Настоящее изобретение относится к новым аминоалкиламидометилзамещенным производным 2-(4-сульфониламино)-3-гидрокси-3,4-дигидро-2H-хромен-6-ила, блокирующим калиевые каналы, в частности, воздействующих на сердечно-сосудистую систему, а также к лекарственным средствам, содержащим эти соединения. Кроме того, настоящее изобретение относится к способу получения новых соединений и к промежуточным продуктам этого способа.
Инданы, бензопираны и аналоги таких соединений, которые блокируют калиевые каналы и в особенности благоприятно воздействуют на сердечно-сосудистую систему, уже известны из описания WO 00/12077 А1.
В документе WO 00/58300 раскрыты производные хромана, которые применимы в качестве лекарственных средств, в особенности антиаритмически эффективных лекарственных средств.
В опубликованной заявке WO 2005/037780 описаны новые амидометилзамещенные производные 2-(4-сульфониламино)-3-гидрокси-3,4-дигидро-2H-хромен-6-ила, которые блокируют калиевые каналы, в частности, воздействующие на сердечно-сосудистую систему, а также лекарственные средства, содержащие эти соединения.
В основу настоящего изобретения положена задача получения новых активных соединений, в особенности предназначенных для лечения сердечно-сосудистых заболеваний, предпочтительно - аритмий сердца, которые отличаются высокой эффективностью при хорошей совместимости и в случае антиаритмического воздействия также заметным избирательным воздействием на предсердия.
Согласно изобретению неожиданно было установлено, что предлагаемая в настоящем изобретении группа новых аминоалкиламидометилзамещенных производных 2-(4-сульфониламино)-3-гидрокси-3,4-дигидро-2H-хромен-6-ила способна блокировать калиевые каналы и пригодна для лечения сердечно-сосудистых заболеваний, предпочтительно - для лечения аритмий сердца. Соединения, предлагаемые в настоящем изобретении, отличаются высокой эффективностью при хорошей совместимости и в случае антиаритмического воздействия также заметным избирательным воздействием на предсердия. Кроме того, соединения, предлагаемые в настоящем изобретении, характеризуются относительно хорошей биологической доступностью. Наряду с этим соединения, предлагаемые в настоящем изобретении, обладают такими характеристиками, которые позволяют ожидать дополнительного воздействия на иммунную систему.
Объектом настоящего изобретения являются новые аминоалкиламидометилзамещенные производные 2-(4-сульфониламино)-3-гидрокси-3,4-дигидро-2H-хромен-6-ила общей формулы I,
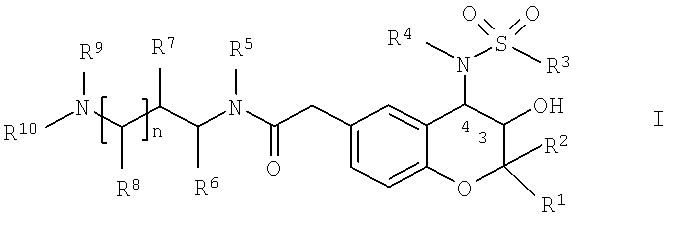
в которой
R1 обозначает С1-С4-алкил;
R2 обозначает С1-С4-алкил;
R3 обозначает фенил, который необязательно содержит 1-3 любых заместителя, выбранных из группы, включающей галоген, C1-С6-алкил и С1-С4-алкоксигруппу;
R4 обозначает водород; C1-С6-алкил или С3-С7-циклоалкил-C1-С4-алкил,
R5 обозначает водород; и
R6 обозначает водород; и
R7 обозначает водород; и
R8 обозначает водород; и
R9 обозначает C1-С4-алкил; и
R10 обозначает C1-С6-алкил; фенил-С0-С4-алкил или пиридинил-С0-С4-алкил; при условии, что R10 не обозначает фенил, если R5 и R9 совместно образуют C2-алкилен; или
R5 и R9 совместно образуют С1-С3-алкилен; или
R6 и R9 совместно образуют С1-С3-алкилен; или
R7 и R9 совместно образуют С2-С4-алкилен или C1-С3-алкиленоксигруппу; или
R8 и R9 совместно образуют С3-С5-алкилен; или
R9 и R10 совместно образуют С4-С6-алкилен; и
n равно 0 или 1,
или их любые физиологически совместимые соли и/или сольваты.
Кроме того, объектом настоящего изобретения являются фармацевтические композиции, содержащие соединения формулы I. Кроме того, объектом настоящего изобретения является способ получения соединений формулы I и промежуточные продукты этого способа.
Если в соединениях формулы I или в других соединениях, описанных в контексте настоящего изобретения, заместители представляют собой C1-С4-алкил или C1-С6-алкил или содержат его, то все они могут обладать линейной или разветвленной цепью.
Все R1 и R2 предпочтительно обозначают метил.
R3 предпочтительно обозначает фенил, который необязательно 1 или 2 раза замещен галогеном, C1-С4-алкилом или С1-С4-алкоксигруппой. Особенно предпочтительно, если R3 обозначает фенил, однократно замещенный С1-С4-алкилом. Если R3 обозначает галогензамещенный фенил, то галогенами могут являться фтор, хлор, бром или йод. Особенно предпочтительно, если R3 обозначает 4-этилфенил.
R4 предпочтительно обозначает водород; С1-С6-алкил или циклопропил-C1-С4-алкил, предпочтительно - циклопропилметил. Если R4 обозначает С1-С6-алкил, то предпочтительно, если он является разветвленным и более предпочтительно, если он обозначает неопентил, 2,2-диметилбутил, 2-этилбутил, 3-метилбутил или 2-метилпропил.
Предпочтительно, если R5 и R9 совместно образуют C1-С3-алкилен.
R10 предпочтительно обозначает C1-С4-алкил; бензил или фенил. Более предпочтительно, если R10 обозначает фенил-С1-С4-алкил или пиридинил-С1-С4-алкил, например, пиридинилметил, в особенности 2-пиридинилметил, 3-пиридинилметил или 4-пиридинилметил; или R9 и R10 совместно образуют С4-С6-алкилен.
Особенно предпочтительные соединения формулы I выбраны из группы, включающей N-{6-[2-(4-бензилпиперазин-1-ил)-2-оксоэтил]-3-гидрокси-2,2-диметилхроман-4-ил}-4-этилбензолсульфонамид; 4-этил-N-{3-гидрокси-2,2-диметил-6-[2-оксо-2-(4-пиридин-3-илметилпиперазин-1-ил)-этил]-хроман-4-ил}-бензолсульфонамид; 4-этил-N-{3-гидрокси-2,2-диметил-6-[2-оксо-2-(4-пиридин-2-илметилпиперазин-1-ил)-этил]-хроман-4-ил}-бензолсульфонамид и 4-этил-N-{3-гидрокси-2,2-диметил-6-[2-оксо-2-(4-пиридин-4-илметилпиперазин-1-ил)-этил]-хроман-4-ил}-бензолсульфонамид. 4-Этил-N-{3-гидрокси-2,2-диметил-6-[2-оксо-2-(4-пиридин-4-илметилпиперазин-1-ил)-этил]-хроман-4-ил}-бензолсульфонамид являются особенно предпочтительным соединением формулы I.
В контексте настоящего изобретения новые соединения формулы I получают с помощью
а) реакции соединения общей формулы II,
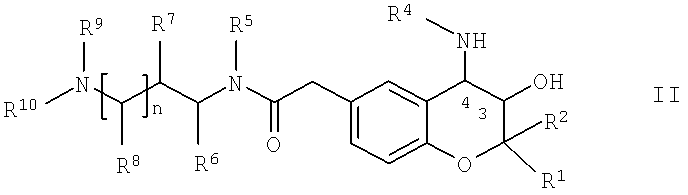
в которой R1, R2, R4, R5, R6, R7, R8, R9, R10 и n имеют указанные выше значения, с соединением общей формулы III,
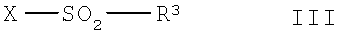
в которой R3 имеет указанные выше значения и Х означает способную к отщеплению удаляемую группу,
b) реакции соединения общей формулы IV
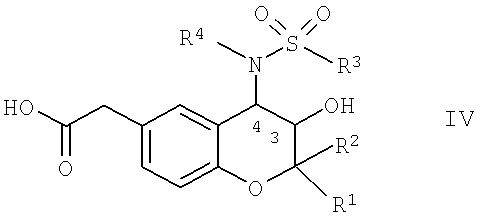
в которой R1, R2, R3 и R4 имеют указанные выше значения, с соединением общей формулы V,
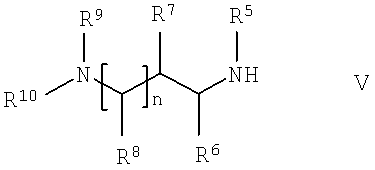
в которой R5, R6, R7, R8, R9, R10 и n имеют указанные выше значения.
Реакцию по варианту а) способа можно провести по обычной мокрой химической методике в органическом растворителе, который является инертным при условиях проведения реакции, предпочтительно - в дипольном апротонном растворителе, таком как дихлорметан, или в смеси таких растворителей и в присутствии основания. Подходящими основаниями являются ненуклеофильные органические азотистые основания, такие как третичные низш. алкиламины, например триэтиламин. В качестве растворителей также можно использовать взятые в избытке. При необходимости реакцию можно катализировать с помощью известного реагента сочетания, такого как 4-N,N-диметиламинопиридин (= ДМАП). Подходящая температура для проведения реакции находится в диапазоне от комнатной температуры до 80°С, например 65°С. Подходящее давление для проведения реакции находится в диапазоне от нормального давления до примерно 200 бар, например, равно 180 бар. Если использующееся соединение формулы III является жидким, то может оказаться предпочтительным удаление растворителя из реакционной смеси после прибавления соединения формулы III к соединению формулы II, растворенному в растворителе, проводимое известным образом, например, при пониженном давлении. Если в исходных соединениях формулы II R4 обозначает водород, то целесообразно использовать эквимолярные количества соединения формулы III. Обычно в соединениях формулы III в качестве отщепляющейся группы Х используют галоген, предпочтительно - хлор, бром или йод. Кроме того, реакцию соединения формулы II с соединением формулы III также можно проводить по известным методикам в твердой фазе, предпочтительно - на реакционноспособной смоле, такой как аминометилполистирол (АМПС). Этот вариант реакции предпочтительно можно использовать для получения небольших количеств вещества, например, порядка от 1 до 10 ммолей. Если синтез проводят в твердой фазе, то в качестве основания предпочтительно можно использовать легко отфильтровывающееся основание, такое как известный метилпиперидин на полимерной подложке (= ПП метилпиперидин) или пиперидин на полимерной подложке (= ПП пиперидин). Подходящая температура проведения реакции при твердофазном синтезе составляет от 10 до 40°С, предпочтительной является комнатная температура. Соединения формулы I можно выделить из реакционной смеси по известным методикам и при необходимости очистить по известным методикам. Если в соединениях формулы I группы R9 и/или R10 не являются частью ароматической или гетероароматической кольцевой системы, то возможно образование соли. Таким образом, подходящие свободные соединения формулы I можно превратить в их физиологически совместимые соли или соли соединений формулы I можно превратить в свободные соединения формулы I.
Реакцию по варианту b) способа можно провести по методике, применяющейся для аминоацилирования. В качестве реагентов для ацилирования можно использовать карбоновые кислоты формулы IV или их реакционноспособные производные, такие как галогенангидриды кислот, предпочтительно - хлорангидриды кислот или бромангидриды кислот. Если кислоты формулы IV сами используются в качестве ацилирующих реагентов, то их реакцию с аминами формулы V также можно с успехом проводить в присутствии одного или большего количества известных реагентов сочетания, применяющихся в реакциях аминоацилирования, например, 1,1-карбонилдиимидазол (КДИ); этилхлорформиат; N-гидроксибензотриазол (= ГОБТ); алкилкарбодиимид, например, N'-(3-диметиламинопропил)-N-этилкарбодиимид (= ЭКД) или N,N'-диизопропилкарбодиимид (= ДИК), или циклоалкилкарбодиимид, такой как дициклогексилкарбодиимид. Ацилирование можно проводить в органическом растворителе, который является инертным при условиях проведения реакции при температуре от -30 до +50°С, предпочтительно - при комнатной температуре. Подходящими растворителями являются галогенированные углеводороды, такие как дихлорметан, или циклические простые эфиры, такие как тетрагидрофуран или диоксан или смеси этих растворителей.
Физиологически совместимые соли соединений формулы I представляют собой их обычные соли с неорганическими кислотами, например серной кислотой, фосфорными кислотами или галогенводородными кислотами, предпочтительно - хлористоводородной кислотой; или с органическими кислотами, например, низшими алифатическими монокарбоновыми, дикарбоновыми или трикарбоновыми кислотами, такими как малеиновая кислота, фумаровая кислота, молочная кислота, винная кислота, лимонная кислота; или с сульфоновыми кислотами, например низшими алкансульфоновыми кислотами, такими как метаносульфоновая кислота или трифторметаносульфоновая кислота, или бензолсульфоновыми кислотами, необязательно замещенными по бензольному кольцу галогеном или низш. алкилом, такими как п-толуолсульфоновая кислота. Предпочтительными являются гидрохлориды соединений формулы I.
Соединения формулы II являются новыми соединениями, которые прекрасно подходят для использования в качестве промежуточных продуктов для получения новых фармакологически активных веществ, например, для получения соединений формулы I.
Соединения формулы II, в которой R4 обозначает водород, можно получить по известным методикам путем проводимого в кислых средах отщепления любой содержащейся защитной группы PG1 от соединений общей формулы VI,
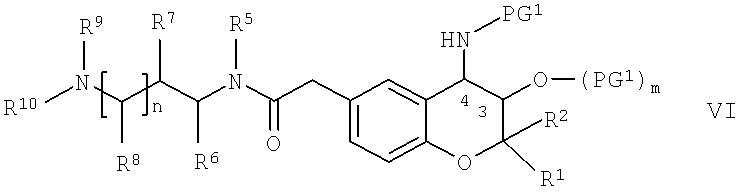
в которой R1, R2, R5, R6, R7, R8, R9, R10 и n обладают указанными выше значениями, PG1 обозначает защитную группу аминогруппы, которую можно отщепить в кислых средах, предпочтительно - трет-бутоксикарбонил (= boc), и m равно 0 или 1. Отщепление защитной группы, например, можно проводить путем прибавления кислоты, такой как неорганическая кислота, предпочтительно - хлористоводородная кислота, например, 4 М хлористоводородная кислота, к соединению формулы VI. Кислоту можно растворить в полярном протонном растворителе, таком как диоксан. Если в соединениях формулы VI или любом соединении, содержащем защитные группы PG1 и указанном ниже в настоящем изобретении, m равно 0, то в каждом случае заместителем в положении 3 пиранового кольца является гидроксигруппа.
Подходящие защитные группы PG1 и другие защитные группы, указанные в настоящей заявке, известны в данной области техники и их обычным образом может выбрать специалист в данной области техники, например, из приведенных в публикации T.W.Greene, P.G.M.Wuts Защитных групп in Organic Synthesis, John Wiley & Sons, последнее издание.
Если необходимы соединения формулы I, в которой R4 обозначает C1-С6-алкил или С3-С7-циклоалкил-C1-С4-алкил, то соединение формулы I, в которой R4 обозначает водород, или предшественник соединения формулы I, в которой R4 обозначает водород, а именно, предшественник соединения формулы II или IV, можно алкилировать по известным методикам. Алкилирование предпочтительно можно провести, как аминоалкилирование, проводимое сначала по реакции соединения формулы I, II или IV, в которой R4 в каждом случае обозначает водород, с альдегидом общей формулы VII,
 ,
,
в которой R401 обозначает водород, С2-С5-алкил или С3-С7-циклоалкил-С0-С3-алкил, с последующим восстановлением полученного промежуточного имина путем прибавления восстановительного реагента к алкиламину формулы I, II или IV. Подходящими восстановительными реагентами являются комплексные борогидриды, такие как NaBH3CN, или известный борогидрид на полимерной подложке (= ПП-ВН4). В первом варианте реакцию можно провести в полярном протонном органическом растворителе, который является инертным при условиях проведения реакции, предпочтительно - в метаноле, восстановление имина проводят in situ без выделения, в том же растворителе. Подходящая температура проведения реакции для этого варианта находится в диапазоне от комнатной температуры до 60°С, например, равна 50°С. Во втором варианте реакцию соединения формулы I, II или IV, в которой R4 обозначает водород, с альдегидом формулы V с получением промежуточного имина можно провести в дипольном апротонном растворителе, предпочтительно - тетрагидрофуране (= ТГФ). В этом случае для ускорения реакции предпочтительно прибавить каталитическое количество гидрофильного реагента, например, гидрофильного реагента, например, ортоэфира, предпочтительно - триметилортоформиата (= ТМОФ). Затем промежуточный имин можно выделить и растворить в полярном протонном растворителе, указанном выше для первого варианта, и провести восстановление в этом растворителе. Этот второй вариант предпочтительно можно проводить при комнатной температуре.
Соединения формулы VI можно получить по реакции производного карбоновой кислоты общей формулы VIII,
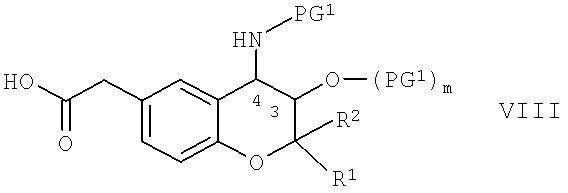
в которой R1, R2, PG1 и m обладают указанными выше значениями, с амином формулы V по известной для аминоацилирования методике, которая подробно описана выше. В качестве ацилирующих реагентов можно использовать карбоновые кислоты формулы VIII или их реакционноспособные производные, такие как галогенангидриды, предпочтительно - хлорангидриды или бромангидриды. Если в качестве ацилирующих реагентов используют сами кислоты формулы VIII, то их реакцию с аминами формулы V также можно с успехом проводить в присутствии одного или большего количества известных реагентов сочетания для реакций аминоацилирования, например, 1,1-карбонилдиимидазола; этилхлорформиата; N-гидроксибензотриазола (= ГОБТ); алкилкарбодиимида, например, N'-(3-диметиламинопропил)-N-этилкарбодиимида (= ЭКД) или N,N'-диизопропилкарбодиимид (= ДИК), или циклоалкилкарбодиимида, такого как дициклогексилкарбодиимид. Ацилирование можно проводить в органическом растворителе, который является инертным при условиях проведения реакции, при температуре от -30 до +50°С, предпочтительно - при комнатной температуре. Подходящими растворителями являются галогенированные углеводороды, такие как дихлорметан, или циклические простые эфиры, такие как тетрагидрофуран или диоксан, или смеси этих растворителей.
Соединения формулы V и соединения формулы VII известны или их можно получить по известным методикам из известных соединений.
Соединения формулы VIII можно получить по известным методикам путем проводимого в щелочных средах отщепления любой содержащейся защитной группы PG2 от соединения общей формулы IX,
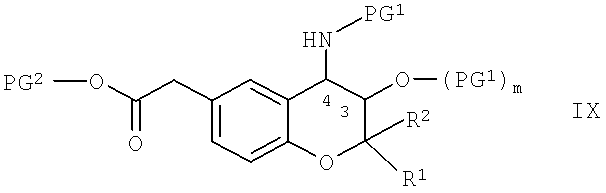
в которой R1, R2, PG1 и m обладают указанными выше значениями и PG2 обозначает защитные группы карбоксигрупп, которые можно отщепить в щелочных средах.
PG2 обычно обозначает защитные группы карбоксигрупп, которые можно отщепить в щелочных средах или в кислых средах. Если PG2 обозначает защитные группы карбоксигрупп, которые можно отщепить в щелочных средах, то подходящими являются обладающие линейной цепью или разветвленные C1-C4-алкильные радикалы, предпочтительно - изопропил или метил. Отщепление защитных групп PG2, которые можно отщепить в щелочных средах, обычно можно проводить путем прибавления основания, такого как гидроксид щелочного металла, например гидроксид лития. Подходящими растворителями в этом случае являются вода или полярные протонные органические растворители, такие как ТГФ, или, предпочтительно, смеси таких органических растворителей с водой. Если PG2 обозначает защитные группы карбоксигрупп, которые можно отщепить в кислых средах, то подходящими являются разветвленные C1-С4-алкильные радикалы, предпочтительно - трет-бутил. Отщепление защитных групп PG2, которые можно отщепить в кислых средах, обычно можно проводить путем прибавления кислоты, такой как трифторуксусная кислота. Подходящими растворителями в этом случае являются полярные апротонные органические растворители, такие как толуол или ксилол, или смеси указанных органических растворителей.
Соединения формулы IX можно получить по известным методикам путем защиты производных аминогидроксихромана общей формулы X,
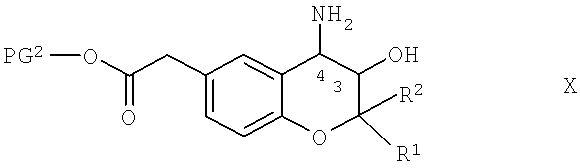
в которой R1, R2 и PG2 обладают указанными выше значениями, приведенными для соединений формулы IX, с помощью защитной группы аминогруппы, которую можно отщепить в кислых средах, предпочтительно - группы boc. При получении защищенных по аминогруппе группой boc соединений формулы Х в качестве реагента можно использовать boc-ангидрид и использовать известные методики. Обычно в этом случае получают смесь содержащего одну защитную группу соединения формулы Х и содержащего две защитные группы соединения формулы X. Обычно продукты находятся в соотношении 2:1 с преобладанием продукта с одной защищенной группой. Обычно последующие реакции получения соединений формулы I, которые являются исходными веществами для получения соединений формулы X, можно выполнять без затруднений, если в каждом случае в качестве исходных веществ использовать смесь защищенных соединений.
Соединения формулы Х можно получить по реакции эпоксида общей формулы XI,
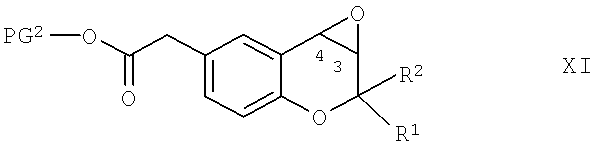
в которой R1, R2 и PG2 обладают указанными выше значениями, приведенными для соединений формулы X, по известным методикам с использованием нуклеофильного органического азотсодержащего соединения, предпочтительно - аммиака в водном растворе, такого как гидроксид аммония, в дипольном протонном растворителе, таком как низш. алкиловый спирт, предпочтительно - этанол. Подходящая температура проведения реакции находится в диапазоне от комнатной температуры до 70°С.
Соединения формулы XI можно получить по реакции соединения общей формулы XII,
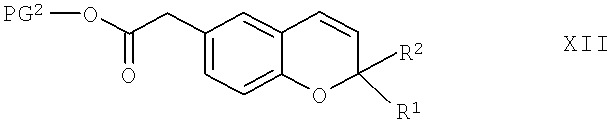
в которой R1, R2 и PG2 обладают указанными выше значениями, приведенными для соединений формулы XI, по известным методикам с пероксидом, способным образовывать эпоксид, предпочтительно - с м-хлорпербензойной кислотой (МХПБК), в полярном апротонном органическом растворителе, который является инертным при условиях проведения реакции, предпочтительно - в дихлорметане, и в присутствии основания. Подходящим основанием предпочтительно является водный раствор гидрокарбоната натрия. Реакцию предпочтительно можно проводить при комнатной температуре.
Соединения формулы XII можно получить по реакции соединения общей формулы XIII,
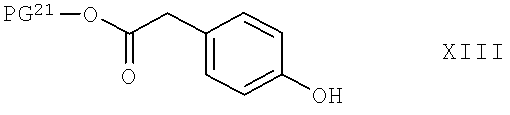
в которой PG21 обладает значением, указанным выше для PG2 в соединениях формулы XII, и предпочтительными альтернативами для PG21 являются неразветвленные низш. алкильные радикалы, такие как C1-С4-алкил, предпочтительно - метил, с соединением общей формулы XIV,
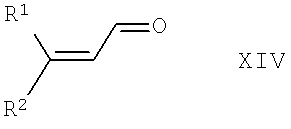
в которой R1 и R2 обладают указанными выше значениями, по известным методикам, и при необходимости с проводимой по известным методикам последующей заменой защитных групп PG21 на любые необходимые защитные группы PG2. Реакцию можно провести в органическом растворителе, который является инертным при условиях проведения реакции, таком как толуол или ксилол, и в присутствии кислоты с отделением воды посредством азеотропной отгонки. Подходящими кислотами являются, например, уксусная кислота или пропионовая кислота. Предпочтительно проводить реакцию с прибавлением катализатора, такого как кислота Льюиса, например, фенилбороновая кислота. Подходящая температура проведения реакции находится в диапазоне от комнатной температуры до температуры кипения растворителя или смеси растворителей, например, равна примерно 120°С.
Соединения формулы XIII и формулы XIV известны или их можно получить по известным методикам из известных соединений.
Соединения формулы IV являются новыми соединениями, которые прекрасно подходят для применения в качестве промежуточных продуктов при получении новых фармакологически активных веществ, например, при получении соединений формулы I.
Соединения формулы IV, в которой R4 обозначает водород, можно получить по известным методикам, например, путем отщепления защитных групп PG3 из соединения общей формулы XV,
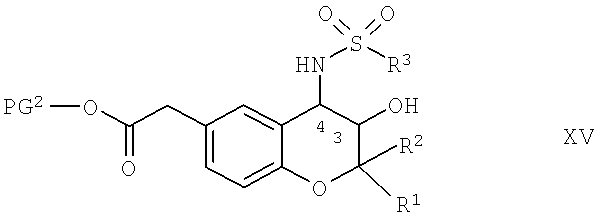
в которой R1, R2 и R3 обладают указанными выше значениями и PG2 обозначает защитные группы карбоксигрупп, которые можно отщепить в кислых средах, такие как разветвленные или неразветвленные C1-С4-алкильные радикалы, предпочтительно - трет-бутил.
Соединения формулы XV можно получить по известным методикам, например, по реакции соединения формулы X, в которой PG2 обладает указанным выше значением, приведенным для соединений формулы XV, с соединением формулы III. Реакцию можно провести так, как это описано выше для варианта а) способа для реакции соединения формулы I с соединением формулы III.
Соединения формулы I по меньшей мере по концевым атомам углерода в положениях 3 и 4 пиранового кольца содержат хиральные центры и поэтому могут находиться в разных изомерных формах. Объектами настоящего изобретения являются и изомерно чистые соединения формулы I, и смеси этих изомеров. Оптически активные соединения формулы I можно получить, например, из смесей изомеров соединений формулы I или из смесей изомеров соединений формулы II или IV по известным методикам, например, путем хроматографического разделения на хиральных разделяющих материалах. Смеси изомеров соединений формулы I, в которой R9 и/или R10 не являются частями ароматической или гетероароматической кольцевой системы, или смеси изомеров соединений формулы II также можно получить по реакции с подходящими оптически активными кислотами, например, камфорсульфоновой кислотой или D- или L-винной кислотой с последующим фракционированием на соответствующие оптические антиподы путем фракционной кристаллизации полученных солей. Смеси изомеров соединений формулы IV также можно получить по реакции с подходящими оптически активными основаниями с последующим фракционированием на соответствующие оптические антиподы путем фракционной кристаллизации полученных солей. Соединения формулы I также могут содержать хиральные центры на атомах углерода, содержащих заместители R5, R6, R7 и/или R8. Эти хиральные центры можно заменять путем выбора подходящих соединений формулы VIII, в которых уже имеются соответствующие хиральные центры или проводимого по подходящим методикам синтеза.
Некоторые оптически активные соединения формулы I также можно получить непосредственно с помощью хирального синтеза. Если необходимо получить соединения формулы I, в которой гидроксигруппа в положении 3 пиранового кольца и заместитель R4NSO2R3 в положении 3 пиранового кольца находятся в трансположении друг к другу, то в каждом случае исходными веществами могут быть эпоксиды формулы XI, уже обладающие определенной стереохимической конфигурацией. Эпоксиды формулы XI, уже обладающие определенной стереохимической конфигурацией, например, можно получить эпоксидированием алкенов формулы XII по известным методикам с использованием хирального катализатора, например, (S,S)-(+)-N,N'-бис(3,5-ди-трет-бутилсалицилиден)-1,2-циклогександиаминомарганец(III)хлорида (=("катализатор Якобсона"; "(S,S)-марганец(III)-сален") по методике Якобсона (см., например, WO 91/14694 А1). Если, например, необходимо получить соединение формулы I, в которой хиральный центр в положении 3 пиранового кольца находятся в S-конфигурации и в которой хиральный центр в положении 4 пиранового кольца находятся в R-конфигурации, то промежуточный продукт формулы XII можно ввести в реакцию в присутствии хирального катализатора, предпочтительно - (S,S)-марганец(III)-салена и в присутствии донора кислорода, предпочтительно - гипохлорита натрия в водном растворе, в органическом растворителе, который является инертным при условиях проведения реакции, предпочтительно - дихлорметане. Реакцию целесообразно проводить при значении рН в диапазоне от 9,5 до 11,5. Для установления подходящего значения pH, предпочтительно прибавить к реакционной смеси буфер, содержащий Na2HPO4, и пиридин-N-оксид. Подходящая температура проведения реакции равна от -10°С до комнатной температуры, предпочтительно - 0°С. Если необходимо получить соединения формулы I, в которой хиральный центр в положении 3 пиранового кольца находятся в R-конфигурации и в которой хиральный центр в положении 4 пиранового кольца находятся в S-конфигурации, то методика может быть аналогичной описанной выше, но вместо (S,S)-марганец(III)-салена используют (R,R)-марганец(III)-сален.
По реакции нуклеофильного раскрытия цикла эпоксидов формулы XI, описанной выше в двух вариантах, как правило, получают соединения формулы X, в которых концевые заместители в положении 3 и в положении 4 пиранового кольца, а именно, гидроксигруппа и аминогруппа находятся в транс-положении друг к другу.
Благоприятные воздействия соединений формулы I, как фармакологически активных веществ, станут понятными из следующих соображений: уже известно, что вещества, которые блокируют эндогенные калиевые каналы сердца, можно использовать в качестве активных веществ для борьбы с сердечно-сосудистыми заболеваниями, в особенности с аритмиями сердца. Путем блокирования направленных наружу потоков калия в сердце можно пролонгировать потенциал сердечной деятельности, что оказывает благоприятное воздействие при аритмии сердца. Примерами этих известных лекарственных средств являются антиаритмические лекарственные средства класса III. Одним недостатком этих неспецифических блокаторов калиевых каналов является их низкая избирательность по отношению к разным тканям сердца. По этой причине в течение длительного времени полагали, что в особенности антиаритмические лекарственные средства класса III могут привести к нежелательному увеличению интервала QT на электрокардиограмме (= ЭКГ) и полиморфным желудочковым тахикардиям ("трепетание - мерцание"), вследствие чего в конечном счете могут возникнуть нежелательные осложнения, такие как, например, фибрилляция желудочков. По этой причине необходимы блокаторы калиевых каналов, способные селективно влиять на потоки калия в предсердии, но не в желудочке. Поскольку недавно открытые калиевые каналы Kv1.5 сердца расположены исключительно в предсердии, а не в желудочке, можно предположить, что эти соединения, блокирующие калиевые каналы Kv1.5, пригодны для использования в качестве избирательно воздействующих на предсердия антиаритмических лекарственных средств. Однако калиевые каналы Kv1.5 и другие калиевые каналы расположены не только в сердце, но и в сосудах организма. Поэтому никогда нельзя исключить того, что соединения, блокирующие калиевые каналы Kv1.5, могут привести к повышению артериального давления вследствие блокирования калиевых каналов сосудов. Поэтому предпочтительны соединения, блокирующие калиевые каналы Kv1.5, которые не вызывают побочных эффектов, приводящих к повышению артериального давления. Другими нежелательными побочными эффектами, которые могут возникнуть при введении многих соединений, блокирующих калиевые каналы Kv1.5, являются дополнительные антиаритмические побочные эффекты класса I, а также негативные инотропные эффекты.
Соединения формулы I характеризуются способностью заметно и избирательно блокировать калиевые каналы Kv1.5 сердца. В дополнение к особенно хорошей эффективности и заметному избирательному воздействию на предсердия соединения формулы I приводит самое большее к небольшим нежелательным побочным эффектам, таким как повышение артериального давления, антиаритмические побочные эффекты класса I и негативные инотропные эффекты. Поэтому соединения формулы I показаны для лечения и/или профилактики сердечно-сосудистых заболеваний, в особенности фибрилляция желудочков, трепетания предсердий и других аритмий сердца у крупных млекопитающих и людей.
Соединения формулы I также характеризуются сравнительно хорошей растворимостью в воде, в особенности те соединения формулы I, в которой заместитель R10 обозначает C1-С6-алкил; фенил-C1-С4-алкил или пиридинил-C1-С4-алкил, и атом азота, непосредственно связанный с R10 не является частью ароматической или гетероароматической кольцевой системы. Предполагается, что повышенная растворимость в воде приведет к улучшенной биологической доступности, что позволит приготовить фармацевтические композиции, содержащие уменьшенное количество органических растворителей и/или веществ, улучшающих растворимость, или даже не содержащие этих компонентов.
Кроме того, соединения формулы I обладают выраженной способностью блокировать калиевые каналы Kv1.3. Калиевые каналы Kv1.3 расположены преимущественно в клетках иммунной системы. Установлена взаимосвязь между блокированием калиевых каналов Kv1.3 и, в частности, антипролиферативным и/или иммунодепрессивным воздействием (см. С.Beeton et al., The Journal of Immunology  (2001) 936-944). Поэтому можно предположить, что соединения, которые способны блокировать калиевые каналы Kv1.3 - например, соединения формулы I, предлагаемые в настоящем изобретении - также применимы для лечения и/или профилактики пролиферативных, хронических воспалительных и аутоиммунных заболеваний. Такие аутоиммунные заболевания могут включать, например, болезнь Аддисона, гнездную алопецию, анкилоз, спондилит, антифосфолипидный синдром, аутизм, аутоиммунный атеросклероз, аутоиммунный диабет, потерю вкусовых ощущений, аутоиммунный эндометриоз, аутоиммунные заболевания глаз, аутоиммунную гемолитическую анемию, аутоиммунную гемофилию, аутоиммунный гепатит, аутоиммунный интерстициальный цистит, аутоиммунный лимфопролиферативный синдром, аутоиммунную миелопатию, аутоиммунный миокардит, аутоиммунные невропатии, аутоиммунный оофорит, аутоиммунный орхит, аутоиммунную тромбоцитопению, аутоиммунные заболевания щитовидной железы, аутоиммунную уртикарию, аутоиммунный увеит, аутоиммунный васкулит; болезнь Бехчета, паралич Белла, буллезный пемфигоид; глютеновую болезнь, синдром хронического переутомления, болезнь Крона; герпетиформный дерматит, дерматомиозит, дискоидную красную волчанку; синдром Гудпасчера, болезнь Грейвса, синдром Гийена - Барре, тиреоидит Хашимото, герпес беременных, болезнь Гентингтона, IgA нефропатию, иммунную тромбоцитопению, пурпуру, интерстициальный цистит; волчанку, болезнь Лима; синдром Миллера-Фишера, смешанное заболевание соединительной ткани; рассеянный склероз, миастению gravis; паранеопластические аутоиммунные синдромы, листовидную пузырчатку, обыкновенную пузырчатку, пернициозную анемию, болезнь Пейрони, полиэндокринный синдром, первичный биллиарный цирроз, первичный гломерулонефрит, первичный склерозирующий холангит, псориаз, псориатический артрит; энцефалит Расмуссена, рецидивирующую полихондрию, ревматоидный артрит; саркоидоз, склеродермию, синдром Шегрена, синдром Штиффа-Персона; хорею Сиденгама, симпатическую офтальмию, темпоральный артериит, диабет типа 1, язвенный колит; витилиго; гранулематоз Вегенера. Кроме того, установлена взаимосвязь между блокированием калиевых каналов Kv1.3 и метаболическими заболеваниями (см. J. Xu et al., Human Molecular Genetics 2003. Vol.12. No, 5, 551-559). Поэтому можно предположить, что соединения, которые способны блокировать калиевые каналы Kv1.3 - например, соединения формулы I, предлагаемые в настоящем изобретении или соединения, раскрытые и заявленные в опубликованной заявке WO 2005/037780 (= US 2005/0148659) - также могут быть пригодны для лечения и/или профилактики метаболических нарушений или заболеваний, таких как центральное ожирение; гипертензия, в особенности артериальная гипертензия; резистентность к инсулину, в особенности сахарный диабет типа II; непереносимость глюкозы или нарушенная непереносимость глюкозы; дислипопротеинемия, в особенности гипертриглицеридемия, сопровождающаяся дислипопротеинемией, происходящей при пониженном уровне ЛВП-холестерина (ЛВП - липопротеины высокой плотности); и гиперурикемия.
(2001) 936-944). Поэтому можно предположить, что соединения, которые способны блокировать калиевые каналы Kv1.3 - например, соединения формулы I, предлагаемые в настоящем изобретении - также применимы для лечения и/или профилактики пролиферативных, хронических воспалительных и аутоиммунных заболеваний. Такие аутоиммунные заболевания могут включать, например, болезнь Аддисона, гнездную алопецию, анкилоз, спондилит, антифосфолипидный синдром, аутизм, аутоиммунный атеросклероз, аутоиммунный диабет, потерю вкусовых ощущений, аутоиммунный эндометриоз, аутоиммунные заболевания глаз, аутоиммунную гемолитическую анемию, аутоиммунную гемофилию, аутоиммунный гепатит, аутоиммунный интерстициальный цистит, аутоиммунный лимфопролиферативный синдром, аутоиммунную миелопатию, аутоиммунный миокардит, аутоиммунные невропатии, аутоиммунный оофорит, аутоиммунный орхит, аутоиммунную тромбоцитопению, аутоиммунные заболевания щитовидной железы, аутоиммунную уртикарию, аутоиммунный увеит, аутоиммунный васкулит; болезнь Бехчета, паралич Белла, буллезный пемфигоид; глютеновую болезнь, синдром хронического переутомления, болезнь Крона; герпетиформный дерматит, дерматомиозит, дискоидную красную волчанку; синдром Гудпасчера, болезнь Грейвса, синдром Гийена - Барре, тиреоидит Хашимото, герпес беременных, болезнь Гентингтона, IgA нефропатию, иммунную тромбоцитопению, пурпуру, интерстициальный цистит; волчанку, болезнь Лима; синдром Миллера-Фишера, смешанное заболевание соединительной ткани; рассеянный склероз, миастению gravis; паранеопластические аутоиммунные синдромы, листовидную пузырчатку, обыкновенную пузырчатку, пернициозную анемию, болезнь Пейрони, полиэндокринный синдром, первичный биллиарный цирроз, первичный гломерулонефрит, первичный склерозирующий холангит, псориаз, псориатический артрит; энцефалит Расмуссена, рецидивирующую полихондрию, ревматоидный артрит; саркоидоз, склеродермию, синдром Шегрена, синдром Штиффа-Персона; хорею Сиденгама, симпатическую офтальмию, темпоральный артериит, диабет типа 1, язвенный колит; витилиго; гранулематоз Вегенера. Кроме того, установлена взаимосвязь между блокированием калиевых каналов Kv1.3 и метаболическими заболеваниями (см. J. Xu et al., Human Molecular Genetics 2003. Vol.12. No, 5, 551-559). Поэтому можно предположить, что соединения, которые способны блокировать калиевые каналы Kv1.3 - например, соединения формулы I, предлагаемые в настоящем изобретении или соединения, раскрытые и заявленные в опубликованной заявке WO 2005/037780 (= US 2005/0148659) - также могут быть пригодны для лечения и/или профилактики метаболических нарушений или заболеваний, таких как центральное ожирение; гипертензия, в особенности артериальная гипертензия; резистентность к инсулину, в особенности сахарный диабет типа II; непереносимость глюкозы или нарушенная непереносимость глюкозы; дислипопротеинемия, в особенности гипертриглицеридемия, сопровождающаяся дислипопротеинемией, происходящей при пониженном уровне ЛВП-холестерина (ЛВП - липопротеины высокой плотности); и гиперурикемия.
Можно ожидать благоприятных эффектов, если аминоалкиламидометилзамещенные производные 2-(4-сульфониламино)-3-гидрокси-3,4-дигидро-2H-хромен-6-ила, предлагаемые в настоящем изобретении, или амидометилзамещенные производные 2-(4-сульфониламино)-3-гидрокси-3,4-дигидро-2H-хромен-6-ила, раскрытые в WO 2005/037780, вводят в комбинации (в фиксированной комбинации или последовательно в любом порядке) по меньшей мере с одним другим обладающим сердечно-сосудистой активностью лекарственным средством, выбранным из группы, включающей:
антагонисты альфа-адренорецептора (неселективные), например, толазолин или феноксибензамин; антагонисты альфа-адренорецептора (селективные), например, доксазосин (мезилат), празосин (гидрохлорид) (и политиазид), теразосин (гидрохлорид) или урапидил;
антагонисты альфа-2-адренорецептора (включая антагонисты альфа-2-адренорецептора центрального действия), например, клонидин, гванфацин, гванабенз, метилдопа и моноксидин;
антиангинальные средства, например, бепридил, бета-блокаторы, дилтиазем, никардипин, нифедипин, нитраты; антикоагулянты, например, далтепарин, данапароид, эноксапарин, гепарин, тинзапарин, варфарин;
антитромбоцитарные лекарственные средства, например, абциксимаб, аспирин, аспирин и дипиридамол (аггренокс), цилостазол, клодопигрел, димиридамол, эптифибатид, тиклодимин, тирофибан;
противоаритмические лекарственные средства, такие как противоаритмические средства класса I, например, блокаторы натриевых каналов, дисопирамид, флекаинид, лидокаин, мексилетин, морицизин, прокаинамид, пропафенон, хинидин, токаинид; или противоаритмические средства класса II, например, бета-блокаторы, ацебутолол, атенолол, бетаксолол, бисопролол, карведиол, эсмолол, метопролол, надолол, пропранолол, сотолол, тимолол; или противоаритмические средства класса III, например, блокаторы калиевых каналов, амиодарон, азимилид, бепридил, дофетилид, ибуталид, соталол, тедисамил; или противоаритмические средства класса IV, например, блокаторы кальциевых каналов, дилтиазем, верапамил;
антагонисты бета-адренорецептора (бета-блокаторы), например, ацебутолол, алпренолол, атенолол, бетаксолол, бисопролол, бупранолол, каразолол, картеолол, цеоипролол, мепиндолол, метипранолол, метопролол, надолол, окспренолол, пенбутолол, пиндолол, пропранолол, соталол и тимолол;
средства, блокирующие кальциевые каналы (= антагонисты кальция), например, амлодипин, бепридил, фелодипин, исрадипин, никардипин, нифедипин, нилвадипин, нимодипин, нисолдипин, нитрендипин, галлопамил, верапамил, дилтиазем и фендилин;
диуретики, например, антагонисты аденозина А1, тиазидные диуретики, аналоги тиазидов, петлевые диуретики, калийсберегающие диуретики, ингибиторы карбоангидразы и/или этакриновой кислоты. Подходящие антагонисты аденозина А1 можно выбрать из группы, включающей 1,3-дипропил-8-циклопентилксантин (ДПЦПК); 4-[(2-фенил-7Н-пирроло[2,3-d]пиримидин-4-ил)амино]-trans-cyclohexanol; (4S)-4-гидрокси-1-(2-фенил-7Н-пирроло[2,3-d]пиримидин-4-ил)-L-пролинамид; 8-циклопентил-3-N-[3-((3-(4-фторсульфонил)бензоил)-окси)-пропил]-1-N-пропилксантин (ФСЦПК); BG-9928 (CAS No. 340021-17-2); СРХ (CAS No. 102146-07-6); FK-352 (CAS No. 143881-08-7); FK-453 (CAS No. 121524-18-3); FK-838 (CAS No. 131185-37-0); FR-166124 (CAS No. 171050-45-6); KW-3902 (CAS No. 136199-02-5); N-0861 ([+/-]N6-эндо-норборнан-2-ил-9-метиладенин, CAS No. 141696-90-4); WRC-0342 (CAS No. 175097-37-7); WRC-0571 (8-(N-метилизопропил)амино-N6-(5'-эндогидроксиэндонорборнил)-9-метиладенин, CAS No. 175097-35-5); наксифиллин (CAS Nos. 166374-48-7 и 166374-49-8) и их любые физиологически совместимые таутомеры, соли, сольваты, пролекарства или сложные эфиры. Подходящие тиазидные диуретики можно выбрать из группы, включающей алтиазид, беметизид, бендрофлуметиазид, бензилгидрохлортиазид, бензтиазид, бутиазид, хлортиазид, циклотиазид, цикопентиазид, этиазид, гидрохлортиазид, гидрофторметиазид, метилклотиазид, парафлутизид, политиазид, теклотиазид, трихлорметиазид и их любые физиологически совместимые таутомеры, соли, сольваты, пролекарства или сложные эфиры. Подходящие диуретики - аналоги тиазидов можно выбрать из группы, включающей хлораминофенамид, хлорталидон, клофенамин, клопамид, клорексолон, фенквизон, индапамид, мефрусид, метолазон, квинэтазон, трипамид и ксипамид. Подходящие петлевые диуретики можно выбрать из группы, включающей азосемид, буметанид, фуросемид, пиретанид, торсемид и их любые физиологически совместимые таутомеры, соли, сольваты, пролекарства или сложные эфиры. Подходящие калийсберегающие диуретики можно выбрать из группы, включающей амилорид, канреноат калия, спиронолактон, триамтерен и их любые физиологически совместимые таутомеры, соли, сольваты, пролекарства или сложные эфиры. Подходящие диуретики - ингибиторы карбоангидразы можно выбрать из группы, включающей ацетазоламид, бринзоламид, дихлорфенамид, дорзоламид, этоксзоламид, индисулам, метазоламид, зонисамид и их любые физиологически совместимые таутомеры, соли, сольваты, пролекарства или сложные эфиры; или из группы, включающей смешанные антагонисты альфа- и бета-адренорецепторов, например карведилол или лабетолол. Другие средства - аденозин, дигоксин.
Описание методик фармакологических исследований
Указанные номера примеров соответствуют номерам примеров получения, приведенных ниже.
1. Исследование in vitro способности соединений блокировать калиевые каналы Kv1.5
Способность соединений блокировать калиевые каналы Kv1.5 продемонстрирована с помощью известной модели исследования или модели, аналогичной этой модели исследования (см. W.Hu et al., J. Pharmacol. Toxicol. Methods 34 (1995) 1-7). В этой модели исследования используют клеточную линию клеток яичника китайского хомячка (= "клетки яичника китайского хомячка", "СНО"), полученные из одной клетки и стабильно экспрессирующие каналы Kv1.5. При инкубировании в течение ночи в питательной среде, содержащей RbCl, или в "нагружающем буфере" (все значения в мМ: RbCl 5, NaCl 140, CaCl2 2, MgSO4 1, буфер HEPES (Н-2-гидроксиэтилпиперазин-Н-2-этансульфоновая кислота) 10, глюкоза 5) указанные выше клетки яичника нагружаются ионами Rb+ под влиянием Na+/K+-АТФазы. Затем порцию клеток яичника, использующуюся в качестве эталонного стандарта, инкубируют при отсутствии ингибитора, а другую порцию клеток яичника инкубируют в присутствии соответствующего ингибирующего исследуемого соединения формулы I. Затем клетки яичника подвергают деполяризации путем повышения внеклеточной концентрации ионов калия, что приводит к открытию калиевых каналов Kv1.5 клеток яичника. При отсутствии ингибитора ионы Rb+ проходят через калиевые каналы Kv1.5 в окружающую среду. С другой стороны, в присутствии ингибирующего исследуемого соединения формулы I ионы Rb+ остаются заблокированными в клетках яичника. Блокирующее воздействие исследуемых соединений формулы I на калиевые каналы Kv1.5 определяют путем измерения концентрации ионов Rb+ в окружающей жидкости с помощью атомной абсорбционной спектроскопии и сопоставления с данными для эталонного стандарта.
Клетки яичника китайского хомячка (см. выше) выращивают в известной содержащей RbCl питательной среде для СНО-клеток и помещают в лунки 96-луночного планшета. Клеткам яичника дают расти в течение ночи, так чтобы образовались монослои клеток яичника. После этого сначала пипеткой удаляют всю питательную среду, а затем каждую лунку с образцом трижды промывают порциями по 100 мкл прединкубационного буфера, обладающего низкой концентрацией ионов калия (все значения в мМ: KCl 5, NaCl 140, CaCl2 2, MgSO4 1, буфер HEPES 10, глюкоза 5). Затем в каждую лунку с образцом прибавляют 50 мкл раствора соответствующего исследуемого соединения (исходный раствор в ДМСО (диметилсульфоксид), разведенный прединкубационным буфером, конечная концентрация исследуемых соединений 10 мкМ) или растворителя (в качестве отрицательных контролей) и инкубируют в течение 10 мин во всех случаях при комнатной температуре. Затем в каждую лунку с образцом прибавляют 50 мкл стимулирующего буфера, обладающего повышенной концентрацией ионов калия (KCl 145 мМ, NaCl 0 мМ, в остальном состав соответствует прединкубационному буферу) и затем образцы инкубируют в течение еще 10 мин при комнатной температуре. Затем во всех случаях из каждой лунки с образцом по 80 мкл жидкости, окружающей клетки яичника, переносят в лунки планшета для проведения анализа и определения концентрации ионов Rb+ в жидкостях с помощью атомной абсорбционной спектроскопии. Все исследуемые соединения изучают дважды. Часть сигнала, которая характеризует компонент Kv1.5 выходящего потока Rb+, определяют путем использования известного блокатора калиевых каналов 4-АР при высокой концентрации (100 × IC50 для канала Kv1.5) в качестве положительного контроля. Это делает возможным определение того, какая часть выходящего потока Rb+ зависит от влияния 4-АР, и тем самым может быть отнесена на счет канала Kv1.5. Для соединений, которые при использовании в концентрации, равной 10 мкМ, приводили к уменьшению выходящего потока Rb+ не менее, чем на 50%, проводят дополнительные исследования при меньших эффективных концентрациях исследуемых соединений и определяют эффективные концентрации, соответствующие половинному максимальному ингибированию. В каждом случае концентрации, соответствующие половинному максимальному ингибированию исследуемыми соединениями формулы I (IC50), приводят в качестве характеристической переменной.
В этой модели исследования исследуемые соединения формулы I, указанные в представленной ниже таблице 1, обладают приведенными ниже значениями IC50:
Таблица 1. Способность исследуемых соединений блокировать калиевые каналы Kv1.5 in vitro
2. Исследование in vitro способности соединений блокировать калиевые каналы Kv1.3
Способность соединений блокировать калиевые каналы Kv1.3 продемонстрирована с помощью известной модели исследования (например, Genion, Hamburg) или модели, аналогичной этой модели исследования (см. J.Plásek and K.Sigler, J. Photochem. Photobiol. 33 (1996) 101-124). В этой модели исследования используют известные клетки яичника китайского хомячка (=СНО), которые стабильно трансфицированы калиевыми каналами Kv1.3. Блокирование активности собственных калиевых каналов Kv1.3 трансфицированных клеток сопровождается положительным смещением мембранного потенциала примерно от -40 до -30 мВ, тогда как для параллельно исследованных клеток СНО дикого типа значительное смещение мембранного потенциала не происходит. Таким образом, изменение мембранного потенциала связано о снижением активности калиевых каналов Kv1.3. При блокировании калиевых каналов Kv1.3, например, соединениями формулы I происходит накопление флуоресцентного красителя, чувствительного к мембранному потенциалу, внутри клеток яичника и в конечном счете усиление флуоресценции. Таким образом, изменение мембранного потенциала клеток яичника измеряют косвенно по усилению флуоресценции красителей, чувствительных к мембранному потенциалу.
Клетки трансфицируют плазмидой Kv1.3 по известным методикам с помощью имеющегося в продаже реагента для трансфекции (DMRIE-C, выпускающегося фирмой Gibco BRL, Germany). Результативность трансфекции определяют с помощью иммунофлуоресцентного анализа и по методике фиксации потенциала потока ионов калия. Флуоресценцию исследуют с помощью устройства считывания флуоресценции Tecan Safire, выпускающегося фирмой Tecan, Germany. В каждом случае интенсивность флуоресценции, обусловленной блокированием калиевых каналов Kv1.3 в клетках яичника соединениями формулы I, при концентрации, равной 10 мкМ, определяют в качестве характеристической переменной. Во всех случаях увеличение интенсивности флуоресценции приведено в процентах (%) при сравнении с увеличением интенсивности флуоресценции, вызванной эталонным соединением маргатоксином. Маргатоксин является известным селективным блокатором калиевых каналов Kv1.3 (см., например, М. Garcia-Calvo et al., J. Biol. Chem. 268 (1993) 18866-18874).
В этой модели исследования исследуемые соединения формулы I, указанные в представленной ниже таблице 2, обладают приведенными ниже выраженными в процентах значениями:
3. Исследование in vitro функциональной эффективности воздействия соединений на предсердие крыс
Функциональная эффективность антиаритмического воздействия соединений продемонстрирована с помощью описанной ниже модели. В этой модели исследования определяют, в какой степени блокирующие Kv1.5 соединения формулы I увеличивают функциональный рефракторный период левого предсердия крыс. Рефракторный период определяют, как минимальный возможный период времени, прошедший от основного раздражения до дополнительного раздражения, по истечение которого может произойти новое сокращение. Степень увеличения функционального рефракторного периода является мерой эффективности антиаритмического воздействия соединений, предлагаемых в настоящем изобретении. Функциональный рефракторный период определяют с помощью воздействия на препарат электрическим раздражителем, устанавливая, через какой период времени, прошедший после предыдущего сокращения, повторное электрическое раздражение может вызвать новое сокращение.
Извлекают сердца у только что умерщвленных крыс (Sprague-Dawley, Charles-River, Germany). Извлекают левое предсердие и закрепляют, чтобы ввести датчики в ванне для органов, в которой поддерживают постоянную температуру (30°С) и газовую атмосферу (О2 95%, CO2 5%) и которую заполняют модифицированным раствором Тироде (все значения в мМ: NaCl 137; KCl 2,7; CaCl2 1,8; MgCl2 0,8; NaHCO3 11,9; NaH2PO4 0,6; глюкоза 5). Для инициирования регулярных сокращений на препарат воздействуют электрическими импульсами (прямоугольные импульсы с амплитудой, равной 3,5хпороговый импульс, ширина импульса 1,5 мс, частота 1 Гц). В самом начале начальное значение функционального рефракторного периода определяют путем подачи импульсов дополнительно к основному импульсу и период времени, прошедший после предыдущего основного импульса, уменьшают пока не перестает происходить дополнительное сокращение. Затем с промежутками в 20 мин последовательно прибавляют соединения формулы I в увеличивающихся концентрациях (0,1-32 мкМ) и через 18 мин после прибавления повторно определяют рефракторный период. Перед измерением готовят исходные растворы исследуемых соединений (3,2 и 0,32 мМ в 100% ДМСО). После этого для обеспечения необходимых конечных концентраций соединений (0,1-32 мкМ) в ванне для органов (объемом 25 или 100 мл) в ванну для органов наливают эти исходные растворы.
В каждом случае в качестве характеристической переменной приводят выраженное в миллисекундах увеличение функционального рефракторного периода (ФРП) для левого предсердия крыс, наблюдающееся после прибавления 10 или 32 мкМ соответствующего соединения формулы I к препаратам предсердия.
В этой модели исследования исследуемые соединения формулы I, указанные в представленной ниже таблице 3, характеризуются увеличением рефракторного периода, приведенным ниже, и более значительные показатели соответствуют большей эффективности антиаритмического воздействия:
4. Исследование in vivo функциональной эффективности воздействия соединений на сердце морских свинок
С помощью описанной ниже модели показано, что соединения, предлагаемые в настоящем изобретении, оказывают самое большее слабое проаритмическое воздействие с реполяризацией желудочка. Для этого in vivo исследовано влияние соединений формулы I на эффективный рефракторный период (ЭРП) и другие изменяющиеся характеристики сердца морской свинки. В этой модели исследования неселективные блокаторы калиевых каналов, не входящие в объем настоящего изобретения, которые также блокируют каналы HERG и/или KvLQT1, приводят к нежелательному увеличению ЭРП и временного интервала QT на электрокардиограмме (= ЭКГ). Временной интервал QT также является мерой реполяризации сердца. Увеличение ЭРП и временного интервала QT, обусловленные соединениями, независимо интерпретируются, как указания на опасность возникновения нежелательных аритмий типа трепетание - мерцание. Кроме того, в каждом случае временной интервал QRS определяют по ЭКГ путем измерения скорости распространения раздражения в желудочке. Даже увеличение временного интервала QRS, обусловленное исследуемым соединением, связывают с повышением опасности нежелательных проаритмических побочных эффектов. Поэтому в этой модели исследования отсутствие увеличения ЭРП и временного интервала QT указывает на незначительную опасность, но, с другой стороны, наличие соответствующего увеличения ЭРП и временного интервала QT указывает на повышение опасности нежелательных проаритмических побочных эффектов. Кроме того, отсутствие увеличения временного интервала QRS, обусловленное исследуемыми соединениями формулы I, указывает на незначительную опасность нежелательных проаритмических побочных эффектов, поскольку отсутствие увеличения временного интервала QRS указывает на неискаженное распространение раздражения в желудочке. Напротив, увеличение временного интервала QRS, к которому обычно приводят антиаритмические лекарственные средства класса I, указывает на уменьшение скорости прохождения раздражения и может способствовать проявлению желудочковой тахикардии или фибрилляции желудочков.
Самцов морских свинок (Dunkin-Hartley, фирмы Charles River) анестезируют (кетамин 50 мг/кг, ксилазин 10 мг/кг) и каждой из них через яремную вену вводят соединения формулы I или растворитель. Через другую яремную вену в правый желудочек морских свинок вводят катетер для биполярной стимуляции (частота стимуляции равна 5 Гц). Артериальное давление измеряют с помощью катетера, введенного в сонную артерию и соединенного с манометром Стэтхема. ЭКГ снимают с помощью игольчатых электродов. Данные измерений оцифровывают с помощью аналого-цифрового преобразователя и регистрируют с помощью компьютера и соответствующего программного обеспечения (Ponemah Physiology Platform, выпускающееся фирмой Gould, USA). После равного 45 мин периода доведения до равновесия с 12-минутными интервалами морским свинкам внутривенно (= ВВ) вводят увеличивающиеся дозы соединений формулы I или растворитель. Перед первым введением и каждый раз через 1 мин после введения увеличивающихся доз (0,1 - максимально 30 мкмоль/кг) соединений формулы I определяют эффективный рефракторный период. Для этого после 5 обычных импульсов в каждом случае подают дополнительный импульс и период времени, прошедший после предыдущего импульса, увеличивают, пока не произойдет сокращение. Наблюдающийся период времени соответствует ЭРП желудочкового миокарда.
Для изучения возможного влияния исследуемых соединений на артериальное давление в этой же модели исследования после каждого введения соединения измеряют систолическое и диастолическое артериальное давление и их сопоставляют с предыдущими значениями давления. Эти характеристики регистрируются автоматически через 1 и 8 мин после каждого введения соединения. В таблице 4 также приведены изменения систолического артериального давления, обусловленные указанными ниже соединениями формулы I (за вычетом воздействия растворителя). Ни одно из указанных соединений не привело к повышению артериального давления.
В этой модели исследования исследуемые соединения формулы I, перечисленные в представленной ниже таблице 4, приводят к указанным ниже воздействиям. Отмечены только статистически значимые воздействия, при статистической обработке использован t-критерий с пределом значимости Р<0,05. В приведенной ниже таблице 4 аббревиатура "СН" (= "статистически незначимо") показывает, что соединение соответствующего примера не приводит к статистически значимому воздействию на указанную измеряемую величину.
Особенно хорошую совместимость соединений, предлагаемых в настоящем изобретении, также можно продемонстрировать с помощью дополнительных фармакологических моделей исследования. Так, например, с помощью исследования in vitro препаратов сердечной мышцы морских свинок можно показать, что соединения формулы I приводят самое большее к небольшим антиаритмическим побочным эффектам класса I. Кроме того, с помощью модели in vitro сердца крыс и другой модели in vitro сердца морских свинок можно показать, что соединения формулы I приводят самое большее к небольшим негативным инотропным эффектам.
Соединения формулы I можно вводить в обычных фармацевтических композициях. В каждом конкретном случае можно указать специальные дозированные формы. Использующиеся дозы могут меняться в зависимости от конкретного пациента и, разумеется, будут меняться в зависимости от типа подвергающегося лечению патологического состояния и использующегося соединения. Однако обычно для введения людям и крупным млекопитающим являются подходящими лекарственные формы, содержащие от 0,2 до 500 мг, предпочтительно - от 10 до 200 мг активного вещества в разовой дозе.
Соединения, предлагаемые в настоящем изобретении, совместно с обычными фармацевтическими вспомогательными веществами и/или носителями могут содержаться в твердых или жидких фармацевтических композициях, пригодных для введения. Указанные фармацевтические композиции можно получить по обычным методикам с использованием вспомогательных веществ, таких как жидкий или твердый носитель. Типы фармацевтических композиций, которые можно использовать специалисту в данной области техники из описания и/или его общей подготовки в данной области техники.
Примерами твердых композиций являются таблетки, включая таблетки с покрытием, микротаблетки и жевательные таблетки; капсулы, включая микрокапсулы; порошки или гранулы; суппозитории или мази, включая кремы и гели. Для приготовления твердых лекарственных форм активные вещества можно, например, обычным образом смешать со вспомогательными веществами и/или носителями и можно подвергнуть мокрому или сухому гранулированию. Гранулы или порошки можно обычным образом поместить непосредственно в капсулы или спрессовать в ядра таблеток. При необходимости по известным методикам на них можно нанести покрытие.
Жидкие композиции, такие как растворы, растворы для парентерального введения, суспензии или эмульсии активных веществ могут содержать обычные разбавители, такие как воду, масла, и/или суспендирующие агенты, такие как полиэтиленгликоль и т.п. Можно дополнительно прибавить другие вспомогательные вещества, такие как консерванты, вещества, меняющие вкус, и т.п.
Фармацевтические композиции, предлагаемые в настоящем изобретении, можно вводить в твердой или жидкой форме, например энтерально, перорально, парентерально (внутримышечно или внутривенно), ректально или локально (местно). Инертными наполнителями, подходящими для таких препаратов, являются обычные фармацевтические жидкие или твердые носители, наполнители, растворители, эмульгаторы, смазывающие вещества, агенты, обеспечивающие распадаемость таблеток, ароматизаторы, красители и/или буферные вещества. Часто применяющимися вспомогательными веществами, которые можно отметить, являются карбонат магния, диоксид титана, лактоза, маннит и другие сахара или гидроксисахара, тальк, белок молока, желатин, крахмал, целлюлоза и ее производные, животные и растительные масла, такие как рыбий жир, подсолнечное, арахисовое или кунжутное масло, полиэтиленгликоль и растворители, такие как, например, стерильная вода и одно- или многоатомные спирты, такие как глицерин.
Соединения, предлагаемые в настоящем изобретении, обычно вводят в виде фармацевтических композиций, которые являются важными и новыми вариантами осуществления настоящего изобретения вследствие наличия соединений, более предпочтительно - конкретных соединений, раскрытых в настоящем изобретении. Варианты осуществления настоящего изобретения относятся к фармацевтической упаковке или набору, содержащему один или большее количество контейнеров, заполненных одним или большим количеством ингредиентов фармацевтической композиции, предлагаемой в настоящем изобретении. К такому контейнеру (контейнерам) могут прилагаться различные письменные материалы, такие как инструкции по применению или сообщения в форме, установленной правительственным агентством, регулирующим изготовление, применение или продажу фармацевтических продуктов, в котором содержатся утвержденные агентством правила изготовления, применения или продажи для использования в медицине или ветеринарии.
Приведенные ниже примеры предназначены для дополнительного разъяснения настоящего изобретения без ограничения его объема.
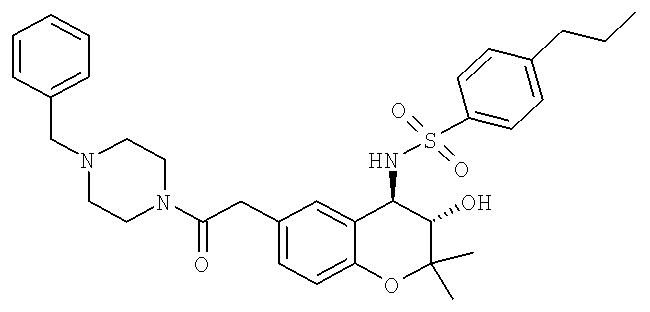
Пример 1:
(3S,4R)-N-{6-[2-(4-Бензилпиперазин-1-ил)-2-оксоэтил]-3-гидрокси-2,2-диметилхроман-4-ил}-4-н-пропилбензолсульфонамид
А) В колбу объемом 5 л помещали метил-4-гидроксифенилацетат (175,6 г), фенилбороновую кислоту (128,9 г) и м-ксилол (3,5 л). К этой смеси прибавляли 3-метилбут-2-еналь (88,9 г) и ледяную уксусную кислоту (130 мл). Полученную смесь нагревали при 140°С в атмосфере азота в аппарате Дина-Штарка. За протеканием реакции следили с помощью ВЭЖХ-NC (высокоэффективная жидкостная хроматография-масс-спектроскопия) и реакцию останавливали, когда больше было невозможно обнаружить ее протекание (примерно 72 ч). Затем реакционную смесь охлаждали до комнатной температуры, фильтровали и растворитель удаляли в вакууме. Остаток растворяли в 1:1 об./об. смеси ТГФ/гидроксид аммония и перемешивали в течение 2 ч. ТГФ удаляли в вакууме и прибавляли этилацетат. Органический слой отделяли и промывали 1 М гидроксидом натрия, рассолом, сушили над Na2SO4 и растворитель удаляли в вакууме. Неочищенный продукт (155 г) очищали с помощью флэш-хроматографии на колонке с использованием градиентного элюирования от 15:1 до 10:1 об./об. смесью гексан/этилацетат и получали 106 г метилового эфира 2,2-диметил-2Н-хромен-6-ил)уксусной кислоты. 1Н-ЯМР (δ мас.част./млн, CDCl3): 7,00 (dd, 1H, J=8,16, 2,32 Гц), 6,89 (d, 1H, J=2,32 Гц), 6,72 (d, 1H, J=8,24 Гц), 6,29 (d, 1H, J=9,80 Гц), 5,60 (d, 1H, J=9,80 Гц), 3,69 (s, 3H), 3,51 (s, 2H), 1,42 (s, 6H).
ВЭЖХ-МC (ИЭ+ (ионизация электрораспылением, положительная), 10 эВ): 233,25 (М+, 10%), 173,16 ([М-C2H3O2]+, 100%).
B) В колбу объемом 1 л помещали метиловый эфир (2,2-диметил-2Н-хромен-6-ил)уксусной кислоты (получение описано выше) (50 г), 500 мл изопропанола и Ti(OEt)4 (0,7 эквивалентов; экв.). Полученный раствор кипятили с обратным холодильником в течение 16 ч. За протеканием реакции до ее завершения следили с помощью жидкостной хроматографии/масс-спектроскопии (= ЖХМС). После вступления в реакцию всего исходного вещества (образование 5% этилового эфира) реакционную смесь охлаждали до комнатной температуры. По каплям прибавляли воду (50 мл) и растворитель удаляли в вакууме. Полученное твердое вещество отфильтровывали и промывали этилацетатом. Раствор этилацетата фильтровали через диоксид кремния и выпаривали в вакууме и получали 56 г изопропилового эфира (2,2-диметил-2Н-хромен-6-ил)уксусной кислоты, который использовали на следующей стадии без дополнительной очистки.
1Н-ЯМР (δ мас. част./млн, CDCl3): 7,00 (dd, 1H, J=8,08, 2,20 Гц), 6,89 (d, 1H, J=1,96 Гц), 6,71 (d, 1H, J=8,08 Гц), 6,28 (d, 1H, J=9,76 Гц), 5,60 (d, 1H, J=9,80 Гц), 5,00 (септет, 1H, J=6,12 Гц), 3,46 (s, 2H), 1,42 (s, 6H), 1,22 (d, 6H, 7=6,12 Гц). ВЭЖХ-NC (ИЭ+): 261,03 ([М+Н]+, 11%), 218,91 ([М-C3H7]+, 100%), 172,84 ([М-C4H7O2]+, 97%).
C) Катализатор (S,S)-(+)-N,N'-бис(3,5-ди-трет-бутилсалицилиден)-1,2-циклогександиаминомарганец(III)хлорид ("катализатор Якобсона"; 5 мол.%) и пиридин-N-оксид (0,5 экв.) прибавляли к раствору хромена (1 экв.) в дихлорметане при 0°С. К смеси прибавляли охлажденный водный раствор NaHPO4 (0,05 М) и свежеприготовленный NaOCl (0,6 М). Реакционную смесь перемешивали при 0°С в течение 6 ч. К реакционной смеси прибавляли дихлорметан и целит® и фильтровали через пористый фильтр, покрытый целитом®. Органический слой фильтрата отделяли от водного слоя, промывали рассолом, сушили над MgSO4 и выпаривали при пониженном давлении. Полученное черное масло перекристаллизовывали из смеси гептан/этилацетат (сначала прибавляли гептан, а затем этилацетат до полного растворения эпоксида). Получали изопропиловый эфир ((3S,4R)-2,2-диметил-1а,7b-дигидро-2Н-1,3-диоксациклопропа[а]нафталин-6-ил)уксусной кислоты в виде белых иголок.
ВЭЖХ-МC (ИЭ+): Rt=1,26 мин, 235,26 ([М-C3H7O]+, 100%), 277,40 (М+, 40%), 312,51 (13%), 317,49 (15%).
D) Изопропиловый эфир ((3S,4R)-2,2-диметил-1а,7b-дигидро-2Н-1,3-диоксациклопропа[а]нафталин-6-ил)уксусной кислоты, полученный выше, обрабатывали смесью EtOH:NH4OH (6:5, об./об.) и получали 0,2 М раствор эпоксида. Раствор нагревали при 50°С в течение 16 ч. После охлаждения растворитель удаляли в вакууме. Полученный неочищенный продукт можно очистить с помощью хроматографии на колонке с использованием градиентного элюирования смесью этилацетат: дихлорметан: МеОН. Получали 7,3 г чистого изопропилового эфира ((3S,4R)-4-амино-3-гидрокси-2,2-диметилхроман-6-ил)-уксусной кислоты.
1Н-ЯМР (δ мас.част./млн, CDCl3): 7,28 (s, 1Н), 7,05 (dd, 1H, J=1,72, 8,28 Гц), 6,74 (d, 1H, J=8,32 Гц), 5,00 (септет, 1H, J=6,36 Гц), 3,70 (d, 1H, J=9,76 Гц), 3,51 (s, 2H), 3,30 (d, 1H, J=9,52 Гц), 2,90 (широкий, s, 3Н), 1,47 (s, 3H), 1,23 (d, 6H, J=6,36 Гц), 1,18 (s, 3H).
ВЭЖХ-МC (ИЭ+): Rt=1,09 мин 235,29 ([М-С3Н7О]+, 100%), 263,36 (16%), 277,42 ([M-NH2]+, 22%), 294,48 (М+, 38%) ([M+Na]+, 35%).
E) Изопропиловый эфир ((3S,4R)-4-амино-3-гидрокси-2,2-диметилхроман-6-ил)-уксусной кислоты (54 г), полученный выше, растворяли в дихлорметане (10 объемов), а затем прибавляли boc-ангидрид (100 г), триэтиламин (78 мл) и ДМАП (22,5 г). Полученный раствор встряхивали в течение ночи. Растворитель удаляли в вакууме и остаток очищали с помощью хроматографии на колонке с использованием смеси гептан: этилацетат 6:1 и получали 66,7 г продукта. Изопропиловый эфир (3S,4R)-(4-трет-бутоксикарбониламино-3-гидрокси-2,2-диметилхроман-6-ил)уксусной кислоты (продукт с одной защищенной группой) и изопропиловый эфир (3S,4R)-(4-трет-бутоксикарбониламино-3-трет-бутоксикарбонилокси-2,2-диметилхроман-6-ил)уксусной кислоты (продукт с двумя защищенными группами) выделяли в виде смеси 2:1 с преобладанием продукта с одной защищенной группой.
1Н-ЯМР (δ мас.част./млн, CDCl3): 7,1 (сложный, 3Н), 6,85 (d, 1H, J=9,04 Гц), 6,75 (d, 0,5N, J=8,32 Гц), 5,0 (сложный, 2H), 4,90 (d, 1H, 11,7 Гц), 4,78 (сложный, 1H), 3,92 (d, 1H, J=11,7 Гц), 3,50 (s,), 3,49 (s,), 1,63 (s, 9H), 1,48 (сложный), 1,22 (сложный, 12Н).
ВЭЖХ-МC (ИЭ+): продукт с одной защищенной группой Rt=1,71 не интерпретирован; продукт с двумя защищенными группами Rt=1,86 не интерпретирован.
F) Смесь (66,7 г) изопропилового эфира (3S,4R)-(4-трет-бутоксикарбониламино-3-гидрокси-2,2-диметилхроман-6-ил)уксусной кислоты и изопропилового эфира (3S,4R)-(4-трет-бутоксикарбониламино-3-трет-бутоксикарбонилокси-2,2-диметилхроман-6-ил)уксусной кислоты, полученную выше, перемешивали в течение 16 ч в растворе ТГФ: Н2О (1:1, 1,4 л) и LiOH (14,7 г). За протеканием реакции следили с помощью ВЭЖХ-МC. Затем прибавляли дополнительное количество LiOH (0,26 г) и реакционную смесь встряхивали в течение еще 4 ч, пока анализ с помощью атомной абсорбционной спектроскопии с индуктивно связанной плазмой не показывал, что реакция завершилась. Раствор по каплям подкисляли с помощью 1М HCl и экстрагировали этилацетатом. Объединенные органические фазы сушили над сульфатом магния, а затем концентрировали в вакууме и получали в виде белой твердой смеси 59 г (3S,4R)-(4-трет-бутоксикарбониламино-3-гидрокси-2,2-диметилхроман-6-ил)уксусной кислоты (продукт с одной защищенной группой) и (3S,4R)-(4-трет-бутоксикарбониламино-3-трет-бутоксикарбонилокси-2,2-диметилхроман-6-ил)уксусной кислоты (продукт с двумя защищенными группами).
ВЭЖХ-МC (ИЭ+): продукт с одной защищенной группой Rt=1,13, 374,19 ([M+Na]+, 70%), 296,07 ([M-C4H8]+, 30%), 234,95 ([M-C5H10NO2]+, 55%), 146,82 (100%); продукт с двумя защищенными группами Rt=1,57, 925,46 ([2M+Na+H]+, 20%), 474,28 ([M+Na+H]+, 40%), 320,10 (50%), 232,93 (100%).
G) Смесь (4,0 г) (3S,4R)-(4-трет-бутоксикарбониламино-3-гидрокси-2,2-диметилхроман-6-ил)уксусной кислоты и (3S,4R)-(4-трет-бутоксикарбониламино-3-трет-бутоксикарбонилокси-2,2-диметилхроман-6-ил)уксусной кислоты, полученную выше, растворяли в дихлорметане (50 мл). Прибавляли ДИК (1,68 мл), ГОБТ (1,46 г) и N-бензилпиперазин (1,90 г) и реакционную смесь встряхивали при комнатной температуре в течение 16 ч. Раствор концентрировали в вакууме и полученную смесь трет-бутилового эфира (3S,4R)-{6-[2-(4-бензилпиперазин-1-ил)-2-оксоэтил]-3-гидрокси-2,2-диметилхроман-4-ил}карбаминовой кислоты (продукт с одной защищенной группой) и 6-[2-(4-бензилпиперазин-1-ил)-2-оксоэтил]-4-трет-бутоксикарбониламино-2,2-диметилхроман-3-илового эфира трет-бутилового эфира (3S,4R)-карбоновой кислоты (продукт с двумя защищенными группами) очищали с помощью хроматографии на колонке с использованием градиента растворителя от дихлорметан: этилацетат (4:1) до дихлорметан: этилацетат (1:1) и затем до этилацетат: МеОН (1:1).
ВЭЖХ-МC (ИЭ+): продукт с одной защищенной группой Rt=1,12 мин, 454,37 ([М-C4H9]+, 100%), 510,41 (М+, 26%), 532,39 ([M+Na]+, 31%); продукт с двумя защищенными группами Rt=1,52 мин, 498,34 ([М-С8Н18]+, 54%), 554,38 ([М-С4Н9]+, 100%), 610,43 (М+, 67%), 632,40 ([M+Na]+, 43%).
H) Смесь (6,0 г) трет-бутилового эфира (3S,4R)-{6-[2-(4-бензилпиперазин-1-ил)-2-оксоэтил]-3-гидрокси-2,2-диметилхроман-4-ил}карбаминовой кислоты и 6-[2-(4-бензилпиперазин-1-ил)-2-оксоэтил]-4-трет-бутоксикарбониламино-2,2-диметилхроман-3-илового эфира трет-бутилового эфира(3S,4R)-карбоновой кислоты, полученную выше, растворяли в 4 М HCl в диоксане (12,6 мл) и встряхивали в течение 16 ч при комнатной температуре. За протеканием реакции следили с помощью ВЭЖХ-МC и прибавляли дополнительное количество реагента, необходимое для завершения реакции. После завершения реакции раствор концентрировали в вакууме и остаток повторно растворяли в смеси дихлорметан: МеОН (1:1). Прибавляли АМПС (2,5 экв.) и суспензию встряхивали при комнатной температуре в течение 5 ч. Раствор фильтровали и концентрировали в вакууме и получали (3S,4R)-2-(4-амино-3-гидрокси-2,2-диметилхроман-6-ил)-1-(4-бензилпиперазин-1-ил)этанон, который использовали без дополнительной очистки. ВЭЖХ-МC (ИЭ+): Rt=0,65 мин, 819,43 ([2M+H]+, 20%), 410,28 ([М+Н]+, 50%), 393,25 ([M-NH2]+, 100%).
I) (3S,4R)-2-(4-Амино-3-гидрокси-2,2-диметилхроман-6-ил)-1-(4-бензилпиперазин-1-ил)этанон, полученный выше, (15 мг) растворяли в дихлорметане (0,6 мл). Прибавляли ПП-пиперидин (20 мг), а затем 4-пропилбензолсульфонилхлорид (1 экв.). Реакционную смесь встряхивали при комнатной температуре в течение 2 дней. Смолу отфильтровывали и прибавляли ПП-АМПС (30 мг), а также необходимое дополнительное количество дихлорметана. Реакционную смесь встряхивали при комнатной температуре в течение еще 16 ч, а затем фильтровали и концентрировали в вакууме и получали искомое соединение. ВЭЖХ-МC (ИЭ+): 592,27 ([М+Н]+, 100%)
Пример 2:
(3S,4R)-2-{4-[(4-Хлор-3-метилбензолсульфонил)-(2-этилбутиламино]-3-гидрокси-2,2-диметилхроман-6-ил}-N-[2-(1-метилпирролидин-2-ил)этил]ацетамид
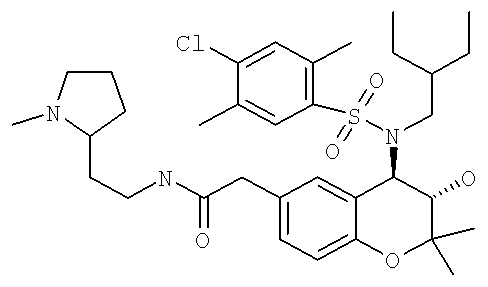
A) Смесь (4,0 г) (3S,4R)-(4-трет-бутоксикарбониламино-3-гидрокси-2,2-диметилхроман-6-ил)уксусной кислоты и (3S,4R)-(4-трет-бутоксикарбониламино-3-трет-бутоксикарбонилокси-2,2-диметилхроман-6-ил)уксусной кислоты (получение описано выше в примере 1F) растворяли в дихлорметане (50 мл). Прибавляли ДИК (1,68 мл), ГОБТ (1,46 г) и 2-(2-аминоэтил)-1-метилпирролидин (1,37 г) и реакционную смесь встряхивали при комнатной температуре в течение 16 ч. Раствор концентрировали в вакууме и полученную смесь трет-бутилового эфира (3S,4R)-(3-гидрокси-2,2-диметил-6-{[2-(1-метилпирролидин-2-ил)этилкарбамоил]метил} хроман-4-ил)карбаминовой кислоты (продукт с одной защищенной группой) и 4-трет-бутоксикарбониламино-2,2-диметил-6-{[2-(1-метилпирролидин-2-ил)этилкарбамоил]метил}хроман-3-илового эфира трет-бутилового эфира (3S,4R)-карбоновой кислоты (продукт с двумя защищенными группами) очищали с помощью хроматографии на колонке с использованием градиента растворителя от дихлорметан: этилацетат (4:1) до дихлорметан: этилацетат (1:1) и затем до этилацетат: МеОН (1:1).
ВЭЖХ-МC (ИЭ+): продукт с одной защищенной группой Rt=1,05 мин, 462,51 (М+, 100%); продукт с двумя защищенными группами Rt=1,46 мин, 562,47 (М+, 100%).
B) Смесь (6,0 г) трет-бутилового эфира (3S,4R)-(3-гидрокси-2,2-диметил-6-{[2-(1-метилпирролидин-2-ил)этилкарбамоил]метил} хроман-4-ил)карбаминовой кислоты и 4-трет-бутоксикарбониламино-2,2-диметил-6-{[2-(1-метилпирролидин-2-ил)этилкарбамоил]метил}хроман-3-илового эфира трет-бутилового эфира(3S,4R)-карбоновой кислоты, полученную выше, растворяли в 4 М HCl в диоксане (12,6 мл) и встряхивали в течение 16 ч при комнатной температуре. За протеканием реакции следили с помощью ВЭЖХ-МC и прибавляли дополнительное количество реагента, необходимое для завершения реакции. После завершения реакции раствор концентрировали в вакууме и остаток повторно растворяли в смеси дихлорметан: МеОН (1:1). Прибавляли АМПС (2,5 экв.) и суспензию встряхивали при комнатной температуре в течение 5 ч. Раствор фильтровали и концентрировали в вакууме и получали (3S,4R)-2-(4-амино-3-гидрокси-2,2-диметилхроман-6-ил)-N-[2-(1-метилпирролидин-2-ил)этил]ацетамид, который использовали без дополнительной очистки.
ВЭЖХ-МC (ИЭ+): Rt=0,72 мин, 345,49 ([M-NH2]+, 84%), 362,53 (М+, 100%), 384,53 ([M+Na]+, 15%)
C) (3S,4R)-2-(4-Амино-3-гидрокси-2,2-диметилхроман-6-ил)-N-[2-(l-метилпирролидин-2-ил)этил]ацетамид, полученный выше, растворяли в метаноле (20 мл) и прибавляли ТМОФ (0,22 мл), а затем молекулярные сита. Прибавляли 2-этилмасляный альдегид и реакционную смесь встряхивали при комнатной температуре в течение 16 ч. После завершения образования имина, подтвержденного посредством ВЭЖХ-МC и/или 1Н ЯМР, прибавляли ПП-ВН4 (5 экв.) и реакционную смесь встряхивали в течение еще 16 ч. Прибавляли дополнительное количество ПП-ВН4, необходимое для завершения реакции. Неочищенный вторичный амин растворяли в дихлорметане и последовательно прибавляли ПП-СНО (0,4 экв.) и АМПС (0,6 экв.). Реакционную смесь встряхивали при комнатной температуре в течение еще 16 ч. Затем смолу отфильтровывали, промывали с помощью ТГФ и фильтраты объединяли и концентрировали в вакууме. Неочищенный продукт очищали с помощью хроматографии на колонке с использованием градиента от дихлорметана до смеси дихлорметан: МеОН (20:80) и получали (3S,4R)-2-[4-(2-этилбутиламино)-3-гидрокси-2,2-диметилхроман-6-ил]-N-[2-(1-метилпирролидин-2-ил)этил]ацетамид.
D) (3S,4R)-2-[4-(2-Этилбутиламино)-3-гидрокси-2,2-диметилхроман-6-ил]-N-[2-(1-метилпирролидин-2-ил)этил]ацетамид, полученный выше, (15 мг) растворяли в дихлорметане (0,6 мл). Прибавляли ПП-пиперидин (20 мг), а затем раствор 4-хлор-2,5-диметилбензолсульфонилхлорида (3 экв.) в дихлорметане (0,4 мл). Реакционную смесь встряхивали при комнатной температуре в течение 2 дней. Смолу отфильтровывали и прибавляли ПП-АМПС (30 мг) и при необходимости дополнительное количество дихлорметана. Реакционную смесь встряхивали при комнатной температуре в течение еще 16 ч, а затем фильтровали и концентрировали в вакууме и получали искомое соединение.
ВЭЖХ-МC (ИЭ+): 648,53/650,53 ([М+Н]+, 100%)
Пример 3:
(3S,4R)-2-{3-Гидрокси-4-[(4-йодбензолсульфонил)-(3-метилбутил)амино]-2,2-диметилхроман-6-ил}-N-(2-пиперидин-1-илэтил)ацетамид
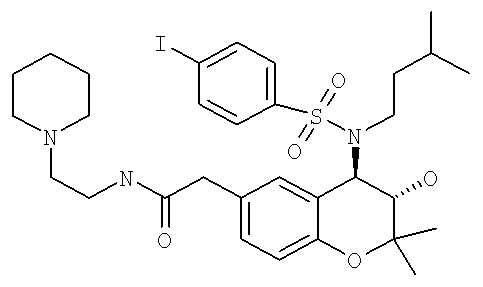
A) Смесь (4,0 г) (3S,4R)-(4-трет-бутоксикарбониламино-3-гидрокси-2,2-диметилхроман-6-ил)уксусной кислоты и (3S,4R)-(4-трет-бутоксикарбониламино-3-трет-бутоксикарбонилокси-2,2-диметилхроман-6-ил)уксусной кислоты (получение описано выше в примере 1F) растворяли в дихлорметане (50 мл). Прибавляли ДИК (1,68 мл), ГОБТ (1,46 г) и N-(2-аминоэтил)пиперидин (1,37 г) и реакционную смесь встряхивали при комнатной температуре в течение 16 ч. Раствор концентрировали в вакууме и неочищенные продукты очищали с помощью хроматографии на колонке с использованием градиента растворителя от дихлорметан: этилацетат (4:1) до дихлорметан: этилацетат (1:1) для удаления реагентов и побочных продуктов, а затем использовали смесь этилацетат: МеОН (1:1) для элюирования смеси трет-бутилового эфира (3S,4R)-{3-гидрокси-2,2-диметил-6-[(2-пиперидин-1-илэтилкарбамоил)метил]хроман-4-ил}карбаминовой кислоты (продукт с одной защищенной группой) и 4-трет-бутоксикарбониламино-2,2-диметил-6-[(2-пиперидин-1-илэтилкарбамоил)метил]хроман-3-илового эфира трет-бутилового эфира (3S,4R)-карбоновой кислоты (продукт с двумя защищенными группами). ВЭЖХ-МC (ИЭ+): продукт с одной защищенной группой Rt=1,03 мин, 406,40 ([М-С4Н9]+, 30%), 462,49 (М+, 100%), 484,46 ([M+Na]+, 14%); продукт с двумя защищенными группами Rt=1,44 мин, 506,46 ([М-С4Н9]+, 14%), 562,50 (М+, 100%), 584,47 ([M+Na]+, 15%).
B) Смесь (6,0 г) трет-бутилового эфира (3S,4R)-{3-гидрокси-2,2-диметил-6-[(2-пиперидин-1-илэтилкарбамоил)метил]хроман-4-ил}карбаминовой кислоты и 4-трет-бутоксикарбониламино-2,2-диметил-6-[(2-пиперидин-1-илэтилкарбамоил)метил]хроман-3-илового эфира трет-бутилового эфира (3S,4R)-карбоновой кислоты, полученную выше, растворяли в 4 М HCl в диоксане (12,6 мл) и встряхивали в течение 16 ч при комнатной температуре. За протеканием реакции следили с помощью ВЭЖХ-МC и прибавляли дополнительное количество реагента, необходимое для завершения реакции. После завершения реакции раствор концентрировали в вакууме и остаток повторно растворяли в смеси дихлорметан: МеОН (1:1). Прибавляли АМПС (2,5 экв.) и суспензию встряхивали при комнатной температуре в течение 5 ч. Раствор фильтровали и концентрировали в вакууме и получали (3S,4R)-2-(4-амино-3-гидрокси-2,2-диметилхроман-6-ил)-N-(2-пиперидин-1-илэтил)ацетамид, который использовали без дополнительной очистки. ВЭЖХ-МC (ИЭ+): Rt=0,75 мин, 361,57 ([M-NH2]+, 41%), 378,61 (M+, 100%)
C) (3S,4R)-2-(4-Амино-3-гидрокси-2,2-диметилхроман-6-ил)-N-(2-пиперидин-1-илэтил)ацетамид (1 экв., 2 ммоля), полученный выше, растворяли в метаноле (20 мл) и прибавляли ТМОФ (0,22 мл), а затем молекулярные сита. Прибавляли изовалериановый альдегид и реакционную смесь встряхивали при комнатной температуре в течение 16 ч. После завершения образования имина, подтвержденного посредством ВЭЖХ-МC и 1Н-ЯМР, прибавляли ПП-ВН4 (5 экв.) и реакционную смесь встряхивали в течение еще 16 ч. Прибавляли дополнительное количество ПП-ВН4, необходимое для завершения реакции. Неочищенный вторичный амин растворяли в дихлорметане и последовательно прибавляли ПП-СНО (0,4 экв.) и АМПС (0,6 экв.). Реакционную смесь встряхивали при комнатной температуре в течение еще 16 ч. Затем смолу отфильтровывали, промывали с помощью ТГФ и фильтраты объединяли и концентрировали в вакууме. Остаток очищали с помощью хроматографии на колонке с использованием градиента от дихлорметана до дихлорметан: МеОН (20:80) и получали (3S,4R)-2-[3-гидрокси-2,2-диметил-4-(3-метилбутиламино)хроман-6-ил]-N-(2-пиперидин-1-илэтил)ацетамид.
D) (3S,4R)-2-[3-Гидрокси-2,2-диметил-4-(3-метилбутиламино)хроман-6-ил]-N-(2-пиперидин-1-илэтил)ацетамид (15 мг), полученный выше, растворяли в дихлорметане (0,6 мл). Прибавляли ПП-пиперидин (20 мг), а затем раствор 4-йодбензолсульфонилхлорида (3 экв.) в дихлорметане (0,4 мл). Реакционную смесь встряхивали при комнатной температуре в течение 2 дней. Смолу отфильтровывали и прибавляли ПП-АМПС (30 мг) и при необходимости дополнительное количество дихлорметана. Реакционную смесь встряхивали при комнатной температуре в течение еще 16 ч, а затем фильтровали и концентрировали в вакууме и получали искомое соединение.
Пример 4:
(3S,4R)-N-(1-Бензилпирролидин-3R-ил)-2-{4-[(2-этилбутил)-(3-метоксибензолсульфонил)амино]-3-гидрокси-2,2-диметилхроман-6-ил}ацетамид
A) Смесь (4,0 г) (3S,4R)-(4-трет-бутоксикарбониламино-3-гидрокси-2,2-диметилхроман-6-ил)уксусной кислоты и (3S,4R)-(4-трет-бутоксикарбониламино-3-трет-бутоксикарбонилокси-2,2-диметилхроман-6-ил)уксусной кислоты растворяли в дихлорметане (50 мл). Прибавляли ДИК (1,68 мл), ГОБТ (1,46 г) и (3R)-(-)-1-бензил-3-аминопирролидин (1,89 г) и реакционную смесь встряхивали при комнатной температуре в течение 16 ч. Раствор концентрировали в вакууме и неочищенные продукты очищали с помощью хроматографии на колонке с использованием градиента растворителя от дихлорметан: этилацетат (4:1) до дихлорметан: этилацетат (1:1) и затем до этилацетат: МеОН (1:1) для элюирования смеси трет-бутилового эфира (3S,4R)-{6-[(1-бензилпирролидин-3R-илкарбамоил)метил]-3-гидрокси-2,2-диметилхроман-4-ил}карбаминовой кислоты (продукт с одной защищенной группой) и 6-[(1-бензилпирролидин-3R-илкарбамоил)метил]-4-трет-бутоксикарбониламино-2,2-диметилхроман-3-илового эфира трет-бутилового эфира (3S,4R)-карбоновой кислоты (продукт с двумя защищенными группами). ВЭЖХ-МC (ИЭ+): продукт с одной защищенной группой Rt=1,13 мин, 454,45([М-С4Н9]+, 63%), 510,51 (М+, 100%), 532,49 ([M+Na]+, 46%); продукт с двумя защищенными группами Rt=1,52 мин, 554,49 ([М-С4Н9]+, 32%), 610,54 (М+, 100%), 632,50 ([M+Na]+, 16%).
B) Смесь (6,0 г) трет-бутилового эфира (3S,4R)-{6-[(1-бензилпирролидин-3R-илкарбамоил)метил]-3-гидрокси-2,2-диметилхроман-4-ил}карбаминовой кислоты и 6-[(1-бензилпирролидин-3R-илкарбамоил)метил]-4-трет-бутоксикарбониламино-2,2-диметилхроман-3-илового эфира трет-бутилового эфира (3S,4R)-карбоновой кислоты, полученную выше, растворяли в 4 М HCl в диоксане (12,6 мл) и встряхивали в течение 16 ч при комнатной температуре. За протеканием реакции следили с помощью ВЭЖХ-МC и прибавляли дополнительное количество реагента, необходимое для завершения реакции. После завершения реакции раствор концентрировали в вакууме и остаток повторно растворяли в смеси дихлорметан: МеОН (1:1). Прибавляли АМПС (2,5 экв.) и суспензию встряхивали при комнатной температуре в течение 5 ч. Раствор фильтровали и концентрировали в вакууме и получали (3S,4R)-2-(4-амино-3-гидрокси-2,2-диметилхроман-6-ил)-N-1-бензилпирролидин-3R-ил)ацетамид, который использовали без дополнительной очистки.
ВЭЖХ-МC (ИЭ+): Rt=0,67 мин, 361,56 ([М-ОН]+, 100%), 378,59 (М+, 72%), 400,60 ([M+Na]+, 35%).
C) (3S,4R)-2-(4-Амино-3-гидрокси-2,2-диметилхроман-6-ил)-N-1-бензилпирролидин-3R-ил) ацетамид растворяли в метаноле (20 мл) и прибавляли ТМОФ (0,22 мл), а затем молекулярные сита. Прибавляли 2-этилмасляный и реакционную смесь встряхивали при комнатной температуре в течение 16 ч. После завершения образования имина, подтвержденного посредством ВЭЖХ-МC и 1Н-ЯМР, прибавляли ПП-BH4 (5 экв.) и реакционную смесь встряхивали в течение еще 16 ч. Прибавляли дополнительное количество ПП-ВН4, необходимое для завершения реакции. Неочищенный вторичный амин растворяли в дихлорметане и последовательно прибавляли ПП-СНО (0,4 экв.) и АМПС (0,6 экв.). Реакционную смесь встряхивали при комнатной температуре в течение еще 16 ч. Затем смолу отфильтровывали, промывали с помощью ТГФ и фильтраты объединяли и концентрировали в вакууме. Остаток очищали с помощью хроматографии на колонке с использованием градиента от дихлорметана до дихлорметан: МеОН (20:80) и получали (3S,4R)-N-(1-бензилпирролидин-3R-ил)-2-[4-(2-этилбутиламино)-3-гидрокси-2,2-диметилхроман-6-ил]ацетамид.
D) (3S,4R)-N-(l-Бензилпирролидин-3-ил)-2-[4-(2-этилбутиламино)-3-гидрокси-2,2-диметилхроман-6-ил]ацетамид (15 мг), полученный выше, растворяли в дихлорметане (0,6 мл). Прибавляли ПП-пиперидин (20 мг), а затем раствор 4-йодбензолсульфонилхлорида (3 экв.) в дихлорметане (0,4 мл). Реакционную смесь встряхивали при комнатной температуре в течение 2 дней. Смолу отфильтровывали и прибавляли ПП-АМПС (30 мг), а также необходимое дополнительное количество дихлорметана. Реакционную смесь встряхивали при комнатной температуре в течение еще 16 ч, а затем фильтровали и концентрировали в вакууме и получали искомое соединение.
ВЭЖХ-МC (ИЭ+): 664,73 ([М+Н]+, 100%)
Пример 5:
(3S,4R)-N-[2-(Бутилэтиламино)этил]-2-{4-[(2-этилбутил)-(4-йодбензолсульфонил)амино]-3-гидрокси-2,2-диметилхроман-6-ил)ацетамид
A) Смесь (4,0 г) (3S,4R)-(4-трет-бутоксикарбониламино-3-гидрокси-2,2-диметилхроман-6-ил)уксусной кислоты и (3S,4R)-(4-трет-бутоксикарбониламино-3-трет-бутоксикарбонилокси-2,2-диметилхроман-6-ил)уксусной кислоты (получение описано выше в примере 1F) растворяли в дихлорметане (50 мл). ДИК (1,68 мл), ГОБТ (1,46 г) и прибавляли 2-(этил-N-бутиламино)этиламин (1,55 г) и реакционную смесь встряхивали при комнатной температуре в течение 16 ч. Раствор концентрировали в вакууме и остаток очищали с помощью хроматографии на колонке с использованием градиента растворителя от дихлорметан: этилацетат (4:1) до дихлорметан: этилацетат (1:1) и затем до этилацетат: МеОН (1:1) для элюирования смеси трет-бутилового эфира (3S,4R)-(6-{[2-(бутилэтиламино)этилкарбонил]метил}-3-гидрокси-2,2-диметилхроман-4-ил)карбаминовой кислоты (продукт с одной защищенной группой) и 4-трет-бутоксикарбониламино-6-{[2-(бутилэтиламино)этилкарбамоил]метил}-2,2-диметилхроман-3-илового эфира трет-бутилового эфира (3S,4R)-карбоновой кислоты (продукт с двумя защищенными группами).
ВЭЖХ-МC (ИЭ+): продукт с одной защищенной группой Rt=1,12 мин, 478,58 (М+, 100%); продукт с двумя защищенными группами Rt=1,53 мин, 578,56 (М+, 100%).
B) Смесь (6,0 г) трет-бутилового эфира (3S,4R)-(6-{[2-(бутилэтиламино)этилкарбонил]метил}-3-гидрокси-2,2-диметилхроман-4-ил)карбаминовой кислоты и 4-трет-бутоксикарбониламино-6-{[2-(бутилэтиламино)этилкарбамоил]метил}-2,2-диметилхроман-3-илового эфира трет-бутилового эфира (3S,4R)-карбоновой кислоты, полученную выше, растворяли в 4 М HCl в диоксане (12,6 мл) и встряхивали в течение 16 ч при комнатной температуре. За протеканием реакции следили с помощью ВЭЖХ-МC и прибавляли дополнительное количество реагента, необходимое для завершения реакции. После завершения реакции раствор концентрировали в вакууме и остаток повторно растворяли в смеси дихлорметан: МеОН (1:1). Прибавляли АМПС (2,5 экв.) и суспензию встряхивали при комнатной температуре в течение 5 ч. Раствор фильтровали и концентрировали в вакууме и получали (3S,4R)-2-(4-амино-3-гидрокси-2,2-диметилхроман-6-ил)-N-[2-(бутилэтиламино)этил]ацетамид, который использовали без дополнительной очистки.
ВЭЖХ-МC (ИЭ+): Rt=0,84 мин, 407,43 ([М-ОН]+, 100%), 424,47 (М+, 44%).
C) (3S,4R)-2-(4-Амино-3-гидрокси-2,2-диметилхроман-6-ил)-N-[2-(бутилэтиламино)этил]ацетамид, полученный выше, растворяли в метаноле (20 мл) и прибавляли ТМОФ (0,22 мл), а затем молекулярные сита. Прибавляли 2-этилмасляный альдегид и реакционную смесь встряхивали при комнатной температуре в течение 16 ч. После завершения образования имина, подтвержденного посредством ВЭЖХ-МC и 1Н-ЯМР, прибавляли ПП-ВН4 (5 экв.) и реакционную смесь встряхивали в течение еще 16 ч. Прибавляли дополнительное количество ПП-ВН4, необходимое для завершения реакции. Неочищенный вторичный амин растворяли в дихлорметане и последовательно прибавляли ПП-СНО (0,4 экв.) и АМПС (0,6 экв.). Реакционную смесь встряхивали при комнатной температуре в течение еще 16 ч. Затем смолу отфильтровывали, промывали с помощью ТГФ и фильтраты объединяли и концентрировали в вакууме. Остаток очищали с помощью хроматографии на колонке с использованием градиента от дихлорметана до дихлорметан: МеОН (20:80) и получали (3S,4R)-N-[2-(бутилэтиламино)-этил]-2-[4-(2-этилбутиламино)-3-гидрокси-2,2-диметилхроман-6-ил]ацетамид.
D) (3S,4R)-N-[2-(Бутилэтиламино)-этил]-2-[4-(2-этилбутиламино)-3-гидрокси-2,2-диметилхроман-6-ил]ацетамид (15 мг), полученный выше, растворяли в дихлорметане (0,6 мл). Прибавляли ПП-пиперидин (20 мг), а затем раствор 4-йодбензолсульфонилхлорида (3 экв.) в дихлорметане (0,4 мл). Реакционную смесь встряхивали при комнатной температуре в течение 2 дней. Смолу отфильтровывали и прибавляли ПП-АМПС (30 мг), а также необходимое дополнительное количество дихлорметана. Реакционную смесь встряхивали при комнатной температуре в течение еще 16 ч, а затем фильтровали и концентрировали в вакууме и получали искомое соединение.
ВЭЖХ-МC (ИЭ+): 728,68 ([М+Н]+, 100%).
Пример 6:
(3S,4R)-N-(4-Бензилморфолин-2-илметил)-2-{4-[(3-метоксибензолсульфонил)-3-метилбутил)амино]-3-гидрокси-2,2-диметилхроман-6-у}ацетамид
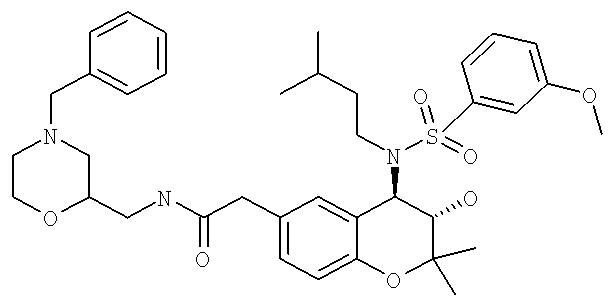
A) Смесь (4,0 г) (3S,4R)-(4-трет-бутоксикарбониламино-3-гидрокси-2,2-диметилхроман-6-ил)уксусной кислоты и (3S,4R)-(4-трет-бутоксикарбониламино-3-трет-бутоксикарбонилокси-2,2-диметилхроман-6-ил)уксусной кислоты (получение описано выше в примере 1F) растворяли в дихлорметане (50 мл). Прибавляли ДИК (1,68 мл), ГОБТ (1,46 г) и N-бензил-3-аминометилморфолин (2,21 г) и реакционную смесь встряхивали при комнатной температуре в течение 16 ч. Раствор концентрировали в вакууме и остаток очищали с помощью хроматографии на колонке с использованием градиента растворителя от дихлорметан: этилацетат (4:1) до дихлорметан: этилацетат (1:1) и затем до этилацетат: МеОН (1:1) для элюирования смеси трет-бутилового эфира (3S,4R)-(6-{[(4-бензилморфолин-2-илметил)карбамоил]метил}-3-гидрокси-2,2-диметилхроман-4-ил)карбаминовой кислоты (продукт с одной защищенной группой) и 6-{[(4-бензилморфолин-2-илметил)карбамоил]метил}-4-трет-бутоксикарбониламино-2,2-диметилхроман-3-илового эфира трет-бутилового эфира (3S,4R)-карбоновой кислоты (продукт с двумя защищенными группами).
ВЭЖХ-МC (ИЭ+): продукт с одной защищенной группой Rt=1,15 мин, 484,38 ([М-С4Н9]+, 17%), 540,37 (М+, 100%), 562,34 ([M+Na]+, 12%); продукт с двумя защищенными группами Rt=1,51 мин, 584,33 ([М-С4Н9]+, 8%), 640,43 (М+, 100%), 662,40 ([M+Na]+, 10%).
B) Смесь (6,0 г) трет-бутилового эфира (3S,4R)-(6-{[(4-бензилморфолин-2-илметил)карбамоил]метил}-3-гидрокси-2,2-диметилхроман-4-ил)карбаминовой кислоты и 6-{[(4-бензил morpholin-2-илметил) карбамоил]метил}-4-трет-бутоксикарбониламино-2,2-диметил хроман-3-илового эфира трет-бутилового эфира (3S,4R)-карбоновой кислоты, полученную выше, растворяли в 4 М HCl в диоксане (12,6 мл) и встряхивали в течение 16 ч при комнатной температуре. За протеканием реакции следили с помощью ВЭЖХ-МC и прибавляли дополнительное количество реагента, необходимое для завершения реакции. После завершения реакции раствор концентрировали в вакууме и остаток повторно растворяли в смеси дихлорметан: МеОН (1:1). Прибавляли АМПС (2,5 экв.) и суспензию встряхивали при комнатной температуре в течение 5 ч. Раствор фильтровали и концентрировали в вакууме и получали (3S,4R)-2-(4-амино-3-гидрокси-2,2-диметилхроман-6-ил)-N-(4-бензилморфолин-2-илметил)ацетамид, который использовали без дополнительной очистки.
ВЭЖХ-МC (ИЭ+): Rt=0,84 мин, 423,59 ([М-ОН]+, 100%), 440,62 (М+, 18%), 462,61 ([M+Na]+, 16%).
C) (3S,4R)-2-(4-Амино-3-гидрокси-2,2-диметилзхроман-6-ил)-N-(4-бензилморфолин-2-илметил) ацетамид (1 экв., 2 ммоля), полученный выше, растворяли в метаноле (20 мл) и ТМОФ (0,22 мл) прибавляли, а затем молекулярные сита. Прибавляли изовалериановый альдегид (1,0 экв., 2 ммоля) и реакционную смесь встряхивали при комнатной температуре в течение 16 ч. После завершения образования имина, подтвержденного посредством ВЭЖХ-МC и/или 1Н-ЯМР, прибавляли ПП-ВН4 (5 экв.) и реакционную смесь встряхивали в течение еще 16 ч. Если реакция не завершилась, то можно прибавить дополнительное количество ПП-ВН4. Неочищенный вторичный амин растворяли в дихлорметане и последовательно прибавляли ПП-СНО (0,4 экв.) и АМПС (0,6 экв.). Реакционную смесь встряхивали при комнатной температуре в течение еще 16 ч. Затем смолу отфильтровывали, промывали с помощью ТГФ и фильтраты объединяли и концентрировали в вакууме. Остаток очищали с помощью хроматографии на колонке с использованием градиента от дихлорметана до дихлорметан: МеОН (20:80) и получали (3S,4R)-N-(4-бензилморфолин-2-илметил)-2-[4-(3-метилбутиламино)-3-гидрокси-2,2-диметилхроман-6-ил]ацетамид.
D) (3S,4R)-N-(4-Бензилморфолин-2-илметил)-2-[4-(3-метилбутиламино)-3-гидрокси-2,2-диметилхроман-6-ил]ацетамид (15 мг), полученный выше, растворяли в дихлорметане (0,6 мл). Прибавляли ПП-пиперидин (20 мг), а затем раствор 3-метоксибензолсульфонилхлорида (3 экв.) в дихлорметане (0,4 мл). Реакционную смесь встряхивали при комнатной температуре в течение 2 дней. Смолу отфильтровывали и прибавляли ПП-АМПС (30 мг), а также необходимое дополнительное количество дихлорметана. Реакционную смесь встряхивали при комнатной температуре в течение еще 16 ч, а затем фильтровали и концентрировали в вакууме и получали искомое соединение.
ВЭЖХ-МC (ИЭ+): 702,56 ([M+Na]+, 21%), 680,57 ([M+H]+, 100%), 256,27 ([M-C25H31N2O4]+, 40%).
Пример 7:
(3S,4R)-N-{6-[2-(4-Бензилпиперазин-1-ил)-2-оксоэтил]-3-гидрокси-2,2-диметилхроман-4-ил}-N-циклопропилметил-4-метилбензолсульфонамид
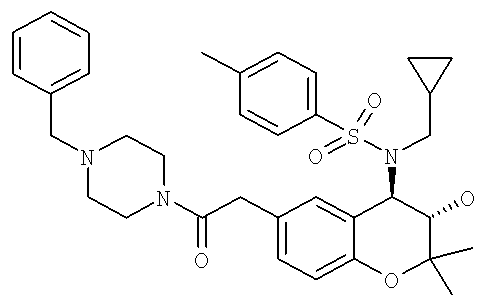
A) (3S,4R)-2-(4-Амино-3-гидрокси-2,2-диметилхроман-6-ил)-1-(4-бензилпиперазин-1-ил)этанон (получение описано выше в примере 1Н) растворяли в метаноле (20 мл) и прибавляли ТМОФ (0,22 мл), а затем молекулярные сита. Прибавляли циклопропанкарбоксальдегид и реакционную смесь встряхивали при комнатной температуре в течение 16 ч. После завершения образования имина, подтвержденного посредством ВЭЖХ-МC и 1Н-ЯМР, прибавляли ПП-ВН4 (5 экв.) и реакционную смесь встряхивали в течение еще 16 ч. Прибавляли дополнительное количество ПП-ВН4, необходимое для завершения реакции. Неочищенный вторичный амин растворяли в дихлорметане и последовательно прибавляли ПП-СНО (0,4 экв.) и АМПС (0,6 экв.). Реакционную смесь встряхивали при комнатной температуре в течение еще 16 ч. Затем смолу отфильтровывали, промывали с помощью ТГФ и фильтраты объединяли и концентрировали в вакууме. Остаток очищали с помощью хроматографии на колонке с использованием градиента от дихлорметана до дихлорметан: МеОН (20:80) и получали (3S,4R)-1-(4-бензилпиперазин-1-ил)-2-[4-(циклопропилметиламино)-3-гидрокси-2,2-диметилхроман-6-ил]этанон.
B) (3S,4R)-1-(4-Бензилпиперазин-1-ил)-2-[4-(циклопропилметиламино)-3-гидрокси-2,2-диметилхроман-6-ил]этанон, полученный выше, (15 мг) растворяли в дихлорметане (0,6 мл). Прибавляли ПП-пиперидин (20 мг), а затем раствор 4-метилбензолсульфонилхлорида (3 экв.) в дихлорметане (0,4 мл). Реакционную смесь встряхивали при комнатной температуре в течение 2 дней. Смолу отфильтровывали и прибавляли ПП-АМПС (30 мг), а также необходимое дополнительное количество дихлорметана. Реакционную смесь встряхивали при комнатной температуре в течение еще 16 ч, а затем фильтровали и концентрировали в вакууме и получали искомое соединение.
ВЭЖХ-МC (ИЭ+): 618,65 ([М+Н]+, 100%).
Пример 8:
4-Этил-N-((3S,4R)-3-гидрокси-2,2-диметил-6-{2-оксо-2-[4-(пиридин-3-илметил)пиперазин-1-ил]этил}-3,4-дигидро-2Н-хромен-4-ил)бензолсульфонамид
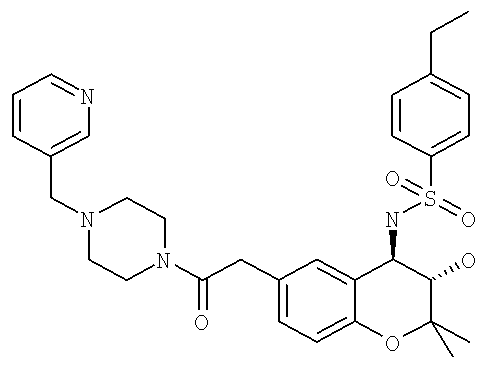
A) Метил-4-гидроксифенилацетат(25,0 г), 3,3-диметилакролеин (14,5 мл) и фенилбороновую кислоту (18,3 г) кипятили с обратным холодильником в течение 7 ч в 1,0 л безводного толуола. Затем прибавляли ледяную уксусную кислоту (60 мл) и полученную смесь кипятили с обратным холодильником в течение еще 7 ч и за протеканием реакции следили с помощью тонкослойной хроматографии (= ТСХ). Затем смесь охлаждали, в основном выпаривали в вакууме и остаток выливали в 300 мл 1:1 смеси этилацетат/вода. Значение рН доводили до 5 карбонатом натрия и содержащий этилацетат слой отделяли и концентрировали в вакууме. Хроматография остатка на колонке (подвижная фаза: петролейный эфир/этилацетат 10:1) давала 16 г метил (2,2-диметил-2Н-хромен-6-ил)ацетата в виде бледно-желтого масла.
B) Метил (2,2-диметил-2Н-хромен-6-ил)ацетат (18,3 г) суспендировали в 125 мл этилового спирта. Прибавляли 150 мл 15% раствора гидроксида натрия и смесь перемешивали в течение 30 мин при комнатной температуре. Затем прибавляли 300 мл воды и 150 мл этилацетата и полученную смесь энергично перемешивали в течение 10 мин. Органический слой отделяли отбрасывали. Щелочной водный слой один раз промывали с помощью 100 мл этилацетата. Слои разделяли и водный слой подкисляли до рН 2,0 водным раствором хлористоводородной кислоты. Затем прибавляли 200 мл этилацетата и полученную смесь энергично перемешивали в течение 10 мин. Органический слой отделяли, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Желтый остаток охлаждали и затем прибавляли петролейный эфир и перемешивали в течение 30 мин. Полученные кристаллы отфильтровывали и сушили в вакууме при 50°С и получали 6,2 г (2,2-диметил-2Н-хромен-6-ил)уксусной кислоты. Маточный раствор концентрировали в вакууме и после охлаждения повторно прибавляли петролейный эфир. Полученные кристаллы сушили в вакууме и получали еще 2,9 г (2,2-диметил-2H-хромен-6-ил)уксусной кислоты.
C) (2,2-Диметил-2H-хромен-6-ил)уксусную кислоту, полученную выше, (31 г, суммарный выход для нескольких партий) растворяли в дихлорметане (450 мл) и прибавляли концентрированную H2SO4 (1,5 мл). К этому раствору при -10°С прибавляли 2-метилпропен (21,0 г) и затем реакционную смесь перемешивали в течение 6 ч при комнатной температуре. Затем прибавляли воду (500 мл) и смесь перемешивали в течение 10 мин. Органический слой экстрагировали водным раствором NaHCO3, промывали рассолом, сушили над Na2SO4, фильтровали и выпаривали. Сушка остатка в вакууме давала трет-бутил(2,2-диметил-2Н-хромен-6-ил)ацетат (30 г) в виде коричневого масла.
D) Трет-бутил(2,2-диметил-2Н-хромен-6-ил)ацетат, полученный выше, (30,0 г) растворяли в дихлорметане (600 мл) и прибавляли (S,S)-(+)-N,N'-Бис-(3,5-ди-трет-бутилсалицилиден)-1,2-циклогександиаминомарганец(III)хлорид (4,25 г) и пиридин-N-оксид (5,25 г). При охлаждении льдом в течение 45 мин прибавляли имеющийся в продаже водный раствор NaOCl (555 мл; выпускающийся фирмой Fluka; по данным анализа ~10% при комнатной температуре) и 9% водный раствор Na2HPO4 (75 мл). Затем полученную смесь перемешивали в течение 4 ч при 0°С. Органический слой фильтровали (целит® типа 503), промывали дихлорметаном и объединенные органические слои сушили над Na2SO4. Фильтрование и выпаривание в вакууме давало коричневое масло, которое растворяли в диэтиловом эфире. Прибавляли лигроин, пока не начиналась кристаллизация. Кристаллы отфильтровывали отсасыванием и сушили и получали трет-бутил-[(1aS,7bS)-2,2-диметил-1а,7b-дигидро-2Н-оксирено[с]хромен-6-ил]ацетат (18,4 г).
Е) Трет-бутил-[(1aS,7bS)-2,2-диметил-1а,7b-дигидро-2Н-оксирено[с]хромен-6-ил]ацетат (18,4 г), полученный выше, растворяли в этаноле (300 мл). Концентрированный NH4OH (300 мл) прибавляли к этому раствору и полученную смесь перемешивали в течение 1 ч и затем выдерживали в течение ночи при комнатной температуре. Прибавляли дихлорметан (400 мл) и перемешивание продолжали в течение еще 15 мин. Органический слой сушили над Na2SO4, фильтровали и в основном выпаривали в вакууме и получали неочищенное масло. Масло растворяли в диэтиловом эфире, экстрагировали водой и органический слой сушили над Na2SO4. Фильтрование, выпаривание и сушка давали трет-бутил-[(3S,4R)-4-амино-3-гидрокси-2,2-диметил-3,4-дигидро-2Н-хромен-6-ил]ацетат (16,3 г) в виде коричневого масла.
F) К раствору трет-бутил-[(3S,4R)-4-амино-3-гидрокси-2,2-диметил-3,4-дигидро-2Н-хромен-6-ил]ацетата (12,0 г), полученному выше, в дихлорметане (280 мл) сначала прибавляли триэтиламин (10,8 мл) и затем 4-этилсульфонилхлорид (80 г). Полученную суспензию перемешивали в течение 5 ч при комнатной температуре, а затем экстрагировали водой. Органический слой промывали водным раствором NaHCO3, сушили над Na2SO4, фильтровали и в заключение выпаривали в вакууме и получали трет-бутил-((3S,4R)-4-{[(4-этилфенил)сульфонил]амино}-3-гидрокси-2,2-диметил-3,4-дигидро-2H-хромен-6-ил)ацетат (19,6 г) в виде коричневого масла. Для дополнительной очистки 1,8 г маслообразного продукта хроматографировали (жидкостная хроматография среднего давления, ЖХСД; стационарная фаза Sili Tech® (32-63, 60 А), подвижная фаза циклогексан/этилацетат 3:1).
G) К раствору трет-бутил ((3S,4R)-4-{[(4-этилфенил)сульфонил]амино}-3-гидрокси-2,2-диметил-3,4-дигидро-2H-хромен-6-ил)ацетата (17,2 г), полученного выше, в толуоле (170 мл) прибавляли трифторуксусную кислоту (17 мл). Реакционную смесь перемешивали в течение 5,5 ч при 40°С, а затем экстрагировали водой (200 мл). Органический слой экстрагировали водным раствором Na2CO3. Значение рН 6 водного слоя доводили до (HCl) и затем дважды экстрагировали этилацетатом. Объединенные органические слои сушили над Na2SO4, фильтровали и выпаривали в вакууме и получали неочищенное коричневое масло. Масло растворяли в диэтиловом эфире и прибавляли лигроин. Смесь перемешивали при комнатной температуре до завершения кристаллизации. Полученные кристаллы отсасывали и сушили и получали ((3S,4R)-4-{[(4-этилфенил)сульфонил]амино}-3-гидрокси-2,2-диметил-3,4-дигидро-2Н-хромен-6-ил)уксусную кислоту (9,3 г).
H) К раствору 1,3 г ((3S,4R)-4-{[(4-этилфенил)сульфонил]амино}-3-гидрокси-2,2-диметил-3,4-дигидро-2Н-хромен-6-ил)уксусной кислоты в 45 мл ТГФ в круглодонной колбе объемом 250 мл прибавляли 550 мг КДИ. Суспензию перемешивали при комнатной температуре в течение 0,5 ч. Прибавляли 600 мг 3-пиридилметилпиперазина и перемешивали в течение 4 ч. Затем смесь выдерживали в течение ночи при комнатной температуре. На следующий день смесь выпаривали досуха и растворяли в смеси 2:1 этилацетат/Н2О. Раствор экстрагировали водным раствором гидроксида натрия при рН 10 и затем водным раствором HCl при рН 1. Значение рН слоя, содержащего HCl, доводили до 8 водным раствором NaOH и экстрагировали этилацетатом. Органический слой сушили над Na2SO4, фильтровали и выпаривали в вакууме и получали 0,85 г желтого вспененного вещества. Вспененное вещество растворяли в изопропаноле и прибавляли 3 капли МеОН. Прибавляли 6 н. раствор HCl в изопропаноле прибавляли и сразу же начиналась кристаллизация гидрохлорида. Кристаллы отсасывали, промывали диэтиловым эфиром и сушили и получали 0,75 г 4-этил-N-((3S,4R)-3-гидрокси-2,2-диметил-6-{2-оксо-2-[4-(пиридин-3-илметил)-пиперазин-1-ил]этил}-3,4-дигидро-2Н-хромен-4-ил)бензолсульфонамидгидрохлорида, температура плавления: 159°С.
13С-ЯМР (101 МГц, МеОН) δ мас. част./млн 15,7 (q, 1 С), 19,0 (q, 1 С), 27,0 (q, 1 С), 29,7 (t, 1 С), 39,7 (t, 1C), 40,0 (t, 1 С), 44,2 (t, 1 С), 52,6 (t, 1 С), 53,0 (t, 1 С), 56,0 (d, 1 С), 56,9 (t, 1 С), 74,8 (d, 1 С), 79,9 (s, 1 С), 118,6 (d, 1 С), 124,2 (s, 1 С), 127,8 (s, 1 С), 128,4 (d, 2 С), 128,9 (d, 1 С), 129,3 (d, 3 С), 130,7 (s, 1 С), 130,9 (d, 1 С), 140,6 (s, 1 С), 144,3 (d, 1 С), 145,9 (d, 1 С), 150,6 (s, 1 С), 151,0 (d, 1 С), 153,3 (s, 1 С), 172,3 (s, 1 С).
Соединения формулы I, указанные ниже в таблице 5, также можно получить по методикам, описанным в приведенных выше примерах, или по аналогичным методикам.
Получены следующие данные спектров 13С-ЯМР:
Пример 10 (соль с HCl): 13С-ЯМР (101 МГц, ДМСО-D6) δ мас. част./млн 15,1 (q, 1 С), 18,7 (q, 1 С), 26,4 (q, 1 С), 27,9 (t, 1 С), 37,8 (t, 1 С), 38,3 (t, 1 С), 41,9 (t, 1 С), 50,1 (t, 1 С), 50,5 (d, 1 С), 54,1 (d, 1 С), 58,6 (t, 1 С), 72,3 (d, 1 С), 78,5 (s, 1 С), 116,4 (d, 1 С), 122,8 (s, 1 С), 126,7 (d, 2 С), 126,8 (s, 1 С), 127,9 (d, 2 С), 128,7 (d, 2 С), 129,1 (d, 1 С), 129,2 - 129,6 (2d, s, 3 С), 131,3 (d, 2 С), 140,0 (s, 1 С), 147,9 (s, 1 C), 151,1 (s, I C), 169,0 (s, 1 C)
Пример 11
Капсулы, содержащие 4-этил-N-{3-гидрокси-2,2-диметил-6-[2-оксо-2-(4-пиридин-4-илметилпиперазин-1-ил)-этил]-хроман-4-ил}-бензолсульфонамид:
Готовили капсулы, содержащие в одной капсуле следующие вещества:
4-Этил-N-{3-гидрокси-2,2-диметил-6-[2-оксо-2-(4-пиридин-4-илметилпиперазин-1-ил)-этил]-хроман-4-ил}-бензолсульфонамид 20 мг
Активное вещество, кукурузный крахмал и лактозу с использованием этилацетата превращали в однородную пастообразную смесь. Пасту измельчали и полученные гранулы помещали на подходящий поддон и для удаления растворителя сушили при 45°С. Высушенные гранулы пропускали через измельчающее устройство и в смесителе смешивали со следующими вспомогательными веществами:
и затем по 400 мг помещали в капсулы (капсулы размера 0).
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ АНТАГОНИСТЫ NK И NK | 2006 |
|
RU2419609C2 |
| ПРОИЗВОДНЫЕ 4-АМИНОПИПЕРИДИНА | 2005 |
|
RU2396257C2 |
| 2-2-ДИАЛКИЛ- ИЛИ ТРАНС-2,2-ДИАЛКИЛ-3,4-ДИГИДРО-3-ГИДРОКСИ-6-(ПИРИДИН-4-ИЛ)-2Н-1-БЕНЗОПИРАН | 1992 |
|
RU2104277C1 |
| СУЛЬФОНАМИДЫ И ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ | 1995 |
|
RU2162084C2 |
| СТЕРЕОИЗОМЕРНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ЛЕЧЕНИЯ ЖЕЛУДОЧНО-КИШЕЧНЫХ РАССТРОЙСТВ И РАССТРОЙСТВ ЦЕНТРАЛЬНОЙ НЕРВНОЙ СИСТЕМЫ | 2005 |
|
RU2374244C2 |
| БЕНЗОПИРАНЫ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ | 1994 |
|
RU2122000C1 |
| МАКРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ МОЧЕВИНЫ И СУЛЬФАМИДА В КАЧЕСТВЕ ИНГИБИТОРОВ TAFIa | 2009 |
|
RU2502736C2 |
| РЕТИНОИДНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ ОБСТРУКТИВНОГО ЗАБОЛЕВАНИЯ ВОЗДУШНЫХ ПУТЕЙ, РАКА ИЛИ ДЕРМАТОЛОГИЧЕСКОГО НАРУШЕНИЯ ИЛИ РАССТРОЙСТВА | 2002 |
|
RU2275367C2 |
| ПРОИЗВОДНЫЕ КАРБАПЕНЕМА И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1992 |
|
RU2091381C1 |
| МОДУЛЯТОРЫ ПРОТЕОЛИЗА И СООТВЕТСТВУЮЩИЕ СПОСОБЫ ПРИМЕНЕНИЯ | 2019 |
|
RU2805511C2 |
Изобретение относится к соединениям общей формулы I,
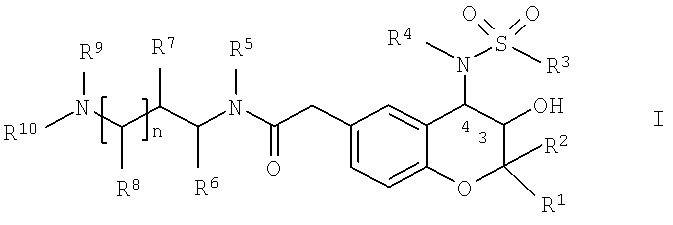 в которой R1 обозначает С1-С4-алкил; R2 обозначает С1-С4-алкил; R3 обозначает фенил, который необязательно содержит 1-3 любых заместителя, выбранных из группы, включающей галоген, C1-С6-алкил и С1-С4-алкоксигруппу; R4 обозначает водород; C1-С6-алкил или С3-С7-циклоалкил-С1-C4-алкил, R5 обозначает водород; и R6 обозначает водород; и R7 обозначает водород; и R8 обозначает водород; и R9 обозначает С1-С4-алкил; и R10 обозначает C1-С6-алкил, фенил-С0-С4-алкил или пиридинил-С0-С4-алкил; при условии, что R10 не обозначает фенил, если R5 и R9 совместно образуют С2-алкилен; или R5 и R9 совместно образуют C1-С3-алкилен; или R6 и R9 совместно образуют C1-С3-алкилен; или R7 и R9 совместно образуют С2-С4-алкилен или C1-С3-алкиленоксигруппу; или R8 и R9 совместно образуют С3-С5-алкилен; или R9 и R10 совместно образуют С4-С6-алкилен; и n равно 0 или 1, или его любые физиологически совместимые соли. Кроме того, изобретение относится к фармацевтической композиции, содержащей соединения формулы I и предназначенной для лечения сердечно-сосудистых заболеваний, к применению этих соединений, для приготовления лекарственного средства, а также к способу получения соединения формулы I. Технический результат: получены и описаны новые соединения, обладающие сердечно-сосудистой активностью. 7 н. и 9 з.п. ф-лы, 5 табл.
в которой R1 обозначает С1-С4-алкил; R2 обозначает С1-С4-алкил; R3 обозначает фенил, который необязательно содержит 1-3 любых заместителя, выбранных из группы, включающей галоген, C1-С6-алкил и С1-С4-алкоксигруппу; R4 обозначает водород; C1-С6-алкил или С3-С7-циклоалкил-С1-C4-алкил, R5 обозначает водород; и R6 обозначает водород; и R7 обозначает водород; и R8 обозначает водород; и R9 обозначает С1-С4-алкил; и R10 обозначает C1-С6-алкил, фенил-С0-С4-алкил или пиридинил-С0-С4-алкил; при условии, что R10 не обозначает фенил, если R5 и R9 совместно образуют С2-алкилен; или R5 и R9 совместно образуют C1-С3-алкилен; или R6 и R9 совместно образуют C1-С3-алкилен; или R7 и R9 совместно образуют С2-С4-алкилен или C1-С3-алкиленоксигруппу; или R8 и R9 совместно образуют С3-С5-алкилен; или R9 и R10 совместно образуют С4-С6-алкилен; и n равно 0 или 1, или его любые физиологически совместимые соли. Кроме того, изобретение относится к фармацевтической композиции, содержащей соединения формулы I и предназначенной для лечения сердечно-сосудистых заболеваний, к применению этих соединений, для приготовления лекарственного средства, а также к способу получения соединения формулы I. Технический результат: получены и описаны новые соединения, обладающие сердечно-сосудистой активностью. 7 н. и 9 з.п. ф-лы, 5 табл.
1. Соединения общей формулы I,
в которой
R1 обозначает С1-С4-алкил;
R2 обозначает С1-С4-алкил;
R3 обозначает фенил, который необязательно содержит 1-3 любых заместителя, выбранных из группы, включающей галоген, C1-С6-алкил и С1-С4-алкоксигруппу;
R4 обозначает водород; С1-С6-алкил или С3-С7-циклоалкил-С1-С4-алкил,
R5 обозначает водород; и
R6 обозначает водород; и
R7 обозначает водород; и
R8 обозначает водород; и
R9 обозначает С1-С4-алкил; и
R10 обозначает С1-С6-алкил, фенил-С0-С4-алкил или пиридинил-С0-С4-алкил; при условии, что R10 не обозначает фенил, если R5 и R9 совместно образуют С2-алкилен; или
R5 R9 совместно образуют C1-С3-алкилен; или
R6 и R9 совместно образуют C1-С3-алкилен; или
R7 и R9 совместно образуют С2-С4-алкилен или C1-С3-алкиленоксигруппу;
или
R8 и R9 совместно образуют С3-С5-алкилен; или
R9 и R10 совместно образуют С4-С6-алкилен; и
n равно 0 или 1,
или его любые физиологически совместимые соли.
2. Соединения по п.1, в которых R1 и R2 оба обозначают метил.
3. Соединения по п.1, в которых R3 обозначает 4-этилфенил.
4. Соединения по п.1, в которых R4 обозначает водород, C1-С6-алкил или циклопропилметил.
5. Соединения по п.1, в которых R5 и R9 совместно образуют C1-С3-алкилен.
6. Соединения по п.1, в которых R10 обозначает С1-С6-алкил; фенил-С1-С4-алкил или пиридинил-С1-С4-алкил.
7. Соединения по п.1, в которых R10 обозначает бензил или пиридинилметил.
8. Соединения формулы I по любому из предыдущих пунктов, которые выбраны из группы, включающей
N-{6-[2-(4-бензилпиперазин-1-ил)-2-оксоэтил]-3-гидрокси-2,2-диметилхроман-4-ил}-4-этилбензолсульфонамид;
4-этил-N-{3-гидрокси-2,2-диметил-6-[2-оксо-2-(4-пиридин-3-илметилпиперазин-1-ил)этил]хроман-4-ил}бензолсульфонамид;
4-этил-N-{3-гидрокси-2,2-диметил-6-[2-оксо-2-(4-пиридин-2-илметилпиперазин-1-ил)этил]хроман-4-ил}бензолсульфонамид, и
4-этил-N-{3-гидрокси-2,2-диметил-6-[2-оксо-2-(4-пиридин-4-илметилпиперазин-1-ил)этил]хроман-4-ил}бензолсульфонамид.
9. Соединение формулы I по любому из предыдущих пунктов, которое представляет собой 4-этил-N-{3-гидрокси-2,2-диметил-6-[2-оксо-2-(4-пиридин-3-илметилпиперазин-1-ил)этил]хроман-4-ил}бензолсульфонамид.
10. Фармацевтическая композиция, предназначенная для лечения сердечно-сосудистых заболеваний у млекопитающих, содержащая фармакологически активное количество соединения формулы I по п.1 и обычные вспомогательные вещества и/или носители.
11. Применение соединений формулы I по п.1 для приготовления лекарственного средства, предназначенного для лечения сердечно-сосудистых заболеваний у млекопитающих.
12. Применение по п.11, где сердечно-сосудистым заболеванием является аритмия.
13. Способ получения соединений формулы I,
в которой R1 обозначает С1-С4-алкил;
R2 обозначает С1-С4-алкил;
R3 обозначает фенил, который необязательно содержит 1-3 любых заместителя, выбранных из группы, включающей галоген, С1-С6-алкил и С1-С4-алкоксигруппу;
R4 обозначает водород; C1-С6-алкил или С3-С7-циклоалкил-С1-С4-алкил,
R5 обозначает водород; и
R6 обозначает водород; и
R7 обозначает водород; и
R8 обозначает водород; и
R9 обозначает С1-С4-алкил; и
R10 обозначает С1-С6-алкил; фенил-С0-С4-алкил или пиридинил-С0-С4-алкил; при условии, что R10 не обозначает фенил, если R5 и R9 совместно образуют С2-алкилен; или
R5 и R9 совместно образуют C1-С3-алкилен; или
R6 и R9 совместно образуют С1-С3-алкилен; или
R7 и R9 совместно образуют С2-С4-алкилен или C1-С3-алкиленоксигруппу;
или
R8 и R9 совместно образуют С3-С5-алкилен; или
R9 и R10 совместно образуют С4-С6-алкилен; и
n равно 0 или 1,
или их любых физиологически совместимых солей,
характеризующийся тем, что соединение общей формулы II,
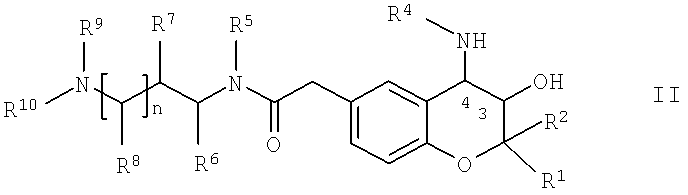
в которой R1, R2, R4, R5, R6, R7, R8, R9, R10 и n имеют указанные выше значения, взаимодействует с соединением общей формулы III,
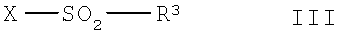
в которой R3 имеет указанные выше значения и Х означает способную к отщеплению удаляемую группу,
и при необходимости полученные свободные соединения формулы I превращают в их физиологически совместимые соли, или соли соединений формулы I превращают в свободные соединения формулы I.
14. Способ получения соединений формулы I,
в которой R1 обозначает С1-С4-алкил;
R2 обозначает С1-С4-алкил;
R3 обозначает фенил, который необязательно содержит 1-3 любых заместителя, выбранных из группы, включающей галоген, С1-С6-алкил и С1-С4-алкоксигруппу;
R4 обозначает водород; С1-С6-алкил или С3-С7-циклоалкил-С1-С4-алкил,
R5 обозначает водород; и
R6 обозначает водород; и
R7 обозначает водород; и
R8 обозначает водород; и
R9 обозначает С1-С4-алкил; и
R10 обозначает C1-С6-алкил; фенил-С0-С4-алкил или пиридинил-С0-С4-алкил; при условии, что R10 не обозначает фенил, если R5 и R9 совместно образуют С2-алкилен; или
R5 и R9 совместно образуют C1-С3-алкилен; или
R6 и R9 совместно образуют C1-С3-алкилен; или
R7 и R9 совместно образуют С2-С4-алкилен или C1-С3-алкиленоксигруппу;
или
R8 и R9 совместно образуют С3-С5-алкилен; или
R9 и R10 совместно образуют С4-С6-алкилен; и
n равно 0 или 1,
или их любых физиологически совместимых солей,
характеризующийся тем, что соединение общей формулы IV
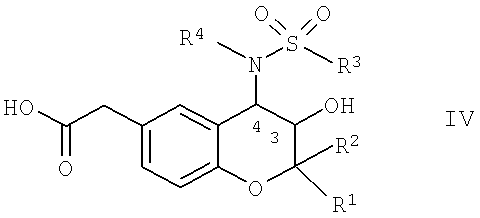
в которой R1, R2, R3 и R4 имеют указанные выше значения, взаимодействует с соединением общей формулы V,
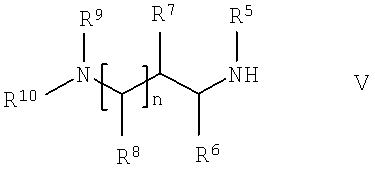
в которой R5, R6, R7, R8, R9, R10 и n имеют указанные выше значения, и при необходимости полученные свободные соединения формулы I превращают в их физиологически совместимые соли или соли соединений формулы I превращают в свободные соединения формулы I.
15. Соединения общей формулы II,
в которой
R1 обозначает С1-С4-алкил;
R2 обозначает С1-С4-алкил;
R4 обозначает водород; C1-С6-алкил или С3-С7-циклоалкил-С1-С4-алкил,
R5 обозначает водород; и
R6 обозначает водород; и
R7 обозначает водород; и
R8 обозначает водород; и
R9 обозначает С1-С4-алкил; и
R10 обозначает C1-С6-алкил; фенил-С0-С4-алкил или пиридинил-С0-С4-алкил; при условии, что R10 не обозначает фенил, если R5 и R9 совместно образуют С2-алкилен; или
R5 и R9 совместно образуют C1-С3-алкилен; или
R6 и R9 совместно образуют C1-С3-алкилен; или
R7 и R9 совместно образуют С2-С4-алкилен или С1-С3-алкиленоксигруппу;
или
R8 и R9 совместно образуют С3-С5-алкилен; или
R9 и R10 совместно образуют С4-С6-алкилен; и
n равно 0 или 1
или их любые соли.
16. Соединения общей формулы IV,
в которой R1 обозначает С1-С4-алкил;
R2 обозначает С1-С4-алкил;
R3 обозначает фенил, который необязательно содержит 1-3 любых заместителя, выбранных из группы, включающей галоген, C1-С6-алкил и С1-С4-алкоксигруппу и
R4 обозначает водород; C1-С6-алкил или С3-С7-циклоалкил-С1-С4-алкил или их любые соли.
| Приспособление для присоединения катодных ламп с баком охлаждения | 1929 |
|
SU12077A1 |
| Щупло к автоматическим ткацким станкам | 1939 |
|
SU58300A1 |
| RU 92016587 A, 20.03.2000 | |||
| RU 9811955 A, 10.09.2000 | |||
| RU 22008611 C2, 20.07.2000 | |||
| СУЛЬФОНАМИДОЗАМЕЩЕННЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ НА ИХ ОСНОВЕ | 1998 |
|
RU2220965C2 |
| RU 20004101964, 27.03.2005. | |||

