Изобретение относится к методам технологического контроля производства в масложировой промышленности, а именно, к способам количественного определения фосфолипидов в вырабатываемых растительных маслах.
В процессе добывания растительных масел значительная часть фосфолипидов, содержащихся в масличном сырье и локализующихся преимущественно в гелевой части семян, растворяется в масле и извлекается вместе с ним.
В зависимости от природы и качества семян, способов и режимов получения и переработки масел содержание фосфолипидов в них колеблется от 0,05 до 3,5% в пересчете на стеароолеолецитин [1].
Обладая важными физиологическими свойствами, фосфолипиды в то же время осложняют ведение последующих этапов переработки растительных масел и отрицательно влияют на технико-экономические показатели рафинации и гидрирования в целом.
В мировой и отечественной практике существуют различные методы удаления фосфолипидов - гидратация водой, паром, растворами электролитов, минеральными и органическими кислотами и другие [2, 3, 4].
Поэтому для оценки эффективности и оперативного управления технологическим процессом получения и переработки растительных масел, а также для оперативного контроля за качеством вырабатываемых масел и жиров необходим экспресс-метод количественного определения фосфолипидов в растительных маслах.
В нашей стране существуют два варианта стандартного метода определения фосфоросодержащих веществ в нерафинированных, гидратированных и рафинированных растительных маслах: весовой и колориметрический [5].
Весовой метод основан на сухом сжигании пробы масла с окисью магния, последующей обработке смесью азотной и серной кислот, осаждения образующегося осадка с помощью молибденовокислого аммония и сушке осадка на фильтре до постоянного веса при температуре 100oC.
Указанный метод достаточно трудоемок и длителен (10-12 ч).
Кроме того, как показано в работе Койфман Т.Ш. [6], при определении содержания фосфолипидов весовым методом вместе с фосфором под действием молибденовокислого реагента соосаждаются присутствующие в масле ионы железа, что приводит к завышенным и недостоверным результатам анализа.
В настоящее время в промышленной практике наибольшее применение получили фотометрические методы определения массовой доли фосфолипидов в растительных маслах, отличающиеся подготовкой и минерализацией пробы масла, типами применяемых окислителей и восстановителей.
Известен способ определения массовой доли фосфора в растительных маслах, основанный на сухом сжигании масла с окисью магния и последующем определении фосфора фотометрическим методом с использованием молибденовокислого натрия или аммония [7]. Недостатком указанного способа является обязательная минерализация пробы путем сухого сжигания в муфельной печи при температуре 800oC в течение 0,3-1,0 ч.
Существует хроматографический способ определения группового состава фосфолипидов [8], основанный на способности отдельных групп фосфолипидов различно адсорбироваться на бумаге, пропитанной кремневой кислотой. Существенным недостатком способа является то, что он предназначен прежде всего для качественной характеристики фосфолипидов и дает данные лишь о приблизительном содержании их в масле.
Известен также метод тонкослойной хроматографии [9] для определения фосфолипидов в растительных маслах и фосфатидных концентратах. Он состоит из операций выделения и обезжиривания фосфолипидов с помощью диализа, последующего разделения их хроматографированием в тонких слоях, минерализации пробы, проведения колориметрической реакции и измерения оптической плотности окрашенного комплекса при аналитической длине волны.
Диализ длится в течение 3-х суток при смене растворителя каждые 24 часа при комнатной температуре.
Наиболее близким к предлагаемому изобретению по технической сущности и достигаемому результату является способ ускоренного определения массовой доли фосфора в растительных маслах [10], заключающийся в жидкость-жидкостной экстракции пробы масла ледяной уксусной кислотой с последующим колориметрическим определением массовой доли фосфора в экстракте с использованием молибденовокислого аммония.
Этот способ и был выбран нами в качестве ближайшего аналога изобретения.
Согласно этому способу [10] в пробирке вместимостью 5 мл на весах 2 кл взвешивают (1-2) ± 0,01 г масла с записью результата до второго десятичного знака. К содержимому пробирки пипеткой добавляют 1 мл ледяной уксусной кислоты. Пробирку плотно закрывают пробкой и интенсивно встряхивают в течение 1 минуты, после чего жировую и уксуснокислую фазы разделяют центрифугированием в течение 5 минут при факторе разделения 210-650.
Затем пробирку открывают, пипеткой отбирают аликвотную часть уксуснокислого экстракта: 50 мкл - для нерафинированного и гидратированного масел и 1 мл - для рафинированных масел и переносят в другую пробирку вместимостью 10 мл.
При анализе рафинированных масел аликвотную часть экстракта упаривают в 1,5-2,0 раза на водяной бане с помощью водоструйного насоса при температуре (50±2)oC и остаточном давлении (3,9±0,1) кПа или в вакуумсушильном шкафу при той же температуре и давлении.
К содержимому пробирки приливают пипеткой 0,4 мл хлороформа и 0,1 мл хромогенного реагента, содержащего молибдат аммония, солянокислое олово, серную кислоту, метиловый спирт и дистиллированную воду.
Пробирку помещают в водяную баню и кипятят с обратным холодильником в течение 1 мин, затем охлаждают до комнатной температуры и вновь добавляют пипеткой еще 5 мл хлороформа.
Образующийся хлороформенный слой отбирают пипеткой и измеряют его оптическую плотность относительно контрольного раствора при длине волны 670 нм в слое 1 см.
Массовую долю фосфора (Ф) в процентах для нерафинированных и гидратированных масел рассчитывают по формуле: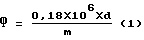 ,
,
рафинированных масел рассчитывают с учетом коэффициента упаривания по формуле: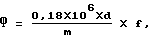 ,
,
где
d - оптическая плотность экстракта;
m - масса масла, взятого для анализа, мкг;
f - соотношение объемов экстракта до и после упаривания.
Для пересчета полученного значения содержания фосфора на фосфолипиды используется коэффициент 26,03.
Хромогенный реагент готовится следующим образом:
1,6±0,0001 г молибдата аммония, взвешенного на весах 2 кл с записью результата до четвертого десятичного знака, растворяют в 12 мл горячей дистиллированной воды (раствор 1).
К 8 мл полученного раствора добавляют несколько капель концентрированной соляной кислоты и 0,1±0,0001 г солянокислого олова с записью результата до четвертого десятичного знака (раствор 2). К оставшейся части раствора 1 осторожно добавляют 20 мл концентрированной серной кислоты и полученный раствор смешивают с раствором 2 (раствор 3).
Отбирают 2,5 мл раствора 3, добавляют 4,5 мл метилового спирта, 2,0 мл дистиллированной воды и получают хромогенный реагент.
Для построения градуировочного графика используют модельные образцы масел с известным содержанием фосфолипидов, которые готовят следующим образом.
Подсолнечное масло подвергают двух-трехкратной нейтрализации и отбелке. Отдельно готовят раствор фосфолипидов, выделенных из соевого фосфатидного концентрата, в гексане из расчета 10 мкг фосфора в 1,0 мл раствора.
В пробы рафинированного и отбеленного масла массой по 10 г вводят раствор фосфолипидов в количествах, соответствующих конечному содержанию фосфолипидов в пробе в пределах 1oC12 мкг. Гексан удаляют под вакуумом и по известному содержанию фосфолипидов в каждом модельном образце и измеренной оптической плотности строят градуировочный график и определяют градуировочный коэффициент для постоянного объема колориметрируемого раствора. Недостатками этого способа являются:
необходимость применения в качестве восстановителя дефицитного реагента солянокислого олова;
необходимость использования специального реагента - метилового спирта, на применение и хранение которого нужно иметь особое разрешение;
сложность приготовления хромогенного реагента;
необходимость применения специального оборудования (центрифуги) для разделения фаз;
необходимость и сложность приготовления модельных образцов масла с известным содержанием фосфолипидов для построения градуировочного графика.
Задачей изобретения является упрощение и интенсификация способа количественного определения массовой доли фосфора в растительных маслах за счет сокращения количества операций, продолжительности проведения анализа и количества используемых реагентов.
Для достижения этой задачи при осуществлении способа количественного определения фосфора в растительных маслах путем жидкость-жидкостной экстракции, кислотной обработки, нагревания и нахождения искомой величины спектрофотометрическим измерением, жидкость-жидкостную экстракцию пробы масла проводят спирто-эфирным раствором в соотношении 1:1 - 1:2 к массе исследуемой пробы с последующим введением в полученную мисцеллу 1,5 - 3,0 мл 10 N раствора серной кислоты, нагревание ведут при температуре 20 - 50oC в течение 30 - 90 сек.
Предлагаемый способ осуществляется следующим образом.
В пробирку вместимостью 10 - 15 мл на весах 2 кл точности взвешивают 1 - 2 г масла с записью результата до второго десятичного знака. К содержимому пробирки последовательно приливают пипеткой 1-2 мл спирто-эфирного раствора из расчета 1:1 - 1:2 и 1,5 - 3,0 мл 10 N раствора серной кислоты. Спирто-эфирный раствор содержит 2 части этилового эфира и 1 часть этилового спирта. Пробирку плотно закрывают пробкой и интенсивно встряхивают в течение 30-90 сек. При анализе твердых жиров пробирку помещают в водяную баню с температурой воды 40-50oC и при необходимости выдерживают в ней в течение 30-90 сек, периодически встряхивая содержимое пробирки. После разделения фаз пробирку осторожно открывают и пипеткой отбирают 0,3-0,6 мл водно-спиртового раствора, предварительно отметив его общий объем, который должен быть в пределах 1,8-3,6 мл. Аликвотную часть экстракта осторожно переносят в другую пробирку и далее известными спектрофотометрическими методами в экстракте анализируют содержание фосфора.
Отличительным признаком способа является проведение жидкость-жидкостной экстракции пробы масла спирто-эфирным раствором в соотношении 1:1 - 1:2 к массе исследуемой пробы с последующей обработкой полученной мицеллы 10 N раствором серной кислоты при температуре 20-50oC в течение 30-90 сек.
Как известно, растворы фосфолипидов в неполярных растворителях, таких как глицериды природных растительных масел, образуют коллоидную мицеллярную структуру, полярные группы которых направлены внутрь мицеллы, а гидрофобные цепи - наружу.
Жидкость-жидкостная экстракция пробы масла спирто-эфирным раствором приводит к изменению структуры, формы, характера и энергии связей внутри и между мицеллами фосфолипидов с образованием мономеров. Поляризующее соединение, как спирт вклинивается в ядро мицеллы фосфолипидов и способствует проникновению водного раствора серной кислоты внутрь мицеллы. В результате снижается энергия связи фосфолипидов с неполярной средой и наоборот возрастает энергия связи их с водной фазой. Это позволяет свести в одну стадию извлечение фосфора из анализируемой пробы масла, получение водорастворимых комплексов и перевод их в водно-спиртовую фазу.
Установлено, что в мицеллах концентрацией менее 25% извлечение фосфолипидов осуществить практически невозможно. В мицеллах концентрацией выше 50% резко повышается вязкость масляных растворов, сильнее проявляются гидрофобные свойства фосфолипидов, в результате чего также резко снижается эффективность извлечения последних.
Показано, что оптимальная концентрация полученной мицеллы, при которой значительно снижается диффузное сопротивление массопереноса, должна находиться в пределах 25-50%, что соответствует соотношению масло:спирто-эфирный раствор 1:1 - 1:2. Оптимальное значение температуры на стадии жидкость-жидкостной экстракции находилось в пределах 20-50oC. При температуре ниже 20oC резко повышается вязкость жидких масел, в результате значительно осложняется процесс растворения пробы масла в спирто-эфирной смеси, сильно замедляется скорость извлечения фосфолипидов из анализируемой жировой фазы в кислую водно-спиртовую. Вместе с тем, при температуре выше 50oC начинается интенсивное испарение жира. Это приводит к нарушению системы и ухудшению условий взаимодействия реагирующих компонентов.
Продолжительность обработки, обеспечивающая требуемую глубину извлечения фосфолипидов, определяется скоростью взаимодействия реагирующих компонентов. Опытным путем также установлено, что при интенсивном смешении реагирующих фаз минимальное время контактирования составляет 30 сек. При продолжительности обработки менее 30 сек степень извлечения фосфолипидов из анализируемой пробы масла существенно снижается. Дальнейшее увеличение продолжительности контактирования фаз (свыше 90 с) не оказывает влияние на эффективность процесса экстракции. Исходя из этих данных продолжительность обработки масла спирто-эфирным раствором и 10 N раствором серной кислоты должна находиться в пределах 30-90 с.
Что касается количества (1,5-3,0 мл) раствора серной кислоты, оно соответствует известному положению о том, что для полного отсечения кислой водно-спиртовой фазы критический избыток водного раствора кислоты должен пятикратно превышать количество спирта в реакционной среде. Граничные условия определялись экспериментально.
Опыты проводили на нерафинированных, гидратированных и рафинированных растительных маслах (подсолнечного и соевого), результаты которых нашли отражение в табл. 1.
Пример 1 (ближайший аналог).
В пробирку вместимостью 5 мл на весах 2 кл точности отвешивают 1,0 г гидратированного подсолнечного масла с записью результата до второго десятичного знака. К содержимому пробирки пипеткой добавляют 1 мл ледяной уксусной кислоты. Пробирку плотно закрывают пробкой и интенсивно встряхивают в течение 1 мин, после чего жировую уксуснокислую фазы разделяют центрифугированием в течение 5 мин. при факторе разделения.
Затем пробирку открывают, пипеткой отбирают 50 мкл уксуснокислого экстракта и аликвотную часть переносят в другую пробирку вместимостью 10 мл.
К содержимому пробирки приливают пипеткой 0,4 мл хлороформа и 0,1 мл хромогенного реагента, содержащего молибдат аммония, солянокислое олово, серную кислоту, метиловый спирт и дистиллированную волу.
Пробирку помещают в водяную баню и кипятят с обратным холодильником в течение 1 мин. Затем пробирку охлаждают до комнатной температуры и к содержимому пробирки вновь добавляют пипеткой 5 мл хлороформа.
Далее пипеткой отбирают образующийся хлороформенный слой и измеряют его оптическую плотность относительно контрольного раствора при длине волны 630 нм в слое 1 см.
Массовую долю фосфора в % рассчитывают по формуле: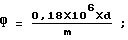 ,
,
где
d - оптическая плотность экстракта;
m - масса пробы масла, взятого на анализ, мкг.
Пример 2 (предлагаемый способ).
В пробирку вместимостью 15 мл на весах 2 кл точности отвешивают 1 г гидратированного соевого масла с записью результата до второго десятичного знака.
К содержимому пробирки последовательно приливают пипеткой 1 мл спирто-эфирного раствора, что соответствует соотношению 1:1 и 1,5 мл 10 N раствора серной кислоты. Пробирку плотно закрывают пробкой и интенсивно встряхивают в течение 60 сек.
После разделения фаз пробирку осторожно открывают и пипеткой отбирают 0,3 мл водно-спиртового раствора.
Аликвотную часть переносят в другую пробирку, добавляют пипеткой 0,3 мл 2;5% водного раствора молибденовокислого аммония, 0,5 мл 0,25% водного раствора гидразинсульфата и дистиллированную воду до метки 5 мл.
Смесь нагревают на кипящей водяной бане в течение 10 мин, охлаждают и измеряют ее оптическую плотность в кювете толщиной 1 см при длине волны 670 нм по отношению к контрольной пробе.
Контрольная проба состоит из 0,3 мл 10 N раствора серной кислоты, 0,3 мл 2,5% водного раствора молибденовокислого аммония, 0,5 мл 0,25% водного раствора гидразинсульфата и дистиллированной воды, добавляемой до объема 5 мл.
Массовую долю фосфора в % рассчитывают по формуле: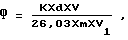 ,
,
где
K - градуировочный коэффициент для постоянного объема колориметрируемого раствора, г, или объема 5 мл Kх100=0,055 г%;
m - масса пробы масла, взятого для анализа, г;
d - оптическая плотность экстракта;
V - объем 10 N раствора H2SO4, взятый для экстракции;
V1 - аликвотная часть спирто-водной кислой фазы, взятой для проведения колориметрической реакции.
Данный способ позволяет исключить применение специальных и дефицитных реагентов, интенсифицировать и упростить существующую методику за счет сокращения количества операций и продолжительности проведения анализа.
Литература.
1. Ксандопуло С. Ю., Копейковский В.М. "Исследование динамики переноса фосфатидов в масло из семян подсолнечника различных классов". Труды ВНИИЖ, 1980 год, Ленинград, стр. 36-41.
2. Болотовская С.Н., Стерлин Б.Я., Игнатьева Л.М. "Влияние режима смешения масла с водой на степень выведения фосфоросодержащих веществ". Масложировая промышленность, 1973 гол, Москва, N 1, стр. 16-18.
3. Корнева Е.П., Арутюнян Н.С. "Исследования в области количественного выведения фосфорсодержащих веществ". Труды ВНИИЖ, 1980 год, Ленинград, стр. 57-63.
4. Паронян В. Х. , Аскинази А. И., Губман И.И., Гапоненко В.Г. и др. "Подготовка масел к дистилляционной нейтрализации". Масложировая промышленность, 1985 год, Москва, N 2, стр. 12-13.
5. ГОСТ 7824-80 "Масла растительные". Методы определения фосфорсодержащих веществ. Москва, 1981 год.
6. Койфман Т.Ш., Сидорова В.П., Эстрина Ф.Б. "Оценка методов определения содержания фосфора в растительных маслах". Труды ВНИИЖ, 1980 год, Ленинград, стр. 81-85.
7. Руководство по методам исследования, технохимическому контролю и учету производства в масложировой промышленности, том VI, выпуск III, ВНИИЖ, Ленинград, 1982 год, стр. 51-55.
8. Руководство по методам исследования, технохимическому контролю и учету производства в масложировой промышленности, том 1, книга 1, ВНИИЖ, Ленинград, 1967 год, стр. 261-266.
9. Руководство по методам исследования, технохимическому контролю и учету производства в масложировой промышленности, том VI, выпуск II, ВНИИЖ, Ленинград, 1974 год, стр. 19-23.
10. Губман И.И., Аскинази А.И., Гапоненко В.Г. "Ускоренный метод определения фосфорсодержащих веществ". Масложировая промышленность, 1985, N 10, стр. 21-23.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ НИКЕЛЯ В КАТАЛИЗАТОРАХ ГИДРИРОВАНИЯ | 1994 |
|
RU2103683C1 |
| Способ определения количества никеля в жирах | 1990 |
|
SU1730579A1 |
| ПИЩЕВОЙ ЖИРОВОЙ ПРОДУКТ | 1994 |
|
RU2091032C1 |
| МАЙОНЕЗ | 1993 |
|
RU2040908C1 |
| СПОСОБ ОЧИСТКИ ЖИРНОГО КОРИАНДРОВОГО МАСЛА | 1993 |
|
RU2101336C1 |
| Способ определения количества никеля в гидрированных жирах | 1987 |
|
SU1441306A1 |
| СПОСОБ НЕЙТРАЛИЗАЦИИ МАСЕЛ И ЖИРОВ | 1992 |
|
RU2008330C1 |
| Майонез | 2017 |
|
RU2682438C1 |
| ПИЩЕВОЙ ЭМУЛЬСИОННЫЙ НИЗКОКАЛОРИЙНЫЙ ЖИРОВОЙ ПРОДУКТ 40%-НОЙ ЖИРНОСТИ | 2004 |
|
RU2268604C1 |
| АРОМАТИЗИРОВАННОЕ МАСЛО РАСТИТЕЛЬНОЕ - СМЕСЬ | 2016 |
|
RU2632000C2 |

Использование: изобретение относится к методам технологического контроля производства в масложировой промышленности, а именно, к способам количественного определения фосфолипидов в растительных маслах. Сущность изобретения: осуществляют жидкость-жидкостную экстракцию пробы масла спирто-эфирным раствором в соотношении 1: 1 - 1:2 к массе пробы с последующей кислотной обработкой путем введения в полученную мисцеллу 1,5 - 3,0 мл 10 N раствора серной кислоты, причем нагревание ведут при температуре 20 - 50oC в течение 30-90 с. 1 табл.
Способ количественного определения фосфора в растительных маслах, предусматривающий жидкость-жидкостную экстракцию, кислотную обработку, нагревание и нахождение искомой величины спектрофотометрическим измерением, отличающийся тем, что жидкость-жидкостную экстракцию пробы масла проводят спиртоэфирным раствором в соотношении 1 1 1 2 к массе исследуемой пробы с последующим введением в полученную мисцеллу 1,5 3,0 мл 10 N раствора серной кислоты, нагревание ведут при температуре 20 50oС в течение 30 - 90 с.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Губман И.И., Аскинази А.И., Гапоненко В.Г | |||
| Ускоренный метод определения фосфорсодержащих веществ | |||
| - Масложировая промышленность, 1985, N 10, с | |||
| Выбрасывающий ячеистый аппарат для рядовых сеялок | 1922 |
|
SU21A1 |

