Изобретение относится к новым соединениям общей формулы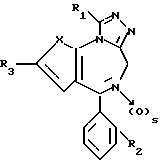
в которой: X является -CH=CH- или S;
R1 низший алкил или трифторметил;
R2 хлор или фтор;
R3 радикал формулы R4-(CH2)n-C≡C- или R5-O-CH2-C≡C-,
где n целое число 0, 1 или 2, s 0 или 1;
R4 фенил или моно-, ди- или трициклический 5-7-членный гетероциклический радикал, содержащий в качестве гетероатомов 0 или S и/или 1-3 атома азота, незамещенный или замещенный низшим алкокси, оксо, оксигруппой или хлором;
R5 фенил или пиридил-радикал, при условии, что когда n равно 0, радикал R4 должен быть присоединен через углерод к кислородной связи, и что R5 всегда присоединен через углерод к кислородной связи, и в случае наличия по меньшей мере одного асимметрического центра их энантиомеры и рацематы, и фармацевтически-приемлемые соли присоединения кислот.
Соединения формулы I обладают свойствами антагонистов фактора активации тромбоцитов (PAF) и поэтому являются полезными при болезненных состояниях, характеризующихся избытком фактора активации тромбоцитов, или для профилактики и лечения сердечно-сосудистых заболеваний, легочных заболеваний, иммунологических нарушений, воспалительных заболеваний, дерматологических нарушений, шоков или отторжений трансплантатов.
Структурные аналоги подобного действия не известны. Наиболее близким аналогом является патент США 4,621,083, в котором описываются бензо- и тиенотриазолбензодиазепины с PAF-антагонистическими свойствами. В упомянутом патенте США конкретно не раскрыто ни одно соединение, замещенное алкинильной группой на конденсированном бензольном или тиофеновом кольце.
Таким образом, в упомянутом патенте США раскрыты соединения одинакового действия, но структурные аналоги в нем не раскрыты. Алкинильная группа R4-(CH2)n-C≡C- или R5-O-CH2-C≡C- в полученных согласно изобретению соединениях является существенной для фармакологического действия и отличается от раскрытых в упомянутом патенте США заместителей в конденсированном бензольном или тиофеновом кольце настолько, что свойства соединений с этой группой нельзя считать очевидными.
Используемый в данном описании термин "низший алкил" обозначает неразветвленную или разветвленную насыщенную углеводородную группу, содержащую от 1 до 7 атомов углерода, предпочтительно от 1 до 4 атомов углерода, например метил, этил, пропил, изопропил, бутил, трет. -бутил, неопентил, пентил, гептил и тому подобные.
Термин "низшая алкоксигруппа" обозначает алкильную эфирную группу, в которой алкильная группа является такой, как это описано выше, и означает, например, метокси, этокси, пропокси, пентокси и тому подобные.
Термин "гетероциклический радикал" обозначает моноциклический 5-, 6- или 7-членный гетероциклический или би- или трициклический гетероциклический радикал, содержащий один или более гетероатомов, выбираемых из азота, кислорода и серы, и который может быть замещенным, предпочтительно моно- или дизамещенным, низшим алкилом, низшей алкоксигруппой, оксогруппой, оксигруппой, хлором. Термин "гетероциклический" относится к карбоциклическому остатку, в котором один или более атомов углерода заменены независимо друг от друга кислородом, азотом или серой.
Типичными моноциклическими 5- или 6-членными ароматическими гетероциклическими радикалами являются пиридинил, имидазолинил, тиенил, 2-хлортиенил, фурил, пиримидинил, оксазолинил или тому подобные.
Типичными моноциклическими 5-, 6- или 7-членными неароматическими гетероциклическими радикалами являются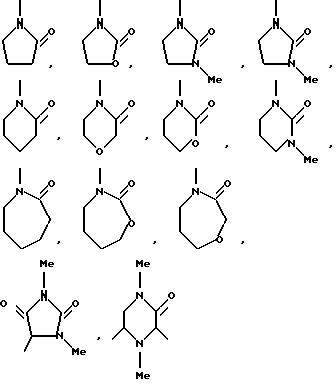
и тому подобные.
Типичными бициклическими гетероциклическими радикалами являются
(а) 5,5-циклические системы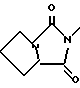
и тому подобные;
(б) 6,5-циклические системы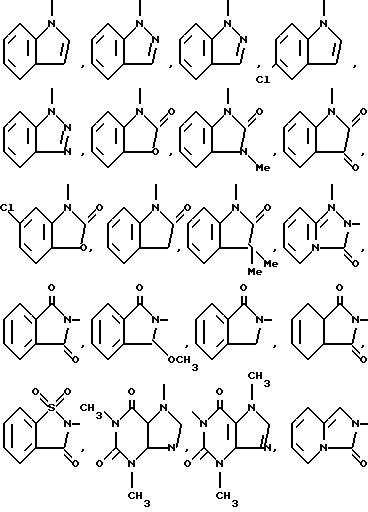
и тому подобные;
(в) 6,6-циклические системы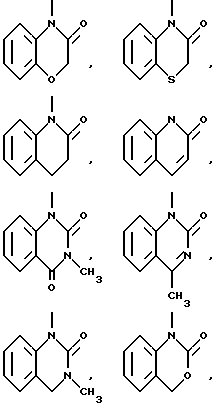
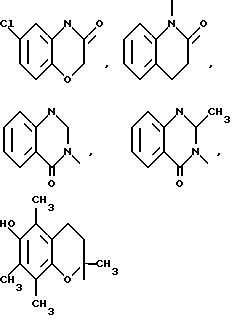
и тому подобные; и
(г) 6,7-циклические системы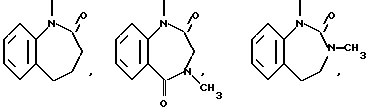
и тому подобные.
Типичными трициклическими гетероциклическими радикалами являются 5,5,6-, 5,6,6-, 6,6,6- и 6,6,7-циклические системы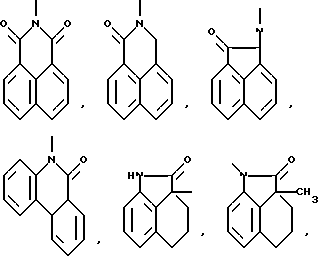
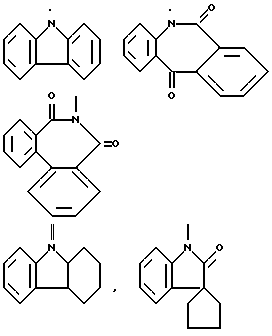
и тому подобные.
Используемое в данном описании и являющееся очевидным из номенклатуры и структур, структурное изображение L□ представляет -C≡CH: →□, а представляет -C≡C-.
Предпочтительной группой соединений формулы I являются соединения, в которых X является -CH=CH- или S, R1 является низшим алкилом, R2 является хлором, фтором; R3 является радикалом формулы R4-(CH2)n-C≡C- или R5-O-CH2-C≡C-,, R4 является бициклическим или трициклическим радикалом и R5 является фенилом при условии, что, когда n=0, R4 должен быть присоединен через углерод к углеродной связи, и что R5 всегда присоединяется через углерод к кислородной связи.
Другой предпочтительной группой соединений формулы I являются соединения, где R1 метил или этил, R2 является фтором или хлором, R3 имеет значения, указанные выше, n 1, 2, s 0.
Более предпочтительной группой соединений формулы I являются соединения, где R1 метил, R2 фтор, хлор, s 0, n 1, R4 би- или трициклический гетероциклический радикал, указанный выше, и R5 - фенил или пиридил.
Самой предпочтительной группой соединений формулы I являются соединения, где X является S, R1 метил, R2 хлор, s=0, R3 является R4-(CH2)n-C≡C- n=1, а R4 является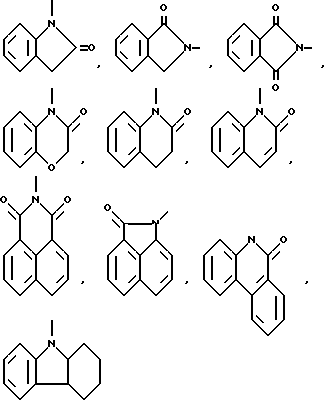
Предпочтительными соединениями изобретения являются
5-{ 3-[4-(2-хлорфенил)-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло [4,3-a] [1,4]диазепин-2-ил]-2-пропинил}-фенантридин-6(5Н)-он;
4-(2-хлорфенил)-2-[3-(1,2,3,4-тетрагидро-9Н-карбазол-9-ил)-1- пропинил] -9-метил-6Н-тиено-[3,2-f][1,2,4]триазоло[4,3-a][1,4]диазепин;
1-{ 3-[4-(2-хлорфенил)-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло [4,3-a] [1,4]диазепин-2-ил]-2-пропинил}-3,4-дигидро-2(1Н)-хинолинон;
2-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло [4,3-a] [1,4]диазепин-2-ил]-2-пропинил]-1H-бенз[de]изохинолин -1,3-(2Н)-дион;
1-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло [4,3-a] [1,4]диазепин-2-ил]-2-пропинил]-бенз[cd]индол-2(1Н)-он;
4-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло [4,3-a] [1,4]диазепин-2-ил]-2-пропинил]-2H-1,4-бензоксазин-3(4Н)-он.
Другими предпочтительными соединениями изобретения являются
1-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло [4,3-a] [1,4]диазепин-2-ил]-2-пропинил]-1Н-индол-2,3-дион;
1-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло [4,3-a] [1,4]диазепин-2-ил]-2-пропинил]-1,3-дигидро-2Н-индол-2-он;
2-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло [4,3-a] [1,4]диазепин-2-ил]-2-пропинил]-1,2,4-триазоло[4,3-a]-пиридин -3(2Н)-он;
1,1-диокись 2-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло [4,3-a][1,4]диазепин-2-ил]-2-пропинил]-1,2-бензизотиазол-3-(2Н)-она;
4-(2-хлорфенил)-2-[3-(1H-индазол-1-ил)-1-пропинил] -9-метил-6Н-тиено [3,2-f][1,2,4]триазоло [4,3-a][1,4]диазепин;
2-[3-(1H-бензимидазол-1-ил)-1-пропинил] -4-(2-хлорфенил)-9-метил-6Н-тиено[3,2-f][1,2,4]триазоло [4,3-a][1,4]диазепин;
2-[3-[6-(2-фторфенил)-1-метил-4Н-[1,2,4] триазоло[4,3-a][1,4]бензодиазепин-8-ил]-2-пропинил]-1Н-изоиндол-1,3(2Н)-дион и
4-[3-[6-(2-фторфенил)-1-метил-4Н-[1,2,4] триазоло[4,3-a][1,4]бензодиазепин-8-ил]-2-пропинил]-2Н-1,4-бензоксазин-3(4Н)-он.
Предметом изобретения также являются соединения общей формулы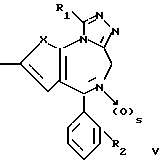
в которых X, R1, R2 и s имеют значение, указанное выше, которые являются промежуточными продуктами для получения соединения формулы I. Кроме того, изобретение относится к соединениям, указанным выше, для использования в качестве терапевтически активного вещества или в качестве антагонистов фактора активизации тромбоцитов (PAF).
Соединения формулы I и их фармацевтически приемлемые соли присоединения кислот могут быть получены в соответствии с изобретение путем
а) взаимодействия соединения общей формулы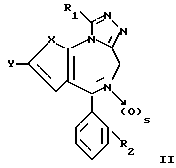
в которой X, R1, R2 и s имеют значение, указанное выше, и Y является бромом или иодом,
с соединением формулы
R4-(CH2)n-C≡C-H (IIIa)
или
R5-O-CH2-C≡C-H (IIIb)
в которой R4, R5, n имеют значение, указанное выше, или
б) взаимодействия соединения общей формулы
в которой X, R1, R2 и s имеют значение, указанное выше, с соединением общей формулы
R4-Y
в которой R4 и Y имеют значение, указанное выше, или
в) для получения соединения формулы I, в которой s является 1 и R4 не содержит остатка, содержащего основной атом азота, обрабатывают соединение формулы I, в которой s является 0 и R4 не содержит остатка, содержащего основной атом азота, надкислотой, или
г) при желании, превращают полученное соединение общей формулы I в фармацевтически приемлемую соль присоединением кислоты.
Более конкретно, соединения формулы I могут быть получены, как это описывается ниже по схемам реакций I и II.
Схема реакции I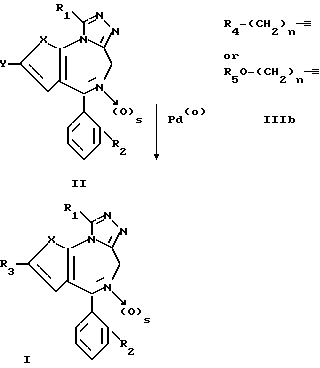
в которой R1, R2, R3, R4, R5, X, n и s являются такими, как это описано ранее, а Y является бромом или иодом.
По схеме I триазолотиено- или триазолобензодиазепин формулы II, в которой Y является бромом или йодом, подвергают взаимодействию с ацетиленом формулы IIIa или IIIb с использованием в качестве катализатора переходного металла, согласно методикам, известным в данной области, чтобы получить соответствующее соединение формулы I согласно варианту а) данного способа.
Реакция бромистого или предпочтительно иодистого соединения структуры II с ацетиленом формулы IIIa или IIIb проводится в инертном растворителе; предпочтительными растворителями являются ацетонитрил, тетрагидрофуран и диметилформамид, при температуре в пределах от комнатной температуры до приблизительно 100oC, в зависимости от природы Y и X в формуле II, в присутствии палладиевого катализатора, например дихлорида или диацетата бис-(трифенилфосфин)-палладия, необязательно в присутствии каталитического количества иодистой меди(I) и избытка акцептора протонов, такого как триэтиламин.
С другой стороны, соединение формулы I, в которой s равно 0, а R3 не содержит остатка, содержащего основный атом азота, может быть превращено согласно варианту в) данного способа, если необходимо, до соответствующего N-оксида путем обработки надкислотой, такой как мета-хлорнадбензойная кислота, надуксусная кислота и тому подобные, в инертном растворителе, таком как хлористый метилен, хлороформ, уксусная кислота и тому подобном, при температуре в пределах примерно от 0 до 80oС.
Получающееся соединение реакции формулы I может быть выделено обычными методами, например хроматографией или кристаллизацией.
Исходные вещества формулы II являются известными соединениями или могут быть получены по аналогии с опубликованными методиками. Это относится также к ацетиленовым соединениям формулы IIIa и IIIb. Ацетилены формулы IIIa, в которой n является 1, удобно получать путем алкилирования соответствующей гетероциклической системы с помощью бромистого пропаргила по известным методам. Соединения формулы IIIa, в которой n является 2, могут быть получены путем алкилирования соответствующей гетероциклической системы, например, 4-тозилокси-1-бутином.
Замечено, что когда соединение формулы I и/или формулы II имеет асимметрический атом углерода, может быть удобным использовать энантиомер вместо рацемической смеси в качестве исходных веществ.
Схема реакции II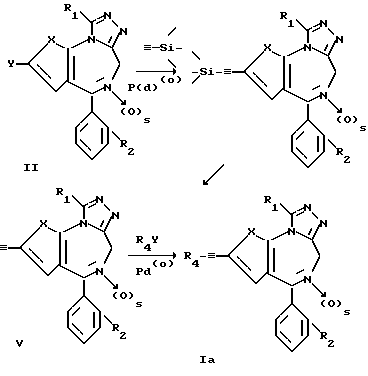
в которой R1, R2, R4, Y и s являются такими, как это описано ранее.
По схеме реакции II соединение формулы I, в которой n равно 0, может быть получено иначе, чем изложено выше. Соединение формулы II, в которой Y является бромом или йодом, подвергается реакции в присутствии палладиевого катализатора с триметилсилилацетиленом, чтобы получить соответствующий продукт реакции формулы IY. Параметры реакции являются в основном такими же, как это описано выше для реакции по схеме I. Далее, соединение формулы IY десилилируется путем обработки водным щелочным раствором, чтобы получить соответствующее этинильное соединение формулы Y. Превращение соединения формулы IY в соединение формулы Y проводится путем гидролиза, предпочтительно путем обработки водным раствором гидроокиси щелочного металла в смешивающемся в воде растворителе, таком как спирт, тетрагидрофуран, диоксан или им подобном, исключая присутствие кислорода. Температура, при которой проводится реакция, не является критической, но предпочтительной является температура в пределах примерно от 0 до 100oC. Получающееся соединение формулы Y подвергается дальнейшей реакции, каталлизируемой палладием, с арилгалогенидом или гетероарилом R4Y, в которой Y является либо бромом, либо иодом, а R4 является арилом или гетероциклическим радикалом, согласно варианту б) способа.
Получающееся соединение формулы Ia может быть выделено по известным методикам, например кристаллизацией или хроматографией.
Соединения формулы I могут образовывать соли присоединения кислот обработкой соединений I сильными неорганическими или органическими кислотами. В частности, они образуют фармацевтически приемлемые соли присоединения кислот с помощью фармацевтически приемлемых как органических, так и неорганических кислот, например с помощью галоидводородных кислот, таких как хлористоводородная кислота, бромистоводородная кислота, иодистоводородная кислота; других неорганических кислот, таких как серная кислота, фосфорная кислота, хлорная кислота или тому подобные; алкил- и моно-арил-сульфоновых кислот, таких как этансульфоновая кислота, толуолсульфоновая кислота, бензолсульфоновая кислота или тому подобные. Фармацевтически неприемлемые соли присоединения кислот соединений формулы I могут быть превращены в фармацевтически приемлемые соли присоединения кислот посредством обычных обменный реакций, при которых фармацевтически неприемлемый анион заменяется фармацевтически приемлемым анионом; или иначе: путем нейтрализации фармацевтически неприемлемой соли присоединения кислоты, а затем взаимодействием полученного таким образом свободного основания с реагентом, приводящим к фармацевтической приемлемой соли присоединения кислоты.
Соединения формулы I проявляют активность антагонистов фактора активации тромбоцитов (PAF) и поэтому являются полезными при болезненных состояниях, характеризующихся избытком фактора активации тромбоцитов, или для профилактики и лечения сердечно-сосудистых заболеваний, легочных заболеваний, иммунологических нарушений, шоков или отторжений трансплантатов.
Активность соединений формулы I может быть продемонстрирована следующим образом:
Количественный анализ связывания
а) Анализ
Количественный анализ связывания был проведен в полиэтиленовых микроцентрифужных пробирках Бэкмана объемом 400 мкл, содержащих 50 мкл масляной смеси из 2 частей Силиконола AR200 (Serva) и 1 части Силиконовой жидкости (Arthur H. Thoma). В пробирки были добавлены буфер, стандарты или аналоги (общий объем 150 мкл). Затем в пробирки добавляют меченый радиоактивным изотопом 3H-PAF (50 мкл). Реакция начинает протекать при добавлении 50 мкл тромбоцитов собаки (2•107 тромбоцитов). Пробирки закупоривают колпачками, переворачивают несколько раз для перемешивания и инкубируют в течение 10 мин при комнатной температуре. Тромбоциты были отделены из инкубационной смеси путем центрифугирования в течение 1 мин в центрифуге типа Микрофуги В Бэкмана. Верхний конец микроцентрифужной пробирки отделяется и тромбоциты смываются с верхнего конца с помощью 200 мкл 50%-ного метанола (Бурдик и Джексон). Добавляют аквазол (NEN, 10 мл) и определяют радиоактивность в образцах, используя жидкий сцинтилляционный счетчик типа Бэкман LS 8100, связанный с самописцем типа Techran. Данные подвергаются обработке с помощью домашней компьютерной системы, либо радиоактивность определяют, используя жидкий сцинтилляционный счетчик типа Searl Merk III, связанный с микрокомпрессором Iso-Data. Результаты представлены в табл. 1 и 2.
б) Получение тромбоцитов
Кровь собирается из анестезированных и неанестезированных собак в пластмассовые центрифужные пробирки объемом 50 мл, содержащие 3,8% цитрата натрия в качестве антикоагулянта (1 объем цитрата/9 объемов крови). Эритроциты удаляются центрифугированием в течение 15 мин при 600 об/мин (100-125 г) при комнатной температуре. Аликвотную часть отстоявшейся плазмы, обогащенной тромбоцитами (PRP), оставляют для определения количества форменных элементов крови, а остающуюся часть подкисляют до рН 6,5 с помощью 0,15М лимонной кислоты. Осадок тромбоцитов в пробирке получают после 10-минного центрифугирования при 2000 об/мин (1000 г) при комнатной температуре. Промытые тромбоциты получают путем пересуспендирования осадка тромбоцитов в пробирке один раз с помощью фосфатно-солевого буферного раствора (PBS), содержащего 1 мМ этилендиаминтетрауксусной кислоты (ЭДТА); центрифугирования так, как это отмечено выше, а затем пересуспендирования тромбоцитов в 0,1%-ном бычьего сывороточного альбумина в фосфатно-солевом буферном растворе (BSA-PBS). Аликвотная часть промытых тромбоцитов отсчитывается. Тромбоциты, используемые для количественных определений связывания, разбавляются до концентрации 2•107 тромбоцитов/импытуемая пробирка (4•103 тромбоцитов/мл). Отсчет тромбоцитов проводится с использованием Ройко Селл-Крит 921 (Royco Cell-Crit 921).
Испытание бронхостеноза, индуцированного фактором активации тромбоцитов (PAF)
Самцы морских свинок (Линия Хартли, весом 400-500 г) анестезируются уретаном (2 г/кг, внутрибрюшинно). Трахею каждого из животных канюлируют и морские свинки снабжаются аппаратом искусственного дыхания, используя респиратор Гарварда для маленьких животных-грызунов (3,0 см3 ударного (систолического) объема крови, 40 вдохов в мину). Трахеальное давление регистрируется из канюли, введенной в трахею и соединенной с датчиком давления Стэтема.
Яремная вена канюлируется для ввода соединения. Самопроизвольное дыхание создается с помощью сукцинилхолина (1,2 мг/кг, внутривенно), вводимого за 2 мин до внутренней инъекции фактора активации тромбоцитов (PAF). Поскольку пропранолол, как было показано, повышает ответные реакции на бронхостеноз, все животные заранее обрабатываются за 5 мин до введения пропранолола (0,1 мг/кг, внутривенно).
Для внутреннего испытания морская свинка подвергается предварительной обработке пропранололом при дозе 0,1 мг/кг внутривенно за мину до инъекции. Испытуемое соединение вводится за мину до внутривенного заражения с помощью PAF. Затем животное заражается внутривенным введением дозы 1 мкг/кг PAF и регистрируется изменение трахеального давления.
Для перорального испытания предварительное введение испытуемого соединения, вводимого через пероральную трубку для зондового питания, проводят за 1 ч до инъекции. Пропранолол или сукцинилхолин и PAF вводятся внутривенно, а изменение трахеального давления регистрируется.
Изменение в трахеальном давлении определяется путем вычитания установившегося состояния базисной линии, достигаемого после введения сукцинилхолина, от пика бронхостеноза, появляющегося после заражения с помощью PAF. Среднее значение вычисляется для каждого испытуемого соединения и сравнивается со средним значением контрольных животных, чтобы получить процент ингибирования бронхостеноза. Стандартная ошибка вычисляется в виде стандартной ошибки среднего значения.
Полученные результаты представлены в табл.1 и 2.
Соединения формулы I включают рацематы и энантиомеры, если один или более асимметрических атомов углерода присутствуют в соединении формулы I.
Соединения формулы I и их фармацевтически приемлемые соли могут быть введены с помощью методов, хорошо известных в данной области. В частности, соединение формулы I или его соль могут быть введены либо отдельно, либо вместе с другими фармацевтическими средствами, например антигистаминами, ингибиторами медиаторного высвобождения, метил-квантинами, бета-агонистами (веществами, обладающими сродством к рецептору) или противоастматическими стероидами, такими как преднизон и преднизолон, перорально, парентерально, прямокишечно (ректально) или путем ингаляции, например, в форме аэрозоля, микроразмельченного порошка или распыляемого раствора. Для перорального ввода они могут быть введены в форме таблеток или капсул, например, в смеси с тальком, крахмалом, молочным сахаром или с другими инертными ингредиентами, то есть с фармацевтически приемлемыми носителями, или в форме водных растворов, суспензий, эликсиров или водных спиртовых растворов, например, в смеси с сахаром или с другими подслащивающими веществами, ароматизирующими веществами, красителями, загустителями и другими обычными фармацевтическими инертными наполнителями. Для парентерального введения они могут быть введены в растворе или суспензии, например, в виде водного раствора или раствора арахисового масла или суспензии, используя инертные наполнители и носители, обычно применяемые для этого способа введения. Для введения в виде аэрозолей они могут быть растворены в подходящем фармацевтически приемлемом растворителе, например этиловом спирте или в смеси смешиваемых растворителей, или могут быть смешаны вместе с приемлемым фармацевтически активным пропеллантом. Такие аэрозольные композиции упаковываются для использования в герметический сосуд, снабженный аэрозольным клапаном, пригодным для высвобождения сжиженной композиции. Предпочтительно чтобы аэрозольный клапан являлся мерным клапаном, то есть клапаном, который при воздействии выделяет заранее определенную эффективную дозу аэрозольной композиции.
На практике доза соединения формулы I или его соли, которая должна быть введена, и частота введения должны зависеть от эффективности и длительности сохранения активности определенного соединения формулы I или соли, которые должны быть введены, и от способа введения, а также остроты состояния, возраста млекопитающего, которое должно быть вылечено, и тому подобных. Пероральные дозы соединения формулы I или его соли, предполагаемого для использования при практическом применении изобретения, находятся в пределах от примерно 0,5 до примерно 1000 мг в день, предпочтительно от около 0,5 до примерно 100 мг в день, предпочтительно примерно от 0,5 до около 10 мг в день, либо в виде единичной дозы, либо в разделенных дозах.
Кроме того, поскольку те соединения формулы I, которые имеют асимметрические центры, и обычно получаются в виде рацемических смесей. В частности, когда R4 и R5 являются гетероциклической группой, последняя может иметь также один или более асимметрических центров, и получающиеся рацематы, энантиомеры и диастереомеры также являются частью изобретения. Разделение таких рацематов на оптически активные изомеры может быть проведено по известным методикам. Некоторые рацемические смеси могут быть подвергнуты осаждению в виде эвтектики и могут быть после этого разделены. Химическое разделение является, однако, предпочтительным. По этому методу диастереомеры образуются из рацемической смеси соединения формулы I вместе с оптически активным разделяющим реагентом. Образуемые диастереомеры разделяются путем селективной кристаллизации или хроматографии и превращаются в соответствующий оптический изомер. Таким образом, изобретение охватывает рацематы соединений формулы I, а также их оптически активные изомеры (энантиомеры).
Примеры, которые следуют ниже, иллюстрируют изобретение. Все температуры представляются в градусах Цельсия, если не указано иное.
Пример 1.
а) Смесь 31 г (0,08 моль) 1,3-дигидро-5-(2-фторфенил)-7-иод-1,4-бензодиазепин-2(2Н)-она (см. G.F.Field и L.A.Sternbach, Швейцарские патенты 561, 706, май 1975 г. и 562, 222, апрель 1975 г.), 20 г (0,09 моль) пентасульфида фосфора, 20 г бикарбоната натрия и 300 мл диглима перемешивают и нагревают до 80-85oC в течение 3 ч. Реакционная смесь выливается затем на лед и разбавляется водой. После перемешивания в течение 30 мин твердый желтый продукт реакции отфильтровывают, промывают водой, 2-пропанолом и малым количеством эфира. Отсасывают досуха в воронке и дополнительно высушивают под вакуумом с получением 26 г (80%) 1,3-дигидро-5-(2-фторфенил)-7-иод-1,4-бензодиазепин-2(2H)-тиона, который далее превращается так, как это описано ниже. Чистое вещество получают путем перекристаллизации из смеси тетрагидрофурана и этанола и имеет т.пл. 242-244oC.
б) К суспензии 8 г вышеуказанного тиона в 40 мл 2-пропанола и 100 мл тетрагидрофурана добавляют 3 мл гидразина. После перемешивания в течение 15 мин при комнатной температуре реакционную смесь фильтруют через 20 г силикагеля, используя тетрагидрофуран для элюирования. Фильтрат выпаривают и остаток кристаллизуют из эфира, чтобы получить 6,7 г (83%) 5-(2-фторфенил)-2-гидразино-7-иод-3H-1,4-бензодиазепина с т.пл. 179-181oC.
в) Смесь 4 г вышеуказанного гидразинового соединения, 20 мл триэтилового эфира ортоуксусной кислоты, 30 мл толуола и 4 г силикагеля нагревают до кипячения с обратным холодильником с перемешиванием в течение 3 ч. Силикагель отфильтровывают и промывают этанолом. Фильтрат выпаривают и остаток кристаллизуют из смеси хлористого метилена и этилацетата, чтобы получить 3,9 г (92%) 6-(2-фторфенил)-8-иод-1-метил-4Н-[1,2,4]триазоло[4,3-a][1,4] -бензодиазепина с т.пл. 235-238oC.
г) Смесь 1,68 г (4 ммоль) 6-(2-фторфенил)-8-иод-1-метил-4Н-[1,2,4]триазоло[4,3-a] [1,4]-бензодиазепина, 0,88 г (4,8 ммоль) N-пропаргилфталимида, 2 мл триэтиламина, 0,36 г трифенилфосфина, 0,08 г иодистой меди (I) и 40 мл диметилформамида перемешивают и дегазируют с помощью медленного тока аргона в течение 15 мин. Тотчас же добавляют 0,12 г ацетата палладия и смесь перемешивают под аргоном в течение 16 ч при комнатной температуре. Реакционную смесь разделяют на части между 200 мл хлористого метилена и 100 мл насыщенного водного раствора бикарбоната натрия. Органическую фазу разделяют, сушат над сульфатом натрия и выпаривают при пониженном давлении, под конец азеотропно с помощью ксилола. Сырой продукт реакции хроматографируют через 120 г силикагеля (Мерк, 70-230 меш), используя 5 об. этанола в хлористом метилене. Очищенные фракции продукта реакции объединяют и выпаривают. Остаток кристаллизуют из этилацетата, чтобы получить 1,6 г (84%) 2-[3-[6-(2-фторфенил)-1-метил-4Н-[1,2,4] триазоло[4,3-a] [1,4] бензодиазепин-8-ил]-2-пропинил] -1Н-изоиндол-1,3(2Н)-диона с т.пл. 253-255oC.
Пример 2
а) 15 мл монохлористого иода (21 г) добавляют к раствору 23 г (0,1 моль) (2-аминофенил)-(2-хлорфенил)-метанола (см. E.Peeder и L.H.Sternbach, патент США 3,371,085, февраль 1968 г.) в 500 мл хлористого метилена, охлажденного до -60oC. После перемешивания с охлаждением в течение 5 ч, охлаждающую баню удаляют и температуре реакционной смеси позволяют достичь 0oC. Вслед за добавлением 300 мл водного раствора бисульфита натрия двухфазную систему перемешивают 10 мин. Органическую фазу разделяют, высушивают над сульфатом натрия и выпаривают. Остаток кристаллизуют из смеси эфира и гексана, чтобы получить 20 г (56% ) (2-амино-5-иодфенил)-(2-хлорфенил)-метанона с т.пл. 120-122oC.
б) К раствору 52 г (0,145 моль) (2-амино-5-иодфенил)-(2-хлорфенил)-метанона в 300 мл хлористого метилена, охлажденного до 0oC, добавляют 15 мл бромистого бромацетила. Медленно с перемешиванием добавляют 10%-ный водный раствор карбоната натрия (150 мл) и двухфазную систему перемешивают при охлаждении в течение 30 мин. Органический слой отделяют, промывают водой и высушивают над сульфатом натрия. Раствор фильтруют и выпаривают. Кристаллизация остатка из смеси хлористого метилена и эфира дает 61 г (90%) 2-бром-N-[2-(2-хлорбензоил)-4-иодфенил] -ацетамида с т. пл. 150-152oC. Раствор 50 г этого вещества в 1 л хлористого метилена добавляют к 800 мл жидкого аммиака, охлаждая с помощью сухого льда. После кипячения с обратным холодильником в течение 16 ч охлаждение прерывают и аммиаку позволяют выпариться. Остающийся раствор промывают водой, высушивают над сульфатом натрия и выпаривают при пониженном давлении. Остаток растворяют в 1 л этанола и раствор нагревают до температуры кипения с обратным холодильником в течение 30 мин после добавления 15 мл уксусной кислоты. Кристаллы, отделяющиеся из охлаждаемой реакционной смеси, собирают, чтобы получить 38 г (89%) 5-(2-хлорфенил)-1,3-дигидро-7-иод-2Н-1,4-бензодиазепин-2-она, который плавится при 260-262oC после перекристаллизации из смеси тетрагидрофурана и этанола.
в) Раствор 15,7 г (0,04 моль) 5-(2-хлорфенил)-7-иод-1,3-дигидро-2H-1,4-бензодиазепин-2-она в 350 мл тетрагидрофурана охлаждают до -30oC. Трет. -бутоксид калия, 4,9 г (0,044 моль) добавляют и перемешивание под азотом продолжают в течение 30 мин при температуре от -10 до -5oC. Затем добавляют 6,6 мл диэтилхлорфосфата и смесь перемешивают при этой температуре в течение дополнительных 30 мин. После добавления 3,4 г ацетилгидразина перемешивание без охлаждения продолжают в течение 1 ч и добавляют 150 мл н-бутанола. Тетрагидрофуран отгоняют из реакционной смеси в течение 45 мин. Остаток разделяют на части между водой и толуолом. Органическую фазу промывают рассолом, высушивают над сульфатом натрия и выпаривают вплоть до малого объема. Выпадающие в осадок кристаллы собирают, чтобы получить 14 г сырого продукта реакции, который очищают хроматографией над 250 г силикагеля, используя 5 об. этанола в хлористом метилене. Очищенные фракции собирают и выпаривают. Кристаллизация из смеси тетрагидрофурана и этанола дает 8,5 г (49% ) 6-(2-хлорфенил)-8-иод-1-метил-4Н-[1,2,4] триазоло[4,3-a][1,4]-бензодиазепина с т.пл. 290-292oC.
г) Реакция 6-(2-хлорфенил)-8-иод-1-метил-4H-[1,2,4]триазоло[4,3-a][1,4] -бензодиазепина с N-пропаргилфталимидом, как это описано в примере 1 г, дает целевой полугидрат 2-[3-[6-(2-хлорфенил)-1-метил-4H-[1,2,4]триазоло[4,3-a] [1,4] бензодиазепин-8-ил] -2-пропинил] -1H-изоиндол-1,3(2H)-диона, который очищают хроматографией и кристаллизуют из смеси метанола и этилацетата, чтобы получить не совсем белые кристаллы с т.пл. 248-250oC.
Пример 3
а) Смесь 1 г 5-(2-фторфенил)-7-иод-2-гидразино-7-иод-3H-1,4-бензодиазепина (см. пример 1б), 5 мл триэтилового эфира ортоприоновой кислоты и 10 мл ксилола нагревают до кипячения с обратным холодильником в течение 1 ч. Растворители частично отгоняют и остаток разбавляют гексаном. Выпадающие в осадок кристаллы собирают и перекристаллизовывают из смеси метанола и этилацетата, чтобы получить 1,05 г (97%) 1-этил-6-(2-фторфенил)-8-иод-4H-[1,2,4]триазоло[4,3-a][1,4] бензодиазепина с т.пл. 209-211oC.
б) Смесь 435 мг (1 ммоль) 1-этил-6-(2-фторфенил)-8-иод-4H-[1,2,4]триазоло[4,3-a] [1,4] бензодиазепина, 220 мг N-пропаргилфталимида, 80 мг трифенилфосфина, 20 мг иодистой меди (I), 0,5 мл триэтиламина и 10 мл диметилформамида перемешивают и дегазируют током аргона в течение 10 мин. Затем добавляют 30 мг ацетата палладия и перемешивание под аргоном продолжают в течение 48 ч. Реакционную смесь разделяют на части между хлористым метиленом и насыщенным водным раствором бикарбоната натрия. Органическую фазу высушивают и выпаривают при пониженном давлении, под конец азеотропно с помощью ксилола. Остаток хроматографируют над 30 г силикагеля (Мерк 70-230 меш), используя 5 об. этанола в хлористом метилене. Кристаллизация объединенных очищенных фракций из этилацетата дает 0,41 г полугидрата 2-[3-[1-этил-6-(2-фторфенил)-4H-[1,2,4] триазоло[4,3-a][1,4]-бензодиазепин-8-ил]-2-пропинил]-1H-изоиндол-1,3(2H)-диона с т.пл. 216-219oC.
Пример 4
6-(2-Хлорфенил)-8-иод-1-метил-4H-[1,2,4] триазоло[4,3-a] [1,4]-бензодиазепин подвергают взаимодействию с 1-(2-пропинил)-1H-индол-2,3-дионом [см F. Lindquist, P.Lagerstrom и R.Dahlbom, Acta Pharm. Suecica, 9, 99 (1972)] как это описано в примере 3б. Сырой продукт реакции очищают хроматографией над 40-кратным количеством силикагеля, используя 5 об. этанола в хлористом метилене. Кристаллизация из этилацетата дает желтые кристаллы 1-[3-[6-(2-хлорфенил)-1-метил-4H-[1,2,4] триазоло[4,3-a] [1,4]-бензодиазепин-8-ил]-2-пропинил] -1H-индол-2,3-диона с т.пл. 210-212oС. Эти кристаллы содержат согласно данным микроанализа и ПМР-спектра 0,25 молярные количества этилацетата.
Пример 5
6-(2-Хлорфенил)-8-иод-1-метил-4H-[1,2,4] триазоло[4,3-a] [1,4]-бензодиазепин подвергают взаимодействию с 2-(2-пропинил)-1H-бенз[de]-изохинолин-1,3(2H)-дионом, как это описано в примере 3б. Сырой продукт реакции хроматографируют над 40-кратным количеством силикагеля, используя 4 об. этанола в хлористом метилене для элюирования. Кристаллизация из смеси хлористого метилена и эфира и перекристаллизация из смеси тетрагидрофурана и этанола дает 2-[3-[6-(2-хлорфенил)-1-метил-4H-[1,2,4] триазоло[4,3-a] [1,4] -бензодиазепин-8-ил-2-пропинил] -1H-бенз [de]-изохинолин-1,3(2H)-дион в виде бесцветных кристаллов с т. пл. 213-215oC, содержащий 0,66 молярное количество воды, согласно ПМР спектру и анализу.
Ацетиленовый компонент реакции получают следующим образом:
6,2 г (0,055 моль) трет. -бутоксида калия добавляют к раствору 9,9 г (0.05 моль) нафталимида в 50 мл диметилформамида, охлажденному до -20oC. После перемешивания при охлаждении в течении 1 ч добавляют 5 мл (0,055 моль) пропаргилбромида в 20 мг диметилформамида и смесь оставляют нагреваться до комнатной температуры. Затем смесь нагревают до 45oC в течение 45 мин. После охлаждения добавляют 15 мл ледяной уксусной кислоты и продукт реакции выпадает в осадок при добавлении воды. Твердые частицы собирают и перекристаллизовывают из этилацетата, чтобы получить 10 г (84%) бесцвестных кристаллов 2-(2-пропинил)-1H-бенз[de]-изохинолин-1,3(2H)-диона с т.пл. 235-237oC.
Пример 6
6-(2-Хлорфенил)-8-иод-1-метил-4H-[1,2,4] триазоло[4,3-a] [1,4] бензодиазепин подвергают взаимодействию с 1-(2-пропинил)-1H- бензимидазолом [см.И.И. Попов, П. В. Ткаченко и А. М. Симонов. Химия гетероциклич.соединений 551, (1973)] как это описано в примере 3б. Продукт реакции выделяют с помощью хроматографии над 40-кратным количеством силикагеля, используя 5 об. этанола в хлористом метилене. Кристаллизация из этанола дает полугидрат 8-[3-(1H-бензимидазол-1-ил)-1-пропинил] -6-(2-хлорфенил)-1-метил-4H-[1,2,4] триазоло[4,3-a] [1,4] -бензодиазепина в виде бесцветных кристаллов с т.пл. 165-168oC. Аналитические и спектроскопические данные показывают наличие полугидрата.
Пример 7
Реакция 6-(2-хлорфенил)-8-иод-1-метил-4H-[1,2,4] триазоло[4,3-a][1,4]-бензодиазепина с 3-(2-пропинил)-2,3-дигидро-1,3-бензоксазол-2-оном [см.A. Lindquist et al. Acta Pharm. Suecica, 9, 99 (1972)] дает после хроматографии над 40-кратным количеством силикагеля с помощью 3 об. этанола в хлористом метилене и кристаллизации из этанола бесцветные кристаллы гидрата 3-[3-[6-(2-хлорфенил)-1-метил-4H-[1,2,4] триазоло[4,3-a] [1,4]-бензодиазепин-8-ил]-2-пропинил]-2,3-дигидро-1,3- бензоксазол-2-она с т.пл. 158-160oC.
Пример 8
6-(2-Хлорфенил)-8-иод-1-метил-4H-[1,2,4] триазоло[4,3-a] [1,4]-бензодиазепин подвергают взаимодействию с 1,3-дигидро-1-(2-пропинил)-2Н-индол-2-оном [см. A. Lindquist et.al.Acta Pharm. Suecica, 9, 99 (1972)] как это описано в примере 3б. Продукт реакции выделяют и очищают с помощью хроматографии над 40-кратным количеством силикагеля, используя 5 об. этанола в хлористом метилене. Кристаллизация из этанола дает 1-[3-[6-(2-хлорфенил)-1-метил-4H- [1,2,4] триазоло[4,3-a] [1,4] -бензодиазепин-8-ил]-2-пропинил]-1,3-дигидро-2H-индол-2-он с т.пл. 141-143oC и содержащего 0,33 моль этанола и 0,66 моль воды.
Пример 9
Смесь 0,84 г (2 ммоль) 6-(2-хлорфенил)-8-иод-1-метил-4H-[1,2,4]триазоло[4,3-a][1,4]-бензодиазепина, 0,5 г (2,4 ммоль) 1-(2-пропинил) -бенз[cd]-индол-2(1H)-она, 90 мг трифенилфосфина, 20 мг иодистой меди (I), 1 мл триэтиламина и 20 мл диметилформамида дегазируют медленным током аргона в течение 15 мин. Затем добавляют 30 мг ацетата палладия и смесь перемешивают под аргоном в течение 5 ч при комнатной температуре. Реакционную смесь разделяют на части между хлористым метиленом и насыщенным водным раствором бикарбоната натрия. Органическую фазу высушивают и выпаривают. Остаток хроматографируют на 40 г силикагеля, используя 5 об. этанола в хлористом метилене для элюирования. Кристаллизация очищенных фракций из смеси метанола и этилацетата дает 1-[3-[6-(2-фторфенил)-1-метил-4H-[1,2,4] триазоло[4,3-a] [1,4] -бензодиазепин-8-ил] -2-пропинил]-бенз[cd]-индол-2(1H)-он в виде светло-желтых кристаллов с т.пл. 224-226oC.
Исходное ацетиленовое соединение получают следующим образом.
К раствору 8,46 г (5 ммоль) бенз[cd]-индол-2(1Н)-она в 100 мл диметилформамида добавляют 6,17 г (0,055 моль) трет.-бутоксида калия. После перемешивания в течение 10 мин при комнатной температуре добавляют 4,9 мл (0,055 моль) бромистого пропаргила и смесь перемешивают в течение 1 ч при комнатной температуре. Реакционную смесь подкисляют уксусной кислотой и разделяют на части между хлористым метиленом и насыщенным водным раствором бикарбоната натрия. Органический слой высушивают и выпаривают и остаток кристаллизуют из смеси тетрагидрофурана и этанола, чтобы получить 8 г (77%) 1-(2-пропинил)-бенз[cd]-индол-2(1H)-она с т.пл. 183-186oC. Соединение перекристаллизовывают дважды для анализа из смеси хлористого метилена и этилацетата и оно имеет т. пл. 185-187oC.
Пример 10
Реакция 0,84 г 6-(2-фторфенил)-8-иод-1-метил-4H-[1,2,4]триазоло [4,3-a] [1,4] -бензодиазепина с 0,53 г (2,6 ммоль) 4-(2-пропинил)-2Н-1,4-бензотиазин-3(4H)-она [см.R.N.Prasad и K.Tietje, Can.J.Chem.44, 1247 (1966)] как это описано в примере 9, дает после хроматографической очистки (5%-ный этанол в хлористом метилене) на силикагеле и кристаллизации из этилацетата 0,5 г (51% ) желтоватых кристаллов 4-[3-[6-(2-фторфенил)-1-метил-4H-[1,2,4] триазоло[4,3-a] [1,4]-бензодиазепин-8-ил]-2-пропинил]-2H-1,4-бензотиазин-3(4H)-она с т. пл. 203-206oC. Эти кристаллы содержат согласно ПМР-спектру и анализу 0,166 молярные количества этилацетата.
Пример 11
4-[3-[6-(2-Фторфенил)-1-метил-4H-[1,2,4] триазоло[4,3-a] [1,4]-бензодиазепин-8-ил]-2-пропинил]-2H-1,4-бензоксазин-3(4H)-он получают подобным образом, используя реакцию 0,84 г 6-(2-фторфенил)-8-иод-1-метил-4H-[1,2,4]триазоло[4,3-a] [1,4]-бензодиазепина с 4-(2-пропинил)-2H-1,4-бензоксазин-3(4H)-оном [см. A. Lindquist et. al, Acta Pharm. Suecica, 9, 99 (1972)] как это описано в примере 9. Продукт реакции выделяют и очищают с помощью хроматографии и кристаллизуют из этилацетата, чтобы получить 0,55 г (56%) светло-желтых кристаллов с т.пл. 238-240oC. Эти кристаллы содержат 0,166 моль этилацетата согласно спектральным и аналитическим данным.
Пример 12
Реакция 0,84 г (2 ммоль) 6-(2-фторфенил)-8-иод-1-метил-4H-[1,2,4] триазоло[4,3-a] [1,4]-бензодиазепина с 0,48 г (2,6 ммоль) 3-(2-пропинил)-4(3H)-хиназолинона [см. J. Maillard et al. Chimie There, 3, 202 (1967)] как это описано в примере 9, дает 0,6 г (59%) не совсем белого продукта реакции, кристаллизуемого из этилацетата. Кристаллы 3-[3-[6-(2-фторфенил)-1-метил-4H-[1,2,4]триазоло[4,3-a] [1,4]-бензодиазепин-8-ил]-2-пропинил]-4(3H)-хиназолинона с т.пл. 199-201oC содержит 1 моль воды и 0,166 моль этилацетата.
Пример 13
3-[3-[6-(2-Фторфенил)-1-метил-4H-[1,2,4] триазоло[4,3-a] [1,4]-бензодиазепин-8-ил] -2-пропинил] -2-метил-4(3H)-хиназолинон получают реакцией 6-(2-фторфенил)-8-иод-1-метил-4H-[1,2,4] триазоло [4,3-a] [1,4]-бензодиазепина с 2-метил-3-(2-пропинил)-4(3H)-хиназолиноном [см. B. Danielson, L. Kronberg и B. Akerman, Acta Pharm. Suecica, 6, 379, (1969)] как это описано в примере 9. Он был выделен с 57%-ным выходом и перекристаллизован из этилацетата, т. пл. 241-244oC с разложением. Кристаллы содержат 0,66 моль воды.
Пример 14
Реакция 6-(2-фторфенил)-8-иод-1-метил-4H-[1,2,4]триазоло[4,3-a] [1,4]-бензодиазепина с 2,3-дигидро-2-(2-пропинил)-1H-изоиндол-1-оном [см. J.I. Neumeyer, U. V. Mayer, J.A. Richman, F.J. Rosenberg и D.G. Teiger, J. Med. Chem. 10, 615 (1967)] дает после хроматографической очистки, как это описано в примере 9, и кристаллизации из этилацетата бесцветные кристаллы 2-[3-[6-(2-фторфенил)-1-метил-4H-[1,2,4] триазоло[4,3-a] [1,4]-бензодиазепин-8-ил]-2-пропинил] -2,3-дигидро-1H-изоиндол-1-она с т. пл. 165-168oC. Согласно спектральным и аналитическим данным эти кристаллы содержат 0,5 моль воды и следы этилацетата.
Пример 15
Рацемический 2,3-дигидро-2-[3-[6-(2-фторфенил)-1-метил-4H-[1,2,4] триазоло[4,3-a] [1,4] -бензодиазепин-8-ил]-2-пропинил]-3-метокси-1H- изоиндол-1-он получают так, как это описано в примере 1г, путем взаимодействия 6-(2-фторфенил)-8-иод-1-метил-4H-[1,2,4]триазоло[4,3-a] [1,4]-бензодиазепина с рацемическим 2,3-дигидро-3-метокси-2-(2-пропинил)-1H-изоиндол-1-оном. Продукт реакции не получается в кристаллическом состоянии и был охарактеризован спектроскопически. Для анализа соединение подвергают осаждению из тетрагидрофурана путем добавления гексана и получающийся аморфный порошок высушивают под вакуумом. ЯМР (CDCL3):2,64 (s, 3, CH3); 2,96 (s, 3, OMe); 4,1 (d, 1) и 5,54 (d, 1); (AB система J=7 Гц, CH2); 4,2 (d,1) и 4,88 (d, 1) (АВ-система, J= 9 Гц, CH2 пропинила); 6,07 (s, 1, C3-H); 6,9-8,0 (m, 11, ароматический H) м.д.
Ацетиленовый компонент реакции получают следующим образом.
Раствор 2 г 2,3-дигидро-3-окси-2-(2-пропинил)-1H-изоиндол-1-она в 20 мл хлористого тионила оставляют стоять при комнатной температуре в течение ночи. Реагент выпаривают азеотропно с помощью толуола при пониженном давлении. Остаток растворяют в 20 мл метанола и раствор обрабатывают с помощью 5 мл триэтиламина. После нагревания на паровой бане в течение 5 мин смесь выпаривают и остаток разделяют на части между хлористым метиленом и насыщенным водным раствором бикарбоната натрия. Органический слой отделяют, высушивают и выпаривают. Остаток кристаллизуют из смеси эфира и гексана, чтобы получить 0,8 г рацемического 2,3-дигидро-3-метокси-2-(2-пропинил)-1H-изоиндол-1-она в виде бесцветных кристаллов с т.пл.85-87oC.
Исходное вещество получают следующим образом.
Смесь 10 г N-пропаргилфталимида и 2 г боргидрида натрия в 100 мл этанола нагревают на паровой бане в течение 15 мин с перемешиванием. Получающийся раствор концентрируют при пониженном давлении до одной трети объема и продукт реакции кристаллизуют добавлением льда и насыщенного раствора бикарбоната натрия. Выпадающие в осадок кристаллы рацемического 2,3-дигидро-3-окси-2-(2-пропинил)-1H-изоиндол-1-она отделяют фильтрацией, промывают водой и отсасывают досуха. После высушивания под вакуумом эти кристаллы имеют т.пл. 157-159oC.
Пример 16
2-[3-[6-(2-Фторфенил)-1-метил-4H-[1,2,4] триазоло[4,3-a] [1,4]- бензодиазепин-8-ил] -2-пропинил] -1,2,4-триазоло[4,3-a]-пиридин-3(2H)- он получают, как это описано в примере 9, реакцией 6-(2-фторфенил)-8-иод-1-метил-4H-[1,2,4] триазоло[4,3-a] [1,4]-бензодиазепина с 2-(2-пропинил)-1,2,4-триазоло[4,3-a]-пиридин-3(2H)-оном. Продукт реакции очищают хроматографией обычным образом и кристаллизуют из смеси этилацетата и этанола. Перекристаллизация из этанола дает светло-желтые кристаллы с т.пл. 170-173oC. Эти кристаллы содержат 0,66 моль воды.
Целевой продукт получают следующим образом.
К раствору 3,25 г (2,4 ммоль) 1,2,4-триазоло[4,3-a]-пиридин-3(2H)-она в 75 мл диметилформамида добавляют 3 г (2,6 ммоль) трет.-бутоксида калия. После перемешивания под азотом в течение 15 мин добавляют 2,35 мл (2,6 ммоль) бромистого пропаргила и перемешивание при комнатной температуре продолжают в течение 1 ч. Растворитель выпаривают при пониженном давлении, под конец азеотропно с помощью ксилола. Остаток экстрагируют хлористым метиленом и раствор выпаривают. Хроматографическая очистка остатка на силикагеле (5% -ным метанолом в хлористом метилене) и кристаллизация очищенных фракций из метанола дает 1,7 г бесцветных кристаллов 2-(2-пропинол)-1,2,4-триазоло[4,3-a]-пиридин-3(2H)-она с т.пл. 126-128oC.
Пример 17
6-(2-Фторфенил)-1-метил-8-[3-(1H-индазол-1-ил)-1-пропинил] -4H-[1,2,4] триазоло[4,3-a] [1,4] -бензодиазепин получают, как это описано в примере 9, путем взаимодействия 6-(2-фторфенил)-8-иод-1-метил-4H-[1,2,4]триазоло[4,3-a] [1,4] -бензодиазепина с 1-(2-пропинил)-1H-индазолом [см. П.В. Ткаченко, И.И. Попов, А. М. Симонов и Ю.В. Медведев, Химия гетероциклич. соедин. 11, 1542 (1975)] Хроматографически выделенный продукт реакции кристаллизуют из этилацетата, чтобы получить желтоватые кристаллы с т.пл. 148-151oC.
Пример 18
Реакция 6-(2-фторфенил)-8-иод-1-метил-4H-[1,2,4]триазоло[4,3-a] [1,4]-бензодиазепина с 1,3-дигидро-1-(2-пропинил)-2H-индол-2-оном [см. A. Lindquist et.al, Acta Pharm. Suecica, 9, 99 (1972)] как это описано в примере 9, дает бесцветные кристаллы 1-[3-[6-(2-фторфенил)-1-метил-4H-[1,2,4]триазоло[4,3-a] [1,4] - бензодиазепин-8-ил]-2-пропинил]-1,3-дигидро-2H-индол-2-он гидрат из этилацетата, которые имеют т.пл. 233-235oC.
Пример 19
Полугидрат 1,1-диокиси 2-[3-[6-(2-фторфенил)-1-метил-4H-[1,2,4] триазоло[4,3-a] [1,4] -бензодиазепин-8-ил]-2-пропинил]-1,2-бензизотиазол-3(2H)-она получают согласно методике примера 9 путем взаимодействия 6-(2-фторфенил)-8-иод-1-метил-4H-[1,2,4] триазоло[4,3-a][1,4]-бензодиазепина с 1,1-диоксидом 2-(2-пропинил)-1,2-бензизотиазол-3(2H)-она [см. R. Granger и J. Giroux, патент Франции 1,273,867, февраль 1962; С.А.57, 7285i (1963)] Продукт реакции выделяют и очищают хроматографией и кристаллизуют из смеси хлористого метилена и этилацетата, чтобы получить бесцветные кристаллы с т.пл. 238-240oC.
Пример 20
Реакция 6-(2-фторфенил)-8-иод-1-метил-4Н-[1,2,4]триазоло[4,3-a] [1,4]-бензодиазепина с 2-(2-пропинил)-тетрагидро-1Н-пирроло[1,2-c] -имидазол-1,3(2Н)-дионом при условиях, описанных в примере 9, дает после хроматографической очистки и кристаллизации из этанола не совсем белые кристаллы полугидрата 2-[3-[6-(2-фторфенил)-1-метил-4Н-[1,2,4]триазоло[4,3-a][1,4]-бензодиазепин-8-ил] -2-пропинил] -тетрагидро-1Н-пирроло-[1,2-c] -имидазол-1,3(2Н)-диона с т.пл. 158-161oC.
Ацетиленовый компонент реакции получают следующим образом:
1,23 г (11 ммоль) трет.-бутоксида калия добавляют к раствору 1,4 г (10 ммоль) тетрагидро-1H-пирроло-[1,2-c] -имидазол-1,3(2H)-диона (L-пролингидантоин) [см. T. Suzuki, K. Igarashi, K. Hase и T.K. Tuzimura, Agr. Biol. Chem. 37, 411 (1973)] в 20 мл диметилформамида. После перемешивания в течение 10 мин при комнатной температуре добавляют 1 мл (11 ммоль) бромистого пропаргила и перемешивание под азотом продолжают в течение 2 ч. Реакционную смесь подкисляют уксусной кислотой и выпаривают при пониженном давлении. Остаток растворяют в хлористом метилене и фильтруют. Фильтрат выпаривают и остаток хроматографируют через 45 г силикагеля, используя 5 об. этанола в хлористом метилене. Очищенные фракции объединяют и выпаривают, чтобы получить 2-(2-пропинил)-тетрагидро-1H-пирроло[1,2-c]-имидазол-1,3(2H)-дион в виде бесцветного вязкого масла.
ЯМР (CDCl3): 1,72 (m, 1, C6-H); 1,9-2,4 (m, 3, C6-H); 2,22 (t, 1, J=1,5 Гц, ацетиленовый H); 3,24 (m, 1, C5-H); 3,70 (m, 1, C5-H); 4,11 (dd, 1, J=4 Гц и 3,5, С7а-H); 4,23 (d, 2, J=1,5 Гц, CH2) м.д.
Пример 21
2-[3-[6-(2-Фторфенил)-1-метил-4H-[1,2,4]триазоло[4,3-a][1,4]-бензодиазепин-8-ил]-2-пропинил]-3а,4,7,7а-тетрагидро-1H-изоиндол-1,3 (2Н)-дион получают по реакции 6-(2-фторфенил)-8-иод-1-метил-4H-[1,2,4] триазоло[4,3-a][1,4] -бензодиазепина с N-пропаргил-тетрагидрофталимидом [см. W.E. Hahn, A. Sokolowska, Soc. Sci. Lodz. Acta Chim. 18, 187 (1974)] следуя методике, описанной в примере 1. Продукт реакции выделяют хроматографически и кристаллизуют из этанола, чтобы получить бесцветные кристаллы с т.пл. 259-261oC. Согласно аналитическим данным эти кристаллы содержат 0,33 моль воды.
Пример 22
Реакция 6-(2-фторфенил)-8-иод-1-метил-4H-[1,2,4]триазоло[4,3-a] [1,4]-бензодиазепина с 3,4-дигидро-4-метил-1-(2-пропинил)-1H-1,4- бензодиазепин-2,5-дионом, как это описано в примере 9, дает после хроматографии и кристаллизации из этилацетата бесцветные кристаллы 1-[3-[6-(2- фторфенил)-1-метил-4H-[1,2,4] триазоло[4,3-a] [1,4] - бензодиазепин-8-ил]-2-пропинил]-3,4-дигидро-4-метил-1H-1,4- бензодиазепин-2,5-диона с т.пл. 179-182oC. Кристаллы содержат согласно аналитическим и ЯМР-данным 0,16 моль этилацетата и 0,66 моль воды.
Необходимый ацетилен получают следующим образом.
2,6 г (22 ммоль) бромистого пропаргила добавляют к смеси 3,8 г (20 ммоль) 3,4-дигидро-4-метил-1H-1,4-бензодиазепин-2,5-диона (см. M. Uskokavic и W. Wenner, патент США 3,261,828, июль 1966 г.) 3,4 окиси бария и 100 мл диметилформамида. После перемешивания при комнатной температуре в течение 2 ч реакционную смесь разделяют на части между водой и хлористым метиленом. Органическую фазу отделяют, промывают водой, высушивают и выпаривают. Остаток растворяют в этилацетате и продукт реакции кристаллизуют добавлением гексана, чтобы получить 3,4-дигидро-4-метил-1-(2-пропинил)-1Н-1,4-бензодиазепин-2,5(2Н)-дион в виде бесцветных кристаллов с т.пл. 148-150oC.
Пример 23
Реакция 6-(2-фторфенил)-8-иод-1-метил-4Н-[1,2,4]триазоло[4,3-a] [1,4]-бензодиазепина с 1-(3-пиридинилокси)-2-пропином [см. J.Bruhn, J. Zsindely, H. Schmid и G. Frater, Helv. Chim. Acta, 61, 2542 (1978)] как это описано в примере 1, дает после хроматографии и кристаллизации из смеси этанола и эфира бесцветные кристаллы 6-(2-хлорфенил)-1-метил-8-[3-(3-пиридинилокси)-1-пропинил] -4H-[1,2,4] триазоло[4,3-a] [1,4]-бензодиазепина с т.пл. 128-130oC. Эти кристаллы содержат 0,66 моль воды согласно аналитическим данным.
Пример 24
6-(2-Хлорфенил)-1-метил-8-(3-фенокси-1-пропинил)-4Н-[1,2,4] триазоло[4,3-a] [1,4] -бензодиазепина получают реакцией 6-(2-фторфенил)-8- иод-1-метил-4Н-[1,2,4] триазоло[4,3-a][1,4]-бензодиазепина с 1-фенокси-2-пропином, как это описано в примере 1. Хроматографическое выделение и кристаллизация из этилацетата дает бесцветные кристаллы с т.пл. 160-162oC.
Пример 25
а) Раствор 54,8 г 5-(2-хлорфенил)-1,3-дигидро-2Н-тиено-[2,3-e] [1,4]-диазепин-2-она (патент Нидерландов 7,205,730, ноябрь 1972, Хоффманн-Ля Рош энд Комп. АГ, Базель) в 350 мл уксусной кислоты и 350 мл метанола обрабатывают с помощью 64,4 г монохлористого иода и 16,2 г ацетата натрия. Смесь перемешивают в течение 15 мин при комнатной температуре. Затем добавляют раствор 65 г бисульфита натрия в 350 мл воды и перемешивание продолжают в течение 10 мин. Смесь нейтрализуют добавлением 500 мл концентрированного аммиака и 1 кг льда. Выпадающий в осадок продукт реакции отфильтровывают и промывают водой и этанолом. Перекристаллизация из смеси тетрагидрофурана и этанола дает не совсем белые кристаллы 5-(2-хлорфенил)-1,3-дигидро-7-иод-2Н-тиено-[2,3-e][1,4]-диазепин-2-она с т.пл.229-231oC.
б) Смесь 70 г 5-(2-хлорфенил)-1,3-дигидро-7-иод-2Н-тиено-[2,3-e] [1,4] -диазепин-2-она, 43,3 г пентасульфида фосфора, 45 г бикарбоната натрия и 700 мл диглима перемешивают и нагревают до 70-80oC в течение 2 ч. После охлаждения до комнатной температуры смесь воды и раздробленного льда добавляют и перемешивание продолжают в течение 15 мин. Выпадающий в осадок 5-(2-хлорфенил)-1,3-дигидро-7-иод-2Н- тиено-[2,3-e] [1,4]-диазепин-2-он собирают фильтрацией, промывают водой и отсасывают досуха.
в) Смесь 64,4 г 5-(2-хлорфенил)-1,3-дигидро-7-иод-2Н-тиено-[2,3- e][1,4] -диазепин-2-тиона, 650 мл тетрагидрофурана и 65 мл гидразина перемешивают при комнатной температуре в течение 30 мин. Растворитель выпаривают при пониженном давлении и остаток перемешивают вместе с 275 мл хлористого метилена и 275 мл воды в течение 15 мин. Выпадающее в осадок кристаллическое вещество отфильтровывают и промывают водой и эфиром. Сырой продукт реакции - 5-(2-хлорфенил)-2-гидразино-7-иод-2Н-тиено-[2,3-e] [1,4]-диазепин объединяют вместе с 375 мл этилацетата, 170 мл триэтилового эфира ортоуксусной кислоты и несколькими кристаллами паратолуолсульфоновой кислоты и смесь нагревают на паровой бане в течение 30 мин. Продукт реакции кристаллизуется во время этого процесса и собирается после охлаждения. Перекристаллизация из смеси хлористого метилена и этанола дает бесцветные кристаллы 4-(2-хлорфенил)-2-иод-9-метил-6Н-тиено-[3,2-f] [1,2,4] триазоло[4,3-a] [1,4]-диазепина с т.пл. 254-256oC.
г) Смесь 0,88 г (2 ммоль) 4-(2-хлорфенил)-2-иод-9-метил-6Н-тиено-[3,2-f] [1,2,4] триазоло[4,3-a] [1,4]-диазепина, 565 мг (2,4 ммоль) 2-(2-пропинил)-1Н-бенз[oe]-изохинолин-1,3(2Н)-диона, 1 мл триэтиламина, 20 мг иодистой меди (I), 90 мг трифенилфосфина и 20 мл диметилформамида дегазируют медленным током аргона в течение 15 мин. Затем добавляют 30 мг ацетата палладия и смесь перемешивают при комнатной температуре под аргоном в течение 20 ч. Тонкослойная хроматография показывает практически полное завершение реакции после 5 ч. Реакционную смесь разделяют на части между хлористым метиленом и насыщенным водным раствором бикарбоната натрия. Органический слой отделяют, высушивают над сульфатом натрия и выпаривают при пониженном давлении, под конец азеотропно с помощью ксилола, чтобы удалить остаточный диметилформамид. Остаток хроматографируют над 40 г силикагеля, используя 5 об. этанола в хлористом метилене для элюирования. Очищенные фракции объединяют и выпаривают. Кристаллизация из смеси метанола и этилацетата дает 0,58 г (53%) 2-[3-[4-(2-хлорфенил)-9-метил -6Н-тиено[3,2-f][1,2,4]триазоло[4,3-a][1,4]-диазепин-2-ил] -2-пропинил] -1Н-бенз[oe] -изохинолин-1,3(2Н)-диона с т.пл. 188-192oC. Присутствует также другая кристаллическая модификация с т.пл. 252-254oC.
Пример 26
1-[3-[4-(2-Хлорфенил)-9-метил-6Н-тиено[3,2-f][1,2,4]триазоло[4,3-a][1,4] -диазепин-2-ил] -2-пропинил]-1Н-индол-2,3-дион получают так, как это описано в примере 25 г, реакцией 4-(2-хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло[4,3-a] [1,4]-диазепина с 1-(2-пропинил)-1Н-индол-2,3-дионом [см. A. Lindquist et al, Acta Pharm. Suecica, 9, 99 (1972)] Хроматографическое выделение и кристаллизация из смеси метанола и этилацетата дает оранжевые кристаллы с т.пл. 185-190oC со вспениванием при 130-140oC. Эти кристаллы содержат согласно аналитическим и спектральным данным молярные количества этилацетата.
Пример 27
Реакция 4-(2-хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло[4,3-a] [1,4] -диазепина с 1-(2-пропинил)-бенз[cd]-индол-2(1Н) -оном, как это описано в примере 25г, дает после хроматографической очистки и медленной кристаллизации из этилацетата желтые кристаллы с т.пл. 202-205oC. Эти кристаллы содержат согласно аналитическим данным 0,75 моль воды. Обработка этого продукта этанольным раствором хлористого водорода и добавление этилацетата дает кристаллический хлоргидрат 1-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло[4,3-a][1,4]-диазепин-2-ил]-2-пропинил]-бенз[cd]-индол-2(1Н) -она с т.пл. 219-222oC.
Пример 28
1-[3-[4-(2-Хлорфенил)-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло [4,3-a] [1,4] -диазепин-2-ил] -2-пропинил] -1,3-дигидро-2Н-индол-2-он получают взаимодействием 4-(2-хлорфенил)-2-иод-9-метил-6Н-тиено [3,2-f] [1,2,4] триазоло[4,3-a] [1,4] -диазепина с 1,3-дигидро-1-(2-пропинил)-2Н-индол-2-оном [ссылка A. Lindquist et al, Acta Pharm. Suecica, 9, 99 (1972)] как это описано в примере 25г. Этот продукт реакции выделяют с помощью хроматографии и кристаллизуют из этилацетата, чтобы получить желтоватые кристаллы с т.пл. 203-206oC. Эти кристаллы содержат согласно ЯМР-спектральным и аналитическим данным 0,66 моль воды и следы этилацетата.
Пример 29
Реакция 4-(2-хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло[4,3-a] [1,4]-диазепина с 1-(2-пропинил)-1Н-бензимидазолом [см. И.И. Попов и др. Химия гетероциклических соединений, 551 (1973)] дает после хроматографического выделения и кристаллизации из смеси этанола и гексана не совсем белые кристаллы с т.пл. 215-217oC. Кристаллы 2-[3-(1Н-бензимидазол-1-ил)-1-пропинил] -4-(2-хлорфенил)-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло[4,3-a] [1,4] -диазепина содержат 0,66 моль воды, что показано аналитическими данными.
Пример 30
2-[3-[4-(2-Хлорфенил)-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло [4,3-a] [1,4]-диазепин-2-ил]-2-пропинил]-1,2,4-триазоло[4,3-a]пиридин-3(2Н)-он получают реакцией 4-(2-хлорфенил)-2-иод-9-метил-6Н-тиено [3,2-f][1,2,4]триазоло[4,3-a] [1,4] -диазепина с 2-(2-пропинил)-1,2,4-триазоло[4,3-a]-пиридин-3(2Н)-оном, как это описано в примере 25г. Продукт реакции выделяют с помощью хроматографии, используя 7,5 об.-ный этанол в хлористом метилене для элюирования. Кристаллизация из этанола дает 0,55 г (55%) желтых кристаллов с т. пл. 220-223oC. Согласно аналитическим и ЯМР-спектральным данным эти кристаллы содержат 0,25 моль этанола.
Пример 31
1,1-Диокись 2-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло[4,3-a] [1,4]-диазепин-2-ил]-2-пропинил]-1,2-бензизотиазол-3(2Н)-она получают взаимодействием 4-(2-хлорфенил)-2-иод-9-метил-6Н-тиено [3,2-f][1,2,4] триазоло[4,3-a] [1,4] -диазепина с 1,1-диокисью 2-(2-пропинил)-1,2-бензизотиазол-3(2Н)-она (см. R. Granger и J. Giroux, патент Франции 1,273,867, февраль 1962), как это описано в примере 25г. Продукт реакции выделяют с помощью хроматографии и кристаллизуют из этилацетата, чтобы получить бесцветные кристаллы с т.пл. 232-234oC.
Пример 32
Реакция 4-(2-хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло[4,3-a] [1,4]-диазепина с 1-(3-пиридинилокси)-2-пропином [см. J. Bruhn, J. Zsindely, H. Schmid и G. Frater, Helv. Chim. Acta, 61, 2542 (1978)] при условиях, описанных в примере 25г, дает после обычного хроматографического выделения смолистое вещество, содержащее 4-(2-хлорфенил)-9-метил-2-[3-(3-пиридинилокси)-1-пропинил] -6Н-тиено[3,2-f] [1,2,4]триазоло[4,3-a][1,4]-диазепин, который не кристаллизуется и поэтому был охарактеризован только лишь спектроскопически.
ЯМР (CDCl3): 2,72 (s, 3,CH3); 4,95 (s, 4, CH2; C6-H); 6,8 (s, 1, C3-H); 7,2-7,6 (m, 6, ароматический Н); 8,26 (m, 1) и 8,36 (уширенный s, 1), пиридин С2 и С6-Н м.д.
Пример 33
4-(2-Хлорфенил)-9-метил-2-[3-(1H-индазол-1-ил)-1-пропинил] -6Н-тиено[3,2-f] [1,2,4] триазоло[4,3-a][1,4]-диазепин получают, как описано в примере 25г, взаимодействием 4-(2-хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло[4,3-a][1,4]-диазепина с 1-(2-пропинил)-1Н-индазолом [см. П. В. Ткаченко и др. Химия гетероциклических соединений, 1542 (1975)] Продукт реакции выделяют и очищают с помощью хроматографии и кристаллизуют из смеси этилацетата и эфира, чтобы получить не совсем белые кристаллы с т.пл. 170-173oC.
Пример 34
Реакция 6-(2-фторфенил)-8-иод-1-метил-4Н-[1,2,4]триазоло[4,3-a] [1,4]-бензодиазепина с 1-(2-пропинил)-1Н-индолом [см. A.J. Hubert и H. Reimlinger, J. Chem. Soc. c.606 (1968)] как это описано в примере 9, дает после хроматографии и кристаллизации из смеси этилацетата и эфира не совсем белые кристаллы 6-(2-фторфенил)-8-[3-(1H-индол-1-ил)-1-пропинил] -1-метил-4Н-[1,2,4] триазоло[4,3-a] [1,4]-бензодиазепина с т.пл. 167-169oC.
Пример 35
Реакция 6-(2-фторфенил)-8-иод-1-метил-4Н-[1,2,4]триазоло[4,3-a] [1,4]-бензодиазепина с 3,7-дигидро-3,7-диметил-1-(2-пропинил)-1Н-пурин-2,6-дионом [см. J. W. Daly, W.L. Padgett и M.T. Shamin, J. Med. Chem. 29, 1305 (1986)] дает после хроматографии и кристаллизации из смеси хлористого метилена и этанола не совсем белые кристаллы 1-[3-[6-(2-фторфенил)-1-метил-4Н-[1,2,4] триазоло[4,3-a] [1,4] -бензодиазепин-8-ил]-2-пропинил]-3,7-дигидро-3,7-диметил-1Н-пурин-2,6-диона с т.пл. 290-292oC. Эти кристаллы содержат 0,75 моль воды согласно аналитическим данным.
Пример 36
2-[4-[6-(2-Фторфенил)-1-метил-4Н-[1,2,4] триазоло[4,3-a] [1,4]-бензодиазепин-8-ил]-3-бутинил]-1Н-изоиндол-1,3(2Н)-дион получают реакцией 6-(2-фторфенил)-8-иод-1-метил-4Н-[1,2,4] триазоло[4,3-a] [1,4]-бензодиазепина с 2-(3-бутин-1-ил)-1Н-изоиндол-1,3(2Н)-дионом [см. K.J. Hoffmann, P. Steuberg, C. Ljunggren, V. Svensson, J.L.G. Nilsson, A. Erikson, A. Hartkoorn и R. Lunden, J. Med. Chem. 18, 278 (1975)] как это описано в примере 1. Продукт реакции выделяют с помощью хроматографии и кристаллизуют из этанола, чтобы получить бесцветные кристаллы с т. пл. 128-130oC (со вспениванием). Эти кристаллы содержат согласно аналитическим и спектральным данным молярные количества этанола.
Пример 37
4-[3-[4-(2-Хлорфенил)-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло [4,3-a] [1,4] -диазепин-2-ил] -2-пропинил]-2Н-1,4-бензоксазин-3(4Н)-он получают взаимодействием 4-(2-хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло[4,3-a] [1,4] -диазепина с 4-(2-пропинил)-2Н-1,4-бензоксазин-3(4Н)-оном [см. A. Lindquist et al, Acta Pharm. Suecica, 9, 99 (1972)] как это описано в примере 25г. После хроматографического выделения продукт реакции кристаллизуют из этилацетата, чтобы получить желтоватые кристаллы с т.пл. 190-192oC. Обработка этанольным раствором хлористого водорода дает кристаллический хлоргидрат с т.пл. 215-218oC.
Пример 38
1-[3-[4-(2-Хлорфенил)-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло [4,3-a] [1,4] -диазепин-2-ил] -2-пропинил]-3,7-дигидро-3,7-диметил-1Н-пурин-2,6-дион получают реакцией 4-(2-хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f][1,2,4]триазоло[4,3-a] [1,4] -диазепина с 3,7-дигидро-3,7-диметил-1-(2-пропинил)-1Н-пурин-2,6-дионом [см. J.W. Daly, W.L. Padgett и M.T. Shamin, J. Med. Chem. 29, 1305 (1986)] как это описано в примере 25г. Продукт реакции выделяют с помощью хроматографии и кристаллизуют из этилацетата, чтобы получить желтые кристаллы с т.пл. 277-280oC.
Пример 39
Реакция 4-(2-хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло[4,3-a] [1,4] -диазепина с 3,7-дигидро-3,7-диметил-7-(2-пропинил)-1Н-пурин-2,6-дионом [см. J.W.Daly, W.L.Padgett и M.T. Shamin, J. Med. Chem. 29, 1305 (1986)] с последующим хроматографическим разделением и кристаллизацией из смеси этилацетата и этанола дает желтые кристаллы 7-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло [4,3-a][1,4]-диазепин-2-ил]-3,7-дигидро-1,3-диметил-1Н-пурин-2,6-диона с т.пл. 229-232oC. Эти кристаллы содержат 0,125 моль этилацетата согласно аналитическим и спектральным данным.
Пример 40
Реакция 4-(2-хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло[4,3-a] [1,4] -диазепина с 2-(3-бутин-1-ил)-1Н-бенз[de] -изохинолин-1,3(2Н)-дионом, как это описано в примере 25г, дает после хроматографии и кристаллизации из этилацетата светложелтые кристаллы 2-[4-[4-(2-хлорфенил)-9-метил-6Н-тиено[3,2-f] [1,2,4]триазоло[4,3-a] [1,4]-диазепин-2-ил]-3-бутинил]-1Н-бенз[de]изохинолин-1,3(2Н)-диона с т.пл. 175-179oC. Присутствует также более высокоплавящаяся кристаллическая модификация с т.пл. 227-229oC.
Ацетиленовый компонент реакции получают следующим образом.
Смесь 6 г (0,03 моль) 1Н-бенз[de]изохинолин-1,3(2Н)-диона, 4 г (0,0355 моль) трет. -бутоксида калия, 9 г (0,04 моль) 4-тозилокси-1-бутина [см. G. Eglinton и M.C. Whiting, J. Chem. Soc. 3650 (1950)] и 150 мл диметилформамида нагревают на паровой бане с перемешиванием в течение 1,5 ч. Массу растворителя затем удаляют при пониженном давлении и остающуюся суспензию фильтруют. Фильтрат разбавляют водой и выпадающий в осадок продукт реакции собирают фильтрацией и растворяют в хлористом метилене. Раствор высушивают и пропускают над запрессованной силикагелью, используя хлористый метилен для элюирования. Кристаллизация из смеси хлористого метилена и этанола дает 2-(3-бутин -1-ил)-1Н-бенз[de] изохинолин-1,3(2Н)-дион в виде бесцветных игл с т.пл. 191-193oC.
Пример 41
а) Смесь 2,52 г 6-(2-фторфенил)-8-иод-1-метил-4Н-{1,2,4]триазоло [4,3-a] [1,4] -бензодиазепина, 270 мг трифенилфосфина, 60 мг иодистой меди (I), 1,5 мл триэтиламина и 60 мл диметилформамида перемешивают и дегазируют медленным током аргона в течение 10 мин. Триметилсилилацетилен 1,2 мл добавляют затем в смесь и дегазирование продолжают в течение 2 мин. В это время добавляют 90 мг ацетата палладия и смесь перемешивают под аргоном в течение 4 ч при комнатной температуре. Реакционную смесь расслаивают между хлористым метиленом и насыщенным раствором бикарбоната натрия. Органический слой промывают водой, высушивают и выпаривают, под конец азеотропно с помощью ксилола. Остаток хроматографируют над 60 г силикагеля (Мерк, 70-230 меш), используя 5 об.-ный этанол в хлористом метилене для элюирования. Кристаллизация объединенных очищенных фракций из смеси этилацетата и гексана дает 2,05 г (86%) бесцветных кристаллов 6-(2-фторфенил)-1-метил-8-[(триметилсилил)-этинил]-4Н-[1,2,4] триазоло[4,3-a][1,4]-бензодиазепина с т.пл. 218-220oC.
б) К раствору 2,3 г 6-(2-фторфенил)-1-метил-8-[(триметилсилил)- этинил] -4Н-[1,2,4] триазоло[4,3-a] [1,4]-бензодиазепина в 50 мл этанола добавляют 1 мл 10N раствора гидроокиси натрия. Смесь перемешивают под аргоном при комнатной температуре в течение 1 ч, а затем разделяют на части между хлористым метиленом и насыщенным водным раствором бикарбоната натрия. Органическую фазу отделяют, высушивают и выпаривают. Остаток фильтруют над мягкой набивкой силикагеля, используя 5%-ный этанол в хлористом метилене для элюирования. Фильтрат выпаривают и остаток кристаллизуют из смеси этилацетата и гексана, чтобы получить бесцветные кристаллы 8-этинил-6-(2-фторфенил)-1-метил-4Н-[1,2,4] триазоло[4,3-a][1,4]-бензодиазепина с т.пл.258-260oC.
в) Смесь 316 мг (1 ммоль) 8-этинил-6-(2-фторфенил)-1-метил-4Н-[1,2,4] триазоло[4,3-a] [1,4] -бензодиазепина, 200 мг (1,25 ммоль) 5-бромпиримидина, 45 мг трифенилфосфина, 10 мг иодистой меди (I), 0,5 мл триэтиламина и 10 мл диметилформамида перемешивают и дегазируют в течение 10 мин медленным током аргона. Затем добавляют 15 мг ацетата палладия и перемешивание под аргоном продолжают в течение 24 ч. Реакционную смесь расслаивают между водным раствором бикарбоната натрия и хлористым метиленом. Органический слой промывают водой, высушивают и выпаривают, под конец азеотропно с помощью ксилола. Остаток хроматографируют над 20 г силикагеля (Мерк, 70-230 меш), используя 5% -ный этанол в хлористом метилене. Объединенные очищенные фракции выпаривают и остаток кристаллизуют из этилацетата, чтобы получить не совсем белые кристаллы 6-(2-фторфенил)-1-метил-8-[(5-пиримидинил)-этинил]-4Н-[1,2,4]триазоло[4,3-a][1,4]-бензодиазепина с т.пл. 143-146oC.
Пример 42
а) Подобным образом получают 6-(2-хлорфенил)-1-метил-8-[(триметилсилил)-этинил] -4Н-[1,2,4] триазоло[4,3-a][1,4]-бензодиазепин, используя реакцию 6-(2-хлорфенил)-8-иод-1-метил-4Н-[1,2,4]триазоло[4,3-a][1,4] -бензодиазепина с триметилсилилацетиленом, как это описано в примере 41а. Продукт реакции выделяют с помощью хроматографии и кристаллизуют из смеси этилацетата и гексана, чтобы получить бесцветные кристаллы с т.пл. 243-245oC.
б) 6-(2-Хлорфенил)-8-этинил-1-метил-4Н-[1,2,4] триазоло[4,3-a] [1,4]-бензодиазепин получают, как это описано в примере 41б, обработкой 6-(2-хлорфенил)-1-метил-8-[(триметилсилил)-этинил]-4Н-[1,2,4]триазоло[4,3-a][1,4] -бензодиазепина гидроокисью натрия в этаноле. Продукт реакции кристаллизуют из этанола, чтобы получить бесцветные кристаллы с т.пл. 304-306oC.
в) 6-(2-Хлорфенил)-1-метил-8-(2-тиенилэтинил)-4Н-[1,2,4]триазоло[4,3-a] [1,4] -бензодиазепин получают, как это описано в примере 41в, реакцией 6-(2-хлорфенил)-8-этинил-1-метил-4Н-[1,2,4] триазоло[4,3-a] [1,4]-бензодиазепина с 2-иодтиофеном. Продукт реакции выделяют и очищают с помощью хроматографии и кристаллизуют из этилацетата, чтобы получить кристаллы с т.пл. 160-163oC. Эти кристаллы содержат согласно аналитическим и спектральным данным 0,25 моль этилацетата и моль воды.
Пример 43.
а) Реакция 4-(2-хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f][1,2,4]триазоло[4,3-a][1,4]-диазепина c триметилсилилацетиленом, как это описано в примере 41а, дает после хроматографического выделения и кристаллизации из этилацетата и гексана не совсем белые кристаллы 4-(2-хлорфенил)-9-метил-2-[(триметилсилил)-этинил] -6Н-тиено[3,2-f] [1,2,4]триазоло[4,3-a][1,4]-диазепина c т.пл. 135-138oC.
б) Обработка 4-(2-хлорфенил)-9-метил-2-[(триметилсилил)-этинил]-6Н-тиено[3,2-f][1,2,4]триазоло[4,3-a][1,4]-диазепина гидроокисью натрия в этаноле дает после хроматографической очистки и кристаллизации из смеси метанола и этилацетата бесцветные кристаллы 4-(2-хлорфенил)-2-этинил-9-метил-6Н-тиено[3,2-f][1,2,4]триазоло[4,3-a] [1,4]-диазепина c т.пл.232-233oC.
в) Смесь 0,34 г 4-(2-хлорфенил)-2-этинил-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло[4,3-a] [1,4] -диазепина, 45 мг трифенилфосфина, 10 мг иодистой меди (I), 1 мл триэтиламина, 0,38 г 1-иоднафталина и 10 мл диметилформамида дегазируют в течение 10 мин медленным током аргона. После добавления 15 мг ацетата палладия смесь перемешивают под аргоном в течение 2 ч при комнатной температуре. Реакционную смесь выливают в насыщенный водный раствор бикарбоната натрия и лед. Выпадающий осадок отфильтровывают, промывают водой и растворяют в хлористом метилене. Раствор высушивают и выпаривают, а остаток хроматографируют на 15 г силикагеля, используя 25 об. гексана в тетрагидрофуране. Объединенные очищенные фракции, содержащие продукт реакции, выпаривают и остаток кристаллизуют из смеси этилацетата и гексана, чтобы получить не совсем белые кристаллы 4-(2- хлорфенил)-2-[(1-нафтил)-этинил]-9-метил-6Н-тиено[3,2-f][1,2,4]триазоло[4,3-a][1,4]-диазепина с т.пл. 197-199oC.
Пример 44
4-(2-Хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f][1,2,4]триазоло[4,3-a][1,4] -диазепин подвергают взаимодействию с 1-(2-пропинил)-2(1Н)-хинилиноном [см. A.Lindquist et al, Acta Pharm. Suecica, 9, 99 (1972)] как это описано в примере 25г. Продукт реакции выделяют хроматографически, пропуская раствор над 50 г силикагеля, используя 5 об. этанол в хлористом метилене и дополнительно очищают с помощью повторной хроматографии над 50 г силикагеля, используя тетрагидрофуран. Кристаллизация из смеси этилацетата и метанола дает не совсем белые кристаллы 1-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено[3,2-f][1,2,4]триазоло[4,3-a] [1,4] -диазепин-2-ил] -2-пропинил] -2(1Н)-хинолинона с т.пл. 162-165oC. Эти кристаллы содержат согласно аналитическим данным 0,5 молярные количества воды.
Пример 45
Смесь 33 г (0,075 моль) 4-(2-хлорфенил)-2-иод-9-метил-6Н-тиено [3,2-f] [1,2,4] триазоло[4,3-a][1,4]-диазепина, 21 г (0,113 моль) 3,4-дигидро-1-(2-пропинил)-2(1Н)-хинолинона [см.A.Lindquist et al, Acta Pharm. Suecica, 9, 99 (1972)] 0,75 г трифенилфосфина, 0,2 г иодистой меди (I), 60 мл триэтиламина и 600 мл диметилформамида перемешивают и дегазируют током аргона в течение 30 мин. За это время добавляют 0,225 г ацетата палладия и смесь перемешивают при комнатной температуре под аргоном в течение 3 дн. Смесь выливают в 2,5 насыщенного водного раствора бикарбоната натрия и льда. После перемешивания в течение 15 мин, выпадающий осадок отфильтровывают, промывают водой и отсасывают досуха. Это вещество растворяют в хлористом метилене и раствор промывают бикарбонатным раствором и высушивают над сульфатом натрия. Растворитель удаляют при пониженном давлении и остаток растворяют путем нагревания в этилацетате. После затравки и охлаждения кристаллический продукт реакции собирают и перекристаллизовывают из смеси метанола и этилацетата, чтобы получить не совсем белые кристаллы 1-[3-[4-(2-хлорфенил)-9-метил-6Н- тиено[3,2-f] [1,2,4] триазоло[4,3-a][1,4]-диазепин-2-ил]-2-пропинил]-3,4-дигидро-2(1Н)-хинолинона с т.пл. 180-182oC.
Пример 46
4-(2-Хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f][1,2,4]триазоло[4,3-a][1,4] -диазепин подвергают взаимодействию с 2,3-дигидро-2-(2-пропинил)-1Н-бенз[de] -изохинолин-1-оном при условиях, описанных в примере 25г. Продукт реакции выделяют с помощью хроматографии над 40-кратным количеством силикагеля, используя тетрагидрофуран для элюирования. Фракции, содержащие названное выше соединение, объединяют и повторно хроматографируют над 30-кратным количеством силикагеля, используя 5 об. этанола в хлористом метилене. Объединенные, очищенные фракции кристаллизуют из смеси метанола и этилацетата, чтобы получить желтоватые кристаллы 2-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено[3,2-f][1,2,4]триазоло[4,3-a][1,4]-диазепин-2-ил]-2-пропинил]-2,3-дигидро-1Н-бенз[de]-изохинолин-1-она с т.пл. 205-210oC с разложением. Эти кристаллы содержат 0,66 моль воды согласно аналитическим и спектральным данным.
Требуемое пропаргиловое производное получают следующим образом.
Смесь 2 г 2-(2-пропинил)-1Н-бенз[de]-изохинолин-1,3(2Н)-диона, 0,75 г боргидрида натрия, 50 мл этанола и 50 мл тетрагидрофурана нагревают на паровой бане до тех пор, пока не завершится растворение. Дополнительную порцию 0,25 г боргидрида добавляют и тетрагидрофуран отгоняют на паровой бане в пределах 30 мин. Оставшуюся смесь охлаждают, разбавляют ледяной водой, создают буфер с помощью уксусной кислоты и разбавляют водным раствором бикарбоната натрия. Выпадающий в осадок продукт реакции отфильтровывают после перемешивания над льдом, отсасывают досуха и растворяют примерно в 250 мл хлористого метилена. Раствор высушивают и выпаривают, а остаток превращают в шлам с помощью смеси хлористого метилена и гексана. Кристаллы собирают и промывают эфиром, чтобы получить 0,67 г 2,3-дигидро-3-окси-2-(2-пропинил)-1Н-бенз[de]-изохинолин-2-она, который восстанавливают в дальнейшем следующим образом.
К перемешиваемому раствору 0,6 г вышеуказанного интермедиата в 6 мл трифторуксусной кислоты малыми порциями добавляют 0,3 г боргидрида натрия. После 15 мин реакционного времени смесь разделяют на части между льдом, гидроокисью аммония и хлористым метиленом. Органический слой отделяют, высушивают и выпаривают, чтобы получить кристаллический остаток, который хроматографируют над 30 г силикагеля, используя 10 об. этилацетата в хлористом метилене. Кристаллизация объединенных очищенных фракций из смеси этилацетата и гексана дает бесцветные кристаллы 2,3-дигидро-2-(2-пропинил)-1Н-бенз[de] -изохинолин-1-она с т.пл. 139-140oC.
Пример 47
4-(2-Хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло [4,3-a] [1,4] -диазепин подвергают взаимодействию с 1,3-дигидро-1-метил-3-(2-пропинил)-2Н-бензимидазол-2-оном при условиях, описанных в примере 25г. Продукт реакции выделяют с помощью хроматографии над 50-кратным количеством силикагеля, используя тетрагидрофуран. Кристаллизация объединенных гомогенных фракций из этанола дает светложелтые кристаллы 1-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено[3,2-f] [1,2,4]триазоло[4,3-a][1,4]-диазепин-2-ил]-2-пропинил]-1,3-дигидро-3-метил-2Н-бензимидазол-2-она с т.пл. 188-191oC.
Требуемый ацетилен получают следующим образом.
К раствору 2,5 г (16,9 ммоль) 1,3-дигидро-1-метил-2Н-бензимидазол-2-она в 25 мл диметилформамида добавляют 2,1 г (18,5 ммоль) трет.-бутоксида калия. После перемешивания под азотом в течение 15 мин добавляют 2,21 г (18,5 ммоль) бромистого пропаргила и смесь перемешивают при комнатной температуре в течение 30 мин. Смесь разбавляют ледяной водой и выпадающий осадок отфильтровывают, промывают водой и отсасывают досуха. Сырой продукт реакции пропускают через силикагель, используя 10 об. этилацетата в хлористом метилене для элюирования. Кристаллизация из смеси этилацетата и гексана дает бесцветные кристаллы 1,3-дигидро-1-метил-3-(2-пропинил)-1Н- бензимидазол-2-она с т.пл. 110-112oC.
Пример 48
4-(2-Хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f][1,2,4]триазоло[4,3-a][1,4] -диазепин подвергают взаимодействию с рацемическим 2а,3,4,5-тетрагидро-2а-[(2-пропинил)-бенз[cd]-индол-2(1Н)-оном при условиях, описанных в примере 9. Продукт реакции выделяют с помощью хроматографии над 50-кратным количеством силикагеля, используя 5 об.-ного этанола в хлористом метилене для элюирования. Объединенные очищенные фракции выпаривают и остаток кристаллизуют из смеси этилацетата и эфира, чтобы получить бесцветные кристаллы рацемического 2а-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено[3,2-f] [1,2,4]триазоло[4,3-a][1,4]-диазепин-2-ил] -2-пропинил]-2а,3,4,5-тетрагидробенз[cd]-индол-2(1Н)-она с т. пл. 215-217oC.
Требуемое пропаргиловое соединение получают следующим образом.
К раствору 6,92 г (0,04 моль) рацемического 2а,3,4,5-тетрагидробенз[cd] -индол-(1Н)2-она в 50 мл диметилформамида добавляют 4,94 г (0,044 моль) трет.-бутоксида калия. После перемешивания в течение 15 мин под азотом добавляют 5,23 г 3,9 мл бромистого пропаргила и перемешивание продолжают в течение 30 мин. Смесь разбавляют водой и льдом и выпадающий осадок собирают фильтрацией, промывают водой и отсасывают досуха. Твердые частицы растворяют в хлористом метилене и раствор высушивают и выпаривают. Кристаллизация остатка из этилацетата дает сырой продукт реакции - рацемический 2а,3,4,5-тетрагидро-2а-(2-пропинил)-бенз[cd]-индол-2(1Н)-он с т.пл.174-177oC, который перекристаллизовывают из этилацетата при 177-180oC.
Пример 49
4-(2-Хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f][1,2,4]триазоло[4,3-a][1,4] -диазепин подвергают реакции с 5-(2-пропинил)-5Н-карбазолом [см. J.L. Dumont et al. Bull. Soc Chim. Fr. 1197 (1967)] при условиях, описанных в примере 25г. Продукт реакции очищают хроматографией над 60-кратным количеством силикагеля, используя 5 об. этанола в хлористом метилене. Объединенные очищенные фракции не кристаллизуются и вязкая смола превращается до кристаллического дихлоргидрата обработкой избытком этанольного раствора хлористого водорода в смеси этанола и этилацетата. Желтые кристаллы дихлоргидарата 2-[3-(9Н-карбазол-9-ил)-1-пропинил] -4-(2-хлорфенил)-9-метил-6Н-тиено[3,2-f] [1,2,4]триазоло[4,3-a][1,4]-диазепина имеют т.пл. 180-184oC.
Пример 50
Смесь 44 г (0,1 моль) 4-(2-хлорфенил)-2-иод-9-метил-6Н-тиено [3,2-f] [1,2,4] триазоло[4,3-a] [1,4] -диазепина, 28 г (0,12 моль) 5-(2-пропинил)-6(5Н)-фенантридинона [см. R. F. Cookson et al. Heterocyclic Chem. 9, 475 (1972)] 1 г трифенилфосфина, 0,25 г иодистой меди (I), 80 мл триэтиламина и 800 мл диметилформамида дегазируют медленным током аргона в течение 30 мин. Затем добавляют 0,3 г ацетата палладия и смесь перемешивают под аргоном в течение 4 дн при комнатной температуре. Нерастворимое вещество удаляют фильтрацией через цеолит и фильтрат концентрируют примерно до 400 мл при пониженном давлении. Этот раствор выливают на 2 л насыщенного водного раствора бикарбоната натрия с перемешиванием. Выпадающий осадок собирают фильтрацией после 10 мин и промывают водой и отсасывают досуха. Твердые частицы разделяют на части между 2 л хлористого метилена, содержащего 5 об. этанола, и раствором бикарбоната натрия. Органический слой высушивают над сульфатом натрия, фильтруют и частично выпаривают. После разбавления с помощью 500 мл этилацетата раствор концентрируют на паровой бане с кристаллизацией продукта реакции. После охлаждения кристаллы собирают и промывают этилацетатом и эфиром, чтобы получить 56,5 г продукта реакции. Аналитический образец перекристаллизовывают сначала из этанола, а затем из смеси тетрагидрофурана и этилацетата, чтобы получить не совсем белые кристаллы 5-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено[3,2-f] [1,2,4]триазоло[4,3-a][1,4]-диазепин-2-ил]-2- пропинил]-фенантридин-6(5Н)-она с т.пл. 247-249oC.
Пример 51
4-(2-Хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f][1,2,4]триазоло[4,3-a][1,4] -диазепин подвергают взаимодействию с 6-(2-пропинил)-5Н-дибенз[c,e]-азепин-5,7(6Н)-дионом [см. J.R. Grunder et al. J. Pharm. Sci.62, 1204 (1973)] при условиях, используемых в примере 25г. Продукт реакции очищают хроматографией через 50-кратное количество силикагеля, используя 5 об. этанола в хлористом метилене. Объединенные очищенные фракции выпаривают и остаток кристаллизуют из смеси этанола и этилацетата, чтобы получить не совсем белые кристаллы 6-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло[4,3-a] [1,4] -диазепин-2-ил]-2-пропинил]-5Н-дибенз[c,e]-азепин-5,7(6Н)-диона с т.пл.220-223oC. Эти кристаллы содержат 0,166 молярные количества этилацетата согласно спектральным и аналитическим данным.
Пример 52
4-(2-Хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f][1,2,4]триазоло[4,3-a][1,4] -диазепин подвергают реакции с рацемическим 2а,3,4,5-тетрагидро-2а-метил-1-(2-пропинил)-бенз[cd] -индол-2(1Н)-оном при условиях, используемых в примере 25г. Продукт реакции очищают с помощью хроматографии через 50-кратное количество силикагеля, используя 5 об. этанола в хлористом метилене для элюирования. Объединенные очищенные фракции выпаривают, чтобы получить смолистое вещество, которое не кристаллизуется, но дает кристаллический дихлоргидрат при обработке избытком этанольного раствора хлористого водорода и этилацетата. Эти кристаллы содержат 0,33 моль этанола согласно спектральным и аналитическим данным. Бледно-желтые кристаллы дихлоргидрата 1-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло[4,3-a][1,4]-диазепин-2-ил] -2-пропинил] -2а, 3,4,5-тетрагидро-2а -метилбенз[cd]-индол-2(1Н)-она с т.пл. 175-178oC.
Исходное пропаргиловое вещество синтезируют следующим образом.
К раствору 6,92 г (0,04 моль) рацемического 2а,3,4,5-тетрагидробенз[cd] -индол-2(1Н)-она в 50 мл диметилформамида добавляют 4,94 гш (0,044 моль) трет. -бутоксида калия. После перемешивания в течение 15 мин под азотом добавляют 6,24 г или 2,75 мл (0,044 моль) иодистого метила и перемешивание продолжают в течение 30 мин. После разбавления водой и льдом смесь экстрагируют хлористым метиленом. Экстракты высушивают над сульфатом натрия и выпаривают. Остаток хроматографируют через силикагель, используя 10 об. этилацетата в хлористом метилене. Объединенные очищенные фракции выпаривают и остаток кристаллизуют из эфира, чтобы получить 3,2 г бесцветного рацемического 2а, 3,4,5-тетрагидро-2а-метилбенз[cd]-индол-2(1Н)-она с т.пл. 148-150oC.
Это вещество, 2,5 г (13,3 ммоль), растворяют в 20 мл диметилформамида. Раствор обрабатывают с помощью 1,65 г (14,7 ммоль) трет.-бутоксида калия и перемешивают под азотом в течение 15 мин. Затем добавляют 1,75 г бромистого пропаргила (1,31 мл, 14 ммоль) и перемешивание при комнатной температуре продолжают в течение 30 мин. Реакционную смесь разбавляют льдом и насыщенным раствором бикарбоната натрия. Выпадающий осадок собирают фильтрацией и твердые частицы промывают водой и отсасывают досуха. Его растворяют в хлористом метилене и высушенный раствор фильтруют через плотный слой силикагеля. Фильтрат выпаривают и остаток кристаллизуют из этилацетата и гексана (смесь), чтобы получить бесцветные кристаллы рацемического 2а,3,4,5-тетрагидро-2а-метил-1-(2-пропинил)-бенз[cd]-индол-2(1Н)-она с т.пл. 127-130oC.
Пример 53
4-(2-Хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло [4,3-a] [1,4] -диазепин подвергают реакции с 3-фенокси-1-пропином при условиях, описанных в примере 25г. Продукт реакции очищают с помощью хроматографии через 50-кратное количество силикагеля, используя 5 об. этанола в хлористом метилене. Выпаренные очищенные фракции дают в остатке вязкое масло, которое не кристаллизуется, но дает кристаллический дигидрохлорид при обработке избытком этанольного раствора хлористого водорода и этилацетата. Бледно-желтые кристаллы дигидрохлорида 4-(2-хлорфенил)-9-метил-2-[3-(фенокси)-1-пропинил] -6Н-тиено[3,2-f][1,2,4]триазоло [4,3-a][1,4]-диазепина плавятся со вспениванием при 148-151oC и по анализу являются гидратом.
Пример 54
Смесь 0,88 г 4-(2-хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f][1,2,4] триазоло[4,3-a][1,4]-диазепина, 0,65 г 3-метил-1(2-пропинил)-2,4 (1Н,3Н)-хиназолиндиона [см. B.Danielsson et al. Acta Pharm. Suecica 2, 167 (1965)] 90 мг трифенилфосфина, 20 мг иодистой меди (I), 2 мл триэтиламина и 50 мл диметилформамида дегазируют с помощью аргона в течение 10 мин. Затем добавляют 30 мг ацетата палладия и смесь нагревают вплоть до 90-95oC в течение 30 мин с перемешиванием под аргоном. Температуру поддерживают при 90-95oC в течение 15 мин и диметилформамид выпаривают при пониженном давлении. Остаток расслаивают хлористым метиленом и водным раствором бикарбоната натрия. Органический слой высушивают и выпаривают, под конец азеотропно с помощью ксилола. Остаток хроматографируют через 30 г силикагеля, используя смесь тетрагидрофурана и гексана в соотношении 4:1 для элюирования. Объединенные очищенные фракции выпаривают и полученную смолу кристаллизуют из этанола, чтобы получить бесцветные кристаллы 1-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено [3,2-f] [1,2,4] триазоло[4,3-a][1,4]-диазепин-2-ил]-2-пропинил]-3-метил-2,4(1Н,3Н)-хиназолиндиона с т. пл. 167-170oC. Эти кристаллы содержат 0,5 моль воды согласно аналитическим и спектральным данным.
Пример 55
4-(2-Хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло [4,3-a] [1,4] -диазепин подвергают реакции с 5-(2-пропинил)-5Н-дибенз[b,e]-азепин-6,11-дионом при условиях, описанных в примере 25г. Продукт реакции очищают с помощью хроматографии через 50-кратное количество силикагеля, используя смесь тетрагидрофурана и гексана в соотношении 4:1 для элюирования. Смола, оставшаяся после выпаривания объединенных очищенных фракций, кристаллизуется из этилацетата, чтобы получить бесцветные кристаллы 5-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено [3,2-f] [1,2,4] триазоло[4,3-a][1,4]-диазепин-2-ил]-2-пропинил] -5Н-дибенз[b, e] -азепин-6,11-диона с т.пл. 245-247oC. Эти кристаллы содержат 0,33 моль этилацетата согласно аналитическим и спектральным данным.
Исходное ацетиленовое вещество получают следующим образом.
К суспензии 4,5 г 5Н-дибенз[b,e]-азепин-6,11-диона в 50 мл диметилформамида добавляют 2,5 г трет.-бутоксида калия. После перемешивания под азотом в течение 30 мин добавляют 2 мл бромистого пропаргила и перемешивание продолжают в течение 1 ч при комнатной температуре. Реакционную смесь подкисляют уксусной кислотой и разбавляют водой. Выпадающий осадок отфильтровывают и отсасывают досуха. Твердые частицы растворяют в хлористом метилене. Раствор высушивают и выпаривают и остаток хроматографируют через 120 г силикагеля, используя хлористый метилен. Объединенные очищенные фракции, содержащие продукт реакции, выпаривают и остаток кристаллизуют из смеси эфира и гексана, чтобы получить бесцветные кристаллы 6-(2-пропинил)-5Н-дибенз[b,e]-азепин-6,11-диона с т.пл. 117-118oC.
Пример 56
4-(2-Хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло [4,3-a] [1,4] -диазепин подвергают реакции с 6-хлор-4-(2-пропинил)-2Н-1,4-бензоксазин-3(4Н)-оном [см. P. Rao et al. Indian J.Chem.24B, 1120 (1985)] как это описано в примере 25г, но продолжительность реакции увеличивают до 72 ч. Продукт реакции выделяют с помощью хроматографии через 100-кратное количество силикагеля (Мерк, 230-400 меш), используя 10 об. этанола в хлористом метилене. Кристаллизация остатка, полученного после выпаривания объединенных очищенных фракций, из этилацетата дает бесцветные кристаллы 6-хлор-4-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено [3,2-f] [1,2,4] триазоло[4,3-a][1,4]-диазепин-2-ил]-2-пропинил]-2Н-1,4-бензоксазин-3(4Н)-она с т.пл. 202-205oC.
Пример 57
4-(2-Хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло [4,3-a] [1,4] -диазепин подвергают реакции с 1,4-дигидро-1-(2-пропинил) -3,1-бензоксазин-2-оном при условиях, описанных в примере 25г. Продукт реакции выделяют с помощью хроматографии через 50-кратное количество силикагеля, используя 5 об. этанола в хлористом метилене, и дополнительно очищают путем повторной хроматографии через 30-кратное количество силикагеля, используя смесь тетрагидрофурана и гексана в соотношении 4:1 для элюирования. Очищенные фракции выпаривают и остаток кристаллизуют из смеси метанола и этилацетата, чтобы получить не совсем белые кристаллы 1-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло[4,3-a] [1,4]-диазепин-2-ил]-2- пропинил] -2Н-3,1-бензоксазин-2-она с т. пл.173-176oC. Эти кристаллы содержат 0,33 моль воды, согласно аналитическим данным.
Необходимый ацетилен синтезируют следующим образом.
К раствору 4,7 г (31 ммоль) 1,4-дигидро-3,1-бензоксазин-2-она в 30 мл диметилформамида добавляют 3,9 г (34,6 ммоль) трет.-бутоксида калия. После перемешивания в течение 15 мин под азотом добавляют 4,1 г или 3,1 мл бромистого пропаргила и перемешивание при комнатной температуре продолжают в течение 30 мин. Реакционную смесь разбавляют насыщенным водным раствором бикарбоната натрия и выпадающий в осадок продукт реакции отфильтровывают, промывают водой и отсасывают досуха. Остаток растворяют в хлористом метилене и раствор пропускают через плотный слой силикагеля. Фильтрат выпаривают и остаток кристаллизуют из метанола, чтобы получить бесцветные кристаллы 1,4-дигидро-1-(2-пропинил)-3,1-бензоксазин-2-она с т.пл. 123-125oC.
Пример 58
4-(2-Хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло [4,3-a] [1,4] -диазепин подвергают реакции с 2-этинилпиридином при условиях, используемых в примере 25г. Продукт реакции выделяют с помощью хроматографии через 50-кратное количество силикагеля, используя 5 об. этанола в хлористом метилене. Фракции, гомогенные по данным ТСХ, объединяют и выпаривают. Кристаллизация остатка из этилацетата и перекристаллизация из смеси тетрагидрофурана и метанола дает не совсем белые кристаллы 4-(2-хлорфенил)-9-метил-2-[2-(2- пиридилинил)-этинил] -6Н-тиено[3,2-f] [1,2,4]триазоло[4,3-a] [1,4]-диазепина с т.пл. 153-155oC.
Пример 59
4-(2-Хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло [4,3-a] [1,4] -диазепин подвергают реакции с 2-(2-пропинил)-1(2Н)-изохинолиноном при условиях, описанных в примере 25г. Продукт реакции выделяют с помощью хроматографии через 50-кратное количество силикагеля, используя смесь тетрагидрофурана и гексана в соотношении 4:1. Фракции, очищенные согласно данным ТСХ, объединяют и выпаривают, а остаток кристаллизуют из смеси этанола и этилацетата, чтобы получить бесцветные кристаллы 2-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено[3,2-f][1,2,4]триазоло[4,3-a][1,4]-диазепин-2-ил]-2- пропинил] -1(2Н)-изохинолинона с т. пл. 148-150oC. Эти кристаллы содержат согласно аналитическим данным 0,66 моль воды.
Необходимый ацетилен получают следующим образом.
Раствор 1 г (7 ммоль) изокарбостирила в 40 мл диметилформамида обрабатывают с 0,86 г (7,7 моль) трет.-бутоксида калия. После перемешивания под азотом в течение 30 мин добавляют 0,7 мл бромистого пропаргила и перемешивание при комнатной температуре продолжают в течение 1 ч. Реакционную смесь подкисляют уксусной кислотой и разбавляют водой. Выпадающий в осадок продукт реакции отфильтровывают, промывают водой и отсасывают досуха. Остаток очищают с помощью хроматографии через 30 г силикагеля, используя 5 об. этанола в хлористом метилене и кристаллизуют из эфира, чтобы получить бесцветные кристаллы 2-(2- пропинил)-1(2Н)-изохинолинона с т.пл. 104-105oC.
Пример 60
4-(2-Хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло [4,3-a] [1,4] -диазепин подвергают реакции с 1,3-дигидро-1-фенил-3-(2-пропинил)-2Н-бензимидазол-2-оном при условиях, используемых в примере 25г. Продукт реакции выделяют с помощью хроматографии через 50-кратное количество силикагеля, используя 5 об. этанола в хлористом метилене. Очищенные фракции объединяют и выпаривают, а остаток кристаллизуют из смеси метанола и этилацетата и перекристаллизовывают из метанола, чтобы получить не совсем белые кристаллы 1-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено[3,2-f] [1,2,4]триазоло[4,3-a][1,4]-диазепин-2-ил] -2-пропинил] -1,3-дигидро-3-фенил-2Н-бензимидазол-2-она с т.пл. 176-179oC. Эти кристаллы содержат 0,66 моль воды согласно аналитическим данным.
Требуемое пропаргиловое производное синтезируют следующим образом.
К раствору 3 г (14,2 ммоль) 1,3-дигидро-3-фенил-2Н-бензимидазол-2-она в 30 мл диметилформамида добавляют 1,76 г (15,7 ммоль) трет.-бутоксида калия и смесь перемешивают под азотом в течение 15 мин. Затем добавляют 1,4 мл (15 ммоль) бромистого пропаргила и перемешивание продолжают в течение 30 мин при комнатной температуре и 15 мин на паровой бане. Продукт реакции высаживают разбавлением насыщенным водным раствором бикарбоната натрия и отфильтровывают, промывают водой и отсасывают досуха. Его растворяют в хлористом метилене и раствор пропускают через плотный слой силикагеля, используя хлористый метилен. Фильтрат выпаривают и остаток кристаллизуют из смеси хлористого метилена и этилацетата, чтобы получить бесцветные кристаллы 1,3-дигидро-1-фенил-3-(2-пропинил)-2Н-бензимидазол-2-она с т.пл. 145-147oC.
Пример 61
4-(2-Хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло [4,3-a] [1,4] -диазепин подвергают реакции с 6,8-дихлор-3,4-дигидро-1-(2-пропинил)-2(1Н)-хинолиноном при условиях, используемых в примере 25г. Продукт реакции очищают с помощью хроматографии через 50-кратное количество силикагеля, используя смесь тетрагидрофурана и гексана в соотношении 4:1. Фракции, гомогенные по данным ТСХ, объединяют и выпаривают. Остаток кристаллизуют из этилацетата, чтобы получить бесцветные кристаллы 1-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено[3,2-f] [1,2,4]триазоло[4,3-a][1,4]-диазепин-2-ил]-2-пропинил] -6,8-дихлор-3,4-дигидро-2(1Н)-хинолинона с т.пл. 163-165oC.
Требуемое пропаргиловое производное получают следующим образом:
Смесь 1,5 г 6,8-дихлор-3,4-дигидро-2(1Н)-хинолинона, 1,3 г окиси бария, 40 мл диметилформамида и 0,8 мл бромистого пропаргила нагревают на паровой бане в течение 45 мин и перемешивают в течение дополнительного часа без нагревания. Продукт реакции выпадает в осадок при добавлении льда и воды и его собирают фильтрацией. Твердые частицы растворяют в хлористом метилене и раствор высушивают и выпаривают. Остаток кристаллизуют из смеси эфира и гексана, чтобы получить бесцветные кристаллы 6,8-дихлор-3,4-дигидро-1-(2-пропинил)-2(1Н)-хинолинона, с т.пл. 92-95oC.
Исходное вещество получают следующим образом.
Раствор 6 г 3,4-дигидро-2(1Н)-хинолинона в 50 мл муравьиной кислоты и 50 мл концентрированной соляной кислоты добавляют к 75 мл 1,2-дихлорэтана, который был насыщен хлором. Смесь перемешивают над льдом в течение 30 мин. Хлор вводят в течение 5 мин и перемешивание на холоду продолжают в течение 2 ч. Затем смесь выливают на лед, подщелачивают с помощью гидроокиси аммония и экстрагируют хлористым метиленом. Экстракты высушивают и выпаривают, а остаток хроматографируют через 300 г силикагеля, используя 8 об. этилацетата в хлористом метилене. Кристаллизация объединенных очищенных фракций из эфира дает бесцветные кристаллы 6,8-дихлор-3,4-дигидро-2(1Н)-хинолинона с т.пл. 145-146oC.
Пример 62
4-(2-Хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло [4,3-a] [1,4] -диазепин подвергают реакции с 3,4-дигидро-2-(2-пропинил)-1(2Н)-изохинолиноном [см. W.Schneider et al. Arch. Pharm. 291, 561 (1958)] при условиях, описанных в примере 25г. Продукт реакции выделяют с помощью хроматографии с помощью 50-кратного количества силикагеля, используя смесь тетрагидрофурана и гексана в соотношении 4:1 для элюирования. Очищенные фракции объединяют и выпаривают, а остаток кристаллизуют из этилацетата, чтобы получить бесцветные кристаллы 2-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло[4,3-a] [1,4] -диазепин-2-ил]-2-пропинил]-3,4-дигидро-1(2Н)-изохинолинона с т.пл. 164-166oC.
Пример 63
4-(2-Хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло [4,3-a] [1,4] -диазепин подвергают реакции с 7-фтор-4-(2-пропинил)-2Н-1,4-бензоксазин-3(4Н)-оном при условиях, используемых в примере 25г. Продукт реакции выделяют с помощью хроматографии через 50-кратное количество силикагеля, используя 4 об. этанола в хлористом метилене. Хорошие фракции объединяют и выпаривают, а остаток кристаллизуют из этилацетата, чтобы получить бесцветные кристаллы 4-[3-[4-(2- хлорфенил)-9-метил-6Н-тиено[3,2-f][1,2,4]триазоло[4,3-a] [1,4] -диазепин-2-ил] -2-пропинил] -7-фтор-2Н-1,4-бензоксазин-3(4Н)-она с т.пл. 215-217oC.
Пропаргиловое соединение получают следующим образом.
Смесь 1,7 г 7-фтор-2,4-дигидро-1,4-бензоксазин-3-она, 2 г окиси бария, 1,3 мл бромистого пропаргила и 40 мл диметилформамида нагревают на паровой бане в течение 45 мин. Охлажденную реакционную смесь разбавляют водой и выпадающий в осадок продукт реакции собирают фильтрацией. Выпадающий осадок растворяют в хлористом метилене и раствор промывают водой, высушивают и выпаривают. Остаток кристаллизуют из смеси эфира и гексана, чтобы получить бесцветные кристаллы 7-фтор-4-(2-пропинил)-2Н-1,4-бензоксазин-3(4Н)-она с т. пл. 98-100oC.
Пример 64
4-(2-Хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло [4,3-a] [1,4] -диазепин подвергают реакции с 1,2,3,4-тетрагидро-9-(2-пропинил)-9Н-карбазолом при условиях, используемых в примере 25г. Продукт реакции выделяют с помощью хроматографии через 50-кратное количество силикагеля, используя 8 об. этанола в хлористом метилене для элюирования. Остаток, полученный после выпаривания объединенных очищенных фракций, содержащих продукт реакции, кристаллизуют из этанола, чтобы получить бесцветные кристаллы 4-(2-хлорфенил)-2-[3-(1,2,3,4-тетрагидро-9Н-карбазол-9-ил)-1-пропинил] -9-метил-6Н-тиено [3,2-f][1,2,4]триазоло[4,3-a][1,4]-диазепина с т.пл. 164-166oC. Эти кристаллы содержат 0,66 моль этанола согласно аналитическим и спектральным данным.
Пропаргиловое соединение синтезируют следующим образом.
К раствору 5 г (30 ммоль) 1,2,3,4-тетрагидро-9Н-карбазола в 50 мл диметилформамида добавляют 3,6 г (33 ммоль) трет.-бутоксида калия. После перемешивания в течение 45 мин под азотом, добавляют 3,9 г или 2,9 мл (33 ммоль) бромистого пропаргила и перемешивание продолжают в течение 30 мин при комнатной температуре, а затем 45 мин на паровой бане. Охлажденную реакционную смесь разбавляют водой и продукт реакции экстрагируют эфиром. Экстракт промывают водой, высушивают над сульфатом натрия и выпаривают. Остаток пропускают через плотно набитый слой силикагеля, используя смесь хлористого метилена и гексана в соотношении 1:1. Фракции, содержащие наименее полярный продукт реакции, объединяют и выпаривают. Остаток кристаллизуют из гексана, чтобы получить бесцветные кристаллы 1,2,3,4-тетрагидро-9-(2-пропинил)-9Н-карбазола с т.пл. 74-76oC.
Пример 65
Смесь 0,88 г (2 ммоль) 4-(2-хлорфенил)-2-иод-9-метил-6Н-тиено [3,2-f] [1,2,4] триазоло[4,3-a][1,4]-диазепина, 0,6 г (2,6 ммоль) 10-(2- пропинил)-9(10Н)-акридинона [см. A.R.Katritzky et al. J.Org. Chem. 50, 852 (1985)] 80 мг трифенилфосфина, 20 мг иодистой меди (I), 5,6 мл триэтиламина и 50 мл диметилформамида дегазируют аргоном в течение 10 мин. Затем добавляют 25 мг ацетата палладия и смесь нагревают до 80-90oC в течение 30 мин. Продукт реакции выпадает в осадок при добавлении насыщенного водного раствора бикарбоната натрия и его собирают фильтрацией. Твердые частицы растворяют в хлористом метилене и раствор высушивают и выпаривают. Остаток хроматографируют через 50 г силикагеля, используя 5 об. этанола в хлористом метилене для элюирования. Объединенные очищенные фракции выпаривают и остаток кристаллизуют из этанола, чтобы получить желтые кристаллы 10-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло[4,3-a][1,4]-диазепин-2-ил]-2- пропинил] -9(10Н)-акридинона с т.пл. 175-180oC со вспениванием. Эти кристаллы содержат моль воды.
Пример 66
4-(2-Хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f][1,2,4]триазоло[4,3-a][1,4] -диазепин подвергают реакции с 3,8-дихлор-5-(2-пропинил)-6(5Н)-фенантридиноном при условиях, используемых в примере 25г, но под конец реакционную смесь нагревают в течение 5 мин при 90-95oC. Продукт реакции выделяют с помощью хроматографии через 50-кратное количество силикагеля, используя смесь тетрагидрофурана и гексана в соотношении 4:1 для элюирования. Кристаллизация объединенных очищенных фракций из смеси тетрагидрофурана и этилацетата дает бесцветные кристаллы 3,8-дихлор-5-[3-[4-(2-хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f] [1,2,4]триазоло[4,3-a][1,4]-диазепин-2-ил]-2-пропинил] -6(5Н)-фенантридинона с т.пл. 218-220oC.
Пропаргиловое производное, используемое для этого опыта, получают следующим образом.
Смесь 5 г (19 ммоль) 3,8-дихлор-6(5Н)-фенантридинона, 3,2 г (21 ммоль) окиси бария и 1,9 мл (21 ммоль) бромистого пропаргила в 40 мл диметилформамида нагревают на паровой бане в течение 1 ч. После охлаждения продукт реакции высаживают в осадок добавлением воды, отфильтровывают и промывают водой. Его растворяют в хлористом метилене и раствор высушивают и выпаривают. Остаток хроматографируют на 150 г силикагеля, используя 10 об. гексана в хлористом метилене. Фракции, содержащие продукт реакции, объединяют и выпаривают. Твердый остаток перекристаллизовывают из этанола, чтобы получить бесцветные кристаллы 3,8-дихлор-5-(2-пропинил)-6(5Н)-фенантридинона с т.пл. 248-250oC.
Пример 67
4-(2-Хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло [4,3-a] [1,4] -диазепин подвергают реакции с фенилацетиленом при условиях, описанных в примере 25г. Продукт реакции выделяют с помощью хроматографии через 50-кратное количество силикагеля, используя 5 об. этанола в хлористом метилене для элюирования. Объединенные очищенные фракции выпаривают и остаток кристаллизуют из этилацетата, чтобы получить бесцветные кристаллы 4-(2-хлорфенил)-2-иод-9-метил-6Н-тиено[3,2-f][1,2,4]триазоло[4,3-a][1,4]-диазепина с т. пл. 215-217oC.
Пример 68
Смесь 0,84 г (2 ммоль) 6-(2-фторфенил)-8-иод-1-метил-4Н-[1,2,4] триазоло[4,3-a] [1,4] -бензодиазепина, 0,48 г (2,6 ммоль) 3,4-дигидро-1-(2-пропинил)-2(1Н)-хинолинона, 80 мг трифенилфосфина, 20 мг иодистой меди (I), 1,5 мл триэтиламина и 50 мл диметилформамида дегазируют аргоном в течение 10 мин. Затем добавляют 25 мг ацетета палладия и перемешивание продолжают в течение 18 ч. Растворитель выпаривают при пониженном давлении и остаток разделяют на части между хлористым метиленом и насыщенным водным раствором бикарбоната натрия. Органический слой отделяют, высушивают и выпаривают и остаток хроматографируют через 50 г силикагеля, используя 5 об. этанола в хлористом метилене для элюирования. Кристаллизация объединенных очищенных фракций из этилацетата дает бесцветные кристаллы 1-[3-[6-(2- фторфенил)-1-метил-4Н-[1,2,4] триазоло[4,3-a][1,4]-бензодиазепин-8-ил]- 2-пропинил]-3,4-дигидро-2(1Н)-хинолинона с т.пл. 236-239oC.
Пример 69
6-(2-Фторфенил)-8-иод-1-метил-4Н-[1,2,4] триазоло[4,3-a] [1,4]- бензодиазепин подвергают реакции с 4 фенил-1-бутином при условиях, используемых в примере 3б. Продукт реакции выделяют с помощью хроматографии с 40-кратным количеством силикагеля, используя 5 об. этанола в хлористом метилене для элюирования. Объединенные очищенные фракции выпаривают и остаток кристаллизуют из этилацетата с охлаждением сухим льдом и перекристаллизовывают из смеси этилацетата, эфира и гексана, чтобы получить бесцветные кристаллы 6-(2-фторфенил)-1-метил-8-(4-фенил-1-бутинил)-4Н-[1,2,4] триазоло[4,3-a][1,4]- бензодиазепина с т.пл. 125-128oC.
Пример 70
а) Смесь 5 г 5-(2-фторфенил)-2-гидразино-7-иод-3Н-1,4-бензодиазепина, 20 мл трифторуксусной кислоты, 5 мл ангидрида трифторуксусной кислоты и 100 мл хлористого метилена нагревают на паровой бане под током азота, чтобы отогнать хлористый метилен. Затем добавляют 100 мл толуола и нагревание на паровой бане продолжают в течение 30 мин. Смесь расслаивают хлористым метиленом и насыщенным водным раствором бикарбоната натрия. Органическую фазу отделяют, высушивают и выпаривают. Остаток кристаллизуют из эфира и перекристаллизовывают из этанола, чтобы получить бесцветные кристаллы 6-(2-фторфенил)-1-(трифторметил)-8-иод-4Н-[1,2,4]триазоло[4,3-a][1,4] -бензодиазепина с т.пл. 202-204oC.
б) Смесь 0,94 г (2 ммоль) 6-(2-фторфенил)-8-иод-1-(трифторметил)- 4Н-[1,2,4]триазоло[4,3-a][1,4]-бензодиазепина, 0,55 г (3 ммоль) N- пропаргилфталимида, 80 мг трифенилфосфина, 20 мг иодистой меди (I), 0,6 мл триэтиламина и 50 мл диметилформамида дегазируют аргоном в течение 10 мин. Затем добавляют 25 мг ацетата палладия и смесь перемешивают в течение 3 дн при комнатной температуре. Продукт реакции осаждают разбавлением раствором бикарбоната натрия и собирают фильтрацией. Его растворяют в хлористом метилене и раствор промывают бикарбонатным раствором, высушивают и выпаривают. Остаток хроматографируют через 40 г силикагеля, используя 5 об. этанола в хлористом метилене. Объединенные хорошие фракции выпаривают и остаток кристаллизуют из этилацетата, чтобы получить бесцветные кристаллы 2-[3-[6-(2-фторфенил)-1-(трифторметил)-4Н-[1,2,4] триазоло[4,3-a] [1,4] -бензодиазепин-8-ил]-2-пропинил]-1Н-изоиндол-1,3(2Н)-диона с т.пл. 204-206oC.
Пример 71
а) Раствор 2,9 г (13 ммоль) N-бензилоксикарбонил-1-аланина в 15 мл тетрагидрофурана охлаждают до -40oC. Затем добавляют 2,7 г (13 ммоль) пятихлористого фосфора и смесь перемешивают в течение 30 мин при -30oC. Затем добавляют раствор 3,41 г (10 ммоль) 2-(2- фторбензоил)-4-иоданилина в 50 мл хлористого метилена и перемешивание продолжают в течение 15 мин при 0-10oC. После добавления 10%-ного водного раствора карбоната натрия двухфазную смесь перемешивают при этой температуре в течение 30 мин. Затем ее экстрагируют эфиром. Экстракты промывают раствором карбоната натрия и водой, высушивают и выпаривают. Остаток пропускают через плотный слой силикагеля вместе с хлористым метиленом. Фильтрат выпаривают и остаток кристаллизуют из этанола, чтобы получить бесцветные кристаллы сложного бензилового эфира (S)-[2-[2-(2-фторбензоил)-4-иодфенил] -амино]-1-метил-2-оксоэтилкарбаминовой кислоты с т. пл. 159-161oC; [α]D= - 10,35° (c 0,985 в CH2Cl2).
б) Смесь 11 г бензилового эфира (S)-[2-[2-(2-фторбензоил)-4-иодфенил]-амино] -1-метил-2-оксоэтилкарбаминовой кислоты и 30 мл уксусной кислоты, содержащей 30% бромистого водорода, перемешивают при комнатной температуре в течение 3 ч. Реакционную смесь расслаивают водой и подщелачивают добавлением льда и аммиака. Выпадающее в осадок вещество экстрагируют хлористым метиленом и экстракты высушивают и выпаривают. Остаток нагревают в 50 мл этанола, содержащего 5 мл уксусной кислоты, на паровой бане в течение 15 мин. Растворитель выпаривают при пониженном давлении и остаток расслаивают хлористым метиленом и 10%-ным водным раствором карбоната натрия. Органическую фазу высушивают и выпаривают и остаток кристаллизуют из этилацетата и перекристаллизовывают из смеси хлористого метилена и этилацетата, чтобы получить бесцветные кристаллы (S)-5-(2-фторфенил)-1,3-дигидро-7-иод-3-метил-2Н-1,4- бензодиазепин-2-она с т.пл. 223-225oС; [α]D= + 100,79° (с 0,9891 в CH2Cl2).
в) Раствор 2 г (5,07 ммоль) (S)-5-(2-фторфенил)-1,3-дигидро-7-иод-3-метил-2Н-1,4-бензодиазепин-2-она в 60 мл тетрагидрофурана охлаждают до 30oC и добавляют 0,57 г (5,7 ммоль) трет.-бутоксида калия. Смесь перемешивают под азотом в течение 30 мин, в то время как температуре позволяют повыситься до 5oС. Затем добавляют 1,03 г (6,5 ммоль) диэтилхлорфосфата и перемешивание продолжают в течение 30 мин без охлаждения. После добавления 0,54 г (7,2 ммоль) ацетилгидразида смесь перемешивают в течение дополнительных 30 мин при комнатной температуре. Затем добавляют 75 мл бутанола и тетрагидрофуран отгоняют. Реакционную смесь выпаривают при пониженном давлении и остаток разделяют на части между хлористым метиленом и насыщенным водным раствором бикарбоната натрия. Органический слой отделяют, высушивают и выпаривают, а остаток хроматографируют через силикагель, используя 5 об. этанола в хлористом метилене для элюирования. Кристаллизация вещества, полученного из объединенных очищенных фракций, из смеси этилацетата и гексана дает бесцветные кристаллы (S)-6-(2-фторфенил)-8-иод-1,4-диметил-4Н-[1,2,4] триазоло[4,3-a] [1,4]-бензодиазепина с т.пл. 142-145oС; [α]D= + 50,38° (c 0,9964 в CH2Cl2).
г) (S)-6-(2-фторфенил)-8-иод-1,4-диметил-4Н-[1,2,4]триазоло [4,3-a][1,4] -бензодиазепин подвергают реакции с 3,4-дигидро-1-(2- пропинил)-2(1Н)-хинолиноном при условиях, используемых в примере 68. Продукт реакции выделяют с помощью хроматографии с 50-кратным количеством силикагеля, используя 5 об. этанола в хлористом метилене для элюирования. Объединенные очищенные фракции выпаривают и остаток кристаллизуют очень медленно из смеси этанола и эфира, чтобы получить бесцветные кристаллы (S)-1-[3-[6-(2-фторфенил)-1,4-диметил-4Н-[1,2,4] триазоло[4,3-a][1,4]-бензодиазепин-8-ил]-2-пропинил]-3,4-дигидро-2(1Н) -хинолинона с т.пл. 155-160o со вспениванием. Эти кристаллы содержат этанол и воду согласно аналитическим и спектральным данным. [α]D= + 87,5° (c 1,0091 в CH2Cl2).
Пример 72
а) Раствор 29 г N-бензилоксикарбонил-L-аланина в 100 мл тетрагидрофурана охлаждают до -40oС. Затем добавляют 27 г пятихлористого фосфора и перемешивание продолжают в течение 30 мин при -30oС. Добавляют раствор 23,7 г (0,1 моль) 2-амино-3-(2-хлорбензоил)-тиофена в 400 мл хлористого метилена и смесь перемешивают в течение 15 мин при 0-10oС. Затем ее расслаивают с помощью 300 мл 10%-ного водного раствора карбоната натрия и двухфазную смесь перемешивают при 0-10oС в течение 30 мин. После разбавления эфиром органический слой отделяют, промывают водным раствором карбоната натрия, высушивают и выпаривают. Остаток кристаллизуют из этанола с помощью затравки. Затравки получают путем хроматографирования 2 г образца через 60 г силикагеля, используя 10 об. этилацетата в хлористом метилене для элюирования. Очищенные фракции дают кристаллы из смеси эфира и гексана, которые перекристаллизовывают из этанола, чтобы получить бесцветные кристаллы бензилового эфира (S)-[2-[3-(2-хлорбензоил)-2-тиенил] -амино] -1-метил-2 -оксоэтилкарбаминовой кислоты с т. пл. 133-135oС; [α]D= - 26,92° (c 0,9546 в CH2Cl2).
б) Раствор 22 г бензилового эфира (S)-[2-[3-(2-хлорбензоил)-2-тиенил]-амино] -1-метил-2-оксоэтилкарбаминовой кислоты в 75 мл уксусной кислоты, содержащей 30% бромистого водорода, оставляют стоять при комнатной температуре в течение 3 ч. Реакционную смесь разделяют на части между водой и эфиром. Водную фазу промывают эфиром и подщелачивают добавлением льда и гидроокиси аммония. Выпадающее в осадок вещество экстрагируют хлористым метиленом. Экстракты высушивают и выпаривают и остаток растворяют в 500 мл толуола. После добавления 66 г силикагеля смесь перемешивают и нагревают до кипения с обратным холодильником в течение 3 ч с отделением образующейся среды. Силикагель отфильтровывают и промывают хорошо метанолом. Фильтрат выпаривают и остаток пропускают через 200 г силикагеля, используя смесь этилацетата и хлористого метилена в соотношении 1:1 для элюирования. Продукт реакции кристаллизуют из смеси хлористого метилена и гексана и перекристаллизовывают из эфира для анализа, чтобы получить (S)-5-(2-хлорфенил)-1,3-дигидро-3-метил-2Н-тиено- [2,3-e] [1,4] -диазепин-2-он с т. пл. 200-203o; [α]D= - 0,4° (c 1,0185 в CH2Cl2).
в) Смесь 7,7 г (26,5 ммоль) (S)-5-(2-хлорфенил)-1,3-дигидро- 3-метил-2Н-тиено-[2,3-e] [1,4] -диазепин-2-она, 60 мл метанола, 60 мл уксусной кислоты, 8,61 (53 ммоль) монохлористого иода и 2,17 г (26,5 ммоль) ацетата натрия перемешивают при обычной температуре в течение 15 мин. Раствор 9 г бисульфита натрия в воде добавляют, чтобы понизить количество избыточного реагента. Реакционную смесь подщелачивают добавлением льда и гидроокиси аммония. Выпадающее в осадок вещество собирают, промывают водой и отсасывают досуха. Его перекристаллизовывают из смеси метанола и этилацетата, чтобы получить бесцветные кристаллы (S)-5-(2-хлорфенил)-1,3-дигидро-7-иод-3-метил-2Н-тиено-[2,3-e][1,4]-диазепин-2-она с т.пл. 235-237oС.
г) Смесь 2,08 г (5 ммоль) (S)-5-(2-хлорфенил)-1,3-дигидро-7-иод-3-метил-2Н-тиено-[2,3-e] [1,4]-диазепин-2-она, 1,25 г пентасульфида фосфора, 1,3 г бикарбоната натрия и 40-50 мл диглима перемешивают под азотом при 80-90o в течение 6 ч. Добавляют воду и лед и смесь перемешивают в течение 15 мин. Твердые частицы отфильтровывают, промывают водой и отсасывают досуха. Получающийся тион дополнительно высушивают под вакуумом при 50oС, чтобы получить 2,6 г сырого вещества, которое далее подвергают реакции следующим образом.
Сырой тион перемешивают с 1,3 мл безводного гидразина в 30 мл тетрагидрофурана в течение 30 мин при комнатной температуре. Растворитель выпаривают при пониженном давлении и остаток перемешивают с 15 мл воды и 15 мл хлористого метилена. Кристаллическое гидразиновое производное собирают фильтрацией, промывают водой и эфиром и добавляют к 13 мл этилацетата и 6,5 мл триэтилового эфира ортоуксусной кислоты. Эту смесь нагревают на паровой бане в течение 30 мин и образующиеся кристаллы отфильтровывают после охлаждения. Продукт реакции перекристаллизовывают из смеси хлористого метилена и метанола, чтобы получить бесцветные кристаллы рацемического 4-(2-хлорфенил)-2-иод-6,9-диметил-6Н-тиено[3,2-f][1,2,4]триазоло[4,3-a][1,4]-диазепина с т.пл. 262-264oС. Эти кристаллы не имеют угла вращения, что указывает на полную рацемизацию.
д) Рацемический 4-(2-хлорфенил)-2-иод-6,9-диметил-6Н-тиено[3,2-f] [1,2,4] триазоло[4,3-a][1,4]-диазепин подвергают реакции с 5-(2- пропинил)-6(5Н)-фенантридиноном при условиях, описанных в примере 25г. Продукт реакции выделяют с помощью хроматографии через 50-кратное количество силикагеля, используя 5 об. этанола в хлористом метилене. Продукт реакции кристаллизуют из этанола, чтобы получить желтоватые кристаллы рацемического 5-[3-[4-(2-хлорфенил)-6,9-диметил-6Н-тиено[3,2-f] [1,2,4] триазоло[4,3-a][1,4]-диазепин-2-ил] -2-пропинил]-6(5Н)- фенантридинона с т.пл.182-186oС со вспениванием. Эти кристаллы содержат согласно аналитическим данным молярные количества воды.
Пример 73
Смесь 1,1 г (2 ммоль) 5-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено [3,2-f] [1,2,4]триазоло[4,3-a][1,4]-диазепин-2-ил]-2-пропинил]-6(5Н)- фенантридинона и 0,68 г мета-хлорнадбензойной кислоты в 100 мл хлористого метилена оставляют стоять при комнатной температуре в течение 20 ч. Раствор промывают 10%-ным водным раствором карбоната натрия, высушивают и выпаривают. Остаток кристаллизуют из смеси метанола и этилацетата, чтобы получить не совсем белые кристаллы 5-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло [4,3-a] [1,4] -диазепин-2-ил]-2-пропинил]-6(5Н)-фенантридинона с т.пл. 260-270oС с разложением.
Пример 74
Смесь 0,7 г 5-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено [3,2-f][1,2,4]триазоло[4,3-a] [1,4] -диазепин-5-окись-2-ил]-2-пропинил] 6(5Н)- фенантридинона, 25 мл ангидрида уксусной кислоты и 30 мл пиридина нагревают на паровой бане в течение 3 ч под аргоном. Реагенты удаляют при пониженном давлении и остаток хроматографируют через 30 г силикагеля, используя 3 об. этанола в хлористом метилене для элюирования. Очищенные фракции, содержащие 5-[3-[6-ацетилокси-4-(2-хлорфенил)-9-метил-6Н-тиено[3,2-f][1,2,4]триазоло[4,3-a][1,4] -диазепин-2-ил] -2-пропинил] -6(5Н)-фенантридинон, объединяют и выпаривают. Остаток не кристаллизуется и характеризуется только спектроскопически.
ЯМР (CDCl3): 2,36 (s, 3, OAc); 2,68 (s,3, 9-Me); 5,43 (s, 2, -CH2-); 6,78 (s, 2, C6-H и тиенил-Н); 7,2-8,6 (m, 12, ароматический Н) м.д.
Пример 75
Раствор 0,25 г 5-[3-[6-ацетилокси-4-(2-хлорфенил)-9-метил-6Н- тиено[3,2-f][1,2,4]триазоло[4,3-a][1,4]-диазепин-2-ил]-2-пропинил]- 6(5Н)-фенантридинона в 30 мл теплого метанола обрабатывают 2 мл 3N раствора гидроокиси натрия. После 15 мин реакционную смесь подкисляют уксусной кислотой и разделяют на части между хлористым метиленом и водным раствором бикарбоната натрия. Органическую фазу отделяют, высушивают и выпаривают. Остаток хроматографируют через 25 г силикагеля (Мерк, 230-400 меш), используя 5 об. этанола в хлористом метилене для элюирования. После элюирования некоторого количества перегруппированного рацемического 4-(2-хлорфенил)-4,5-дигидро-9-метил- 2-[3-(5,6-дигидро-6-оксо-5-фенантридинил)-1-пропинил]-6Н-тиено[3,2-f] [1,2,4] триазоло[4,3-a] [1,4] -диазепин-6-она с т.пл.225-230oС (из смеси метанола и этилацетата), фракции, содержащие рацемический 5-[3-[4-(2-хлорфенил)-6-окси-9-метил-6Н-тиено[3,2-f] [1,2,4] триазоло[4,3-a] [1,4]-диазепин-2-ил] -2-пропинил] -6(5Н)-фенантридинон, объединяют и выпаривают. Остаток кристаллизуют из смеси метанола и этилацетата, чтобы получить бесцветные кристаллы с т.пл. 255-258oС.
Пример 76
6-(2-Фторфенил)-8-иод-1-метил-4Н-[1,2,4] триазоло[4,3-a] [1,4]-бензодиазепин подвергают реакции с рацемическим 2-этинил-3,4-дигидро-2,5,7,8-тетраметил-2Н-2-бензопиран-6-олом [см. H. Meyer et al. Helv. Chim. Acta, 67, 650 (1963)] при условиях, описанных в примере 25г. Продукт реакции выделяют с помощью хроматографии с 50-кратным количеством силикагеля, используя 5 об. этанола в хлористом метилене. Очищенные фракции, содержащие продукт реакции, объединяют и выпаривают. Остаток кристаллизуют из смеси этанола и этилацетата, чтобы получить бесцветные кристаллы рацемического 2-[[6-(2-фторфенил)-1-метил-4Н-[1,2,4] триазоло[4,3-a] [1,4] -бензодиазепин-8-ил] -этинил]- 3,4-дигидро-2,5,7,8-тетраметил-2Н-1-бензопиран-6-ола с т.пл. 265-267oС.
Пример 77
4-(2-Хлорфенил)-2-иод-9-метил-6Н-тиено-[3,2-f] [1,2,4] триазоло [4,3-a] [1,4] -диазепин подвергают реакции с рацемическим 2-этинил-3,4-дигидро-2,5,7,8-тетраметил-2Н-1-бензопиран-6-олом при условиях, описанных в примере 25г. Продукт реакции выделяют с помощью хроматографии через 50-кратное количество силикагеля, используя 5 об. этанола в хлористом метилене. Объединенные очищенные фракции выпаривают и остаток кристаллизуют из этилацетата, чтобы получить бесцветные кристаллы рацемического 2-[[4-(2-хлорфенил)-9-метил-6Н-тиено-[3,2-f] [1,2,4] триазоло[4,3-a][1,4]-диазепин-2-ил]-этинил]-3,4-дигидро-2,5,7,8- тетраметил-2Н-1-бензопиран-6-ола с т.пл. 155-160oС со вспениванием. Эти кристаллы содержат 1,5 моль воды, что следует из аналитических и спектральных данных.
Пример 78
а) Раствор 0,8 г трет.-бутоксида калия в 20 мл тетрагидрофурана и 15 мл трет. -бутанола и 0,6 мл триэтилфосфита охлаждают до -30oС с перемешиванием под аргоном. Раствор 0,8 г 6-(2-фторфенил)-8-иод-1- метил-4Н-[1,2,4]триазоло[4,3-a] [1,4] -бензодиазепина в 5 мл диметилформамида добавляют и перемешивание продолжают в течение 1 ч при температуре от -20 до -10oС. Во время перемешивания смеси пропускают кислород в течение дополнительного ч при этой температуре. Реакционную смесь подкисляют добавлением уксусной кислоты и разделяют на части между раствором карбоната натрия и хлористым метиленом, содержащим 10 об. этанола. Органический слой высушивают и выпаривают и остаток кристаллизуют из смеси хлористого метилена и этилацетата и перекристаллизовывают из этанола, чтобы получить бесцветные кристаллы рацемического 6-(2-фторфенил)-4-окси-8-иод-1-метил-4Н-[1,2,4]триазоло[4,3-a][1,4]-бензодиазепина с т.пл. 258-260oС.
б) Смесь 0,435 г (1 ммоль) рацемического 6-(2-фторфенил)-4-окси-8-иод-1-метил-4Н-[1,2,4] триазоло[4,3-a] [1,4]-бензодиазепина, 3 мл хлористого тионила и 20 мл хлористого метилена перемешивают при комнатной температуре в течение 2 ч. После выпаривания при пониженном давлении остаток растворяют в 20 мл метанола и раствор обрабатывают с 3 мл триэтиламина. После нагревания на паровой бане в течение 5 мин смесь выпаривают досуха и остаток разделяют на части между хлористым метиленом и водным раствором бикарбоната натрия. Органический слой высушивают и выпаривают и остаток кристаллизуют из этилацетата, чтобы получить бесцветные кристаллы рацемического 6-(2- фторфенил)-8-иод-4-метокси-1-метил-4Н-[1,2,4] триазоло[4,3-a] [1,4]-бензодиазепина с т. пл. 240-242oС. Аналитический образец перекристаллизовывают из смеси метанола и этилацетата и получают т.пл. 243-244oС.
в) Рацемический 6-(2-фторфенил)-8-иод-4-метокси-1-метил-4Н-[1,2,4]триазоло[4,3-a] [1,4] -бензодиазепин подвергают реакции с 3,4-дигидро-1-(2-пропинил)-2(1Н)-хинолиноном при условиях, используемых в примере 25г. Продукт реакции выделяют с помощью хроматографии через 50-кратное количество силикагеля, используя 5 об. этанола в хлористом метилене для элюирования. Объединенные очищенные фракции выпаривают и остаток кристаллизуют из смеси этилацетата и эфира, чтобы получить бесцветные кристаллы рацемического 1-[3-[6-(2-фторфенил)-4-метокси-1-метил-4Н- [1,2,4] триазоло[4,3-a] [1,4]-бензодиазепин-8-ил]-2-пропинил]-3,4- дигидро-2(1Н)-хинолинона с т.пл. 155-160oС. Эти кристаллы содержат 0,33 моль воды.
Пример 79
6-(2-Фторфенил)-1-(трифторметил)-8-иод-4Н-[1,2,4] триазоло[4,3-a] [1,4] -бензодиазепин подвергают реакции с 3,4-дигидро-1-(2-пропинил)- 2(1Н)-хинолиноном при условиях, используемых в примере 25г. Продукт реакции очищают с помощью хроматографии через 50-кратное количество силикагеля, используя 5 об. этанола в хлористом метилене. Очищенные фракции объединяют и выпаривают и остаток кристаллизуют из этилацетата, чтобы получить бесцветные кристаллы 1-[3-[6-(2-фторфенил)-1-(трифторметил)-4Н-[1,2,4]триазоло[4,3-a][1,4]-бензодиазепин-8-ил]-2-пропинил]-3,4-дигидро-2(1Н)-хинолинона с т.пл. 193-196oС.
Пример 80
Рацемический 6-(2-фторфенил)-4-окси-8-иод-1-метил-4Н-[1,2,4] триазоло[4,3-a] [1,4] -бензодиазепин подвергают реакции с 3,4-дигидро-1-(2-пропинил)-2(1Н)-хинолиноном при условиях, используемых в примере 25г. Продукт реакции выделяют с помощью хроматографии над 50-кратным количеством силикагеля, используя 5 об. этанола в хлористом метилене. Объединенные хорошие фракции выпаривают и остаток кристаллизуют из смеси метанола и этилацетата, чтобы получить бесцветные кристаллы рацемического 1-[3-[6-(2-фторфенил)-4-окси-1-метил-4Н-[1,2,4]триазоло[4,3-a][1,4]- бензодиазепин-8-ил]-2-пропинил] -3,4-дигидро-2(1Н)-хинолинона с т.пл. 253-255oС с разложением. Эти кристаллы содержат молярные эквиваленты воды.
Пример 81
4-(2-Хлорфенил)-2-иод-9-метил-6Н-тиено-[3,2-f] [1,2,4] триазоло [4,3-a] [1,4] -диазепин подвергают реакции с 1'-(2-пропинил)-спиро-[циклопентан-1,3'-(3Н)-индол] -2'-(1'H)-оном при условиях, описанных в примере 25г. Продукт реакции выделяют с помощью хроматографии с 50-кратным количеством силикагеля, используя 5 об. этанола в хлористом метилене для элюирования. Объединенные хорошие фракции выпаривают и остаток превращают в кристаллический дигидрохлорид путем обработки избытком этанольного раствора хлористого водорода в смеси этанола и этилацетата. Светло-желтые кристаллы дигидрохлорида 1-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено-[3,2-f] [1,2,4]триазоло[4,3-a][1,4]-диазепин-2-ил] -2-пропинил] -циклопентан-1,3'-(3Н)-индол-2(1'H)-она имеют т. пл. 173-176oС со вспениванием и содержат 2 моль воды и 0,66 моль этанола согласно аналитическим и спектральным данным.
Исходное пропаргиловое производное, используемое в этом опыте, получают следующим образом.
К раствору 0,56 г (3 ммоль) спиро-[циклопентан-1,3'-(3Н)-индол]-2'-(1'H)-она [см. R.J. Owellen, J. Org. Chem.39, 69 (1974)] в 10 мл диметилформамида добавляют 0,37 г (3,3 ммоль) трет.-бутоксида калия. Смесь перемешивают в течение 15 мин и добавляют 0,3 мл бромистого пропаргила и перемешивание продолжают в течение 30 мин. Реакционную смесь расслаивают между ксилолом и насыщенным раствором бикарбоната натрия. Органическую фазу высушивают и выпаривают и остаток хроматографируют с 20 г силикагеля, используя хлористый метилен для элюирования. Объединенные очищенные фракции выпаривают и остаток кристаллизуют из смеси эфира и гексана, чтобы получить бесцветные кристаллы 1'-(2-пропинил)-спиро-[циклопентан-1,3'-(3Н)-индол] -2'- (1'H)-она с т.пл. 109-111oС.
Пример 82
Смесь 0,5 г (1 ммоль) 1-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено-[3,2-f] [1,2,4] триазоло[4,3-a] [1,4]-диазепин-2-ил]-2-пропинил]-3,4-дигидро-2(1Н)-хинолинона и 0,34 (2 ммоль) мета-хлорнадбензойной кислоты в 30 мл хлористого метилена оставляют стоять при комнатной температуре в течение ночи. Затем ее промывают 10%-ным водным раствором карбоната натрия, высушивают и выпаривают. Остаток очищают с помощью хроматографии через 20 г силикагеля (230-400 меш), используя 5 об. этанола в хлористом метилене. Объединенные очищенные фракции выпаривают и остаток кристаллизуют из этилацетата и перекристаллизовывают из смеси этилацетата и метанола, чтобы получить бесцветные кристаллы 1-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено-[3,2-f] [1,2,4]триазоло[4,3-a] [1,4] -диазепин-5-окись-2-ил] -2-пропинил]-3,4-дигидро-2(1Н)-хинолинона с т. пл. 225-230oС с разложением. Эти кристаллы содержат согласно спектральным и аналитическим данным 0,166 моль этилацетата.
Пример 83
а) К раствору 4,4 г 4-(2-хлорфенил)-2-иод-9-метил-6Н-тиено-[3,2-f] [1,2,4]триазоло[4,3-a][1,4]-диазепина в 200 мл хлористого метилена добавляют 3,4 г мета-хлорнадбензойной кислоты. После перемешивания при комнатной температуре в темноте в течение 18 ч смесь промывают 10%-ным водным раствором карбоната натрия. Органическую фазу высушивают над сульфатом натрия, фильтруют и выпаривают. Остаток кристаллизуют из смеси тетрагидрофурана, метанола и этилацетата, чтобы получить 3,3 г сырого продукта реакции. Его очищают путем пропускания через плотный слой силикагеля, используя 10 об. метанола в хлористом метилене. Элюат выпаривают и остаток кристаллизуют из смеси тетрагидрофурана и метанола, чтобы получить не совсем белые кристаллы 4-(2-хлорфенил)-2-иод-9-метил-6Н-тиено-[3,2-f] [1,2,4]триазоло[4,3-a][1,4] -диазепин-5-окиси с т.пл. 280-283oС с разложением.
б) Продукт реакции из примера 73 получают реакцией 4-(2-хлорфенил)-2-иод-9-метил-6Н-тиено-[3,2-f] [1,2,4]триазоло[4,3-a][1,4]- диазепин-5-окиси с 5-(2-пропинил)-6(5Н)-фенантридиноном, используя условия, описанные в примере 25г.
Пример 84
а) Смесь 1 г 4-(2-хлорфенил)-2-иод-9-метил-6Н-тиено-[3,2-f] [1,2,4] триазоло[4,3-a] [1,4]-диазепин-5-окиси, 25 мл пиридина и 15 мл ангидрида уксусной кислоты нагревают на паровой бане в течение 4 ч. Смесь выпаривают при пониженном давлении, под конец азеотропно с помощью ксилола. Остаток хроматографируют через 25 г силикагеля, используя 5 об. этанола в хлористом метилене для элюирования. Объединенные очищенные фракции выпаривают и продукт реакции кристаллизуют из этилацетата и перекристаллизовывают из смеси тетрагидрофурана, метанола и этилацетата, чтобы получить бесцветные кристаллы рацемического 6-ацетилокси-4-(2-хлорфенил)-2-иод-9-метил-6Н- тиено-[3,2-f] [1,2,4]триазоло[4,3-a][1,4]-диазепина с т.пл. 248-250oС.
б) К раствору 0,3 г рацемического 6-ацетилокси-4-(2-хлорфенил)-2-иод-9-метил-6Н-тиено-[3,2-f][1,2,4]триазоло[4,3-a][1,4]-диазепина в 30 мл метанола добавляют 1 мл 3N раствора гидроокиси натрия и 10 мл воды. После стояния при комнатной температуре в течение 30 мин раствор подкисляют уксусной кислотой и расслаивают хлористым метиленом и водным раствором бикарбоната натрия. Органическую фазу отделяют, высушивают и выпаривают. Остаток кристаллизуют из смеси метанола и этилацетата, чтобы получить бесцветные кристаллы рацемического 4-(2-хлорфенил)-2-иод-9-метил-6Н-тиено-[3,2-f] [1,2,4] триазоло[4,3-a][1,4]-диазепин-6-ола с т.пл.240-243oС (разл).
в) Продукт реакции из примера 75 получают путем взаимодействия 4-(2-хлорфенил)-2-иод-9-метил-6Н-тиено-[3,2-f] [1,2,4] триазоло[4,3-a] [1,4]-диазепин-6-ола с 5-(2-пропинил)-6(5Н)-фенантридиноном при условиях, описанных в примере 25г.
Пример 85
а) Раствор 23 г (2-аминофенил)(4-хлорфенил)-метанола в 500 мл хлористого метилена охлаждают до -50oС. Затем добавляют 21 г или 15 мл монохлористого иода и смесь перемешивают на холоду в течение 4 ч. Затем ей позволяют нагреться до 0oС и реакцию прекращают добавлением раствора бисульфита натрия в воде. После перемешивания в течение 10 мин органический слой отделяют, высушивают и выпаривают. Продукт реакции кристаллизуют из смеси толуола и гексана, чтобы получить желтые кристаллы (2-амино-5-иодфенил)(4-хлорфенил)-метанона с т.пл. 137-139oС.
б) К раствору 18 г (2-амино-5-иодфенил)(4-хлорфенил)-метанона в 250 мл хлористого метилена добавляют 5 мл бромистого бромацетила и смесь перемешивают вместе с мелкораздробленным льдом в течение 15 мин. Органический слой отделяют, промывают раствором бикарбоната натрия, высушивают и выпаривают. Остаток кристаллизуют из смеси хлористого метилена и эфира, чтобы получить 18 г бромацетил-производного. Это вещество растворяют в 200 мл хлористого метилена и добавляют к 300 мл жидкого аммиака. Аммиаку позволяют постепенно выпариваться в течение ночи, а остающийся хлористый метилен промывают водой, высушивают и выпаривают. Остаток растворяют в 300 мл этанола и нагревают до кипения с обратным холодильником в течение 30 мин после добавления 10 мл уксусной кислоты. Растворитель частично выпаривают и продукт реакции кристаллизуют путем охлаждения. Перекристаллизация из смеси хлористого метилена и этанола дает бесцветные кристаллы 5-(4-хлорфенил)-1,3-дигидро-7-иод-2Н-1,4-бензодиазепин-2-она с т.пл. 246-248oС.
в) Смесь 12 г 5-(4-хлорфенил)-1,3-дигидро-7-иод-2Н-1,4-бензодиазепин-2-она, 8 г пентасульфида фосфора, 8 г бикарбоната натрия и 100 мл диглима перемешивают и нагревают до 80-85oС в течение 3 ч. После охлаждения добавляют воду и мелкораздробленный лед и перемешивание продолжают в течение 10 мин. Выпадающий в осадок продукт реакции собирают фильтрацией и промывают водой, 2-пропанолом и эфиром. Для анализа его перекристаллизовывают из смеси тетрагидрофурана и этанола, чтобы получить 5-(4-хлорфенил)-1,3-дигидро-7-иод-2Н-1,4-бензодиазепин-2-тион, который плавится при 260-262oС.
Смесь 4 г 5-(4-хлорфенил)-1,3-дигидро-7-иод-2Н-1,4-бензодиазепин-2-тиона, 50 мл тетрагидрофурана, 20 мл 2-пропанола и 1,5 мл гидразина перемешивают при комнатной температуре в течение 30 мин. Эту смесь фильтруют через плотный слой силикагеля (10 г), используя тетрагидрофуран для элюирования. Фильтрат выпаривают и остаток кристаллизуют из эфира. Перекристаллизация этого вещества из смеси тетрагидрофурана и этанола дает бесцветные кристаллы 5-(4-хлорфенил)-2-гидразино-7-иод-3Н-1,4-бензодиазепина с т.пл. 250-252oС.
д) Смесь 2,8 г 5-(4-хлорфенил)-2-гидразино-7-иод-3Н-1,4-бензодиазепина, 40 мл ксилола и 10 мл триэтилового эфира ортоуксусной кислоты нагревают до кипения с обратным холодильником в течение 1,5 ч. Кристаллы, которые выделяются из охлаждаемой реакционной смеси, отфильтровывают и перекристаллизовывают из смеси тетрагидрофурана и этанола, чтобы получить бесцветные кристаллы 6-(4-хлорфенил)-8-иод-1-метил-4Н-[1,2,4] триазоло[4,3-a][1,4]-бензодиазепина с т.пл. 358-360oС.
е) 6-(4-хлорфенил)-8-иод-1-метил-4Н-[1,2,4]триазоло[4,3-a][1,4]-бензодиазепин подвергают реакции с 3,4-дигидро-1-(2-пропинил)-2(1Н)-хинолиноном при условиях, используемых в примере 25г. Продукт реакции выделяют с помощью хроматографии через 50-кратное количество силикагеля, используя 20 об. гексана в тетрагидрофуране для элюирования. Кристаллизация объединенных очищенных фракций из этилацетата и перекристаллизация из того же растворителя дает бесцветные кристаллы 1-[3-[6-(4-хлорфенил)-1-метил-4Н-[1,2,4] триазоло [4,3-a] [1,4]-бензодиазепин-8-ил]-2-пропинил]-3,4-дигидро-2(1Н)-хинолинона с т.пл. 215-217oС. Эти кристаллы содержат 0,33 моль воды согласно аналитическим данным.
Пример 86
а) Смесь 3,4 г 5-(4-хлорфенил)-2-гидразино-7-иод-3Н-1,4-бензодиазепина, 75 мл хлористого метилена, 5 мл ангидрида трифторуксусной кислоты и 15 мл трифторуксусной кислоты нагревают под азотом на паровой бане, чтобы удалить хлористый метилен в течение приблизительно 30 мин. Затем добавляют 100 мл толуола и нагревание на паровой бане продолжают в течение 30 мин. Охлажденную смесь промывают насыщенным водным раствором бикарбоната натрия, высушивают и выпаривают. Остаток хроматографируют через 200 г силикагеля, используя 10 об. этилацетата в хлористом метилене для элюирования. Фракции, содержащие продукт реакции, объединяют и выпаривают. Кристаллизация из этилацетата дает бесцветные кристаллы 6-(4-хлорфенил)-1-трифторметил-8-иод-4Н-[1,2,4]триазоло [4,3-a][1,4]-бензодиазепина с т.пл. 243-245oС.
б) 6-(4-Хлорфенил)-1-трифторметил-8-иод-4Н-[1,2,4]триазоло [4,3-a][1,4] -бензодиазепин подвергают реакции с 3,4-дигидро-1-(2-пропинил)-2(1Н)-хинолиноном при условиях, используемых в примере 25г, но повышая продолжительность реакции до 48 ч. Продукт реакции выделяют с помощью хроматографии через 50-кратное количество силикагеля, используя хлористый метилен и этилацетат в соотношении 1:1. Очищенные фракции, содержащие продукт реакции, объединяют и выпаривают и остаток кристаллизуют из смеси этилацетата и гексана, чтобы получить не совсем белые кристаллы 1-[3-[6-(4-хлорфенил)-1- трифторметил-4Н-[1,2,4]триазоло[4,3-a][1,4]-бензодиазепин-8-ил]-2-пропинил]-3,4-дигидро-2(1Н)-хинолинона с т.пл. 220-222oС. Эти кристаллы содержат 0,5 молярные количества воды согласно аналитическим данным.
Пример А
Суспензия (для перорального использования), г:
2-[3-[4-(2-Хлорфенил)-9-метил-6Н-тиено-[3,2-f] [1,2,4] триазоло [4,3-a] [1,4] -диазепин-2-ил] -2-пропинил] -1Н-бенз[d, e] -изохинолин-1,3(2Н)-дион (микрофайн)(Соединение А) 5,0
Оксипропилметил-целлюлоза 8,0
Полисорбат 80 0,5
Дистиллированная вода Количество доводят до 100,0 мл
Методика приготовления
1. Добавить соединение А к раствору полисорбата 80 и диспергировать.
2. В дистиллированной воде приготовить раствор оксипропилметил-целлюлозы.
3. Смешать вещества стадий 1 и 2, затем добавить дистиллированную воду, чтобы довести до 100 мл.
Пример Б
Капсулы, мг/капсула
2-[3-[4-(2-Хлорфенил)-9-метил-6Н-тиено-[3,2-f] [1,2,4] триазоло [4,3-a] [1,4] -диазепин-2-ил] -2-пропинил] -1Н-бенз[d, e] -изохинолин-1,3(2Н)-дион (микрофайн)(Соединение А) 50,00
Поливинилпирролидон К-90 0,50
Полисорбат 80 0,25
Микрокристаллическая целлюлоза 99,00
Дистиллированная вода До полного растворения
Стеарат магния 0,25
Общий вес 150,00 мг
Методика приготовления
1. К соединению А добавляют достаточное количество водного раствора поливинилпирролидона К-90 и полисорабата 80.
2. Размельчают до консистенции, высушивают и просеивают через сито 40 меш.
3. Добавляют микрокристаллическую целлюлозу и стеарат магния, затем смешивают и заполняют капсулы.
Пример В
Таблетки, мг/таблетка
2-[3-[4-(2-Хлорфенил)-9-метил-6Н-тиено-[3,2-f] [1,2,4] триазоло [4,3-a] [1,4] -диазепин-2-ил] -2-пропинил] -1Н-бенз[d, e] -изохинолин-1,3(2Н)-дион (микрофайн)(Соединение А) 50,00
Поливинилпирролидон (очищенный) 14,25
Поливинилпирролидон К-90 0,50
Микрокристаллическая целлюлоза 35,00
Крахмальный гликолят натрия 10,00
Дистиллированная вода Достаточное для растворения
Стеарат магния 0,25
Общий вес 110,00
Методика приготовления
1. Приготовить смесь соединения А, крахмального гликолята натрия и поливинилпирролидона, затем добавить достаточное количество водного раствора поливинилпирролидона К-90.
2. Размельчить до консистенции, высушить и просеять через сито 40 меш.
3. Добавить микрокристаллическую целлюлозу и стеарат магния, затем смешать и запрессовать в таблетки.
Пример Г
Аэрозольная суспензия, 0,5 мг/готовая к использованию
2-[3-[4-(2-Хлорфенил)-9-метил-6Н-тиено-[3,2-f] [1,2,4] триазоло [4,3-a] [1,4] -диазепин-2-ил] -2-пропинил] -1Н-бенз[d, e] -изохинолин-1,3(2Н)-дион (микрофайн)(Соединение А) 120,00 мг
Триолеат сорбита 40,00 мг
Трихлормонофторметан 1,80 мл
Дихлордифторметан 10,20 мл
Методика приготовления
1. К соединению А добавить раствор триолеата сорбита и трихлормонофторметана.
2. Гомогенизировать и добавить суспензию в алюминиевый сосуд.
3. Ввинтить 50-микролитровый мерный клапан в сосуд и под давлением заполнить дихлордифторметан.
Пример Д
1%-ный раствор для наружного применения, г:
2-[3-[4-(2-Хлорфенил)-9-метил-6Н-тиено-[3,2-f] [1,2,4] триазоло [4,3-a] [1,4] -диазепин-2-ил] -2-пропинил] -1Н-бенз[d, e] -изохинолин-1,3(2Н)-дион (микрофайн)(Соединение А) 1,0
Полиэтиленгликоль 400 99,0
Методика приготовления
1. Добавить соединение А к полиэтиленгликолю 400 и перемешивать хорошо до тех пор, пока не растворится.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ТРИАЗОЛО/4,3-А/ /1,4/ДИАЗЕПИНОВ | 1988 |
|
RU2071962C1 |
| Способ получения производных тиенотриазолодиазепина или их солей | 1978 |
|
SU747429A4 |
| ЛЕКАРСТВЕННАЯ КОМПОЗИЦИЯ ДЛЯ РЕЛИЗКОНТРОЛЯ | 1994 |
|
RU2174832C2 |
| Способ получения производных имидазо/1,5-а//1,4/диазепина или их солей | 1977 |
|
SU725563A3 |
| Способ получения производных тиенотриазолодиазепина или их кислотно-аддитивных солей | 1978 |
|
SU1060115A3 |
| Способ получения производных имидазо (1,5-а) /1,4/- диазепина или их солей | 1976 |
|
SU730308A3 |
| АРИЛ- /ГЕТЕРОАРИЛ - ЦИКЛОГЕКСЕНИЛ - ТЕТРААЗАБЕНЗО[е]АЗУЛЕНЫ В КАЧЕСТВЕ АНТАГОНИСТОВ ВАЗОПРЕССИНА | 2011 |
|
RU2568642C2 |
| Способ получения соединенийиМидАзО (1,5-A)(1,4)диАзЕпиНАили иХ фАРМАцЕВТичЕСКи пРиМЕНи-МыХ СОлЕй | 1975 |
|
SU814278A3 |
| СПИРО-5,6-ДИГИДРО-4Н-2,3,5,10В-ТЕТРААЗА-БЕНЗО[Е]АЗУЛЕНЫ | 2009 |
|
RU2514429C2 |
| ИМИДАЗОДИАЗЕПИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 1995 |
|
RU2139873C1 |
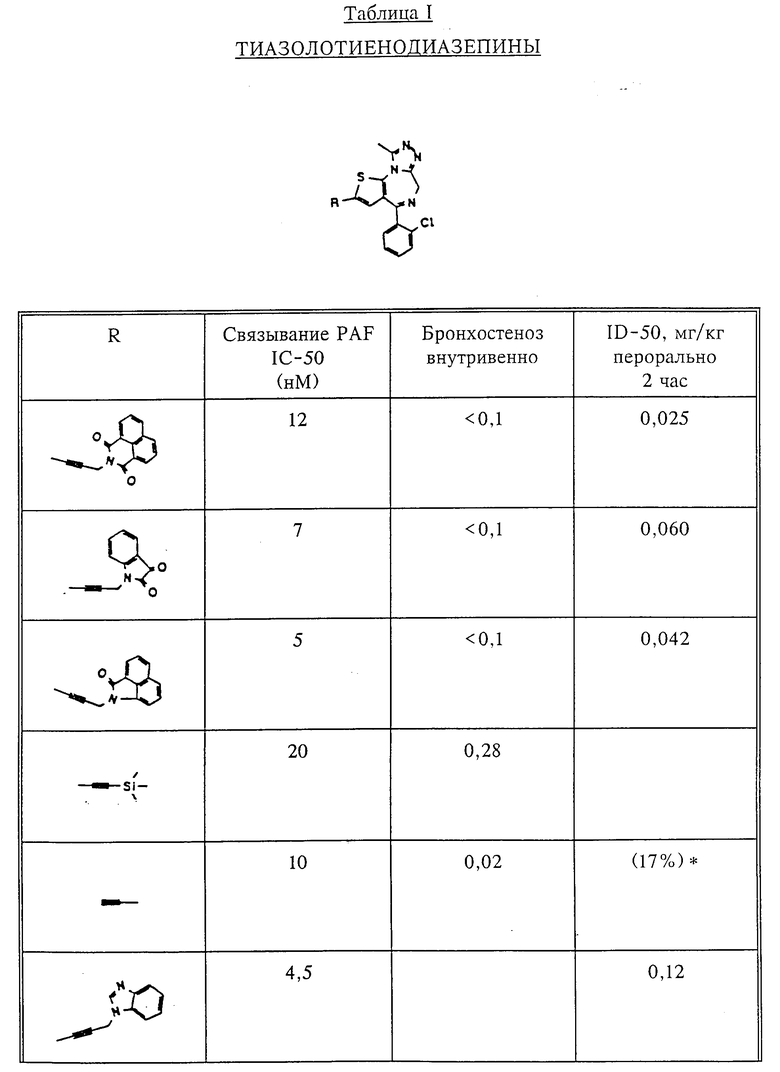
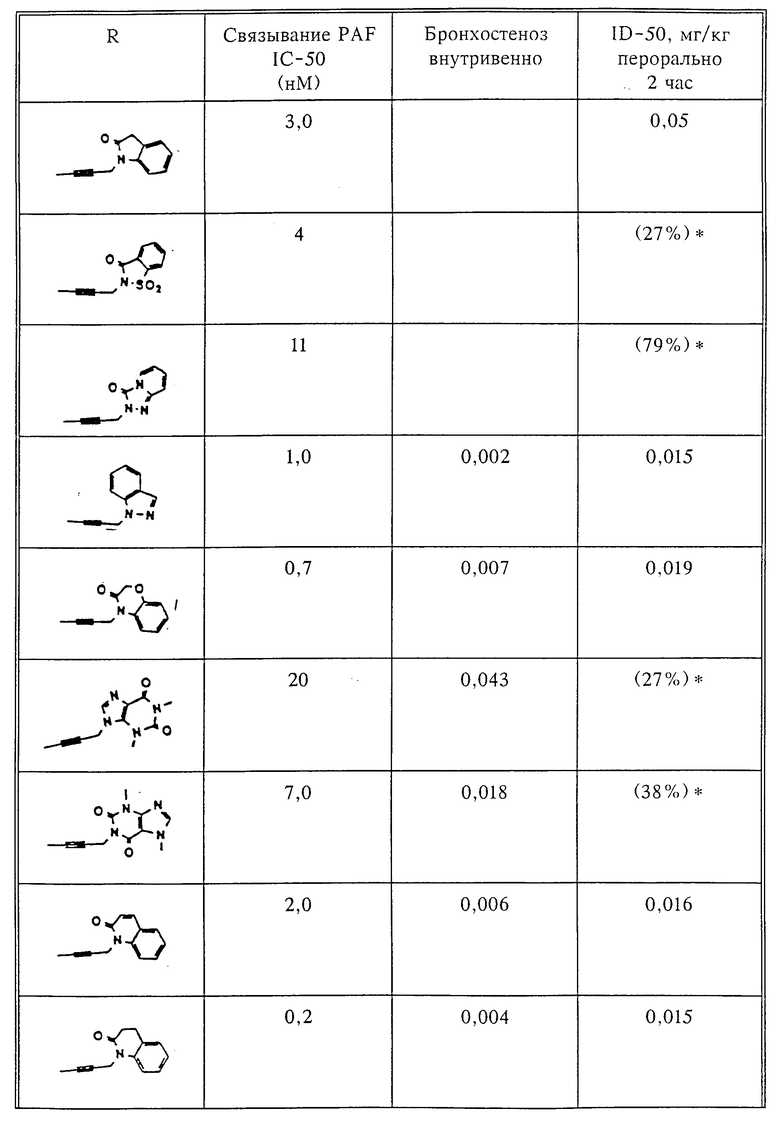
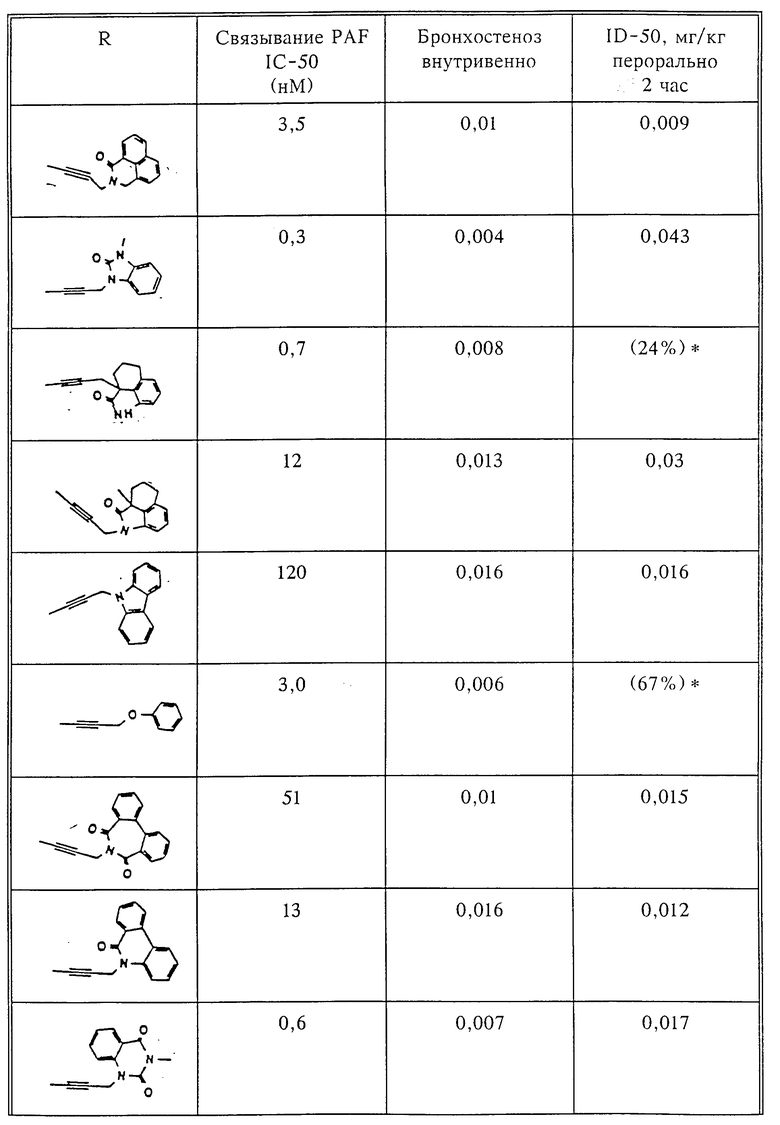
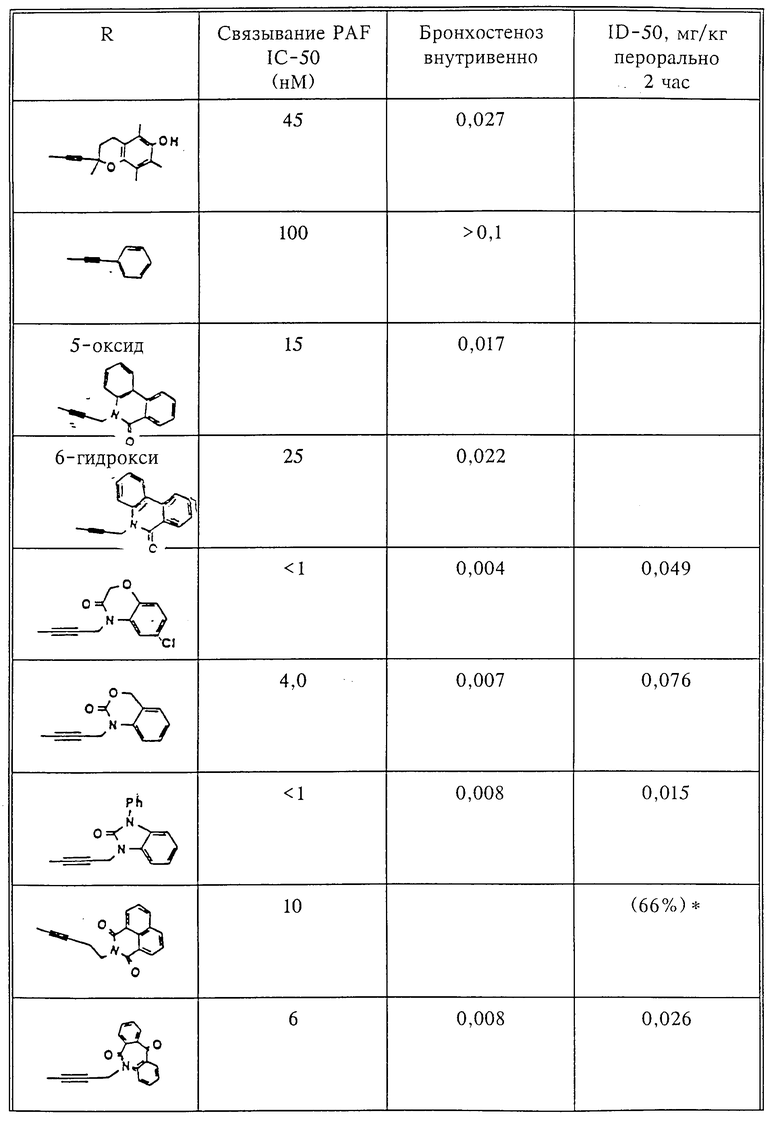
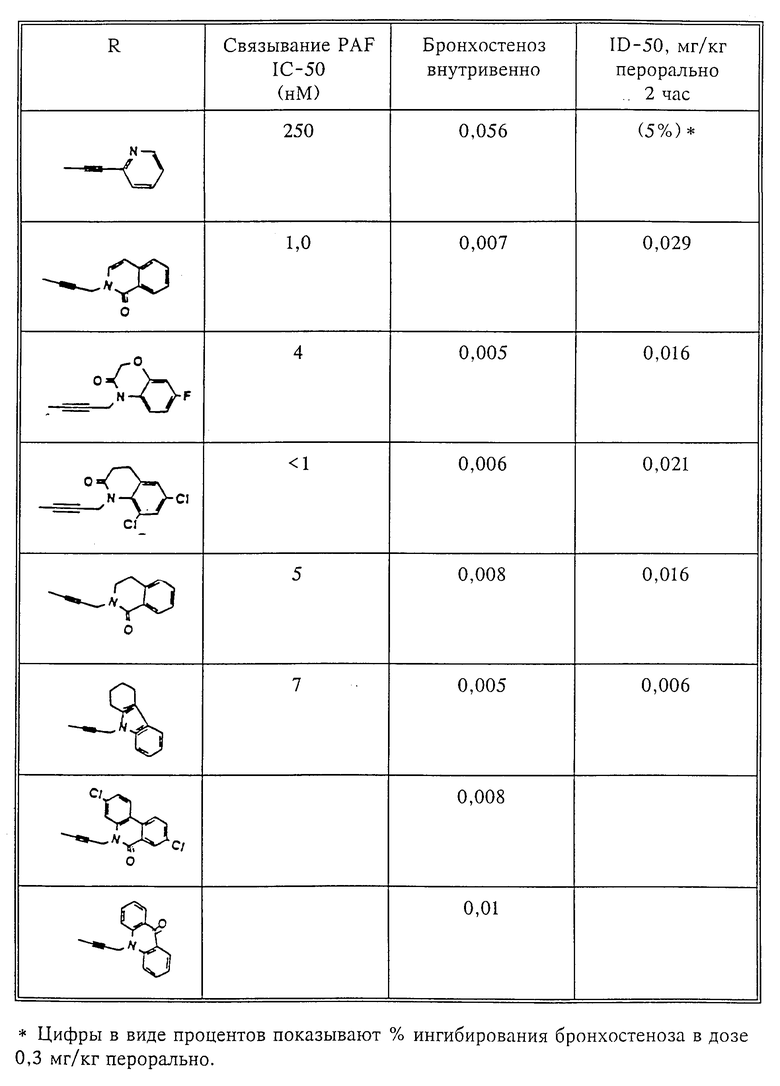
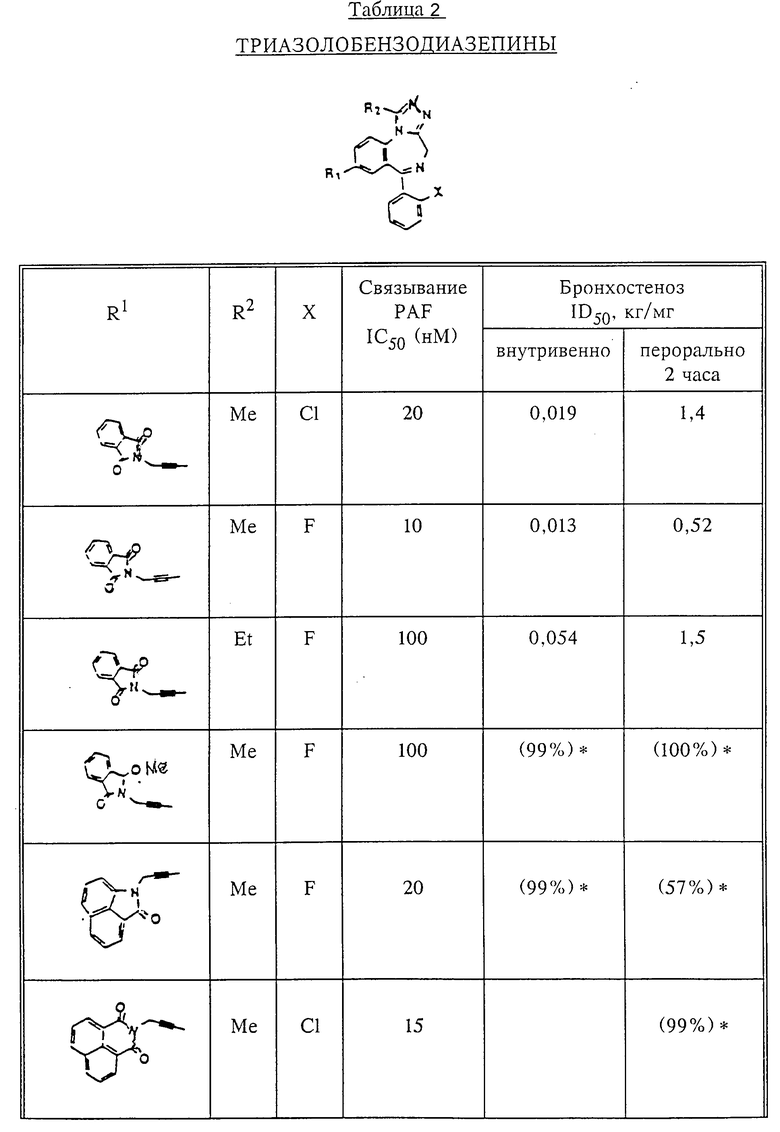
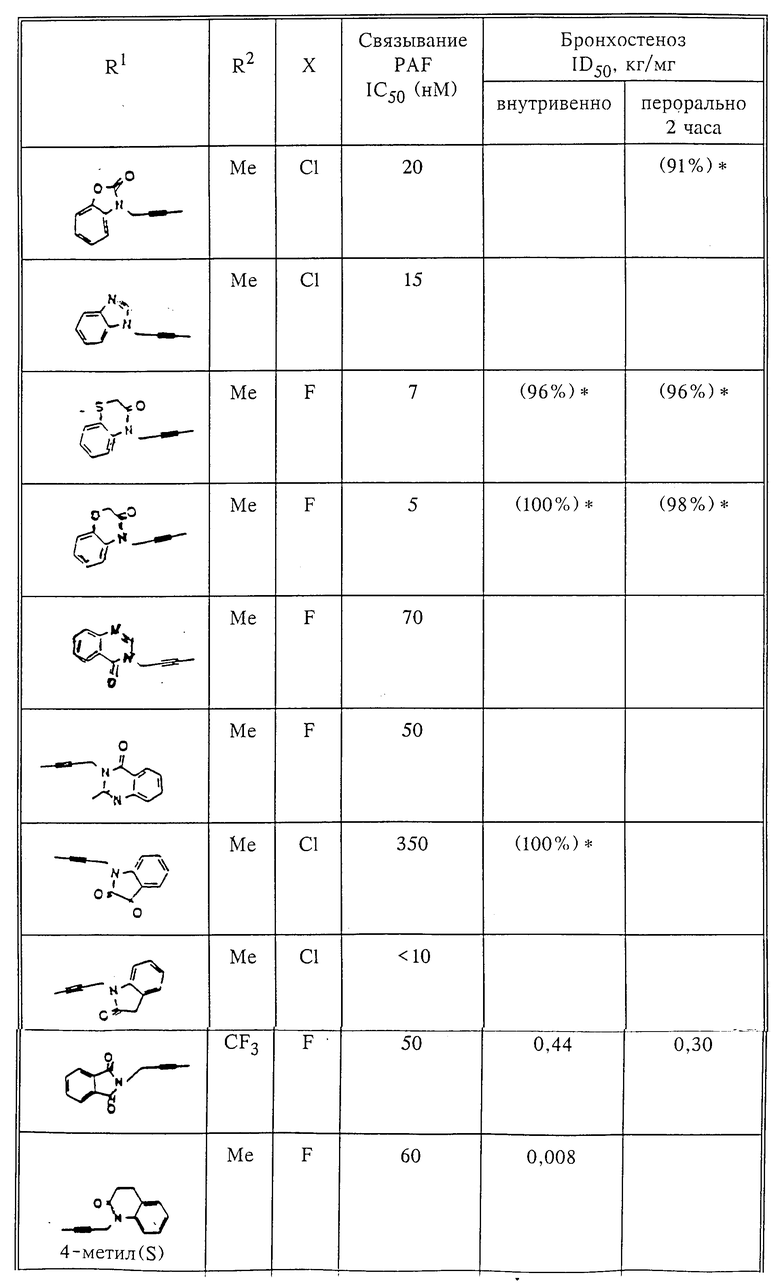
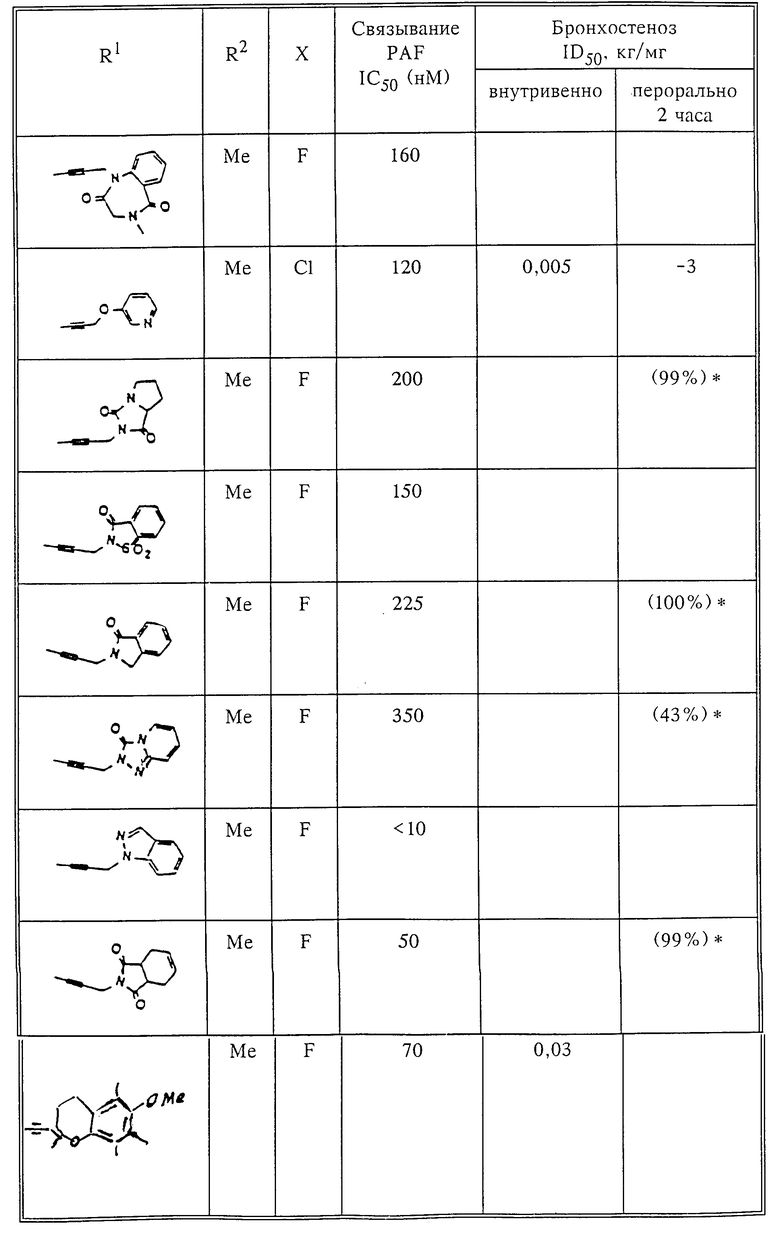
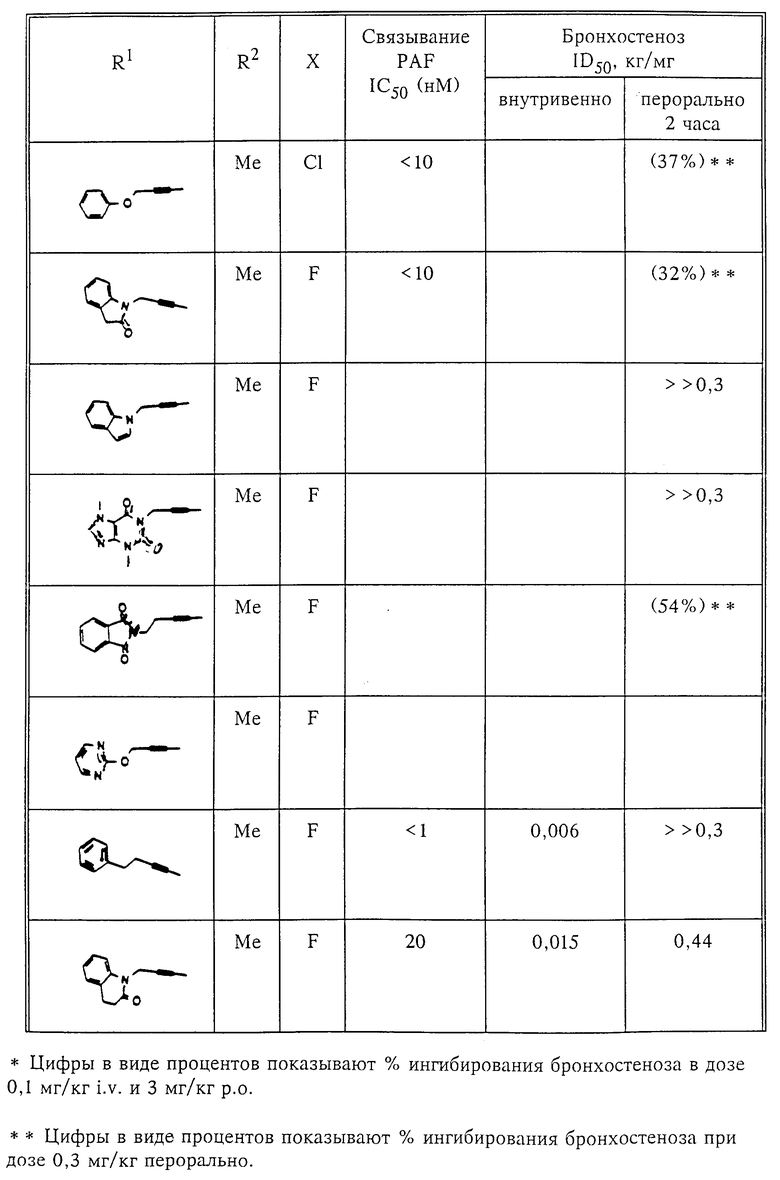
Изобретение относится к новым триазоло[4,3-a][1,4] бензодиазепинам или тиено[3,2-f][1,2,4]триазоло[4,3-а]диазепинам общей формулы I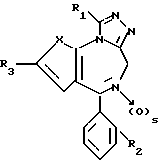
где X является -CH=CH- или S; R1 - низший алкил или трифторметил; R2 - хлор или фтор; R3 - радикал формулы R4-(CH2)n-C≡C- или R5-O-CH2-C≡C-, , где n - целое число 0,1 или 2; s - 0 или 1; R4 - фенил или моно-, ди- или трициклический 5-7-членный гетероциклический радикал, содержащий в качестве гетероатомов О или S и/или 1-3 атома азота, незамещенный или замещенный низшим алкокси, оксо, оксигруппой или хлором; R5 - фенил или пиридил-радикал, при условии, что, когда n равно 0, радикал R4 должен быть присоединен через углерод к углеродной связи, и что R5 всегда присоединяется через углерод к кислородной связи, и в случае наличия по меньшей мере одного асимметрического центра, их энантиомерам и рацематам и фармацевтически-приемлемым солям присоединения кислот, проявляющим свойства антагонистов фактора активации тромбоцитов (PAF) и, соответственно, обладающим ангиопротекторными, иммунологическими, антишоковыми свойствами, а также свойствами, препятствующими отторжению трансплантируемых органов, и фармакомпозициям на их основе. Новыми являются также соединения формулы I, где R3 означает группу - C≡CH. Получают соединения формулы I или их фармацевтически-приемлемые соли присоединения кислот взаимодействием соединения формулы II с соединение R4 - Y, в которой R4 означает указанное в п.1, и Y - бром или иод и, при необходимости, для получения соединения формулы I, в котором s является 1 и R4 отличается от остатка, содержащего основной атом азота, обрабатывают соединение формулы I, в которой s является 0 и R4 отличается от остатка, содержащего основной атом азота, надкислотой, и/или, при желании, превращают полученное соединение формулы I в фармацевтически-приемлемую соль присоединения кислот. 3 с. и 14 з. п.ф-лы, 2 табл.
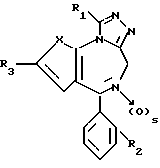
где Х -СН=СН- или S;
R1 низший алкил или трифторметил;
R2 хлор или фтор;
R3 радикал формулы R4-(CH2)n-C≡C- или R5-O-CH2-C≡C-, где n 0, 1 или 2;
s 0 или 1;
R4 фенил или моно-, ди- или трициклический 5 7-членный гетероциклический радикал, содержащий в качестве гетероатомов О или S и/или 1 3 атома азота, незамещенный или замещенный низшим алкокси, оксо, оксигруппой или хлором;
R5 фенил или пиридил при условии, что когде n 0, R4 должен быть присоединен через углерод к углеродной связи и R5 всегда присоединен через углерод к кислородной связи, и в случае наличия по меньшей мере одного асимметрического центра, их энантиомеры и рацематы,
и фармацевтически приемлемые соли присоединения кислот.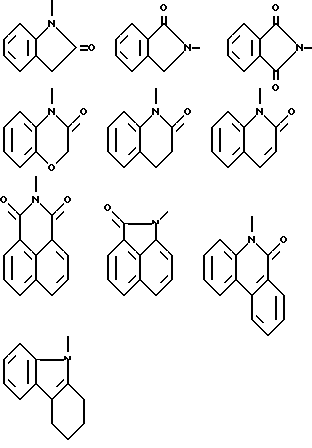
6. Соединение по п. 1, отличающееся тем, что оно представляет 5-[3-[4-(2-хлорфенил)-9-метил-6Н-тиено[3,2-f] [1,2,4]тиазоло[4,3-а][1,4]диазепин-2-ил]-2-пропинил -фенантридин-6(5Н)-он.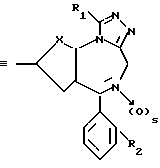
где X, R1, R2 и s имеют значения, указанные в п.1.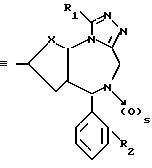
где X, R1, R2 и s имеют значения, указанные в п.1, с соединением общей формулы
R4 Y,
где R4 имеет значение, указанное в п.1;
Y бром или иод,
и при необходимости для получения соединения формулы I, в которой s 1 и R4 отличается от остатка, содержащего основной атом азота, обрабатывают соединение формулы I, в которой s 0 и R4 отличается от остатка, содержащего основной атом азота, над кислотой, и/или при желании превращают полученное соединение общей формулы I в фармацевтически приемлемую соль присоединения кислот.
Приоритет по признакам:
03.08.88 при R1 трифторметил, s 1;
18.12.87 все другие значения радикалов.
| J | |||
| Heterocyclic Chem | |||
| ПРИБОР ДЛЯ ЗАПИСИ И ВОСПРОИЗВЕДЕНИЯ ЗВУКОВ | 1923 |
|
SU1974A1 |
| J | |||
| Org | |||
| Chem | |||
| Планшайба для точной расточки лекал и выработок | 1922 |
|
SU1976A1 |
