Изобретение относится к новым замещенным 3-аминохроманам и тиохроманам, их энантиомерам и солям; способам их получения, фармацевтическим композициям, содержащим такие терапевтически активные соединения, а также к новым интермедиатам, применяемым при получении этих терапевтически активных соединений, и к использованию указанных активных соединений в терапии.
Целью этого изобретения является предоставление соединений для терапевтического использования, особенно соединений, имеющих терапевтическую активность через центральную нервную систему (ЦНС). Дополнительной целью является предоставление соединений, имеющих селективное действие на 5-гидрокси-триптаминовые рецепторы у млекопитающих, включая человека.
Терапевтически полезные 3-амино-дигидро(1)-бензопиран и бензотиопиран, обладающие эффектом в отношении 5-гидркоситриптаминовых неронов у млекопитающих, раскрыты в Европатенте 0222996.
Эти соединения определены формулой: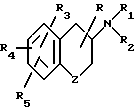
в которой Z является атомом кислорода или серы;
R атом водорода, или низший алкил;
R1 атом водорода, низший алкил или арил-низший алкил;
R2 атом водорода, низший алкил или арил низший алкил;
или R1 и R2 вместе образуют кольцо из 4-6 атомов углерода;
R3 атом водорода, гидрокси-, низший алкокси-, арил-низший алкокси-, ацилокси- или арилокси-группа, когда Z является атомом серы, и R3 является гидрокси-, низший алкокси-, арилнизший алкокси-; оцилокси- или арилокси-, когда Z это атом кислорода, причем R3 находится в положении 5- или 8-, когда Z атом кислорода;
R4 и R5 независимо являются атомом водорода, низшим алкилом или галогеном, и их моно- или ди-S-оксидами, когда Z является атомом серы, и фармацевтически приемлемыми их солями.
Гидрохлориды 3-хроманамина с двумя алкильными группами в ароматическом кольце, имеющие центральную стимулирующую активность, описаны в журнале J. Med. Chem. 1972, том 15, с. 863-865.
Целью настоящего изобретения является получение новых соединений, которые обладают высоким сродством к 5-гидрокситриптаминовым рецепторам в центральной нервной системе и в то же время они действуют как агонисты, парциальные агонисты или антагонисты в отношении серотониновых рецепторов.
Таким образом, группа новых веществ формулы I настоящего изобретения, а также их энантиомеры и соли используются при терапевтическом лечении состояния, которые лечатся 5-гидрокситриптамином, и нарушений, таких как депрессия, беспокойство, потеря аппетита, старческое слабоумие, мигрень, заболевание Альцхаймера, гипертония, нарушений терморегуляции и сексуальных нарушений. Дополнительные аспекты этого изобретения связаны с использованием указанных веществ их энантиомеров и солей для подавления болевых ощущений и регулирования сердечно-сосудистой системы.
Таким образом, изобретение предоставляет вещества формулы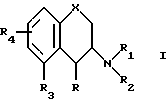
в которой X это атом кислорода или группа S=(O)p;
p целое число: 0, 1 или 2; R является атомом водорода фтора или алкилом C1-C6; R1 является атомом водорода, алкилом C1-C6 или алкенилом C2-C6; R2 является атомом водорода, алкилом C1-C6, алкенилом C2-C6; алкил (C1-C4)арилом, в котором арил может содержать 1 или 2 гетероатома, выбранных из азота, кислорода или серы, и необязательно замещен атомом галогена, циано-, трифторметильной, C1-C6-алкильной, C2-C6-алкенильной или C1-C4-алкоксильной группой; или
R1 и R2 вместе могут образовать пяти- или шестичленный цикл, который может содержать 1 или 2 гетероатома, выбранных из азота, кислорода или серы;
R3 является атомом галогена, циано-, трифторметильной, группой SO3CF3, N3, NO2, C1-C6 алкильной, C2-C6 алкенильной, амино-группой, NR5R6, COR7, пяти- или шестичленным арилом, который может содержать 1 или 2 гетероатома, выбранных из азота, кислорода или серы и который является: а) необязательно замещенным одним или несколькими заместителями, которые независимо выбраны из галогена, циано-, трифторметильной, C1-C6 алкильной, C2-C6 алкенильной или C1-C4 алкоксильной группы, либо б) сконденсирован по двум соседним атомам углерода с арильным кольцом, причем упомянутый арил необязательно замещен одним или несколькими заместителями, которые независимо выбраны из галогена, циано-, трифторметильной, C1-C6 алкильной, C2-C6 алкенильной или C1-C4 алкоксильной группы;
R4 является атомом водорода, галогена или алкильной группой C1-C6;
R5 является атомом водорода, алкилом C1-C6, алкенилом C2-C6; R6 является алкилом C1-C6 или алкенилом C2-C6 или
R5 и R6 вместе могут образовать пяти- или шестичленный цикл, который может содержать 1 или 2 гетероатома, выбранных из азота, кислорода или серы;
R7 является атомом водорода, хлора, брома, гидроксильной группой, C1-C6 алкильной, C2-C6 алкенильной, C1-C4 алкоксильной, NR8 R9 или пяти-, или шестичленным арилом, который может содержать 1 или 2 гетероатома, выбранных из азота, кислорода или серы и необязательно замещен одним или несколькими атомами галогена, циано-, трифторметильной, C1-C6 алкильной, C2-C6 алкенильной или C1-C4 алкоксильной группы;
R8 и R9 каждый независимо является атомом водорода, C1-C6 алкильной, C2-C6 алкенильной группой пяти- или шестичленным арилом, который может содержать 1 или 2 гетероатома, выбранных из азота, кислорода или серы и который необязательно замещен атомом галогена, циано-, трифторметильной, C1-C6 алкильной, C2-C6 алкенильной или C1-C4 алкоксильной группой или могут вместе образовать пяти- или шестичленное кольцо, содержащее 1 или 2 гетероатома, выбранных из азота, кислорода или серы; или его энантиомеры, или соли.
Дополнительным аспектом этого изобретения является фармацевтический препарат, содержащий в качестве активного компонента вещество формулы I,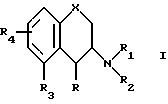
в которой X это атом кислорода или группа S=(O)p;
p целое число: 0, 1 или 2; R является атомом водорода, фтора или алкилом C1-C6; R1 является атомом водорода, алкилом C1-C6 или алкенилом C2-C6; R2 является атомом водорода, алкилом C1-C6 алкенилом C2-C6; алкил (C1-C4) арилом, в котором арил может содержать 1 или 2 гетероатома, выбранных из азота, кислорода или серы, и необязательно замещен атомом галогена, циано-, трифторметильной, C1-C6 алкильной, C2-C6 алкенильной или C1-C4 алкоксильной группой; или
R1 и R2 вместе могут образовать пяти- или шестичленный цикл, который может содержать 1 или 2 гетероатома;
R3 является атомом галогена, трифторметильной группой C1-C6 алкильной, C2-C6 алкенильной, -NR5R6, COR7, пяти- или шестичленным арилом, который может содержать 1 или 2 гетероатома, выбранных из азота, кислорода или серы и который является: a) необязательно замещенным одним или несколькими заместителями, которые независимо выбраны из галогена, циано-, трифторметильной, C1-C6 алкильной, C2-C6 алкенильной или C1-C4 алкоксильной группы, либо б) сконденсирован по двум соседним атомам углерода с арильным кольцом, причем упомянутый арил необязательно замещен одним или несколькими заместителями, которые независимо выбраны из галогена, циано-, трифторметильной, C1-C6 алкильной, C2-C6 алкенильной или C1-C4 алкоксильной группы;
R4 является атомом водорода или галогена;
R5 является атомом водорода, алкилом C1-C6 алкенилом C2-C6;
R6 является алкилом C1-C6 или алкенилом C2-C6 или
R5 и R6 вместе могут образовать пяти- или шестичленный цикл, который может содержать 1 или 2 гетероатома, выбранных из азота, кислорода или серы;
R7 является атомом водорода, C1-C6 алкильной, C2-C6 алкенильной, C1-C4 алкоксильной группой NR8R9 или пяти-, или шестичленным арилом, который может содержать 1 или 2 гетероатома, выбранных из азота, кислорода или серы и необязательно замещен одним или несколькими атомами галогена, циано-, трифторметильной, C1-C6 алкильной, C2-C6 алкенильной или C1-C4 алкоксильной группы;
R8 и R9 каждый независимо является атомом водорода, C1-C6 алкильной, C2-C6 алкенильной группой, пяти- или шестичленным арилом, который может содержать 1 или 2 гетероатома, выбранных из азота, кислорода или серы и который необязательно замещен атомом галогена, циано-, трифторметильной, C1-C6 алкильной, C2-C6 алкенильной или C1-C4 алкоксильной группой или могут вместе образовать пяти- или шестичленное кольцо, содержащее 1 или 2 гетероатома, выбранных из азота, кислорода или серы; или его энантиомеры, или соли.
Предпочтительной группой терапевтически активных веществ формулы I являются те, в которых R1 и R2 каждый независимо является атомом водорода, н-пропилом, изопропилом или циклопропилом и R3 является карбонильной группой COR7. Среди этих групп имеются такие определения R7 как алкил, аминоалкил, например метил, этил, н-пропил, изопропил, циклопропил, н-бутил, изобутил, трет-бутил и циклобутил или арил, аминоарил, например, фенил, тиенил, фторфенил и фуранил. Другой предпочтительной группой является такая, когда R3 является арилом, например фенилом, тиенилом, фторфенилом или фуранилом. Другой предпочтительной группой является такая, когда R3 является алкилом, например н-пропилом, изопропилом или алкенилом, например изопропенилом и аллилом. Другой предпочтительной группой активных соединений являются такие, в которых R4 является атомом галогена в положении 8, а также его энантиомеры.
Вещества формулы I, в которой R3 является циано-, карбокси-, COCl, COBr, NH2 или SO3CF3 группой, являются новыми промежуточными соединениями для приготовления терапевтически активных веществ формулы I.
Алкил C1-C6 в формуле I представляет собой нормальные, разветвленные и циклические алкильные группы, имеющие от 1 до 6 атомов углерода, например, метил, этил, н-пропил, изопропил, циклопропил, н-бутил, изобутил, трет-бутил, циклобутил, н-пентил, изопентил, трет-пентил, нео-пентил, циклопентил, н-гексил, изогексил, циклогексил, метилциклопропил, этилциклопропил, метилциклобутил. Предпочтительные алкильные группы имеют от 1 до 4 атомов углерода.
Алкенил C2-C6 в формуле I представляет собой нормальные или разветвленные алкенильные группы, имеющие от 2 до 6 атомов углерода и содержащие одну или две двойных связи, например алкил, пропенил, изопропенил, бутенил, изобутенил, пентенил, изопентенил. Предпочтительные алкенильные группы имеют от 2 до 4 атомов углерода и одну двойную связь.
Алкокси C1-C4 в формуле I представляет собой нормальные алкоксильные группы, имеющие от 1 до 4 атомов углерода, например метокси, этокси, пропокси, бутокси, предпочтительно метокси- и этокси.
Алкил C1-C4 арил, когда арил может содержать 1 или 2 гетероатома, выбранных из азота, кислорода или серы, в определении R2 в формуле I представляет собой арильный остаток, имеющий от 3 до 12 атомов углерода в ароматическом кольце и необязательно 1 или 2 гетероатома, выбранных из азота, кислорода или серы, в ароматическом кольце, связанный нормальной или разветвленной алкиленовой цепочкой, имеющей от 1 до 4 атомов углерода в алифатической цепочке. Ароматическое кольцо может быть замещено одной или несколькими группами: нитрильной, трифторметильной, галоидной, такой как фтор, хлор, бром, иод, алкил C1-C6, например метил, этил, пропил, алкенил C2-C6, например аллил, пропенил, или алкокси C1-C4 предпочтительно в мета и/или пара-положении. Примерами подходящих арильных групп в алкил C1-C4 ариле являются фенил, нафтил, дифенил, тиенил, фурил, пирил, пиримидил и пирридинил. Предпочтительными алкил C1-C4 арильными группами являются незамещенные и замещенные фенилалкильные группы, в которых алкильная группа является нормальным или разветвленным алкилом, имеющим от 1 до 4 атомов углерода, а ароматическое кольцо может быть замещено одной или несколькими группами, такой как фтор, хлор, бром, иод, нитрил, трифторметил, метил или этил в мета- и/или пара-положении. Например, бензил, фенетил и фенилпропил, особенно предпочтительным является фенилпропил.
Атом галогена в формуле I представляет собой фтор, хлор, бром, иод, предпочтительно фтор, хлор и бром.
Пяти- или шестичленный арил, который может содержать 1 или 2 гетероатома, выбранных из азота, кислорода или серы и который является: а) необязательно замещенным одним или несколькими заместителями, которые независимо выбраны из галогена, циано-, трифторметильной, C1-C6 алкильной, C2-C6 алкенильной или C1-C4 алкоксильной группы, либо б) сконденсирован по двум соседним атомам углерода с арильным кольцом, причем упомянутый арил необязательно замещен одним или несколькими заместителями, которые независимо выбраны из галогена, циано-, трифторметильной, C1-C6 алкильной, C2-C6 алкенильной или C1-C4 алкоксильной группы; в определении R3 в формуле I, представляет собой либо а) замещенный или незамещенный фенил, тиенил, фурил, пиридил, пиримидил, пиразинил, пирадазинил, тиазолил, изотиазолил, оксазолил, изоксазолил, имидазолил, пиразолил, пиперазинил или морфолинил, либо б) замещенный или незамещенный хинолил, изохинолил, хиназолил, хинаксазолил или индолил.
Пяти- или шестичленный арил, который может содержать 1 или 2 гетероатома, выбранных из азота, кислорода или серы, в определении R7, R8 и R9 в формуле I представляет собой фенил, тиенил, фурил, пиридил, пиримидил, пиразинил, пиридазинил, тиазолил, изотиазолил, оксазолил, изоксазолил, имидазолил, пиразолил, пиперазинил или морфолинил.
Примерами подходящих пяти- или шестичленных циклических структур, которые могут быть образованы радикалами R1 и R2 или R5 и R6, или R7 и R8 и которые могут содержать дополнительный гетероатом, выбранный из азота, кислорода или серы, являются пиперазин, морфолин, пирролидин, пиррол, пирролин, имидазол, имидазолин, имидазолидин, пиридин, пиразол, пиримидин, пиразин, пиридазин.
Вещества этого изобретения имеют один или два асимметричных атома углерода. Когда R является атомом водорода, вещества имеют асимметричный атом углерода, смежный с атомом азота, то есть C3, и когда R является алкилом C1-C6, вещества имеют асимметричный атом углерода, смежный с атомом азота, и асимметричный атом углерода, смежный с алкильной группой, то есть C4. Таким образом, эти вещества существуют в виде двух или четырех оптических изомеров, т. е. энантиомеров. В объем защиты этого изобретения входят как чистые энантиомеры, так и рацемические смеси. Терапевтические свойства этих веществ могут в большей или меньшей степени быть приписаны рацемату или имеющимся энантиомерам.
Для образования нетоксичных, фармацевтически приемлемых кислотных аддитивных солей соединений этого изобретения могут использоваться как органические, так и неорганические кислоты. Примерами кислот являются серная, азотная, фосфорная, щавелевая, соляная, муравьиная, бромистоводородная, лимонная, уксусная, молочная, винная, памовая, этандисульфокислота, сульфаминовая, янтарная, метилсульфоновая, пропионовая, гликолиевая, яблочная, глюконовая, пировиноградная, фенилуксусная, 4-аминобензойная, антраниловая, салициловая, 4-аминосалициловая, 4-гидроксибензойная, никотиновая, метансульфоновая, этансульфоновая, гидроксиэтансульфоновая, бензолсульфокислота, пара-толуолсульфокислота, сульфениловая, нафталинсульфокислота, аскорбиновая, циклогексилсульфаминовая, фумаровая, малеиновая и бензойная кислоты. Эти соли легко получаются методами, которые известны в этой области химии.
Вещества формулы I могут быть получены с помощью следующих процессов, составляющих дополнительный аспект этого изобретения.
а. Превращение вещества формулы II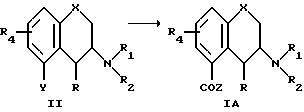
в которой Y является отщепляемой группой, такой как трифторметансульфонат (OSO2CF3), атом галогена, например хлор или бром, и X, R, R1, R2 и R4, такие как определено выше, в результате замещения группы V в карбоксигруппу COZ, в которой Z является хлором, бромом, гидроксилом, группой ORp, где Rp является алкилом C1-C6, с образованием вещества формулы I, в которой R3 является группой COZ (IA).
Соединение формулы II может быть превращено в вещество формулы IA в результате следующего каталитического цикла. Металл M0 должен быть нульвалентным переходным металлом (M), таким как палладий или никель, которые способны подвергаться окислительному присоединению по связям арил-Y, например, связям арил-SO3CF3. Этот металл может генерироваться на месте, из двухвалентного металла M'' Арил-CO-M''-Y образуется за счет обработки монооксидом углерода (CO).
Дополнительными реагентами являются спирт, например, такой как метанол, этанол; аминное основание, такое как триалкиламин, например триэтиламин; в инертном органическом растворителе, предпочтительно в полярном апротонном растворителе, таком как диметилформамид (ДМФ), диметилсульфоксид (ДМСО), ацетон, ацетонитрил и др. Обычно реакцию осуществляют при температуре между +40 и +120 oC и давлении между 100 и 500 кПа. Затем необязательно следует гидролиз и обработка тионилгалогенидом, например тионилхлоридом, для того чтобы получить соответствующую производную галогенида кислоты.
b) Вещество формулы I, в которой R3 является группой COZ (IA), также может быть получено с помощью обратного процесса. Взаимодействие в виде каталитического цикла с использованием нульвалентного переходного металла M0, такого как палладий или никель, который может подвергаться окислительному присоединению по связи Z-Y, где Z является атомом хлора, брома, группой OH, ORp, причем ORp является алкилом C1-C6 и Y отщепляемой группой, такой как OSO2CF3, атом галогена, обработка монооксидом углерода с последующим добавлением соединения формулы III, в которой X, R, R1, R2 и R4 такие, как определено в формуле I.
Соединение Z-CO-M''-Y также может образоваться непосредственно из Z-CO-Cl. Условия реакции и реагенты такие же самые, как описано выше в методе a). Гидролиз подходящего сложного эфира карбоновой кислоты дает свободную кислоту, которая может быть превращена в производную галогенида кислоты.
c. Превращение соединения формулы II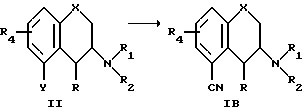
в которой X, R, R1, R2 и R4 такие, как определено выше, и Y является отщепляемой группой, такой как SO3CF3, хлор или бром, посредством обработки цианидным реагентом, таким как цианид меди, с образованием соединения формулы I, в которой R3 является группой CN (IB). Взаимодействие с цианидным реагентом осуществляют в инертном органическом растворителе, таком как диметилформамид, гексаметилентриамид фосфорной кислоты и др. при температуре между +20 и +200 oC, предпочтительно между 50 и 150 oC и нормальном давлении.
d. Аминирование соединения формулы IA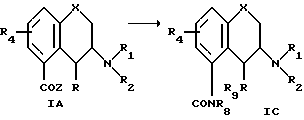
в которой X, R, R1, R2 и R4, такие как определено выше, и Z является атомом хлора, брома, группой OH или ORp, причем ORp является алкилом C1-C6.
Если соединение формулы IA является сложным эфиром карбоновой кислоты, то его необходимо сначала подвергнуть гидролизу до свободной кислоты. Затем свободную кислоту переводят в амид формулы IC через производную хлорида кислоты посредством взаимодействия с соответствующим амином NHR8R9, где R8 и R9 такие, как определено в формуле I, в неполярном апротонном растворителе, например толуоле, бензоле с обратным холодильником при температуре между 0 и 100 oC.
e. По реакции Виттига с образованием соединения формулы I, в которой R3 является алкенильной группой C2-C6 (IE)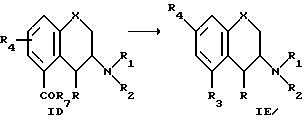
5-карбокси-хроман/тиохроман-производную, в которой X, R, R1, R2 и R4, такие как определено в формуле I, и R7 является алкилом, определенным выше (ID), превращают с использованием диполярного реагента, такого как галогенид алкилтрифенилфосфония, с образованием соответствующей алкенильной группы (IE).
f. Каталитическое гидрирование 5-алкен-хроман/тиохроман-производной формулы I, в которой R3 является алкенильной, группой C2-C6, с использованием водорода и палладия, водорода и платины или водорода и никеля Ренея с получением соответствующей производной формулы I, в которой R3 является алкильной группой C1-C6 (IF).
g. Замещение 5-бромхроман/тиохроман-производной посредством обработки триалкилоловянным реагентом в присутствии нульвалентного металла, предпочтительно палладия, с образованием вещества формулы I, в которой R3 является алкилом C1-C6 алкенилом C1-C4 или арилом, в присутствии монооксида углерода дает соединение формулы I, в которой R3 является группой COR7, в которой R7 определен как алкил C1-C6, алкилен C2-C6 или арил.
Замещение может быть осуществлено одним из следующих способов:
h. Превращение 5-карбокси-хроман/тиохроман-производной формулы I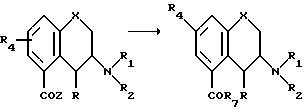
в которой X, R, R1, R2 и R4 такие, как определено выше, и Z является хлором или бромом, с использованием R7Zi, в котором R7 определен как алкил C1-C6, алкилен C2-C6 или арил, чтобы получить соответствующую 5-кето-хроман/тиохроман-производную. Подходящим используемым алкиллитиевым соединением является, например метиллитий, алкениллитием виниллитий и ариллитием фениллитий. Взаимодействие осуществляют в инертном органическом растворителе, предпочтительно неполярном апротонном растворителе, таком как простые эфиры, например диэтиловый эфир, тетрагидрофуран при температуре между -50 и +50 oC.
i. Гидролизом соединения формулы I, в которой R3 является циано-группой (IB)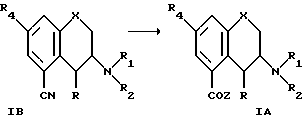
в которой X, R, R1, R2 и R4 такие, как определено выше, с необязательной последующей обработкой тионилгалогенидом, например тионилхлоридом, тионилбромидом, для того чтобы получить соединение формулы (I), в которой R3 является группой COZ, где Z гидроксил, хлор или бром.
j. Замещением вещества формулы I, в которой R3 является циано-группой (IB)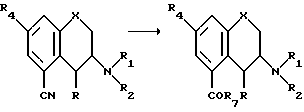
в которой X, R, R1, R2 и R4 такие, как определено выше, посредством обработки подходящим металлоорганическим реагентом, предпочтительно литийорганическим, таким как R7Zi или реактив Гриньяра, такой как R7Mg-галогенид, в инертном органическом растворителе, предпочтительно неполярном апротонном растворителе, таком как бензол, простые эфиры, например диэтиловый эфир, тетрагидрофуран, с последующим гидролизом промежуточного комплекса, чтобы получить соединение формулы I, в которой R3 является группой COR7, где R7 является алкилом C1-C6, алкенилом C2-C6 или арилом.
k. Гидрирование 5-алкен-тиохроман/хромановой производной формулы I, в которой R3 является алкенильной группой C2-C6, с использованием водорода и палладия, водорода и платины или водорода и никеля Ренея или азодикарбоксилата калия, с образованием соответствующей тиохроман/хромановой производной формулы I, в которой R3 является алкилом C1-C6.
l. Превращение соединения формулы II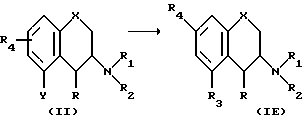
в которой Y является отщепляемой группой, такой как трифторметансульфонат, фосфонат, галогенид, такой как бром или иод, и R, R1, R2 такие как определено выше, посредством замещения группы Y на R3, которая является алкенильной группой C2-C6 (IE).
Соединение (II) может быть превращено в (IE) путем взаимодействия с переходным металлом, таким как палладий или никель, которые могут образовывать лигандный комплекс и подвергаться окислительному присоединению. Подходящий алкенильный заместитель может быть введен в результате обработки триалкилалкенилстаннаном.
Дополнительными реагентами являются амин, такой как триэтиламин, и литиевая соль, например, хлорид лития. Взаимодействие предпочтительно осуществляют в полярном апротонном растворителе, таком как диметилформамид, диоксан, ацетонитрил или диметилсульфоксид при температуре между +40 и 120 oC.
m. Превращение соединения формулы II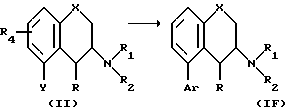
в которой Y является отщепляемой группой, такой как трифторметансульфонат, фосфонат, галогенид, такой как бром или иод, и R, R1, R2 такие как определенно выше, посредством замещения группы Y на пяти- или шестичленный арил, который может содержать 1 или 2 гетероатома, выбранных из азота, кислорода или серы и который или замещен, или сконденсирован по двум соседним атомам углерода с арильным кольцом, которое определено выше, с образованием вещества формулы IF.
Соединение (II) может быть превращено в (IF) посредством взаимодействия с переходным металлом, таким как палладий или никель, которые способны образовать лигандный комплекс и подвергаться окислительному присоединению. Подходящий арильный заместитель может быть введен в результате обработки триалкиларилстаннаном или арилборной кислотой.
Дополнительными реагентами являются амин, такой как триэтиламин, и литиевая соль, например хлорид лития. Взаимодействие предпочтительно осуществляют в полярном апротонном растворителе, таком как диметилформамид, диоксан, ацетонитрил или диметилсульфоксид при температуре между +40 и 120 oC.
Следующий метод описывает один из путей получения промежуточного соединения формулы IB, в которой R1, R2 и R4 такие, как определенно в формуле I.
В соответствии с изобретением соединения формулы I обычно назначаются перорально, ректально или посредством инъекции в виде фармацевтических препаратов, включающих активный компонент или в виде свободного основания, или фармацевтически приемлемой нетоксичной аддитивной кислотной соли, например гидрохлорида, гидробромида, лактата, ацетата, фосфата, сульфата, сульфамата, цитрата, тертрата, оксалата и тому подобных в фармацевтически приемлемых формах дозировки. Форма дозировки может быть твердой или жидким препаратом. Обычно активное вещество может составлять от 0,1 до 99 мас. от веса препарата, более конкретно между 0,5 и 20 от веса препарата, предназначенного для инъекции, и между 0,2 и 50 от веса препарата, пригодного для перорального назначения.
Для того чтобы получить фармацевтические препараты, содержащие вещество формулы I в единичной форме дозировки для перорального использования, выбранное вещество может быть смешано с твердой средой для лекарства, например лактозой, сахарозой, сорбитолом, маннитолом, крахмалами, такими как картофельный крахмал, кукурузный крахмал или амидопектин, производными целлюлозы, связующим, таким как желатин или поливинилпирролидон, и смазывающим веществом, таким как стеарат магния, стеарат кальция, полиэтиленгликоль, воски, парафин и тому подобные, и затем смесь прессуют в таблетки. Если требуются таблетки с покрытием, то основа, приготовленная как описано выше, может быть покрыта концентрированным раствором сахара, который может содержать арабскую камедь, желатин, тальк, диоксид титана и т.п. Альтернативно таблетка может быть покрыта полимером, который известен специалистам в этой области химии, растворенным в легко испаряющемся органическом растворе или смеси органических растворителей. В эти покрытия могут быть добавлены красящие вещества, для того чтобы легко различать таблетки, содержащие различные активные вещества или различные количества активных веществ.
Для приготовления мягких желатиновых капсул активное вещество может быть смешано, например, с растительным маслом или полиэтиленгликолем. Твердые желатиновые капсулы могут содержать гранулы активного вещества с использованием либо вышеупомянутых сред для таблеток, например лактозы, сахарозы, сорбита, маннитола, крахмалов (например картофельного, кукурузного или амилопектина), производных целлюлозы или желатина. Кроме того, твердые желатиновые капсулы могут быть заполнены жидкими или полутвердыми лекарствами.
Единичные дозировки для ректального применения могут быть растворами или суспензиями, или могут быть приготовлены в форме свеч, включающих активное вещество в смеси с нейтральным жидким основанием, или желатиновые ректальные капсулы, включающие активное вещество в смеси с растительным или парафиновым маслом.
Жидкие препараты для перорального применения могут быть в форме сиропов или суспензий, например растворов, содержащих приблизительно от 0,2 до 20 от массы активного вещества, описанного здесь, остальное приходится на сахар и смесь этилового спирта, воды, глицерина и пропиленгликоля. Такие жидкие препараты необязательно могут содержать красящие агенты, вкусовые вещества, сахарин и карбоксиметилцеллюлозу в качестве загущающего агента или другие среды для лекарств, которые известны специалисту в этой области.
Растворы для парэнтеральных применений посредством инъекции могут быть приготовлены в водном растворе водорастворимой фармацевтически приемлемой соли активного вещества, предпочтительно в концентрации примерно от 0,5 до 10 мас. Эти растворы также могут содержать стабилизирующие агенты и/или буферные агенты, причем они могут быть удобно предоставлены в различных ампулах единичной дозировки.
Подходящие суточные дозы веществ этого изобретения при терапевтическом лечении людей составляют примерно от 0,01 до 100 мг/кг веса тела при пероральном назначении и от 0,001 до 100 мг/кг веса тела при парэнтеральном назначении.
Пример 1. 3-дипропиламино-5-трифторметансульфонилхроман.
3-дипропиламино-5-гидроксихроман (Торберг и с отр. Acta Phasm. Suec. 1987, 24) 1,4 г (4,0 ммоль) и 0,1 г (0,75 ммоль) N,N-диметиламинопиридина растворяют в 50 мл хлористого метилена и охлаждают до -30 oC. Добавляют 0,75 мл (5,7 ммоль) 2,4,6-коллидина и затем 1,0 мл (6,0 ммоль) ангидрида трифторметансульфокислоты. Раствор перемешивают 3 ч при -20 oC и затем дают ему нагреться до комнатной температуры. Этот раствор промывают водным раствором кислого карбоната натрия, сушат над сульфатом натрия и выпаривают досуха. Светложелтое масло окончательно очищают методом флэш-хроматографии (силикагель) посредством элюирования смесью этилацетат/гексан 1:9. Выход равен 55 Т.пл. 125-127 oC (оксалат).
Пример 2. 3-дипропиламино-5-метилоксикарбонилпроман.
3-дипропиламино-5-трифторметансульфонилхроман (пример 1: 4,43 г, 11,6 ммоль) растворяют в 80 мл смеси диметилформамид/метанол 6:2 и раствор дегазируют 15 мин при 20 oC при давлении 10 тор. Затем добавляют ацетат палладия (76 мг, 0,34 ммоль), 141 мг (0,34 ммоль) 1,3-бис-дифенилфосфинопропана и 3,5 мл (25 ммоль) триэтиламина. Смесь нагревают до 70 oC в атмосфере CO и перемешивают в течение 5 ч. Раствор охлаждают, разбавляют 200 мл толуола, промывают водным раствором бикарбонатом натрия, сушат сульфатом натрия и выпаривают досуха, масло очищают методом флэш-хроматографии на силикагеле при элюировании смесью этилацетат/гексан 1:8. Выход равен 76 Т.пл. 150-152 oC (гидрохлорид).
Пример 3.
3-дипропиламино-5-карбамоилхроман. 3-дипропиламино-5-метилоксихроман (пример 2: 400 мг, 1,37 ммоль) растворяют в 10 мл метанола и добавляют 60 мл (1,5 ммоль гидроксида натрия в 2 мл воды. Смесь кипятят с обратным холодильником 5 ч, охлаждают, фильтруют через Целит и выпаривают досуха. Остаток кипятят с хлористым сульфуридом SOCl2 (5 мл, 68 ммоль) в течение 30 мин. Избыток хлористого сульфурида затем удаляют в вакууме, чтобы получить гидрохлорид 3-дипромиламино-5-хлорформилхромана в виде смолы. Эту светлокоричневую смолу растворяют в 50 мл хлористого метилена и пропускают через раствор поток газообразного аммиака в течение 2 мин. Раствор промывают водным раствором гидрокарбоната натрия, сушат сульфатом натрия и выпаривают досуха. Масло очищают методом флэш-хроматографии (на силикагеле) путем элюирования смесью этилацетат/гексан 1:4. Выход 80 Спектр ЯМР C13: 172,0; 154,9; 136,5; 126,9; 120,4; 119,1; 118,6; 67,8; 53,0; 52,6; 26,1; 22,4; 21,9; 14,1; 11,7.
Пример 4. 3-дипропиламино-5-N,N-диметилкарбамоилхроман.
Указанное в заголовке вещество было приготовлено по методике, аналогичной использованной в примере 3, исходя из 3-дипропиламино-5-метилоксикарбонилхроман и используя вместо аммиака газообразный диметиламин. Спектр ЯМР 13С: 189,3; 170,3; 149,9; 137,4; 126,7; 126,1; 124,9; 65,8; 64,7; 48,2; 47,7; 30,7; 26,0; 15,1; 10,9.
Пример 5. 3-Дипропиламино-5-N,N-диизопропилкарбамоилхроман.
Указанное в заголовке вещество было приготовлено по методике, аналогичной использованной в примере 3, исходя из 3-дипропиламино-5-метилоксихромана. Т. пл. 228-230 oC (гидрохлорид).
Пример 6. 3-дипропиламино-5-N-метилкарбамоилхроман.
Указанное в заголовке вещество было приготовлено аналогично методике, использованной в примере 3, исходя из 3-дипропиламино-5-метилоксикарбонилхромана и используя вместо аммиака газообразный метиламин. Т. пл. 95-97 oC (оксалат).
Пример 7. 3-диипропиламино-5-ацетилхроман.
Гидрохлорид 3-дипропиламино-5-хлорформилхромана (4,42 г, 13,4 ммоль), приготовленный из 3-дипропиламино-5-метилоксикарбонилхромана (пример 2) по методике, аналогичной использованной в примере 3, в 20 мл сухого тетрагидрофурана добавляют к предварительно полученному раствору диметилкупрата лития, полученного из метиллития и иодистой меди, в 200 мл тетрагидрофурана при -78 oC. Этот раствор перемешивают в течение 15 мин при -78 oC и затем дают ему нагреваться до комнатной температуры в течение 10 мин. Затем медленно добавляют 30 мл воды. Органическую фазу декантируют, сушат сульфатом натрия и выпаривают досуха. Остаток очищают методом флэш-хроматографии (на силикагеле) путем элюирования смесью этилацетат/гексан 1:8. Указанное в заголовке соединение кристаллизуется в виде соли из этилацетата. Т. пл. 106-108 oC (оксалат).
Пример 8. 3-дипропиламино-5-циклопропилкарбонилхроман.
Указанное в заголовке соединение было приготовлено по методике, аналогичной использованной в примере 7, используя вместо диметилкупрата лития дициклопропилкупрат лития (J. Org. Chem. том 41, с. 22, 1976), Т. пл. 100- 102 oC (оксалат).
Пример 9. 3-дипропиламино-5-трет-бутилкарбонилхроман.
Указанное в заголовке соединение было приготовлено по методике, аналогичной использованной в примере 7, используя вместо диметилкупрата лития, ди-трет-бутилкупрат лития (из трет-бутиллития и комплекс бромид меди диметилсульфид). Т. пл. 118-120 oC (оксалат).
Пример 10. 3-дипропиламино-5-трет-бутилкарбонилхроман.
Указанное в заголовке соединение было приготовлено по методике, аналогичной использованной в примере 7, с использованием вместо диметилкупрата лития диизопропилкупрата магния (из пропилмагний хлорида и комплекса CuBr* диметилсульфид). Т. пл. 60-62 oC (оксалат).
Пример 11. 3-дипропиламино-5-(4-торфенилкарбонил)хроман.
Указанное в заголовке соединение было приготовлено по методике, аналогичной использованной в примере 7, с использованием вместо диметилкупрата лития ди-(4-фторфенил) купрата магния (из 4-фторфенилмагний бромида и иодида меди). Т. пл. 98,3-98,4 oC (оксалат).
Пример 12. 3-дипропиламино-5-(тиенилкарбонил)хроман.
Указанное в заголовке соединение было приготовлено по методике, аналогичной в примере 7, с использованием вместо диметилкупрата лития ди-(2-тиенил)купрата лития (из 2-тиениллития и иодистой меди). Т. пл. 87-88,5 oC (оксалат).
Пример 13. 3-дипропиламино-5-изопропенилкарбонилхроман.
Раствор 0,62 г (1,74 ммоль) метилтрифенилфосфонийбромида в осушенном диэтиловом эфире (20 мл) в атмосфере азота при комнатной температуре, добавляют 1,74 ммоль н-бутиллития (0,7 мл 2,5 молярного раствора) и раствор перемешивают 4 ч. Растворяют 0,4 г (1,45 ммоль) 3-дипропиламино-5-ацетилхромана (пример 7) в 2,0 мл осушенного диэтилового эфира и этот раствор добавляют к предварительно приготовленному реактиву Виттига. Смесь перемешивают при комнатной температуре в течение ночи. Раствор разбавляют толуолом и промывают водой. Высушивание органической фазы сульфатом натрия и выпаривание ее досуха дает твердое вещество, которое окончательно было очищено методом флэш-хроматографии при элюировании смесью этилацетат/гексан 1:4. Собранные фракции были выпарены и дали указанное в заголовке соединение в виде бесцветного масла. Спектр ЯМР 13C: 11,82, 21,94 24,28 26,69 52,79 53,64 67,70 115,03 118,73 120,07 126,83, 144,88 145,27 154,03.
Пример 14. 3-дипропиламино-5-аминохроман.
Растворяют 1,0 г (3,4 ммоль) 3-дипропиламино-5-метилоксикарбонилхромана (пример 2) в метаноле (20 мл). Добавляют 0,16 г (4,1 ммоль) гидроксида натрия в 1,0 мл воды и этот раствор кипятят с обратным холодильником в азоте в течение ночи. Раствор выпаривают досуха, добавляют 20 мл толуола и снова выпаривают досуха. Остаток растворяют в 20 мл толуола, добавляют 1,87 г (6,8 ммоль) дифенилфосфорилазида и раствор кипятят с обратным холодильником в течение 2 ч. Добавляют 2 мл метанола и кипячение продолжают еще 4 ч. Раствор охлаждают, промывают водой и экстрагируют разбавленной соляной кислотой. Кислую водную фазу нейтрализуют водным раствором гидроксида натрия и экстрагируют толуолом. Высушивают толуольную фазу сульфатом натрия и выпаривают толуолом. Высушивают толуольную фазу сульфатом натрия и выпаривают ее досуха. Остаток растворят в этаноле, содержащем 10-ный гидроксид натрия (20 мл) и раствор кипятят с обратным холодильником в течение ночи. Этот раствор охлаждают и разбавляют толуолом. Промывка водой, высушивание органической фазы и выпаривание ее досуха дает указанное в заголовке вещество в виде масла, которое превращается в дихлористоводородную соль. Т. пл. 173-174 oC.
Пример 15. 3-дипропиламино-5-нитрохроман.
3-дипропиламино-5-аминохроман (пример 14, 0,05 г, 0,2 ммоль) растворяют в смеси 0,08 мл (1 ммоль) трифторуксусной кислоты и 5 мл воды. Прозрачный раствор охлаждают до 0-4 oC. При хорошем перемешивании по каплям добавляют нитрит натрия (0,017 г, 2,5 ммоль) в 1 мл воды. Раствор перемешивают 15 мин и нейтрализуют карбонатом кальция. Добавляют раствор нитрита натрия (0,5 г, 7,2 ммоль) в 1 мл воды и затем смесь сульфата меди (0,01 г, 0,62 ммоль) и закиси меди в 1 мл воды. Раствор перемешивают при 0 oC в течение 20 мин и затем при комнатной температуре 2 ч. Раствор экстрагируют диэтиловым эфиром. Органическую фазу сушат сульфатом натрия и выпаривают досуха. Остаток очищают методом флэш-хроматографии на силикагеле при элюировании смесью этилацетат/гексан 1:9. Получают указанное в заголовке соединение с т. пл. 150-151 oC (гидрохлорид).
Пример 16. 3-дипропиламино-5-азидохроман.
3-дипропиламино-5-аминохроман (пример 14, 0,05 г, 0,2 ммоль) диазотируют по методике примера 15. После перемешивания в течение 15 мин добавляют 0,026 г (0,4 ммоль) азида натрия в 1 мл воды. После перемешивания при 5 oC в течение ночи раствор обрабатывают и очищают по методике примера 15, получая указанное в заголовке соединение с т. пл. 167-168 oC (оксалат).
Пример 17. 3-дипропиламино-5-(пиррол-1-ил)хроман.
3-дипропиламино-5-аминохроман (пример 14, 0,6 г, 2,42 ммоль) растворяют в 10 мл уксусной кислоты и добавляют 0,4 г (3 ммоль) 2,5-диметокситетрагидрофурана. Раствор кипятят с обратным холодильником в течение 1 ч. Этот раствор нейтрализуют водным гидроксидом натрия и экстрагируют толуолом. Органическую фазу сушат сульфатом натрия и выпаривают досуха. Остаток очищают методом флэш-хроматографии на силикагеле при элюировании смесью этилацетат/гексан 1:9 и получают указанное в заголовке соединение. Спектр ЯМР 13C: 11,75 21,89 24,81 52,69, 53,15 67,94 108,93 115,67 118,22 118,44 121,87 127,22 141,47 155,27.
Пример 18. 3-метил-3-фенилпропил)амино-5-гидроксихроман.
3-амино-5-метоксихроман (Торберг и сотр. Acta Phasm. Snec. 1987, p.24) (2 г, 9,28 ммоль) растворяют в 50 мл метанола и подкисляют уксусной кислотой до pH, равного 6,0. Раствор охлаждают до 0 oC и добавляют 0,87 г (13,8 ммоль) цианоборгидрида натрия вместе с 3-фенилпропаналем (1,22 мл, 9,28 ммоль). Удаляют охлаждение и раствор перемешивают при комнатной температуре 4 ч. Добавляют 0,42 г (14 ммоль) параформальдегида и 0,87 г (13,8 ммоль) цианоборгидрида натрия и перемешивание продолжают в течение ночи при комнатной температуре. Раствор разбавляют толуолом и промывают водой. Высушивание сульфатом натрия и выпаривание досуха дает масло. Это масло очищают методом флэш-хроматографии на силикагеле при элюировании смесью этилацетат/гексан 1: 4. Отобранные фракции выпаривают и получают масло, которое обрабатывают водным бромистым водородом (47) при 120 oC 1 ч. Раствор охлаждают и нейтрализуют гидроксидом натрия и экстрагируют толуолом. Органическую фазу сушат сульфатом натрия и выпаривают, получая указанное в заготовке соединение в виде масла. Спектр ЯМР 13C: 22,502 29,09 33,47 38,19 53,66 67,76 102,004 109,2 110,46 125,78 127,05 128,36 142,2 155,29 158,28.
Пример 19. 3-(метил-3-фенилпропил)амино-5-метилоксикарбонилхроман.
3-(метил-3-фенилпропил)амино-5-гидроксихроман (пример 18, 1 г, 3,37 ммоль) растворяют в 20 мл хлористого метилена при 20 oC. Добавляют 0,32 мл (4 ммоль) пиридина, 0,65 мл (5,9 ммоль) ангидрида трифторметансульфокислоты и 0,041 г (0,59 ммоль) диметиламинопиридина (ДМАП) при 20 oC и в атмосфере азота. Раствор перемешивают 3 ч при -20 oC. Удаляют охлаждение и раствор разбавляют толуолом, промывают водным раствором бикарбоната натрия, сушат сульфатом натрия, фильтруют через силикагель и выпаривают досуха. Оставшееся масло растворяют в 13 мл дегазированной смеси метанол/диметилформамид 3:10. Добавляют 0,056 г (0,25 ммоль) ацетата палладия, 0,103 г (0,25 ммоль) 1,3-бис (дифенилфосфино) пропана и 0,76 мл (5 ммоль) триэтиламина и раствор продувают газообразным оксидом углерода при интенсивном перемешивании. Давление в реакционном сосуде повышают до 20,2 кПа с помощью баллона с оксидом углерода, снабженном регулятором. Перемешивание продолжают в течение ночи при 75 oC. Давление и температуру доводят до нормальных значений и раствор разбавляют толуолом и промывают водой. Высушивают органическую фазу и выпаривают досуха. Оставшееся масло очищают методом флэш-хроматографии на силикагеле при элюировании смесью этилацетат/гексан 1:4. Отобранные фракции выпаривают и получают указанное в заготовке соединение в виде бесцветного масла. Спектр ЯМР 13C: 26,88 29,0 33,2 37,85 51,64 53,37 55,44 67,24 120,6 123,06 123,4 125,59 126,47 128,17 128,17 128,24 130,36 142,01 154,93 167,29.
Пример 20. 3-(метил-3-фенилпропил)амино-5-N-метилкарбамоилхроман.
3-(метил-3-фенилпропил)амино-5-метилоксикарбонилхроман (пример 19, 0,32 г, 0,94 ммоль) растворяют в 10 мл метанола. Добавляют 0,08 г гидроксида натрия (2 ммоль) в 1 мл воды и раствор кипятят в течение ночи в атмосфере азота. Раствор выпаривают досуха и снова выпаривают досуха совместно с толуолом (10 мл). Оставшееся твердое вещество кипятят с обратным холодильником в хлористом сульфуриле в течение 30 мин и выпаривают досуха. Светлокоричневую смолу растворяют в 20 мл тетрагидрофурана, обрабатывают газообразным метиламином 1 мин при интенсивном перемешивании. Раствор разбавляют толуолом и промывают водным раствором бикарбоната натрия. Высушивание и выпаривание дает смолу, которую окончательно очищают методом флэш-хроматографии на силикагеле при элюировании смесью этилацетат/гексан 1:2. Отобранные фракции выпаривают и получают указанное в заголовке соединение в виде бесцветной смолы. При кристаллизации из этилацетата щавелекислая соль образует белые иголочки. Т. пл. 150-151 oC (оксалат).
Пример 21. 3-дипропиламино-5-трифторметансульфонилтиохроман.
3-дипропиламино-5-гидроксибензотиопиран (Европейский патент 0222 996; 420 мг, 1,58 ммоль) и 0,27 г (0,29 мл) коллидина растворяют в 15 мл хлористого метилена и охлаждают до -30 oC. По каплям добавляют 0,54 г (0,32 мл) ангидрида трифторметансульфокислоты и дают смеси нагреться до комнатной температуры. Спустя 20 мин разбавляют смесь хлористым метиленом. Раствор промывают насыщенным раствором бикарбоната натрия, сушат сульфатом натрия и выпаривают в вакууме. Хроматография на силикагеле посредством элюирования хлороформом дает 0,62 г указанного в заголовке соединения в виде основания. Выход равен 98 Т. пл. 37-38 oC. Спектр ЯМР 133C (200 МГц - CDCl3), м.д. 148,3 136,7 128,4 127,2 126,3 122,0 117,1 115,2 55,6 52,5 28,0 26,6-22,6 11,8.
Пример 22. 3-дипропиламино-5-метилоксикарбонилтиохроман.
3-дипропиламино-5-гидроксибензотиопиран (Европатент 0222 996, 620 мг, 1,6 ммоль) растворяют в 11 мл смеси (6:2) диметилформамид/метанол и раствор дегазируют при 10 торах при 22 oC 15 мин. В реакционную смесь добавляют 11 мг ацетата палладия, 19 мг 1,3-бис-дифенилфосфинопропана и 0,48 мл (0,35 г) триэтиламина. Смесь нагревают до 70 oC в атмосфере монооксида углерода и перемешивают 5 часов. Раствор охлаждают, разбавляют 30 мл толуола, промывают насыщенным раствором бикарбоната натрия, сушат сульфатом натрия и выпаривают в вакууме. Хроматографирование на силикагеле посредством элюирования с использованием градиента хлороформа от 10 этилацетата в хлороформе дает 310 мг указанного в заголовке соединения (основание) в виде светложелтого масла. Выход 64 Спектр ЯМР 13C (200 МГц CDCl3), м.д. 168,2 136,6 134,8 131,6 130,1 126,5 125,7 56,7 52,5, 52,1, 30,4, 28,022,3 11,9.
Пример 23. 3-дипропиламино-5-ацетилтиохроман.
3-дипропиламино-5-метилоксикарбонилтиохроман (например 22, 310 мг, 1,01 ммоль) растворяют в 8 мл метанола и добавляют 60 мл гидроксида натрия в 2 мл воды. После кипячения в течение 5 ч смесь охлаждают и выпаривают в вакууме. Остаток растворяют в тионилхлориде (5 мл) и повторно кипятят в течение 1 ч. Избыток тионилхлорида выпаривают в вакууме, получая смолу. Остаточную смолу растворяют в минимальном количестве тетрагидрофурана и по каплям добавляют к охлажденному до -78 oC раствору диметилкупрата лития (2,02 ммоль) в 20 мл тетрагидрофурана. Реакционную смесь перемешивают 15 мин при -78 oC, затем ей дают нагреться до комнатной температуры и спустя 10 мин реакцию прерывают, добавляя 0,9 мл воды. Реакционную смесь фильтруют через "Целит" и выпаривают досуха. Остаток растворяют в эфире, промывают насыщенным раствором бикарбоната натрия, обрабатывают рассолом, сушат сульфатом натрия и выпаривают в вакууме, получая неочищенное основание в виде масла. Сырой остаток подвергают хроматографированию на силикагеле посредством элюирования с использованием градиента от хлороформа до 5 этилацетата в хлороформе. Хлористоводородная соль была получена путем растворения чистого основания в эфире и прикапывания избытка эфирного раствора HCl. Перекристаллизация в смеси трихлорметан/диэтиловый эфир дает 92 мг указанного в заголовке соединения в виде белого твердого вещества. Выход 27 т. пл. 141-142 oC. Спектр ЯМР 13C (200 МГц CDCl3), м.д. 201,9 138,4 135,9 131,7 131,2 127,2 127,0 59,9 54,1 51,8 29,9 27,9 26,1, 18,6 18,2, 11,6.
Пример 24. 5-аллил-3-(дипропиламино) тиохроман.
К раствору 3-(дипропиламино)-5-трифторметансульфонилтиохрома (пример 21, 1,28 г, 3,22 ммоль), 76 мг (0,06 ммоль) тетракис-(трифенилфосфин) палладия (O) и нескольких кристаллов 2,6-ди-трет-бутил-4-метилфенола в 10 мл безводного толуола добавляют 1,17 г (1,1 мл, 3,53 ммоль) чистого тибутилаллилолова. Образовавшийся раствор кипятят с обратным холодильником 4 ч, затем добавляют 1 мл пиридина к охлажденному раствору и после этого 2,1 мл комплекса фтористый водородный пиридин (Стилл и сотр. J. Org. Chem. 1987, т. 52, с.422). После перемешивания 1 ч при комнатной температуре реакционную смесь добавляют 50 мл диэтилового эфира и последовательно обрабатывают 50 мл 1-молярного раствора гидроксида натрия водой (2 раза), промывают насыщенным раствором хлористого натрия и сушат сульфатом натрия. После фильтрации и удаления растворителя в вакууме получают неочищенное вещество в виде темного масла. Хроматографирование на силикагеле посредством элюирования с использованием градиента от гексана до 5 этилацетата в гексане дает 0,85 г указанного в заголовке вещества в виде (основания) светложелтого масла. Выход 91 Спектр ЯМР 13C (200 МГц CDCl3), м.д. 139,0 136,5 134,0 133,0 126,1 125,9 125,0 116,0 57,0 52,6 37,7 29,5 27,7 22,5. Часть основания и переводят в хлористоводородную соль посредством растворения числового основания в эфире и прикапывания избытка эфирного раствора HCl. Перекристаллизацию их смеси ацетонитрила-эфира-гексана дает более твердое вещество с т. пл. 164-165 oC.
Пример 25. 3-(дипропиламин)-5-пропилтиохроман-гидрохлорид.
К перемешиваемой суспензии азодикарбоксилата калия (0,76 г, 3,9 ммоль) [свежеприготовлен из диэтилазодикарбоксилата и гидроксида калия] и 5-аллил-3-(N, N-дипропиламино)тиохромана (пример 24, 0,4 г, 1,4 ммоль) в 10 мл безводного метанола добавляют раствор ледяной уксусной кислоты в метаноле (1: 4) до тех пор, пока не исчезнет желтое окрашивание. Спустя 30 мин перемешивания при комнатной температуре добавляют еще 200 мг азодикарбоксилата калия и снова разлагают, как описано выше. Этот процесс продолжают до тех пор, пока по данным анализа (газовая хроматография) не останется исходного материала в смеси. По завершении реакции (2 ч и 4 добавления соли калия) растворитель удаляют в вакууме. К остатку добавляют двухмолярный раствор гидроксида натрия, который дважды экстрагируют диэтиловым эфиром, и объединенные органические экстракты обрабатывают насыщенным раствором хлористого натрия и сушат сульфатом натрия. Неочищенное основание получают в виде легко окрашенного масла после удаления растворителя в вакууме. Хроматографирование на силикагеле посредством элюирования с использованием градиента от гексана до 5 этилацетата в гексане дает указанное в заголовке вещество (основание) в виде прозрачного масла. Хлористоводородную соль получают путем растворения чистого основания в эфире и прикапывания избытка эфирного раствора HCl. Перекристаллизация (хлороформдиэтиловый эфир) дает 0,3 г белого твердого вещества, выход 66 Т. пл. 150-151 oC. Спектр ЯМР 13С (основания; 200 МГц CDCl3), м.д. 141,6 133,7 132,8 125,8 125,6 124,5 57,1, 52,6 35,3 29,5 27,6 23,6 22,4 14,3 11,9.
Фармакологические испытания депрессии у людей.
Имеются свидетельства, что у пациентов с депрессией может быть нарушение трансмиссии в центральной нервной системе. Эти нарушения, как оказалось, заключают нейротрансмиттеры нор-адреналина и 5-гидрокситриптамина (5-ГТ). Считается, что медикаменты, которые наиболее часто используются при лечении депрессии, действуют посредством улучшения нейротрансмиссии или одного или обоих указанных физиологических агонистов. Имеющиеся данные позволяют предположить, что увеличение трансмиссии 5-гидрокситриптамина может главным образом улучшить депрессивное настроение и беспокойство, тогда как увеличение нейротрансмиссии норадреналина скорее дает улучшение симптомов торможения, возникающих у пациентов с депрессией. В последние годы были предприняты многочисленные усилия для того, чтобы разработать новые медикаменты с высокой селективностью для улучшения нейротрансмиссии 5-ГТ в центральной нервной системе.
Механизм действия для медикаментов, которые обычно используются сегодня при терапии мозговой депрессии, является косвенным, то есть они действуют посредством блокирования повторного поглощения нейротрансмиттеров (норадреналина или/и 5-гидрокситриптамина), выделяющихся из нервных окончаний в центральной нервной системе. Таким образом, увеличивается концентрация этих трансмиттеров в синаптических разветвлениях и в результате восстанавливается соответствующая нейротрансмиссия.
Совершенно другим способом улучшения нейротрансмиссии в центральных 5-ГТ-нейронах было бы использование непосредственного агониста 5-ГТ-рецептора. Тогда для того, чтобы минимизировать побочные эффекты, была бы предпочтительна высокая селективность для рецепторов такого типа.
Антагонизм ингибирующих авторецепторов, расположенных на клеточных телах 5-ГТ-нейронов, был бы другим фундаментально отличающимся способом улучшения нейротрансмиссии 5-гидрокситриптамина.
Неожиданно мы обнаружили, что группа веществ формулы I обладает селективным, непосредственно стимулирующим или ингибирующим эффектом на подгруппу центральных 5-ГТ-рецепторов. Другое наблюдение состоит в том, что некоторые из этих веществ обладают особенно хорошей оральной биодоступностью. Для того чтобы оценить стимулирующий эффект и селективность 5-ГТ-рецептора, была измерена аффиность для различных рецепторов в мозгу крысы (вне организма) с использованием рецепторных испытаний (Ki нМ).
Испытание вне организма. Анализ связывания рецептора.
Анализ связывания 5ГТIA. Кору головного мозга гипокамп от каждой крысы рассекают и гомогенизируют в 15 мл охлажденного до 0 oC 0,05-молярного буферного раствора Трис-HCl, 0,004-молярного хлорида кальция и 0,0057-молярного раствора аскорбиновой кислоты, pH 7,5 с помощью "Ультра-Турракс" (фирма Янке и Кункель, г. Штауфен, ФРГ) в течение 10 с. После центрифугирования в течение 12,5 мин при скорости 17000 об/мин (39800 в Бекмановской центрифуге с охлажденным JA-17 ротором / фирма Бекман, г. Пало-Альто, США) таблетки повторно суспендируют в том же самом буферном растворе и повторяют гомогенизирование и центрифугирование. К каждой таблетке добавляют 5 мл 0,32 молярного раствора сахарозы, охлажденного до 0 oC и гомогенизируют в течение 5 с. Эти образцы хранятся замороженными при -70 oC. При использовании они разбавляются буферным раствором до концентрации 8 мг ткани/мл и гомогенизируются в течение 10 с. Гомогенизаты ткани выдерживаются в течение 10 мин при 37 oC и затем в них добавляют 10 мкмоль паргилина с последующим повторным инкубированием в течение 10 мин.
Анализ связывания проводят, как описано Перуоткой в J. Neurochem. 1986, т. 47, с. 529- 540. Смесь после инкубации (2 мл) содержит 3H-8-OH-DPAT (от 0,25 до 8 наномоль), 5 мг/мл тканевого гомогенизата в 50 ммоль/д буферного раствора Трис-HCl, содержащего 4 ммоль/л хлорида кальция и 5,7 ммоль/л аскорбиновой кислоты, pH 7,5. Шесть различных концентраций 3H-8-OH-DPAT были проанализированы. Эксперименты по связыванию начинались с добавления гомогенизата ткани с последующим инкубированием при 37 oC в течение 10 мин. Инкубационные смеси фильтровались через стеклянные фильтры "Ватман Джи-Эф/Би" с устройством для сбора урожая клеток Бранделя (Гайтерсбург, шт. Мериленд, США). Эти фильтры дважды промывали 5 мл буфера 50 ммоль/л Трис-HCl, охлажденного до 0 oC с pH 7,5 и проводили счет импульсов в 5 мл "Реди-солв Эйч-Пи" (фирма Бекман) в бекмановской сцинцилляционной камере Эл-Эс 3801. Неспецифическое связывание измеряли посредством добавления 10 мкмоль/л 5-гидрокситриптамина к реакционной смеси. Данные связывания обрабатывали посредством компьютерного анализа методом наименьших квадратов (нелинейный) / Мунсон и Родбард. Anal. Biochem. 1980, т. 107, с. 220- 239).
Результаты испытания выражены в виде Ki и даны в наномоль/л. Например, 3-дипропиламино-5-ацетилхроман имеет значение Ki 1,0 (наномоль/л), 3-дипропиламино-5-карбамоилхроман имеет Ki 3,1 нмоль/л, 3-дипропиламино-5-N-метилкарбамоилхроман- Ki нмоль/л и 3-дипропиламино-5-(2-тиенилкарбонилхроман) имеет Ki 1,7 нмоль/л.
Пример 26. 5-метокси-3-циклопропиламино-хроман-хлоргидрат.
Целевое соединение получалось согласно известным методам восстановительного аминирования (Cdinton F. Lane Synthesis 1975, том 146, стр. 135) из метокси-3-хроманона и циклопропиламина. Т. пл. 188-189 oC.
Пример 27. 3-(N-Циклопропиламино)-5-гидроксихроман.
3-(N-Циклопропиламино)-5-метоксихроман-хлоргидрат (5,6 г, 22 ммоля) суспендировался в метиленхлориде (140 мл) в атмосфере азота. Смесь охлаждалась на бане из смеси сухой лед/этанол до -20 oС. К перемешиваемой смеси в течение получаса добавлялся BBr3 (4,1 мл, 44 ммоля), растворенный в метиленхлориде (60 мл). Желтый прозрачный раствор медленно подогревался до 0 oC и хранился при этой температуре до тех пор, пока ГХ не указала на завершение реакции (через 3-5 ч). Затем раствор выливался на раздробленный лед (200 г) и добавлялся достаточно конц. аммиак (водн.) для достижения pH порядка 8-9. Смесь экстрагировалась простым эфиром (3•200 мл). Собранные эфирные фазы сушились (сульфатом магния), фильтровались и концентрировались в вакууме, давая белое твердое вещество. Кристаллизация из абсолютного этанола давала 3-(N-циклопропиламино)-5-гидроксихроман (3,9 г, 88 выход) в виде бесцветных игл. Т. пл. 147-148 oC.
Пример 28. 3-(N-Циклопропиламино)-5-трифторметансульфонилоксихроман.
Целевое соединение получалось по аналогии с процедурой, используемой в примере 1, исходя из продукта, образованного в примере 27. Основание характеризовалось в виде хлоргидратной соли. Т. пл. 207-209 oC (разложение).
Пример 29. 3-(N-Циклопропиламино)-5-(N-циклопропил)карбамоилхроман.
3-(N-Циклопропиламино)-5-трифторметансульфонилоксихроман (0,51 г, 1,5 ммоля) и триэтиламин (0,46 мл, 3,3 ммоля) растворялись в ДМФ (7,5 мл) в 200-мл сосуде для гидрирования. Сосуд вакуумировался с последующим вводом моноокиси углерода (повторно три раза). Добавлялись циклопропиламин (2,9 мл, 42 ммоля), 1,3-бис(дифенилфосфино)пропан (0,023 г, 55 мкмолей) и ацетат палладия (II) (0,12 г, 55 мкмолей), а затем смесь встряхивалась при 70 oC в течение 3 ч при давлении CO 2-2,5 бар. После охлаждения до комнатной температуры реакционная смесь распределялась между насыщенным бикарбонатом натрия и эфиром (5•30 мл). Собранные эфирные фазы сушились (сульфатом магния), фильтровались и концентрировались в вакууме. Мгновенная хроматография неочищенного продукта на двуокиси кремния с использованием ТГФ-этилацетата (8: 92) в качестве элюента давала 0,32 г белого твердого вещества. Перекристаллизация из смеси ТГФ-эфира давала 3 -(N-циклопропиламино)-5-(N-циклопропил)карбамоилхроман 0,17 г, 42 выход в виде бесцветных игл. Т. пл. 126-127 oC.
Пример 30. Хлоргидрат-3-циклопропиламино-5-[N-(2,6-ксилидино)карбамоил] -хромана.
Натриевая соль 3-(N-циклопропил-N-трифторацетил)аминохроман-5-карбоновой кислоты (2,1 ммоля), полученная в примере 28, после N-трифторацетилзащиты, сложной этерификации (согласно примеру 2) и последующего гидролиза и тионил хлорид (10 мл) в атмосфере азота нагревалась с обратным холодильником в течение 2 ч. Избыток тионилхлорида выпаривался совместно с сухим толуолом несколько раз на вращающемся испарителе. Полученный таким образом хлорангидрид кислоты растворялся в метиленхлориде (10 мл) и добавлялся по каплям к перемешиваемому раствору 2,6-диметиланилина (0,52 г, 4,3 ммоля) и сухого пиридина (6 мл) в атмосфере азота при комнатной температуре. Когда реакция завершалась (в пределах двух часов согласно данным ТСХ и капиллярной ГХ), летучие вещества выпаривались на роторном испарителе. Остаток повторно растворялся в сухом толуоле и концентрировался в вакууме повторно (четыре раза). Очистка с помощью мгновенной хроматографии (флэш-хроматографии) на двуокиси кремния с использованием смеси ТГФ-н-гексан (1:4) в качестве элюента давала 0,72 г 3-(N-циклопропил-N-трифторацетиламино)-5-[N-(2,6 - ксалидино)карбамоил] -хромана (72 выход) в виде бесцветного твердого вещества. Т. пл. 146-149 oC (кристаллизованное из смеси CHCl3-н-гексан). Часть данного амида (0,44 г, 0,95 ммоля) добавлялась небольшими порциями в течение 15 мин к перемешиваемой суспензии тетрагидридоалюмината лития (0,072 г, 1,9 ммоля) в 20 мл сухого ТГФ (перегнанного из бензофенон-кетил-натрия) в атмосфере азота. Смесь перемешивалась при 45 oC до тех пор, пока ТСХ и капиллярная ГХ не показывала завершение реакции (через 40 ч), и затем гасилась осторожным добавлением водного натрий-калиевого тартрата (0,5 М). После доведения pH до 10 (концентрированным аммиаком) раствор экстрагировался простым эфиром (2 • 50 мл). Собранные эфирные фазы сушились (карбонатом калия), фильтровались и концентрировались в вакууме. Получающееся в результате масло хроматографировалось на двуокиснокремниевой колонке, элюируемой смесью CHCl3 EtOAc (1:1), давая 3-циклопропиламино-5-[N-(2,6-ксилидино)карбамоил]-хроман (0,071 г, 22 общий выход) в виде масла. Небольшой избыток HCl (приблизительно 3 М в эфире) добавлялся по каплям к перемешиваемому и охлажденному (+4 oC) раствору основания из описанной выше стадии и метанола (2 4 мл). Растворители затем роторно выпаривались, добавлялся эфир и повторно осуществлялось выпаривание для отгонки следов HCl. Когда колба, заполненная эфиром, оставлялась в холодильнике на ночь, масло затвердевало. Кристаллизация из абсолютного этанола давала хлоргидрат 3-циклопропиламино-5-[N-(2,6-ксилидино)карбамоил]-хромана в виде бесцветных игл. Т. пл. 189-191 oC (разложение).
Пример 31. 3-(N,N-диаллиламино)-5-метоксихроман и 3-(N-аллиламино)-5-метоксихроман.
Хлоргидрат 3-амино-5-метоксихромана (Acta Pharm. Suec. 24 (1987)) (5,0 г, 23 ммоля), аллилбромид (3,4 мл, 39 ммоля), безводный карбонат калия (9,6 г, 69 ммолей) и ДМФ (8,0 мл) перемешивались в атмосфере азота при комнатной температуре в течение 72 ч. Добавлялся эфир (150 мл), соли отфильтровывались с помощью отсасывания, и прозрачный фильтрат концентрировался в вакууме. Остаток очищался на кремнеземной колонке, элюируемой 1000 мл-ми ТГФ-н-гексан (1: 9) и 1000 мл-им ТГФ-гексан (1:3), давая 3-(N-аллиламино)-5-метоксихроман (1,7 г, 33 выход) и 3-(N,N-диаллиламино)-5-метоксихроман (3,4 г, 56 выход) в виде масел. Диаллиламино производное, указанное выше, выделялось в виде хлоргидратной соли с помощью добавления небольшого избытка HCl (приблизительно 3 М в эфире) к эфирному раствору амина. Неочищенная HCl соль кристаллизовалась при стоянии, с эфиром, на холоде. Т. пл. 139-140 oC.
Пример 32. 3-(N-аллил-N-н-пропиламино(-5-метоксихроман.
3-(N-Аллиламино)-5-метоксихроман (1,3 г, 5,9 ммоля), полученный аналогично описанному в примере 26, н-пропилиодид (2,1 мл, 21 ммоль), безводный карбонат калия (3,0 г, 21 ммоль) и ацетонитрил (5,5 мл) перемешивались в атмосфере азота при 47 oC (температура масляной бани) в течение пяти дней до тех пор, пока ГХ не показывала завершенную реакцию. Добавлялся эфир (40 мл), соли отфильтровывались отсасыванием, и прозрачный фильтрат концентрировался в вакууме, давая 3-(N-аллил-N-н-пропиламино)-5-метоксихроман (1,24 г, 81 выход) в виде масла. Основание осаждалось из эфирного раствора добавлением легкого избытка HCl (приблизительно 3 М в эфире). Кристаллизация неочищенной HCl соли давала бесцветные иглы. Т. пл. 117-118 oC.
Пример 33. 3-(N-аллил-N-н-пропиламино)-5-гидроксихроман.
Целевое соединение получалось по аналогии с процедурой, используемой в примере 27, с использованием продукта, полученного в примере 32. Кристаллизация из смеси CHCl3-н-гексан давала бесцветные иглы. Т. пл. 78-80 oC.
Пример 34. 3-(N-Аллил-N-н-пропиламино)-5-трифторметансульфонилоксихроман.
Целевое соединение получалось по аналогии с процедурой примера 1 с использованием продукта из примера 33 в качестве исходного материала. MC/EI; 70 ev/m/z 379 (M+, 8), 350 (100), 246 (10).
Пример 35. 5-ацетил-3-(N-аллил-N-н-пропиламино)хроман.
3-(N-Аллил-N-н-пропиламино)-5- трифторметансульфонилоксихроман (0,28 г, 0,74 ммоля), хлорид лития (0,097 г, 2,3 ммоля), PdCl2 (dppf)=дихлор[1,1'-бис(дифенилфосфино)ферроцен] -палладий(П) (0,031 г, 0,04 ммоля) т 2,6-ди-трет.-бутил-4-метиленфенол (0,005 г) растворялись в ДМФ (5,0 мл) в трехгорлой круглодонной колбе (50 мл) с магнитной мешалкой. Колба вакуумировалась с последующим вводом CO (три раза). Добавлялось тетраметилолово (0,12 мл, 0,89 ммоля), в затем смесь перемешивалась в атмосфере CO (1 атм.) при 120 oC (темп. масляной бани) в течение 4 ч. Растворитель выпаривался, остаток распределялся между водным аммиаком (2 М) и метиленхлоридом (3 • 15 мл), и органические фазы сушились (сульфатом натрия), фильтровались и концентрировались в вакууме. Хроматография на колонке из кремнезема с использованием ТГФ-н-гексана (1: 9) в качестве элюента давала 5-ацетил-3-(N-аллил-N-н пропиламино)хроман (0,078 г, 39 выход) в виде масла. Основание осаждалось из эфирного раствора добавлением небольшого избытка HCl (приблизительно 3 М в эфире). Неочищенная соль собиралась и сушилась в вакууме при 40 oC, давая белый аморфный порошок. Т. пл. 125-127 oC.
Пример 36. Хлоргидрат 3-изопропиламино-5-метоксихромана.
Целевое соединение получалось аналогично процедуре, используемой в примере 26, исходя из 5-метокси-3-хромана и изопропиламина. Т. пл. 255 oC.
Пример 37. 3-(N, N-изопропил-н-пропил)амино-5-метоксихроман.
Смесь продукта, полученного в примере 36 (14 г), 0,6 моля, 1-иодопропана (15 г, 0,08 моля), карбоната калия и ацетонитрила (250 мл) перемешивалась в условиях дефлегмации в течении 4 дней. После хроматографии целевой продукт выделялся в виде бесцветного масла. ГХ-МС (Cl-mode) М+1 264 (100).
Пример 38. 3-(N, N-изопропил-н-пропил)амино-5-гидроксихроман.
Продукт из примера 37 (10 г, 0,038 моля) диметилировался с использованием трехбромистого бора в дихлорметане. ГХ-МС (Cl-mode) M+1 250 (100).
Пример 39. 3-(N, N-изопропил-н-пропиламино)-5-трифторметансульфоноксихроман.
Целевое соединение получалось аналогично процедуре, используемой в примере 1, с использованием продукта из примера 38 в качестве исходного материала. ГХ-МЦ (Cl-mode) M+1 382 (100).
Пример 40. Хлоргидрат 5-ацетил-3-(N-изопропил-N-н-пропиламино)хромана.
Целевое соединение получалось аналогично примеру 35, исходя из продукта, полученного в примере 39, и с добавлением CO в аппарате Парра при 2 бар. Т. пл. 170 oC.
Пример 41. 3-(N-изопропил-N-н-пропиламино)-5-метилоксикарбонилхроман.
Целевое соединение получалось аналогично процедуре, используемой в примере 2, исходя из продукта, полученного в примере 39 (3 г, 0,016 моля), Pd(OAc)2 (75 мг), 1,3-бис(дифенилфосфин)-пропана (150 мг), ДМФ (50 мл) и метанола (25 мл) в аппарате Парра под давлением CO (2 бара) при 70 oC в течении 6 ч, ГХ-МС (Cl-mode) M+1 292 (100).
Пример 42. Хлоргидрат 3-(N-изопропил-N-н-пропиламино)-5-(N-метил)-карбамоилхромана.
Продукт, полученный из примера 41 (1 г, 0,0034 моля), NaCl (50 мг), метанол (30 мл) и насыщенный водный раствор CH3NH2 (5 мл) подвергались реакции в стальном сосуде при 80 oC на протяжении ночи. После обработки бесцветное масло превращалось в хлоргидратную соль. Т. пл. 123 oC.
Пример 43. Хлоргидрат 3-(N-изопропил-N-н-пропиламино)-5-(N-этил)карбамоилхромана.
Целевое соединение получалось аналогично процедуре, используемой в примере 42, исходя из продукта, полученного в примере 41 (1 г, 0,0034 моля), KCN (50 мг), метанола (40 мл) и этиламина (5 мл, 70-й водный раствор) в стальном сосуде при 80 oC в течении 4 дней. Хроматографическая обработка давала бесцветное масло, которое превращалось в HCl-соль. Т. пл. 198 oC.
Пример 44. 3-(N-Циклопропилметил-N-н-пропиламино)-5-(N-циклопропилметил)-карбамоилхроман.
Целевое соединение получалось из 3-(N-циклопропилметил-N-н-пропиламино)-5-трифторметансульфонилоксихромана (1,08 г, 0,0026 моля), полученного аналогично описанному в примерах 36-39, 1,3-бис(дифенилфосфин)пропана (40 г), ацетата палладия (II (22 мг) и циклопропилметиламина (2,3 г, 0,0264 моля) в 30 мл ДМФ, которые помещались в стеклянный сосуд Парра. Добавлялась CO при 2 бор. и смесь встряхивалась при 60 oC в течение 4 ч. После обработки и хроматографической очистки получалось целевое соединение в виде кристаллов с т. пл. 124 oС (основание) в виде игл. Т. пл. 94-95 oC.
Пример 45. Хлоргидрат 3-(N-циклопропил-N-н-пропиламино)-5-фенихлоромана.
Целевое соединение получалось из смеси 3-(N-циклопропил-N-н-нпропиламино)-5-трифторметилсульфонилоксихромана, полученного аналогично примерам 36-39 (2 г, 5,1 ммоля), триметилфенилстаннана (1,8 г, 4,8 ммоля), тетракис(трифенилфосфин)-палладия (О) (280 мг, 0,24 ммоля). Хлорид лития (600 мг, 14,4 ммоля) и 2,6-ди-трет.-бутил-4-метилфенол в 60 мл диоксана и 6 мл ДМФ перемешивался при 105 oC в стальном сосуде в течении 3 дней. Смесь фильтровалась и экстрагировалась. Хроматографическая очистка на оксиноалюминиевой колонке давала целевое соединение с выходом 55 Соединение выделялось в виде хлоргидратной соли. Т. пл. 160 oC.
Пример 46. 3-(N, N-дипропиламино)-5-(N-циклопропил)-карбамоилтиохроман
3-(N, N-Дипропиламино)-5-трифторметансульфонилокситиохроман (0,70 г, 1,76 ммоля), триэтиламин (0,39 г, 0,54 мл, 3,9 ммоля) и ДМФ (5 мл) смешивались вместе, и раствор дегазировался (10 мм рт. ст. комн.темп. 15 мин), затем подвергался действию CO атмосферы (•3). Затем добавлялись ацетат палладия (II) (12 мг), 1,3-бис-дифенилфосфинопропан (22 мг) и циклопропиламин (3,0 г, 3,7 мл, 52,8 ммоля). Получающаяся смесь снова подвергалась действию атмосферы CO и нагревалась до 70 oC и перемешивании в течение 4 ч. Раствор охлаждался, выпаривался в вакууме (вакуумный насос), затем разбавлялся этилацетатом. Смесь промывалась раствором бикарбоната (три раза), обрабатывалась солевым раствором, сушилась (сульфатом натрия) и выпаривалась в вакууме, давая сырой продукт. Хроматография на двуокиси кремния (кремнеземе) (элюент:50 этилацетат/гексан) давала 0,38 г вещества в виде белых твердых кристаллов (64 выход). Т. пл. 109-110 oC. 13C ЯМР (200 МГц CDCl3) мин. дол. 171,1, 137,8, 134,5, 133,2, 128,0, 126,0, 122,7, 56,5, 52,5, 29,9, 28,1, 23,0, 22,4, 11,9, 7,0.
Пример 47. 3-(N, N-Дипропиламино)-5-метилоксикарбонилтиохроман.
3-(N, N-Дипропиламино)-5-трифторметансульфонилтиохроман (620 мг, 1,6 ммоля) растворяли в 11 мл смеси ДМФ/метанол 6:2, и раствор дегазировался в течение 15 мин. К реакционной смеси добавлялись Pd(OAc)2 (11 мг), 1,3-бис-дифенил-фосфинопропан (19 мг) и триэтиламин (0,48 мл, 0,35 г). Смесь нагревалась до 70 oC в атмосфере моноокиси углерода и перемешивалась в течение 5 ч. Раствор охлаждался, разбавлялся 30 мл толуола, промывался насыщенным бикарбонатом натрия, сушился (сульфонатом натрия) и выпаривался в вакууме. Хроматография на двуокиси кремния (элюент: градиент CHCl3 __→ 10 EtOAc/CHCl3) давала 310 мг (64 выход) целевого соединения (основание) в виде слегка желтого масла. 13C ЯМР: (200 мГц, CDCl3) млн.дол. 168того масла. 3C ЯМР: (200 мГц, CDCl3) млн.дол. 168,2, 136,6, 134,8, 131,6, 130,1, 126,5, 125,7, 56,7, 52,5, 52,1, 30,4, 28,0, 22,3, 11,9.
Пример 48. 3-(N-Изопропил-N-н-пропиламино)-5-фенил-1-оксотиохроман.
Хлоргидрат 3(N-изопропил-N-н-пропиламино)-5-фенилтиохромана (310 мг, 0,86 ммоля) растворялся в 6 мл хлороформа, и к охлажденному раствору (ледяная баня) добавлялась м-хлорнадбензойная кислота (348 мг, 1,72 ммоля) в виде одной порции. Реакционной смеси давали возможность перемешиваться при комнатной температуре в течение 18 ч. Растворитель удалялся в вакууме, и остатки экстрагировались смесью эфир/2 M NaOH, обрабатывались солевым раствором и сушились (натрием). Растворитель выпаривался в вакууме, давая сырую смесь. Хроматография на двуокиси кремния (элюент: 25 этилацетат/метиленхлорид) дала 79 мг (27 выход) желаемого сульфоксида в виде диастереомерной смеси (18,82 по данным ГХ) получаемой в виде не совсем белого твердого вещества. Т. пл. 109-112 oC.
Пример 49. 5-(2-фуранил)-3-(N-изопропил-N-н-пропиламино)-1-оксотиохроман.
Хлоргидрат 5-(2-фуранил)-3-(N-изопропил-N-н-пропиламино)-тиохромана (341 мг, 0,97 ммоля) растворялся в 7 мл метиленхлорида и охлаждался до -20 oC. Добавлялась м-хлорнадбензойная кислота (258 мг, 1,27 ммоля) одной порцией, и реакционная смесь оставлялась перемешиваться при комнатной температуре в течение 18 ч. Растворитель удалялся в вакууме, и остатки экстрагировались смесью эфир/2 M гидроокись натрия, обрабатывались солевым раствором и сушились (сульфатом натрия). Растворитель выпаривался в вакууме, давая сырую смесь. Хроматография на двуокиси кремния (элюент:25 этилацетат/метиленхлорид) давала 35 мг неполярного диастереомера целевого соединения, 75 мг полярного (основного) диастереомера целевого соединения и 92 мг диастереомерной смеси (25: 75 по ГХ), давая объединенный выход 63 Неполярный (второстепенный) изомер (не совсем белое твердое вещество): т. пл. 63-65 oC. Полярный (главный) изомер (не совсем белое твердое вещество): т. пл. 83-85 oC.
Пример 50. 5-(2-фуранил)-3-(N-изопропил-N-н-пропиламино)-1,1-диоксотиохроман.
5-(2-фуранил)-3-(N-изопропил-N-н-пропиламино)-1-оксотиохроман (в виде диастереомерной смеси (25: 75 по ГХ)) (92 мг, 0,29 ммоля) растворялся в эфире, и по каплям добавлялся HCl в эфире до тех пор, пока раствор не становился кислым. Растворитель удалялся в вакууме. Полученное белое твердое вещество растворялось в 3 мл метиленхлорида, и раствор охлаждался до -15 oC. Добавлялась м-хлорнадбензойная кислота (1018 мг, 0,58 ммоля) одной порцией, и реакционная смесь оставлялась перемешиваться при комнатной температуре в течение 18 ч. Реагировал только второстепенный диастереомер, давая желаемый сульфон вследствие стерических причин. Растворитель удалялся в вакууме, а остатки экстрагировались смесью эфир/2 M гидроокись натрия, обрабатывались солевым раствором и сушились (сульфатом натрия). Растворитель выпаривался в вакууме, давая сырую смесь. Препаративная ТСХ (элюент:20 этилацетат/метиленхлорид) давала 5 мг (5 выход) целевого соединения. МС 347. 13C ЯМР: (200 МГц, CDCl3) млн.дол. 151,7, 143,0, 139,3, 132,3, 131,6, 127,5, 125,6, 123,5, 111,7, 110,5, 54,3, 51,3, 49,4, 47,4, 34,4, 23,3, 21,7, 20,0, 11,8.
Пример 51. Хлоргидрат 5-(5-фуранил)-3-N-изопропил-N-н-пропиламино)тиохромана.
К раствору 3-(N-изопропил-N-н-пропиламино)-5-трифторметансульфонилтиохромана (0,92 г, 2,31 ммоля), этанола (10,2 мл), хлористого лития (0,20 г, 4,8 ммоля), 2 A карбоната натрия (3,4 мл) и тетракис(трифенилфосфин)палладия(O) (49,0 мг, 0,042 ммоля), растворенных в толуоле (23 мл), добавлялась 2-фуранилборная кислота (0,62 г, 4,6 ммоля) в виде одной порции в атмосфере азота. Получающийся в результате раствор нагревался до 95 oC в течение 2 ч, затем реакционная смесь оставлялась охлаждаться. Реакционная смесь фильтровалась, и растворитель выпаривался в вакууме. Остаток брался в эфир, промывался 2 M аммиаком, обрабатывался солевым раствором и сушился (сульфатом натрия). Удаление растворителя в вакууме давало неочищенное соединение. Хроматография на двуокиси кремния (элюент:3 этилацетат/хлороформ) давала 0,68 г (выход 93) целевого соединения (основание) в виде слегка желтого масла. Хлоргидратная соль получалась с помощью растворения чистого основания в эфире и добавления по каплям избытка эфирного раствора HCl, затем перекристаллизации (этилацетат/эфир), давая бесцветные кристаллы. Т. пл. 145-147 oC.
Пример 52. 5-изопропиламидо-3-(N-изопропил-N-н-пропиламино)тиохроман
3-(N-Изопропил-N-н-пропиламино)-5-трифторметансульфонилтиохроман (0,99 г, 2,49 ммоля) растворялся в диоксане (15 мл), и раствор дегазировался (10 мм рт.ст. комн.темп.), затем подвергался действию CO атмосферы (•3). Добавлялись изопропиламин (1,1 мл, 12,5 ммолей), ацетат палладия (15 мг) и 1,3-бис-ди-фенилфосфинопропан (29 мг). Получающаяся смесь снова подвергалась действию атмосферы CO и нагревалась до 80 oC при перемешивании в течение 4 ч. Раствор охлаждался, выпаривался в вакууме (вакуумный насос), затем разбавлялся эфиром. Смесь промывалась 2 M раствором аммиака (•2), обрабатывалась солевым раствором, сушилась (сульфатом натрия) и выпаривалась в вакууме, давая сырое вещество. Хроматография на двуокиси кремния (элюент:30 этилацетат/гексан) давала 0,76 г масла (выход 87), которое кристаллизовалось и перекристаллизовывалось (из гексана), давая белое твердое вещество. Т. пл. 90- 91 oC.
Пример 53. 3-(N-Изопропил-N-н-пропиламино)-5-трифторметансульфонилтиохроман.
5-Гидрокси-3-(N-изопропил-N-н-пропиламино)тиохроман (3,13 г, 11,9 ммоля) и коллидин (2,02 г, 2,2 мл, 16,7 ммоля) растворяли в 100 мл метиленхлорида и охлаждали до -30 oC. По каплям добавлялся трифторметансульфоновый ангидрид (4,03 г, 2,4 мл, 14,3 ммоля), и смесь оставлялась подогреваться до температуры окружающей среды, а спустя 20 мин разбавлялась метиленхлоридом. Раствор промывался насыщенным бикарбонатом натрия, сушился (сульфатом натрия) и выпаривался в вакууме. Хроматография на двуокиси кремния (элюент:3 этилацетат/гексан) давала 3,9 г (83 выход) целевого соединения в виде слегка желтого масла. МС 397. 13C ЯМР: (200 МГц CDCl3) млн. дол. 148,3, 136,7, 128,8, 127,2, 126,3, 121,9, 117,0, 115,5, 52,8, 48,8, 47,1, 30,2, 28,8, 23,7, 21,2, 20,9, 11,7.
Пример 54. 3-(N-Изопропил-N-н-пропиламино)-5-метилоксикарбонилтиохроман.
3-(N-Изопропил-N-н-пропиламино)-5- трифторметансульфонилтиохроман (0,97 г, 2,44 ммоля) растворялся в 17 мл смеси ДМФ/метанол 6:2, и раствор дегазировался в течение 15 мин. К реакционной смеси добавлялись Pd(OAc)2 (17 мг), 1,3-бис-дифенилфосфинопропан (29 мг) и триэтиламин (0,54 г, 0,75 мл, 5,4 ммоля). Смесь нагревалась до 70 oC в атмосфере моноокиси углерода и перемешивалась в течение 4 ч. Раствор охлаждался, разбавлялся толуолом, промывался насыщенным бикарбонатом натрия, сушился (сульфатом натрия) и выпаривался в вакууме. Хроматография на двуокиси кремния (элюент:5 этилацетат/гексан) давала 0,69 г (выход 92%) целевого соединения (основание) в виде прозрачного масла. МС 307. 13С ЯМР: (200 МГц, CDCl3) млн. дол. 168,3, 137,0, 134,8, 131,6, 130,1, 126,5, 125,7, 53,9, 52,2, 48,9, 47,3, 32,7, 30,1, 23,8, 21,1, 11,8.
Пример 55. Хлоргидрат 3-(N-изопропил-N-н-пропиламино)-5-фенилтиохромана.
К раствору 3-(N-изопропил-N-н-пропиламино)-5- трифторметансульфонилтиохромана (0,95 г, 2,39 ммоля), этанола (10,5 мл), хлорида лития (0,20 г, 4,8 ммоля), 2 М карбоната натрия (3,5 мл) и тетракси(трифенилфосфин)палладия(О) (50 г, 0,043 ммоля), растворенному в толуоле (23 мл), добавлялась фенилборная кислота (0,35 г, 2,9 ммоля) в виде одной порции в атмосфере азота. Получающийся в результате раствор нагревался до 95 oC в течение 5 ч, затем реакционная смесь оставлялась охлаждаться. Реакционная смесь фильтровалась; и растворитель выпаривался в вакууме. Остаток брался в эфир, промывался 2 М аммиаком, обрабатывался солевым раствором и сушился (сульфатом натрия). Удаление растворителя в вакууме давало сырое соединение. Хроматография на двуокиси кремния (элюент: метиленхлорид) давала 0,73 г (выход 94) целевого соединения (основание) в виде масла. Хлоргидратная соль получалась с помощью растворения чистого основания в эфире и добавления по каплям избытка эфирного раствора HCl, затем перекристаллизовывалась (этилацетат/эфир), давая слегка желтое твердое вещество. Т. пл. 110- 112 oC.
Пример 56. (R)-3-(N-Изопропил-N-н-пропиламино)-5-(3-тифен)хроман.
(R)-3-(N-изопропил-N-н-пропиламино)-5- трифторметансульфонилоксихроман (0,3 г, 0,8 ммоля), 3-тиофенборная кислота (0,2 г, 1,6 ммоля), хлористый литий (0,07 г, 1,6 ммоля), карбонат натрия (2 М, 3 мл), этанол (7 мл) и толуол (15 мл) смешивались в трехгорлой круглодонной колбе в атмосфере азота. Добавлялся катализатор Pd(PPh3)4, и реакционная смесь перемешивалась при 90 oC в течение 4 ч. Растворитель удалялся в вакууме до тех пор, пока не оставалось примерно 15 мл, перед тем как разбавлялся диэтиловым эфиром, смесь промывалась (NH3, 2 М) и сушилась (сульфатом магния). Растворитель удалялся в вакууме, давая коричневатый маслянистый остаток, который очищался с помощью флэш-хроматографии (двуокись кремния, метиленхлорид/этилацетат, 10:1), давая целевое соединение с выходом 80 (0,2 г). α (основание) -40 oC (MeOH, 0,1 М, 22 oC). Хлоргидратная соль осаждалась из диэтилового эфира при 0 oC. Т. пл. 174-175 oC.
Пример 57. (R)-3-(N-Изопропил-N-н-пропиламино)-5- трифторметансульфонилоксихроман (0,4 г, 1,1 ммоля), 2-тиофенборная кислота (0,27 г, 2,1 ммоля), хлорид лития (0,09 г, 2,1 ммоля), карбонат натрия (2 М, 3 мл), этанол (7 мл) и толуол смешивались в атмосфере азота, затем добавлялся Pd(PPH3)4 (0,03 г, каталитическое количество). Смесь затем перемешивалась в течение 8 ч при 90 oC. Растворитель удалялся в вакууме до тех пор, пока не оставалось 10 мл. Остаток разбавлялся диэтиловым эфиром, промывался аммиаком (2 М) и сушился сульфатом магния. Растворитель удалялся в вакууме, давая коричневатый маслянистый остаток, который очищался с помощью флэш-хроматографии (двуокись кремния, метиленхлорид/этилацетат, 10: 1), давая целевое соединение с выходом 94 (0,3 г). a (основание) -36 oC (метанол, 0,1 М, 22 oC). Хлоргидратная соль осаждалась из диэтилового эфира при 0 oC. Т. пл. 189-191 oC.
Пример 58. (R)-5-Изопропоксикарбонил-3-(N-изопропил-N-н-пропиламино)хроман.
(R)-3-(N-изопропил-N-н-пропиломино)-5- трифторметансульфонилоксихроман (0,4 г, 1,1 ммоля), триэтиламин (0,2 г, 2,2 ммоля), ДМФ (6 мл) и изопропанол (2 мл) смешивались в трехгорлой 50 мл круглодонной колбе. Колба вакуумировалась с последующим вводом CO газа (повторялось три раза, CO в бюретке с водой), затем добавлялись 1,3-бис(трифенилфосфин)пропан (0,02 г, каталитическое количество) и ацетат палладия (П) (0,08 г, каталитическое количество). Смесь перемешивалась при 80 oC в течение 7 ч. Растворитель удалялся в вакууме, и остаток растворялся в диэтиловом эфире, промывался аммиаком (2 М) и сушился (сульфатом магния). Растворитель удалялся в вакууме, давая желтоватый маслянистый остаток, который очищался с помощью флэш-хроматографии (двуокись кремния, метиленхлорид/этилацетат, 10:1), давая целевое соединение с выходом 66 (0,2 г). a (основание) -110,5 oС (метанол, 0,1 М, 22 oC). Хлоргидратная соль осаждалась из диэтилового эфира при 0 oC и перекристаллизовывалась из смеси этилацетат/диэтиловый эфир. Т. пл. 153-155 oC.
Пример 59. (R)-3-(N-изопропил-N-н-пропиламино)-5-(2-N-тиазоламинокарбонил)хроман.
(R)-5-Хлоркарбонил-3-(N-изопропил-N-н-пропиламино)хроман (0,65 г, 2,3 ммоля) и 2-аминотиазол (0,68 г, 6,8 ммоля), растворенные в метиленхлориде (50 мл), перемешивались при комнатной температуре в течение 2 ч. Удаление растворителя в вакууме давало коричневатый маслянистый остаток, который очищался с помощью флэш-хроматографии (двуокись кремния, метиленхлорид/этилацетат; 10:1), давая целевое соединение с выходом 43 (0,35 г). Хлоргидратная соль осаждалась из диэтилового эфира при 0 oC, а затем перекристаллизовывалась из смеси этилацетат/диэтиловый эфир. a (HCl-соль) -20,0 oC. Спекается при менее 140 oC.
Пример 60. (R)-3-(N-Изопропил-N-н-пропиламино)-5-(3-пиридин)хроман.
(R)-3-(N-Изопропил-N-н-попиламино)-5- трифторметансульфонилоксихроман (0,37 г, 0,96 ммоля) растворялся в толуоле в атмосфере азота. Добавлялись этанол (7 мл), 2 М карбонат натрия (3 мл), хлористый литий (0,08 г, 1,9 ммоля), 3-пиридинборная кислота (0,7 г, 0,5 ммоля) и наконец Pd(PPH3)4 (0,04 г), и реакционная смесь нагревалась с обратным холодильником в течение 6 ч. Растворитель удалялся в вакууме до тех пор, пока не оставалась 10 мл. Остаток разбавлялся диэтиловым эфиром, промывался 2 М аммиаком и сушился (сульфатом магния). Удаление растворителя давало желтоватый маслянистый остаток, который очищался с помощью флэш-хроматографии (двуокись кремния, метиленхлорид/этилацетат, 5:1), давая целевое соединение с выходом 94 (0,28 г) a (основание ) -47,6 oC (метанол, 0,1 М, 21 oC). Диоксалат осаждался с помощью добавления щавельной кислоты (2,2 экв.), растворенной в диэтиловом эфире, к раствору основания в диэтиловом эфире. Затем соль перекристаллизовывалась из смеси этанол/диэтиловый эфир. Спекается при менее 135 oC.
Пример 61. (R)-3-(N-Изопропил-N-н-пропиламино)-5-фенилхроман.
(R)-3-(N-Изопропил-N-н-пропиламино)-5-трифторметансульфонилоксихроман (1 г, 2,6 ммоля) растворялся в толуоле (25 мл) в атмосфере азота. Добавлялись этанол (11,5 мл, хлористый литий (0,22 г, 5,2 ммоля), карбонат натрия (2 М, 3,8 мл), Pd (PPh3)4 (0,054 г, 0,047 ммоля) и фенилборная кислота (0,38 г, 3,1 ммоля), и реакционная смесь перемешивалась при 90 oС в течение 7 ч. Растворитель удалялся в вакууме до тех пор, пока не оставалось 15 мл. Остаток разбавлялся диэтиловым эфиром, промывался аммиаком (2 М) дважды и сушился (сульфатом магния). Удаление растворителя в вакууме давало желтовато-коричневый маслянистый остаток, который очищался с помощью флэш-хроматографии (двуокись кремния, метиленхлорид), давая целевое соединение с выходом 86 (0,7 г). a 50,7 oС (MeOH, 0,1 М, 22 oC). HCl - соль осаждалась из эфира медленным добавлением HCl в эфире к охлажденному льдом раствору основания. Сырая соль перекристаллизовывалась из смеси этилацетат/диэтиловый эфир, давая 650 мг иглообразных кристаллов. Т. пл. 141-142 oC.
Пример 62. 5-аминокарбонил-3-(N-изопропил-N-н-пропиламсино)хроман.
Раствор (R)-5-хлоркарбонил-3-(N-изопропил-N-н-пропиламино)хромана (0,37 г, 1,3 ммоля) в метиленхлориде (30 мл) осторожно продувался аммиаком в течение 30 с. Немедленно образовывался белый осадок. Реакционная смесь затем перемешивалась в течение 30 мин при комнатной температуре, затем она промывалась водой и сушилась (сульфатом магния). Растворитель удалялся в вакууме, давая сырое масло, которое очищалось с помощью флэш-хроматографии (двуокись кремния, метиленхлорид/этилацетат, 10: 1). Целевое соединение получалось в выходом 58 (0,21 г) в виде бесцветных кристаллов из смеси диэтиловый эфир/гексан. a 115,8 oС (MeOH, 0,1 М, 21 oC). Т. пл. 120,6- 122 oC.
Пример 63. (R)-3-(N-Изопропил-N-н-пропиламино)-5-N-фениламинокарбонилхроман.
(R)-5-хлоркарбонил-3-(N-изопропил-N-н-пропиламино)хроман (0,65 г, 2,3 ммоля) и анилин (0,86 г, 9,2 ммоля), растворенные в метиленхлориде (30 мл), перемешивались при комнатной температуре в течение 1 ч. Реакционная смесь затем промывалась 2 М аммиаком и сушилась (сульфатом натрия). Растворитель удалялся в вакууме, давая коричневатый маслянистый осадок, который очищался с помощью флэш-хроматографии (двуокись кремния, метиленхлорид/этилацетат, 5: 1), давая чистое целевое соединение с выходом 91 (0,75 г). a (основание) -91,3 oС (MeOH, 0,1 M, 21 oC). Хлоргидратная соль осаждалась из диэтилового эфира при 0 oC, а затем перекристаллизовывалась из смеси этилацетат/диэтиловый эфир. Спекается при менее 120 oC.
Пример 64. (R)-5-(2-ферил)-3-(N-Изопропил-N-н-пропиламинохроман.
(R)-3-(N-изопропил-N-н-пропиламино)-5- трифторметансульфонилоксихроман (0,4 г, 1 ммоль), 2-фурилборная кислота (0,3 г, 2,6 ммоля), хлористый литий (0,09 г, 2 ммоля), карбонат натрия (2 М, 3 мл), этанол (7 мл) и толуол (15 мл) смешивались в атмосфере азота в трехгорлой 100 мл круглодонной колбе, снабженной конденсором. Наконец добавлялся Pd (PPh3)4 (0,03 г, каталитическое количество), и реакционная смесь нагревалась с обратным холодильником в течение 2 ч, затем она разбавлялась диэтиловым эфиром, промывалась (2 М аммиаком) и сушилась (сульфатом натрия). Растворитель удалялся в вакууме, давая коричневатый маслянистый остаток, который очищался с помощью флэш-хроматографии (двуокиси кремния, метилехлорид/этилацетат, 10:1), давая чистое целевое соединение с выходом 95 (0,3 г). a (основание) - 57,4 oС (MeOH, 0,1 M, 21 oC). Хлоргидратная соль осаждалась из диэтилового эфира при 0 oC, а затем перекристаллизовывалась из смеси этилацетат/диэтиловый эфир. Т. пл. 151-152 oC.
Пример 65. 5-формил-3-(N-изопропил-N-пропиламино)хроман.
a) 5-Гидроксиметил-3-(N-изопропил-N-пропиламино)хроман. 5-Метоксикарбонил-3-(N-изопропил-N-пропиламино)хроман (0,69 г, 2,36 ммоля) растворялся в 25 мл сухого метиленхлорида и охлаждался до -78 oC. По каплям добавлялся 1 М раствор диизобутилалюминийгидрида в гексане (6,6 мл), и реакционная смесь оставлялась подогреваться до комнатной температуры в течение 50 мин. По каплям добавлялся 1 М раствор KOH, а затем реакционная смесь помещалась в холодильник на ночь. Раствор выливался через слой сульфата натрия, сушился (сульфатом натрия), фильтровался, и растворитель удалялся в вакууме, давая 0,60 г (97 выход) целевого соединения в виде прозрачного масла. 13C ЯМР: (200 МГц CDCl3) млн. дол. 154,6, 140,1, 127,0, 120,5, 119,8, 116,3, 69,0, 62,9, 50,9, 48,9, 47,8, 27,2, 24,3, 21,0, 20,1, 11,7.
b) 5-формил-3-(N-изопропил-N-пропиламино)хроман (0,60 г, 2,27 ммоля) растворялся в 50 мл метиленхлорида, и к раствору добавлялась в виде одной порции двуокись марганца (1,98 г, 22,7 ммоля), и реакционная смесь оставлялась перемешиваться при комнатной температуре в течение 8 дней. Реакционная смесь фильтровалась через слой целита, промывалась теплым метиленхлоридом, и растворитель удалялся в вакууме. Хроматография на двуокиси кремния (элюент:5 этилацетат/гексан) давала 0,33 г (56 выход) целевого соединения (основание) в виде слегка желтого масла. 13C ЯМР: (200 МГц, CDCl3) млн. дол. 193,0, 155,1, 135,0, 127,0, 126,8, 124,0, 69,2, 50,3, 48,9, 47,7, 28,1, 24,3, 21,1, 19,9, 11,6.
Пример 66. (R)-3-(N-Изобутил)амино-5-(N-метил)-карбамоилхроман.
К перемешиваемому раствору (R)-3-амино-5-(N-метил)карбамоилхромана в метаноле (25 мл) добавлялся изомасляный альдегид (0,33 г, 4,60 ммоля). Смесь охлаждалась до 0 oC, и порциями добавлялся цианоборгидрат натрия (0,34 г, 5,44 ммоля). Величина pH доводилась приблизительно до pH 5 с помощью уксусной кислоты. Смесь перемешивалась при комнатной температуре в течение 3,5 ч. Добавлялось небольшое количество изобутиральдегида (15 мг, 0,21 ммоля) вследствие неполной реакции, и смесь перемешивалась в течение еще 20 мин. Растворитель выпаривался, и остаток распределялся между эфиром (100 мл) и 1М раствором аммиака (20 мл). Слои разделялись, водная фаза экстрагировалась эфиром (6•50 мл). Объединенные органические слои промывались 1 М раствором аммиака (20 мл), сушились (сульфатом магния), фильтровались и выпаривались. Остаток хроматографировался на короткой колонке из силикагеля (элюент:этилацетат + 0,5 конц. аммиак). Чистые фракции сливались и выпаривались, давая 0,81 г (74 ) целевого соединения в виде белого твердого вещества: т. пл. 111-112,8 oC (α)21 -33,1, (c 2,6, MeOH).
Пример 67. Хлоргидрат (R)-3-(N-изобутил-N-пропил)амино-5-(N-метил)-карбамоилхромана.
Пропионовый альдегид (0,11 г, 131 мкл, 1,82 ммоля) добавлялся к перемешиваемому охлажденному льдом раствору (R)-3-(N-изобутил)амино-5-(N-метил)-карбамоилхромана (0,43 г, 1,65 ммоля) в метаноле (10 мл). Добавлялся порциями цианоборгидрид натрия (0,14 г, 2,15 ммоля), и величина pH доводилась до 5 с помощью уксусной кислоты. Реакционная смесь перемешивалась при комнатной температуре. Для завершения реакции необходимо было несколько раз добавить пропиональдегид. Всего к реакционной смеси добавлялось 0,20 г, 248 мкл, 3,44 ммоля. Растворитель удалялся в вакууме, и остаток распределялся между эфиром (100 мл) и 1 М раствором аммиака (20 мл). Слои разделялись, и водный слой экстрагировался эфиром (50 мл). Объединенные органические слои промывались солевым раствором (20 мл), сушились (сульфатом магния), фильтровались и выпаривались. Неочищенный продукт фильтровался через короткую колонку из силикагеля (элюент: этилацетат/гексан 1:1 + 0,5 конц. аммиак). Элюент удалялся в вакууме, давая неокрашенное масло, которое превращалось в хлоргидрат в смеси HCl/эфир.
Пример 68. (R)-3-(N-(2-пиридил)метил)амино-5-(N-метил)-карбамоилхроман.
(R)-3-амино-5-(N-метил)-карбамоилхроман (1,02 г, 4,96 ммоля) растворялся в метаноле (30 мл). Реакционный сосуд накрывался алюминиевой фольгой для предотвращения подвержения воздействию света. Раствор охлаждался до 0 oC и добавлялся пиколинальдегид (0,69 г, 617 мкл, 6,45 ммоля). Затем добавлялся цианоборгидрид натрия (0,53 г, 8,44 ммоля) порциями, и величина pH доводилась до 5,5 с помощью уксусной кислоты. Реакционная смесь перемешивалась при комнатной температуре в течение 2 ч. Добавлялось еще 50 мкл, 0,52 ммоля пиколинальдегида для того, чтобы завершить реакцию, и смесь перемешивалась в течение 1 ч. Растворитель удалялся в вакууме, и остаток распределялся между эфиром (100 мл) и 1 М раствором аммиака (25 мл). Слои разделялись, и водный слой экстрагировался эфиром (2•50 мл). Объединенные эфирные экстракты не содержали какого-либо существенного количества желаемого продукта. Поэтому водная фаза с дополнительным количеством NaCl экстрагировалась этилацетатом (4•100 мл). Объединенные органические слои сушились (сульфатом натрия), фильтровались и выпаривались, давая желтое твердое вещество. Перекристаллизация из смеси этилацетат:гексан (35:65) давала белое твердое вещество, которое необходимо было далее очищать с помощью хроматографии на колонке (этилацетат:этанол, 1:1 + 1 конц. аммиак), давая 0,64 г (43) целевого соединения в виде белого твердого вещества. Т. пл. 141,2-141,6 oC; (α)21 -26,2 (c 3,0, MeOH).
Пример 69. Хлоргидрат (R)-3-(N-гексил)амино-5-фенилхромана.
К раствору (R)-3-амино -5-фенилхромана (1,00 г, 4,44 ммоля) в метаноле (25 мл) добавлялся при перемешивании гексаналь (0,49 г, 0,60 мл, 4,91 ммоля). Смесь охлаждалась до 0 oC, и порциями добавлялся цианоборгидрид натрия (0,36 г, 5,78 ммоля). Величина pH доводилась до 5,5 с помощью уксусной кислоты. Реакционная смесь перемешивалась при комнатной температуре в течение 2,5 ч. Вследствие неполного превращения исходного материала добавлялось порциями еще 300 мкл (2,46 ммоля) гексаналя. Растворитель удалялся при пониженном давлении, и остаток распределялся между эфиром (100 мл) и 1 М раствором аммиака (25 мл). Слои разделялись, и эфирный экстракт промывался 1 М раствором аммиака (25 мл). Водная фаза удалялась, а эфирная фаза встряхивалась с 1 М раствором HCl (10 мл). Хлоргидратная соль кристаллизовалась непосредственно в разделительной воронке и отфильтровывалась и промывалась охлажденным льдом гексаном. После сушки получалось 0,86 г (56) целевого соединения в виде белых кристаллов: т. пл. 171,6-174,2 oC; (α)21 +82,8 (c 1, MeOH).
Пример 70 Хлоргидрат (R)-3-(N-метил-N-пропил)амино-5-фенилхромана.
(R)-3-(N-пропил)амино-5-фенилхроман (0,46 г, 1,74 ммоля) растворялся в метаноле (15 мл). Добавлялся формальдегид (0,70 г, 8,70 ммоля), и реакционный сосуд охлаждался на ледяной бане. Порциями добавлялся цианоборгидрид натрия (0,58 г, 9,23 ммоля), который после этого доводился до pH 6 с помощью нескольких капель концентрированной уксусной кислоты. Реакционная смесь перемешивалась при комнатной температуре в течение 3 ч. Растворитель выпаривался, а остаток распределялся между эфиром (50 мл) и 1 М раствором аммиака (10 мл). Слои разделялись, и водная фаза экстрагировалась эфиром (50 мл). Объединенные органические слои промывались 1 М раствором аммиака (10 мл), сушились (сульфатом магния), фильтровались и выпаривались. Остаток очищался с помощью колоночной хроматографии (элюент:гексан: этилацетат 70:30 + 0,1 конц. NH3), давая 0,40 г (81) соответствующего основания целевого соединения в виде неокрашенного масла, которое затвердевало после стояния: (α)21 -35,9 (с 1,0, MeOH). Превращение в хлоргидрат осуществлялось в смеси HCl/эфир, давая 0,4 г (общий выход: 75) целевого соединения. Т. пл. 162-164 oC.
Пример 71. Хлоргидрат (R)-3-N-[2-(2-тиенил)этил]амино-5-фенилхромана.
К раствору (R)-3-амино-5-фенилхромана (0,41 г, 1,83 ммоля) в сухом ДМФ (2,0 мл) добавлялся карбонат калия (0,28 г, 2,02 ммоля) и 2-(2-тиенил)этилбромид (0,36 г, 1,80 ммоля). Реакционная смесь перемешивалась в атмосфере азота при 65 oC в течение 6 ч. Растворитель удалялся в вакууме, а остаток распределялся между эфиром (50 мл) и 1 М раствором аммиака (10 мл). Слои разделялись, и водная фаза насыщалась NaCl и экстрагировалась дополнительным количеством эфира (25 мл). Органические слои объединялись, сушились (сульфатом магния), фильтровались и концентрировались. Остаток очищался с помощью хроматографии на колонке (элюент:этилацетат: гексан 35:65 + 0,2 конц. NH3), давая 0,19 г (31) соответствующего основания (целевого соединения) в виде светло-желтого масла: (α)21 +8,2 в смеси HCl/эфир, давая 0,20 г (общий выход: 29) целевого соединения: т. пл. 191,6-193,2 oC.
Пример 72. 3-Дипропиламино-8-фтор-5-трифторметансульфонилоксихроман.
a) 2-фтор-5-метоксибензойная кислота. Целевое соединение приготавливалось согласно методу, описанному авторами Hay и Blanchard (Canad. J. Chem. 43, 1306 (1965)). В трехгорлой круглодонной колбе (1000 мл), оборудованной магнитной мешалкой, термометром, обратным холодильником и трубкой для газа, растворялся тетрагидрат ацетата кобальта (II) (12,6 г, 51 ммоль) в ледяной уксусной кислоте (404 мл) при перемешивании. Добавлялись промышленный 4-фтор-3-метиланизол (35,4 г, 252 ммоля) и 33 HBr в уксусной кислоте (10,1 мл, 51 ммоль), и затем смесь нагревалась на масляной бане до 90-95 oC. Все время через раствор пропускался поток кислорода со скоростью примерно 660 мл/мин. Спустя 3 ч реакционная смесь охлаждалась до комнатной температуры, а затем выпаривалась в роторных условиях досуха. Окрашенное в фиолетовый цвет твердое вещество, полученное таким образом, переносилось в колбу Эрлен-Мейера, растворялось в кипящей воде (350 мл), содержащей концентрированную соляную кислоту (5-10 мл), а затем оставлялось кристаллизоваться в холодном месте в течение ночи. Неочищенная кислота отфильтровывалась путем отсасывания, промывалась небольшими порциями ледяной воды до тех пор, пока промывная жидкость не становилась бледно-желтой, растворялась в 2 М гидрокиси натрия (120 мл) и промывалась толуолом (2•100 мл). Органические экстракты отбрасывались. Водная фаза фильтровалась отсосом, охлаждалась в химическом стакане на ледяной бане и осторожно подкислялась концентрированной соляной кислотой до тех пор, пока вся кислота не осаждалась. Содержимое химического стакана нагревалось до точки кипения, посредством чего наибольшая часть кислоты растворялась. Раствор охлаждался при комнатной температуре и наконец в холодильнике. Маточная жидкость отсасывалась. Кристаллическая масса промывалась ледяной водой (3•50 мл), а затем сушилась в вакуумной печи при 40 oC, давая 31,5 г целевого соединения в виде серо-белого твердого вещества. Т. пл. 146-148 oC.
b) 2-Фтор-5-метоксибензамид. В трехгорлой круглодонной колбе (500 мл), снабженной магнитной мешалкой и обратным холодильником, нагревались в условиях дефлегмации в атмосфере азота в течение 2 ч 2-фтор -5-метоксибензойная кислота (33,8 г, 189 ммоля) и тионилхлорид (200 мл). Избыток тионилхлорида выпаривался, оставляя маслянистый остаток, который растворялся в метиленхлориде (100 мл) и выпаривался. Данная процедура повторялась 3 раза. Неочищенный хлорангидрид кислоты, полученный таким образом, растворялся в метиленхлориде (100 мл) и добавлялся по каплям в течение 10 мин к холодному (-40.-50 oC) и механически перемешиваемому раствору сухого ТГФ (300 мл) и жидкого аммиака (100 мл) в трехгорлой круглодонной колбе (1000 мл) в атмосфере азота. Когда добавление завершалось, охлаждающая баня удалялась, и смесь медленно нагревалась до комнатной температуры, при этом перемешивание продолжалось. Добавлялась вода (50 мл) для растворения выпавшей в осадок соли, полученный двухфазный раствор концентрировался досуха с помощью роторного испарения. Неочищенный амид промывался повторно небольшими порциями разбавленного водного аммиака, а затем водой до тех пор, пока промывные жидкости не были бесцветными. Сушка при пониженном давлении на протяжении ночи давала 32,0 г целевого соединения в виде светло-коричневых кристаллов. Т. пл. 122- 124 oC.
c) 2-Фтор-5-метоксианилин. В круглодонной колбе (500 мл), снабженной обратным холодильником и магнитной мешалкой, бром (11,4 мл, 223 ммоля) добавлялся по каплям к холодному (+4 oC) и перемешиваемому раствору гидроокиси натрия (35,5 г, 887 ммолей) и воды (322 мл). Затем порциями добавлялся 2-фтор-5-метоксибензамид (31,9 г, 189 ммолей). Смесь нагревалась на масляной бане, нагревалась с обратным холодильником в течение 1 ч, охлаждалась до комнатной температуры и экстрагировалась простым эфиром (3•300 мл). Сушка объединенных эфирных фаз (карбонатом калия), фильтрование, выпаривание растворителя и вакуумная перегонка маслянистого остатка давала 20,1 г целевого соединения в виде бледно-желтого масла (т. кип. 113-114 oC) 14 мм рт.ст.), которое затвердевало на холоде (т. пл. 27-28 oC).
d) 2-Фтор-5-метоксифенол. Способ, используемый для получения фенола, представляет собой в основном процедуру, описанную авторами Claudi и др. (Med. Chem, 33, 2408 2421 (1990) с видоизменением согласно методике Lambooy (J. Amer. Chzem. Soc. 72, 5327 5328 (1950)). В трехгорлой круглодонной колбе (1000 мл), снабженной капельной воронкой и магнитной мешалкой, смешивались 2-фтор-метоксианилин (11,3 г, 80 ммолей), концентрированная серная кислота (35 мл) и вода (200 мл), а затем охлаждалась до 3- 5 oC. К перемешиваемой смеси по каплям при температуре выше 2-4 oC добавлялся раствор нитрита натрия (6,07 г, 88 ммолей) в воде (20 мл). Когда добавление завершалось, раствор перемешивался в течение пяти минут, а затем добавлялась мочевина (0,48 г) с последующим добавлением воды (100 мл). Между тем подготавливался аппарат, состоящий из трехгорлой круглодонной колбы (1000 мл), термометра, электрического нагревательного кожуха, устройства для паровой отгонки и длинного (40 см) водяного холодильника, на верху которого была присоединена капельная воронка (500 мл). В колбу помещались CuSO4•5H2O (56 г), концентрированная серная кислота (160 мл) и вода (160 мл). Смесь нагревалась до 150 oC, по каплям из капельной воронки добавлялся раствор диазониевой соли, охлаждаемый все время путем добавления к ней мелких кусочков льда, и через систему пропускался поток пара для удаления фенола. Дистиллят экстрагировался эфиром (3• 400 мл), и объединенные эфирные экстракты сушились (сульфатом магния), фильтровались и концентрировались в вакууме. Неочищенный фенол очищался на колонке из двуокиси кремния, элюируемой смесью эфир/н-гексан (15:85), давая 4,03 г целевого соединения в виде кристаллического твердого вещества. Т. пл. 25-26 oC.
e) 3-Циано-8-фтор-5-метокси-2-H -хромен. Синтез названного в заголовке соединения основан на методе, описанном авторами Thorberg и др. в Acta Chem. Scand. 24, 169-182 (1987). Трихлоруксусная кислота (приблизительно 0,04 г) добавлялась к перемешиваемому раствору 2-фтор-5-метоксифенола (6,8 г, 48 ммолей) и этилвинилового эфира (9,1 мл, 95 ммолей) в атмосфере азота в круглодонной колбе (50 мл) при +4 oC. Смесь перемешивалась при комнатной температуре на протяжении ночи. Избыток этилвинилового эфира подвергался роторному испарению, а остаток растворялся в эфире (100 мл). Эфирный раствор промывался насыщенным карбонатом натрия (2•5 мл), сушился (карбонатом калия), фильтровался и концентрировался в вакууме. Маслянистый остаток растворялся в сухом толуоле (100 мл), и раствор концентрировался сначала с использованием роторного испарителя, а затем вакуумного насоса (0,1 мм рт.ст.), давая 10,0 г защищенного фенола в виде желтого масла. В сухой трехгорлой круглодонной колбе (100 мл), снабженной магнитной мешалкой и каучуковой пробкой, 8,8 г (41 ммоль) защищенного фенола, полученного выше, растворялось в сухом ТГФ (30 мл) в атмосфере азота. Раствор охлаждался до -25 oC на бане из сухого льда, затем в течение 15 мин к перемешиваемому раствору с помощью шприца по каплям добавлялся 1,6 M н-бутиллитий в гексане (32 мл, 51 ммоль). Когда добавление завершалось, смесь подогревалась до -15 oC за 1 ч, затем охлаждалась до -20 oC. Сухой диметилформамид (4,7 мл, 60 ммолей) добавлялся по каплям в течение 5 мин при поддержании температуры при -25.-20 oC (реакция экзотермическая). Когда добавление завершалось, раствор подогревался до -5 oC на протяжении 2 ч, а затем выливался в охлажденную льдом 2 M соляную кислоту (200 мл). После перемешивания в течение 40 мин гидролиз завершался, как можно было видеть по данным ТСХ. Получающаяся смесь промывалась эфиром (3•250 мл). Объединенные эфирные фазы сушились (сульфатом магния), фильтровались и концентрировались в вакууме. Желтый твердый остаток растворялся в эфире (50 мл) и концентрировался, давая 7,12 г неочищенного 3-фтор-2-гидрокси-6-метокси бензальдегида в виде желтого кристаллического твердого вещества. Неочищенный 3-фтор-гидрокси-6-метоксибензальдегид (8,0 г), полученный выше, акрилонитрил (13 мл, 193 ммоля) и 1,4-диазабицикло(2,2,2)октан (0,66 г, 5,9 ммоля) нагревались с обратным холодильником в трехгорлой круглодонной колбе (50 мл) в атмосфере азота в течение 4 ч. Роторное испарение летучих веществ оставляло густое красное масло, которое подвергалось флэш-хроматографии на колонке из двуокиси кремния, элюируемой смесью метиленхлорида и н-гексана (4: 5). Концентрирование соответствующих фракций давало неочищенный продукт (91 ГХ), который кристаллизовался из этилацетата, давая 1,31 г целевого соединения в виде бесцветных палочек. Т. пл. 133-135 oC.
f) 8-Фтор-5-метоксихроман-3-ил-карбоновая кислота. 3-Циано-8-фтор -5-метокси-2-H-хромен (1,23 г, 6,0 ммолей) и 2 M раствор гидроокиси натрия (25 мл) нагревались с обратным холодильником в атмосфере азота в течение 7,5 часов, а затем перемешивался при комнатной температуре на протяжении ночи. К раствору добавлялся избыток концентрированной соляной кислоты при перемешивании. После охлаждения суспензии на ледяной бане осажденная карбоновая кислота отсасывалась насухо, переносилась в 250 мл колбу для гидрирования (Парра) и растворялась в ледяной уксусной кислоте (110 мл). Добавлялся палладий (5) на активированном угле (0,104 г). Гидрирование в аппарате Парра при 1 атм. давлении водорода и 40- 50 oC (ИК-лампа) в течение 10 ч завершало реакцию. Суспензия фильтровалась через целит, и фильтрат концентрировался в вакууме. Остаток растворялся в толуоле (25 мл), и раствор концентрировался с помощью роторного испарения. Данная процедура повторялась один раз. Выпаривание с помощью вакуумного насоса (0,05 мм рт.ст.) давало 1,12 г целевого соединения в виде серо-белого твердого вещества. Т. пл. 140-145 oC (разлож.)
g) Бензиловый эфир 8-фтор-5-метоксихроман-3-ил-карбаминовой кислоты. 8-Фтор-5-метоксихроманил-3-карбоновая кислота (1,12 г, 85 ммолей), дифениилфосфорилазид (1,3 мл, 5,9 ммоля) и толуол (10 мл) смешивались в трехгорлой круглодонной колбе (50 мл) в атмосфере азота. Добавлялся триэтиламин (0,83 мл, 5,9 ммоля), и прозрачный раствор перешивался при 100 oC в течение 2 ч. Добавлялся бензиловый спирт (0,61 мл, 5,9 ммоля) в виде одной порции, и перемешивание продолжалось при 90 oC в течение 17 ч. Летучие вещества выпаривались, и остаток брался в толуол (25 мл). После промывки 10-й уксусной кислотой (1•30 мл) и 2М аммиаком (1 •30 мл) раствор сушился (сульфатом магния), фильтровался и концентрировался в вакууме, давая коричневое масло. Флэш-хроматогрфия неочищенного продукта на колонке из двуокиси кремния, элюируемой смесью этилацетата и толуола (3:97) давала соединение в виде масла (загрязненного 2 непрореагировавшего изоцианата по данным ГХ).
h) 3-Амино-8-фтор-5-метоксихроман. Бензиловый эфир 8-фтор-5-метоксихромат-3-ил-карбаминовой кислоты (1,37 г, 4,0 ммоля), абсолютный этанол (90 мл), ледяная уксусная кислота (10 мл) и 5-й палладий на активированном угле (0,090 г) смешивались в 250 мл колбе гидрирования (Парра). Гидрирование в аппарате Парра при давлении водорода 1 атм и 45 oC (ИК-лампа) на протяжении ночи завершало реакцию (потребление водорода 80 мл при 20 oC). Суспензия фильтровалась через целит, и фильтрат концентрировался с помощью роторного испарения. Остаток растворялся в толуоле (25 мл) и снова концентрировался. Данная процедура повторялась один раз. Выпаривание с помощью вакуумного насоса (0,05 мм рт.ст.) давало 1,20 г влажной кристаллической массы, состоящей главным образом (93 ГХ) из названного в заголовке соединения (в виде ацетата).
i) 3-Дипропиламино-8-фтор-5-метоксихроман. В круглодонную колбу, снабженную магнитной мешалкой, помещались неочищенный ацетат 3-амино-8-фтор-5-метоксихромана (1,18 г, приблизительно 3,9 ммоля), полученный на предыдущей стадии, сухой метанол (10 мл), пропаналь (2,8 мл, 39 ммолей), ледяная уксусная кислота (pH 3-4) и боргидрит натрия (0,25 г, 3,9 ммоля), но без молекулярных сит. Реакционная смесь перемешивалась в атмосфере азота при комнатной температуре в течение 1 ч. Раствор фильтровался через Целит, и растворитель выпаривался, оставляя жидкий остаток, который разбавлялся водой (25 мл) и экстрагировался эфиром (2•100 мл), после доведения pH до 10-11 с помощью 5 М гидроокиси натрия. Собранные эфирные фазы сушились (C2CO3), фильтровались и концентрировались в вакууме. Флэш-хроматография сырого вещества на двуокисно-кремниевой колонке, элюируемой этилацетатом (2-м и 10-м) в н-гексане давала 0,235 г целевого соединения в виде бесцветного масла.
j) Хлоргидрат 3-дипропиламино-8-фтор-5-гидроксихромана. Хлоргидратная соль 3-дипропиламино-8-фтор-5-метоксихромана получалась с помощью добавления избытка хлоргидрата в эфире к перемешиваемому и охлажденному (+4 oC) раствору основания (0,235 г, 0,84 ммоля) в эфире (10 мл), затем выделения осажденной соли и сушки ее вакуумной печи при 50 oС. В трехгорлой круглодонной колбе (25 мл), снабженной магнитной мешалкой и каучуковой пробкой, полученная выше соль растворялась в метиленхлориде (7 мл) в атмосфере азота. Раствор охлаждался на бане из сухого льда до -40 oC, затем медленно добавляют трехбромистый бор (0,158 г, 1,7 ммоля) в метиленхлориде (1 мл) к перемешиваемому раствору с помощью шприца. Когда добавление завершалось (2 мин), температура раствора медленно поднималась до +4 oC и поддерживалась на данном уровне с помощью ледяной бани. По истечении всего периода реакции (7 ч) раствор выливался в насыщенный бикарбонат натрия (20 мл). Смесь экстрагировалась эфиром (3•30 мл), и собранные эфирные фазы промывались солевым раствором, сушились (сульфатом натрия), фильтровались и концентрировались в вакууме. Флэш-хроматография неочищенного продукта на колонке из двуокиси кремния, элюируемой смесью этилацетата и н-гексана (15:85) давала 0,195 г 3-дипропиламино-8-фтор-5-гидроксихромана в виде масла. Целевое соединение получалось с помощью осаждения основания избытком хлоргидрата в эфире и сушки полученной таким образом соли в вакуумной печи при 50 oC в течение 5 ч. Выход 0,220 г (99 из основания), белое аморфное твердое вещество. Т. пл. 190-192 oC.
k) 3-Дипропиламино-8-фтор -5- трифторметансульфонилоксихроман. В сухой трехгорлой круглодонной колбе (25 мл), оборудованной магнитной мешалкой и каучуковой пробкой, 3-дипропиламино-8-фтор-5-гидроксихроман-хлоргидрат (0,211 г, 0,70 ммоля), 2,4,6-коллидин (0,110 мл, 0,84 ммоля) и метиленхлорид (7,3 мл) смешивались в атмосфере азота. Светлый раствор осаждался до -40 oC на сухой ледяной бане, затем медленно (5 мин) к перемешиваемой смеси с помощью шприца добавлялся трифторметансульфоновый ангидрид (0,230 мл, 1,37 ммоля) в метиленхлориде (0,3 мл). Когда добавление заканчивалось, раствор перемешивался в течение 1 ч (температура увеличивалась до 0 oC), затем выливался в холодный (+4 oC) насыщенный бикарбонат натрия (20 мл) и экстрагировался эфиром (2•40 мл). Собранные эфирные фазы промывались водой (1• 30 мл), сушились (сульфатом натрия), фильтровалдись и концентрировались в вакууме. Сырой продукт подвергался мгновенной хроматографии на колонке из двуокиси кремния, элюируемой смесью этилацетат/н-гексан (1:19), давая 0,213 г названного в заголовке соединения в виде масла.
Пример 73. Хлоргидрат 3-дипропиламино-8-фтор-5-(2-фурил)хромана.
В трехгорлой круглодонной колбе емкостью 25 мл, снабженной магнитной мешалкой и обратным холодильником, в атмосфере азота смешивались следующие реагенты: 3-дипропиламино-8-фтор-5-трифторметансульфонилхроман (0,104 г, 0,25 ммоля), абсолютный этанол (1,7 мл), толуол (3,6 мл), 2-фурилборная кислота (0,071 г, 0,64 ммоля), хлористый литий (0,022 г, 0,51 ммоля), 2М карбонат натрия (0,7 мл) и тетракис(трифенилфосфин)палладий (0) (0,0073 г). Смесь нагревалась на масляной бане до температуры дефлегмации (75-80 oC). Спустя 3 ч ГХ-анализ показал частичную реакцию (20 продукта и 66 исходного материала). Поэтому добавлялись еще 2-фурилборная кислота (0,031 г, 0,28 ммоля), абсолютный этанол (0,2 мл) и тетракис (трифенилфосфин)палладий(0) (0,009 г). Смесь перемешивалась при 75-80 oC на протяжении ночи, затем выливалась в 2М аммиак (40 мл) и экстрагировалась эфиром (2•40 мл). Собранные эфирные фазы сушились сульфатом натрия, фильтровались и концентрировались в вакууме. Неочищенный продукт очищался с помощью флэш-хроматографии на колонке из двуокиси алюминия, элюируемой смесью этилацетата и н-гексана (2: 98). Соответствующие фракции сливались и концентрировались, давая масло, состоящее из нечистого продукта (72 ГХ). Данный материал очищался с помощью еще одного цикла флэш-хроматография на колонке из двуокиси кремния, элюируемой смесью этилацетата и метиленхлорида (1:99), давая 0,042 г (51 выход) 3-дипропиламино-8-фтор-5-(2-фурил)хромана в виде масла. Целевое соединение получалось из основания, как описано для хлоргидрата 3-дипропиламино-8-фтор-5-гидроксихромана выше. Белое кристаллическое твердое вещество. Т. пл. 162-164 oC.
Пример 74. Хлоргидрат 3-дипропиламино-8-фтор-5-N-изопропилкарбамоилхромана.
В трехгорлой круглодонной колбе (25 мл), снабженной магнитной мешалкой и вводом для моноокиси углерода от бюретки для гидрирования, смешивались диоксан (1,3 мл), 3-дипропиламино-8- фтор-5-трифторметансульфонилхромаан (пример 63k) (0,110 г, 0,20 ммоля) и изопропиламин (0,118 мл, 1,4 ммоля). Колба вакуумировалась и заполнялась моноокисью углерода. Данная процедура повторялась дважды. Добавлялись 1,3-бис(дифенилфосфино)пропан (0,0032 г) и ацетат палладия (II) (0,0016 г), а затем смесь перемешивалась при 75o C в атмосфере моноокиси углерода при 1 атм. ГХ-анализ показал, что реакция оказалась неожиданно медленной. Перемешивание на протяжении ночи не улучшило выход желаемого продукта (59 ГХ), и все еще оставалось много трифлата (27 ГХ). Соответственно добавлялись еще 1,3-бис(дифенилфосфино)пропан (0,0065 г) и ацетат палладия (0,0032 г). К сожалению, перемешивание на протяжении ночи не улучшило выход, а привело в результате к некоторому разложению продукта, о чем свидетельствовал ГХ. В данный момент реакционная смесь обрабатывалась путем добавления насыщенного бикарбоната натрия (5 мл) и экстрагирования смеси этилацетатом (2•10 мл). Объединенные органические фазы сушились (сульфатом натрия), фильтровались и концентрировались в вакууме. Сырой продукт подвергался флэш-хроматографии на колонке из двуокиси кремния, элюируемой этилацетатом (15 и 33) в н-гексане, давая 0,021 г исходного материала (трифлата) и 0,030 г (40 выход в расчете нарегенерированный трифлат) 3 - дипропиламино-8-фтор-5-N-изопропилкарбамоилхромана в виде масла. Целевое соединение получалось из основания, как описано для 3-дипропиламино-8-фтор-5-гидроксихроман-гидрохлорида выше. Белое твердое вещество. Т. пл. (70 eV); m/z (отн. инт. ), 337 (4,4, M + 1), 336 (17, M), 308 (21), 307 (100), 236 (36), 194 (6), 177 (6), 43 (19).
Пример 75. (R)-8-фтор-3-(N-изопопил-N-пропиламино)-5- трифторметансульфонилхроман.
(R)-8-Фтор-3-(N-изопропил-N-пропиламино)-5- гидроксихроман (0,71 г, 2,66 ммоля) и коллидин (0,49 мл, 3,72 ммоля) растворялись в 25 мл метиленхлорида и охлаждались до -40 oC. По каплям добавлялся трифторметансульфоновый ангидрид (0,54 мл, 3,2 ммоля), и смесь оставлялась подогреваться до температуры окружающей среды, и после достижения 0 oC проходила реакция. Реакционная смесь разбавлялась метиленхлоридом и промывалась насыщенным бикарбонатом натрия, сушилась (сульфатом натрия), фильтровалась и выпаривалась в вакууме, давая сырое вещество. Хроматография на двуокиси кремния (элюент: метиленхлорид) давала 0,82 г (77 выход) целевого соединения в виде светлого масла. (α)21 81,5 oC (C=0,1, CHCl3).
Пример 76. Хлоргидрат (R)-5-ацетил-8-фтор-3-(N-изопропил-N-пропиламино)-хромана.
(R)-8-фтор-3-(N-изопропил-N-пропиламино)-5- трифторметансульфонилхроман (403 мг, 1,01 ммоля) растворялся в 3 мл ДМФ, а затем добавлялись триэтиламин (0,28 мл, 2,02 ммоля), бутилвиниловый эфир (0,78 мл, 6,06 ммоля), ацетат палладия (7 мг) и 1,3-бис-дифенилфосфинопропан (14 мг). Получающаяся в результате смесь нагревалась до 80- 90 oC при перемешивании в течение 3,5 ч. Раствор охлаждался, и добавлялось 5 мл 2 M HCl раствора, и реакционная смесь перемешивалась при комнатной температуре в течение 1 ч. Смесь разбавлялась эфиром и промывалась 2 M раствором аммиака. Водная фаза повторно экстрагировалась эфиром, и объединенные эфирные фазы обрабатывались солевым раствором, сушились (сульфатом магния), фильтровались и выпаривались в вакууме, давая коричнево-оранжевое масло в виде сырого вещества. Хроматография на двуокиси кремния (элюент:15 этилацетат/гексан) давала 201 мг в виде желтого масла (68 выход). (α)21 165,1 o (C=0,1, CHCl3). HCl соль осаждалась из эфира и перекристаллизовывалась из смеси этилацетат/эфир, давая белое твердое вещество. Т. пл. 148 oC.
Пример 77, Хлоргидрат (R)-5-фенил-8-фтор-3-(N-изопропил-N-пропиламино)-хромана.
К раствору (R)-8-фтор-3-(N-изопропил-N-пропиламино)-5-трифторметансульфонилхромана (0,58 г, 1,45 ммоля), этанола (7 мл), хлорида лития (124 мг, 2,9 ммоля), 2M карбоната натрия (2,2 мл) и тетракис(трифенилфосфин) палладия (0) (31 мг, 0,026 ммоля), растворенного в толуоле (15 мл) добавлялась фенилборная кислота (0,22 г, 1,74 ммоля) в виде одной порции в атмосфере азота. Получающийся в результате раствор нагревался до 95 oC в течение 5 ч, затем реакционная смесь оставлялась охлаждаться. Реакционная смесь фильтровалась, и растворитель выпаривался в вакууме. Остатки брались в эфир, промывались 2 M аммиаком, обрабатывались солевым раствором и сушились (сульфатом магния). Удаление растворителя в вакууме давало неочищенное соединение. Хроматография на двуокиси кремния (элюент: метиленхлорид + метиленхлорид + аммиак) давала 0.42 г (89 выход) целевого соединения (основания) в виде масла. (α)21 45,0 o (C=0,1, CHCl3). Хлоргидратная соль получалась путем растворения чистого основания в эфире и добавления по каплям избытка эфирного раствора HCl, давая белое твердое вещество. Т. пл. 202-203 oC.
Пример 78. Хлоргидрат (R)-5-[карбонил(2,6-диметилфенил)]-8-фтор-3-(N, N-дипропиламино)хромана.
2-Бром-м-ксилол(0,5 мл, 3,6 ммоля) растворялся в 4 мл безводного ТГФ в атмосфере азота и охлаждался до -78 oC. Добавлялся по каплям н-бутиллитий (1,6 M в гексане, 2,0 мл, 3,2 ммоля), и реакционная смесь оставлялась перемешиваться при -78 oC в течение 1 ч. (R)-8-фтор-5-формил-3-(N,N-дипропиламино)хроман (0,30 г, 1,07 ммоля) растворялся в 5 мл безводного ТГФ и добавлялся по каплям к реакционной смеси, а затем она оставлялась перемешиваться при -78 oC в течение 1 ч. Реакционная смесь гасилась добавлением 2 мл воды и оставлялась подогреваться до комнатной температуры. Растворитель удалялся в вакууме, остатки брались в 2 M раствор аммиака, а затем экстрагировались трижды эфиром. Объединенные эфирные порции обрабатывались солевым раствором, сушились (сульфатом магния), фильтровались, и растворитель удалялся в вакууме, давая неочищенный спирт в виде белого твердого вещества. Неочищенное твердое вещество на протяжении ночи подвергалось мягкому продуванию струей азота для удаления избытка 2-бром-м-ксилола. Неочищенный спирт окислялся непосредственно на следующей стадии реакции без дальнейшей очистки. Оксалилхлорид (122 мкл, 1,4 ммоля) растворялся в 5 мл безводного метиленхлорида в атмосфере азота и охлаждался до -78 oC. На протяжении 5 мин добавлялся ДМСО (0,24 мл, 3,4 ммоля), затем реакционная смесь оставлялась перемешиваться при -78 oC в течение 5 мин. Неочищенный спирт (из предыдущей стадии) растворялся в 10 мл безводного метиленхлорида и добавлялся на протяжении 10 мин к реакционной смеси. После перемешивания при -78 oC в течение 25 мин добавлялся триэтиламин (0,75 мл, 5,4 ммоля), и спустя 5 мин реакционная смесь оставлялась подогреваться до комнатной температуры. К реакционной смеси добавлялся 2 M раствор аммиака в воде, и смесь экстрагировалась трижды метиленхлоридом. Объединенные метиленхлоридные фазы сушились (сульфатом магния), фильтровались, и растворитель удалялся в вакууме, давая неочищенный кетон. Хроматография на двуокиси кремния (элюент:8,7 этилацетат/гексан) давали 345 мг (84 выход) целевого соединения в виде слегка желтого масла. (α)21 57,0 o (C= 0,1, CHCl3). Масс спектр (70 eV) m/z (относительная интенсивность) 383 (56M+), 354 (100), 283 (26), 355 (36). Хлоргидратная соль приготавливалась путем растворения чистого основания в эфире и добавления по каплям избытка эфирного HCl. Соль сушилась в вакуумной печи при 50 oC, давая белое твердое вещество в виде полугидрата, который спекается при 80 oC.
Пример 79. Хлоргидрат (R)-8-фтор-3-(N-изопропил-N-н-пропиламино)-5-(5-оксазолил)хромана.
(R)-8-фтор-5-формил-3-(N-изопропил-N-н-пропиламино)хроман (0,48 г, 1,7 ммоля), тозилметилизоцианид (0,37 г, 1,9 ммоля) и карбонат калия (0,28 г, 2,0 ммоля) суспендировались в метаноле (50 мл, высушенный 3 ангстрем) в атмосфере азота. Реакционная смесь нагревалась с обратным холодильником в течение 1 ч. Добавлялись диэтиловый эфир и 2 M аммиак. Слои разделялись, и водная фаза экстрагировалась дважды диэтиловым эфиром. Органические слои объединялись и сушились (сульфатом натрия). Растворитель удалялся в вакууме, давая бесцветный маслянистый остаток, который очищался с помощью мгновенной хроматографии (двуокись кремния, метиленхлорид/этилацетат, 10: 1), давая целевое соединение с выходом 60 (320 мг). (α)
Пример 80. (R)-8-фтор-5-метоксикарбонил-3-(N,N-ди-н-пропиламино)хроман.
(R)-8-фтор-3-(N, N-ди-н-пропиламино)-5-трифторметансульфонилксихроман (0,95 г, 2,4 ммоля), триэтиламин (0,53 г, 5,2 ммоля), 1,3-бис(дифенилфосфино)пропан (100 мг, каталитическое количество), ацетат палладия (II) (50 мг, каталитическое количество) и ДМФ/MeOH (18 мл, 6:2) смешивались в 50 мл трехгорлой круглодонной колбе. Колба вакуумировалась с последующим вводом CO (повторялось три раза). Реакционная смесь перемешивалась при 70 oC в течение 7 ч. Растворитель удалялся в вакууме, и остаток растворялся в диэтиловом эфире/2M аммиаке. Слои разделялись, и водная фаза экстрагировалась один раз эфиром. Объединенные эфирные слои сушились (сульфатом магния), и растворитель удалялся в вакууме, давая коричневый маслянистый остаток, который очищался с помощью флэш-хроматографии (двуокись кремния, метиленхлорид/этилацетат, 20: 1), давая 650 мг целевого соединения в виде светлого масла (87 выход). (α)
Пример 81. (R)-8-фтор-5-формил-3-(N,N-ди-н-пропиламино)хроман.
(R)-8-фтор-5-метоксикарбонил-3-(N,N-ди-н-пропиламино)хроман (0,65 г, 2,1 ммоля) растворялся в ТГФ (30 мл, высушенный натрием) в атмосфере азота. Раствор охлаждался до -78 oC, затем на протяжении 10 мин добавлялся диизобутилалюмнийгидрид (DIBAL-H, 1,0 M, 8,4 мл, 8,4 ммоля). Охлаждающая баня удалялась, и температуре реакционной смеси давали возможность медленно подниматься до комнатной (20 oC). Реакционная смесь перемешивалась в течение 3 ч перед тем, как реакционная смесь гасилась добавлением 2 M гидроокиси натрия (2 мл). Добавлялись насыщенный тартрат натрия и диэтиловый эфир, и смесь перемешивалась до тех пор, пока "гель" не растворялся. Слои разделялись, и водная фаза экстрагировалась дважды эфиром. Объединенные органические слои сушились (сульфатом магния), и растворитель удалялся в вакууме, давая спирт. Оксалилхлорид (0,30 г, 2,3 ммоля) растворялся в метиленхлориде в атмосфере азота. Раствор охлаждался до -78 oC, затем к нему по каплям на протяжении 5 мин добавлялся ДМСО (0,4 г, 5,0 ммолей). После перемешивания в течение 5 мин добавлялся спирт, растворенный в метиленхлориде, на протяжении 30-минутного периода. Реакционная смесь перемешивалась в течение 30 мин, затем на протяжении 5-минутного периода добавлялся триэтиламин (1,1 г, 10,5 ммолей). Спустя 30 мин охлаждающая баня удалялась, и реакционная смесь медленно подогревалась до комнатной температуры, а затем перемешивалась в течение дополнительных 30 мин перед тем, как добавлялся бикарбонат натрия. Слои разделялись, и водная фаза экстрагировалась дважды эфиром. Объединенные органические слои сушились (сульфатом магния), и растворитель удалялся в вакууме, давая маслянистый остаток, который очищался с помощью флэш-хроматографии (двуокись кремния, метиленхлорид кремния, метиленхлорид/этилацетат, 20:1), давая 350 мг целевого соединения (60 выход). (α)
Пример 82. Хлоргидрат (R)-5-[Карбонил-(3,5-диметилизоксазол)] -8-фтор-3-(N,N-ли-н-пропиламино)хромана.
4-Бром-3,5-диметилизоксазол (1,9 г, 10,7 ммолей) растворялся в ТГФ (20 мл, высушенном натрием) в атмосфере азота. Раствор охлаждался до -90 oC перед тем, как на протяжении 10 мин добавлялся н-бутиллитий (1,6 M в гексане, 9,7 мл, 9,7 ммоля). Реакционная смесь перемешивалась в течение 15 мин, затем на протяжении 10 мин добавлялся (R)-8-фтор -5- формил-3-(N,N-ди-н-пропиламино)хроман (0,6 г, 2,2 ммоля), растворенный в ТГФ (10 мл). Реакционная смесь перемешивалась при -90 oC в течение дополнительных 20 мин, а затем при -78 oC в течение 15 мин перед тем, как она гасилась добавлением хлористого аммония. Добавлялись диэтиловый эфир и 2 M аммиак. Слои разделялись, и водная фаза экстрагировалась один раз эфиром. Объединенные органические слои сушились (сульфатом натрия). Растворитель удалялся в вакууме, давая желтый маслянистый остаток, который продувался на протяжении ночи струей азота. Оксалилхлорид (0,28 г, 2,2 ммоля) растворялся в метиленхлориде в атмосфере азота. Раствор охлаждался до -78 oC. По каплям на протяжении 5-минутного периода добавлялся ДМСО (0,38 г, 4,8 ммоля). Реакционная смесь перемешивалась в течение 20 мин, затем в течение 15 мин добавлялся неочищенный спирт, растворенный в метиленхлориде. Реакционная смесь перемешивалась в течение 30 мин. Добавлялся триэтиламин (1,01 г, 10 ммолей). Перемешивание продолжалось в течение 1 ч, охлаждающая баня удалялась, и реакционная смесь оставлялась подогреваться до комнатной температуры (21 oC). Добавлялись диэтиловый эфир и бикарбонат натрия. Слои разделялись, и водная фаза экстрагировалась один раз эфиром. Объединенные органические слои сушились (сульфатом натрия), и растворитель удалялся в вакууме, давая желтый маслянистый остаток, который очищался с помощью мгновенной хроматографии (двуокись кремния, метиленхлорид/гексан/этилацетат, 10:10:1), давая 340 мг целевого соединения (выход 42). Масс. спектр (70 eV)m/z (относительная интенсивность) 374 (11, M+), 346 (21), 345 (100), 274 (15), 43 (11). HCl - соль осаждалась из диэтилового эфира при 0 oC, а затем перекристаллизовывалась из смеси метиленхлорид/диэтиловый эфир. Т. пл. 202-205 oC.
Пример 83. Хлоргидрат (R)-5-N-Морфолинил -3- (N-изопропил-N-н-пропиламино)-хромана.
Дигликолевая кислота (0,15 г, 1,1 ммоля) и N,N-карбонилдиимидазол (CDI, 0,28 г, 1,8 ммоля) растворялись в ТГФ (высушенном над натрием) в атмосфере азота. Смесь нагревалась с обратным холодильником в течение 15 мин и добавлялся (R)-5-амино-3- (N-изопропил-N-н-пропиламино)хроман (NAE 277) (0,2 г, 0,8 ммоля). Нагревание с обратным холодильником продолжалось в течение 18 ч. Реакционная смесь затем объединялась с аналогичной реакционной смесью от идентичной реакции (исходящей из 0,1 г анилина). Добавлялись диэтиловый эфир и 2 М аммиак. Слои разделялись, и водная фаза экстрагировалась один раз эфиром. Объединенные органические слои сушились (сульфатом натрия), и растворитель удалялся в вакууме, давая маслянистый остаток. Остаток растворялся в диэтиловом эфире в атмосфере азота. Добавлялся литийалюминийгидрид (0,23 г, 6 ммолей), и смесь нагревалась с обратным холодильником в течение 18 ч. При энергичном перемешивании добавлялась вода (0,3 мл) с последующим добавлением 0,3 мл 15-й гидроокиси натрия и наконец 1 мл воды. Раствор декантировался с белого твердого остатка, и остаток промывался дважды эфиром. Органические слои объединялись, и растворитель удалялся в вакууме, давая неочищенный остаток, который очищался с помощью мгновенной хроматографии (двуокись кремния, метиленхлорид/этилацетат, 5:1, содержащая также 0,5 аммиака), давая 180 мг целевого соединения (47 выход). (α)
Пример 84. (R)-5-Карбамоил-2-аминохроман.
а) (R)-5-Карбамоил-2-N-трифторацетиламинохроман. (R)-2-N-Трифторацетиламино-5-трифторметансульфонилоксихроман (2,0 г, 5,0 ммолей) растворялся в ДМФ (20 мл) в трехгорлой круглодонной колбе. Колба вакуумировалась с последующим вводом CO-газа (повторялось три раза). Добавлялись 1,3-бис(дифенилфосфино)-пропан (0,2 г, каталитическое количество), ацетат палладия (II) (0,1 г, каталитическое количество) и диоксан, насыщенный аммиаком (20 мл), а затем реакционная смесь перемешивалась при 70 oC в течение 8 ч. Растворитель удалялся в вакууме. Остаток растворялся в смеси диэтиловый эфир/аммиак (2М). Слои разделялись, и водная фаза экстрагировалась два раза. Растворитель удалялся в вакууме, давая оранжевый маслянистый остаток, который очищался с помощью флэш-хроматографии (двуокись кремния, метиленхлорид: этилацетат, 4:1(+0,5 аммиака)), давая целевое соединение с выходом 21 (0,30 г). Т. пл. 178-180 oC. Масс спектр (70 eV) m/z (относительная интенсивность) 288 (5, M+), 176 (11), 175 (100), 174 (22), 158 (13), 131 (25), 130 (42), 51 (19), 44 (16).
b) (R)-5-Карбамоил-2-аминохроман. (R)-5-Карбамоил-2-N-трифторацетиламинохроман (0,30г, 1,04 ммоля) растворялся в метиленхлориде (50 мл). Добавлялся гидроксид натрия (5 мл 15-го раствора в воде), и смесь перемешивалась в течение 4 ч. Слои разделялись, и водная фаза экстрагировалась метиленхлоридом четыре раза. Органические слои объединялись, и растворитель удалялся в вакууме, давая твердый остаток, который суспендировался в диэтиловом эфире (300 мл) и фильтровался. Эфир удалялся в вакууме, давая целевое соединение с выходом 90 (180 мг). Т. пл. 194-196 oC. (α)
Пример 85. (R)-2-N,N-Дибензиламино-5-метоксикарбонилхроман.
a) (R)-2-N,N-Дибензиламино -5-метокси-хроман (R)-5-Метокси -2-аминохроман (2,6 н, 14 ммолей), карбонат калия (7,0 г, 51 ммоль), бензилбромид (6,0 г, 35 ммолей) и каталитическое количество иодистого калия смешивались в ацетонитриле (100 мл) в атмосфере азота. Реакционная смесь перемешивалась при 85 oC в течение примерно 72 ч. Растворитель удалялся в вакууме, и остаток растворялся в смеси диэтилового эфира и аммиака (2 М). Слои разделялись, и водная фаза экстрагировалась дважды диэтиловым эфиром. Эфирные слои объединялись и сушились (сульфатом магния). Растворитель удалялся в вакууме, давая желтый маслянистый остаток, который очищался с помощью флэш-хроматографии (двуокись кремния, метиленхлорид), давая целевое соединение с выходом 64 (3,2 г). HCl соль осаждалась из диэтилового эфира при 0 oC, а затем перекристаллизовывалась из смеси этанол/диэтиловый эфир. Кристаллы являются гигроскопичными и начинают плавиться очень медленно, начиная со 100 oC и плавятся наконец между 118 и 120 oC. (α)
b) (R)-2-N,N-Дибензиламино-5-гирокиси-хроман. Хлоргидрат (R)-2-N,N-дибензиламино-5-метоксихромана (1,6 г, 4,0 ммоля) растворялся в метиленхлориде (40 мл) в атмосфере азота. Раствор затем охлаждался до -70 oC. Добавлялся по каплям на протяжении 5-минутного периода трехбромистый бор, растворенный в метиленхлориде (25 мл). Реакционная смесь затем подогревалась медленно до 0 oC и перемешивалась при этой температуре на протяжении ночи. Реакционная смесь медленно выливалась в перемешиваемый насыщенный раствор бикарбоната натрия. Слои разделялись, и водная фаза экстрагировалась три раза метиленхлоридом. Органические слои объединялись и сушились (сульфатом магния). Растворитель удалялся в вакууме, давая коричневатый маслянистый остаток, который очищался с помощью флэш-хроматографии (двуокись кремния, метиленхлорид), давая целевое соединение с выходом 98 (135) мг. (α)
c) (R)-2-N, N-Дибензил-5-трифторметансульфонилоксихроман. (R)-2-N,N-Дибензиламино-5-гидроксихроман (1,5 г, 4,3 ммоля) и коллидин (0,6 г, 5,2 ммоля) растворялись в метиленхлориде (30 мл) в атмосфере азота. Раствор охлаждался до -70 oC, и в течение 10-минутного периода по каплям добавлялся трифторметансульфоновый ангидрид (1,3 г, 4,7 ммоля), растворенный в метиленхлориде (20 мл). Охлаждающая баня удалялась, и реакционная смесь медленно подогревалась до комнатной температуры (21 oC). Реакционная смесь промывалась аммиаком (2М) м сушилась (сульфатом магния). Растворитель удалялся в вакууме, давая оранжевый твердый остаток, который очищался с помощью флеш-хроматографии (двуокись кремния, метиленхлорид), давая целевое соединение с выходом 90 (1,85 г). (α)
d) (R)-2-N,N-Дибензиламино-5-метоксикарбонилхроман.
(R)-2-N, N-Дибензиламино-5-трифторметансульфонилоксихроман (1,8 г, 3,8 ммоля) и триэтиламин (0,8 г, 8,3 ммоля) растворялись в растворе ДМФ/MeOH (20 мл, 6: 2) в трехгорлой круглодонной колбе. Колба откачивалась с последующим вводом CO газа (повторялось три раза). Добавлялись ацетат палладия (II) (28 мг, каталитическое количество) и 1,3-бис(дифенилфосфино)-пропан (60 мг, каталитическое количество), и реакционная смесь перемешивалась при 75 oC в течение 6 ч. Растворитель удалялся в вакууме, давая коричневатый маслянистый остаток. Масло растворялось в смеси диэтиловый эфир/аммиак (2 М). Слои разделялись, и водная фаза экстрагировалась один раз диэтиловым эфиром. Объединенные эфирные слои сушились (сульфатом магния). Растворитель удалялся в вакууме, давая коричневый маслянистый остаток, который очищался с помощью мгновенной хроматографии (двуокись кремния, метиленхлорид/гексан, 1:1), давая целевое соединение с выходом 95 (1,4 г). (α)
Пример 86. (R)-5-Карбамоил-2-N,N-бензилхроман.
(R)-2-N, N-Дибензиламино-5-метоксикарбонилхроман (1,4 г, 3,6 ммоля) растворялся в метаноле (20 мл). Добавлялся раствор гидроокиси натрия (0,16 г, 4,0 ммоля) в воде (6 мл), и реакционная смесь нагревалась с обратным холодильником на протяжении ночи. Растворитель удалялся в вакууме. Добавлялся толуол, и растворитель удалялся снова (повторялось дважды для того, чтобы удалить воду). Остаток растворялся в SOCl2 (6 мл) и нагревался с обратным холодильником в течение 1 ч. Растворитель удалялся в вакууме. Остаток растворялся в метиленхлориде (20 мл) и через раствор продувался аммиак-газ в течение 2 мин. Реакционная смесь перемешивалась при комнатной температуре в течение 1 ч перед промывкой (2 М аммиаком) и сушилась (сульфатом магния). Растворитель удалялся в вакууме, давая неочищенный коричневатый маслянистый остаток, который очищался с помощью флэш-хроматографии (двуокись кремния, метиленхлорид/этилацетат, 5: 1 (+ 0,5 аммиака)), давая целевое соединение с выходом 41 (0,55 г). (α)
Пример 87. (R)-5-[1-Метил(-имидазолил)] -3-(N-изопропил-N-н-пропиламино)-хроман.
(R)-5-Формил-3-(N-изопропил-N-н-пропиламино)хроман (0,4 г, 1,5 ммоля) растворялся в метаноле (15 мл), насыщенном метиламином. Добавлялось некоторое количество молекулярных сит (3 ангстрем). Растворитель удалялся в вакууме. Остаток растворялся в метаноле (30 мл) в атмосфере азота. Добавлялись тозилметилизоцианид 0,36 г, 1,8 ммоля) и карбонат калия (0,25 г, 1,8 ммоля), и реакционная смесь нагревалась с обратным холодильником в течение 1 ч. Добавлялись дополнительно тозилметилизоцианид (0,36 г, 1,8 ммоля) и карбонат калия (0,25 г, 1,8 ммоля), и нагревание с обратным холодильником продолжалось в течение 3 ч. Добавлялся еще раз тозилметилизоцианид (0,1 г, 5,2 ммоля), и нагревание с обратным холодильником продолжалось в течение 12 ч. Растворитель удалялся в вакууме. Остаток растворялся в смеси диэтиловый эфир/2 М аммиак. Слои разделялись, и водная фаза экстрагировалась еще раз эфиром. Органические слои объединялись и сушились (сульфатом натрия). Растворитель удалялся в вакууме, давая коричневый маслянистый остаток, который очищался с помощью флэш-хроматографии (двуокись кремния, сначала метиленхлорид/ацетат, 5: 1, содержащая также 0,5 аммиака, а затем этилацетат/этанол, 50:1 + 0,5 аммиака), давая 200 мг целевого соединения (42 выход). (α)
Пример 88. a) (R)-3-(N-Изопропил-N-н-пропиламино)-5-метоксикарбонилхроман.
(R)-3-(N-Изопропил-N-н-пропиламино)- 5-трифторметилсульфонилоксихроман (4,0 г, 10,5 ммолей), триэтиламин (2,3 г, 23,1 ммоля) и смесь ДМФ/метанол (18 мл, 6:2) смешивались в трехгорлой круглодонной колбе. Колба откачивалась с последующим вводом газа CO (повторялось два раза). Наконец добавлялся 1,3-бис(дефинилфосфино) пропан (0,11 г, каталитическое количество) и ацетат палладия (II) (0,07 г, каталитическое количество), и смесь перемешивалась при 70 oC в течение 17 ч. Реакционная смесь разбавлялась диэтиловым эфиром, промывалась 2 М аммиаком и сушилась (сульфатом магния). Удаление растворителя в вакууме давало коричневый маслянистый остаток, который очищался с помощью флэш-хроматографии (двуокись кремния, метиленхлорид/этилацетат, 10:1), давая целевое соединение с выходом 82 (2,5 г). (α) -131,6 o (MeOH, 0,1 М, 21 oC).
Анализ C17H25O3N:
Вычислено: C 70,1; H 8,6; N 4,8.
Найдено: C 70,2; H 8,6; N 5,1.
13C ЯМР (CDCl3) 300 МГц, δ 167,6, 155,0, 130,5, 126,5, 124,2, 123,0, 120,6, 69,1, 51,8, 50,6, 48,8, 47,6, 29,3, 24,3, 21,0, 19,9, 11,5
Масс. спектр (70 eV) (относительная интенсивность) 291 (48, М+), 276 (34), 262 (100), 191 (18), 18 (27), 70 (15), 56 (23), 43 (24).
HCl-соль осаждалась из диэтилового эфира при 0 oC, а затем перекристаллизовывалась из смеси этанол/диэтиловый эфир. Т. пл. 125,5-127,4 oC.
Анализ для C17H26O3N1Cl1:
Вычислено: C 62,3; H 8,0; N 4,3.
Найдено: C 62,5; H 7,9; N 4,4.
b) (R)-3-(N-Изопропил-N-н-пропиламино)-5-(N-изопропил)-карбамоилхроман. (R)-3-(N-Изопропил-N-н-пропиламино)-5-метоксикарбонилхроман (0,46 г, 1,6 ммоля), растворенный в метаноле (10 мл), смешивался с раствором гидроокиси натрия (0,06 г, 1,6 ммоля) в воде (3 мл). Реакционная смесь затем нагревалась с обратным холодильником 3,5 ч, а затем растворитель удалялся в вакууме. Остаток растворялся в толуоле. Толуол удалялся в вакууме (повторялось два раза) для того, чтобы образовать азеотроп для удаления воды. Карбоновая кислота затем растворялась в сульфонилхлориде (5 мл) и нагревалась с обратным холодильником в течение 1 ч. Избыток SOCl2 удалялся в вакууме. Хлорангидрид кислоты затем растворялся в метиленхлориде (20 мл, высушенном молекулярными ситами 3 ангстрем), и затем добавлялось 4 мл изопропиламина. Реакционная смесь перемешивалась в течение 1 ч при комнатной температуре (21 oC), разбавлялась диэтиловым эфиром, промывалась (2 М аммиак) и сушилась (сульфатом магния). Удаление растворителя в вакууме давало желтый маслянистый остаток, который очищался с помощью флэш-хроматографии (двуокись кремния, метиленхлорид/этилацетат, 5: 2), давая чистое целевое соединение с выходом 88 (0,46 г). a -90,4 o (MeOH, 0,1 M, 21 oC). Т. пл. 92,5-94 oC.
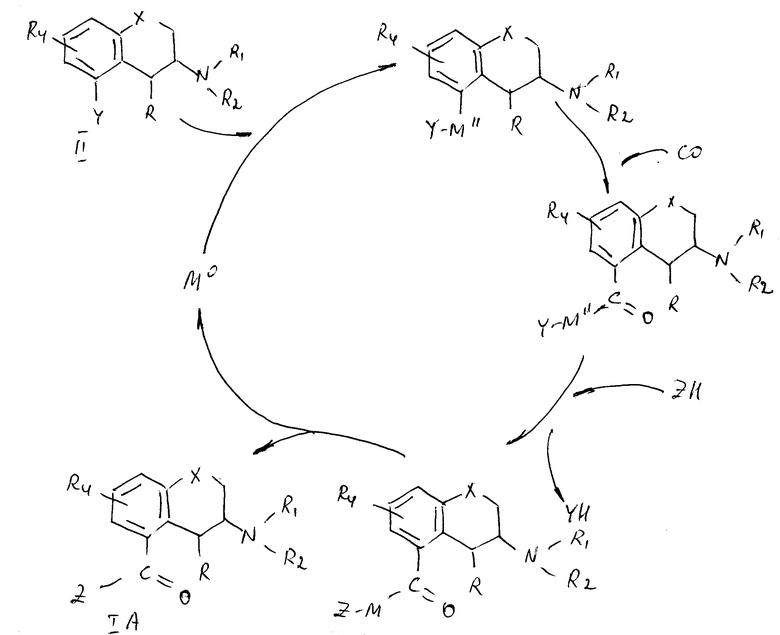

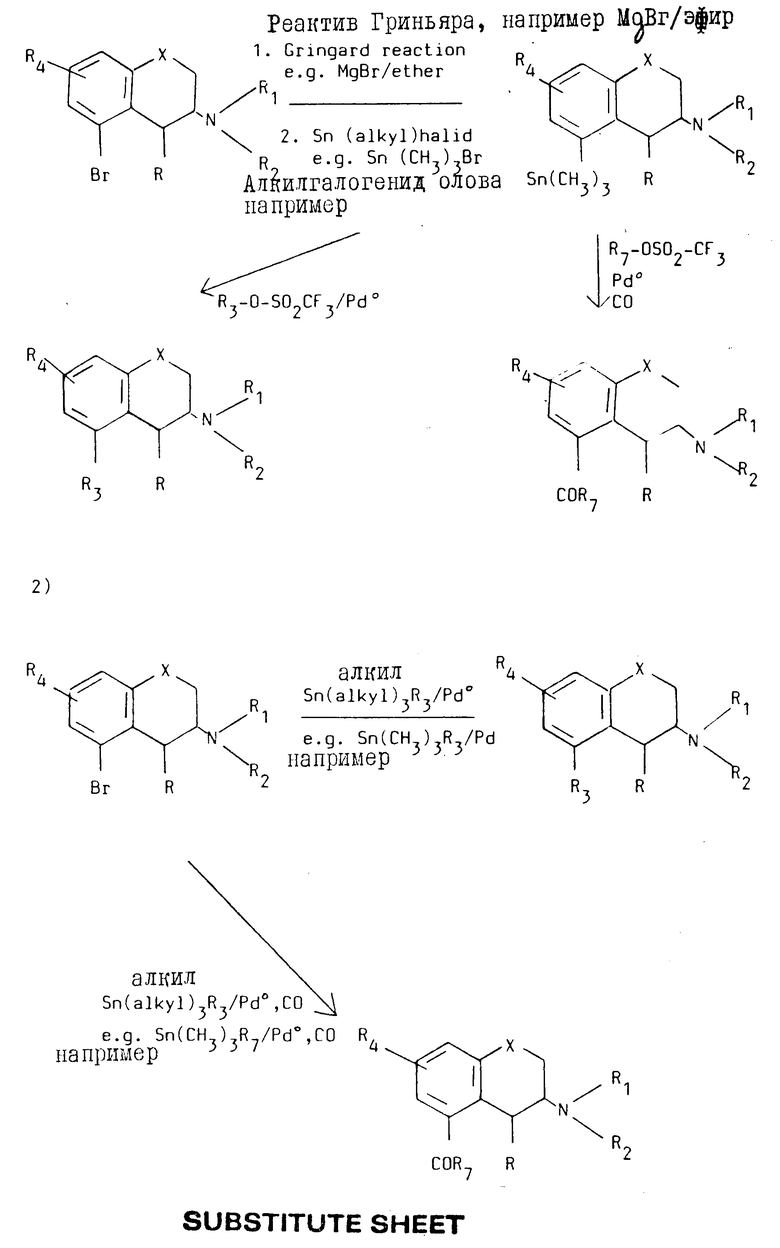
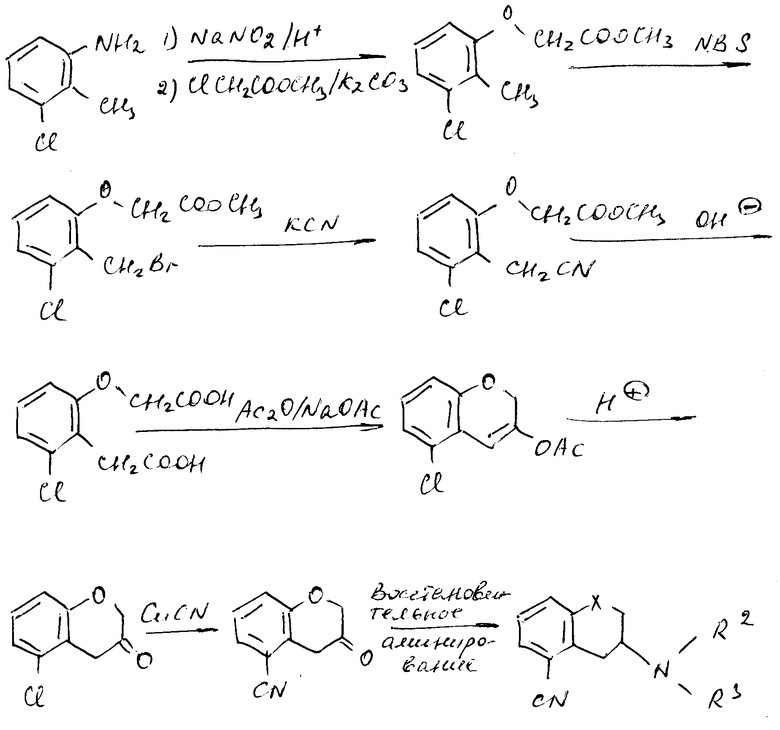
Использование: в медицинской практике для подавления болевых ощущений и регулирования сердечно-сосудистой системы. Продукт: 3-(дипропиламин)-5-пропилтиохромангидрохлорид, т. пл. 150-151 oC, выход 66 %: 3-(метил-3-фенилпропил)амино-5-N-метилкарбамоилхроман, т.пл. 150-151 oC (оксалат). 5 с. и 38 з.п. ф-лы.
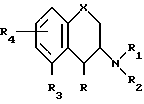
где Х кислород или 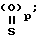
где p 0, 1 или 2;
R водород;
R1 водород, С1 С6- алкил или С2 - С6-алкенил;
R2 водород, С1 С6-алкил, С2 С6- алкенил, С1 С4-алкиларил, где арил может содержать 1 или 2 гетероатома, выбранных из N или S, и возможно замещен С1 - С6-алкилом или С1 С4- алкоксигруппой;
R3 группа NR5R6, COR7, 5- или 6-членный арил, который может содержать 1 или 2 гетероатома, выбранных из N, O или S, и возможно имеет один или несколько заместителей, выбранных из галогена, С1 С6-алкила или С1 С4-алкоксигруппы;
R4 водород или галоген;
R5 водород, С1 С6-алкил или С2 - С6-алкенил;
R6-C1 C6-алкил или C2 C6-алкенил или R5 и R6 могут вместе образовать 5- или 6-членное кольцо, которое может содержать 1 или 2 гетероатома, выбранных из N, O или S;
R7 С1 С6-алкил, С1 - С4-алкоксигруппа, NR8R9 или 5- или 6- членный арил, который может содержать 1 или 2 гетероатома, выбранных из N, O или S, и возможно моно- или полизамещен галогеном, CN, CF3, С1 - С6-алкилом или С1 С4-алкоксигруппой;
R8 или R9 независимо водород, С1 - С6-алкил, С2 С6-алкенил, 5- или 6-членный арил, который может содержать 1 или 2 гетероатома, выбранных из N, O или S, и возможно замещен галогеном, СN, CF3, С1 С6-алкилом, С1 - С4- алкоксигруппой, или могут вместе образовать 5- или 6-членное ядро, содержащее 1 или 2 гетероатома, выбранных из N, O или S,
энантиомер или соль этого соединения.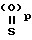 и p 0, 1 или 2.
и p 0, 1 или 2.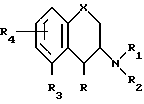
где X кислород или 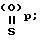
p 0, 1 или 2;
R водород;
R1 водород, С1 С6-алкил или С2 - С6-алкенил;
R2 водород, С1 С6-алкил или С2 - С6-алкенил, С1 С4- алкиларил, где арил может содержать 1 или 2 гетероатома, выбранных из N или S, и возможно замещен С1 С6-алкилом или С1 С4-алкоксигруппой;
R3 COOH, COCl, COBr, N3 или SO3CF3;
R4 водород или галоген;
R5 водород, С1 С6-алкил или С2 - С6-алкенил;
R6 C1 C6-алкил или C2 C6-алкенил или
R5 и R6 могут вместе образовывать 5- или 6-членное ядро, которое может содержать 1 или 2 гетероатома, выбранных из N, O или S, энантиомер или соль его.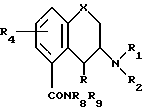
где Х кислород или 
p 0, 1 или 2;
R водород;
R1 водород, С1 С6-алкил или С2 - С6-алкенил;
R2 водород, С1 С6-алкил или С2 - С6-алкенил, С1 С4- алкиларил, где арил может содержать 1 или 2 гетероатома, выбранных из N или S, и возможно замещен С1 С6-алкилом или С1 С4-алкоксигруппой;
R4 водород или галоген;
R8 и R9 водород, С1 С6-алкил, С2 - С6-алкенил, 5- или 6-членный арил, который может содержать 1 или 2 гетероатома, выбранных из N, O или S, и возможно замещен галогеном, СN, CF3, С1 С6-алкилом, С1 - С4-алкоксигруппой, или могут вместе образовать 5- или 6-членное ядро, содержащее 1 или 2 гетероатома, выбранных из N, O или S,
отличающийся тем, что аминируют соединение общей формулы IА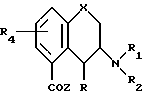
где X, R, R1, R2 и R4 имеют значения, указанные в п. 1;
Z Сl, OH или ORp, где Rp С1 С6-алкил,
с соединением формулы
NHR8R9,
для образования амида формулы I с последующим возможным превращением полученного свободного основания в соль с кислотой, или превращением полученной соли в основание или другую соль с кислотой, или возможным разделением полученной смеси изомеров для выделения чистого энантиомера. и p 0, 1 или 2.
и p 0, 1 или 2.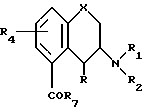
где Х кислород или 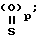
p 0,1 или 2
R водород;
R1 водород, С2 С6-алкил или С2 - С6-алкенил;
R2 водород, С2 С6-алкил или С2 - С6-алкенил, С1 С4- алкиларил, где арил может содержать 1 или 2 гетероатома, выбранных из N или S, и возможно замещен С1 С6-алкилом или С1 С4-алкоксигруппой;
R4 водород или галоген;
R7 С1 С6-алкил, С1 С4-алкоксигруппа, 5- или 6-членный арил, который может содержать 1 или 2 гетероатома, выбранных из N, O или S, и возможно моно- или полизамещен галогеном, СN, СF3, С1 С6-алкилом или С1 - С4-алкоксигруппой,
энантиомера или соли его, отличающийся тем, что производное 5-карбоксихроман-тиохромана общей формулы IА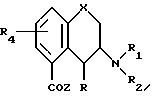
где Х, R, R1, R2 и R4 имеют значения, указанные в п. 1;
Z хлор, бром,
обрабатывают
R7Li,
где R7 алкил, алкенил или арил,
в присутствии соединения меди с последующим возможным превращением полученного основания в соль с кислотой или превращением полученной соли в основание или другую соль с кислотой или возможным разделением полученной смеси изомеров для выделения чистого энантиомера.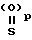 и p 0, 1 или 2.
и p 0, 1 или 2.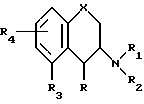
где Х кислород или 
p 0, 1 или 2;
R водород;
R1 водород, С1 С6-алкил или С2 - С6-алкенил;
R2 водород, С1 С6-алкил или С2 - С6-алкенил, С1 С4- алкиларил, где арил может содержать 1 или 2 гетероатома, выбранных из N или S, и возможно замещен С1 С6-алкилом или С1 С4-алкоксигруппой;
R3 5- или 6-членный арил, который может содержать 1 или 2 гетероатома, выбранных из N, O или S, и возможно имеет один или несколько заместителей, выбранных из галогена, С1 С6-алкила и С1 С4-алкоксигруппы;
R4 водород или галоген,
его энантиомера или соли, отличающийся тем, что превращают соединение общей формулы II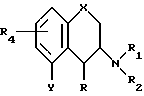
где Y триметилсульфонат, фосфонат или галогенид;
X, R, R1, R2 и R4 имеют указанные значения,
реакцией его с переходным металлом для образования комплекса лиганда, который подвергают окислительному присоединению путем обработки триалкиларилстаннаном или арилборной кислотой для образования соединения формулы I с последующим возможным превращением полученного основания в соль его с кислотой, или превращением полученной соли в основание или другую соль с кислотой, или возможным разделением полученной смеси изомеров для выделения чистого анантиомера.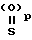 и p 0, 1 или 2.
и p 0, 1 или 2.
| US, патент, 4782083, кл | |||
| Устройство для сортировки каменного угля | 1921 |
|
SU61A1 |

