Изобретение относится к новым 2,2-диалкил- и 2,2-диалкил-3,4-дигидро-3- гидрокси-2Н-1-бензопиранам и их солям, сложным эфирам и N-оксидам и к способам их получения, также к их использованию в качестве фармацевтических средств и к содержащим их фармацевтическим композициям.
Более конкретно изобретение в своем самом широком аспекте включает:
1. 2,2-ди-/C1-C5-алкил/- или транс-2,2-ди-/C1-C5-алкил/-3,4-дигидро- 3-гидрокси-6-/пиридин-4-ил/- 2Н-1-бензопиран, содержащий карбоксамидный фрагмент в положении 4 и где пиридин-4-ильная группа замещена в положении 2 и/или 3 одним или двумя членами, выбираемыми из группы, включающей C1-C5алкил, C1-C5-гидроксиалкил и /C1-C5/ алкоксиалкил;
или его N-оксид;
или физиологически гидролизуемый и физиологически приемлемый сложный эфир такого бензопирана или N-оксида, или соли присоединения кислоты, или четвертичная аммониевая соль такого бензопирана, N-оксида, или сложный эфир.
Алкильные группы и фрагменты соединений, как определенные в п. 1 могут представлять собой линейную или разветвленную цепь. Предпочтительными значениями заместителей в положении 2 ядра бензопирана также, как в положении 2 и/или 3 пиридинильной группы являются значения, указанные ниже в отношении формулы /I/ для R6 и R7 и R1 и R2.
Как описано в дальнейшем, соединения по изобретению, например, как определенные в п. 1, обладают раскрывающей калиевые /K+/ каналы активностью [см. , например, Cook и др., "Potassium Channels: Structure, Classification. Function and Therapeutic Potential" ed. N.S. Cook, Ellis Horwood, Chechester /1990/, с. 181-255]. Бензопирановые производные, которые в положении 4 содержат замещенный карбоксамидный фрагмент и обладают раскрывающей калиевые каналы активностью, широко описаны в уровне техники и относятся к классу важных и признанных соединений. 4-Карбоксамидный фрагмент в соединениях изобретения может представлять собой любой из таких известных и описанных в уровне техники в связи с раскрывающими K+-каналы бензопиранами, в том числе N-замещенных, например, циклических, карбоксамидных фрагментов. Предпочтительными карбоксамидными фрагментами в отношении соединений изобретения являются такие формулы -N/ R9/-COR10, как описано ниже.
Следует принимать во внимание, что бензопирановое ядро определенных в п. 1 соединений может содержать заместители в дополнение и таковым, специфически определенным. В особенности, они могут быть, например, 7-/C1-C5/-алкил-замещенными, особенно 7-метил-замещенными, например, как указано далее в связи с формулой I.
В соответствии с изобретением предпочтительны 2,2-ди-/C1-C5/-алкил- 3,4-дигидро-3-гидрокси-6-/пиридин-4-ил/-2Н-1- бензопираны и/или их -оксиды, сложные эфиры и соли, как описано в п.1. 3-Гидроксильная группа и 4-карбоксамидный фрагмент в таких соединениях находятся в транс-конфигурации, как описано в п. 1. Для этой группы соединений, как правило, должны быть предпочтительны /3S,4R/-энантиомеры в чистой или в основном в чистой форме, или в виде смеси изомеров, например в виде рацемической смеси, как описано далее в связи с соединениями формулы /I/.
В более специфическом аспекте данное изобретение включает:
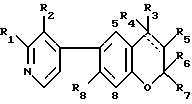
2. соединение формулы /I/: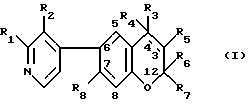
где R1 и R2 независимо друг от друга обозначают водород, C1-C5-алкил, C1-C5-гидроксиалкил или C1-C5-/алкоксиалкил/, причем по меньшей мере один из R1 и R2 является отличным от водорода;
R3 обозначает группу формулы - N/R9/-COR10, где R9 обозначает водород и R10 обозначает пиридил или R9 и R10 вместе обозначают 1,3-бутадиенилен или обозначает группу формулы -/CH2/n- или -/CH2/m , в которой "n" обозначает целое число от 3 до 5 и m обозначает 1 или 2;
, в которой "n" обозначает целое число от 3 до 5 и m обозначает 1 или 2;
R4 обозначает водород и R5 обозначает гидроксил в транс-положении по отношению к R3, или
R4 и R5 вместе образуют дополнительную связь, как показано пунктирной линией;
R6 и R7 независимо друг от друга обозначают C1-C5-алкил и
R8 обозначает водород или C1-C5 - алкил, или его N-оксид, или физиологически гидролизуемый и физиологически приемлемый сложный эфир такого соединения, или N-оксид, или соль присоединения кислоты или четвертичная аммониевая соль такого соединения, N-оксид или сложный эфир.
Алкильные группы, такие как R1, R2, R6, R7 и R8, а также алкильные фрагменты гидроксиалкила и алкоксиалкила, а также R1 и R2, могут быть с линейной или разветвленной цепью. Алкоксиалкильными группами предпочтительно являются /C1-C4-алкокси/-метил, в особенности метоксиметил. Предпочтительной гидроксиалкильной группой является гидроксиметил. R6 и R7 оба предпочтительно обозначают метил. R8 предпочтительно представляет собой водород или метил наиболее предпочтительно водород.
В предпочтительной группе соединений формулы /I/, R1 имеет любое из указанных для формулы /I/ значений и R2 обозначает водород или C1-C5-алкил [особенно метил], предпочтительно водород.
В дальнейшей предпочтительной группе соединений формулы /I/, R3, R9 обозначает водород и R10 обозначает пиридил [особенно 3-пиридил] или R9 и R10 вместе обозначают 1,3-бутадиенилен, триметилен или тетраметилен. Наиболее предпочтительно R9 и R10 вместе обозначают тетраметилен.
Предпочтительно R4 обозначает водород, а R5 обозначает гидроксил. Бензопираны согласно изобретению, например соединения формулы /I/, образуют N-оксиды, например на атоме азота 6-пиридильной группы. Такие N-оксиды обладают активностью /как описано далее/ и переносимостью, сравнимыми в основном соединениями, и также составляют часть данного изобретения.
Под термином "физиологически гидролизуемый и физиологически приемлемый сложный эфир", как он используется здесь, понимают сложный эфир, в котором гидроксильная группа [например, в отношении формулы /I/, гидроксильные группы R5 и/или гидроксильные фрагменты любой гидроксиалкильной группы, обозначаемой как R1 и/или R2] этерифицирована до сложно-эфирной и который гидролизуется при физиологических условиях с образованием кислоты, которая сама физиологически переносима во вводимых дозах. Следует отметить, что такие сложные эфиры являются пролекарственными формами обычного типа и обладают активностью и переносимостью, сравнимыми с основными соединениями. Пример.ы таких сложных эфиров включают, например, ацетаты.
Соли присоединения кислоты, например, соединений формулы /I/, их N-оксидов и их определенных сложных эфиров, включают соли как неорганических, так и органических кислот. Такие соли также имеют активность, сравнимую со свободными соединениями, N-оксидами и сложными эфирами. Фармацевтически приемлемые соли присоединения кислот для фармацевтического использования согласно изобретению, как описано далее, представляют собой, например, соли соляной, серной и фумаровой кислот.
Четвертичные аммониевые соли, например соединений формулы /I/, их N-оксидов и их определенных сложных эфиров, включают, например, соли с галоидорганическими соединениями, например с алкилгалогенидами. Фармацевтически приемлемые четвертичные аммониевые соли для фармацевтического использования согласно изобретению, включают, например, такие соли с метилиодидом.
Для фармацевтического использования согласно изобретению сложно-эфирные формы, как указано выше, обычно менее предпочтительны.
Соединения формулы /I/, в которой R4 обозначает водород и R5 обозначает гидроксил, также как их N-оксиды, сложные эфиры и соли, как указано, имеют конфигурацию /3S*, 4R*/, т.е. конфигурация групп R3 и R5 в 3- и 4- положениях - транс-конфигурация. Соединения по изобретению существуют в энантиомерной форме, т. е. в виде оптически активных антиподов, имеющих /3S, 4R/- или /3R, 4S/-конфигурацию. Изобретение охватывает как оба индивидуальных энантиомера [оптические активные /3S, 4R/ или /3R, 4S/ - антиподы], так и также их смеси, например рацемические смеси.
Для такого фармацевтического использования согласно изобретению полагают, что эти соединения предпочтительно должны находиться при преобладать в виде /3S,4R/-энантиомеров. Соответственно упомянутые /3S,4R/-энантиомеры должны быть, или должны использоваться, согласно изобретению в очищенной форме, т. е. включающей менее, чем 50% загрязняющего [другого] энантиомера, более пригодно, в чистой или в основном в чистой форме, например, содержащей менее, чем 10%, предпочтительно 5% или менее, например 1 или 2% или менее, загрязняющего /3R,4S/-энантиомера.
В дополнение к сказанному настоящее изобретение также включает:
3. способ получения бензопирана такого, как описанный в п. 1., например соединения формулы /I/, и такого, как описанное в п.2 или его N-оксида, или физиологически гидролизуемого и физиологически приемлемого сложного эфира такого бензопирана или N-оксида, или соли присоединения кислоты или четвертичной аммониевой соли такого бензопирана, N-оксида или сложного эфира, причем этот способ включает:
1/ для получения описанного бензопирана:
i11/ взаимодействие Ia,7b-дигидро-2,2-ди[C1-C5-алкил]-6-[пиридин-4-ил] -2Н-оксирено /c/ /1/-бензопирана, где пиридин-4-ильная группа замещена в 2- и/или 3-положении одним или двумя членами, выбираемыми из группы, включающей C1-C5-алкил, C1-C5-гидроксиалкил и C1-C5-/алкоксиалкил/, например, соединения формулы /II/: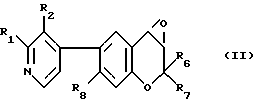
где R1, R2, R6, R7 и R8 имеют указанные для формулы /I/ значения с солью щелочного металла карбоксамида, например с соединением формулы /III/: R10 - CO - N⊖ - R9 M⊕ /III/,
где R9 и R10 имеют указанные для формулы /I/ значения и M+ обозначают ион лития, натрия или калия; или
i2/ ацилирование и, если необходимо, алкилирование амино-группы 2,2-ди-/C1-C5 - алкил/- или транс-2,2-ди-/C1-C5-алкил/-3,4-дигидро-3-гидрокси-4-амино-6-/пиридин-4-ил/-2Н-1-бензопирана, где пиридин-4-ильная группа замещена в 2- и/или 3-положении одним или двумя членами, выбираемыми из группы, включающей C1-C5-алкил, C1-C5-гидроксиалкил и C1-C5-/алкоксиалкил/, например, взаимодействие соединения формулы /IV/: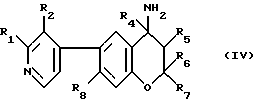
где R1, R2 и R4-R8 имеют указанные для формулы /I/ значения с соединением формул /V/, /V'/ или /V''/: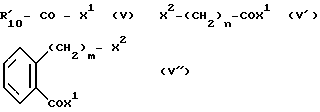
где 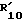 обозначает пиридил и X1 и X2 обозначают удаляемые группы;
обозначает пиридил и X1 и X2 обозначают удаляемые группы;
ii/ для получения N-оксида бензопирана или физиологически гидролизуемого и физиологически приемлемого сложного эфира бензопирана или N-оксид-бензопирана, как указано выше,
этерифицирование до сложного эфира бензопирана или бензопиран-N-оксида, как определено в п.1 содержащего свободную гидроксильную группу или фрагмент, для введения соответствующей сложно-эфирной группы, например взаимодействие соединения формулы /I/, как указанное выше, где R5 обозначает гидроксил и/или по меньшей мере один из R1 и R2 обозначает C1-C5-гидроксиалкил, или его N-оксида с соответствующим галоидом кислоты или ангидридом, и/или окисление бензопирана или его физиологически гидролизуемого и физиологически приемлемого сложного эфира, как определено в п.1, например окисление соединения формулы /I/, как определено выше, или его физиологически гидролизуемого и физиологически приемлемого сложного эфира;
и регенерирование полученного бензопирана, бензопиран-N-оксида или его физиологически гидролизуемого и физиологически приемлемого сложного эфира в свободной форме или в форме соли присоединения кислоты или четвертичной аммониевой соли.
Способ стадии i1/ может быть осуществлен согласно известным в уровне техники методом, например путем реакции при температурах от комнатной до температуры кипения с обратным холодильником в присутствии инертного растворителя или разбавителя, такого как тетрагидрофуран или диметилсульфоксид. Соответственно, необходимую соль щелочного металла, например соединение формулы /III/, получают предварительно in situ, например, как описано в примерах 1-11 и примере 21. Путем соответствующего использования, например натриевых солей, могут быть получены как бензопираны, так и дигидро-бензопираны согласно изобретению, например, как проиллюстрировано в примерах 11 и 12. Использование литиевых солей приводит первоначально или только к предпочтительным дигидро-бензопиранам согласно изобретению, как проиллюстрировано в примерах 1-10.
Способ стадии i2/ также может быть осуществлен согласно известным методам уровня техники. Пригодными удаляемыми группами X1 являются галогены и активированные сложно-эфирные группы, а пригодными удаляемыми группами X2 является галогены. Реакцию пригодным образом осуществляют при 0-100oC в инертном растворителе или разбавителе, таком, как ацетонитрил или дихлорметан, предпочтительно в присутствии кислотосвязующего агента, например, как триалкиламины или карбонаты щелочных металлов. Способ проиллюстрирован в примерах 13-20.
Способ стадии ii/ может быть осуществлен согласно обычным способам ацилирования /N-окисления, например для получения N-оксидов, путем обработки пероксидом водорода, м-хлорнадбензойной кислотой или реактивом Collin /CrO3•Pу2/, как проиллюстрировано в примере 23.
Первоначально полученные свободные основания могут быть превращены в соли присоединения кислот или четвертичные аммониевые соли путем реакции с кислотами или, например, алкил-галогенидами, например как метилгалогениды и наоборот.
При использовании рацематов соединений формулы /II/ и формулы /IV/ полученные 4-карбоксамидо-3,4-дигидро-3-гидрокси-бензопираны должны быть в форме транс-рацематов [т. е. включая /3S,4R/ - плюс /3R,4S/, - изомеры]. Полученные рацематы могут быть разделены с получением индивидуального /3R,4S/ - или [предпочтительно] /3S,4R/ - энантиомера, например, хроматографически при использовании хиральной стационарной фазы. Когда требуются индивидуальные /3S,4R/-энантиомеры, однако это предпочтительно достигается путем использования соответствующего изомера в качестве исходного материала, т.е. в отношении формулы /II/, 3S,4S-антипода и в отношении формулы /IV/, 3S,4R-антипода. Подходящим способом получения является способ, представленный на схеме реакций A.
Как следует заметить, могут быть использованы известные в уровне техники в качестве вариантов или альтернатив указанных способов, например, для взаимопревращения первоначально полученных соединений или для введения альтернативных карбоксамидных групп в 4-положение. Лабильные группы могут быть защищены, например, путем ацилирования, использованием обычной защиты, например, с помощью защитных для гидроксила групп. В дополнение первоначально полученные бензопираны, если желательно, можно превращать в соответствующие бензопираны путем дегидратации по 3,4-связи, опять в соответствии со стандартными способами. Дальнейшие альтернативы должны быть известны специалисту из уровня техники.
Соединения формулы /II/ могут быть получены согласно следующей общей реакционной последовательности A: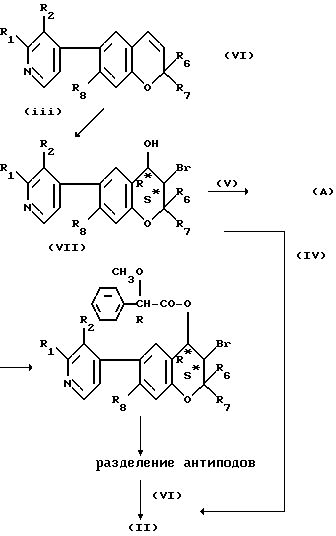
Стадии /III/-/VI/ могут быть осуществлены обычным образом, например, в соответствии с обычными способами, проиллюстрированными в примерах 24.A.4; A.5; A.5'a; и A.5'b.
Исходные материалы формулы /IV/ могут быть получены из соответствующих соединений формулы /II/ путем введения во взаимодействие с аммиаком, например, в соответствии с общими способами, проиллюстрированными в примере 24. A.6'.
Полученное через стадии /III/ и /IV/ соединение формулы /II/ представляет собой цис-рацемат, т.е. включает /3R,4R/- и /3S,4S/-антиподы. Стадия /V/ включает введение соответствующей хиральной ацильной группы [на схеме A, например, за счет /R/-α-метоксифенилацетила]. Хиральный рацемат /VIII/ может быть легко разделен путем колоночной хроматографии или фракционной перекристаллизации на его индивидуальные /3R,4S/- и /3R,4S/- антиподы. Путем использования /3R, 4S/-антипода и осуществляя стадию /VI/, соединение /II/ в качестве исходного материала может быть получено в чистой или в практически чистой /3S,4S/-энантиомерной форме.
Соединения формулы /VI/ могут быть получены в соответствии со следующей общей реакционной схемой Б: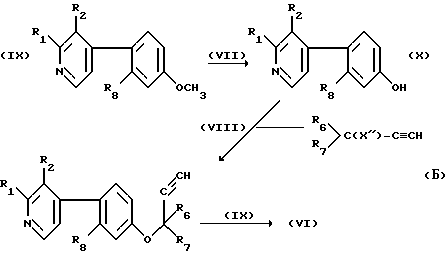
в которой X'' обозначает удаляемую группу, обычно атом галогена, например хлор. Стадии /VII/-/IX/ могут быть осуществлены обычным образом, например, в соответствии с общими способами, проиллюстрированными в примерах 24. А. 2 и А.3, причем стадии /VIII/ и /IX/ осуществляются в примере 24.А.3 без очистки промежуточных продуктов. Как указано в примере 24.А.2, стадию /VII/ обычно осуществляют в апротонном растворителе, таком, как ацетон, в присутствии основания, такого, как K2CO3 и катализатора такого, как KI.
Соединения формулы /IX/ могут быть получены разными возможными путями, например, как показано в следующей общей реакционной схеме В: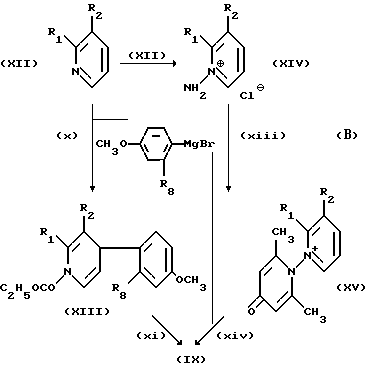
Способы стадий /X/ и /XI/ и /XII/-/XIV/, могут быть осуществлены обычным образом, например, в соответствии с общими способами, проиллюстрированными в примерах 24. А1а и 24.А.1а' и в', соответственно. Обычно методика получения через стадии /X/ и /XI/ будет предпочтительна для синтезов в широком масштабе.
Когда желательно получать соединения изобретения, в которых R1 и/или R2 обозначают гидроксиалкил или алкоксиалкил, то это также может быть достигнуто путем конверсии алкильных заместителей, таких как R1/R2. Подобным образом метильные заместители, как R1/R2, если желательно, могут быть превращены в высшие алкильные заместители. Обычно такие реакции превращения осуществляют на стадии синтеза соединения формулы /VI/, используя обычные приемы, например, в соответствии с обычными способами, проиллюстрированными в примерах 24.E.3; F.3; G3.a- G3.b и H.3.a-H.3.b.
Соединения формулы /VI/, где R8 отличен от водорода, обычно можно получать в соответствии со следующей реакционной схемой Г:
в которой Hal обозначает галоген, например, бром, и R8 обозначает C1-C5-алкил. Способы стадий /XV/-/XXI/ могут быть осуществлены обычным образом, например, в соответствии со способами, проиллюстрированными в примере 24.I.
Промежуточные соединения, проиллюстрированные выше и в сопутствующих примерах, особенно промежуточные соединения формул /II/, /IV/ и /VI/- /VIII/, являются новыми. Такие промежуточные соединения, в особенности промежуточные соединения формул /II/ и /IV/ и способы их получения также составляют часть изобретения.
Следующие примеры иллюстрируют способы изобретения. Все ЯМР-спектры снимаются при 360 мГц. Все температуры даются в градусах Цельсия и без поправки.
Пример. 1. Получение /-/-/3S,4R/-1-/3,4-дигидро-2,2-диметил-3-гидрокси- 6-/2-метилпиридин-4-ил/-2Н-1-бензопиран-4-ил/-2-пиперидинона /формула /I/:
R1 = R6 = R7 = CH3; R2 = R4 = R8 = H; R5=-OH; 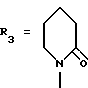 : чистый или практически чистый /3S,4R/- энантиомер/.
: чистый или практически чистый /3S,4R/- энантиомер/.
Перемешиваемый раствор 2-пиперидинона /19,8 г/ в безводном тетрагидрофуране /400 мл/ при 10oC и в атмосфере аргона обрабатывают раствором би/триметилсилил/амида лития [200 мл / 1.0 М] в тетрагидрофуране и перемешивают при комнатной температуре в течение 2 ч. Полученную суспензию обрабатывают раствором /-/-/3S, 4S/-4-[1a, 7b - дигидро-2,2-диметил-2Н-оксирено /c/1/-бензопиран-6-ил] -2-метилпиридина {26,7 г; см. пример 24.А.5'} в безводном тетрагидрофуране /150 мл/ и кипятят с обратным холодильником в течение 31 ч. Смесь охлаждают до 15oC, обрабатывают насыщенным водным раствором хлорида аммония /300 мл/ и экстрагируют этилацетатом /2 раза по 150 мл/. Объединенные экстракты промывают рассолом /300 мл/, сушат /Na2SO4/ и отфильтровывают. Растворитель выпаривают при пониженном давлении, получая сырой продукт, который очищают путем хроматографии [силикагель, 5% C2H5OH в CH2Cl2] и перекристаллизуют из смеси этанола с пентаном с получением целевого соединения; т. пл. = 192oC, (α)
Следующие соединения формулы /Ia/: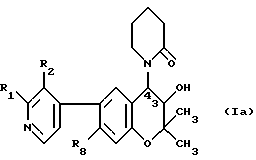
получают аналогичным образом, причем получение необходимого оксирено-исходного материала проиллюстрировано в примере, указанном в правой колонке табл.1.
Все перечисленные в табл.1 соединения находятся в виде трансрацематов.
Пример. 11. Получение 1-[2,2-диметил-6-{2-метилпиридин-4-ил}-2Н-1-бензопиран- 4-ил]-2-пиперидинона [формула /I/: R1 = R6 = R7 = CH3; R2 = R8 = H; R4- R5 = дополнительная связь; 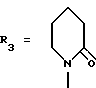 ].
].
Перемешиваемый раствор 2-пиперидинона /0,90 г/ в безводном диметилсульфоксиде /20 мл/ обрабатывают с помощью NaH [0,48 г 55 %-ной дисперсии в масле] и перемешивают при 50oC в течение 30 мин в атмосфере аргона. Смесь охлаждают до 10oC и обрабатывают раствором /±/-4-[1a, 7b - дигидро-2,2-диметил-2Н-оксирено-/c/ /1/-бензопиран-6-ил]-2-метилпиридина /2,67 г, см. пример 24. А. 5/ и перемешивают при комнатной температуре в атмосфере аргона в течение 14 ч. Растворитель выпаривают при пониженном давлении и остаток обрабатывают насыщенным водным раствором NH4Cl /200 мл/ и экстрагируют дихлорметаном /3 раза по 100 мл/. Объединенные экстракты сушат /сульфат натрия/, отфильтровывают и растворитель выпаривают при пониженном давлении, получая смесь. Ее разделяют хроматографически [силикагель, 2% C2H5OH в CH2Cl2], получая менее полярный продукт /А/, который перекристаллизовывают из смеси этилового эфира с пентаном, получая целевое соединение и более полярное соединение /Б/, которое перекристаллизовывают из смеси дихлорметана с диэтиловым эфиром, получая такой же продукт, как и в примере 2. Т.пл. целевого соединения 95-98oC.
Пример. 12. Получение 1-[2,2-диметил-6-/3-метилпиридин-4-ил/-2Н-1-бензопиран- 4-ил]-2-пиперидинона [формула /I/: R2 = R6 = R7 = CH3; R1 = R8 = H; R-4- R5 = дополнительная связь; 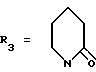 ]
]
Целевое соединение получают одновременно с продуктом примера 3, приведенного выше, исходя из продукта примера 24.Д.5., приведенного ниже, и поступая аналогично примеру 11. Физические характеристики целевого соединения: 1H-ЯМР / δ -d6 ДМСО/: 1,41 /с., 3Н/; 1,45 /с., 3Н/; 1,77-1,96 /м., 3Н/; 2,26 /с. , 3Н/; 2,27-2,68 /м., 3Н/; 3,37-3,58 /м., 2Н/; 5,83 /с., 1Н/; 6,92 /д., 1Н/; 7,14-7,20 /м., 2Н/; 7,36 /м., 1Н/; 8,39 /д., 1Н/; и 8,45 /с., 1Н/.
Пример. 13. Получение /+/-/3S,4R/-[3,4-дигидро-2,2-диметил-3-гидрокси-6-/2-метилпиридин- 4-ил/-2Н-1-бензопиран] -3-пиридин-карбоксамида {формула /I/ R1 = R6 = R7 = CH3; R2 = R4 = R8 = H; 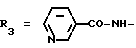 чистый или в практически чистый /3S,4R/ - энантиомер.
чистый или в практически чистый /3S,4R/ - энантиомер.
Перемешиваемый раствор /+/-/3S, 4R/-4-амино-3,4-дигидро-2,2-диметил-6- /2-метилпиридин-4-ил/-2Н-1-бензопиран-3-ола [0,65 г, см. пример 24.А.6' ниже] , триэтиламина /0,50 г/ и 4-диметиламинопиридина /0,002 г/ в безводном дихлорметане /30 мл/ при 2oC и в атмосфере аргона обрабатывают гидрохлоридом никотиноилхлорида /0,445 г/. Смесь перемешивают в течение 2 ч при комнатной температуре, после чего смесь обрабатывают водным раствором карбоната натрия - [100 мл/2М] и экстрагируют смесью 3:1 CH2Cl2/CH3OH {3 раза по 100 мл}. Объединенные экстракты промывают рассолом, сушат /сульфат натрия/, отфильтровывают и растворитель выпаривают при пониженном давлении с получением масла. Масло очищают путем хроматографии [силикагель, 2% C2H5OH в CH2Cl2] и перекристаллизуют из смеси этанола с диэтиловым эфиром, получая целевое соединение в энантиомерно чистой или в практически чистой форме; т.пл. 247-248oC; (α)
Следующие соединения формулы /Ib/: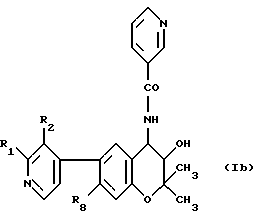
получают аналогично, причем получение необходимого 4-амино-бензопиран-3-ола в качестве исходного материала проиллюстрировано в примере, указанном в правой колонке табл.2.
Все перечисленные в табл.2 соединения находятся в виде трансрацематов.
Пример. 21. Получение транс-/+/-1/[3,4-дигидро-2,2-диметил-3-игдрокси-6-/2-метилпиридин-4-ил/-2Н-1- бензопиран-4-ил]-2Н-1-пиридинона {формула /I/: R1 = R6 = R7 = CH3-; R2 = R4 R8 = H; 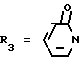 ; в виде транс-рацемата}.
; в виде транс-рацемата}.
Перемешиваемый раствор 2-гидроксипиридина /0,42 г/ в безводном этаноле /20 мл/ обрабатывают с помощью NaH {0,21 г 55 %-ной дисперсии в масле} и перемешивают при комнатной температуре в течение 15 мин в атмосфере аргона. Смесь затем охлаждают до 2oC и обрабатывают раствором /±/-4-[1a,7b-дигидро-2,2-диметил-2Н-оксирено/c/ /1/-бензопиран-6-ил]-2-метилпиридина {1,07 г, см. пример 24.А.5.} и перемешивают 96 ч при комнатной температуре. Растворитель выпаривают при пониженном давлении и остаток обрабатывают насыщенным водным раствором хлорида аммония /100 мл/ и экстрагируют метиленхлоридом /3 раза по 100 мл/. Объединенные экстракты сушат /сульфат натрия/, отфильтровывают и растворитель выпаривают при пониженном давлении, получая сырой продукт, который очищают путем хроматографии [силикагель, 2% C2H5OH в CH2Cl2] и перекристаллизуют из смеси этанола с диэтиловым эфиром с получением целевого соединения. Т.пл. 213-214oC.
Пример. 22. Получение транс-/+/-2-[3,4-дигидро-2,2-диметил-3-гидрокси-6- /2-метилпиридин-4-ил/-2Н-1-бензопиран 4-ил] -2,3-дигидро-1Н-изоиндол-1-она [формула /I/: R1 = R6 = R7 = CH3; R2 = R4 = R8 = H; 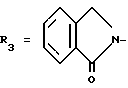 в виде транс-рацемата.
в виде транс-рацемата.
Перемешиваемый раствор транс -/±/-4-амино-3,4-дигидро-2,2-диметил-6-/2-метилпиридин-4-ил/-2Н-1-бензопиран- 3-ола /2,84 г/ [см. пример 24.А.6] и метилового эфира 2-бромметилбензойной кислоты /2,30 г/ в безводном ацетонитриле /100 мл/ обрабатывают с помощью KI /0,84 г/ и затем K2CO3 /4,20 г/, в атмосфере аргона. Реакционную смесь перемешивают в течение 1 ч при 20oC, в течение 1 ч при 60oC и затем в течение 15 ч при 85oC. Растворитель выпаривают при пониженном давлении, и остаток обрабатывают водой /300 мл/ и экстрагируют с помощью 2% CH3OH в метиленхлориде /4 раза по 150 мл/. Объединенные экстракты промывают раствором тиосульфата натрия /100 мл/2%/, сушат [сульфат натрия] и отфильтровывают. Растворитель выпаривают при пониженном давлении, получая сырой продукт, который очищают путем хроматографии /силикагель, 5% C2H5OH в CH2Cl2/ и перекристаллизуют из смеси этанола с ацетоном, получая целевое соединение. Т.пл. 222-224oC.
Пример. 23. Получение /-/-/3S,4R/-4-[3,4-дигидро-2,2-диметил-3-гидрокси-4-/2- оксо-пиперидин-1-ил/-2Н-1-бензопиран-6-ил]-2-метилпиридин-N-оксида.
Раствор продукта примера 1 /2,6 г/ в CH2Cl2 /40 мл/ обрабатывают 3-хлорпероксибензойной кислотой /1,92 г, 90 %-ная/ и перемешивают при комнатной температуре в течение 18 ч. Растворитель выпаривают при пониженном давлении с получением неочищенного продукта, который очищают путем хроматографии /силикагель, 5% C2H5OH в CH2Cl2/ и перекристаллизуют из смеси ацетона с диэтиловым эфиром, получая целевое соединение. Т.пл. 202-205oC.
Пример. 24. Получение исходных материалов примеров 1-23
А.1. Получение 4-/4-метоксифенил/-2-метилпиридина.
А. 1. а Этиловый эфир-4-/4-метоксифенил/-2-метил-1/4Н/-пиридинкарбоновой кислоты.
Этилхлорформиат /10,85 г/ добавляют к перемешиваемой смеси 2-пиколина /9,30 г/ и иодида меди-/I/ /0,77 г/ в безводном тетрагидрофуране /150 мл/, при -20oC и в атмосфере аргона. Все перемешивают в течение 3 ч при -20oC и затем обрабатывают путем добавления по каплям с помощью раствора 4-метоксифенилмагнийбромида, полученного из 4-броманизола /18,7 г/ и магниевой стружки /2,64 г/ в безводном тетрагидрофуране /100 мл/, причем прикапывают с такой скоростью, чтобы температура оставалась в пределах (-15)-(-20)oC. Смесь перемешивают при -15oC в течение 1 ч и в течение 16 ч при 20oC, и после этого обрабатывают насыщенным водным раствором NH4Cl /300 мл/ и экстрагируют этилацетатом /4 раза по 200 мл/. Объединенные экстракты сушат /сульфат натрия/, отфильтровывают и растворитель выпаривают при пониженном давлении, получая сырой продукт. Его очищают путем хроматографии /силикагель, 50% толуола в гексане/, получая целевое соединение в виде бледно-желтого масла.
А.1.в. 4-/4-Метоксифенил/-2-метилпиридин.
Продукт стадии А.1.а. /16,9 г/ и серу /2,0 г/ в декагидронафталине /100 мл/ перемешивают в течение 3 ч при 200oC. Растворитель выпаривают при пониженном давлении и остаток растворяют в этилацетате /500 мл/ и экстрагируют соляной кислотой /3 раза по 200 мл, 2М/. Объединенные экстракты промывают этилацетатом /2 раза по 100 мл/, подщелачивают при охлаждении льдом до pH=11 с помощью NaOH и экстрагируют с помощью CH2Cl2 /3 раза по 200 мл/. Объединенные экстракты сушат /сульфат натрия/, отфильтровывают и растворитель выпаривают при пониженном давлении, получая сырой продукт. Его очищают хроматографически /силикагель, 2% C2H5OH в CH2Cl2/ и перекристаллизуют из смеси этанола с этилацетатом с получением целевого соединения. Т.пл. 88-91oC.
Целевое соединение альтернативно может быть получено следующим путем.
А.1.а', 2,2',6'-Триметил-4-оксо-1,1'-/4'Н/-бипиридиний-тетрафторборат.
2-Метилпиридин /11,7 г/ добавляют к свежеприготовленному раствору гидроксиламин-0-сульфокислоты /45,2 г, 90 %-ная/ в воде /260 мл/ при 0oC. Смесь нагревают до 95oC, перемешивают в течение следующих 45 мин, охлаждают до 10oC и осторожно обрабатывают с помощью карбоната калия /55 г/. Смесь промывают диэтиловым эфиром /2 раза по 100 мл/ и воду выпаривают при 40oC при пониженном давлении. Остаток обрабатывают этанолом /600 мл/ и осадившийся K2SO4 удаляют фильтрованием. Фильтрат обрабатывают соляной кислотой /120 мл, 18 М/ и выпаривают досуха при 50oC и пониженном давлении, получая остаток, который обрабатывают дегидроуксусной кислотой /68,6 г/ и HCl /150 мл, 18 М/ и кипятят с обратным холодильником в течение 90 мин. Раствор выпаривают досуха при 50oC при пониженном давлении и остаток в течение 15 мин перемешивают с этанолом /200 мл/, отфильтровывают и осадок промывают этанолом /200 мл/. Объединенные фильтрат и промывные растворы обрабатывают тетрафторборной кислотой в диэтиловом эфире /50 мл, 50%/, разбавляют диэтиловым эфиром /250 мл/. Оставляют выкристаллизовываться целевое соединение и его отфильтровывают и сушат в вакууме при 20oC. Т.пл. 206-208oC.
А.1.в'. 4-/4-Метоксифенил/-2-метил-пиридин
Перемешиваемый раствор 4-метоксифенилмагнийбромида, полученный из 4-броманизола /56,1 г/ и магниевых стружек /7,92 г/ в безводном тетрагидрофуране /400 мл/, при 0-5oC, обрабатывают продуктом стадии /А.1.а'/ (30,2) в атмосфере аргона. Смесь перемешивают при комнатной температуре в течение 48 ч, промывают насыщенным водным раствором хлорида аммония /300 мл/ и водную фазу экстрагируют с помощью CH2Cl2 /3 раза по 100 мл/. Объединенные тетрагидрофурановый раствор и CH2Cl2-экстракты сушат /сульфат натрия/, отфильтровывают и растворители выпаривают при пониженном давлении, получая сырой 4'-[4-метоксифенил] -2,2', 6'-триметил- {1,1'/4Н,4'Н/-бипиридин}-4-он. Сырой продукт растворяют в безводном диметилформамиде /400 мл/ и кипятят с обратным холодильником в течение 4 ч. Растворитель выпаривают при пониженном давлении, получая остаток, который очищают путем хроматографии /силикагель, 2% C2H5OH в CH2Cl2/ и перекристаллизуют из этанола с получением целевого соединения. Т.пл. 88-91oC.
А.2. Получение 4-/4-гидроксифенил/-2-метилпиридина.
Раствор продукта примера /А.1./ /6,45 г/ в HBr /100 мл, 48%/ нагревают при 135oC в течение 3 ч. Избыток HBr выпаривают при пониженном давлении, получая остаток, который нейтрализуют водным раствором NaHCO3 и экстрагируют с помощью смеси 3:1 CH2Cl2/C2H5OH [3 раза по 150 мл]. Объединенные экстракты сушат /сульфат натрия/, отфильтровывают и растворитель выпаривают при пониженном давлении с получением сырого продукта. Его перекристаллизуют из смеси этанола с диэтиловым эфиром, получая целевое соединение. Т.пл.203-204oC.
А.3. Получение 2,2-диметил-6-/2-метилпиридин-4-ил/-2Н-1-бензопирана.
Перемешиваемую смесь продукта примера А.2. /4,07 г/, безводного K2CO3 /6,9 г/ и KI /1,0 г/ в безводном ацетоне, в атмосфере аргона, обрабатывают с помощью 3-хлор-3-метилбутина /5,56 г/ и кипятят с обратным холодильником в течение 120 ч. Смесь отфильтровывают и растворитель выпаривают при пониженном давлении, получая сырой 4-[4-/1,1-диметил-2-пиропинил/оксифенил]-2-метилпиридин. Последний растворяют в 1,2-дихлорбензоле /50 мл/ и нагревают при 170oC в течение 1 ч. Растворитель выпаривают при пониженном давлении с получением сырого целевого соединения, которое очищают путем хроматографии [силикагель, 2% C2H5OH в CH2Cl2], получая целевое соединение. Т.пл. 37-40oC.
Следующие соединения могут быть получены аналогично примерам А.1.-А.3., осуществляя или стадии /А.1.а./-/А.1.в./ или /А.1.а'/-/А.1.в'/; все соединения получаются в виде масел.
В.3. 2,2-Диметил-6-/2-этилпиридин-4-ил/-2Н-1-бензопиран.
С.3. 2,2-Диметил-6-/2,3-диметилпиридин-4-ил/-2Н-1-бензопиран.
Д.3. 2,2-Диметил-6-/3-метилпиридин-4-ил/-2Н-1-бензопиран.
Е.3. Получено 2,2-диметил-6-/2-пропилпиридин-4-ил/-2Н-1-бензопирана.
К перемешиваемому раствору продукта примера А.3. /5,02 г/ в безводном тетрагидрофуране /50 мл/, при -25oC и в атмосфере аргона, добавляют раствор н-бутиллития [12,5 мл/ /1,6 М] в гексане. Полученную смесь перемешивают при 10oC в течение 40 мин, охлаждают до -5oC и обрабатывают этилиодидом /2,4 мл/. Смесь оставляют нагреваться до комнатной температуры, перемешивают в течение дополнительных 2 ч и затем обрабатывают с помощью насыщенного водного раствора NH4Cl /100 мл/ и экстрагируют этилацетатом /2 раза по 100 мл/. Объединенные экстракты сушат /сульфат натрия/, отфильтровывают и растворитель выпаривают при пониженном давлении, получая сырой продукт, который очищают путем хроматографии /силикагель, 10% ацетона в гексане/, получая целевое соединение в виде масла.
F.3. Получение 2,2-диметил-6-/2-изобутилпиридин-4-ил/-2Н-1-бензопирана.
Целевое соединение, приготовляемое аналогично примеру E.3. При использовании изопропилиодида вместо этилиодида получают в виде масла.
G. 3. Получение 2,2-диметил-6-/2-гидроксиметилпиридин-4-ил/-2Н-1-бензопиран-ацетата.
G.3.а. 4-/2,2-Диметил-2Н-1-бензопиран-6-ил/-2-метилпиридин-N-оксид.
Раствор продукта примера A.3. /13,4 г/ в CH2Cl2 /200 мл/ обрабатывают 3-хлорпероксибензойной кислотой /13,5 г, 70 %-ная/ и перемешивают при комнатной температуре в течение 1 ч. Растворитель выпаривают при пониженном давлении с получением остатка, который очищают путем хроматографии /силикагель, 5% C2H5OH в CH2Cl2/, получая целевое соединение в виде желтой смолы.
G. 3. в. /2,2-Диметил-6-/2-гидроксиметилпиридин-4-ил/-2Н-1-бензопиранацетат.
Смесь продукта стадии G. 3. a. /4,8 г/ и уксусного ангидрида /50 мл/ нагревают при 80oC в атмосфере аргона в течение 1 ч. Растворитель выпаривают при пониженном давлении с получением сырого продукта, который очищают путем хроматографии /силикагель, 10% этилацетата в толуоле/, получая целевое соединение в виде масла.
H. 3. Получение 2,2-диметил-6-/2-метоксиметилпиридин-4-ил/-2Н-1-бензопирана.
H.3.a. /2,2-Диметил-2Н-1-бензопиран-6-ил/-2-пиридинметанол.
Смесь продукта примера G.3. /69,5 г/, Na2CO3 /95,4 г/, H2O /160 мл/ и C2H5OH /600 мл/ перемешивают в течение 22 ч при комнатной температуре. Смесь отфильтровывают и фильтрат выпаривают досуха при пониженном давлении с получением остатка, который обрабатывают водой /400 мл/ и экстрагируют дихлорметаном /3 раза по 150 мл/. Объединенные экстракты сушат /сульфат натрия/ и растворитель выпаривают при пониженном давлении, получая сырой продукт. Его очищают путем хроматографии /силикагель, этилацетат/ и перекристаллизуют из пентана, получая целевое соединение. Т.пл. 86-88oC.
H.3.b. /2,2-Диметил-2Н-1-бензопиран-6-ил/-2-метоксиметил-пиридин.
Перемешиваемый раствор продукта стадии H.3.a. /8,0 г/ в безводном тетрагидрофуране /150 мл/ при 15oC и в атмосфере аргона обрабатывают с помощью NaH /0,90 г 80 %-ной дисперсии в масле/ и перемешивают при комнатной температуре в течение 45 мин. Добавляют метилиодид /4,26 г/ и смесь перемешивают при комнатной температуре в течение 18 ч. Смесь обрабатывают насыщенным водным раствором NH4Cl /200 мл/ и экстрагируют этилацетатом /2 раза по 200 мл/. Объединенные экстракты сушат /сульфат натрия/, отфильтровывают и растворитель выпаривают при пониженном давлении, получая сырой продукт. Его очищают путем хроматографии /силикагель, 20% ацетона в гексане/, получая целевое соединение в виде масла.
l. Получение 6-/2-метилпиридин-4-ил/-2,2,7-триметил-2Н-1-бензопирана.
a. 4-Бром-3-метилфенолацетат.
Перемешиваемую смесь 4-бром-3-метилфенола /184,1 г/ и водного раствора NaOH /850 мл, 2М/ при 20oC обрабатывают уксусным ангидридом /136 мл/ и перемешивают при комнатной температуре в течение 1 ч. Суспензию экстрагируют диэтиловым эфиром /3 раза по 300 мл/, и объединенные экстракты промывают водным раствором NaOH /2 раза по 100 мл, 2М/, сушат /сульфат натрия/ и отфильтровывают. Растворитель выпаривают при пониженном давлении, получая целевое соединение в виде масла.
b. 1-/3-Бром-2-гидрокси-5-метилфенил/-этанон.
Смесь продукта стадии /a/ /195,6 г/ и хлорида алюминия /152,6 г/ перемешивают в атмосфере аргона при 165oC в течение 45 мин. Охлажденную смесь обрабатывают охлажденной льдом соляной кислотой /2000 мл, 2М/ и экстрагируют с помощью CH2Cl2 /4 раза по 600 мл/. Объединенные экстракты промывают рассолом, сушат /сульфат натрия/, отфильтровывают и растворитель выпаривают при пониженном давлении, получая сырой продукт. Его очищают путем хроматографии /силикагель, 10% толуола в гексане/ и перекристаллизуют из смеси диэтилового эфира с гексаном, получая целевое соединение. Т.пл. 81-82oC.
c. 6-Бром-3,4-дигидро-2,2,7-триметил-2Н-1-бензопиран-4-он.
Смесь продукта стадии /b/ /48 г/, ацетона /31 мл/ и пирролидина /21 мл/ в безводном бензоле /500 мл/ перемешивают при комнатной температуре в течение 3 ч и затем кипятят с обратным холодильником в течение 6 ч, причем образующуюся воду удаляют через насадку Дина-Старка. Охлажденную смесь обрабатывают соляной кислотой /200 мл, 2М/, перемешивают в течение 10 мин, подщелачивают водным раствором NaOH /1 М/ и экстрагируют с помощью CH2Cl2 /3 раза по 300 мл/. Объединенные экстракты сушат /K2CO3/, отфильтровывают и растворитель выпаривают при пониженном давлении, получая сырой продукт. Его очищают путем хроматографии /силикагель, 50% толуола в гексане/ и перекристаллизуют из смеси диэтилового эфира с пентаном с получением целевого соединения. Т. пл. 95-96oC.
d. 6-Бром-3,4-дигидро-4-гидрокси-2,2,7-триметил-2Н-1-бензопиран.
Перемешиваемый раствор продукта стадии /c/ /26,9 г/ в этаноле /200 мл/ при 5oC обрабатывают боргидридом натрия /1,95 г/ и перемешивают при комнатной температуре в течение 12 ч. Растворитель выпаривают при пониженном давлении с получением остатка, который обрабатывают водой /500 мл/ и экстрагируют диэтиловым эфиром /3 раза по 200 мл/. Объединенные экстракты сушат /сульфат натрия/, отфильтровывают и растворитель выпаривают при пониженном давлении, получая сырой продукт. Его очищают путем хроматографии /силикагель, 10% ацетона в гексане/ и перекристаллизуют из смеси диэтилового эфира с пентаном, получая целевое соединение. Т.пл. 92-93oC.
e. 6-Бром-2,2,7-триметил-2Н-1-бензопиран.
Перемешиваемый раствор продукта стадии /d/ /27,1 г/ в безводном толуоле /300 мл/ обрабатывают п-толуолсульфокислотой /1,15 г/ и кипятят с обратным холодильником в течение 2 ч, причем образующуюся воду удаляют через насадку Дина-Старка. Охлажденный раствор промывают водным раствором карбоната натрия /100 мл, 2М/, сушат /сульфат натрия/, отфильтровывают и растворитель выпаривают пир пониженном давлении, получая сырой продукт. Его очищают путем хроматографии /силикагель, гексан/, получая целевое соединение в виде масла.
f. 6-/2-Метилпиридин-4-ил/-2,2,7-триметил-2Н-1-бензопиран.
Раствор продукта стадии /e/ /7,60 г/ в безводном тетрагидрофуране /30 мл/ добавляют в течение 15 мин к перемешиваемой смеси стружек магния /0,85 г/ и иода /0,06 г/ в безводном тетрагидрофуране /25 мл/ при 45oC и в атмосфере аргона. Смесь кипятят с обратным холодильником в течение 3 ч, охлаждают до 5oC, обрабатывают бис-/трифенилфосфин/-хлоридом никеля /II/ /0,32 г/ и раствором 4-бромпиколина /4,8 г/ в безводном тетрагидрофуране /50 мл/ и перемешивают при комнатной температуре в течение 18 ч. Смесь обрабатывают соляной кислотой /140 мл, 1М/ и экстрагируют диэтиловым эфиром /2 раза по 60 мл/. Объединенные эфирные экстракты промывают соляной кислотой. Объединенные кислотные растворы подщелачивают до pH=10 с помощью карбоната калия, экстрагируют диэтиловым эфиром /3 раза по 100 мл/, сушат /сульфат натрия/, отфильтровывают и растворитель выпаривают при пониженном давлении, получая сырой продукт. Его очищают путем хроматографии /силикагель, 10% ацетона в гексане/, получая целевое соединение в виде масла, которое обладает следующими физическими характеристиками:
1H-ЯМР / δ CDCl3/: 1,44 /с., 6Н/; 2,21 /с., 3Н/; 2,59 /с., 3Н/; 5,60 /д. , 1Н/; 6,31 /д., 1Н/; 6,70 /с., 1Н/; 6,82 /с., 1Н/; 7,04 /дд., 1Н/; 7,09 /д. , 1Н/; и 8,49 /д., 1Н/.
A. 4. Получение транс-/+/-3-бром-3,4-дигидро-2,2-диметил-6-/2-метилпиридин- 4-ил/-2Н-1-бензопиран-4-ола.
N-Бромсукцминимид /2,20 г/ порциями добавляют в перемешиваемому раствору продукта A.3. /2,50 г/ в диметилсульфоксиде /6 мл/ и воде /0,36 мл/ при 0oC. После прекращения экзотермической реакции, непрерывно перемешивают в течение дополнительного 1 ч и реакцию прерывают за счет насыщенного водного раствора NH4Cl /200 мл/ и смесь экстрагируют с помощью CH2Cl2 /3 раза по 100 мл/. Объединенные экстракты сушат /сульфат натрия/, отфильтровывают и растворитель выпаривают при пониженном давлении, получая сырой продукт. Его очищают путем хроматографии [силикагель, 2% C2H5OH в CH2Cl2] и перекристаллизуют из смеси этанола с диэтиловым эфиром, получая целевое соединение. Т.пл. 212-214oC.
A.5. Получение /+/-4-[1a,7b-дигидро-2,2-диметил-2Н-оксирено/c/ /1/-бензопиран-6-ил]-2-метилпиридина.
Раствор продукта примера A.4. /3,5 г/ в безводном тетрагидрофуране /80 мл/ обрабатывают с помощью NaH {0,90 г 55 %-ной дисперсии в масле} и перемешивают при комнатной температуре в атмосфере аргона в течение 1 ч. Реакцию прекращают с помощью насыщенного водного раствора NH4Cl /150 мл/ и экстрагируют реакционную смесь с помощью диэтилового эфира /3 раза по 100 мл/. Объединенные экстракты сушат /сульфат натрия/, отфильтровывают и растворитель выпаривают при пониженном давлении, получая остаток, который очищают путем хроматографии /силикагель, 2% C2H5OH в CH2Cl2/, получая целевое соединение в виде масла, которое имеет следующие характеристики:
1H-ЯМР / δ -d6 ДМСО/: 1,23 /с., 3Н/; 1,50 /с., 3Н/; 2,52 /с., 3Н/; 3,75 /д. , 1Н/; 4,14 /д., 1Н/; 6,89 /д., 1Н/; 7,47 /дд, 1Н/; 7,56 /с., 1Н/; 7,19 /дд., 1Н/; 7,96 /д., 1Н/; и 8,47 /д., 1Н/.
Следующие соединения формулы /II/, в которой R6 и R7, каждый обозначает метил и R1, R2 и R8 имеют указанные значения, могут быть получены аналогично примерам A4 и A5, но осуществляя реакцию путем one-pot-методики. Это осуществляют путем разбавления смеси, полученной после экзотермической реакции и перемешивания согласно примеру A.4. диоксаном, обработки водным раствором NaOH /0,6 М/ и перемешивания при комнатной температуре в течение еще примерно 1 ч. Очистку затем осуществляют аналогично примеру A.5., следующую за выпариванием диоксана. Исходные материалы указаны в колонке 2 табл. 3. Все продукты получают в форме /±/-рацематов в виде масла.
Характеристика:
B. 5. 1H-ЯМР/ δ -CDCl3/: 1,31 /с., 3Н/; 1,37 /т., 3Н/; 1,62 /с., 3Н/; 2,89 /л., 2Н/; 3,56 /д., 1Н/; 3,99 /д., 1Н/; 6,91 /дщ., 1Н/; 7,28 /дд, 1Н/; 7,34 /м., 1Н/; 7,54 /дд., 1Н/; 7,63 /д., 1Н/; и 8,54 /дд., 1Н/.
C. 5. 1H-ЯМР/ δ -CDCl3/: 1,32 /с., 3Н/; 1,61 /с., 3Н/; 2,20 /с., 3Н/; 2,61 /с. , 3Н/; 3,54 /д., 1Н/; 3,39 /д., 1Н/; 6,87 /д., 1Н/; 6,98 /д., 1Н/; 7,17 /дд., 1Н/; 7,26 /д., 1Н/; и 8,32 /д., 1Н/.
M. 5. 1H-ЯМР/ δ -CDCl3/: 1,33 /с., 3Н/; 1,62 /с., 3Н/; 2,31 /с., 3Н/; 3,55 /д., 1Н/; 3,95 /д., 1Н/; 6,89 /д., 1Н/; 7,13 /д., 1Н/; 7,23 /дд., 1Н/; 7,31 /с., 1Н/; 8,45 /д., 1Н/; и 8,49 /с., 1Н/.
E. 5. 1H-ЯМР/ δ -CDCl3/: 1,01 /т., 3Н/; 1,30 /с., 3Н/; 1,62 /с., 3Н/; 1,82 /секстет, 2Н/; 2,82 /т., 2Н/; 3,54 /д., 1Н/; 3,98 /д., 1Н/; 6,91 /д., 1Н/; 7,28 /дд. , 1Н/; 7,32 /д., 1Н/; 7,53 /дд., 7,63 /д., 1Н/; и 8,54 /д., 1Н/.
F. 5. 1H-ЯМР/ δ -CDCl3/: 0,96 /д., 6Н/; 1,30 /с., 3Н/; 1,62 /с., 3Н/; 2,15 /дт., 1Н/; 2,70 /д., 2Н/; 3,54 /д., 1Н/; 3,93 /д., 1Н/; 6,90 /д., 1Н/; 7,22-7,30 /м., 2Н/; 7,53 /дд., 1Н/; 7,63 /д., 1Н/; и 8,54 /д., 1Н/.
G. 5. 1H-ЯМР/ δ -CDCl3/: 1,31 /с., 3Н/; 1,63 /с., 3Н/; 3,55 /д., 1Н/; 3,98 /д., 1Н/; 4,83 /с., 2Н/; 6,92 /д., 1Н/; 7,35-7,45 /м., 2Н/; 7,54 /дд., 1Н/; 7,64 /д., 1Н/; и 8,57 /д., 1Н/.
I. 5. 1H-ЯМР/ δ -CDCl3/: 1,29 /с., 3Н/; 1,59 /с., 3Н/; 2,21 /с., 3Н/; 2,60 /с., 3Н/; 3,50 /д., 1Н/; 3,89 /д., 1Н/; 6,74 /с., 1Н/; 7,04 /дд., 1Н/; 7,10 /д., 1Н/; 7,18 /с., 1Н/; и 8,50 /д., 1Н/.
A. 5' Получение /-/-/3S,4S/4-{1a,7b-дигидро-2,2-диметил-2Н- оксирено/c/ /1/ бензопиран-6-ил}-2-метил-пиридина.
A. 5'. a. 3-Бром-3,4-дигидро-2,2-диметил-6-/2-метилпиридин-4-ил/-2Н-1-бензопиран-4-иловый эфир -[1R-/1α {3R,4S}, β ]- и [1R-/1 α {3S,4R}, 4 β ]- α -метоксибензолуксусной кислоты.
Раствор продукта примера A.4 /9,80 г/, /-/-/R/- α -метоксифенилуксусной кислоты /5,65 г/ и 4-диметиламинопиридина /0,45 г/ в безводном CH2Cl2 /330 мл/ обрабатывают N, N-дициклогексилкарбодиимидом /6,81 г/ и перемешивают в течение 90 мин при комнатной температуре. Смесь отфильтровывают и растворитель выпаривают при пониженном давлении, получая сырую смесь дистереоизомеров, которую очищают путем хроматографии {силикагель, 5% ацетона в CH2Cl2} , получая мерее полярный продута /A/, который перекристаллизуют из смеси ацетона с пентаном с получением [1R-{1 α /3R,4S/,4 β }-изомера целевого соединения, т.пл. 137-138oC, (α)
(α)
A. 5'.б. /-/-/3S,4S/-4-[1a,7b-дигидро-2,2-диметил-2Н-оксирено /c/ /1/ -бензопиран-6-ил]-2-метилпиридин.
Раствор [1R-{ 1 α /3R,4S/,4 β } - изомера из стадии /A.5'.а/ /3,16 г/ в диоксане /75 мл/ при 20oC обрабатывают водным раствором NaOH /45 мл, 0,58М/ и перемешивают в течение 10 мин при 20oC. Диоксан выпаривают при пониженном давлении и остаток обрабатывают водой /100 мл/ и экстрагируют с помощью CH2Cl2 /3 раза по 100 мл/. Объединенные экстракты сушат /сульфат натрия/, отфильтровывают и растворитель выпаривают при пониженном давлении, получая остаток, который очищают путем колоночной хроматографии /силикагель, 5% C2H5OH в CH2Cl2 /с получением целевого соединения в чистой или в основном чистой энантиомерной форме в виде бесцветного масла. (α)
A. 6'. Получение /+/-/3S,4R/-4-амино-3,4-дигидро-2,2-диметил-6-{2- метилпиридин-4-ил}-2Н-1-бензопиран-3-ола.
Раствор продукта примера A.5' /0,64 г/ обрабатывают насыщенным раствором аммиака в этаноле /15 мл/ и нагревают при 80oC в автоклаве в течение 15 ч. Растворитель выпаривают при пониженном давлении, получая сырой продукт, который очищают путем хроматографии /силикагель, 5% C2H5OH в CH2Cl2/ с получением целевого соединения в виде пены: (α)
Следующие соединения формулы /IV/, в которой R4 обозначает водород, R5 обозначает гидроксил, R6 и R7, каждый обозначает метил и R1, R2 и R8 имеют указанные значения, могут быть получены аналогично из указанного исходного материала. Все соединения, перечисленные в табл. 4, находятся в форме транс-рацематов и за исключением продукта примера В6, получаются в виде масла.
Физическая характеристика:
A. 6. 1H-ЯМР/ δ6 -ДМСО/: 1,12 /с., 3Н/; 1,38 /с., 3Н/; 2,06 /широкий, 2Н/; 2,50 /с., 3Н/; 3,23 /дд., 1Н/; 3,59 /д., 1Н/; 5,47 /д., 1Н/; 6,81 /д., 1Н/; 7,43 /дд. , 1Н/; 7,51 /с., 1Н/; 7,55 /дд., 1Н/; 7,99 /м., 1Н/; и 8,44 /д., 1Н/;
B.6. Т.пл. 140-141oC.
C. 6. 1H-ЯМР/ δ -CDCl3/: 1,27 /с., 3Н/; 1,54 /с., 3Н/; 2,15 /с., 3Н/; 2,2-2,5 /ш. с., 3Н/; 2,57 /с., 3Н/; 3,41 /д., 1Н/; 3,71 /д., 1Н/; 6,85 /д., 1Н/; 6,99 /д., 1Н/; 7,10 /дд., 1Н/; 7,31 /дд., 1Н/; и 8,31 /д., 1Н/.
E. 6. 1H-ЯМР/ δ -CDCl3/: 1,00 /т., 3Н/; 1,27 /с., 3Н/; 1,55 /с., 3Н/; 1,80 /с. , 2Н/; 2,0-2,4 /ш.с., 3Н/; 2,80 /т., 2Н/; 3,41 /д., 1Н/; 3,74 /д., 1Н/; 6,89 /д. , 1Н/; 7,25-7,36 /м., 2Н/; 7,47 /дд., 1Н/; 87,70 /д., 1Н/; и 8,51 /д., 1Н/.
F. 6. 1H-ЯМР/ δ -ДМСО/: 0,90 /д., 6Н/; 1,13 /с., 3Н/; 1,41 /с., 3Н/; 2,0-2,2 /ш.с., 2Н/; 2,09 /дт., 1Н/; 2,65 /д., 2Н/; 3,24 /дд, 1Н/; 3,63 /д., 1Н/; 5,45 /ш.д., 1Н/; 6,84 /д., 1Н/; 7,48 /м., 2Н/; 7,58 /дд., 1Н/; 8,00 /д. , 1Н/; и 8,46 /д., 1Н/.
I. 6. 1H-ЯМР/ δ -CDCl3/: 1,24 /с., 3Н/; 1,53 /с., 3Н/; 2,1-2,4 /ш.с., 3Н/; 2,20 /с., 3Н/; 2,60 /с., 3Н/; 3,38 /д., 1Н/; 3,67 /д., 1Н/; 6,73 /с., 1Н/; 7,05 /д., 1Н/; 7,10 /с., 1Н/; 7,21 /с., 1Н/ и 8,49 /д., 1Н/.
Бензопираны и дигидробензопираны, как определенные в п.1, например соединения формулы /I/, и их N-оксиды, и их физиологически гидролизуемые и физиологически приемлемые сложные эфиры также как фармацевтически приемлемые соли присоединения кислот и четвертичные аммониевые соли указанных бензопиранов, дигидробензопиранов, N-оксидов, сложных эфиров [ниже все вместе называются как Агенты изобретения] пригодны в качестве фармацевтических средств.
Агенты изобретения обладают релаксирующей гладкие мышцы активностью и проявляют открывающую калиевые каналы активность в отношении клеточной мембраны, как демонстрируется путем их влияния в концентрациях в пределах 1-500 нМ на напряжение В, и на выделение Rb+ из различных препаратах гладких мышц в соответствии или по аналогии с методами, описанными в Quast. Brit. J. Pharmac., 91, 569-578 /1987/. Агенты изобретения в связи с этим характеризуются как открывающие K+-каналы агенты.
Агенты изобретения таким образом пригодны для лечения состояний или расстройств, для которых показана терапия с использованием открывающего K+-каналы агента. Терапевтическая пригодность в качестве открывающих K+-каналы агентов далее может быть продемонстрирована в стандартных фармакологических тестах, например сердечно-сосудистой активности, ин витро и ин виво. Это влияние на кровяное давление может быть показано на анестезированной, нормально напряженной, канюлированной (с введенной канюлей) крысе после интрадуоденального введения спустя 1 ч после канюлирования. Антиишемическая активность может быть продемонстрирована в соответствии с методами, описанными Hof и др. , Circ. Res., 62, 679 /1988/. Агенты изобретения проявляют гипотоническую активность в стандартном тест-методе при пороговых дозах примерно 0,03-1,0 мг/кг интрадуоденально и антиишемическую активность в последнем тест-методе в дозах примерно 0,001-0,03 мг/кг внутривенно.
Агенты изобретения соответственно пригодны, например, в качестве релаксантов гладких мышц, в особенности для использования в качестве сосудорасширяющих агентов, например, для лечения гипертонии или хронической сердечной недостаточности. Далее они пригодны в качестве антиишемических и антивазоспатических агентов, например для использования при лечении нарушенного кровоснабжения, например, в отношении сердца, скелетных мышц или мозга. Они также пригодны, например, для лечения грудной жабы, ишемии миокарда или инфаркта миокарда; в качестве антифибрилляторных агентов; для лечения расстройств периферического круга кровообращения, например, как периодическое прихрамывание, венозная язва или язва Morbus Raynaud, также как для лечения, включающего профилактику, церебральной ишемии, старческого слабоумия, паралича, субарахноидального кровотечения и других родственных или логически вытекающих заболеваний или расстройств.
Агенты изобретения еще показаны для использования в качестве антиспастических агентов желудочно-кишечного, мочевого и утробного тракта, например для лечения язвы желудка и двенадцатиперстной кишки, раздражения ободочной (толстой) кишки, дивертикулита, опасности выкидыша с последующими преждевременными родами и недержания мочи.
Агенты изобретения кроме того показаны для использования в качестве стимулирующих рост волос агентов, например для лечения выпадения волос вследствие старения, например, облысевших или частично облысевших мужчин, или заболевания, связанного с выпадением волос, например вследствие инфекции или расстройства иммунной системы.
Пригодные для такого использования дозы могут изменяться, например, в зависимости от индивидуального состояния пациента, вида используемого агента изобретения, способа введения и желательного эффекта. Однако обычно достаточная суточная оральная доза, например, для использования против гипертонии составляет примерно 0,03-2,0 мг/кг, а, например, для антиишемического использования примерно 0,015-0,3 мг/кг. Для крупных млекопитающих, например людей, суточная оральная доза составляет примерно 2-150 мг при использовании против гипертонии или около 1-20 мг для антиишемического использования, причем введение осуществляют один раз в день или в виде разделенных доз 2 раза в день. Оральные дозировочные формы, используемые по указанным показаниям, соответственно содержат примерно от 0,5 или 1,0 до примерно 20 или 150 мг агента изобретения вместе с фармацевтически приемлемым разбавителем или носителем. Для использования в качестве стимулятора роста волос агенты изобретения соответственно применяют топически, например в виде соответствующего крема, геля или эмульсии или т.п., которые известны из уровня техники.
Согласно изобретению, в качестве наиболее важного свойства найдено, что агенты изобретения обладают бронхорасширяющей активностью и уменьшают или изменяют гиперреактивность дыхательных путей. Эти активности также могут быть продемонстрированы на фармацевтической тест-модели ин виво и ин витро, например, следующим образом.
Тест 1: Бронхорасширяющая активность.
1.1. На морской свинке.
Морских свинок (Dunkin - Hartley, самцы, весом 400-600 г) анестезируют с помощью фенобарбитала (100 мг/кг интраперитонеально) и пентобарбитала (30 мг/кг интраперитонеально) и парализуют с помощью галламина (8 мг-кг внутримышечно) и вентилируют с помощью смеси воздуха с кислородом (45:55 по объему). Животных вентилируют через трахеальную канюлю (10 мл/кг, 1 Гц). Величину кровяного давления и сердечной недостаточности регистрируют через сонную артерию. Вентилирование контролируют с помощью проточного датчика. Когда производят измерения потока, соответствующие изменениям давления в грудной клетке, то контроль осуществляют прямо через интраторакальный троакар, позволяющий видеть дифференциальное давление, относящееся к трахее. Из этой информации и резистентности и изменении рассчитывают каждый вдох.
Внутривенное вливание бомбезина (100 нг/кг/ч) вызывает длительный спазм бронхов. Способность испытуемого вещества аннулировать реакцию, когда его вводят интратрахеальным путем, служит мерой эффективности аннулирования возникшего спазма бронхов. Бронхорасширяющее действие выражается в количественном уменьшении максимальной реакции на бомбезин, измеряемой в регулярные интервалы.
В указанном модельном тесте агенты изобретения эффективны, в отношении аннулирования спазма бронхов, в дозах примерно 0,001-1,0 мг/кг.
1.2. На макаке резус.
Макак резус (самец и самка, весом 6,8-11,8 кг), как известно нормально реагирующих на метахолин (MeCH), анестезируют (сначала: кетамин 20 мг/кг внутримышечно; для поддержания: тиопентал 8 мг/кг/г внутривенно). Затем в трахею вставляют, слегка ударяя рукой; педиатрическую эндотрахеаальную трубку (5,0 см) (ксилокаин: топическое введение в надгортанник) и измеряют базовую (основную) резистентность легкого.
После осуществления этих манипуляций, в киль (carina) с помощью педиатрического фиброоптического бронхоскопа вводят 2 мл ксилокаина (1 мас.% раствор). Спустя 10 мин снова измеряют легочную резистентность. Ксилокаин не оказывает влияния на базовую резистентность. Испытуемое вещество вводят подобным предобработке ксилокаином образом, в виде суспензии с лактозой в качестве эксципиента (1 мг/мл; 1 мл нагнетаемого объема) кумулятивным образом в интервале 30 мин. В момент времени 15 мин осуществляют введение одного MeCH (количество 0,6-2,5 мг/мл раствора, подсчитанное примерно для достижения приблизительно 50-100%-ного изменения базовой линии) и рассчитывают % ингибирования из реакции после введения эксципиента.
Агенты изобретения оказывают сильное, зависящее от дозы бронхорасширяющее воздействие в указанном тест-методике при дозах примерно 10 нг/кг-10 мкг/кг.
Тест: 2 Подавление гиперреактивности дыхательных путей.
2.1. Индуцированная PAF гиперреактивность.
Морских свинок подготавливают для регистрации легочной функции, как описано в тесте 1.1. Внутривенная инъекция гистамина (1,8-3,2 мкг/кг) делает дыхательные пути чувствительными к спазмогенности. После вливания PAF (тромбоцитного активирующего фактора) более 1 ч (полная доза 600 нг/кг) повторная инъекция гистамина вызывает развитие гиперреактивности дыхательных путей, которая для удобства может быть выражена в виде парного различия между прежней ответной амплитудой и таковой после воздействия PAF.
При введении агентов изобретения интратрахеально, после воздействия PAF, в дозах около 0,1-100 мгк/кг наблюдают аннулирование индукции гиперреактивности дыхательных путей.
2.2. Индуцированная иммунным комплексом гиперреактивность.
Морских свинок подготавливают для регистрации легочной функции, как описано в тесте 1.1. Аллергическую реакцию вызывают за счет внутривенной инъекции заранее сформированного иммунного комплекса (приготовленного путем добавления 30 мкг бычьего гамма-глобулина в 0,05 мл солевого раствора к 0,05 мл анти-сыворотки антибычьего гамма-глобулина морской свинки) в регулярные (10 мин) интервалы в течение 30 мин. Внутривенные инъекции гистамина (1,0-3,2 мкг/кг в 10-минутные интервалы) используют для определения чувствительности прежних дыхательных путей до и после последнего введения иммунного комплекса. Гиперреактивность дыхательных путей выражается как парное отличие максимального значения легочной резистентности в ответ на введенный перед этим гистамин и после повторной инъекции иммунного комплекса. Испытуемые соединения вводятся интратрахеально.
Индуцированная гиперреактивность дыхательных путей заметно уменьшается в указанном тест-методе за счет предварительной обработки с помощью агентов изобретения в дозах примерно от 10 нг/кг до примерно 10.0 мкг/кг.
Агенты изобретения, следовательно, в особенности пригодны в качестве бронхорасширяющих агентов и в качестве агентов для лечения гиперреактивности дыхательных путей, например, в качестве агентов для симптоматического, так и также профилактического лечения обструктивного или воспалительного заболевания дыхательных путей, в особенности астмы. В качестве бронхорасширяющих агентов агенты изобретения могут быть использованы в особенности в качестве избавляющей от недуга терапии, для лечения приступа вследствие сужения бронхов, например, при астме. Дополнительно путем непрерывного введения агенты изобретения могут быть использованы для контроля, ограничения или аннулирования гиперреактивности дыхательных путей или для осуществления успешной защиты против рецидива приступа сужения бронхов вследствие обструктивного или воспалительного заболевания дыхательных путей, особенно астмы. Термины "лечение" и "обработка" в качестве используемых в описании и в формуле изобретения в отношении применения агентов изобретения для лечения обструктивного или воспалительного заболевания дыхательных путей, в особенности астмы, следовательно, нужно понимать как профилактические, так и также симптоматические (т. е. бронхорасширяющие) способы лечения, если не указано ничего другого.
В соответствии с упомянутым изобретение также включает:
4. способ лечения любого, описанного здесь заболевания или состояния;
4.а. Cпособ лечения обструктивного или воспалительного заболевания дыхательных путей, включающему:
4. а.1. cпособ симптоматического лечения обструктивного или воспалительного заболевания дыхательных путей, например, путем осуществления расширения бронхов или
4. а. 2. cпособ профилактики воспалительного или обструктивного заболевания дыхательных путей, например, для лечения гиперреактивности дыхательных путей;
в случае нуждающегося в таком лечении субъекта, причем способ состоит во введении указанному субъекту эффективного количества агента изобретения или
альтернативно:
5. агент изобретения для фармацевтического использования, например, для применения при лечении любого описанного здесь заболевания или состояния, в особенности для использования при лечении обструктивного или воспалительного заболевания дыхательных путей, например, как указано в п.п. 4.а.1 или 4.а.2, или
6. фармацевтическая композиция, содержащая агент изобретения, или использование агента изобретения для приготовления фармацевтической композиции, использующейся для лечения любого, описанного здесь заболевания или состояния, в особенности для применения, как указано в п.5.
Воспалительные или обструктивные заболевания дыхательных путей, к которым применимо изобретение, включают астму любого типа или генезиса, включая как внутреннюю, так и особенно наружную астму. Агенты изобретения пригодны для лечения аллергической астмы, как атопической (т.е. Ig E-косвенной), так и неатопической, также как и, например, бронхиальной астмы, индуцированной за счет бактериальной инфекции, или других типов неаллергического происхождения астм. Лечение астмы также было изучено на больных, страдающих астмой менее 4 или 5 лет, в особенности ночью, и имеющих диагноз или диагносцируемых как "младенческое сопение", как установленную категорию пациентов, привлекающую основное внимание врачей, и более конкретно в настоящее время идентифицируемых как начальные или находящиеся в ранней фазе астматики. (Для удобства это особое астматическое состояние относят к "синдрому начального страдания астмой").
Профилактическая эффективность при лечении астмы может быть доказана по уменьшению частоты или тяжести симптоматического приступа, например, острого астматического или бронхосуживающего приступа.
Далее эта эффективность может быть обнаружена по уменьшенной потребности в другой, симптоматической терапии, т.е. терапии во время приступа или подразумевающей ограничение или прекращение симптоматического приступа, когда он происходит, например, как противовоспалительная (например, кортикостероидная) или бронхорасширяющая (например, β2 -адренергическая) терапия.
Полезность профилактики астмы в особенности может быть наглядно показана на субъектах, склонных к "утреннему погружению". "Утреннее погружение" представляет собой известный астматический синдром, распространенный у значительного количества астматиков и характеризующийся приступом астмы, например, в части примерно 4-6, т.е. во время, обычно значительно удаленное от любой, заранее осуществленной симптоматической терапии астмы.
Воспалительные или обструктивные заболевания дыхательных путей, к которым применимо изобретение, также включают пневмокониоз (воспалительное, обычно профессиональное, хроническое или острое, заболевание легких, часто сопровождающееся обструкцией дыхательных путей и случающееся время от времени за счет повторяющегося вдыхания пыли) любого типа или генезиса, включающий, например, алюминоз, антракоз, асбестоз, халикоз, птилоз, сидероз, силикоз, табакоз и в особенности биссиноз.
Далее воспалительные или обструктивные заболевания и состояния дыхательных путей, к которым применимо изобретение, включают синдром "зрелого" респираторного недомогания /ARDS/, хроническое обструктивное легочное или дыхательных путей заболевание (COPD или COAD) и бронхит также, как обострение гиперреактивности дыхательных путей вследствие другой лекарственной терапии, в особенности другой, за счет ингаляции лекарства, терапии, например, как β -агонистическая бронхорасширяющая терапия, включающая в особенности использование агентов изобретения в качестве бронхорасширяющих средств для лечения хронической или острой обструкции дыхательных путей также, как одышки, ассоциированной с любым из указанных заболеваний или состояний.
Для использования при лечении воспалительного или обструктивного заболевания дыхательных путей агенты изобретения могут вводиться любым обычным образом, в особенности кишечно, например перорально, например в виде таблеток или капсул, или парентерально, например, в форме таблеток или капсул, или парентерально, например в форме растворов или суспензий для инъекций. Однако предпочтительно их можно вводить легочным путем, например путем ингаляции из соответствующего распылителя, ингалятора или тому подобного приспособления, известного из уровня техники.
Используемые для лечения воспалительного или обструктивного заболевания дыхательных путей дозы изменяются в зависимости, например, от индивидуального состояния излечиваемого субъекта, используемого отдельного агента изобретения, способа введения и желательного эффекта.
Однако обычно для легочного введения в случае крупных млекопитающих, например людей, пригодная суточная доза, вводимая в легкие, должна быть порядка примерно 0,1-100 мкг, в особенности 1,0- 50.0 мкг, причем введение осуществляют соответственно через ингаляторное устройство один или 2-4 раза в день порциями от 1 до 4 вдуваний на каждое введение.
Для орального введения пригодная суточная доза обычно порядка примерно 0,1-30 мкг/кг. Пригодная суточная оральная доза в случае крупных млекопитающих, например людей, порядка примерно 7 мкг-2,1 мг, вводимая в виде одноразовой дозы, в виде разделенных доз, которые вводят 2-4 раза в день или в пролонгированной форме. Оральные единичные дозировочные формы для такого использования соответственно включают около 1,75 мкг-2,1 мг агента изобретения вместе с фармацевтически приемлемым разбавителем или носителем.
В этой связи, в особенности следует заметить, что агенты изобретения обычно активны в качестве бронхорасширяющих средств или в качестве агентов для лечения гиперреактивности дыхательных путей в дозах, в особенности ингаляционных дозах, при которых кардиоваскулярные эффекты, которые могут быть нежелательны в связи с такой терапией, например гипотонически/тахикардиальный эффект, незначительны или находятся в приемлемых пределах переносимости в отношении осуществляемой терапии.
В соответствии с вышесказанным, настоящее изобретение также включает:
7. фармацевтическую композицию, содержащую агент изобретения вместе с фармацевтически приемлемым разбавителем или носителем.
Такие композиции могут быть приготовлены обычным образом, например, для легочного введения, путем смешения агента изобретения в тонкоизмельченной, высокодиспергированной форме, например, вместе с тонкоизмельченной лактозой в качестве носителя/разбавителя для получения вводимого путем ингаляции порошка.
Как было указано, терапевтическая дозировка, необходимая для осуществления изобретения, может изменяться в зависимости от разных факторов. Дозировка для любого отдельного агента изобретения также зависит от его относительной эффективности действия. Для предпочтительного агента изобретения, а именно продукта примера 1, в чистой или в основном чистой (3S,4R)-энантиомерной форме, ID50, установленная в первом продемонстрированном тесте, осуществленном в соответствии с указанным тестом 1.1, составляет 0,02 мг/кг интратрахеально. Установленная по тому же самому тест-методу для известного, вводимого путем ингаляции, бронхорасширяющегося лекарства Сальбутамола [α1 -{ /(1,1-диметилэтил)-амино/-метил}-4-гидрокси-1,3-бензолметанола] ID50 составляет 0,001 мг/кг интратрахеально. Соответствующие дозировки соединения примера 1 для введения путем ингаляции, например, для бронхорасширяющего действия при лечении астмы, таким образом могут быть примерно в 20 раз выше таковых, обычно требующихся при использовании Сальбутамола.
| название | год | авторы | номер документа |
|---|---|---|---|
| БЕНЗОПИРАНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1996 |
|
RU2160735C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ БЕНЗОПИРАНА ПРОТИВ АРИТМИИ | 2002 |
|
RU2291867C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПИРАНИЛ-ЦИАНОГУАНИДИНА | 1990 |
|
RU2057129C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ БЕНЗОЦИКЛОАЛКЕНИЛДИГИДРООКСИАЛКАНОВЫХ КИСЛОТ | 1990 |
|
RU2012554C1 |
| БЕНЗОПИРАНЫ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ | 1994 |
|
RU2122000C1 |
| СПОСОБ ЭПОКСИДИРОВАНИЯ ПРОХИРАЛЬНОГО ОЛЕФИНА | 1995 |
|
RU2204562C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ БЕНЗОПИРАНА В КАЧЕСТВЕ ПРОТИВОАРИТМИЧЕСКИХ АГЕНТОВ | 2005 |
|
RU2380370C2 |
| БЕНЗОПИРАНЫ И БЕНЗОКСЕПИНЫ, СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СПОСОБ ИХ ПОЛУЧЕНИЯ | 1999 |
|
RU2228333C2 |
| АМИНОАЛКИЛАМИДОМЕТИЛЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 2-(4-СУЛЬФОНИЛАМИНО)-3-ГИДРОКСИ-3,4-ДИГИДРО-2Н-ХРОМАН-6-ИЛА И ЛЕКАРСТВЕННЫЕ СРЕДСТВА, СОДЕРЖАЩИЕ ЭТИ СОЕДИНЕНИЯ | 2006 |
|
RU2413725C2 |
| ПРОИЗВОДНЫЕ ИНДОЛА | 1992 |
|
RU2061694C1 |
Сущность:2,2-ди-/C1-C5/-итранс-2,2-ди-/C1-C5/-3,3-дигидро-3-гидрокси-6-/пиридин/-4-ил/2Н-1- бензопираны, например, формулы /I/:
в которой R1 и R2 = H, алкил, гидроксиалкил или алкоксиалкил, причем по меньшей мере один из них отличен от водорода, а R3= обычно 2-пиперидиноновая группа; R4= H и R5=-OH, или R4 + R5 = дополнительная связь и R6, R7=алкил и R3=H или алкил; их N-оксиды, сложные эфиры и соли; способы их получения и их использование в качестве фармацевтических средств, например в качестве открывающих K+-каналы агентов, бронхорасширяющих средств и профилактических средств при астме. 2 с. и 3 з. п. ф-лы, 4 табл.
где R1 и R2 независимо друг от друга водород, C1 - C5-алкил, C1 C5-гидроксиалкил или C1 - C5-алкоксиалкил, причем по меньшей мере один из R1 и R2 отличен от водорода;
R3 группа формулы -N(R9)-COR1 0, где R9 водород, R1 0 пиридил или R9 и R1 0 вместе 1,3-бутадиенилен, или группа формулы -(CH2)n- или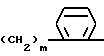
в которой n 3 5, целое число, m 1 или 2;
R4 водород;
R5 гидроксил в трансположении к R3 или R4 и R5 вместе обозначают дополнительную связь, как указано пунктирной линией;
R6 и R7 независимо друг от друга C1 - C5-алкил;
R8 водород или C1 C5-алкил,
или его N-оксид, или физиологически гидролизуемый и физиологически приемлемый сложный эфир такого соединения или N-оксида, или соль присоединения кислоты, или четвертичная аммониевая соль такого соединения, N-оксида или сложного эфира.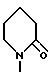
и R1 метил, этил, н-пропил, изобутил, гидроксиметил или метоксиметил, R2 и R8 каждый водород или R1 и R8 каждый водород и R2 метил, или R1 и R8 каждый метил и R2 водород, или Б) R3 группа формулы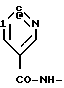
R1 метил, этил, н-пропил, изобутил или метоксиметил или R2 и R8 каждый водород, или R1 и R2 каждый - метил и R8 водород, или R1 и R8 каждый метил и R2 водород, или В) R3 группа формулы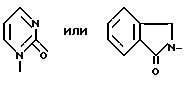
R1 метил и R2 и R8 каждый водород, или R1 метил и R2 водород или R1 водород и R2 метил, R6 и R7 каждый метил, R8 водород, R4 и R5 вместе дополнительная связь и R3 группа формулы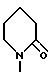
или его N-оксид, или физиологически гидролизуемый и физиологически приемлемый сложный эфир такого соединения или его N-оксида, или соль присоединения кислоты или четвертичная аммониевая соль такого соединения, N-оксида или сложного эфира.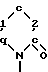
или его N-оксид, или соль присоединения кислоты такого соединения или N-оксида.
гидроксил в трансположении.
| EP, патент, 0346724, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Brit | |||
| J.Pharmac | |||
| Кузнечная нефтяная печь с форсункой | 1917 |
|
SU1987A1 |
