Настоящее изобретение относится к новому способу получения 3-N,N-дициклобутиламино-8-фтор-3,4-дигидро-2Н-1-бензо-пиран-5-карбоксамида, в особенности (R)-3-N, N-дициклобутиламино-8-фтор-3,4-дигидро-2Н-1-бензопиран-5-карбоксамида и к новым промежуточным продуктам, получаемым в ходе этого процесса.
(R)-3-N, N-Дициклобутиламино-8-фтор-3,4-дигидро-2Н-1-бензопиран-5-карбоксамид раскрыт в WO 95/11891, где также описан способ получения рассматриваемого соединения. Указанный способ включает ряд стадий реакции. Атом фтора вводят в бензопирановое кольцо селективным бромированием в 8-е положение с последующим N, N-дибензилированием, с последующей реакцией галогенлитиевого обмена в полученном бромсодержащем соединении и реакции с соответствующим фторирующим агентом. Полученный (R)-3-N,N-дибензиламино-8-фтор-5-метокси-3,4-дигидро-2Н-1-бензопиран затем дибензилируют, N,N-диалкилируют реакцией восстановительного алкилирования с циклобутаноном, деметилируют и каталитически превращают, используя переходный металл, моноокись углерода и соответствующий спирт в промежуточный алкил (R)-3-N,N-дициклобутиламино-8-фтор-3,4-дигидро-2Н-1-бензопиран-5-карбоксилат. Гидролиз сложного эфира до карбоновой кислоты с последующей обработкой кислоты тионилхлоридом дает хлорангидрид кислоты, который при обработке аммиаком дает желаемый (R)-3-N, N-дициклобутиламино-8-фтор-3,4-дигидpo-2H-1-бeнзопиpaн-5-кapбoкcaмид.
Однако, несмотря на наличие указанных методов и другой информации, все еще имеется потребность в новых, более удобных и эффективных способах производства (R)-3-N, N-дициклобутиламино-8 -фтор- 3,4- дигидро-2Н-1-бензопиран-5-карбоксамида.
Способ получения 3-N, N-дициклобутиламино-8-фтор-3,4-дигидро-2Н-1-бензопиран-5-карбоксамида настоящего изобретения имеет преимущества с технической точки зрения по сравнению со способом, описанным в WO 95/11891. В заявленном способе используют исходный материал, который имеет фтор-замещение в нужном положении, что не требует проведения нежелательной стадии фторирования. Введение фтора в соответствии с традиционным процессом требует низких температур для обработки литием и проведения реакции с дорогим, вредным и, возможно, даже токсичным фторгующим агентом. Кроме того, реакция приводит к получению в качестве побочного продукта значительного количества (R)-3-N, N-дибензиламино-5-метокси-3,4-дигидро-2Н-1-бензопирака, который должен быть отделен от желаемого фторсодержащего конечного продукта методом дорогой и технически сложной хроматографии. Способ настоящего изобретения является коммерчески более выгодным, чем способ, известный из WO 95/11891.
Настоящее изобретение относится к новому способу получения рацемического соединения 3-N, N-дициклобутиламино-8-фтор-3,4-дигидро-2Н-1-бензопиран-5-карбоксамида, имеющего формулу (I), его R-энантиомера (формула R-(I) и его S-энантиомера (формула S-(I)):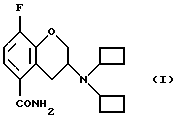
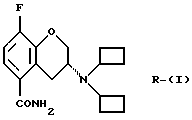
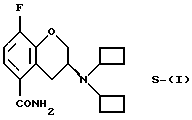
и их фармацевтически приемлемых солей и/или сольватов.
Новый путь синтеза для производства соединений, имеющих формулы (I), R-(I) и S-(I) приводится ниже. Основным важным способом является способ производства (R)-3-N,N-дициклобутиламино-8-фтор-3,4-гидро-2Н-1-бензопиран-5-карбоксамида.
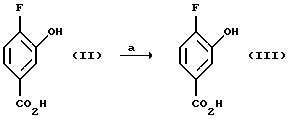
Исходный материал соединения (II) может быть приобретен, например, в компании Frinton laboratories. Inc. США. В соответствии со способом настоящего изобретения, соединение (III), в котором R представляет собой C1-C4 алкил, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, т-бутил получают (а) этерификацией соединения (II) триалкилортоформиатом в безводном растворителе, таком как соответствующий алкиловый спирт. Этерификация катализируется кислотой, такой, как H2SО4, при 0 - 100oС. Реакция может быть также проведена с использованием других методов этерификаиии, таких как нагревание соединения (II) до 40 - 100oС в соответствующем спирте, таком, как метанол, этанол или пропанол, в присутствии кислоты, такой, как Н2SO4. Карбоновая кислота (III) может быть также защищена с помощью других защитных групп, известных специалистам в данной области, см., например: Protective Groups in Organic Synthesis; Second Edition; Theodora W. Green and Peter G.H. Wuts; John W-ley&Sons, Inc.; 1991.
Соединение (IV, получают (b) алкилированием соединения (III) галогенидами пропалгила, например, бромидами, хлоридами или иодидами, или пропалгиловым спиртом, активированным в виде сульфоната, например, п-толуолсульфоната в органическом растворителе в присутствии основания при 20 - 100oC. Примерами оснований, которые могут использоваться, является карбонаты, такие как карбонат натрия и карбонат калин, или амины, такие как триалкиламины, например триэтиламин, хотя специалистам в данной области известны другие приемлемые основания. Предпочтительно используют карбонат калия. Органический растворитель может быть выбран из ацетона, изобутилметилкетона, ацетонитрила и толуола, но могут быть также применимы и другие органические растворители, известные специалистам в данной области. Предпочтительно используют ацетон.
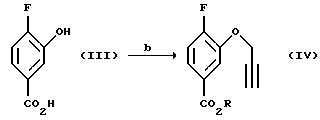
Соединение (V) получают (с) разрезанием соединения (IV) в чистом виде или в подходящем ароматическом растворителе, таком как диэтиланилин, диметиланилин, дифениловый эфир или в ароматическом растворителе, например в толуоле или ксилоле, при повышенном давлении или в насыщенном высшем углеводороде, например ундекане или додекане, при 150 - 250oС, предпочтительно 210 - 230oС.
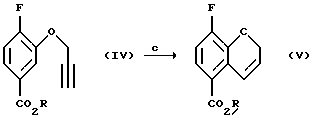
Соединение (VI) получают (d) гидролизом в присутствии основания или кислоты в смеси органического растворителя и воды при 20 - 100oС. Органический растворитель может быть выбран из метанола, этанола, этиленгликоля или их смесей, однако могут быть использованы и другие растворители или смеси растворителей, известные специалистам в данной области. Предпочтительно используют метанол. Могут быть использованы различные основания, такие как гидроксид натрия, гидроксид калия или гидроксид лития, или кислота, такая, как соляная кислота, серная кислота или трифторметансульфоновая кислота.
Соединение (VII) получают (e(i)) взаимодействием соединения (VI) при 0 - 100oС с оксалилхлоридом или тионилхлоридом в присутствии или без органического растворителя или смеси органических растворителей, с проведением затем реакции с аммиаком или гидроксидом аммония. Органическим растворителем, используемым в данной реакции может быть метиленхлорид, этилацетат или толуол, или их смеси. Соединение (VII) может быть также получено (e(ii)) взаимодействием соединения (V) с аммиаком в соответствующем растворителе при 20 - 200oС под давлением или без повышения давления. Реакция может быть проведена в присутствии или в отсутствие каталитических количеств кислот или оснований, например гидроксида натрия, гидроксида калия, гидроксида лития, серной кислоты, соляной кислоты или сульфоновой кислоты. Могут использоваться также другие катализаторы, известные специалистам в данной области. Растворитель может быть выбран из спирта, воды или толуола, или их смеси, а также из других приемлемых растворителей, известных специалистам в данной области. Соединение (VII) может быть также получено взаимодействием соединения (V) с соответствующим амидом, например, формамидом, по реакции трансамидирования в присутствии подходящего катализатора, например, цианида.
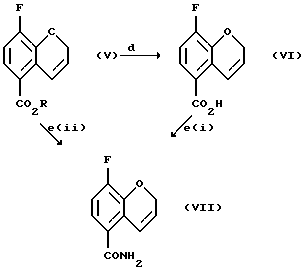
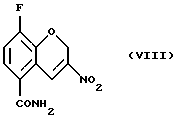
Соединение (VIII) получают (f) взаимодействием соединения (VII) с йодом или другими йодирующими агентами, например монохлоридом йода и нитритной солью, такой как нитрит серебра, нитрит натрия, или нитрит тетрабутиламмония в органическом растворителе, таком как этилацетат, этанол, этиленгликоль, тетрагидрофуран, моно- или диглим или метанол и их смесь, в присутствии или в отсутствие воды, при 0 - 100oС.
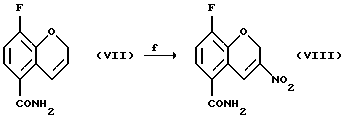
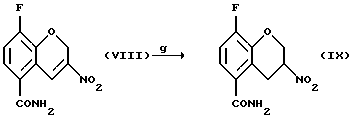
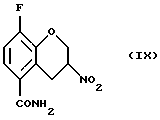
Соединение (IX) получают (g) взаимодействием соединения (VIII) с восстановителем, таким как боргидрид натрия, цианборгидрид натрия, алюмогидрид лития или другой подходящий восстановитель, в присутствии этиленгликоля или силикатов и органического растворителя, такого как этилацетат, метиленхлорид, метанол или этанол, в присутствии или в отсутствие воды и/или уксусной кислоты при температуре -20 - 100oС, предпочтительно при 0 - 20oС.
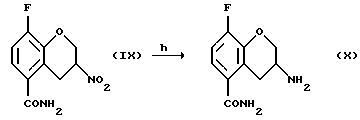
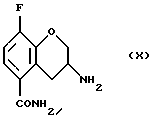
Соединение (X) получают (h) восстановлением соединения (IX), например цинком и соляной кислотой в уксусной кислоте при 20 - 150oС или гидрогенированием в присутствии катализатора, такого как платина, палладий или никель Ренея и газообразного водорода в органическом растворителе, таком как тетрагидрофуран, этил ацетат, низший спирт или их смесь, предпочтительно в присутствии кислоты, например, соляной кислоты в диапазоне температур от -20 до 100oС. Могут использоваться также другие подходящие восстановители, известные специалистам в данной области.
(R)-энантиомер соединения (XI) получают в соответствии с известными методами, такими как фракционная кристаллизация диастереомерных солей. Диастереомерную соль получают (i) обработкой соединения (X) чистым энантиомером хиральной кислоты, такой как карбоновая кислота или сульфоновая кислота, в соответствующем растворителе, таком как метанол, этанол, этилацетат или вода, хотя могут использоваться и другие растворители и/или смеси растворителей, известные специалистам в данной области. Кислота может быть выбрана из чистых энантиомеров винной кислоты, миндальной кислоты и камфановой кислоты, но могут использоваться и другие кислоты, известные специалистам в данной области. Предпочтительно используют L-(+)-винную кислоту. Чистое соединение R-(XI) получают обработкой соли основанием, таким как карбонат натрия, карбонат калия, гидроксид кальция, гидроксид натрия или аммиак, и экстрагируют соответствующим органическим растворителем, например диэтиловым эфиром.
(S)-Энантиомер соединения (ХI) получают по методике (i), описанной для соединения R-(XI), используя кислоту, энантиомерно противоположную применяемой для получения R-(XI). Предпочтительно используют D-(-)-винную кислоту.
Соединения (I), R-(I) и S-(I) получают (j) алкилированием соединений (X), R-(XI) и S-(XI) с помощью соответствующих известных методов, таких как восстановительное алкилирование с использованием циклобутанона, в присутствии восстановителя, такого как боргидрид натрия, цианборгидрид натрия или катализатора гидрирования, такого как палладий или платина, в присутствии водорода, в органическом растворителе, таком, как метанол, этанол, толуол, уксусная кислота или этилацетат, или их смесь.
Альтернативно (см. схему I в конце описания), соединения (I), R-(I) и S-(I) могут быть получены (j) алкилированием соединений (X), R-(XI) и S-(XI), соответственно, алкилирующим агентом, таким как циклобутил галогенид или мезилат или тозилат циклобутанола, в подходящем органическом растворителе в присутствии основания и/или катализатора. Органический растворитель может быть выбран из ацетонитрила или этанола, а также других приемлемых растворителей, которые известны специалистам в данной области. Основание может быть выбрано из карбоната натрия, карбоната калия или триэтиламина, а также из других возможных оснований, которые известны специалистам в данной области. Катализатор представляет собой йод, предпочтительно, иодид натрия.
Соль соединений (I), R-(I) и S-(I) может быть получена с использованием традиционных методов.
Промежуточные продукты
Настоящее изобретение относится также к новым промежуточным продуктам, а именно к промежуточным продуктам формул (III)-(X), R-(XI) и S-(Xl).
Особенно предпочтительными промежуточными продуктами являются следующие промежуточные продукты:
соединение формулы (V):
где R - C1-C4 алкил;
соединение формулы (VII):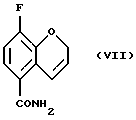
соединение формулы: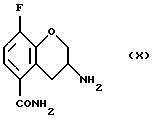
Настоящее изобретение поясняется более детально с помощью следующих примеров.
Пример 1. Получение метил 4-фтор-3-гидроксибензоата (Соединение (III))
4-Фтор-3-гидроксибензойную кислоту (20,0 г, 0,13 моль) растворяют в безводном метаноле (160 мл), смешивают с триметилортоформиатом (25 мл) и затем добавляют концентрированную H2SO4 (3 мл) и реакционную смесь нагревают до 40-55oС в течение ночи. Половину растворителя удаляют в вакууме, полученный раствор выливают в смесь лед/Н2О и продукт экстрагируют дважды диэтиловым эфиром. Объединенные эфирные фазы промывают дважды Н2О, обрабатывают холодным раствором насыщенного NaHCО3, обрабатывают насыщенным солевым раствором, высушивают MgSО4, фильтруют и растворитель удаляют в вакууме с получением 21,6 г (99% выход) белого твердого вещества в качестве целевого соединения (т. пл. 93,5-94,5oC).
Масс-спектр (70eV) m/z (относительная интенсивность) 170 (44, М+), 139 (100), 111 (83), 83(83), 82(16), 81(11), 63(11), 57(24).
Пример 2. Получение метил 4-фтор-3-пропаргилоксибензоата (Соединение (IV)
Метил 4-фтор-3-гидроксибензоат (20,0 мл, 0,118 моль) растворяют в безводном ацетоне (450 мл), смешивают с пропаргилбромидом (26,2 г, 0,177 моль) и затем добавляют порошкообразный К2СО3 (32,4 г, 0,236 моль) и реакционную смесь перемешивают в течение ночи при комнатной температуре. Реакционную смесь фильтруют и растворитель удаляют в вакууме. Остаток растворяют в диэтиловом эфире, промывают 4 раза Н2О, обрабатывают насыщенным солевым раствором, высушивают МgSО4, фильтруют и растворитель удаляют в вакууме с получением в качестве целевого соединения 25,5 г (100% выход) окрашенного в светло-абрикосовый цвет твердого вещества (т.пл.60,5-61,5oС).
Масс-спектр (70eV) m/z (относительная интенсивность) 208 (27, М+), 207 (100), 193 (22), 177 (20), 149 (50), 82 (21), 81 (10).
Пример 3. Получение метил 8-фтор-2Н-1-бензопиран-5-карбоксилата (Соединение (V))
Метил 4-фтор-3-пропаргилоксибензоат (140,0 г, 67,2 ммоль) смешивают с N, N-диэтиланилином и реакционную смесь нагревают до 220oС в течение 5 часов. Окрашенную в черный цвет реакционную смесь охлаждают, растворяют в диэтиловом эфире (600 мл) и промывают 2М НСl, порциями по 1 л. Промывные воды экстрагируют повторно диэтиловым эфиром, объединенные эфирные фазы промывают H2O до нейтральной реакции, обрабатывают насыщенным солевым раствором, высушивают МgSО4, фильтруют и растворитель удаляют в вакууме с получением темно-коричневого неочищенного остатка. Неочищенное твердое вещество хроматографируют на силикагеле (элюент: метиленхлорид/четыреххлористый углерод 1: 1) с получением 11,9 г (85% выход) буровато-желтого твердого вещества в качестве целевого соединения (т. пл. 73,5-74,5oС).
Масс-спектр (70еV) m/z (относительная интенсивность) 208 (65, М+), 207 (42), 194 (12), 193 (100), 177 (32), 149 (10), 148 (12).
Пример 4. Получение 8-фтор-3Н-1-бензопиран-5-карбоновой кислоты (Соединение (VI))
Meтил 8-фтор-2Н-1-бензопиран-5-карбоксилат (7,36 г, 35,4 ммоль) растворяют в абсолютном этаноле (220 мл), добавляют NaOH (2,0 г, 49, 6 ммоль) в Н2О (25 мл) и реакционную смесь кипятят с oбpaтным холодильником в течение 1,5 часов. Затем реакционную смесь охлаждают и растворитель удаляют в вакууме. Желтое твердое вещество растворяют в Н2О (150 мл), добавляют активированный уголь и cмесь фильтруют. Полученную слегка окрашенную жидкость промывают диэтиловым эфиром, водный раствор подкисляют с помощью 2М НСl, и продукт дважды экстрагируют этилацетатом. Объединенные органические порции обрабатывают насыщенным солевым раствором, высушивают МgSO4, фильтруют и растворитель удаляют в вакууме с получением 6,64 г (97% выход) желтоватого твердого вещества (высушивают в эксикаторе над Р2О5) в качестве целевого соединения (т.пл.224-226oС).
Масс-спектр (70еV) m/z (относительная интенсивность) 194 (93, М+), 193 (100), 149 (56), 148 (68), 120 (13), 88 (25), 75 (28), 74 (21), 60 (12).
Пример 5. Получение 3-фтор-2Н-1-бензопиран-5-карбоксамида (Соединение (VII))
К 8-фтор-2Н-1-бензопиран-5-карбоновой кислоте добавляют тионилхлорид (60 мл) и раствор перемешивают при комнатной температуре в течение ночи. Избыток тионилхлорида удаляют в вакууме, добавляют безводный толуол и растворитель удаляют в вакууме. Хлорангидрид кислоты растворяют в метиленхлориде (60 мл) и добавляют по каплям к охлажденному раствору (ледяная баня) концентрированного аммиака (60 мл). Реакционную смесь перемешивают при комнатной температуре в течение 30 минут. К реакционной смеси добавляют этилацетат и отделяют органическую фазу. Водную фазу повторно экстрагируют смесью метиленхлорида/этилацетата и объединенные органические фазы высушивают MgSО4, фильтруют, и растворитель удаляют в вакууме с получением 1,91 г (98% выход) белого твердого вещества в качестве целевого соединения (т. пл. 194,5-195,0oС).
Масс-спектр (70eV) m/z (относительная интенсивность) 193 (51, М+), 192 (19), 176 (11), 175 (33), 174 (100), 149 (20), 148 (38), 101 (14), 75 (17).
Пример 6. Получение 8-фтор-2Н-1-бензопиран-5-карбоксамида (Соединение (VIII))
К раствору 8-фтор-3-нитро-2Н-1-бензопиран-5-карбоксамида (4,46 г, 23,1 ммоль) в этилацетате (220 мл) добавляют этиленгликоль (4,0 мл) и раствор нитрита натрия (6,52 г, 92,4 ммоль) в Н2О (11 мл) и затем йод (9,0 г, 34,7 ммоль). Реакционную смесь кипятят с обратным холодильником в течение 24 часов и в этот период добавляют Н2О (22 мл) и этиленгликоль (4,0 мл) порциями. Реакционную смесь охлаждают, разбавляют этилацетатом, промывают 5% раствором NaS2O3, обрабатывают насыщенным солевым раствором, высушивают МgSO4, фильтруют и растворитель удаляют в вакууме с получением неочищенного желтого твердого вещества. Твердое вещество повторно кристаллизуют из абсолютного этанола с получением 1,3 г (24% выход) блестящих желтых кристаллов в качестве целевого соединения (т. пл. 227,8-228,2oС).
Масс-спектр (70eV) m/z (относительная интенсивность) 238 (57, М+), 221 (100), 192 (71), 191 (46), 190 (10), 148 (14), 109 (16), 94 (12).
Пример 7. Получение 8-фтор-3-нитро-3, 4-дигидро-2Н-1-бензопиран-5-карбоксамида (Соединение (IX))
8-Фтор-3-нитро-2Н-1-бензопиран-5-карбоксамид (730 мг, 3,1 ммоль) перемешивают в виде суспензии в хлороформе (75 мл) и изопропиловом спирте (25 мл). К перемешиваемой смеси добавляют силикагель (2,2 г, 230-400 меш АSТМ) и затем порциями в течение 15 минутного периода - порошкообразный бор-гидрид натрия (255 мг, 6,2 ммоль). После завершения добавления реакционную смесь, перемешивают в течение 20 минут и реакцию гасят добавлением уксусной кислоты (2 мл) и перемешивают еще 30 минут. Отфильтровывают нерастворимый материал и растворитель удаляют в вакууме. Остаток распределяют между этилацетатом и водой. Водную фазу экстрагируют этилацетатом, объединенные этилацетатные фазы обрабатывают насыщенным солевым раствором, высушивают MgSО4, фильтруют и растворитель удаляют в вакууме с получением 0,67 г (91% выход) желтовато-белого твердого вещества в качестве целевого соединения (т.пл.191,0-191,5oС).
Масс-спектр (70eV) m/z (относительная интенсивность) 240 (1, М+), 195 (17), 194 (100), 193 (17), 177 (26), 151 (44), 149 (27), 148 (18), 123 (23), 103 (48), 102 (11), 101 (29), 96 (13), 95 (15), 94 (11), 88 (41), 83 (14), 77 (25), 76 (11), 75 (39), 74 (23), 70 (10), 63 (11), 60 (11), 51 (37), 50 (10).
Пример 8. Получение 3-амино-8-фтор-3,4-дигидро-2Н-1-бензопиран-5-карбoксамида (Соединение (X))
8-Фтор-3-нитро-3,4-дигидро-2Н-1-бензопиран-5-карбоксамид (9,0 г, 37,5 ммоль) растворяют в тетрагидрофуране (100 мл) и абсолютном этаноле (400 мл) и помещают в условия гидрогенирования при атмосферном давлении с использованием никеля Ренея (W-2,9 г) при комнатной температуре. Реакцию завершают через 48 часов, по прошествии указанного времени отфильтровывают катализатор, промывают смесь горячим этанолом и объединенные растворители удаляют в вакууме с получением 7,8 г (99% выход) желтовато-белого твердого вещества. Часть его повторно кристаллизуют из этилацетата с получением белых кристаллов в качестве целевого соединения (т.пл.187-188oС).
Масс-спектр (70eV) m/z (относительная интенсивность) 210 (6, М+), 194 (30), 193 (100), 192 (20), 178 (12).
Пример 9. Получение(R)-3-амино-8-фтор-3,4-дигидро-2Н-1-бензопиран-5-карбоксамида (Соединение R-(XI))
L-(+)-Винную кислоту (7 г, 47 ммоль) растворяют в смеси 30% этанола в воде (300 мл) и нагревают до кипения. Добавляют рацемический амин формулы (X) (8 г, 38 ммоль). Раствор медленно охлаждают до комнатной температуры. Осадок фильтруют и промывают этанолом с получением 4,7 г (65%) слегка коричневых кристаллов (т. пл. 175oС).
[α]
Свободное основание получают добавлением раствора Na2CO3 к суспензии тартрата в этаноле. Смесь фильтруют и растворитель удаляют в вакууме. Остаток растворяют в 200 мл кипящего этилацетата/этанола (95:5) и фильтруют через целит. Раствор выпаривают до тех пор, пока не начинается кристаллизация продукта и с этого момента медленно доводят до комнатной температуры. Осадок отфильтровывают, промывают этилацетатом и сушат на воздухе с получением 1,4 г свободного основания в виде белых кристаллов (т.пл.196oС разлож.).
[α]
Масс-спектр (70еV) m/z (относительная интенсивность) 210 (5, М+), 194 (31), 193 (100), 192 (13), 178 (17), 148 (11).
Пример 10. Получение (S)-3-амино-8-фтор-3,4-дигидро-2Н-1-бензопиран-5-карбоксамида (Соединение S-(XI))
(S)-Энантиомер, полученный из маточного раствора после предыдущего разделения и удаления основания (4 г, 19 ммоль), растворяют в метаноле (20 мл) и добавляют раствор D-(-)-винной кислоты (3 г, 20 ммоль), растворенной в 20 мл метанола (20 мл). Полученное кристаллическое твердое вещество фильтруют и повторно кристаллизуют из раствора 40% этанола в воде (100 мл). Получают 3 г бесцветных кристаллов (т.пл.173oС разлож.).
[α]
[α]
Масс-спектр (70eV) m/z (относительная интенсивность) 210 (4, М+), 194 (32), 193 (100), 192 (12), 178 (16).
Пример 11. Получение (R)-3-N,N-дициклобутиламино-8-фтор-3,4-дигидро-2Н-1-бензопиран-5-карбоксамида (Соединение R-(I))
(R)-3-Амино-8-фтор-3,4-дигидро-2Н-1-бензопиран-5-карбоксамид (0,5 г, 2,4 ммоль) растворяют в безводном метаноле (10 мл), и к указанному раствору добавляют перемешиваемый раствор НОАс (140 кг, 2,4 ммоль), циклобутанона (0,5 г, 7 ммоль) и NaCNBH3 (0,3 г, 5 ммоль). Смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь нагревают до 60oС и добавляют порциями в течение 6 дней дополнительное количество циклобутанона (0,8 г, 11 ммоль), NaCNBH3 (200 мг, 3,2 ммоль) и НОАс (100 мг, 1,4 ммоль). Раствор выпаривают в вакууме, остаток перемешивают с 2М раствора NН3 и затем экстрагируют дважды этилацетатом. Объединенные этилацетатные порции сушат над Na2SO4, фильтруют и растворитель удаляют в вакууме, с получением неочищенного остатка. Хроматография на силикагеле (элюент: этилацетат) дает 0,5 г (82%) целевого соединения в виде белых кристаллов (т.пл.138-139oС).
[α]
Масс-спектр (70eV) m/z (относительная интенсивность) 318 (3, М+), 193 (55), 177 (11), 176 (21), 149 (18), 148 (31), 98 (54), 70 (100), 69 (40), 68 (11), 55 (40), 54 (34), 44 (17), 42 (19), 41 (59), 39 (29).

Изобретение относится к новому способу получения 3-N,N-дициклобутиламино-8-фтор-3,4-дигидро-2Н-1-бензопиран-5-карбоксамида в виде рацемического соединения или R- или S-энантиомеров формулы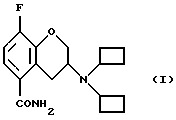
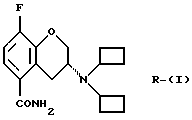

и их фармацевтически приемлемых солей, а также промежуточным продуктам, получаемым и используемым в указанном способе формул V-X: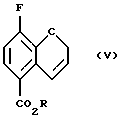

Технический результат - упрощение процесса за счет исключения стадии фторирования, а также за счет того, что этот способ позволяет исключить получение побочного продукта. Этот результат достигается благодаря тому, что в качестве исходного используют соединение, имеющее фтор-заместитель в нужном положении. 6 с. и 10 з.п.ф-лы.

его R-энантиомера (формула R-(I))
и его S-энантиoмера (формула S-(I))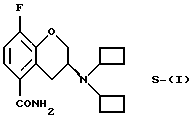
и их фармацевтически приемлемой соли и/или сольвата, включающий следующие реакционные стадии а) эстерификации соединения (II) с получением соединения (III)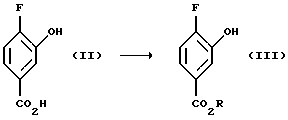
где R - С1-С4алкил
b) пропаргилирования соединения (III) в присутствии основания с получением соединения (IV)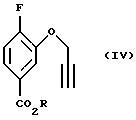
с) нагревания соединения (IV) с получением соединения (V)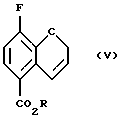
d) гидролиз соединения (V) в присутствии основного или кислотного катализатора с получением соединения (VI)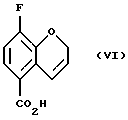
е) взаимодействия соединения (VI) с оксалилхлоридом или тионилхлоридом с последующей обработкой аммиаком с получением соединения (VII)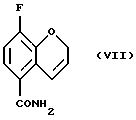
f) взаимодействия соединения (VII) с йодом или нитритной солью с получением соединения (VIII)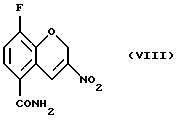
g) взаимодействия соединения (VIII) с восстановителем с получением соединения (IX)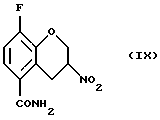
h) восстановление соединения (IX) с получением соединения (X)
i) если желателен (R)- или (S)-энантиомер соединения (I), взаимодействия соединения (X) с соответствующим чистым энантиомером хиральной кислоты с последующей фракционной кристаллизацией и обработкой полученной соли основанием с получением соединения (R)-(XI)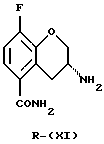
или S-(XI)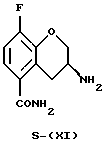
j) алкилирования соединения R-(XI) или S-(XI) с получением соединений формулы R-(I) или S-(I) соответственно или алкилирования соединения (X) с получением рацемического соединения формулы (I), k) необязательной обработки соединений, полученных на стадии j) традиционным способом, с получением их соли или сольвата.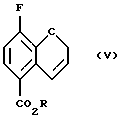
где R - С1-С4 алкил.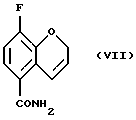
14. Соединение формулы (X)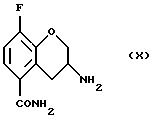
15. Соединение формулы (VIII)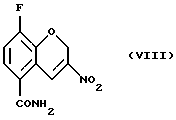
16. Соединение формулы (IX)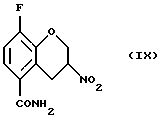
| WO 9511891 A1, 04.05.1995 | |||
| КОЛОДКА ДЛЯ РЕМОНТА ОБУВИ | 0 |
|
SU280269A1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ АДДИТИВНЫЕ СОЛИ КИСЛОТЫ, ИЛИ N-ОКСИД ГЕТЕРОЦИКЛИЧЕСКОГО СОЕДИНЕНИЯ, ИЛИ ЕГО АДДИТИВНАЯ СОЛЬ КИСЛОТЫ | 1992 |
|
RU2047614C1 |

