Данное изобретение относится к получению катализатора или сокатализатора тримеризации, олигомеризации и/или полимеризации олефинов. Катализаторы используют в процессах тримеризации, олигомеризации и/или полимеризации олефинов.
Нанесенные хромоксидные катализаторы были определяющим фактором для получения олефиновых полимеров, например, таких как полиэтилен или сополимер этилена и гексана. Эти катализаторы можно использовать в различных процессах полимеризации. Однако большинство известных соединений хрома должно быть нанесено на носитель для того, чтобы они проявляли каталитическую активность. Более того, большинство соединений хрома на носителе можно использовать только для полимеризации олефинов. Если необходимы сополимеры олефина, то процесс полимеризации становится более сложным, поскольку в реактор полимеризации должны подаваться два различных мономера.
Катализаторы тримеризации и олигомеризации олефинов также хорошо известны в данной области техники. Но обычно они имеют недостаточную селективность по целевому продукту и, кроме того, обеспечивают получение продукта с низким выходом. Однако тримеризация и/или олигомеризации олефинов, если она протекает эффективно, представляет собой процесс, с помощью которого получают необходимые олефины. Такие олефиновые продукты могут быть дополнительно тримеризованы, олигомеризованы и/или возможно введены в процесс полимеризации.
В Европейской заявке EP-A-41-6304 описаны хромсодержащие соединения формулы:
Cr5(C4H4N)10(C4H3O)4;
[Cr(C4H4N)4] [NaI2] • 2(OC4H3);
[Cr(C4H4N)5(OC4H3)] [NaI2] • 4(OC4H3);
или
Cr(NC4H4)3Cl(O2C2H4 (CH3)2)3Na.
В соответствии с предметом вышеуказанной заявки, такие новые хромсодержащие соединения готовятся из реакционной смеси, содержащей соль хрома, амид металла и любой электронодонорный растворитель, такой как, например, эфир. Каталитические системы могут быть использованы или нанесенными, или без подложки при тримеризации и/или полимеризации олефинов.
Солью хрома может быть одна или более органических или неорганических солей хрома, в которых степень окисления хрома изменяется от 0 до 6. Используемое в этом изобретении определение "соль хрома" включает и металлический хром. В общем случае соль хрома будет иметь формулу CrXn, где X могут быть одинаковыми или различными и могут быть любыми органическими или неорганическими остатками, а n представляет собой целое число от 1 до 6. Органические остатки могут содержать от приблизительно от 1 до приблизительно 20 атомов углерода в одном остатке и выбираются из группы, включающей алкильную или алкоксигруппы, эфирный, кетонный остатки и/или амидный фрагмент. Органические остатки могут быть линейными или разветвленными, циклическими или ациклическими, ароматическими и/или алифатическими группами и могут состоять из смешанных алифатических, ароматических и/или циклоалифатических групп. Примерами неорганических остатков являются, но ими не ограничиваются, галогениды, сульфаты и/или оксиды.
Предпочтительно соль хрома представляет собой галогенид, например, фторид хрома (II), фторид хрома (III), хлорид хрома (II), хлорид хрома (III), бромид хрома (II), бромид хрома (III), иодид хрома (II), иодид хрома (III) и их смеси. Из-за простоты отделения побочных продуктов реакции, таких как хлорид натрия, а также относительно низкой стоимости, наиболее предпочтительной солью хрома является хлорид, например, хлорид хрома (II) и/или хлорид хрома (III).
Амид металла может быть любым амидом, который взаимодействует с солью хрома с образованием хромамидного комплекса. В более широком смысле амид металла может быть любым гетеролептическим или гомолептическим металлическим комплексом или солью, в которых амидный заместитель представляет собой любой азотсодержащий органически остаток. Амид металла может быть добавлен или непосредственно в реакционную смесь, или генерироваться in situ. В общем случае амид металла будет содержать от приблизительно 1 до приблизительно 20 атомов углерода.
В число предпочтительных амидов металлов входят, но не ограничиваются только ими, диметиламид лития, диэтиламид лития, диизопропиламид лития, дициклогексиламид лития, бис(триметилсилил)амид натрия, индолид натрия, пирролиды щелочных или щелочно-земельных металлов, или смеси из двух или более указанных соединений. Вследствие высокой реакционной способности и активности по отношению к другим реагентам наиболее предпочтительны вышеуказанные пирролиды металлов, например, пирролид лития, пирролид натрия, пирролид калия и пирролид цезия. Примеры замещенных пирролидов включают, но не ограничиваются только ими, 2,5-диметилпирролид и/или 3,4-диметил пирролид натрия. Когда амид металла представляет собой пирролидиновый лиганд, конечным соединением хрома является пирролид хрома.
В соответствии с первым вариантом заявляемого изобретения сам пиррол (пирролид водорода) или замещенный пиррол рассматриваются как приемлемые пирролиды. Пиррол является предпочтительным пирролидом водорода. Замещенные пирролы иллюстрируются ниже.
Эфир в реакционной смеси может представлять собой одно или несколько эфирных соединений, которые оказывают влияние на взаимодействие соли хрома с амидом металла. Хотя теоретически эфир не должен образовывать химическую связь, полагают, что он может быть растворителем, а также возможным реагентом. В качестве эфира может использоваться любое алифатическое и/или ароматическое соединение, содержащее фрагмент ROR, где заместители R могут быть одинаковыми или разными, но, предпочтительно, не должны быть атомами водорода. Предпочтительными эфирами являются алифатические эфиры, так как ароматические эфиры токсичны для человека. Более того, предпочтительными эфирами являются те, которые ускоряют реакцию между галогенидом хрома и пирролидами металлов группы IA или IIA, и которые также могут быть легко удалены из реакционной смеси. Примерами таких соединений являются, но не ограничиваются ими, соединения из числа тетрагидрофурана, диоксана, диэтилового эфира, диметоксиэтана (глима), диглима, триглима и смеси одного или более указанных соединений. Наиболее предпочтительно эфиры выбираются из группы, включающей тетрагидрофуран, производные тетрагидрофурана, диметоксиэтан, производные диметоксиэтана и их смеси, по причинам, указанным выше, а также из-за того, что предпочтительные соли аминов растворимы в этих эфирах.
Три реагента могут быть смешаны любым способом при условиях, подходящих для образования раствора, содержащего одно или более заявляемых соединений хрома. Реакция предпочтительно протекает в отсутствии кислорода и влаги, следовательно, в инертной атмосфере, например, в атмосфере азота и/или аргона. Реакционное давление должно быть достаточным, чтобы реагенты находились в жидком состоянии. В общем случае приемлемо давление в интервале от приблизительно атмосферного до приблизительно 3 атмосферы. Для упрощения работы обычно используется атмосферное давление.
Выпадающие в осадок заявляемые соединения хрома могут быть выделены любым известным в этой области техники способом. Наиболее простой методикой выделения осадка хромсодержащих соединений является фильтрация.
В соответствии с вторым вариантом данного изобретения пирролиды металлов, используемые в составе катализаторов и/или сокатализаторов, могут быть также приготовлены из солей металлов, отличных от хрома. Примерами таких металлов являются никель, кобальт, железо, молибден и медь. Как и в случае солей хрома, степень окисления металла может быть любой, включая нулевую. Пирролиды этих металлов могут быть получены по методике, аналогичной методике, ранее описанной для пирролидов хрома.
Соединения хрома или других металлов, приготовленные по вышеописанному способу, могут быть использованы в качестве каталитической системы или нанесенными на носитель и/или без нанесения для тримеризации, олигомеризации и/или полимеризации олефинов. Каталитическая система на носителе может быть приготовлена с любым носителем, пригодным для нанесения хромсодержащего катализатора. Примерами таких носителей для катализатора являются, но не ограничиваются только ими, цеолиты, неорганические оксиды, или сами по себе, или в виде смешанных окислов, фосфатированные неорганические оксиды и их смеси. Особенно предпочтительными являются носители, выбираемые из группы, состоящей из двуокиси кремния, алюмосиликата, окиси алюминия, фторированной окиси алюминия, силилированной окиси алюминия, окиси тория, алюмофосфата, фосфата алюминия, фосфатированной двуокиси кремния, фосфатированной окиси алюминия, титаносиликата, соосажденных двуокиси кремния/двуокиси титана, фосфатированной/силилированной окиси алюминия и их смесей, а также любой один или более из тех носителей, которые могут содержать хром. В настоящее время наиболее предпочтительным носителем для катализатора, вследствие наиболее высокой активности при тримеризации, является алюмофосфат, описанный в Пат. США 4364855 (1982).
Количество пирролида хрома на грамм носителя может быть выражено различными, тем не менее, эквивалентными понятиями, например, в молях хрома на 1 г носителя. По причинам, указанным выше, обычно достаточно менее приблизительно 8.6•10-3 моля хрома на 1 г носителя. Предпочтительно используется от приблизительно 1.7•10-6 - 1.7•10-5 до 8.6•10-4 молей хрома на 1 г носителя.
После добавления носителя и тщательного его перемешивания с пирролидом хрома носитель может быть отфильтрован, высушен в вакууме, а затем к смеси носитель/пирролид хромаю добавляют активирующее соединение, обычно в виде раствора одной или более кислот Льюиса и/или металл-алкилов, предпочтительно используя в качестве растворителя углеводород. Затем с помощью фильтрации может быть получена активированная каталитическая система на носителе. В соответствии с данным описанием кислота Льюиса определяется как любое соединение, которое представляет собой акцептор электронов. Предпочтительно активирующее соединение представляет собой соединение, которое можно рассматривать и как кислоту Льюиса и как металл-алкил. В соответствии с более широким определением в данной заявке активирующее соединение может содержать любое количество атомов углерода. Однако, вследствие коммерческой доступности и простоты использования, активирующие соединения будут обычно содержать менее приблизительно 70 атомов углерода в молекуле металл-алкила и предпочтительно менее приблизительно 20 атомов углерода.
Предпочтительными активирующими соединениями, которые представляют собой как металл-алкилы, так и кислоты Льюиса, являются, но не ограничиваются только ими, алюминий-алкилы, бор-алкилы, магний-алкилы, цинк-алкилы и/или литий-алкилы. Примерами таких металл-алкилов являются, но не ограничиваются только ими, металл-алкилы из числа бутиллития, втор.-бутиллития, трет.-бутиллития, диэтилмагния, дибутилмагния, диэтилцинка, триэтилалюминия, триметилалюминия, триизобутилалюминия и их смеси. Наиболее предпочтительно активирующие соединения выбираются из группы, включающей негидролизованные, то есть не контактировавшие с водой, алкил-алюминиевые соединения, производные алкилалюминиевых соединений, галогенированные алкилалюминиевые соединения и их смеси для улучшения селективности по продукту, а также для повышения реакционной способности, активности и/или производительности каталитической системы. Примерами таких соединений являются, но не ограничиваются только ими, триэтилалюминий, трипропилалюминий, трибутилалюминий, диэтилалюминийхлорид, диэтилалюминийбромид, диэтилалюминийэтоксид, полуторный этилалюминийхлорид или их смеси для получения каталитической системы с наиболее высокой активностью и селективностью по продукту. Наиболее предпочтительным соединением является триэтилалюминий, так как при его использовании достигаются наилучшие результаты по активности каталитической системы и селективности по продукту.
Любое количество активирующего соединения, такого как металл-алкил и/или кислота Льюиса, достаточно для активирования и/или взаимодействия с пирролидхлоромовым катализатором. Обычно может быть использовано около 200 г активирующего соединения, то есть металл-алкила и/или кислоты Льюиса, на 1 г хрома. Предпочтительно для получения наиболее высокой каталитической активности используется от приблизительно одного до приблизительно 100 г активирующего соединения, такого, как металл-алкил и/или кислота Льюиса, на 1 г пирролида хрома, наиболее предпочтительно приблизительно от 5 до 30 г активирующего соединения, такого как металл-алкил и/или кислота Льюиса на 1 г пирролида хрома. Однако, используемое количество активирующего соединения, такого как металл-алкил и/или кислота Льюиса, может изменяться в зависимости от используемого носителя. Например, если носитель представляет собой двуокись кремния и/или окись алюминия, слишком большое количество такого активирующего соединения, как металл-алкил или кислота Льюиса, может уменьшать каталитическую активность. Однако такое же количество активирующего соединения, такого как металл-алкил и/или кислота Льюиса, при использовании с алюмофосфатным носителем не снижает значительно каталитическую активность.
Углеводород, используемый в качестве растворителя, может представлять собой любое сочетание одного или более ароматических или алифатических ненасыщенных углеводородов. Хотя теоретически углеводород не должен вступать в химическое взаимодействие, полагают, что он может выступать в качестве реагента и/или стабилизирующего компонента во время и/или после образования заявляемой каталитической системы. Примерами ненасыщенных углеводородов, таких как например, растворитель, могут быть любые ненасыщенные углеводороды, которые способны растворять активирующее соединение, когда активирующим соединением является кислота Льюиса и/или металл-алкил. Соответственно в третьем варианте данного изобретения помимо ароматических соединений, содержащих от 6 до 50 атомов углерода в одной молекуле, используемых в качестве растворителя, могут быть использованы ненасыщенные алифатические углеводороды, содержащие в молекуле менее приблизительно 20 атомов углерода.
Специфическими примерами ненасыщенных алифатических соединений являются, но не ограничиваются ими, этилен, 1-гексан, 1,3-бутадиен и их смеси. Наиболее предпочтительным алифатическим углеводородом является этилен, так как при его использовании исключается стадия приготовления каталитической системы, и в связи с тем, что этилен может быть реагентом для тримеризации и/или олигомеризации. Специфическими примерами ненасыщенных ароматических углеводородов являются, но не ограничиваются только ими, толуол, бензол, ксилол, мезитилен, гексаметилбензол и их смеси. Наиболее предпочтительным ненасыщенным ароматически углеводородным растворителем является толуол, который может быть легко удален и оказывает минимальное влияние на получаемую каталитическую систему.
Ненасыщенный углеводород может присутствовать или во время начального контактирования пирролида хрома и активирующего соединения, то есть до введения в реактор тримеризации, олигомеризации и/или полимеризации, или ненасыщенный углеводород может быть введен непосредственно в реактор. Как указывалось выше, в качестве ненасыщенных углеводородов могут рассматриваться один или более олефиновых реагентов. Предпочтительно ненасыщенный углеводород присутствует при начальном контактировании пирролида хрома и активирующего соединения для того, чтобы стабилизировать получаемую каталитическую систему. В отсутствие ненасыщенного углеводорода получаемая каталитическая система через какое-то время может дезактивироваться и потерять активность.
Хотя может быть использовано любое количество ненасыщенного углеводорода, слишком большое или слишком маленькое его количество может отрицательно влиять на активность каталитической системы. Следовательно, предпочтительно, чтобы из получаемой каталитической системы некоторый избыток ненасыщенного ароматического углеводорода был десорбирован. Десорбция избытка ненасыщенного ароматического углеводорода может быть проведена любым известным в этой области техники методом, например, таким как методы удаления растворителя. Примерами методов удаления растворителя являются, но не ограничиваются только ими, такие методы, как фильтрация, вакуумная сушка, сушка в инертной атмосфере и сочетание этих методов. Хотя теоретически ненасыщенный углеводород не вступает в химическое взаимодействие, полагают, что остающийся ненасыщенный углеводород может стабилизировать получаемую каталитическую систему. Если ненасыщенный углеводород отсутствует, то полагают, что каталитическая система потеряет активность.
Альтернативный и поэтому предпочтительный способ, в соответствии с изобретением, получения каталитической системы на носителей состоит в объединении одного или более твердых заявляемых пирролидов хрома с ненасыщенным углеводородным растворителем, более широко описанного здесь, с тем, чтобы включить, например, толуол и/или этилен, и активирующим соединением, описанным ранее, таким как металл-алкил и/или кислота Льюиса, например, триэтилалюминий.
В соответствии с четвертым вариантом настоящего изобретения каталитическая система может быть приготовлена путем объединения источника металла, пирролсодержащего соединения и металл-алкила и предпочтительно ненасыщенного углеводорода в общем растворителе без стадии предварительного взаимодействия с использованием электронодонорного растворителя (для взаимодействия между источником металла и пирролсодержащим соединением, как это описано выше). Вероятный (необязательный) ненасыщенный углеводород, например, толуол, может использоваться в качестве общего растворителя. В отсутствие такого ненасыщенного растворителя, выступающего в качестве общего растворителя, может быть использован циклогексан. Эти каталитические системы могут дополнительно содержать носитель для катализатора. Предпочтительно источник металла является источником хрома, но может быть источником любого другого металла, описанного выше.
Источником хрома, аналогично ранее описанной соли хрома, может быть одно или более органических или неорганических соединений, в которых хром имеет степень окисления от 0 до 6. В этом описании металлический хром включается в это определение солей хрома. В общем случае источник хрома будет иметь формулу CrXn, где заместители X могут быть одинаковыми или различными и могут представлять собой любой органический или неорганический остаток, а n принимает целые значения от 1 до 6. Органические остатки могут содержать от приблизительно 1 до приблизительно 20 атомов углерода в одном остатке и выбираются из группы, включающей алкильную группу, алкоксигруппу, эфирный, кетонный остатки и/или амидный фрагмент.
Органические заместители могут быть линейными или разветвленными, циклическими или ациклическими, ароматическими или алифатическими, могут состоять из смешанных алифатических, ароматических и/или циклоалифатических групп. Неорганические остатки в качестве примеров включают галогениды, сульфаты и/или оксиды, но не ограничиваются только ими.
Предпочтительно источник хрома представляет собой хром (II)- и/или хром (III)-содержащие соединения, которые могут образовывать каталитическую систему с улучшенной активностью в реакциях тримеризации. Наиболее предпочтительный источник хрома представляет собой соединение хрома (III), так как они удобны при использовании, доступны и образуют каталитическую систему с повышенной каталитической активностью. В качестве примеров соединения хрома (III) включают, но не ограничиваются только ими, хромкарбоксилаты, нафтенаты хрома, галогениды хрома, пирролиды хрома и/или хелатные хромовые соединения на основе дикетонов (дионаты). Конкретные примеры соединений хрома (III) включают 2,2,6,6-тетраметилгептадионат хрома (III), [Cr (ТМ ГД)3], 2-этилгексаноат хрома (III) [Cr (ДГ)3], нафтенат хрома [Cr (НФ)3], хлорид хрома (III), трис(2-этилгексаноат)хрома (III), бромид хрома (III), хлорид хрома (III), фторид хрома (III), окси-2-этилгексаноат хрома (III), ацетилацетонат хрома (III), дихлорэтилгексаноат хрома (III), ацетат хрома (III), бутират хрома (III), неопентаноат хрома (III), лаурат хрома (III), стеарат хрома (III), пирролид(ы) хрома (III) и/или оксалат хрома (III), но не ограничиваются только ими.
Конкретные примеры соединений хрома (II) включают, но не ограничиваются только ими, фторид хрома (II), бромид хрома (II), хлорид хрома (II), иодид хрома (II), бис(2-этилгексаноат) хрома (II), ацетат хрома (II), бутират хрома (II), неопентаноат хрома (II), лаурат хрома (II), стеарат хрома (II), пирролиды хрома (II) и/или оксалат хрома.
Пирролсодержащим соединением может быть любое пирролсодержащее соединение, которое будет реагировать с солью хрома с образованием хромпирролидного комплекса. Используемое в этом описании понятие "пирролсодержащее соединение" относится к пирролиду водорода, то есть к пирролу (C4H5N), производным пирролида водорода, а также к металпирролидным комплексам. "Пирролидом" (или "пирролом", как указывалось для первого варианта изобретения) может быть любое соединение, содержащее 5-ти членный, азотсодержащий гетероцикл, например, такой как пиррол, производные пиррола и их смеси. В более широком смысле, пирролсодержащим соединением может быть пиррол и/или любой гетеролептический или гомолептический комплекс металла или соль, содержащие пирролидный остаток или лиганд. Пирролидсодержащее соединение может быть или непосредственно добавлено к реакционной смеси, или генерируется in situ. В общем случае пирролсодержащее соединение будет содержать от приблизительно 1 до приблизительно 20 атомов углерода в одной молекуле.
Примерами пирролидов (или пирролов) являются пирролид водорода (пиррол), производные пиррола, замещенные пирролиды (или пирролы), пирролид лития, пирролид натрия, пирролид калия, пирролид цезия и/или соли замещенных пирролидов из-за их высокой реакционной способности и активности по отношению к другим реагентам. Примеры замещенных пирролидов (или пирролов) включают, но не ограничиваются только ими, пиррол-2-карбоновую кислоту, 2-ацетилпиррол, пиррол-2-карбальдегид, тетрагидроиндол, 2,5-диметилпиррол, 2,4-диметил-3-этилпиррол, 3-ацетил-2,4-диметилпиррол, этил-2,4-диметил-5-этоксикарбонил-3-пирролпропионат, этил-3,5-диметил-2-пирролкарбоксилат. Когда пирролсодержащее соединение содержит хром, получаемое соединение может быть названо пирролидом хрома.
Наиболее предпочтительные пирролсодержащие соединения для использования в каталитических системах для тримеризации выбираются из группы, включающей пирролид водорода, то есть пиррол (C4H5N) и/или 2,5-диметилпиррол. Хотя все пирролсодержащие соединения могут давать каталитические системы с высокой активностью и продуктивностью, использование пиррола и/или 2,5-диметилпиррола может приводить к образованию каталитических систем с повышенной активностью и селективностью по целевому продукту тримеризации, например, по продукту тримеризации этилена 1-гексену, а также может уменьшить выход полимеров.
Метал-акрилом, также упоминавшимся ранее как активирующее соединение, может быть любой гетеролептический или гомолептический металл-алкил. Могут быть использованы один или более металл-алкилов. Лиганд(ы) при металле может (могут) быть ароматическим и/или алифатическим. Предпочтительно лиганд(ы) представляют собой ненасыщенный алифатический остаток. Металл-алкил сможет содержать любое число атомов углерода. Однако из-за коммерческой доступности и простоты применения обычно используется металл-алкилы, содержащие менее приблизительно 790 атомов углерода в одной молекуле, более предпочтительно менее приблизительно 20 атомов углерода. Предпочтительными металл-алкилами являются алкилалюминиевые соединения, алкилборные соединения, алкилмагниевые соединения, алкилцинковые соединения и/или алкиллитиевые соединения, но не ограничиваются только ими. Примеры металл-алкилов включают, но не ограничиваются ими, н.-бутиллитий, втор.-бутиллитий, трет.-бутиллитий, диэтилмагний, диэтилцинк, триэтилалюминий, триметилалюминий, триизобутилалюминий и их смеси.
Наиболее предпочтительно для улучшения селективности по продукту, а также реакционной способности, активности и/или производительности каталитической системы активирующее соединение выбирается из группы, включающей негидролизованные, то есть не контактировавшие с водой, алкилалюминиевые соединения, производные алкилалюминиевых соединений, галогенированные алкилалюминиевые соединения и их смеси. Примеры указанных соединений включают, но не ограничиваются ими, триэтилалюминий, трипропилалюминий, трибутилалюминий, диэтилалюминийхлорид, диэтилалюминийбромид, диэтилалюминийэтоксид, полуторный этилалюминийхлорид и их смеси, вследствие того, что они образуют лучшие каталитические системы по активности и селективности. Наиболее предпочтительно алкилалюминиевым соединением является триэтилалюминий, который образует наилучшие каталитические системы с точки зрения активности и селективности, а также из-за коммерческой доступности.
Если желаемым продуктом является каталитическая система для тримеризации, активизирующее соединение должно быть по меньшей мере одним негидролизованным алкилалюминиевым соединением, представленным общими формулами AlR3, AlR2X, AlRX2, AlR2OR, AlRXOR и/или Al2R3X3, где R представляет собой алкильную группу, а X - атом галогена. Примеры таких соединений включают, но не ограничиваются только ими, триэтилалюминий, трипропилалюминий, трибутилалюминий, диэтилалюминийхлорид, диэтилалюминийбромид, диэтилалюминийэтоксид, диэтилалюминийфеноксид, триэтилалюминийметоксихлорид и/или полуторный этилалюминийхлорид. Предпочтительно активирующее соединение для каталитической системы для тримеризации представляет собой триалкилалюминиевое соединение формулы AlR3 по причинам, указанным выше. По тем же причинам наиболее предпочтительным триалкилалюминиевым соединением является триэтилалюминий.
Образование стабильных и активных каталитических систем может осуществлять в присутствии ненасыщенного углеводорода. Как обсуждалось выше, ненасыщенный углеводород может присутствовать или во время первоначального контактирования источника хрома, пирролсодержащего соединения и металл-алкила, или может быть введен непосредственно в реактор тримеризации, олигомеризации и/или полимеризации. Кроме того, один или несколько олефиновых реагентов могут рассматриваться как ненасыщенный углеводород.
Может быть использован любой ненасыщенный ароматический или алифатический углеводород. Предпочтительно ненасыщенный углеводород изначально присутствует в реакционной смеси и наиболее предпочтительно ароматический углеводород и/или этилен присутствуют на начальной стадии для того, чтобы получить высокоактивный катализатор с точки зрения активности и селективности, а также стабильную каталитическую систему. Ненасыщенный углеводород может иметь в молекуле любое число атомов углерода. Обычно промышленно доступны и просты в обращении ненасыщенные углеводороды, которые содержат в молекуле менее чем приблизительно 70 атомов углерода, предпочтительно менее 20 атомов углерода.
Незамещенный углеводород может быть газообразным, жидким или твердым. Предпочтительно для достижения полного контактирования и смешения соли хрома, пирролсодержащего соединения, металл-алкила ненасыщенный углеводород должен быть жидким и/или должен находиться в растворенном состоянии. Примеры ненасыщенных алифатических углеводородов включают, но не ограничиваются ими, этилен, 1-гексан, 1,3-бутадиен и их смеси. Наиболее предпочтительным ненасыщенным алифатическим углеводородом является этилен, так как он может быть и реагентом в процессе тримеризации, олигомеризации и/или полимеризации. Примеры ненасыщенных ароматических углеводородов включают, но не ограничиваются ими, толуол, бензол, ксилол, мезитилен, гексаметилбензол и их смеси. Ненасыщенные углеводороды предпочтительны для улучшения стабильности каталитической системы, а также для улучшения активности каталитической системы. Для получения наиболее высокой стабильности и активности каталитической системы наиболее предпочтительным ненасыщенным ароматическим углеводородом является толуол.
Если ненасыщенный ароматический углеводород добавляется до введения соединения(ий) хрома в реактор тримеризации, олигомеризации и/или полимеризации, удаление или десорбирование ненасыщенного ароматического углеводорода до введения в реактор соединения(ий) хрома может улучшить активность и/или селективность каталитической системы по продукту. Удаление ненасыщенного ароматического углеводорода может быть осуществлено любым способом, известным в данной области техники, например, с помощью упаривания или испарения. Получаемый продукт представляет собой концентрированный или ненасыщенный раствор заявляемой каталитической системы.
Если ненасыщенный ароматический углеводород удаляется до введения в реактор, концентрированный или насыщенный раствор предлагаемой каталитической системы может быть растворен в растворителе, совместимом с процессом тримеризации, олигомеризации и/или полимеризации, для того, чтобы облегчить работу с предлагаемой каталитической системой. В общем случае используется тот же растворитель, который используется в качестве разбавителя в реакторе. Предпочтительными растворителями являются, но не ограничиваются только ими, циклогексан, изобутан, гексан, пентан или их смеси.
Реакция при необходимости также может протекать в присутствии источника галогенида. Наличие источника галогенида в реакционной смеси может увеличивать активность каталитической системы и ее производительность, а также селективность по продукту. Примерами галогенидов являются, но не ограничиваются только ими, фторид, хлорид, бромид и/или иодид. Из-за простоты использования и доступности предпочтительны хлориды. С точки зрения улучшения активности, производительности и/или селективности бромиды являются более предпочтительными галогенидами.
Источником галогенида может быть любое соединение, содержащее атом галогена. Примеры таких соединения включают, но не ограничиваются только ими, соединения общей формулы RmXn, где R может быть любым органическим и/или неорганическим остатком, а X может быть галогенидом, выбираемым из группы, включающей фторид, хлорид, бромид и/или иодид, а m+n принимает любое значение больше 0. Если R представляет собой органический остаток, то он предпочтительно содержит от приблизительно 1 до приблизительно 70 атомов углерода, наиболее предпочтительно от 1 до 20 атомов углерода для обеспечения лучшей совместимости и активности каталитической системы. Если R представляет собой неорганический остаток, то он предпочтительно выбирается из группы, включающей алюминий, кремний, германий, бор, литий, олово, галлий, индий, свинец и их смеси. Конкретные примеры таких соединений включают, но не ограничиваются только ими, метиленхлорид, хлороформ, бензилхлорид, четыреххлористый кремний, хлорид олова (II), хлорид олова (IV), четыреххлористый германий, трихлорид бора, трехбромистый алюминий, треххлористый алюминий, 1,4-дибромбутан и/или 1-бромбутан.
Кроме того, источник хрома, металл-алкил и/или ненасыщенный углеводород могут содержать и обеспечивать галогенид для реакционной смеси. Предпочтительно источником галогенида является алкилалюминийгалогенид и используется вместе с алкилалюминиевыми соединениями, вследствие простоты их использования и совместимости, а также благодаря улучшенной активности и селективности получаемой каталитической системы. Примерами алкилалюминийгалогенидов являются, но не ограничиваются только ими, диизобутилалюминийхлорид, диэтилалюминийхлорид, полуторный этилалюминийхлорид, этилалюминийдихлорид, диэтилалюминийбромид, диэтилалюминийиодид и их смеси.
Когда целевым продуктом является каталитическая система для тримеризации, реакционная смесь предпочтительно содержит источник галогенида. Более того, наиболее предпочтительно источник галогенида выбирается из группы, включающей галогениды олова (IV), галогениды германия и их смеси. Источник галогенида наиболее предпочтительно смешивается в источником хрома и пирролсодержащим соединением до добавления металлалкила, то есть источник хрома и пирролсодержащее соединение предварительно подвергается обработке источником галогенида с целью увеличения продуктивности каталитической системы.
Количество каждого реагента, используемое для приготовления каталитической системы для тримеризации, может быть любым количеством, достаточным для того, чтобы при смешении с одним или более олефинами протекала реакции тримеризации. Обычно, чтобы получить каталитическую систему для тримеризации, приблизительно один моль хрома из расчета на элементарный хром (Cr) может быть смешан с приблизительно 1 - 50 молями пирролсодержащего соединения и с приблизительно 1 - 75 молями алюминия из расчета на элементарный алюминий в избытке ненасыщенного углеводорода. Если присутствует необязательный источник галогенида, то его количество обычно составляет 1 - 75 молей галогенида из расчета на элемент. Предпочтительно около 1 моля хрома из расчета на элементарный хром (Cr) может быть смешан с приблизительно 1 - 15 молями пирролсодержащего соединения и с приблизительно 5 - 40 молями алюминия из расчета на элементарный алюминий (Al) в избытке ненасыщенного углеводорода. Если присутствует необязательный источник галогенида, то предпочтительно его количество составляет приблизительно 1 - 30 молей галогенида из расчета на элементарный галоген (X). Наиболее предпочтительно около 1 моля хрома из расчета на элементарный (C ) смешивается с 2 - 4 молями пирролсодержащего соединения и с 10 - 20 молями алюминия из расчета на элементарный (Al) в избытке ненасыщенного углеводорода. Если присутствует необязательный источник галогенида, то наиболее предпочтительно его количество составляет 2 - 15 молей галогенида из расчета по элементу X.
Избыток пирролсодержащего соединения, как оказалось, не улучшает активность, производительность и/или селективность каталитической системы. Ненасыщенный углеводород может улучшать стабильность, активность и/или селективность каталитической системы. Избыток ненасыщенного углеводорода может привести к ухудшению селективности и/или активности каталитической системы. Слишком большое количество алкилалюминия может привести к уменьшению активности каталитической системы и ее селективности по продукту. Слишком маленькое количество алкилалюминия может привести к неполному образованию каталитической системы, что, в свою очередь, приводит к низкой активности каталитической системы и увеличению образования нежелательных полимерных побочных продуктов. Избыток добавляемого необязательного источника галогенида может дезактивировать каталитическую систему, и, следовательно, может привести к уменьшению ее активности. Как указывалось ранее, присутствие источника галогенида повышает активность каталитической системы и ее селективность по продукту.
В соответствии с четвертым вариантом изобретения предпочтительно пирролсодержащее соединение находится в реакционной смеси вместе с источником металла до введения металлалкила. Если следовать этому порядку добавления, может быть получена более качественная с точки зрения активности и продуктивности каталитическая система.
Реакция предпочтительно протекает в отсутствие кислорода, который может дезактивировать катализатор, в безводных условиях, то есть в условиях изначального отсутствия воды. Поэтому наиболее предпочтительная сухая, инертная атмосфера, например, атмосфера азота и/или аргона. Кроме того, металлалкил является негидролизованным металалкилом.
Реакционное давление может быть любым давлением, которое не оказывает отрицательного воздействия на реакцию. В общем случае приемлемо давление в пределах от приблизительно атмосферного до приблизительно трех атмосфер. С целью упрощения работы реакцию обычно проводят при атмосферном давлении.
Температура реакции может быть любой. Для обеспечения более эффективного протекания реакции предпочтительны температуры, при которых реакционная смесь находится в жидком состоянии.
Время реакции может представлять собой любой промежуток времени, необходимый для осуществления реакции. Реакцию можно рассматривать как процесс растворения; достаточно любого времени, в течение которого могут раствориться практически все реагенты. В зависимости от реагентов, а также от температуры и давления реакции, время реакции может изменяться. Обычно достаточно менее приблизительно одних суток. Обычно время реакции составляет менее 60 мин. При оптимальных условиях время реакции изменяется в интервале от приблизительно 1 с до приблизительно 15 мин. Более длительный промежуток времени реакции не приводит к дополнительным преимуществам, а более короткого времени реакции может оказаться недостаточным для ее завершения.
Гетерогенная система, т.е. каталитическая система на носителе, может быть приготовлена в соответствии с четвертым воплощением изобретения in situ в реакторе путем добавления твердого носителя непосредственно в реактор. Как указывалось ранее, примерами каталитических носителей являются, но не ограничиваются ими, цеолиты, неорганические оксиды в отдельности или в смеси, фосфатированные неорганические оксиды и их смеси. Особенно предпочтительны носители, выбираемые из группы, включающей двуокись кремния, алюмосиликат, окись алюминия, фторированную окись алюминия, силилированную окись алюминия, двуокись тория, алюмофосфат, фосфат алюминия, фосфатированную двуокись кремния, фосфатированную окись алюминия, двуокись кремния - двуокись титана, соосажденные двуокись титана /двуокись кремния, фосфатированно/силилированную окись алюминия и их смеси, а также один или более из тех носителей, которые могут содержать хром. В настоящее время наиболее предпочтительным носителем для катализатора, благодаря самой высокой активности в реакции тримеризации, является алюмофосфат, описанный в Пат. США 4364855. Получение гетерогенной каталитической системы in situ, используемой в реакциях тримеризации или олигомеризации, может привести к уменьшению нежелательного образования полимера.
Гетерогенные каталитические системы для тримеризации, олигомеризации и/или полимеризации могут быть также приготовлены в соответствии с этим вариантом изобретения путем образования реакционной смеси, содержащей источник хрома, пирролсодержащие соединения, металлалкил, ненасыщенный углеводород и неорганический оксид, описанный выше. При необходимости, как описано ранее, может быть добавлен источник галогена. Стехиометрия и условия реакции аналогичны стехиометрии и условиям второго варианта изобретения.
Достаточен любой избыток источника хрома по отношению к носителю катализатора на основе неорганического оксида. Однако обычно достаточно менее 5 г хромпирролидного соединения на 1 г носителя. Предпочтительно для наилучшего наполнения носителя и более эффективного использования реагентов рекомендуется приблизительно от 0.001 - 0.01 до 0.5 г хромпирролидного соединения или источника хрома на 1 г носителя. Количество хромпирролидного соединения или источника хрома на 1 г носителя может быть представлено другими, но эквивалентными понятиями, такими, например, как количество молей хрома на 1 г носителя. Обычно достаточно менее приблизительно 8.6 • 10-3 молей хрома на 1 г носителя. Предпочтительно по причинам, указанным выше, используется приблизительно от 1.7 • 10-6 - 1.7 • 10-5 до 8.6 • 10-4 молей хрома на 1 г носителя.
Получаемая гетерогенная каталитическая система может быть выделена путем фильтрации, чтобы отделить твердую каталитическую систему. Для сохранения химической стабильности и реакционной способности твердая каталитическая система предпочтительно хранится в сухой инертной атмосфере.
В Евр. заявке EP-A-417477 описывается катализатор полимеризации и сокаталитическая система, причем сокатализатор включает хромсодержащие соединения Евр. заявки EP-A-416304. В общем случае каталитической системой полимеризации считается или хромсодержащий катализатор (также известный как "катализатор Филлипса") или титан-, цирконий- и/или ванадийсодержащие катализаторы.
Может быть использована любая известная в этой области техники хромсодержащая каталитическая система. Выпускаемые промышленностью хромсодержащие каталитические системы обычно содержат хром, по меньшей мере часть которого находится в шестивалентном состоянии, нанесенный на неорганический оксид; при необходимости, каталитическая система для полимеризации может дополнительно содержать металл-алкил в качестве сокатализатора. Примерами хромсодержащих каталитических систем являются, но не ограничиваются ими, системы, раскрытые в пат. США N 3887494, 3900457, 4053436, 4151122, 4294724, 4392990 и 4405501.
Могут также использоваться любые титан-, цирконий- и/или ванадийсодержащие каталитические системы, известные в этой области техники. Выпускаемые промышленностью титан-, цирконий- и/или ванадийсодержащие каталитические системы обычно содержат комплексы галогенидов переходных металлов с металлорганическими соединениями. Примеры магний/титансодержащих катализаторов включают, но не ограничиваются ими, катализаторы, описанные в пат. США 4394291, 4326988 и 4347158.
Количество новых сокаталитических систем для тримеризации и/или олигомеризации, включая заявляемые хромсодержащие соединения, используемое в качестве сокатализатора, может быть любым количеством, достаточным для генерирования сомономера, который может быть введен в полимерный продукт. Хромсодержащие каталитические системы, полученные в соответствии с различными воплощениями данного изобретения, могут служить в качестве сокатализаторов в сочетании с титан-, цирконий- и/или ванадийсодержащими катализаторами, описанными выше.
По способу в соответствии с вышеуказанным четвертым вариантом изобретения при смешении с источником металла, пирролсодержащим соединением и металлалкилом и предпочтительно ненасыщенным углеводородом, исключается необходимость выделения первого продукта реакции, как в способах, описанных в наших упоминавшихся выше заявках. Более того, каталитические системы, получаемые по способу в соответствии с четвертым вариантом изобретения, обладают улучшенной производительностью и селективностью по целевому продукту тримеризации, например, при получении 1-гексана из этилена.
Реагенты реакции полимеризации.
Реагенты, которые используются при полимеризации на каталитических системах и сокаталитических системах в соответствии с настоящим изобретением, представляют собой олефиновые соединения, которые могут полимеризоваться, то есть взаимодействовать с тем же или с другими олефиновыми соединениями. Каталитические системы данного изобретения могут использоваться для полимеризации по меньшей мере одного линейного или разветвленного моно-1-олефина, содержащего приблизительно 2 - 8 атомов углерода. Примерами таких соединений являются, но не ограничиваются ими, этилен, пропилен, 1-бутен, 1-пентен, 1-гексен, 1-октен и их смеси.
Каталитические системы данного изобретения также применимы в процессах олигомеризации с использованием олефиновых соединений, содержащих от приблизительно 2 до приблизительно 30 атомов углерода и имеющих по меньшей мере одну двойную связь. Примерами таких моноолефиновых соединений являются, но не ограничиваются ими, ациклические и циклические олефины, такие, как, например, этилен, пропилен, 1-бутен, 2-бутен, изобутен, 1-пентен, 2-пентен, 1-гексен, 2-гексен, 3-гексен, 1-гептен, 2-гептен, 3-гептен, четыре нормальных октена, четыре нормальных нонена и смеси любых двух или более указанных соединений. Примерами диолефиновых соединений являются, но не ограничиваются ими, 1,3-бутадиен, изопрен, 1,4-пентадиен, и 1,5-гексадиен. Если используются в качестве реагентов линейные и/или циклические олефины, то, хотя теоретически они не вступают в химическую реакцию, полагают, что протеканию процесса тримеризации могут препятствовать стерические затруднения. Поэтому разветвленный и/или циклический фрагмент(ы) олефина должен быть удален от двойной углерод-углеродной связи.
Процесс тримеризации, используемый в настоящем изобретении, определяется как соединение любых двух, трех и более олефинов, в которых число двойных связей ограничено двумя. Реагенты, которые могут использоваться в процессе тримеризации в соответствии с настоящим изобретением представляют собой олефиновые соединения, которые могут а) реагировать сами с собой, то есть тримеризоваться с образованием необходимых продуктов, так, например, взаимодействие этилена с этиленом может привести к 1-гексену, а взаимодействие 1,3-бутадиена с самим собой - к 1,5-циклооктадиену; и/или в) олефиновые соединения, которые могут реагировать с другими олефиновыми соединениями, то есть стримеризоваться, давая необходимые продукты, как например, сотримеризация этилена и гексена может привести у к 1-децену и/или 1-тетрадецену, сотримеризация этилена и 1-бутадиена дает 1-октен, сотримеризация 1-децена и этилена может привести к 1-тетрадецену и/или 1-докозену, или сотримеризация, 1-8-бутадиена и, 1,5-гексадиена может привести к 1,5-циклооктадекадиену. Например, число олефиновых связей в комбинации трех этиленовых фрагментов уменьшается вдвое до одной олефиновой связи в 1-гексене. В другом примере число олефиновых связей при соединении двух 1,3-бутадиеновых фрагментов уменьшается вдвое до двух олефиновых связей. Используемый в описании термин "тримеризация" подразумевает включение в это понятие димеризации олефинов также, как и термин "сотримеризация".
Олефиновые соединения, которые могут тримеризоваться, содержат от приблизительно 2 до приблизительно 30 атомов углерода и по меньшей мере одну олефиновую двойную связь. Примерами таких моноолефиновых соединений являются, но ими не ограничиваются, ациклические и циклические олефины, такие, как, например, этилен, пропилен, 1-бутен, 2-бутен, изобутилен, 1-пентан, 2-пентен, 1-гексен, 2-гексен, 3-гексен, 1-гептен, четыре нормальных октена, четыре нормальных нонена и смеси любых двух и более названных соединений. Примерами диолефиновых соединений являются, но не ограничиваются ими, 1,3-бутадиен, 1,4-пентадиен и 1,5-гексадиен. Если в качестве реагентов используются разветвленные и/или циклические олефины, то, хотя теоретически они не вступают в химическую реакцию, полагают, что процессу тримеризации могли мешать стерические затруднения. Поэтому разветвленный и/или циклический фрагмент(ы) олефина предпочтительно должен находиться на расстоянии от углерод-углеродной двойной связи.
Каталитические системы, получаемые в соответствии с настоящим изобретением, предпочтительно используются в качестве каталитических систем для тримеризации.
Условия реакции.
Продукты реакции, то есть тримеры и/или полимеры, могут быть получены на каталитических системах настоящего изобретения путем реакций в растворе, реакций в суспензии и/или по методикам газофазной реакции с использованием обычного оборудования и обычных процессов контактирования. На контактирование мономеров с каталитической системой или с каталитической системой для полимеризации и сокаталитической системой тримеризации/олигомеризации можно влиять любым способом, известным в области гомогенных (жидких) или гетерогенных (твердых) каталитических систем. Одним из удобных методов являются суспендирование каталитической системы в органической среде и перемешивание смеси с целью сохранения каталитической системы в суспензии в течение всего процесса тримеризации, олигомеризации и/или полимеризации. Также могут быть использованы другие методы контактирования, например, кипящий слой, гравитационный слой или неподвижный слой. Один из подходящих методов для систем катализатор/сокатализатор заключается в суспендировании каталитической системы для полимеризации в органической среде и перемешивании смеси для сохранения каталитической системы в виде суспензии в течение всего процесса тримеризации и/или полимеризации. Затем может быть добавлена заявляемая сокаталитическая система. Каталитическая система для полимеризации и заявляемая сокаталитическая система предпочтительно могут одновременно подаваться в реактор полимеризации в виде одного или более питающих потоков каталитической и/или сокаталитической системы. Другие методы контактирования, такие как кипящий слой, гравитационный слой или неподвижный слой, также могут быть использованы для всех этих каталитических систем.
Температура и давление реакции могут иметь любое значение, при которых происходит тримеризация, олигомеризация и/или полимеризация олефиновых компонентов. Обычно температура реакции лежит в интервале от приблизительно 0oC до приблизительно 250oC. Предпочтительно температура реакции составляет от приблизительно 60oC до приблизительно 200oC и наиболее предпочтительно от 80oC до 150oC. Реакционное давление обычно лежит в интервале от приблизительно атмосферного до приблизительно 175 атм (2500 фунтов/кв.дюйм). Предпочтительно давление составляет от приблизительно атмосферного до приблизительно 70 атм (1000 фунтов/кв. дюйм) и наиболее предпочтительно от 21 до 49 атм (300 до 700 фунтов/кв.дюйм).
Слишком низкая температура реакции может привести к образованию слишком большого количества нежелательных нерастворимых продуктов, а слишком высокая температура может вызвать разложение каталитической системы и продуктов реакции. При слишком низком реакционном давлении может снизиться активность каталитической системы. Слишком высокое давление может привести к образованию слишком большого количества нежелательных нерастворимых продуктов.
При необходимости в реактор может быть добавлен водород, чтобы ускорить реакцию и/или повысить каталитическую активность системы.
Каталитические системы в соответствии с настоящим изобретением особенно приемлемы для использования в процессах тримеризации и/или олигомеризации. Процесс суспендирования обычно осуществляют в инертном разбавителе (среде), таком как парафин, циклопарафины или ароматический углеводород. Примеры разбавителей для реактора включают, но не ограничиваются только ими, изобутан и циклогексан. Изобутан может уменьшать набухание полимерного продукта. Однако гомогенная сокаталитическая система для тримеризации/олигомеризации больше растворима в циклогексане. Поэтому предпочтительным разбавителем для гомогенного процесса тримеризации и олигомеризации является циклогексан, а предпочтительным разбавителем для гетерогенного процесса тримеризации или олигомеризации является изобутан. Если реагентом является преимущественно этилен, то используется температура реакции в интервале от приблизительно 0oC до приблизительно 300oC. Предпочтительно, если преимущественным реагентом является этилен, температура реакции составляет приблизительно 60-150oC.
Для того, чтобы получить полимер с желаемым набором оптимальных свойств, таких, например, как плотность, индекс расплава, индекс расплава при повышенном напряжении сдвига и молекулярный вес, в реакторе полимеризации может присутствовать любое количество каталитической системы. Обычно может присутствовать до приблизительно 40 мас. ч. нанесенной, то есть гетерогенной сокаталитической системы из расчета на каждую весовую часть каталитической системы. Для получения полимера с необходимыми физическими и рабочими характеристиками присутствует предпочтительно приблизительно от 1 до приблизительно 25 мас.ч. сокаталитической системы из расчета на 1 мас.ч. каталитической системы для полимеризации, и наиболее предпочтительно 3 - 15 мас.ч. сокаталитической системы на 1 мас.ч. каталитической системы для полимеризации.
Продукты. Олефиновые и/или полимерные продукты данного изобретения находят широкое применение, например, в качестве мономеров при получении гомополимеров, сополимеров и/или трехзвенных полимеров (терполимеров). Полимерные продукты данного изобретения также находят широкое применение, например, полиэтилен.
Дополнительное понимание данного изобретения и его преимуществ может быть получено из следующих примеров.
Примеры.
В описании и в примерах используются различные аббревиатуры. Ниже дан их список:
Триэтилалюминий - ТЭА, Al(C2H5)3
Диэтилалюминийхлорид - ДЭАХ, Al(C2H5)2Cl;
2-Этилгексаноат хрома (III) - Cr(ЭГ)3, CrЭГ,CrЭГ3
Пирролид водорода - пиррол, Py, PyH, C4H5N
Ацетилацетонат хрома (III) - Cracac, Cracac3, Cr(acac)3, Cr(C5H7O2)3
Пирролид хрома (III) - CrPy2, [Na(C4H10O2)2][Cr(C4H4N)3 Cl(C4 H10O2)] [Na(ДМЭ)2] , [Cr(C4H4N)3Cl(ДМЭ)], [Na(ДМЭ)2] [Cr(Py)3Cl (ДМЭ)], продукт V, соединение V.
Трис-тетрагидрофуран-хлорид хрома (III) - CrCl3ТГФ3, CrCl3(ТГФ)3.
2,5-Диметилпиррол - 2,5-диметилпирролид водорода, C6H9N, 2,5-ДМП.
Бутен C4 =
1-Гексен - 1-C6 =
Гексен - C6 =
Октен - C8 =
Децен - C10 =
Додецен - C12 =
Тетрадецен -C14 =
Пример 1.
Цикл 1001. В толстостенной трубке на 25 мл взвешивали 0.14 г (0,29 ммоля) 2-этилгексаноата хрома (III) (CrЭГ3), [Cr(C8H15O2)3]. Трубку закрывали самозапирающейся круглой крышкой. Добавляли с помощью шприца 0.062 мл (0.89 ммоля) пиррола (PyH), [C4H5N] и циклогексан, который используется в качестве разбавителя, и получали раствор с общим объемом приблизительно 8 мл.
Затем для образования каталитической системы в автоклав на 1 л, содержащий 300 мл циклогексана, при противотоке этилена (химически чистого) добавляли 0.9 мл 1.1 М раствора (0.99 ммоля) триэтилалюминия (ТЭА) в гептане и аликвоту в 0.9 мл раствора CrЭГ3/Py. Реактор закрывали и прекращали подачу этилена до тех пор, пока температура в реакторе не поднималась до 80oC. Давление этилена поднимали до общего давления реакции 38,5 атм (550 фунтов/кв. дюйм). Затем подавали по потребности этилен в течение 30 мин, которые составляли время цикла. По окончании цикла пробу получаемой жидкой реакционной смеси анализировали с помощью капиллярной газовой хроматографии. Оставшуюся жидкую реакционную смесь упаривали и определяли количество твердого продукта.
Результаты представлены ниже в табл.1.
Цикл 1002. Следовали методике цикла 1001 за исключением того, что для образования раствора с общим объемом 10 мл было добавлено 8 мл 1.1 М раствора (8.8 ммолей) ТЕЭ в нептане непосредственно к раствору CrЭГ3/PyH,а не в реактор. Аликвоту в 0.7 мл раствора CrЭГ3/PyH/ТЭА добавляли в автоклав. Дополнительно ТЭА в реактор не вводили.
Результаты представлены ниже в табл.1.
Цикл 1003. Следовали методике цикла 1002 за исключением того, что вместо CrЭГ3 использовали 0.10 г (0.29 ммоля) ацетилацетонана хрома (III) (Cracac3), [Cr(C5H7O2)3] и 6 мл 1.1 М раствора (6.6 ммоля) ТЭА в гептане для приготовления раствора Crocac3/PyH/ТЭА (общий объем 8 мл). В автоклав добавляли 1.4 мл раствора Cracac3/PyH/ТЭА.
Результаты представлены ниже в табл.1.
Цикл 1004. Следовали методике, описанной в цикле 1001 за исключением того, что 0.9 мл 1 М раствора (0.9 ммоля) диэтилалюминийхлорида (ДЭАХ), [AlCl(C2H5)2] в гексане добавляли к раствору CrЭГ3/PyH и получали раствор CrЭГ3/PyH/ДЭАХ. В автоклав добавляли 0,65 мл раствора CrЭГ3/PyH/ДЭАХ и 0.9 мл 1.1 М раствора (0.99 ммолей) ТЭА в гептане.
Результаты представлены ниже в табл.1.
Цикл 1005. Следовали методике цикла 1001 за исключением того, что 0.9 мл 1 М раствора (0.9 ммолей) ДЭАХ в гексане добавляли к раствору CrЭГ3/PyH и получаемый раствор CrЭГ3/PyH/ДЭАХ выдерживали 1 сутки в атмосфере сухого азота при комнатной температуре и атмосферном давлении. Затем 0.65 мл выдержанного раствора CrЭГ3/PyH/ДЭАХ и 0.9 мл 1.1 М раствора (0.99 ммолей) ТЭА в гептане добавляли в автоклав.
Результаты представлены ниже в табл.1.
Цикл 1006. Следовали методике цикла 1001 за исключением того, что раствор был приготовлен с использованием 0.13 мл пиррола. Дополнительно, наряду с ТЭА, в реактор добавляли 1.0 мл 0.1 М раствора (0.1 ммоля) ДЭАХ в гексане. Использовали 0.9 мл раствора CrЭГ3/PyH.
Результаты представлены ниже в табл.1.
Цикл 1007. Следовали методике цикла 1003 за исключением того, что использовали 3 мл 0.9 М раствора (5.7 ммоля) ТЭА в толуоле и толуол также использовали вместо гексана в качестве разбавителя при приготовлении раствора CrЭГ3/PyH/ТЭА. В реакторе, следовательно, находился избыток толуола. Использовали 0.9 мл раствора CrЭГ3/PyH/ТЭА.
Результаты представлены ниже в табл.1.
Цикл 1008. Следовали методике цикла 1002 за исключением того, что вместо CrЭГ3 использовали 0.10 г пирролида хрома (III), (CrPy3), [Cr(C4H4N)3ClNa(C4H10O2)3] (0.17 ммоля) и раствор готовили с использованием 0.04 мкл (0.52 ммоля) PyH и 3.5 мл 1.1 М раствора (3.85 ммоля) ТЭА в гептане. Конечный объем раствора составлял приблизительно 5 мл. Использовали 1.0 мл раствора CrPy3/PyH/ТЭА.
Результаты представлены ниже в табл.1.
Цикл 1009. Следовали методике цикла 1008 за исключением того, что использовали 1.8 мл 1.9 М раствора (3.42 ммоля) ТЭА в толуоле и вместо циклогексана использовали толуол при получении раствора CrPy3/PyH/ТЭА. Следовательно, в реакторе находился избыток толуола. Использовали 1.4 мл раствора CrPy3/PyH/ТЭА.
Результаты представлены ниже в табл.1.
Цикл 1010. Следовали методике цикла 1008 за исключением того, что при приготовлении раствора CrPy3/ТЭА не добавляли чистый PyH. Использовали 1.4 мл раствора CrPy3/ТЭА в циклогексане.
Результаты представлены ниже в табл.1.
Цикл 1011. Следовали методике цикла 1009 за исключением того, что при приготовлении раствора CrPy3/ТЭА не добавляли чистый PyH. Использовали 1.4 мл раствора CrPy3/ТЭА в толуоле. Следовательно, в реакторе присутствовал избыток толуола.
Результаты представлены ниже в табл.1.
Пример II.
Следует отметить, что результаты, представленные в таблице XXIII из примера X, и результаты в таблице XXIV из примера XI нельзя сравнивать непосредственно, так как реакции проводили в различных реакторах при различных условиях, используя в качестве сырья различные этилен и циклогексан, а также различные разбавители. Однако прямое сравнение в пределах каждого примера может быть осуществлено.
Цикл 2001. Смешивали 0.30 г (0.62 ммоля) 2-этилгексаноата хрома (III) (CrЭГ3) (10.15 мас.% Cr) с 0.12 мл (1.73 ммоля) чистого пиррола (PyH) в 10 мл толуола. Добавляли 2.8 мл 1.9 М раствора (5.32 ммоля) триэтилалюминия (ТЭА) в толуоле и раствор CrЭГ3/PyH/ТЭА перемешивали в течение 30 мин в атмосфере сухого азота при комнатной температуре и атмосферном давлении. Темно-коричневый раствор CrЭГ3/PyH/ТЭА фильтровали и избыток толуола удаляли с помощью вакуумной десорбции. Получали 1.0 мл темно-коричневого масла, которое использовали в качестве каталитической системы. В автоклав на 2 л, содержащий 1.2 л циклогексана, при 80oC добавляли при противотоке этилена 0.5 мл (0.15 г CrЭГ3; 15.2 мг Cr) каталитической системы и 4.0 мл нонана (внутренний стандарт реактора). Давление в реакторе поднимали до 38.5 атм (550 фунтов/кв. дюйм) с помощью этилена. Время цикла 30 мин, причем по мере потребности подавали этилен.
Результаты представлены в табл.2.
Цикл 2002. Следовали методике цикла 2001 за исключением того, что кроме раствора CrЭГ3/PyH/ТЭА добавляли диэтилалюминийхлорид.
Смешивали 0.53 г (1.10 ммоля) CrЭГ3 (10.15 мас.% Cr) с 0.52 мл (7.5 ммоля) чистого PyH в 15 мл толуола и перемешивали 5 мин. Добавляли 9.0 мл 1.9 М раствора (17.1 ммоля) ТЭА в толуоле и раствор CrЭг3/PyH/ТЭА перемешивали в течение ночи в атмосфере сухого азота при комнатной температуре и атмосферном давлении. Из полученного темно-коричневого раствора, удаляли избыток толуола путем вакуумной десорбции и получали 2.5 мл темно-коричневого масла. Смешивали 0.5 мл (10.8 мг, 0.2 ммоля Cr) темно-коричневого масла с 1.0 мл 0.87 М раствора (0.87 ммоля) диэтилалюминийхлорида (ДЭАХ) в нонане и раствор CrЭГ/PyH/ТЭА/ДЭАХ перемешивали в течение ночи в атмосфере сухого азота при комнатной температуре и атмосферном давлении. Полученный продукт использовали в качестве каталитической системы.
Непосредственно в реактор на 2 л, содержащий 1.2 л циклогексана, при 80oC при противотоке этилена загружали 1.3 мл (9.4 мг, 0.18 ммоля Cr) каталитической системы и 4.0 мл нонана (внутренний стандарт реактора). Давление в реакторе с помощью этилена поднимали до 38,5 атм (550 фунтов/кв.дюйм), время реакционного цикла составляло 30 мин, причем по мере необходимости подавали этилен.
Результаты представлены ниже в табл.2.
Цикл 2003. Смешивали 0.33 г (0.68 ммоля) CrЭГ3 (10.15 мас.% Cr) с 13 мл (1.87 ммоля) чистого PyH в 10 мл толуола и перемешивали в течение 5 мин. Добавляли 1.9 мл 1 М раствора (1.9 ммоля) ДЭАХ в гексане и раствор CrЭГ3/PyH/ДЭХА, перемешивали в течение 30 мин в атмосфере сухого азота при комнатной температуре и атмосферном давлении. Получали светлый желто-зеленый раствор. Добавляли 5.1 мл 1.9 М раствора (9.7 ммоля) диэтилалюминий хлорида (ДЭАХ) в толуоле и раствор CrЭГ3/PyH/ДЭАХ/ТЭА перемешивали в течение 0.5 ч, получали темный желто-коричневый раствор. Избыток толуола и гексана удаляли из темного желто-коричневого раствора CrЭГ3/PyH/ДЭАХ/ТЭА с помощью вакуумной десорбции, получали темное желто-коричневое масло. Темное желто-коричневое масло растворяли в циклогексане и доводили общий объем до 25 мл, использовали в качестве каталитической системы (1.32 мг Cr/мл). В реактор на 2 л, содержащий 1.2 л циклогексана, при 80oC при противотоке этилена загружали 7.0 мл (9.2 мг Cr, 0.178 ммоля Cr) каталитической системы и 4.0 мл нонана (внутренний стандарт реактора). Затем давление в реакторе поднимали до 38,5 атм (550 фунтов/кв.дюйм) с помощью этилена. Время реакции - 30 мин, причем в этот период при необходимости подавали этилен.
Результаты приведены ниже в табл.2.
Цикл 2004. Следовали методике, описанной в цикле 2002, за исключением того, что раствор CrЭГ3/PyH/ТЭА/ДЭАХ разбавляли циклогексаном до загрузки в реактор, а в реактор добавляли водород (H2)/0,35 атм = 50 фунтов/кв.дюйм/ до того, как давление в реакторе поднимали до необходимого с помощью этилена.
Смешивали 0,30 г (0.62 ммоля) CrЭГ3 (10.15% С) с 0.12 мл (1.73 ммоля) чистого PyH в 10 мл толуола. Добавляли 1.7 мл 1 М раствора ДЭАХ (1.7 ммоля) в гексане и раствор CrЭГ3/PyH/ДЭАХ перемешивали в течение 5 мин в атмосфере сухого азота и при комнатной температуре и атмосферном давлении. Добавляли 1.8 мл 1.9 М раствора (3.42 ммоля) ТЭА в толуоле и раствор CrЭГ3/PyH/ДЭАХ/ТЭА перемешивали в течение 30 мин в атмосфере сухого азота при комнатной температуре и атмосферном давлении. Получаемый темно-коричневый раствор фильтровали и избыток толуола и гексана удаляли с помощью вакуумной десорбции, получали 0.8 мл темно-желтого-коричневого масла, которое использовали в качестве каталитической смеси. В реактор на 2 л, содержащий 1.2 л циклогексана, при 80oC при противотоке этилена загружали 0.4 мл (15.2 мг Cr, 0.29 ммоля Cr) каталитической системы и 4.0 мл нонана (внутренний стандарт реактора). В реактор подавали водород (H2) под давлением 3.5 атм (50 фунтов/кв. дюйм), а затем с помощью этилена поднимали давление до 38,5 атм (550 фунтов/кв.дюйм). Время реакции - 30 мин, в течение которого при необходимости подавали этилен.
Результаты приведены ниже в табл.2.
Цикл 2005. В колбе Шленка на 500 мл смешивали 1.98 г (3.4 ммоля) CrPy3 (11.1 мас. % Cr) с 40 мл толуола и 54 мл 1.9 М раствора (102.6 ммоля) ТЭА в толуоле. Получаемую темно-коричневую реакционную смесь перемешивали в течение 1 ч в атмосфере сухого азота при комнатной температуре и атмосферном давлении. Избыток толуола удаляли с помощью вакуумной десорбции, при этом получали 13 мл темного желто-коричневого масла и небольшое количество осадка. Темное желто-коричневое масло отделяли, собирая с осадка с помощью шприца, и использовали в качестве каталитической системы. Разбавляли 2.0 мл каталитической системы 27 мл циклогексана и перед использованием выдерживали в течение 3 дней в атмосфере сухого азота при комнатной температуре и атмосферном давлении.
Непосредственно в автоклав на 2 л, содержащий 1.2 л циклогексана, при 80oC при противотоке этилена загружали 3.0 мл (9.3 мг, 0.18 ммоля Cr) каталитической системы в виде раствора в циклогексане и 4.0 мл нонана (внутренний стандарт реактора). Затем давление в реакторе поднимали с помощью этилена до 38,5 атм (550 фунтов/кв.дюйм). Время реакции - 30 мин, причем по мере надобности осуществляли подачу этилена.
Результаты приведены ниже в табл.2
Цикл 2006. Следовали методике цикла 2005 за исключением того, что использовали меньше реагентов и меньшее время выдерживания.
В колбе Шленка на 500 мл смешивали 0.25 г (0.432 ммоля) CrPy3 (11.1 мас. % Cr) с 10 мл толуола и 3.4 мл 1.9 М раствора (6.46 ммоля) ТЭА в толуоле. Получаемую темно-коричневую реакционную смесь перемешивали 30 мин в атмосфере сухого азота при комнатной температуре и атмосферном давлении. Избыток толуола удаляли с помощью вакуумного десорбирования, получали при этом темно-коричневое масло. Все темно-коричневое масло разбавляли циклогексаном до общего объема 25 мл и получали раствор, содержащий 1.11 мг Cr/мл, который использовали в качестве каталитической системы.
Непосредственно в автоклав на 2 л, содержащий 1.2 л циклогексана, при 80oC при противотоке этилена загружали 8.0 мл (8.88 мг, 0.171 ммоля Cr) каталитической системы в виде раствора в циклогексане и 4.0 мл нонана (внутренний стандарт реактора). Затем давление в реакторе поднимали с помощью этилена до 38,5 атм (550 фунтов/кв.дюйм), время реакции составляло 30 мин, причем при необходимости осуществляли подачу этилена.
Результаты приведены ниже в табл.2.
Цикл 2007. Следовали методике цикла 2005 за исключением того, что в реакторе тримеризации находился избыток толуола.
В колбе Шленка на 500 мл смешивали 1.98 г (3,4 ммоля) CrPy3 (11.1 мас.% Cr) с 40 мл толуола и 54 мл 1.9 М раствора (102 ммоля) ТЭА в толуоле. Получаемую темно-коричневую реакционную смесь перемешивали в течение 1 ч в атмосфере сухого азота при комнатной температуре и атмосферном давлении. Избыток толуола удаляли с помощью вакуумной десорбции, получали 13 мл темного желто-коричневого масла и небольшое количество слабоокрашенного осадка. Темное желто-коричневое масло выделяли, собирая его с осадка с помощью шприца, и использовали в качестве каталитической системы. Разбавляли 2.0 мл каталитической системы 27 мл циклогексана и перед применением выдерживали в течение 3-х дней в атмосфере сухого азота при комнатной температуре и атмосферном давлении.
Непосредственно в автоклав на 2 л, содержащий 1.2 л циклогексана, при 80oC при противотоке этилена загружали 0.5 мл (8.5 мг, 0.163 ммоля Cr) каталитической системы в виде раствора в циклогексане, 4.5 мл толуола и 4.0 мл нонана (внутренний стандарт реактора). Давление в реакторе поднимали до 38,5 атм (550 фунтов/кв.дюйм) с помощью этилена, время реакции - 30 мин, причем при необходимости осуществляли подачу этилена.
Результаты приведены ниже в табл.2.
Цикл 2008.
Смешивали 0.28 г (0.802 ммоля) Cracac3 с 0.17 мл (2,45 ммоля) чистого пиррола в 10 мл толуола и перемешивали в атмосфере сухого азота при комнатной температуре и атмосферном давлении в течение 5 мин. Затем добавляли 6.3 мл 1.9 М раствора (12.0 ммолей) ТЭА в толуоле. Получаемую темно-коричневую реакционную смесь перемешивали в течение 30 мин в атмосфере сухого азота при комнатной температуре и атмосферном давлении. Избыток толуола удаляли с помощью вакуумной десорбции и получали темное желто-коричневое масло. Все темное желто-коричневое масло разбавляли циклогексаном до объема 25 мл, получали раствор, содержащий 0.0112 г Cracac3/мл, который использовали в качестве каталитической системы.
В автоклав на 2 л, содержащий 1.2 л циклогексана, при 80oC при противотоке этилена загружали 7.0 мл (15.2 мг, 0.293 ммоля Cr) каталитической системы в виде раствора в циклогексане и 4.0 мл нонана (внутренний стандарт реактора). Давление в реакторе поднимали до 38,5 атм (550 фунтов/кв. дюйм), время реакции - 30 мин, причем при необходимости осуществляли подачу этилена.
Результаты приведены ниже в табл.2.
Цикл 2009. Следовали методике цикла 2008 за исключением того, что в качестве источника хрома использовали нафтенат хрома (III).
Смешивали 0.33 г (0.508 ммоля) CrНф3 (8.0 мас.% Cr) с 0.12 мл (1.73 ммоля) чистого пиррола в 10 мл толуола и перемешивали в атмосфере сухого азота при комнатной температуре и атмосферном давлении в течение 5 мин. Затем добавляли 4.6 мл 1.9 М раствора (8.74 ммоля) ТЭА в толуоле. Полученную темно-коричневую реакционную смесь перемешивали в течение 30 мин в атмосфере сухого азота при комнатной температуре и атмосферном давлении. Избыток толуола удаляли с помощью вакуумной десорбции, получали темное желто-коричневое масло. Все темное желто-коричневое масло разбавляли циклогексаном до объема 25 мл, получали раствор, содержащий 1.056 мг Cr/мл, который использовали в качестве каталитической системы.
В автоклав на 2 л, содержащий 1.2 л циклогексана, при 80oC при противотоке этилена загружали 7.0 мл 7.39 мл, 0.142 ммоля Cr) каталитической системы в виде раствора в циклогексане и 4.0 мл нонана (внутренний стандарт реактора). давление в реакторе с помощью этилена поднимали до 38,5 атм (550 фунтов/кв. дюйм), время реакции - 30 мин, причем при необходимости осуществляли подачу этилена.
Результаты приведены ниже в табл.2.
Цикл 2010. Следовали методике цикла 2008 за исключением того, что в качестве источника хрома использовали хлорид хрома (III).
Смешивали 0.41 г (1.09 ммоля) CrCl3ТГФ3 с 0,23 мл (3.32 ммоля) чистого пиррола в 10 мл толуола и перемешивали в атмосфере азота при комнатной температуре и атмосферном давлении в течение 5 мин. Затем добавляли 8,8 мл 1.9 М раствора (16.3 ммоля) ТЭА в толуоле. Полученную темно-коричневую реакционную смесь перемешивали в атмосфере сухого азота при комнатной температуре и атмосферном давлении в течение 30 мин. Избыток толуола удаляли с помощью вакуумной десорбции, получали темное желто-коричневое масло. К темному желто-коричневому маслу добавляли 7.5 мл нонана и полученный раствор разбавляли циклогексаном до общего объема 25 мл, получали раствор, содержащий 0.0164 г CrCl3ТГФ3/мл. Раствор фильтровали и фильтрат использовали в качестве каталитической системы.
Непосредственно в автоклав на 2 л, содержащий 1.2 л циклогексана, при 80oC при противотоке этилена загружали 5.0 мл (11.38 мг, 0.219 ммоля Cr) каталитической системы в виде раствора в циклогексане и нонане и 2.5 мл нонана (внутренний стандарт реактора). Давление в реакторе повышали до 38,5 атм (550 фунтов/кв. дюйм), время реакции - 30 мин, причем по мере необходимости осуществляли подачу этилена.
Результаты приведены ниже в табл.2.
Цикл 2011. Следовали методике цикла 2005 за исключением того, что в реактор тримеризации загружали избыток гексана.
В колбе Шленка на 500 мл смешивали 1.98 г (3.4 ммоля) CrPy3 (11.1 мас.% Cr) с 40 мл толуола и 54 мл 1.9 М раствора (102.6 ммоля) ТЭА в толуоле. Получаемую темно-коричневую реакционную смесь перемешивали в течение 1 ч в атмосфере сухого азота при комнатной температуре и атмосферном давлении. Избыток толуола удаляли с помощью вакуумной десорбции, получали 13 мл темно желто-коричневого масла и небольшое количество слабо окрашенного осадка. Темное желто-коричневое масло отделяли, собирая его с осадка с помощью шприца, и использовали в качестве каталитической системы. Разбавляли 2 мл каталитической системы 27 мл циклогексана и перед использованием выдерживали 3 дня в атмосфере сухого азота при комнатной температуре и атмосферном давлении.
В автоклав на 2 л, содержащий 1.2 л циклогексана, при 80oC при противотоке этилена загружали 1.0 мл (16.9 мг, 0.325 ммоля Cr) каталитической системы в виде раствора в циклогексане, 55 мл 1-гексена и 4.0 мл нонана (внутренний стандарт реактора). Давление в реакторе поднимали до 38,5 атм (550 фунтов/кв. дюйм) с помощью этилена, время реакции - 30 мин, причем при необходимости осуществляли подачу этилена.
Результаты приведены ниже в табл.2.
Цикл 2012. Следовали методике цикла 2005 за исключением того, что в качестве источника хрома использовали пирролид хрома (II) (соединение I).
Смешивали 0.30 г (приблизительно 0.85 ммоля) соединения I (CrPy10ТГФ4) с 10 мл толуола и 6.7 мл 1.9 М раствора (12.7 ммоля) ТЭА в толуоле. Получаемую темно-коричневую реакционную смесь перемешивали в течение 30 мин в атмосфере сухого азота при комнатной температуре и атмосферном давлении. Избыток толуола удаляли вакуумной десорбцией, получали темное желто-коричневое масло и небольшое количество слабо окрашенного осадка. Темное желто-коричневое масло фильтровали и фильтрат разбавляли до общего объема 25 мл циклогексаном, получали раствор, содержащий 0.012 г соединения I (CrPy10ТГФ4), который использовали в качестве каталитической системы.
В автоклав на 2 л, содержащий 1.2 л циклогексана, при 80oC при противотоке этилена загружали 7.0 мл каталитической системы в виде раствора в циклогексане и 4.0 мл нонана (внутренний стандарт реактора). Давление в реакторе поднимали до 38.5 атм (550 фунтов/кв.дюйм) с помощью этилена, время реакции - 30 мин, причем при необходимости осуществляли подачу этилена.
Результаты представлены ниже в табл.2.
Пример III.
Цикл 3001. Смешивали 0,21 г (0.601 ммоля) Cracac3 с 0,12 мл (1,73 ммоля) чистого пиррола и 15 мл толуола. Полученный раствор перемешивали в атмосфере сухого азота при комнатной температуре и атмосферном давлении в течение 5 мин. Затем добавляли 6 мл 1.9 М раствора (11.4 ммоля) ТЭА в толуоле. Получаемую темно-коричневую реакционную смесь перемешивали 5 мин в атмосфере сухого азота при комнатной температуре и атмосферном давлении. Затем добавляли 2 г алюмофосфатного носителя (мольное соотношение P/Al 0,4, активирован при 700oC), приготовленного в соответствии с Пат. США 4364855 (1982), который уже упоминался, и полученную суспензию перемешивали в течение 12 ч. Продукт отфильтровывали, по меньшей мере дважды промывали аликвотами по 10 мл толуола и пентана до тех пор, пока фильтрат не переставал окрашиваться, осадок сушили в вакууме. Высушенный продукт использовали в качестве твердой каталитической системы на носителе.
В автоклав на 2 л, содержащий 1 л изобутана, добавляли при противотоке этилена 2.1022 г твердой каталитической системы. До загрузки катализатора в реактор добавляли 0.25 мл (16.5 мас.%) раствора ТЭА в нонане для того, чтобы нейтрализовать какие-либо яды в этиленовом сырье. Реактор закрывали и прекращали подачу этилена до тех пор, пока температура реактора не достигнет необходимого значения, например, 90oC. Затем давление этилена повышали до 38,5 атм (550 фунтов/кв.дюйм) и в течение 30 мин этилен подавали до потребности. В конце реакции отбирали пробу жидкой реакционной смеси и анализировали с помощью газовой хроматографии. Оставшуюся реакционную смесь упаривали и определяли количество твердого продукта.
Результаты представлены ниже в табл.3.
Цикл 3002. Следовали методике цикла 3001 за исключением того, что добавляли диэтилалюминийхлорид к раствору Cracac3/PyH наряду с ТЭА до добавления алюмофосфатного неорганического оксида.
В пробирке на 30 мл с завинчивающейся крышкой взвешивали 0.21 г Cracac3 (0,6 ммоля). Добавляли 0.12 мл PyH (1,73 ммоля) и 15 мл толуола, получаемый раствор закрывали и перемешивали в течение 5 мин. Затем продолжали перемешивание, добавляли 6 мл 1.9 М раствора (11.4 ммоля) ТЭА в толуоле. Затем раствор Cracac3/PyH/ТЭА перемешивали в течение 5 мин, добавляли 2.4 мл 1М раствора (2.4 ммоля) ДЭАХ в гексане и раствор Cracac3/PyH/ТЭА/ДЭАХ перемешивали 5 мин. Добавляли 2 г алюмофосфатного носителя (мольное соотношение P/Al 0,4, активирован при 700oC), приготовленного по методике Пат. США 4364855 (1982), а получаемую суспензию перемешивали приблизительно 12 ч. Продукт отфильтровывали и промывали по меньшей мере дважды аликвотами толуола и пентана по 10 мл, пока получаемый фильтрат не переставал окрашиваться. Осадок сушили в вакууме. Высушенный продукт использовали в качестве твердой каталитической системы на носителе.
В автоклав на 2 л, содержащий 1 л изобутана, при противотоке этилена добавляли 0.5048 г твердой каталитической системы. До загрузки катализатора добавляли 3.0 мл 1.6 %-ного раствора ТЭА в нонане для того, чтобы нейтрализовать яды в этиленовом сырье. Реактор закрывали и прекращали подачу этилена, пока температура в реакторе не достигнет необходимой температуры, например, 90oC. Давление этилена в реакторе поднимали до 38,5 атм (550 фунтов/кв.дюйм), время реакции 30 мин, при этом по потребности подавали этилен. По окончании реакции отбирали небольшую пробу жидкой реакционной смеси и анализировали с помощью газовой хроматографии. Полученную реакционную смесь упаривали и определяли количество твердого продукта. Расход этилена определяли с помощью калиброванного расходомера.
Результаты представлены ниже в табл.3.
Цикл 3003. Следовали методике цикла 3002 за исключением того, что в качестве источника хрома использовали CrЭГ3, а ароматический растворитель не использовали в процессе приготовления каталитической системы. Кроме того, каталитическая система на носителе была приготовлена в реакторе in situ.
Раствор CrЭГ3/PyH готовили путем смешения 0.33 г (0.69 ммоля) CrЭГ3 с 0.26 мл (3.75 ммоля) PyH в 16 мл пентана и выдерживали перед употреблением в течение 4-х дней в атмосфере сухого азота при комнатной температуре и атмосферном давлении. В автоклав на 2 л при комнатной температуре и противотоке этилена загружали 0.40 г алюмофосфатного носителя (мольное соотношение P/Al 0.9, активирован при 700oC), который приготовлен в соответствии с методикой Пат. США 4364855 (1982) и 2.0 мл 1М раствора (2.0 ммоля) ТЭА в гексане. Затем в реактор загружали 1 л циклогексана, 2.1 мл (4.32 мг, 0.083 ммоля Cr) раствора CrЭГ3/PyH и водород (H2) под давлением 3,5 атм (50 фунтов/кв.дюйм).
Результаты приведены ниже в табл.3.
Пример IV.
Цикл 4001. В толстостенной трубке на 100 мл взвешивали 3,5 г (10 ммолей Cracac3). В трубку помещали брусок для перемешивания и закрывали самозапирающейся крышкой. Шприцом добавляли 40 мл толуола и 2.1 мл (30 ммолей) PyH. Затем медленно добавляли 12 мл 2.5 М раствора (30.0 ммолей) н.-бутиллития в гексане. Образующийся осадок выделяли, промывали толуолом (2 х 10 мл) и циклогексаном (2 х 10 мл) до неокрашенного промывного фильтрата. Всего получали 5.59 г твердого продукта. Пир условиях, описанных в цикле 1001, использовали 0.5 мл 1.1 М раствора (0.55 ммолей) ТЭА в гептане и суспензию 38 мг твердого продукта в циклогексане.
Результаты представлены ниже в табл.4.
Цикл 4002. Следовали методике цикла 4001 за исключением того, что твердый катализатор готовили (всего 88 г) в толстостенной трубке на 25 мл, используя 0,349 г (1 ммоль) Cracac3, к мл толуола, 0,14 мл (2 ммоля) PyH и 0.8 мл 2.5 М раствора (2.0 ммоля) н.-бутиллития в гексане.
При условиях, описанных в цикле 1001, использовали 0.5 мл 1.1М раствора (0.55 ммоля) ТЭА и суспензию 16 г твердого продукта в циклогексане.
Результаты представлены ниже в табл.4.
Цикл 4003. В толстостенной трубке на 25 мл взвешивали 1.0 г алюмофосфатного носителя (мольное соотношение P/Al 0.4, активирован при 700oC), полученного по методике Пат. США 4364855 (1982), и 93 мг твердого продукта, описанного в цикле 4001. Трубку закрывали, шприцом добавляли 5 мл толуола и 3 мл 1.9 М раствора (5.7 ммоля) ТЭА в толуоле. Полученную суспензию выдерживали в течение суток. Твердый продукт выделяли, промывали толуолом и циклогексаном порциями по 10 мл, пока промывной раствор не переставал окрашиваться.
При условиях цикла 1001 использовали 0.5 мл 1.1 М раствора (0.55 ммоля) раствора ТЭА в гептане и суспензию 80 мг твердого продукта в циклогексане.
Результаты представлены ниже в табл.4.
Цикл 4004. В толстостенной трубке на 25 мл взвешивали 0.7 г алюмофосфатного носителя (мольного соотношение P/Al 0.4, активирован при 700oC), полученного по методике патента США 4364855 (1982), и 53 мг твердого продукта, описанного в цикле 4002. Трубку закрывали, шприцом добавляли 3.5 мл толуола и 2 мл 1.9 М раствора (3.8 ммоля) ТЭА в толуоле. Полученную суспензию выдерживали в течение суток. Твердый продукт выделяли, промывали толуолом и циклогексаном порциями по 10 мл, пока промывной раствор не переставал окрашиваться.
При условиях цикла 1001 использовали 0.5 мл 1.1 М раствора (0.55 ммоля) ТЭА в гептане и суспензию 78 мг твердого продукта в циклогексане.
Результаты представлены в табл.4.
Пример V.
Цикл 5001. В толстостенной трубке на 25 мл взвешивали 0.17 г 2,2,6,6-тетраметил-3,5-гептадионата хрома (III) (Cr(III)ТМГД) (0.28 ммоля). Трубку закрывали самозапирающейся круглой крышкой. Шприцем добавляли 0.06 мл пиррола (0.89 ммоля) и 0.17 ммоля (0.87 ммоля) чистого диизобутилалюминийхлорида (DuBAlCl), получали раствор Cr (III)ТМГД/DuBAlCl/Py, который разбавляли циклогексаном до общего объема 8 мл. В стеклянный сосуд, содержащий 100 мл циклогексана и 10 г бутадиена, добавляли 0.25 мл 1.1 М раствора (0.28 ммоля) ТЭА в гептане и 0.75 мл раствора Cr (III)NVUL/DuBAlCl/Py. Стеклянный сосуд помещали в термостатируемую баню с температурой 70oC при атмосферном давлении и перемешивали 16 ч. Через 16 ч отбирали небольшую пробу жидкой реакционной смеси и анализировали с помощью газовой хроматографии. Оставшуюся жидкую реакционную смесь упаривали и определяли количество твердого продукта. Результаты представлены ниже в табл.5.
Цикл 5002. Следовали методике цикла 5001 за исключением того, что в реакторе отсутствовал избыток алкилалюминиевого соединения, а катализатор был получен с использованием методики, описанной в цикле 3001, следующим образом.
Смешивали 0.21 г (0.601 ммоля) Cracac3 с 0.12 мл (1,73 ммоля) чистого пиррола и с 15 мл толуола. Полученный раствор перемешивали в атмосфере сухого азота при комнатной температуре и атмосферном давлении в течение 5 мин. Затем добавляли 6.0 мл 1.9 М раствора (11.4 ммоля) ТЭА в толуоле. Полученную темно-коричневую реакционную смесь перемешивали 5 мин в атмосфере сухого азота при комнатной температуре и атмосферном давлении. Затем добавляли 2 г алюмофосфатного носителя (мольное соотношение P/Al 0.4, активирован при 700oC), полученного по методике Пат. США 4364855 (1982), и полученную суспензию перемешивали в течение приблизительно 12 ч. Продукт отфильтровывали, промывали по меньшей мере дважды порциями толуола и пентана по 10 мл, пока промывной раствор не переставал окрашиваться, и сушили в вакууме. Высушенный продукт использовали в качестве каталитической системы на носителе.
Использовали загрузку катализатора 0.28 г в реакции бутадиена.
Пример VI.
В следующих циклах все каталитические системы были приготовлены в боксе, в атмосфере сухого азота, при комнатной температуре и атмосферном давлении. Соединения переходных металлов взвешивали и смешивали с тремя эквивалентами (0.062 мл) пиррола, 2 мл циклогексана в качестве растворителя и 6 мл 1.1 М раствора триэтилалюминия (ТЭА) в гептане. Полученный продукт встряхивали в течение от 5 мин до 16 ч.
Все циклы проводили в автоклаве на 1 л, содержащий 300 мл циклогексана. Разбавляли 1 мл жидких каталитических систем циклогексаном и добавляли при противотоке этилена (химически чистого) в реактор. Реактор закрывали, а подачу этилена прекращали до тех пор, пока температура в реакторе не поднимается до температуры реакции 80oC. Затем давление этилена в реакторе поднимали до 550 фунтов/кв.дюйм. Этилен подавали по потребности в течение 30 мин. При необходимости реактор нагревали так, чтобы температура оставалась без изменения, то есть 80oC.
В конце цикла отбирали пробу жидкой реакционной смеси и анализировали с помощью капиллярной газовой хроматографии на газовом хроматографе HP-5880, снабженном пламенно-ионизационным детектором и ДВ-1 колонкой (60 м, внутренний диаметр 0.25 мм, толщина пленки 0.25 мкм). Газовый хроматограф постепенно разогревали от 40 до 272oC при скорости нагрева 10 o/мин в течение 20 мин. В качестве внутреннего стандарта использовали циклогексан. Оставшуюся реакционную смесь упаривали и определяли количество твердого продукта.
Результаты представлены ниже в табл.6.
Данные в табл. показывают, что соединения других металлов могут использоваться при тримеризации, олигомеризации и/или полимеризации 1-олефинов. Из этих соединений металлов IINiacac2, цикл 6001 показал наилучшую активность и селективность при тримеризации.
Пример VII.
В следующем примере циклы 7001 - 7005 иллюстрируют влияние гидролиза металлалкилов до их использования, а также влияние присутствия или отсутствия пирролсодержащего соединения. Циклы 7006-7009 в сравнении с циклами 7010 - 7013, показывают влияние использования ненасыщенного углеводорода при приготовлении каталитической системы.
Циклы 7001- 7005.
В циклах 7001 - 7005 мольное соотношение элементарного хрома к элементарному алюминия и к лиганду (Cr:Al:Л) каталитических компонентов, добавляемых в реактор, составляет 1:30:10. В циклах 7001 - 7003 в качестве соединения хрома использовали Cr(ЭГ)3, а в циклах 7004 и 7005 - Cr(Py)3. В качестве алюминиевого соединения использовали триизобутилалюминий (Al (изо-Bu)3), который подвергался следующей обработке. К приблизительно 10%-ному (мас.) раствору триизобутилалюминия в гексане добавляли 1.0 молярный эквивалент дистиллированной воды, медленно, но в одну порцию, охлаждая при этом колбу, содержащую раствор, водой со льдом так, чтобы температура находилась в интервале от приблизительно 10oC до приблизительно 20oC. Раствор энергично перемешивали до и после добавления воды до прекращения выделения газа. В качестве лиганда использовали диметоксиэтан (ДМЭ).
Циклы 7001 - 7005 проводили в автоклаве на 2 л. Соединение хрома растворяли в 400 - 500 мл безводного н.-гептана и добавляли в реактор в атмосфере сухого азота. Затем добавляли требуемый объем перемешанного, обработанного как описано выше, 0.31 С раствора Al(изо-Bu)3 в гептане. Затем добавляли требуемый объем ДМЭ вместе с 5 мл нонана (внутренний стандарт реактора). Реактор закрывали и поднимали температуру до 80oC в цикле 7001 и до 95oC в циклах 7002 - 7005. Давление этилена поднимали до 38,5 атм (550 фунтов/кв. дюйм). Этилен при необходимости подавали в реактор в течение всего цикла - 25 мин в цикле 7001, 30 мин в цикле 7002 и 45 мин в циклах 7003 - 7005.
В конце каждого цикла отбирали пробу жидкой реакционной смеси и анализировали с помощью капиллярной газовой хроматографии на газовом хроматографе HP-5880, снабженном пламенно-конизационным детектором и ДВ-1 колонкой (60 м, внутренний диаметр 0.25 мм, толщина пленки 0.25 мкм). Газовый хроматограф постепенно разогревали от 40 до 272oC при скорости нагрева 10 o/мин в течение 20 мин. В качестве внутреннего стандарта использовали циклогексан. Оставшуюся реакционную массу упаривали и определяли количество твердого продукта.
Каталитические системы, используемые в циклах 7006 - 7013, готовили в соответствии со следующими методиками. Каталитические системы в циклах 7006 - 7009 готовили в присутствии толуола - ненасыщенного ароматического углеводорода. Каталитические системы в циклах 7010 - 7013 готовили в присутствии 1-гексена - ненасыщенного алифатического углеводорода.
Цикл 7006. Смешивали 3.72 г /Na(ДМЭ2/ /CrCl(Py)3 с 50 мл толуола. Медленно добавляли 26.4 мл чистого (93%) ТЭА и перемешивали в течение 30 мин. Получали суспензию темно-коричневого цвета. Избыток растворителя удаляли в вакууме, получали темное желто-коричневое масло и твердый продукт. Добавляли приблизительно 70 мл циклогексана. Полученный продукт фильтровали, фильтрат разбавляли 200 мл циклогексана и 8 мл раствора загружали в реактор. Продукт содержал 1.67 мг Cr/мл.
Цикл 7007. Смешивали 0.35 г этилгексаноата хрома (III) (CrЭГ3) с приблизительно 15 мл толуола, получали интенсивно окрашенный зеленый раствор. Добавляли 0.22 мл 2,5-диметилпиррола (2,5-ДПМ) и 0.20 мл 1-бромбутана. Медленно добавляли 5.7 мл 1.9М раствора ТЭА в толуоле и перемешивали в течение 30 мин, получали зеленовато-коричневый раствор и твердый продукт. Избыток растворителя удаляли под вакуумом и жидкость экстрагировали приблизительно 15 мл циклогексана. Полученный продукт фильтровали, фильтрат разбавляли 25 мл циклогексане и получали золотистый раствор, 7.0 мл которого загружали в реактор. Продукт содержал 0.014 CrЭГ3/мл.
Цикл 7008. Следовали методике, описанной в цикле 7007, за исключением того, что использовали 0.22 г CrЭГ3 и 0.13 мл 2,5-ДМП. Кроме того, вместо 1-бромбутана использовали 0.10 мл GeCl4. Добавляли 3.4 мл 1.9 М раствора ТЭА в толуоле и получали раствор от коричневого до желто-коричневого цвета и осадок. Конечный продукт после фильтрации и разбавления 25 мл циклогексана имел яркий золотисто-зеленый цвет и содержал 0.0088 г CrЭГ3/мл. В реактор загружали 3.0 мл раствора.
Цикл 7009. Добавляли 2.070 г CrPy3Cl и 70 мл толуола и 62 мл 1.9 М раствора ТЭА в толуоле, перемешивали и фильтровали. Объем фильтрата уменьшали до приблизительно 20 мл упариванием в вакууме. Вязкий коричневый раствор снова фильтровали, затем к фильтрату добавляли приблизительно 30 мл пентана. Через сутки из раствора в вакууме удаляли избыток растворителя. Затем добавляли 38.1 г алюмофосфатного носителя. (мольное соотношение P/Al 0.9, активирован при 700oC), полученного по методике Пат. США 4364855 (1982). Суспензию перемешивали приблизительно 30 ч. Твердый продукт отфильтровывали, промывали толуолом, циклогексаном, а затем пентаном. В реактор загружали 0.4388 г твердой каталитической системы.
Цикл 7010. Смешивали 0.21 г [Na(ДМЭ)2] [CrCl(Py)3ДМЭ] с приблизительно 15 мл 1-гексена. Медленно добавляли 0.75 мл чистого (93%) ТЭА, получали коричневый раствор и малозаметный осадок, смесь перемешивали 30 мин. Избыток растворителя удаляли в вакууме. Остаток экстрагировали приблизительно 15 мл циклогексана, фильтровали, фильтрат разбавляли циклогексаном до 25 мл. В реактор загружали 0.8 мл (0.067 г) раствора.
Цикл 7011. Следовали методике цикла 7010 за исключением того, что получаемую в циклогексане каталитическую систему перед использованием выдерживали в течение приблизительно 24 ч. В реактор загружали 0.8 мл (0,067 г) раствора.
Цикл 7012. Растворяли 0.26 г CrЭГ3 в приблизительно 15 мл 1-гексена. Добавляли 0.15 мл 2,5-ДМП и 0.13 мл 1-бромбутана. Медленно добавляли 1.0 чистого (93%/ ТЭА и перемешивали в течение 30 мин. Избыток растворителя удаляли в вакууме, а жидкость экстрагировали приблизительно 15 мл циклогексана. Полученный продукт фильтровали, фильтрат разбавляли циклогексаном до 25 мл. В реактор загружали 7.0 мл раствора.
Цикл 7013. Смешивали 0.21 г [Na(ДМЭ)2] [CrCl(Py)3ДМЭ] с 15 мл 1-гексена. Медленно добавляли 1.0 мл чистого (93%) ТЭА, получали темно-коричневый раствор и осадок, которые перемешивали в течение приблизительно 1 ч. Раствор декантировали и добавляли к 1.5 г алюмофосфата (мольное соотношение P/Al 0.4, активирован при 700oC), полученного по методике Пат. США 4364855 (1982).
Каталитическую систему на носителе отфильтровывали, промывали 1-гексаном и сушили в атмосфере азота. В реактор загружали 0.6328 г твердой каталитической системы.
Циклы 7006 - 7013 проводили в автоклаве на 1,2 л, содержащем циклогексан. Гетерогенную высушенную каталитическую систему на носителе (циклы 7009 и 7013) суспендировали в циклогексане для облегчения добавления в реактор полимеризации и суспензию добавляли в реактор полимеризации при противотоке этилена (химически чистого). Гомогенные жидкие каталитические системы (без нанесения на носитель) (циклы 7006 - 7008 и 7010 - 7012) разбавляли циклогексаном и при противотоке этилена (химически чистого) добавляли в реактор полимеризации. Реактор закрывали, а подачу этилена прекращали до тех пор, пока температура в реакторе не поднимается до температуры 80oC. Затем давление этилена в реакторе поднимали до 38,5 атм (550 фунтов/кв.дюйм). Этилен подавали по потребности в течение 30 мин.
В конце цикла отбирали пробу жидкой реакционной смеси и анализировали с помощью капиллярной газовой хроматографии на газовом хроматографе HP-5880, снабженном пламенно-иодинационным детектором и ДВ-1 колонкой (60 м, внутренний диаметр 0.25 мм, толщина пленки 0.25 мкм). Газовый хроматограф постепенно разогревали от 40 до 275oC при скорости нагрева 10o/мин, в течение 20 мин. Оставшуюся реакционную смесь упаривали и определяли количество твердого продукта.
Результаты представлены ниже в табл.7.
Данные в табл. 7 показывают, что присутствие воды (циклы 7001 - 7005) оказывает отрицательное влияние на образование жидких продуктов, например, таких как 1-гексен. В действительности, наличие в реакторе воды приводит к образованию большого числа твердых продуктов. Циклы 7006 - 7013 показывают, что каталитическая система, приготовленная в присутствии любого ненасыщенного углеводорода, эффективно содействует процессу тримеризации. Однако сравнение каталитических систем циклов 7006 - 7009, приготовленных в толуоле, с циклами 7010-7013, приготовленных в 1-гексена, показывает, что ненасыщенный ароматический углеводород является предпочтительной средой для приготовления каталитической системы.
Пример VIII.
В следующем примере, циклы 8001 - 8017, иллюстрируется влияние используемых пирролсодержащего соединения, галогена и добавок металла.
Все каталитические системы, используемые в циклах 8001 - 8017, были приготовлены по одной общей методике. В соответствии с этой методикой 2-этилгексаноат хрома (III) растворяли в толуоле, затем к раствору добавляли 3 эквивалента 2,5-диметилпиррола (или пирролида водорода в случае циклов 8014 - 8017). Добавляли необходимое количество галогенида (2 - 3 мольных эквивалента), а затем 15 - мольных эквивалентов триэтилалюминия (ТЭА). Реакционную смесь перемешивали 5 - 10 мин, толуол удаляли в вакууме. Жидкий остаток разбавляли циклогексаном до общего объема 10 мл и соответствующую аликвоту загружали в реактор в качестве каталитической системы.
Опыты по тримеризации проводили в автоклаве на 2 л, содержащем 1.2 л 85 %-ного циклогексана в качестве разбавителя. Каталитическую систему загружали в реактор после добавления циклогексана. Температуру реактора повышали до 80oC и вводили этилен. Давление в реакторе поддерживали 38,5 атм 550 фунтов/кв.дюйм, подавая при необходимости этилен. Каждый цикл до стравливания этилена продолжали 30 мин. В конце цикла отбирали пробы и анализировали с помощью газовой хроматографии, как это описано в других примерах. Результаты циклов и данные анализов приведены в табл.8.
Данные в табл.8 показывают, что селективность по 1-гексену увеличивается в следующем порядке I<Cl<B. Бромсодержащие добавки устойчиво обладают самой высокой селективностью для образования 1-гексена в сравнении с соответствующими хлоридами и иодидами. Увеличение образования 1-гексана также означает уменьшение образования биопродуктов (C4=, C8=, C10=). Соотношение 1-гексена и гексенов с неконцевой олефиновой связью также имеют тенденцию к увеличению в порядке I<Cl<B.
Таким образом, использование галогенидов приводит не только к получению большого количества продукта, но также и к более чистому тримеру. Активность каталитической системы увеличивается в порядке I<Cl<Br. Однако различия в активности между Br- и Cl - аналогами, как оказалось, являются непредсказуемыми. В случае некоторых добавок (SnX4 или AlX3SiX) бромиды являются более активными.
Данные в табл. 8 также показывают, что тенденция селективности по 1-гексену и активности каталитической системы могут быть перенесены на каталитические системы, содержащие другие пирролы, как это показано в циклах 8014 - 8017.
В целом наилучшее сочетание активности и селективности получается при использовании в качестве добавок галогенида GeCl4 или SnCl4. Однако показано, что на селективность по 1-гексену также влияет отношение галогенида к триэтилалюминию, что делает возможным получать высокую селективность при использовании добавок других галогенидов.
Пример IX.
В следующем примере в циклах 9001 - 9004 показано, что избыток ненасыщенного ароматического углеводорода может отрицательно влиять на процесс тримеризации и/или олигомеризации. Следовательно, когда каталитическая система готовится в присутствии ароматического углеводорода, например, такого, как толуол, избыток ароматического углеводорода должен быть предпочтительно удален. Полученную жидкость затем экстрагировали или растворяли в необходимом растворителе, например, таком как циклогексан или гептан. Хотя теоретически ароматический углеводород не вступает в химическую реакцию, полагают, что он может конкурировать с мономером, который подвергается тримеризации и/или олигомеризации, например, с этиленом, за активные центры каталитической системы. Полагают, что такая конкуренция может привести к уменьшению активности каталитической системы.
Каталитическую систему, используемую в циклах 9001 - 9004, готовили с использованием растворов 1.35 г 2-этилгексаноата хрома (III) в толуоле. К раствору добавляли 0.86 мл (3.2 ммольных эквивалента) 2,5-диметилпиррола. Затем добавляли 0.90 мл (3.2 мольных эквивалента) н.-бутилбромида с последующим добавлением 7.60 мл (21 мольный эквивалент) 93%-ного триэтилалюминия. Смесь перемешивали в течение 5 - 10 мин и удаляли в вакууме толуол. Жидкий остаток растворяли в 30 мл циклогексана, фильтровали и затем разбавляли циклогексаном до общего объема 50 мл. В реактор загружали 4 мл полученного раствора вместе с необходимым количеством безводного, не содержащего газа толуола (0,5, 10 или 15 мл).
Реакцию тримеризации проводили в автоклаве на 2 л, содержащем 1.2 л 85%-ного циклогексана в качестве разбавителя. Каталитическую систему загружали в реактор после добавления циклогексана. Температуру реактора поднимали до 80oC и вводили этилен. Давление в реакторе поднимали до 38,5 атм (550 фунтов/кв. дюйм) и поддерживали на этом уровне, подавая по потребности этилен. Каждый цикл до стравливания этилена продолжался 30 мин. Общий расход этилена замеряли.
Результаты циклов 9001-9004 приведены в табл.9.
Данные в табл.9 показывают, что наличие ароматического углеводорода, то есть толуола, может привести к значительному уменьшению активности каталитической системы, о чем свидетельствует замеряемый расход этилена. Это уменьшение пропорционально количеству ароматического углеводорода, добавленного в реактор.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ И СТАБИЛИЗИРОВАННАЯ КАТАЛИТИЧЕСКАЯ СИСТЕМА ДЛЯ ПОЛУЧЕНИЯ ОЛЕФИНОВ | 1995 |
|
RU2154046C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОЛЕФИНОВ | 1995 |
|
RU2131405C1 |
| СОЕДИНЕНИЯ ХРОМА, СПОСОБ ИХ ПОЛУЧЕНИЯ, СОСТАВ КАТАЛИЗАТОРА ДЛЯ ТРИМЕРИЗАЦИИ И/ИЛИ ПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1990 |
|
RU2102142C1 |
| СПОСОБ ОЛИГОМЕРИЗАЦИИ ОЛЕФИНОВ | 1996 |
|
RU2171248C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИОЛЕФИНОВ | 1990 |
|
RU2107696C1 |
| СПОСОБ ПОЛУЧЕНИЯ ОЛЕФИНОВ | 2000 |
|
RU2260578C2 |
| СПОСОБ ОЛИГОМЕРИЗАЦИИ (ВАРИАНТЫ) И СПОСОБ ПРЕДОТВРАЩЕНИЯ КОРРОЗИИ В ПРОЦЕССЕ ОЛИГОМЕРИЗАЦИИ | 2000 |
|
RU2249585C2 |
| СПОСОБ ПОЛУЧЕНИЯ КАТАЛИТИЧЕСКОЙ СИСТЕМЫ ДЛЯ ОЛИГОМЕРИЗАЦИИ ОЛЕФИНОВ И СПОСОБ ОЛИГОМЕРИЗАЦИИ ОЛЕФИНОВ | 2009 |
|
RU2412002C1 |
| СПОСОБ ПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ И КАТАЛИТИЧЕСКАЯ СИСТЕМА | 1997 |
|
RU2193042C2 |
| СПОСОБ ВЫДЕЛЕНИЯ ПРОДУКТОВ ОЛИГОМЕРИЗАЦИИ ОЛЕФИНОВ И РАЗЛОЖЕНИЯ ОСТАТКОВ КАТАЛИЗАТОРА ОЛИГОМЕРИЗАЦИИ | 2011 |
|
RU2471762C1 |

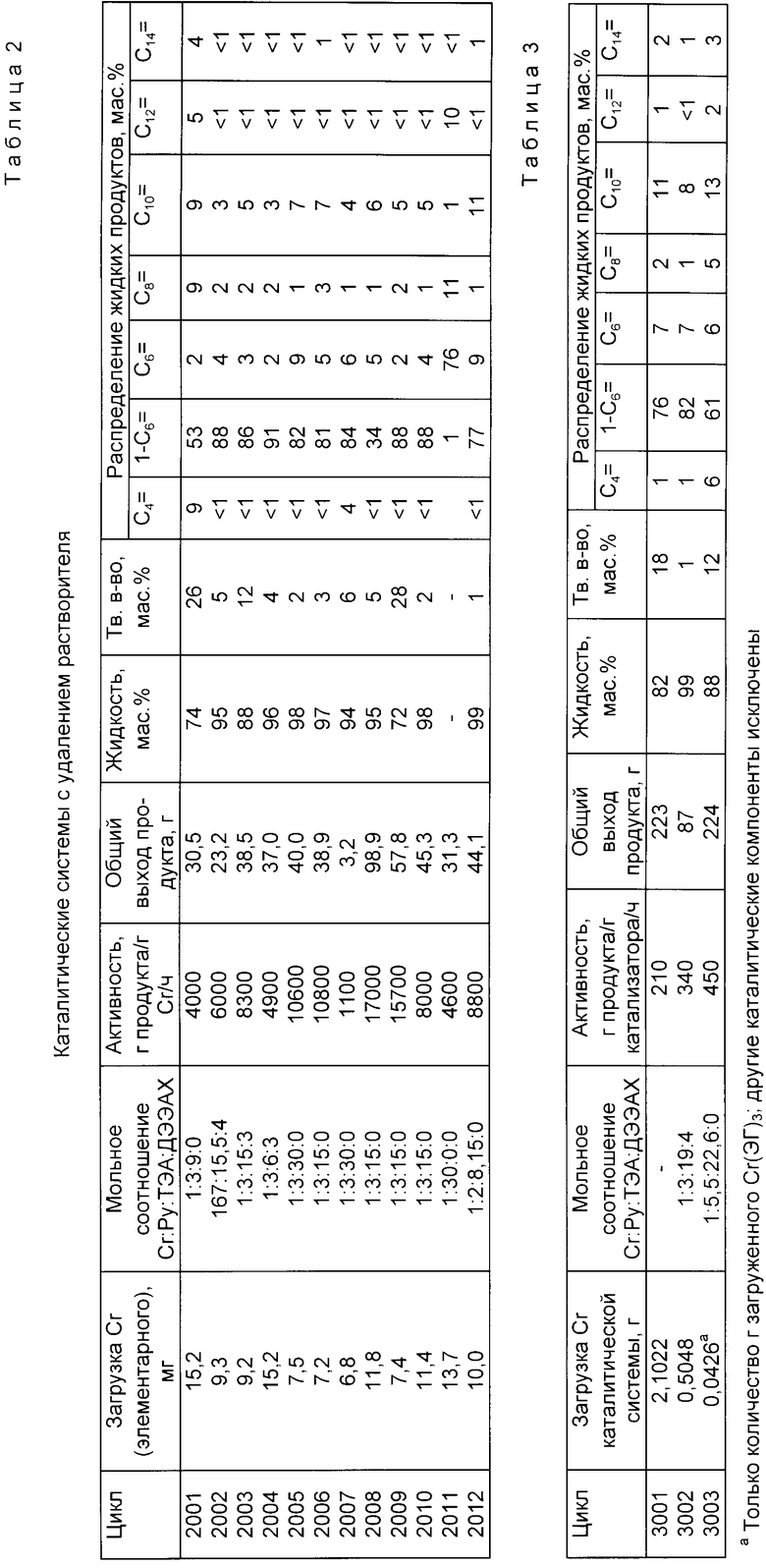
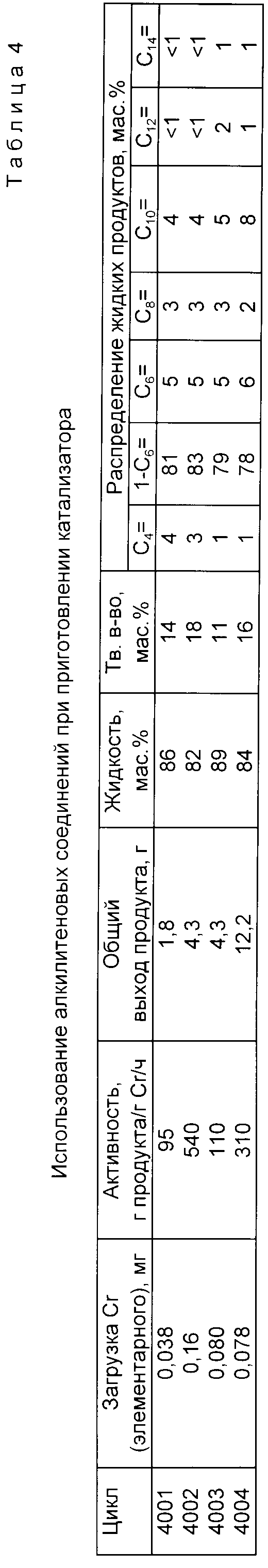
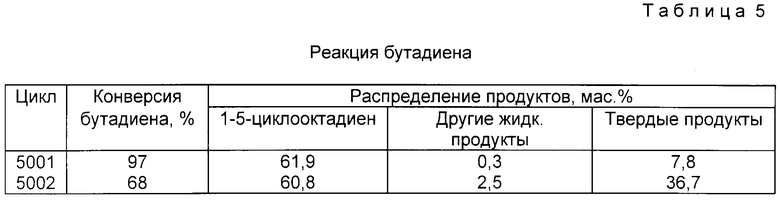
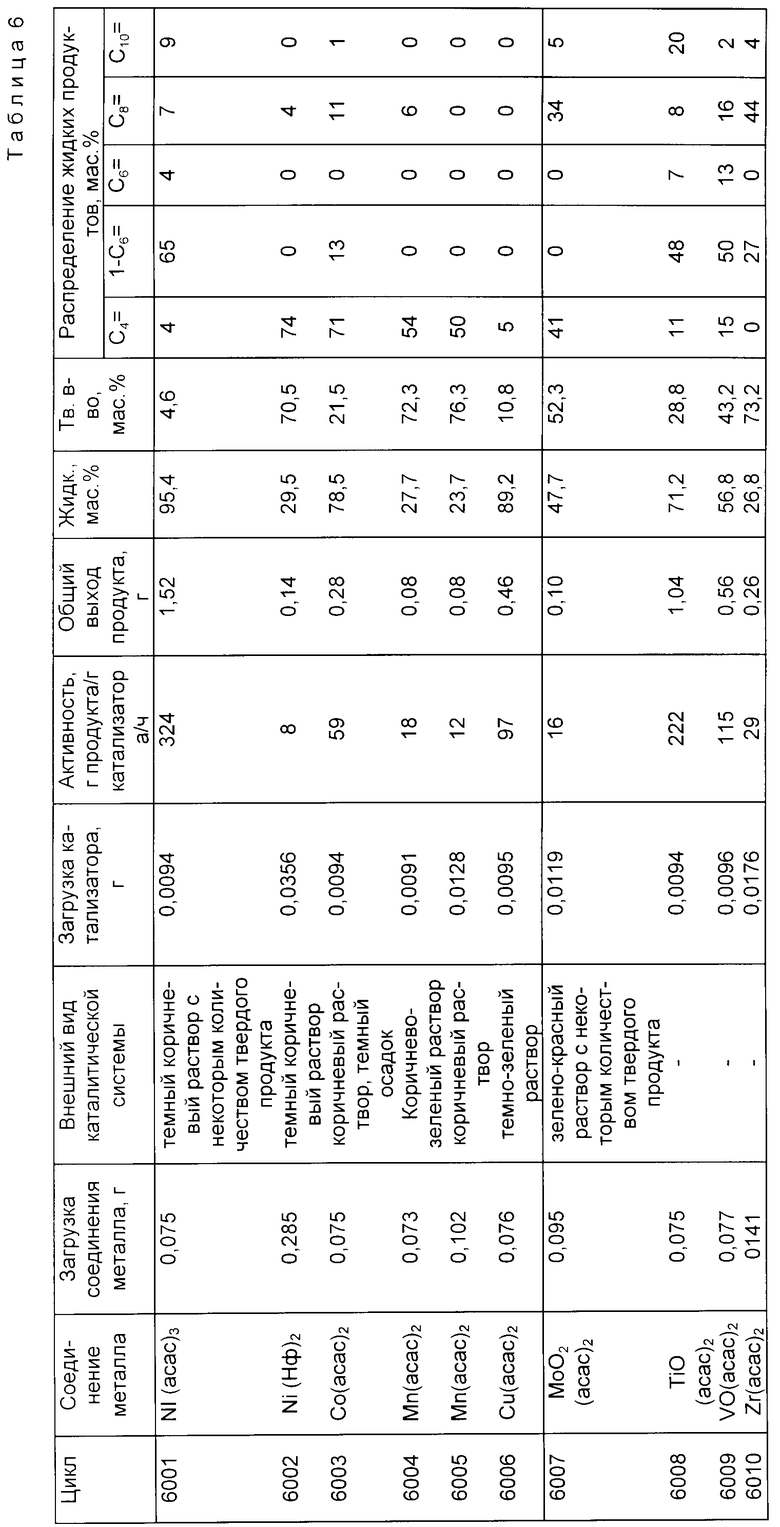
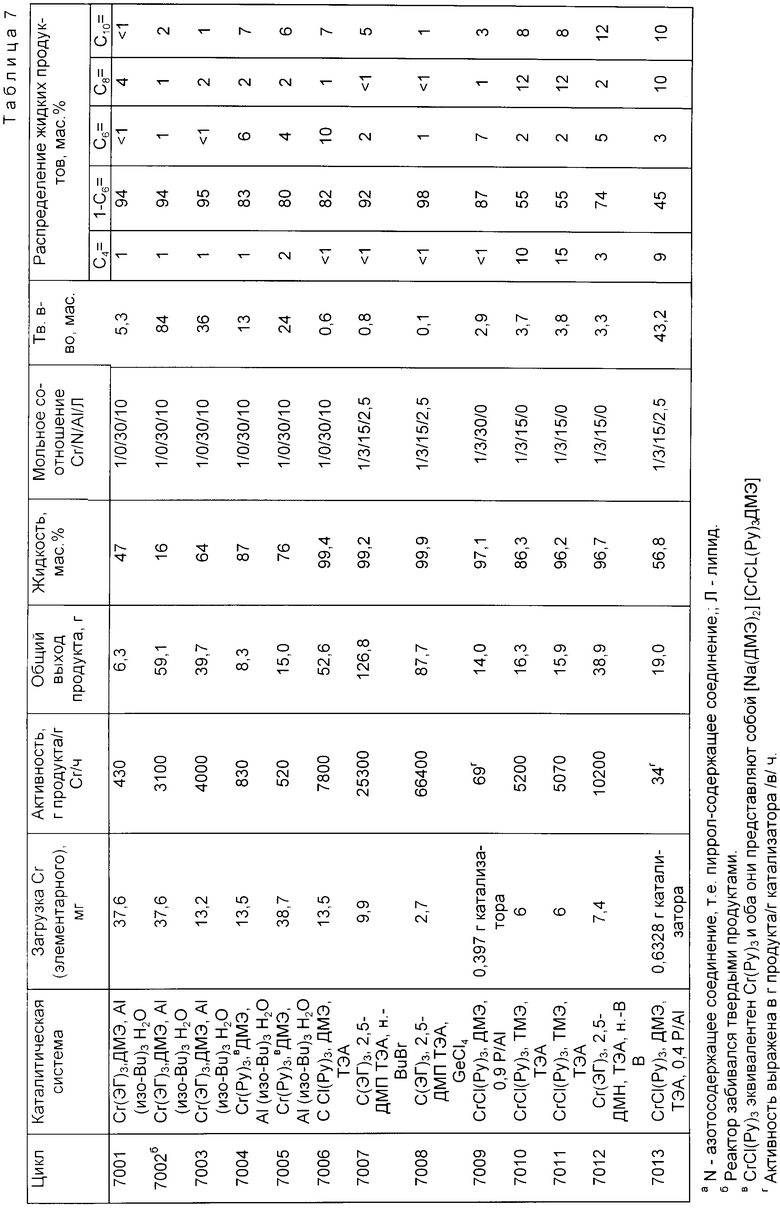
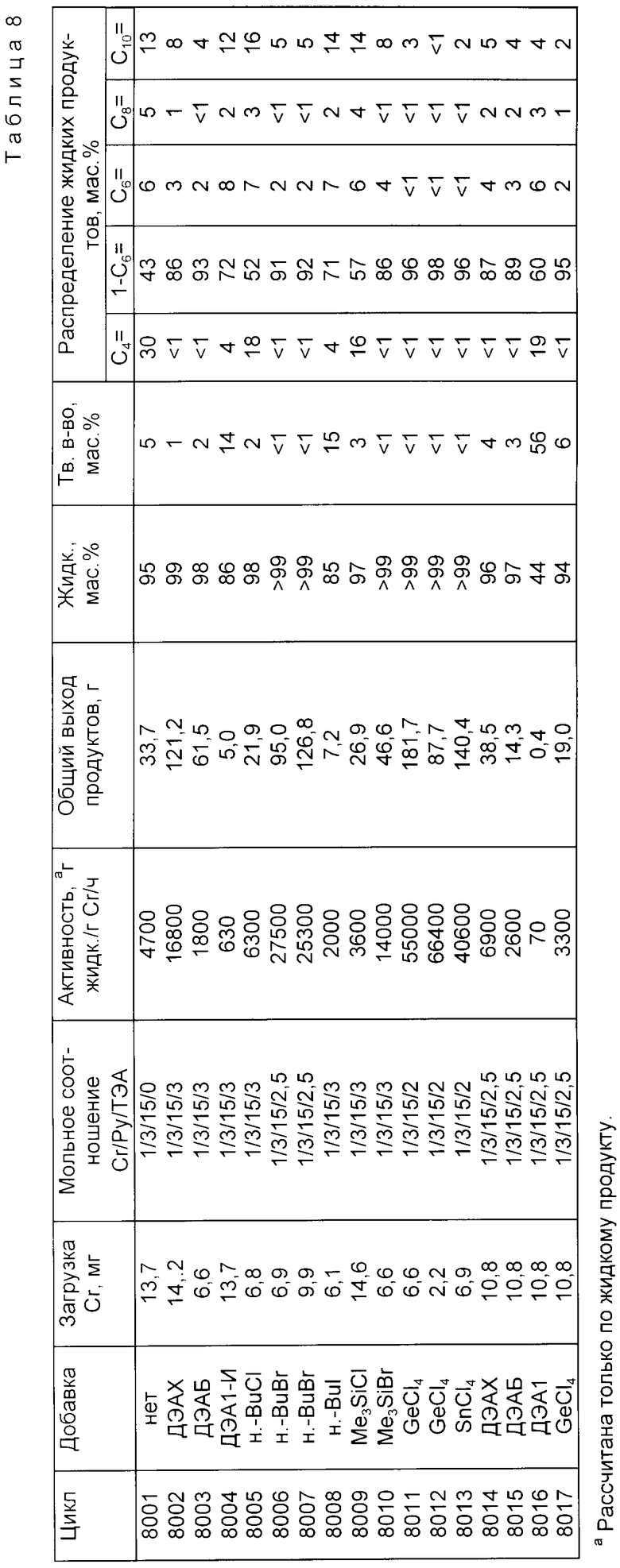

Ранее известный способ получения хромсодержащих соединений, таких, например, как пирролиды хрома, путем приготовления смеси соли хрома, амида металла, в особенности пирролида металла, и электронодонорного растворителя такого, например, как эфир, и взаимодействия с ненасыщенным углеводородом, усовершенствован использованием пиррола или его производных в качестве пирролида, и алифатических соединений в качестве ненасыщенного углеводорода. Новый способ приготовления каталитической системы включает смешение источника металла, пирролсодержащего соединения и металлалкила без стадии предварительного взаимодействия между источником металла и пирролсодержащим соединением в присутствии электронодонорного растворителя. Эти каталитические системы и хромсодержащие соединения без нанесения или с нанесением на носитель на основе неорганического оксида могут быть использованы при необходимости в качестве сокатализатора в сочетании с другим катализатором полимеризации, например, таким как хром- или титансодержащий катализатор, для тримеризации, олигомеризации и полимеризации олефинов. 4 с. и 28 з.п.ф-лы, 9 табл.
RmXn,
где R органический или неорганический остаток;
X галоген;
m + n > 0.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| EP, патент, 416304, кл | |||
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| US, патент, 4364855, кл | |||
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| EP, патент, 417477, кл | |||
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |

