Изобретение относится к новым соединениям, способам их получения, содержащим их композициям и их применению в качестве нейрозащитных агентов.
Производные тиомочевины и изотиомочевины были описаны ранее для ряда терапевтических применений. WO 94/12165 (Wellcome) описывает простые производные изотиомочевины, применяемые для лечения, в том числе, системной гипотензии, септического шока и воспалительных состояний; в WO 95/09619 (Wellcome) (опубликован после даты приоритета этой заявки) описаны замещенные производные мочевины и изотиомочевины, применяемые для лечения церебральной ишемии; в патенте Великобритании N1178242 (Wellcome) описаны бисизотиомочевины, имеющие противовоспалительную активность; в заявке на Европейский патент N 411615 (Warner Lambert) описаны производные тиомочевины, применяемые для лечения симптомов ухудшения познавательной способности; в заявке на Европейский патент N 392802 (Beecham) описаны производные тиомочевины, применяемые для лечения бронхиальных, цереброваскулярных или нейронных нарушений.
Производные изотиомочевины известны также в качестве химических промежуточных продуктов для получения производных гуанидина (смотри патент Соединенных Штатов 4211867 (McNeil Laboratories) и Synthesis (1988) 6, 460-466 (Rasmussen), в которых описан метиловый эфир 4-диметиламинофенилкарбамимидотиоевой кислоты, и патент Соединенных Штатов 5223498 (Boots).
Производные N-алкоксифенил-N'-хинолинилтиомочевины, пригодные в качестве туберкулостатических агентов, описаны в патенте Германии B-1157626 (Hoechst).
В заявке на Международный патент WO 95/05363 (Fisons) (опубликована после даты приоритета этой заявки) описаны производные N-фениламидина, которые предназначаются для лечения, в том числе, нейродегенеративной болезни.
В настоящем изобретении предложена новая и полезная группы производных изотиомочевины.
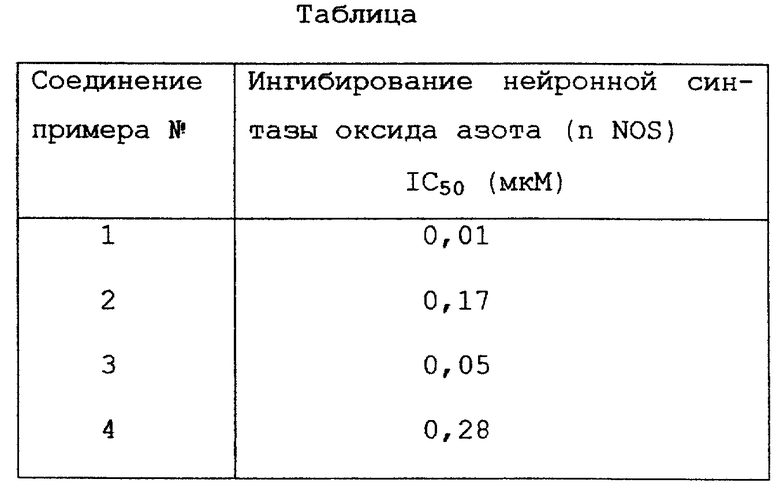
В соответствии с изобретением представляются соединения формулы I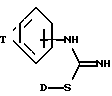 , (I)
, (I)
где D представляет алкил от C1 до C6,
T представляет C3-5 насыщенную или ненасыщенную алкиленовую цепь, замещенную - (CH2)m-NXY; -O-(CH2)2-NH-, замещенную -(CH2)m-NXY, или -U-(CH2)a-N(X)-(CH2)b-;
U представляет NH, O или CH2;
a и b, которые могут быть одинаковыми или разными, равны целому числу от 0 до 3, при условии, что сумма a + b равна от 1 до 3;
X и Y, которые могут быть одинаковыми или разными, представляют водород, C1-C6-алкил или группу -(CH2)nQ,
или NXY вместе представляют пиперидинил, пирролидинил, морфолинил или тетрагидроизохинолинил;
Q представляет фенил или фенил, замещенный одним или несколькими заместителями, выбранными из группы, включающей C1-C6-алкил, C1-C6-алкокси, трифторметил, галоген, нитро и циано; m и n независимо равны целому числу от 0 до 5;
и их фармацевтически приемлемые соли.
Предпочтительно, чтобы T представлял -U-(CH2)a-N (X)-(CH2)b-. В частности, предпочтительно, чтобы T представлял -U-(CH2)a-N(X)-(CH2)b- и U представлял CH2. Наиболее предпочтительно, чтобы T представлял -U-(CH2)a-N(X)-(CH2)b-, U представлял CH2 и каждый из a и b был равен 1.
Предпочтительно, чтобы D представлял C1-C3-алкил, в частности метил или этил, в особенности этил.
Когда T представляет -U-(CH2)a-N(X)- (CH2)a-, предпочтительно, чтобы X представлял водород, метил или группу -CH2Q.
Когда T представляет C3-5 насыщенную или ненасыщенную алкиленовую цепь, замещенную -(CH2)m-NXY, или -O-(CH2)2-NH-, замещенную -(CH2)m-NXY, предпочтительно, чтобы X и Y независимо представляли водород, метил или группу -CH2Q, хотя не является предпочтительным, чтобы X и Y оба представляли группу -CH2Q. В частности предпочтительно, чтобы один из X и Y представлял водород или метил и другой представлял группу -CH2Q.
Предпочтительно, чтобы n равнялся 1.
Предпочтительно, чтобы Q представлял фенил или фенил, замещенный заместителем, выбранным из группы, включающей C1-C6-алкил, C1-C6-алкокси, трифторметил, галоген, нитро и циано. В частности предпочтительно, чтобы представлял фенил или фенил, замещенный C1-C6-алкилом или галогеном.
В настоящем изобретении также представлен способ получения соединений формулы I и их фармацевтически приемлемых солей, который включает:
(а) получение соединения формулы I, где X или по меньшей мере один из X и Y представляет C1-C6-алкил или группу -(CH2)nQ, реакцией соответствующего соединения формулы 1, где X или один или оба из X и Y представляют водород, с соединением формулы II
R'-L, (II)
где R' представляет C1-C6-алкил или группу -(CH2)nQ и L представляет уходящую группу, или
(б) получение соединения формулы I, где T представляет C3-5 насыщенную или ненасыщенную алкиленовую цепь, замещенную -(CH2)m- NXY, или -O-(CH2)2-NH-, замещенную -(CH2)m-NXY, реакцией соответствующего соединения, где T представляет C3-5 насыщенную или ненасыщенную алкиленовую цепь, замещенную -(CH2)m-L, или -O-(CH2)2-NH-, замещенную -(CH2)m-L, и L представляет уходящую группу,
с соединением формулы III
XYNH, (III)
где X и Y принимают значения, определенные выше,
или
(в) реакцию соединения формулы IV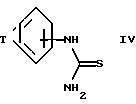 ,
,
где T принимает значения, определенные выше,
с соединением формулы V
D-L, (V)
где D определен выше, и L представляет уходящую группу, и где необходимо или желательно превращение полученного соединение в его фармацевтически приемлемую соль или, наоборот, как описано ниже.
В способах (а) и (б) реакции будут протекать в стандартных условиях, например, реакцией двух исходных реагентов в инертном растворителе в основных условиях при комнатной температуре в течение периода времени вплоть до 12 часов. Обнаружено, что часто амин до реакции его с другим соединением желательно обработать NaH. Подходящие уходящие группы L включают тиоалкил, сульфокислоту, трифторуглеродсульфокислоту, галоген, алкиловые и ариловые спирты и тозиловые группы; другие такие группы перечислены в "Alvanced Organic Chemistry", J.March (1985), 3rd Edition, McGraw-Hill, на странице 315 и хорошо известны в данной области. Предпочтительно, чтобы представлял галоген, в особенности бром.
В способе (в) реакция будет протекать при смешивании двух реагентов в инертном растворителе, например ацетоне. Подходящие уходящие группы, которые может представлять L, включают тиоалкил, сульфокислоту, трифторуглеродсульфокислоту, галоген, алкиловые и ариловые спирты и тозиловые группы; другие такие группы перечислены в "Alvanced Organic Chemistry", J.March (1985), 3rd Edition, McGraw-Hill, на странице 315 и хорошо известны в этой области. Предпочтительно применять иод, толуолсульфонатное или метансульфонатное производное.
Соединения формулы IV можно получить по методу Rasmussen et al. in Synthesis (1988) 456-459. Соединения формулы III можно, таким образом, получить реакцией соединения формулы VI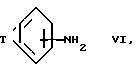
где T такой, как определен выше, с бензоилизотиоцианатом с последующим водно-щелочным расщеплением получаемого производного бензоилтиомочевины.
Соединения формулы VI можно получить восстановлением соответствующего соединения формулы VII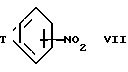 ,
,
где T такой, как определен выше.
Реакцию восстановления можно проводить в различных условиях, например в условиях, описанных в J. March "Advanced Organic Chemistry", 3rd Edition (1985), на странице 1103-1104. Они включают каталитическое гидрирование, применение металла Zn, Sn или Fe, AlH3-AlCl3, сульфидов и других реагентов. Предпочтительно проводить реакцию гидрированием при атмосферном давлении в присутствии катализатора, палладия и угля, в течение обычно 1-4 часов или до завершения восстановления.
Соединения формулы VII, где T такой, как определен выше, или X или, по меньшей мере, один из X и Y представляет C1-C6-алкил или группу -(CH2)nQ, можно получить реакцией соответствующего соединения формулы VII, где X и/или Y представляет водород, с соединением формулы II.
Реакцию можно проводить в условиях, аналогичных условиям, описанным выше для способа (а).
Соединения формулы VII, где T представляет C3-5 насыщенную или ненасыщенную алкиленовую цепь, замещенную - (CH2)m-NXY, или -O-(CH2)2-NH-, замещенную -(CH2)m-NXY, можно получить реакцией соответствующего соединения, где T представляет C3-5 насыщенную или ненасыщенную алкиленовую цепь, замещенную -(CH2)m-L, или -O-(CH2)2-NH-, замещенную -(CH2)m-L, и L представляет уходящую группу, с соединением формулы III.
Реакцию можно проводить в условиях, аналогичных условиям, описанным выше для способа (б).
Соединения формулы VII, где X представляет водород, либо известны, либо их можно получить известными способами. Например, их можно получить нитрованием ненитрованного производного. Эту реакцию нитрования обычно проводят реакцию ненитрованного ароматического соединения, либо только с азотной кислотой или с азотной кислотой в воде, уксусной кислоте, уксусном ангидриде или серной кислоте. Дальнейшие подробности проведения этих реакций и дополнительные альтернативные реагенты указаны в J.March "Advanced Organic Chemistry", 3-е издание (1985), на страницах 468-470.
Соединения формул II, III и V либо известны, либо их можно получить обычными способами, известными per se.
Соединение формулы I можно получить как таковые или в виде кислотно-аддитивных солей типа, описанного выше. Альтернативно, их можно получить в виде фармацевтически неприемлемой аддитивной соли, например соли щавелевой кислоты, и любого продукта, которые можно затем превратить в фармацевтически приемлемую соль обычными способами.
Соли соединений формулы I можно получить реакцией свободной кислоты, основания или его соли с одним или несколькими эквивалентами подходящего основания или кислоты. Реакцию можно проводить в растворителе или среде, в которой соль нерастворима, или в растворителе, в котором соль растворима, например воде, диоксане, этаноле, тетрагидрофуране или диэтиловом эфире, или смеси растворителей, которые можно удалить в вакууме или сушкой вымораживанием. Реакцию можно проводить способом метатезиса или ее можно проводить на ионообменной смоле.
Если необходимо, амин или другие реакционно-способные группы можно защитить с применением защитной группы, как описано в стандартном руководстве "Protecting group in Organic Synthesis", 2nd Edition (1991), by Greene and Wuts. Аминозащитные группы, которые можно упомянуть в частности, включают C2-C7-алкилоксикарбонил, например трет-бутилоксикарбонил, C8-C13-фенилалкилоксикарбонил, например бензилоксикарбонил. Тем не менее предпочтительно защищать аминогруппы обработкой трйфторуксусным ангидридом в подходящем растворителе (например, метиленхлориде, метаноле) при комнатной температуре. Удаление защитной группы можно осуществить гидролизом в воде.
Соединения данного изобретения и промежуточные продукты можно выделить из реакционных смесей стандартными способами.
Термин "C1-C6-алкил" включает неразветвленный, разветвленный насыщенный, ненасыщенный алифатический и циклический алкил, содержащий от 1 до 6 атомов углерода. Подобным образом можно интерпретировать "C1-C3-алкил".
Соединения формулы I могут существовать в энантиомерных формах. Различные оптические изомеры можно выделить разделением рацемической смеси соединений с использованием обычных способов, например фракционной кристаллизацией или ВЭЖХ. Альтернативно, индивидуальные энантиомеры можно получить реакцией подходящих оптически активных исходных материалов в условиях реакции, которые не приводят к рацемизации.
Промежуточные соединения также могут существовать в энантиомерных формах, их можно применять в виде очищенных энантиомеров, диастереомеров, рацематов или смесей.
Соединения общей формулы I обладают полезной фармакологической активностью для животных. В частности, они обладают полезной, ингибирующей синтазу оксида азота активностью, и предполагается, что они полезны при лечении или профилактике болезней или состояний человека, в которых синтез или избыточный синтез оксида азота играет роль в их развитии, таких как гипоксия, например в случаях остановки сердца, удара и неонатальной гипоксии, нейродегенеративные состояния, включающие дегенерацию нервов и/или некроз нервов в таких нарушениях, как ишемия, гипоксия, гипогликемия, эпилепсия и внутренние раны (например, повреждение спинного мозга и тепловой удар), судороги и токсичность, вызванные сверхатмосферным кислородом, и слабоумие, например пресенильное слабоумие, болезнь Альцгеймера и связанное со СПИДом слабоумие, хорея Сиденгама, болезнь Паркинсона, болезнь Гентингтона (Хантингтона), боковой амиотрофический склероз, корсаковский психоз, имбецильность, связанная с нарушением церебральных сосудов, нарушение сна, шизофрения, тревожные состояния, депрессия, сезонное аффективное нарушение, расстройство нормального циркадного ритма, депрессия и другие симптомы, связанные с предменструальным синдромом (ПМС), тревожным состоянием и септическим шоком. Можно также предположить, что соединения формулы I проявляют активность в предупреждении и обратном развитии до предыдущего уровня толерантности к опиатам и диазепинам, лечении привыкания к чрезмерному употреблению лекарственных средств, ослаблении боли и лечении мигрени и других сосудистых головных болей. Соединения настоящего изобретения могут также проявлять полезную иммуносупрессивную активность; могут быть пригодными для лечения или профилактики воспаления, для лечения нарушений перистальтики желудочно-кишечного тракта и для вызывания родов. Соединения могут также быть полезными при лечении раковых болезней, которые экспрессируют синтазу оксида азота.
Предполагается, что соединения формулы I, в частности, полезны для лечения или профилактики нейродегенеративных состояний или мигрени, или для предупреждения или обратного развития до предыдущего уровня толерантности к опиатам и диазепинам, или лечения привыкания к чрезмерному употреблению лекарственных средств, и в особенности для лечения или профилактики нейродегенеративных нарушений. В частности, в настоящем изобретении рассмотрены состояния, выбранные из группы, включающей гипоксию, ишемию, удар и боковой амиотрофический склероз.
Таким образом, в соответствии со следующим аспектом изобретения предложено применение соединения формулы I или его фармацевтически приемлемой соли в качестве фармацевтического препарата.
В соответствии с другим отличительным признаком изобретения предложены применение соединения формулы I или его фармацевтически приемлемой соли для приготовления лекарственного средства для лечения или профилактики вышеупомянутых болезней или состояний; и способ лечения или профилактики любой из вышеупомянутых болезней или состояний, который включает введение терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли человеку, страдающему от такой болезни или состояния или восприимчивому к нему.
Для вышеупомянутых терапевтических показаний вводимая доза будет, конечно, изменяться в зависимости от применяемого соединения, способа введения и желаемого результата лечения. Тем не менее в общем удовлетворительные результаты получают, когда соединения вводят человеку при суточной дозе в виде твердой формы от 1 до 2000 мг в день.
Соединения формулы I и их фармацевтически приемлемые соли можно применять как таковые или в форме подходящих лекарственных препаратов для энтерального или парентерального введения.
В соответствии с изобретением предложена фармацевтическая композиция, содержащая предпочтительно менее 80% и более предпочтительно менее 50% соединения формулы I или его фармацевтически приемлемой соли в смеси с фармацевтически приемлемым разбавителем или носителем.
Предложен также способ получения такой фармацевтической препаративной формы, который заключается в смешивании ингредиентов.
Примерами таких разбавителей и носителей являются: для таблеток и драже: лактоза, крахмал, тальк, стеариновая кислота; для капсул: винная кислота или лактоза; для инъецируемых растворов: вода, спирты, глицерин, растительные масла; для суппозиториев: природные или гидрогенизированные масла или воски.
Композиции в форме, подходящей для перорального введения, например через пищевод, включают: таблетки, капсулы и драже; композиции с пролонгированным действием, включающие композиции, в которых активный ингредиент связан с ионообменной смолой, которую необязательно покрывают диффузионным барьером для модификации высвобождающих свойств смолы.
Предпочтительно, чтобы композиция содержала вплоть до 50% и более предпочтительно вплоть до 25% по массе соединения формулы I или его фармацевтически приемлемого производного.
Фермент синтеза оксида азота имеет ряд изоформ, и для соединения формулы I и его фармацевтически приемлемых солей можно провести скрининг по их активности, ингибирующей синтазу оксида азота, способами, основанными на способах, описанных в Bredt and Snyder in Proc. Natl. Acad. Sci. (1990) and Forstermann et al. , Eur. J.Pharm. (1992) 225, 161-165, следующим образом. Синтаза оксида азота превращает 3H-L-apгинин в 3H-L-цитруллин, который можно отделить катионообменной хроматографией и количественно определить сцинтилляционным счетом.
Скрининг A
(A) Скрининг на ингибирующую активность в отношении нейронной синтазы оксида азота
Фермент выделяют из крысиного гиппокампа или мозжечка. Мозжечок или гиппокамп удаляют у самцов крыс Sprague-Dawlew (250-275 г) после анестезии животного CO2 и декапитации. Супернатант мозжечка или гиппокампа получают гомогенизацией его в 50 мМ Трис-HCl с 1 мМ буфера ЭДТК (этилендиаминтетрауксусная кислота) (pH 7,2 при 25oC) и центрифугированием в течение 15 минут при 20000 g. Остаточный L-аргинин удаляют из супернатанта хроматографией последовательно через колонки с натриевой формой и водородной формой Dowex AG-50W-X8 и дальнейшим центрифугированием при 1000 g в течение 30 секунд.
Для проведения анализа 25 мл конечного супернатанта добавляют в каждую из 96 лунок (фильтрационного планшета на 96 лунок), содержащих 25 мкл раствора L-аргинина (концентрация 1H-L-арнинина 18 мкМ, 3H-L-аргинина 96 нМ) и либо 25 мкл буфера для анализа (50 мМ ГЕПЕС, 1 мМ ЭДТК, 1,5 мМ CaCl2, pH 7,4), либо 25 мкл испытуемого соединения в буфере, при 22oC. В каждую пробирку для испытания для инициирования реакции добавляют 25 мкл полного буфера для анализа (50 мМ ГЕПЕС, 1 мМ ЭДТК, 1,5 нМ CaCl2, 1 мМ ДТТ (дитиотрентол), 100 мкМ НАДФН (никотинамидаденин-динуклеотидфосфат, восстановленный), 10 мкг/мл калмодулина, pH 7,4), и реакцию останавливают через 10 минут добавлением 200 мкл суспензии буфера для остановки реакции (20 мМ ГЕПЕС, 2 мМ ЭДТК, pH 5,5) и Dowex AG-50W-X8 200-400 меш.
Меченый L-цитруллин отделяют от меченого L-аргинина фильтрованием каждого фильтрационного планшета и 75 мкл каждой реакционной смеси с завершенной реакцией добавляют к 3 мл коктейля для сцинтилляционного счета. L-цитроллин затем количественно определяют сцинтилляционным счетом.
В типичном эксперименте с использованием церебрального супернатанта базальная активность пробы повышается на 20000 распадов в минуту/мл относительно контроля на реагенты, который имеет активность 7000 распадов в минуту/мл. Для проверки методики в данном анализе испытывали контрольный стандарт, N-нитро-L-аргинин, который дает 80% ингибирование синтазы оксида азота при концентрации 1 мкМ.
Скрининг Б
(Б) Скрининг на ингибирующую активность в отношении индуцирующей синтазы оксида азота
Фермент получают после индуцирования из культурированной клеточной линии карциномы ободочной и прямой кишки человека, DLD-1 (получена из Европейской коллекции клеточных культур животных). Клетки DLD-1 культурируют в среде RPMI 1640, дополненной 10%-ной фетальной бычьей сывороткой, 4 мМ L-глутамина и антибиотиками (100 единиц/мл пенициллина G, 100 мкг/мл стрептомицина и 0,25 мкг/мл амфотерицина В) и 100 мкг/мл канамицина. Клетки выращивают, как обычно, в колбах объемом 225 см3, содержащих 35 мл среды, которую выдерживают при 37oC, и в увлажненной атмосфере, содержащей 5% CO2.
Синтаза оксида азота продуцируется клетками в ответ на интерферон - около 250 Е/мл IL- 1, 1000 Е/мл IFNγ, 200 Е/мл IL-6 и 2-00 Е/мл TNF-альфа. После периода культивирования 17-20 часов сбор клеток выполняют соскребанием клеточного пласта с поверхности колбы в культуральную среду. Клетки собирают центрифугированием (1000 г, в течение 10 минут) и лизат получают добавлением к клеточному осадку раствора, содержащего 50 нМ Трис-HCl (pH 7,5 при 20oC), 10% (об./об.) глицерина, 0,1% (об./об.) Тритона-X-100, 0,1 мкМ дитиотреитола и смеси ингибиторов протеазы, содержащей лейпептин (2 мкг/мл), ингибитор трипсина соевых бобов (10 мкг/мл), апротинин (5 мкг/мл) и фенилметилсульфонилфторид (50 мкл/мл).
Для проведения анализа 25 мл субстратной смеси (50 мМ Трис-HCl (pH 7,5 при 20oC), 400 мкМ НАДФН, 20 мкМ флавинаденин-динуклеотида, 20 мкМ флавинмононуклеотида, 4 мкМ тетра-гидробиоптерина, 12 мкМ L-аргинина и 0,025 мкКюри L-[3H] - аргинина) добавляют в лунки фильтрационного планшета на 96 лунок (размер пор 0,45 мкМ), содержащие 25 мкл раствора испытуемого соединения, в 50 мМ Трис-HCl. Реакцию начинают добавлением 50 мкл лизата клеток (получен, как указано выше) и после инкубации в течение 1 часа при комнатной температуре останавливают добавлением 50 мкл водного раствора 3 мМ нитроангинина и 21 мМ ЭДТК.
Меченый L-цитруллин отделяют от меченого L-аргинина при помощи Dowex AG-50W. К анализируемой смеси добавляют 150 мкл 25%-ной водной суспензии DOWEX 50W (Na+-форма), после чего ее фильтруют в планшеты на 96 лунок. Отбирают пробы по 70 мкл фильтрата и добавляют в лунки планшета на 96 лунок, содержащие твердый сцинтиллянт. После сушки проб L-цитруллин количественно определяют сцинтилляционным счетом.
В типичном эксперименте базальная активность составляет 300 распадов в минуту на 70 мкл пробу, которая возрастает до 1900 распадов в минуту в контроле на реагент. Аминогуанидин, который дает IC50 (концентрация 50% ингибирования) при 10 мкМ, испытывали в качестве стандарта для проверки методики.
Скрининг В
(В) Скрининг на ингибирующую активность в отношении эндотелиальной синтазы оксида азота
Фермент можно выделить из эндотелиальных клеток умбиликальной вены человека (HUVECs) методикой, основанной на методике Pollock et al. (1991) Proc. Nat. Acad. Sci., 88, 10480-10484. HUVECs покупали у Clonetics Corp. (San Diego, CA, USA) и культурировали до конфлюэнции. Клетки можно хранить для пассирования 35-40 раз без значительной потери выхода синтазы оксида азота. Когда клетки достигают конфлюэнции, их ресуспендируют в физиологическом растворе, забуференном фосфатом Дульбекко, центрифугируют при 800 об./минуту в течение 10 минут, клеточный осадок гомогенизируют в охлажденном льдом 50 мМ Трис-HCl, 1 мМ ЭДТУ, 10% глицерина, 1 мМ фенилметилсульфонилфторида, 2 мкМ лейпептина при pH 4,2. После центрифугирования при 34000 об./минуту осадок солюбилизируют в буфере для гомогенизации, который содержит также 20 мМ CHAPS. После 30 мин инкубирования на льду суспензию центрифугируют при 34000 об. /мин в течение 30 минут. Получаемый супернатант сохраняют при -80oC до использования.
Для проведения анализа 25 мкл полученного супернатанта добавляют в каждую лунку фильтрационного планшета на 96 лунок, содержащую 25 мкл раствора L-аргинина (концентрация 1H-L-аргинина 12 мкМ, 3H-L-аргинина 64 нМ) и либо 25 мкл буфера для анализа (50 мМ ГЕПЕС, 1 мМ ЭДТК, 1,5 мМ CaCl2, pH 7,4), либо 25 мкл испытуемого соединения в буфере, при 22oC. В каждую лунку для инициирования реакции добавляли 25 мкл полного буфера для анализа (50 мМ ГЕПЕС, 1 мМ ЭДТК, 1,5 мМ CaCl2, 1 мМ ДТТ, 100 мкМ НАДФН, 10 мкг/мл калмодулина, 12 мкМ тетрагидробиоптерина, pH 7,4) и реакцию останавливали через 30 минут добавлением 200 мкл 50%-ной суспензии буфера для остановки реакции (20 мМ ГЕПЕС, 2 мМ ЭДТК, pH 5,5) и Dowex AG-50W-X8 200-400 меш.
Меченый L-цитруллин отделяют от меченого L-аргинина фильтрованием в другой планшет на 96 лунок и 75 мкл каждой реакционной смеси с завершенной реакцией добавляют к 3 мл коктейля для сцинтилляционного счета. L-Цитруллин затем определяют количественно сцинтилляционным счетом.
В типичном эксперименте базальная активность образца повышается на 5000 распадов в минуту/мл относительно контроля на реагент, который имеет активность 1500 распадов в минуту/мл. Контрольный стандарт, N-нитро-L-аргинин, который дает 70-90% ингибирование синтазы оксида азота при концентрации 1 мкМ, испытывали в анализе для проверки методики.
Для определения степени проникновения в мозг соединения можно также испытывать в анализе ex vivo.
Скрининг Г
(Г) Анализ ex vivo на ингибирующую активность в отношении нейронной синтазы оксида азота
Самцам крыс Sprague-Dawley (250-275 г) внутривенно вводили дозу испытуемого соединения 10 мг/кг, растворенную в 0,9%-ном солевом растворе, или только солевой раствор в качестве контрольного опыта. Через заданное время (обычно 2-24 часа) после обработки животных умерщвляли, мозжечок удаляли, приготовляли супернатант и анализировали его для определения активности синтазы оксида азота, как описано в Скрининге А.
В качестве дополнительного подтверждающего испытания фракцию супернатанта мозжечка вводили в колонку с сефаразой 2',5'-ADP (которая связывает синтазу оксида азота) и затем элюировали НАДФН. Элюент испытывали на активность синтазы оксида азота по методике Скрининга А.
Соединения, которые проникают в головной мозг крыс и ингибируют нейронную синтазу оксида азота, приводят к снижению активности синтазы оксида азота как в препарате супернатанта, так и в элюенте из колонки с сефарозой 2', 5'-ADP.
В скринингах на ингибирующую активность в отношении синтазы оксида азота активность соединения выражают как IC50 (концентрация лекарственного вещества, которая дает 50% ингибирование фермента в анализе). Величины IC50 для испытуемых соединений сначала оценивали по ингибирующей активности растворов соединений с концентрацией 1, 10 и 100 мкМ. Соединения, которые ингибировали фермент по меньшей мере на 50% при 10 мкМ, испытывали повторно с применением более подходящих концентраций так, чтобы можно было определить IC50.
В приведенном выше Скрининге А (скрининг на активность в отношении нейронной изоформы синтазы оксида азота) соединение Примера 1, приведенного ниже, показало величину IC50 менее 10 мкМ, которая доказывает, что соединение, как ожидалось, проявляет полезную терапевтическую активность. В Скринингах Б и В (скрининги на активность в отношении макрофаговых и эндотелиальных изоформ синтазы оксида азота) соединение Примера 1 показало величины IC50, более чем в 10 раз превышающие величину, полученную в Скрининге А, которые доказывают, что оно проявляет желательную селективность.
Соединение Примера 2 также испытывали в Скрининге А, и оно имело величину IC50 менее 10 мкМ. Таким образом, также ожидают, что это соединение проявляет полезную терапевтическую активность.
При сравнении с соединениями, известными из уровня техники, соединения формулы I и их фармацевтически приемлемые соли имеют преимущество, заключающееся в том, что они менее токсичны, более эффективны, имеют более длительный период действия, имеют более широкий диапазон активности, имеют большую потенцию, более селективны для нейронных изоформ фермента синтеза оксида азота, вызывают меньшее побочное действие, легче абсорбируются или могут иметь другие полезные фармакологические свойства.
Изобретение иллюстрируется, но никоим образом не ограничивается следующими примерами.
Пример 1
Этиловый эфир N-(1,2,3,4-тетрагидроизохинолин-7-ил) карбамимидотиовой кислоты
(а) 2-(7-Амино-1,2,3,4-тетрагидроизохинолин)-2,2,2-трифторацетамид
К раствору 4,21 г (23,6 ммоль) 7-нитро-1,2,3,4- тетрагидроизохинолина и 3,6 мл (26 ммоль) триэтиламина в 100 мл метиленхлорида при 0oC добавляли 3,5 мл (25 ммоль) трифторуксусного ангидрида. Реакционную смесь перемешивали в течение ночи. Реакционную смесь экстрагировали разбавленной соляной кислотой. Водную фазу подщелачивали и экстрагировали метиленхлоридом. Высушенная органическая фаза (сульфат магния) давала N-(7-нитро-1,2,3,4-тетрагидроизохинолин)трифторацетамид в виде желтого твердого продукта. Соединение сразу обрабатывали 200 мл этанола, добавляли 0,50 г 5%-ного палладия на угле и смесь гидрировали в аппарате для гидрирования Парра при 3,164 (45 psi) атм в течение 1,5 часа. Катализатор удаляли фильтрованием и растворитель выпаривали. Остаток растирали со 100 мл петролейного эфира, получая 5,45 г (95%) соединения, указанного в заглавии в виде серого твердого продукта, т.пл. 61-3oC.
(б) 1,2,3,4-тетрагидроизохинолин-7-тиомочевина
К раствору 1,3 мл (9,7 ммоль) бензоилизотиоцианата в 13 мл ацетона при кипячении с обратным холодильником быстро добавляли 1,25 г (5,12 ммоль) 2-(7-амино-1,2,3,4-тетрагидроизохинолин) трифторацетамида с такой скоростью, чтобы регулировать кипячение с обратным холодильником. После завершения добавления реакционную смесь перемешивали в течение 3 ч. После охлаждения твердую часть собирали и промывали 30 мл ацетона, получая 1,74 г (83%) промежуточной 1-бензоил-3-[2-(2,2,2- трифторацетамид)-1,2,3,4-тетрагидроизохинолин-7-тиомочевины в виде не совсем белого твердого продукта. Это соединение сразу добавляли к 20 мл 5%-ного раствора гидроксида натрия и получаемый раствор нагревали при 80oC в течение 1 часа. После охлаждения до комнатной температуры раствор фильтровали, получая 0,78 г (74%) соединения, указанного в заглавии, т.пл. 198-203oC.
(в) Этиловый эфир N-(1,2,3,4-тетрагидроизохинолин-7ил) карбамимидотиовой кислоты
К суспензии 0,75 г (3,61 ммоль) 1,2,3,4-тетрагидроизохинолин-7- тиомочевины в 10 мл изопропанола добавляли 0,35 г (3,7 ммоль) метансульфокислоты в 2 мл изопропанола. Реакционную смесь перемешивали в течение 0,25 часа с последующим добавлением 0,85 мл (8,4 ммоль) этилметансульфоната. Реакционную смесь нагревали при кипячении с обратным холодильником в течение 4 ч. Растворитель выпаривали в вакууме, получая масло, которое растворяли в 100 мл воды. Водную фазу подщелачивали насыщенным бикарбонатом натрия и водный слой экстрагировали 8 раз 100 мл метиленхлорида. Объединенные экстракты сушили над сульфатом магния и концентрировали, получая 0,61 г масла, которое затвердевало при стоянии. Колоночная хроматография на силикагеле с применением 10%-ного метанола в хлороформе, насыщенном аммиаком, давала 0,45 г (53%) соединения, указанного в заглавии в виде белого твердого продукта, МС 236 (M+H).
Пример 2
Этиловый эфир N-5(-2-(((3-хлорфенил)метил)(метил)амино)инданил) карбамимидотиовой кислоты
(a) 2- ((3-Хлорфенил) карбонил) амино-5-нитроиндан
К гидрохлориду 2-амино-5-нитроиндана (1,5 г, 7,0 ммоль) в метиленхлориде (50 мл) при 0oC добавляли триэтиламин (2,1 мл, 15,0 ммоль), затем 3-хлорбензоилхлорид (1,0 мл, 7,5 ммоль) Смесь сразу выливали в воду, слои разделялись. Водный слой экстрагировали метиленхлоридом (2 х 20 мл) и объединенные экстракты промывали водой, сушили над MgSO4, фильтровали и концентрировали до образования масла, которое было гомогенным при разделении ТСХ и которое сразу использовали в следующей стадии: МС (M+H)+ = 317.
(б) 2-((3-Хлорфенил)метил)амино-5-нитроиндан
2-((3-Хлорфенил)карбонил)амино-5-нитроиндану (2,2 г, 7,0 ммоль) в ТГФ (75 мл) добавляли по каплям BH3•ТГФ (1,0 М, 35 мл, 35 ммоль). Смесь кипятили с обратным холодильником в течение 12 ч, охлаждали до 0oC, прекращали реакцию 4 н. HCl (60 мл) и кипятили с обратным холодильником в течение 1 ч. Полученный раствор выпаривали до образования масла, подщелачивали 50%-ным NaOH и экстрагировали метиленхлоридом (3 х 20 мл). Объединенные экстракты промывали водой, сушили над MgSO4, фильтровали и концентрировали до масла. Обработкой ИПС (изопропиловый спирт)/HCl получали 2-((3-хлорфенил)-метил)амино-5-нитроиндан (2,1 г, выход 88% двух стадий); т.пл. 234-237oC.
(в) 2-((3-Хлорфенил)метил)(метил)амино-5-нитроиндан
К 2-((3-хлорфенил)метил)амино-5-нитроиндану (4,4 г, 14,5 ммоль) в муравьиной кислоте (5,5 мл) добавляли формальдегид (12 мл). Смесь нагревали для кипячения с обратным холодильником в течение 30 минут, охлаждали, нейтрализовали 2 н. NaOH и экстрагировали этилацетатом (3 х 70 мл). Объединенные экстракты промывали водой, сушили над MgSO4, фильтровали и концентрировали до образования масла: (4,2 г, 91%); МС (M+H)+ = 317.
(г) 2-((3-Хлорфенил)метил)(метил)амино-5-аминобензол
К 2-((3-хлорфенил)метил)(метил)амино-5-нитроиндану (4,3 г, 13,6 ммоль) в 85% AcOH/H2O (100 мл) добавляли металлический цинк (7,1 г, 109,0 ммоль). Смесь перемешивали в течение 5 мин, фильтровали через целит и выпаривали до образования масла. Масло выливали в основную воду и экстрагировали этилацетатом (3 х 100 мл). Объединенные экстракты промывали водой, сушили над MgSO4, фильтровали и концентрировали до образования масла (3,6 г, 92%); МС (M+H)+ = 287.
(д) 5-(2-(((3-Хлорфенил)метил)(метил)амино)инданил)-1-бензоил- 2-тиомочевина
К раствору бензоилизотиоцианата (2,7 г, 16,5 ммоль) в 15 мл сухого ацетона, предварительно нагретого до очень спокойного кипячения с обратным холодильником, быстро добавляли 2-((3- хлорфенил)метил)(метил)амино-5-аминобензол (3,6 г, 12,4 ммоль), растворенный в 10 мл сухого ацетона, с такой скоростью, чтобы можно было регулировать энергичное кипячение с обратным холодильником. Реакционную смесь кипятили с обратным холодильником в течение 30 минут, выливали при энергичном перемешивании в лед и экстрагировали этилацетатом (3 х 100 мл). Объединенные экстракты промывали водой, сушили над MgSO4, фильтровали и концентрировали до образования твердого продукта, который перекристаллизовали из ИПС (изопропиловый спирт): (3,12 г, 58%); т.пл. 128-130oC.
(е) 5-(2-(((3-Хлорфенил)метил)(метил)амино)инданил)-2- тиомочевина
Смесь 5-(2-(((3-хлорфенил)метил)(метил)амино)инданил)- 1-бензоил-2-тиомочевины (3,1 г, 7,12 ммоль) и 40 мл 2,5 н. водного гидроксида натрия нагревали при 90o в течение 35 минут с перемешиванием. Теплую реакционную смесь выливали в 60 мл воды при перемешивании. Продукт экстрагировали тремя порциями метиленхлорида. Объединенные экстракты промывали водой, сушили над сульфатом магния и концентрировали досуха. Остаток хроматографировали на силикагеле (этилацетат/ гексан, 8:1) и концентрировали до образования масла: (2,2 г, 93%); МС (M+H)+ = 246.
(ж) Этиловый эфир N-5(-2-(((3-хлорфенил)метил)(метил)амино)- инданил)карбамимидотиовой кислоты
5-(2-(((3-Хлорфенил)метил)(метил)амино)инданил-2-тиомочевину (2,2 г, 6,33 ммоль) суспендировали в 20 мл этанола и смесь обрабатывали 0,41 мл метансульфокислоты и 1,35 мл этилметансульфоната. Смесь кипятили с обратным холодильником в течение 4 ч, выпаривали, подщелачивали насыщенным бикарбонатом и экстрагировали метиленхлоридом (3 х 30 мл). Объединенные экстракты промывали водой, сушили над MgSO4, фильтровали и концентрировали до образования масла, которое растворяли в этилацетате и обрабатывали смесью ИПС/HCl. Твердую часть отделяли фильтрованием и промывали ИПС: (2,40 г, 83%); т.пл. > 150oC с разложением.
Пример 3
Дигидрохлорид этилового эфира N-(2-метил-1,2,3,4-тетрагидроизохинолин-7ил)карбамимидотиовой кислоты
(а) Гидрохлорид 7-нитро-2-метил-1,2,3,4-тетрагидроизохинолина
Раствор 4,00 (18,7 ммоль) 7-нитроизохинолина в 10 мл муравьиной кислоты и 17 мл 38%-ного водного формальдегида нагревают при кипячении с обратным холодильником в течение 1 ч. Реакционную смесь охлаждали, выливали в лед и подщелачивали водным аммиаком. Смолистый остаток, который осаждался, экстрагировали два раза метиленхлоридом. Высушенную (сульфатом магния) органическую фазу концентрировали, получая сырой 7-нитро-2-метил-1,2,3,4- тетрагидроизохинолин в виде густого масла. Это масло сразу обрабатывали этанолом (50 мл) и добавляли раствор соляной кислоты в этаноле до тех пор, пока раствор не был отчетливо кислотным по лакмусу. Для индуцирования осаждения добавляли эфир и полученный твердый продукт собирали, получая 3,99 г (93%) соединения, указанного в заглавии в виде желтого твердого вещества, т.пл. 236-8oC (разложение).
(б) Гидрохлорид 7-амино-2-метил-1,2,3,4-тетрагидроизохинолина
Суспензию 3,98 г (17,5 ммоль) гидрохлорида 7-нитро-2-метил-1,2,3,4-тетрагидроизохинолина и 0,4 г 10%-ного палладия на угле в 200 мл этанола гидрировали при 3,515 атм в течение 2 ч. Катализатор отделяли фильтрованием и промывали небольшим количеством воды. Фильтрат концентрировали, получая водный раствор. Добавляли абсолютный этанол и выпаривали, удаляя избыток воды, до образования твердого продукта. Этот твердый продукт растворяли в горячем этаноле (60 мл) и для инициирования кристаллизации медленно добавляли эфир. Продукт собирали, получая 3,38 г (97%) соединения, указанного в заглавии в виде не совсем белого твердого вещества, т.пл. 114-9oC.
(в) 2-Метил-1,2,3,4-тетрагидроизохинолин-7-тиомочевина
Раствор 3,88 г (19,5 ммоль) гидрохлорида 7-амино-2-метил-1,2,3,4-тетрагидроизохинолина в 100 мл воды подщелачивали раствором карбоната калия и экстрагировали дважды метилен-хлоридом. Высушенную (сульфатом магния) органическую фазу концентрировали, получая 3,13 г (99%) свободного основания в виде масла. Это масло обрабатывали ацетоном (75 мл) и добавляли 2,21 г (19,4 ммоль) трифторуксусной кислоты в 100 мл ацетона. Раствор нагревали для кипячения с обратным холодильником, по каплям добавляли 5,2 мл (39 ммоль) бензилизоцианата. Реакционную смесь нагревали в течение 1 ч до охлаждения до комнатной температуры. Растворитель удаляли в вакууме и получаемое масло обрабатывали метанолом (150 мл) и 2,5 М гидроксидом натрия (50 мл). Раствор нагревали при 65oC в течение 1 ч до охлаждения до комнатной температуры. Метанол выпаривали в вакууме и водный раствор охлаждали для осаждения продукта. Твердый продукт собирали, получая 2,22 г соединения, указанного в заглавии в виде бледно-желтого твердого продукта, т.пл. 184-6oC. Получали также вторую порцию соединения, указанного в заглавии (0,79 г, общий выход 69%).
(г) Дигидрохлорид этилового эфира N-(2-метил- 1,2,3,4-тетрагидроизохинолин-7-ил)карбамимидотиовой кислоты
К суспензии 0,88 г (4,0 ммоль) 2-метил-1,2,3,4-тетрагидроизохинолин- 7-тиомочевины в 8 мл изопропанола добавляли 0,39 г (3,9 ммоль) метансульфокислоты. Раствор нагревали при кипячении с обратным холодильником в течение 0,5 ч для обеспечения образования метансульфонатной соли, где как соль, так и свободное основание нерастворимы в изопропаноле. К этому раствору добавляли 1,5 мл (14 ммоль) этилметансульфоната и нагревание продолжали в течение ночи, что позволяло получить прозрачный раствор. Растворитель удаляли в вакууме и полученное масло обрабатывали водой, подщелачивали карбонатом калия и экстрагировали два раза метиленхлоридом. Высушенную (сульфатом магния) органическую фазу концентрировали, получая масло. Это масло обрабатывали этанолом и подкисляли соляной кислотой в этаноле до достижения отчетливо кислотной реакции по лакмусу. Добавляли эфир и соль выделяли в виде вязкого масла. Растворитель декантировали и масло промывали несколько раз эфиром. Масло обрабатывали водой (250 мл) и раствор обрабатывали обесцвечивающим углем. Раствор фильтровали и фильтрат разбавляли водой до объема 500 мл. Этот раствор сушили вымораживанием, получая 1,06 г (78%) соединения, указанного в виде моногидрата. МС (Cl) 250 (M+H); ЯМР (ДМСО/D2O) 7,33 (д, 1H), 7,21 (д, 1H), 7,17 (с, 1H), 4,36 (широкий с, 2H), 3,0-3,6 (м, 6H), 3,17 (с, 3H), 1,30 (т, 3H).
Пример 4
Дигидрохлорид метилового эфира N-(2-метил-1,2,3,4- тетрагидроизохинолин-7-ил)карбамимидотиовой кислоты
К суспензии 1,00 г (4,52 ммоль) 2-метил-1,2,3,4-тетрагидроизохинолин-7-тиокарбамида (Пример 3, стадия (в)) в 10 мл изопропанола добавляли 0,44 г (4,5 ммоль) метансульфокислоты. Раствор перемешивали при комнатной температуре в течение 2 ч для обеспечения образования метансульфонатной соли, где как соль, так и свободное основание были нерастворимы в изопропаноле. К этому раствору добавляли 6,7 г (47 ммоль) метилиодида и реакционную смесь перемешивали в течение ночи. Растворитель выпаривали в вакууме и остаток растворяли в воде, обрабатывали обесцвечивающим углем и фильтровали, что давало прозрачный, бесцветный водный раствор. Этот раствор подщелачивали карбонатом калия и экстрагировали дважды метиленхлоридом. Органические фазы объединяли, сушили (сульфатом магния) и концентрировали в вакууме, получая 1,02 г (96%) продукта в виде свободного основания. Это масло обрабатывали этанолом и подкисляли до получения явной кислой реакции добавлением соляной кислоты в этаноле. Добавление избытка эфира вызывает выделение соли в виде масла. Растворитель декантировали и масло промывали несколько раз эфиром. Масло обрабатывали 250 мл воды и снова обрабатывали обесцвечивающим углем. Раствор фильтровали и фильтрат разбавляли водой до объема 500 мл. Раствор сушили вымораживанием, получая 0,70 г соединения заглавия в виде белого твердого продукта. МС (Cl) 236 (M+H). ЯМР (ДМСО/D2O) 11,6-11,9 (широкий, 1H), 9,4-9,7 (широкий, 1H), 7,36 (д, 1H), 7,24 (д, 1H), 7,18 (с, 1H), 4,2-4,6 (широкий м, 2H), 3,0-3,7 (широкий м, 4H), 2,87 (с, 3H), 2,70 (с, 3H).
Данные по биологической активности
Тест проводили по методике, описанной выше (см. Скрининг А).
Полученные данные представлены в таблице.
Описываются новые бициклические производные изотиомочевины общей формулы I, где D представляет C1-C6-алкил; Т представляет C3-C5 насыщенную алкиленовую цепь, замещенную - (CH2)m-NXY или U-(CH2)a-N(X)-(CH2)b; U представляет CH2, a и b, которые могут быть одинаковыми или разными, равны целому числу от 0 до 3, при условии, что сумма a + b равна от 1 до 3; X и Y, которые могут быть одинаковыми или разными, представляют водород, C1-C6-алкил или группу - (CH2)nQ; Q представляет фенил, необязательно замещенный C1-C6-алкилом, C1-C6-алкокси, трифторметилом, галогеном; m и n независимо равны целому числу от 0 до 5, и его фармацевтически приемлемые соли. Описывается также способ их получения и композиции, содержащие их. Соединения формулы I будут пригодны, в частности, для лечения нейродегенеративных нарушений. 3 с. и 5 з.п. ф-лы, 1 табл.
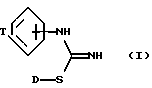
где D представляет C1 - C6-алкил;
T представляет C3 - C5 насыщенную алкиленовую цепь, замещенную -(CH2)m-NXY или U-(CH2)a-N(X)-(CH2)b;
U представляет CH2;
a и b, которые могут быть одинаковыми или разными, равны целому числу от 0 до 3, при условии, что сумма a + b равна от 1 до 3;
X и Y, которые могут быть одинаковыми или разными, представляют водород, C1 - C6-алкил или группу -(CH2)nQ;
Q представляет фенил, необязательно замещенный C1 - C6 алкилом, C1 - C6 алкокси, трифторметилом, галогеном;
m и n независимо равны целому числу от 0 до 5,
и его фармацевтически приемлемые соли.
где T имеет значения, определенные в п.1,
подвергают взаимодействию с соединением формулы V
D - L,
где D такой, как определен в п.1;
L представляет уходящую группу,
с последующим, в случае необходимости или по желанию, превращением полученного соединения в его фармацевтически приемлемую соль или наоборот.
| 0 |
|
SU266949A1 | |
| Галоидгидраты @ -метиларалкилизотиомочевины,проявляющие вазоактивное действие | 1976 |
|
SU599502A1 |
| СПОСОБ ОБЖИГА ПОДИНЫ АЛЮМИНИЕВОГО ЭЛЕКТРОЛИЗЕРА | 0 |
|
SU358416A1 |
