Настоящее изобретение относится к новым соединениям, содержащим замещенную сульфамоильную группу и амидиногруппу, к способу их получения и содержащим их фармацевтическим композициям.
Эти соединения обладают сродством к биологическим рецепторам нейропептида Y (NPY), имеющимся в центральной нервной и периферической системах.
Нейропептид Y обнаружен только десяток лет тому назад и в настоящее время очень мало известно об агонистах или антагонистах его рецепторов, которые бы не были полипептидами, поскольку использование последних в терапии не является простым, особенно, из-за их разложения в желудочно-кишечном тракте; в обзоре, опубликованном в Drugs of the Future, 17 /1/ 39-45 /1992/, назван бенекстрамин, фосфат инозитола и антигистаминное производное гуанидиноалкилимидазола.
Соединения со структурой, близкой к структуре соединений данного изобретения, описаны в европейских патентах A-0236163 и A-0236164; они отвечают формуле A: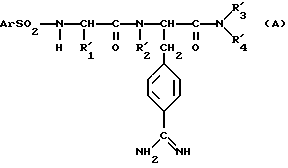
в которой R'1, R'2, R'3, R'4 являются в особенности алкилами или фенилами. Эти соединения обладают антикоагулирующей и антитромботической активностью, поэтому этот документ не предвосхищает активность соединений согласно изобретению.
Соединения согласно изобретению отвечают формуле /I/:
в которой Ar1 означает нафтил, фенил, хинолил или изохинолил, возможно замещенные хлором, фтором, (C1-C4)алкилом, (C1-C4)алкоксигруппой, гидроксилом или (C1-C4)диалкиламином;
Ar2 означает фенил или тиенил, возможно замещенные хлором, фтором, (C1-C4)алкилом, (C1-C4)алкоксигруппой или гидроксилом;
R1, R2 и R'2 независимо друг от друга, обозначают водород или (C1-C4)алкил; или R1 ничего не обозначает и N связан с Ar2, и в известных случаях R2 и R'2 образуют двойную связь; или R1, или R2 соединен с Al2 и обозначает C1-, C2- или C3-алкилен;
R3 и R4, одинаковые или разные, обозначают водород, алкил или вместе с атомом азота, с которым они связаны, образуют насыщенный гетероцикл C5-C7, выбираемый среди пирролидина, пиперидина и гексагидроазепина;
Z1 обозначает C1-C12-алкилен, в известных случаях прерываемый или продленный C5-C7 - циклоалкилом или фенилом;
Q1 обозначает метил; в известных случаях замещенный фенил; амино; алкоксикарбониламино; алкиламино; диалкиламино; гетероциклическую насыщенную амино-группу C5-C7; амидино; алкиламидино; гуанидино; алкилгуанидино; пиридил; имидазолил; пиримидинил; индолил; гидроксил; алкоксил; алкоксикарбонил C2-C8; амино-(C1-C4)-алкил- (C1-C4)-алкиламино или карбамоил;
Q2 обозначает водород или алкил;
Q3 обозначает водород или (C1-C4)-алкил;
или Q1 и Q3 связаны с образованием гетероцикла и вместе обозначают C2 - или C3 - алкилен, при этом Z1 ничего не обозначает; и соли присоединения этих соединений к кислотам.
Алкильные и алкоксильные, линейные или разветвленные группы содержат 1-4 C-атома, за исключением противоположного указания; насыщенными гетероциклическими аминогруппами могут быть пирролидинил, пиперидино, морфолино, пиперазинил, 4-алкилпиперазинил, фенильные группы, за исключением противоположного указания, могут быть замещены хлором, фтором, (C1-C4)-алкилом или (C1-C4)-алкоксилом.
Соли обычно получают с фармацевтически приемлемыми кислотами, но соли других кислот, используемые для очистки или выделения продуктов формулы /I/, также составляют часть изобретения.
Соединения формулы /I/ обычно содержат 2 асимметрических атома углерода и 4 чистых энантиомера, а также их смеси в любых пропорциях.
Соединения согласно изобретению могут быть получены из соединений формулы /II/:
способами, принципы которых известны и которые специалист в состоянии приспособить к реакционноспособности и растворимости используемых продуктов.
Многочисленные способы синтеза амидинов описываются в работе "The chemistry of amidines and imidates"["Химия амидинов и имидатов"] D.G. Neilson, Ed. Saul Patai; Wiley и Sons - с. 389-394/1975/. Обычно нитрил превращают в соль имидата путем воздействия спирта в сильно кислой среде по так называемой реакции Пиннера, и этот сложный имидоэфир, в известных случаях в свободной форме, вводят во взаимодействие с амином формулы /III/:
в полярном инертном растворителе, предпочтительно в спирте, при температуре от 0oC до температуры рефлюкса растворителя.
Большинство аминов формулы /III/ известно, и новые продукты могут быть получены при использовании хорошо известных специалисту принципов и методов. Например, в случае получения производных, в которых Q1 обозначает имидазолил, можно сослаться на патент США 3881016 и на Synth. Communic. 17, 223-227 /1987/, или, когда Q1 обозначает трет. -бутоксикарбониламиногруппу - на Synth. Communic. 20/16/, 2559-2564 /1990/.
Соединения формулы /I/, в которой Q1 обозначает NH2 или алкиламиногруппу, могут быть получены путем гидролиза соединений формулы /I/, в которой Q1 обозначает трет.-бутоксикарбониламино-группу.
Соединения формулы /I/, в которых Q1 обозначает гуанидиногруппу, замещенную или нет, можно получать путем взаимодействия соединения, в котором Q1= NH2 с соединением формулы:  , в которой R обозначает водород или алкил и Z обозначает нуклеофуг, такой как SO3H, например, в условиях, описанных в Tetrahedron Letters, 3183-3186/1988/, с аминоиминометансульфокислотой; N-метиламиноиминометансульфокислота может быть получена, как описано в F.Org.Chem 51, 1882/1986/.
, в которой R обозначает водород или алкил и Z обозначает нуклеофуг, такой как SO3H, например, в условиях, описанных в Tetrahedron Letters, 3183-3186/1988/, с аминоиминометансульфокислотой; N-метиламиноиминометансульфокислота может быть получена, как описано в F.Org.Chem 51, 1882/1986/.
Соединения формулы /I/, в которых амидинная функция включена в гетероцикл, могут быть получены само по себе известным образом путем воздействия диамина H2N-/CH2/n-N2H, в котором "n" обозначает 2 или 3, на сложный имидоэфир, в известных случаях путем воздействия диамина одна из функций которого защищена лабильной группой, которую удаляют перед циклизацией.
Некоторое число способов получения нитрилов формулы /II/, в которой Ar1 обозначает нафтил; R1=R2=R'2 обозначают водород, описывается в европейском патенте A-0236163, в частности, получение чистых энантиомеров, исходя из каждого чистого стереоизомера 4-цианофенилаланина, функция карбоновой кислоты которого блокирована, в известных случаях, в форме амида, замещенного с помощью R3 и R4, как в формуле /I/; с этим соединением вводят во взаимодействие альфа-аминокислоту формулы /IV/,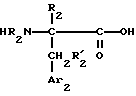
аминогруппа которой предварительно защищена либо в сульфомоильной форме  как в формуле /I/, либо с помощью лабильной группы, такой как трет. -бутоксикарбонил, которую удаляют после конденсации классическим образом путем воздействия сильной безводной кислоты.
как в формуле /I/, либо с помощью лабильной группы, такой как трет. -бутоксикарбонил, которую удаляют после конденсации классическим образом путем воздействия сильной безводной кислоты.
Также в многочисленных публикациях, касающихся химии пептидов и особенно в "The Peptides", изд. E.Cross и F.Meienhofer, т. 1, 65-104 /1979/ - Acad. Press, описываются способы получения амидов путем реакции карбоксильной группы и аминогруппы, которые находятся у 2-х асимметрических атомов углерода, без рацемизации у какого-либо из этих атомов углерода.
Обычно эти реакции протекают при температурах 0-40oC, в инертном растворителе, таком как дихлорметан, ацетонитрил, тетрагидрофуран или диметилформамид, в присутствии по крайней мере одного эквивалента третичного амина, такого как триэтиламин, или предпочтительно в присутствии N-этилморфолина.
Сульфамоильная группа  может быть получена классическим способом путем воздействия сульфохлорида Ar1-SO2-Cl в присутствии основания, в известных случаях в двухфазной среде в присутствии катализатора переноса фаз, либо на аминокислоту /IV/ или соответствующий сложный алкиловый эфир, либо на нитрил формулы /V/:
может быть получена классическим способом путем воздействия сульфохлорида Ar1-SO2-Cl в присутствии основания, в известных случаях в двухфазной среде в присутствии катализатора переноса фаз, либо на аминокислоту /IV/ или соответствующий сложный алкиловый эфир, либо на нитрил формулы /V/:
Когда R1 отлично от водорода, то нитрилы формулы /II/ могут быть получены путем воздействия R1X на сульфонамид /II/, в котором R1 обозначает водород, в присутствии основания, причем X обозначает атом галогена или сульфонатную группу.
Альфа-аминокислоты формулы /IV/ или соответствующие сложные алифатические эфиры представляют собой известные соединения или соединения, которые могут быть получены способами, аналогичными таковым, которые используют в отношении известных производных. Можно, например, сослаться на J.P. Greenstein и M. Winitz в "Chemistry of Amino Acides, J.Willey и Sons, Jnc. ed., 1961, стр. 697-714, стр. 2693-2770; и на G.C. Barrett в Chemistry and Biochemistry of the Amino Acides, Chapman и Hall Ltd ed., 1985, стр. 246-353.
Например, если R'2 = H, то используют основание Шиффа (см. Synthesis 313-315 (1984)) согласно следующей реакционной схеме: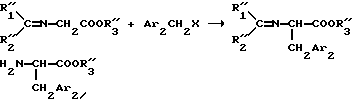
в которой R''1 и R''2 обозначают фенильные ядра; R"3 обозначает алкил и Ar2 имеет указанное в формуле /1/ значение.
Для получения альфа-аминокислот формулы IV, где R2 не является водородом, можно получить вариантом предыдущего способа. Основание Шиффа последовательно алкилируют с помощью Ar2CH2X, затем R2X, работая в ТГФ с основанием, например с алкоголятом щелочного металла при температуре от -70oC до 25oC.
Для получения аминокислот формулы /IV/ или соответствующих сложных эфиров в форме одного из чистых энантиомеров, можно осуществлять фракционные кристаллизации рацемической соли с оптически активной кислотой или оптически активным основанием согласно способу, принцип которого хорошо известен; также можно выделить один из энантиомеров рацемического сложного эфира аминокислоты формулы /IV/ в виде соответствующей аминокислоты, осуществляя ферментативный гидролиз рацемической смеси с помощью стереоселективного фермента, такого как альфа-химотрипсин. Этот способ описан, в частности, в Journal of Biochemistry 19, 877-881 /1971/.
Соли соединений формулы /I/ получают путем взаимодействия кислот с амидином формулы /I/ в растворителе; полученные соли выделяют путем отгонки растворителя или добавления нерастворителя для их осаждения.
Соединения формулы /I/ и их фармацевтически приемлемые соли способны фиксироваться на биологических рецепторах нейропептида Y /NPY/, пептида из 36 аминокислот с широким спектром физиологической активности, в частности в отношении центральной нервной системы или сердечно-сосудистой системы. NPY контролирует психомоторную активность, беспокойство, успокоение, он является стимулятором приема пищи, он принимает участие в депрессии, процессах запоминания некоторых сексуальных поведений и в эпилепсии; он ингибирует выделение инсулина, глюкагона и лютеинизирующего гормона; он действует на уровне почек и особенно воздействует на систему ренин-ангиотензин; наконец, он является сильным сосудосуживающим средством. Можно сослаться на обзор, опубликованный в Drugs of Future, 17 /1/, 39-45 /1992/, где также указываются потенциальные терапевтические активности антагонистов NPY.
Сродство соединений изобретения к рецепторам NPY можно продемонстрировать, ин витро, используя опыт, описанный Unden и сотр. в Eur. F.Biochem. 145, 525-530 /1984/, на мембранах коры крысы; в этих условиях соединения изобретения, приведенные в качестве примеров ниже, имеют ИК50 [концентрация, ингибирующая 50% связи NPY его рецептором] составляет 10 нмоль - 10 мкмоль, тогда как таковая NPY составляет 0,5 нмоль.
Соединения могут быть агонистами или антагонистами действия пептида NPY на его рецептор.
Антагонистическая активность NPY может быть продемонстрирована при использовании опыта, описанного в Proc. Soc. Exp. Biol. Med., 64 453-455 /1947/, на амиелированной крысе; в этих условиях введение NPY вызывает гипертензорный эффект, который уменьшается, даже ликвидируется, когда животным вводят антагонисты изобретения.
Для соединений, обладающих сильным сродством к рецепторам, измеряют DI50 в несколько мкг/кг во время перфузии внутривенно 10 мкг/кг NPY.
В настоящее время неизвестен специфичный, с большим сродством и конкурентноспособный антагонист, и поэтому соединения согласно изобретению представляют особый интерес; их предпочтительно можно использовать в качестве антигипертензоров или для лечения стенокардии, особенно из-за их сосудорасширяющей активности, или для борьбы против коронарных и церебральных спазмов сосудов, так же, как при лечении атеросклероза и сердечной недостаточности. Эти соединения также могут быть использованы в качестве анорексигенных, антидепрессорных, успокаивающих агентов для уменьшения беспокойства или регуляции некоторых расстройств сексуального поведения. Они также представляют реальный интерес при лечении воспаления, аллергии, некоторых желудочно-кишечных расстройств, таких как болезнь Crohn, или при лечении ожирения, проявляя свою липолитическую активность; они также являются иммуномодуляторами. Соединения могут использоваться при всех NPY-зависимых патологиях или нарушениях.
Изобретение также относится к фармацевтическим композициям, содержащим в качестве действующего начала один из энантиомеров соединений формулы /I/, одну из их смесей или их солей с фармацевтически приемлемой кислотой, а также эксципиент, пригодный для введения перорально путем инъекции или чрескожно. Суточные дозы зависят от излечиваемой патологии и заболевания.
В фармацевтической композиции активный ингредиент содержится в эффективном количестве.
Вышеуказанные эксципиенты выбирают в зависимости от фармацевтической формы и желательного способа введения.
В фармацевтических композициях настоящего изобретения для орального, подъязычного, подкожного, внутримышечного, внутривенного, топического, внутритрахеального, внутриносового, чрескожного или ректального введения, действующие начала вышеприведенной формулы /I/ или их возможные соли могут быть введены в единичных дозах в смеси с классическими фармацевтическими носителями животным и среди них людям для профилактики или лечения вышеуказанных расстройств или вышеуказанных заболеваний. Соответствующие единичные дозы введения могут находиться в виде таблеток, желатиновых капсул с лекарством, порошков, гранул, растворов или суспензий для орального введения.
Для топического применения можно использовать соединения согласно изобретению в кремах, помадах или лосьонах.
Для того, чтобы получить желаемый профилактический или терапевтический эффект, доза действующего начала может изменяться в пределах 0,01-50 мг на кг веса тела и в день.
Каждая единичная доза может содержать 0,5-1000 мг, предпочтительно 1-500 мг, активных ингредиентов в комбинации с фармацевтическим носителем. Эта доза может вводиться 1-5 раз в день для того, чтобы вводить суточную дозировку 0,5-5000 мг, предпочтительно 1-2500 мг.
Когда получают твердую композицию в виде таблеток, то смешивают основной активный ингредиент с фармацевтическим эксципиентом, таким как желатина, крахмал, лактоза, стеарат магния, тальк, гуммиарабик или аналогичные материалы. Таблетки можно покрывать сахарозой, целлюлозным производным или другими соответствующими веществами или можно их обрабатывать таким образом, чтобы они обладали пролонгированной или замедленной активностью и непрерывно высвобождали заранее определенное количество действующего начала.
Препарат в виде желатиновых капсул с лекарством получают путем смешения активного ингредиента с разбавителем и внесения полученной смеси в мягкие или жесткие желатиновые капсулы.
Препарат в виде сиропа или эликсира или для введения в форме капель может содержать активный ингредиент вместе с подслащивающим веществом, предпочтительно некалорийным, метилпарабеном и пропилпарабеном в качестве антисептиков, так же, как с агентом, придающим соответствующий вкус и цвет.
Диспергируемые в воде порошки или гранулы могут содержать активный ингредиент в смеси с диспергаторами, или смачивателями, или суспендирующими агентами, как поливинилпирролидон, точно так же, как с подслащивающими веществами или улучшающими вкус веществами.
Для ректального введения прибегают к свечам, которые готовят со связующими, плавящимися при ректальной температуре, например как масло какао или полиэтиленгликоли.
Для парентерального введения используют водные суспензии, изотонические солевые растворы или стерильные и инъекцируемые растворы, которые содержат диспергаторы и/или смачиватели, которые фармакологически приемлемы, например как пропиленгликоль или бутиленгликоль.
Действующее начало может быть заключено в микрокапсулу в известных случаях с одним или несколькими носителями или добавками.
Композиции настоящего изобретения могут содержать вместе с вышеуказанными продуктами формулы /I/ или одной из их фармацевтически приемлемых солей другие действующие начала, которые могут быть пригодны для лечения вышеуказанных расстройств или заболеваний.
Особенно предпочтительны соединения, в которых Z обозначает C4-C9 - алкилен и Q1 фиксирован на Z1 на атоме азота и обозначает амино-, гуанидино- или амидиногруппу, замещенную или незамещенную с другой стороны, предпочтительны соединения, в которых NR3R4 обозначает пирролидинил.
Особенно предпочтительны соединения формулы /I/, в которых Z1 обозначает метилен-циклогексилметилен; Q1 обозначает амино-, алкиламинот диалкиламиногруппу; R3 и R4 вместе с атомом азота, с которым они связаны, образуют пирролидинил; Ar2 обозначает фенил или метоксифенил; Ar1 обозначает нафтил, и Q2, Q3, R1, R2 и R'2 имеют указанное для формулы /I/ значение.
Ниже описываются примеры соединений изобретения и способы получения. Предварительно в качестве иллюстрации указывается получение некоторого числа промежуточных соединений синтеза.
Соединения формулы /I/ обычно содержат два асимметрических атома углерода и могут быть выделены в виде смеси двух пар рацемических диастереоизомеров, относительные соотношения которых зависят от рабочих условий, учитывая их различные физические свойства. Когда исходные продукты, которые содержат асимметрический углерод, не являются рацемическими смесями, а обогащены тем или другим из энантиомеров, то целевой продукт обычно не является смесью двух рацематов за исключением того случая, если рабочие условия вызывают рацемизацию.
Для продуктов формулы /I/, описанных ниже, измеряют относительное содержание двух пар рацематов классическими методами, такими как высокоэффективная жидкостная хроматография или ЯМР-спектроскопия.
За исключением противоположного указания выделяемые нитрилы формулы /II/ представляют собой эквимолекулярную смесь диастереоизомеров.
А - Получение сульфонамидов
N-/2-Нафтил-сульфонил/-1,2,3,4-тетрагидроизохинолин-3-карбоновая кислота
В суспензию из 5 г хлоргидрата 1,2,3,4-тетрагидроизохинолин-3-карбоновой кислоты в 150 мл диоксана вводят 46,8 мл водного 1н раствора NaOH, затем медленно добавляют 5,3 г 2-нафталинсульфонилхлорида и 1н раствора NaOH для поддержания pH-значения, близкого к 10. По окончании добавления реакционную смесь выдерживают 3 часа при перемешивании при температуре около 20oC перед добавлением 150 мл метиленхлорида. После подкисления водной фазы вплоть до pH 2, органическую фазу отделяют и водную фазу снова экстрагируют с помощью 150 мл метиленхлорида. Органические фазы сушат, концентрируют и остаток очищают путем хроматографии на силикагеле, элюируя смесью метиленхлорида с метанолом /80/20 по объему/. Получают 7 г сульфонамида в виде полугидрата. Т.пл. 110oC.
N-/3,4-дихлорфенилсульфонил/-O-метил-тирозин
При перемешивании вводят 5 г 3,4-дихлорфенилсульфонилхлорида в смесь 3,8 г этил-O-метил-тирозината, в 35 мл метиленхлорида и 50 мл водного насыщенного раствора карбоната калия. По истечении ночи, твердое вещество удаляют, органическую фазу отделяют, а водную фазу снова экстрагируют метиленхлоридом. Высушенные органические фазы концентрируют и остаток хроматографируют на колонке с диоксидом кремния, элюируя смесью метиленхлорида с метанолом /95/5 по объему/. Получают 6,8 г рацемического этил-N-/3,4-дихлорфенилсульфонил/-O-метилтирозината, который плавится при 99oC. Этот сложный эфир гиролизуют в 100 мл этанола, содержащего 9,5 мл водного 5н раствора KOH, получая после подкисления 5,5 г соответствующей кислоты, которая плавится при 183oC.
Таким же образом получают:
N-/2-нафтилсульфонил/-фенилаланин: т. пл. = 146oC /сложный метиловый эфир: т.пл. = 144oC/
N-/2-нафтилсульфонил/-O-метилтирозин; т. пл. = 174oC /сложный этиловый эфир: т.пл. = 138oC/
N-метил-N-/2-нафтилсульфонил/-фенилаланин; т. пл. = 122oC /сложный метиловый эфир: т.пл. = 106oC/
N-/2-нафтилсульфонил/-2-амино-индан-2-карбоновая кислота; т.пл. = 264oC
N-/5-изохинолилсульфонил/-альфа-метил-фенилаланин; сложный этиловый эфир; т.пл. = 60oC
N-/8-хинолинилсульфонил/-O-метил-тирозин; т.пл. = 228oC
N-/2-нафтилсульфонил/-O-бензил-тирозин; т.пл. = 182oC
N-/1-нафтилсульфонил/-2,4-диметил-фенилаланин; т. пл. = 220oC /сложный этиловый эфир: т. пл. 134oC/
N-/4-толил-сульфонил/-4-хлор-фенилаланин; т. пл. = 164oC, сложный этиловый эфир, т.пл. = 144oC.
Исходный этил 2-амино-индан-2-карбоксилат может быть получен из этил-N-дифенилметиленглицината, при -70oC, 25 г этил-N-дифенилметиленглицината вводят в 1500 мл тетрагидрофурана, содержащего 10,5 г трет.-бутилата калия, затем медленно добавляют 12,6 мл α, α- (дибром)-орто-ксилола и, спустя 12 часов, 10,5 г трет. -бутилата калия. Оставляют температуру повышаться до комнатной и, спустя 12 часов, в среду вводят водный насыщенный раствор NH4Cl. Растворители удаляют путем отгонки при пониженном давлении и остаток экстрагируют диэтиловым эфиром. Отделенную органическую фазу перемешивают в течение 16 часов при комнатной температуре вместе с 150 мл 1н водного раствора HCl. Водный раствор после трехкратной промывки с помощью диэтилового эфира, доводят до pH 8 за счет добавления NaHCO3 и 13,4 г сложного целевого эфира экстрагируют метиленхлоридом. Т.пл. = 56oC.
Б - Получение /4-циано/-фенил-аланиламилов
1. Сложный этиловый эфир /4-циано/-фенилаланина
В раствор 40,4 г 4-/бромметил/-бензонитрила в 460 мл безводного CH3 CN вводят 7,23 г тетрабутиламмонийбромида, 93 г карбоната натрия, затем 60 г этил-N-/дифенилметилен/-глицината. Среду выдерживают 4 часа при температуре ее кипения с обратным холодильником, затем твердые вещества отделяют и растворители удаляют путем отгонки при пониженном давлении. Остаток обрабатывают с помощью 1 л диэтилового эфира, затем после фильтрации концентрируют до объема 500 мл перед тем, как добавить туда 300 мл водного 1н раствора HCl. После перемешивания в течение 16 часов смесь декантируют, и pH-значение отделенной водной фазы доводят до примерно 8. Целевой продукт экстрагируют метиленхлоридом. Получают 34,8 г сложного эфира в маслянистой форме, хлоргидрат которого плавится при 170oC.
2. Разделение левовращающего и правовращающего изомеров вышеполученного сложного эфира путем ферментативного гидролиза
В течение 16 часов при 25oC перемешивают 10 г рацемического сложного эфира, 20 мг альфа-химотрипсина и 0,9 г сывороточного бычьего альбумина в 1 л водного 0,1 М раствора CH3COONa, pH-значение которого доведено до 6,5 - 6,8 путем добавления водного 0,1н раствора NaOH. После отфильтровывания среды через тальк, затем активный уголь, половину растворителя удаляют путем отгонки при пониженном давлении примерно при 35oC и остаточный водный раствор доводят до pH 8 путем добавления NaOH, затем экстрагируют метиленхлоридом. После обычных обработок органической фазы получают 4,5 г левовращающего, маслянистого этил-2-амино-3-/4-цианофенил/-пропионата с [α]
Водный раствор основного характера содержит натриевую соль соответствующей кислоты в виде другого энантиомера. После подкисления вплоть до 4-х, затем лиофилизации выделяют порошок белого цвета, который содержит левовращающую аминокислоту.
3. N-/трет.-бутоксикарбонил/-4-циано-фенилаланин
При 5oC, 20 мл водного 1н раствора NaOH и 4,34 г ди-/трет.- бутил/-карбоната вводят в раствор 4,34 г сложного этилового эфира 4-циано-фенилаланина в 70 мл диоксана. После возвращения к комнатной температуре и перемешивания в течение 3-х часов реакционную среду выпаривают досуха, затем на остаток выливают 100 мл воды и после промывки этилацетатом водный раствор доводят до pH 2 путем добавления раствора KHSO4, после этого целевой продукт экстрагируют метиленхлоридом.
4. 1-[2-Амино-3-/4-цианофенил/]-пропионил-пирролидин
При 0oC 1,93 г пирролидина и 3 г гидроксибензотриазола вводят в 70 мл метиленхлорида, содержащего 5,4 г N-/трет.-бутоксикарбонил/-4-цианофенилаланина, затем при температуре около -5oC добавляют раствор 4 г дициклогексилкарбодиимида в 30 мл метиленхлорида. После перемешивания в течение 16 часов при 20oC отфильтровывают и органическую фазу промывают водным насыщенным раствором карбоната натрия, затем раствором KHSO4 с pH 2 и, наконец, водой. После обычных обработок получают 4,73 г целевого производного, первичная аминная функция которого защищена трет.-бутоксикарбонильной группой; эта группа может быть удалена путем воздействия кислоты, соединение растворяют в 50 мл этилацетата и примерно при 0oC добавляют 50 мл насыщенного при 15oC хлороводорода в растворе этилацетата, после перемешивания в течение 2-х часов при 20oC растворитель удаляют и получают хлоргидрат целевого продукта. Рацемический хлоргидрат плавится при 224oC. Энантиомер, полученный из левовращающего сложного эфира, является левовращающим: [α]
5. 1-[2-Амино-3-/4-цианофенил/]-пропионил-пиперидин
Рацемический хлоргидрат плавится при 218oC; промежуточное N-/тет.-бутоксикарбонильное/ соединение плавится при 132oC.
6. /N-Метил-N-этил/-2-амино-3-/4-цианофенил/-пропионамид
Рацемический хлоргидрат плавится при 228oC.
В- Получение соединений формулы /II/:
1. Путем взаимодействия сульфонамида с /4-циано/-фенилаланиламидом.
1-[2-{ 2-/3,4-Дихлорфенилсульфамоил/-3-/4-метоксифенил/ -пропионамидо}-3-/4-цианофенил/-пропионил]-пирролидин (соединение 1):
В 100 мл ацетонитрила при 0oC вводят 2,5 г N-/3,4-дихлорфенилсульфонил/-0-метилтирозина, 1,82 г хлоргидрата 1-[2-амино-3- -/4-циано/-фенилпропионил] -пирролидина и 2,87 г бензотриазолил-1-окси-трис/диметиламино/-фосфоний-гексафторфосфата /БОФ/, затем 1,75 мл триэтиламина при температуре ниже 5oC. После перемешивания в течение 16 часов при комнатной температуре /около 20oC/ растворитель удаляют при пониженном давлении и остаток растворяют в 80 мл этилацетата. После промывки органической фазы водным раствором с pH 2, насыщенным раствором гидрокарбоната натрия, затем водой, растворитель удаляют путем отгонки и остаток хроматографируют на колонке с силикагелем, элюируя смесью метиленхлорида с метанолом /98:2 по объему/. Получают 2,9 г смеси диастереоизомеров нитрила, которая плавится при 101oC.
Таким же образом получают
N-этил-N-метил-2-[2-/3,4-дихлорфенилсульфамоил/-3-/4-метоксифенил/- пропионамидо] -3-/4-цианофенил/-пропионамид (соединение 2), т. пл. 192oC; кристаллизованный с 1,5 H2O;
1-[2-{ 2-/2-нафтилсульфамоил/-3-/4-метоксифенил/-пропионамидо} -3- /4-цианофенил/-пропионил] -пирролидин /соединение 3/, получают из рацемического сульфонамида и левовращающего хлоргидрата
1-[2-амино-3-/4-цианофенил/-пропионил] -пирролидина, т.пл. 135oC, кристаллизованный с 1,5 H2O;
1-/2-{ 2-/2-нафтилсульфамоил/-3-фенил-пропионамидо} -3-/4- цианофенил/пропионил]-пирролидин /соединение 4/, т.пл. 206oC, кристаллизованный с 1 H2O;
1-[2-{ 2-/2-нафтилсульфамоил/-3-фенил-пропиоамидо} - 3-/4-цианофенил/-пропионил]-пиперидин /соединение 5/, т.пл. 210oC, кристаллизованный с 1 H2O;
N-этил-N-метил-2-[2-/1-нафтилсульфамоил/-3-/3,4-дихлорфенил/ пропионамидо] -3-/4-цианофенил/-пропионамид/соединение 6/ т. пл. 182oC, кристаллизованный с 0,5 H2O;
N-/2-нафтилсульфонил/-3-[1-/пирролидинилкарбонил/-2-/4- цианофенил/-этиламинокарбонил] -тетрагидроизохинолин/соединение 7/, т.пл. 232oC, кристаллизованный с 0,75 H2O;
1-[2-{ [2-(2-нафтилсульфамоил)-2-инданил] -карбоксамидо}-3-/4- -цианофенил/-пропионил]-пиперидин (соединение 8), т.пл. 224oC, кристаллизованный с 1 H2O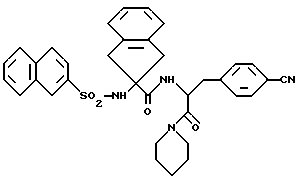
1-[2{ 2-/8-хинолилсулъфамоил/-3-/4-метоксифенил/-пропионамидо} - 3-/4-цианофенил/пропионил] -пирролидин /соединение 9/ получают из рацемического сульфонамида и левовращающего хлоргидрата 1-[2-амино-3-/4-цианофенил/]-пропионил-пирролидина, т.пл. 175oC, кристаллизованный с 1,5 H2O;
1-[2-{ 2-/2-нафтил-сульфамоил/-3-/4-бензилоксифенил/- пропионамидо}-3-/4-цианофенил/-пропионил] -пирролидин/соединение 10/, т.пл. 110oC, кристаллизованное с 1 H2O соединение.
2. Путем конденсации альфа-аминокислоты с /4-цианофенил/- аланил-амидом с последующей реакцией с сульфохлоридом.
2.1. 1[2-{ 2-/2-Нафтилсульфамоил/-3-фенил-пропионамидо} -3-/4- цианофенил/-пропионил]-пирролидин /соединение 4 и соединение 4-бис/
а/ При 0oC медленно вводят в 20 мл ацетонитрила 1,75 г N-/трет.-бутоксикарбонил/-фенилаланина, 0,95 мл N-этил-морфолина, 3,34 г БОФ и 1,75 г хлоргидрата 1-[2-амино-3-/4-цианофенил/-пропионил]-пирролидина, затем 1,6 мл П-этилморфолина. После перемешивания в течение 16 часов при комнатной температуре растворитель удаляют путем отгонки при пониженном давлении и остаток растворяют в этилацетате в присутствии насыщенного раствора гидрокарбоната натрия. Органическую фазу, промытую и высушенную обычным образом, концентрируют при пониженном давлении и остаток очищают путем колоночной хроматографии на силикагеле, элюируя смесью метиленхлорида с метанолом /98/2 по объему/.
Полученный продукт растворяют в 50 мл метиленхлорида и при 0oC добавляют 50 мл CF3 COOH. Когда температура среды повысится до комнатной, перемешивают еще 30 минут, после чего летучие продукты удаляют путем отгонки при пониженном давлении, после добавления 40 мл воды смесь лиофилизируют, получая 2,9 г трифторацетата.
б/ При 0oC медленно 1,75 мл N-этилморфолина вводят в 35 мл раствора 2,3 г вышеполученного трифторацетата в метиленхлориде, затем добавляют 1,1 г 2-нафталинсульфонилхлорида в виде раствора в 10 мл метиленхлорида. После перемешивания в течение 4-х часов при комнатной температуре органическую фазу промывают 0,1 н водным раствором HCl, затем водой. Полученный после отгонки растворителя остаток хроматографируют на силикагеле, элюируя смесью метиленхлорида с метанолом /95/5 по объему/, получая 1,95 г целевого соединения 4.
Когда оба исходных продукта представляют собой чистые энантиомеры, то в этих условиях получают один из четырех стереоизомеров соединения 4.
Продукт /соединение 4-бис/, полученный из двух левовращающих энантиомеров, описанных выше, и кристаллизованный с 0,25 H2O, плавится при 118oC [α]
Согласно такой же последовательности реакций, исходя из N-(трет.-бутоксикарбонил)-L-фенилаланина и левовращающего хлоргидрата 1-[2-амино-3-(4-цианофенил)-пропионил]-пирролидина, получают соединение 4 (трет) [α]
Исходя из N-(трет. -бутоксикарбонил)-D-фенилаланина и правовращающего хлоргидрата 1-[2-амино-3-(4-цианофенил)-пропионил] -пирролидина, получают 1-(2-[2-(5-диметиламино-1-нафтилсульфамоил)-3-фенилпропионамидо] -3-(4-цианофенил)-пропионил)-пирролидин (соединение 11), т.пл. 116oC; [α]
Исходя из рацемических смесей, получают 1-(2-[2-(1- нафтилсульфамоил)-3-(2-тиенил)пропионамидо] -3-(4- цианофенил)пропионил)пирролидин (соединение 12) т.пл. 121oC.
исходя из N-(трет.-бутоксикарбонил)-O-этил-D-тирозина и левовращающего энантиомера хлоргидрата 1-[2-амино-3-(4- цианофенил)пропионил]пирролидина и используя соответствующий сульфонилхлорид, получают соединение 5А.
Соединение 5А: гидратированный 1-(2-[2-(8-хинолинилсульфамоил)-3-(4-этоксифенил)пропионамидо]- 3-(4-цианофенил)пропионил)пирролидин.
Температура плавления 122oC
[α]
2.2. 1-(2-[(N-(2-нафтил-сульфамоил)-5-метокси-2-индолил)- карбоксамидо] -3-(4-цианофенил)-пропионил)-пирролидин (соединение 13).
а) При 0oC в 100 мл CH3CN вводят 1,3 г 5-метоксииндол-2-карбоновой кислоты, 3,16 г БОФ и 2 г хлоргидрата 1-[2-амино- -3-/4-цианофенил/-пропионил]-пирролидина, затем 2,5 мл триэтиламина.
После перемешивания в течение 16 часов при комнатной температуре отфильтровывают, промывают эфиром и высушивают 1-[2-{/5-метокси-2-индолил/-карбоксамидо}-3-/4-цианофенил/-пропионил] -пирролидин. Т.пл. 204oC.
б/ При 0oC к суспензии 2,2 г вышеполученного продукта в 100 мл ТГФ добавляют 0,23 г 60%-ной масляной суспензии гидрида натрия. После перемешивания в течение 1 часа при 5oC при температуре около 0oC добавляют 1,3 г 2-нафталинсульфонилхлорида в виде раствора в 20 мл ТГФ и перемешивают при комнатной температуре в течение 16 часов, затем при 50oC в течение 3-х часов. Отфильтровывают от осадка и фильтрат выпаривают при пониженном давлении. Остаток хроматографируют на колонке с диоксидом кремния, элюируя смесью метиленхлорида с циклогексаном /70/30 по объему/, получая полугидрат целевого продукта, который плавится при 186oC.
Таким же образом, исходя из 2-индол-карбоновой кислоты, выделяют 1-[2-[/N-(2-нафтилсульфамоил)-2-индолил/-карбоксамидо} - -3-/4-цианофенил/-пропионил]-пирролидин /соединение 14/. Т.пл.180oC.
3. Путем замещения сульфонамида формулы /II/ для получения соединения, в котором R1 отлично от водорода.
1-[2-{2-/N-метил-2-нафтилоульфамоил-3-фенил/-пропионамидо}-3-/4- -цианофенил/-пропионил]-пирролидин /соединение 15/.
2 г Соединения 4 растворяют в 20 мл диметилформамида при 0oC и добавляют 0,475 г карбоната калия и 0,214 мл метилиодида. Спустя 24 часа при 5oC, добавляют 20 мл воды и 40 мл метиленхлорида. Декантированную органическую фазу промывают, сушат и выпаривают досуха. Остаток перекристаллизуют из /CH3/2CHOH, получая 1,76 г целевого продукта с т.пл. 186oC.
Полученный из соединения 4-бис, описанного в В-2, энантиомер, является правовращающим. [α]
4. Путем реакции сульфонамида со сложным эфиром 4-цианофенилаланина с последующим омылением и ацилированием.
1-[2-{ 2-/4-Метилфенилсульфамоил/-3-/4-хлорфедил/-пропионамидо}- -3-/4-цианофенил/-пропионид]-гексагидроазепин /соединение 16/.
а/ В условиях, описанных в /В-1/, 4 г N-/4-толил-сульфонил/-4-хлорфенил/-аланина вводят во взаимодействие с 4,1 г хлоргидрата сложного этилового эфира 4-цианофенилаланина и получают путем обычных обработок 3,7 г этил - 2-[2-/4-толилсульфамоил/-3-/4-хлорфенил/-пропионамидо] -3-/4- цианофенил/-пропионата, который плавится при 82oC.
Этот сложный эфир гидролизуют с помощью раствора 1 г KOH в смеси 10 мл воды и 20 мл этанола, получая, после подкисления 2,5 г соответствующей кислоты, которая плавится при 104oC.
б/ При 0oC к раствору 2,5 г вышеполученной кислоты в 80 мл CH3CN добавляют 2 г БОФ, 1,6 мл N-этилморфолина, затем 0,5 г гексаметиленимина. После перемешивания в течение 16 часов при 20oC растворитель выпаривают. Остаток растворяют в метиленхлориде. Органическую фазу промывают обычным образом, сушат, затем концентрируют при пониженном давлении.
После хроматографии на силикагеле, элюируя смесью метиленхлорида с метанолом /95/5 по объему/, получают 1,3 г целевого продукта. Т.пл. 194 oC.
Таким же образом получают полугидрат [2-{2-/1-нафтилсульфамоил/-3-/2,4-диметилфенил/пропионамидо} -3- /4-цианофенил/-пропионил]- диметиламина /соединение 17/. Т.пл. 140oC.
Г. Получение промежуточных сложных имидоэфиров при получении амидинов из нитрилов
1. Из соединения 5 и этанола
Быстро вводят 2 г соединения 5 в 20 мл безводного этанола, насыщенного при 0oC HCl. После перемешивания в течение ночи при температуре 0-5oC растворитель удаляют путем отгонки при температуре ниже 25oC, получая хлоргидрат целевого продукта.
2. Из соединения 4 и метанола
Быстро вводят 5 г соединения 4 в 100 мл безводного метанола, насыщенного при 0oC HCl. После перемешивания в течение ночи при 0oC растворитель удаляют путем отгонки при температуре ниже 22oC, получая хлоргидрат сложного имидоэфира.
Для получения соответствующего основания хлоргидрат растворяют в 100 мл метиленхлорида, затем при 5oC добавляют триэтиламин до достижения pH 7,5 /измерено в водной среде/. Органическую фазу после этого промывают 5 раз по 30 мл воды с температурой около 20oC, сушат и концентрируют, получая 5,2 г сложного имидоэфира.
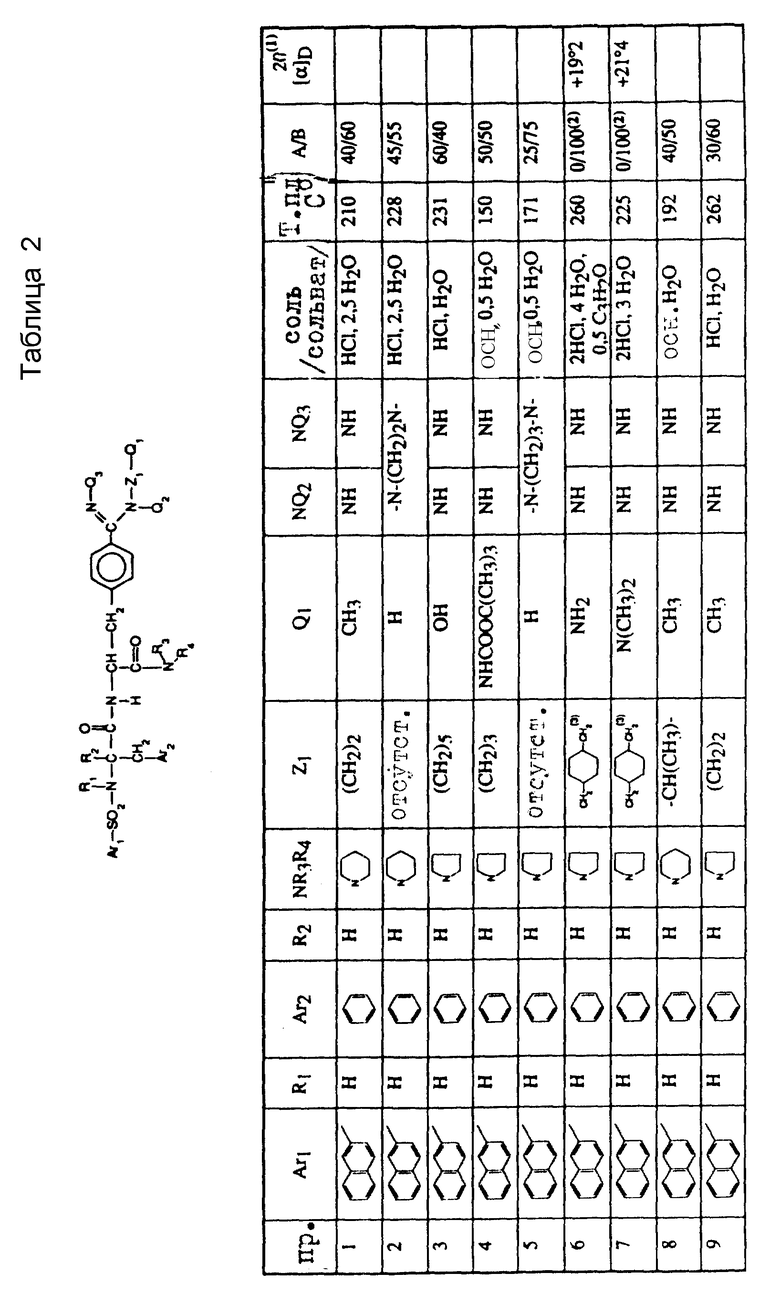
Развернутые формулы и физико-химические характеристики продуктов, подученных как описано в нижеследующих примерах, представлены в таблице I; А/В обозначает относительные пропорции 2-х рацематов.
Пример I
2 г Хлоргидрата сложного имидоэфира соединения 5, подученного согласно Г-1, растворяют в 20 мл безводного изопропанола и в раствор добавляют 1,6 мл н-пропиламина. После перемешивания в течение 2-х часов растворитель удаляют и остаток хроматографируют на силикагеле, элюируя смесью метиленхлорида с метанолом /9/1 по объему/. Таким образом получают 1,7 г хлоргидрата целевого продукта.
Пример 2
1 г Хлоргидрата сложного имидоэфира соединения 5, полученного согласно Г-1, растворяют в 10 мл безводного этанола. Добавляют 0,14 мл 1,2-этандиамина и среду выдерживают около 70oC в течение 1,5 часов. Растворитель удаляют при пониженном давлении и остаток очищают путем хроматографии на силикагеле, элюируя смесью метиленхлорида с метанолом /92/8 по объему/.
Пример 3
К раствору 2 г хлоргидрата сложного имидоэфира соединения 4, полученного согласно Г-1, в 60 мл безводного этанола при 5oC добавляют 2,1 мл триэтиламина и 0,33 мл 1-аминопентанола. После перемешивания в течение 16 часов при комнатной температуре растворитель выпаривают при пониженном давлении и остаток хроматографируют на силикагеле, элюируя смесью метиленхлорида с метанолом /9/1 по объему/ и получая 1,5 г хлоргидрата.
Пример 4
При 5oC в раствор 4,5 г хлоргидрата сложного имидоэфира соединения 4 вводят 3,5 мл триэтиламина и 2,2 г 1,3-диамин-N-/трет.-бутилокси-карбонил/-пропана. После выдерживания при комнатной температуре в течение 20 часов растворитель удаляют путем отгонки при пониженном давлении и остаток растворяют в 50 мл метиленхлорида. Органический раствор промывают водным 0,1 н раствором NaOH, затем водой и высушивают. Полученный после концентрирования остаток кристаллизуют из смеси этилацетата с [(CH3)2CH]2O, получая 2,5 г целевого продукта.
Пример 5
При 5oC в раствор 4 г полученного в примере 4 продукта в 40 мл этилацетата вводят 40 мл этилацетата, насыщенного HCl. Спустя 2 часа выдержки при 20oC, выделяют осадок, промывают его этилацетатом, затем растворяют в смеси 40 мл метиленхлорида и 20 мл водного 0,5 н раствора NaOH. После перемешивания в течение 16 часов при 20oC органическую фазу отделяют и обрабатывают ее обычным образом, получая 2,1 г целевого продукта, после перекристаллизации из смеси этилацетата с метанолом.
Пример 6
2 г Сложного имидоэфира соединения 4-бис, полученного согласно Г-2, вводят в 30 мл безводного метанола и добавляют 0,82 г транс-4-[N-трет.-бутоксикарбонил/-аминометил] -циклогексилметиламина и затем несколько капель насыщенного с помощью HCl метанола вплоть до получения значения pH, равного 8 /измерено в воде/.
Спустя 52 часа выдержки при комнатной температуре, метанол выпаривают, добавляют в среду 30 мл метиленхлорида, затем удаляют метиленхлорид при пониженном давлении; при 5oC полученный остаток вносят в 30 мл этилацетата, насыщенного при 15oC с помощью HCl. После возвращения к комнатной температуре перемешивают еще 0,5 часа, после чего растворитель удаляют перед очисткой остатка путем хроматографии на силикагеле, элюируя смесью метиленхлорида с метанолом /80/20/ по объему/. После перекристаллизации из 1-пропанола выделяют 1,8 г указанного в таблице 1 продукта.
Пример 7
а/. Транс-/N-,N-диметил-/4-/аминометил/-циклогексилметидамин
При 0oC в 50 мл диоксана, содержащего 5 г транс-4-аминометил- циклогексилкарбоновой кислоты, вводят 63,6 мл водного 1н раствора NaOH и 1,28 г MgO, затем медленно добавляют 6,94 г ди-/трет.-бутил/-карбоната в виде раствора в 20 мл диоксана. Спустя 20 часов выдержки при комнатной температуре, отфильтровывают, растворитель удаляют и остаток обрабатывают с помощью 100 мл воды и водную фазу промывает диэтиловым эфиром перед подкислением до pH 2 за счет добавления KHSO4; затем экстрагируют этилацетатом, получая 7,3 г транс-N-/трет. -бутоксикарбонил/-4-аминометилциклогексил-карбоновой кислоты, которая плавится при 125oC.
Это соединение затем растворяют в смеси 20 мл метиленхлорида и 25 мл /CH3/2NCHO, в которую вводят 4,8 г гидроксибензотриазола, затем 6,15 г N, N'-дициклогексилкарбодиимида в виде раствора в 50 мл метиленхлорида. После перемешивания в течение 2-х часов добавляют 4 г безводного /CH3/2NH и оставляют при перемешивании в течение 16 часов. Осадок затем отделяют, органическую фазу промывают несколько раз водным насыщенным раствором NaOH3, затем водой. После высушивания, концентрирования и хроматографии остатка на силикагеле, элюируя этилацетатом, выделяют 5,8 г транс-N,N-диметил-N'-/трет.-бутоксикарбонил/-4- аминометилциклогексилкарбоксамида, который плавится при 94oC.
Это соединение растворяют в 50 мл этилацетат, насыщенного HCl, и, спустя 1 час, отфильтровывают появившийся осадок хлоргидрата, который, обработанный основанием, дает 3,5 г транс-N,N- -диметил-4-аминометилциклогексилкарбоксамида в виде масла.
Это масло растворяют в 10 мл тетрагидрофурана, в который вводят затем при 0oC 25 мл 1М раствора литийалюминийгидрида в том же растворителе. После перемешивания в течение 16 часов при комнатной температуре охлаждают до 0oC и добавляют 0,9 мл ледяной воды, затем 2,7 мл водного 15%-ного раствора NaOH /вес/объем/ и, наконец, выпаривают путем отгонки при пониженном давлении, получая целевой диамин, который перегоняется при 60oC при давлении 1 Па.
б/. Вводя во взаимодействие, согласно способу работы, описанному в примере 1, транс-/N,N-диметил/-4-аминометилциклогексилметиламин со сложным имидоэфиром соединения 4-бис, после перекристаллизации из изопропанола получают чистый энантиомер, описанный в таблице 2.
Получение соединения примера 46 из соединения примера 38
В инертной атмосфере к раствору 0,5 г соединения 38 в 10 мл безводного метанола добавляют 0,08 г аминоиминометансульфокислоты и 0,1 мл триэтиламина. Спустя 16 часов при температуре около 20oC, растворитель выпаривают и остаток обрабатывают 20 мл водного 1н раствора NaOH при температуре около 0oC экстрагируют дихлорметаном. Органическую фазу сушат, концентрируют и остаток хроматографируют на колонке с диоксидом кремния /элюирующее средство: дихлорметан/метанол = 9/1 по объему/, затем с помощью смеси метанол/ водный концентрированный раствор NH4OH /7/3 по объему/. После перекристаллизации из смеси этанола с этилацетатом (в соотношении 8/2 до объему) выделяют целевой продукт в виде основания, из которого получают дихлоргидрат путем воздействия HCl в этаноле. Т.пл. 185oC /2 HCl, H2O/.
Согласно общему способу работы, описанному в примере 3, с адекватными аминами получают:
соединение примера 20 из соединения 4-бис; соединение примера 60 из соединения 4з; соединение примера 58 из соединения 3-бис; соединение примера 64 из соединения 10.
Согласно описанному в примере 6 способу работы с адекватными аминами получают соединения примеров 45, 47 и 48 из соединения 4-бис.
Пример 61
а/. Цис-N-/бутоксикарбонил/-4-/аминометил/-циклогексилметиламин
При 0oC порциями 1,6 г трет.бутилата калия добавляют к раствору 2 г цис-1,4-/диметиламино/-циклогексан-дихлоргидрата [получен согласно методике, описанной в Ber. 71 B, 759 /1938/] в 70 мл безводного метанола, затем добавляют раствор 2,1 г ди-трет.-бутилдикарбоната в 100 мл метанола.
Нагревают 16 часов при 36oC, осадок отфильтровывают и растворитель выпаривают при пониженном давлении. Целевой продукт выделяют путем хроматографии на колонке с диоксидом кремния, элюируя смесью метиленхлорида с метанолом в соотношении 95/5, затем 80/20 /по объему/. Т.пл. 201oC.
б/ Вводя во взаимодействие вышеполученный амин с 2 г сложного имидоэфира соединения 4-бис, полученного согласно Г-2, в условиях, описанных в примере 6, получают целевой продукт, который растворяют в 15 мл HCl, отфильтровывают, затем водную фазу экстрагируют 3 раза по 7 мл бутанола. Растворитель выпаривают при пониженном давлении. Остаток обрабатывают водой и лиофилизуют, получая чистый энантиомер, описанный в таблице 2.
Пример 67
а/ Цис-4-/N,N-диметиламинометил/циклогексилметиламин -цис-циклогексил-1,4-диметанол
При 0oC, медленно, добавляют 328 мл 1М раствора в ТГФ литийалюминийгидрида к раствору 66 г(цис)этилциклогексил-1,4-дикарбоксилата в 500 мл ТГФ.
Перемешивают 16 часов при комнатной температуре и добавляют, примерно при 0oC, 13 мл воды, 39 мл водного 15%-ного раствора NaOH /по весу/, затем снова 13 мл воды. Соли отфильтровывают, растворитель выпаривают при пониженном давлении, остаток перегоняют при 120-124oC при давлении 45•10-6 бар /4,5 Па/. Получают 37 г целевого продукта.
-Цис-циклогексил-1,4-ди-паратолуол-сульфонат
При 0oC к раствору 41 г пара-толуол-сульфонилхлорида и 28 мл триэтиламина в 35 мл ТГФ добавляют раствор 10 г вышеполученного спирта в 70 мл ТГФ. Перемешивают в течение 24-х часов при температуре около 25oC и в течение 3-х часов нагревают примерно при 50oC.
После охлаждения добавляют тогда 50 мл насыщенного раствора NaCl и 50 мл примерно 1н раствора соляной кислоты. Растворитель выпаривают при пониженном давлении и остаток обрабатывают с помощью 300 мл эфира и 200 мл 2н раствора NaOH и перемешивают 16 часов при комнатной температуре.
Декантируют и водную фазу экстрагируют дихлорметаном. Органические фазы сушат и растворитель отгоняют при пониженном давлении. Выделяют 29 г целевого продукта. Т.пл. 91oC.
Цис- 4-/N,N-Диметиламинометил/циклогексилметиламин
При 25oC в автоклаве в течение 48 часов перемешивают 14 г вышеполученного дитозилата в насыщенном аммиаком метанольном растворе. После выпаривания при пониженном давлении остаток обрабатывают с помощью метиленхлорида и 1н соляной кислоты, декантируют, водную фазу подщелачивают с помощью 2н NaOH и экстрагирует метиленхлоридом. После выпаривания растворителя при пониженном давлении остаток хроматографируют на диоксиде кремния, элюируя смесью метиленхлорида с метанолом = 70:30 /по объему/ и выделяют 6 г 4-аминометилциклогексил-пара-толуол-сульфоната/цис/.
К насыщенному раствору диметиламина в метаноле добавляют вышеполученный тозилат и нагревают в автоклаве при 70oC в течение 20 часов.
После выпаривания при пониженном давлении остаток обрабатывают метиленхлоридом, 2 мл воды и 2 г твердого KOH.
Органическую фазу сушат и концентрируют в вакууме. После хлоргидратации и перекристаллизации из этанола получают 3 г дихлоргидрата целевого продукта с т. пл. 252oC.
б/ Соединение 67
0,9 г Вышеполученного дихлоргидрата вводят во взаимодействие с 2,4 г хлоргидрата сложного имидоэфира соединения 4-бис и 1,2 мл N-этилморфолина в 100 мл метанола. После перемешивания в течение 16 часов при 40oC растворитель выпаривают и остаток обрабатывают согласно способу работу, описанному в примере 61, 6, для получения чистого энантиомера, описанного в таблице 2.
Пример 69
/Случай, где R2 и R'2 образуют двойную связь/.
1-[2-/N-{ 2-Нафтилсульфамоил} -2-индолил-карбоксамидо /-3-/4-{ N-[4-(диметиламинометил)транс-циклогексилметил] -амидино} фенил/пропионил]-пирролидин-дихлоргидрат. 4 H2O.
Это соединение получают из соединения 14 согласно общему способу работы, описанному в примере 1. Т.пл. 230oC.
Пример 70
1-[2-[N-/2-Нафтилсульфамоил/-5-метокси-2-индолил] - карбоксамидо/-3-/4{ N-[4-(диметиламинометил)транс-циклогексилметил] амидино}фенил/-пропионил]-пирролидин.
Это соединение получают таким же образом из соединения 13.
Т.пл. 230oC.
Соединение 18
1-[2-[2-{ 5-Изохинолилсульфамоил}-2-метил-3-фенил-пропион- амидо]-3-/4-цианофенил/-пропионил]-пирролидин, Т.пл. 264oC.
Это соединение получают таким же образом, как соединение 16 (см. В-4).
Пример 71
Работают согласно примеру 67, но исходя из 4- (пирролидинометил)циклогексилметиламина (транс) и нитрила 5А, полученного, как описано выше в п. 2.1, получают соединение, указанное в таблице 2.
4-(Пирролидинометил)циклогексилметилмалин(транс) получали таким же образом, как и в примере 7а, исходя из пирролидина.
Пример 83
Работают согласно примеру 67, исходя из 4- (пирролидинометил)циклогексилметиламина и нитрила 4бис.
4-(Пирролидинометил)циклогексилметиламин получен согласно методу, описанному в примере 67а, исходя из пирролидина.
1-Фармакологическая ценность заявленных соединений была установлена на основании теста, выявляющего сродство заявленных соединений к рецепторам нейропептида Y, осуществленного в соответствии с протоколом, описанным в Eur. J. Biochem. 145, 525-530 (1984) Унденом и его сотрудниками, на коре головного мозга крысы. Полученные результаты выражены параметром C1 50, который представляет собой концентрацию соединения, обеспечивающего ингибирование 50% связи NPY с его рецепторами.
Эти результаты приводятся в таблице 2.
При дозах до 10 мг/кг не было отмечено никакого летального исхода или аномального явления у крысы без спинного мозга при проведении опыта in vivo для оценки антагонистической активности NPY.
Таким же образом, на примере соединения, полученного в примере 67 в тесте in vivo, осуществленном на других видах животных, подтвердилось отсутствие токсичности.
Таким образом, проведенные испытания доказывают безвредность заявленных соединений.
Ниже приведены некоторые из возможных препаративных форм фармацевтических композиций:
Заявитель направляет примеры фармацевтических композиций:
Таблетка с дозой 50 мг:
Активное начало - 50 мг
Микрокристаллическая целлюлоза - 200 мг
Модифицированный крахмал кукурузы - 47,5 мг
Стеарат магния - 2,5 мг
Желатиновая капсула с дозой 15 мг:
Активное начало - 15 мг
Лактоза - 150 мг
Модифицированный крахмал кукурузы - 30 мг
Стеарат магния - 5 мг
Инъецируемые препараты с дозой 10 мг
Активное начало - 10 мг
Инозитол - 100 мг
Бензиловый спирт - 20 мгн









Соединения общей формулы 1, где Аr1 - фенил, нафтил, хинолил, изохинолил, возможно замещенный хлором, фтором, алкилом, алкоксилом или диалкиламино; Аr2 - фенил, возможно замещенный хлором, фтором, алкилом или алкоксилом, или тиенил; R1 и R2 - Н или алкил; R'2 - водород или R2 и R'2 образуют двойную связь, или же R1 ничего не обозначает, тогда N соединен с Аr2; R3 и R4 обозначают алкил или образуют с атомом азота, с которым они связаны, пирролидин, пиперидин или гексагидроазепин; Z1 - C1-12 алкилен, возможно прерываемый С5-7-циклоалкилом или фенилом; Q1 - Н, метил, амино, гидроксил, диалкиламино, гуанидино, алкилгуанидино, пиридил, индолил, алкил-4-пиперазинил, аминоалкил-N-алкиламино, алкоксикарбониламино, карбамоил, фенил, возможно замещенный алкоксилом, или пиридил, имидазолил, индолил или пирролидинил; Q2 и Q3 - Н или алкил, или Q2 и Q3 образуют вместе С2-3 -алкилен, в виде чистых энантиомеров или их смесей, а также их фармацевтически приемлемых солей с кислотами. Соединения формулы 1 и их фармацевтически приемлемые соли способны фиксироваться на биологических рецепторах нейропептида Y (NРY), пептида из 36 аминокислот с широким спектром физиологической активности, в частности в отношении центральной нервной системы или сердечно-сосудистой системы .
 3 с. и 4 з.п. ф-лы, 2 табл.
3 с. и 4 з.п. ф-лы, 2 табл.

в которой Ar1-фенил, нафтил, хинолил, изохинолил, возможно замещенный Cl, F, (C1-С4)-алкилом, (С1-С4)-алкоксилом или (С1-С4) диалкиламино;
Ar2 - фенил, возможно замещенный Cl, F, (C1-С4)-алкилом или (С1-С4)-алкоксилом, или тиенил;
R1 и R2 независимо друг от друга обозначают Н или (С1-С4)-алкил; - атом водорода или же R2 и
- атом водорода или же R2 и  образуют вместе двойную связь, или же R1 ничего не обозначает, тогда N соединен с Ar2;
образуют вместе двойную связь, или же R1 ничего не обозначает, тогда N соединен с Ar2;
R3 и R4 - одинаковые или разные, обозначают (C1-С4)-алкил или образуют с атомом азота, с которыми они связаны, насыщенный (C5-С7) гетероцикл, выбираемый среди пирролидина, пиперидина или гексагидроазепина;
Z1 - (C1-С12)-алкилен, возможно прерываемый (C5-С7) циклоалкилом или фенилом;
Q1 - атом водорода, метил, амино, гидроксил, ди (C1-С4) алкиламино, гуанидино, (C1-С4)-алкилгуанидино, пиридил, индолил, (C1-С4-алкил-4-пиперазинил, амино- (C1-С4)-алкил-N-(C1-С4) алкиламино, (C1-С4)-алкоксикарбониламино,
карбамоил, фенил, возможно замещенный (C1-С4)-алкоксилом, (C2-С8)-алкоксикарбонил или пиридил, имидазолил, индолил или пирролидинил;
Q2 - Н или (C1-С4)-алкил;
Q3 - атом водорода (C1-С4)-алкил или же Q2 и Q3 образуют вместе (C2-С3)-алкилен, когда Z1 означает связь, а Q1 обозначает атом водорода,
в виде чистых энантиомеров или их смесей, а также их фармацевтически приемлемых солей с кислотами.
Ar1 обозначает фенил, нафтил, возможно замещенный C1, (C1-С3)-алкилом или (C1-С3) алкоксилом;
Ar2 обозначает фенил, возможно замещенный C1, (C1-С3)-алкилом или (C1-С3)-алкоксилом;
R1 и R2, независимо друг от друга, обозначают Н или (C1-С4)-алкил; - атом водорода, или же R2 и
- атом водорода, или же R2 и  образуют вместе двойную связь, или же R1 ничего же обозначает, а N связан при этом с Ar2;
образуют вместе двойную связь, или же R1 ничего же обозначает, а N связан при этом с Ar2;
R3 и R4, одинаковые или разные, обозначают (C1-С4)-алкил или образуют с атомом азота, с которым они связаны, насыщенный гетероцикл, выбираемый среди пирролидина, пиперидина и гексагидроазепина;
Z1 - (C1-С12)-алкилен, возможно (C5-С7)-циклоалкилом или фенилом;
Q1 - метил, амино, (C1-С4)-алкоксикарбониламино, (C1-С4)-диалкиламино, (C1-С4)-алкил-4-пиперазин, гуанидино, (C1-С4)-алкилгуанидино, пиридил, гидроксил, амино-(C1-С4)-алкил-N-(C1-С4)-алкиламино, карбамоил или фенил, возможно замещенный (C1-С3)-алкоксилом;
Q2 - Н или (C1-С4)-алкил;
Q3 - Н или (C1-С4)-алкил, или Q2 и Q3 связаны для образования (C2-С3)-алкилена, когда Z1 обозначает связь, а Q1 обозначает атом водорода, в виде чистого энантиомера или смеси энантиомеров в любой пропорции,
а также их фармацевтически приемлемые соли с кислотами.
в которой Ar1 - фенил, нафтил или хинолин, возможно замещенный Cl, F (C1-С4)-алкилом, (C1-С3)-алкоксилом;
Ar2 - фенил, возможно замещенный Cl, F, (C1-С4)-алкилом, (C1-С4)-алкоксилом;
R1 ничего не обозначает, а N связан с Ar2, или же R1 обозначает Н;
R2 - Н, или же R2 и  образуют вместе двойную связь, или же
образуют вместе двойную связь, или же  обозначает Н;
обозначает Н;
R3 и R4, одинаковые или разные, обозначают (C1-С4)-алкил, или же образуют вместе с атомом азота, с которым они связаны, насыщенный (C5-С7) гетероцикл, выбираемый среди пирролидина, пиперидина;
Q1 - амино, метил, (C1-С4)-алкоксикарбониламино, гидроксил, ди (C1-С4)-алкиламино или пирролидинил;
Q2 - Н;
Q3 - Н;
Z1 - (C1-С12)-алкилен, прерываемый (C5-С7)-циклоалкилом или фенилом, отличающийся тем, что нитрил формулы II
в которой Ar1, R1, R2, Ar2,  R3 и R4 имеют вышеуказанные значения,
R3 и R4 имеют вышеуказанные значения,
в виде чистого энантиомера или смесь изомеров в любом соотношении вводят во взаимодействие с низшим спиртом ROH в кислой среде с получением промежуточного сложного имидоэфира, который затем вводят во взаимодействие с амином формулы
в которой Z1, Q1, Q2 имеют значения, указанные выше.
| СОЕДИНЕНИЕ ГИБКОГО ШЛАНГА, ИМЕЮЩЕГО МЕТАЛЛИЧЕСКУЮ ОПЛЕТКУ, С НИППЕЛЕМ | 0 |
|
SU236163A1 |
| РАЗЪЕМНОЕ КОНЦЕВОЕ СОЕДИНЕНИЕ ГИБКИХ | 0 |
|
SU236164A1 |
| Drugs of the Future | |||
| Пуговица для прикрепления ее к материи без пришивки | 1921 |
|
SU1992A1 |
| Способ получения @ -арилсульфонил- @ -аргининамида или его солей | 1978 |
|
SU1181539A3 |
