Изобретение относится к производным имидазола, способу их получения и их применению.
Разработке новых ангиотензин-11-рецептор-антагонистов придают возрастающее значение, принимая во внимание получение новых активных веществ. Из европейского патент A-28834 известны, например, 1-бензил-замещенные производные имидазола, из европейского патента A-253 310 известны производные имидазола с функцией диарилкарбоновой кислоты и из европейского патента A-324 377 производные имидазола с группой диарил-тетразолила и их применение в качестве антагонистов ангиотензин-11-рецепторов.
Кроме того, в европейском патенте A-0503162 представлены 2-n-бутил-замещенный производные имидазола, имеющие структуру бифенилсульфонилмочевины или бифенилсульфонилуретана, и их применение в качестве антагонистов ангиотензин-11-рецепторов.
В данном изобретении описываются новые производные имидазола с боковой цепью бифенилсульфонилмочевины или бифенилсульфонилуретана, которые в положении 2 имидазолового кольца имеют специальный заместитель R1 и неожиданно высокоэффективных ангиотензин-11-рецептор-антагонистов в пробирке и в живом организме.
Изобретение относится к соединениям формулы I: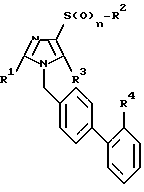
в которой символы имеют следующее значение:
а) R1 обозначает (C4-C3)-алкил, преимущественно n-пропил или этил, но особенно n-пропил;
б) R2 - 1. (C1-C6)-алкил, преимущественно метил,
2. (C3-C7)-циклоалкил,
3. фенил или
4. бензил;
c) R3 - 1. водород,
2. CH2OR5,
3. CO-R6 или
4. O-R7;
d) R4 - 1. CO2NR7R8.
2. SO2-NR8-CO-NR7R9,
3. SO2-NH-COO-R7,
4. SO2-NH-SO2-NR7R9,
5. SO2-NH-CO-R7,
6. SO2-NH-SO2-R7 или
7. SO2N = CH-N(CH3)2;
e) R5 - 1. водород или
2. (C1-C6)-алкил;
f) R6 - 1. водород или
2. OR7;
g) R7, R9 - одинаковые или различные
1. водород,
2. (C1-C6)-алкил, преимущественно метил, этил или пропил,
3. (C3-C8)-циклоалкил,
4. (C3-C6)-циклоалкил-(C1-C3)-алкил,
5. (C6-C12)-арил, преимущественно фенил,
6. (C6-C10)-арил-(C1-C4)-алкил, преимущественно бензил,
7. (C1-C9)-гетероарил, который может быть частично или полностью гидрирован,
8. (C1-C9)-гетероарил-(C1-C3)-алкил, причем гетероарильная часть может быть частично или полностью гидрирована,
9. определенный выше в пунктах 5, 6, 7 и 8 остаток, замещенный 1 или 2 одинаковыми или различными остатками из ряда галогена, гидрокси, (C1-C4)-алкила, метокси, нитро и циано,
10. (C2-C6)-алкенил или (C3-C6)-алкеноил,
11. (C3-C8)-циклоалкенил,
12. (C3-C8)-циклоалкенил-(C1-C3)-алкил,
13. (C6-C10)-арил-(C3-C6)-алкенил,
14. (C1-C9)-гетероарил-(C3-C6)-алкенил и
15. (C3-C6)-алкинил;
h) R8 обозначает водород;
i) n обозначает 0, 1 или 2,
а также к их физиологически совместимым солям.
Предпочитают соединение формулы (I), в котором
R1 обозначает этил- или n-пропил, или их физиологически совместимые соли. R1 обозначает особенно n-пропил.
Предпочитают также соединение формулы I, в которой R2 обозначает (C1-C6)-алкил; R3 обозначает COR6; n равно 0; R4 обозначает SO2-NH-CO-OR7, SO2NHCO-NHR7 или SO2-NH-CO-R7; R6 обозначает водород или R7 и R7 обозначает водород или (C1-C6)-алкил, а также их физиологически совместимые соли.
Алкил, алкенил и алкинил могут быть с прямой или разветвленной цепью.
Под циклоалкилом подразумевают также алкил-замещенные циклы.
(C6-C12)-арил представляет, например, фенил, нафтил или бифенил, преимущественно фенил.
Под (C1-C9) гетероарилом подразумевают, в частности, остатки, которые образуются от фенила или нафтила, в которых одна или несколько CH-групп замещены через N и/или в которых по меньшей мере две соседние CH-группы (при образовании пятичленного ароматического кольца) замещены через S, NH или O. Далее один или оба атома места конденсации бициклических остатков (как в индолизиниле) могут быть также N-атомами.
В качестве гетероарила действуют в частности фуранил, тиенил, пирролил, имидазолил, пиразолил, триазолил, тетразолил, оксазолил, изоксазолил, тиазолил, изотиазолил, пиридил, пиразинил, пиримидинил, пиридазинил, индолил, индазолил, хинолил, изохинолил, фталазинил, хиноксалинил, хиназолинил, циннолинил.
В случае необходимости существующие стереоцентры могут иметь конфигурацию как (R)-, так и (S)-.
Под физиологически переносимыми солями соединений формулы I понимают как их органические, так и их неорганические соли, как они описаны в Almington's Pharmaceutical Scinces (17-е издание, стр. 1418 (1985)).
На основании физической и химической стабильности и растворимости для кислых групп предпочитают среди прочего соли натрия, соли калия, соли кальция и соли аммония; для основных групп предпочитают среди прочего соли соляной кислоты, серной кислоты, фосфорной кислоты или карбоновых кислот или сульфокислот, как например, уксусной кислоты, лимонной кислоты, бензойной кислоты, малеиновой кислоты, фумаровой кислоты, винной кислоты и p-толуолсульфокислоты.
Изобретение относится также к способу получения новых соединений формулы I, а также к их физиологически переносимым солям, который отличается тем, что соединения формулы II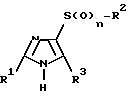
где
R1, R2, R3 и n имеют указанное определение, алкилируют с соединениями формулы III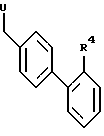
где
R4 имеет указанное определение и U обозначает летучую группу, в случае необходимости снова отщепляют временно введенные защитные группы, переводят полученные сульфонамиды формулы I в случае необходимости в уретаны формулы I, переводят полученные сульфонамиды формулы I в случае необходимости в сульфонилмочевины формулы I и полученные соединения формулы I переводят в случае необходимости в их физиологически переносимые соли.
Подходящими летучими группами U являются преимущественно нуклеонепроницаемые группы (см. Angew. Chem. 72 [1960] 71), как галоген, о-толуолсульфонат, мезилат или трифлат.
Способы получения предварительных стадий формулы I, известны среди прочего из патента США 4 355 044 и из уже названных европейских патентов A-324 377 и A-323841.
Другие способы описывают G. L'affe (Chem. Rev. 69, 345 1969)), T. Srodsky ("The Chemistry of the Azido Groug", Нью-Йорк, 1971, стр. 331) H. Wamhoff ("Comprehensive Hetera cyclic Chemistry") и S. Katritzky (издательство Пергамон Пресс, Нью-Йорк (1984)).
Другой способ получения соединений формулы II исходит из производных 2-оксима 1-цианоглиоксиловой кислоты и после восстановления оксима при помощи известных из литературных восстановителей и присоединения меркаптосоединений к группе нитрила при применении подходящих защитных групп дает предварительные стадии, которые в условиях отщепления воды можно циклизировать до имидазолов. Для стадии циклизации можно применять между прочим смеси из PCl5 и диметиламинопиридина DMAP, POCl3 и SOCl2 и их смеси с DMAP.
Для алкилирования соединений формулы II применяют напр. соответствующие бензилгалогениды, бензилтозилаты, бензилмезилаты или бензилтрифлаты или соответствующие алкилгалогениды, алкилтозилаты, алкилмезилаты или алкилтрифлаты.
Получение этих соединений осуществляют известным образом, например, галогенированием соответствующих метиловах предварительных стадий. Для этого применяют преимущественно N-бромсукцинимид, см. напр. J. Org. Chem. 44, 4733 (1979) и Hel Chim. Acta 62, 2661 (1979).
Производные бифенила можно синтезировать напр. исходя из производных арилборной кислоты сопряжением с замещенными арилгалогенидами с катализаторами переходных металлов, особенно палладием. Соответствующие реакции описывают R. B. Miller et al. al. (Organometallics 1984, 3, 1261) или A. Zuzuki et al. (Synthetic Commun. 11(7), 513 (1981)).
Производные сульфонилуретана формулы I можно получать из соответствующих сульфонамидов формулы I превращением со сложными эфирами хлоругольной кислоты и основаниями, как напр. карбонат калия, в инертных растворителях, преимущественно при температурах до точки кипения соответствующего растворителя.
Производные сульфонилмочевины формулы I можно получать на выбор или из соответствующих сульфонамидов формулы I превращением с изоцианатами или с производными 2,2,2-трихлорацетамида подходящего амина в инертных, высококипящих растворителях, как напр. DMSO (диметилсульфоксид) или из сульфонилуретанов формулы I реакцией соответствующего амина в инертных, высококипящих растворителях, как напр. толуол, при температурах до точки кипения соответствующего растворителя.
Аналогично можно получать сульфонил-сульфонамиды из соответствующих сульфонамидов превращением с хлорангидридами сульфокислоты или с сульфамоилхлоридами.
Остаток сульфонамида можно, если требуется, получать из аминогруппы посредством перегруппировки Меервайна. Для этого сначала диазотируют гидрохлорид амина и затем превращают его в присутствии медного катализатора с двуокисью серы в ледяной уксусной кислоте. Последующее воздействие аммиака приводит к сульфонамидной группе.
Сульфонамидную группу защищают например временно переводом в группу 2N, N-диметиламиноформилсульфонамида. Этот перевод осуществляют при этом альтернативно или превращением соответствующего сульфонамидного соединения с N, N-диметилформамиддиметилацеталем или превращением соответствующего сульфонамидного соединения с N,N-диметилформамидом в присутствии отщепляющих воду средств, как SOCl3, POCl3, PCl3 или сложные этиловые эфиры хлормуравьиной кислоты.
Эту защитную группу можно отщеплять как в присутствии основания, так и в присутствии кислоты.
В качестве альтернативы можно соответствующий тиофенол окислением с хлором и последующей реакцией с аммиаком превращать в сульфонамид.
Алкилирование осуществляют, в принципе, известными способами аналогичным образом.
Имидазолы формулы (II) металлизируют например, в присутствии основания.
Предпочтительными основаниями являются гидриды металлов, как гидрид лития, гидрид натрия или гидрид калия, например, в диметилформамиде ДМФ или диметилсульфоксиде ДМСО в качестве растворителей или алкоголяты металлов формулы MOR, причем R обозначает метил, этил, трет.-бутил, и реакцию осуществляют в соответствующем спирте, диметилформамиде или диметилсульфоксиде. Образованные таким путем соли производных имидазола растворяют в апротонном растворителе, как диметилформамид или диметилсульфоксид и смешивают с подходящим количеством агента алкилирования.
Альтернативную возможность для депротонирования производных имидазола представляет напр. превращение карбоната калия в диметилсульфоксиде.
Окисление тиосоединений формулы (I) с n = 0 до соответствующих сульфонов (n: = 1) и сульфоксидов (n = 2) осуществляют преимущественно над кислотами в соответствующих растворителях, как например, CH2Cl2.
Взаимодействие проводят при температурах ниже комнатной температуры до точки кипения реакционной смеси, преимущественно между +20oC и точкой кипения реакционной смеси в течение приблизительно 1 - 10 ч.
Соединения формулы I согласно изобретению оказывают антагонистическое действие на ангиотензин-II-рецепторы и поэтому могут применяться для лечения гипертензии, которая зависит от ангиотензина-II. Далее, их можно применять при сердечной недостаточности, кардиозащите, инфаркте миокарда, гипертрофии сердца, артериосклерозе, нефропатии, почечной недостаточности, а также при сердечно-сосудистых заболеваниях головного мозга, как транзисторные ишемические приступы и кровоизлияние в мозг.
Ренин представляет собой протеолитический фермент из класса аспартил-протеаз, который выделяется в результате различных стимулов (уменьшение объема, дефицит натрия, стимуляция β-рецепторов) смежных агломерированных клеток почек в кровообращение. Там от выделенного из печени ангиотензиногена отщепляют декапептид ангиотензин I. Его "ферментативным превращением ангиотензина" (ACE) переводят в ангиотензин II. Ангиотензин II играет существенную роль при регулировании кровяного давления, так как он непосредственно повышает кровяное давление в результате ангиоспазма. Дополнительно он стимулирует секрецию альдостерона из надпочечника и повышает таким образом, через торможение выделения натрия внеклеточный объем жидкости, что со своей стороны способствует повышению кровяного давления.
Пострецепторными действиями являются, среди прочего стимуляция превращения фосфоинозитола (Ca2+ - освобождение), активация протеинкиназы C и возможности зависимых от c-AMP гормональных рецепторов.
Сродство соединения формулы I к ангиотензин-II-рецептору можно определить измерением 125J-ангиотензин-II- или 3H-ангиотензин-II-вытеснения рецепторов на мембранах гломерулозной зоны надпочечников крупного рогатого скота. Для этого препарированные мембраны суспендируют в буферном растворе при pH 7,4.
Чтобы препятствовать деградации радиоактивных лиганд во время инкубирования, добавляют ингибитор пептидазы. Дополнительно применяют около 14.000 срм радиоактивного индикатора с удельной активностью 74 TBg/ммол (продается фирмой Амерохам Бухлер, Брауншвайг, ФРГ) и количество рецепторного протеина, которое связывает 50% радиоактивного индикатора. Реакцию начинают добавкой 50 мл мембранной суспензии в смесь 100 мл буферного раствора + апротинин; 50 мл буферного раствора с ангиотензином-II или без ангиотензина-II или антагонист рецептора и 50 мл радиоактивного индикатора. После продолжительности инкубационного периода 60 мин при 25oC связанный и свободный радиоактивный лиганд отделяют осаждением фильтрованием (напр. с Whatman®GFIC фильтрами на Скатрон® - клеточном сборнике).
Неспецифические связи предотвращают обработкой фильтров при помощи 0,3% полиэтиленимина pH 10 (напр. Сигма, N 3143). Измерением радиоактивности в гамма-сцинтилляционном счетчике определяют вытеснение радиоактивных лиганд рецептором.
IC50 - величины, которые обозначают концентрацию ингибитора, чтобы вытеснить 50% лиганда, определяют по J. Theor. Biol. ol. 59, 253 (1970). Они составляют для соединений формулы I в области 1 - 10-4 - 1 - 10-9 М.
В качестве альтернативы можно определять сродство соединений формулы I к ангиотензин-II-рецептору измерением 125Н-ангиотензин-II или 3H-ангиотензин-II-вытеснения рецепторных препарирований из различных органов (печень, легкое, надпочечник, головной мозг и т.д.).
Для этого препарированные мембраны суспендируют в инкубационном буферном растворе (напр. 20 ммол Трис, pH 7,4, содержащий 135 ммол NaCl, 10 ммол KCl, 10 ммол MgCl2, 5 ммол глюкозы, 0,2% сывороточного альбумина крупного рогатого скота, а также ингибиторы протеазы PMSF 0,3 ммол и бацитрацин 0,1 ммол) и вместе с меченым изотопным индикатором ангиотензином-II и различными концентрациями испытываемых соединений инкубируют 90 мин при 25oC. Затем связанный и свободный радиоактивный лиганд отделяют фильтрованием через фильтр из микростекловолокна (напр. , GF51, Шляйхер Шюлл на клеточном сборнике (СКАТРОН).
Измерением связанной рецептором радиоактивности на фильтрах при помощи бета- или гамма-спектрометра определяют степень вытеснения радиоактивного лиганда рецептора испытываемыми соединениями. Силу вытеснения радиоактивного лиганда с рецептора испытываемыми соединениями определяют как IC50, то есть концентрация ингибитора, которая вытесняет 50% связанного радиоактивного лиганда с рецептора. Вычисление IC50 - величин производят при помощи математического обеспечения пропорциональным счетчиком (наприм. LIGAND, G.A. Mc Pherson 1985, Elsevier - BIOSOFT, 68 Hills Road, Кембридж CB 2 ILA, U.K.). Измеренные для соединений формулы I IC50 - величины лежат в области 1 • 10-5 - 1 • 10-11 М.
Для определения антагонистического действия соединений формулы I на живом организме можно измерить их тормозящий эффект на вызванное ангиотензином-II повышение кровяного давления на немеченых радиоактивным изотопом Spragul - Dawley-крысах (Меллегард, Дания). Внутривенное применение в вену полового члена.
После препарирования животного и 20-минутного времени выжидания для стабилизирования гемодинамических параметров назначают 3 следующих друг за другом инъекции по 10, 30 и 100 мг ангиотензина-II в 0,1 мл водного раствора с интервалами в 5 мин. Соединения формулы (I) растворяют в дистиллированной воде, при известных обстоятельствах при добавке 10%-ного этанола и/или оснований (pH < 10) или кислот - (pH > 3), и при дозах 1 - 300 мг/кг вводят внутривенно или 5 - 1000 мг/кг интрадуоденально.
При интрадуоденальном приеме инъекцию ангиотензина-II производят через 20, 40 и 60 мин, в то время как при внутривенном приеме "ответная реакция на лекарственное вещество, повышающее кровяное давление" происходит с интервалами в 10 мин.
Соединения формулы (I) являются особенно эффективными в области 1 - 300 мг/кг при внутривенном назначении или 5 - 300 мг/кг при интрадуоденальном назначении.
Изобретение относится также к фармацевтическим лекарственным формам, состоящим в основном из соединения формулы I и в случае необходимости из других активных веществ, как напр. мочегонные средства или нестероидальные противовоспалительные активные вещества. Соединения формулы I можно применять также в качестве диагностических средств для системы ренинангиотензин.
Фармацевтические препараты содержат эффективное количество активного вещества формулы I и в случае необходимости другие активные вещества вместе с неорганическим или органическим, применяемым в фармации веществом-носителем и в случае необходимости другие дополнительные или вспомогательные вещества. Применение может быть внутрь носа, внутривенное, подкожное или через рот. Дозировка активного вещества зависит от рода теплокровных животных, от веса тела, от возраста и от вида назначения.
Фармацевтические препараты настоящего изобретения получают известными способами растворения, смешивания, гранулирования или дражирования.
Для оральной формы применения активные соединения смешивают с обычными для этой цели добавками, как вещества-носители, стабилизаторы или инертные растворители, и приводят в подходящие формы введения, как таблетки, драже, капсули в оболочке, водные, спиртовые или масляные суспензии или водные, спиртовые или масляные растворы. В качестве инертных носителей можно применять напр. гуммиарабик, магнезию, карбонат магния, фосфат калия, молочный сахар, глюкозу, стеарилфумарат магния или крахмал, особенно кукурузный крахмал. При этом можно получать лекарственную форму как сухой или влажный гранулят. В качестве масляных веществ-носителей или растворителей принимают во внимание, например, растительные или животные масла, как подсолнечное масло и рыбий жир.
Для подкожного или внутривенного применения активные соединения или их физиологически переносимые соли, в случае необходимости с обычными для этой цели веществами, как агенты растворения, эмульгаторы или другие вспомогательные вещества, переводят в растворы, суспензии или эмульсии. В качестве растворителей принимают во внимание напр. воду, физиологический раствор хлористого натрия или спирты, как этанол, пропандиол или глицерин, а также растворы сахара, как растворы глюкозы или маннита или смеси из названных растворителей.
Список сокращений:
Ac ацетил
DMF - N,N-диметилформамид
DME - диметоксиэтан
EE - этилацетат
DCl - химическая ионизация при десорбции
FAB - бомбардировка быстрыми атомами
RT - комнатная температура
mp - точка плавления
h - ч (часы)
Min. - мин (минуты)
Изобретение поясняют следующими примерами.
Пример 1. Сложный этиловый эфир 1-[(2'-n-пропиламинокарбониламиносульфонил-бифенил-4-ил)метил] -2-n- пропил-4-метилтио-имидазол-5-карбоновой кислоты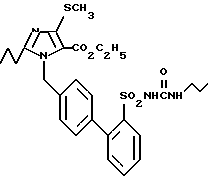
а) Сложный этиловый эфир 2-амино-2-циано-уксусной кислоты.
К 70 г (0,492 мол) сложного этилового эфира 2-цианоглиоксаловой кислоты-2-оксима в 700 мл воды и 560 мл насыщенного раствора NaHCO3 добавляют при комнатной температуре частями в течение 20 мин 228 г (1,3 мол) дитионита натрия. Перемешивают 12 ч при 35oC и после охлаждения и насыщения NaCl экстрагируют с CH2Cl2. Сушка с Na2SO4 и концентрирование досуха дает 30 г заглавного соединения как масло, коэффициент Rf(CH2Cl2/CH3OH 9/ = 0,6.
b) Сложный этиловый эфир 2-циано-2-n-пропилкарбониламиноуксусной кислоты.
В 30 г (0,233 мол) соединения из Ia) в 250 мл абсолютного CH2Cl2 и 18,9 мл (0,233 мол) пиридина закапывают при 0 - 5oC раствор 24,2 мл (0,23 мол) хлорангидрида масляной кислоты в 25 мл CH2Cl2. Затем перемешивают 12 ч при комнатной температуре. Органическую фазу промывают 3 • H2O и 1 • насыщенным раствором NaCl, сушат с Na2SO4 и концентрируют. Кристаллизация из простого диизопропилового эфира дает 29,5 г заглавного соединения. Rf(CH Cl /MCO 9/1) = 0,7, точка плавления: 106oC.
c) Сложный этиловый эфир 3-амино-2-n-пропилкарбониламино-3- метилтиоакриловой кислоты.
К 29,5 г (0,149 мол) соединения из Iб) и 2,05 мл (0,0145 мол) триэтиламина в 500 мл этанола добавляют при комнатной температуре 14,3 г (0,297 мол) конденсированного метилмеркаптана. После 4 дней выдерживания при комнатной температуре растворитель удаляют и остаток кристаллизуют из простого диизопропилового эфира, причем получают 36,2 г заглавного соединения. Rf(CH2Cl2/минута 9:1) = 0,4.
Точка плавления: 119oC.
d) Сложный этиловый эфир 2-n-пропил-4-метилтио-имидазол-5-карбоновой кислоты.
В 29,16 г (0,142 мол) пентахлорида фосфора в 240 мл CH2Cl2 закапывают при -78oC 19,3 г (0,157 мол) 4-диметиламинопиридина в 120 мл CH2CH2. Через 10 мин закапывают 17,6 г (0,071 мол) соединения из 1c) в 200 мл CH2Cl2. Нагревают до комнатной температуры и перемешивают 2,5 ч при комнатной температуре. Затем при охлаждении льдом добавляют 1,6 л 1н. раствора NaHCO3, перемешивают 1 ч. и выдерживают в течение ночи. После разделения фаз водную фазу экстрагируют 3 х с EE, соединенные органические фазы сушат с Na2SO4 и концентрируют. Хроматография на SiO2 с CH2Cl2/EE 4/1 дает 5,6 г заглавного соединения как масло.
Rf(CH2Cl2/EE 4:1) = 0,4, MS (DCl) : 229 (M + H).
e) Сульфонамидобромбензол.
6 г (0,3 мол) о-броманилина в атмосфере аргона добавляют к раствору из 100 мл концентрированной HCl и 30 мл ледяной уксусной кислоты, при -10oC закапывают раствор 22,4 г нитрита натрия в 30 мл воды и перемешивают реакционный раствор 60 мин при -5oC. Полученный раствор закапывают в насыщенный SO2 раствор 7 г CuCl2 • 2H2O и 0,5 г CuCl в 300 мл ледяной уксусной кислоты, смесь после 60 мин перемешивания при комнатной температуре выливают в смесь льда/воды, экстрагируют простым эфиром, промывают эфирные экстракты насыщенным раствором NaHCO3 и водой, сушат над MgSO4 и концентрируют. Полученные 67,8 г сульфонилхлоридного соединения смешивают в 500 мл ацетона при охлаждении 300 мл концентрированного аммиака. После отвода ацетона получающуюся суспензию разбавляют водой, осаждающиеся белые кристаллы отсасывают, промывают H2O и сушат в высоком вакууме. Заглавное соединение без дальнейшей очистки применяют в следующей реакции.
Точка плавления: 190oC.
f) 2-N,N-диметиламиноформилсульфонамидобромбензол.
0,236 мол соединения из примера 1e) перемешивают в 150 мл абсолютного диметилформамида с 40 мл N,N-диметилформамиддиметилацеталя 2 ч при комнатной температуре. Реакционный раствор выливают на 200 мл 5%-ного раствора NaHSO4/лед (1: 1), отсасывают осаждающийся осадок, промывают H2O и сушат в вакууме. Получают 67 г заглавного соединения.
Точка плавления: 148oC. Rf(SiO2), EE/гептан 1: 1) = 0,1, MS (DCl)6: 291/293 (M + H).
g) 4'-Метил-бифенил-2-N,N-диметиламиноформилсульфонамид.
К 11 г (37,9 ммол) соединения из примера 1f), 1 г трифенил фосфина, 8 г Na2CO3 в 150 мл толуола и 40 мл H2O добавляют в атмосфере аргона сначала 420 мг Pd(OAc)2 и затем 5,66 г (41,9 ммол) толилборной кислоты в 100 мл этанола. Теперь нагревают 4 ч до кипения, потом концентрируют и поглощают в 500 мл EE и 500 мл H2O. Образующийся осадок отфильтровывают их характеризуют как заглавное соединение. EE-фазу отделяют, сушат над Na2SO4 и концентрируют. Хроматография на SiO2 с EE дает другую часть заглавного соединения; общий выход 7,6 г, точка плавления: 181 - 184oC, Rf (SiO2, EE/гептан 1:1) = 0,2, MS (DCl): 303 (M + H).
h) 4'-Бромметилбифенил-2-N,N-диметиламиноформилсульфонамид.
7,4 г (0,025 мол) соединения из 1g) нагревают с 4,6 г (0,026 мол) N-бромсукцинимида в 130 мл хлорбензола в присутствии 300 мг перекиси бензоила 2 ч до флегмы. После охлаждения добавляют 50 мл насыщенного раствора Na2SO3, органическую фазу отделяют, промывают насыщенным раствором Na2CO3, водой и насыщенным раствором NaCl, сушат над Na2SO4 и концентрируют. Остаток размешивают с EE и отсасывают 6,7 г заглавного соединения.
Точка плавления: 168 - 171oC, Rf (SiO2, EE) = 0,5, MS (DCl) 381/383 (M + H).
i) Сложный этиловый эфир 1-[(2'-N,N-диметиламиноформилсульфонамидо-бифенил-4-ил)метил]-2-n- пропил-4-метилтио-имидазол-5-карбоновой кислоты.
2,0 г (8,75 ммол) соединения из 1d), 4,15 г (8,75 ммол) соединения из 1h) (75%-ного) и 1,25 г (9,0 ммол) K2CO3) перемешивают в 50 мл абсолютного диметилформамида в течение ночи при комнатной температуре. Концентрируют, остаток растворяют в 400 мл EE, EE-раствор промывают 3 х водой, сушат над Na2SO4 и концентрируют. Остаток перемешивают с этанолом и простым диизопропиловым эфиром и отсасывают 4,14 г заглавного соединения как осажденный осадок.
Точка плавления: 169 - 171oC, Rf (SiO2, EE) = 0,4, MS (FAB): 529 (M + H).
j) Сложный этиловый эфир 1-[(2'-сульфонамидобифенил-4-ил)метил]- 2-n-пропил-4-метилтио-имидазол-5-карбоновой кислоты.
4 г (7,60 ммол) соединения из 1i) кипятят в 40 мл метанола с 20 мл концентрированной соляной кислоты 3 ч при флегме. Охлаждают до комнатной температуры, отгоняют растворитель, устанавливают величину pH водного раствора при помощи 6н. раствора NaOH до 5-6 и экстрагируют несколько раз с EE. Соединенные EE-фазы сушат над Na2SO4 и концентрируют. Получающуюся пену перемешивают с этанолом и простым диизопропиловым эфиром и отсасывают осадок. Получают 3 г заглавного соединения.
Точка плавления: 125 - 127oC, Rf (SiO2, EE) = 0,7, MS (FAB): 474 (M + H).
k) Сложный этиловый эфир 1-[(2'-n-пропиламинокарбониламиносульфонил-бифенил-4-ил)метил]-2-n-пропил-4-метилтио-имидазол-5-карбоновой кислоты.
2 г (4,22 ммол) соединения из 1j) в 50 мл абсолютного ацетона смешивают с 1,75 г (12,66 ммол) безводного K2CO3. После 30 мин нагревания до флегмы этот раствор смешивают с 395 л (4,22 ммол) пропилизоцианата и перемешивают 1 ч на флегме. Затем охлаждают, смешивают с 15 мл 2н. HCl, сгущают в вакууме и экстрагируют несколько раз с CH2Cl2. Сушка над Na2SO4, концентрирование и кристаллизация из EE дает 1,9 г заглавного соединения.
Точка плавления: 160 - 165oC, Rf (SiO2, EE/гептан 2:1) = 0,25, MS (FAB): 559 (M + H).
Пример 2. 1-[(2'-n-Пропиламинокарбониламиносульфонил-бифенил-4- ил)метил]-2-n-пропил-4-метилтио-имидазол-5-карбоновая кислота.
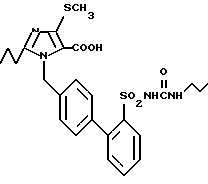
700 мг (1,25 ммол) соединения из 1k) перемешивают в 40 мл метанола с 10 мл 2н. раствора NaOH 3 дня при комнатной температуре. Затем растворитель удаляют в вакууме, водный раствор при помощи 2 н. соляной кислоты доводят до pH 6, осаждающийся осадок отсасывают и сушат в высоком вакууме. Получают 600 мг заглавного соединения.
Точка плавления: 133 - 135oC, Rf (SiO2, CH2Cl2/метанол 10/1) = 0,19, MS (FAB): 531 (M + H).
Пример 3. Сложный этиловый эфир 1-[(2'-этоксикарбониламиносульфонилбифенил-4-ил)метил]-2-n-пропил-4-метилтио-имидазол-5-карбоновой кислоты.
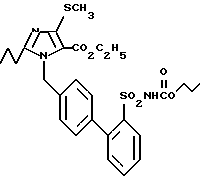
1,5 г (3,17 ммол) соединения из 1j) кипятят в атмосфере аргона в 25 мл абсолютного DME с 0,876 г (6,34 ммол) К2CO3 и 0,35 мл (3,17 ммол) сложного этилового эфира хлормуравьиной кислоты 3 ч при флегме. Затем отводят в значительной степени растворитель, устанавливают величину pH остающегося раствора при помощи 10%-ного раствора KH2PO4 приблизительно до 4 и экстрагируют несколько раз с EE. Соединенные экстракты промывают насыщенным раствором NaCl, сушат над MgSO4, концентрируют и сушат остаток в высоком вакууме. Получают 1,79 г заглавного соединения как желтую пену.
Rf(SiO2, EE) = 0,5, MS (FAB): 546 (M + H).
Пример 4. 1-[(2'-Этоксикарбониламиносульфонил-бифенил-4-ил)метил]-2-пропил-4-метилтио-имидазол-5-карбоновая кислота.
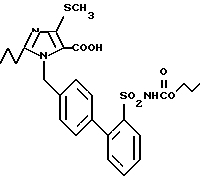
500 мг (0,92 ммол) соединения из примера 3) перемешивают в 3 мл этанола с 2,5 мл 2н. раствора NaOH 24 ч при комнатной температуре. Растворитель отгоняют в вакууме, остающийся водный раствор при помощи 2н. соляной кислоты доводят приблизительно до pH 5 и отсасывают осаждающийся осадок. Получают 380 мг заглавного соединения в форме окрашенного в слабый желтый цвет твердого продукта.
Точка плавления: 156 - 160oC, Rf(SiO2), CH2Cl2/CH3OH 9/1/ - 0,3, MS (FAB): 518 (M + H).
Пример 5. Сложный этиловый эфир 1-[(2'-метиламинокарбониламиносульфонил-бифенил-4-ил)метил]-2-этил-4-метилтиоимидазол-5-карбоновой кислоты.
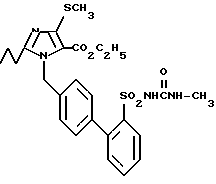
a) Сложный этиловый эфир 2-циано-2-этилкарбониламиноуксусной кислоты.
Заглавное соединение получают по указанному в примере 1б) способу, причем здесь вместо хлорангидрида масляной кислоты превращают хлорангидрид пропионовой кислоты с соединением из 1а). Из 12,8 г (0,1 мол) соединения из 1а) получают 11,4 г заглавного соединения.
Точка плавления: 111 - 113oC, Rf(SiO2, EE) = 0,6, MS (DCl) : 185 (M + H).
b) Сложный этиловый эфир 3-амино-2-этилкарбониламино-3-метил-тиоакриловой кислоты.
Это соединение получают аналогично указанному в примере 1c) способу.
Точка плавления: 127oC, Rf(SiO2, EE) = 0,18, MS (DCl): 233 (M + H).
c) Сложный эииловый эфир 2-этил-4-метилтио-имидазол-5-карбоновой кислоты.
Это соединение получают аналогично приведенному в 1d способу.
Точка плавления: 141 - 143oC, Rf(SiO2, EE) = 0,4, MS (DCl): 215 (M + H).
d) Сложный этиловый эфир 1-[(2'-N,N-диметиламиноформилсульфонамидо-бифенил-4-ил)-метил]-2-этил-4-метилтио-имидазол-5-карбоновой кислоты.
Заглавное соединение получают из соединения 5c) и соединения 1h) по описанному в примере 1) способу.
Точка плавления: 189 - 194oC, Rf(SiО2, EE) = 0,3, MS (FAB): 515 (M + H).
e) Сложный этиловый эфир 1-[(2'-сульфонамидобифенил-4-ил)метил]-2-этил-4-метилтио-имидазол-5-карбоновой кислоты.
Заглавное соединение получают по способу примера 1) из соединения из примера 5d).
Точка плавления: 153 - 155oC, Rf(SiO2, EE), = 0,5, MS (FAB) : 460 (M + H).
f) 2,2,2-Трихлор-N-метил-ацетамид.
1,6 г (51,5 ммол) метиламина конденсируют и смешивают в 20 мл абсолютного диоксана с 7,14 мл (51,5 ммол) триэтиламина и 5,7 мл (51,5 ммол) трихлорацетилхлорида, растворенного в 10 мл абсолютного диоксана, и получающийся раствор перемешивают 3 ч при комнатной температуре. Затем смешивают с водой, устанавливают величину pH раствора при помощи 2н. соляной кислоты приблизительно 1 и отсасывают осаждающееся заглавное соединение (7,6 г).
Точка плавления: 90 - 95oC.
g) Сложный этиловый эфир 1-[(2'-метиламинокарбониламиносульфонил-бифенил-4-ил)-метил]-2-этил-4-метилтио-имидазол-5-карбоновой кислоты.
135 мг (0,316 ммол) соединения из 5e) перемешивают в 2 мл абсолютного DMSO в атмосфере аргона с 38 мг (0,1 ммол) измельченной в порошок гидроокиси натрия NaOH и с 61 мг (0,348 ммол) соединения из 5f) 30 мин при 80oC. Реакционный раствор выливают на ледяную воду, подкисляют 2н. соляной кислотой и экстрагируют несколько раз с EE. После промывки соединенных EE-фаз насыщенным раствором NaCl, сушки над MgSO4 и концентрирования полученный кристаллический остаток размешивают с небольшим количеством EE. Отсасывание осадка дает 97 мг заглавного соединения.
Точка плавления: 220 - 223oC, Rf(SiO2, CH2Cl2/CH3OH 9/1) = 0,6, MS (FAB): 517 (M + H).
Пример 6. 1-[(2'-Метиламинокарбониламиносульфонил-бифенил-4-ил)-метил] -2-этил-4-метилтио-имидазол-5-карбоновая кислота.

Заглавное соединение получают по способу примера 2) из соединения из 5g). Из 50 мг (0,1 ммол) соединения 5g) получают 40,5 мг заглавного соединения.
Точка плавления: 155oC, Rf(SiO2, CH2Cl2/CH3OH/ уксусная кислота 9/1/0,2) = 0,46, MS (FAB): 489 (M + H).
Пример 7. Сложный этиловый эфир 1[(2'-этоксикарбониламиносульфонилбифенил-4-ил)метил]-2-этил-4-метил-тио-имидазол-5-карбоновой кислоты.
1,4 г (3 ммол) соединения из примера 5e) и 825 мг (6 ммол) K2CO3 кипятят в 25 мл абсолютного диметоксиэтана кипятят в 25 мл абсолютного диметоксиэтана с 0,3 мл (3,05 ммол) сложного этилового эфира хлормуравьиной кислоты 3 ч на флегме. Концентрируют, при помощи 10%-ного раствора KH2PO4 устанавливают pH приблизительно 5 и экстрагируют с EE. После сушки над Na2SO4 и концентрирования получают 1,45 г заглавного соединения.
Rf(SiO2), EE = 0,3, MS (FAB): 532 (M + H).
Пример 8. Сложный этиловый эфир 1-[(2'-n-пропиламино-карбониламиносульфонил-бифенил-4-ил)метил] -2-этил-4-метилтио-имидазол-5-карбоновой кислоты.
300 мг (0,56 ммол) соединения из примера 7 кипятят в 7 мл абсолютного толуола с 1,5 мл n-пропиламина 3 ч при флегме. После концентрирования и хроматографии на SiO2 и EE в качестве растворителя получают 130 мг заглавного соединения.
Точка плавления: 202 - 203oC, Rf(SiO2), EE) = 0,24, MS (FAB): 545 (M + H).
Пример 9. 1[(2'-n-Пропиламинокарбониламиносульфонил-бифенил-4-ил)-метил] -2-этил-4-метилтио-имидазол-5-карбоновая кислота.
Заглавное соединение получают по способу примера 2) из соединения из примера 8). Из 100 мг (0,18 ммол) соединения 8) получают 70 мг заглавного соединения.
Точка плавления: 135 - 140oC, Rf(SiO2, EE) = 0,1, MS (FAB): 517 (M + M).
Пример 10. Сложный этиловый эфир 1[(2'-n-пропиламинокарбониламиносульфонил-бифенил-4-ил)метил] -2-n-пропил-4-метилсульфонил-имидазол-5-карбоновой кислоты.

95,0 мг (0,17 ммол) соединения из примера 1) перемешивают в 10 мл абсолютного CH2Cl2 при -78oC с 59 мг (0,17 ммол) m-хлорнадбензойной кислоты (50%-ной) 20 мин. Смешивают с 10 мл 10%-ного раствора бисульфита натрия, нагревают до комнатной температуры и после разделения фаз экстрагируют с EE. Соединенные органические фазы промывают насыщенным раствором Na2CO3, сушат над Na2SO4 и концентрируют. Получают 110 мг заглавного соединения.
Точка плавления: 65 - 68oC, Rf(SiO2, EE) = 0,1, MS (FAB): 575 (M + M).
Пример 11. 1[(2'-n-Пропиламинокарбониламиносульфонил-бифенил-4-ил)метил] -2-n-пропил-4-метилсульфоксил-имидазол-5-карбоновая кислота
Заглавное соединение получают из соединения примера 10) по способу примера 2). Из 100 мг (0,17 ммол) соединения из примера 10) получают 83 мг заглавного соединения.
Точка плавления: 105 - 108oC, Rf(SiO2), EE) = 0,1, MS (FAB): 547 (M + M).
Пример 12. Сложный этиловый эфир 1-[(2'-этоксикарбониламиносульфонил-бифенил-4-ил)метил] -2-n-пропил-4-метилсульфонил-имидазол-5-карбоновой кислоты.

350 мг (0,64 ммол) соединения из примера 3) нагревают с 443 мг (1,28 ммол) m-хлорбензойной кислоты (50%-ной) в 20 мл абсолютного CH2Cl2 1 ч при флегме. Обработки осуществляют аналогично обработке примера 10) и получают 364 мг заглавного соединения как бесцветную пену.
Rf(SiO2), CH2Cl2 (MCOH 9:1) = 0,74, MS (FAB): 578 (M + H).
Пример 13. 1-[(2'-Этоксикарбониламиносульфонил-бифенил-4-ил)метил]-2-n-пропил-4-метилсульфонил-имидазол-5-карбоновая кислота.
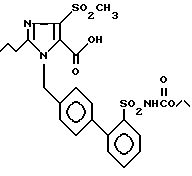
Из 120 мг (0,2 ммол) соединения из примера 12) получают по способу примера 2) 84 мг заглавного соединения.
Точка плавления: 156 - 159oC, Rf(SiO2), CH2Cl2(MCOH 9:1:0,2) = 0,5, MS (FAB): 550 (M + H).
Пример 14. Сложный этиловый эфир 1[(2'-этиламинокарбониламиносульфонил-бифенил-4-ил)метил] -2-n-пропил-4-метилсульфонил-имидазол-5-карбоновой кислоты.
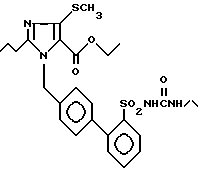
Превращение 200 мг (0,42 ммол) соединения из примера 1j) с 34 мл (0,42 ммол) этилизоцианата по способу примера 1k) дает 170 мг заглавного соединения.
Точка плавления: 161 - 162oC, Rf(SiO2, EE) = 0,43, MS (FAB): 545 (M + H).
Пример 15. 1-[(2'-Этиламинокарбониламиносульфонил-бифенил-4-ил)метил]-2-n-пропил-4-метилсульфонил-имидазол-5-карбоновая кислота.
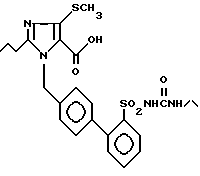
Из 61 мг (0,11 ммол) соединения из примера 14) получают по способу примера 2) 56 мг заглавного соединения.
Точка плавления: 131oC, Rf(SiO2, CH2Cl2, M2OH 10:1) = 0,2, MS (FAB): 517 (M + H).
Пример 16. Сложный этиловый эфир 1-[(2'-аллиламинокарбониламиносульфонил-бифенил-4-ил)метил]-2-n-пропил-4-метилтио-имидазол-5-карбоновой кислоты.

Превращение 200 мг (0,42 ммол) соединения из примера 1j) с 38 мл (0,42 ммол) алкилизоцианата по способу примеру 1k) дает 150 мг заглавного соединения.
Точка плавления: 184oC, Rf(SiO2, EE) = 0,43, MS (FAB): 557 (M + H).
Пример 17. 1-[(2'-Аллиламинокарбониламиносульфонил-бифенил-4-ил)метил] -2-n-пропил-4-метилтио-имидазол-5-карбоновая кислота.
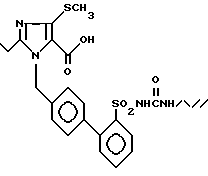
Из 60 мг (0,1 ммол) соединения из примера 16) получают по способу примера 2) 54 мг заглавного соединения.
Точка плавления: 148oC, Rf(SiO2, CH2Cl2/MCOH 10:1) = 0,3, MS (FAB): 529 (M + H).
Пример 18. Сложный этиловый эфир 1-[(2'-метоксикарбониламиносульфонил-бифенил-4-ил)метил]-2-n-пропил-4-метилтио-имидазол-5-карбоновой кислоты.
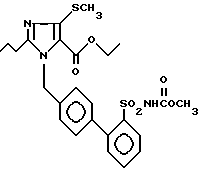
500 мг (1,06 ммол) соединения из примера 1j) кипятят с 293 мг (2,12 ммол) K2CO3, 106 мл (1,06 ммол) диметилдикарбоната и 53 мг ДМАР в 20 мл простого диэтиленгликольдиметилового эфира 2 ч при флегме. Вращают, остаток смешивают с EE/раствором KH2PO2, органическую фазу отделяют и после сушки над Na2SO4 концентрируют.
Хроматография на SiO2 (EE/гептан 2:1) дает 225 мг заглавного соединения.
Точка плавления: 146oC, Rf(SiO2, EE) = 0,37, MS (FAB): 532 (M + H).
Пример 19. 1-[(2'-Метоксикарбониламиносульфонил-бифенил-4-ил)метил]-2-n-пропил-4-метилтио-имидазол-5-карбоновая кислота.

Перемешивание 150 мг (0,263 ммол) соединения из примера 18) аналогично соединению примера 2) дает 110 мг заглавного соединения.
Точка плавления: 131oC, Rf(SiO2, CH2Cl2/MCOH 10:1) = 0,15, MS (FAB): 504 (M + H).
Пример 20. Сложный этиловый эфир 1-[(2'-циклопропилметиламинокарбониламиносульфонил-бифенил-4-ил)метил] -2-n-пропил-4-метилтио-имидазол-5-карбоновой кислоты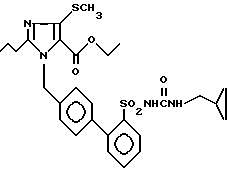
500 мг (1,06 ммол) соединения из примера 1j) перемешивают с 408 мг (1,06 ммол) дигидроксибензотриазолилкарбоната (70%-ного) и 85 мл (1,06 ммол) пиридина в 20 мл абсолютного CH2Cl2 2 ч при комнатной температуре. Затем реакционный раствор снова перемешивают 2 ч с 114 мг (1,06 ммол) циклопропилметиламин-гидрохлорида и 170 мл (2,12 ммол) пиридина. Вращают, поглощают остаток в EE, промывают EE-фазу раствором NaHCO3, раствором NaHSO4, сушат над Na2SO4 и концентрируют. Хроматография на SiO2 (EE/гептан 1:3) дает 72 мг заглавного соединения.
Точка плавления: 125oC, Rf(SiO2, EE) = 0,47, MS (FAB): 571 (M + H).
Пример 21. 1-[(2'-Циклопропилметиламинокарбониламиносульфонил-бифенил-4-ил)метил]-2-n-пропил-4-метилтио-имидазол-5-карбоновая кислота.
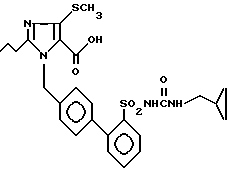
Аналогично примеру 2) омыление 45 мг (0,08 ммол) соединения из примера 20) дает 34 мг заглавного соединения.
Точка плавления: 138oC, Rf(SiO2, CH2Cl2/MCOH 9:1) = 0,1, MS (FAB): 543 (M + H)
Пример 22. Сложный этиловый эфир 1-[(2'-пропилоксикарбониламиносульфонил-бифенил-4-ил)метил]-2-n-пропил-4-метилтио-имидазол-5-карбоновой кислоты.
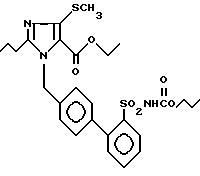
Аналогичное примеру 3) превращение 200 мг (0,42 ммол) соединения из примера 1j) с 70 мл (0,63 ммол) сложного пропилового эфира хлормуравьиной кислоты дает после хроматографии на SiO2 (EE/гептан 2:1) 200 мг заглавного соединения.
Точка плавления: 144oC, Rf(SiO2, EE) = 0,54, MS (FAB): 560 (M + H).
Пример 23. 1-[(2'-пропилоксикарбониламиносульфонил-бифенил-4-ил)метил] -2-n-пропил-4-метилтио-имидазол-5-карбоновая кислота.
Это соединение получают из соединения примера 22) по способу примера 2).
Точка плавления: 124oC, Rf(SiO2, CH2Cl2/MCOH 10:1) = 0,1, MS (FAB): 532 (M + H).
Пример 24. Сложный этиловый эфир 1-[(2'-бензилоксикарбониламиносульфонил-бифенил-4-ил)метил]-2-n-пропил-4-метилтио-имидазол-5-карбоновой кислоты.
Аналогичное примеру 3) превращение 200 мг (0,42 ммол) соединение из примера 1j) с 89 мл (0,633 ммол) сложного бензилового эфира хлормуравьиной кислоты дает после хроматографии на SiO2 (EE/гептан 2:1) 250 мг заглавного соединения.
Точка плавления: 158oC, Rf(SiO2, EE) = 0,55, MS (FAB): 608 (M + H).
Пример 25. 1-[(2'-Бензилоксикарбониламиносульфонил-бифенил-4-ил)метил] -2-n-пропил-4-метилтио-имидазол-5-карбоновая кислота.
Это соединение получают из соединения примера 24 по способу примера 2.
Точка плавления: 115oC, Rf(SiO2, CH2Cl2/MCOH 15:1) = 0,1, MS (FAB): 580 (M + H).
Данные по активности конкретных соединений на основе значений IC50 по примерам с 1 по 25. Причем определение значений IC50 осуществлялось на основе описанного тексте A - по рецепторной связи, (см. таблицу).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПОНИЖАЮЩАЯ КРОВЯНОЕ ДАВЛЕНИЕ | 1992 |
|
RU2104272C1 |
| ЗАМЕЩЕННЫЕ АЗОЛЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2047604C1 |
| ИМИДАЗО-АННЕЛИРОВАННЫЕ ИЗО- И ГЕТЕРОЦИКЛЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2076105C1 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА | 1995 |
|
RU2092482C1 |
| ЧЕТЫРЕХЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2174513C2 |
| ЗАМЕЩЕННЫЕ ОСНОВАНИЕМ БЕНЗОИЛГУАНИДИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ ИНГИБИРОВАНИЯ КЛЕТОЧНОГО NA/H-АНТИПОРТЕРА | 1996 |
|
RU2161604C2 |
| БЕНЗОИЛГУАНИДИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ ДЛЯ ИХ ПОЛУЧЕНИЯ, СПОСОБ ИНГИБИРОВАНИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 1995 |
|
RU2160727C2 |
| Способ получения производных дипептидов или их фармацевтически приемлемых солей | 1990 |
|
SU1836382A3 |
| СЛОЖНЫЙ 17-ДЕЗОКСИ-КОРТИКОИД-21-ЭФИР КАРБОНОВОЙ КИСЛОТЫ, СПОСОБЫ ЕГО ПОЛУЧЕНИЯ, ЛЕКАРСТВЕННОЕ СРЕДСТВО | 1995 |
|
RU2161624C2 |
| ПРИМЕНЕНИЕ ПРОИЗВОДНЫХ ИМИДАЗОЛА ДЛЯ ПРИГОТОВЛЕНИЯ ФАРМКОМПОЗИЦИЙ, АКТИВНЫХ К РЕЦЕПТОРАМ АТ1 И АТ2 АНГИОТЕНЗИНА, НЕКОТОРЫЕ ИЗ ЭТИХ СОЕДИНЕНИЙ, СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ИМИДАЗОЛА И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ | 1995 |
|
RU2141321C1 |
Сущность: производные имидазола с боковой цепью бифенилсульфонилмочевины или бифенилсульфонилуретана, способ их получения и их применение. Соединения формулы I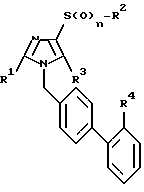
где R1 - напр. этил, R2 - напр. метил, N - напр. O, R3 - напр. COOH и R4 - напр. SO2NHCONHCH3, представляют высокоактивные антагонисты ангиотензин-II-рецепторов. 4 с. и 3 з.п. ф-лы, 1 табл.
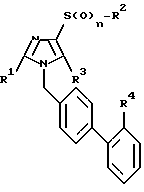
где R1 - C1 - C3-алкил;
R2 - C1 - C6-алкил, C3 - C7-циклоалкил;
R3 - водород, CH2OR5, COR6;
R4 - SO2NHR7, SO2NH-CO-NR7R9, SO2NHCOOR7, SO2NH-COR7;
R5 - водород, (С1 - С6)-алкил;
R6 - водород или OR7;
R7 и R9 - одинаковые или различные и обозначают водород, С1 - С3-алкил, С3 - С8-циклоалкил, С3 - С8-циклоалкил-С1 - С3-алкил, фенил, фенил-С1 - С4-алкил, С2 - С6-алкенил;
n = 0, 1 или 2,
или их физиологически совместимые соли.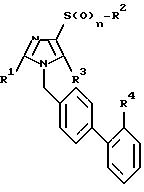
где R1, R2, R3, R4 и n указанные значения,
или их физиологически совместимых солей, отличающийся тем, что соединение формулы II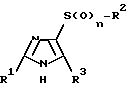
где R1, R2, R3 и n имеют указанные значения,
подвергают взаимодействию с соединением общей формулы III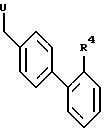
где R4 имеет указанные значения и U обозначает отщепляемую группу и, в случае необходимости, удаляют имеющиеся защитные группы, переводят полученные сульфонамиды общей формулы I в уретаны общей формулы I или переводят полученные сульфонамиды общей формулы I или полученные уретаны общей формулы I в сульфонилмочевины общей формулы I с выделением целевого продукта в свободном виде или в виде физиологическим совместимой соли.
| SU, авторское свидетельство, 1518949, A 61 K 31/415, 1982 | |||
| WO, патент, 92 /00068, A1, A 61 K 31/415, 09.01.92 | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| GB, патент, 2142533, A, A 61 K 31/ 415, 23.01.88. | |||
