Изобретение относится к применению группы 2-фенил-3-ароилбензотиофенов для предупреждения и лечения остеопороза.
Механизм разрежения кости еще не совсем изучен, но из практики известно, что указанное нарушение возникает вследствие дисбаланса в образовании новой здоровой кости и ресорбции старой кости, причем этот дисбаланс имеет тенденцию к потере костной ткани. Указанное разрежение кости заключается в уменьшении содержания как минеральных компонентов, так и компонентов белковой матрицы в кости и приводит к увеличению частоты переломов преимущественно бедренных костей, костей предплечья и позвоночника. Указанные переломы, в свою очередь, приводят к повышению общей заболеваемости, значительному снижению роста и подвижности, а во многих случаях к увеличению смертности из-за последующих осложнений.
Остеопорозу подвержена широкая категория людей, включая женщин, переживающих постклимактерический период, пациентов, принимающих или долгое время принимающих кортикостероиды, пациентов, страдающих синдромом Кушинга, и пациентов, страдающих гонадной дисплазией.
Имеются предположения, хотя и непроверенные, что разрежение костей может приводить к остеопорозу, значительному осложнению заболевания, в основе которого лежит потеря костной массы (уменьшение плотности кости и ее пористости) при сохранении общего объема кости, увеличению пористости и хрупкости кости.
Было установлено, что чаще всего остеопорозом страдают женщины в климактерическом периоде, и лишь в США число женщин с этим заболеванием составляет от 20 до 25 миллионов. Основной особенностью постклимактерического остеопороза является значительная и быстрая потеря костной массы вследствие прекращения продуцирования яичниками эстрогена. Действительно, имеющиеся данные полностью подтверждают тот факт, что эстрогены способны подавлять развитие остеопороза, и в США, а также во многих других странах лечение постклимактерического остеопороза проводят в основном путем восполнения нехватки эстрогена в организме. Однако, хотя эстрогены оказывают благоприятное воздействие на кости даже при очень низких уровнях их содержания в организме, все же длительная эстрогеновая терапия может вызвать осложнение ряда заболеваний, включая увеличение риска заболевания раком матки или молочной железы, и это обстоятельство вынуждает многих женщин отказываться от лечения эстрогенами. Недавно были предложены схемы лечения, позволяющие снизить риск заболевания раком и предусматривающие введение комбинации прогестерона и эстрогена, которая вызывает у пациентов регулярные абстинентные кровотечения, что является неприемлемым для очень пожилых женщин. Значительные нежелательные явления, связанные с эстрогеновой терапией, и ограниченная способность эстрогенов к реверсии имеющегося разрежения кости подтверждают необходимость разработки альтернативной терапии для остеопороза, которая могла бы благоприятно воздействовать на костную ткань и не вызывала бы при этом нежелательных побочных явлений.
В целях устранения недостатков имеющихся способов терапии были предприняты попытки использовать соединения, известные как антиэстрогены, которые взаимодействуют с рецептором эстрогена, однако этот способ не имел большого успеха, вероятно, вследствие того, что указанные соединения обнаруживали в основном смешанный агонистический / антагонистический эффект. То есть, хотя эти соединения могут препятствовать взаимодействию эстрогена с рецептором, однако сами по себе они могут вызывать эстрогенные ответы в тех тканях, которые имеют рецепторы эстрогенов. Поэтому некоторые антиэстрогены вызывают те же самые нежелательные эффекты, которые обычно ассоциируются с применением эстрогеновой терапии.
Настоящее изобретение относится к способам ингибирования разрежения кости, которые при этом не вызывают неблагоприятных побочных явлений, обычно связанных с эстрогеновой терапией, а поэтому являются эффективными и приемлемыми способами лечения остеопороза.
2-Фенил-3-ароилбензотиофеновые соединения, которые являются активными компонентами композиций и способов настоящего изобретения, были впервые разработаны C.David Jones и Tilio Suarez в качестве противозачаточных средств (см. [1]). Было обнаружено, что некоторые соединения этой группы могут быть использованы для подавления роста опухолей молочной железы.
Позже Джоунсом была получена группа родственных соединений, которая предназначалась для использования в антиэстрогеновой и антиандрогеновой терапии, в частности, для лечения опухолей молочной железы и предстательной железы (см. [2] ). Одно из этих соединений, а именно соединение формулы I, в котором n=0 и R1 является гидроксилом, а R2 является пиперидиновым кольцом, было подвергнуто клиническому испытанию в течение небольшого периода времени на лечение рака молочной железы. Это соединение сначала было названо кеоксифеном, а затем оно получило название ралоксифен. При лечении ралоксифен предпочтительно вводить в виде соли, а более предпочтительно - в виде гидрохлоридной соли.
Настоящее изобретение относится к новым способам лечения разрежения кости, заключающимся во введении человеку, нуждающемуся в таком лечении, эффективного количества соединения формулы I: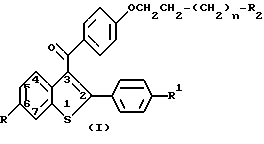
где n=0, 1 или 2;
R и R1 независимо представляют собой водород, гидроксил, C1-C6-алкокси, C1-C6-ацилокси, C1-C6-алкокси-C2-C6-ацилокси, R3-замещенный арилокси, R3-замещенный ароилокси, R4-замещенный карбонилокси, хлоро или бромо;
R2 является гетероциклическим кольцом, выбранным из группы, включающей в себя пирролидино, пиперидино и гексаметиленимино;
R3 представляет собой C1-C3-алкил, C1-C3-алкокси, водород или галоген и
R4 представляет собой C1-C6-алкокси или арилокси, или его фармацевтически приемлемой соли.
Настоящее изобретение также относится к фармацевтической композиции, способствующей ингибированию разрежения кости и содержащей соединение формулы I, в котором R, R1, R2 и n являются такими, как они были определены выше, в количестве, эффективном для повышения или сохранения плотности кости, и фармацевтически приемлемому носителю.
Настоящее изобретение относится к обнаружению того факта, что группа 2-фенил-3-ароилбензотиофенов (бензотиофены) формулы I могут быть использованы для лечения остеопороза. Бензотиофены формулы I способствуют снижению потери костной ткани, обусловленной недостатком эндогенного эстрогена, который имеет место у женщин после прекращения менструаций, вследствие климакса, хирургических операций или других причин. Снижение плотности кости и ее массы, которое редко наблюдается у мужчин, также тесно связано с нарушением гормональной регуляции, а поэтому является объектом терапии в соответствии со способами настоящего изобретения.
Бензотиофены формулы I представляют собой серию нестероидных соединений, обладающих высокой степенью аффинности по отношению к обычным рецепторам эстрогена в первичных половых тканях-мишенях. Однако они вызывают минимальный эстрагенозный ответ в этих тканях и фактически служат в качестве сильных антагонистов природных эстрогенов, таких как эстрадиол. Бензотиофены формулы I обладают способностью подавлять классические эстрогеновые ответы в первичных половых тканях-мишенях без значительного снижения плотности кости при их введении интактным или эстрогенобработанным животным, и эти соединения способны предупреждать разрежение кости у животных с дефицитом эстрогена. Такая дихотомия свидетельствует о селективном агонистическом / антагонистическом действии на специфические клетки-мишени и очевидно именно такое действие было бы весьма желательным при лечении климактерического синдрома. В соответствии с этим реальное преимущество настоящего изобретения заключается в том, бензотиофены формулы I ингибируют разрежение кости, но не вызывают при этом значительного эстрогенового ответа в первичных половых тканях-мишенях. Таким образом, настоящее изобретение относится к способу ингибирования разрежения кости, заключающемуся во введении человеку, нуждающемуся в таком лечении, определенного количества соединения формулы I, которое ингибирует разрежение кости, но не оказывает значительного воздействия на первичные половые ткани-мишени. Указанная комбинация свойств рассматриваемых соединений позволяет проводить длительное лечение хронических заболеваний при минимальном риске возникновения нежелательных побочных эффектов, обычно связанных с традиционной эстрогеновой заместительной терапией.
Биологическое действие бензотиофенов формулы I является сложным и может быть не связанным с обнаружимым присутствием исходного соединения в крови. После перорального введения предпочтительного бензотиофена настоящего изобретения, а именно ралоксифена (вводимого в виде гидрохлоридной соли), пациентам в клинике исходное соединение не было обнаружено в сыворотке этих пациентов. Было установлено, что после перорального введения это соединение было экстенсивно конъюгировано до глюкуронидированной формы и быстро выводилось из кровотока. Хотя у людей-реципиентов не были определены биологические конечные концентрации, однако это не означает, что рассматриваемое соединение не является биологически доступным.
Для выяснения степени биодоступности соединения были осуществлены эксперименты на лабораторных животных, где биологическая активность может быть проанализирована. Исследования на животных показали, что ралоксифен обнаруживал максимальную активность в ингибировании поглощения маткой меченного тритием эстрадиола и нормального устройства утеротропного ответа на эстрадиол даже при условиях, когда ралоксифен является экстенсивно связанным в плазме животных. Кроме того, конъюгат, выделенный из мочи людей, которым был введен ралоксифен, обнаруживал значительную антиэстрогенную / антиуретотропную активность при внутривенном введении крысам и ингибировал взаимодействие меченного тритием эстрадиола с рецепторами маточного эстрогена крыс таким же образом, как и исходное соединение. Описанные исследования позволяют предположить, что конъюгированное соединение может быть превращено в свою первоначальную форму in situ и вероятно с помощью β-глюкуронидазы. Такое превращение, очевидно, способствует активности данного соединения. β-Глюкуронидаза, будучи довольно распространенным ферментом, очевидно, обладает активностью в ресорбционном процессе восстановления кости и по всей вероятности участвует в превращении конъюгированного соединения в его первоначальную форму, если это необходимо для активности. Поэтому конъюгация бензотиофенов формулы I не должна непрерывно рассматриваться как неблагоприятный фактор для их биологической доступности в качестве ингибиторов разрежения кости.
Таким образом, способ лечения, рассматриваемый в настоящей заявке, осуществляют путем введения человеку, нуждающемуся в лечении от разрежения кости, некоторой дозы соединения формулы I или его фармацевтически приемлемой соли, которая является эффективной для ингибирования разрежения кости. Особое преимущество данного способа заключается в том, что он позволяет избежать потенциально вредных и неприемлемых побочных эффектов, связанных с традиционной эстрогеновой терапией. Способ настоящего изобретения, предназначенный для ингибирования разрежения кости, предусматривает консервативное терапевтическое и/или профилактическое лечение.
Указанный способ также предусматривает введение соединения формулы I в сочетании с эстрогеном. Термин "эстроген", используемый в настоящем описании, относится к любому соединению, которое обладает приблизительно таким же спектром активности, как и природная молекула, известная под названием 17β-эстрадиол. Примерами таких соединений могут служить эстриол, эстрон, этинил-эстрадиол, Премарин (Premarin) (коммерческий препарат конъюгированных эстрогенов, выделенных из натуральных источников - Ayevst) и т.п. Кроме того, благодаря избирательным агонистическим / антагоническим свойствам соединений формулы I указанный выше комбинированный метод обладает всеми преимуществами эстрогеновой терапии, но при этом позволяет избежать неблагоприятных побочных эффектов, связанных с применением лишь одной эстрогеновой терапии.
Основные химические термины, используемые в настоящем описании при определении соединения формулы I, имеют свои обычные значения. Например, термин "C1-C3-алкил" относится к таким группам, как метил, этил, пропил и изопропил.
Термин "C1-C6-алкокси" относится к таким группам, как метокси, этокси, пропокси, бутокси, пентилокси и гексилокси, а также к структурам с разветвленной цепью, таким как, например, изопропокси и изобутокси.
Термин "C1-C6-ацилокси" включает в себя метаноилокси, этаноилокси, пропаноилокси, бутаноилокси, пентаноилокси, гексаноилокси и т.п., а также структуры с разветвленной цепью, такие как 2,2-диметилпропаноилокси и 3,3-диметилбутаноилокси.
Термин "C1-C6-алкокси-C2-C6-ацилокси" включает в себя, например, метоксиэтаноилокси, метоксипропаноилокси, метоксибутаноилокси, метоксипентаилокси, метоксигексаноилокси, этоксиэтаноилокси, этоксипропаноилокси, этоксибутаноилокси, этоксипентаноилокси, этоксигексаноилокси, пропоксиэтаноилокси, пропоксипропаноилокси, пропоксибутаноилокси и т.п.
При этом следует отметить, что используемые в настоящем описании ссылки на алкильные или алкокси-структуры также относятся к циклоалкильным и циклоалкокси-группам, причем число атомов углерода в этих структурах составляет по крайней мере 3.
Термины "R3-замещенный арилокси" и R3-замещенный ароилокси" относятся к таким группам, как фенилокси, тиенилокси, фурилокси, нафтилокси, бензоилокси, тиеноилокси, фуроилокси, нафтоилокси и т.п., где группа замещения R3 может быть водородом, гидроксилом, C1-C3-алкилом, C1-C3-алкокси или галогеном.
Термин "R4- замещенный карбонилокси", где группа замещения R4 может быть C1-C6-алкокси или арилокси, относится к карбонатным структурам, таким как метоксикарбонилокси, этоксикарбонилокси, пропоксикарбонилокси, бутоксикарбонилокси, пентилоксикарбонилокси, гексилоксикарбонилокси, фенилоксикарбонилокси, тиенилоксикарбонилокси, фурилоксикарбонилокси и нафтилоксикарбонилокси.
Предпочтительные варианты осуществления настоящего изобретения относятся к использованию соединений формулы I, в которых R и R1 не являются водородом, алкокси, арилокси, хлором или бромом, а представляют собой сложно-эфирные или карбонатные конфигурации. В других предпочтительных вариантах настоящего изобретения используются соединения формулы I, в которых R и R1 имеют аналогичные значения. При осуществлении настоящего изобретения некоторые группы R2 также имеют свои предпочтительные значения. Например, в предпочтительном варианте настоящего изобретения группа R2 в соединениях формулы I представляет собой пиперидино или пирролидино, а предпочтительно пиперидино. Еще более предпочтительная подгруппа пиперидино- и пирролидино-соединений включает в себя соединения, где R и R2 не являются водородом, а предпочтительны такие соединения, где R и R1 являются гидроксилом. Наиболее предпочтительные к варианту осуществления настоящего изобретения относятся к использованию ралоксифена, особенно, если он вводится в виде гидрохлоридной соли.
Все соединения, используемые в способах настоящего изобретения, могут быть получены в соответствии с разработанными процедурами, которые подробно описаны в патентах США NN 4133814 и 4418068. В общих чертах эти процедуры осуществляют исходя из бензо [b] тиофена, имеющего 6-гидроксильную группу и 2-(4-гидроксифенил)ьную группу. Это исходное соединение подвергают блокированию, алкилированию, а затем разблокированию с образованием соединений формулы I, в которых R и R1 оба являются гидрокси-группой. Если необходимо, то затем могут быть получены соединения формулы I, представляющие собой простые эфиры, сложные эфиры и карбонаты. Примеры получения таких соединений описаны в указанных выше патентах США. Конкретные процедуры получения других дериватизированных соединений, используемых в настоящем изобретении, представлены ниже в главах "Получения". Для введения реактивных функциональных групп конкретных заместителей может оказаться необходимой модификация вышеуказанных методов. Очевидность таких модификаций может быть легко установлена любым специалистом.
Соединения, используемые в способах настоящего изобретения, образуют фармацевтически приемлемые кислые и основные аддитивные соли с широким рядом органических и неорганических кислот и оснований, включающие в себя физиологически приемлемые соли, которые часто используются в фармацевтической химии. Такие соли также являются частью настоящего изобретения. Типичными неорганическими кислотами, используемыми для получения этих солей, являются соляная, бромистоводородная, иодистоводородная, азотная, серная, фосфорная, фосфорноватая и т. п. Могут быть также использованы соли, происходящие от органических кислот, таких как алифатические моно- и дикарбоновые кислоты, фенил-замещенные алкановые кислоты, гидроксиалкановая и гидроксиалкандионовая кислоты, ароматические кислоты, алифатические и ароматические сульфоновые кислоты. Примерами таких фармацевтически приемлемых солей являются ацетат, фенилацетат, трифторацетат, акрилат, аскорбат, бензоат, хлоробензоат, гидроксибензоат, метоксибензоат, метилбензоат, o-ацетоксибензоат, нафталин-2-бензоат, бромид, изобутират, фенилбутират, β-гидроксибутират, бутин-1,4-диоат, гексин-1,4-диоат, капрат, каприлат, хлорид, циннамат, цитрат, формат, фумарат, гликоллат, гептаноат, гиппурат, лактат, малат, малеат, гидроксималеат, малнат, манделат, мезилат, никотинат, изоникотинат, нитрат, оксалат, фталат, терафталат, фосфат, кислый монофосфат, кислый дифосфат, метафосфат, пирофосфат, пропионат, фенилпропионат, салицилат, себацат, сукцинат, суберат, сульфат, бисульфат, пиросульфат, сульфит, бисульфит, сульфонат, бензол-сульфонат, п-бромофенилсульфонат, хлоро-бензолсульфонат, этансульфонат, 2-гидроксиэтансульфонат, метан-сульфонат, нафталин-1-сульфонат, нафталин-2-сульфонат, п-толуолсульфонат, ксилолсульфонат, тартарат и т.п.
Кроме того, некоторые из соединений формулы I могут образовывать сольваты с водой или органическими растворителями, такими как этанол. Указанные сольваты также могут быть использованы в способах настоящего изобретения.
Фармацевтически приемлемые кислые аддитивные соли обычно получают с помощью реакции соединения формулы I с эквимолярным или избыточным количеством кислоты. Эти реагенты обычно смешивают в общем растворителе, таком как диэтиловый эфир или бензол. Как правило, соль осаждается из раствора в течение периода времени, составляющего примерно от 1 ч до 10 дн и может быть выделена путем фильтрации или путем отгонки растворителя стандартными методами.
Примерами оснований, обычно используемых для получения солей, могут служить гидроксид аммония, гидроксиды щелочных и щелочноземельных металлов, карбонаты и бикарбонаты, а также алифатические и ароматические амины, алифатические диамины и гидроксиалкиламины. Особенно предпочтительными основаниями для получения аддитивных солей являются гидроксид аммония, карбонат калия, бикарбонат натрия, гидроксид кальция, метиламин, диэтиламин, этилендиамин, циклогексиламин и этаноламин.
Фармацевтически приемлемые соли обычно имеют более высокую растворимость по сравнению с соединениями, от которых они происходят, а поэтому их чаще всего используют для изготовления препаратов в виде жидкостей или эмульсий.
Настоящее изобретение также относится к фармацевтическим препаратам, предназначенным для ингибирования разрежения кости и содержащим соединение формулы I или его фармацевтически приемлемую соль в сочетании с одним или несколькими фармацевтически приемлемыми наполнителями. Эти фармацевтические препараты могут быть получены в соответствии со стандартными процедурами, хорошо известными специалистам. Например, рассматриваемые препараты могут содержать указанные соединения в сочетании с общеизвестными наполнителями, разбавителями или носителями и могут быть изготовлены в виде таблеток, капсул, суспензий, порошков и т.п. Примерами наполнителей, разбавителей и носителей, которые могут быть использованы для изготовления указанных препаратов, являются такие наполнители, как крахмал, сахара, маннит, кремниевые производные, связующие агенты, такие как карбоксиметилцеллюлоза и другие производные целлюлозы, альгинаты, желатин и поливинилпирролидон, смачивающие агенты, такие как глицерин, дезинтегрирующие агенты, такие как агар-агар, карбонат кальция и бикарбонат натрия, добавки, замедляющие растворение, такие как парафин, добавки, ускоряющие ресорбцию, такие как четвертичные аммониевые основания, и поверхностно-активные вещества, такие как цетиловый спирт, моностеарат глицерина, адсорбирующие носители, такие как каолин или бентонит, и замасливающие агенты, такие как тальк, стеарат кальция и магния и твердые полиэтиленгликоли.
Композиции, содержащие вышеуказанные соединения, могут быть также изготовлены в виде эликсиров или растворов, пригодных для перорального введения, либо в виде растворов, предназначенных для парентерального введения, например, путем внутримышечных, подкожных или внутривенных инъекций. Кроме того, эти соединения обладают свойствами, позволяющими с успехом использовать их для получения лекарственных форм с пролонгированным высвобождением активного компонента и т.п. При этом могут быть изготовлены такие препараты, в которых высвобождение активного ингредиента будет происходить только или предпочтительно в определенной части кишечного тракта и, возможно, в течение определенного периода времени. Покрытия, оболочки и защитные матрицы могут быть изготовлены, например, из полимеров или восков.
Конкретные дозы соединения формулы I, необходимые для лечения или ингибирования разрежения кости в соответствии с настоящим изобретением, зависят от способа его введения, тяжести заболевания и от других факторов, которые могут быть конкретно учтены лечащим врачом. В основном эффективная доза соединения формулы I будет составлять от около 0,1 до около 1000 мг, предпочтительно от около 50 до около 400 мг, а наиболее предпочтительно от около 50 до около 200 мг. Указанные дозы могут быть введены пациенту, нуждающемуся в лечении, от одного до около трех раз в день или более часто, если это необходимо для эффективного ингибирования процесса разрежения кости.
Соединение формулы I в основном предпочтительно вводить в виде кислой аддитивной соли, как это обычно практикуется специалистами при введении фармацевтических препаратов, имеющих основную группу, такую как пиперидиновое кольцо. Если указанное соединение вводится пожилому человеку (например, женщине, переживающей постклимактерический период, или мужчине, у которого был диагностирован остеопороз с помощью рентгеновского анализа), то это соединение предпочтительно вводить перорально. В этих целях могут быть использованы пероральные лекарственные формы, описанные ниже.
Препараты.
В представленных ниже рецептах препаратов термин "активный ингредиент" означает соединение формулы I.
Препарат 1. Желатиновые капсулы.
Жесткие желатиновые капсулы получали с использованием следующих ингредиентов:
Ингредиент - Количество (мг/капсула)
Активный ингредиент - 0,1-1000
Крахмал, NF - 0-650
Текучий порошок крахмала - 0-650
Кремнийорганическая жидкость, 350 сантистокс - 0-15
Ингредиенты смешивали, пропускали через сито N 45 меш. (США), и этой смесью заполняли жесткие желатиновые капсулы.
Конкретные примеры указанных препаратов, содержащих ралоксифен в виде гидрохлоридной соли, представлены ниже.
Препарат 2. Капсула, содержащая ралоксифен.
Ингредиент - Количество (мг/капсулы)
Ралоксифен, гидрохлорид - 1
Крахмал, NF - 112
Текучий порошок крахмала - 225,3
Кремнийорганическая жидкость, 350 сантистокс - 1,7
Препарат 3. Капсула, содержащая ралоксифен.
Ингредиент - Количество (мг/капсула)
Ралоксифен, гидрохлорид - 5
Крахмал, NF - 108
Текучий порошок крахмала - 225,3
Кремнийорганическая жидкость, 350 сантистокс - 1,7
Препарат 4. Капсула, содержащая ралоксифен.
Ингредиент - Количество (мг/капсула)
Ралоксифен, гидрохлорид - 10
Крахмал, NF - 103
Текучий порошок крахмала - 225,3
Кремнийорганическая жидкость, 350 сантистокс - 1,7
Препарат 5. Капсула, содержащая ралоксифен.
Ингредиент - Количество (мг/капсула)
Ралоксифен, гидрохлорид - 50
Крахмал, NF - 150
Текучий порошок крахмала - 397
Кремнийорганическая жидкость, 350 сантистокс - 3
Указанные выше конкретные препараты могут варьироваться в соответствии с возможными и допустимыми вариантами.
Препараты в виде таблеток получали с использованием следующих ингредиентов.
Препарат 6. Таблетки.
Ингредиент - Количество (мг/таблетка)
Активный ингредиент - 0,1-1000
Микрокристаллическая целлюлоза - 0-650
Коллоидный диоксид кремния - 0-650
Стеарат - 0-15
Эти компоненты смешивали и спрессовывали в таблетки.
Альтернативно таблетки, каждая из которых содержит 0,1-1000 мг активного ингредиента, получали следующим образом.
Препарат 7. Таблетки.
Ингредиент - Количество (мг/таблетка)
Активный ингредиент - 0,1-1000
Крахмал - 45
Микрокристаллическая целлюлоза - 35
Поливинилпирролидон (в виде 10%-ного раствора в воде) - 4
Натриевая карбоксиметилцеллюлоза - 4,5
Стеарат магния - 0,5
Тальк - 1
Активный ингредиент, крахмал и целлюлозу пропускали через сито N 45 меш. (США) и тщательно перемешивали. Раствор поливинилпирролидона смешивали с полученными порошками, которые затем пропускали через сито N 14 меш. (США). Полученные таким образом гранулы осушали при 50-60oC и пропускали через сито N 18 меш. (США). Натрийкарбоксиметиловый крахмал, стеарат магния и тальк, предварительно пропущенный через сито N 60 меш. (США), затем добавляли к гранулам, которые после размешивания спрессовывали в таблетировочной машине, и получали таблетки.
Суспензии, каждая из которых содержит 0,1-1000 мг лекарственного средства на 5-мл дозу, получали следующим образом.
Препарат 8. Суспензии.
Ингредиент - Количество (мг/5 мл)
Активный ингредиент - 0,1-1000 мг
Натрийкарбоксиметилцеллюлоза - 50 мг
Сироп - 1,25 мг
Раствор бензойной кислоты - 0,10 мл
Отдушка - любое кол-во
Краситель - любое кол-во
Очищенная вода - до 5 мл
Лекарственное средство пропускали через сито N 45 меш. (США) и смешивали с натрийкарбоксиметилцеллюлозой и сиропом, в результате чего получали однородную пасту. Раствор бензойной кислоты, отдушку и краситель разводили некоторым количеством воды и добавляли к полученной пасте, перемешивая при этом. Затем добавляли достаточное количество воды до получения требуемого объема.
Характерные соединения, которые могут быть использованы в препаратах и методах настоящего изобретения, представлены в табл. 1.
В представленных ниже примерах получения цифры, сопровождающие соединения, соответствуют цифрам, данным в табл. 1.
Получение 1. Получение соединения 1: 6-(4-фторобензил окси)-2-[4-(4 -фторобензил окси)-фенил] бен зо[b]ти ен-3-ил-[4-[2-(пипери дин-1-ил)этокси] фенил]метанон.
Ралоксифена гидрохлорид (также называемый 6-гидро кси-2-(4-гидрокси фенил)-бензо[b] тиен-3-ил-4-[2-(пипери дин-1-ил) этоксифенил] -метанона) гидрохлоридом (5,1 г, 10 мМ) суспендировали в 250 мл сухого тетрагидрофурана (ТГФ) и 7,1 г (70 мМ) триэтиламина, а затем добавляли приблизительно 10 мг 4-(N,N-диметиламино)пиридина. Эту суспензию охлаждали в ледяной бане и помещали в атмосферу азота. Затем медленно в течение 20 мин добавляли 4-фторобензоилхлорид (4,75 г, 30 мМ), растворенный в 20 мл сухого ТГФ. После того реакционную смесь размешивали и медленно нагревали до комнатной температуры в течение 18 ч. После этого смесь фильтровали, а фильтрат выпаривали в вакууме и получали продукт в виде камеди. Полученный таким образом неочищенный продукт растворяли в небольшом количестве хлороформа и хроматографировали (ВЖХР) на колонке с силикагелем, элюируя линейным градиентом растворителя, начиная с хлороформа и заканчивая смесью хлороформа-метанола (19:1, по объему). Фракции, содержащие нужный продукт, проанализированный с помощью тонкослойной хроматографии (двуокись кремния, хлороформ-метанол, 9:1), объединяли и выпаривали, в результате чего получали смолистый продукт в виде камеди. Конечный продукт кристаллизовали из эфира и получали 21 г соединения 1.
ПМР: Данные соответствуют нужной структуре.
Масс-спектроскопия FD MS: m/e = 717 (M+).
Элементный анализ для C42H33F2NO6S:
Вычислено: C 70,20; H 4,60; N 1,95.
Найдено: C 70,05; H 4,60; N 1,89. Мол.
Мол. масса: 717.
Получение 2. Получение соединения 2: 5-(4-фторобензоил окси)-2-[4-(4-фторобензоилокси)-фе нил]-бензо[b]тиен-3-ил-[4-[2-(пипери дин-1-ил)-этокси] -фенил]-метанон, гидрохлорид.
Соединение 1 (5,15 г, 7,18 мМ) растворяли в 15 мл ТГФ и добавляли 150 мл простого эфира. Сухой газообразный гидрохлорид барботировали в раствор, в результате образовывался белый смолистый осадок. Эту жидкость удалили путем декантирования, а остаток кристаллизовали из этилацетата с небольшим количеством добавленного этанола и получали раствор. Полученный продукт фильтровали, промывали эфиром и высушивали, в результате чего получали 4,41 г соединения 2 в виде белого порошка.
ПМР: Данные соответствуют нужной структуре.
Элементный анализ для C42H34ClF2NO6S:
Вычислено: C 66,88; H 4,54; N 1,86.
Найдено: C 66,59; H 4,39; N 1,60.
Мол. масса: 753,5.
Получение 3. Получение соединения 3: 6-(циклопропилкарбонил окси)-2-[4 -циклопропилкарбонилокси)фенил] -бен зо[b] тиен-3-ил-[4-2-(пипери дин-1-ил)этокси]фенил метанон.
Целевое соединение получали в соответствии с процедурой, аналогичной описанной в Получении 1, но с использованием циклопропилкарбонилхлорида и за исключением того, что полученный продукт не кристаллизовали. Выход 2,27 г.
ПМР: Данные соответствуют нужной структуре.
FD MS: m/e= 610 (M+).
Получение 4. Получение соединения 4: 6-(циклопропилкарбонил окси)-2-[4 -(циклопропилкарбонилокси)фенил] бензо[b] тиен-3-ил-[4-[2-(пипери дин-1-ил)этокси] фенил]метанон, гидрохлорид.
Соединение 4 получали из соединения 3, как описано в Получении 2.
Получение 5. Получение соединения 5: 6-(н-бутаноил окси)-2-[4-(н-бутаноилокси)фе нил] -бензо[b]тиен-3-ил-[4-[2-(пипери дин-1-ил)этокси]фенил]метанон.
В соответствии со способом, описанным в Получении 1, за исключением того, что в качестве исходного соединения использовали н-бутаноилхлорид, получали 4,12 г соединения 5 в виде маслообразного продукта.
ПМР: Данные соответствуют нужной структуре.
FD MS: m/e = 614 (M+1).
Получение 6. Получение соединения 6: 6-(н-бутаноил окси)-2-[4-(н-бутаноилокси)фе нил]-бензо[b]тиен-3-ил-[4-[2-(пипери дин-1-ил)этокси]-фенил]метанон, гидрохлорид.
Соединение 5 (4,12 г) растворяли в этилацетате (50 мл) и затем добавляли раствор HCl в простом эфире до тех пор, пока не прекратится осаждение. Полученную жидкость отгоняли, а белый смолистый остаток перетирали с диэтиловым эфиром и фильтровали. После этого остаток осушали и получали 1,33 г соединения 6.
ПМР: Данные соответствуют нужной структуре.
Элементный анализ для C36H40ClNO6S:
Вычислено: C 66,50; H 6,20; N 2,15.
Найдено: C 66,30; H 6,28; N 1,98.
Мол. масса: 650,24.
Получение 7. Получение соединения 7: 6(2,2-(диметилпропаноил окси)-2-[4-(2,2-диметилпропаноил окси)фенил] бензо[b]ти ен-3-ил-[4-(2-(пипери дин-1-ил)этокси]фенил]метанон.
Соединение 7 получали с использованием процедуры, описанной в Получении 1, за исключением того, что использовали 2,2-диметилпропаноилхлорид.
Получение 8. Получение соединения 8: 6-(2,2-диметилпропаноил окси)-2-[4-(2,2-диметилпропаноил окси)-фенил]бензо[b]ти ен-3-ил-[4-[2-(пипери дин-1-ил)этокси]фенил]-метанон, гидрохлорид.
Соединение 8 получали из соединения 7, как описано в Получении 2.
FD MS: m/e = 641 (M-HCl-I).
Элементный анализ для C38H44ClNO6S:
Вычислено: C 67,29; H 6,54; N 2,07.
Найдено: C 67,02; H 6,54; N 1,90.
Мол. масса: 678,29.
Получение 9. Получение соединения 9: 6-(3,3-диметилбутаноил окси)-2-[4-(3,3-диметилбутаноил окси)-фенил]бензо[b]ти ен-3-ил-[4-2-(пипери дин-1-ил)этокси]фенил]-метанон.
Соединение 9 получали в соответствии с процедурой, описанной в Получении 1, но с использованием 3,3-диметилбутаноилхлорида.
Получение 10. Получение соединения 10: 6-(3,3-диметилбутаноил окси)-2-[4-(3,3 -диметилбутаноилокси)фенил]бензо[b]ти ен-3-ил-[4-[2-(пипери дин-1-ил)этокси]фенил]метанон, гидрохлорид.
Соединение 10 получали из соединения 9, как описано в Получении 2.
FD MS: m/e = 669 (M-HCl-I).
Элементный анализ для C40H48ClNO6S:
Вычислено: C 68,02; H 6,85; N 1,98.
Найдено: C 67,75; H 6,83; N 2,04.
Мол. масса: 706,35.
Получение 11. Получение соединения 11: 6-(4-метилбензил окси)-2-[4-(4-метилбензил окси)-фенил]бензо[b]ти ен-3-ил[4-[2-(пипери дин-1-ил)этокси]фенил]метанон, гидрохлорид.
Соединение 11 получали из свободного основания с использованием процедуры, описанной в Получении 2.
FD MS: m/e = 710 (M-HCl-I).
Элементный анализ для C44H40ClNO6S:
Вычислено: C 70,81; H 5,39; N 1,88.
Найдено: C 71,10; H 5,39; N 1,94.
Мол. масса: 746,33.
Получение 12. Получение соединения 12: 6-бензоилокси-2-[4-бензоилокси фенил]бензо[b]-ти ен-3-ил [4-[2-(пипери дин-1-ил)этокси]фенил]метанон.
Соединение 12 получали из соответствующего хлорангидрида, как описано в Получении 1.
FD MS: m/e = 682 (M+1).
Элементный анализ для C42H35NO6S:
Вычислено: C 73,80; H 5,14; N 2,05.
Найдено: C 73,27; H 5,27; N 1,94.
Мол. масса: 681,9.
Получение 13. Получение соединения 13: 6-(н-бутоксиоил окси)-2-[4-(н-бутоксиоил окси)-фенил] бензо[b]ти ен-3-ил-[4-[2-(пипери дин-1-ил)-этокси]фенил]метанон.
Соединение 13 получали способом, аналогичным описанному в Получении 1, за исключением того, что вместо хлорангидрида использовали н-бутилхлороформат. Выход 6,13 г в виде маслообразного вещества.
ПМР: Данные соответствуют нужной структуре.
FD MS: m/e = 674 (M+1).
Получение 14. Получение соединения 14: 6-(н-бутоксикарбонил окси)-2-[4-(н -бутоксикарбонилокси)фенил] бензо[b] ти ен-3-ил[4-[2-(пипери дин-1-ил)-этокси]фенил]метанон, гидрохлорид.
Соединение 13 превращали в гидрохлоридную соль способом, аналогичным описанному в Получении 6.
ПМР: Данные соответствуют нужной структуре.
Элементный анализ для C33H44ClNO8S:
Вычислено: C 64,26; H 6,24; N 1,97.
Найдено: C 63,97; H 6,34; N 1,98.
Мол. масса: 710,29.
Получение 15. Получение соединения 15: 6-(фенилоксикарбонил окси)-2-[4 -(фенилоксикарбонилокси)фенил] бен зо[b] тиен-3-ил 4-[2-(пипери дин-1-ил)этокси]фенил]метанон.
Это соединение получали способом, аналогичным описанному в Получении 13, но с использованием соответствующего ацильного сложного эфира. Выход 3,59 г конечного продукта в виде рыжевато-коричневого аморфного порошка.
ПМР: Данные соответствуют нужной структуре.
FD MS: m/e = 713 (M+).
Получение 16. Получение соединения 16: 6-(фенилоксикарбонил окси)-2-[4-(н -фенилоксикарбонилокси)фенил] бен зо[b]тиен-3-ил[4-[2-(пипери дин-1-ил)этокси]фенил]метанон, гидрохлорид.
Соединение 15 превращали в гидрохлоридную соль способом, описанным в Получении 6.
ПМР: Данные соответствуют нужной структуре.
Элементный анализ для C38H44ClNO8S:
Вычислено: C 67,24; H 4,84; N 1,87.
Найдено: C 66,94; H 4,96; N 1,84.
Мол. масса: 750,27.
Получение 17. Получение соединения 17: 6-(нафтоил окси)-2-[4-(1-нафтоилокси)фенил]бен зо[b]тиен-3-ил[4-[2-(пипери дин-1-ил)этокси]фенил]-метанон.
Соединение 17 получали способом, описанным в Получении 1 с использованием соответствующего галогенангидрида. Выход 3,5 г белого аморфного порошка.
ПМР: Данные соответствуют нужной структуре.
FD MS: m/e = 781 (M+).
Элементный анализ для C50H39NO6S:
Вычислено: C 76,80; H 5,03; N 1,79.
Найдено: C 76,53; H 5,20; N 1,53.
Мол.масса: 781,94.
Получение 18. Получение соединения 18: 6-(метоксиэтаноил окси)-2-[4-(метоксиэтаноил окси)-фенил]бензо[b]ти ен-3-ил[4-[2-(пипери дин-1-ил)этокси] фенил]метанон.
Соединение 18 получали в соответствии с Получением 1, но с использованием соответствующего галогенангидрида. Выход 3,61 г смолистого твердого вещества.
ПМР: Данные соответствуют нужной структуре.
FD MS: m/e = 618 (M+1).
Получение 19. Получение соединения 19: 6-(метоксиэтаноил окси)-2-[4-(метоксиэтаноил окси)-фенил]бензо[b]ти ен-3-ил[4-[2-(пипери дин-1-ил)этокси] фенил]метанон, гидрохлорид.
Соединение 19 получали из 3,5 г соединения 18, как описано в Получении 2. Выход 1,65 г аморфного белого порошка.
ПМР: Данные соответствуют нужной структуре.
FD MS: m/e = 618 (M+1).
Элементный анализ для C34H36NO8S:
Вычислено: C 62,43; H 5,55; N 2,14.
Найдено: C 62,23; H 5,63 N 2,15.
Ниже представлены примеры, иллюстрирующие, но не ограничивающие способы и композиции настоящего изобретения.
Пример 1.
В примерах, иллюстрирующих способы настоящего изобретения, была использована модель постклимактерического остеопороза, с помощью которой исследовали воздействие различных способов обработки на плотность бедренной кости.
Самки крыс в возрасте 75 дн и весом 225-274 г (Sprague Dawley) поставлялись из Chorles River Laboratories (Portage, M1). Этих животных разделили на 3 группы, и давали пищу (содержание кальция составляло приблизительно 1%) и воду ad libitum. Комнатную температуру поддерживали при 22,2 ± 1,7oC с минимальной относительной влажностью 40%. Фотопериод в помещении, в котором содержались животные, составлял 12 ч свет и 12 ч темнота.
Через 1 нед после получения животных подвергали двусторонней овариэктомии под наркозом (44 мг/кг Кетамина и 5 мг/кг ксилазина) (Butler, Indianapolis, 1N), вводимом внутримышечно. Обработку наполнителем, эстрогеном или соединением формулы I начинали в день хирургической операции после выхода из наркоза. Пероральные дозы вводили через желудочный зонд в 0,5 мл 1% карбоксиметилцеллюлозы (СМС). Вес тела определяли в день хирургической операции, а затем каждую неделю после операции, причем вводимую дозу корректировали в соответствии с весом тела. Обработанные наполнителем или эстрогеном крысы, подвергнутые удалению яичников (оперированные) крысы и непрооперированные (интактные) крысы параллельно сравнивались с каждой экспериментальной группой и служили в качестве негативного и позитивного контроля.
Крыс обрабатывали ежедневно в течение 35 дн (6 животных на каждую обрабатываемую группу), а на 36-ой день животных умерщвляли путем декапитирования. Периода в 35 дн было вполне достаточно, чтобы добиться максимального снижения плотности кости, измеряемой описываемыми в заявке способами. После умерщвления матки крыс удаляли, отсекали от посторонних тканей, содержащуюся жидкость удаляли, а затем определяли мокрый вес для подтверждения дефицита эстрогена, обусловленного полной овариэктомией. В результате овариэктомии вес матки обычно снижается на 75%. После этого матки помещали в 10%-ный нейтральный забуференный формалин для проведения последующего гистологического анализа.
Правые бедренные кости удаляли и сканировали у дистального метафиза в 1 мм от наклонной борозды с помощью фотонной абсорбиометрии. В результате денситометрических измерений вычисляли плотность кости как функции содержания костного минерального вещества и ширины кости.
Влияние ралоксифена на плотность кости.
Результаты контрольной обработки, полученные из пяти отдельных экспериментов, систематизированы в табл. 2. В целом овариэктомия крыс вызывала снижение плотности бедренной кости примерно на 25% по сравнению с интактной контрольной группой, обработанной наполнителем. Эстроген, вводимый в перорально активной форме этинилэстрадиола (EE2), способствовал предупреждению разрежения кости в зависимости от дозы, но при этом он также оказывал стимулирующее действие на матку, в результате чего вес матки приближался к весу матки интактной крысы при введении дозы 100 мкг/кг. Полученные данные представляли собой средние величины, полученные для 30 крыс ± ср. квадр. ошибка.
В этих исследованиях ралоксифен, вводимый в виде гидрохлорида, также способствовал предупреждению разрежения кости в зависимости от дозы, однако при этом он вызывал лишь минимальное увеличение веса матки у животных по сравнению с овариэктомизированной контрольной группой. Результаты, полученные из пяти оценок с использованием ралоксифена, систематизированны в табл. 3. В соответствии с этим каждая точка отражает ответы от 30 крыс и иллюстрирует кривую зависимости доза - ответ для ралоксифена в этой модели. Представленные данные представляют собой средние величины ± ср. квадр. ошибка.
Пример 2. Ралоксифен в виде гидрохлорида вводили отдельно или в сочетании с этинилэстрадиолом. Крысы, обработанные только одним ралоксифеном, имели вес матки, который маргинально превышал данные, полученные для овариэтомизированной контрольной группы, но был значительно меньше, чем данные, полученные для крыс, обработанных этинилэстрадиолом, которые приближались к данным, полученным для интактной контрольной группы. И наоборот, обработка ралоксифеном способствовала значительному снижению разрежения кости у овариэктомизированных крыс, а обработка ралоксифеном в сочетании с этинил-эстрадиолом не показала заметного снижения протективного действия эстрогена на плотность кости. Результаты экспериментов представлены в табл. 4.
Пример 3. Способность ралоксифена к ингибированию разрежения кости сравнивали с такой способностью тамоксифена (SIGMA, St, Lonis, Mo). Было установлено, что тамоксифен, хорошо известный как антиэстроген и используемый в настоящее время для лечения некоторых раковых заболеваний, обладает способностью ингибировать разрежение кости (см., например, Love R. и др., 1992, Effects of tamoxifen on bone mineral density in postmenopan women with breast cancer. N. Eng. I. Med. 326:852; Turner R. и др. 1988, Tamoxifen inhibits osteoclast - mediated resorption of trabecular bonein ovarian hormone-defirient rats, Endo 122:1146). Ралоксифен и тамоксифен в относительно узком диапазоне доз перорально вводили овариэктомизированным крысам как было описано в предыдущем примере. Хотя оба из этих лекарственных препаратов обнаруживали способность к предупреждению снижения плотности бедренной кости, вызывая при этом лишь очень незначительную утеротропную активность, что было установлено благодаря приросту веса матки (табл. 5), однако сравнение некоторых гистологических параметров показало заметное различие между крысами, обработанными этими средствами (табл. 6).
Увеличение высоты эпителия является признаком эстрогенности терапевтического средства и может быть связано с повышенным риском заболевания раком матки. При введении ралоксифена в соответствии с описанием в примере 1 лишь при одной дозе наблюдалось статистически значимое увеличение высоты эпителия по сравнению с овариэктомизированной контрольной группой. Эти результаты явно контрастируют с результатами, полученными для тамоксифена и эстрогена. При всех вводимых дозах тамоксифена наблюдалось увеличение высоты эпителия, равное высоте эпителия интактных крыс и в шесть раз превышающее величину, полученную в результате обработки ралоксифеном. Обработка эстрадиолом приводила к увеличению высоты эпителия до величины, превышающей толщину эпителия интактных крыс.
Эстрогенность также определяли путем оценки побочной реакции в виде эозинофильной инфильтрации в стромальный слой матки (табл. 6). При введении ралоксифена не наблюдалось какого-либо увеличения числа эозинофилов в стромальном слое овариэктомизированных крыс, тогда как введение тамоксифена приводило к его значительному увеличению. При обработке эстрадиолом, как и ожидалось, наблюдали значительное возрастание эозинофильной инфильтрации.
Влияние ралоксифена и тамоксифена на толщину стромы и миометрия не обнаруживали заметного различия либо это различие было очень незначительным. Увеличение этих параметров, обусловленное обработкой указанными двумя агентами, было значительно меньше, чем эффект, полученный при обработке эстрогеном.
Общая оценка эстрогенности, полученная по всем четырем параметрам, показала, что ралоксифен обладает значительно меньшей эстрогенностью, чем тамоксифен.
Пример 4. В соответствии с процедурой, описанной в примере 1, крысам были перорально введены и другие соединения формулы I. В табл. 7 представлены результаты воздействия дозы 1 мг/кг каждого соединения, выраженные в виде процента ингибирования разрежения кости и процента увеличения веса матки.
Пояснения к табл. 7.
a) Процент ингибирования разрежения кости = (плотность кости обработанных овариэктомизированных крыс - плотность кости необработанных овариэктомизированных крыс) + (плотность кости эстрогенобработанных овариэктомизированных крыс - плотность кости необработанных овариэктомизированных крыс) х 100.
b) Процент увеличения веса матки = (вес матки обработанных овариэктомизированных крыс - вес матки овариэктомизированных крыс) + (вес матки эстрогенобработанных овариэктомизированных крыс - вес матки овариэктимизированных крыс) х 100.
Пример 5. Частота переломов костей вследствие остеопороза обратно пропорциональна плотности минерализованной костной ткани. Однако изменения плотности костной ткани происходят крайне медленно, и получить значимые результаты ее измерения можно лишь через многие месяцы или годы. И все же, продемонстрировать положительное воздействие соединений формулы I, таких как ралоксифен, на плотность кости и разрежение кости можно путем измерения различных быстро реагирующих биохимических параметров, изменения которых отражают изменения скелетного метаболизма. В этих целях в экспериментах по испытанию ралоксифена (вводимого в виде гидрохлорида) участвовали по крайней мере 160 пациентов, которые были произвольно разделены на четыре группы: группу, получившую эстроген, две группы, получавшие две различные дозы ралоксифена, и группу, получившую плацебо. Пациенты получали вышеуказанные средства ежедневно в течение восьми недель.
Кровь и мочу брали перед, во время и после завершения испытания. Помимо этого в начале и после завершения исследования проводили оценку эпителия матки. Введение эстрогена и плацебо служило в качестве положительного и отрицательного контроля соответственно.
Пациентами были здоровые женщины, переживающие постклимактерический период (после операции или естественный) в возрасте 45-60 лет, которым была рекомендована эстрогеновая заместительная терапия для лечения остеопороза. В этом эксперименте участвовали женщины с неповрежденной маткой, у которых последняя менструация была не позднее, чем 6 мес назад и не ранее, чем 6 лет назад.
Из экспериментов исключались пациенты, которые перед их началом систематически принимали следующие лекарственные средства: витамин D, кортикостероиды, гиполипидемические средства, тиазиды, средства против подагры, салицилаты, фенотиазины, сульфонаты, тетрациклины, неомицин и противоглистные препараты. Из испытаний также были исключены пациенты, прошедшие курс эстрогеновой, прогестиновой или андрогеновой терапии позднее, чем за три месяца до начала испытаний; пациенты, подвергавшиеся когда-либо терапии с использованием кальцитонина, фторида или бифосфоната; пациенты с сахарным диабетом; пациенты, имеющие в истории болезни за последние пять лет раковое заболевание; пациенты с любыми недиагностированными аномальными генитальными кровотечениями; пациенты с прогрессирующими тромбоэмболическими нарушениями (зафиксированными в истории болезни); пациенты с нарушениями функций печени или почек; пациенты с нарушенной функцией щитовидной железы; пациенты, относящиеся к группам аллергического или психического риска; или пациенты с чрезмерным употреблением алкоголя или злоупотребления лекарственными средствами.
Пациенты эстрогеновой группы получали дозу 0,625 мг/день, а пациенты двух ралоксифеновых групп получали дозы 200 мг/день и 600 мг/день, причем все указанные группы получали пероральные препараты в виде капсул. В качестве кальциевой добавки использовали карбонат кальция в таблетках (648 мг), причем все пациенты в течение курса испытания принимали по 2 таблетки каждое утро.
Исследования проводили двойным слепым методом. Ни исследователи, ни пациенты не знали, к какой именно группе принадлежал данный пациент.
Начальное обследование каждого пациента включало в себя количественное определение содержания кальция, креатинина, гидроксипролина и пиридинолиновых поперечных сшивок в моче. Пробы крови анализировали на сывороточные уровни остеокальцина, костоспецифической щелочной фосфатазы, ралоксифена и метаболитов ралоксифена. Начальное обследование также включало в себя исследование матки, включая ее биопсию.
В последующие визиты к врачу-исследователю измерения вышеуказанных параметров проводили как описано выше, но уже в ответ на вводимые лекарственные препараты. Было установлено, что все биохимические маркеры, перечисленные выше и ассоциирующиеся резорбцией кости, ингибировались при введении эстрогена по сравнению с данными, полученными для необработанной группы. Ралоксифен, как и ожидалось, также ингибировал указанные маркеры у пациентов с дефицитом эстрогена, что свидетельствует о том, что ралоксифен является эффективным ингибитором разрежения кости, действие которого начинается непосредственно со времени его введения.
Последующие длительные исследования дадут возможность непосредственно измерить плотность кости с помощью фотонной абсорбциометрии, а также провести измерения частоты переломов кости после проведения терапии.
Изобретение относится к фармацевтической композиции на основе бензотиофенов и к применению последних для лечения или предупреждения остеопороза путем ингибирования разрежения кости. Препараты согласно изобретению могут быть использованы без нежелательных побочных явлений, обычно связанных с применением традиционной эстрогеновой терапии, а поэтому они являются эффективными для предупреждения или лечения остеопороза. 2 с. и 4 з.п. ф-лы, 7 табл.
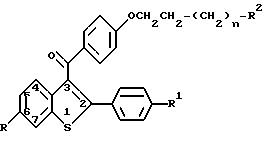
где n 0, 1 или 2;
R и R1 независимо водород, гидроксил, С1-С6-алкокси, С1-С6-ацилокси, С1-С6-алкокси- С2-С6-ацилокси, R3- замещенная арилокси-, R3-замещенная ароилокси-, R4 замещенная карбонилокси-, хлор- или бромгруппа;
R2 гетероциклическое кольцо, выбранное из группы, включающей пирролидино, пиперидино и гексаметиленимино;
R3 представляет собой С1-С3-алкил, С1-С3-алкокси, водород или галогеновую группу;
R4 С1-С6-алкокси или арилокси,
или его фармацевтически приемлемой соли для ингибирования разрежения кости у человека.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| US, патент N 4133814, 1979, C 07 D 333/52 | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| US, патент N 4418068, 1983, C 07 D 333/52. | |||
