Изобретение относится к новому способу получения некоторых замещенных бензопирановых соединений, промежуточным соединениям, полезным в этом способе, и к способу получения промежуточных соединений.
Замещенные бензопирановые соединения известны из уровня техники. Например, EP 0 173 516-A раскрывает класс замещенных бензопирановых соединений, описываемых как соединения, имеющие активность в качестве лейкотриеновых антагонистов и полезные в терапии для лечения, например, заболеваний, вызываемых лейкотриенами и 5 α редуктазой.
Изобретение относится к новому способу получения некоторых из бензопирановых соединений, описанных в EP 0 173 516-A, и в частности предлагает эффективный путь получения соединений, включающий гораздо меньше стадий реакции, чем описано до сих пор. Уменьшение числа стадий реакции при получении целевых продуктов обеспечивает в результате гораздо более эффективный и экономичный путь, чем путь, предусматривающий большое число стадий.
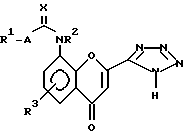
Таким образом, изобретение предлагает в первом аспекте способ получения соединения структуры (I):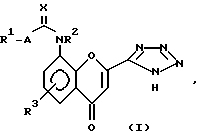
в которой:
R1 представляет собой группу структуры: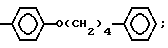
R2 представляет собой водород или C1-6 алкил;
R3 представляет собой водород;
A представляет собой одинарную связь; и
X представляет собой кислород;
или его соли, сольвата или гидрата, который включает циклизацию соединения структуры (II):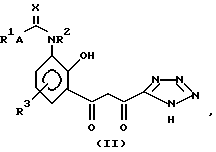
или его соли, гидрата или сольвата, где R1, R2, R3, A и X имеют те же значения, что и описанные для структуры (I), и полученные затем в случае необходимости, его соли, сольвата или гидрата.
R2 представляет собой предпочтительно водород.
Циклизацию соединения структуры (II) проводят подходящим образом в присутствии кислоты. Например, циклизация может быть проведена в присутствии серной кислоты, в метаноле или в уксусной кислоте в качестве растворителя. Предпочтительно, реакцию проводят в смеси растворителей метанол/тетрагидрофуран в присутствии хлористоводородной кислоты. Альтернативные условия кислота/растворитель очевидны специалистам и включают, например, кислоты, такие как бромистоводородную или иодистоводородную кислоту, хлорную кислоту или толуолсульфокислоту и кислоты Льюиса, например трихлорид алюминия, в подходящих растворителях, таких как вода, C1-4алканолы, такие как этанол или метанол, и ненасыщенные карбоциклические углеводороды, такие как бензол или толуол.
Следует отметить, что, хотя для удобства структура (II) представлена как "ди-кето"-форма, соединения структуры (II) могут также существовать в "кето-энольной" форму и в "циклической гидроксихромановой" форме (IIB):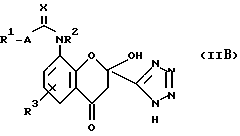
Имеется в виду, что структура (II) охватывает все таутомерные формы соединений структуры (II).
В предпочтительном аспекте, следовательно, предлагается способ получения соединения структуры (IA) или его соли, сольвата или гидрата: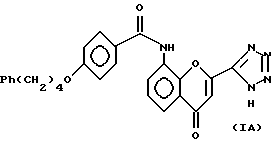 включающий циклизацию соединения структуры (IIA) или его соли, сольвата или гидрата:
включающий циклизацию соединения структуры (IIA) или его соли, сольвата или гидрата: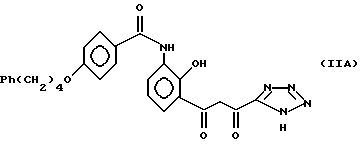 и после этого необязательно получение его соли, сольвата или гидрата. Наиболее предпочтительно, циклизацию проводят в присутствии хлористоводородной кислоты в смеси метанол/тетрагидрофуран в качестве растворителя.
и после этого необязательно получение его соли, сольвата или гидрата. Наиболее предпочтительно, циклизацию проводят в присутствии хлористоводородной кислоты в смеси метанол/тетрагидрофуран в качестве растворителя.
Что касается соединений структуры (II), соединения структуры (IIA) могут, конечно, существовать в соответствующих кето-энольной и циклической хромановой формах, каждая из которых предполагается охваченной структурой (IIA).
Соединения структуры (II) (и, в частности, структура (IIA)) являются новыми и образуют дальнейший аспект изобретения. Соединения структуры (II) могут быть получены по реакции соединения структуры (III):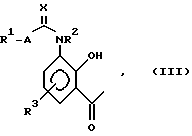
в которой R1, R2, R3, A и X имеют значения, определяемые для структуры (I) в п. 1 формулы изобретения, с соединением структуры (IV) или его солью: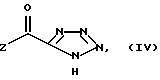
в которой Z представляет собой группу R6O, в частности, C1-C6 алкокси.
Соответственно, реакцию проводят в органическом растворителе, таком, как, например, диметилформамид, эфирные растворители, такие как тетрагидрофуран, толуол или бензол, гексан или C1-6алканолы, такие как метанол или этанол, в присутствии основания, например алкоксида щелочного металла, такого как т-бутоксид калия, метоксид натрия или метоксид калия, гидридов, таких как гидрид натрия, или амидного основания, такого как амид калия или амид натрия. Предпочтительно, реакцию проводят в тетрагидрофуране, в качестве растворителя в присутствии метоксида натрия в качестве основания.
Способ получения соединений структуры (II) является новым и представляет собой еще один аспект изобретения. В частности, для использования в получении соединений структуры (IIA) способ проводится путем реакции следующих соединений структуры (IIIA) и соединения структуры (IVA): или его соли в присутствии метоксида натрия в тетрагидрофуране в качестве растворителя: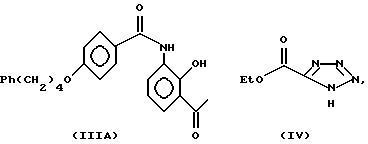
Соединения структуры (III) и (IV) получают из промышленно доступных исходных материалов по стандартным методикам, как описано ниже. Например, получение соединений структуры (III) описано в EP 0173516-A. Соединения структуры (IV), например, в которой Z является R6O, могут быть получены из азида натрия и подходящего алкилового эфира цианомуравьиной кислоты, такого как этиловый эфир цианомуравьиной кислоты, по известным методам или из динатриевой соли тетразол-5-карбоновой кислоты (имеющейся в продаже) путем реакции с подходящим алкиловым, ариловым или арилалкиловым эфиром галоидмуравьиной кислоты, например, этилхлорформиатом или изобутилхлорформиатом. Получение соединений структуры (IV) из соответствующей динатриевой соли тетразол-5-карбоновой кислоты является новым и составляет еще один аспект изобретения. Следует отметить, что соединения структуры (IV) могут быть получены и затем выделены перед реакцией с подходящими соединениями структуры (III) или могут быть получены "in situ" и дальше приведены во взаимодействие с соединениями структуры (III) без предварительного выделения.
Настоящее изобретение, в частности, полезно для получения соединений структуры (IA), исходя из соединений (IIIA) и (IVA) для получения промежуточных соединений структуры (IIA), которые подвергаются циклизации в описываемых здесь условиях с образованием целевого продукта. Соединения структуры (II) могут быть выделены из реакционной смеси перед циклизацией соединений структуры (I) или, альтернативно, как описано в примерах, реакция между соединениями структур (III) и (IV) с последующей циклизацией соединений структуры (II), образовавшихся таким образом, может быть доведена до конца "в одном сосуде", так сказать, без выделения промежуточных соединений, образовавшихся при этом.
Следующие примеры служат иллюстрацией изобретения. Температуры даны в градусах Цельсия (oC).
Примеры
1. Получение этилового эфира 1Н-тетразол-5-карбоновой кислоты
Трифторуксусную кислоту (24,47 г, 0,21 М) добавляют по каплям в течение 0,5 ч под азотом к перемешанной суспензии азида натрия (12,59 г, 0,19 М) в 2,6-лутидине (100 мл) при 8 12o. После перемешивания в течение 7 мин добавляют этиловый эфир цианомуравьиной кислоты (20,4 г, 0,20 М) одной порцией. Смесь нагревают и перемешивают при 75o в течение 6 ч и затем, после охлаждения, перемешивают при 20o в течение 16 ч. После охлаждения до 10o смесь добавляют ко льду (250 г) и II-мольной хлористоводородной кислоте (100 мл), поддерживая температуру ниже 20o. Продукт экстрагируют в этилацетат (1•250, 1•200, 2•100 мл), и объединенные экстракты сушат над сульфатом магния. После испарения растворителя при пониженном давлении маслянистый продукт (38,22 г) собирают в эфир (50 мл), и добавляют гексан (25 мл). Хранение при 4o в течение 2 3 дн дает кристаллический этиловый эфир 1Н-тетразол-5-карбоновой кислоты, который отфильтровывают, промывают охлажденным эфиром и сушат воздухом, 14,12 г (выход 52,6%), т. пл. 88 93o.
ЯМР: (270 МГц, раствор в CDCl3): δ 13,6 13,8 (с, 1Н); 4,6 - 4,5 (к, 2Н); 1,5 1,4 (т, 3Н).
Видоизменение методики
В более крупном масштабе (84,4 г азида натрия) реакцию проводят иначе, чтобы предупредить какое-либо выделение азотоводородной кислоты.
После перемешивания при 75o и 20o добавляют нитрат натрия (63 г) в воде (300 мл) в течение 10 мин при 20 30o. Смесь перемешивают при 20 - 25o в течение 20 мин и затем добавляют охлажденную смесь воды (1,5 л) и II-мольной хлористоводородной кислоты (690 мл), поддерживая температуру между 25 и 30o. Затем продукт экстрагируют в этилацетат и кристаллизуют, как описано выше.
2. Получение изобутилового эфира 1Н-тетразол-5-карбоновой кислоты
К перемешанной суспензии динатриевой соли тетразол-5-карбоновой кислоты (15,8 г, 0,1 моль) в диметилформамиде (100 мл) в азотной атмосфере при 5o добавляют изобутиловый эфир хлормуравьиной кислоты (13,6 г, 13 мл, 0,1 моль) по каплям в течение 15 мин. Смесь перемешивают при 5 10o в течение 2 ч, затем при 20o в течение 2 ч. Смесь добавляют к воде (500 мл) и экстрагируют этилацетатом (2•200 мл). Водную фазу затем подкисляют до pH 1 концентрированной HCl и далее экстрагируют этилацетатом (2•200 мл). Этилацетатные экстракты промывают водой (2•200 мл), сушат (MgSO4) и упаривают, получая целевое соединение в виде смолы (8,6 г, 50,5%).
1H ЯМР (CDCl3): d 0,95 (д, 6Н, CH3), 2,08 (тк, 1Н, CH), 4,25 (д, 2Н, CH2).
3. Получение метилового эфира 4-(4-фенилбутокси)бензойной кислоты
Раствор метилового эфира 4-гидроксибензойной кислоты (13,4 кг, 88 моль) в диметилформамиде (52 л) добавляют по каплям к смеси aOMe (4,8 кг, 89 моля) и диметилформамида (50 л) при комнатной температуре при мягком потоке азота. Реакционную смесь нагревают при 60 70o в течение 1 ч с перемешиванием и затем охлаждают до комнатной температуры. К этой смеси по каплям добавляют раствор 4-фенилбутилбромида (16,92 кг, 79,4 моль) в диметилформамиде (5 л). Получающуюся смесь нагревают при 60 70o в течение 1 ч с постоянным перемешиванием и охлаждают до комнатной температуры. После добавления 1 н. NaOH (110 л) продукт экстрагируют дважды этилацетатом (50 л и 80 л). Экстракты последовательно промывают 1 н. NAOH (110 л) и насыщенным рассолом (20 л) и затем концентрируют досуха под вакуумом, получая целевое соединение с количественным выходом.
4. Получение 4-(4-фенилбутокси)бензойной кислоты
К раствору соединения из примера 3 в MeOH (50 л) добавляют 3 н. NaOH (46 л). Смесь нагревают с обратным холодильником в течение 1,5 ч. По окончании реакции MeOH удаляют перегонкой в вакууме. К остатку добавляют воду со льдом (120 л) и нейтральные вещества экстрагируют эфиром (30 л•3). Объединенные эфирные экстракты промывают 2 н. NAOH (25 л). Водные слои объединяют и доводят до pH 2 3 концентрированной HCl (16 л). Выпавшие в осадок твердые вещества собирают фильтрованием в центробежном поле, промывают водой и сушат нагреванием при 70 80o в токе воздуха, получая целевой продукт (17,67 кг, 65,4 моль, выход 82% из 4-гидроксибензоата).
5. Получение 3-/4-(4-фенилбутокси)бензоиламино/-2-гидроксиацетофенона
К раствору соединения из примера 4 (18,1 г, 67 ммоль) в CH2Cl2 (45 мл) добавляют каталитическое количество диметилформамида (0,45 мл) с последующим добавлением тионилхлорида (6,26 мл, 85,8 ммоль) при комнатной температуре в атмосфере азота. После кипячения с обратным холодильником в течение 2 ч смесь охлаждают до комнатной температуры и добавляют к раствору хлоргидрата 3'-амино-2'-гидроксиацетофенона (12 г, 64 ммоль) и пиридина (15,5 мл, 192 ммоль) в CH2Cl2 (90 мл), поддерживая температуру между 0o и 3o. Смесь перемешивают при температуре 0 3o в течение 2 ч и выливают в 2 н. HCl (200 мл). Водные слои отделяют. Продукт в водных слоях экстрагируют дважды CH2Cl2 (150 Мл и 100 мл). Метиленхлоридные слои объединяют, промывают последовательно водой, насыщенным NaHCO3 (150 мл) и насыщенным рассолом (150 мл), сушат над MgSO4. Получающийся в результате раствор концентрируют в вакууме, пока не выпадут в осадок несколько кристаллов. К остатку добавляют этилацетат (150 мл), и раствор концентрируют в вакууме, пока не будет отогнана приблизительно половина этилацетата. Смесь охлаждают до приблизительно 0o. Выпавшие кристаллы собирают и сушат в вакууме, получая целевое соединение (21,6 г, 53,6 ммоль, 90%-ный выход).
6. Получение 2-/4-(4-фенилбутокси)бензоиламино/- 6-/1,3-диоксо-3-(тетразол-5-ил)пропилфенола
Под атмосферой азота, растворяют при перемешивании трет.-бутоксид калия (31,36 г, 0,28 моль) в сухом диметилформамиде (160 мл). К полученному раствору добавляют соединение гидроксиацетофенона из примера 5 (16,12 г, 0,04 моль), а затем 5-этоксикарбрнилтетразол из примера 1 (1,39 г, 0,052 моль, 1,3 экв.) при комнатной температуре. Температура реакции поднимается до ≈ 45o. Смесь перемешивают в течение 3 ч при 40o (масляная баня), затем охлаждают до 30o и выливают в холодную 1 н. HCl (800 мл). Получающийся осадок отфильтровывают, промывают водой (500 мл) и затем сушат при 70o в печи с вентилятором, получая целевое соединение (19,4 г, 97%). Проводят очистку, используя любую из следующих процедур.
Процедура 1
Перемешанную суспензию сырого продукта (10 г) в этилацетате (150 мл) нагревают при 60o в течение 2 ч. После охлаждения до комнатной температуры смесь переносят в холодильник и оставляют на 2 ч. Продукт затем фильтруют, промывают холодным этилацетатом (15 мл) и сушат при 70o в печи с вентилятором, получая очищенный продукт (8,5 г, 85%).
Процедура 2
Перемешанную суспензию сырого продукта (5 г) в ацетоне (50 мл) нагревают с обратным холодильником в течение 2 ч. После охлаждения до комнатной температуры смесь переносят в холодильник и оставляют на 2 ч. Продукт затем фильтруют, промывают холодным ацетоном (5 10 мл) и сушат при 70o в печи с вентилятором, получая целевой продукт (4,1 г, 82%).
7. Получение 4-оксо-8-/4-(4-фенилбутокси)бензоиламино/- 2-тетразол-5-ил-4Н-1-бензопирана полугидрата
К перемешанной суспензии очищенного продукта из примера 6 (7,984 г, 0,016 моль) в метаноле (72 мл) добавляют концентрированную серную кислоту (0,6 мл) и реакционную смесь нагревают с обратным холодильником в течение 3 ч. Смеси дают остыть до комнатной температуры, и затем переносят ее в холодильник на 2 ч. Густую смесь затем фильтруют, промывают холодным метанолом (40 мл) и водой (90 мл) и затем опять холодным метанолом (30 мл). Продукт сушат при 0o в печи с вентилятором и затем оставляют стоять на 24 ч при комнатной температуре, получая целевое соединение (7,36 г, 96%).
Примеры 8 и 9
Эти два примера иллюстрируют способ получения соединений (I) из промежуточных соединений структур (III) и (IV) "в одном сосуде".
8. Получение 4-оксо-8-/4-(4-фенилбутокси)бензоиламино/- 2-тетразол-5-ил-4Н-1-бензопирана полугидрата
К перемешанной суспензии метоксида натрия (15 г, 0,28 моль) в сухом тетрагидрофуране в атмосфере азота добавляют порциями соединение гидроксиацетофенона из примера 5 (16 г, 0,04 моль) при приблизительно 25o. Затем добавляют раствор этилового эфира тетразол-5-карбоновой кислоты из примера 1 (7,3 г, 0,05 моль) в тетрагидрофуране, поддерживая температуру реакции приблизительно 25oC. Реакционную смесь перемешивают в условиях дефлегмации в течение приблизительно 100 мин для обеспечения полного образования соединения дикетона из примера 6. К реакционной смеси добавляют метанол, а затем добавляют концентрированную хлористоводородную кислоту (28 мл, 0,34 моль) и последующее нагревание реакционной смеси с обратным холодильником в течение приблизительно 2 ч приводит к образованию целевого соединения, которое кристаллизуется из раствора. После охлаждения до приблизительно 20o продукт отделяют фильтрованием и промывают метанолом. Выделенное твердое вещество очищают путем превращения в натриевую соль в метаноле и повторного осаждения целевого соединения хлористоводородной кислотой. Повторно осажденный продукт выделяют фильтрацией, промывают водным раствором метанола, сушат и затем повторно гидратируют при комнатной температуре, получая целевое соединение (18,56 г, 94%).
9. Получение 4-оксо-8-/4-(4-фенилбутокси)бензоиламино/- 2-тетразол-5-ил-4Н-1-бензопирана полугидрата
К перемешанной суспензии метоксида натрия (14,1 кг, 261 моль) в сухом тетрагидрофуране под атмосферой азота порциями добавляют соединение гидроксиацетофенона из примера 5 (15,0 кг, 37,2 моль) при приблизительно 25o. Затем добавляют раствор этилового эфира тетразол-5-карбоновой кислоты из примера 1 (6,8 кг, 47,9 моль) в тетрагидрофуране, поддерживая температуру реакции при приблизительно 25o. Реакционную смесь перемешивают при дефлегмации в течение приблизительно 100 мин для обеспечения полного образования соединения дикетона из примера 6. К реакционной смеси добавляют метанол и затем концентрированную хлористоводородную кислоту (31,4 кг, 314 моль), и последующее нагревание реакционной смеси с обратным холодильником в течение приблизительно 2 ч приводит к образованию целевого соединения, которое кристаллизуется из раствора. После охлаждения до приблизительно 20oC продукт отделяют фильтрацией и промывают метанолом. Отделенное твердое вещество очищают превращением в натриевую соль в метаноле и повторным переводом в осадок с помощью хлористоводородной кислоты. Повторно выпавший в осадок продукт отделяют фильтрацией, промывают водным раствором метанола, сушат и затем повторно гидратируют при комнатной температуре с получением целевого соединения (15,5 кг, 85%).
Описывается способ получения замещенных бензопирановых производных, включающий на последней стадии циклизацию соответствующих промежуточных соединений с открытой цепью. 7 с. и 1 з. п. ф-лы.
где R1 группа
R2 водород или С1 С6-алкил;
R3 водород;
А одинарная связь;
Х кислород,
или их солей, гидратов или сольватов, отличающийся тем, что осуществляют циклизацию соединения общей формулы II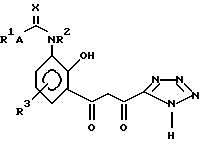
где R1, R2, R3, А и Х имеют указанные значения,
или его соли, гидрата или сольвата и при необходимости целевой продукт выделяют в виде соли, гидрата или сольвата.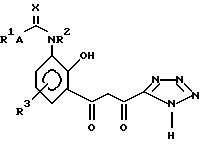
где R1, R2, R3, А и Х имеют указанные в п. 1 значения,
или его соль, гидрат или сольват.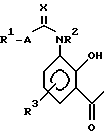
где R1, R2, R3, А и Х имеют указанные в п. 1 значения,
с соединением формулы IV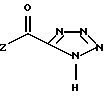
где Z C1 C6-алкоксигруппа,
или его солью.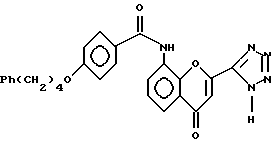
или его соли, гидрата или сольвата, отличающийся тем, что соединения IIIA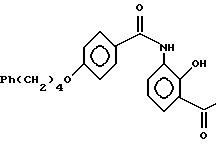
подвергают взаимодействию с соединением формулы IVA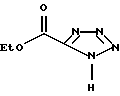
или его солью с последующей циклизацией полученного промежуточного соединения формулы IIA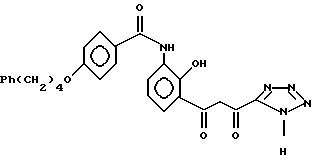
или его соли, гидрата или сольвата.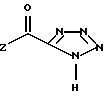
где Z С1 С6-алкоксигруппа,
отличающийся тем, что динатриевую соль тетразол-5-карбоновой кислоты подвергают взаимодействию с соответствующим алкиловым эфиром галоидмуравьиной кислоты.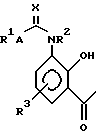
где R1, R2, R3, Х и А имеют указанные в п. 1 значения.
| ЕР, патент, 0173516, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

