Настоящее изобретение относится к новому способу получения акрил-пиперидинкарбинолов.
Патент США 4007196 описывает определенные соединения, которые обладают антидепрессантной активностью.
Промежуточным соединением в получении вышеупомянутых соединений является соединение формулы (A):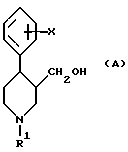
в которой R1 представляет водород, трифтор(C1-4)-алкил, алкил или алкинил, и X представляет водород, алкил с 1-4 атомами углерода, алкокси, трифторалкил, гидрокси, галоген, метилтио или аралкилокси.
Соединения формулы (A) раскрываются как обладающие фармакологическими свойствами, которые делают их полезными в качестве антидепрессантов.
Было найдено, что одно из соединений формулы (A) особенно эффективно как антидепрессант. Это соединение известно под названием пароксетин и имеет следующую формулу: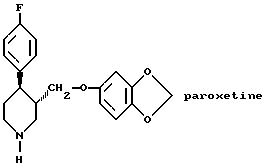
В патенте США 4902801 описывается получение соединений формулы (B):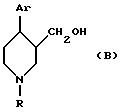
в которой Ar представляет арильную или замещенную арильную группу, и R представляет водород, алкильную или аралкильную группу; восстановлением соединения формулы (C):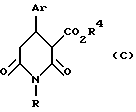
в которой Ar и R имеют те же значения, что определены для формулы (B), и Ra - алкильная группа.
Такой способ описывается как приемлемый для получения соединений-предшественников пароксентина формулы (B).
Единственными известными восстанавливающими агентами для использования в способе, описанном в патенте США 4902801, являются литийалюмогидрид или алюмогидрид. Эти восстанавливающие агенты являются дорогостоящими, трудны в обращении и их использование связано с большим выделением тепла, что создает проблемы контроля процесса при проведении реакции в большом объеме.
Настоящее изобретение решает эти проблемы использованием диборана в качестве восстанавливающего агента. Кроме того, этот способ дает лучший выход и более экономичен.
Соответственно настоящее изобретение предлагает способ получения соединения формулы (I):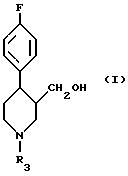
в которой R3 - водород, C1-6-алкил или C1-6-алкиларил, восстановлением с использованием диборана соединения формулы (II):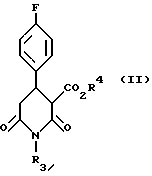
в которой R3 имеет то же значение, что в формуле (I) и R4 - C1-6-алкил.
Предпочтительно R3 - метил.
Предпочтительно R4 - этил или метил или смесь этила/метила.
Реакция проводится в инертном растворителе, таком как тетрагидрофуран или диметоксиэтан (ДМЭ).
Диборан обычно получают in situ добавлением эфирата трехфтористого бора к борогидриду натрия в присутствии соединения формулы (II) при пониженной температуре, такой как -10 - 20oC, предпочтительно от 0 до 5oC. В другом случае, что более предпочтительно по причинам удобства и безопасности, получают диборан добавлением газообразного хлороводорода (который можно растворить в инертном растворителе, таком как ДМЭ) к борогидриду натрия в присутствии соединения формулы (II) при пониженной температуре, такой как от -10 до 20oC, предпочтительно от 0 до 5oC. Когда добавление эфирата трехфтористого бора или газообразного хлороводорода завершено, температуру реакции доводили до температуры окружающей среды или повышенной температуры, например от 20 до 60oC, предпочтительно от 20 до 40oC.
Затем реакцию можно завершить или загасить добавлением реакционной смеси к минеральной кислоте, такой как водная хлористоводородная кислота, или добавлением минеральной кислоты, такой как хлористоводородная кислота, к реакционной смеси. Затем любое полученное твердое вещество можно отфильтровать, а продукт - соединение формулы (I) можно выделить отгонкой растворителя реакции растворением сконцентрированного раствора продукта в подходящем растворителе, таком как толуол, из которого можно осадить продукт, и осаждением продукта добавлением подходящего осаждающего растворителя, такого как н-гептан.
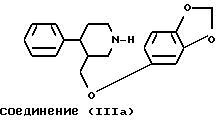
Настоящее изобретение также предлагает способ получения пaроксетина или его фармацевтически приемлемой соли, в частности полугидрата гидрохлорида, который включает образование соединения формулы (I), как описано выше, и затем последующее превращение его в пароксетин или его фармацевтически приемлемую соль, с использованием традиционных методик, в частности, описанных в патентах США 4902801 и 4721723.
Конкретно пароксетин может быть получен из соединения формулы (I), где R3 представляет метил (Ia), взаимодействием указанного соединения с бензолсульфонилхлоридом в присутствии N, N-диметилэтиламина с получением соответствующего фенилсульфоната (Ib), который затем взаимодействует in situ с сезамолом в присутствии метоксида натрия с получением соединения (IIIb)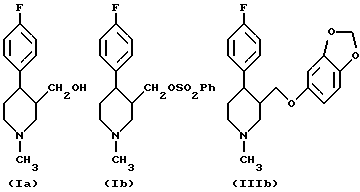
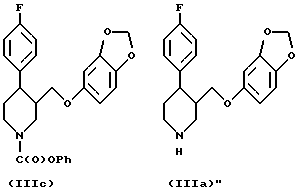
Соединение (IIIb) затем взаимодействует с фенилхлорформиатом с получением N-феноксикарбонильного соединения (IIIc). Наконец, соединение (IIIc) взаимодействует с гидроксидом калия в толуоле с получением пароксетина (IIIa), который может быть затем превращен in situ в гидрохлоридную соль.
Следующие примеры иллюстрируют настоящее изобретение.
Пример 1.
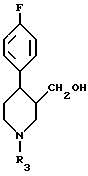
(±)-Транс-4-(4'-фторфенил)-3-гидроксиметил-N-метилпиперидин
Загрузка:
*(±)-транс-3-этокси/метоксикарбонил-4-(4'-фторфенил)-N-метилпиперидин-2,6-дион.
15,3 г 93,7%-ной чистоты
Борогидрид натрия 6,3 г
Эфират трехфтористого бора 18 мл
Тетрагидрофуран (ТГФ) 75 мл
Толуол 200 мл
3N HCl 40 мл
Гептан 70 мл
40%-ный раствор гидроксида натрия 25 мл
Способ - Методика проведения следующая:
1. К 50 мл ТГФ добавить 6,3 г борогидрида натрия.
2. Охладить раствор до 0 - 5oC.
3. Растворить 15,3 г (±)-транс-3-этокси/метоксикарбонил-4-(4'-фторфенил)-N-метилпиперидин-2,6-дион I в 25 мл ТГФ. Добавить приблизительно в течение 5 мин к раствору борогидрида, сохраняя температуру от 0 до 5oC.
4. Медленно добавить к раствору 18 мл эфирата в течение 15 мин, сохраняя температуру от 0 до 5oC.
5. Дать подняться температуре до 20oC в течение около 1 ч.
6. Подогреть раствор до 35 - 40oC за 2 ч.
7. Охладить раствор до 0 - 5oC.
8. Теперь медленно добавить раствор к 40 мл 3N HCl, позволяя температуре подняться до 20 - 25oC.
9. Охладить раствор до 5oC и отфильтровать твердую борную кислоту.
10. Промыть фильтр 20 мл воды.
11. Кипятить реакционную массу с обратным холодильником при 65oC, чтобы собрать ТГФ.
12. Позволить подняться температуре раствора до 100oC.
13. Добавить 50 мл воды/75 мл толуола, чтобы охладить раствор до 60oC.
14. Отделить нижний водный слой.
15. Добавить еще 50 мл воды к толуолу с поддержанием температуры при 60oC.
16. Отделить и собрать водные фракции.
17. Добавить 75 мл толуола к водной фракции. Довести pH до 12 - 12,5 и разделить слои.
18. Добавить еще 50 мл толуола к водной фракции и разделить.
19. Соединить фазы толуола и выпаривать приблизительно до 20 г.
20. Добавить 50 мл гептана, охладить до 5oC и фильтровать.
21. Промыть фильтр 20 мл гептана.
22. Сушить в вакуумной печи в течение ночи при 40oC.
Полученный вес 9,6 г.
Чистота 97%.
Выход 85%.
Анализы проводились с использованием ВЭЖХ.
*Получен по методике, описанной в патенте США N 4902801.
Пример 2.
Синтез (±)-транс-4-(4'-фторфенил)-3-гидроксиметил-N-метилпиперидина.
Загрузка:
**(±)-транс-3-этокси/метоксикарбонил-4-(4'-фторфенил)-N-метилпиперидин - 15,3 г.
Борогидрид натрия - 8,0 г
Газ хлороводород - 6,5 г
Диметоксиэтан (ДМЭ) - 150 мл
Толуол - 50 мл
Раствор 3 N хлористоводородной кислоты - 60 мл
Гептан - 20 мл
40%-ный раствор гидроксида натрия - 25 мл
Способ - Методика проведения следующая:
1. Добавить борогидрид натрия (8,0 г) к ДМЭ (75 мл).
2. Охладить раствор до 0 - 5oC.
3. Растворить (±)-транс-3-этокси/метоксикарбонил-4-(4'-фторфенил)-N-метилпиперидин (15,3 г) в ДМЭ (25 мл) и добавить к суспензии борогидрида натрия с поддержанием температуры при 0 - 5oC.
4. Растворить газообразный хлороводород (6,5 г) в ДМЭ (50 мл).
5. Добавить раствор хлороводорода/ДМЭ к суспензии борогидрида с поддержанием температуры при 0 - 5oC. В течение этого периода реакция проходит под слоем азота и высвобождается водород.
6. Перемешивать реакционную смесь при 0 - 5oC 30 мин.
7. Нагреть смесь до 35 - 40oC в течение 2 ч.
8. Охладить реакционную смесь до 0 - 5oC.
9. Погасить реакцию добавлением 3 N раствора хлористоводородной кислоты (60 мл) с поддержанием температуры ниже 20oC.
10. Добавить воду (50 мл) в реакционную смесь с поддержанием температуры ниже 20oC.
11. Отогнать раствор при температуре до 95oC и собрать влажный раствор ДМЭ (приблизительно 150 мл).
12. Добавить толуол (50 мл) и дать температуре упасть до 80oC.
13. Разделить фазы.
14. Охладить водную фазу до 50 - 55oC и добавить гептан (20 мл).
15. Добавить раствор гидроксида натрия до величин pH 11,0 - 11,5 с поддержанием температуры 50 - 55oC.
16. Охладить смесь до 5 - 10oC в течение по меньшей мере 30 мин.
17. Отфильтровать продукт.
18. Промыть продукт водой (2 х 20 мл).
19. Высушить продукт приблизительно при 40oC.
Обычно вес выделенного продукта 9,1 г.
Чистота 90 - 95%.
Выход 78 - 80%.
**Получено по методикам, описанным в патенте США 4902801.
Пример 3.
Синтез транс-4-(4'-фторфенил)-3-(3', 4'-метилендиоксифеноксиметил)-N-метилпиперидина (IIIb).
Загрузка:
транс-4-(4'-фторфенил)-3-гидроксиметил-N-метилпиперидин (Ia) - 125 г
дихлорметан - 920 мл
N,N-диметилэтиламин - 84 мл
бeнзолсульфонилхлорид - 82 мл
сезамол - 81 г
диметилформамид - 660 мл
метоксид натрия - 45 г
вода - 1860 мл
сульфат магния (безводный) - 15 г
Способ.
Раствор транс-4-(4'-фторфенил)-3-гидроксиметил-N-метилпиперидина (Ia) в дихлорметане и N,N-диметилэтиламина охлаждают до 0oC. Добавляют бензолсульфонилхлорид в дихлорметане. Затем добавляют воду и смесь перемешивают в течение примерно 30 мин. Слой дихлорметана отделяют, водный слой промывают дихлорметаном и объединенные органические растворы фильтруют через сульфат магния. Слои дихлорметана выпаривают и растворитель замещают диметилформамидом. Добавляют сазамол либо в виде твердого вещества, либо в виде раствора в DMF. Раствор охлаждают до примерно 20oC, затем постепенно добавляют метоксид натрия с небольшим количеством воды и смесь перемешивают в течение 2 - 4 ч при примерно 50oC. Смесь разбавляют водой и охлаждают до примерно 20oC. Продукт выделяют, промывают водой и сушат.
Типичный выход: 70 - 90%.
Синтез транс-4-(4'-фторфенил)-3-(3', 4'- метилендиоксифеноксиметил)-N-феноксикарбонилпиперидина (IIIc).
Загрузка:
транс-4-(4'-фторфенил)-3-(3', 4'-метилендиоксифеноксиметил)-N-метилпиперидин (IIIb) - 180 г
толуол - 1600 мл
фенилхлорформиат - 72 мл
серная кислота - 25 мл
пропан-2-ол - 2250 мл
вода - 815 мл
ускоритель фильтрования (Целит) - 5 г
Способ.
Фенилхлорформиат медленно добавляют к раствору транс-4-(4'-фторфенил)-3-(3', 4'-метилендиоксифеноксиметил)-N-метилпиперидина (IIIb) в толуоле, поддерживая температуру при 60 - 65oC, и смесь перемешивают в течение дополнительных 1 - 2 ч. Смесь промывают разбавленной серной кислотой и затем водой. Водные растворы отделяют и промывают толуолом. Объединенные толуольные экстракты фильтруют через ускоритель фильтрования, такой как Целит. Раствор концентрируют и растворитель замещают пропан-2-олом. Смесь охлаждают до ниже 5oC, продукт выделяют, промывают пропан-2-олом и сушат.
Типичный выход: 70 - 90%
Синтез транс-4-(4'-фторфенил)-3-(3',4'-метилендиоксифеноксиметил)пиперидин гидрохлорида (IIIa гидрохлорид, пароксетин гидрохлорид).
Загрузка:
транс-4-(4'-фторфенил)-3-(3', 4'-метилендиоксифеноксиметил)-N-феноксикарбонилпиперидин (IIIc) - 90 г
толуол - 1670 мл
гидроксид калия - 173 г
вода - 2600 мл
хлористоводородная кислота 32% вес/вес - 52 мл
промышленный этиловый спирт - 80 мл
пароксетин гидрохлорид (затравочные кристаллы) - 100 мг
Способ.
Гидроксид калия добавляют к раствору транс-4-(4'-фторфенил)-3-(3',4'-метилендиоксифеноксиметил)-N-феноксикарбонилпиперидина (IIIc) в толуоле. Смесь нагревают до флегмы в течение 2 - 3 ч и затем охлаждают, после чего промывают водным раствором гидроксида калия и затем водой. После отделения слоя толуола добавляют промышленный этиловый спирт. Смесь затравливают кристаллами пароксетин гидрохлорида и добавляют хлористоводородную кислоту. Смесь перемешивают в течение примерно 2 ч при примерно 20oC, затем охлаждают до 0 - 5oC и перемешивают в течение дополнительных 2 ч. Продукт выделяют, промывают толуолом и сушат.
Типичный выход: 90 - 95%.
Изобретение относится к усовершенствованному способу получения 4-(4'-фторфенил)-3-гидроксиметилпиперидинов ф-лы I, где R3 представляет водород; С1-С6-алкил или С1-С6-алкиларил, восстановлением соответствующего пиперидин 2,6-диона ф-лы II, где R3 имеет вышеуказанные значения, R4 - С1-С6-алкил, в присутствии восстанавливающего агента - диборана, в среде инертного растворителя, такого как тетрагидрофуран или диметоксиэтан. Способ позволяет повысить выход целевого продукта и упростить процесс за счет использования более доступного восстанавливающего агента. Изобретение относится также к усовершенствованному способу получения 4-(4'-фторфенил)-3-(3',4'-метилендиоксифеноксиметил)пиперидина ф-лы IIIa взаимодействием соединения ф-лы I, полученного восстановлением соединения ф-лы II в присутствии диборана, с бензосульфонилхлоридом в присутствии N,N-диметилэтиламина и затем in situ с сезамолом, полученное соединение подвергают взаимодействию с фенилхлорформиатом, а затем с гидроксидом калия в толуоле. Процесс получения соединения IIIa значительно упрощается за счет использования на первой стадии восстанавливающего агента-диборана, а также повышается выход целевого продукта. 2 с. и 11 з.п. ф-лы.
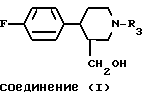
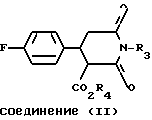
где R3 представляет водород, C1-C6-алкил или C1-C6-алкиларил, восстановлением соответствующего пиперидин-2,6-диона формулы II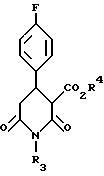
где R3 имеет значения, указанные для формулы I, и R4 представляет C1-C6-алкил,
в присутствии восстанавливающего агента в среде инертного органического растворителя, отличающийся тем, что в качестве восстанавливающего агента используют диборан.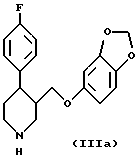
отличающийся тем, что 4-(4'-фторфенил)-3-гидроксиметил-N-метилпиперидин формулы Ia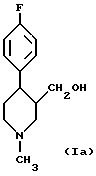
полученный по п.1 восстановлением соответствующего пиперидин-2,6-диона формулы IIa, где R4 представляет C1-6-алкил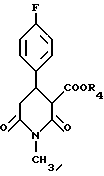
в присутствии диборана в среде инертного органического растворителя, подвергают взаимодействию с бензолсульфонилхлоридом в присутствии N,N-диметилэтиламина и затем in situ с сезамолом с получением соединения формулы IIIb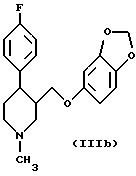
которое, в свою очередь, взаимодействует с фенилхлорформиатом с получением соединения формулы IIIc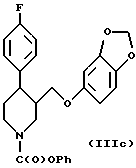
которое затем взаимодействует с гидроксидом калия в толуоле с получением пароксетина формулы IIIa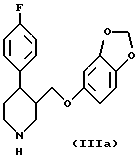
8. Способ по п.7, отличающийся тем, что R4 в соединении IIa представляет этил или метил.
| US 4902801 A, 1990 | |||
| US 4007196 A, 1977 | |||
| СПОСОБ УДАЛЕНИЯ ОСТАТКОВ КОМПЛЕКСНЫХ МЕТАЛЛООРГАНИЧЕСКИХ КАТАЛИЗАТОРОВ ИЗ УГЛЕВОДОРОДНЫХ РАСТВОРОВ СТЕРЕОРЕГУЛЯРНЫХ СИНТЕТИЧЕСКИХ КАУЧУКОВ | 1966 |
|
SU223334A1 |
| Способ получения производных пиперидина или их солей | 1976 |
|
SU633473A3 |
