Изобретение относится к новому способу получения эпоксидов из олефинов, в частности хиральнообогащенных эпоксидов, к определенным новым катализаторам, используемым в таком способе, и к соединениям, связанным с этим способом.
В заявке WО 91/14694 описаны некоторые катализаторы, имеющие следующую формулу: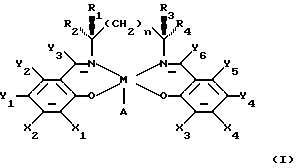
в которой М является ионом переходного металла, А представляет анион и n равно или 0, или 1, или 2. По крайней мере, один из Х1 или Х2 выбирают из группы, состоящей из силилов, арилов, вторичных алкилов и третичных алкилов; и, по крайней мере, один из X3 или Х4 выбирают из такой же группы. Y1, Y2, Y3, Y4, Y5 и Y6 независимо выбирают из группы, состоящей из водорода, галогенидов, алкилов, арильных групп, силильных групп и алкильных групп, имеющих гетероатомы, таких как алкокси и галогенид. Кроме того, по крайней мере, один из R1, R2, R3 и R4 выбирают из первой группы, состоящей из Н, CH3, C2H5 и первичных алкилов. Кроме того, если R1 выбирают из первой группы, тогда R2 и R3 выбирают из второй группы, состоящей из арильных групп, имеющих гетероатом ароматических групп, вторичных алкилов и третичных алкилов. Если R2 выбирают из упомянутой первой группы, тогда R1 и R4 выбирают из упомянутой второй группы. Если R3 выбирают из упомянутой первой группы, тогда R1 и R4 выбирают из упомянутой второй группы. Если R4 выбирают из упомянутой первой группы, тогда R2 и R3 выбирают из упомянутой второй группы. Такие катализаторы описаны в качестве пригодных при энатиоселективном эпоксидировании прохиральных олефинов.
Кроме того, в заявке WO 91/14694 описаны некоторые катализаторы, имеющие нижеприведенную формулу, обозначенную здесь (IА)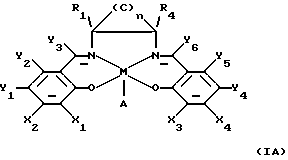
в которой М определен как ион переходного металла и А является анионом; n равно 3, 4, 5 или 6; по крайней мере, один из Х1 или Х2 выбирают из группы, состоящей из арилов, первичных алкилов, вторичных алкилов, третичных алкилов и гетероатомов; по крайней мере, один из Х3 или Х4 выбирают из группы, состоящей из арилов, первичных алкилов, вторичных алкилов, третичных алкилов и гетероатомов; по крайней мере, один из Y1 или Y2 выбирают из группы, состоящей из арилов, первичных алкилов, вторичных алкилов, третичных алкилов и гетероатомов; по крайней мере, один из Y4 или Y5 выбирают из группы, состоящей из арилов, первичных алкилов, вторичных алкилов, третичных алкилов и гетероатомов; Y3 и Y6 независимо выбирают из группы, состоящей из водорода и первичных алкильных групп; R1 и R4 находятся в транс-положении по отношению друг к другу, и, по крайней мере, один из R1 и R4 выбирают из группы, состоящей из первичных алкилов и водорода; и углероды в (С)n части имеют заместители, выбранные из группы, состоящей из водорода, алкила, арила и гетероатомов.
Такие катализаторы описаны как пригодные при энантиоселективном эпоксидировании прохиральных олефинов. Эти катализаторы принадлежат к классу катализаторов, известных в данной области как "солевые катализаторы".
В находящейся на совместном рассмотрении международной заявке на патент РСТ/GВ 93/01666 (в настоящее время международная заявка на патент номер публикации WO 94/03271) также представлен ряд солевых катализаторов, по структуре отличающихся от катализаторов формулы (I) и имеющих общую формулу (II)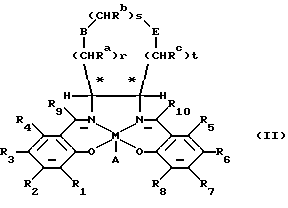
в которой М представляет ион переходного металла;
А, если необходимо, является противоином;
r, s и t составляют независимо от 0 до 3, при этом сумма r+s+t должна находиться в диапазоне от 1 до 3;
Ra, Rb, Rc независимо являются водородом или CH2OR', где R' является водородом или органической группой;
В и Е независимо являются кислородом, СН2, NRd, где Rd является алкилом, водородом, алкилкарбонилом или арилкарбонилом или SОn, где n равно 0 или целому числу 1 или 2, при условии, что В и Е не являются одновременно СН2 и что, когда В является кислородом, NRd или SОn, тогда r не может быть 0, и когда Е является кислородом, NRd или SOn, тогда t не может быть равно нулю;
R1, R2, R3, R4, R5, R6, R7, R8, R9 и R10 независимо являются водородом, алкилом или алкокси.
Соединения формулы (II) также катализируют энантиоселективное эпоксидирование определенных прохиральных олефинов.
В данной области предполагалось, что применение соединений, таких как пиридиноксид и 2-метилимидазол, в сочетании с определенными хиральными (солевыми) марганцевыми (III) комплексными катализаторами, улучшает химический выход таких реакций (Syn. Lett. , April, 1991, 265-266), хотя воздействие на энантиоселективность катализируемых реакций в настоящее время является неясным (Tetrahedron. Vol. 50, N15, р.4323-4334, 1994). В этом контексте пиридиноксид и 2-метилимидазол относят к "донорным лигандам", поскольку считают, что они связаны донорной связью с ионом металла солевого катализатора.
Одной важной проблемой, связанной с применением таких донорных лигандов, является полное удаление донорного лиганда из конечного эпоксидного продукта, особенно в крупномасштабных реакциях и, главным образом, когда используют двухфазные реакционные системы.
Было обнаружено, что одно конкретное соединение, изохинолин-N-оксид, о котором ранее не сообщалось как о донорном лиганде, является в особенности эффективным в качестве донорного лиганда, так как он усиливает оборот катализатора и, кроме того, он обладает очень хорошей растворимостью для использования в качестве донорного лиганда, что дает возможность использовать его в реакциях эпоксидирования, катализируемых комплексом металла, и впоследствии легко удалять из эпоксидных продуктов реакции. Было также обнаружено, что конкретная группа солевых катализаторов является в особенности пригодной для использования с донорными лигандами, потому что присутствие донорных лигандов последовательно дает не только увеличение скорости реакции, но также увеличение энантиоселективной специфичности реакций эпоксидирования.
Кроме того, был получен еще один ряд солевых катализаторов, которые по структуре отличаются от катализаторов формул (I), (IA) и (II) и которые, к удивлению, также обладают способностью катализировать энантиоселективное эпоксидирование определенных прохиральных олефинов.
Соответственно по первому аспекту изобретение обеспечивает способ энантиоселективного эпоксидирования прохиральных олефинов, включающий взаимодействие прохирального олефина с источником кислорода в присутствии солевого катализатора и источника лиганда, отдающего электрон, отличающийся тем, что донорным лигандом является изохинолин-N-оксид или соединение, имеющее активность донорного лиганда и имеющее по существу те же самые свойства растворимости, что и изохинолин-N-оксид.
Подходящим солевым катализатором является соединение формулы (I), (IA), (II) или соединение формулы (III). Соединение формулы (III) будет определено в дальнейшем.
Изобретение обеспечивает также в качестве донорного лиганда изохинолин-N-оксид или соединение, имеющее активность донорного лиганда, и имеющее по существу те же самые свойства растворимости, что и изохинолин-N-оксид.
По еще одному аспекту изобретение обеспечивает способ этантиоселективного эпоксидирования прохирального олефина, включающий взаимодействие прохирального олефина с источником кислорода в присутствии солевого катализатора - источника лиганда, отдающего электрон, отличающийся тем, что солевым катализатором является соединение формулы (II).
Источник лиганда, отдающего электрон, подходяще обеспечивают посредством соединения, которое способно образовывать донорную связь с переходным металлом М упомянутого солевого катализатора, для того, чтобы при использовании возрастала скорость реакции эпоксидирования и можно было также повысить энантиоселективную специфичность полученного продукта.
Источник лиганда, отдающего электрон, подходяще обеспечивают посредством соединения, которое способно образовывать донорную связь с переходным металлом М солевого катализатора, для того, чтобы при использовании увеличилась энантиоселективная специфичность соединения формулы (I).
Подходящий источник лиганда, отдающего электрон, может быть выбран из списка, состоящего из пиридин-N-оксида, 2-метилпиридин-N-оксида, 4-метилпиридин-N-оксида, 4-фенилпиридин-N-оксида или изохинолин-N-оксида, особенно подходящим является изохинолин-N-оксид.
В соединениях формул (I) и (IА) предпочтительные значения для М, A, n, X1, Х2, X3, X4, Y1, Y2, Y3, Y4, Y5, Y6, R1, R2, R3, R4 определены в WO 91/14694.
Подходящими катализаторами являются катализаторы формулы (IА), которая определена выше.
Предпочтительной подгруппой катализаторов являются катализаторы формулы (IВ), которая определена ниже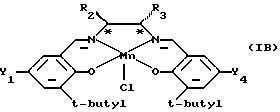
в которой Y1 и Y2 являются одинаковыми и их выбирают из группы, состоящей из метила, трет-бутила или метокси, и R2 и R3 оба являются фенилом или вместе с атомами углерода, к которым они присоединены, образуют гексильное кольцо.
В катализаторах формулы (IВ) наиболее предпочтительно оба Y1 и Y4 являются трет-бутилом и R2 и R3 вместе с углеродными атомами, к которым они присоединены, образуют гексильное кольцо.
В соединениях формулы (II) подходящие, пользующиеся преимуществом и предпочтительные значения переменных величин А, В, Е, Ra, Rc, Rd, R', Rb, R1, R2, R3, R4, R5, R6, R7, R8, R9, R10, n, r, s и t являются такими, как они описаны в WO 94/03271, если здесь не указано иным образом.
Подходящие органические группы R' включают алкильные, алкилкарбонильные, арилкарбонильные или арильные производные.
Отдельные примеры R' включают замещенные алкильные группы.
Одним примером R' является трифенилметильная группа.
Предпочтительные значения s и t равны нулю, r равен 1 и Ra является водородом, В является кислородом и Е представляет СН2, или r, s, t равны 1, Ra, Rb и Rc являются водородом и оба В и Е являются кислородом или s равно 0, оба r и t равны 1, Ra является водородом или трифенилметилоксиметиленом и Rc является водородом, В является кислородом и Е предсталвляет -СН2-; или оба r и t равны 1, s равно О, Ra и Rc являются водородом, В представляет NRd, где Rd является фенилкарбонилом и Е представляет СН2.
Каждый из R2, R4, R5, и R7 подходяще независимо представляет водород.
Каждый из R1, R3, R6 и R8 подходяще независимо представляет C1-6 алкил.
R1 и R8 предпочтительно представляют разветвленные алкильные группы, например третичные алкильные группы.
R3 и R6 также выгодно представляют разветвленные алкильные группы.
Одним предпочтительным примером любого из R1 и R8 является третичный бутил.
Особыми примерами R3 и R6 являются третичный бутил и метил.
Примеры R2, R4, R5 и R7 представляют водород.
Примеры соединений формулы (II) включают такие, которые приведены в качестве примеров в WO 94/03271, и в особенности такие соединения, на которые ссылаются здесь.
Как указывалось выше, одним аспектом изобретения является обнаружение нового ряда солевых катализаторов.
Соответственно по одному аспекту настоящее изобретение обеспечивает соединение формулы (III)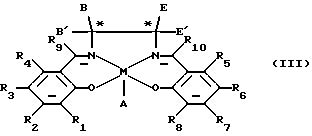
в которой М является ионом переходного металла;
А, если он необходим, представляет противоион;
В, В', Е и Е' независимо выбирают из группы, состоящей из водорода, арила, С1-6 алкила, силила или арил-C1-6 алкила, у которых арильная или алкильная составляющая необязательно является замещенной, или В' и В или Е' и Е вместе образуют C2-6 полиметиленовую связь, при условии, что только один из углеродов, помеченных звездочкой, представляет хиральный центр;
R1, R2, R3, R4, R5, R6, R7, R8, R9 и R10 независимо являются водородом, алкилом или алкокси.
В соединениях формулы (III)
Каждый из R2, R4, R5 и R7 подходяще независимо представляет водород.
Каждая из R1, R3, R6 и R8 подходяще независимо представляет С1-6 алкил.
R1 и R8 предпочтительно представляют разветвленные алкильные группы, например третичные алкильные группы.
R3 и R6 также выгодно представляют разветвленные алкильные группы.
Одним предпочтительным примером любого из R1 и R8 является третичный бутил.
Особыми примерами R3 и R6 являются третичный бутил и метил.
Примерами R2, R4, R5 и R7 является водород.
Одним из примеров В и Е является фенил, метил или изопропил, а другим - водород. Одним из наиболее предпочтительных примеров В и Е является фенил, а другим - водород.
Соединения формулы (III) также являются подходящими солевыми катализаторами для применения в способе изобретения.
В соединениях формулы (I), (IA), (IВ), (II), и (III)
Связь между М и А имеет изменяющиеся степени ионного характера, зависящие от используемого аниона.
Подходящие ионы переходного металла М включают Мn, Сr, Fе, Ni, Со, Ti, V, Ru и 0s в соответствующей степени окисления.
Предпочительным ионом переходного металла М является Мn в степени окисления (II) или (III).
Следует учитывать, что в некоторых случаях, например, когда М является Мn (II), противоион не является необходимым.
Подходящие противоионы А включают такие анионы, которые упоминаются в WO 91/14694 и WO 94/03271.
А предпочтительно является хлоридом.
В способе изобретения
Подходящие прохиральные олефины включают соединения, которые содержат в качестве части своей структуры следующие соединения: циклогексен, 5,6-дигидро-2Н-пиран, 1,2,5,6-тетрагидропиридин, 1,2,3,4-тетрагидропиридин и 5,6-дигидро-2Н-тиопиран.
Пользующиеся предпочтением прохиральные олефины включают такие соединения, которые содержат в качестве части своей структурной формы следующие группы: 1,2-дигидронафталин, 2Н-хромен, 1,2-дигидроизохинолин и 2Н-тиохромен. Такие соединения хорошо известны в области активаторов калиевого канала.
Предпочтительные прохиральные олефины включают такие, которые упоминаются в ЕР-А-0376254, например соединения формулы (ХIV), в частности 2,2-диметил-6-пентафторэтил-2Н-1-бензопиран.
Подходящие источники кислорода включают окислители, например гипохлорит натрия.
Для обеспечения необходимого эпоксида реакцию эпоксидирования можно осуществлять с использованием соответствующей методики, при которой взаимодействуют прохиральный олефин, источник кислорода, соединение формулы (I) и источник лиганда, отдающего электрон.
Реакцию подходяще осуществляют в двухфазной системе, особенно, когда источник кислорода и/или один из компонентов реакции растворим в воде, и особенно, когда источником кислорода является гипохлорит натрия.
Подходящими двухфазными системами являются такие системы, которые обычно используют в данной области, при этом принимают во внимание характер конкретных реагентов, примером двухфазной системы является метиленхлорид и вода.
Солевые катализаторы, например соединение формулы (I), (IА), (II) или (III), прохиральный олефин и источник лиганда, отдающего электрон, в инертном, водонесмешивающемся растворителе, например дихлорметане, могут взаимодействовать с источником кислорода в воде.
Обычно реакция происходит при рН в диапазоне между 10 и 13, предпочтительно между 10,5 и 12, наиболее предпочтительно между 11 и 11,5, рН удобно регулируют посредством присутствия буфера, например первичного кислого фосфата натрия.
Реакцию можно осуществлять при подходящей температуре, обеспечивая пригодную скорость образования требуемого продукта. Вследствие увеличения скорости реакции, вызванной присутствием источника лиганда, отдающего электрон, реакцию можно осуществлять при температуре более низкой, чем та температура, при которой реакцию осуществляют без лиганда, например в диапазоне между 0 и 40oС. Обычно ее осуществляют при окружающей или слегка повышенной температуре, но предпочтительно при окружающей температуре.
Подходящее мольное процентное отношение соединения формулы (I) к прохиральному олефину находится в диапазоне от 0,01 до 10, предпочтительно в диапазоне от 0,1 до 0,05, от 0,5 до 5, от 1 до 5, от 1 до 3, от 0,5 до 2, наиболее предпочтительно в диапазоне от 0,2 до 2.
Подходящее мольное отношение источника лиганда, отдающего электрон, к прохиральному олефину находится в диапазоне от 0,05 до 3, например от 0,1 до 2,0 или от 1 до 2, предпочтительной диапазоне от 0,1 до 2. Подходящее мольное отношение для N-пиридиноксида находится, например, в диапазоне от 0,5 до 2. Подходящее мольное отношение для изохинолин-N-оксида находится, например, в диапазоне от 0,1 до 0,5.
Настоящее изобретение также распространяется на получение всех эпоксидов, которые являются предшественниками соединений формулы (I) и ЕР-А-0376524, в особенности конкретных примеров этой заявки.
Настоящее изобретение также распространяется на получение всех эпоксидов, которые являются предшественниками соединений формулы (I) в WO 92/22293, в особенности конкретных примеров этой заявки.
Настоящее изобретение распространяется также на последующее превращение любого из упомянутых эпоксидов в соответствующие соединения формулы (I) ЕР-А-0376524, в особенности на превращение релевантных предшествующих эпоксидов в соответствующий конкретный пример соединения ЕР-А-0376524 и в особенности на превращение 2,2-диметил-6-пентафторэтил-3,4-эпокси-2Н-1-бензофурана в (-)-транс-3,4-дигидро-2,2-диметил-4-(2-оксопиперидин-1-ил)-6-пентафторэтил-2Н-1-бензофуран-3-ол или в его (+)-транс-изомер.
Настоящее изобретение распространяется также на последующее превращение любого из упомянутых эпоксидов в соответствующие соединения формулы (I) WO 92/22293, в частности на превращение релевантных предшествующих эпоксидов в соответствующий конкретный пример соединения WO 92/22293, в особенности на превращение (3R, 4R)-6-ацетил-2,2-диметил-3,4-эпокси-2Н-1-бензопирана в транс-6-ацетил-4S-(4-фторбензоиламино)-3,4-дигидро-2,2-диметилл-2Н-1-бензопиран-3R-ол или в его 3S, 4R-изомер.
Настоящее изобретение распространяется также на продукт, образованный между солевым катализатором, например соединением формулы (I), (IA), (IВ), (II) или (III), и лигандом, отдающим электрон.
При этом термин "хиральный солевой катализатор" относится к солевым катализаторам, в которых преобладает один конкретный энантиомер и которые при использовании обеспечивают преобладание одного конкретного энантиомера в эпоксиде, полученном на основе прохирального олефина.
Термин "алкил", когда используется только он или когда алкил образует часть других групп (например, алкоксигруппы или алкилкарбонильные группы), включает алкильные группы с прямой или разветвленной цепью, содержащие от 1 до 12 атомов углерода, подходяще от 1 до 6 атомов углерода, примеры алкила включают метильную, этильную, н-пропильную, изопропильную, н-бутильную, изобутильную или трет-бутильную группу.
Когда здесь используется термин "арил", он включает фенил и нафтил, необязательно замещенные группами в количестве до пяти, предпочтительно до трех, выбранными из галогена, алкила, фенила, алкокси, галогеналкила, алкилкарбонила и фенилкарбонила.
Предпочтительной арильной группой является замещенная или незамещeнная фенильная группа.
Переходные металлы М включают такие металлы, которые имеют степень окисления, равную (II) или более.
Подходящие заместители арила включают алкил, галоген и алкокси.
Необязательные заместители арила включают такие, которые упомянуты здесь для арильных групп, особым примером является фенил.
Следует учитывать, что атомы углерода, помеченные звездочкой, представляют хиральные центры и что настоящее изобретение распространяется на каждый отдельный энантиомер и на его любые смеси.
Соединения формул (I), (IA) и (IВ) могут быть получены в соответствии с методиками, описанными в WO 91/14694, или посредством аналогичных им методик.
Соединения формулы (II) могут быть получены в соответствии с методиками, представленными в международной заявке WO 91/14694, или в соответствии с аналогичными им методиками.
Содержание WO 91/14694 и WO 94/03271, включая конкретные описания и примеры, приведены здесь для ссылки.
Что касается соединений формулы (III), то настоящее изобретение обеспечивает также способ получения соединений формулы (III), который включает образование комплекса переходного металла следующего соединения формулы (IV):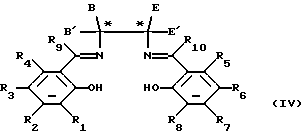
в которой переменные величины R1, R2, R3, R4, R5, R6, R7, R8, R9 и R10, B, B', E и E' являются такими, как они определены в отношении формулы, в которой только один из углеродов, помеченных звездочкой, является хиральным центром, и в дальнейшем, если необходимо, разделение энантиомеров.
Комплекс иона переходного металла может быть подходяще образован путем добавления соответствующей соли переходного металла, например ацетата марганца (II) или (III), предпочтительно ацетата марганца (III), к соединению формулы (IV) в соответствующем растворителе, например этаноле или метилендихлориде, при повышенной температуре. Необязательное замещение или взаимопревращение противоиона можно осуществить путем добавления подходящего источника желательного противоиона, такого как соль щелочного металла, например LiCI.
Разделение энантиомеров можно осуществить общепринятыми методами, например кристаллизацией производных или хроматографией. Однако следует учитывать, что разделение энантиомеров осуществляют предпочтительно перед образованием комплекса переходного металла.
Изобретение, кроме того, обеспечивает способ получения соединений формулы (IV), который включает последовательную конденсацию, в любом порядке, соединения формулы (V)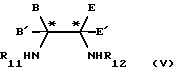
в которой В, В', Е и Е' являются такими, как они определены в формуле (III), и R11 и R12 независимо представляют водород или защитную группу для амина, при условии, что, по крайней мере, один из R11 и R12 является водородом, с
(i) соединением формулы (VI)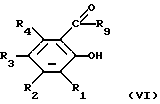
в которой R1, R2, R3, R4 и R9 являются такими, как они определены в отношении формулы (III), и
(ii) соединением формулы (VII)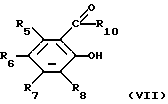
в которой R5, R6, R7, R8 и R10 являются такими, как они определены в отношении формулы (III), и в дальнейшем, когда необходимо, удаление защитной группы R11 и R12, выделение требуемого соединения, включая, если это является необходимым, разделение энантиомеров.
Предпочтительно, чтобы соединение формулы (IV) получали из оптически чистых соединений формулы (V), которые сами предпочтительно получают из оптически чистых исходных материалов. Альтернативно рацематы или смеси энантиомеров формулы (VI) или (VII) могут сами растворяться при применении обычных методов, известных в данной области, например кристаллизации производных или хроматографии.
Когда являются необходимыми соединения формулы (IV), в которых один или несколько из R1, R2, R3, R4 и R9 неодинаковые, соответственно, когда являются неодинаковыми один или несколько из R8, R7, R6, R5 и R10, тогда соединения формулы (V) можно последовательно конденсировать с соединениями формулы (VI) и формулы (VII) в любом порядке, путем нагревания соответствующим образом защищенного соединения формулы (V) с соединением формулы (VI) или (VII) (при мольном соотношении 1:1) в инертном растворителе, например этаноле, если необходимо, очистки образованного промежуточного соединения формулы (VIII) или (IX)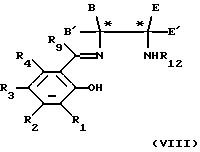
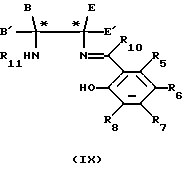
в которых переменные от R1 до R12, Е, Е', В и В' являются такими, как они определены в отношении формул (V), (VI) или (VII), с использованием обычных методов разделения, например хроматографии; удаления защитной группы R11 или R12 и затем, когда это необходимо, повторения реакции с использованием соединения формулы (VI) или (VII).
Подходящие защитные группы R11 или R12, включают обычные защитные группы для амина, внедрение и удаление которых совместимо с природой молекул, подлежащих защите, например бензильные группы, силильные группы или ацильные группы, например бензоильные группы, предпочтительно силильные группы.
Удаление R11 или R12, когда они представляют защитные группы, можно осуществлять с использованием методов, общепринятых в данной области, зависящих от природы защитной группы.
Следует учитывать, что когда каждый из R1, R2, R3, R4 и R9 является одинаковым, как и каждый из R8, R7, R6, R5 и R10, соответственно соединения формул (VI) и (VII) являются одинаковыми, поэтому предпочтительно используют соединения формулы (V), в которой R11 и R12 являются водородом и две молекулы соединения формулы (VI) или (VII).
Реакцию подходяще осуществляют в инертном растворителе, например этаноле, при повышенной температуре, например при температуре перегонки выбранного растворителя.
Соединения формулы (V) являются или известными соединениями, или могут быть получены в соответствии с известными способами, или в соответствии со способами, аналогичными известным, или в соответствии со способами, аналогичными описанным здесь.
Соединения формулы (VI) и (VII) являются коммерчески доступными известными соединениями или могут быть получены в соответствии с известными способами или в соответствии со способами, аналогичными известным, например в соответствии с такими, которые описаны в источнике: G. Casiraghi et al. J. Chem. Soc. Perkin Transactions I, 1980, p. 1862-1865.
Новые соединения формул (IV), (VI), (VII), (VIII) и (IX) образуют аспект настоящего изобретения.
Как указывалось здесь выше, соединения формулы (II) могут быть получены с использованием способов, представленных в находящейся на совместном рассмотрении международной заявке на патент РСТ/GB 93/01666 (теперь международная заявка на патент WO 94/03271). Во избежание сомнений эти способы включают следующее.
Соединения формулы (II) могут быть получены путем образования комплекса переходного металла следующего соединения формулы (X):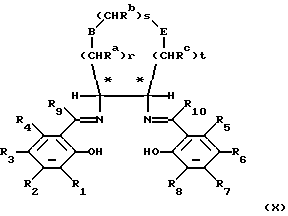
в которой переменные от R1 до R10, В, Е, r, s, t, Ra, Rb, Rc являются такими, как они определены в отношении формулы (II), и затем, если необходимо, разделения энантиомеров.
Комплекс переходного металла может быть подходяще образован путем добавления соответствующей соли переходного металла, например ацетата марганца (II) или (III), предпочтительно ацетата марганца (III), к соединению формулы (II) в соответствующем растворителе, например этаноле или метилендихлориде, при повышенной температуре. Необязательное замещение или взаимопревращение противоиона может быть осуществлено путем добавления соответствующего источника желательного противоиона, такого как соль щелочного металла, например LiСl.
Разделение энантиомеров может быть осуществлено общепринятыми методами, например кристаллизацией производных или хроматографией. Однако следует учитывать, что разделение энатиомерсв предпочтительно осуществляют перед образованием комплекса переходного металла.
Соединения формулы (X) могут быть также получены путем последовательной конденсации, в любом порядке соединения формулы (XI)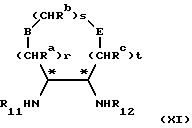
в которой r, s, t, Ra, Rb и Rc, E, В являются такими, как они определены в формуле (II), и R11 и R12 независимо представляют водород или защитную группу для амина, при условии, что, по крайней мере, один из R11 и R12 является водородом, с соединением вышеопределенных формул (VI) и (VII). Условия реакции аналогичны условиям, приведенным выше в отношении реакции между соединением формулы (V) и соединениями формул (VI) и (VII).
Предпочтительно, чтобы соединение формулы (X) получали из оптически чистых соединений формулы (XI), которые сами предпочтительно получают из оптически чистых исходных материалов. Альтернативно рацематы или смеси энантиомеров формулы (X) или (XI) могут сами раствориться при использовании общепринятых методов в данной области, например кристаллизации производных или хроматографии.
Когда необходимы соединения формулы (X), в которых один или несколько из R1, R2, R3, R4 и R9 неодинаковые, соответственно, когда являются неодинаковыми один или несколько из R8, R7, R6, R5 и R10, тогда соединения формулы (XI) можно последовательно конденсировать с соединениями формулы (VI) и формулы (VII), в любом порядке, путем нагревания соответствующим образом защищенного соединения формулы (XI) с соединением формулы (VI) или (VII) (при мольном отношении 1:1) в инертном растворителе, например этаноле, если необходимо, очистки образованного промежуточного соединения формулы (ХII) или (ХIII)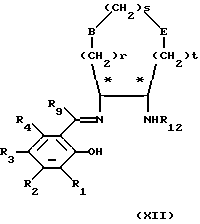
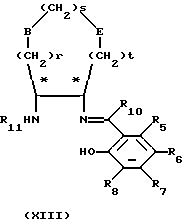
в которых переменные величины от R1 до R12, r, s, t, Ra, Rb и Rc, Е и В являются такими, как они определены в формулах (XI), (VI) и (VII), с использованием общепринятых методов, например хроматографии, удаления защитных групп R11 или R12 и затем, когда необходимо, повторения реакции с использованием соединения формулы (VI) или (VII).
Подходящие защитные группы R11 и R12 и способы удаления таких групп являются такими, как они описаны выше.
Следует учитывать, что, когда каждый из R1, R2, R3, R4 и R9 является одинаковым, как и каждый из R8, R7, R6, R5 и R10, соответственно соединения формул (VI) и (VII) являются одинаковыми, поэтому предпочтительно используют соединения формулы (XI), в которой R11 и R12 являются водородом, и применяют две молекулы соединения формулы (VI) или (VII) в инертном растворителе, например этаноле, при повышенной температуре, например при температуре перегонки.
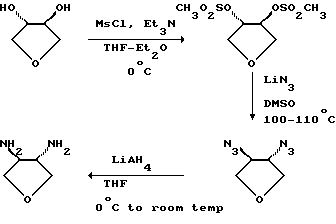
Соединения формулы (XI) являются известными соединениями или могут быть получены в соответствии с известными способами или в соответствии со способами, аналогичными известным, или в соответствии со способами, аналогичными описанным здесь способам, например, когда соединением формулы (XI) является 3,4-диаминотетрагидрофуран, такое соединение может быть получено в соответствии со следующей схемой, например, как описано в описаниях 1 и 2.
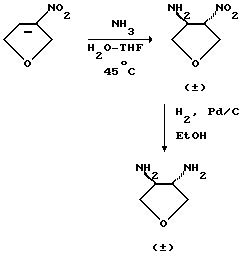
Альтернативно (S,S)-транс- 3,4-диаминотетрагидрофуран может быть получен в соответствии со следующей схемой, например, как описано в описаниях 4-6.
5R, 6R-Диамино-1,3-диоксепан может быть получен в соответствии с методиками, которые описаны в описаниях 8-13.
3R, 4S-ДиаминотетрагидроФфран может быть получен в сответствии с методиками, которые описаны в описаниях 15-17.
3R,4S-Диамино-(2S)(трифенилметоксиметил)тетрагидрофуран может быть получен в соответствии с методиками, которые описаны в описаниях 21-24.
(+)-транс-1-Бензоил-3,4-диаминопиперидин может быть получен в соответствии с методиками, которые описаны в описаниях 25-27.
Катализаторы формулы (III) получают предпочтительно в хиральной форме путем использования растворенного соединения формулы (XI), которое может быть растворено с применением общепринятых методов. Само соединение формулы (XI) может быть получено из соответствующих предшествующих соединений, например таких, которые обрисованы выше в общих чертах и которые могут быть растворены при использовании общепринятых методов или могут быть приобретены в растворенном виде. Альтернативно связанное соединение формулы (X) можно растворить с использованием общепринятых методов.
Настоящее изобретение обеспечивает также способ получения соединений формулы (А), которая определена в WO 93/17026, или, когда это является подходящим, их фармацевтически приемлемых солей или их фармацевтически приемлемого сольвата, который включает взаимодействие соединения формулы (I), источника кислорода, соединения формулы (С) и источника лиганда, отдающего электрон, и затем превращение полученного соединения формулы (В) в соединение формулы (А) или, когда это является подходящим, в его фармацевтически приемлемую соль или в его фармацевтически приемлемый сольват.
Настоящее изобретение распространяется также на продукт, образованный между соединением формулы (I) и лигандом, отдающим электрон, обеспеченным посредством вышеупомянутого источника.
Соединения формулы (С) коммерчески доступны или могут быть получены в соответствии с методиками, на которые ссылаются или которые обрисованы в общих чертах в ЕР-А-0376524.
Настоящее изобретение поясняют последующие описания и примеры.
(А) Примеры с использованием катализаторов, представленных в WO 91/14694.
Пример 1. Получение (2R,4R)-6-ацетил-2,2-диметил-3,4-эпокси-2Н-1-бензопирана с использованием пиридин-N-оксида в качестве лиганда, отдающего электрон.
С помощью 8N NaOH установили рН раствора гипохлорита натрия (54 мл, 13,7% вес./об.), 0,05 М Na2HPO4 (50 мл) и воды (70 мл), равным 11,3.
Смешали вместе 6-ацетил-2,2-диметилхромен (10 г, 0,049 моля) и R,R-[1,2-бис(3,5-ди-трет-бутилсалицилидамино)циклогексан] марганец(III)хлоридный катализатор (320 мг, 1 мол.%), пиридин-N-оксид (9,5 г, 2 экв) и дихлорметан (50 мл) и смесь перемешивали в течение 1 часа.
Раствор разбавили DСМ (дихлорметаном) (200 мл) и фильтровали через целит, слои разделили. Водный слой реэкстрагировали DCM (200 мл), затем органические слои соединили. Органическую фазу промыли водой (2х400 мл) и выпарили досуха до получения коричневого масла, 12 г, е.е. 95% (хиральная HPLC (высокоэффективная жидкостная хроматография)). Масло выкристаллизовали из IРЕ (2 1/2 объема), затравленного эпоксидом, до получения названого соединения в виде не совсем белого/коричневого твердого вещества (6,45 г, 60%), е. е. 99%.
Для проведения такой же реакции без добавления лиганда, отдающего электрон, например пиридин-N-оксида, обычно необходим 1 мол. % катализатора для достижения полного превращения при комнатной температуре в течение около 4 часов (сырой эпоксид, е.е. 92%).
Пример 2. Получение (3R,4R)-2,2-диметил-3,4-эпокси-6-пентафторэтил-2Н-1-бензопирана с использованием изохинолин N-оксида в качестве лиганда, отдающего электрон.
С помощью разбавленной ортофосфорной кислоты установили рН раствора гипохлорита натрия (44 мл, 17% вес./об.), воды (70 мл) и 0,05 М NaH2PO4 (50 мл), равным 11,3. Добавили 2,2-диметил-6-пентафторэтил-2Н-1-бензопиран (13,6 г, 50 ммолей), дихлорметан (100 мл), изохинолин-N-оксид (0,725 г, 10 мол.%) и R,R-[1,2-бис(3,5-ди-трет-бутилсалицилидамино)циклогексан]марганец(III)хлорид (64 мг, 0,2 мол.%) и смесь быстро перемешали при комнатной температуре. Через 2 часа анализ HPLC (высокоэффективной жидкостной хроматографией) показал 95% превращение хромена в эпоксид. Реакционную смесь перемешивали в течение еще 3 часов при комнатной температуре, но дополнительного превращения хромена в эпоксид не произошло. Измерили е.е. сырого (3R, 4R)-эпоксида, который составил в соответствии с хиральной HPLC 92,5%. Смесь разбавили дихлорметаном (200 мл), фильтровали через целит и слои разделили. Органическую фазу промыли водой (3х100 мл), затем выпарили дослуха до получения сырого названного соединения (15,0 г) в виде желтого твердого вещества. Сырой продукт перекристаллизовали из гексана (3 объема) до получения чистого названного соединения (8,0 г, 54%) в виде бесцветных игольчатых кристаллов (е.е. > 99%).
Для проведения такой же реакции без лиганда, отдающего электрон, например изохинолин-N-оксида, обычно необходимо 2 мол.% катализатора для получения полного превращения.
Пример 3. Получение (3R,4R)-6-ацетил-2,2-диметил-3,4-эпокси-2Н-1-бензопирана с использованием изохинолин-N-оксида в качестве лиганда, отдающего электрон.
Повторили методику примера 1 с использованием 10 мол.% изохинолин-N-оксида вместо пиридин-N-оксида. Количество катализатора также уменьшили до 0,1 мол. %. Полного превращения в желательный эпоксид (е.е. 96%) достигли в течение менее 15 минут.
Пример 4. Получение (2R,4R)-6-ацетил-2,2-диметил-3,4-эпокси-8-иодо-2Н-1-бензопирана с использованием изохинолин-N-оксида в качестве лиганда, отдающего электрон.
Повторили методику примера 1 с использованием 0,2 эквивалента изохинолин-N-оксида вместо пиридин-N-оксида. Количество катализатора уменьшили до 0,2 мол.%. Через 2 часа наблюдали полное превращение в эпоксид (е.е. 98%). Сырой продукт перекристаллизовали из IРЕ (3 объема) до получения энантиомерно чистого названного соединения, точка плавления 123,6-125,4oC, выход 72%.
(В) Примеры с использованием катализаторов, представленных в WO 94/03271.
Пример 5. Получение (3R,4R)-6-ацетил-2,2-диметил-3,4-эпокси-2Н-1-бензопирана с использованием пиридин-N-оксида в качестве лиганда, отдающего электрон.
С помощью 8N NаОН установили рН гипохлорита натрия (21,5 мл, 17,3% вес. /об. ), воды (34 мл) и 0,05 М Nа2НРО4 (25 мл), равным 13. К дихлорметану (50 мл) добавили 6-ацетил-2,2-диметил-2Н-1-бензопиран (5,0 г, 25 ммолей), пиридин-N-оксид (5,0 г, 52 ммоля) и S,S-Mn-солевой катализатор - (3S, 4S)-бис(3,5-ди-трет-бутилсалицимидамино)тетрагидрофуран марганец(II)хлорид (D34, 152 мг, 1 мол.%), и смесь перемешали при комнатной температуре. Через 2 часа в соответствии с анализом HPLC реакция закончилась. Смесь разбавили дихлорметаном и фильтровали через целит. Две фазы разделили и органическую фазу промыли водой (200 мл), затем выпарили досуха под пониженным давлением до получения сырого названного соединения в виде коричневого масла (5,0 г). Как показала хиральная HPLC, е.е. составил 94%.
Названное соединение получили энантиомерно чистым (е.е. > 99.8%) путем перекристаллизации сырого продукта из диизопропилового эфира, степень извлечения составила 44%.
Пример 6. Получение (3S,4S)-6-ацетил-2,2-диметил-3,4-эпокси-2Н-1-бензопирана без применения лиганда, отдающего электрон.
С помощью 8N NaOH установили рН раствора гипохлорита натрия (8,0 мл, 17,3% вес. /об. ), воды (14 мл) и 0,05 М Na2HPO4 (10 мл), равным 13. К дихлорметану (20 мл) добавили 6-ацетил-2,2-диметил-2Н-1-бензопиран (2,0 г, 10,0 ммолей), R, R-Mn-солевой катализатор - R,R-[5,6-бис(3,5-ди-трет-бутилсалицилидамино)-1,3-диоксепан] марганец(III)хлорид (D31, 63 мг, 1 мол.%) и смесь перемешивали при комнатной температуре в течение всей ночи. Анализ HPLC показал, что около 13% хромена все же осталось.
Смесь разбавили дихлорметаном (50 мл) и фильтровали через целит. Органическую фазу отделили, затем промыли водой (100 мл) и выпарили досуха до получения сырого названного соединения в виде масла (2,1 г, выход 96 вес.%). Анализ этой пробы хиральной HPLC показал е.е. 86%.
Пример 7. Получение (3S,4S)-6-ацетил-2,2-диметил-3,4-эпокси-2Н-1-бензопирана с использованием пиридин-N-оксида в качестве лиганда, отдающего электрон.
Повторили методику примера 6, но с добавлением пиридин-N-оксида (1,9 г, 20 ммолей). Анализ HPLC показал, что после перемешивания в течение всей ночи при комнатной температуре реакция завершилась. Тем же самым путем выделили сырой продукт, получив при этом 2,3 г названного соединения с е.е. 95%. Названное соединение получили энантиомерно чистым (е.е. > 98%) посредством перекристаллизации сырого продукта из диизопропилового эфира, степень извлечения составила 50%.
Пример 8. Получение (3R,4R)-6-ацетил-2,2-диметил-3,4-эпокси-2Н-1-бензопирана с использованием изохинолин-N-оксида в качестве лиганда, отдающего электрон.
Повторили методику примера 5, но с добавлением вместо пиридин-N-оксида изохинолин-N-оксида (1,74 г, 12 ммолей). Анализ HPLC показал, что после перемешивания в течение 30 минут при комнатной температуре реакция завершилась. Тем же самым путем выделили сырой продукт, получив названное сырое соединение в виде коричневого масла (5,1 г). Хиральная HPLC показала, что е.е. составил 94%. Названное соединение получили энантиомерно чистым (е.е. > 99,8%) путем перекристаллизации из диизопропилового эфира, степень извлечения составила 48%.
Пример 9. Получение (3S,4S)-6-ацетил-2,2-диметил-3,4-эпокси-2Н-1-бензопирана.
С помощью 8N NаОН установили рН раствора гипохлорита натрия (31 мл, 12,1% вес. /об. , 50 ммолей), воды (34 мл) и 0,05 М Nа2НРО4 (25 мл), равным 13. К дихлорметану (50 мл) добавили 6-ацетил-2,2-диметил-2Н-1-бензопиран (5,0 г, 25 ммолей), изохинолин-N-оксид (0,362 г, 5 ммолей, 0,2 экв) и (3R, 4S)-бис(3,5-ди-трет-бутилсалицилидамино)гидропиранмарганец(III)хлорид (0,032 г, 0,05 ммоля, 0,2 мол.%) и смесь перемешали при 15-20oС. Через 4 часа в соответствии с анализом HPLC реакция завершилась.
Смесь разбавили дихлорметаном и фильтровали через целит. Разделили две фазы и органическую фазу промыли водой (2х200 мл), затем выпарили досуха под пониженным давлением до получения сырого названного соединения в виде бледно-коричневого масла (5,3 г). Хиральная хроматография показала, что е.е. составил 92%.
Названное соединение получили энантиомерно чистым (е.е. > 99,8%), точка плавления 51oС, путем перекристаллизации сырого продукта из диизопропилового эфира, степень извлечения составила 41%.
Пример 10. Получение (3S,4S)-6-циано-2,2-диметил-3,4-эпокси-2Н-1-бензопирана.
Повторили методику, описанную в примере 9, с использованием в качестве хроменовой основы 6-циано-2,2-диметил-2Н-1-бензопирана (4,63 г, 25 ммолей). Образованный сырой эпоксид (3S,4S) имел е.е. 93%. Его перекристаллизовали из 2-пропанола до получения названного соединения (е.е.> 99%), точка плавления 144-145oC, степень извлечения составила 75%.
Пример 11. Получение (3S,4S)-6-бром-2,2-диметил-3,4-эпокси-2Н-1-бензопирана.
Повторили методику, описанную в примере 9, с использованием 6-бром-2,2-диметил-2Н-1-бензопирана (5,98 г, 25 ммолей) в качестве хроменовой основы. Образованный сырой эпоксид (3S,4S) имел е.е. 95%. Его перекристаллизовали из гексана/этилацетата до получения названного соединения (е.е. 99%), точка плавления 101-102oC, степень извлечения 65%.
(С) Примеры с использованием в качестве катализатора соединения формулы (III).
Пример 12. (R)-1-Фенил-1,2-бис(3-трет-бутил-5-метилсалицилидамино)этан марганец(III)хлорид (Е12).
В этаноле (50 мл) растворили (R)-1-фенил-1,2-бис(3-трет-бутил-5-метилсалицилидамино)этан (D 37, 2,42 г, 5,0 ммолей) и добавили твердый тетрагидрат ацетата марганца(II) (2,45 г, 10,0 ммолей). Раствор нагревали в колбе с обратным холодильником в течение 2 часов, затем добавили хлорид лития (безводный) (0,64 г, 15,0 ммолей) и раствор нагрели в колбе с обратным холодильником в течение еще 30 минут. Затем к перемешиваемому раствору добавили охлаждающую воду (1 мл). Осадок удалили фильтрацией, промыли 90% водным раствором этанола (10 мл), затем сушили в вакууме над Р2О5 до получения названного соединения в виде коричневого твердого вещества, 2,73 г, выход 95%.
Пример 13. Хиральное эпоксидирование 2,2-диметил-6-пентафторэтилхромена с использованием (Е12) для получения 2,2-диметил-6-пентафторэтил-1-Н-бензопиран-(3R, 4R)-эпоксида.
Водный раствор гипохлорита натрия (8,5 вес./об., 17,5 мл, 20,0 ммолей) разбавили до 25 мл водой, после чего добавили 0,05 М NаН2РО4 (водный раствор) (10 мл). Установили рН равным 11,3 и раствор охладили до 0oС, затем добавили к раствору 2,2-диметил-6-пентафторэтилхромена (2,78 г, 10,0 ммолей) и (Е12) (0,115 г, 0,20 ммолей) в метиленхлориде (10 мл) при 0oС. Реакционную смесь перемешивали в течение 1 часа при 0oС, затем при комнатной температуре в течение всей ночи.
Добавили гексан (100 мл) и воду (50 мл) и отделили органический слой. Водный слой экстрагировали дополнительной порцией гексана (100 мл), смешанный органический слой сушили над МgSO4, удалили в вакууме растворитель, получив названное соединение в виде коричневого масла, 2,7 г (выход 94%).
Масло очистили тонкослойной (флэш) хроматографией (силикагель 60, MERCK 9385, 230-400 меш) (30 г) при элюировании 0,5% диэтиловым эфиром в гексане до получения названного соединения в виде бледно-желтого, частично кристаллического твердого вещества, 2,11 г, выход 72%, одинаковый с достоверной пробой (1Н ЯМР, TLC (тонкослойная хроматография), HPLC)), как показала хиральная HPLC, е.е. 63%.
Пример 14. (R)-1-Фенил-1,2-бис(3,5-ди-трет-бутилсалицилидамино)этанмарганец(III)хлорид (Е14).
(R)-1-Фенил-1,2-бис(3,5-ди-трет-бутилсалицилидамино)этан (D38, 1,70 г, 3,0 ммоля) растворили в этаноле (30 мл) и добавили тетрагидрат ацетата марганца(II) (1,47 г, 6,0 ммолей). Раствор нагрели в колбе с обратным холодильником в течение 16 часов, затем добавили хлорид лития (0,38 г, 9,0 ммолей), реакционную смесь нагрели в колбе с обратным холодильником в течение еще 30 минут, затем охладили до комнатной температуры.
К перемешиваемому раствору добавили воду (1 мл) и полученный осадок удалили фильтрацией, получив при этом продукт в виде коричневого твердого вещества, которое сушили в вакууме над Р2О5 до получения 2,56 г названного соединения, выход 78%.
Пример 15. Хиральное эпоксидирование 2,2-диметил-6-пентафторэтилхромена с использованием (Е14) для получения 2,2-диметил-6-пентафторэтил-1Н-бензопиран-(3R, 4R)-эпоксида.
Водный раствор гипохлорита натрия (8,5% вес./об., 17,5 мл, 20,0 ммолей) разбавили до 25 мл водой, после чего добавили 0,05 М NaH2PO4 (водный раствор) (10 мл). Установили рН равным 11,3 и раствор охладили до 0oС, затем добавили к раствору 2,2-диметил-6-пентафторэтилхромена (2,78 г, 10,0 ммолей) и (R)-1-фенил-1,2-бис(3,5-ди-трет-бутилсалицилидамино)этанмарганец(III)хлорида (E14) (0,131 г, 0,20 ммолей) в метиленхлориде (10 мл) при 0oС. Реакционную смесь перемешивали в течение 2 часов при 0oС, затем всю ночь при комнатной температуре.
Добавили гексан (100 мл) и воду (50 мл) и отделили органический слой. Водный слой экстрагировали дополнительной порцией гексана (100 мл), смешанные органические слои сушили над MgSO4, удалили в вакууме растворитель, получив названное соединение в виде желтого масла, 2,91 г (выход 99%).
Масло очистили тонкослойной (флэш) хроматографией (силикагель 60, MERCK 230, 400 меш) (40 г) при элюировании 0,5% диэтиловым эфиром в гексане до получения названного соединения в виде бледно-желтого кристаллического твердого вещества, 1,81 г, выход 62%, одинаковый с достоверной пробой (1H ЯМР, TLC, HPLC), как показала хиральная HPLC, е.е. 68%.
Пример 16. (S)-1-Метил-1,2-бис(3-трет-бутил-5-метилсалицилидамино)этанмарганец(III)хлорид (Е16).
(S)-1-Метил-1,2-бис(3-трет-бутил-5-метилсалицилидамино)этан (D39) (338 мг, 0,8 ммолей) растворили в EtOH (8 мл) и добавили тетрагидрат ацетата марганца(II) (392 мг, 1,6 ммоля). Смесь нагревали в колбе с обратным холодильником в течение 2 часов, затем добавили хлорид лития (102 мг, 2,4 ммоля) и после дополнительного нагрева в колбе с обратным холодильником в течение одного часа смесь охладили до окружающей температуры. Добавили несколько капель воды, полученный осадок отфильтровали и сушили в вакууме над Р2О5 до получения названного соединения в виде коричневого порошка, 270 мг (выход 66%).
Пример 17. Хиральное эпоксидирование 2,2-диметил-5-пентафторэтилхромена с использованием (Е16) для получения 2,2-диметил-6-пентафторэтилхромен-(3S, 4S)-эпоксида.
Водный раствор гипохлорита натрия (16,75% вес./об., 8,9 мл, 20 ммолей) разбавили до 25 мл водой, после чего добавили 0,05 М NаН2РО4 (водный раствор) (10 мл). Установили рН равным 11,3 и раствор охладили до 0oС, затем добавили к раствору 2,2-диметил-6-пентафторэтилхромена (2,78 г, 10,0 ммолей) и (S)-1-метил-1,2-бис(3-трет-бутил-5-метилсалицилидамино)этанмарганец(III)хлорида (Е16, 102 мг, 0,20 ммолей) в метиленхлориде (10 мл) при 0oС. Реакциционную смесь перемешивали в течение одного часа при 0oС, затем всю ночь при комнатной температуре.
Добавили гексан (100 мл) и воду (50 мл) и отделили органический слой. Водный слой экстрагировали дополнительной порцией гексана, объединенные органические слои сушили над МgSО4 и растворитель удалили в вакууме, получив при этом сырое названное соединение в виде коричневого масла, 2,78 г (выход 95%). Количественный анализ (HPLC) показал, что оно содержит 2,27 г (выход 77%) названного соединения, одинаковый с достоверной пробой (TLC, HPLC), как показала хиральная HPLC, е.е. 32%.
Пример 18. (S)-1-Изопропил-1,2-бис(3-трет-бутил-5-метилсалицилидамино)этанмарганец(III)хлорид (Е18).
В этаноле (10 мл) растворили (S)-1-изопропил-1,2-бис(3-трет-бутил-5-метилсалицилидамино)этан (D40, 240 мг, 0,53 ммоля) и добавили дигидрат ацетата марганца(III) (0,14 г, 0,53 ммоля). Смесь нагревали в колбе с обратным холодильником в течение 2 часов, затем добавили хлорид лития (34 мг, 0,8 ммоля). После дополнительного нагрева в колбе с обратным холодильником в течение одного часа раствор охладили, удалили в вакууме растворитель и остаток подвергли хроматографии на диоксиде кремния (MERCK 9385, 20 г, при элюировании 0,6% метанолом в хлороформе) до получения названного соединения в виде коричневого порошка 60 мг (выход 21%).
Пример 19. Хиральное эпоксидирование 2,2-диметил-6-пентафторэтилхромена с использованием (Е18) для получения 2,2-диметил-6-пентафторэтилхромен-(3S, 4S)-эпоксида.
Водный раствор гипохлорита натрия (15,24%, вес./об., 2 мл, 4 ммоля) разбавили до 5 мл водой. Добавили 0,05 М NаН2РО4 (водный раствор) (2 мл) и установили рН равным 11,3. Раствор охладили до 0oС, затем добавили к раствору (0,56 г, 2 ммоля) и катализатору - (S)-1-изопропил-1,2-бис(3-трет-бутил-5-метилсалицилидамино)этанмарганец(III)хлориду (Е18, 21,5 мг, 0,04 ммоля) в метиленхлориде (6 мл). Смесь перемешивали при 0oС в течение 1 часа, затем при комнатной температуре всю ночь.
Добавили гексан (20 мл) и воду (10 мл) и отделили органический слой. Водную фазу экстрагировали дополнительной порцией гексана (20 мл), смешанную органическую фазу сушили (MgSO4) и растворитель удалили в вакууме, получив при этом названное соединение в виде желтого масла (0,51 г). Количественный анализ (НРLС) показал, что оно содержало 0,42 г (выход 71%) названного соединения, одинакового с достоверной пробой (TLC, HPLC), как показала хиральная НРLС, е.е. 23%.
Пример 20. Хиральное эпоксидирование 6-ацетил-2,2-диметилхромена с использованием (Е14) для получения 6-ацетил-2,2-диметилхромен-(3R, 4R)-эпоксида.
С помощью 8N NaОН установили рН раствора гипохлорита натрия (8,6 мл, 17,3% вес./об.), воды (14 мл) и Na2HPO4 (0,05 М, 10 мл) равным 11,3. Добавили 6-ацетил-2,2-диметилхромен (2 г), (Е14) (65,6 ммг, 1 мол.%) и дихлорметан (20 мл) и смесь быстро перемешивали при комнатной температуре всю ночь.
Смесь разбавили дихлорметаном (50 мл) и фильтровали через целит. Два слоя разделили и органическую фазу промыли водой (100 мл), высушили досуха, получив при этом названное соединение (2,0 г, 92%), е.е. 67%, как показала хиральная НРLС.
Пример 21. Хиральное эпоксидирование 6-ацетил-2,2-диметилхромена с использованием Е14 для получения 6-ацетил-2,2-диметилхромен-(3R,4R)эпоксида при использовании пиридин-N-оксида в качестве катализатора, отдающего электрон.
Повторили реакцию примера 20 при добавлении пиридин-N-оксида (1,9 г, 2 экв). При использовании хиральной НРLС было найдено, что е.е. названного продукта составляет 79%.
Описания промежуточных соединений для получения соединений формулы (II) (которые описаны в WO 94/03271)
Описание 1.
(+)-2,5-Дигидро-3-нитрофуран (D1).
Смесь (±)-транс-3-хлормеркурио-4-нитро-2,5-дигидрофурана 1 (38,54 г, 109,6 ммолей) и Et3N (11,07 г, 109,6 ммолей) в CH2Cl2 (2,2п) при 25oС перемешивали в течение 1,25 часа. Добавили 5% водный раствор лимонной кислоты (1,1 л) и перемешивание продолжили в течение еще 5 минут. Смесь фильтровали через целит, отделили органическую фазу, которую промыли 5% водным раствором лимонной кислоты (220 мл), сушили над Nа2SО4 и концентрировали в вакууме. После проведения хроматографии остатка на диоксиде кремния (MERCK 9385 г, 300 г) при элюировании CHCl3-гексаном (1:1-->1:0) получили (D1) в виде бледно-желтого масла, которое выкристаллизовали в холодной установке, 5,45 г (43,2%).
δ (CDCl3): 4,95 (4H, s) и 7,10 (1Н, s).
1. P.Bitha и Y.-I. Lin. J. Heterocyclic Chem., 1988, 25, 1035-1036.
Описание 2.
(±)-3,4-Диаминотетрагидрофуран (D2).
Раствор (±) 4-амино-3-нитротетрагидрофурана, полученный из (D1) с помощью способа Битца и Лина 1, (4,66 г, 35,3 ммоля) в ЕtОН (100 мл), содержащий 10% палладия на углероде (2,5 г), гидрировали в качающейся колбе Парра при давлении 35 рsi (2,4609 кг/см2) в течение 65 часов при 20oС. Суспензию фильтровали, твердое промыли EtOH (100 мл) и смешанный фильтрат выпарили в вакууме, получив при этом (±)-(D2) в виде бесцветного масла, 3,26 г (81,5%).
δ (CDCl3): 1,40 (4H, bs), 3,20 (2H, m), 3,50 (2H, dd) и 4,08(2H, dd).
Описание 3.
(±)-3,4-бис(3-трет-бутил-5-метилсалицилидамино)тетрагидрофуран (D3).
Раствор рацемического диамина (D2) (8,55 мг, 8.38 ммоля) и 3-трет-бутил-5-метилсалицилового альдегида (3,22 г, 16,76 ммолей) в ЕtОН (50 мл) нагрели в колбе с обратным холодильником в течение 1,5 часа. Растворитель удалили в вакууме и остаток подвергли хроматографии на диоксиде кремния (MERCK 9385, 300 г) с использованием в качестве элюента CHCl3, получив при этом (±)-(D3) в виде бледно-желтых игольчатых кристаллов, 1,35 г (35,8%).
δ (CDCl3): 1,42 (18H, s), 2,25 (6H, s) 3,95-4,10 (2Н, m), 4,43 (2Н, q), 6,90 (2H, d), 7,15 (2H, d), 8,30 (2H, s) и 13,10 (2Н, bs).
Описание 4.
(S,S)-транс-3,4-бис(Метансульфонилокси)тетрагидрофуран (D4).
Раствор 1,4-ангидро-L-треита (2,45 г, 23,5 ммоля), например, Аldrich Сhemical Company (Олдрич Кемикал Компани) в смеси ТНF (75 мл) и Еt2О (75 мл) при 0oС последовательно обработали триэтиламином (7,2 мл, 51,7 ммоля, 2,2 экв) и метансульфонилхлоридом (3,82 мл, 49,35 ммоля, 2,1 экв). Смесь перемешивали в течение 4 часов, затем хранили при 0oС с вечера и всю ночь (~ 16 часов).
Реакционную смесь фильтровали и твердое промыли THF (20 мл). Смешанный фильтрат выпарили в вакууме и разделили между 10% водным раствором лимонной кислоты (60 мл) и ЕtОАс (150 мл). Органическую фазу сушили (MgSO4) и выпарили до получения (D4) в виде бесцветного масла, 5,82 г (95%).
δ (CDCl3): 3,12 (6H, bs), 4,00 (2H, dd), 4,18 (2H, dd) и 5,25 (2H, dd).
Описание 5.
(S,S)-транс-3,4-Диазидотетрагидрофуран (D5).
Смесь димезилата (D4) (5,80 г, 22,3 ммоля) и азида лития (5,46 г, 111,5 ммоля, 2,5 экв) в DMSO (60 мл) нагрели при 100-110oС в течение 40 часов. После охлаждения до окружающей температуры реакционную смесь разбавили водой (1 л) и экстрагировали ЕtОАс (1 л, 2х0,75 л). Объединенную органическую фазу промыли водой (0,5 л) и рассолом (0,5 л), сушили над МgSO4 и выпарили в вакууме до получения названного соединения в виде бледно-желтого масла, 2,18 г (61,5%).
δ (CDCl3): 3,75 (2H, dd) и 3,90-4,05 (4Н, m).
Описание 6.
(S,S)-транс-3,4-Диаминотетрагидрофуран.
К алюмогидриду лития (2,05 г, 54 ммоля) в сухом ТНF (тетрагидрофуране) (150 мл) при 0oС по каплям в течение 10 минут добавили диазид (D5) (2,08 г, 13,5 ммолей) в ТНF (50 мл). Через 15 минут раствор нагрели до окружающей температуры, затем перемешивали в течение 16 часов.
Реакционную смесь повторно охладили до 0oС, и затем резко последовательно охладили Н2О (2 мл), 15% водным раствором NаОН (2 мл) и дополнительно водой (6 мл) и нагрели до окружающей температуры. После перемешивания в течение 1 часа смесь фильтровали через целит, промыли THF (2х150 мл) и объединенный фильтрат выпарили в вакууме, получив при этом (D6) в виде бледно-желтого масла, 1,28 г (93%).
δ (CDCl3): 1,30 (4H, bs), 3,30 (2H, dd), 3,50 (2Н, dd) и 4,08 (2Н, dd).
Описание 7.
(S, S)-транс-3,4-бис(3-трет-Бутил-5-метилсалицилидамино)тетрагидрофуран (D7).
Раствор (S, S)-диамина (D6) (1,26 г, 12,35 ммолей) и 3-трет-бутил-5-метилсалицилового альдегида (4,74 г, 24,70 ммолей) в EtOH (75 мл) нагрели в колбе с обратным холодильником в течение 3,5 часа. Раствор охладили и растворитель удалили в вакууме, получив при этом сырое соединение (5) в виде желтого масла, 5,50 г (99%).
Пробу сырого материала (4,55 г) подвергли хроматографии на диоксиде кремния (MERCK 9385, градиент CHCl3 в гексане), получив при этом (D7) в виде желтой пены, 4,39 г (выход 95,5%).
δ (CDCl3): 1,42 (18Н, s), 2,25 (6Н, s), 3,95-4,10 (4H, m), 4,33 (2Н, q), 6,90 (2H, d), 7,15 (2Н, d), 8,30 (2Н, s) и 13,15 (2Н, bs).
Описание 8.
(2R,3R)-1,4-Дибензилокси-2,3-диметансульфонилоксибутан.
К раствору (2R, 3R)-(+)-1,4-дибензилокси-2,3-бутандиола (25,3 г, 83,7 ммоля, например, Олдрич Кемикал Компани) в дихлорметане (165 мл), охлажденному в солевой ванне со льдом, добавили метансульфонилхлорид (13,0 мл, 167,4 ммоля). Затем медленно добавили триэтиламин (23,3 мл, 167,4 ммоля) так, чтобы температура не превысила 5oС. После завершения добавления реакционную смесь перемешивали в ванне со льдом в течение 3 часов. Затем добавили воду (600 мл) и отделили органическую фазу. Водную фазу реэкстрагировали дихлорметаном (200 мл) и объединенные органические фазы промыли водой (400 мл) и рассолом (400 мл), сушили (МgSO4) и растворитель выпарили, получив при этом бледно-желтое твердое вещество. При растирании в порошок с диэтиловым эфиром получили названное соединение (28,2 г, 74%) в виде бесцветных кристаллов, точка плавления 72-73oС.
1Н ЯМР (CDCl3): δ 3,03 (s, 6H, 2xCH3), 3,76 (m, 4Н, 2xCH2O), 4,48 (d, 2H, CH2Ph), 4,57 (d, 2H, CH2Ph), 5,00 (m, 2H, 2xCH), 7,27-7,39 (m,10H, 2xPh).
13C ЯМР (СDCl3): δ 38,8 (2xCH3), 68,7 (2xCH2), 73,7 (2xCH2), 78,7 (2xCH), 128,1, 128,2, 137,0 (2xPh).
EI-MS: m/e 459 (MH+), 367 (M+-CH2Ph).
C20H26O8S2. Вычислено,%: С 52,39; Н 5,72.
Найдено,%: С 52,36; Н 5,59.
Описание 9.
(2R, 3R)-1,4-Диметансульфонилоксибутан-1,4-диол.
В ацетоне (500 мл) растворили (2R,3R)-1,4-дибензилокси-2,3-диметансульфонилоксибутан (27,6 г, 60,3 ммоля) (D8), добавили суспензию 10% Рd/С (29,9 г) в ацетоне (300 мл) и смесь гидрировали при давлении 1 атм в течение двух часов при окружающей температуре. Затем смесь три раза фильтровали через прокладку из кремнезема и целита и растворитель выпарили, получив при этом названное соединение в виде масла цвета соломы (14,7 г, 87%), которое при стоянии затвердело.
1Н ЯМР (DMSO-d6): δ 3,24 (s, 6H, 2xCH3), 3,69 (m, 4H, 2хСН2), 4,76 (m, 2Н, 2хСН), 5,33 (t, 2Н, 2хОН).
13С ЯМР (DМSО-d6): δ 38,1 (2xCH3), 59,7 (2xCH2), 80,3(2хСН).
EI-MS: m/e 279 (MH+), 261 (MH+-H2O), 183 (M+-OMs), 165 (М+-OMS, H2O).
Описание 10.
(6R,7R)-Диметансульфонилокси-2,4,9,11-тетраоксадодекан.
(2R, 3R)-Диметансульфонилоксибутан-1,4-диол (14,7 г, 52,9 ммоля) (D9) растворили в диметоксиметане (89,5 мл) и дихлорметане (30 мл) при 40oС. Добавили бромид лития (0,91 г) и моногидрат п-толуолсульфоккслоты (1,01 г, 5,29 ммоля), и смесь нагревали в колбе с обратным холодильником в течение 3-х часов. Реакционную смесь охладили до окружающей температуры и затем влили в насыщенный раствор бикарбоната натрия (200 мл), экстрагировали этилацетатом (2х200 мл), сушили (МgSО4) и выпарили, получив при этом бесцветное масло (8,2 г, 42%). Его очистили колоночной хроматографией на диоксиде кремния при элюировании 0,1% метанолом в дихлорметане, получив при этом названное соединение в виде бесцветного масла (8,2 г, 42%).
1Н ЯМР (CDCl3): δ 3,13 (s, 6H, 2xCH3), 3,39 (s, 6H, 2xOCH3), 3,87 (m, 4H, 2xCH2), 4,66 (m, 4H, 2xOCH2O), 5,02 (m, 2H, 2xCH).
13C ЯМР (СDСl3): δ 38,8 (2xSCH3), 55,8 (2xOCH3), 66,1 (2xCH2), 78,4 (2xCH), 96,8 (2xOCH2O).
CI-MS: m/e 384 (MNH4 +).
C10H22O10S2. Вычислено,%: С 32,78; Н 6,05.
Найдено,%: С 32,22; Н 5,62.
Описание 11.
(5R,6R)-Диметансульфонилокси-1,3-диоксепан.
Раствор (6R,7R)-диметансульфонилокси-2,4,9,11-тетраоксадодекана (8,2 г, 22,4 ммоля) (D10) и моногидрата п-толуолсульфокислоты (0,26 г, 1,34 ммоля) в толуоле (165 мл) нагревали в колбе с обратным холодильником всю ночь. Растворитель выпарили и коричневый остаток растерли в порошок с диэтиловым эфиром, получив при этом названное соединение в виде не совсем белого твердого вещества (5,9 г, 91%), точка плавления 133-134oС.
1Н ЯМР (CDCl3): δ 3,13 (s, 6H, 2xCH3), 3,84 (m, 2H, 2CH2), 4,06 (m, 2H, CH2), 4,77 (s, 2Н, ОСН2О), 4,81 (m, 2Н, 2хСН).
13С ЯМР (СDCl3): δ 38,8 (2xSCH3), 64,1 (2хСН2), 78,3 (2хСН), 94,6 (ОСН2О).
EI-MS: m/e 291(MNH4 +), 195(М+-ОМs).
С7Н14O8S2. Вычислено,%: С 28,96; Н 4,86.
Найдено,%: С 29,22; Н 4,61.
Описание 12.
(5R,6R)-Диазидо-1,3-диоксепан.
Смесь (5R, 6R)-Диметансульфонилокси-1,3-диоксепана (5,0 г, 17,2 ммоля) (D11) и азида лития (4,2 г, 86 ммолей) в диметилсульфоксиде (60 мл) перемешивали и нагревали до 110-120oС всю ночь. Затем реакционную смесь охладили, влили в воду (200 мл) и экстрагировали этилацетатом (2х150 мл). Объединенные органические фазы промыли водой (2х150 мл) и рассолом (150 мл), сушили (МgSO4) и выпарили, получив при этом названное соединение в виде коричневого масла (2,7 г, 85%).
1Н ЯМР (CDCl3): δ 3,49 (m, 2Н, 2хСН), 3,74 (m, 2Н, 2хСН2), 3,93 (m, 2Н, СН2), 4,73 (s, 2Н, ОСН2О).
13С ЯМР (CD Cl3): δ 64,3 (2xCH), 64,6 (2хСН2), 94,3 (ОСН2О).
ЕI-МS: м/е 185 (МН+), 157(МН+-N2), 142(М+-N3).
C5H8N6O2. Вычислено,%: С 32,61; Н 4,38; N 45,63.
Найдено,%: С 32,33; Н 4,67; N 45,38.
Описание 13.
(5R,6R)-Диамино-1,3-диоксепан.
К суспензии алюмогидрида лития (2,1 г, 55,3 ммоля) в сухом тетрагидрофуране (70 мл) при 0oС в атмосфере аргона по каплям добавили раствор (5R, 6R)-диазидо-1,3-диоксепана (2,6 г, 14,1 ммоля) (D12) в сухом тетрагидрофуране (50 мл). Во время добавления температуру реакции поддерживали ниже 10oС с помощью солевой ванны со льдом. После этого реакционную смесь нагрели до окружающей температуры и перемешивали в течение еще 1,5 часов. Затем реакционную смесь повторно охладили, после чего резко охладили путем добавления воды (2 мл), 2 M NаОН (2 мл) и воды (4 мл), температуру опять поддерживали ниже 0oС посредством солевой ванны со льдом. Резко охлажденную реакционную смесь нагрели до окружающей температуры, перемешивали в течение еще 2 часов, затем фильтровали через целит и фильтрованную прокладку хорошо промыли тетрагидрофураном. Объединенные фильтраты выпарили, получив при этом названное соединение в виде бледно-желтого масла (1,3 г, 70%).
1Н ЯМР (CDCl3): δ 1,56 (brs, 4H, 2xNH3), 2,62 (m, 2Н, 2хСН), 3,58 (m, 2Н, СН2), 3,77 (m, 2Н, 2хСН2), 4,72 (s, 2Н, ОСН2О).
13С ЯМР (СDСl3): δ 57,9 (2хСН), 67,5 (2хСН2), 93,8 (ОСН2О).
C5H12N2O2. Вычислено,%: С 45,44; Н 9,15; N 21,20.
Найдено,%: С 45,13; Н 8,76; N 19,58.
EI-MS: m/e 133(MH+), 116(М+-NН2)+.
Описание 14.
(5R,6R)-Ди-(3,5-ди-трет-бутил)салицилидамино-1,3-диоксепана.
(5R, 6R)-Диамино-1,3-диоксепан (1,0 г, 7,6 ммоля) (D13) и 3,5-ди-трет-бутилсалициловый альдегид (3,6 г, 15.4 ммоля, 2 экв) растворили в этаноле (100 мл) и раствор перемешивали при нагревании в колбе с обратным холодильником в течение 3 часов. Затем реакционную смесь охладили, выпарили растворитель и остаток очистили колоночной хроматографией на диоксиде кремния при элюировании 4% диэтиловым эфиром в гексане. Получили названное соединение в виде светло-желтой пены (3,5 г, 82%).
1Н ЯМР (CDCI3): δ 1,23 (s, 18H, 6xCH3), 1,41 (s,18H, 6xCH3), 3,85 (m, 2H, CH2), 4,07 (m, 2H, CH2), 4,87 (s, 2Н, ОСН2О), 6,99 (d, 2Н, Аr), 7,33 (d, 2Н, Аr), 8,33 (s, 2H, 2xCH=N), 13,20 (brs, 2Н, 2хОН).
13С ЯМР (СDCI3): δ 29,4 (6xCH3), 31,4 (6xCH3), 34,1 
35,0  , 67,7 (2xCH), 73,8 (2хСН2), 94,2 (ОСН2О), 117,6, 126,4, 127,4, 136,6, 140,3, 157,9 (Аr), 168,4 (2хС=N).
, 67,7 (2xCH), 73,8 (2хСН2), 94,2 (ОСН2О), 117,6, 126,4, 127,4, 136,6, 140,3, 157,9 (Аr), 168,4 (2хС=N).
C35H52N2O4. Вычислено,%: С 74,43; H 9,28; N 4,96.
Найдено,%: С 74,56; Н 9,15; N 4,92.
СI-МS: m/е 566 (МН+).
Описание 15.
(5R, 6R)-Диацетокситетрагидропиран (D15).
Раствор 3,4-ди-О-ацетил-D-ксилаля2 (11,6 г) в 50% водном растворе этанола (400 мл), содержащем РtO2 (400 мг), гидрировали при атмосферном давлении в течение 3,5 часов при 25oС. Суспензию фильтровали через целит, промыли 50% водным раствором этанола (50 мл) и водой (50 мл), и объединенный фильтрат выпарили в вакууме, получив при этом названное соединение в виде бесцветного масла, 9,6 г (85%).
δ (CDCl3): 1,30-1,50 (1H, m), 2,10 (6Н, s), 2,10-2,20 (1Н, m), 3,35-3,60 (2Н, m), 3,80-4,00 (2Н, m) и 4,80-5,00 (2Н, m).
2. Dictionary of Organic Compounds, 5th Edition, 1982, Chapman & Hall, London, 579 и ссылки, приведенные в нем.
Описание 16.
(3R,4R)-Диаметансульфонилокситетрагидропиран (D16).
Натрий (~50 мг) растворили в метаноле (100 мл) при окружающей температуре. К полученному раствору добавили раствор эфира двухосновной кислоты (D15) (9,56 г, 47,3 ммоля) в метаноле (100 мл) и смесь перемешивали в течение 72 часов. Добавили смолу Amberlite IR 12 OH+ (20 г) и смесь фильтровали. После концентрирования фильтрата в вакууме получили диол в виде бесцветного масла. Его растворили в смеси тетрагидрофурана (220 мл) и диэтилового эфира (220 мл). Добавили триэтиламин (10,86 г, 107,5 ммоля), раствор охладили до 0oС. При 0oС по каплям добавили метансульфонилхлорид (11,76 г, 102,7 ммоля), раствор перемешивали в течение еще одного часа, затем хранили при 4oС в течение 16 часов. Полученную суспензию фильтровали и твердое промыли тетрагидрофураном (2х95 мл) и диэтиловым эфиром (2х180 мл). Объединенный фильтрат выпарили в вакууме и остаток разделили между этилацетатом (200 мл) и 10% водным раствором лимонной кислоты (200 мл). Органическую фазу сушили (МgSO4), фильтровали в вакууме до получения названного соединения в виде бесцветной пены, 12,07 г (93%).
δ (CDCl3): 3,10 (6Н, s), 2,00-2,40 (2Н, m), 3,40-4,20 (4Н, m), 4,55-4,65 (1H, m) и 4,70-4,85 (1Н, m).
Описание 17.
(3R,4S)-Диаминотетрагидропиран (D17).
В диметилсульфоксиде (88 мл) растворили димезилат (D16) (12,07 г, 44 ммоля) и обработали азидом лития (10,8 г, 220 ммолей). Смесь нагревали при 100oC в течение 40 часов, затем охладили до окружающей температуры, влили в воду (1,03 л) и экстрагировали этилацетатом (1,03 л, 2х0,59 л). Объединенную органическую фазу промыли водой (300 мл) и рассолом (300 мл), сушили над MgSO4 и концентрировали в вакууме, получив при этом сырой диазид в виде коричневого масла 3,7 г. Его растворили в тетрагидрофуране (45 мл) и по каплям добавили к холодной (0oС) суспензии алюмогидрида лития (3,34 г, 88 ммолей) в тетрагидрофуране (220 мл), поддерживая температуру ниже 10oС. После завершения добавления суспензию перемешивали в течение 0,5 часов при 0oС, затем нагрели до окружающей температуры и перемешивали в течение 16 часов.
Смесь опять охладили до 0oС, затем резко последовательно охладили водой (3,34 мл) в тетрагидрофуране (5 мл), 15% водным раствором гидроксида натрия (3,34 мл) и дополнительно водой (10 мл). Смесь нагрели до окружающей температуры, перемешивали в течение 1 часа, затем фильтровали через целит, промыли тетрагидрофураном (2х400 мл). Смешанный фильтрат концентрировали в вакууме, получив при этом названный диамин в виде бесцветного масла, 2,62 г (51%).
δ (CDCl3): 1,20-1,90 (6H, m), 2,40-2,50 (2H, m), 2,90-3,40 (2Н, m) и 3,80-4,00 (2Н, m).
Описание 18.
(3R,4S)-бис(3,5-Ди-трет-бутилсалицилидамино)тетрагидропиран (D18).
К диамину (D17) (2,55 г, 22 ммоля) в этаноле (220 мл) добавили 3,5-ди-трет-бутилсалициловый альдегид (10,3 г, 44 моля). Смесь нагрели в колбе с обратным холодильником в течение 2 часов, охладили до окружающей температуры, фильтровали и кристаллический продукт сушили в вакууме до получения названного соединения в виде желтых кристаллов, 4,81 г (40%).
δ (CDCl3): 1,29 (18Н, s), 1,40 (18Н, s), 1,50-2,20 (2Н, m), 3,50-3.70 (4H, m), 4,00-4,15 (2H, m), 7,00 (2Н, bs), 7,35 (2Н, bs), 8,33 (1Н, s), 8,37 (1Н, s) и 13,20 (2Н, bs).
Описание 19.
(3R,4S)-бис(3-трет-Бутил-5-метилсалицилидамино)тетрагидропиран (D19).
Раствор диамина (D17) (0,62 г, 5,35 ммоля) и 3-трет-бутил-5-метилсалицилового альдегида (2,05 г, 10,7 ммоля) в этаноле (40 мл) нагревали в колбе с обратным холодильником в течение 2 часов. Раствор охладили, затем хранили при 4oС в течение 70 часов до получения желтого осадка. Его отфильтровали, промыли холодным 95% водным раствором этанола (5 мл) и сушили в вакууме до получения названного соединения, 1,22 г (49%).
δ (CDCl3): 1,40 (18Н, s), 1,80-2,20 (2H, m), 2,20 (6H, s), 3,40-3,70 (4Н, m), 4,00-4,20 (2H, m), 6,80 (2H, bs), 7,05 (2Н, bs), 8,27 (1Н, s), 8,30 (1H, s) и 13,30 (2H, bs).
Описание 20.
(3S,4S)-бис(3,5-Ди-трет-бутилсалицилидамино)тетрагидрофуpaн (D20).
Раствор (S, S)-диамина (D6) (0,96 г, 9,4 ммоля) и 3,5-трет-бутилсалицилового альдегида (4,4 г, 18,8 ммоля) в этаноле (90 мл) нагрели в колбе с обратным холодильником в течение 2 часов. Смесь охладили до 0oС, затем фильтровали, твердое промыли холодным этанолом и сушили, получив при этом названное соединение в виде желтых кристаллов, 3,07 г (61%).
δ (CDCl3): 1,27 (18H, s), 1,45 (18Н, s), 3,95-4,10 (4Н, m), 4,30-4,40 (2Н, m), 7,05 (2Н, d), 7,40 (2Н, d), 8,35 (2Н, s) и 13,20 (2H, s).
Описание 21.
(3S,4R)-Дигидрокси-(2R)-(гидроксиметил)тетрагидрофуран (D21).
Раствор D-глюкаля3 (16,0 г, 0,11 моля) в 50% водном растворе этанола (500 мл) обработали оксидом платины (0,75 г) и гидрировали при окружающей температуре и атмосферном давлении в течение 5 часов. Суспензию обработали углем (50 г), фильтровали через целит (200 г) и твердое промыли 50% водным раствором этанола (300 мл). Объединенные фильтраты выпарили в вакууме и сушили над Р2О5 до получения названного соединения в виде бесцветного масла, 16,0 г (99%).
δ (CD3OD): 1,50-1,70 (1Н, m), 1,80-2,20 (1H, m), 3,00-3,20 (2H, m), 3,30-3,70 (3H, m), 3,80-4,00 (2Н, m) и 4,90 (3H, bs).
3. Dictionary of Organic Compounds, 5th Edition, 1982, Chapman and Hall, London, 2754 и ссылки, приведенные в нем.
Описание 22.
(3S,4R)-Дигидрокси-(2R)-(трифенилметоксиметил)тетрагидропиран (D22).
Раствор триола (D21) (1,76 г, 11,9 ммолей) в пиридине (20 мл) обработали тритилхлоридом (3,31 г, 11,9 ммолей) и 4-(диметиламино)пиридином (50 мг). Добавили диизопропилэтиламин (1,92 г, 14,8 ммоля, 1,25 экв) и раствор перемешивали в течение 4 часов при окружающей температуре.
Смесь влили в воду (200 мл) и экстрагировали диэтиловым эфиром (2х200 мл). Объединенную органическую фазу промыли 10% водным раствором лимонной кислоты (100 мл) и рассолом (100 мл), сушили над MgSO4 и концентрировали в вакууме до получения масла. Остаток подвергали хроматографии на диоксиде кремния (элюент: градиент метанола в хлороформе) до получения названного соединения в виде бесцветной пены, 3,70 г (79,7%).
δ (CDCl3): 1,60-1,80 (1H, m), 1,90-2,00 (1Н, m), 2,70 (2Н, bs, D2O обм. ), 3,25-3,50 (5Н, m), 3,60-3,70 (1Н, m), 3,90-4,00 (1Н, m) и 7,20-7,50 (15Н, m).
Описание 23.
(3R, 4R)-Диметансульфонилокси-(2R)-трифенилметоксиметил)тетрагидропиран (D23).
К диолу (D22) (3,10 г, 7,95 ммоля) в смеси диэтилового эфира и тетрагидрофурана (2: 1, 150 мл) добавили триэтиламин (1,76, 17,5 ммоля). Смесь охладили до 0oС и добавили метансульфонилхлорид (1,91 г, 16,7 ммоля). Через 2 часа суспензию фильтровали и фильтрат концентрировали в вакууме, затем заново растворили в этилацетате (200 мл). Раствор промыли 10% водным раствором лимонной кислоты (100 мл) и рассолом (50 мл), затем сушили над МgSО4. Растворитель удалили в вакууме и остаток сушили до получения (12) в виде бесцветного твердого вещества, 4,26 г (95%).
δ (CDCl3): 2,20-2,50 (2H, m), 2,50 (3Н, s), 3,10 (3Н, s), 3,20-3,30 (1Н, m), 3,40-3,60 (3H, m), 3,95-4,10 (1Н, m), 4,70-4,80 (2Н, m) и 7,20-7,50 (15Н, m).
Описание 24.
(3R, 4S)-бис(3,5-Ди-трет-бутилсалицилидамино)-(2S)-трифенилметоксиметил)тетрагидропиран (D24).
Смесь димезилата (D23) (2,85 г, 5,22 ммоля) и азида лития (1,28 г, 26,1 ммоля) в диметилсульфоксиде (20 мл) нагревали при 100-110oC в течение 24 часов. Раствор охладили, влили в воду (200 мл) и экстрагировали этилацетатом (2х300 мл). Объединенную органическую фазу промыли водой (2х300 мл) и рассолом (300 мл) и сушили над МgSО4. После удаления растворителя получили промежуточное соединение диазид в виде желтой пены (1,52 г).
К суспензии алюмогидрида лития (470 мг, 12,4 ммоля) в тетрагидрофуране (30 мл) при 0oС добавили 1,40 г диазида в тетрагидрофуране (30 мл). После перемешивания при 0oС в течение 1 часа смесь нагрели до окружающей температуры и перемешивали в течение 16 часов. Суспензию опять охладили до 0oС и затем резко последовательно охладили водой (0,5 мл), 15% водным раствором гидроксида натрия (0,5 мл) и затем водой (1,5 мл). После нагревания до окружающей температуры и перемешивания в течение 1 часа смесь фильтровали, твердое промыли тетрагидрофураном (2х20 мл) и объединенный фильтрат выпарили, получив при этом сырой диамин в виде пены (1,28 г).
Часть диамина (1,18 г) и 3,5-ди-трет-бутилсалицилового альдегида (1,42 г, 6,08 ммолей) в этаноле (30 мл) нагрели в колбе с обратным холодильником в течение 4 часов, затем охладили до окружающей температуры. Растворитель удалили в вакууме и остаток подвергли хроматографии на диоксиде кремния (элюент: градиент хлороформа в гексане) до получения названного соединения в виде желтого порошка, 210 мг, общий выход из (D23) 8,4%.
δ (CDCl3): 1,25 (9Н, m), 1,30-1,60 (2H, m), 1,32 (9H, s), 1,40 (9Н, s), 1,50 (9Н, s), 2,40-2,55 (1H, s), 2,70-2,80 (1H, s), 3,30-3,60 (2H, m), 3,90-4,39 (3H, m), 6,85 (1H, bs), 7,00-7,35 (16Н, m), 7,38 (1Н, bs), 8,30 (1Н, s), 8,50 (1Н, s), 13,25 (1Н, s) и 13,50 (1Н, s).
Описание 25.
(±)-транс-1-Бензоил-3,4-бис(метансульфонилокси)пиперидин (D25).
(±)-транс-1-Бензоилпиперидин-3,4-диол 4 (3 г, 13,6 ммолей) суспендировали в дихлорметане (70 мл) и добавили триэтиламин (5,74 мл, 43 ммоля). Смесь охладили до -10oС и в течение 5 мин добавили метансульфонилхлорид (2,6 мл, 34 ммоля). Через 15 мин смесь влили в смесь льда и воды (50 мл) и органический слой промыли 5% водным раствором лимонной кислоты (30 мл). Раствор сушили над МgSО4 и концентрировали в вакууме до получения пены, 5,3 г (100%).
δH (CDCl3): 1,95 (2H, m), 2,30 (2Н, m), 3,15 (6Н, s), 4,70 (2Н, m), 4,85 (2Н, m) и 7,45 (5H, m).
V. Petrow and O. Stephenson, J.Pharm. Pharmacol., 1962, 14, 306-314.
Описание 26.
(±)-транс-1-Бензоил-3,4-диазидопиперидин (D26).
Смесь димезилата (D25) (5,3 г, 14 ммолей) и азида лития (3,4 г, 69 ммолей) в диметилсульфоксиде (36 мл) нагревали при 100oС в течение 18 часов. После охлаждения реакционную смесь разделили между дихлорметаном (200 мл) и водой (50 мл). Водную фазу отделили и затем экстрагировали дихлорметаном (100 мл, 50 мл) и смешанные органические экстракты промыли водой (3х50 мл), сушили (Na2SO4) и концентрировали в вакууме. Остаток подвергли хроматографии на диоксиде кремния (элюент: градиент метанола в дихлорметане), получив при этом названное соединение в виде бесцветного твердого вещества, 90 мг (24%).
δH (CDCl3): 1,60 (2H, m), 2,10 (2Н, m), 3,05, 2H, s), 3,20 (2H, m) и 7,40 (5Н, m).
Описание 27.
(±)-транс-1-Бензоил-3,4-диаминопиперидин (D27).
Раствор диазида (D26) (450 мг, 1,7 ммоля) в этаноле (30 мл) обработали катализатором Линдлара (5% Pd/CaCO3, 250 мг) и перемешивали под водородом (1 атм) в течение 24 часов. Смесь фильтровали и растворитель удалили в вакууме, получив при этом названное соединение в виде масла, 350 мг (94%).
δH (DMSO): 1,20 (1H, m), 1,65-1,80 (2Н, m), 2,20 (2Н, m), 2,70 (1Н, m), 3,00 (1H, m), 3,30 (1Н, m), 4,40 (1Н, m) и 7,40 (5Н, m).
Описание 28.
(-)-транс-1-Бензоил-3,4-бис(3,5-ди-трет-бутилсалицилидамино)пиперидин (D28).
Раствор амина (D27) (350 мг, 1,6 ммоля) и 3,5-ди-трет-бутилсалицилового альдегида (960 мг, 4,1 ммоля) в этаноле (40 мл) нагревали в колбе с обратным холодильником в течение 3 часов. Смесь охладили и фильтровали до получения рацемического бис-имина, 6,52 мг (63%).
Отделили 100 мг пробу хиральной HPLC (CHIRALPAK AD, элюент 2% этанол в гексане), получив при этом названное соединение в виде одиночного энантиомера, [α]
δH(CDCl3): 1,20 (18H, s), 1,45 (18H, s), 2,00 (2Н, m), 3,25 (2H, m), 3,45 (1H, m), 3,55 (1H, m), 4,35 (2H, m), 6,95 (2H, s), 7,40 (7Н, m), 8,30 (2Н, s) и 13,15 (2Н, bs).
Описание 29.
(±)-3,4-бис(3-трет-Бутил-5-метилсалицилидамино)тетрагидрофуранмарганец(III)хлорид (D29).
Суспензию рацемического лиганда (D3) (690 мг, 1,53 ммоля) в ЕtОН (25 мл) нагрели с Мn(ОАс)2•4Н2О (750 мг, 3,06 ммолей) в колбе с обратным холодильником в течение 18 часов. Добавили LiСl (195 мг, 4,49 ммоля) и нагревание в колбе с обратным холодильником продолжили в течение еще 0,5 часа. Растворитель удалили в вакууме и остаток подвергли хроматографии на диоксиде кремния (MERCK 9385, 100 г) при элюировании градиентом МеОН в CHCl3 до получения названного соединения в виде коричневого порошка (90 мг, 11%) вместе с непрореагировавшим (D3), 420 мг, степень извлечения 61%.
Описание 30.
(S,S)-транс-3,4-бис(3-трет-Бутил-5-метилсалицилидамино)тетрагидрофуранмарганец(III)хлорид (D30).
Раствор (D7) (0,95 г, 2,11 ммоля) и Мn(ОАс)2•4Н2О (1,03 г, 4,22 ммоля) в EtOH (40 мл) нагрели в колбе с обратным холодильником в течение 17 часов. Добавили хлорид лития (268 мг, 6,33 ммоля) и нагревание в колбе с обратным холодильником продолжили в течение еще 0,5 часа. После охлаждения до окружающей температуры растворитель удалили в вакууме и остаток подвергли хроматографии на диоксиде кремния (MERCK 9385, градиент МеОН в CHCl3) до получения (Е3) в виде коричневого порошка, 26 мг (2,3%) вместе с непрореагировавшим (D7), 683 мг (72%).
Способ В, с использованием ацетата марганца(III) 5.
Раствор (D7) (1,53 г, 3,4 ммоля) в смеси CH2Cl2 (17 мл) и МеОН (17 мл) обработали Mn(OAc)3•2H2O (0,01 г, 3,4 ммоля). Смесь нагревали в колбе с обратным холодильником в течение 3 часов, охладили до окружающей температуры и обработали хлоридом лития (0,21 г, 5,1 ммоля). После перемешивания в течение 16 часов содержание растворителя уменьшили в вакууме до около 8 мл, добавили Еt2О (70 мл) и суспензию перемешивали в течение 1 часа. Смесь фильтровали, твердое промыли Еt2О (3х20 мл) и сушили в вакууме, получив при этом (Е3) в виде коричневого порошка, 1,57 г (86%).
5. T. Matsushita and T.Shono, Bull. Chem. Soc. Japan., 1981, 54, 3743-3748.
Описание 31.
Получение (R,R)-[5,6-бис(3,5-ди-трет-бутилсалицилидамино)-1,3-диоксепан] марганец(III)хлорида (D31).
(5R, 6R)-Ди-(3,5-ди-трет-бутил)салицилидамино-1,3-диоксепан (1,0 г, 1,77 ммоля) (D14) и тетрагидрат ацетата марганца(II) (2,17 г, 8,87 ммоля) суспендировали в 95% водном растворе этанола (50 мл) и смесь нагревали в колбе с обратным холодильником при перемешивании всю ночь. Затем добавили хлорид лития (0,38 г, 8,96 ммоля) и нагревание продолжили в течение еще 30 минут. Затем реакционную смесь охладили, добавили воду (60 мл) и фильтровали через целит. Темный осадок промыли водой, затем растворили в дихлорметане (80 мл), сушили (МgSO4) и растворитель выпарили для получения названного соединения в виде темно-коричневого твердого вещества (0,9 г, 78%).
С35Н50N2О4MnСl. Вычислено,%: С 64,36; Н 7,72; N 4,29.
Найдено,%: С 64,57; Н 7,57; H 4,09.
CI-MS: m/e 565 (МН-Мn,Сl)+, 235 (3,5-ди-трет-бутилсалициловый альдегид Н)+
Описание 32.
(3R,4S)-бис(3,5-ди-трет-Бутилсалицилидамино)тетрагидропиранмарганец(III)хлорид (D32).
Раствор лиганда (D18) (4,81 г, 8,8 ммолей) в дихлорметане-метаноле (1:1, 88 мл) обработали дигидратом триацетата (2,35 г, 8,8 ммоля) и смесь нагревали в колбе с обратным холодильником в течение 4 часов. Добавили хлорид лития (0,56 г, 13,2 ммоля) и нагревание в колбе с обратным холодильником продолжили в течение еще 1 часа. Смесь охладили, концентрировали в вакууме и остаток растерли в порошок с диэтиловым эфиром (220 мл). Твердый продукт отфильтровали, промыли диэтиловым эфиром (2х65 мл) и сушили до получения (5) в виде коричневого порошка, 5,3 г (94%).
Описание 33.
(3R,4S)-бис(3-трет-Бутил-5-метилсалицилидамино)тетрагидропиранмарганец(III)хлорид (D33).
Раствор лиганда (D19) (9,28 г, 2 ммоля) в дихлорметане-метаноле (1:1, 20 мл) обработали дигидратом триацетата марганца (536 мг, 2 ммоля) и смесь нагревали в колбе с обратным холодильником в течение 3 часов. Смесь охладили до окружающей температуры, добавили хлорид лития (128 мг, 3 ммоля) и раствор перемешивали в течение 1 часа. Реакционную смесь концентрировали в вакууме и остаток растерли в порошок с диэтиловым эфиром (40 мл). Твердый продукт отфильтровали, промыли диэтиловым эфиром (2х15 мл) и сушили в вакууме до получения названного соединения в виде коричневого порошка, 1,09 г (98%).
Описание 34.
(3S,4S)-бис(3,5-Ди-трет-бутилсалицилидамино)тетрагидрофуранмарганец(III)хлорид (D34).
Раствор лиганда (D20) (1,07 г, 2 ммоля) и дигидрат триацетата марганца (536 мг, 2 ммоля) в смеси дихлорметана и метанола (1:1, 20 мл) нагревали в колбе с обратным холодильником в течение 6,5 часов. Раствор охладили до окружающей температуры, добавили хлорид лития (128 мг, 3 ммоля) и смесь перемешивали в течение 16 часов. Реакционную смесь концентрировали в вакууме и остаток растерли в порошок с диэтиловым эфиром (50 мл). Твердый продукт отфильтровали, промыли диэтиловым эфиром (2х15 мл) и сушили в вакууме до получения названного соединения в виде коричневого порошка, 1,12 г (89%).
Описание 35.
(3R,4S)-бис(3,5-ди-трет-Бутилсалицилидамино)-(2R)-(трифенилметоксиметил)тетрагидропиранмарганец(III)хлорид (D35).
К лиганду (D24) (160 мг, 195 ммолей) в дихлорметане-метаноле (3:2, 5 мл) добавили NaОН (0,93 мл 0,417 M в метаноле, 390 ммолей) и дигидрат триацетата марганца (52,5 мг, 195 ммолей). Раствор нагревали в колбе с обратным холодильником в течение 3 часов, добавили хлорид лития (12,5 мг, 300 ммолей) и смесь перемешивали в течение 15 часов. Раствор удалили в вакууме и остаток растерли в порошок с диэтиловым эфиром (10 мл). Твердый продукт отфильтровали, промыли диэтиловым эфиром (2х2 мл) и сушили в вакууме до получения названного соединения в виде коричневого порошка, 136 мг (77%).
Описание 36.
(-)-транс-1-Бензоил-3,4-бис(3,5-ди-трет-бутилсалицилидамино)пиперидинмарганец(III)хлорид (D36).
Смесь (-)-лиганда (D28) (28 мг, 0,013 ммоля) и дигидрата триацетата марганца (10 мг, 0,037 ммоля) в дихлорметане-метаноле (3:2, 5 мл) нагревали в колбе с обратным холодильником в течение 4 часов. Добавили хлорид лития (1,6 мг, 0,038 ммоля) и нагревание в колбе с обратным холодильником продолжили в течение еще 1 часа.
Растворитель удалили в вакууме и остаток подвергли хроматографии на диоксиде кремния (элюент: 10% метанол в дихлорметане) до получения названного соединения в виде коричневого порошка, 22 мг (97%).
Описания промежуточных соединений для получения соединений формулы (III).
Описание 37. (R)-1-Фенил-1,2-бис(3-трет-бутил-5-метилсалицилидамино)этан (D37).
(R)-1,2-Диамино-1-фенилэтан, полученный из (R)-2-аминофенилацетамида 6 посредством восстановления в диамин методом Брауна и Гейма 7 (1,36 г, 10,0 ммолей) растворили в этаноле (50 мл) и добавили твердый 2-гидрокси-3-трет-бутил- 5-метилбензальдегид, полученный из 2-трет-бутил-4-метилфенола посредством метода Кейсираджи и др.8 (3,84 г, 20,0 ммолей). Через 90 минут нагрева в колбе с обратным холодильником реакционную смесь охладили и добавили воду (1 мл). Образованный желтый твердый продукт удалили фильтрацией, промыли 95% водным раствором этанола (10 мл) и сушили в вакууме над Р2O5 до получения названного соединения в виде желтого твердого вещества, 3,33 г, выход 69%.
δ (CDCl3): 1,41 (9Н, s), 1,43 (9Н, s), 2,22 (3Н, s), 2,23 (3Н, s), 3,93 (1Н, dd), 4,12 (1H, dd), 4,68 (1H, dd), 6,84 (2Н, s), 7,09 (2Н, s), 7,30-7,50 (5Н, m), 8,25 (1Н, s), 8,37 (1H, s) и 13,50 (2Н, bs).
6. C.G.Nielson and D.F.Ewing, J.Chem. Soc., (С) 1966, pp.393-397.
7. H.С.Brown and P.Heim, J.Org. Chem., 1973, 38, pp.912-916.
Описание 38. (R)-1-Фенил-1,2-бис(3,5-ди-трет-бутилсалицилидамино)этан (D38).
(R)-1,2-Диамино-1-фенилэтан (0,68 г, 5,0 ммолей) растворили в этаноле (50 мл) и добавили 2-гидрокси-3,5-ди-трет-бутилбензальдегид, полученный из 2,4-ди-трет-бутилфенола методом Кейсираджи и др.8 (2,34 г, 10,0 ммолей). Реакционную смесь нагрели в колбе с обратным холодильником в течение 2 часов, охладили до комнатной температуры и к перемешиваемому раствору добавили воду (1 мл). Продукт выделили фильтрацией, промыли 95% водным раствором этанола (5 мл) и сушили в вакууме над Р2О5 до получения названного соединения в виде желтого твердого вещества, 2,11 г, выход 74%.
δ (CDCl3): 1,24 (9H, s), 1,27 (9Н, s), 1,41 (9H, s), 1,45 (9Н, s), 3,95 (1H, dd), 4,15 (1H, dd), 4,70 (1H, dd), 7,05 (2H, bs), 7,30-7,50 (7H, m), 8,34 (1H, s), 8,42 (1H, s) и 13,60 (2H, bs).
8. G. Casiraghi, G.Casnati, G.Puglia, G.Sartori and G.Terenghi, J.Chem. Soc. Perkin l, 1980, pp. 1862-1865.
Описание 39. (S)-1-Метил-1,2-бис(3-трет-бутилсалицилидамино)этан (D39).
Суспензию (S)-1,2-диаминопропандихлорида (290 мг, 2 ммоля) в ЕtОН (5 мл) обработали 1M NаОН в этаноле (4 мл, 4 ммоля). Добавили 2-гидрокси-5-трет-бутил-3-метилбенpальдегид (770 мг, 4 ммоля) и смесь нагрели в колбе с обратным холодильником в течение 1,5 часа. Суспензию отфильтровали, частично выпарили и к осадку названного соединения в виде желтого твердого вещества добавили небольшое количество воды. Его отфильтровали, промыли 95% водным раствором ЕtОН и сушили в вакууме над Р2О5 до получения названного соединения, 730 мг (выход 86%).
δ (CDCl3): 1,33 (3H, s), 1,36 (18H, d), 2,25 (6H, s), 3,32 (2H, m), 3,76 (1H, m), 6,80 (2H, s), 7,03 (2H, s), 8,20 (1H, s), 8,25 (1H, s), 13,50 (2H, bs).
Описание 40. (S)-1-Изопропил-1,2-бис(3-трет-бутил-5-метилсалицилидамино)этан (D40).
К боргидрату натрия (1,13 г, 30 ммолей) в глиме (30 мл) под азотом при перемешивании добавили (S)-валинамидгидрохлорид (1,53 г, 10 ммолей), суспендированный в глиме (35 мл). Раствор охладили до 10oС и по каплям в течение 20 минут добавили эфирное соединение трехфтористого бора (4,9 мл, 40 ммолей) в глиме (10 мл), затем смесь нагревали в колбе с обратным холодильником в течение 16 часов. После охлаждения до окружающей температуры добавили воду (7,5 мл), затем 3М NаОН (15 мл) и полученный прозрачный раствор нагрели в колбе с обратным холодильником в течение 2 часов. Раствор удалили в вакууме, получив при этом белый твердый продукт, который экстрагировали хлороформом (3х10 мл), смешанный экстракт выпарили, получив диамин (0,34 г). Его растворили в этаноле (15 мл) и обработали 2-гидрокси-3-трет-бутил-5-метилбензальдегидом (1,28 г, 6,6 ммоля). Раствор нагрели в колбе с обратным холодильником в течение 2 часов, охладили, концентрировали в вакууме и остаток подвергли хроматографии на диоксиде кремния (МЕRСК 9385, при элюировании 0,6% МеОН в хлороформе), получив при этом названное соединение, 0,73 г (выход 16%).
δ (CDCl3): 1,04 (6Н, m), 1,39 (18Н, 2s), 2,10 (1H, m), 2,24 (6H,s), 3,3-4,0 (3H, bm), 6,85 (2H, m), 7,09 (2H, m), 8,24 (2H, s), 13,60 (2H bs).
Изобретение относится к способу эпоксидирования прохирального олефина, который включает взаимодействие прохирального олефина с источником кислорода в присутствии солевого катализатора. Эпоксидирование проводят в присутствии изохинолин-N-оксида, являющегося донорным лигандом. В качестве катализатора используют соединение формулы (IB) или (II), охарактеризованное в п.2 формулы изобретения. Кроме того, предложен вариант способа, согласно которому взаимодействие прохирального олефина с источником кислорода проводят в присутствии в качестве солевого катализатора соединения формулы (II) и источника лиганда, отдающего электрон. 2 с. и 5 з.п.ф-лы.

в которой YI и Y4 являются одинаковыми и их выбирают из группы, состоящей из метила, трет-бутила или метокси;
R2 и R3 оба являются фенилом или вместе с атомами углерода, к которым они присоединены, образуют гексильное кольцо;
(ii) соединение формулы (II)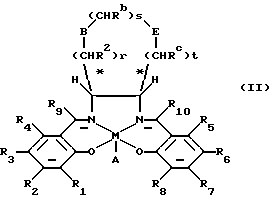
в которой М представляет ион переходного металла;
А, если необходимо, является противоионом;
r, s и t составляют независимо от 0 до 3 и этом сумма r+s+t должна находиться в диапазоне от 1 до 3;
Ra, Rb, Rc независимо являются водородом или СН2ОR', где R' является водородом или органической группой;
В и Е независимо являются кислордом, CН2, NRd, где Rd является алкилом, водородом, алкилкарбонилом или арилкарбонилом или SОn, где n равно 0 или целому числу 1 или 2, при условии, что В и Е не являются одновременно СН2 и что, когда В является кислородом, NRd или SОn, тогда не может быть 0 и, когда Е является кислородом, NRd или SОn, тогда t не может быть равно нулю;
R1, R2, R3, R4, R5, R6, R7, R8, R9 и R10 независимо являются водородом, алкилом, алкокси;
3. Способ по п. 1 или 2, отличающийся тем, что солевой катализатор выбирают из группы, состоящей из:
R, R-[1,2-бис(3,5-ди-трет-бутилсалицилидамино)циклогексан] марганец (III)-хлорида;
(3S, 4S)-бис-(3,5-ди-трет-бутилсалицилидамино)тетрагидрофуранмарганец (III) хлорида;
(R, R)-5,6-бис(3,5-ди-трет-бутилсалицилидамино)-1,3-диоксепан] -марганец (III)-хлорида;
(R)-1-фенил-1,2-бис-(3-трет-бутил-5-метилсалицилидамино) этанмарганец (III) хлорида;
(R)-1-фенил-1,2-бис-(3,5-ди-трет-бутилсалицилидамино) этанмарганец (III) хлорида;
(S)-1-метил-1,2-бис-(3-трет-бутил-5-метилсалицилидамино) этанмарганец -(III) хлорида;
(S)-1-изопропил-1,2-бис-(3-трет-бутил-5-метилсалицилидамино) этанмарганец (III)-хлорида;
4. Способ по любому из пп. 1-3, отличающийся тем, что прохиральным олефином является 2,2-диметил-6-пентафторэтил-2Н-1-бензопиран или 6-ацетил-2,2-диметил-2Н-1-бензопиран.
Приоритет по пунктам и признакам:
04.02. , 1994 по п. 2, где катализатор имеет формулу (II), пп. 5 и 6;
15.06.1994 по п. 1 и 2, где катализатор имеет формулы (IB), пп. 4 и 7.
| Экономайзер | 0 |
|
SU94A1 |
| Огнетушитель | 0 |
|
SU91A1 |
| Часы на многоустойчивых элементах с цифровой индикацией | 1976 |
|
SU588527A1 |
| DE 4238076 С1, 16.09.1993. | |||

