Изобретение относится к медицине и ветеринарии, конкретно - к иммуномодулирующим лекарственным средствам на основе гидрофобных производных индукторов интерферона, применяемых в противовирусной терапии.
Предшествующий уровень техники
Известны применяемые в противовирусной терапии иммуномодулирующие лекарственные средства на основе индукторов интерферона [1] - [8]:
природные высокомолекулярные индукторы (полирибонуклеиновые кислоты), например, ларифан (ридостин) [1];
синтетические высокомолекулярные индукторы (полирибонуклеиновые кислоты), например, полигуаниловая (poly (G), полиинозиловая (poly(I)) и другие кислоты [2];
природные (растительные) полифенолы, например, госсипол [3];
синтетические аналоги растительных полифенолов, например, мегасин [4];
синтетические низкомолекулярные индукторы:
на основе 10-акридонуксусной кислоты или ее натриевой соли [5];
на основе 3,6-бис(гетероаминоэтокси) акридинов, а именно - 3,6-бис(2-диметиламиноэтокси) акридин [6].
Перечисленные лекарственные средства дают определенный профилактический и терапевтический эффект. Их используют растворенными в воде, на мазевой основе или в других фармацевтически приемлимых носителях.
Известно также, что вышеуказанные средства имеют ряд недостатков. В частности, природные и синтетические полирибонуклеиновые кислоты высокотоксичны и быстро разрушаются нуклеазами организма. Природным и синтетическим полифенолам свойственна невысокая интерферониндуцирующая активность и токсичность. Синтетические низкомолекулярные индукторы интерферона на основе 10-акридонуксусной кислоты или ее натриевой соли при высокой эффективности и малой токсичности гидрофильны, что затрудняет преодоление лекарственным средством липидного слоя клеточных мембран и, как следствие, ослабляет воздействие лекарственного средства на клетки-мишени иммунной системы, что затрудняет проникновение препарата через гистогематические (в том числе гемато-энцефалитический) барьеры. Кроме того, вследствие гидрофильности, лекарство быстро выводится из организма, что требует многократного его введения для достижения терапевтического эффекта.
Синтетический низкомолекулярный индуктор интерферона на основе 3,6-бис(2-диэтиламиноэтокси) акридина, в частности - 3,6-бис (2-этиламиноэтокси)-4,5-дихлор-2-нитро акридина гидрофобен не имеет недостатков указанного выше индуктора на основе 10-акридонуксусной кислоты или ее натриевой соли. Это средство наиболее близко к заявляемому по его сущности и механизму воздействия на клетки иммунной системы, поэтому выбрано в качестве прототипа. Более подробно прототип описан в патенте США N 4314061, МКИ C 07 D 413/14, НКИ 544-80, заявлено 01.08.77, опубликовано 02.02.82 (авторы - K.C. Murdock, M.R. Damiani, F.E. Durr).
Наряду с большими достоинствами прототип, как и другие аналоги, имеет ряд слабых мест:
сравнительно узкая область эффективного действия (в основном активность в отношении вируса простого герпеса-1);
высокая токсичность;
недостаточная эффективность, особенно при местном применении;
трудность получения.
Раскрытие изобретения
Задачей настоящего изобретения является уменьшение указанных недостатков, т.е. расширение области эффективного действия, повышение эффективности, снижение токсичности и упрощение производства.
Эта задача решается тем, что в известное лекарственное средство, содержащее низкомолекулярный индуктор интерферона и фармацевтически приемлемый носитель, внесены существенные изменения, а именно: в качестве индуктора интерферона выбран эфир 10-метиленкарбокси-9- акридона.
Кроме того, при водном носителе, для обеспечения растворимости, в раствор могут быть введены pH-стабилизирующие добавки, например, цитратный буфер и гидроксиды щелочных и щелочноземельных металлов в эквивалентных количествах к эфирам.
Кроме того, при липидном носителе, для повышения эффективности раствор может быть микрокапсулирован.
Указанный выше выбор основан на результатах исследований, проведенных авторами данного изобретения, обнаружившими новые физико-химические свойства сложных эфиров 10-метиленкарбокси-9-акридона, основные из которых - высокая фармакологическая активность и новые фармакологические свойства, способность растворяться в воде в присутствии гидроксидов щелочных и щелочноземельных металлов, способность включаться в искусственные липидные слои при нейтральных или близких к нейтральным значениях pH. Именно эти особенности и свойства сложных эфиров 10-метиленкарбокси-9-акридона, неизвестные авторам из доступных источников информации, позволили реализовать поставленную задачу.
Лекарственная форма на основе сложных эфиров 10-метиленкарбокси-9-акридона включает в себя оптимальные для заданного фармакологического эффекта количество сложного эфира и стабилизирующие добавки, растворенные в апирогенной воде (другом фармацевтически приемлимом носителе) или включенные в искусственные липидные структуры (другие фармацевтически приемлимые носители). Заявляемая структура лекарственной формы позволила получить набор биологически активных и фармацевтически приемлимых лекарственных форм - парентеральных, пероральных, ингаляционных, дермальных и интраназальных. Другими словами, новое иммуномодулирующее средство построено на основе фармацевтически приемлимых комплексов эфиров 10-метиленкарбокси-9-акридона с макромолекулярными носителями. Полученные макромолекулярные интерфероногенные комплексы обладают повышенной эффективностью индукции интерферонов, расширенным спектром противовирусной активности, сниженной частотой побочных эффектов и улучшенными фармацевтическими свойствами. Использованы водные растворы и липидные смектические мезофазы (липосомы), содержащие эфиры 10-метиленкарбокси-9-акридона. В частности, используют указанные эфиры и амфифильные липидные молекулы, которые первоначально образуют сложный раствор в приемлемых органических растворителях, а затем, при поэтапной замене органического растворителя на фармацевтически приемлемый водный раствор, происходит образование липидных смектических мезофаз (липосом), представляющих собой искусственные сферические мембраны размерами 60 - 7000 нанометров, состоящие из одного и более слоев, в которых эфиры 10-метиленкарбокси-9-акридона связаны с липидами нековалентными (водородными и гидрофобными) связями.
Заявляемое лекарственное средство получают, например, следующим образом. Смесь липидов (0,5 - 0,9 экв.) и сложного эфира 10-метиленкарбокси-9-акридона (0,3 - 0,1 экв.) прямо или косвенно растворяют в приемлемом органическом растворителе - хлорированных углеводородах, простых алифатических эфирах, спиртах или других приемлемых растворителях. Подобное соотношение является оптимальным, так как при увеличении доли сложного эфира более 0,1 - 0,3 экв. (в зависимости от строения эфирного радикала) стабильность формируемого комплекса снижается и наблюдается кристаллизация сложного эфира 10-метиленкарбокси-9-акридона. Уменьшение доли сложного эфира приводит к снижению интерферониндуцирующей активности и фармацевтической приемлемости комплекса. Производится замена органического растворителя на водный, либо путем предварительного удаления растворителя с последующей гидратацией сухого вещества, либо путем удаления органической фазы из эмульсии органический растворитель - водная фаза. При этом процедуры по замене органического растворителя на водную фазу должны протекать при температуре большей, чем температура фазового перехода наиболее тугоплавкого из присутствующих в смеси липидов. Объем органической фазы определяется растворимостью компонентов в приемлемом растворителе, а объем водного раствора - количеством используемых для синтеза комплекса компонентов.
Анализ получаемых комплексов с помощью сканирующей и трансмиссионной электронной микроскопии, динамического рассеивания лазерного света, проточной цитофлюорометрии, жидкостной хроматографии высокого разрешения и колоночной хроматографии на гидрофобных колонках POLY-PREP показал, что выход гомогенных стабильных липосом составляет не менее 95% и что не менее 89% применяемого эфира стабильно интегрированы в составе липосом.
Возможность получения заявляемых лекарственных средств иллюстрируется следующими практическими примерами.
Пример 1.
100 г этилового эфира 10-метиленкарбокси-9-акридона смешивают с 500 мл апирогенной воды, вносят 1,05 экв. гидроксида натрия. Полученную смесь выдерживают при температуре 90 - 95oC в течение 1 ч. За это время отгоняют 80 мл растворителя. Охлаждают до комнатной температуры. К полученной смеси добавляют 400 мл апирогенной воды и фильтруют. В фильтрате спектрофлюрометрически определяют содержание этилового эфира 10-метилкарбокси-9-акридона. Добавляют апирогенной воды до получения раствора нужной концентрации и вносят лимонную кислоту до pH = 7,75 (примерно 1,5 г кислоты). Полученную лекарственную форму фильтруют через бактериальный фильтр и разливают по флаконам с последующей герметизацией и стерилизацией. Получают готовую к парентеральному применению лекарственную форму с выходом около 98% (считая на этиловый эфир 10-метиленкарбокси-9-акридона). Описанная парентеральная лекарственная форма в качестве добавок, стабилизирующих pH, может содержать: замещенные фенолы, карбоновые кислоты, соли слабых оснований и сильных кислот.
Пример 2.
500 мг липидов (дипальмитоилфоофатидилхолин, фоофатидилсерин в молярном отношении 10 : 0,1) и 100 мг лаурилового эфира 10-метиленкарбокси-9-акридона растворяют в 500 мл хлороформа при 22oC и помещают в толстостенную круглодонную колбу объемом 4 л. Колбу помещают на роторный испаритель и удаляют растворитель при 65oC. На стенках колбы образуется тонкая полупрозрачная пленка с зеленоватым оттенком. Колбу помещают в сушильный шкаф, оборудованный ловушкой влаги, и досушивают в вакууме в течение 1 ч. В колбу вносят 10 мл дважды дистилированной деиононизированной воды и после продувки колбы гелием регидрируют пленку путем интенсивного встряхивания при 65oC в течение 1 ч. Образуется 10 мл густого геля с зеленоватым оттенком.
Гель разводят 90 мл фосфатного буфера (pH=7,4; 22oC) и обрабатывают ультразвуком при 44 кГц 30 раз по 30 с с минутными интервалами. Обработанную ультразвуком суспензию продавливают под давлением (дважды, последовательно) азота через поликарбонатные мембраны с диаметром пор сначала 500, и далее 100 нм. Образуется опалесцирующая суспензия с зеленоватым оттенком, представляющая собой водную взвесь маленьких однослойных липосом размером 80 - 130 нм, содержащих 500 мг липидов и 100 мг лаурилового эфира 10-метиленкарбокси-9-акридона. Взвесь в присутствии стабилизатора (трехалоза) замораживают в жидком азоте, после чего подвергают лиофильному высушиванию в течение 24 ч. Образовавшийся порошок подвергают микрокапсулированию в кислотоустойчивое покрытие. Микрокапсулы, содержащие 500 мг липидов и 100 мг лаурилового эфира 10-метиленкарбокси-9-акридона, прессуют в таблетки, содержащие по 20 мг производного акридона и 100 мг липида.
Пример 3.
500 мг липидов (фосфатидилхолин, фосфатидилэтаноламин, холестерин в молярном соотношении 1 : 0,001 : 0,5) растворяют в 400 мл диэтилового эфира при температуре 22oC. 50 мг этилового эфира 10-метиленкарбокси-9-акридона растворяют в 1 мл диметилсульфоксида при 60oC, затем охлаждают до 22oC и приливают по каплям к 400 мл раствора липидов в диэтиловом эфире при интенсивном перемешивании. К полученному в результате раствору добавляют 100 мл дважды дистилированной воды и образовавшуюся двухфазную систему обрабатывают ультразвуком до исчезновения границы раздела фаз и получения дисперсной системы (эмульсии). Органическую фазу удаляют на роторном испарителе при температуре, превышающей температуру фазового перехода наиболее тугоплавкого из присутствующих в смеси фосфолипидов (60oC) при постепенном усилении вакуума до 0,1 тор (к концу упаривания). Образуется 100 мл густого геля бледно-зеленого цвета, который после замораживания в жидком азоте подвергается лиофильному высушиванию.
Образуется порошок бледно-зеленого цвета общей массой 550 мг. Порошок затем смешивают с 10 мл фосфатного буфера (pH = 7,0 и 22oC). Образуется суспензия бледно-зеленого цвета, представляющая собой водную взвесь многослойных липосом размером 400 - 800 нм, содержащих 500 мг липидов и 50 мг этилового эфира 10-метиленкарбокси-9-акридона. Далее - аналогично примеру 2.
Промышленная применимость
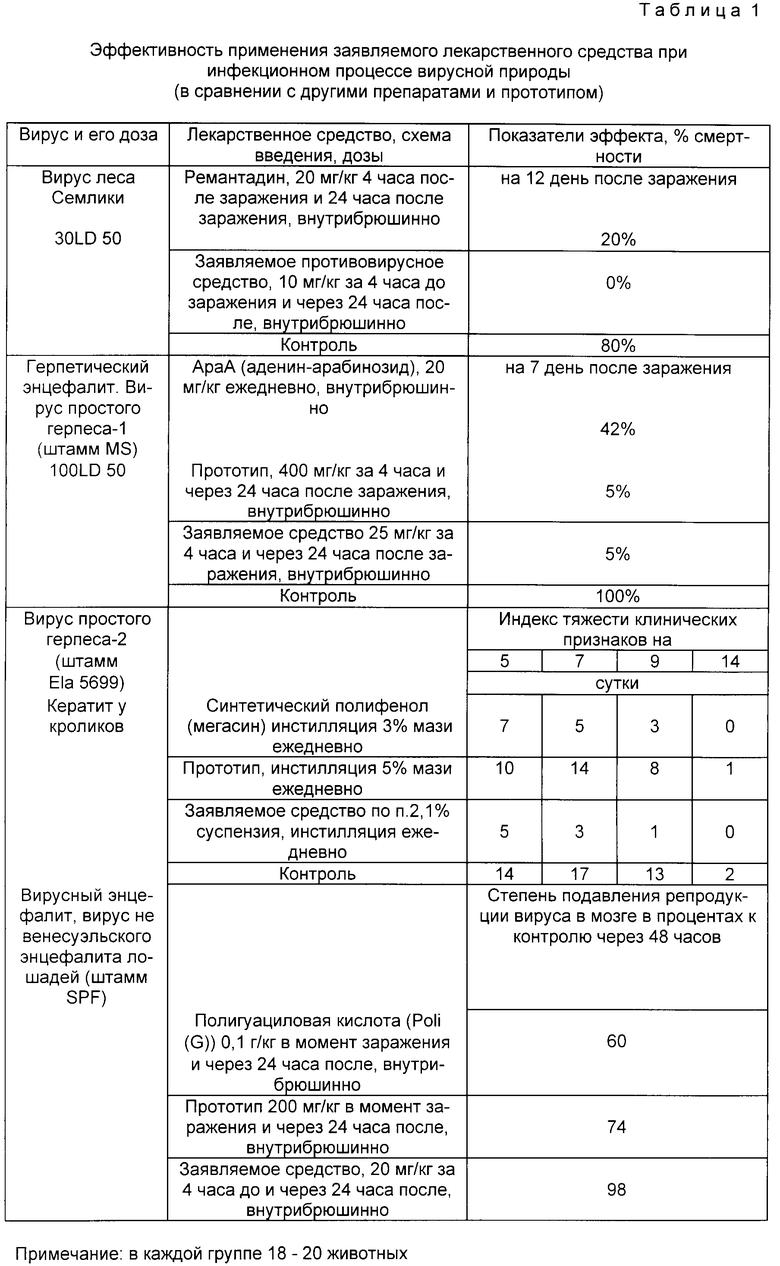
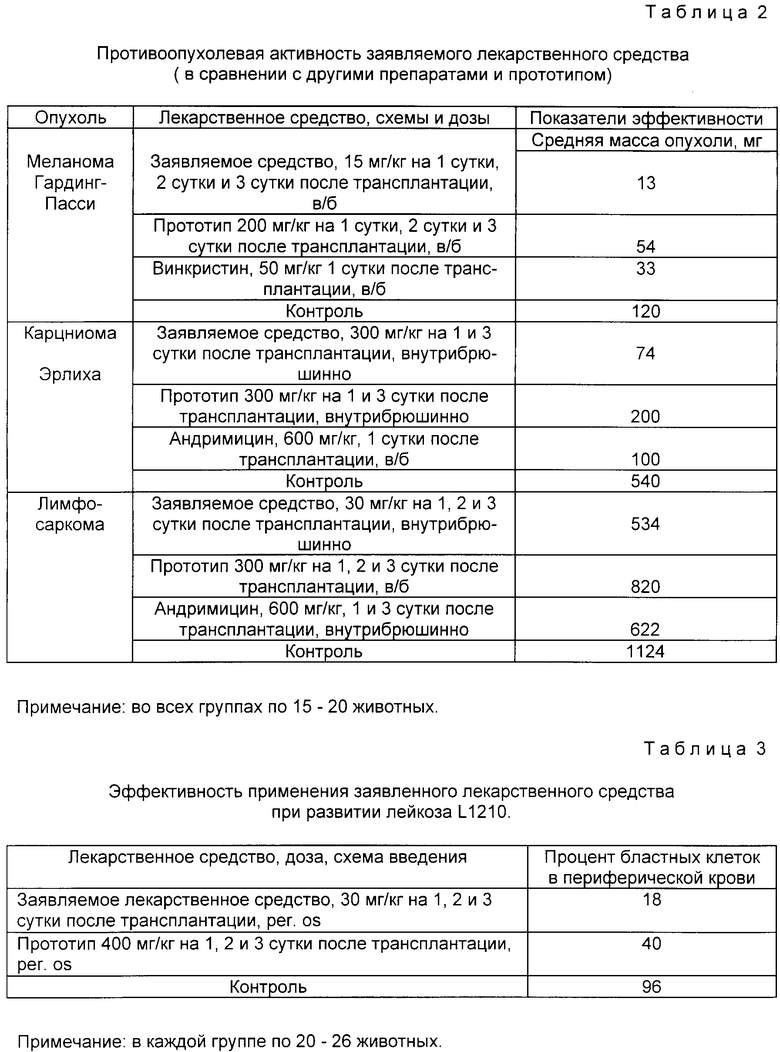
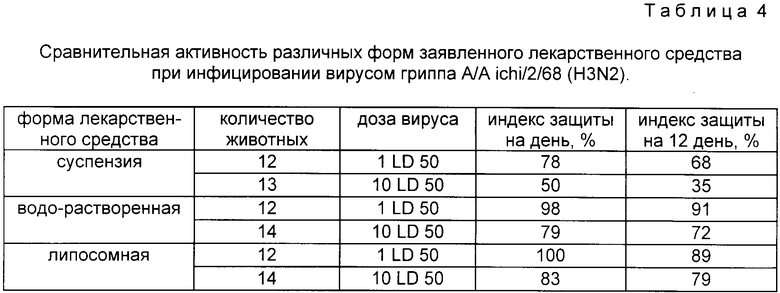
В прилагаемых таблицах 1 - 4 приведены результаты сравнительных испытаний заявленного лекарственного средства с аналогами, прототипом и контрольной группой.
Таблица 1 - эффективность применения заявленного лекарственного средства при инфекционном процессе вирусной природы (в сравнении с другими препаратами и прототипом).
Таблица 2 - противоопухолевая активность заявленного лекарственного средства (в сравнении с другими препаратами и прототипом).
Таблица 3 - эффективность применения заявленного лекарственного средства при развитии лейкоза L 1210.
Таблица 4 - сравнительная активность разных форм заявленного лекарственного средства при инфицировании вирусом гриппа A/Aichi/2/68(H3N2).
Из таблицы 1 следует, что заявленное средство имеет более широкую область эффективного применения при инфекционных процессах вирусной природы, причем его эффективность в несколько раз выше, чем у прототипа и еще более в сравнении с другими широкоприменяемыми препаратами. Из таблицы 2 видно, что противоопухолевая активность заявляемого препарата в несколько раз больше, чем у прототипа и сравнима с активностью противоопухолевых препаратов. Из таблицы 3 следует, что применение заявленного средства при развитии лейкоза L 1210 снижает процент опухолевых клеток в крови в 2 раза эффективнее прототипа. Таблица 4 показывает, что индекс противовирусной активности водорастворимой формы заявляемого средства выше, чем суспензии, а липосомной формы - выше, чем водорастворимой (на примере однократного введения препарата за 1 ч до инфицирования вирусом A/Aichi/2/68 (H3N2).
Кроме целевых экспериментов в процессе исследований постоянно контролировалось наличие побочных и токсических эффектов - раздражений кожи, слизистых оболочек, расстройства пищеварительной и сердечно-сосудистой систем, аллергических реакций и т.п. Токсичность (терапевтический индекс) составил для заявляемого средства 100 - 500, в то время как для прототипа он был 10 - 15.
Из приведенных примеров получения лекарственного средства ясно, что его производство достаточно просто, а трудоемкость по крайней мере в полтора раза ниже, чем у прототипа.
Таким образом, по нашему мнению, заявленное лекарственное средство является новым, имеет изобретательский уровень (неочевидно), промышленно применимо и успешно решает важную для человечества задачу - получено новое высокоэффективное, фармацевтически приемлемое средство для парентерального, энтерального, наружного и других способов применения
Литература.
1. Государственный реестр лекарственных средств, разрешенных для применения в медицинской практике и к промышленному производству, М.: Министерство здравоохранения СССР, 1990.
2. Gordon G., Minks M.A. The imterferone renaissance: molekular aspects of induction and action. Microbiol. Rev., vol. 45, p. 244, 1981.
3. Hudson J.B. Antiviral compounds from plants. CRC Press, 1990.
4. Ершов Ф.А. Интерферон, в книге Общая и частная вирусология, М.: Медицина, 1982, с. 323 - 341.
5. Патент США N 3681360, Antiviral substitute acridones. Заявлен 09.04.71, опубликован 01.06.72.
6. Патент США N 4314061, МКИ C 07 D 413/14, НКИ 544-80. Некоторые 3,6-бис(гетероаминоалкокси) акридины, K.C. Murdock, R.Damiani, F.E. Durr. Заявлен 01.08.77, опубликован 02.02.82.
7. Gamage I., A. Swarha, W. Gordon. Structural-activity relationship for substituted 9-oxo-9,10-dihidracridine-4-acetic-acid in J.Anti-Cancer Drug. Des., p. 403-414, 7(5), 1992.
Tareporewala J., B. Zrach et al. Synthesis and structure-activity relationship of anti-inflammatory 9,10-dihidro-oxo-2-acridinealkanoic acids and 4(2-carboxyphenyl), J.Pharm. Sci. p. 173-178, 79(2), 1990.
Использование: изобретение относится к медицине и ветеринарии, конкретно - к иммуномодулирующим лекарственным препаратам на основе гидрофобных производных индукторов интерферона, применяемых в противовирусной терапии. Задача изобретения - расширить область эффективного применения, повысить эффективность, снизить токсичность и упростить производство лекарственного средства. Сущность изобретения: заключается применения сложных эфиров 10 -метиленкарбокси-9-акридона в качестве низкомолекулярного индуктора интерферона совместно с фармацевтически приемлимым носителем, выбор средств для обеспечения его растворимости в воде и для микрокапеулирования при липидном носителе. Технический результат заключается в усилении иммуномодулирующего действия. 2 з.п. ф-лы, 4 табл.
| US, патент, 4314061, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

