Изобретение относится к области синтеза органических соединений и касается новых производных имидазола, являющихся промежуточными продуктами в синтезе производных имидазола, обладающих ценными биологическими свойствами. Указанные биологически активные производные имидазола являются объектом основной заявки N 48944882/04, по которой принято решение Роспатента от 27 февраля 1995 г. о выдаче патента России и из которой выделена данная заявка.
Объектом данной заявки являются новые производные имидазола общей формулы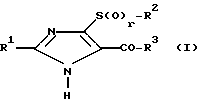
где R1 бутил;
R4 атом водорода или алкил (C1-C6);
R2 алкил (C1-C4);
R3 атом водорода или группа -OR4;
r 0, 1 или 2.
Получение новых соединений согласно изобретению описано в приведенном ниже примере.
Эти соединения участвуют в синтезе производных имидазола общей формулы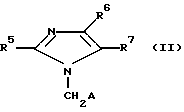
гду R5 алкил (С3-С7);
R7 группа формила, карбоксила, (C1-C4)-алкоксикарбонила или гидроксиметила;
R6 хлор или группа S(O)r-R8;
R8 алкил (C1-C6);
A гетероцеклический остаток из группы: имидазо-(1,2a)-пиридил, имидазо-(1,2a)-пиримидил, бензоксазолил, бензо-(b)-тиофенил, причем этот остаток может быть замещен одним или двумя одинаковыми или различными остатками, выбранными из группы: хлор, тетразолил, карбоксил, карбокси-(C1-C4)-алкил, (C1-C4)-алкоксикарбонил и фенил, возможно замещенный на фтор;
r 0, 1 или 2.
Эти целевые производные имидазола оказались неожиданно высокоэффективными антагонистами рецепторов ангиотензина II, как in vitro, так и in vivo.
Их синтез осуществляется тем, что (в общем случае) соединение общей формулы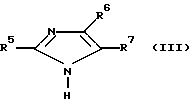
где R5 и R7 имеют указанные выше значения;
R6 хлор или группа S-R8, где R8 имеет вышеуказанное значение, причем, когда заместитель R7 содержит карбоксильную группу, то она может быть защищена,
подвергают взаимодействию с соединением формулы
UCH2A1 (IY)
где A1 вышеуказанный гетероциклический остаток, замещенный одним или двумя одинаковыми или различными остатками, выбранными из ряда, содержащего хлор, циано, карбоксил, карбокси-(C1-C4)-алкил, (C1-C4)-алкоксикарбонил и фенил, возможно замещенный фтором, причем, когда заместитель A1 содержит карбоксильную группу, то она может быть защищена;
U отщепляемая группа,
с получением соединений общей формулы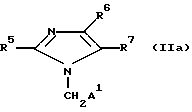
где R5, R7 и A1 имеют вышеуказанные значения;
R6- хлор или группа S-R8, где R8 имеет вышеуказанное значение, причем, когда R7 содержит карбоксильную группу, то она может быть защищена,
и в случае получения соединений формулы II, в которой R6 означает группу S(O)rR8, где R8 имеет вышеуказанное значение, r 1 или 2, соединение формулы IIa, где R6 означает группу S-R8, где R8 имеет вышеуказанное значение, подвергают взаимодействию с окислителем, или в случае получения соединений общей формулы I, в которой заместитель A содержит тетразольный остаток, соединение формулы IIa, где заместитель A1 содержит цианогруппу, подвергают взаимодействию с циклизующим агентом с последующим удалением при необходимости карбоксизащитной группы и выделением целевого соединения в свободном виде или в виде физиологически переносимой соли.
Под физиологически переносимыми солями соединений формулы II понимают их органические и неорганические соли.
Алкилирование происходит согласно известным способам аналогичным образом.
В азолпроизводном формулы I замещают атомом металла атом водорода, соединенный с атомом углерода, например в присутствии основания. Предпочтительными основаниями являются гидриды металлов формулы МН, такие как, например, гидрид лития, натрия или калия, например в ДМФ или ДМСО в качестве растворителей, или металлические оксиды формулы МОR, причем R означает метил, этил, трет-бутил, а реакция проводится в соответствующем спирте ДМФ или ДМСО. Образованные таким образом соли азолов растворяются в апротонном растворителе, как ДМФ или ДМСО, и смешиваются с необходимым количеством агента алкилирования.
Альтернативной возможностью депротонизации азолпроизводных представляется, например, реакция с карбонатом калия в ДМФ или ДМСО.
Реакции проводятся при температурах от ниже комнатной до температуры кипения реакционной смеси, преимущественно между +20oC и температурой кипения реакционной смеси в течение приблизительно 1-10 ч.
Соответствующие изобретению соединения формулы I антагонистически влияют на ангиотензин-II-рецепторы, поэтому могут применяться для лечения зависимой гипертензии ангиотензина II. Применение возможно далее при сердечной недостаточности, кардиозащите, инфаркте миокарда, гипертрофии сердца, артериосклерозе, нефропатии, почечной недостаточности, а также сосудистых заболеваниях мозга, таких как транзиторные ишемические приступы и апоплексия. Ниже представлены примеры на получение соединений формулы I, являющихся объектом данной заявки, и конечных соединений формулы II.
Пример 1 (получение соединений формулы I).
К 35 г (0,246 моль) 2-оксима этилового эфира 1-цианоглиоксиаловой кислоты в 350 мл воды и 280 мл насыщенного раствора бикарбоната натрия добавляют при комнатной температуре порционно (15 мин) 119 г дитионита натрия. Вслед затем нагревают 1 ч до 35oC; затем насыщают NaCI и 5 раз экстрагируют с дихлорметаном. После высушивания с хлоридом кальция органическая фаза сгущается. Получают 11,8 г этилового эфира 2-амино-2-циано-уксусной кислоты в виде масла с
Rf(CH2CI2/CH3OH 9/1)=0,6
К 3,6 г указанного выше соединения (28,09 ммоль) в 50 мл сухого CH2CI2 и 2,3 мл (28,09 ммоль) пиридина накапывают при температуре от -5o до 0oC 3,39 мл (28,09 ммоль) валероилхлорида в 5 мл CH2CI2. Затем 1 ч перемешивают при комнатной температуре. Органическая фаза промывается затем 3 раза водой, 1 раз насыщенным раствором NaCI, сушится с хлоридом кальция и сгущается. Кристаллизация из диизопропилового эфира дает 1,7 г этилового эфира 2-циано-2-н.бутил-карбоний-аминоуксусной кислоты.
Rf(CH2CI2/CH3OH 9/1)=0,35
Тпл= 87oC.
К 2,9 г (13,67 ммоль) вышеуказанного соединения и 0,19 мл (1,36 ммоль) триэтиламина в 60 мл абсолютного этанола добавляют при комнатной температуре 2 мл ( 27,26 ммоль) конденсированного метилмеркаптана. Через 3 дня добавляют еще раз 0,5 мл метилмеркаптана. Через следующие 24 ч при комнатной температуре еще раз впрыскивают 0,5 мл метиленмеркаптана и 0,19 мл триэтиламина, перемешивают следующие 24 ч при комнатной температуре. Затем удаляют растворитель и кристаллизуют осадок из диизопропилового эфира. Получают 2,4 г этилового эфира 3-амино-2-н.бутил-карбонил-аминометил-тиоакриловой кислоты.
Rf(CH2CI2/этилацетат 4/1)=0,3
Тпл=120oC.
К 4,17 г (20,0 ммоль) пентахлорида фосфора в 20 мл CH2CI2 прикапывают при -78oC 2,44 г (20 ммоль) 4-диметиламинопиридина в 12 мл CH2CI2. Через 5 мин прикапывают 2,42 г (10,0 ммоль) указанного выше соединения в 25 мл CH2CI2. Затем поднимают температуру до комнатной температуры и разбавляют 30 мл CH2CI2. Через 2 ч при охлаждении льдом добавляют 300 мл Iн. раствора бикарбоната натрия и перемешивают 1 ч. Затем фазы разделяют, водную фазу 3 раза экстрагируют EE, а объединенные органические фазы сушат с хлоридом кальция. Хроматографией на SiO2 с CH2CI2/этилацетат (9/1) получают в виде масла 1,4 г этилового эфира 2-н.бутил-4-метилтиоимидазол-5-карбоновой кислоты.
Rf(CH2CI2/EE 9/1)=0,6
MS (DCJ)= 243 (M++H)
Пример 2 ( получение соединения общей формулы II).
180 мг (0,38 ммоль) соединения общей формулы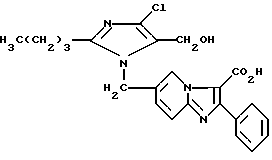
в 2 мл этанола смешивают с 0,5 мл водного Iн. NaOH и перемешивают 20-60 ч при температуре 25oC. Затем раствор сгущают, а осадок очищают хроматографией на SiO2 с CH2CL2/MeOH (8/2) в качестве жидкости-носителя, причем кислота образуется в виде аморфного порошка.
Пример 3 ( получение соединения формулы II).
0,2 г (1,07 ммоль) 2-бутил-4-хлоро-5-формилимидазола, 0,38 г (1,07 ммоль) 6-бромметила-3-этоксикарбонил-2-фенилимидазо-[1,2-a] пирдина, 0,15 г K2CO3 (1,07 ммоль) и 0,5 г порошкообразного молекулярного сита перемешивают 5 ч при температуре 60oC в 5 мл ДМФ. Далее добавляют 100 мл этилацетата и 3 раза промывают водой. После высушивания с Na2SO4 сгущают, а осадок хроматографируют на SiO2 с этилацетат/н.гептан (1/1) в качестве элюента.
MS (DXV) 465 (M+ H+)
Rf[SiO2; этилацетат/н.гептан (1:1)]0,18л
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ АЗОЛЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2047604C1 |
| ИМИДАЗО-АННЕЛИРОВАННЫЕ ИЗО- И ГЕТЕРОЦИКЛЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2076105C1 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПОНИЖАЮЩАЯ КРОВЯНОЕ ДАВЛЕНИЕ | 1992 |
|
RU2104272C1 |
| ПРОИЗВОДНЫЕ ЦИКЛОГЕКСАНКАРБОНОВОЙ КИСЛОТЫ, СМЕСЬ ИХ ИЗОМЕРОВ ИЛИ ОТДЕЛЬНЫЕ ИЗОМЕРЫ И ФИЗИОЛОГИЧЕСКИ ПЕРЕНОСИМЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2140416C1 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА ИЛИ ИХ ФИЗИОЛОГИЧЕСКИ СОВМЕСТИМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, СРЕДСТВО ДЛЯ СНИЖЕНИЯ ВЫСОКОГО ДАВЛЕНИЯ КРОВИ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ СНИЖЕНИЯ КРОВЯНОГО ДАВЛЕНИЯ | 1993 |
|
RU2116300C1 |
| НОВЫЕ ЦИКЛОАЛКИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ РЕЗОРБЦИИ КОСТИ И АНТАГОНИСТОВ РЕЦЕПТОРА ВИТРОНЕКТИНА | 1997 |
|
RU2180331C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2-ЗАМЕЩЕННЫХ 4-(3-ТРЕТ-БУТИЛ-4-ГИДРОКСИФЕНИЛ)-ТИАЗОЛОВ | 1990 |
|
RU2021264C1 |
| ПРОИЗВОДНЫЕ ГУАНИДИНАЛКИЛ-1,1-БИС-ФОСФОНОВЫХ КИСЛОТ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2114855C1 |
| ПРИМЕНЕНИЕ АМИДОВ ИЗОКАЗОЛ-4-КАРБОНОВОЙ КИСЛОТЫ И АМИДОВ ГИДРОКСИАЛКИЛИДЕНЦИАНУКСУСНОЙ КИСЛОТЫ | 1990 |
|
RU2084223C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-ЗАМЕЩЕННЫХ 4-(3-ТРЕТ-БУТИЛ-4-ГИДРОКСИФЕНИЛ)-ТИАЗОЛОВ | 1990 |
|
RU2017739C1 |
Предлагаются новые производные имидазола, являющиеся промежуточными соединениями в синтезе ценных биологически активных лекарственных веществ. 1 з.п. ф-лы.
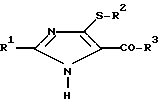
где R1 бутил;
R2 С1 С4-алкил;
R3 группа (OR4), где R4 водород или С1 - С 4-алкил.
| SU, патент, 640662, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
