 100 до 600 мг, которая может применяться или в виде единственной дозы, или в виде разделенных на несколько доз.
| название | год | авторы | номер документа |
|---|---|---|---|
| КРЕМНИЙСОДЕРЖАЩИЕ СОЕДИНЕНИЯ ИЛИ ИХ СОЛИ, ОБЛАДАЮЩИЕ ФУНГИЦИДНОЙ АКТИВНОСТЬЮ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2069213C1 |
| МАКРОЛИДНЫЕ СОЕДИНЕНИЯ ИЛИ ИХ СОЛИ, ИЛИ ИХ СЛОЖНЫЕ ЭФИРЫ | 1993 |
|
RU2049785C1 |
| ПРОИЗВОДНЫЕ КАРБАПЕНЕМА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2097383C1 |
| α,ω ДИАРИЛАЛКАНЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2105752C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОТИВООПУХОЛЕВОГО КОМПЛЕКСА ПЛАТИНЫ | 1988 |
|
RU2007413C1 |
| ЧЕТЫРЕХКООРДИНАЦИОННЫЕ КОМПЛЕКСЫ ДВУХВАЛЕНТНОЙ ПЛАТИНЫ | 1992 |
|
RU2039064C1 |
| ПРОИЗВОДНЫЕ АЗЕТИДИНОНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2047602C1 |
| ПРОИЗВОДНЫЕ БЕНЗОПИРАНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2038354C1 |
| СПОСОБ ПОЛУЧЕНИЯ ОПТИЧЕСКИ АКТИВНОГО ПРОИЗВОДНОГО ИНДОЛОБЕНЗОХИНОЛИНА ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ | 1990 |
|
RU2045528C1 |
| ПРОИЗВОДНЫЕ ТИАЗОЛИДИН-2,4-ДИОНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2103265C1 |

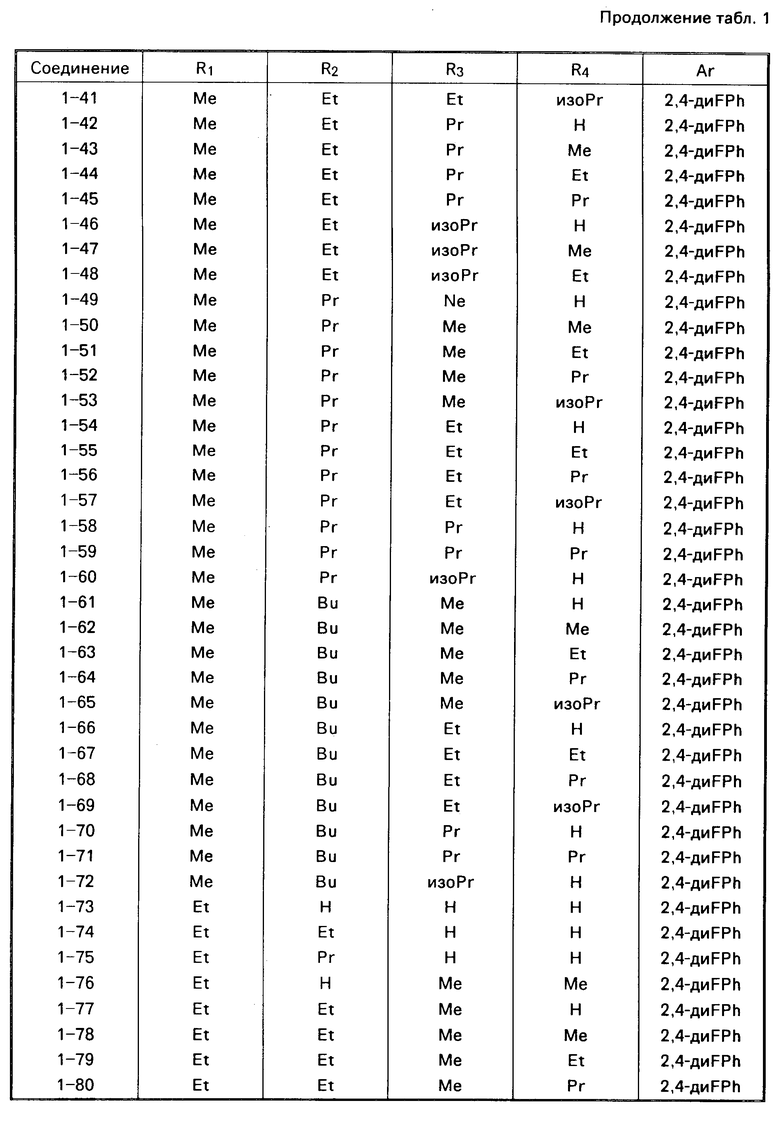
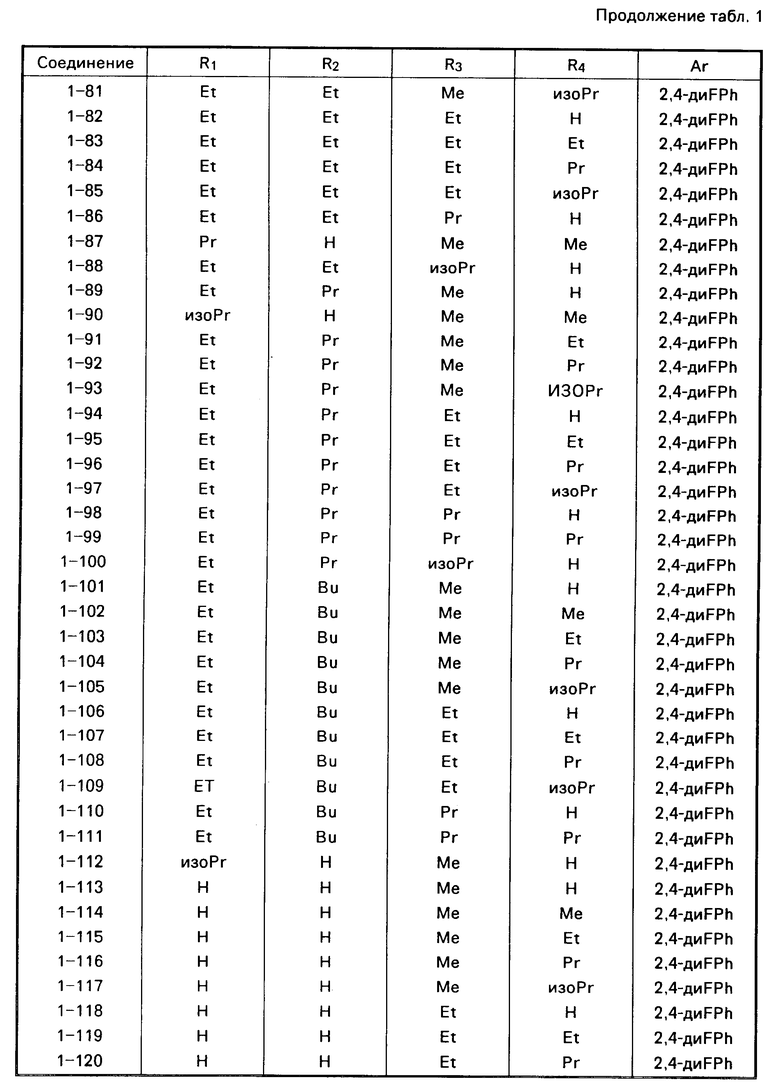
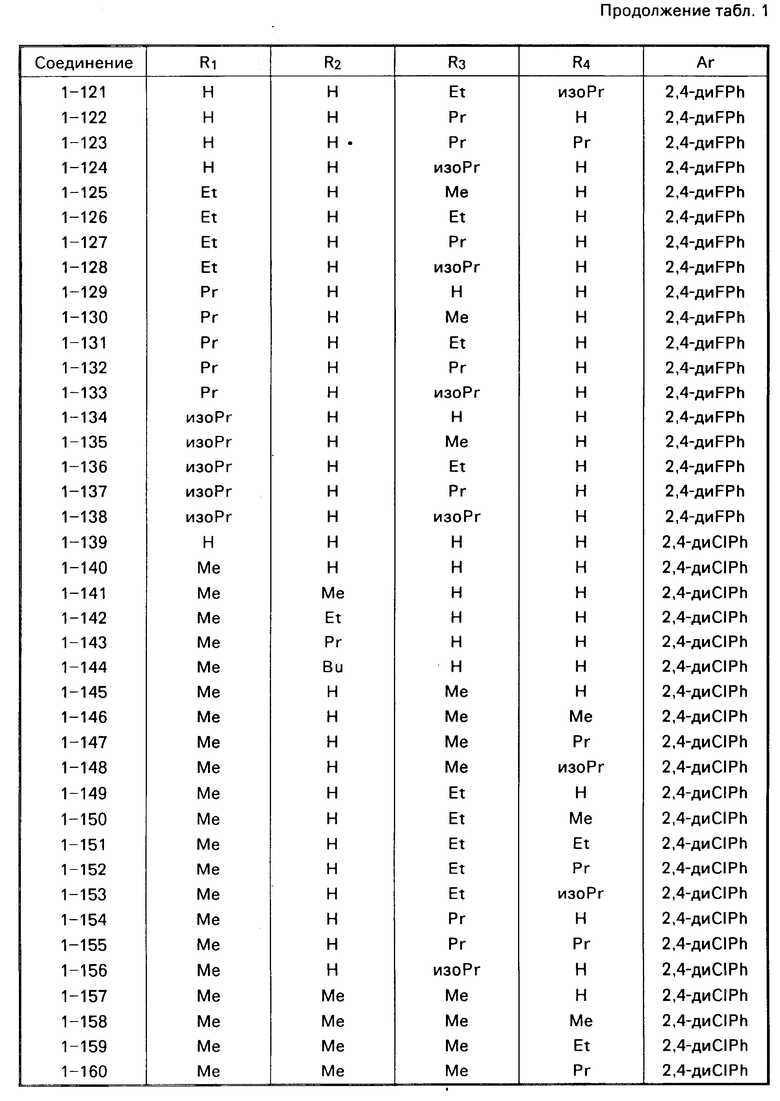
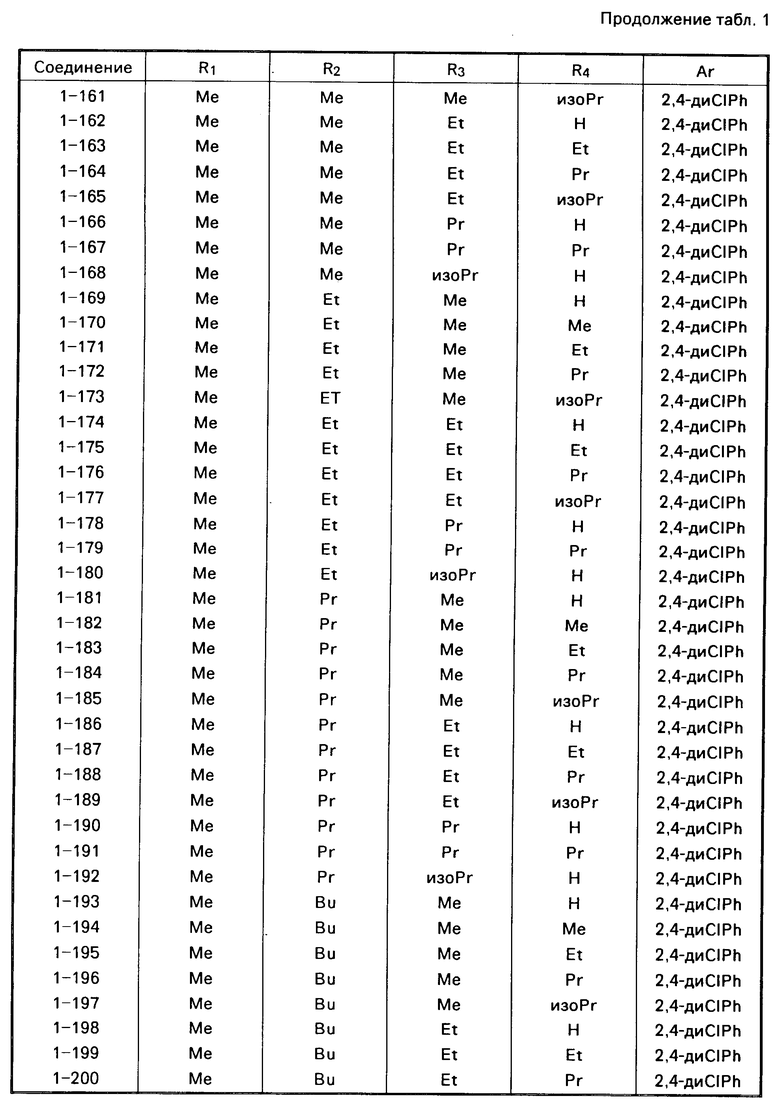
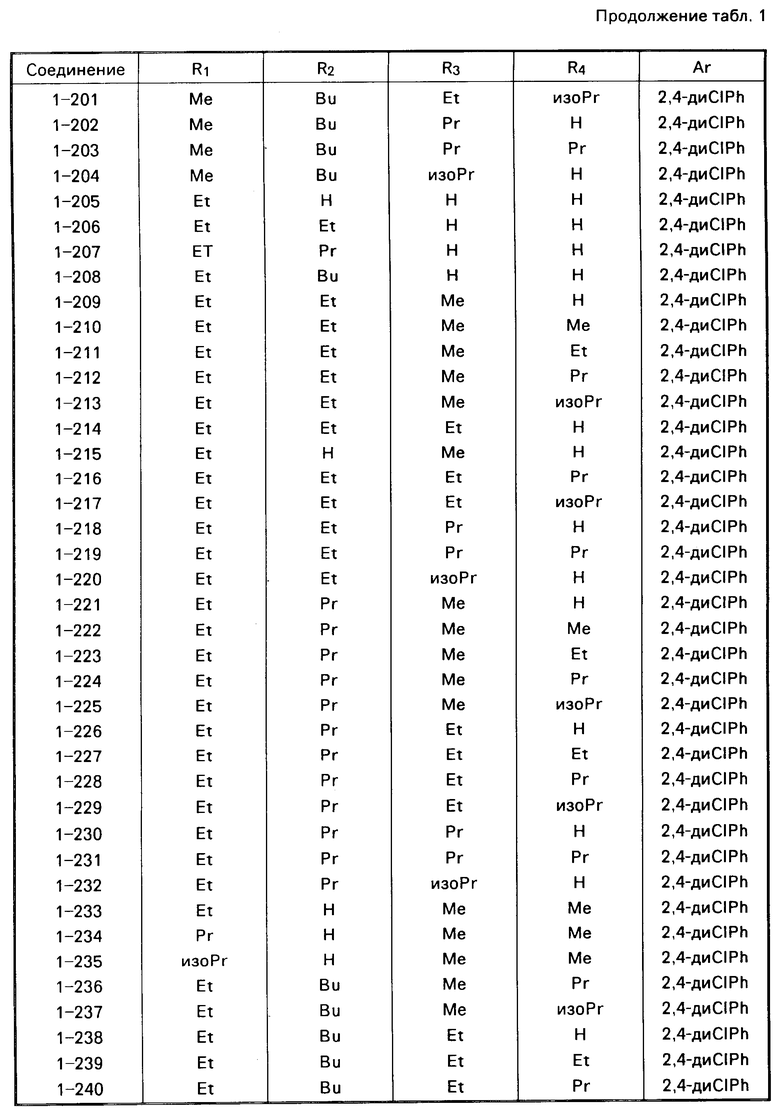
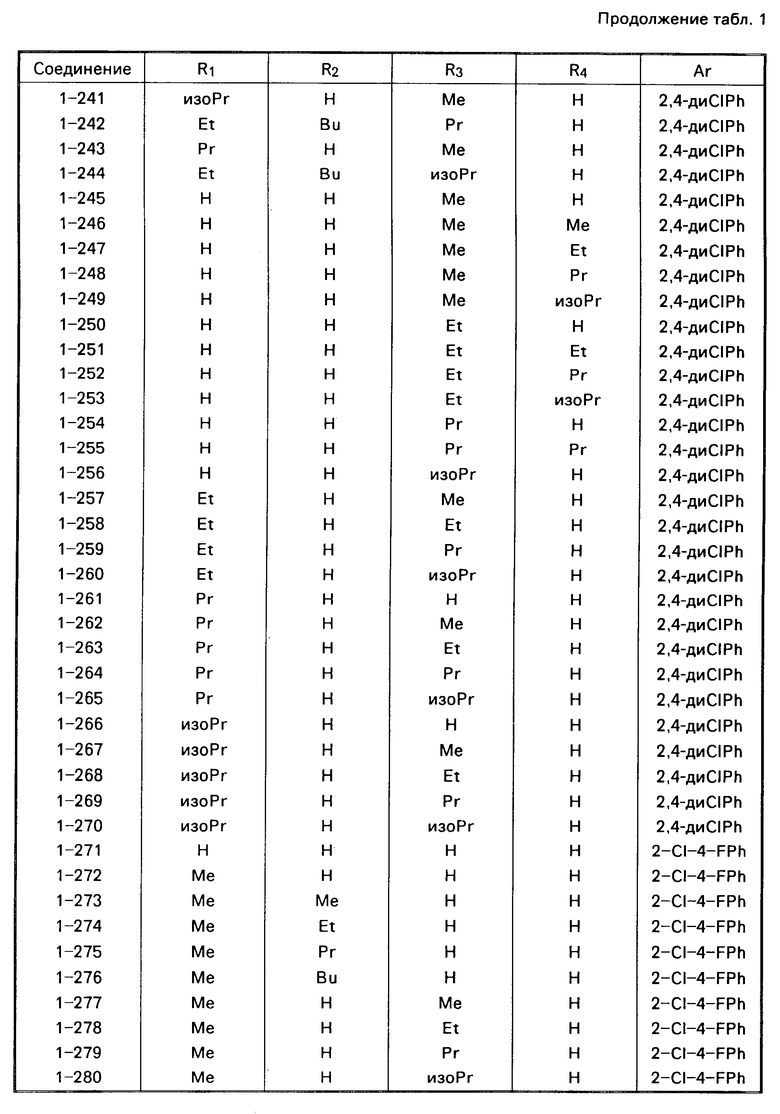
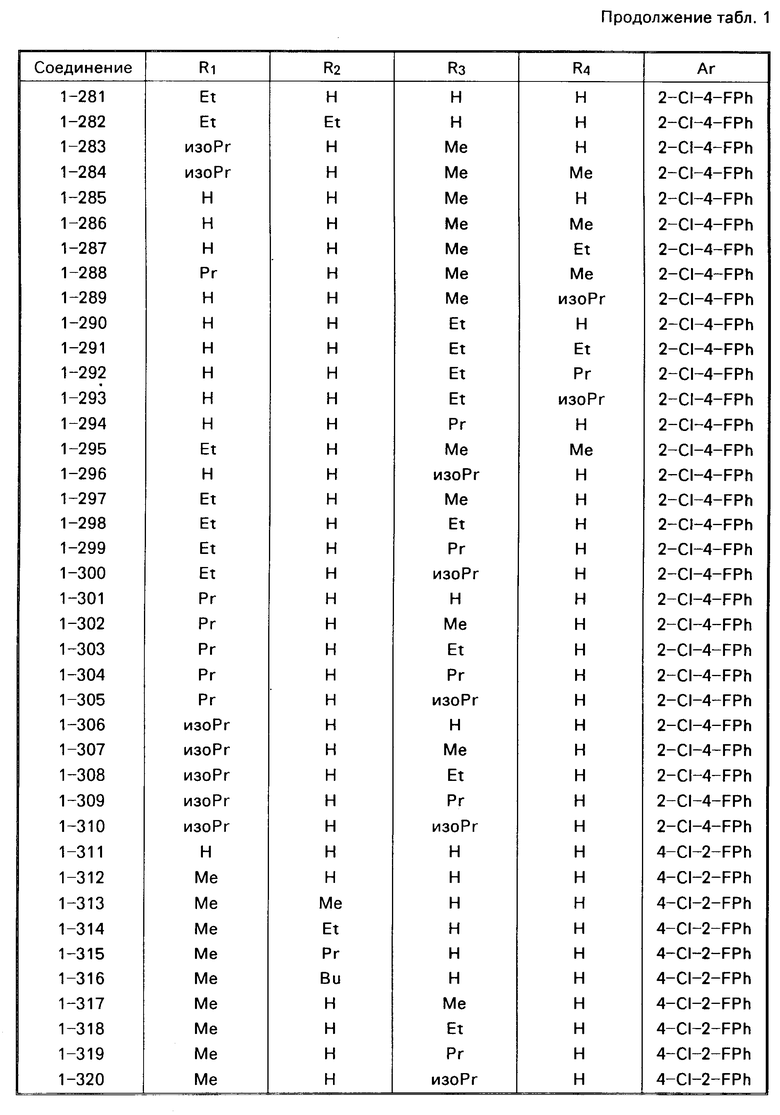
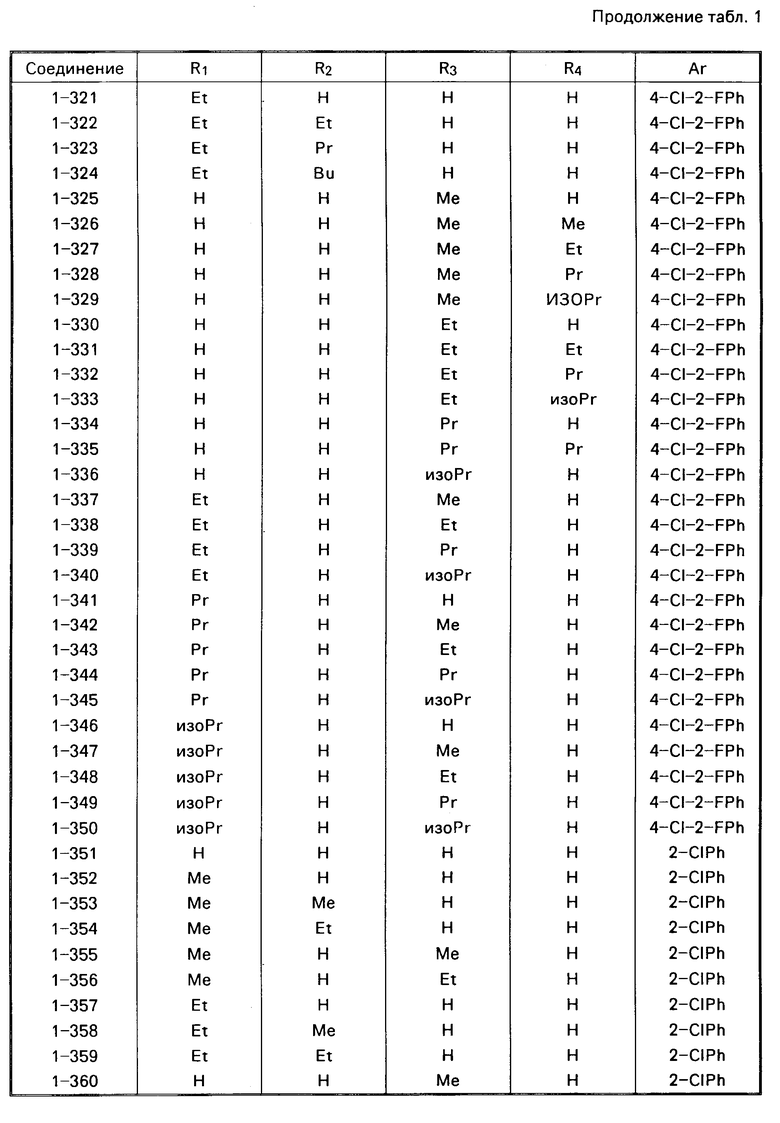
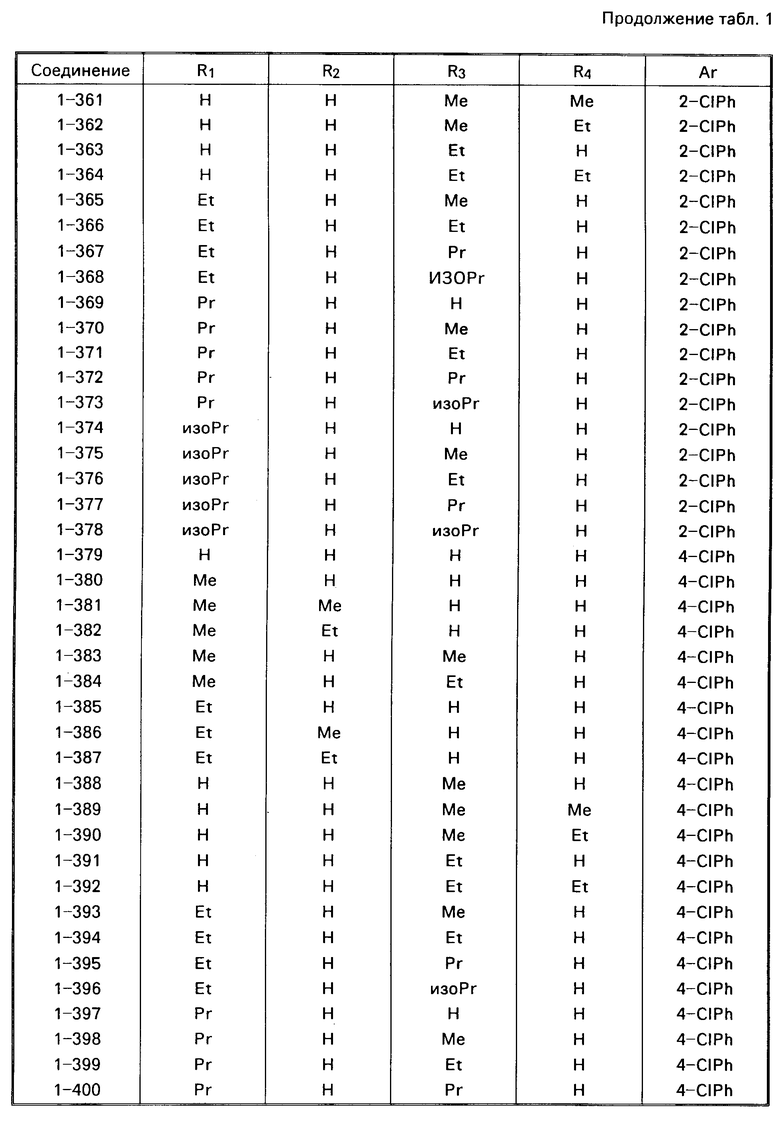
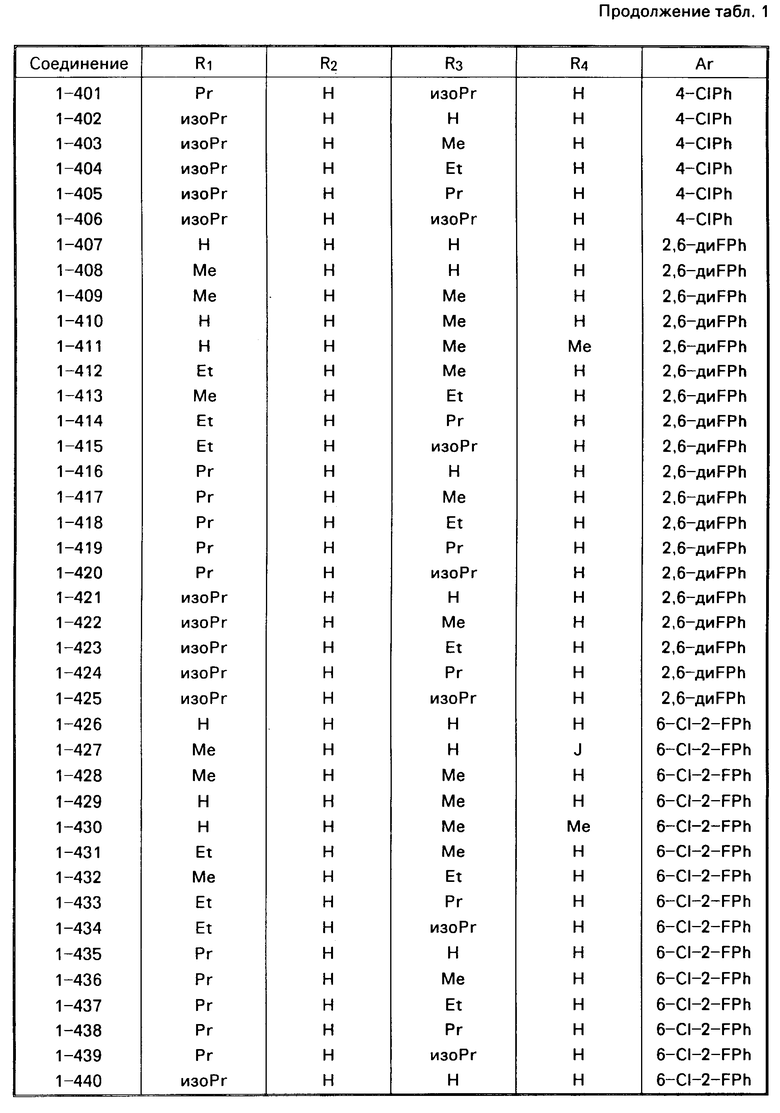
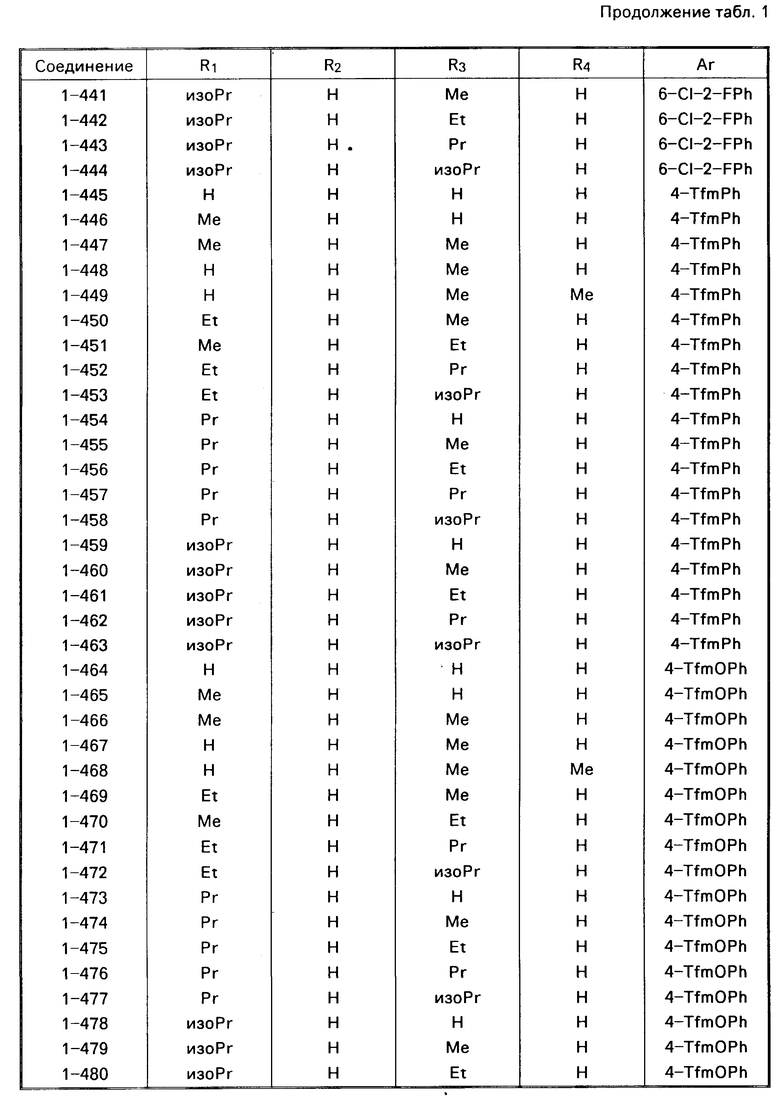
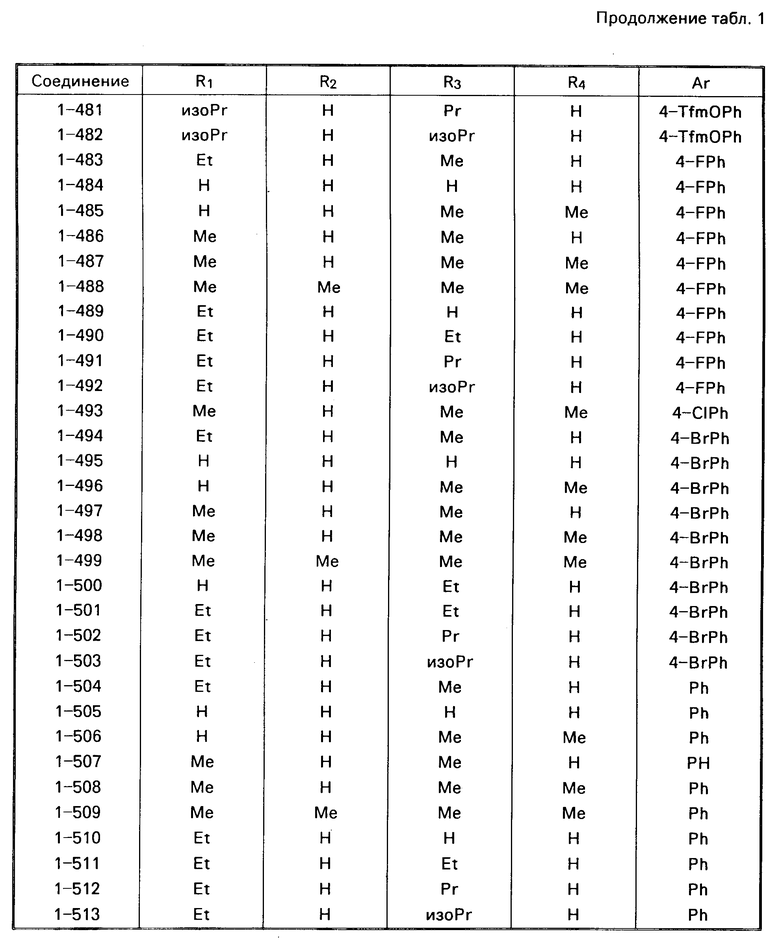
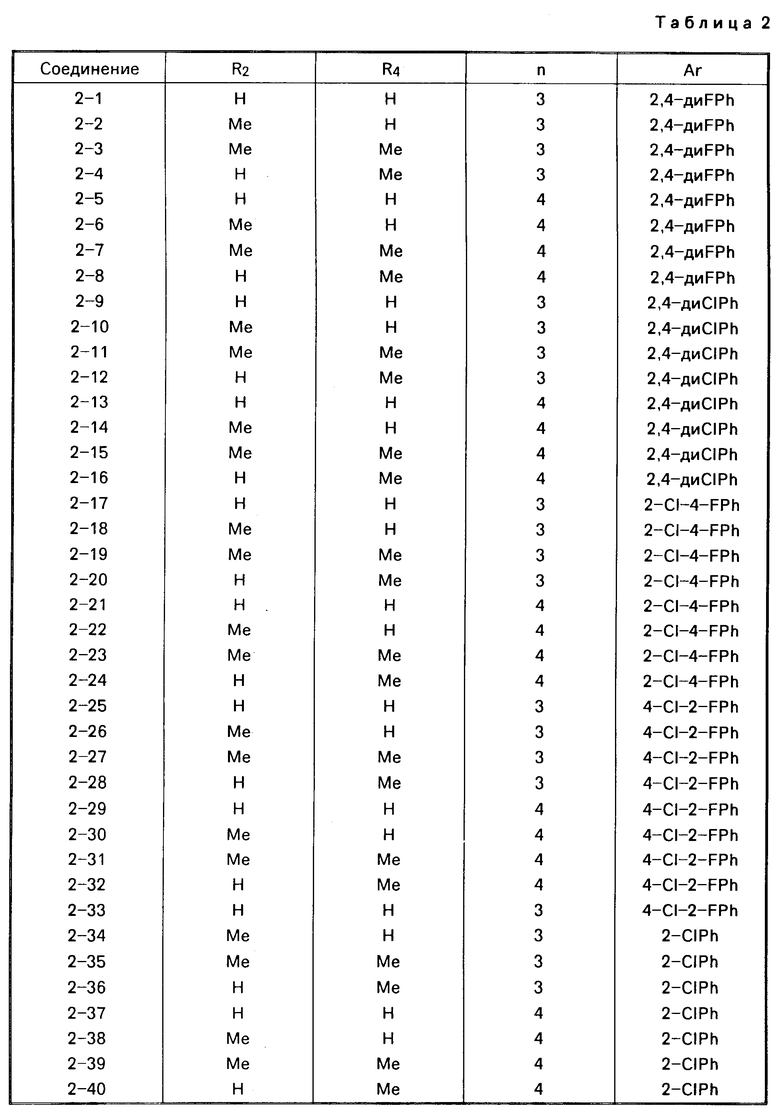
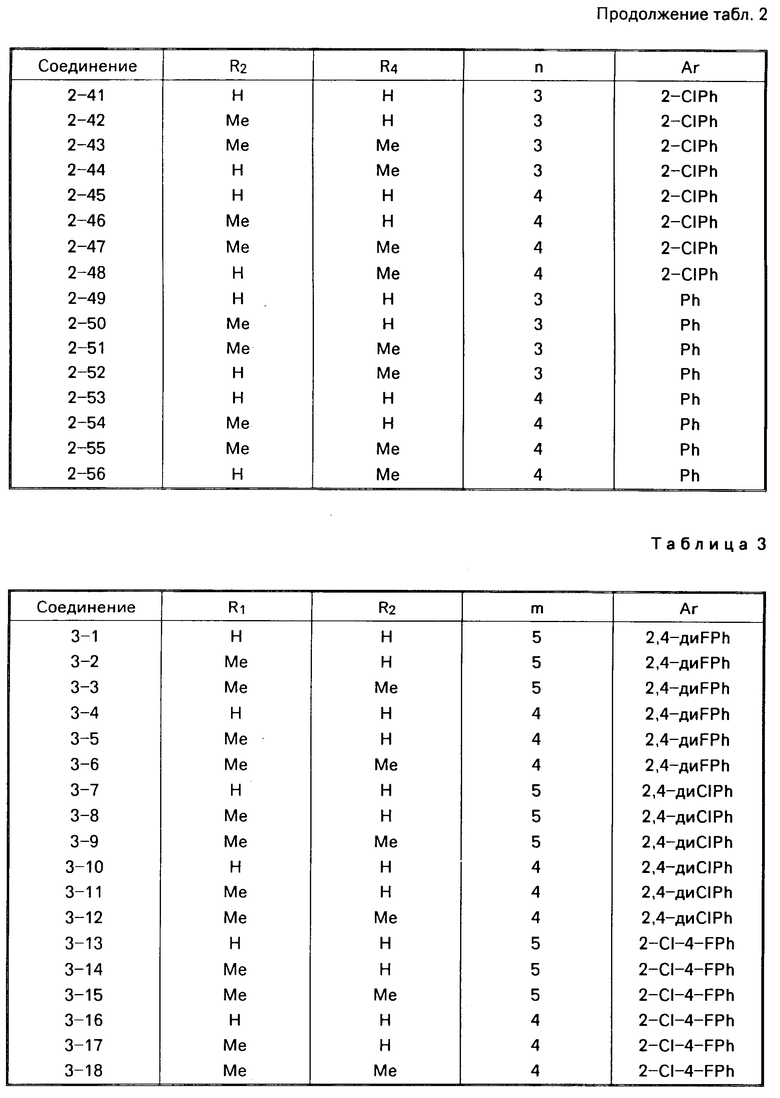
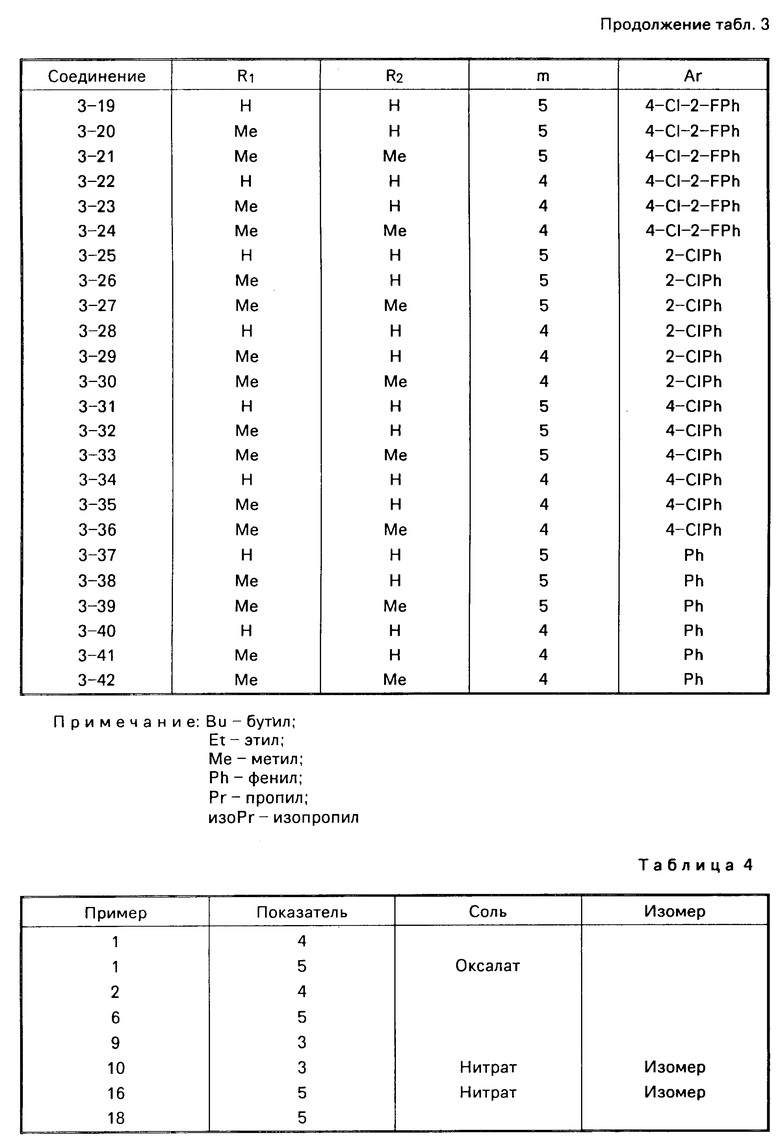
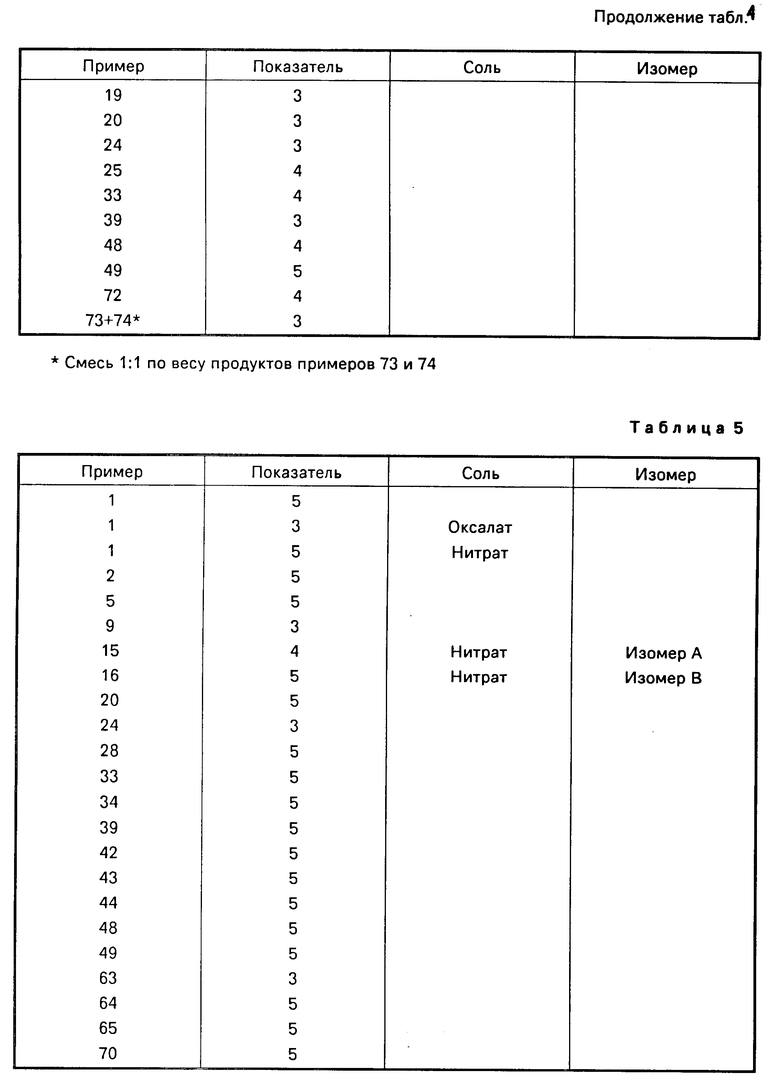


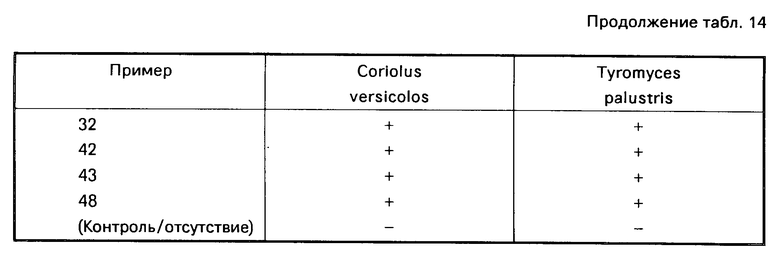
Использование: в сельском хозяйстве в качестве фунгицида. Сущность изобретения: продукт-производные октана ф-лы I, где R1-R9 имеют соответствующие значения, и их соли. Эти соединения являются высокоэффективными противогрибковыми агентами, не причиняя вреда растениям, по отношению к которым они применяются. Структура соединения ф-лы I 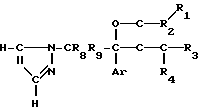 27 з. п. ф-лы.
27 з. п. ф-лы.
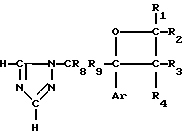
где R1 и R2 одинаковые или различные, каждый водород или С1-С6 -алкильная группа, или R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют С3-С6 -циклоалкильную группу;
R3 и R4 одинаковые или различные, каждый водород, С1-С6 -алкильная группа или фенил, или R1, R3 и атомы углерода, к которым они присоединены, вместе образуют циклоалкильную группу, имеющую 5 или 6 кольцевых атомов и сконденсированную с оксетановым кольцом;
Ar фенил, замещенный радикалами R5, R6 и R7, где R5, R6 и R7 одинаковые или различные, каждый водород, галоген, С1-С6 -алкильная группа, С1-С6 -алкоксигруппа, С1-С6 -галоидалкильная группа или С1-С6 -галоалкоксигруппа;
R8 и R9 одинаковые или различные, каждый водород или С1-С4 -алкильная группа,
и их соли.
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| Guen et al"Protective Groups in Organic Synthesis", 1981, опубл | |||
| Niley - jnterscience, гл | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
