Изобретение относится к некоторым производным тетрагидрокарбазола для применения в лечении расстройств, характеризуемых избыточным расширением просвета сосудов, в частности лечения мигрени.
Мигрень - несмертельное заболевание, которым страдает один из десяти человек; основной симптом - головная боль, другие симптомы включают рвоту, светобоязнь. В настоящее время наиболее широко используемым лечением является назначение эрготамина, дигидроэрготамина или метисергида, которые также используются как профилактические средства. Эти лекарства, между прочим, являются агонистами 5-HT1-подобного рецептора, но обладают и другим действием; лечение ими связано с рядом неблагоприятных побочных эффектов. Кроме того, некоторые пациенты испытывают "головные боли при снятии лекарств", которые появляются после прекращения лечения продуктом спорыньи, таким как эрготамин, заставляя их повторять лечение, и приводит к привыканию. Совсем недавно для потенциального использования в лечении мигрени были предложены различные производные триптамина.
Ввиду вышесказанного существует необходимость получить эффективные и безопасные медикаменты для лечения мигрени.
В патентах США N 4257952, 4172834, 4062864 и 3059309 раскрыт большой класс тетрагидрокарбазолов с формулой: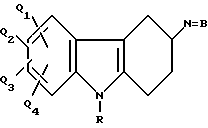
в которой N = B является, помимо прочего, -NHR' или -NR'R'', где R' и R'' - низший алкил, арил - низший алкил или вместе образуют гетероциклическое кольцо; R - помимо прочего, водород;
Q1 - помимо прочего, водород, галоген, низший алкокси, циано, -CO2R1 или -CONR2R3, где R1 может быть водородом, низшим алкилом или -CH2Ar, и R2 и R3 - водород, низший алкил или вместе образуют гетероциклическое кольцо;
Q2 - помимо прочего, водород, арил-(низший алкокси), гидрокси, тригалометил, нитро или алканоиламино; и
Q3 и Q4 могут каждый быть, помимо прочего, водородом.
Эти соединения известны как обладающие обезболивающим, психотропным и антигистаминным действием.
Неожиданно было выявлено, что некоторые тетрагидрокарбазолы противодействуют и частично противодействуют 5-окситриптамин-подобным рецепторам и могут найти применение в лечении состояний, в которых показан 5-HT1-подобный агонист или частичный агонист, в особенности состояний, связанных с головной болью, таких как мигрень, общая головная боль и боль, связанная с изменениями в сосудах. В этом описании термин "5-HT1-подобный агонист" включает также и частичные агонисты этого рецептора.
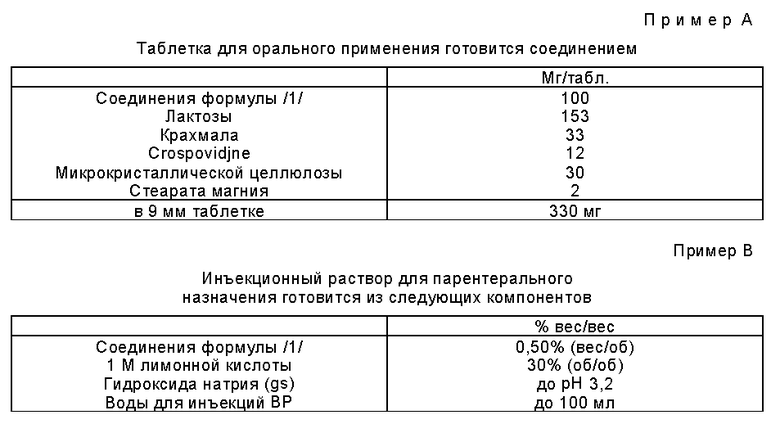
Поэтому настоящее изобретение предусматривает испольование соединений общей формулы I: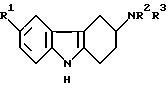
в которой
R1 представляет водород, галоген, трифторметил, нитро, гидрокси, C1-6-алкил, C1-6-алкокси, арил С1-6-алкокси, -CO2R4, -/CH2/nCN, -/CH2/nCONR5R6, -/CH2/nSO2NR5R6, C1-6-алканоиламино /CH2/n, или C1-6-алкилсульфониламино /CH2/;
R4 представляет водород, C1-6-алкил или арил C1-6-алкил;
R5 и R6 каждый независимо представляет водород или C1-6-алкил, или R5 и R6 вместе с атомом азота, к которому они присоединены, образуют кольцо;
n представляет 0, 1 или 2; и
R2 и R3 каждый независимо представляет водород, C1-6-алкил или бензил или вместе с атомом азота, к которому они присоединены, образуют кольцо пирролидино, пиперидино или гексагидроазепино; и их физиологически приемлемых солей в производстве лекарства для лечения состояния, где показан 5-HT1-подобный антагонист, в частности, мигрени, а также ее профилактики.
Изобретение также предусматривает способ лечения состояния, в котором показан 5-HT1-подобный агонист, в частности мигрени, который включает назначение субъекту, нуждающемуся в этом, эффективного количества соединения формулы (I) или его физиологически приемлемой соли.
R1 представляет водород, галоген, циано, гидрокси, C1-6-алкокси, арил C1-6-алкокси, -CO2R4, -/CH2/nCONR5R6 или -/CH2/nSO2NR5R6, и R2 и R3 каждый независимо представляют водород или C1-6-алкил.
Следует сказать, что соединения формулы I могут содержать один или несколько асимметричных центров, и такие соединения существуют как оптические изомеры /энантиомеры/. Итак, изобретение включает все такие энантиомеры и смеси, включая их рацемические смеси.
В соединениях формулы I атомом галогена может быть фтор, хлор, бром или йод. Алкильная группа или половина может иметь прямую или разветвленную цепь. Подходящие ариловые группы включают, например, ненасыщенные моноциклические или бициклические кольца и частично насыщенные бициклические кольца с 1 до 12 атомами углерода, такие, как фенил, нафтил и тетрагидронафтил. Если R5 и R6 вместе с атомом азота образуют кольцо, это предпочтительно 5 - 7-членное насыщенное гетероциклическое кольцо, которое может содержать другой гетероатом, выбранный из кислорода, серы или азота. Подходящие кольца включают пирролидино, пиперидино, пиперазино и морфолино.
В вышеуказанных соединениях R1 предпочтительно представляет галоген /напр. бром/, CF3, C1-6-алкокси /напр. метокси/, /CH2/nCN, -/CH2/nCONR5R6, -/CH2/nSO2NR5R6 или C1-6-алканоиламино. Наиболее предпочтительно R1 представляет группу -/CH2/nCONR5R6, в которой n представляет 0, и R5 и R6 каждый независимо представляют водород, метил, этил или пропил. Желательно, чтобы R5 и R6 независимо представляли водород или метил.
Если R1 представляет -CO2R4, R4 предпочтительно представляет C1-6-алкил.
R2 и R3 каждый предпочтительно представляет водород, метил или этил. Наиболее предпочтительно NR2R3- -NH2.
Для использования в соответствии с настоящим изобретением соединение формулы I является предпочтительно частичным антагонистом.
Подходящие физиологически приемлемые соли очевидны для специалистов данной области и включают, например, соли кислотного присоединения, такие, как те, которые образованы с неорганическими кислотами, например, хлористоводородной, серной или фосфорной кислотами и органическими кислотами, например, янтарной, малеиновой, уксусной или фумаровой кислотами. Другие неприемлемые физиологически соли, например, оксалаты, могут использоваться, например, в изоляции соединений формулы I, и включены в объем этого изобретения.
В объем изобретения также включены сольваты и гидраты соединений формулы IA.

в которой R1 - как определено выше, с оговоркой, что R1 - не водород, гидрокси, метокси или бензилокси, и их соли.
Настоящее соединение далее предусматривает следующие специфические соединения, которые также считаются новыми:
3-амино-6-циано-1,2,3,4-тетрагидрокарбазол гидрохлорид,
/+/-3-амино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол гидрохлорид,
/-/-3-амино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол гидрохлорид,
3-амино-6-бромо-1,2,3,4-тетрагидрокарбазол гидрохлорид,
3-амино-6-метил-1,2,3,4-тетрагидрокарбазол оксалат,
3-амино-6-этоксикарбнил-1,2,3,4-тетрагидрокарбазол оксалат,
3-амино-6-/N-метил карбоксамидо/-1,2,3,4-тетрагидрокарбазол полуоксалат,
3-амино-6-цианометил-1,2,3,4-тетрагидрокарбазол оксалат,
3-амино-6-/N-метилсульфонамидометил/-1,2,3,4-тетрагидрокарбазол оксалат,
3-амино-6-хлоро-1,2,3,4-тетрагидрокарбазол оксалат,
3-амино-6-трифторметил-1,2,3,4-тетрагидрокарбазол оксалат,
3-амино-6-н-бутилокси-1,2,3,4-тетрагидрокарбазол оксалат,
3-амино-6-сульфонамидо-1,2,3,4-тетрагидрокарбазол оксалат,
3-амино-6-нитро-1,2,3,4-тетрагидрокарбазол оксалат,
3-амино-6-/N, N-диметилкарбоксамидо/ -1,2,3,4-тетрагидрокарбазол полуоксалат,
3-амино-6-нитро-1,2,3,4-тетрагидрокарбазол оксалат,
3-амино-6-/N, N-диметилкарбоксамидо/ -1,2,3,4-тетрагидрокарбазол полуоксалат,
3-амино-6-/пиперидин-1-илкарбонил/ -1,2,3,4-тетрагидрокарбазол гидрохлорид,
3-амино-6-/пирролидин-1-илкарбонил/ -1,2,3,4-тетрагидрокарбазол гидрохлорид,
3-амино-6-/N, N-диэтилкарбоксамидо/ -1,2,3,4-тетрагидрокарбазол гидрохлорид,
3-амино-6-/ацетамидо/-1,2,3,4-тетрагидрокарбазол оксалат,
3-амино-6-метансульфонамидо-1,2,3,4-тетрагидрокарбазол оксалат,
3-амино-6-карбоксамидометил -1,2,3,4-тетрагидрокарбазол гидрохлорид,
3-метиламино-6-карбоксамидо -1,2,3,4-тетрагидрокарбазол оксалат,
3-этиламино-6-карбоксамидо -1,2,3,4-тетрагидрокарбазол оксалат,
3-н-пропиламино-6-карбоксамидо -1,2,3,4-тетрагидрокарбазол оксалат,
3-i-пропиламино-6-карбоксамидо -1,2,3,4-тетрагидрокарбазол оксалат,
3-диметиламино-6-карбоксамидо -1,2,3,4-тетрагидрокарбазол оксалат,
3-бензиламино-6-карбоксамидо -1,2,3,4-тетрагидрокарбазол оксалат,
3-пирролидинил-6-карбоксамидо -1,2,3,4-тетрагидрокарбазол оксалат, и
3-/N-/метил/этиламино/-6-карбоксамидо -1,2,3,4-тетрагидрокарбазол оксалат,
3-амино-6-/2-карбоксамидоэтил/ -1,2,3,4-тетрагидрокарбазол оксалат.
В другом аспекте настоящее изобретение предусматривает новое соединение формулы I, т.е. соединение IA или какое-либо из упомянутых соединений /в виде свободного основания или физиологически приемлемой соли/ для использования в качестве терапевтического агента, в частности, как 5-HT1 агониста или частичного агониста, например, в лечении мигрени.
Изобретение также предусматривает способ получения новых соединений формулы I.
Соединения формулы I можно получить методами, известными в области техники для получения тетрагидрокарбазолов, например:
A) Реакцией соединения формулы II: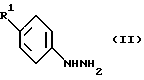
в которой R1 - как определен выше /или его соли кислотного присоединения с соединением формулы III: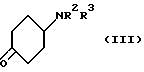
/в которой R2 и R3 - как определены выше/ или его N-защищенным производным; или
B) Реакцией соединения формулы IV: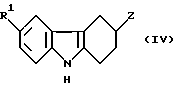
/в которой R1 имеет то же значение, как определено для формулы I и Z - уходящая группа/, с соединением формулы HNR2R3;
C) Реакцией соединения формулы V:
с ацилирующим или сульфонилирующим агентом;
D) Преобразованием одного соединения формулы I в другое соединение формулы I, например
/i/ чтобы получить соединение формулы I, в котором R1 представляет -/CH2/nCONH2 или CO2R4, проводят гидролиз соединения формулы I, в котором R1 представляет -/CH2/nCN, или его N-защищенного производного;
/ii/ чтобы получить соединение формулы I, в котором R1 представляет -CONR5R6, аминируют соединение формулы I, в котором R1 представляет -CO2H, или его N-защищенного производного; или
/iii/ чтобы получить соединение формулы I, в котором один из R2 и R3 - водород, а другой - C1-6-алкил, алкилируют соединение I, в котором R2 и R3 - оба водороды;
/iv/ чтобы получить соединение формулы I, в котором R1 представляет гидрокси, расщепляют соединение, в котором R1 представляет алкокси или аралкокси; при необходимости с последующим освобождением от защиты любых защитных атомов азота и образованием соли при желании.
Способ (A), который является формой синтеза индола Фишера, может проводиться с использованием методов, хорошо известных в химии. Так, реакцию можно провести в растворителе, например в спирте, таком как этанол или бутанол; или уксусной кислоте, и при температуре в пределах 0 - 150oC.
Гидразины формулы II, которые обычно используются в качестве соли гидрохлорида, - соединения известные и их можно получить известными способами.
Циклогексанон формулы III можно получить окислением соответствующего циклического спирта с использованием окисляющего агента, такого, как пиридин хлорхромат, пиридин дихромат, дипиридин Cr /VI/ оксид, гипохлорит натрия, гипохлорит кальция или диоксид марганца.
Уходящей группой Z в соединениях формулы /IV/ может быть, например, атом галогена или сульфонилокси группа, например, п-толуолсульфонилокси или метансульфонилокси. Процесс (B) можно провести в инертном органическом растворителе, таком, как спирт, например, метанол или эфир, например, тетрагидрофуран, и при температуре в пределах 0 - 150oC. Соединения формулы IV можно получить реакцией гидразина формулы II с соответствующе замещенным соединением циклогексанона. Если Z - ацилокси или сульфонилокси, его можно получить из соединения IV, в котором Z - гидрокси, с использованием стандартных процедур. Подходящие ацилирующие и сульфонилирующие агенты, которые могут использоваться в способе (C), включают хлориды карбоновой и сульфокислоты /например, ацетилхлорид или метансульфонилхлорид/, алкилэстеры, активированные эстеры и симметричные и смешанные ангидриды. Реакцию можно проводить в органическом растворителе, таком, как галоалкан /например, дихлорметане/, амиде /например, N,N-диметилформамиде/; эфире /например, тетрагидрофуране/ или третичном амине, таком, как пиридин. В общем также используется основание, например, триэтиламин, диметиламинопиридин или карбонат или бикарбонат щелочного металла. Реакция проводится при температуре в пределах от -10 до 100oC.
Соединения формулы V можно получить способами, аналогичными процессам (A) и (B), описанным выше.
Еще соединение V можно получить восстановлением соединения формулы I, в котором R1 - нитро, например, каталитическим гидрированием.
В химии хорошо известно, что гидролиз нитрила первоначально дает амид, который затем можно гидролизовать в кислоту. Поэтому точный продукт способа (Di) будет зависеть от условий реакции, выбранных для гидролиза. Чтобы получить соединение, в котором R1 представляет H2NCO-, гидролиз предпочтительно проводится с использованием перекиси водорода в присутствии гидроксида щелочного металла, например, гидроксида натрия, в растворителе, таком, как спирт, метаноле. Другие подходящие средства гидролиза включают уксусную кислоты и BF3; или муравьиную кислоту и бромистоводородную или хлористоводородную кислоты. Чтобы получить соединение, в котором R1 представляет -COOH кислоту или основание, можно использовать каталитический гидролиз.
Способ (Dii) можно осуществить реакцией соединения формулы I, в котором R1 - -CO2H, с амидом HNR5R6, в присутствии связующего агента, например, дициклогексилкарбодиимида или N, N-карбонилдиимидазола. В другом варианте исходный материал карбоновой кислоты можно вначале подвергнуть реакции для получения активированного производного карбоксильной группы, например, кислотного хлорида, кислотного ангидрида или активированного эстера, которое затем непосредственно реагирует с амином HNR5R6. Карбоновую кислоту можно также активировать на месте обработкой гексаметилфосфоротриамидом.
Алкилирование в соответствии со способом (Diii) можно осуществить реакцией амина формулы I с ацилирующим агентом, например, ангидридом, таким, как уксусный или пропионовый ангидрид, чтобы получить промежуточное соединение, в котором один из R2 или R3 является -C/O/C1-6-алкилом, с последующим восстановлением данного промежуточного соединения для получения нужного продукта. Специалистам также известны другие реагенты и условия.
Расщепление по способу (Div) можно провести восстановлением с использованием хорошо известных методов.
Во многих из указанных реакций необходимо защитить группу - R2R3, когда одна или обе из групп представляют водород. Подходящие N-защитные группы хорошо известны в области техники и включают, например, ацильные группы, такие как ацетил, трифтороацетил, бензоил, метоксикарбонил, т-бутоксикарбонил, бензилоксикарбонил или фталоил; и аралкильные группы, такие как бензил, дифенилметил или трифенилметил. Если R2 и R3 оба представляют водород, атом азота предпочтительно защищен как фталимид. Защитные группы должны легко удаляться в конце последовательности реакции. N-защиту можно снять обычными способами, например фталоиловую группу можно убрать реакцией с гидразином; ацильную группу, такую как бензоил, можно расщепить гидролизом, и аралкильную группу, такую как бензил, можно расщепить гидрогенолизом.
Если соединение формулы I получается в виде смеси энантиомеров, их можно разделить обычными методами, например, реакцией смеси с подходящей оптически активной кислотой, такой как д-винно-каменная кислота, л-яблочная кислота, л-миндальная, л-гулоновая или 2,3,4,6-ди-О-изопропилиден-кетогулоновая кислоты, чтобы получить две стереоизомерные соли, которые можно разделить, например кристаллизацией.
Иначе смеси энантиомеров можно разделить хроматографией, например на хиральной ВЭЖХ колонне.
Как было выявлено, соединения формулы I являются агонистами и частичными агонистами на рецепторах, подобных 5-HT1, и найдут применение в лечении и/или профилактике мигрени и других состояний, связанных с головными болями.
Для использования в медицине соединения настоящего изобретения обычно назначаются как стандартные фармацевтические композиции. Поэтому далее настоящее изобретение предусматривает фармацевтические композиции, включающие новое соединение формулы I или его физиологически приемлемую соль и физиологически приемлемый носитель.
Соединения настоящего изобретения формулы I могут назначаться орально, парентерально и приемом за щеку, под язык, в нос, через прямую кишку, через кожу в виде соответствующих фармацевтических композиций.
Соединения формулы I и их фармацевтически приемлемые соли, которые активны при оральном приеме, могут быть в форме жидкостей, например, сиропов, суспензий или эмульсий, или таблеток, капсул и лепешек.
Жидкая лекарственная форма может обычно состоять из суспензии или раствора соединения или его физиологически приемлемой соли в подходящем жидком носителе, например, водном растворителе /вода, этанол, глицерин/, или неводном растворителе, таком, как полиэтиленгликоль или масло. Форма может также содержать суспендирующий агент, консервант, вкусовой или цветовой агент.
Композицию в форме таблетки можно приготовить с использованием любого подходящего фармацевтического носителя, применяемого в приготовлении твердых форм. Примеры таких носителей включают стеарат магния, крахмал, лактозу, сахарозу и целлюлозу.
Композицию в форме капсулы можно получить с использованием традиционных процедур капсулирования. Например, гранулы, содержащие активный ингредиент, можно приготовить с использованием стандартных носителей и затем заполнением в твердую желатиновую капсулу; дисперсию или суспензию можно приготовить с применением подходящих фармацевтических носителей, например, водных смол, целлюлоз, силикатов или масел и заполнением дисперсии или суспензии в мягкую желатиновую капсулу.
Обычные парентеральные композиции состоят из раствора или суспензии соединения или физиологически приемлемой соли в стерильном водном носителе или парентерально приемлемом масле, например, полиэтиленгликоле, поливинил пирролидоне, лецитине, арахисовом масле или кунжутном масле. В другом случае раствор может быть лиофилизирован и затем восстановлен подходящим растворителем перед приемом.
Композиции для носового применения могут иметь форму аэрозолей, капель, гелей и порошков. Форма аэрозоли обычно включает раствор или мелкую суспензию активного вещества в физиологически приемлемом водном или неводном растворителе и предназначена для одноразового или многоразового использования в стерильной форме и герметичном контейнере, который может иметь форму патрона или заполняться в емкость с распылителем. Герметичный контейнер может быть универсальным дозатором в виде одноразового ингалятора или в виде аэрозольного дозатора с измерительным клапаном, который используется, пока не закончится лекарство, а потом выбрасывается. Если дозирующая форма включает аэрозольный дозатор, она содержит пропеллант, который может быть сжатым газом, воздухом, или органический пропеллант, такой как фторохлороуглеводород. Аэрозольная дозирующая форма может также быть насосом-распылителем.
Композиции, предназначенные для применения за щеку или под язык, включают таблетки, лепешки и пастилки, в которых активный ингредиент формуется с носителем, таким, как сахар и аравийская камедь, трагакант или желатин и глицерин.
Композиции для ректального применения обычно в форме шариков, содержащих обычную суппозитарную основу - масло какао.
Композиции, приемлемые для трансдермального применения, включают жидкие мази, гели и пластыри.
Предпочтительна форма дозировки - единичная доза /таблетка, капсула или ампула/.
Каждая единица дозировки содержит для орального применения от 1 до 250 мг /а для парентерального - от 0,1 до 25 мг/ соединения формулы I или его физиологически приемлемой соли, рассчитанного как свободное основание.
Физиологически приемлемые соединения изобретения обычно назначаются в ежедневном режиме /для взрослого пациента/ с оральной дозой между 1 и 500 мг, предпочтительно между 10 и 400 мг, при внутривенном, подкожном применении - 10 - 250 мг, внутримышечная доза составляет 0,1 - 100 мг, предпочтительно 0,1 - 50 мг соединения формулы I или ее физиологически приемлемой соли, рассчитанной как свободное основание, причем соединение назначается от 1 до 4 раз в день на период продолжительной терапии, например, в течение недели или больше.
Биологические данные.
Тестирование 5-HT1-подобного рецептора.
Подкожная вена собаки.
Геликоиды подкожной вены собаки помещались при 37oC в модифицированный раствор Кребса с силой покоя 10 мN. Раствор также содержал 1 мкмоль/л каждого из кетанзерин празозина, атропина и мепирамина, 6 мкмоль/л кокаина и 200 мкмоль/л аскорбата. На полиграфе с датчиками силы измерялись почти изомерные сокращения. Ткани дважды подвергались действию 5-окситриптамина /5-HT/ в количестве 2 мкмоль/л с последующим промыванием. Определялась кривая эффекта кумулятивной концентрации, а затем кривая на 5-HT в присутствии наивысшей используемой концентрации испытуемого соединения. Сокращения, вызванные испытуемым соединением, сравнивались с сокращениями, вызванными 5-HT. Рассчитывалась активность испытуемого соединения как коэффициент максимального действия, вызванного соединением, над действием, вызванным 2 мкмоль/л 5-HT. Из соответствующей кривой действия был вычислен EC50 испытуемого соединения. Затем методом Марано и Кауманна /1976, J.Pharmacol. Exp. Ther. 198, 518-525/ были определены константы Кр соответствующей диссоциации равновесия.
В этом тестировании соединения примеров 2, 4, 5, 6, 9, 10, 11, 13, 17, 18, 21 и 24 имели EC50 в пределах от 0,1 до 15 мкмоль.
Основная артерия кролика.
Методы.
Эксперименты проводились на внутричерепных артериях кролика, на изолированной базилярной артерии таким же методом, как описан выше /Парзон и Уэлли, 1989, Eur. J. Pharmacol. 174, 189-196/.
Вкратце, кролики умерщвлялись большой дозой анeстеризующего вещества /пентобарбитон натрия/.
Весь мозг быстро удалялся и погружался в холодный модифицированный раствор Кребса, и базилярная артерия удалялась с помощью препаровальной лупы. Раствор Кребса имел следующий состав /мМ /Na+ /120/; K+/5/; Ca2+ /2.25/; Mg2+ /0.5/; Cl- /98.5/; SO4 2- /1/; ЭДТК /0.04/, уравновешенный 95% O2 /5%/ CO2. Эндотелий удалялся мягким прикосновением к полости тонкой металлической проволоки. Артерии нарезались в кольцевые сегменты /шириной 4 - 5 мм/ и погружались для записи изометрического напряжения в 50-мл ванночки для тканей с модифицированным раствором Кребса и добавлением /мМ/ Na2+ /20/; фумарата /10/; пирувата /5/; L-глютамата /5/ и глюкозы /10/. Затем артерии помещались в силу покоя 3-4 мN при 37oC и через раствор пузырьками пропускался 95% O2 /5%/ CO2.
После тестов на первоначальную реактивность деполяризующим раствором KCl /90 мМ/ и на отсутствие расслабления, вызванного ацетилхолином, из-за предшествующего сокращения от 5-HT /10 мМ/, в присутствии аскорбата 200 мМ, кокаина 6 мМ, индометацина 2.8 мМ, кетанзерина 1 мМ и празозина 1 мМ были построены кумулятивные кривые концентрации - воздействия /2 нМ - 60 мМ/ на 5-HT.
В этом тестировании соединения примеров 2, 5, 6, 15, 17, 24, 25, 26, 28 и 29 имели EC50 в пределах 0,04 - 15.
Пример 1.
3-Амино-6-циано-1,2,3,4-тетрагидрокарбазол гидрохлорид.
Раствор 4-аминоциклогексанол гидрохлоридa /6.08 г, 0.04 М/ в воде /60 мл/ доводился до pH 8 водным раствором бикарбоната натрия. Добавлялся N-карбэтокси-фталимид /0.76 г, 0.04 М/, а затем тетрагидрофуран /до получения гомогенного раствора/. Чистый раствор перемешивался при комнатной температуре всю ночь. Во время этого выпадало в осадок вещество белого цвета. Тетрагидрофуран удалялся под вакуумом, и оставшийся водный раствор экстрагировался этилацетатом до полного очищения раствора. Экстракты этилацетата соединялись, промывались водой, высушивались /MgSO4/ и концентрировались до получения 4-фталимидо циклогексанола в виде белого вещества /7,1 г/.
Раствор 4-фталимидо циклогексанола /7,1 г, 0,029 М/ в дихлорметане /250 мл/ обрабатывался пиридиний хлорохроматом /8,6 г, 0,04 М/, и полученная темная смесь перемешивалась при комнатной температуре всю ночь. Был добавлен диэтиловый эфир /50 мл/, и смесь фильтровалась через кизельгур. Фильтрат концентрировался в вакууме, и остаток очищался колонной хроматографией /SiO2; CHCl3 /EtOAc/ для получения 4-фталимидо циклогексанона в виде твердого вещества белого цвета /6,4 г/.
4-Цианофенил гидразин гидрохлорид /4,41 г, 0,026 М/ растворялся в уксусной кислоте /100 мл/ и добавлялся ацетат натрия /2 г/. Добавлялся 4-фталимидо циклогексанон /6,4 г, 0.026 М/, и смесь нагревалась с флегмацией всю ночь. Растворитель удалялся в вакууме, и остаток растирался с метанолом для получения 3-фталимидо-6-циано-1,2,3,4-тетрагидрокарбазола в виде твердого вещества бежевого цвета /5,3 г/.
Суспензия указанного продукта /1 г/ в этаноле /40 мл/ обрабатывалась гидразином в воде /10 мл/. Реакционная смесь перемешивалась при комнатной температуре всю ночь, в течение которой реактанты растворялись. Растворитель удалялся в вакууме, и остаток разделялся между водным карбонатом калия и этилацетатом. Раствор этилацетата промывался водой, высушивался и концентрировался в вакууме для получения 3-амино-6-циано-1,2,3,4-тетрагидрокарбазола в виде твердого вещества бежевого цвета /500 мг/. Этот продукт был преобразован в соль гидрохлорида для получения искомого соединения, т. пл. 289oC /разл./.
1H ЯМР [250 МГц, CD3OD] 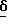 1,98 - 2,18 /1H, м/, 2,25 - 2,40 /1H, м/, 2,77 /1H, дд/, 2,98 /2H, м/, 3,22 /1H, дд/, 3,68 /1H, м/, 7,34 /1H, д/, 7,43 /1H, д/, 7,82 /1H, с/.
1,98 - 2,18 /1H, м/, 2,25 - 2,40 /1H, м/, 2,77 /1H, дд/, 2,98 /2H, м/, 3,22 /1H, дд/, 3,68 /1H, м/, 7,34 /1H, д/, 7,43 /1H, д/, 7,82 /1H, с/.
Пример 2.
3-Амино-карбоксамидо-1,2,3,4-тетрагидрокарбазол гидрохлорид.
Продукт примера 1 /400 мг/ растворялся в тетрагидрофуране, и добавлялся ди-т-бутил дикарбонат /500 мг/. Смесь перемешивалась при комнатной температуре всю ночь. Растворитель удалялся в вакууме и остаток очищался колонной хроматографией /SiO2; CHCl3/EtOAc/ для получения 3-т-бутилоксикарбониламино-6-циано- 1,2,3,4-тетрагидрокарбазола /40 мг/.
Смесь нитрила вышеуказанного продукта /440 мг/, водной перекиси водорода /30%, 0,5 мл/ и гидроксида натрия /вод./ /20%, 0,5 мл/, в метаноле /25 мл/ перемешивались при комнатной температуре всю ночь. Добавлялся метабисульфит натрия /100 мг/, и растворитель удалялся в вакууме. Остаток растворялся в этилацетате, и слой этилацетата отделялся, высушивался и концентрировался в вакууме для получения тягучего твердого вещества, которое очищалось колонной хроматографией /SiO2; CHCl3/EtOAc/ для получения 3-т-бутилоксикарбониламино- 6-карбоксиамидо-1,2,3,4-тетрагидрокарбазола в виде вещества белого цвета /400 мг, т.пл. 270oC /разл./.
Вышеуказанный продукт /400 мг, 0,0012 М/ растворялся в диоксане /100 мл/, и через раствор в течение 20 мин пропускался газ HCl. В течение этого времени осаждалось белое вещество. Избыток хлороводорода удалялся из раствора пропусканием через него N2, и твердый продукт, 3-амино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол гидрохлорид собирался фильтрацией, промывался диэтиловым эфиром и высушивался для получения искомого соединения в виде твердого вещества белого цвета /300 мг/. т.пл. 270oC /разл./.
1H ЯМР [250 МГц, ДМСО-д6] 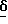 1,96 /1H, м/, 2,16 - 2,30 /1H, м/, 2,74 /1H, дд/, 2,85 /2H, м/, 3,12 /1H, дд/, один сигнал затемнен H2O на 3.6, 7,08 /1H, шир. с/, 7,27 /1H, д/, 7,61 /1H, д/, 7,87 /1H, шир.с/, 7,99 /1H, с/, 8,39 /3H, шир.с/.
1,96 /1H, м/, 2,16 - 2,30 /1H, м/, 2,74 /1H, дд/, 2,85 /2H, м/, 3,12 /1H, дд/, один сигнал затемнен H2O на 3.6, 7,08 /1H, шир. с/, 7,27 /1H, д/, 7,61 /1H, д/, 7,87 /1H, шир.с/, 7,99 /1H, с/, 8,39 /3H, шир.с/.
Пример 3.
3-Амино-6-метокси-1,2,3,4-тетрагидрокарбазол гидрохлорид.
Реакция 4-метоксифенил гидразин гидрохлорида /0,87 г, 5,0 мМ/ с 4-фталимидо-циклогексаноном /1,22 г, 5,0 мМ/ в этаноле /20 мл/ нагревалась с флегмацией 2 ч с последующим охлаждением и удалением осажденного вещества фильтрацией, что дало 3-фталимидо-6-метокси-1,2,3,4-тетрагидрокарбазол /1,62 г/.
Указанный продукт /1,57 г, 4,5 мМ/ суспендировался в метаноле /100 мл/ и обрабатывался гидразин гидратом /23 мл/ с перемешиванием при комнатной температуре. После 30 мин растворитель удалялся в вакууме и остаток делился между K2CO3 /вод./ и EtOAc. Последний слой отделялся, промывался водой, высушивался /MgSO4/ и выпаривался до сухости. Этот остаток растворялся в этаноле и обрабатывался эфирным HCl до помутнения, затем выстаивался всю ночь для получения искомого соединения /0,95 г/, т.пл. > 250oC.
1H ЯМР [250 МГц, ДМСО-д6]  1,81 - 2,02 /1H, м/, 2,10 - 2,28 /1H, м/, 2,65 /1H, дд/, 2,82 /2H, м/, 3,02 /1H, дд/, 1 сигнал затемнен H2O на 3,5, 3,74 /3H, с/, 6,66 /1H, д/, 6,84 /1H, д/, 7,14 /1H, д/, 8,16 /3H, шир.с/.
1,81 - 2,02 /1H, м/, 2,10 - 2,28 /1H, м/, 2,65 /1H, дд/, 2,82 /2H, м/, 3,02 /1H, дд/, 1 сигнал затемнен H2O на 3,5, 3,74 /3H, с/, 6,66 /1H, д/, 6,84 /1H, д/, 7,14 /1H, д/, 8,16 /3H, шир.с/.
Пример 4.
3-Амино-6-бромо-1,2,3,4-тетрагидрокарбазол гидрохлорид.
Реакция 4-бромофенилгидразин гидрохлорида /4,0 г, 18,1 мМ/ с 4-фталимидо циклогексаноном /4,39 г, 18,1 мМ/ в орошающем н-бутаноле в течение 20 мин с последующим охлаждением, фильтрацией и выпариванием фильтрата до сухости дала 3-фталимидо-6-бромо-1,2,3,4- тетрагидрокарбазол в виде твердого вещества оранжевого цвета /7,45 г/.
Этот продукт /0,33 г, 0,83 мМ/ суспендировался в этаноле /13 мл/ и обрабатывался гидратом гидразина /3 мл/, затем перемешивался при комнатной температуре всю ночь. Твердый осадок отфильтровывался и фильтрат выпаривался до сухости и разделялся между K2CO3 /вод/ и этилацетатом. После отделения органического слоя, промывания водой, высушивания /MgSO4/ и выпаривания до сухости остаток растворялся в MeOH и обрабатывался газом HCl. Растворитель удалялся в вакууме и остаток кристаллизовался из этанол/этилацетата для выхода искомого соединения в виде твердого вещества кремового цвета /0,15 г/, т.пл. 308 - 310oC.
1H ЯМР [250 МГц, ДМСО-д6] 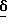 1,91 /1H, м/, 2,10 - 2,26 /1H, м/, 2,63 1H, дд/, 2,84 /2H, м/, 3,04 /1H, дд/, 3,50 /1H, м/, 7,12 /1H, д/, 7,24 /1H, д/, 7,55 /1H, с/, 8,15 /2H, шир.с/, 11,12 /1H, с/.
1,91 /1H, м/, 2,10 - 2,26 /1H, м/, 2,63 1H, дд/, 2,84 /2H, м/, 3,04 /1H, дд/, 3,50 /1H, м/, 7,12 /1H, д/, 7,24 /1H, д/, 7,55 /1H, с/, 8,15 /2H, шир.с/, 11,12 /1H, с/.
Пример 5.
3-Амино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол.
4-Карбоксамидофенилгидразин гидрохлорид /2,87 г/ и 4-фталимидоциклогексанон /3,00 г/ смешивались в уксусной кислоте, и смесь нагревалась с флегмацией 2 ч. После охлаждения смесь нейтрализовалась с использованием водного раствора карбоната калия, и полученное желтое твердое вещество фильтровалось, промывалось водой и высушивалось. Очистка колонной хроматографией дала 3-фталимидо-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол /2,8 г/. (SiO2, CHCl3/CH3OH).
Указанный продукт /1,0 г/ суспендировался в этаноле /10 мл/ и добавлялся гидрат гидразина /5 мл/. Получался чистый раствор, и смесь оставлялась перемешиваться всю ночь до получения осадка. Вся смесь выпаривалась до сухости, промывалась водным раствором K2CO3 и водой. Получалось в виде моногидрата искомое соединение 3-амино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол, т.пл. 146 - 148oC.
1H ЯМР [250 МГц, ДМСО-д6] 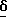 1,49 - 1,77 /1H, м/, 1,83 - 2,03 /1H, м/, 2,17 - 2,40 /1H, м/, 2,62 - 2,80 /1H, м/, 2,90 /1H, дд/, 1 сигнал невидим из-за воды на 3.1, 7,03 /1H, шир.с/, 7,18 /1H, д/, 7,58 /1H, д/, 7,83 /1H, шир.с/, 7,98 /1H, с/.
1,49 - 1,77 /1H, м/, 1,83 - 2,03 /1H, м/, 2,17 - 2,40 /1H, м/, 2,62 - 2,80 /1H, м/, 2,90 /1H, дд/, 1 сигнал невидим из-за воды на 3.1, 7,03 /1H, шир.с/, 7,18 /1H, д/, 7,58 /1H, д/, 7,83 /1H, шир.с/, 7,98 /1H, с/.
Пример 6.
/+/- и /-/-3-Амино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол гидрохлорид.
Способ 1.
/±/-3-т-Бутилоксикарбониламино-6-карбоксамидо-1,2,3,4- тетрагидрокарбазол разделялся на свои энантиомеры с использованием хиральной ВЭЖХ: /chiracel OD 4,6 мм колонна с промыванием гексаном/этанолом 85:15/. /+/-Энантиомер собирался первым и имел т. пл. 150 - 152oC и [α]
Способ 2.
/±/-6-Карбоксамидо-3-амино-1,2,3,4-тетрагидрокарбазол обрабатывался одним эквивалентом 2,3,4,6-ди-O-изопропилен-2-кето-L-гулоновой кислоты в метаноле, чтобы получить соль /+/-энантиомера в 38% выходе /относительно рацемата/ и 34% энантиомерного избытка /эи/. Этот материал рекристаллизовался дважды из метанола для получения соли /+/-энантиомера в 25% общем выходе /относительно рацемата/, и >98% эи. Этот продукт преобразовывался в соль гидрохлорида сначала обработкой водным р-ром щелочного металла, и осажденное свободное основание обрабатывалось 2 М вод. HCl в этаноле, чтобы получить /+/-энантиомер-3-амино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол гидрохлорид.
Пример 7.
3-Амино-6-метил-1,2,3,4-тетрагидрокарбазол оксалат.
Реакция 4-фталимидоциклогексанона /2,16 г/ с 4-толуoлгидразин гидрохлоридом /1,41 г/ и последующее снятие защиты продукта способом, описанном в примере 3, дало свободное основание искомого соединения, которое было преобразовано в соль оксалата /0,23 г/, т. пл. 272 - 5oC.
Пример 8.
3-Амино-6-этоксикарбонил-1,2,3,4-тетрагидрокарбазол оксалат.
Pеакция 4-фталимидоциклогексанона /0,37 г/ с 4-этоксикарбонилфенилгидразин гидрохлоридом /0,33 г/, и последующее снятие защиты способом, описанным в примере 3, дало свободное основание искомого соединения. Оно было преобразовано в соль оксалата /0,11 г/, т. пл. 230 - 240oC, разл.
Пример 9.
3-Амино-6-/N-метил-карбоксамидо/-1,2,3,4-тетрагидрокарбазол полуоксалат.
Реакция 4-фталимидоцикогексанона /1,20 г/ с 4-/N-метилкарбоксамидо/-фенилгидразин гидрохлоридом /1,00 г/, и последующее снятие защиты способом, описанным в примере 3, дало свободное основание искомого соединения. Оно было преобразовано в соль полуоксалата /0,22 г/, т. пл. 227oC, разл.
Пример 10.
3-Амино-6-цианометил-1,263,4-тетрагидрокарбазол оксалат.
Реакция 4-фталимидоциклогексанона /1,05 г/ с 4-цианометилфенилгидразин гидрохлоридом /0,79 г/, и последующее снятие защиты способом, описанным в примере 3, дало свободное основание искомого соединения, которое обрабатывалось щавелевой кислотой для получения соли оксалата /0,49 г/, т. пл. 219 - 224oC, разл.
Пример 11.
3-Амино-6-/N-метилсульфонамидометил/-1,26364-тетрагидрокарбазол оксалат.
Реакция 4-фталимидоциклогексанона /0,42 г/ с 4-/N-метилсульфонамидометил/ фенил гидразин гидрохлоридом /0,44 г/, и последующее снятие защиты способом, описанным в примере 3, дали свободное основание искомого соединения. Оно обрабатывалось щавелевой кислотой, чтобы получить соль оксалата /0,15 г/, т. пл. 218 - 222oC, разл.
Пример 12.
3-Амино-6-хлоро-1,2,364-тетрагидрокарбазол оксалат.
Реакция 4-фталимидоциклогексанона /6,7 г/ с 4-хлорофенил гидразин гидрохлоридом /4,93 г/, и последующее снятие защиты способом, описанным в примере 3, дало свободное основание искомого соединения, которое обрабатывалось щавелевой кислотой, чтобы получить соль оксалата /2,77 г/, разл., 220oC.
Пример 13.
3-Амино-6-трифторметил-1,2,3,4-тетрагидрокарбазол оксалат.
Реакция 4-фталимидоциклогексанона /1,14 г/ с 4-трифторметил фенил гидрохлоридом /1,0 г/ и последующее снятие защиты способом, описанным в примере 3, дало свободное основание искомого соединения /0,40 г/. Оно обрабатывалось щавелевой кислотой, т. пл. 212 - 213oC.
Пример 14.
3-Амино-6-н-бутилокси-1,2,3,4-тетрагидрокарбазол оксалат.
Реакция 4-фталимидоциклогексанона /1,12 г/, и последующее снятие защиты способом, описанным в примере 3, дало свободное основание искомого соединения. Оно обрабатывалось щавелевой кислотой, и получалась соль оксалата /0,47 г/, т.пл. 227 - 229oC.
Пример 15.
3-Амино-6-сульфонамидо-1,2,3,4-тетрагидрокарбазол оксалат.
Реакция 4-фталимидоциклогексанона /1,00 г/ с 4-сульфонамидо фенил гидразин гидрохлоридом /1,08 г/, и последующее снятие защиты способом, описанным в примере 3, дали свободное основание искомого соединения. Оно было преобразовано в соль оксалата /0,090 г/, разл. > 200oC.
Пример 16.
3-Амино-6-нитро-1,2,364-тетрагидрокарбазол оксалат.
Реакция 4-фталимидоциклогексанона /1,28 г/ с 4-нитрофенил гидразин гидрохлоридом /1,00 г/, и последующее снятие защиты способом, описанным в примере 3, дали свободное основание искомого соединения, которое было преобразовано в соль оксалата /0,25 г/, т. пл. 275 - 277oC.
Пример 17.
3-Амино-6-/N,N-диметил карбоксамидо/-1,2,3,4-тетрагидрокарбазол полуоксалат.
3-Амино-6-этоксикарбонил-1,2,3,4-тетрагидрокарбазол /260 мг, 1,0 мМ/ суспендировался в сухом ТГФ /5 мл/ и добавлялся ди-трет-бутил дикарбонат /320 мг, 1,5 мМ/. После 10 мин раствор получался чистым. Смесь перемешивалась 20 ч, затем растворитель удалялся, и остаток растворялся в этилацетате, промывался водным раствором бикарбоната натрия и высушивался /MgSO4/. После удаления этилацетата остаток растирался эфиром и гексаном, чтобы получить 3-т-бутилоксикарбониламино-6-этоксикарбонил-1,2,3,4-тетрагидрокарбазол /310 мг/.
Вышеуказанный продукт /556 мг, 1,55 мМ/ суспендировался в этаноле /5 мл/ и 2 М NaOH /3 мл/ добавлялись к нему. Смесь нагревалась с флегмацией 1 ч и выпаривалась до сухости. Остаток растворялся в воде и нейтрализовался уксусной кислотой, когда выпадал в осадок 3-т-бутилоксикарбониламино-6-карбокси-1,2,3,4-тетрагидрокарбазол в виде твердого вещества белого цвета /425 мг/. Раствор вышеуказанного продукта /400 мг, 1,2 мМ/ в сухом ТГФ /8 мл/ обрабатывался гексаметил фосфористым триамидом /198 мг, 1,2 мМ/, и охлаждался до -10oC.
Газообразный диэтиламин пропускался через смесь 10 мин при этой температуре, затем по каплям добавлялся тетрахлорид углерода /185 мг, 1,2 мМ/ в атмосфере азота. Смесь перемешивалась при комнатной температуре 1 ч, затем ДМС удалялся в вакууме. Остаток разделялся между этилацетатом и водой и органический слой промывался насыщенным раствором бикарбоната натрия, затем рассолом и высушивался /MgSO4/. Растворитель удалялся в вакууме, и остаточное масло растиралось эфиром и гексаном, и твердое вещество рекристаллизовалось из толуола, чтобы получить 3-т-бутилоксикарбониламино-6-/N,N-диметил карбоксамидо/-1,2,3,4-тетрагидрокарбазол /198 мг/.
Этот продукт растворялся в диоксане /5 мл/, и через него пропускался газ HCl, чтобы осадить масло. Растворитель удалялся в вакууме, и масло растворялось в воде и обрабатывалось раствором K2CO3, чтобы довести pH до 12. Затем свободное основание амина экстрагировалось этилацетатом, высушивалось /MgSO4/ и выпаривалось до сухости. Полученное масло растворялось в метаноле и обрабатывалось щавелевой кислотой, чтобы получить искомое соединение в виде твердого вещества бледно-розового цвета /140 мг/ т.пл. = 190 - 195oC.
Пример 18.
3-Амино-6-/пиперидин-1-ил карбонил/-1,2,3,4-тетрагидрокарбазол гидрохлорид.
Реакция 3-т-бутилоксикарбониламино-6-карбокси-1,2,3,4- тетрагидрокарбазола /175 мг/ с пиперидином и продукт снятия защиты способом, описанным для примера 17, дали искомое соединение, т. пл. 246 - 249oC /55 мг/.
Пример 19.
3-Амино-6-/пирролидин-1-ил карбонил/-1,2,3,4-тетрагидрокарбазол гидрохлорид.
Реакция 3-т-бутилоксикарбониламино-6-карбокси-1,2,3,4- тетрагидрокарбазола /140 мг/ с пирролидином, и продукт со снятой защитой с последующим, как описано для примера 17, дали искомое соединение, т.пл. 201 - 212oC /81 мг/.
Пример 20.
3-Амино-6-/N,N-диэтил карбоксамидо/-1,2,3,4-тетрагидрокарбазол гидрохлорид.
Реакция 3-т-бутилоксикарбониламино-6-карбокси-1,2,3,4- тетрагидрокарбазола /105 мг/ с диэтиламином и снятие защиты продукта, как описано в примере 17, дали искомое соединение, т. пл. 200 - 205oC /50 мг/.
Пример 21.
3-Амино-6-/ацетамидо/-1,2,3,4-тетрагидрокарбазол оксалат.
Реакция 4-фталимидо циклогексанона /1,2 г/ с 4-/ацетамидо/-фенил гидразин гидрохлоридом /1,0 г/, и последующее снятие защиты продукта способом примера 3 дало свободное основание искомого соединения /570 мг/. Часть этого продукта /50 мг/ обрабатывалась щавелевой кислотой, в метаноле, чтобы получить соль оксалата, которая размягчается при >170oC /38 мг/.
Пример 22.
3-Амино-6-метансульфонамидо-1,2,3,4-тетрагидрокарбазол оксалат.
3-Фталимидо-6-нитро-1,2,3,4-тетрагидрокарбазол /4,00 г/ растворялся в горячем этилацетате /130 мл/. К охлажденному раствору добавлялся скелетный никелевый катализатор гидрирования, и смесь гидрировалась при первоначальном давлении в 39 ф/д2 /2.74 кг/см2/ 4 ч. После отфильтровывания нерастворимых материалов фильтрат выпаривался до сухости и экстрагировался дважды в 20%-ном водном метаноле, и экстракты соединялись и уменьшались в объеме, чтобы дать 3-фталимидо-6-амино-1,2,3,4-тетрагидрокарбазол /0,31 г/.
Указанный продукт /0,50 г/ растворялся в свежем дистиллированном пиридине /30 мл/, и добавлялись метансульфонилхлорид /0,28 г/ и 4-диметиламинопиридн /46 мг/. Смесь нагревалась с перемешиванием при 50oC 5 ч, затем выпаривалась до сухости. Остаток растворялся в хлороформе, промывался водой, рассолом и водным бикарбонатом натрия, затем высушивался /MgSO4/, и выпаривался до сухости, чтобы получить бледно-желтое вещество, которое рекристаллизовалось из водного этанола для выхода 3-фталимидо-6-метансульфонамидо-1,2,3,4- тетрагидрокарбазола /0,27 г/.
Указанное соединение суспендировалось в этаноле /15 мл/ и добавлялся гидрат гидразина /2,72 г/. После 25-минутного перемешивания при комнатной температуре смесь выпаривалась до сухости, разделялась между водой и этилацетатом, и водный слой повторно экстрагировался этилацетатом. Органические экстракты соединялись, промывались водой, высушивались /MgSO4/ и выпаривались до получения бледно-желтого твердого вещества. Оно растворялось в метаноле и обрабатывалось щавелевой кислотой /89 мг/. Добавление эфира привело к кристаллизации искомого соединения /50 мг/, т. пл. 230 - 233oC.
Пример 23.
3-Амино-6-карбоксамидометил-1,2,3,4-тетрагидрокарбазол гидрохлорид.
3-Амино-6-цианометил-1,2,3,4-тетрагидрокарбазол /2,5 г/ и ди-т-бутил дикарбонат /3,63 г/ перемешивались в ТГФ /56 мл/ 2 ч. ТГФ выпаривался, и остаток разделялся между водным раствором бикарбоната натрия и этилацетатом. Водная фаза повторно экстрагировалась этилацетатом, и соединенные органические экстракты промывались водой, высушивались /MgSO4/, и выпаривались до сухости, чтобы получить твердое вещество, которое растиралось эфиром/гексаном /20%/ для получения 3-т-бутилоксикарбониламино-6- цианометил-1,2,3,4-тетрагидрокарбазола в виде чисто-белого вещества /3,44 г/.
Продукт /7,0 г/ растворялся в ДМСО /70 мл/, и добавлялась перекись водорода /100 об., 3,5 мл/. После перемешивания 1 ч добавлялась еще перекись /8,5 мл/, и смесь перемешивалась 2 ч при комнатной температуре. Добавлялся карбонат калия /0,84 г/, и смесь перемешивалась всю ночь и еще 20 ч. Реакционная смесь выливалась в воду /500 мл/, и полученное твердое вещество белого цвета отфильтровывалось и рекристаллизовалось из метанола, чтобы получить 3-т-бутилоксикарбониламино-6-карбоксамидо-метил-1,2,3,4- тетрагидрокарбазол /5,42 г/.
Указанный продукт /500 мг/ растворялся в сухом диоксане /30 мл/, и газ HCl пропускался через него 20 мин. Полученный раствор и отложившаяся смола выпаривались до сухости и обрабатывались водным раствором карбоната калия. Это экстрагировалось этилацетатом, и экстракты соединялись, высушивались /MgSO4/ и выпаривались до сухости. Остаток растворялся в метаноле и обрабатывался избытком щавелевой кислоты. Добавление эфира давало кристаллизацию искомого соединения /250 мг/, т.пл. 257 - 260oC.
Пример 24.
3-Метиламино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол гидрохлорид.
4-Цианофенил гидразин гидрохлорид /20,2 г/ и 4-бензоилоксициклогексанон /25,9 г/ растворялись в ледяной уксусной кислоте /400 мл/, и смесь нагревалась с флегмацией 1,5 ч. После охлаждения смесь фильтровалась, и фильтрат выпаривался до сухости, и нейтрализовался водным раствором бикарбоната натрия, чтобы получить твердый осадок, который очищался хроматографией /SiO2; гексан/этилацетат/ для выхода 3-бензоилокси-6-циано-1,2,3,4-тетрагидрокарбазола /18,0 г/. Этот продукт /11,6 г/ суспендировался в этаноле /230 мл/ и обрабатывался 2,5%-ным водным раствором гидроокиси калия /120 мл/, и нагревался с флегмацией 1 ч. Охлажденная смесь нейтрализовалась охлажденной уксусной кислотой и выпаривалась до твердого остатка, который промывался водой и высушивался, чтобы получить 3-гидрокси-6-циано-1,2,3,4-тетрагидрокарбазол /6,6 г/.
Указанный продукт /3,57 г/ растворялся в сухом пиридине /35 мл/, и смесь перемешивалась при 100oC 2 ч. После охлаждения раствор выливался в воду /500 мл/. Экстрагировался этилацетатом, и последний экстракт промывался 2 М HCl, высушивался /MgSO4/ и выпаривался до сухости. Очистка хроматографией /SiO2; гексан/этилацетат/ дали 3-тосулокси-6-циано-1,2,3,4-тетрагидрокарбазол /0,53 г/.
Этот продукт /0,40 г/ растворялся в 33%-ном метиламине в спирте /25 мл/ и нагревался при 100oC в герметичном стальном сосуде 1,5 ч. После охлаждения смесь выпаривалась до сухости и очищалась хроматографией /SiO2; хлороформ/метанол/, чтобы получить 3-метиламино-6-циано-1,2,3,4-тетрагидрокарбазол /0,13 г/.
Указанный продукт /0,12 г/ растворялся в ТГФ /10 мл/ и реагировал с ди-трет-бутил дикарбонатом /0,36 г/ в ТГФ /3 мл/ при комнатной температуре всю ночь. Реакционная смесь выпаривалась до сухости, разделялась между 2 М раствора бикарбоната натрия и этилацетатом, и органический слой высушивался и выпаривался до получения вещества белого цвета. Оно растиралось эфиром/гексаном, чтобы получить 3-т-бутилоксикарбонил-метил амино-6-циано-1,2,3,4-тетрагидрокарбазол /0,14 г/.
Этот продукт растворялся в метаноле /15 мл/ и обрабатывался смесью 20%-ного водного гидроксида натрия /0,20 мл/ и 30%-ной перекисью водорода /0,20 мл/, и вся смесь перемешивалась при комнатной температуре всю ночь. Добавлялся метабисульфит натрия /38 мг/, и раствор выпаривался до сухости и хроматографировался /SiO2; хлороформ/10% NH4OH в метаноле/, чтобы получить 3-метиламино-6-карбоксамид-1,2,3,4-тетрагидрокарбазол /0,12 г/. Указанное соединение /0,11 г/ растворялось в метаноле /10 мл/ и обрабатывалось 3 М хлористоводородной кислоты при комнатной температуре. Смесь выпаривалась до сухости, азеотропировалась этанолом, чтобы получить твердое вещество, которое рекристаллизовалось из метанола/эфира для получения искомого соединения, т. пл. 327 - 328oC /80 мг/.
1H ЯМР [250 МГц, MeOH-д4] д 1,98 - 2,20 /1H, м/, 2,29 - 2,49 /1H, м/, 2,75 - 2,90 /5H, с+м/, 2,90 - 3,09 /2H, м/, 3,52 - 3,69 /1H, м/, 7,31 /1H, д/, 7,63 /1H, д/, 8,05 /1H, с/.
Пример 25.
3-Этиламино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол оксалат.
1,4-Циклогександион моно-2',2'-диметил триметилен кетал /2,00 г/ смешивался с безводным этиламином /10,0 г/ и бензолом /10,0 мл/, и смесь охлаждалась до 5oC. По каплям добавлялся раствор титаний тетрахлорида /0,95 г/ в бензоле /10 мл/, затем смесь перемешивалась при комнатной температуре 1 ч. Смесь фильтровалась и выпаривалась до сухости, чтобы получить масло, которое растворялось в этаноле /30 мл/. К этому раствору добавлялся катализатор палладий на углероде /100 мг/, и смесь гидрировалась при 50 ф/д2 /3,5 кг/см2/ давлении всю ночь. Катализатор отфильтровывался, и этанол выпаривался для получения 4-этиламино-циклогексанон 2',2'-диметил триметилен кетала в виде масла /2,0 г/.
Это соединение /0,80 г/ растворялось в муравьиной кислоте /20 мл/, и раствор нагревался до 90oC 1 ч. Муравьиная кислота выпаривалась, и остаток разделялся между хлороформом и 1М хлористоводородной кислоты. Водный слой выпаривался до сухости, чтобы дать выход 4-этиламиноциклогексанону /0,40 г/.
Смесь указанного продукта /0,40 г/ и 4-карбоксамидофенил гидразин гидрохлорида /0,60 г/ в ледяной уксусной кислоте /20 мл/ нагревалась с флегмацией 1 ч. Кислота выпаривалась в вакууме до масла, которое очищалось хроматографией /SiO2; CHCl3/10% NH3 в MeOH/, чтобы получить масло /0,50 г/. Часть этого продукта /150 мг/ растворялась в метаноле и обрабатывалась щавелевой кислотой. Раствор обрабатывался эфиром для получения искомого соединения в виде кристаллического твердого вещества, т.пл. 165-170oC /100 мг/.
1H ЯМР [250 МГц, ДМСО-д6] д 1,25 /3H, т/, 1,81-2,05 /1H, м/, 2,20 - 2,38 /1H, м/, 2,61 - 2,79 /1H, м/, 2,79 - 2,94 /2H, м/, 2,98 - 3,28 /3H, дд+с/, 3,41 - 3,60 /1H, м/, 7,08 /1H, шир.с/, 11,12 /1H, с/.
Пример 26.
3-н-Пропиламино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол оксалат.
Пропиламин /1,81 г/ растворялся в метаноле /12,5 мл/, и с охлаждением добавлялось 1,5 М HCl в метаноле /5,6 мл/. После 1 мин добавлялся 1,4-циклогександион моно-2',2'-диметил триметилен кетал /1,0 г/, а через еще 10 мин - цианоборогидрид натрия /0,23 г/. Смесь перемешивалась при комнатной температуре 3 дня. Полученная смесь фильтровалась и фильтрат выпаривался и обрабатывался 1М HCl /10 мл/ с охлаждением. Остаток выпаривался для образования раствора, который промывался эфиром, доводился основанием до pH 12 с помощью водного гидроксида натрия и экстрагировался дихлорметаном. Этот экстракт промывался насыщенным водным раствором бикарбоната натрия, высушивался /MgSO4/, и выпаривался до сухости. Хроматография /SiO2; хлороформ/метанол/аммиак/ дала 4-н-пропиламино циклогексанон 2',2'-диметил триметилен кетал /0,72 г/.
Этот продукт /0,66 г/ гидролизовался до кетона, который реагировал с 4-карбоксамидофенил гидразин гидрохлоридом и преобразовывался в соль оксалата, как описано в примере 25, чтобы дать выход искомому соединению /0,44 г/, т.пл. >168oC, разл.
Пример 27.
3-i-Пропиламино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол оксалата.
Реакция изопропиламина /9,54 г/ с 1,4-циклогександион моно-2',2'-диметил триметилен кеталом /2,0 г/ способом, описанным для примера 25, дала 4-i-пропиламино циклогексанон 2', 2'-диметил триметилен кетал /2,38 г/. Этот продукт /0,66 г/ гидролизовался и реагировал с 4-карбоксамидофенил гидразин гидрохлоридом /0,45 г/, и смесь обрабатывалась, как описано выше, чтобы получить свободное основание искомого соединения /0,34 г/. Оно преобразовывалось в оксалат, т.пл. >235oC, разл.
Пример 28.
3-Диметиламино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол оксалат.
Диметиламин /10,0 г/ реагировал с 1,4-циклогександион моно-2',2'-диметил триметилен кеталом /2,0 г/ способом, описанным для примера 25, чтобы получить 4-диметиламино-циклогексанон-2',2'-диметил триметилен кетал /0,72 г/. Этот продукт /0,72 г/ гидролизовался и реагировал с 4-карбоксамидофенил гидразин гидрохлоридом /0,47 г/, и продукт преобразовывался в соль оксалата, как описано выше, чтобы получить искомое соединение /0,20 г/, т.пл. 99 - 101oC.
1H ЯМР [250 МГц, ДМСО-д6] д 1,83 - 2,05 /1H, м/, 2,27 - 2,40 /1H, м/, 2,72 - 3,00 /9H, 2м+с/, 2,37 - 3,22 /1H, дд/, 3,50 - 3,68 /1H, м/, 7,05 /1H, шир. с/, 7,27 /1H, д/, 7,60 /1H, д/, 7,81 /1H, шир.с/, 8,00 /1H, с/, 11,11 /1H, с/.
Пример 29.
3-Бензиламино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол оксалат.
Реакция бензиламина /0,59 г/ с 1,4-циклогександион-моно-2',2'-диметил триметил кеталом /1,0 г/ и последующее восстановление имина цианоборогидридом натрия способом, описанным для примера 26, дала 4-бензиламино-циклогексаном 2', 2'-диметил триметилен кетал /0,54 г/. Этот продукт /0,52 г/ реагировал с 4-карбоксамидофенил гидразин гидрохлоридом /0,34 г/, и продукт обрабатывался щавелевой кислотой, чтобы получить искомое соединение, т. пл. >190oC, разл. /0,11 г/.
Пример 30.
3-Пирролидинил-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол оксалат.
Реакция пирролидина /15,6 г/ с 1,4-циклогександион моно-2',2'-диметил триметилен кеталом /2,0 г/ способом, описанным для примера 26, дала 4-пирролидинил-циклогексанон-2',2'-диметил триметилен кетал /1,74 г/. Этот продукт /1,70 г/ гидролизовался и реагировал с 4-карбоксамидофенил гидразин гидрохлоридом /1,70 г/, и продукт обрабатывался щавелевой кислотой, как описано выше, чтобы получить искомое соединение /32 мг/, т. пл. >190oC, разл.
Пример 31.
3-/N-Метил этиламино/-6-карбоксамидо-1,2,3,4-тетрагидрокарбазол оксалат.
Реакция N-метил этиламина /13,0 г/ с 1,4-циклогександион моно-2',2'-диметил триметилен кеталом /2,0 г/ способом, описанным для примера 265, дала 4-/N-метил этиламино/-циклогексанон-2',2'-диметил триметилен кетал /1,71 г/. Этот продукт /0,86 г/ гидролизовался и реагировал с 4-карбоксамидофенил гидразин гидрохлоридом /0,52 г/ и обрабатывался, как описано выше, чтобы получить искомое соединение /76 мг/, т.пл. >130oC, разл.
Пример 32.
3-Амино-6-/2-карбоксамидоэтил/-1,2,3,4-тетрагидрокарбазол оксалат.
Смесь 4-нитрокоричной кислоты /22,5 г/ и тионил хлорида /20,8 г/ в бензоле /160 мл/ нагревалась с флегмацией 4 ч. Полученная оранжевая смесь фильтровалась и выпаривалась для получения кислотного хлорида /22,9 г/. Это растворялось в дихлорметане /1 л/, пропускался аммиак с охлаждением до ниже 20oC и перемешиванием. Растворитель удалялся в вакууме, и остаток растворялся в горячем этилацетате, и раствор встряхивался с 1М раствора гидроксида натрия. Полученная органическая фаза высушивалась, фильтровалась и выпаривалась. Оставался осадок, который шламовался этилацетатом, чтобы получить 4-нитро циннамамид в виде кристаллического вещества /18,6 г/. Этот продукт /18,6 г/ суспендировался в этаноле /1 л/ и гидрировался с использованием Pd-C катализатора /6,6 г/ при давлении 50 фунт/д2 /3,5 кг/см2/ в течение 1 ч. Полученная смесь фильтровалась и выпаривалась до сухости с получением 4-аминофенил пропионамида /17,1 г/.
Медленно добавлялась концентрированная хлористоводородная кислота /4 мл/ к продукту 4-аминофенил пропионамида /0,80 г/, с поддержанием температуры ниже 5oC. К этому шламу добавлялся раствор нитрита натрия в /0,37 г/ в воде /2 мл/ в течение 15 мин по каплям с последующим перемешиванием еще в течение 15 мин. Полученный таким образом мутный раствор порциями добавлялся к охлажденному перемешанному раствору двухлористого олова /2,19 г/ HCl /4 мл/, и полученная смесь перемешивались 1 ч. После фильтрования раствор восстанавливался в объеме, до тех пор, пока не формировался неорганический осадок. Он отфильтровывался, и фильтрат выпаривался до сухости. Остаточная смола кристаллизовалась из уксусной кислоты для получения сырого 4-гидразинофенил пропионамид гидрохлорида /1,05 г/.
Смесь вышеуказанного продукта /1,05 г/ и 4-фталимидоциклогексанона /1,18 г/ в уксусной кислоте /40 мл/ нагревалась с флегмацией 40 мин. Растворитель удалялся в вакууме, и остаток разделялся между водным раствором карбоната калия и этилацетатом. Органическая фаза высушивалась /MgSO4/ и выпаривалась до сухости, и остаток хроматографировался /SiO2; CH2Cl2/MeOH, чтобы дать 3-фталимидо-6-карбоксамидоэтил-1,2,3,4-тетрагидрокарбазол /0,70 г/.
Этот продукт /0,70 г/ растворялся в метаноле /50 мл/, обрабатывался гидразин гидратом /1,0 мл/ и нагревался с флегмацией 30 мин. Смесь выпаривалась до сухости, затем разделялась между этилацетатом и водным раствором карбоната калия. Органическая фаза высушивалась /MgSO4/ и выпаривалась до сухости, и остаток растворялся в этаноле и обрабатывался щавелевой кислотой /83 мг/ в этаноле. Получалось твердое вещество, которое рекристаллизовалось из этанола, чтобы получить искомое соединение /110 мг/, т.пл. 232-5oC.
Лекарственные формы (табл. 1,2 - примеры А,В) см. в конце описания.
Соединение формулы I (из примера В в конце описания) растворяется в лимонной кислоте, и pH медленно доводится до 3,2 раствором гидроксида натрия. Затем раствор дополняется до 100 мл водой, стерилизуется фильтрацией и запаивается в ампулы разных размеров.
Использование соединений общей формулы I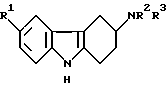
в которой R1 представляет водород, галоген, трифторметил, нитро, гидрокси, C1-6-алкокси, арил-C1-6-алкокси, -CO2R4, -(CH2)nCN, (CH2)nCONR5R6, (CH2)nSO2NR5R6, C1-6-алканоиламино(CH2)n или C1-6-алкилсульфониламино(CH2)n; R4 представляет водород, C1-6-алкил или арил C1-6; R5 и R6 каждый независимо представляют водород или C1-6-алкил или R5 и R6 вместе с атомом азота, к которому они присоединены, образуют кольцо; n представляет 0,1 или 2; R2 и R3 каждый независимо представляют водород, C1-6-алкил или бензил или вместе с атомом азота, к которому они присоединены, образуют кольцо пирролидино, пиперидино или гексагидроазепино, или его физиологически приемлемой соли для лечения состояния, в котором показан 5-HT1-подобный агонист, например мигрени. Описываются также новые соединения формулы I, способы их получения и фармацевтические композиции, содержащие их. 4 с. и 15 з.п. ф-лы, 2 табл.
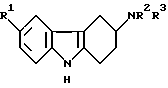
в которой R1 представляет водород, галоген, трифторметил, нитро, гидрокси, C1-6-алкил, C1-6-алкокси, арил-C1-6-алкокси, -CO2R4, -(CH2)nCN, -(CH2)nCONR5R6, -(CH2)nSO2NR5R6, C1-6-алканоиламино(CH2)n или C1-6алкилсульфониламино (CH2)n;
R4 представляет водород, C1-6алкил или арил C1-6алкил; R5 и R6 каждый независимо представляет водород или C1-6алкил или
R5 и R6 вместе с атомом азота, к которому они присоединены, образуют кольцо;
n представляет 0, 1 или 2;
R2 и R3 каждый независимо представляет водород, C1-6-алкил или бензил или вместе с атомом азота, к которому они присоединены, образуют кольцо пирролидино, пиперидино или гексагидроазепино,
или его физиологически приемлемую соль, сольват или гидрат.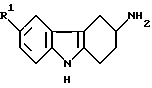
где R1 представляет нитро, CN, -CO2R4, -(CH2)nCONR5R6, C1-6алкилсульфониламино (CH2)n или -(CH2)nSO2NR5R6, где R4 означает арил C1-6алкил, R5 и R6 каждый независимо представляет водород или C1-6-алкил, или R5 и R6 вместе с атомом азота, с которым они соединены, образуют кольцо,
n = 0, 1 или 2,
или его физиологически приемлемая соль, сольват или гидрат этого соединения.
(+)-3-амино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазола, 3-амино-6-циано-1,2,3,4-тетрагидрокарбазола,
(-)-3-амино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазола,
3-амино-6-(N-метилкарбоксамидо)-1,2,3,4-тетрагидрокарбазола,
3-амино-6-(N-метилсульфонамидометил)-1,2,3,4-тетрагидрокарбазола,
3-амино-6-сульфонамидо-1,2,3,4-тетрагидрокарбазола,
3-амино-6-нитро-1,2,3,4-тетрагидрокарбазола,
3-амино-6-(N,N-диметилкарбоксамидо)-1,2,3,4-тетрагидрокарбазола,
3-амино-6-(пиперидин-1-ил-карбонил)-1,2,3,4-тетрагидрокарбазола,
3-амино-6-(пирролидин-1-ил-карбонил)-1,2,3,4-тетрагидрокарбазола,
3-амино-6-(N,N-диэтилкарбоксамидо)-1,2,3,4-тетрагидрокарбазола,
3-амино-6-метансульфонамидо-1,2,3,4-тетрагидрокарбазола,
3-амино-6-карбоксамидометил-1,2,3,4-тетрагидрокарбазола,
3-амино-6-(2-карбоксамидоэтил)-1,2,3,4-тетрагидрокарбазола,
или физиологически приемлемая соль соединения, его сольват или его гидрат.
3-этиламино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазола,
3-н-пропиламино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазола,
3-изопропиламино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазола,
3-диметиламино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазола,
3-бензиламино-6-карбоксамидо-1,2,3,4-тетрагидрокарбазола,
3-пирролидинил-6-карбоксамидо-1,2,3,4-тетрагидрокарбазола,
3-(N-(метил)этиламино)-6-карбоксамидо-1,2,3,4-тетрагидрокарбазола,
или физиологически приемлемая соль соединения, его сольват или его гидрат.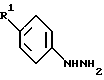
в которой R1 имеет значения, указанные в п.10, или его аддитивную соль кислоты, подвергают взаимодействию с соединением формулы III
в которой R2 и R3 - водород,
или его N-защищенным производным, с последующим, в случае необходимости, превращением соединения формулы IА, где R1=(CH2)nCN, гидролизом в соединение формулы IА, где R1=(CH2)nCONH2 или CO2R4 или его N-защищенное производное, или превращением соединения формулы IА, где R1=CO2H, аминированием в соединение формулы IА, где R1=CONR5R6 или его N-защищенное производное, значения R4, R5, R6 определены в п.10, с последующим, если необходимо, удалением любой группы, защищающей атом азота, и при желании образованием соли полученного соединения.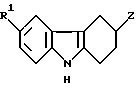
в которой R1 имеет значения, указанные в п.10,
Z - уходящая группа,
с соединением формулы
HNR2R3,
в которой R2 и R3 - водород,
с последующим, в случае необходимости, превращением соединения формулы IА, где R1= -(CH2)nCN, гидролизом в соединение формулы IА, где R1=-(CH2)nCONH2 или CO2R4 или его N-защищенное производное, или превращением соединения формулы IА, где R1=-CO2H, аминированием в соединение формулы IА, где R1= -CONR5R6 или его N-защищенное производное, значения R4, R5 и R6 определены в п.10, с последующим, если необходимо, удалением любой группы, защищающей атом азота, и, при желании, образованием соли полученного соединения.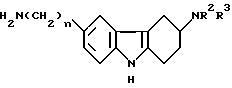
в которой R2, R3 - водород,
с ацилирующим или сульфонилирующим агентом с последующим, в случае необходимости, превращением соединения формулы IА, где R1=-(CH2)nCN, гидролизом в соединение формулы IА, где R1=-(CH2)n-CONH2, или CO2R4 или его N-защищенное производное, или превращением соединения формулы IА, где R1=CO2H, аминированием в соединение формулы IА, где R1=-CONR5R6 или его N-защищенное производное, значения R4, R5 и R6 определены в п.10, с последующим, если необходимо, удалением любой группы, защищающей атом азота, и при желании, образованием соли полученного соединения.
| Способ получения производныхКАРбАзОлил (4)-ОКСи-пРОпАНОлАМиНАили иХ СОлЕй (ЕгО ВАРиАНТы) | 1979 |
|
SU810079A3 |
| СПОСОБ ЗАКРЕПЛЕНИЯ ОСНОВНЫХ КРАСИТЕЛЕЙ В ПЕЧАТИ НА ХЛОПЧАТО-БУМАЖНЫХ ТКАНЯХ | 1924 |
|
SU4342A1 |
| Двухсторонняя автокормушка для полужидких кормов | 1957 |
|
SU115607A1 |
| US 4257952, 1979. | |||

