Изобретение относится к новым соединениям, имеющим фармакологическую активность, способу их получения и применения в качестве фармацевтических препаратов.
В Европейской заявке [1] описаны производные индола, имеющие активность антагониста 5 - HT3 рецептора.
В Европейском журнале "Фармакология", 146 1988,187 - 188 и в Наунин - Шмиедеберг'с Apr. Фармакология,1989,403 - 410 описаны неклассический рецептор 5-кситриптамин, который теперь известен, как рецептор 5 - HT4, и ICS 205 - 930, являющийся также антагонистом рецептора 5-HT3, который действует в качестве антагониста на этом рецепторе.
В международной заявке [2] описано применение антагонистов рецепторов 5-HT4 для лечения сердечной аритмии и паралича.
В европейской заявке N A-501322 (Глаксо Групп Лимитед) описаны производные индола, имеющие активность антагониста рецептора 5-HT4.
Теперь открыт класс новых различных по структуре соединений,которые представляют собой индоловые производные, 1,2-дизамещенные алкиленокси, с азациклической, конденсированной азабициклической или аминоалкиловой составляющей эти соединения имеют активность антагониста рецептора 5-HT4.
Таким образом, в соответствии с настоящим изобретением получено соединение формулы (I) или приемлемая для фармацевтических целей его соль: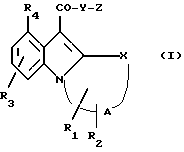
где X - O, S, SO, SO2, CH2, CH или NR,
где R - водород или С1-6 алкил;
A - цель насыщенного или ненасыщенного полиметилена с 2 - 4 атомами углерода;
R1 и R2 - водород или С1-6алкил;
R3 - водород, гало, С1-6алкил, амино, нитро или С1-6алкокси;
R4 - водород, гало, C1-6алкил или С1-6алкокси;
Y - O или NH;
Z выбирают из группы, представленной формулами (a), (b) или (c):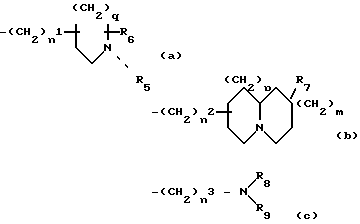
где n1- 1, 2, 3 или 4; n2 - 0, 1, 2, 3 или 4; n3 - 2, 3, 4 или 5;
q - 0, 1, 2 или 3; p = 0, 1 или 2; m = 0, 1 или 2;
R5- водород, С1-12алкил, аралкил или R5 = (CH2)z - 10, где Z равен 2 и 3;
R10 выбран из циано, гидроксила, С1-6алкокси, фенокси, C(O)C1-6алкила, COC6H5, - CONR11R12, NR11COR12, SO2NR11R12 или NR11SO2R12, где R11 и R12- водород или C1-6алкил; и
R6, R7 и R8 - независимо водород или C1-6алкил; и R9 - водород или C1-10алкил;
или соединение формулы (I), в которой CO - Y связь замещена гетероциклическим биоизостерным соединением; имеющим активность рецептора 5 - HT4.
Примеры алкиловых или алкилсодержащих групп включают в себя C1, C2, C3, C4, C5, C6, C7, C8, C9, C10, C11 или C12 с разветвленной, нормальной цепью или циклический алкил в качестве соответствующей группы, C1-4алкиловые группы включают в себя метил, этил, п - и изо - пропил, п -, изо-, сек - трет -бутил. Циклический алкил включает в себя циклопропил, циклобутил, циклопентил, циклогексил, циклопентил и циклооктил.
Арил включает в себя фенил и нафтил, возможно замещенные одним или несколькими заместителями, выбранными из гало, C1-6алкила и C1-6алкокси.
Гало включает в себя фтор, хлор, бром и йод.
Соответствующим биоизостерным соединением для связи амида или сложного эфира, содержащего Y в формуле (I), является соединение формулы (d):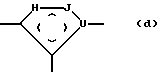
где круг, обозначенный точками, представляет собой одну или две двойные связи в любом положении пятичленного кольца;
H, J, и 1 представляют независимый кислород, серу,азот или углерод при условии, что, по крайней мере,одни из H, J и 1 - другой, а не углерод;
U - азот или углерод.
В европейской заявке N A - 328200 (Мерк Шарп энд Дохм Лтд.) даны соответствующие примеры (d) для X, Y и Z, например, оксадиазоловая составляющая. X часто представляет собой 0.
Значения для A включают в себя - CH2 - (CH2)rCH2 - где r - 0, 1 или 2; - CH2 - CH = CH -; - C(CH3) = CH - или когда x = CH или N, A - может быть - (CH2)2 - CH = или - CH = CH - CH =.
В примерах, которые будут представлены, описаны другие примеры A.
R1 и R2 - часто водород или R1 и R2 - гем - диметил. Значение r часто равно 1;
R3 - предпочтительно водород;
R4 - предпочтительно водород или гало, например, фтор;
Y - предпочтительно 0 или NH.
Когда z выбран из подформулы (c), n3 предпочтительно равно 2, 3 или 4.
R9 и R10 - предпочтительно оба - алкил, особенно один из R9 и R10 представляет (C4 или больше) алкил.
Конкретными значениями для z, которые представляют особый интерес, являются следующие: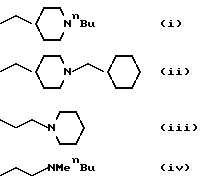
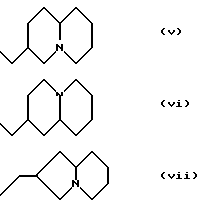
Изобретение также относится к новым соединениям формулы (I) с боковыми цепями (i), (ii), (iii), (iv), (v), (vi) или (vii). Согласно другому аспекту, пиперидиновое кольцо в (i), (ii) или (iii) может быть пирролидинилом или азетидинилом, и/или N-заместителем в (i), либо (ii) может быть замещен C3 или главным образом алкилом или возможно замещенным бензилом.
Согласно другому аспекту, N - заместитель в формуле (i) или в формуле (ii) может быть замещен (CH2)nR4, как показано в формуле (I) и в конкретных примерах, представленных в заявке ЕР - А - 501322.
Фармацевтически приемлемые соли соединений формулы (I) включают в себя соли кислот от реакции присоединения с обычными кислотами, например соляная, бромистоводородная, борная, фосфорная, серная кислоты и с фармацевтически приемлемыми органическими кислотами, например уксусная, винная, малеиновая, лимонная, янтарная, метансульфокислота, а-кетоглутаровая, а-глицерофосфорная и глюкоза - 1 - фосфорная кислоты.
Примеры фармацевтически приемлемых солей включают в себя четвертичные производные соединений формулы (I), например, соединения, кватернизованные соединения Rx - T, где Rx = C1-6алкил, фенил - C1-6алкил или C5-7 циклоалкил, а T - радикал, соответствующий аниону кислоты. Соответствующие примеры Rx включают в себя метил, этил и n - и изо - пропил; и бензил и фенетил. Соответствующие примеры T включают в себя галоид, например хлорид, бромид и йодид.
Примеры фармацевтически приемлемых солей также включают в себя внутренние соли, например N-оксиды.
Соединения формулы (I), их фармацевтически приемлемые соли (включая четвертичные производные и N-оксиды) могут также образовывать фармацевтически приемлемые сольваты, например гидраты, которые включают в указанные здесь соединение формулы (I) или его соль.
Также предусмотрено, что составляющая (CH2)n2 в соединениях формулы (I), где Z = (b), может принимать а, β или конфигурацию, относящуюся к конденсированной азабициклической составляющей.
Соединения формулы (I) можно получить путем обычного соединения индоловой составляющей Z. Соответствующие способы описаны в патентах Великобритании NN 2125398А (Сандоз Лимитед), 1593146А и в патентах ЕР-А-36269 (Бичем Груп p. l. c.), 429984 (Нишин Фор Миллинг Ко.) и 328200 (мерк Шарп энд Дохм Лимитед). Также даем ссылку на ЕР-А-501322 (Глаксо Групп Лимитед). Следует отметить, что ввод/модификацию (CH2)rO - содержащего кольца или R3/R4 можно осуществлять до или после соединения.
Аза(би)циклические промежуточные химические соединения с боковой цепью являются известными соединениями, однако их можно получить в соответствии со способами, описанными в PCT/GB 92/01519 и /01612 (Смитклайн Бичем p.l.c.).
Соединения в соответствии с настоящим изобретением представляют антагонисты рецептора 5-HT4, следовательно, их можно использовать для лечения профилактики желудочно-кишечных расстройств, сердечно-сосудистых заболеваний и расстройств центральной нервной системы (CNS).
Эти соединения представляют собой потенциальный интерес для лечения такого заболевания, как синдром IBS (заболевание кишечника), в частности поноса при расстройствах пищеварительного тракта, то есть эти соединения блокируют способность рецептора 5-HT к побуждению кишок к действию через активацию нейронов. При синдроме расстройства кишечника (IBS) у животных это можно проверить по уменьшению количества кала. Эти соединения также можно применять для лечения такой болезни, как, например, недержание мочи, которое часто связано с синдромом IBS.
Их можно также применять для лечения других желудочно-кишечных заболеваний, например, тех, которые связаны с активностью в верхней части пищеварительного канала, и как противорвотные средства. В частности они могут найти потенциальное применение для устранения симптомов гастрита при желудочно-кишечных заболеваниях и дисперсии. Противорвотную активность определяют на известных животных моделях по рвоте, вызванной цитотоксичным веществом или радиацией.
Также полагают, что конкретные антагонисты рецептора 5-НТ4, которые препятствуют возникновению мерцательной аритмии и других видов аритмии сердца, связанной с 5-НТ, будут уменьшать возможность возникновения паралича (А. Дж. Кауман 1990, Наумин-Шмидеберг'с Apr. Фармакология, 342, 619 - 622. Соответствующий метод испытания на животных).
Полагают, что 5-НТ, полученный из кровяных пластинок (тромбоцитов), вызывает аритмию, которая приводит к возникновению мерцательной аритмии сердца, и что нарушения сердечной деятельности связаны с симптоматической закупоркой кровеносных сосудов головного мозга и ситемической эмболией. Закупорка кровеносных сосудов головного мозга является причиной возникновения ишемической болезни, а сердце является наиболее известным источником материала для закупорки кровеносных сосудов. Особый интерес представляет частота возникновения эмболии, связанной с мерцательной аритмией сердца.
На активность, вызывающую состояние тревоги или беспокойства, вероятно оказывает влияние аммониев рог головного мозга (Думуис и др., 1988, Мол. Фармакол. , 34, 880 - 887). Активность можно продемонстрировать на стандартных животных моделях посредством испытания на социальное взаимодействие и посредством лабиринтного испытания-X.
Те, кто страдают мигренью, часто испытывают ситуации беспокойства и эмоционального стресса, которые предшествуют появлению головной боли (см. Sachs, 1985, Мигрень, Pan Books, Лондон). Также обнаружено, что в течение 48 ч приступов мигрени уровни циклического АМР значительно увеличиваются в спинномозговой жидкости (Welch и др., 1976, Головная боль, 16, 160 - 167). Считают, что мигрень, включая фазу, предшествующую началу приступа, и соответствующее увеличение уровней циклического АМР, связаны с возбуждением рецепторов 5-НТ4, следовательно, назначение антагониста рецептора 5-НТ4 может принести потенциальную пользу в снятии приступа мигрени.
Также предложена фармацевтическая композиция в соответствии с изобретением, содержащая соединение формулы (I) или фармацевтически приемлемую его соль и фармацевтически приемлемый носитель.
Эти композиции получают путем смешения компонентов и обычно они предназначены для приема внутрь, например, через рот, нос, ректально или парэнтерально, причем, как таковые, они могут быть в форме таблеток, капсул, жидких препаратов для орального применения, порошков, гранул, лепешек, порошков, аэрозолей для распыления через нос, свечей, растворов или суспензий для инъекции. Также предусматривается ввод лекарства под язык или через инъекцию в кожу. Составы, предназначенные для приема через рот, предпочтительны, поскольку они более удобны для применения.
Таблетки и капсулы для приема через рот обычно представлены в виде единичной дозы, причем они содержат обычные инертные наполнители, например связующие, наполнители, разбавители, вещества для таблетирования, смазки, диспергаторы, красители, ароматизирующие вещества и увлажнители. На таблетки можно нанести известными способами покрытие, например, против раздражения кишок.
Соответствующие наполнители для применения включают в себя целлюлозу, маннит, лактозу и другие подобные вещества. Соответствующие диспергаторы включают в себя крахмал, поливинилполипирролидон и такие производные крахмала как, например, крахмал гликолят натрия. Соответствующие смазки включают в себя, например, стеарат магния.
Соответствующие фармацевтически приемлемые увлажнители включают в себя лауриловый сульфонат натрия. Жидкие препараты для приема через рот могут быть в форме, например, водных или масляных суспензий, растворов, эмульсий, сиропов или эликсиров либо в форме сухого продукта для смешивания с водой или другим соответствующим носителем до его применения. Такие жидкие препараты могут содержать обычные добавки, например, суспендирующие агенты, например, сорбитол, сироп, метилцеллюлозу, желатин, оксиэтилцеллюлозу, карбоксиметилцеллюлозу, гело стеарата алюминия или гидрогенизированные съедобные жиры, эмульгаторы, например, лецитин, сорбитан-моноолеат или аравийская камедь; неводные носители (которые могут включать в себя съедобные масла), например, миндальное масло, ректифицированное кокосовое масло, масляные сложные эфиры, например, эфиры глицерина, пропиленгликоля или этилового спирта; антикоагуляторы, например, метил или пропил p - оксибензоат или сорбиновая кислота, и, если это требуется, то можно добавлять обычные ароматизирующие вещества или красители.
Жидкие препараты для принятия через рот обычно присутствуют в форме водных для масляных суспензий, растворов, эмульсий, сиропов или эликсиров или в виде сухого продукта для смешения с водой или соответствующим носителем до его применения. Такие жидкие составы могут содержать обычные добавки, например суспендирующие агенты, эмульгаторы, неводные носители (которые могут включать в себя съедобные масла), антикоагуляторы и ароматизирующие или окрашивающие вещества.
Составы для применения через рот можно получить обычными способами смешения, расфасовки или таблетирования. Можно применять операции повторного смешения для распределения активного вещества во всех этих составах с применением большого количества наполнителей. Такие операции, конечно, известны в технике.
Для парэнтерального применения приготавливают формы единичной дозы жидкости, содержащие соединение в соответствии с настоящим изобретением и стерильный растворитель. Соединение в зависимости от растворителя и концентрации можно суспендировать либо растворять. Растворы для парэнтерального применения обычно приготовляют путем растворения соединения в растворителе, причем до его заполнения в соответствующий пузырек или ампулу и герметизации его стерилизуют через фильтр. Преимущественно в растворителе также растворяют фармацевтические препараты, усиливающие действие другого препарата, например средства для местной анестезии, антикоагуляторы и буферные агенты. Для повышения устойчивости состав можно заморозить после его заполнения в пузырек, а воду можно удалить под вакуумом.
Суспензии для парэнтерального применения приготовляют по существу так же за исключением, что соединение суспендируют в носителе вместо его растворения и стерилизуют посредством подвергания его действию окиси этилена до суспендирования в стерильном носителе. Преимущественно для упрощения равномерного распределения соединения в соответствии с изобретением в состав добавляют поверхностно-активное вещество или увлажнитель.
Также в соответствии с изобретением предложен способ лечения синдрома раздражения кишечника (IBS), заболеваний желудка и пищевода, дисперсии, аритмии сердца и паралича, состояния беспокойства и тревоги и/или приступов мигрени у млекопитающих, например, у людей, причем способ заключается в назначении эффективного количества соединения формулы (I) или фармацевтически приемлемой его соли. В частности, способ заключается в лечении синдрома раздражения кишечника (IBS), сердечной аритмии и паралича.
Количество, эффективное для лечения описанных расстройств, зависит от относительной эффективности соединений в соответствии с изобретением, характера и сложности заболевания, а также веса млекопитающего. Однако единичная доза для взрослого человека весом 70 кг будет обычно содержать 0,05 - 1000 мг, например 0,5 - 500 мг соединения в соответствии с изобретением. Единичные дозы можно принимать один или несколько раз в день, например 2, 3 или 4 раза в день, обычно 1 - 3 раза в день, то есть в пределах 0,0001 - 50 мг/кг/день, обычно 0,0002 - 25 мг/день.
Вредные токсикологические эффекты не отмечаются в пределах указанной дозировки.
Изобретение также касается соединения формулы (I) или фармацевтически приемлемой его соли для применения в качестве активного терапевтического вещества, в частности для применения в качестве антагониста рецептора 5-НТ4 для лечения описанных расстройств.
Также изобретение касается применения соединения формулы (I) в приготовлении медикамента для использования в качестве антагониста рецептора 5-НТ4 для лечения описанных болезней.
Примеры иллюстрируют приготовление соединений формулы (I) (табл. 1). Следующие затем описания иллюстрируют получение промежуточных соединений.
Пример 1. (1-п-бутил-4-пиперидил)метил-3,4-дигидро- 2H-[1,3] оксазино[3,2-a]индол-10-карбоксилат (Е1).
a) Суспензию индол-3-карбоновая кислота (500 мг, 0,003 моль) в дихлорметане (50 мл) обработали оксалилхлоридом (0,635 г, 0,005 моль) и двумя каплями диметилформамида. Смесь перемешивали при комнатной температуре в течение 1,5 ч, затем удалили под вакуумом растворитель, оставив кислый хлорид.
Раствор 1-бутил-4-пиперидинметанола, Д6, (513 мг, 0,003 моль) в сухом THF (10 мл) в атмосфере азота охладили в ледяной ванне. По капле добавили n-бутиллитий (1,88 мл 1,6 М раствора в гексане, 0,003 моль), и полученный раствор перемешивали при 0oC в течение 15 мин.
Кислый хлорид растворили в сухом THF (20 мл) и раствор добавляли по капле в раствор алкоголята лития при 0oC.
Реакционной смеси позволили подогреться до комнатной температуры и перемешивали в течение 3 ч. Растворитель удалили под вакуумом, а остаток разделили между хлороформом и водой. Хлороформ отделили, промыли несколько раз водой, высушили и концентрировали, получив (1-бутил-4-пиперидил) метил-1H-индол-3-карбоксилат в виде бледно-коричневой смолы.
1HNMR (250 MHz) CDCl3
δ : 9,90 (brs, 1H), 8,10 - 8,18 (m, 1H), 7,78 (d, 1H), 7,37 - 7,46 (m, 1H), 1,16 - 7,28 (m, 2H), 4,19 (d, 2H), 3,05 - 3,15 (brd, 2H), 2,40 - 2,49 (m, 2H), 0,90 (t, 3H), 1,20 - 2,18 (m, 11H).
b). Суспензию N-хлорсукцинимида (57 мг, 0,48 ммоль) в хлороформе (2 мл) обработали раствором (1-п бутил-4-пиперидил) метилиндол-3-карбоксилата (100 мг, 0,32 ммоль) в хлороформе (2 мл), и смесь перемешивали при комнатной температуре в течение 2 ч. Бледно-желтый раствор обработали 3-бром-1-пропанолом (0,03 мл, 0,32 ммоль), перемешивали при комнатной температуре в течение 16 ч; затем посредством 10%-ного раствора Na2CO3 повысили основность и экстрагировали хлороформом. Экстракт высушили и концентрировали, оставив желтую смолу, которую растворили в ацетоне (6 мл), обработали безводным карбонатом калия (130 мг, 0,94 ммоль) и перемешивали при комнатной температуре в течение 18 ч. Смесь обработали 10%-ным раствором Na2CO3 и экстрагировали этилацетатом. Экстракт высушили и концентрировали, оставив коричневое масло, которое подвергали хроматографии сначала на силикагеле, при этом элюирование осуществляли с хлороформом/метанолом (97:3), затем на основной окиси алюминия с элюированием этилацетатом, получив бесцветное масло. Его кристаллизовали из простого эфира/пентана для получения соединения (Е1) в виде белого твердого вещества (11 мг) с температурой плавления 117 - 119oC.
1HNMR (CDCl3)
δ : 7,97 (d, 1H), 7,10 - 7,30 (m, 3H), 4,55 (t, 2H), 420 (d, 2H), 4,11 (t, 2H), 2,90 - 3,03 (m, 2H), 2,25 - 2,40 (m, 4H), 1,75 - 2,00 (m, 5H), 1,22 - 1,55 (m, 6H), 0,91 (t, 3H)
MS (EI) M+370.
Пример 2. eq-хинолизидин-2-илметил-3,4-дигидро-2H- [1,3]оксазино[3,2-a] индол-10-карбоксалат (Е2).
a) eq-2-оксиметилхинолизидин (Н.Дж. Леонард и др., ж.Органическая химия, 1957, 22, 1445) подвергли реакции с индол-3-хлоридом карбоновой кислоты с использованием способа, описанного в примере 1, для получения eq-хинолизидин-2-yl-метил 1-H-индол-3-карбоксилата с температурой плавления 154-157oC.
1HNMR (CDCl3)
δ : 9,40 (b.r.s 1H), 8,10 - 8,20 (m, 1H), 7,87 (d, 1H), 7,35 - 7,45 (m, 1H), 7,20 - 7,30 (m, 2H), 4,20 (d, 2H), 2,80 - 2,97 (m, 2H), 1,43 - 2,20 (m, 11H), 1,10 - 1,40 (m, 3H).
b) eq-хинолизидин-2-ил-метил 1H-индол-3-карбоксилат обрабатывали сначала N-хлорсукцинимидом (1,5 эквивалентов) в течение 2 часов, затем 3-бром-1-пропанолом (2 эквивалента) в течение 16 ч и после этого безводным карбонатом калия в ацетоне с использованием способа, описанного в примере 1в. Сырой продукт очистили, при этом применяли те же условия хроматографии, что и в примере 1в, для получения соединения в виде бесцветного масла (51%). Его превратили в хлористоводородную соль и кристаллизовали из ацетона при температуре плавления 164 - 167oC.
1HNMR (соль HCl) (d6 DMSO)
δ : 10,35 (b.r.s., 1H), 7,85 (d, 1H), 7,32 (d, 1H), 7,07 - 7,20 (m, 2H), 4,54 (t, 2H), 4,13 (t, 2H), 4,05 (d, 2H), 3,25 - 3,43 (m, 2H), 2,74 - 3,15 (m, 3H), 2,20 - 2,33 (m, 2H), 2,00 - 2,15 (m, 1H), 1,35 - 1,95 (m, 10H).
Пример 3. N-[1-пбутил-4-пиперидил)метил] -3,4-дигидро-2H- [1,3]оксазино[3,2-a]индол-10 карбоксамид (Е3).
Способ 1. Перемешанный раствор N-хлорсукцинимида (57 мг, 0,48 ммоль) в хлороформе (3 мл) обработали раствором N-[(1-пбутил- 4-пиперидил)метил]индол-3-карбоксамида, Д1, (100 мг, 0,32 ммоль) в хлороформе (8 мл) и поддерживали при комнатной температуре в течение 2 ч, затем обработали 3-бромо-1-пропанолом (0,03 мл, 0,32 ммоль). После перемешивания в течение 16 ч добавили еще 3-бром-1-пропанола (0,03 мл, 0,32 ммоль). Смесь перемешивали при комнатной температуре еще в течение 3 ч, затем обработали с избытком 10%-ным раствором Na2CO3 и экстрагировали хлороформом. Экстракт высушивали (Na2SO4) и концентрировали в вакууме, получив желтое масло, которое растворили в ацетоне (10 мл), обработали безводным карбонатом калия (130 мг, 0,96 ммоль) и перемешивали при комнатной температуре в течение 16 ч. Смесь концентрировали под вакуумом, остаток обработали 10%-ным раствором Na2CO3 (10 мл) и экстрагировали хлороформом. Экстракт высушили (Na2SO4) и концентрировали под вакуумом, оставив желтое масло, которое подвергли хроматографии сначала на силикагеле с элюированием хлороформом/метанолом (19:1), затем на основной окиси алюминия с элюированием этилацетатом. Полученное бесцветное масло кристаллизовали из простого эфира, получив соединение (Е3) в виде белого твердого вещества (20 мг, 17%) с температурой плавления 110 - 113oC.
1HNMR(CDCl3)
δ : 8,34 (d, 1H), 7,05 - 7,30 (m, 3H), 6,55 (t, 1H), 4,53 (t, 2H), 4,10 (t, 2H), 3,33 (t, 2H), 2,90 - 3,05 (m, 2H), 2,25 - 2,45 (m, 4H), 1,90 - 2,25 (m, 2H), 1,20 - 1,85 (m, 9H), 0,92 (t, 3H)
M(Cl) MH+370.
Способ 2. Перемешанную суспензию N-[(1-пбутил-4- пиперидил)метил]индол-3-карбоксамид (Д1, 120 г, 0,38 моль) в хлороформе (2 л) в атмосфере азота при комнатной температуре обработали свежим дистиллированным 3-бром-1-пропанолом (69 мл, 0,77 моль), затем по частям добавляли сухой N-хлорсукцинимид (55 г, 0,42 моль) в течение 5 мин. Полученный желтый раствор перемешивали в течение 2,5 ч, затем обрабатывали IMHCl в эфире (15 мл, 0,015 моль). Образовался умеренный экзотерм и цвет реакции изменился на оранжевый. После дополнительных 2 ч смесь обработали 10%-ным раствором Na2CO3 (700 мл), слой хлороформа отделили, высушили (Na2SO4) и концентрировали в вакууме, оставив густое красное масло. Его обработали ацетоном (1,5 л) и безводным карбонатом калия (130 г, 0,95 моль), затем перемешивали при комнатной температуре в течение 18 ч. Реакционную смесь концентрировали в вакууме, остаток обработали водой (1 л) и экстрагировали этилацетатом. Спустя 2 ч при 8oC его профилировали и высушили, получив 51,7 г соединения (Е3) в виде твердого вещества. Маточные жидкости извлекли при помощи IMHCl кислоты (800 мл), затем кислый экстракт превратили в основание с применением K2CO3 и экстрагировали хлороформом (2х700 мл). Объединенные экстракты хлороформа высушили (Na2SO4), концентрировали в вакууме, а остатки подвергли хроматографии на силикагеле с элюированием хлороформом/метанолом (96: 4). Получили желтое масло, которое после растирания в порошок простым эфиром дало дополнительно 21,3г указанного соединения (Е3) в виде белого твердого вещества. Превращение в хлористоводородную соль и рекристаллизация из этанола / 60 - 80 бензил дало твердое вещество с температурой плавления 254 - 256oC.
Соль HCl-1HNMR (D2O)
δ : 7,90 (d, 1H), 6,88 - 7,20 (m, 3H), 4,35 (br.t, 2H), 3,70 (br.t, 2H), 3,40 (br. d. , 2H), 3,20 (br.d., 2H), 2,9 (br.t 2H), 2,65 (br.t, 2H), 2,12 (br.t., 2H) 1,20 - 1,90 (m, 9H), 0,87 (t, 3H).
Пример 4. 2-(1-пиперидил)этил 3,4-дигидро-2H-[1,3] оксазино[3,2-a]индол-10-карбоксилат (E4).
a). 1-пиперидинэтанол подвергли реакции с 1H-индол-3-хоридкарбоновой кислотой, применяя способ, описанный в примере 1a, для получения 2-(1-пиперидил)этил 1H-индол-3-карбоксилата.
1HNMR (CDCl3)
δ : 9,6 (br.s, 1H), 8,03 - 8,12 (m, 1H) 7,43 (d, 1H), 7,30 - 7,40 (m, 1H), 7,13 - 7,25 (m, 2H), 4,48 (t, 2H), 2,82 (t, 2H), 2,82 (t, 2H), 2,50 - 2,65 (m, 4H), 1,35 - 1,70 (m, 6H).
b). 2-(1-пиперидил)этил 1H-индол-3-карбоксилат сначала обрабатывали N-хлорсукцинимидом (1,5 эквивалентов) в течение 2 ч, затем 3-бром-1-пропанолом (3 эквивалента) в течение 21 ч, после этого безводным карбонатом в ацетоне с использованием способа, описанного в описании 1b. Сырой продукт очистили, применяя те же условия хроматографии, как и в описании 1b, для получения указанного соединения (E4) в виде бледно-желтого масла (15%). Его превратили в его оксалатовую соль и кристаллизовали из ацетона с точкой плавления 174 - 177oC.
Свободное основание: 1HNMR (CDCl3)
δ : 8,02 (d, 1H), 7,07 - 7,30 (m, 3H), 4,40 - 4,55 (m, 4H), 4,08 (t, 2H), 2,78 (t, 2H), 2,45-2,65 (m, 4H), 2,25-2,38 (m, 2H), 1,54 - 1,66 (m, 4H), 1,35 - 1, 50 (m, 2H).
MS (Cl) MH+ 329.
Пример 5. N-[2-(1-пиперидил)этил] 3,4-дигидро- 2H-[1,3]оксазино[3,2-a] индол-10-карбоксамил (E5).
N-[2-(1-пиперидил)этил] 1H-индол-3-карбоксамид (D2) обрабатывали сначала N-хлорсукцинимидом, затем 3-бром-1-пропанолом и после этого карбонатом калия в ацетоне в соответствии со способом, описанным в примере 3. Сырой продукт подвергли хроматографии на силикагеле с элюированием хлороформом/метанолом (19: 1), получив бледно-желтое масло, которое кристаллизовали из простого эфира, получив указанное в заглавии соединение (E5) в виде белого твердого вещества (29%) с точкой плавления 124-127oC.
1H NMR (CDCl3)
δ : 8,33 (d, 1H), 7,06-7,28 (m, 3H), 7,02 (Br.t, H), 4,51 (t, 2H), 4,08 (t, 2H), 3,50-3,60 (m, 2H), 2,54 (t, 2H), 2,30-2,60 (m, 6H), 1,40-1,65 (m, 6H)
MS (Cl) MH+ 328.
Пример 6. (1-п бутил-4-пиперидил)метил-2,3-дигидрооксазол[3,2-a] инодол-9-карбоксилат (E6).
(1-п бутил-пиперидил)метил 1H-индол-3-карбоксилат (E1a) обрабатывали сначала N-хлорсукцинимидом (1,5-эквивалентов) в течение 4 ч, затем 2-бром-этанолом (2 эквивалента) в течение 18 ч с последующей обработкой безводным карбонатом калия в ацетоне (18 ч) способом, описанным в примере 1b. Сырой продукт очистили при тех же условиях хроматографии, что и в примере 1b, получив бесцветное масло (26%), которое кристаллизовали из простого эфира, получив указанный продукт (E6) в виде белого твердого вещества с точкой кипячения 128-130oC.
1HNMR (CDCl3)
δ : 7,95-8,03 (m, 1H), 7,07-7,27 (m, 3H), 5,18-5,27 (m, 2H), 4,24-4,33 (m, 2H), 4,19 (d, 2H), 2,92-3,04 (m, 2H), 2,27-2,38 (m, 2H), 1,75-2,05 (m, 5H), 1,25-1,66 (m, 6H), 0,91 (t, 3H).
M (Cl) M+ 356.
Пример 7. (1-п бутил-4-пиперидил)метил-3,3-диметил- 3,4-дигидро-2H-[1,3] оксазино[3,2-a]индол-10-карбоксилат (E7).
(1-п-бутил-4-пиперидил)метил-1H-индол-3-карбоксилат (E1a) обрабатывали сначала N-хлорсукцинимидом (1,5 эквивалентов) в течение 2 ч, затем 3-бром-2,2-диметил-1-пропанолом (2 эквивалента) в течение 20 ч и после этого безводным карбонатом калия в ацетоне (2 1/2 дней) согласно способу, описанному в примере 1b. Сырой продукт подвергали хроматографии на силикагеле с элюированием хлороформом/метанолом (95:5), получив указанное соединение (E7) в виде твердого вещества белого цвета (10%) с точкой плавления 134-135oC.
1HNMR (CDCl3)
δ : 7,98 (d, 1H), 7,08-7,30 (m, 3H), 4,21 (d, 2H), 4,15 (S, 2H), 3,77 (S, 2H), 2,95-3,07 (m, 2H), 2,95-3,07 (m, 2H), 2,32-2,42 (m, 2H), 1,80-2,10 (m, 5H), 1,26-1,60 (m, 6H), 1,20 (S, 6H), 0,93 (t, 3H).
MS (Cl) MH+ 399.
Пример 8. (1-п бутил-4-пиперидил)метил-3,4-дигидро- 2H-[1,3] тиазино[3,2-a]индол-10-карбоксилат (E8).
(1-п бутил-4-пиперидил)метил-1H-индол-3-карбоксилат, E1a (314 мл, 0,0010 моль) обрабатывали сначала N-хлорукцинимидом (180 мг, 0,0015 моль) в течение 2 ч, затем 3-хлор-1-пропантиолом (0,20 мл, 0,0020 моль) в течение 5 дней согласно способу, описанному в примере 1b. У полученного раствора повысили основность посредством 10%-раствора Na2CO3и экстрагировали хлороформом. Экстракт высушили (Na2SO4) и концентрировали под вакуумом, получив темное масло, которое подвергали хроматографии на силикагеле с элюированием хлороформом/метанолом (95: 5), получив (1-п бутил-4-пиперидил)метил-2-(3-хлорпропилмеркапто)- -1H-индол-3-карбоксилат в виде масла серого цвета (220 мг). Его растворили в ацетоне (50 мл), обработали безводным карбонатом калия (220 мг, 0,0015 моль) и иодидом натрия (390 мг, 0,0026 моль) и нагревали под флегмой (орошением) в течение 8 ч. Смесь концентрировали под вакуум, а остаток обработали 10%- ным раствором Na2CO3 и затем экстрагировали этилацетатом. Экстракт высушили (Na2SO4) и концентрировали. Остаток подвергли хроматографии на основной окиси алюминия с элюированием этилацетатом. Полученное бесцветное масло кристаллизовали из простого эфира, получив указанное соединение (E8) в числе белого твердого вещества (80 мг, 21%) с точкой плавления 99-100oC.
1H NMR (CDCl3)
δ : 7,97-0,04 (m, 1H), 7,14-7,30 (m, 3H), 4,22 (d, 2H), 4,15 (t, 2H), 3,05-3,15 (m, 2H), 2,92-3,02 (m, 2H), 2,38-2,50 (m, 2H), 2,27-2,37 (m, 2H), 1,75-2,02 (m, 5H), 1,20-1,55 (m, 6H), 0,91 (t, 3H)
MS (E1) M+ 386.
Пример 9. (1-п бутил-4-пиперидил)метил- 2,3,4,5-тетрагидро[1,3]оксазепин[3,2-a]индол-11-карбоксилат (E9).
(1-п бутил-4-пиперидил)метил 1H -индол-3-карбоксилат (EIa) обрабатывали сначала N-хлорсукцинимидом (1,5 эквивалентов) в течение 2 ч, затем 4-хлор-1-бутанолом (2 эквивалента) в течение 18 ч способом, описанным в примере 1в, полученный продукт изолировали как в примере 8, получив (1-п бутил-4-пиперидил)метил 2-(4-хлорбутокси)-1H-индол-3-карбоксилат в виде желтого масла. Раствор в ацетоне обработали безводным карбонатом калия и иодидом натрия и нагревали под орошением в течение 30 м, затем его очистили, как в примере 8, получив указанное соединение (E9) в виде бледно-желтого масла (21%). Его превратили в его оксалатовую соль и кристаллизовали из ацетона, получив белое твердое вещество с точкой кипения 161-164oC.
Оксалатовая соль: - 1HNMR (d6DMSO)
δ : 7,85 (m, 1H), 7,45 - 7,55 (m, 1H), 7,10 - 7,25 (m, 2H), 4,15 - 4,30(m, 4H), 4,10 (d, 2H), 3,35 - 3,45 (m, 2H), 2,80 - 3,05 (m, 4H), 1,80 - 2,10 (m, 7H), 1,50 - 1,75 (m, 4H), 1,20 - 1,40 (m, 2H), 0,89 (t, 3H)
MS (EI) M+384.
Пример 10. (1-п бутил-4-пиперидил) метил-6,7,8,9-тетрагидропирил [1,2-а] индол-1-карбоксилат (Е10).
Раствор 6,7,8,9-тетрагидропирил [1,2-a] индол-1-карбоновой кислоты, Д3, (400 мг, 0,00186 моль) в дихлорметане (20 мл) обрабатывали оксалилхлоридом (0,20 мл, 0,023 моль) и 2 каплями DMF (диметилформамид) и перемешивали при комнатной температуре в течение 2 ч, затем концентрировали в вакууме, получив кислый хлорид в виде твердого вещества оранжевого цвета.
Раствор (1-пбутил-4-пиперидил)метанола (Д6)(0,32 г, 0,0186 моль) в сухом THF (тетрагидрофуране) (25 мл) при 5oC в атмосфере азота обработали 1,5 М метиллитием в простом эфире (1,24 мл, 0,00186 моль) и оставили для перемешивания в течение 15 мин, затем обработали раствором упомянутого кислотного хлорида в сухом THF (15 мл). После 16 ч при комнатной температуре смесь обработали насыщенным раствором K2CO3 (50 мл) и экстрагировали в этилацетат (2х75 мл), высушили (Na2SO4) и концентрировали в вакууме. Остаток подвергли хроматографии на силикагеле с элюированием хлороформом (95:5), получив указанное соединение (Е10) в виде желтого масла. Его превратили в его хлористоводородную соль для получения белого твердого вещества с точкой плавления 230-232oC.
HCl соль: 1H NMR (d6 DMSO)
δ : (br. s., 1H), 7,92-8,03 (m, 1H),7,43 - 5,53 (m, 1H), 7,16 - 7,26 (m, 2H), 4.18 (d, 2H), 4,11 (t, 2H), 3,43 - 3,56 (m, 2H), 3,23 (t, 2H), 2,82 - 3,05 (m, 4H), 1,85 - 2,12 (m, 7H), 1,60 - 1,80 (m, 4H), 1,25 - 1,40 (m, 2H), 0,90 (t, 3H)
MS (E1) M+368.
Пример 11. (1-п бутил-4-пиперидил) метил-2,3-дигидро-1H-пирроло[1,2-a] индол-9-карбоксилат (Е11).
Указанное соединение (Е11) приготовили из 2,3-дигидро-1H-пирроло[1,2-a] индол-9-карбоновой кислоты (Д4), применяя способ из примера 10, и его изолировали в виде твердого вещества бледно-оранжевого цвета (24%) с точкой плавления 100-102oC.
1H NMR (CDCl3)
δ : 8,03-8,12 (m, 1H), 7,13 - 7,28 (m, 3H), 4,17 (d, 2H), 4,11 (t, 2H), 3,29 (t<2H), 2,95 - 3,08 (m, 2H), 2,57 - 2,72 (m, 2H), 2,30 - 2,41 (m, 2H), 1,92 - 2,07 (m, 2H), 1,73 - 1,90 (m, 3H), 1,40 - 1,60 (m, 4H), 1,22 - 1,39 (m, 2H), 0,92 (t, 3H)
MS (EI) M+354.
Пример 12 (1-пбутил-4-пиперидил)метил 7,8,9,10-тетрагидро-6H-азепин[1,2-f]индол-11-карбоксилат (12).
Указанное соединение (Е12) приготовили из 7,8,9,10-тетрагидро-6H-азепин[1,2-а] индол-11-карбоновой кислоты (Д5) согласно способу из примера 10. Сырой продукт очистили посредством хроматографии на силикагеле с элюированием хлороформом/этанолом (98 : 2), получив желтое масло, которое превратили в его хлористоводородную соль для получения твердого вещества (20%) с точкой плавления 196 - 198oC.
1H NMR (d6 DMSO) - HCl соль
δ : 10,52 (br.s, 1H), 7,93 - 8,00 (m, 1H), 7,55 - 7,62 (m, 1H), 7,13 - 7,25 (m, 2H), 4,25 - 4,40 (m, 2H), 4,17 (d, 2H), 3,35 - 3,55 (m, 4H), 2,80 - 3,10 (m, 4H), 1,55 - 2,15 (m, 13H), 1,24 - 1,40 (m, 2H), 0,88 (t, 3H)
MS (Cl) MH+383.
Пример 13. N-[(1-пбутил-4-пиперидил)метил] -2,3- дигидро-1H-пирроло[1,2-a]индол-9-карбоксамид (Е13).
Раствор 2,3-дигидро-1H-пирроло[1,2-a] индол-9-карбоновой кислоты, Д4, (180 мг, 0,89 ммоль) в дихлорметане (20 мл) обработали оксалилхлоридом (0,096 мл, 1,1 ммоль) и 2 каплями диметилформамида (DMF), перемешивали при комнатной температуре в течение 1 ч, затем концентрировали в вакууме, получив кислый хлорид в виде желтого твердого вещества.
Раствор (1-п бутил-4-пиперидил) метиламина, Д8, (150 мг, 0,89 ммоль) и триэтиламина (0,15 мл, 1,1 ммоль) в дихлорметане (20 мл) в атмосфере азота обрабатывали раствором упомянутого кислого хлорида в дихлорметане (5 мл) и перемешивали при комнатной температуре в течение 3 ч. Раствор обработали 10%-ным раствором Na2CO3, органический слой отделили, высушили (Na2SO4) и концентрировали в вакууме, получив твердое вещество. Его рекристаллизовали из этилацетата, получив соединение (Е13) в виде белого твердого вещества (180 мг, 55%) с точкой плавления 152-154oC.
1H NMR (CDCl3)
δ : 7,75 - 7,84 (m, 1H), 7,13 - 7,33 (m, 3H0, 5,93 (br.t, NH), 4,10 (t, 2H) 3,38 (t<2H), 3,31 (t, 2H), 2,90 - 3,02 (m, 2H), 2,65 (квинтет, 2H), 2,28 - 2,36 (m, 2H), 12,60 - 2,10 (m, 6H), 1,22 - 1,55 (m, 5H), 0,90 (t, 3H)
MS (Cl) MH+354.
Пример 14. N-[(1-п бутил-4-пиперидил)метил]-2,3-дигидрооксазол[3,2-а]индол-9-карбоксамид (E14).
N-[(1-п бутил-4-пиперидил)метил] индол-3-карбоксамид (Д1) обрабатывали сначала N-хлорсукцинимидом (1,5 эквивалентов) в течение 2 ч, затем 2-бромэтанолом (2 эквивалента) в течение 16 ч и после этого карбонатом калия (3 эквивалента) в ацетоне в течение 68 ч согласно способу из примера 1в.
Сырой продукт очистили посредством хроматографии на силикагеле с элюированием хлороформом/этанолом (19:1) для получения указанного соединения (Е14) в виде белого твердого вещества с последующей рекристаллизацией из хлороформа/эфира (14%) при температуре плавления 156 - 158oC.
1H NMR (CDCl3)
δ : 8,19 (d, 1H), 7,00 - 7,30 (m, 3H), 6,00 (t, NT), 5,15 (t, 2H), 4,20 (t, 2H), 3,32 (t, 2H), 2,90 - 3,15 (m, 2H) 2,25 - 2,42 (m, 2H), 1,20 - 2,05 (m, 1H), 0,90 (t, 3H)
MS (Cl) MH+356.
Пример 15. (1-бензил-4-пиперидил) метил-3,4-дигидро-2H-[1,3] оксазино[3,2-a]индол-10-карбоксилат (Е15).
а) Индол-3-карбоновая кислота превратила его в кислый хлорид и затем подвергли реакции с 1-бензил-4-пиперидинметанол (Д7) согласно способу, описанному в примере 1а. Полученное оранжевое масло подвергли хроматографии на силикагеле с элюированием хлороформом/этанолом (9:1) для образования (1-бензил-4-пипердил) метил-индол-3-карбоксилата в виде желтого масла (88%).
1H NMR (CDCl3)
δ : 9,24 (s, 1H), 8,12 - 8,20 (m, 1H), 7,81 (d, 1H), 7,20 - 7,45 (m, 8H), 4,20 (d, 2H), 2,90 - 3,04 (m. 2H), 1,73-2,10 (m, 5H), 1,36-1,58 (m, 2H)
b) (1-бензил-4-пиперидил) метил-индол-3-карбоксилат сначала обрабатывали N-хлорсукцидинимидом (1,5 эквивалентов) в течение 2 ч, затем 3-бром-1-пропанолом (2 эквивалента) в течение 16 ч и после этого безводным карбонатом калия в ацетоне с использованием способа, описанного в примере 1в. Сырой продукт очистили посредством хроматографии из силикагеля с элюированием хлороформом/этанолом (19: 1) для получения указанного соединения (Е15) в виде твердого вещества с последующей рекристаллизацией из хлороформа/эфира (47%) при температуре плавления 158-160oC.
1HNMR (CDCl3)
δ : 7,94-8,00 (m, 1H), 7,10-7,38(m, 8H), 4,48 - 4,56 (m, 2H), 4,19 (d, 2H), 4,19 (d, 2H), 4,05-4,12 (m, 2H), 3,50 (S, 2H), 2,28-2,98 (m, 2H), 2,28-2,39 (m, 2H), 1,75-2,08 (m, 5H), 1,35 - 1,55 (m, 2H)
MS (Cl) MH+405.
Пример 16. (1-п-бутил-4-пиперидил)метил-3,4-дигидро-1-оксо- 2H-[1,3-a] тиазино[3,2-a] индол-10-кароксилат (У16).
Раствор (1-п бутил-4-пиперидил)метил 3,4-дигидро- 2H-[1,3]тиазино[3,2-a] индол-10-карбоксилаита (E8, 80мг, 0,21 ммоль) в ацетоне (5 мл и воде 5 мл) обрабатывали периодатом натрия (100 мг, 0,46 ммоль) и перемешивали при комнатной температуре в течение 24 ч. Затем раствор обрабатывали и насыщали раствором K2CO3 (10мл) и экстрагировали с использованием этилацетата (2 ч 25 мл). Экстракт высушивали (Na2SO4) и концентрировали в вакууме, получив желтое масло, которое подвергали хроматографии на силикагеле с элюированием 5% метанола/хлороформа. Полученное бесцветное масло кристаллизовали из эфира, получив указанное соединение (Е16) в виде белого твердого вещества (27 мг, 32%) с температурой плавления 130 - 135oC.
1H NMR (CDCl3)
δ : 8,24 (d, 1H), 7,30-7,50 (m, 3H), 4,54 (dd, 1H), 4,22-4,38 (m, 2H), 4,05 (dt, 1H), 3,40 (dd, 1H), 3,21(dg, 1H), 2,86-3,08(m,3H), 2,30-2,45 (m, 3H), 1,80-2,10 (m, 4H), 1,20-1,40 (m, 2H), 0,90 (t, 3H)
MS(Cl) MH+403.
Пример 17. (1-п бутил-4-пиперидил)метил 6,7-дигидропирид[1,2-a]индол-10-карбоксилат (Е17).
Указанное соединение получили из 6,7-дигидропирид[1,2-a] индол-10-карбоновой кислоты (Д8) согласно способу, описанному в Примере 10, и подвергли хроматографии на силикагеле с элюированием этилацетатом, получив желтое твердое вещество (18%) с температурой плавления 60-62oC (п-пентан).
1H NMR (CDCl3)
δ : 8,10 - 8,17 (m, 1H), 7,42 (dt, 1H), 7,18-7,33(m, 3H), 6,25 - 6,35 (m, 1H), 4,22 (d, 2H), 4,15 (t, 2H),2,90 - 3,05 (m,2H), 2,63 - 2,75 (m, 6H), 0,91 (t, 3H)
MS(E1) M+366.
Пример 18. (1-п бутил-4-пиперидил)метил-пиридо[1,2-a]индол- 10-карбоксилат (Е18).
Указанное соединение получили из пиридо[1,2-a]индол-10-карбоновой кислоты (Д9) согласно способу из примера 10 и подвергли хроматографии на силикагеле с элюированием этилацетата, получив желтое твердое вещество (10%) с температурой плавления 57-59oC (п-пентан)
1H NMR (CDCl3)
δ : 8,35 - 8,50 (m, 3H), 7,88 (d, 1H), 7,48 - 7,56 (m, 1H), 7,28 - 7,40 (m, 2H), 6,78 - 6,86 (m, 1H), 4,30 (d, 2H), 2,95 - 3,05 (m, 2H), 2,30 - 2,40 (m, 2H), 1,85 - 2,05 (m, 5H), 1,43 - 1,60 (m, 4H) 1,25 - 1,40 (m, 2H), 0,92 (t, 3H)
MS(E1) M+364.
Пример 19. N-[1-п бутил-4-пиперидил)метил]3,4-дигидро -2H-[1,3]тиазин [3,2-a] индол-10-карбоксамии(Е19).
Указанное соединение получили из N-[1-пбутил- 4-пиперидил)метил]индол-3-карбоксамида (Д1в) согласно способу из Примера 8 в виде белого твердого вещества (7%) с температурой плавления 141-142oC.
1H NMR (CDCl3)
δ : 7,70 (d, 1H), 7,13 - 7,30 (m, 3H), 6,0oC7 (t, 1H), 4,16 (t, 2H), 3,38 (t, 2H), 3,08 (t, 2H), 2,90 - 3,02 (m, 2H), 2,38 - 2,50 (m, 2H), 2,25-2,36 (m, 2H), 1,60 - 2,00 (m, 5H), 1,23 - 1,56 (m, 6H), 0,91 (t, 3H)
MS(E1) M+385.
Пример 20. N-[(1-бензил-4-пиперидил]3,4-дигидро- 2H-[1,3]оксазин[3,2-a] индол-10 карбоксамид (Е20).
а). Индол-3-карбоновую кислоту превратили в хлорид этой кислоты и затем подвергли реакции с (1-Бензил-4-пиперидил) метиламином (Д10) согласно способу, раскрытому в Описании 1а, для получения N-[(1-бензил-4-пиперидил)метил]индол-3-карбоксамина в виде белого твердого вещества (60%).
1H NMR (CDCl3).
δ : 9,90 (S, 1H), 7,85 - 7,95 (m, 1H), 7,64 (d, 1H), 7,15 - 7,43 (m, 8H), 6,17 (n, 1H), 3,48 (S, 2H), 3,37 (t, 2H), 2,83 - 2,98 (m, 2H), 1,87 - 2,08 (m, 2 H), 1,54 - 1,82 (m, 3H), 1,23 - 1,50 (m, 2H).
b) Перемешанную суспензию N-[1-бензил-4-пиперидил)метил]индол- 3-карбоксамида (17,5 г, 0,050 моль) в хлороформе (250 мл) обработали 3-бром-1-пропенол (10,1 мл, 0,11 моль) и N-хлорсукцинимидом (8,7 г, 0,065 моль) при комнатной температуре и за 15 мин получили прозрачный раствор.
После 1 ч реакционная смесь потемнела, изменив свой цвет от бледно-желтого до оранжевого, и температура повысилась до 38oC. Спустя еще 1 ч реакционную смесь обрабатывали 10%-ным раствором NAHCO3, слой хлороформа отделили, высушили (NA2SO4) и концентрировали в вакууме, получив желтое масло, которое подвергли хроматографии на силикагеле с элюированием 3% метанол/хлороформом. Промежуточное соединение 2-(3-бромпропокси) индол растворили в ацетоне (400 мл), обработали безводным карбонатом калия (11 г, 0,80 моль) и перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали под вакуумом, а остаток обработали водой (200 мл) и экстрагировали хлороформом (2 ч 250 мл). Объединенные экстракты высушили (Na2SO4), концентрировали под вакуумом, а остаток подвергли хроматографии на силикагель с элюированием 5%-ным метанолом/хлороформом, получив указанное соединение (Е20) в виде бледно-желтого масла (3,1 г, 15%). Его превратили в его оксалатовую соль и кристаллизовали из ацетона, получив белое твердое вещество с температурой плавления 169-170oC.
Свободное основание: 1H NMR (CDCl3)
δ : 8,32 (d, 1H), 7,05 - 7,38 (m, 8H), 6,53 (t, 1H), 4,50 (t, 2H), 4,08 (t, 2H), 4,08 (t, 2H), 3,48 (s, 2H), 3,31 (t, 2H), 2,83 - 2,97 (m, 2H), 2,27 - 2,41 (m, 2H),1,54 - 2,06 (m, 5H), 1,25 - 1,45 (m, 2H).
Пример 21. N-(4-пиперидилметил)3,4-дигидро-2H-[1,3]оксазин[3,2-a]индол- 10 карбоксамид (Е21).
Перемешанную суспензию N -[(1-бензил-4-пиперидил)метил]3,4-дигидро-2H-[1,3] оксазино[3,2-а] индол- 10-карбоксамид-оксалатовой соли (Е20, 2,25 г, 0,0046 моль) в этаноле (100 мл) и ледяной уксусной кислоты (4 мл) гидрогенизировали над 10% Pd-C(0,8 г) при атмосферном давлении и температуре 45oC в течение 18 ч. Смесь профильтровали, фильтрат концентрировали под вакуумом. Большая часть продукта была в виде твердого вещества, который отфильтровали. Этот материал встряхивали с концентрированным раствором карбоната калия (50 мл) и хлороформом (50 мл) вместе с остатком от фильтрата. Смесь профильтровали, отделили слой хлороформа и высушили (Na2SO4), затем концентрировали в вакууме, получив указанное соединение в виде белого твердого вещества (1,52 г, 100%). Его кристаллизовали из хлороформа/ 60 -80 бензина, температура плавления 139 - 141oC.
1H NMR (CDCl3)
δ 8,32 (d, 1H), 7,03 - 7,30 (m, 3H), 6,53 (t, 1H), 4,48 (t, 2H), 4,05 (t, 2H), 3,30 (t, 2H), 3,02 - 3,15 (m, 2H), 2,52 - 2,70 (m, 2H), 2,27 - 2,40 (m, 2H), 1,65 - 1,90 (m, 4H), 1,10 - 1,30 (m, 2H).
MS(EI) M+ 313.
Пример 22. N-[(1-п гексил-4-пиперидил)метил]3,4-дигидро- 2H-[1,3]оксазино[3,2-a]индол-10-карбоксид (E22).
Раствор N-(4-пиперидилметил)3,4-дигидро-2H-[1,3] оксазино[3,2-a] индол- 10-карбоксамида (E21, 250 мг, 0,70 ммоль) в ацетоне (12 мл) обработали 1-бромгексаном (0,14 мл, 1,0 ммоль) и безводным карбонатом калия (280 мг, 2,0 ммоль) и перемешивали при комнатной температуре в течение 10 ч. Смесь концентрировали в вакууме, а остаток обработали 10%-ным раствором Na2CO3 и экстрагировали хлороформом. Экстракт сушили (Na2SO4),концентрировали в вакууме, а остаток подвергли хроматографии на силикагеле с элюированием 5% метанола/ хлороформа, получив желтое масло. Его пропустили через короткую пробку из основной окиси алюминия с элюированием этилацетатом, получив соединение (E22) в виде бесцветного масла (150 мг, 54%). Его превратили в его хлористоводородную соль и кристаллизовали из ацетона/эфира, получив белое твердое вещество с температурой плавления 170 - 171oC.
Свободное основание: 1HNMR (CDCl3)
δ : (d, 2H), 7,02 - 7,30 (m, 3H), 6,53 (t, 1H), 4,48 (t, 2H), 4,04 (t, 2H), 3,32(t, 2H), 2,90 - 3,00 (m, 2H), 2,25 - 2,38 (m, 4H), 1,83 - 1,96 (m, 2H), 1,20 - 1,81 (m, 13H), 0,88 (t, 3H)
MS (EI) M+397.
Пример 23. N-(1-циклогексилметил-4-пиперидил)метил 3,4-дигидро- 2H-[1,3] оксазин[3,2-a] индол-10 карбоксамид (E23). -(4-пиперидилметил) 3,4-дигидро-2H-[1,3] оксазин [3,2-a] индол-10-карбоксамид (E21) алкилировали циклогексилметилбромидом способом из примера 22, при этом продолжительность реакции составила 10 ч при комнатной температуре, после этого реакцию проводили в течение 8 ч при температуре флегмы. Указанное соединение (E23) получили в виде белого твердого вещества (31%), которое превратили в его хлористоводородную соль и кристаллизовали из ацетона/эфира в виде белого твердого вещества с температурой плавления 209-210oC
HCl соль: - 1HNMR (CD3OD)
δ : 8,03 - 8,09 (m, 1H), 7,20 - 7,28 (m, 1H), 7,10 - 7,17 (m, 2H), 4,60 (t, 2H), 4,15 (t, 2H), 3,53 - 3,65 (m, 2H), 3,36 (d, 2H), 2,85 - 3,05 (m, 4H), 2,30 - 2,43 (m, 2H), 1,50 - 2,07 (m, 11H), 1,18 - 1,46 (m, 3H), 0,95 - 1,13 (m, 2H).
MS (EI) M+ 409.
Пример 24. N-[(1-этил-4-пиперидил)метил] 3,4-дигидро- 2H-[1,3] оксазин[3,2-a]индол-10-карбоксамид (E24).
N-(4-пиперидилметил)3,4-дигидро-2H[1,3] оксазин[3,2-a]индол- 10-карбамид (E21) алкилировали йодистым этанолом согласно способу из примера 22. Указанное соединение получили в виде белого твердого вещества (27%), которое превратили в его хлористоводородную соль и кристаллизовали из ацетона/ этанола/эфира, получив твердое вещество белого цвета с температурой плавления 243-245oC.
Свободное основание: 1HNMR (CDCl3)
δ : (d, 1H), 7,05 - 7,23 (m, 3H), 6,55 (t, 1H), 4,52 (t, 2H), 4,07 (t, H), 3,33 (t, 2H), 2,90 - 3,02 (m, 2H), 2,30 - 2,40 (m, 4H), 1,55 - 1,98 (m, 5H), 1,25 - 1,45 (m, 2H)
MS (E1) M+ 341.
Пример 25. N-[(1-(2-метансульфонамидоэтил)- 4-пиперидил)метил]3,4-дигидро-2H-[1,3]оксазин[3,2-a]индол- 10-карбоксамид (E25).
Перемешанный раствор N-(4-пиперидил)3,4-дигидро- 2H-[1,3]оксазин[3,2-a] индол-10-карбоксиламида (E21, 220 мг, 0,70 ммоль) в ацетонитриле (8 мл) обработали диизопропилэтиламином (0,24 мл, 1,4 ммоль) и N-(2-бромэтил) метансульфонамидом (D14, 160 мг, 0,77 ммоль) и смесь нагревали под флегмой в течение 2,5 ч. Реакционную смесь концентрировали в вакууме и остаток подвергли хроматографии на силикагеле с элюированием дихлорметаном/метанолом /0,88 раствором аммиака (200 : 10 : 1).
Полученное бесцветное масло растворили в хлороформе (30 мл) и промыли водой (2х20 мл), затем высушили (Na2SO4) и концентрировали в вакууме. Остаток пропустили через короткую пробку из основной окиси алюминия с элюированием этилацетатом, получив указанное соединение в виде бесцветного масла (34 мг, 11%). Его превратили в его оксалатовую соль и кристаллизовали из ацетата, получив белое твердое вещество с температурой плавления 80 - 85oC.
Свободное основание: 1HNMR (CDCl3)
δ : 8,32 (d, 1H), 7,05 - 7,30 (m, 3H), 6,56 (t, 1H), 4,53 (t, 2H), 4,08 (t, 2H), 3,33 (t, 2H), 3,17 (t, 2H), 2,95 (s, 3H), 2,78 - 2,92 (m, 2H). 2,50 (t, 2H), 2,28 - 2,44 (m, 2H), 1,55 - 2,10 (m, 6H), 1,20 - 1,45 (m, 2H).
Пример 26. N-(eq-хинолизидин-2-илметил), 3,4-дигидро-2H-[1,3] - оксазин-[3,2-a]индол-10-карбоксамид (E26).
а)eq-хинолизидин-2-илметиламин (D12) подвергли реакции с индол-3-хлоридом карбоновой кислоты, применяя способ, раскрытый в примере 1b, для получения соединения N-(eq-хинолизидин -2-илметил)индол-3-карбоксамид в виде белого твердого вещества (55%).
1HNMR (CD3OD)
δ : 8,06 - 8,15 (m, 1H), 7,39 - 7,46 (m, 1H), 7,10 - 7,22 (m, 2H), 3,27 (d, 2H), 2,80 - 2,95 (m, 2H), 2,04 - 2,23 (m, 2H), 1,53 - 1,98 (m, 8H), 1,22 - 1,48 (m, 3H), 0,96 - 1,15 (m, 1H).
b). Перемешанную суспензию N-(eq-хинолизидин-2-илметил)индол- 3-карбоксамид (300 мг, 0,94 ммоль) в хлороформе (16 мл) обработали 3-бром-1-пропанолом (0,17 мл, 1,9 ммоль), затем N-хлорсукцинимидом (140 мг, 1,05 ммоль) и получили прозрачный раствор в течение 30 мин. После 2 ч смесь обработали IH HCl/эфиром (3 капли), окрасив ее в желтый цвет, затем, спустя 1,5 ч смесь обработали с излишком - 10%-ным раствором NaHCO3, слой хлороформа отделили, высушили (Na2SO4) и концентрировали в вакууме, получив желтое масло. Его растворили в ацетоне (20 мл), обработали безводным карбонатом калия (400 мг, 2,9 ммоль) и перемешивали при комнатной температуре в течение 24 ч, затем концентрировали в вакууме. Остаток обработали 10%-ным раствором Na2CO3 и экстрагировали хлороформом. Экстракт высушили (Na2SO4) и концентрировали, оставив желтое масло, которое подвергли хроматографии на силикагеле с элюированием 10%-ным метанолом/хлороформом. Масло пропустили через короткую трубку из основной окиси алюминия с элюированием этилацетатом, получив указанное соединение (E26) в виде бесцветного масла (110 мг, 32%). Его превратили в его хлористоводородную соль и кристаллизовали из метанола/эфира в виде белого твердого вещества с температурой плавления 243-247oC.
Свободное основание: - 1H NMR (CDCl3).
δ : 8,30 (d, 1H), 6,98 - 7,25 (m, 3H), 6,51 (t, 1H), 4,45 (t, 2H), 3,96 (t, 2H), 3,20 - 3,37 (m, 2H), 2,78 - 2,92 (m, 2H), 2,20 - 2,35 (m, 2H), 1,94 - 2,14 (m, 2H), 0,98 - 1,85 (m, 12H)
MS (Cl) MH+ 368.
Пример 27. (I-п бутил-4-пиперидил)метил 8-фтор-3,4- дигидро-2H-[1,3] оксазин[3,2-а]индол-10-карбоксилат (E27).
a) 5-фториндол-3-хлорид карбоновой кислоты подвергали реакции с (I-пбутил-4-пиперидил)метанолом (Д6), применяя способ из Примера 1a, получив (I-пбутил-4-пиперидил)метил 5-фториндо-3-карбоксилат в виде масла оранжевого цвета (30%), затем подвергли хроматографии на силикагеле с проявлением 10% этанолом/хлороформом.
1H NMR (CDCl3)
δ : 9,95 (br.s, 1H), 7,82 (s, 1H), 7,78 (dd, 1H), 7,33 (dd, 1H), 7,00 (dt, 1H), 4,22 (d, 2H), 3,00 - 3,15 (m, 2H), 2,33 - 2,47 (m, 2H), 1,95 - 2,10 (m, 2H), 1,75 - 1,93 (m, 3H), 1,22 - 1,65 (m, 6H), 0,92 (t, 3H).
b) (I-пбутил-4-пиперидин)метил-5-фториндол-3-карбоксилат подвергли реакции с-хлорсукцинимидом и 3-бром-1-пропанолом, затем с карбонатом калия в ацетоне, применяя способ из примера 36b, получив масло бледного цвета, которое подвергли хроматографии на силикагеле с проявлением 10% этанолом/хлороформом.
Указанное соединение (E27) получили в виде бледно-желтого масла (8%), которое превратили в его оксалатовую соль и получили твердое вещество с температурой плавления 118-119oC.
Свободное основание: - 1H NMR (CDCl3).
δ : 7,64 (dd, 1H), 7,04 (dd, 1H), 6,87 (dt, 1H), 4,55 (t, 2H), 4,20 (d, 2H), 4,10 (t, 2H), 2,96 - 3,10 (m, 2H), 2,28 - 2,47 (m, 2H), 1,77 - 2,14 (m, 5H), 1,25 - 1,65 (m, 6H), 0,92 (t, 3H)
MS (Cl) MH+ 389.
Пример 28. N-[I-пбутил-4-пиперидил)метил] 8-фтор-3,4- дигидро-2H-[1,3] оксазин[3,2-a]индол-10-карбоксамид (E28).
a)5-фториндол-3-хлорид карбоновой кислоты подвергли реакции с (I-пбутил-4-пиперидил)метиламином (D1a) согласно способу, раскрытому в Описании 1b, получив соединение N-[I-пбутил-4- пиперидил)метил]5-фториндол-3-карбоксамид в виде небелого твердого вещества (64%).
1H NMR (CD3OD)
δ : 7,92 (S, 7h), 7,78 (DD, 1h), 7,38 (dd, 1H), 6,95 (dt, 1H), 3,28 (d, 2H), 2,93 - 3,07 (m, 2H), 2,30 - 2,42 (m, 2H), 1,60 - 1,87 (m, 3H), 1,22 - 1,60 (m, 6H), 0,94 (t, 3H).
b) N-[(I-пбутил-4-пиперидил)метил]-5-фториндол-3- карбоксамид подвергли реакции с 3-бром-1-пропаном и N-хлорсукцинимидом, затем с карбонатом калия в ацетоне, применяя способ из примера 26b, при этом получили желтое масло, которое подвергли хроматографии на силикагеле с проявлением 20% этанолом/хлороформом, получив указанное соединение в виде бледно-желтого масла 8%). Его превратили в его хлористоводородную соль, которую получили в виде твердого вещества с температурой плавления 90oC.
Свободное основание: - 1H NMR (CDCl3)
δ : 7,98 (dd, 1H), 6,98 (dd, 1H), 6,83 (dt, 1H), 6,56 (t, 1H), 4,56 (t, 2H), 4,08 (t, 2H), 3,33 (t, 2H), 3,05 - 3,20 (m, 2H), 2,30 - 2,58 (m, 4H), 2,10 - 2,26 (m, 2H), 1,25 - 1,90 (m, 9H), 0,92 (t, 3H)
MS (Cl) MH+ 388.
Пример 29. (I-пбутил-4-пиперидил)метил-1-метил-1,2,3,4- тетрагидропиримид[1,2-a]индол-10-карбоксилат (E29).
Раствор (I-nбутил-4-пиперидил)метанола (Д6, 1,7 г, 0,010 моль) в сухом THF (20 мл) в атмосфере азота при температуре 10oC обработали 1,5 M метиллитием в эфире (2,7 мл, 0,004 моль) и перемешивали в течение 15 мин, затем добавили раствор метил-1-метил-1,2,3,4-тетрагидропиримид [1,2-a]индол-10-карбоксилата (Д11, 0,5 г, 0,002 моль) в THF (5 мл), и реакционную смесь нагревали с обратным холодильником в течение 24 ч. Смеси дали охладиться и затем ее обработали 10%-ным раствором Na2CO3 (50 мл) и экстрагировали этилацетатом (2x40 мл). Объединенные экстракты высушили (Na2SO4), концентрировали в вакууме, а остаток подвергли хроматографии на силикагеле с элюированием 2%-ным метанолом/хлороформом, при этом получили указанное соединение (E29) в виде бесцветного масла (0,58 г, 74%). Соединение превратили в его оксалатовую соль и рекристаллизовали из материала, получив белое твердое вещество с температурой плавления 186 - 187oC.
Свободное основание: - 1HMNR (CDCl3).
δ : 7,92 (d, 1H), 7,00 - 7,20 (m, 3H), 4,17 (d, 2H), 3,95 (t, 2H), 3,37 (t, 2H), 3,28 (s, 3H), 2,92 - 3,03 (m, 2H), 2,28 - 2,38 (m, 2H), 2,12 - 2,24 (m, 2H), 1,80 - 2,03 (m, 5H), 1,23 - 1,57 (m, 6H), 0,92 (t, 3H)
MS (E1) M+ 383.
Пример 30. (I-пбутил-4-пиперидил)метил-3-метилтиазол[3,2- a]индол-9-карбоксилат (E30).
Указанное соединение (E30) получили из 3-метилтиазол [3,2-a]индол-9-карбоновой кислоты (Д13) в соответствии со способом из примера 10. Сырой продукт очистили посредством хроматографии на силикагеле с элюированием хлороформом/метанолом (95:5), затем его пропустили через короткую пробку из основной окиси алюминия с элюированием эфиром, получив бледно-желтое масло (35%). Его превратили в его оксалатовую соль и кристаллизовали из метанола, получив белое твердое вещество с температурой плавления 224 - 226oC.
Свободное основание: - 1H MNR (CDCl3)
δ : 8,18 (d, 1H), 7,77 (d, 1H), 7,14 - 7,42 (m, 2H), 6,40 (s, 1H), 4,25 (d, 2H), 2,92 - 3,08 (m, 2H), 2,73 (s, 3H), 2,28 - 222,40 (m, 2H), 1,75 - 2,05 (m, 5H), 1,20 - 1,62 (m, 6H), 0,92 (t, 3H)
MS (Cl) MH+ 385.
Пример 31. (I-пбутил-4-пиперидил)метил-2,3-дигидротиазол [3,2-a]индол-9-карбоксилат (E31).
Указанное соединение получили из 2,3-дигидротиазол[2,3-a]индол- 9-карбоновой кислоты (Д15) согласно способу из примера 10. Сырой продукт очистили посредством хроматографии на силикагеле с элюированием 5%-ным метанолом/хлороформом, получив желтое масло. Его пропустили через пробку из основной окиси алюминия с элюированием этилацетатом, получив указанное соединение в виде бледно-желтого масла (31%), которое превратили в его оксалатовую соль и кристаллизовали из ацетона, получив небелое твердое вещество с температурой плавления 212 - 215oC.
Свободное основание: - 1H MNR (CDCl3)
δ : 7,98 (d, 1H), 7,09 - 7,26 (m, 3H), 4,29 (t, 2H), 4,20 (d, 2H), 3,80 (t, 2H), 2,94 - 3,06 (m, 2H), 2,30 - 2,40 (m, 2H), 1,73 - 2,06 (m, 5H), 1,24 - 1,60 (m, 6H), 0,92 (t, 3H).
Пример 32. (I-пбутил-4-пиперидил)метилиазол[3,2-a]индол- 9-карбоксилат E32).
Указанное соединение (E32) получили из тиазол[3,2-a]индол-9- карбоновой кислоты (Д16), применяя способ из примера 10.Сырой продукт очистили посредством хроматографии на силикагеле с элюированием 3% метанолом/хлороформом, получив бледно-пурпуровое твердое вещество (70%). Его превратили в его оксалатовую соль и рекристаллизовали из метанола, получив бледно-голубое твердое вещество с температурой плавления 217 - 218oC.
Свободное основание: 1HNMR (CDCl3)
δ : 8,18 (d, 1H), 7,79 (d, 1H), 7,65 (d, 1H), 7,33 - 7,43 (m, 1H), 7,20 - 7,30 (m, 1H), 6,91 (d, 1H), 4,27 (d, 2H), 2,95 - 3,07 (m, 2H), 2,30 - 2,40 (m, 2H), 1,79 - 2,08 (m, 5H), 1,40 - 1,62 (m, 4H), 1,33 (секстет, 2H), 0,92 (t, 3H).
Пример 33. (1-пбутил-4-пиперидил)метил- 2,4-диметилпиримид[1,2-а] индол-10-карбоксилат (E33).
Указанное соединение (E33) получили из метил 2,4-диметил-пиримид[1,2-а] индол-10-карбоксилата (D17), применяя способ из примера 29. Сырой продукт очистили посредством хроматографии на силикагеле с элюированием этилацетатом, получив оранжевое масло (21%). Его превратили в оксалатовую соль, при этом получили оранжевое твердое вещество с температурой плавления 195 - 198oC.
Оксалатовая соль: - 1H NMR (d6 DMSO).
δ : 8,45 (d, 1H), 8,35 (d, 1H), 7,59 (t, 1H), 7,41 (t, 1H), 6,97 (s, 1H), 4,90 (brs, 2H). 4,27 (d, 2H), 3,38 - 3,60 (m, 2H), 3,14 (s, 3H), 3,27 - 3,04 (m, 4H), 2,61 (s, 3H), 2,01 - 2,27 (m, 3H), 1,55 - 1,84 (m, 4H), 1,37 (секстет, 2H), 0,97 (t, 3H).
Пример 34. N-[(1-п бутил-4-пиперидил)метил]2,3- дигидротиазол[2,3-а]индол-9-карбоксамид.
Указанное соединение (E34) получили из 2:3-дигидротиазол [3,2-а]индол-9-карбоновой кислоты (D15) через хлорид этой кислоты, применяя способ из примера 13. Сырой продукт очистили посредством хроматографии, получив желтое твердое вещество (63%). Его превратили в его оксалатовую соль и рекристаллизовали из ацетона, получив твердое вещество с температурой плавления 203 - 204oC.
Оксалатовая соль: - 1H NMR (d6 DMSO)
δ : 7,83 - 7,92 (m, 1H), 7,33 - 7,45 (m, 2H), 7,08 - 7,18 (m, 1H), 4,35 (t, 2H), 3,84 (t, 2H), 3,35 - 3,50 (m, 2H), 3,18 - 3,30 (m, 2H), 2,75 0 3,05 (m, 4H), 1,75 - 1,95 (m, 3H), 1,40 = 1,70 (m, 4H), 1,30 (секстет, 2H), 0,88 (t, 3H).
Пример 35. N-[(1-п бутил-4-пиперидил)метил]диазол[3,2-а]индол-9-карбоксамид (E35).
Указанное соединение (E35) получили из тиазол [3,2-а]индол-9-карбоновой кислоты (D16) через хлорид этой кислоты, применяя способ из примера 13. Сырой продукт очистили посредством хроматографии на силикагеле с элюированием 5% метанолом/хлороформом, получив твердое вещество пурпурного цвета (73%). Его превратили в его оксалатовую соль и рекристаллизовали из ацетона, получив пурпуровое твердое вещество с температурой плавления 205 - 207oC.
Оксалатовая соль: -1HNMR (d6 DMSO)
δ : 8,49 (d, 1H), 8,14 (d, 1H), 8,05 (d, 1H), 7,54 (t, 1H), 7,20 - 7,40 (m, 3H), 3,38 - 3,50 (m, 2H), 3,24 - 3,35 (m, 2H), 2,75 - 3,05 (m, 4H), 1,80 - 2,00 (m, 3H), 1,40 - 1,70 (m, 4H), 1,30 (секстет, 2H), 0,88 (t, 3H).
Пример 36. (1-пбутил-4-пиперидил)метил1,2,3,4,-тетрагидропиримид [1,2-а] индол-10-карбаксилал (E36).
Указанное соединение (E36) получили из метил 1,2,3,4-тетрагидропиримид[1,2-а] индол-10-карбоксилата (D21) согласно способу из примера 29, при этом время нахождения под флегмой составило 140 ч. Сырой продукт очистили посредством хроматографии на силикагеле с элюированием сначала этилацетатом, затем 10% метанолом/этилацетатом, получив желтое твердое вещество. Его присутствие через пробку из основной окиси алюминия, при этом элюирование осуществляли этилацетатом, получив указанное соединение в виде твердого вещества (23%), которое превратили в его оксалатовую соль и кристаллизовали из ацетона, получив твердое вещество с температурой плавления 190 - 194oC.
Свободное основание: 1H NMR (CDCl3)
δ : 7,71 (brd, 1H), 6,98 - 7,18 (m, 3H), 7,0 (brs, 1H), 4,17 (d, 2H), 3,98 (t, 2H), 3,46 - 3,57 (m, 2H), 2,92 - 3,06 (m, 2H), 2,30 - 2,40 (m, 2H), 2,22 (квинтет, 2H), 1,75 - 2,09 (m, 5H), 1,23 - 1,60 (m, 6H), 0,92 (t, 3H).
Пример 37. eq-хинолизин-2 илметил 2,3-дигидрооксазол[3,2-а]индол- 9-карбоксилат (E37).
Перемешанную суспензию eq - хинолилин-2-илметил 1H-индол-3-карбоксилата (E2a, 280 мг, 0,94 ммоль) в хлороформе (10 мл) обработали 2-бромэтанолом (0,13 мл), затем N-хлороукцинимидом (135 мг, 1,0 ммоль) и поддерживали 2 ч при комнатной температуре. Затем смесь обработали 1M HCl в простом эфире (0,05 мл, 005 ммоль) и после 2 ч у полученного желтого раствора повысили основность посредством добавления 10%- ного раствора Na2CO3 (10 мл) и экстрагировали хлороформом (2 x 15 мл). Объединенные экстракты высушили (Na2SO4) и концентрировали в вакууме, получив оранжевое масло. Его растворили в ацетоне (20 мл), обработали безводным карбонатом калия (410 мг, 3,0 ммоль) и перемешивали при комнатной температуре в течение 22 ч,затем концентрирования в вакууме, а остаток обработали 10%-ным раствором Na2CO3 (20 мл) и экстрагировали этилацетатом (2 x 20 мл). Объединенные экстракты высушили (Na2SO4), концентрировали в вакууме, а остаток подвергли хроматографии на силикагеле с элюированием 3% метанолом/хлороформом. Полученное желтое масло (145 мг, 44%) пропустили через пробку из основной окиси алюминия с элюированием этилацетатом, получив указанное соединение (E37), которое кристаллизовали в виде белого твердого вещества из этилацетата/эфира с температурой плавления 153 - 155oC.
1H NMR (CDCl3)
δ : 7,95 (d, 1H), 7,00 - 7,25 (m, 3H), 5,14 (t, 2H), 4,10 (t, 2H), 4,15 (d, 2H), 2,73 - 2,98 (m, 2H), 1,02 - 2,18 (m, 14H).
Пример 38. N-[(1-п бутил-4-пиперидил)метил]2,3,4.5-тетрагидро[1,3]оксазепин[3,2-а]индол- 11-карбоксамид (E38).
а) Перемешанную суспензию N-[1-п бутил-4-пиперидил)метил]индол-3-карбоксамид (D1b, 1,0 г, 0,0032 моль) в хлороформе (25 мл) обработали 4-хлорюутанолом (0,69 мл, 0,0064 моль), затем N-хлорсукцинимидом (470 мг, 0,0035 моль) и в пределах 5 мин получили желтый раствор. По истечении дополнительных 40 мин было отмечено, что раствор потемнел до оранжевого цвета. Смесь поддерживали при комнатной температуре в течение 1 ч, затем обработали 10%-раствором Na2CO3 (30 мл) и экстрагировали хлороформом (2 x 30 мл). Объединенные экстракты высушили (Na2SO4) и концентрировали в вакууме, получив оранжевое масло, которое подвергали хроматографии на силикагеле с элюированием 5% метанолом/хлороформом, получив N-[(1-п бутил-4-пиперидил)метил] 2-(4-хлорбутокси)индол-3-карбоксамид (0,67 г, 50%) в виде желтого масла.
1H NMR (CDCl3)
δ : 10,7 (brs, 1H), 8,23 (d, 1H), 7,00 - 7,32 (m, 3H), 6,88 (t, 1H), 4,43 (t, 2H), 3,48 (t, 2H), 3,34 (t, 2H), 2,86 - 3,02 (m, 2H), 2,25 - 2,40 (m, 2H), 1,18 0 2,00 (m, 15H), 0,90 (t, 3H).
b) Раствор N-[(1-п бутил-4-пиперидил)метил]2-(4-хлорбутокси]индол-3-карбоксамида (0,67 г, 0,0016 моль) в ацетоне (25 мл) обработали безводным карбонатом калия (0,74 г, 0,0054 моль) и йодистым натрием (1,34 г, 0,0089 моль) и нагревали с обратным холодильником в течение 24 ч. Смесь концентрировали в вакууме, а остаток обработали 10%-ным раствором Na2CO3 (25 мл) и экстрагировали хлороформом (2 x 30 мл). Объединенные экстракты высушили (Na2SO4), концентрировали в вакууме, а остаток подвергли хроматографии на силикагеле с элюированием 5% метанолом/хлороформом. Полученное бесцветное масло пропустили через пробку из основной окиси алюминия с элюированием этилацетатом, получив указанное соединение (Е38) в виде белого твердого вещества (370 мг, 60%). Его превратили в его оксалатовую соль и кристаллизовали из ацетона в виде белого твердого тела с температурой плавления 210-211oC.
Свободное основание: - 1HNMR (CDCl3)
δ : 8,36-8,44 (m, IH), 7,17-7,25 (m, 3H), 6,94 (t, IH), 4?30 (t, 2H), 4,11-4,20 (m, 2H), 3,35 (t, 2H), 2,90-3,00 (m, 2H), 2,25-2,35 (m, 2H), 2,18 (квинтет, 2H), 1,55-2,02 (m, 7H), 1,23-1,55 (m, 6H), 0,92 (t, 3H).
Пример 39. (I-п бутил-4-пиперидил)метил пиримид[1,2-а]индол-10-карбоксилат (Е39).
Указанное соединение получили из метил-пиримид [1,2-а] индол-10-карбоксилата (Д19), применяя способ из примера 29. Сырой продукт промыли при -78oC п-пентаном, а остаток подвергли хроматографии на силикагеле с элюированием 5% метанолом/хлороформом, получив оранжевое масло.
1HNMR (CDCl3)
δ : 8,68-8,78 (m, 2H), 8,45 (d, IH), 7,87 (d, IH), 7,59 (t, IH), 6,77-6,89 (m, IH), 4,37 (dd, 2H), 2,90-3,12 (m, 2H), 2,25-2,48 (m, 2H), 1,75-2,13 (m, 5H), 1,19-1,70 (m, 6H), 0,92 (t, 3H).
Также приготовили следующие соединения:
eq-хинолизидин-2-илметил 2,3-дигидротиазол[3,2-а] индол-9-карбоксилат (Е40).
Соединение 2,3-дигидротиазол[3,2-а]индол-9-карбоновая кислота превратили в хлорид его кислоты и подвергли реакции с eq-2-гидрометилхинолизидином, применяя способ, аналогичный способу, описанному в примере 10.
Свободное основание: - 1HNMR (CDCl3)
δ : 8,00 (d, IH), 7,15-7,30 (m, 3H), 4,34 (t, 2H), 4,10-4,25 (m, 2H), 3,87 (t, 2H), 2,80-3,00 (m, 2H), 1,05-2,20 (m, 14H).
eq-хинолизидин-2-илметил 2,3-дигидротиазол[3,2-а] индол- 9-карбоксамил (Е41).
Соединение 2,3-дигидротиазол [3,2-а]индол-9-карбоновая кислота превратили в хлорид его кислоты и подвергли реакции с eq -хинолизидин-2-илметиламином (Д13), применяя способ, аналогичный способу, раскрытому в описании Ib.
eq-хинолизидин-2-илметил[тиазол[3,2-а]индол-9-карбоксилат (Е42).
Тиазол[3,2-а] индол-9-карбоновая кислота превратили в хлорид его кислоты и подвергли реакции с eq-2-гидрометилхинолизином, применяя способ, аналогичный способу, описанному в примере 10, получив указанные соединения в виде белого твердого вещества с температурой 129-131oC (простой эфир).
1HNMR (CDCl3)
δ : 8,16 (d, IH), 7,75 (d, IH), 7,61 (d, IH), 7,33-7,42 (m, IH), 7,19-7,30 (m, IH). 6,87 (d, IH), 4,15-4,32 (m, 2H), 2,80-300 (m, 2H), 1,40-2,18 (m, IH), 1,08-1,40 (m, 3H).
eq-хинолизидин-2-илметил тиазол[3,2-а]индол-9-карбоксамид (Е43).
Соединение тиазол [3,2-а]индол-9-карбоновая кислота превратили в хлорид его кислоты и подвергли реакции с eq-хинолизидин-2-илметиламином, применяя способ, аналогичный способу, раскрытому в описании Ib.
eq-хинолизидин-2-илметил 3,4-дигидро- 2H-[1,3]тиазино[3,2-а]индол-10-карбоксилит (Е44).
3,4-дигидро-2H-[1,3] тиазин[3,2-а]индол-1-карбоновая кислота получили из тиоксиндола, применяя способ, аналогичный способу, раскрытому в описании 15. Его превратили в хлорид его кислоты и подвергли с eq-2-оксиметилхинолизидином, применяя способ, аналогичный способу, описанному в примере 10. Получили оксалатовую соль с температурой плавления 130-132oC.
Свободное основание: -1HNMR (CDCl3).
δ : 7,96-8,04 (m, IH), 7,13-7,30 (m, 3H), 4,05-4,30 (m, 4H), 2,90-3,20 (m, 4H), 2,35-2,51 (m, 2H), 1,20-2,32 (m, 14H).
(I-п бутил-4-пиперидил)метил пиримид[1,2-а]индол-10-карбоксамил (Е45).
а) Применяя способ, аналогичный способу, раскрытому в описании 19, приготовили бензил-пиримид [1,2-а]индол-10-карбоксилит, который затем гидрогенизировали на 10% PD/C в этаноле, получив пиримид [1,2-а]индол-1-карбоновая кислота.
b) Соединение пиримид [1,2-а]индол-10-карбоновая кислота превратили в хлорид его кислоты и подвергли реакции (I-пбутил-4-пиперидил)метиламином, применяя способ, раскрытый в Описании Ib. (I-пбутил-4-пиперидил) метил 1,2,3,4-тетрагидропиримид[1,2-а]индол-10-карбоксамид (Е46).
а) 2-хлориндол-3-карбоновая кислота (Л.Марчетти и А.Андреани, Ann Chim. (Рим), 1973, 63, 681) превратили в хлорид его кислоты и подвергли реакции с N-(I-пбутил-4-пиперидил)метил-амином (ДI), применяя способ из Описания Ib, получили N-1[(I-пбутил-4-пиперидил)метил]2-хлориндол-3-карбоксамид.
b) N-[(I-бутил-4-пиперидил)метил] 2-хлоридол-3-карбоксамил подвергли реакции с 3-хлорпропиламином, применяя способ, аналогичный способу, раскрытому в описании 18.
Пример 40. Получение N-[(1-(3-феноксипропил)-4-пиперидинил)метил]3, 4-дигидро-2H-[1,3]оксазино[3,2-а]индол-10-карбоксамида. (Соединение Е47).
а) Раствор изонипекотамида (30,1 г, 0,23 моль) и бензилбромида (27,9 мл, 0,23 моль) в этаноле (250 мл) обрабатывают при перемешивании безводным карбонатом калия (64,9 г, 0,47 моль) и кипятят с обратным холодильником в течение 3 ч. Смесь оставляют охлаждаться, затем фильтруют и фильтрат концентрируют в вакууме. Оставшееся масло растворяют в хлороформе (200 мл) и промывают водой (1 X 150 мл), затем сушат (Na2SO4) и концентрируют в вакууме, получают желтое твердое вещество (41,0 г). Это твердое вещество тщательно смешивают с пентаоксидом фосфора (38,3 г, 0,27 моль) и смесь выдерживают при 180oC в атмосфере азота в течение 2,5 ч при мягком перемешивании. Реакционную смесь оставляют охлаждаться, затем обрабатывают водой (300 мл). После растворения твердой массы раствор подщелачивают путем добавления твердого K2CO3 и экстрагируют этилацетатом (2 x 250 мл). Объединенные экстракты сушат (Na2SO4) и концентрируют в вакууме, получают коричневое масло (35,5 г). Масло растворяют в сухом эфире (250 мл) и добавляют по каплям в течение 30 мин при 0oC в атмосфере азота к перемешиваемой суспензии алюмогидрида лития (10,1 г, 0,26 моль) в эфире (150 мл). После завершения добавления смеси дают нагреться до комнатной температуры и перемешивают в течение 1,5 ч. Смесь повторно охлаждают до 0oC и осторожно обрабатывают водой (10 мл), 10%-ным раствором NaOH (15 мл) и снова водой (25 мл). Смесь фильтруют через кизельгур и фильтрат концентрируют в вакууме, получают коричневое масло, которое перегоняют в вакууме, после перегонки получают (1-бензил-4-пиперединил)метиламин в виде бесцветного масла (27,8 г, 67%), т.кип. 106oC при 0,25 мм рт.ст.
Спектр ПМР (CDCl3, м.д.) 7.20-7.37 (м, 5H), 3,48 (с, 2H), 2,85-2,95 (м, 2H), 2,55 (д, 2H), 1,87-2,00 (м, 2H), 1,60-1,75 (м, 2H), 1,10-1,40 (м, 5H).
б) К раствору индол-3-карбоновой кислоты (15 г, 0,093 моль) в дихлорметане (250 мл) при перемешивании в атмосфере азота добавляют оксалилхлорид (8,7 мл, 0,10 моль) и сухой диметилформамид (6 капель). Через 2 ч растворитель упаривают при пониженном давлении. Оставшийся хлорид кислоты (0,093 моля) растворяют в дихлорметане (100 мл) и добавляют по каплям к перемешиваемому раствору N-(1-бензил-4-пиперидинил)метиламина (D1, 16,4 г, 0,093 моль) и триэтиламина (15,5 мл, 0,11 моль) в дихлорметане (150 мл) при 5oC. После перемешивания при комнатной температуре в течение ночи реакционную смесь промывают 10%-ным Na2CO3 и органическую фазу сушат (Na2SO4). Растворитель упаривают при пониженном давлении и оставшееся твердое вещество перекристаллизовывают из этилацетата, получают N-[(1-бензил-4-пиперидинил)метил]индол-3-карбоксамид в виде белого твердого вещества (17,5 г, 60%).
Спектр ПМР (CDCl3, м.д.) 9,90 (с, 1H), 7,85-7,95 (м, 1H), 7,64 (д, 1H), 7,15-7,43 (м, 8H), 6,17 (т, 1H), 3,84 (с, 2H), 3,37 (т, 2H), 2,83-2,98 (м, 2H), 1,87-2,08 (м, 2H), 1,54-1,82 (м, 3H), 1,23-1,50 (м, 2H).
в) Суспензию N-[(1-бензил-4-пиперидинил)метил] индол-3- карбоксамида (17,5 г, 0,050 моля) в хлороформе (250 мл) обрабатывают при перемешивании и при комнатной температуре 3-бром-1-пропанолом (10,1 мл, 0,11 моль) и N-хлорсукцинимидом (8,7 г, 0,065 моля) и через 15 мин получают прозрачный раствор. Через 1 ч реакционная смесь меняет цвет от светло-желтого до оранжевого, а температура повышается до 38oC. Через 1 ч реакционную смесь обрабатывают 10%-ным раствором Na2CO3, слой хлороформа отделяют, сушат (Na2SO4) и концентрируют в вакууме, получают желтое масло, которое хроматографируют на силикагеле, элюент 3% метанол/хлороформ. Промежуточный 2-(3-бромпропокси)индол растворяют в ацетоне (400 мл), обрабатывают безводным карбонатом калия (11 г, 0,08 моль) и перемешивают при комнатной температуре в течение 20 ч. Реакционную смесь концентрируют в вакууме и остаток обрабатывают водой (200 мл) и экстрагируют хлороформом (2 х 250 мл). Объединенные экстракты сушат (Na2SO4), концентрируют в вакууме и остаток хроматографируют на силикагеле, элюент 5% метанол/хлороформ, получают N-[(1-бензил-4-пиперидинил)-метил] -3,4-дигидро-2H-[1,3] оксазино[3,2-a]индол-10-карбоксамид в виде светло-желтого (3,1 г, 15%). Это соединение переводят в соль щавелевой кислоты и кристаллизуют из ацетона в виде белого твердого вещества, т.пл. 169-170oC.
Спектр ПМР (свободного основания) (CDCl3, м.д.) 8,32 (д, 1H), 7,05-7,38 (м, 8H), 6,53 (т, 1H), 4,50 (т, 2H), 4,08 (т, 2H), 3,48 (с, 2H), 3,31 (т, 2H), 2,83-2,97 (м, 2H), 2,27-2,41 (м, 2H), 1,54-2,06 (м, 5H), 1,25-1,45 (м, 2H).
г) Суспензию оксалата N-[(1-бензил-4-пиперидинил)метил]-3,4- дигидро-2H-[1,3] оксазино3,2-а индол-10-кабоксамида (2,25 г, 0,0046 моль) в этаноле (100 мл) и ледяной уксусной кислоте (4 мл) при перемешивании гидрируют над 10% Pd-C (0,8 г) при атмосферном давлении и 45oC в течение 18 ч. Смесь фильтруют и фильтрат концентрируют в вакууме. Большая часть продукта находится в твердом состоянии, ее отфильтровывают. Этот продукт вместе с оставшимся фильтратом встряхивают с концентрированным раствором карбоната калия (50 мл) и хлороформом (50 мл). Смесь фильтруют, слой хлороформа отделяют и сушат (Na2SO4), затем концентрируют в вакууме, получают N-(4-пиперидинилметил)-3,4-дигидро-2H-[1,3]оксазино[3,2-a]индол-10- карбоксамид (E21) в виде белого твердого вещества (1,52 г, 100%). Его перекристаллизовывают из смеси хлороформ/60-80 бензин, т.пл. 139-141oC.
Спектр ПМР (свободного основания) (CDCl3, м.д.): 8,32 (д, 1H), 7,03-7,30 (м, 3H), 6,53 (т, 1H), 4,48 (т, 2H), 4,05 (т, 2H), 3,30 (т, 2H),02-3,15 (м, 2H), 2,52-2,70 (м, 2H), 2,27-2,40 (м, 2H), 1,65-1,90 (м, 4H), 1,10-1,30 (м, 2H).
д) Раствор N-(4-пиперидинилметил)-3,4-дигидро-2H-[1,3]оксазино [3,2-a] индол-10-карбоксамида (E21) (250 мг, 0,80 ммоль) и триэтиламина (0,25 мл, 1,8 ммоль) в смеси ацетонитрила (15 мл) и N,N-диметилформамида (10 мл) при перемешивании обрабатывают 3-феноксипропилбромидом (0,13 мл, 0,88 ммоль) и раствор кипятят с обратным холодильником в течение 48 ч. Смесь оставляют охлаждаться, концентрируют в вакууме и остаток растворяют в этилацетате (25 мл) и промывают водой (20 мл). Органический раствор сушат (MgSO4), концентрируют в вакууме и остаток очищают быстрой хроматографией на силикагеле, элюент 0-20% метанол/этилацетат, после растирания с эфиром получают N-[(1-феноксипропил)-4-пиперидинил)метил] 3,4-дигидро-2H-[1,3] оксазино [3,2-a]индол-10-карбоксамид (E47, 44 мг) в виде светло-розового твердого вещества, т. пл. 120-126oC.
Спектр ПМР (CDCl3, м.д.) 8,30 (д, 1H), 7,0-7,37 (м, 5H), 6,96 (т, 1H), 6,88 (д, 2H), 6,64 (т, 1H), 4,57 (т, 2H), 4,13 (т, 2H), 4,30 (т, 2H), 3,22-3,43 (м, 4H), 2,78-3,00 (м, 2H), 2,13-2,57 (м, 6H), 1,59-2,03 (м, 5H).
Описания.
Описание 1.Промежуточные соединения для примеров 3, 13, 14, 19 и 28.
а) N-(I-nбутил-4-пиперидил)метиламин).
Перемешанный раствор изонипекотамида (70 г, 0,55 моль) и 1-бромбутана (58,8 мл, 0,55 моль) в этаноле (700 мл) обработали безводным карбонатом калия (152 г, 1,10 моль) и нагревали с обратным холодильником в течение 3 ч. Смеси позволили охладиться, затем профильтровали, и фильтрат концентрировали под вакуумом. Остаточное масло растворили в хлороформе (400 мл) и промыли водой (1 х 300 мг), затем высушили (Na2SO4) и концентрировали под вакуумом, получив желтое масло (77,5 г). Это масло тщательно смешали с фосфорным ангидридом (75 г) и смесь нагревали при 160-180oC в атмосфере азота в течение 2,5 ч при плавном перемешивании. Реакционной смеси позволили охладиться, затем обработали водой (500 мл). Когда твердую массу растворили, основность раствора повысили путем добавки твердого K2CO3 и затем его экстрагировали этилацетатом (2 х 400 мл). Объединенные экстракты высушили (Na2SO4) и концентрировали в вакууме, получив коричневое масло (78 г). Его растворили в сухом эфире (400 мл) и в течение 30 мин добавляли по капле в перемешанную суспензию в алюмогидрид лития (25 г, 0,66 моль) в эфире (200 мл) при 0oC в атмосфере азота. После завершения добавки смеси позволили нагреться до комнатной температуры и перемешивали в течение 18 ч. Ее снова охладили до 0oC и тщательно обработали водой (25 мл), 10%-ным раствором NaOH (25 мл) и снова водой (75 мл). Смесь профильтровали через инфузорную землю и фильтрат концентрировали в вакууме, получив коричневое масло, которое подвергли перегонке под вакуумом, получив указанное соединение в виде бесцветного масла (66 г, 71%) с температурой кипения 96 - 99oC под давлением 3 мм рт. ст.
1H MNR (CDCl3)
δ : 2,90-3,02 (m, 2H), 2,58 (d, 2H), 2,25-2,38 (m, 2H), 1,65-2,00 (m, 4H), 1,08-1,58 (m, 9H), 0,92 (t, 3H).
b) N-[(I-nбутил-4-пиперидил)метил]индол-3-карбоксамид.
В перемешанный раствор индол-3-карбоновая кислота (1 г) в дихлорметане (20 мл) при 0oC под азотом добавили хлорангидрид щавелевой кислоты (0,81 мл) и сухой диметилформамид (3 капли). Через 3 ч растворители испарили при уменьшенном давлении. Часть остаточного кислого хлорида (420 мг) растворили в дихлорметане (12 мл) и добавили по капле в раствор N-(I-пбутил-4-пиперидил/метиламина (400 мг) в дихлорметане (12 мл) с последующей обработкой триэтиламином (0,36 мл). После перемешивания при температуре окружающей среды в течение ночи. Реакционную смесь промыли насыщенным NaHCO3 и органическую фазу высушили (Na2SO4). Растворитель испарили под уменьшенным давлением, а остаток рекристаллизовали из этилацетата, получив указанное соединение (Д1) (467 мг, 64%).
1HMNR (CDCl3) 250 MHz
δ : 9,29 (Brs, 1H), 8,05 - 7,9 (m, 1H), 7,81 (d, 1H), 7,55 - 7,4 (m, 1H), 7,39 - 7,2 (m, 2H), 6,28 (brs, 1H), 3,39 (t, 2H), 3,0 (brd, 2H), 2,45-2,25 (m, 2H), 2,1-1,1 (m, 11H), 0,9 (t, 3H).
Описание 2. Промежуточное соединение для примера 5. N-[2-(1-пиперидинил)этил]1H-индол-3-карбоксамид.
1-пиперидин этиламин подвергли реакции с 1H-индол-3-хлоридом карбоновой кислоты, применяя способ, раскрытый в описании 1, при этом получили указанное соединение (Д2) в виде твердого вещества.
1H MNR (CDCl3)
δ : 9,90 (brs, 1H), 7,97 - 8,07 (m, 1H), 7,97-8,07 (m, 1H), 7,78 (d, 1H), 7,36 - 7,50 (m, 1H), 7,15-7,30 (m, 2H), 7,13-13 (br.t, NH), 3,55-3,68 (m, 2H), 2,60 (t, 2H), 2,80 - 2,55 (m, 4H), 1,40-1.73 (m, 6H).
Описание 3.Промежуточное соединение для примера 10.
a) Этил 2-аминофенилацетат.
Раствор этил 2-нитрофенилацетата (13,6 г, 0,065 моль) в этаноле (150 мл) гидрогенизировали на 10% Pd /с катализатора (1 г) при комнатной температуре и под давлением в течение 18 ч. Реакционную смесь профильтровали через инфузорную землю и концентрировали в вакууме, получив указанное соединение в виде прозрачного масла, которое затвердело при выдержке (10,8 г, 93%).
1H NMR (CDCl3)
δ : 7,05 - 7,15 (m, 2H), 66,68 - 6,8 (m, 2H), 4,13 (q, 2H), 4,05 (br.s, 2H), 3,55 (s, 2H), 1,25 (t, 3H).
b)Этил 2-(5-хлорвалериламино)фенилацетат.
Раствор этил-2-аминофенилацетата (5,60 г, 0,013 моль) и диизопропилэтиламина (7,08 мл, 0,042 моль) в сухом тетрагидрофуране (THF) (75 мл) обработали 5 хлорангидрид - хлорвалериановой кислоты (4,00 мл, 0,031 моль) и оставили для перемешивания в течение 1 ч. Реакционную смесь концентрировали в вакууме, а остаток растворили в этилацетате (200 мл) и промыли кислотой 1M HCl (100 мл), высушили (Na2SO4) и концентрировали в вакууме, получив твердое вещество. Его промыли п-пентаном/эфиром (1:1) и высушили, получив указанное соединение в виде светлого твердого вещества (8,1 г,91%).
1HNMR (CDCl3)
δ : 8,90 (br.s, 1H), 7,88 (d, 1H), 7,05-7,37 (m, 3H), 4,17 (q, 2H), 3,60 (s, 2H), 3,45-3,65 (m, 2H), 2,35-2,55 (m, 2H), 1,68-1,98 (m, 4H), 1,28 (t, 3H).
c)Этил 6,7,8,9-тетрагидропиридо[1,2-a]индол-10-карбоксилат.
Раствор этил 2-(5-хлорвалериламино)фенилацетата (8,10 г, 0,027 моль) в сухом THF (50 мл) добавили в перемешанную суспензию калий-бутилата (7,62 г, 0,068 моль) в сухом THF (200 мл) при комнатной температуре в атмосфере азота. Через 1 ч полученный пурпуровый раствор обработали водой (10 мл) и концентрировали в вакууме. Остаток взбалтывали с этилацетоном (200 мл) и насыщенным раствором хлористого аммония (150 мл), затем отделили органический слой (Na2SO4) и концентрировали в вакууме, получив оранжевое масло. Его подвергли хроматографии на силикагель с элюированием эфира, получив указанное соединение в виде желтого твердого вещества (1,25 г, 20%).
1H MNR (CDCl3)
δ : 8,07 - 8,17 (m, 1H), 7,13 - 7,30 (m, 3H), 4,38 (q, 2H), 4,00 (t, 2H), 3,30 (t, 2H), 1,82-2,12 (m, 4H), 1,43 (t, 3H).
d) 6,7,8,9-тетрагидропирид [1,2-a]-индол-10-карбоновая кислота.
Раствор этил 6,7,8,9-тетригидро-1H-пирид[1,2-a] индол-10- карбоксилата (1,20 г, 0,0047 моль) в этаноле (50 мл) и 10%-ный раствор NaOH (50 мл) нагревали под флегмой в течение 4 ч. Затем реакцию подкислили кислотой 1MHCl (50 мл). Органический слой отделили и экстрагировали 10%-раствором Na2CO3 (120 мл), затем водный раствор снова подкислили кислотой 5M HCl и экстрагировали в этилацетате (2 х 75 мл).Органические экстракты соединили,высушили (NaSO4) и концентрировали в вакууме, получив указанное соединение (Д3) в виде белого твердого вещества (400 мг, 40%).
1H NMR (CDCl3)
δ : 8,23 (d, 1H), 7,20 - 7,35 (m, 3H), 4,10 (t, 2H), 3,40 (t, 2H), 2,00 - 2,15 (m, 2H), 1,85 - 2,00 (m, 2H).
Описание 4.промежуточное соединение для примеров 11 и 13.
a)Этил 2-(4-хлорбутириламинофенилацетат.
Указанное соединение получили из этил 2-аминофенилацетата, применяя способ из описания 3b, и его изолировали в виде твердого вещества (100%).
1H MNR (CDCl3)
δ : 8,90 (br.s., 1H), 7,85 (d, 1H), 7,05 - 7,35 (m, 3H), 4,15 (q, 2H), 3,68 (t, 2H), 3,6 (s, 2H), 2,96 (t, 2H), 2,1 - 2,3 (m, 2H), 1,26 (t, 3H).
b)Этил 2,3-дигидро-1H-пирроло[1,2-a]индол-9-карбоксилат.
Указанное соединение получили из этил 2-(4-хлорбутириламино) фенилацетата согласно способу из описания 3c, и его изолировали в виде оранжевого масла, которое кристаллизовали во время выдержки (15%).
1H MNR (CDCl3)
δ : 8,05 - 8,15 (m, 1H), 7,15 - 7,30 (m, 3H), 4,35 (q, 2H), 4,06 (t, 2H), 3,28 (t, 2H), 2,55 - 2,72 (m, 2H), 1,40 (t, 3H).
c)2,3-дигидро-1H-пиррило[1,2-a]индол-9-карбоновая кислота.
Указанное соединение (Д4) получили из этил 2,3-дигидро-1H- пирроло[1,2-a] индол-9-карбоксилата, применяя способ из Описания 3d, затем его изолировали в виде небелого твердого вещества (42%).
1H MNR (d6DMSO)
δ : 11,85 (br.s, 1H), 7,90 - 8,02 (m, 1H), 7,32 - 7,47 (,, 1H), 7,10 - 7,25 (m, 2H), 4,15 (t, 2H), 3,20 (t, 2H), 2,50 - 2,70 (m, 2H).
Описание 5.Промежуточное соединение для примера 12.
a)Этил 2-(6-хлоргексаноиламино)фенилацетат.
Указанное соединение получили из этил 2-аминофенилацетата и 6-бромгексаноилхлорида согласно способу из описания 3в и его изолировали в виде твердого вещества (100%).
1H NMR (CDCl3)
δ : 8,90 (brs, 1H), 7,90 (d, 1H), 7,05 - 7,35 (m, 3H), 4,17 (q, 2H), 3,60 (s, 2H), 3,42 (t, 2H), 2,45 (t, 2H), 1,45 - 2,00 (m, 6H), 1,28 (t, 3H).
b)Этил 7,8,9,10-тетрагидро-6H-азепин [1,2-а]индол-11-карбоксилат.
Указанное соединение получили из этил 2-(6-хлоргексаноиламино)фенилацетата, применяя способ из описания 3c, затем его очистили посредством хроматографии на силикагеле с элюированием 60 - 80 бензином/эфиром (9: 1), получив более твердое вещество (16%).
1H NMR (CDCl3)
δ : 8,07 - 8,19 (m, 1H), 7,15 - 7,35 (m, 3H), 4,40 (q, 2H), 4,15 - 4,25 (m, 2H), 3,45 - 3,60 (m, 2H), 1,67 - 2,00 (m, 6H), 1,45 (t, 3H).
c)7,8,9,10-тетрагидро-6H-азепин[1,2-а]индол-11-карбоновая кислота.
Указанное соединение (D5) получили из этил 7,8,9,10-тетрагидро-6H-азепин[1,2-а] индол-11-карбоксилата посредством гидролиза с применением едкого натра согласно способу из описания 3d. После 4 ч нагрева с обратным холодильником смесь подкислили кислотой 5M HC, полученное белое твердое вещество отфильтровывали и высушили (82%).
1H NMR (d6DMSO)
δ : 12,05 (s, 1H), 7,95 - 8,04 (m, 1H), 7,48 - 7,60 (m, 1H), 7,05 - 7,20 (m, 2H), 4,24 - 4,36 (m, 2H), 3,38 - 3,53 (m, 2H), 1,54 - 1,90 (m, 6H).
Описание 6.Промежуточное соединение для примеров 1,10, 27 и 29)
(1-п бутил -4-пиперидил)метанол.
Смесь этилизонипекотата (102 г, 0,65 моль) и 1-бромбутана (72 мл, 0,67 моль) в этаноле (1,2 л) обработали безводным карбонатом калия (180 г, 1,3 моль) и нагревали с обратным холодильником в течение 2 ч. Смеси позволили охладиться и затем ее профильтровали через инфузорную землю. Фильтрат концентрировали в вакууме, получив желтое масло, которое растворили в эфире (300 мл) и добавляли по капле в течение 20 мин в перемешанную суспензию алюмогидрида лития (50 г, 1,3 моль) в эфире при 0 oC в атмосфере азота. Смесь перемешивали при комнатной температуре в течение 18 ч, затем ее охладили до 0oC и обработали водой (50 мл), 10%-ным раствором NaOH (50 мл) и водой (150 мл). Смесь профильтровали через инфузорию земли, а фильтрат концентрировали под вакуумом, получив бледно-желтое масло, которое подвергали перегонке и получили указанное соединение в виде бесцветного масла (88,5 г, 80%) с температурой кипения 102 - 108oC под давлением 0,1 мм Hg.
1H NMR (CDCl3)
δ : 3,48 (d, 2H), 2,88 - 3,03 (m, 2H), 2,25 - 2,38 (m, 2H), 2,10 (br.s, 1H), 1,66 - 2,00 (m, 4H),1,17 - 1,60 (m, 7H), 0,90 (t, 3H).
Описание 7.Промежуточное соединение для Примера 15.
(1-бензил-4-пиперидил)метанол.
Этилизонипектоат сначала алкилировали бромистым бензилом и продукт подвергали отгонке алюмогидридом лития в соответствии со способом из описания 6, получив указанное соединение (Д7) в виде бесцветного масла (100%).
1H NMR (CDCl3)
δ : 7,20 - 7,35 (m, 5H), 3,52 (s, 2H), 3,48 (d, 2H), 2,86 - 3,00 (m, 2H), 1,20 - 2,05 (m, 8H).
Описание 8.Промежуточное соединение для примеров 13 и 17.
6,7-дигидропиридо[1,2-а] индол-10-карбоксилата (Т.Тейтей и Л.К. Дальтон, Австралийский журнал "Chem", 1969, 22, 997) (1,0 г, 0,0044 моль) в метаноле (40 мл) обработали раствором едкого калия (3,0 г, 0,054 моль) в воде (50 мл) и нагревали с обратным холодильником в течение 3 ч. Раствору дали охладиться, затем его подкисляли кислотой HCl и экстрагировали этилацетатом. Экстракт высушили (Na2SO4) и концентрировали под вакуумом, получив указанное соединение (ДВ) в виде желтого твердого вещества (600 мг, 64%).
1H NMR (CDCl3)
δ : 8,18 - 8,22 (m, 1H), 7,50 (d, 1H), 7,20 - 7,35 (m, 3H), 6,27 - 6,38 (m, 1H), 4,15 (t, 2H), 2,62 - 2,78 (m, 2H).
Описание 9. Промежуточное соединение для примера 18.
Пиридо[1,2-е]индол-10-карбоновая кислота.
Указанное соединение (Д9) получили из метилпиридо[1,2-а]индол-10-карбокислота (Т. Тейтей и Л.К. Дальтон, Австралийский журнал "Chem", 1969, 22, 997) в соответствии со способом из описания 8, получил светло-желтое твердое вещество (76%).
1H NMR (CDCl3 + CD3OD)
δ : 8,56 (d, 1H), 8,34 - 8,46 (m, 2H), 7,93 (d, 1H), 7,32 - 7,57 (m, 3H), 6,87 (t, 1H).
Описание 10.Промежуточное соединение для примера 20.
(1-бензил-4-пиперидил)метиламин (Д10).
Сначала изонипекотамид алкилировали бромистым бензилом, затем амид дегидратировали пятиокисью фосфора, и полученный нитрил подвергали отгонке алюмогидридом лития в соответствии со способом из Описания а, получив указанное соединение в виде бесцветного масла после перегонки (67%) с температурой кипения 106oC под давлением 0,25 мм H.
1H NMR (CDCl3)
δ : 7,20 - 7,37 (m, 5H), 3,48 (s, 2H), 2,85 - 2,95 (m, 2H), 2,55 (d, 2H), 1,37 - 2,00 (m, 2H), 1,60 - 1,75 (m, 2H), 1,10 - 1,40 (m, 5H).
Описание 11.Промежуточное соединение для примера 29.
a)Метил 2-хлориндол-3-карбоксилат.
Перемешанную суспензию метилиндол-3-карбоксилата (6,0 г, 0,034 моль) в хлороформе (200 мл) обработали N-хлорсукцинимидом (5,04 г, 0,033 моль), получив в течение 15 мин прозрачный раствор. После 2 ч при комнатной температуре его обработали 1M HCl/эфиром (34 мл, 0,034 моль) и позволили ему перемещаться в течение дополнительного 1 ч, затем его обработали избыточным количеством 10%-ного раствора Na2CO3, отделили слой хлороформа, высушили (Na2SO4) и концентрировали в вакууме. Остаточное желтое вещество рекристаллизовали из хлороформа /60 - 50 бензина (петрол), получив указанное соединение (DIIa) в виде твердого вещества (3,4 г, 48%).
1H NMR (CDCl3/d6DMSO)
δ : 11,3 (br.s, 1H), 8,02 - 8,12 (m, 1H), 7,30 - 7,40 (m, 1H), 7,18 - 7,26 (m, 2H), 3,95 (s, 2H).
MS (E1) M+ 209 и 211
b) Метил-1-метил-1,2,3,4-тетрагидропиримид[1,2-а]индол-10-карбоксилат.
Раствор метил 2-хлориндол-3-карбоксилата (3,4 г, 0,016 моль) в сухом THF (70 мл) при 5oC в атмосфере азота обработали порциями гидридом натрия (430 мг 80% масляной дисперсии, 0,016 моль) и затем перемешивали при комнатной температуре в течение 30 мин. Полученный раствор обработали раствором 3,3-диметиламинопропилхлорида (0,020 моль_ в толуоле (30 мл) и нагревали с обратным холодильником в течение 48 ч, затем концентрировали в вакууме, а остаток обработали 10%-ным раствором Na2CO3 (50 мл) и экстрагировали этилацетатом (2 x 70 мл). Объединенные экстракты высушили (Na2SO4) и концентрировали в вакууме, получив желтое масло, которое подвергли хроматографии на силикагеле с элюированием эфиром /60 - 80 бензином (1:1). Указанное соединение (ДII) было получено в виде твердого вещества (1,95 г, 50%).
1H NMR (CDCl3)
δ : 7,92 (d, 1H), 6,97 - 7,19 (m, 3H), 3,92 (t, 2H), 3,88 (s, 3H), 3,36 (t, 2H), 3,27 (s, 3H), 2,10 - 2,22 (m,2H).
Описание 12.Промежуточное соединение для примера 26).
eg-хинозидин-2-илметиламин.
Перемешанную суспензию алюмогидрида лития (400 мг, 0,010 моль) в THF (тетрагидрофуран) (20 мл) при комнатной температуре в атмосфере азота обработали раствором eg-2-цианохинолизидином (Е.Копинака и др. Якугаку Дзасси, 1980, 100, 88) в THF (3 мл) и затем смесь нагревали с обратным холодильником в течение 20 мин. Смеси дали охладиться, затем обработали осторожно водой (0,4 мл), 10%-ным раствором NaOH и водой (1,2 мл). Полученную смесь профильтровали, а фильтрат концентрировали в вакууме. Остаток дистиллировали в устройстве Kugelrohr, получив указанное соединение (Д13) в виде бесцветного масла (700 мг, 97%).
1HNMR (CDCl3)
δ : 2,80 - 2,92 (m, 2H), 2,57 (d, 2H), 1,94 - 2,12 (m, 2H), 1,20 - 1,80 (m, 13H), 0,88 - 1,05 (m, 1H).
Описание 13.Промежуточное соединение для Примера 30).
3 - метилтиазол [3,2-а] индол - 9 - карбоновая кислота.
а) Перемешанный раствор 3-метилтиазол [3,2-а]индол (А.Киприянов и В.Хилиа, Zh "Орган. Хим". 2966, 2, 1474) (270 мг,0,0014 моль) в диметилформамиде (DMF) (3 мл) охладили до 5oC под азотом и обработали трифторуксусным ангидридом (0,23 мл, 0,0017 моль), затем дали ему нагреваться до комнатной температуры в течение 3 ч. Раствор вылили в воду (25 мл) и смесь экстрагировали этилацетатом (2 x 20 мл). Объединенные экстракты высушили (Na2SO4) и затем экстрагировали в вакууме, получив 3-метил-9-трифторацетилтиазол[3,2-а] индол (370 мг, 90%) в виде коричневого твердого вещества.
1HNMR (CDCl3)
δ : 8,10 (br.s, 1H), 7,85 (d, 1H), 7,39 - 7,47 (m, 1H), 7,25 - 7,35 (m, 1H), 6,69 (S, 1H), 2,83 (S, 3H).
b) 3-метил-9-трифторацетилтиазоло[3,2-а] индол (370 мг, 0,0013 моль) обработали 20%-ным раствором NaOH(15 мл) и этанолом (15 мл) и нагревали с обратным холодильником в течение 6 ч. Смесь концентрировали в вакууме до половины ее объема, остаток подкислили кислотой 2M HCl и затем экстрагировали этилацетатом (2 x 30 мл). Объединенные экстракты высушивали (Na2SO4) и концентрировали в вакууме, получив указанное соединение (D13) в виде коричневого твердого вещества (300 мг, 100%).
1HNMR (d6DMSO)
δ : 12,3 (v.br.s, IH), 7,93 - 8,08 (m, 2H), 7,16 - 7,40 (m, 2H), 6,95 (s, 1H), 2,59 (s, 3H).
Описание 14.Промежуточное соединение для примера 25).
N-(2- бромэтил)метансульфонамид.
Перемешанный раствор 2-бромэтиламин-бромгидрата (5,10 г, 0,025 моль) и триэтиламина (6,9 г, 0,050 моль) в дихлорметане (200 мл) при температуре ледяной ванны обработали по капле метансульфохлоридом (1,96 мл, 0,025 моль). Смеси позволили нагреться до комнатной температуры и ее перемешивали в течение 16 ч, затем промывали водой и кислотой 5 MHCl, высушили (Na2SO4) и концентрировали в вакууме, получив указанное соединение (Д14) в виде бесцветного масла, которое затвердело при выдержке, дав белое твердое вещество (3,5 г, 69%).
1HNMR (CDCl3)
δ : 4,92 (s, 1H), 3,62 - 3,48 (m, 4H), 3,05 (s, 3H).
Описание 15. Промежуточное соединение для примера 31.
а) 2,3-дигидротиазол [3,2-а]индол.
Раствор тиоксиндола (400 мг, 0,0027 моль) и 1,2-дибромэтана (0,24 мл, 0,0027 моль) в сухом THF (10 мл) добавляли в перемешанный раствор калий-t-бутилат (760 мг, 0,0068 моль) в сухом THF(тетрагидрофуран) (40 мл) при комнатной температуре в атмосфере азота. Смесь перемешивали в течение 3 ч, затем обработали водой (100 мл) и экстрагировали этилацетатом (2 x 70 мл.) Объединенные экстракты высушили (Na2SO4) и концентрировали в вакууме, получив оранжевое масло, которое подвергли хроматографии на силикагеле с элюированием 10% эфиром/60-80 бензином. Указанное соединение получили в виде белого твердого вещества (135 мг, 29%).
1HNMR (CDCl3)
δ : 7,42 - 7,23 (m, 1H), 7,00 - 7,25 (m, 3H), 6,20 (s, 1H), 4,23 (t, 2H), 3,79 (t, 2H).
в)2,3-дигидротиазол[3,2-а]индол-9-карбоновая кислота.
2,3-дигидротиазол[3,2-а] индол обработали трифторуксусным ангидридом в соответствии со способом из описания 13а, получив 9-трифторацетилтиазол [3,2-а]индол в виде твердого вещества пурпурового цвета (85%).
1HNMR (CDCl3)
δ : 7,93 (br.s, 1H), 7,07 - 7,30 (3H), 4,30 (t, 2H), 3,85 (t, 2H).
Указанное соединение (Д15b) получили из 9-трифторацетил-2,3-дигидротиазол[3,2-а]индола, применяя способ из Описания 13b, при этом получив пурпуровое твердое вещество (95%), которое использовали без очистки.
Описание 16.
а) Тиазол[3,2-а]индол.
Перемешанный раствор тиоксиндола (3,8 г, 0,025 моль) и бромуксусный ангидрид - диэтилацеталя (3,9 мл, 0,026 моль) в ацетоне (200 мл) обработали безводным карбонатом калия (6,9 г, 0,50 моль), и смесь нагревали с обратным холодильником в течение 2 ч и затем в течение 12 ч при комнатной температуре, смесь концентрировали в вакууме, остаток обработали водой (100 мл) и экстрагировали этилацетатом (2 x 100 мл). Объединенные экстракты высушили (Na2SO4), концентрировали в вакууме, а остаток подвергали хроматографии на силикагеле с элюированием 10%)танолом/60-80 бензином, получив 2-(2,2-диэтоксиэтилмеркапто)индол (3,0 г, 44%) в виде желтого масла.
1HNMR (CDCl3)
δ: 9,30 (br.s, 1H) 7,52 (d, 1H), 7,28 (d, 1H), 7,04 - 7,20 (m, 2H), 6,58 (s, 1H), 4,72 (t, 1H), 3,55 - 3,85 (m, 4H), 3,05 (d, 2H), 1,31 (t, 6H).
Хорошо перемешанную смесь 2-(2,2-диэтоксиэтилмеркапто) индола (1,5 г, 0,0057 моль) в полифосфорной кислоте 30 г) нагревали до 130oC в течение 20 мин, затем ее охладили до комнатной температуры и смесь разбавили водой (300 мл). У полученного водного раствора увеличили основность посредством добавки твердого карбоната калия и затем его экстрагировали этилацетатом (2 x 120 мл). Объединенные экстракты высушили (Na2SO4), концентрировали в вакууме, а остаток подвергли хроматографии на силикагеле с элюированием 10% эфиром/60 - 80 бензилом, получив указанное соединение в виде белого твердого вещества (0,56 г, 57%).
1HNMR (CDCl3)
δ: 7,60 - 7,70 (m, 3H), 7,11 - 7,28 (m, 2H), 6,60 (d, 1H), 6,53 (S, 1H).
b)Тиазол[3,2-а]индол-9-карбоновая кислота.
Тиазол[3,2-а] индол обработали трифторуксусным ангидридом, применяя способ из описания 13а, получили 9-трифторацетил-тиазол[3,2-а]индол в виде твердого вещества (95%).
1HNMR (CDCl3)
δ: 8,06 (br. s, 1H), 7,94 (d, 1H), 7,69 (d, 1H), 7,39 - 7,48 (m, 1H), 7,30 - 7,37 (m, 1H), 7,18 (d, 1H).
Указанное соединение (Д16b) получили из 9-трифторацетилтиазол[3,2-а] индола, применяя способ из Описания 13в, и его изолировали в виде светло-пурпурового твердого вещества (84%).
1HNMR (CDCl3/d6DMSO)
δ: 7,98 - 8,08 (m, 2H), 7,73 (d, 1H), 7,10 - 7,31 (m, 2H), 7,00 (d, 1H).
Описание 17.
Метил 2,4-диметилпиримидо[1,2-а]индол-10карбоксилат.
Перемешанный раствор метил 2-аминоиндол-3-карбоксилата (И.Форбес и др., Журнал "Chem. Soc," Перкин 1, 1992, 275)(0,25 г, 0,0013 моль) в кислоте (5 мл) обработали 2,4-пентандионом (0,13 г, 0,0013 моль) и несколькими кристалликами 4-толуолсульфокислоты и нагревали с обратным холодильником в течение 2 ч. Смесь концентрировали в вакууме, а остаток растворили в хлороформе (20 мл), промыли водой (2-х 20 мл), высушили (MgSO4) и концентрировали в вакууме, получив указанное соединение в виде коричневого твердого вещества (0,25 г, 75%).
δ : 8,58 (d, 1H), 8,09 (d, 1H), 7,52 (dt, 1H), 7,34 (dt, 1H), 6,53 (s, 1H), 4,06 (s, 3H), 3,03 (s, 3H), 2,68 (s, 3H).
Описание 18.
Метил 1,2,3,4-тетрагидропиримидо[1,2-а]индол-10-карбоксилат.
Раствор метил 2-хлоиндол-3-карбоксилата (Д11а, 1,5 г, 0,007 моль) в THF (30 мл) в атмосфере азота обработали водородистым натрием (215 мг, от 80% масляной дисперсии, 0, 0071 моль) и перемешивали в течение 20 мин).
Полученный раствор обработали раствором 3-бромпропиламина (0,0093 моль) в толуоле (15 мл), при этом образовался белый студенистый осадок. Эту смесь разбавили еще тетрагидрофураном (THF) (30 мл) и нагревали с обратным холодильником в течение 18 ч, затем концентрировали в вакууме, а остаток достаточно взболтали с этилацетатом (40 мл) и 10%-ным раствором Na2CO3 (30 мл). Органический слой отделили, высушили (Na2SO4) и концентрировали в вакууме, получив твердое вещество. Его подвергли хроматографии на силикагеле с элюированием эфиром/ 60 - 80 бензином (1:1), получив непрореагировавший исходный материал (600 мг) и указанное соединение в виде белого твердого вещества (110 мг, 6%).
1HNMR (d6 DMSO)
δ : 7,58 (d, 1H), 7,26 (br.s, 1H), 7,12 (d, 1H), 6,88 - 7,05 (m, 2H), 3,98 (t, 2H). 3,73 (S, 3H), 3,38 - 3,46 (m, 2H), 2,08 (квинтет, 2H).
Описание 19.
Метил пиримидо[1,2-а]индол-10-карбоксилат.
Перемешанный раствор метил-2-аминоиндол-3-карбоксилата (И.Форбес и др., Журнал "Chem. Soc.", Перкин 1, 1992, 275) (0,5 г, 0,0026 моль) в ксилоле (10 мл) обработали 1,1, 3,3-тетраметоксипропаном (0,43 г, 0,0026 моль) и несколькими кристалликами 4-толуолсульфокислоты и нагревали с обратным холодильником в течение 2,5 ч. Смесь концентрировали в вакууме, а остаток растворили в хлороформе 25 мл), промыли водой (2х10 мл), высушили (MgSO4) и концентрировали в вакууме, получив темно-оранжевое твердое вещество. Его очистили посредством хроматографии на силикагеле с элюированием этилацетатом, получив указанное соединение (ДТ9) в виде оранжевого твердого вещества (0,23 г, 35%).
1HNMR (CDCl3)
δ : 8,68 - 8,78 (m, 2H), 8,57 (d, 1H), 7,89 (d, 1H), 7,59 (dt, 1H), 7,45 (dt, 1H), 6,80 - 6,90 (m, 1H), 4,08 (S, 3H).
Активность антагониста рецептора 5 - HT4.
1) Толстая кишка морской свинки.
Применяли морские свинки - самцы весом 250-400 г. Из более отдельной от центра области толстой кишки брали образцы сплетений кишечной продольной мышцы длиной примерно 3 см. Их суспендировали под нагрузкой 0,5 г в изолированных тканевых ваннах, содержащих раствор Кребса, которые барботируют посредством 5% CO2 в O2 и поддерживают при 37oC. Во всех экспериментах раствор Кребса также содержал метиотепин 10-7M и гранисетрон 10-6M для блокирования эффектов на рецепторах 5 - HT2 и 5 - HT3.
После построения простой кривой концентрация -ответная реакция с 5 - HT и применяя время контакта 30 с и цикл дозирования 15 мин, выбирали такую концентрацию 5 - HT, чтобы получить сокращение мышцы примерно на 40 - 70% максимум (10-9M приблизительно). Затем в ткань попеременно вводили каждые 15 мин эту концентрацию 5 - HT и затем, вводили примерно в равной степени эффективную концентрацию стимулятора никотинового рецептора, диметилфенилпиперазина (DMPP). После получения соответствующих ответных реакций на 5 - HT и DMPP добавляли в раствор ванны повышенные концентрации предполагаемого рецептора 5 - HT4. Затем определяли эффекты этого соединения в значении процента сокращения мышцы, вызываемого 5 - HT или DMPP. По этим данным определяли значения pIC50, выраженные как - log концентрация антагониста, который уменьшал на 50% сокращение мышцы. Считают, что соединение, которое уменьшает ответную реакцию на 5 - HT, а не на DMPP, действует в качестве антагониста рецептора 5 - HT4.
Соединения были эффективными при концентрации обычно в пределах порядка pIC50 = 7 или больше, причем E1, E2, E4, E6, E8, E15, и E27 показывают особенно хорошую активность, когда Y представляет 0, а E3, E20, E23 и E28 демонстрируют особенно хорошую активность, когда Y представляет NH.
2) Предсердие поросенка.
Соединения были испытаны на самопроизвольное биение сердца у поросят (Naunyn - Schmiede berg's Arch. Pharmacol, 342, 619 - 622). Значение pKB ( - Log10KB) для соединения из примера 3 было равно 10,05.
3)Пищевод у крысы.
Слизистую оболочку мышечной рубашки пищевода у крысы исследовали в соответствии со способом, описанным в публикации Бакстера и др., в Naunyn - Schmiedeberg's Arch. Pharmacol, 343, 439 - 446 (1991).
Внутреннюю гладкую мышечную трубку слизистой мышечной оболочки изолировали для получения записи изомерного растяжения в насыщенном кислородом (95% O2 / 5% CO2) растворе Tyrodes при 37oC. Все эксперементы проводили в препаратах, предварительно обработанных паргилином (pargyline), (100 мк в течение 15 мин с последующим вымыванием и в присутствии кокаина (30 мк). Реакцию релаксанта на 5 - HT получают после предварительного сжатия ткани пищевода карбоколом (carbachol) (3 мк).
4)Подвижность сумки желудка у собаки, вызванная рецептором 5 - HT.
Соединения испытали на исключение подвижности желудочной сумки, применяя метод испытания in vivo, описанный в статье "Стимулирование подвижности желудка у собаки посредством BRL 24924, новый агент для исключения подвижности желудка". Бермудез и др. Журнал".1990, 2 (4), 281-286.
Испытание in vivo на активность, вызывающую беспокойство или тревогу.
Испытание на определение взаимоотношений между крысами при их общении.
Крыс (мужская особь, Sprague Dawleys, Charles River массой 250 - 300 г) помещают группами из восьми штук в комнату, где их выдерживают в течение 5 дней. Затем из помещают отдельно в комнату, расположенную смежно с экспериментальной комнатой, на 4 дня до того дня, когда начнут проведение эксперимента. В день проведения эксперимента крысам вводят носитель, испытываемое соединение или бензодиазепиновое возбуждающее средство, хлордиазепоксид, р. о. парами (п = 8-16) с интервалами в 15 мин, начиная с 10 ч утра. Через 30 мин их помещают попарно с самками с одинаковыми весовыми категориями (особи в паре встречаются впервые) в ящик, установленный в отдельной комнате, для исследования их реакции при общении. Ящик изготовлен из материала Perspex белого цвета и имеет размер 54 х 37 см, причем он выполнен с прозрачной передней стенкой из Perspex и у него отсутствует крышка. Пол в ящике разделен на 24 квадрата и ящик ярко освещается (115 лк). В последующие 15 мин на видеомониторе с дистанционным управлением увидели, что у животных появилось активное поведение (прихорашивание, обнюхивание, ползание сверху или снизу, ходьба друг за другом, покусывание, лазание и драка). Во время испытания также отмечают и суммируют количество квадратов, пересеченных каждой крысой. После завершения каждого испытания ящик тщательно протирают.
Соединение E3 увеличивало общее количество отмеченного взаимодействия в пределах интервала дозы 0,01 - 10 мг/кг р.о.
Пример фармацевтического состава (твердая желатиновая капсула).
Ингредиент - Количество на 1 капсулу, мг:
Соединение примера 3 - 1 , 5, 10 или 20
Гидроксипропил - метилцеллюлозы - 12
Гликоллят натриевый крахмал - 12
Стеарат магния - 2,4
Маннитол - До 240,0н
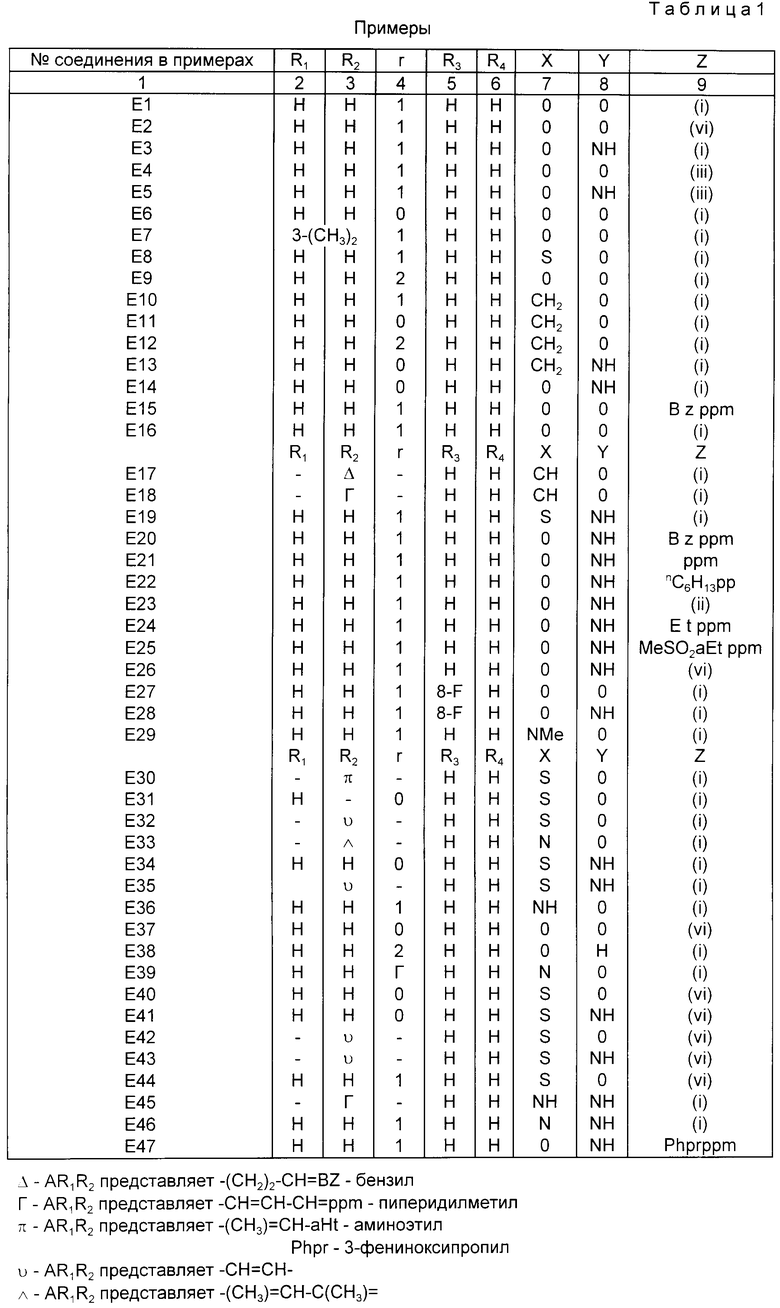

Использование: в химии гетероциклических соединений, проявляющих активность антагониста 5-HT4 рецептора. Сущность изобретения: конденсированное индольное производное формулы I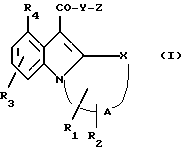 ,
,
где X-O, A- -(CH2)3, R1, R2, R3, R4 - водород, Y-NH, Z представляет группу формулы
где R5 представляет водород, C1-12 алкил, фенил(C1-C6) алкил или R5-(CH2)Z - R10, где Z = 2 или 3, и R10 выбран из фенокси и SO2NR11R12, где R11 и R12 - водород или C1-6 алкил, или его фармацевтически приемлемая соль, а также фармацевтическая композиция, проявляющая активность антагониста 5-HT4 рецептора, содержащая эффективное количество соединения формулы (I). 2 с. и 2 з.п. ф-лы, 2 табл.
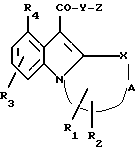
где X кислород;
A (CH2)3;
R1 и R2 водород;
R3 водород;
R4 водород;
Y NH;
Z группа общей формулы
где R5 водород, C1 C1 2-алкил, фенил(C1 C6)алкил или R5 - (CH2)z-R1 0, где Z 2 или 3, R1 0 - фенокси или SO2NR1 1R1 2, где R1 1 и R1 2 водород или C1 - C6-алкил или его фармацевтически приемлемая соль.
Приоритет по признакам:
12.03.92 при R5 водород, C1 C1 0-алкил или фенил(C1-C6)алкил;
25.09.92 при R5 C1 1 C1 2-алкил;
29.12.92 при R5-(CH2)z-R1 0.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| EP, заявка, 429984, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| WO, заявка, 91/16045, кл | |||
| Устройство для сортировки каменного угля | 1921 |
|
SU61A1 |

