Изобретение относится к некоторым имидазохиноксалинам, которые избирательно связываются с ГАМК-рецепторами. Соединения согласно настоящему изобретению могут использоваться при лечении беспокойства или страха, нарушений сна, эпилептических припадков, последствий передозировки бензодиазепиновых лекарственных средств и состояний повышенной тревоги. Описывается взаимодействие имидазохиноксалинов настоящего изобретения с сайтом связывания ГАМК, бензодиазепиновым рецептором. Данное взаимодействие приводят в результате к фармакологической активности указанных соединений.
γ-Аминомасляная кислота (ГАМК) считается одним из главных ингибирующих аминокислотных переносчиков в головном мозге млекопитающих. Наличие этих медиаторов в головном мозге было уже установлено свыше 30 лет назад (Roberts Frankel, J. Biol. Chem. 187: 55-63, 1950; Udenfriend, J. Biol. 187: 65-69, 1950). С того времени множество работ было посвящено исследованиям причастности ГАМК к этиологии эпилептических припадков, расстройств сна, тревожных состояний и расстройств познавательной способности (Tallman и Gallager, Ann. Rev. Neuroscience 8: 21-44, 1985). Считается, что широко распространенная, хотя и неодинаково, по всему головному мозгу млекопитающего ГАМК является переносчиком приблизительно в 30% синапсов головного мозга. В большинстве областей головного мозга ГАМК ассоциирована с локальными ингибиторными нейронами, и лишь в двух областях ГАМК ассоциирована с более длинными отростками. ГАМК медиирует многие из своих действий при помощи комплекса белков, локализованных как на телах клеток, так и на нервных окончаниях, они называются ГАМК-рецепторами. Постсинаптические ответные реакции на ГАМК медиируются посредством изменения хлоридной проводимости, которая обычно, хотя и не всегда, приводит к гиперполяризации клетки. Проведенные недавно исследования показали, что комплекс белков, ассоциированных с постсинаптическими ГАМК-ответными реакциями, является основным участком действия для многих структурно отличающихся соединений, способных к модификации постсинаптических ответных реакций на ГАМК. В зависимости от типа взаимодействия эти соединения способны продуцировать широкий спектр активностей (либо седативную, транквилизирующую и противосудорожную, либо возбуждающую и стимулирующую тревожные состояния и эпилептические припадки активность).
1,4-Бензодиазепины до сих пор остаются одними из наиболее широко применяемых лекарственных средств в мире. Среди них наиболее известными лекарственными препаратами являются хлордиазепоксид, диазепам, флуразепам и триазолам. Эти соединения широко используются в качестве транквилизаторов, седативно-снотворных средств, миорелаксантов и противосудорожных средств. Ряд этих соединений является чрезвычайно сильными лекарственными средствами; и такая активность указывает на наличие сайта высокоаффинного и специфического действия для отдельных рецепторов. Ранние электрофизиологические исследования показали, что основная активность бензодиазепинов направлена на повышение ГАМК эргического ингибирования. Бензодиазепины обладают способностью повышать предсинаптическое ингибирование моносинаптического рефлекса брюшных корешков, медиатором которого является ГАМК (Schmidt и др. 1967, Arch. Exp. Path. Pharmacol. 258: 69-82). Все последующие электрофизиологические исследования (Tallman и др. 1980, Science 207: 274-281, Haefley и др. 1981, Handb. Exp. Pharmacol. 33: 95-102) подтвердили эти наблюдения, а в середине 1970-х годов электрофизиологи пришли к общему соглашению, что бензодиазепины могут усиливать действие ГАМК.
С открытием "рецептора" для бензодиазепинов и последующим определением природы взаимодействия между ГАМК и бензодиазепинами стало очевидным, что функционально важные взаимодействия бензодиазепинов с различными нейротрансмиттерными системами в значительной степени обусловлены повышенной способностью самой ГАМК к модификации этих систем. Каждая модифицированная система, в свою очередь, может быть ассоциирована с экспрессией поведенческой функции.
Исследования природы механизма указанных взаимодействий зависят от обнаружения сайта высокоаффинного связывания бензодиазепина (рецептора). Такой рецептор присутствует в ЦНС всех позвоночных, которые филогенетически являются более поздними видами, чем рыбы Squieres Braestrup 1977, Nature 166: 732-734, Mohler Okada, 1977, Science, 198: 854-861, Mohler Okada, 1977, Br. J. Psychiatry 133: 261-268). С помощью использования диазепама, меченного тритием, и многих других соединений было показано, что указанные сайты связывания бензодиазепина удовлетворяют многим критериям фармакологических рецепторов; и связывание с этими сайтами in vitro является быстрым, обратимым, стереоспецифическим и способным к насыщению. И что более важно, была проиллюстрирована особенно тесная связь между способностью бензодиазепинов вытеснять диазепам из его сайта связывания и активностью ряда животных в поведенческих тестах на эффективность бензодиазепина (Braestrupe Squires, 1978, Br. J. Psychiatry, 133: 249-260, Mohler Okada, 1977, Science 198: 854-5, Mohler Okada, 1977, Br. J. Psychiatry, 133: 261-268). Средние терапевтические дозы указанных лекарственных средств для человека также коррелируют с рецепторной активностью (Tallman и др. 1980, Science, 207: 274-281).
В 1978 году было установлено, что ГАМК и ее аналоги могут взаимодействовать в сайте низкоаффинного связывания (1 мкМ) ГАМК с повышением связывания бензодиазепина с клоназепам-чувствительным сайтом (Tallman и др. 1978, Nature, 274: 383-385). Это повышение может быть вызвано увеличением аффинности сайта связывания бензодиазепина, которое обусловлено занятием сайта ГАМК. Эти данные были интерпретированы как свидетельство того, что сайты ГАМК и бензодиазепина являются аллостерически связанными в мембране как часть белкового комплекса. Для ряда ГАМК-аналогов способность к повышению связывания диазепама максимум на 50% может непосредственно коррелироваться со способностью к ингибированию ГАМК в оболочках головного мозга на 50% Увеличение связывания бензодиазепина ГАМК-агонистами блокируется антагонистом ГАМК-рецептора, (+) бикукуллином; стереоизомер (-) бикукуллин является значительно менее активным (Tallman и др. 1978, Nature 274: 383-385).
Вскоре после открытия сайтов высокоаффинного связывания для бензодиазепинов было установлено, что в ряде областей головного мозга триазолопиридазин может взаимодействовать с бензодиазепиновыми рецепторами способом, согласующимся с вариабельностью рецепторов или негативной кооперативностью. В этих исследованиях, коэффициенты Хилла значительно меньшие, чем единица, наблюдались в ряде отделов головного мозга, включая кору головного мозга, гиппокамп, и полосатое тело. В мозжечке, триазолопиридазин взаимодействовал с сайтами бензодиазепина с коэффициентом Хилла 1 (Squires и др. 1979, Pharmacol. Biochem. Behav. 10: 825-830, Klepner и др. 1979, Pharmacol. Biochem. Behav. 11: 457-462). Таким образом, многие рецепторы бензодиазепина были предсказаны для коры головного мозга гиппокампа и полосатого тела, но не для мозжечка.
На основании этих исследований были осуществлены авторадиографические исследования локализации рецепторов с использованием оптического микроскопа. Хотя и было проиллюстрировано наличие множества рецепторов (Young Kuhar, 1980, J. Pharmacol. Exp. Ther. 212: 337-346, Young и др. 1981, J. Pharmacol. Exp. Ther. 216: 425-430, Niehoff и др. 1982, J. Pharmacol. Exp. Ther. 221: 670-675), однако в ранних исследованиях не было выявлено прямой корреляции между локализацией подтипов рецепторов и поведением, ассоциированным с этой областью. Кроме того, в мозжечке, где при изучении связывания было предсказано наличие одного рецептора, авторадиографически было выявлено разнообразие рецепторов (Niehoff и др. 1982, J. Pharmacol. Exp. Ther. 221: 670-675).
Sieghart Karobath (1980, Nature, 286: 285-287) продемонстрировали физическое обоснование различия в специфичности лекарственных средств для этих двух явных подтипов сайтов бензодиазепина. С помощью гель-электрофореза с додецилсульфатом натрия было установлено наличие нескольких молекулярно-массовых рецепторов для бензодиазепинов. Эти рецепторы идентифицировались путем ковалентного введения радиоактивного флунитразепама, бензодиазепина, который может ковалентно метить все типы рецепторов. Большинство меченных полос имели молекулярную массу от 50000 до 53000, 55000 и 57000, а триазолопиридазины ингибировали мечение форм с несколько более высокими молекулярными массами 53000, 55000, 57000 (Sieghart и др. 1983, Eur. J. Pharmacol. 88: 291-299).
В то же время, было высказано предположение, что многие формы рецепторов представляют собой "изорецепторы" или многочисленные аллельные формы рецептора (Tallman Gallager, 1985, Ann. Rev. Neurosci. 8, 21-44). Хотя вообще для ферментов, в основном, не были описаны генетически отчетливые формы рецепторов. Еще в начале исследований рецепторов, предпринятых авторами настоящего изобретения с использованием специфических радиоактивных зондов и техники электрофореза, стало почти очевидным, что изучение изорецепторов будет важнейшей фазой на пути к пониманию этиологии психиатрических расстройств у человека.
Субъединицы рецептора ГАМК клонировали на бычьих и человеческих кДНК-библиотек (Schoenfield и др. 1988; Duman и др. 1989). Путем клонирования и экспрессии ряд различных кДНК были идентифицированы как субъединицы ГАМК-рецепторного комплекса. Эти субъединицы подразделяются на категории α,β,γ,δ,ε и дают молекулярную основу гетерогенности ГАМК-рецепторов и характерной региональной фармакологии (Shivvers и др. 1980; Levitan и др. 1989). По-видимому, g-субъединица позволяет лекарственному средству подобно бензодиазепину модифицировать ГАМК-ответы (Putchett и др. 1989). Наличие низких коэффициентов Хилла при связывании лигандов с ГАМК-рецептором указывает на уникальные профили специфического фармакологического действия подтипа.
Лекарственные средства, взаимодействующие с ГАМК-рецептором, могут обладать определенным спектром фармакологических активностей в зависимости от их способности модифицировать действия ГАМК. Например, β-карболины были сначала выделены на основании их способности конкурентно ингибировать связывание диазепама с его сайтом его связывания (Nielsen и др. 1979, Life Sci. 25: 679-686). Анализ на связывание рецептора является непредсказуемым в отношении биологической активности таких соединений, ингибировать связывание могут агонисты, частичные агонисты, инверсные агонисты и антагонисты. Когда определялась структура β-карболина, было возможно синтезировать ряд аналогов и проводить поведенческие тесты этих соединений. В результате, было сразу установлено, что β-карболины могут быть антагонистами поведенческой активности диазепама (Tenen Hirsh 1980, Nature, 288: 609-610). В дополнение к этому антагонизму β-карболины обладают активностью, противоположной активности бензодиазепинов, и они стали известными как инверсные агонисты.
Кроме того, на основании способности к ингибированию связывания бензодиазепинов были разработаны и другие специфические антагонисты бензодиазепиновых рецепторов. Из этих соединений лучше всего изучен имидазодиазепин (Hunkeler и др. 1981, Nature, 290: 514-516). Данное соединение является высокоаффинным конкурентным ингибитором связывания бензодиазепина и бета-карболина и обладает способностью блокировать фармакологическое действие соединений этих классов. Само по себе это соединение обладает небольшой присущей ему фармакологической активностью в организме человека и животного (Hunkeler и др. 1981, Nature, 290: 514-516; Darragh и др. 1983, Eur. J. Clin. Pharmacol. 14: 569-570). Изучение радиомеченой формы этого соединения (Mohler и Richards, 1981, Nature, 294: 763-765) показало, что это соединение взаимодействует с тем же числом сайтов, что и бензодиазепины и бета-карболины и что взаимодействия этих соединений являются чисто конкурентными. Рассматриваемое соединение представляет собой оптимальный лиганд для связывания с ГАМК-рецепторами, поскольку оно не обладает рецепторной специфичностью подтипа и определяет каждое состояние рецептора.
Исследование взаимодействия широкого ряда соединений аналогичных описанному выше, позволило разделить эти соединения на категории. В настоящее время соединения, обладающие активностью, аналогичной активности бензодиазепинов, называются агонистами. Соединения, обладающие активностью, противоположной активности бензодиазепинов, называются инверсными агонистами, а соединения, ингибирующие оба типа активности, названы антагонистами. Вышеупомянутое разделение на категории было сделано для того, чтобы подчеркнуть тот факт, что широкий ряд соединений может обнаруживать широкий спектр фармакологического действия, а также показать, что соединения могут взаимодействовать с тем же рецептором, давая противоположные эффекты, и что бета-карболины и антагонисты с присущим им психомоторным действием не являются синонимами. Биохимический тест на фармакологические и психореактивные свойства соединений, взаимодействующих с бензодиазепиновым рецептором, еще раз продемонстрировал их связь с ГАМК эргической системой. В противоположность бензодиазепинам, которые показывают повышение их аффинности, обусловленной ГАМК (Tallman и др. 1978, Nature, 274: 383-385, Tallman и др. 1980, Science, 207: 274-281), соединения с антагонистическими свойствами показывают небольшой ГАМК-сдвиг (т. е. изменение аффинности рецептора, обусловленной ГАМК) (Mohler и Richard, 1981, Nature, 294: 763-765), а инверсные агонисты показывают фактически снижение аффинности, обусловленной ГАМК (Braestrup и Nielson, 1981, Nature, 294: 472-474). Таким образом, ГАМК-сдвиг, в основном, указывает на предполагаемые психореактивные свойства указанных соединений.
В качестве агонистов и антагонистов бензодиазепина были получены различные соединения. Например, в патентах США NN 4312870 и 4713383, и в заявке на европатент EP 181282 описываются некоторые соединения, используемые при лечении тревожных состояний или депрессии. В патенте США N 4713383 раскрываются соединения формулы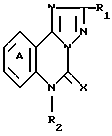
где R1 (не)замещенный Ph, (дигидро)фуранил, тетрагидрофуранил, (дигидро)тиенил, тетрагидротиенил, пиранил, рибофуранозил, все С связанные;
R2 H, алкил;
X O, S, R3N; R3 H, алкенил, алкинил, C3-C20-циклоалкил, (не)замещенный алкил; арил; аралкил, где арилом является Ph, пиридинил, тиенил, фуранил;
кольцо А может быть замещено алкилом, алкокси, галоидом, амино, алкилтио и т.п.
В заявке на европатент EP 181282 раскрываются соединения формулы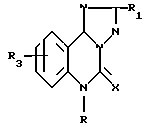
где R1 (замещенный) Ph или гетероцикл;
R2 H, алкил, алкенил, гидроксиалкил, аралкил, аралкенил, арил;
R3 H, алкил, алкокси, HO, галоид, F3C, O3N, H2N, алкилтио, алкилсульфинил, алкилсульфонил, аралкокси;
X O, S, NR4; а R4 H, алкил, аралкил, циклоалкил, алкенил, алкинил, арил, (замещенный)аминоалкил, гидроксиалкил.
В патенте США N 4312870 описываются соединения формулы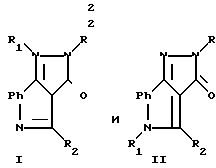
где Ph 1,2-фенилен, незамещенный или замещенный от 1 до 3-х одинаковыми или различными членами, выбранными из группы, включающей низший алкил, низший алкокси, низший алкилтио, гидрокси, галогено, трифторметил, нитро, амино, моно- или ди-низший алкиламино, циано, карбамоил и карбокси;
R незамещенный или замещенный фенил, обозначаемый H-Ph, пиридил, низший алкилпиридил или галогенпиридил;
R1 водород, низший алкил или низший (гидрокси, диалкиламино или H-Ph)-алкил;
R2 водород или низший алкил-алкил; их 3-гидрокситаутомеры; низший алканоил, карбамоил, моно- или ди-низший алкил-карбамоил-производные указанных (гидрокси или амино)-(фенил или фенилен) соединений,
и соединения формулы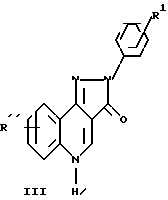
где R'' водород, алкил или алкокси, каждый из которых содержит до 4 атомов углерода, гидрокси, фторо, хлоро, бромо, или трифторметил;
R' водород, о- или м-фторо; или если R'' является хлоро, то R' является п-фторо.
Соединения настоящего изобретения отличаются от описанных выше соединений. Эти соединения не являются имидазохиноксалинами и не имеют различных кольцевых заместителей соединений настоящего изобретения.
Настоящее изобретение представляет новые соединения формулы I, которые взаимодействуют с сайтом связывания ГАМК, бензодиазепиновым рецептором.
Настоящее изобретение представляет также соединения, полезные при лечении невротических состояний, связанных с беспокойством, нарушением сна и страхами, а также при лечении эпилептических припадков и последствий передозировки бензодиазепина. Соответственно, в широком смысле воплощения настоящее изобретение относится к соединениям формулы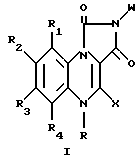
и к их фармацевтически приемлемым нетоксичным солям,
где R водород или метил;
R1 и R4 водород;
X водород;
W фенил; фенил, замещенный галогеном, ацетокси, гидрокси, низшей алкильной группой с прямой или разветвленной цепью, имеющей 1-4 атома углерода, амино, или низшей алкокси группой с прямой или разветвленной цепью, имеющей 1-4 атома углерода; фенил, дизамещенный галогеном и низшим алкилом с прямой или разветвленной цепью, имеющим 1-4 атома углерода; или фенил, дизамещенный галогеном и низшей алкокси группой с прямой или разветвленной цепью, имеющей 1-4 атома углерода; и
R2 и R3 одинаковые или различные и представляют водород, галоген, низший алкил с прямой или разветвленной цепью, имеющий 1-4 атома углерода.
Описанные соединения являются высокоселективными агонистами, антагонистами или инверсными агонистами ГАМК-рецепторов головного мозга, или пролекарствами агонистов. Эти соединения полезны в диагностике и лечении тревожных состояний, нарушений сна, эпилептических припадков, последствий передозировки бензодиазепиновых лекарственных средств и для улучшения памяти.
На фиг. 1 (a d), 2 представлены характерные имидазохиноксалины настоящего изобретения.
Новые соединения согласно настоящему изобретению могут быть представлены следующей общей формулой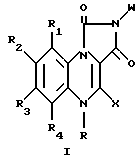
а также их фармацевтически приемлемыми нетоксичными солями,
где R водород или метил;
R1 и R4 водород;
X водород;
W фенил; фенил, замещенный галогеном, ацетокси, гидрокси, низшей алкильной группой с прямой или разветвленной цепью, имеющей 1-4 атома углерода, амино, или низшей алкокси группой с прямой или разветвленной цепью, имеющей 1-4 атома углерода; фенил, дизамещенный галогеном и низшим алкилом с прямой или разветвленной цепью, имеющим 1-4 атома углерода; или фенил, дизамещенный галогеном и низшей алкокси группой с прямой или разветвленной цепью, имеющей 1-4 атома углерода; и
R2 и R3 одинаковые или различные и представляют водород, галоген, низший алкил с прямой или разветвленной цепью, имеющий 1-4 атома углерода.
Настоящее изобретение также относится к соединениям формулы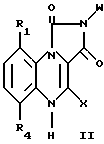
где R1 и R4 водород;
X водород;
W фенил; или фенил, замещенный галогеном, ацетоном, гидрокси, низшей алкильной группой с прямой или разветвленной цепью, имеющей 1-4 атома углерода, или низшей алкокси группой с прямой или разветвленной цепью, имеющей 1-4 атома углерода.
Кроме того, настоящее изобретение относится к соединениям формулы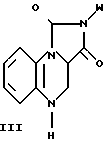
где W фенил, дизамещенный галогеном и низшим алкилом с прямой или разветвленной цепью, имеющим 1-4 атома углерода; фенил, дизамещенный галогеном и низшей алкокси группой с прямой или разветвленной цепью, имеющей 1-4 атома углерода.
Настоящее изобретение охватывает также соединения формулы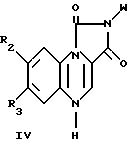
где W фенил, дизамещенный галогеном и низшей алкокси группой с прямой или разветвленной цепью, имеющей 1-4 атома углерода; и
R2 и R3 водород.
Нетоксичными фармацевтически приемлемыми солями настоящего изобретения могут быть соли, образованные кислотами, такими как соляная, фосфорная, бромистоводородная, серная, сульфиновая, муравьиная, сульфоновая, иодистоводородная, уксусная кислота и т.п. Любому специалисту известен широкой ряд нетоксичных фармацевтически приемлемых аддитивных солей.
Характерные представители соединений настоящего изобретения, которые охватываются формулой I, включают, но не ограничиваются соединениями, приведенными на фиг. 1 и 2, и их фармацевтически приемлемыми солями. Настоящее изобретение также включает ацилированные предшественники соединений формулы I. Для получения нетоксичных фармацевтически приемлемых аддитивных солей и ацилированных предшественников соединений формулы I могут быть использованы различные методы синтеза, хорошо известные специалистам.
Используемый в настоящем описании термин "низший алкил" означает алкильные группы с прямой или разветвленной цепью, имеющие 1-4 атома углерода, например, такие как метил, этил, пропил, изопропил, н-бутил, втор-бутил, трет-бутил, пентил, 2-пентил, изопентил, неопентил, гексил, 2-гексил, 3-гексил и 3-метиленпентил.
Используемый в настоящем описании термин "низший алкокси" означает алкокси группы с прямой или разветвленной цепью, имеющие 1-4 атома углерода, например, такие как метокси, этокси, пропокси, изопропокси, н-бутокси, втор-бутокси, трет-бутокси, пентокси, 2-пентил, изопентокси, неопентокси, гексокси, 2-гексокси, 3-гексокси и 3-метилпентокси.
Используемый в настоящем описании термин "галоген" означает фтор, бром, хлор или иод.
Фармацевтическая полезность соединений настоящего изобретения проиллюстрирована с помощью следующего анализа на ГАМК-рецепторную активность.
Анализы проводили в соответствии с описанием, приведенным в работе Thomas и Tallman J. Bio. Chem. 156: 9838-9842, J. Neurocsi. 3: 433-440, 1983. У крысы вырезали ткань коры головного мозга и гомогенизировали в 25 объемах (мас. /об.) 0,05 М Трис-HCl-буфера (pH 7,4 при 4oC). Полученный гомогенат ткани центрифугировали в течение 20 мин при 4oC и при 20000 g. Супернатант декантировали, а осадок снова гомогенизировали в том же объеме буфера и снова центрифугировали при 20000 g. После этого супернатант декантировали, а осадок оставляли на ночь при -20oC. Затем осадок размораживали и гомогенизировали в 25 объемах (мас./об.) буфера, и эту процедуру повторяли дважды. И наконец, полученный осадок повторно суспендировали в 50 объемах буфера (мас./об. 0,05 М Трис-HCl, pH 7,4 при 40oC).
Инкубационная смесь содержала 100 мкл тканевого гомогената, 100 мкл радиоактивно меченного лиганда (0,5 нМ, 3H-R015-1788 [3H-Флумазенил] специфическая активность 80 Ku/мМ), лекарственное средство или ингибитор, и буфер до полного объема 500 мкл. Инкубацию осуществляли в течение 30 мин при 4oC, после чего смесь быстро фильтровали через CFB-фильтры для разделения свободного и связанного лиганда. Фильтры промывали дважды свежим Трис-HCl-буфером (0,05 М, pH 7,4 при 4oC), и проводили подсчет в жидкостном сцинтилляционном счетчике. Затем в те же пробирки добавляли 1,0 мкМ диазепама для оценки неспецифического связывания. Данные, полученные при трехкратных измерениях, усредняли и вычисляли ингибирования полного специфического связывания (полное специфическое связывание все связывание неспец. связывание). В некоторых случаях количества намеченного лекарственного средства варьировали, и в соответствии с этим строили кривые полного замещения связывания. Полученные данные преобразовывали для вычисления IC50 и коэффициента Хилла. Указанные данные представлены ниже.
Соединение N IC50 мкМ
1 0,0095
3 0,015
4 0,0095
5 0,016
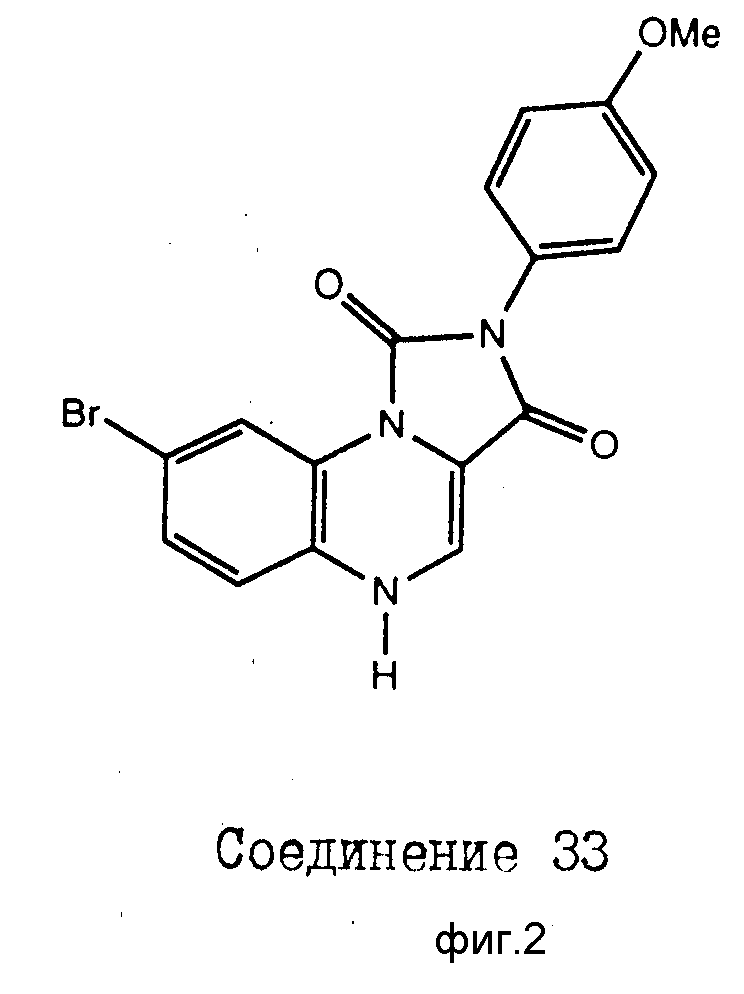
33 0,0024
Номера соединений относятся к соединениям, показанным на фиг. 1, 2.
Соединения 1, 3 и 4 являются особенно предпочтительными соединениями настоящего изобретения.
Соединения общей формулы I могут быть введены перорально, местно, парентерально, путем ингаляции или впрыскивания, или ректально в виде разовых лекарственных форм, содержащих стандартные фармацевтически приемлемые носители, адъюванты и наполнители. Используемый в настоящем описании термин "парентеральное введение" включает в себя подкожные, внутривенные, внутримышечные или внутригрудинные инъекции или вливания. Фармацевтические композиции на основе соединений настоящего изобретения содержат соединение общей формулы I и фармацевтически приемлемый носитель. При этом в указанной композиции может присутствовать одно или более соединений общей формулы I в сочетании с одним или несколькими нетоксичными фармацевтически приемлемыми носителями и/или разбавителями, и/или адъювантами, и, если необходимо, в сочетании с другими активными ингредиентами. Фармацевтические композиции, содержащие соединения общей формулы I, могут быть введены в виде соответствующих форм для перорального применения, например, таких как таблетки, пастилки, водные или масляные суспензии, диспергируемые порошки или гранулы, эмульсии, жесткие или мягкие капсулы, сиропы или эликсиры.
Композиции для перорального использования могут быть получены в соответствии со стандартной техникой, применяемой в фармацевтической промышленности, и указанные композиции могут содержать одно или несколько веществ, выбранных из группы, включающей в себя подслащивающие агенты, ароматизирующие вещества, красители и консерванты, для придания препарату приятных вкусовых качеств и эстетичного вида. Таблетки обычно содержат активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми наполнителями, которые часто используются при изготовлении таблеток. Такими наполнителями могут быть инертные разбавители, например карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; гранулирующие или дезинтегрирующие агенты, например кукурузный крахмал, или альгиновая кислота; связующие вещества, например крахмал, желатин или аравийская камедь; замасливающие агенты, например стеарат магния, стеариновая кислота или тальк. Таблетки могут оставаться непокрытыми, либо они могут быть покрыты стандартными способами для того, чтобы замедлить дезинтеграцию и абсорбцию активного ингредиента в желудочно-кишечном тракте, сообщая тем самым препарату пролонгированное действие. В этих целях, например, может быть использован моностеарат или дистеарат глицерина.
Препараты для перорального введения могут быть также изготовлены в виде твердых желатиновых капсул, в которых активный ингредиент смешан с инертным твердым разбавителем, например, таким как карбонат кальция, фосфат кальция или каолин; или в виде мягких желатиновых капсул, в которых активный ингредиент смешан с водой или масляным наполнителем, например, таким как арахисовое масло, жидкий парафин или оливковое масло.
Жидкие суспензии содержат активные ингредиенты, как правило, в сочетании с наполнителями, обычно используемыми для изготовления водных суспензий. Такими наполнителями могут быть суспендирующие агенты, например натрийсодержащая карбометилцеллюлоза, метилцеллюлоза, гидропропилметилцеллюлоза, альгинат натрия, трагакантовая камедь и аравийская камедь; и диспергирующие или смачивающие агенты, которые могут быть натуральными фосфатидами, например лецитин, или продуктами конденсации алкиленоксида с жирными кислотами, например полиоксиэтиленстеарат, или продуктами конденсации этиленоксида с длинноцепными алифатическими спиртами, например гептадекаэтиленоксицетанол, или продуктами конденсации этиленоксида с неполным эфиром многоатомного спирта, происходящим от жирных кислот и гексита, например полиоксиэтиленсорбитмоноолеат, или продуктами конденсации этиленоксида с неполными эфирами, происходящими от жирных кислот и гекситангидридов, например полиэтиленсорбитанмоноолеат. Водные суспензии могут также содержать один или несколько консервантов, например этил-, или н-пропил-п-гидроксибензоат; один или несколько окрашивающих агентов; один или несколько ароматизирующих агентов; и один или несколько подслащивающих агентов, таких как сахароза или сахарин.
Масляные суспензии могут быть получены путем суспендирования активных ингредиентов в растительном масле, таком как арахисовое масло, оливковое масло, кунжутное масло или кокосовое масло; или в минеральном масле, например в жидком парафине. Масляные суспензии могут содержать загуститель, например пчелиный воск, твердый парафин или цетиловый спирт. Подслащивающие вещества, например, такие как были указаны выше, и ароматизирующие вещества могут быть добавлены в композиции для придания им приятного вкуса и эстетического вида. Для предохранения композиций от воздействия окружающей среды в них могут быть добавлены антиоксиданты, например аскорбиновая кислота.
Диспергируемые порошки и гранулы, предназначенные для изготовления водных суспензий путем добавления воды, представляют собой активный ингредиент в сочетании с диспергирующим или смачивающим агентом и одним или несколькими консервантами. Примеры подходящих диспергирующих или смачивающих и суспендирующих агентов приведены выше. В композиции могут также присутствовать дополнительные ингредиенты, например подслащивающие, ароматизирующие и окрашивающие агенты.
Фармацевтические композиции могут быть также изготовлены в виде эмульсий типа "масло в воде". Масляной фазой в такой эмульсии может быть растительное масло, например оливковое масло, или минеральное масло, например жидкий парафин, или их смеси. Подходящими эмульгирующими агентами являются природные смолы, такие как аравийская камедь или трагакантовая камедь; природные фосфатиды, такие как соя и лецитин; и сложные эфиры или неполные сложные эфиры, происходящие от жирных кислот и гекситов и ангидридов, например, такие как сорбитанмоноолеат, а также продукты конденсации указанных неполных эфиров с этиленоксидом, например, такие как полиоксиэтиленсорбитанмоноолеат. Указанные эмульсии могут также содержать подслащивающие и ароматизирующие агенты.
Сиропы и эликсиры могут быть приготовлены с использованием подслащивающих агентов, таких как глицерин, пропиленгликоль, сорбит или сахароза. Указанные препараты могут также содержать средство, уменьшающее раздражение, консервант, ароматизирующее средство и краситель. Фармацевтические композиции могут быть изготовлены в виде стерильных инъецируемых водных или масляных суспензий. Эти суспензии могут быть получены в соответствии со стандартной техникой с использованием подходящих диспергирующих или смачивающих агентов, указанных выше. Такими инъецируемыми стерильными препаратами могут быть также стерильные инъецируемые растворы или суспензии в нетоксичном парентерально приемлемом разбавителе или растворителе, например раствор в 1,3-бутандиоле. Примерами приемлемых наполнителей или растворителей являются вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды могут быть использованы стерильные жирные масла. Для этих целей может быть использовано любое мягкое жирное масло, включая синтетические моно- или диглицериды. Кроме того, в инъецируемых препаратах могут быть использованы жирные кислоты, например, такие как олеиновая кислота.
Соединения общей формулы I могут быть также введены ректально в виде суппозиториев. Эти композиции могут быть получены путем смешивания активного ингредиента с подходящим не раздражающим наполнителем, который при комнатной температуре является твердым, а попадая в прямую кишку, расплавляется и становится жидким, высвобождая тем самым лекарственное средство. Для этих целей обычно используют какао-масло и полиэтиленгликоли.
Соединения общей формулы I могут быть введены парентерально в стерильной среде. Лекарственное средство в зависимости от используемых наполнителей и концентрации может быть либо суспендировано, либо растворено в наполнителе. Если используются такие адъюванты, как местные анестезирующие средства, консерванты и буферные агенты, то они могут быть растворены в наполнителе.
Используемые для лечения вышеуказанных расстройств дозы составляют ≈ 0,1 140 мг на 1 кг тела пациента в день, что составляет для одного пациента ≈ 0,5 мг 7 г в день. Количество активного ингредиента, комбинируемого с материалом носителя при изготовлении разовой лекарственной формы, может варьироваться в зависимости от состояния пациента и способа введения лекарственного средства. Стандартная разовая форма обычно содержит ≈ 1 500 мг активного ингредиента.
Очевидно понятно, однако, что конкретная доза для каждого конкретного пациента зависит от многих факторов, а именно от активности конкретно используемого средства, возраста, веса, общего состояния здоровья и пола пациента, а также от диеты, схемы введения, способа введения, скорости высвобождения лекарственного средства, его состава и серьезности заболевания.
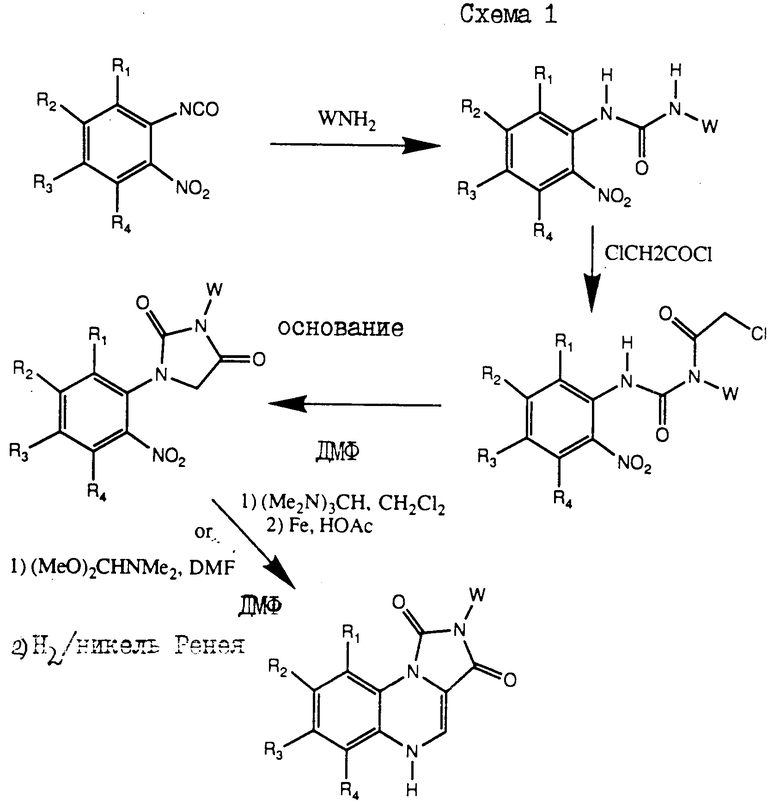
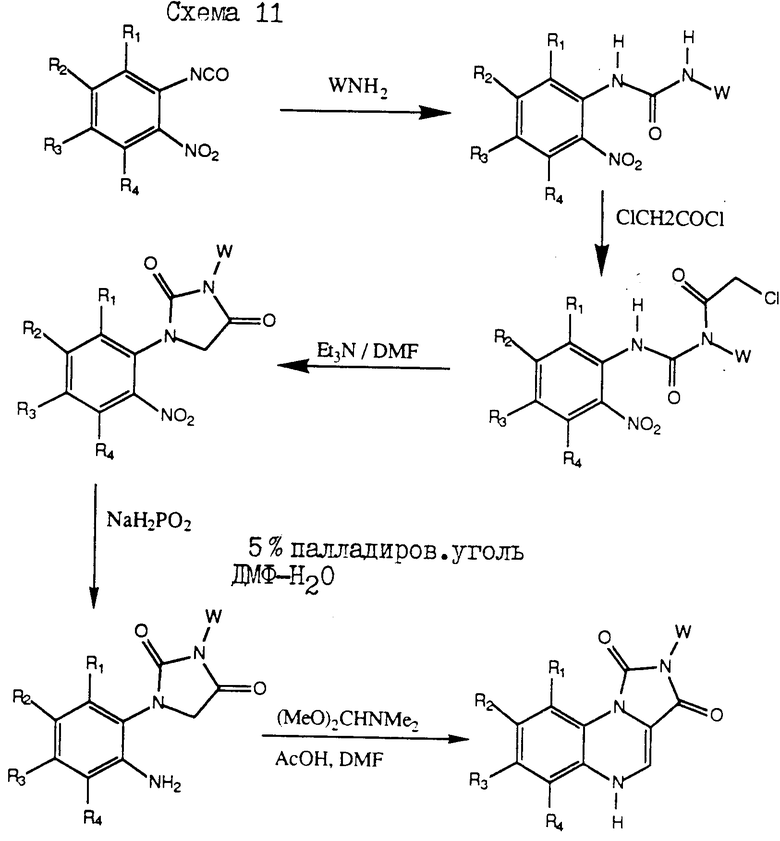
Получение соединений настоящего изобретения иллюстрируется схемами I и II, приведенными ниже. Специалистам в данной области очевидно ясно, что в представленную схему получения соединений настоящего изобретения могут быть введены дополнительные стадии и используемые исходные материалы могут также варьироваться, как это показано в приведенных ниже примерах.
На схемах I и II, приведенных ниже, обозначены: R1, R4 и X представляют собой водород; и R2, R3 и W имеют значения, определенные для формулы I.
В некоторых случаях, для осуществления указанных выше трансформаций может быть необходимо провести блокирование определенных реакционноспособных функциональных групп. Необходимость в таком блокировании, а также условия, необходимые для присоединения и удаления таких групп, в основном могут быть определены специалистами в области органического синтеза.
Пример I.
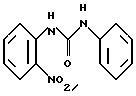
К смеси изоцианата 2-нитрофенила (3,34 г) в 100 мл толуола добавляли анилин (2 г). Смесь перемешивали 30 мин при 20oC. Затем добавляли гексан (300 мл) и полученное твердое вещество фильтровали и высушивали, в результате чего получали N-(2-нитрофенил)-N'-фенил-мочевину в виде светло-желтого твердого вещества.
Пример II.
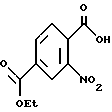
К раствору диэтилнитротерефталата (17,9 г) в 300 мл этанола добавляли 1н. NaOH (70 мл) и перемешивали в течение ночи. Затем добавляли 1н. HCl (70 мл) и реакцию распределяли между метиленхлоридом (200 мл) и водой (200 мл). Водный слой экстрагировали еще 3 раза. Объединенные органические экстракты высушивали, а растворитель удаляли в вакууме, в результате чего получали этил-3-нитро-4-карбоксибензоат в виде белого твердого вещества.
Пример III.
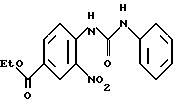
К дифенилфосфорилазиду (5,75 г) в безводном толуоле (50 мл) при 100oC в атмосфере азота по капле добавляли раствор, содержащий этил-3-нитро-4-карбоксибензоат (5 г) и триэтиламин (4 мл) в безводном толуоле (50 мл). Смесь перемешивали в течение 1 ч с последующим добавлением анилина (5 мл), и реакционную смесь оставляли для охлаждения до комнатной температуры (40 мин). Затем добавляли этилацетат (300 мл) и раствор промывали последовательно с 1н. HCl (300 мл) водой (300 мл), 1н. NaOH (300 мл) и водой (300 мл). Органический слой высушивали, а растворитель удаляли в вакууме. К полученному маслянистому веществу добавляли диэтиловый эфир (50 мл), и полученное твердое вещество соединяли и высушивали, в результате чего получали N-(2-нитро-5-метил-фенил)-N'-фенил-мочевину в виде белого твердого вещества.
Пример IV.
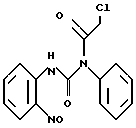
Раствор, содержащий N-(2-нитрофенил)-N'-фенил-мочевину (5,76 г) и хлороацетилхлорид (40 мл) нагревали с обратным холодильником в атмосфере азота в течение 30 мин. После удаления избытка хлороацетилхлорида в вакууме добавляли диэтиловый эфир, а полученное твердое вещество фильтровали и высушивали, в результате чего получали N'-(2-хлороацетил)-N-(2-нитрофенил)-N'-фенил-мочевину в виде белого твердого вещества.
Пример V.
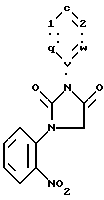
Раствор N'-(2-хлороацетил)-N-(2-нитрофенил)-N-фенил-мочевины (3,7 г), диметилформамида (15 мл) и диизопропилэтиламина (15 мл) нагревали с обратным холодильником в течение 5 мин. Нагретую смесь оставляли для охлаждения до комнатной температуры и осаждали с добавлением смеси в 200 мл воды. Осадок собирали и высушивали, в результате чего получали 1-(2-нитрофенил)-3-фенил-имидазолин-2,4 (1Н, 3Н)-дион.
Пример VI.
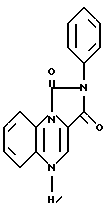
К раствору 1-(2-нитрофенил)-3-фенил-имидазолин-2,4(1Н, 3Н)-диона (2,7 г) в безводном диметилформамиде (2 мл) в атмосфере азота добавляли N,N-диметилформамиддиметилацеталь (2,7 г). Реакционную смесь перемешивали 2 ч при 80oC и растворитель удаляли в вакууме. К полученному маслянистому веществу добавляли железный порошок (5 г) и уксусную кислоту (250 мл). Эту смесь осторожно нагревали с обратным холодильником 3 мин, с последующим перемешиванием реакционной смеси дополнительно 30 мин. Гетерогенную смесь разбавляли 10% метанолметиленхлоридом (200 мл) и фильтровали через силикагель, используя в качестве элюента смесь 10% метанола и метиленхлорида. Растворитель удаляли в вакууме, а затем добавляли нагретый этанол (200 мл). К этой смеси добавляли воду (200 мл) и полученное твердое вещество фильтровали и промывали последовательно с этанолом, этилацетатом, диэтиловым эфиром и высушивали, в результате чего получали 2-фенил-имидазо [1,5-а] хиноксалин-1,3 (2Н, 5Н)-дион в виде желтого твердого вещества с т. пл. 231-234oC (соединение 1).
Пример VII.
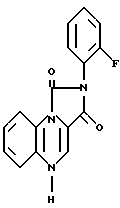
К раствору, содержащему 1-(2-нитрофенил)-3-(2-фторофенил)-имидазолин-2,4 (1Н, 3Н)-дион (1,18 г) в безводном метиленхлориде (5 мл) в атмосфере азота добавляли трис диметиламино метан (1 мл). Реакционную смесь перемешивали 20 мин при комнатной температуре, а растворитель удаляли в вакууме. Полученное твердое вещество растворяли в ДМФ 100 мл и добавляли взвесь никелевого катализатора гидрирования (50% раствор в воде, 1 мл). Смесь гидрогенизировали при давлении 50 фунт/кв.дюйм (344,7 кПа) в течение 45 мин. После фильтрации через целит, растворитель концентрировали в вакууме в количестве 30 мл, а затем добавляли воду (50 мл). Полученное твердое вещество собирали и промывали последовательно с этанолом, этилацетатом и диэтиловым эфиром и проветривали, в результате чего получали 2-(2-фторофенил)-имидазо[1,5-а]квиноксалин-1,3-(2Н, 5Н)-дион в виде желтого твердого вещества (соединение 2), т. пл. 261-264oC.
Пример VIII.
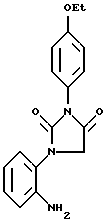
К раствору, содержащему ДМФ (100 мл), H2O (15 мл), 5%-Ро-уголь (1,25 г) и 1-(2-нитрофенил)-3-(4-этоксифенил)-имидазолин-2,4 (1Н,3Н)-дион (25 г) при 60oC, по капле добавляли раствор, содержащий гипофосфит натрия (15 г) в H2O (40 мл). Через 3 ч смесь охлаждали до комнатной температуры и фильтровали через целит. Смесь выливали в 500 мл H2O, фильтровали и высушивали, в результате чего получали 1-(2-аминофенил)-3-4-этоксифенил-имидазолин-2,4(1Н, 3Н)-дион.
Пример IX.
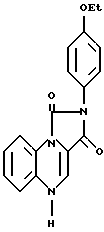
К 1-(2-аминофенил)-3-(4-этоксифенил)-имидазолин-2,4-(1Н,3Н)-диона (2 г) добавляли ДМФ (5 мл), уксусную кислоту (5 мл) и диметилформамиддиметилацеталь (5 мл). Реакционную смесь нагревали при 60oC 16 ч, затем охлаждали и фильтровали. Полученное оранжевое твердое вещество промывали изопропанолом и перекристаллизировали уксусной кислотой, в результате чего получали 2-(4-этоксифенил)-имидазо [1.5, а] хиноксалин-1.3 (2Н,5Н)-дион с т. пл. 268-269oC (соединение 3).
Пример X. Следуя в основном процедуре, описанной в примерах VI, VII и IX, получали следующие соединения:
1. 2-(4-метоксифенил)-имидазо [1.5, а] хиноксалин-1.3 (2Н, 5Н)-дион (соединение 4), т. пл. 240-242oC.
2. 2-(4-метилфенил)-имидазо [1.5,а] хиноксалин-1.3(2Н,5Н)-дион (соединение 5), т. пл. 305-308oC.
3. 2-4-фторофенил-имидазо[1.5, а]хиноксалин-1.3(2Н,5Н)-дион (соединение 6), т. пл. 235-238oC.
4. 2-(2-аминофенил)-имидазо[1.5,а]хиноксалин-1.3 (2Н,5Н)-дион (соединение 7), т. пл. 247-249oC.
5. 2-(3-фторофенил)-имидазо[1,5,а]хиноксалин-1,3(2Н,5Н)-дион (соединение 8), т. пл. 265-266oC.
6. 2-(4-хлорофенил)-имидазо[1.5,а]хиноксалин-1,3(2Н,5Н)-дион (соединение 9), т. пл. 235-238oC.
7. 2-(3-метилфенил)-имидазо[1,5,а]хиноксалин-1,3(2Н,5Н)-дион (соединение 10), т. пл. 263-265oC.
8. 2-(2-фторо-4-этоксифенил)-имидазо [1,5,а] хиноксалин-1,3(2Н,5Н)-дион (соединение 11), т. пл. 264-267oC.
9. 2-(3-хлорофенил)-имидазо[1,5,а]хиноксалин-1,3(2Н,5Н)-дион (соединение 12), т. пл. 235-239oC.
10. 2-(2-метоксифенил)-имидазо[1,5, а] хиноксалин-1,3(2Н, 5Н)-дион (соединение 13), т. пл. 270-272oC.
11. 2-(4-этилфенил)-имидазо[1,5,а]хиноксалин-1,3(2Н,5Н)-дион (соединение 14), т. пл. 215-216oC.
12. 2-(2-фторо-4-метилфенил)-имидазо [1,5,а] хиноксалин-1,3(2Н,5Н)-дион (соединение 15), т. пл. 280-284oC.
13. 2-(3-метоксифенил)-имидазо[1,5, а] хиноксалин-1,3(2Н, 5Н)-дион, (соединение 16), т. пл. 212-214oC.
14. 2-(3-этоксифенил)-имидазо[1,5,а]хиноксалин-1,3(2Н,5Н)-дион (соединение 17), т. пл. 197-200oC.
15. 2-(4-н-пропилоксифенил)-имидазо [1,5,а] хиноксалин-1,3(2Н,5Н)-дион (соединение 18), т. пл. 182-185oC.
16. 2-(4-н-бутоксифенил)-имидазо [1,5, а] хиноксалин-1,3(2Н, 5Н)-дион (соединение 19), т. пл. 155-156oC.
17. 2-(4-изопропоксифенил)-имидазо [1,5, а] хиноксалин-1,3(2Н,5Н)-дион (соединение 20), т. пл. 164-167oC.
18. 7-хлоро-2-(4-метилфенил)-имидазо [1,5,а] хиноксалин-1,3(2Н,5Н)-дион (соединение 21), т. пл. 200-204oC.
19. 7,8-диметил-2-(4-этоксифенил)-имидазо [1,5, а] хиноксалин-1,3(2Н, 5Н)-дион (соединение 22).
20. 8-метил-2-фенил-имидазо[1,5,а]хиноксалин-1,3(2Н,5Н)-дион (соединение 23), т. пл. 240-244oC.
21. 8-карбоэтокси-2-(4-этоксифенил)-имидазо [1,5,а] хиноксалин-1,3(2Н, 5Н)-дион (соединение 24).
22. 7-карбоэтокси-2-(4-этоксифенил)-имидазо [1,5,а] хиноксалин-1,3(2Н, 5Н)-дион (соединение 25).
23. 8-бромо-2-(4-этоксифенил)-имидазо [1,5,а] хиноксалин-1,3(2Н,5Н)-дион (соединение 26), т. пл. 152-155oC.
24. 2-(4-пропилфенил)-имидазо[1,5,а]хиноксалин-1,3(2Н,5Н)-дион (соединение 27), т. пл. 185-186oC.
25. 7-метил-2-(4-этоксифенил)-имидазо [1,5,а] хиноксалин-1,3(2Н,5Н)-дион (соединение 28), т. пл. 200-203oC.
26. 2-(3-бромо-4-этоксифенил)-имидазо [1,5,а] хиноксалин-1,3(2Н,5Н)-дион (соединение 29), т. пл. 147-150oC.
27. 2-(3-тиенил)-имидазо[1,5, а] хиноксалин-1,3(2Н,5Н)-дион (соединение 30).
28. 2-(2-тиенил)-имидазо[1,5, а] хиноксалин-1,3(2Н,5Н)-дион (соединение 31).
29. 2-(4-ацетоксифенил)-имидазо [1,5, а] хиноксалин-1,3(2Н, 5Н)-дион (соединение 32), т. пл. 210-211oC.
Пример XI.
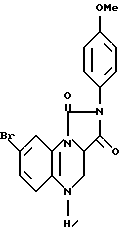
К суспензии 2-4-метоксифенил-имидазо[1,5,а] хиноксалин-1,3(2Н,5Н)-диона (100 мг) в безводном диоксане (4 мл) добавляли бром (200 мг). Реакционную смесь перемешивали 15 мин при 20oC, а затем выливали непосредственно в кипящую уксусную кислоту (50 мл), содержащую цинковый порошок (500 мг). Реакционную смесь нагревали с обратным холодильником в течение 5 мин, а затем охлаждали до комнатной температуры. После разбавления 10% метанолом и метиленхлоридом (100 мл) смесь фильтровали через силикагель, растворитель удаляли в вакууме, а полученное твердое вещество обрабатывали кипящим этанолом (25 мл) с последующим разбавлением водой (100 мл). Смесь охлаждали до 0oC, а твердое вещество фильтровали и высушивали, в результате чего получали 8-бромо-2-(4-метоксифенил)-имидазо[1,5,а]хиноксалин-1,3(2Н,5Н)-дион (соединение 33) в виде желтого твердого вещества с т. пл. 154oC.
Пример XII. Следуя в основном процедуре, описанной в примере XI, получали следующее соединение:
8-бромо-2-(3-бромо-4-этоксифенил)-имидазо [1,5, а] квиноксалин-1,3(2Н, 5Н)-дион (соединение 34), т. пл. 146-149oC.
Пример XIII.
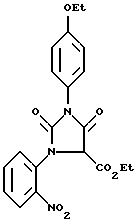
Раствор, содержащий 1-(2-нитрофенил)-3-(4-этоксифенил) имидазолин-2,4(1Н,3Н)-дион (5 г) в безводном тетрагидрофуране (50 мл) добавляли по капле в течение 30 мин к раствору 0,5 М LDA в тетрагидрофуране (29 мл) при -78oC. Через 20 мин добавляли одной порцией этилхлороформат (1,2 мл) в тетрагидрофуране (5 мл). Реакционную смесь нагревали до комнатной температуры 30 мин, а затем гасили насыщенным раствором хлорида аммония. Реакционную смесь распределяли между этилацетатом и водой, органический слой высушивали, а растворитель удаляли в вакууме. Затем проводили колоночную хроматографию на силикагеле, элюируя смесью 50% этил-ацетата и гексана, в результате чего получали 5-карбоэтокси-1-(2-нитрофенил)-3-(4-этоксифенил)-имидазолин-2,4 (1Н,3Н)-дион.
Пример XIV.
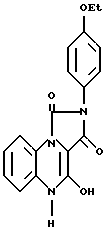
К суспензии, содержащей цинковый порошок (6 г) в уксусной кислоте (250 мл) добавляли 5-карбоэтокси-1-(2-нитрофенил)-3-(4-этоксифенил)-имидазолин-2,4 (1Н, 3Н)-дион (2,5 г). Смесь нагревали 15 мин с обратным холодильником, охлаждали до комнатной температуры, фильтровали, растворитель удаляли в вакууме, а полученное твердое вещество перемешивали с этанолом (50 мл) и фильтровали, в результате чего получали 4-гидрокси-2-(4-этоксифенил)-имидазо [1,5,а] хиноксалин-1,3(2Н,5Н)-дион (соединение 35).
Пример XV.
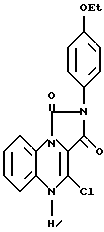
Раствор 4-гидрокси-2-(4-этоксифенил)-имидазо [1,5,а]-квиноксалин-1,3(2Н, 5Н)-дион (3,2 г) в оксихлориде фосфора (40 мл) нагревали с обратным холодильником в течение 16 ч. Растворитель удаляли в вакууме и добавляли воду 15 мл. Затем pH доводили до 7,0 с использованием гидроксида аммония и полученное твердое вещество фильтровали и высушивали, в результате чего получали 4-хлоро-2-(4-этоксифенил)-имидазо[1,5,а]хиноксалин-1,3(2Н,5Н)-дион (соединение 36).
Пример XVI.
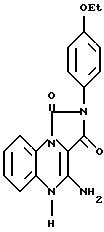
Раствор тетрагидрофурана (10 мл), аммиака (10 мл) и 4-хлоро-2-(4-этоксифенил)-имидазо[1,5, а]хиноксалин-1,3(2Н,5Н)-диона (100 мг) нагревали 4 ч в герметично закрытой пробирке при 100oC. После охлаждения до комнатной температуры растворитель удаляли в вакууме. Твердое вещество суспендировали в 50% EtOH-H2O и фильтровали с получением 4-амино-2-(4-этоксифенил)-имидазо[1,5,а] хиноксалин-1,3(2Н,5Н)-диона (соединение 37).
Пример XVII. Следуя в основном процедуре, описанной в примере XVI, получали следующие соединения:
1. 4-диметиламино-2-(4-этоксифенил)-имидазо [1,5,а] хиноксалин-1,3(2Н, 5Н)-дион (соединение 38).
2. 4-н-пропиламино-2-(4-этоксифенил)-имидазо [1,5,а] хиноксалин-1,3(2Н, 5Н)-дион (соединение 39).
3. 4-N-метиламино-2-(4-этоксифенил)-имидазо [1,5,а] хиноксалин-1,3 (2Н, 5Н)-дион (соединение 40).
Пример XVIII.

2-(4-ацетоксифенил)-имидазо[1,5, а] хиноксалин-1,3(2Н, 5Н)-дион (250 мг) добавляли к раствору этанола (50 мл), насыщенному HCl. Раствор перемешивали 2 ч, а растворитель удаляли в вакууме, в результате чего получали 2-(4-гидроксифенил)-имидазо [1,5, а] хиноксалин-1,3(2Н,5Н)-дион (соединение 41), т. пл. 318-322oC.
Пример XIX.
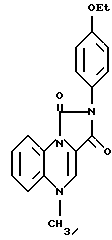
2-(4-этоксифенил)-имидазо[1,5, а] хиноксалин-1,3 (2Н, 5Н)-дион (100 мг) добавляли к раствору диметилформамиддиметилацеталя (10 мл) и ДМФ (5 мл), затем реакционную смесь нагревали 4 ч при 100oC. Раствор охлаждали до комнатной температуры и выливали в воду (200 мл). Полученный осадок собирали и высушивали, в результате чего получали 5-N-метил-2-(4-этоксифенил)-имидазо [1,5,а] хиноксалин-1,3 (2Н,5Н)-дион (соединение 42), т. пл. 263-266oC.
Пример XX.
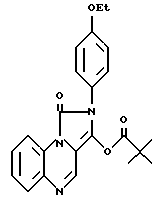
2-(4-этоксифенил)-имидазо[1,5, а]хиноксалин-1,3 (2Н,5Н)-дион добавляли к раствору безводного ДМФ (5 мл) и трет-бутоксида калия (125 мг) при 50oC. Через 5 мин добавляли триметилацетилхлорид (150 мл). Реакционную смесь перемешивали 15 мин и выливали в воду (25 мл). Полученный осадок собирали, промывали EtOH и высушивали, в результате чего получали 3-триметилецетокси-2-(4-этоксифенил)-имидазо [1,5,а] хиноксалин-1,3 (2Н,5Н)-дион (соединение 43).
Пример XXI. Следуя, в основном, процедуре, описанной в примере XX, получали следующие соединения:
1. 3-н-пропокси-2-(4-этоксифенил)-имидазо [1,5, а] хиноксалин-1 (2Н, 5Н)-он (соединение 44).
2. 5-пропионил-2-(4-этоксифенил)-имидазо [1,5, а] хиноксалин-1,3 (2Н, 5Н)-дион (соединение 45).
Несмотря на проиллюстрированные и описанные выше конкретные варианты осуществления изобретения, специалистам в данной области техники очевидны различные его модификации, в связи с чем подразумевается, что настоящее изобретение не ограничивается изложенными вариантами его осуществления или отдельными их деталями, и что возможны отклонения от них в пределах существа и объема настоящего изобретения, определенных в приведенной ниже формуле изобретения.
Использование: в химико-фармацевтической промышленности. Сущность изобретения: соединения: имидазохиноксалины формулы I, в которой R - H, CH3; R1 и R4 - H; X - H; W - фенил или фенил, замещенный галогеном, ацетокси, гидрокси, низшей алкильной группой C1 - C4, амино, или низшей алкоксигруппой C1 - C4, или фенил, дизамещенный галогеном и низшим алкилом C1 - C4, или фенил, дизамещенный галогеном и низшей алкоксигруппой C1 - C4; R2 и R3 одинаковые или различные и являются H, галогеном, алкилом C1 - C4. Соединения обладают активностью связывания ГАМК-рецепторов. Структура соединения формулы I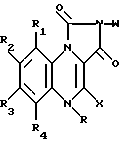
39 з.п. ф-лы, 2 ил.
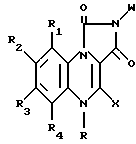
где R водород или метил;
R1 и R4 водород;
Х водород;
W фенил или фенил, замещенный галогеном, ацетокси, гидрокси, низшей алкильной группой с прямой или разветвленной цепью, имеющей 1 4 атома углерода, амино или низшей алкоксигруппой с прямой или разветвленной цепью, имеющей 1 4 атома углерода, или фенил, дизамещенный галогеном и низшим алкилом с прямой или разветвленной цепью, имеющим 1 4 атома углерода, или фенил, дизамещенный галогеном и низшей алкоксигруппой с прямой или разветвленной цепью, имеющей 1 4 атома углерода;
R2 и R3, одинаковые или различные, водород, галоген, низший алкил с прямой или разветвленной цепью, имеющий 1 4 атома углерода.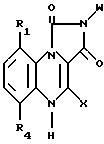
где R1 и R4 водород;
Х водород;
W фенил, фенил, замещенный галогеном, ацетокси, гидрокси, низшим алкилом с прямой или разветвленной цепью, имеющим 1 4 атома углерода, или низшей алкоксигруппой с прямой или разветвленной цепью, имеющей 1 4 атома углерода.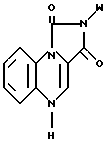
где W фенил, дизамещенный галогеном и низшим алкилом с прямой или разветвленной цепью, имеющим 1 4 атома углерода.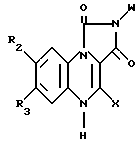
где W фенил, дизамещенный галогеном и низшим алкокси с прямой или разветвленной цепью, имеющим 1 4 атома углерода;
R2 и R3 водород.
| WO, 91/07407, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| US, 5011574, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| US, 5102885, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
