Изобретение относится к способу снижения уровня холестерина в сыворотке с использованием низких доз (малых количеств) конкретных соединений -2-фенил-3-ароилбензотиофенов.
Холестерин необходим для всех клеток млекопитающих в качестве структурного компонента их клеточных мембран и для нестериновых конечных продуктов. Холестерин необходим также для синтеза стероидных гормонов. Однако, именно то свойство, которое делает холестерин необходимым для клеточных мембран, его нерастворимость в воде, делает его также потенциально летальным. Если холестерин накапливается в не предназначенном для этого участке, например, на внутренних стенках артерий, он не может быть мобилизован с легкостью, и его присутствие приводит к образованию атеросклеротических бляшек. Было показано, что повышенные концентрации сывороточного холестерина, связанные с липопротеинами низкой плотности, являются основным фактором, который вносит вклад в развитие и прогрессирование атеросклероза.
Сывороточный липопротеин у млекопитающих состоит из холестерина со сложными эфирами холестерила, триглицеридами, фосфолипидами и апопротеинами. Сывороточные или плазменные липопротеины состоят из нескольких фракций. Основные фракции или классы липопротеинов плазмы являются липопротеинами очень низкой плотности (ЛОНП), липопротеинами низкой плотности (ЛНП), липопротеинами промежуточной плотности (ЛПП) и липопротеинами высокой плотности (ЛВП). Эти классы различаются по размеру, плотности и относительному содержанию триглицероидов и сложных холестериловых эфиров в ядре, и в соответствии с природой апопротеинов - на поверхности.
У млекопитающих сывороточный холестерин образуется из эндогенных источников питания, а также за счет эндогенного синтеза. Эндогенный синтез холестерина включает комплекс энзимкатализируемых реакций и регуляторных механизмов, который обычно называют мевалонатной схемой. Клетки представляют комплексную проблему в регулировании мевалонатного синтеза, так как холестерин, основной конечный продукт мевалонатного метаболизма, получается из плазменного липопротеина низкой плотности, который поступает в клетки за счет осуществляемого с помощью рецептора эндоцитоза, а также за счет синтеза внутри клеток. Каждая клетка должна сбалансировать эти внутренние и внешние источники таким образом, чтобы мевалонатный синтез продолжался, но при этом избегая избыточного накопления стерина. Этот баланс достигается за счет регулирования с помощью обратной связи, по крайней мере, двух последовательных энзимов в мевалонатном синтезе, 3-гидрокси-3-метилглутарилкоэнзим A (HMG-CoA) синтазы и HMG-CoA редуктазы, а также за счет рецепторов ЛНП. В отсутствие ЛНП клетки млекопитающих сохраняют высокие активности этих двух энзимов, обеспечивая за счет этого синтез мевалоната для продуцирования холестерина, а также нестериновых продуктов. В присутствии ЛНП из экзогенных источников, HMC-GoA синтазная и редуктазная активности подавляются, и клетки продуцируют меньшие количества мевалоната для нестериновых конечных продуктов.
Имеются доказательства того, что лечение гиперлипопротеинэмии снижает или предотвращает атеросклеротические осложнения. Помимо диеты, которая поддерживает нормальный вес тела и сводит к минимуму концентрации липидов в плазме, терапевтический подход включает исключение таких факторов, которые обостряют гиперлипопротеинэмию, и введение таких терапевтических агентов, которые снижают концентрацию липопротеинов в плазме, либо за счет уменьшения продуцирования липопротеинов, либо за счет повышения эффективности их выведения из плазмы.
Наиболее обещающий класс лекарств, доступных в настоящее время для лечения гиперхолестеринэмии, действует по принципу НМС-СоА редуктазы, энзима, ограничивающего скорость эндогенного синтеза холестерина. Лекарственные препараты этого класса конкурентно ингибируют активность энзима. В итоге это снижает эндогенный синтез холестерина и, за счет обычных гомеостатических механизмов, плазменный холестерин отбирается рецепторами для сохранения внутриклеточного баланса холестерина.
Что касается других клеток организма, клетки печени играют критическую роль в поддержании гомеостаза сывороточного холестерина, как за счет выделения предшественников ЛНП, так и за счет осуществляемого с помощью рецепторов вывода ЛНП из сыворотки. При проведении исследований как на животных, так и на людях, по-видимому, существует обратная зависимость между рецепторами ЛНП печени и связанными с ЛНП уровнями сывороточного холестерина. Обычно большее количество гепатоцитных рецепторов приводит к снижению связанных с ЛНП уровней содержания холестерина в сыворотке. Холестерин, выделенный в гепатоциты, может храниться в них в виде сложных эфиров холестерина, превращаться в желчные кислоты и выводиться с желчью, либо может попадать в пул оксихолестеринов. Считают, что именно пул оксихолестеринов включен в репрессию конечного продукта как генов рецептора ЛНП, так и энзимов, вовлеченных в схему синтеза холестерина.
Как известно, транскрипция гена рецепторов ЛНП подавляется, если в клетки поступает избыточное количество холестерина, вероятно в форме оксихолестерина. Промоторный участок ДНК последовательности рецептора ЛНП, известный как элемент, ответственный за стерин, по-видимому, участвует в подавлении этого конечного продукта стирола. Этот элемент был подвергнут интенсивному изучению (Brown, Goldstein and Russell, патент США N 4745060 и N 4935363) и, по-видимому, состоит из 16 пар оснований, которые находятся в 5' кодирующем участке рецептора ЛНП. Ответственный за стерин элемент можно встроить в гены, которые обычно не отвечают за холестерин, придавая тем самым химерическому гену свойство подавления конечного стирольного продукта. Точный механизм такого подавления непонятен. Однако, существует доказательство того, что полярные промежуточные биоситеза холестерина, а также как природные, так и синтетические репрессорные гены гидроксистеринов содержат ответственный за стерины элемент.
Было выдвинуто предположение, что рецептором служит протеин, связывающий гидроксихолестерин. Когда такой рецептор связан с оксистерином, он действует на ответственный за стерин элемент таким образом, чтобы регулировать транскрипцию за счет механизма, который аналогичен действию членов семейства генов рецепторов стероидных гормонов.
В популяциях, где коронарные болезни сердца являются основной проблемой здоровья, случаи заболеваний у женщин значительно реже, чем у мужчин. Это особенно справедливо для более молодой возрастной группы, а именно, для мужчин и женщин от 35 до 44 лет.
Обычно на метаболизм липопротеинов в плазме влияет циркулирующая концентрация гонадных стероидов. Изменения концентраций эстрогена и андрогена в сыворотке, обусловленные изменениями гонадного статуса или введением экзогенных гонадных стероидов, связаны с изменениями уровней липопротеинов в сыворотке. Такие изменения за счет эстрогенов и андрогенов в общем подтверждают предположение, что сексуальные различия в липопротеинах связаны с гормональными различиями между мужчинами и женщинами.
Общепринятое соотношение между гонадными стероидами и липопротеинами плазмы состоит в том, что андрогены снижают концентрации ЛВП и повышают ЛНП, что приводит к низким уровням ЛВП и высоким уровням ЛНП, наблюдающимся у мужчин по сравнению с женщинами. Эстрогены же оказывают на липопротеины обратное действие; т. е. уровень ЛВП возрастает, а ЛНП снижается. Эти сексуальные, связанные со стероидами различия в концентрациях липопротеинов, как считают, вносят вклад в снижение случаев сердечно-сосудистых заболеваний у женщин по сравнению с мужчинами. После менопаузы защитное действие эстрогенов у женщин снижается, и число сердечно-сосудистых заболеваний возрастает, приближаясь к числу заболеваний у мужчин. В постклимактерическом периоде число сердечно-сосудистых заболеваний у женщин, которые получают эстрогены, ниже, чем у тех, кто их не получает. Эстроген при оральном приеме снижает уровни содержания в плазме ЛНП и повышает уровни ЛВП.
Механизм, за счет которого эстроген снижает уровни ЛНП и повышает уровни ЛВП, неизвестен. Обычно изменение концентрации липопротеина в плазме происходит за счет изменений скорости синтеза или скорости катаболизма. Так, например, эстроген может снижать ЛНП уровни за счет ускорения выведения ЛНП из плазмы, так как эстроген повышает у животных число ЛНП рецепторов печени.
Хотя эстрогены оказывают благоприятное воздействие на ЛНП сыворотки, при введении в очень малых дозах, длительное лечение эстрогенами осложняется различными расстройствами, включая возрастание риска возникновения рака матки и вероятность рака груди, что приводит к отказу многих женщин от такого лечения. Недавно предложенный терапевтический режим, который был создан для снижения вероятности возникновения рака, включающий введение сочетаний прогестогена и эстрогена, вызывает у пациенток регулярные кровотечения, что неприемлемо для большинства пожилых женщин. Кроме того, объединение прогестерона с эстрогеном, по-видимому, уменьшает снижающее сывороточный холестерин действие эстрогена. С точки зрения значительных нежелательных эффектов, связанных с лечением эстрогенами, остается необходимость в разработке альтернативных способов лечения гиперхолестеринэмии, которые оказывали бы необходимое воздействие на ЛНП сыворотки, но не вызывали бы нежелательных эффектов.
Попытки удовлетворения такой необходимости за счет использования соединений, обычно известных как антиэстрогены, которые взаимодействуют с рецепторами эстрогенов и/или связывают то, что носит название антиэстрогенных связывающих сайтов/АЕВ, имели ограниченный успех, вероятно, из-за того, что эти соединения обычно оказывают смешанное агонист/антагонистическое действие. То есть, хотя эти соединения могут оказывать антагонистическое действие на взаимодействие эстрогена с рецептором, сами соединения могут вызывать эстрогенные реакции в тех тканях, которые имеют эстрогенные рецепторы, например, в тканях матки. Поэтому некоторые антиэстрогены, такие как тамоксифен, также оказывают такое же вредное воздействие как то, которое связано с лечением эстрогенами.
Настоящее изобретение включает также способ снижения уровня холестерина в сыворотке, включающий введение соединения формулы 1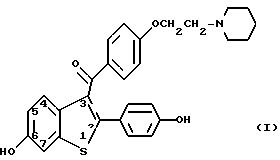
в количестве от около 55 до около 150 мг/день.
Настоящее изобретение включает также композиции в стандартной дозированной форме, содержащие дозу в количестве от около 55 до около 150 мг соединения формулы I.
Настоящее изобретение включает открытие того факта, что соединения формулы I пригодны для снижения уровня холестерина в сыворотке в дозах от около 55 до около 150 мг/день. Способы, предложенные в настоящем изобретении, предлагают введение нуждающемуся в лечении человеку дозы соединения формулы I, или его фармацевтически приемлемой соли, или сольвата в количестве от около 55 до около 150 мг/день для снижения уровня холестерина в сыворотке.
Обычно соединение используют в композиции с обычными наполнителями, разбавителями или носителями и прессуют в виде таблеток или приготавливают в виде эликсиров или растворов для обычного перорального приема, или вводят внутримышечно или внутривенно. Соединения можно вводить через кожу, и они пригодны для использования в лекарственных формах с замедленным высвобождением и т.п.
Способ настоящего изобретения пригоден как для мужчин, так и для женщин. Практическое отсутствие эстрогенной реакции должно позволить мужчинам использовать способ настоящего изобретения без проявления реакции феминизации эстрогена или агонистов эстрогена, такой как гинекомастия. Однако, предпочтительно использовать способы настоящего изобретения для женщин и более предпочтительно для женщин с недостатком эстрогена.
2-фенил-3-ароилбензотиофеновые соединения, которые являются активными компонентами в способе настоящего изобретения, впервые были разработаны C. Dabid Jones and Tulio Suazez, как противозачаточные средства (США патент N 4133814, выданный 9 января 1979). Было обнаружено, что некоторые соединения в этой группе можно использовать для подавления роста опухолей молочной железы.
Позднее Lones обнаружил группу родственных соединений, которые можно было использовать в антиэстрогенной и антиандрогенной терапии, особенно при лечении опухолей молочной железы и простаты (патент США N 4418068, выданный 29 ноября 1983). Одно из этих соединений, гидрохлорид соединения формулы I, было протестировано в клинике (непродолжительное время) для лечения рака груди. Это соединение носит название ралоксифен (предшествующее название - кеоксифен).
В настоящее время ралоксифен проходит клинические испытания при лечении остеопороза и снижения содержания липидов. Dzafer et al. ("Влияние ралоксифена на биохимические маркеры кости и метаболизм липидов у здоровых женщин в постклимактерическом возрасте". Четвертый Международный симпозиум по остеопорозу, Гон-Конг, март 29, 1993) обсуждает некоторые положительные свойства ралоксифена для снижения уровня халестерина в сыворотке. Испытываемые дозы составляли 200 и 600 мг/день. Как следует из ЕРО публикации ЕР-А-584952, опубликованной 2 марта 1994 (соответствует патентной заявке США 07/920933, поданной 28 июля 1992), предпочтительный интервал указан как 200-600 мг/день. Хотя такой интервал доз, как 200-600 мг/день вызывает достаточную реакцию и фармацевтически приемлем, неожиданно было обнаружено, что более низкий интервал доз ралоксифена, от около 55 мг/день до около 150 мг/день, обеспечивает эквивалентные преимущества по сравнению с более высокими дозами.
Было показано, что ралоксифен связывается с рецепторами эстрогена, и вначале считалось, что именно он является молекулой функции, и фармакология которой в качестве антиэстрогена состоит в том, что она блокирует способность эстрогена активировать ткани матки и зависимые от эстрогена раковые заболевания груди. Действительно, ралоксифен блокирует действие эстрогена в некоторых клетках, однако, в других типах клеток ралоксифен активирует те же гены, что и эстроген, и демонстрирует ту же самую фармакологию, то есть, остеопороз, гиперлипидемию. Теперь считают, что уникальный профиль, который демонстрирует ралоксифен и которым он отличается от эстрогена, связан с уникальной активацией и/или подавлением различных генных функций за счет ралоксифен- эстрогенного рецепторного комплекса, в противоположность активации и/или подавлению генов эстроген-эстрогенного рецепторного комплекса. Поэтому, хотя ралоксифен и эстроген используют и конкурируют за одни и те же рецепторы, фармакологический результат генной регуляции этих двух соединений не так легко предсказать, и он различен у каждого из них.
Обычно соединения используют в композиции с обычными наполнителями, разбавителями или носителями, прессуют в таблетки или приготавливают в виде эликсиров или растворов для обычного перорального введения, или вводят внутримышечно, или внутривенно. Соединения можно вводить через кожу или вагинально, и они могут быть приготовлены в виде лекарственных форм с замедленным выделением.
Соединения, используемые в способах настоящего изобретения, можно получить в соответствии с известными процедурами, например по способу патентов США NN 4133814, 4418068 и 4380635, которые включены в описание в качестве ссылок. Вообще, в этих способах исходят из бензо-[b]-тиофена с 6-гидроксильной группой и 2-/4-гидроксильной группой. Гидроксильные группы исходного соединения защищены, три положения ацилированы, и удаляя защиту, получают соединение формулы I. Примеры получения таких соединений приведены в патентах США, обсуждавшихся ранее.
Используемые в способах настоящего изобретения соединения образуют фармацевтически приемлемые соли присоединения кислот и оснований с широким кругом органических и неорганических кислот и оснований, включая физиологически приемлемые соли, которые часто используют в фармацевтической химии. Такие соли также входят в объем изобретения. Типичные неорганические кислоты, которые используют для получения таких солей, включают соляную, бромистоводородную, иодистоводородную, азотную, серную, фосфорную, гипофосфорную и т. п. Соли, полученные из органических кислот, таких как алифатические моно- и дикарбоновая кислоты, фенилзамещенные алкановые кислоты, гидроксиалкановые и гидроксиалкандиоевые кислоты, ароматические кислоты, алифатические и ароматические сульфоновые кислоты, также можно использовать. Такие фармацевтически приемлемые соли включают ацетат, фенилацетат, трифторацетат, акрилат, аскорбат, бензоат, хлорбензоат, ди-нитробензоат, гидроксибензоат, метоксибензоат, метилбензоат, о-ацетоксибензоат, нафталин-2- бензоат, бромид, изобутират, фенилбутират, гидроксибутират, бутин-1,4-диоат, гексин-1,4-диоат, капрат, каприлат, хлорид, циннамат, цитрат, формат, фумарат, гликоллят, гептаноат, гиппурат, лактат, малат, малеат, гидроксималеат, малонат, манделат, мезилат, никотинат, изоникотинат, нитрат, оксалат, фталат, терефталат, фосфат, моно (кислый) фосфат, ди (кислый) фосфат, метафосфат, пирофосфат, пропиолат, фенилпропионат, пропионат, салицилат, себакат, сукцинат, суберат, сульфат, бисульфат, пиросульфат, сульфит, бисульфит, сульфонат, бензол-сульфонат, пара-бромбензолсульфонат, хлорбензолсульфонат, этансульфонат, 2-гидроксиэтаносульфонат, метансульфонат, нафталин-1-сульфонат, нафталин-2-сульфонат, р-толуолсульфонат, ксилолсульфонат, тартрат и т.п. Предпочтительной солью является гидрохлорид.
Фармацевтически приемлемые соли присоединения кислот обычно получают, подвергая взаимодействию соединение формулы I с эквимолярным количеством или избытком кислоты. Обычно реагенты объединяют в таком общем растворителе, как диэтиловый эфир или бензол. Обычно соль осаждается из раствора за промежуток времени от одного часа до 10 дней, и ее можно выделить фильтрованием, или растворитель можно выпарить обычным способом.
Основания, которые обычно используют для получения солей, включают гидроксид аммония и гидроксиды и карбонаты щелочных и щелочноземельных металлов, а также алифатические и первичные, вторичные и третичные амины, алифатические диамины. Основания, которые наиболее подходят для получения солей присоединения, включают гидроксид натрия, гидроксид калия, гидроксид аммония, карбонат калия, метиламин, диэтиламин, этилендиамин и циклогексиламин.
Фармацевтически приемлемые соли обычно отличаются повышенной растворимостью по сравнению с соединениями, из которых они получены, и поэтому они часто более применимы для получения композиций в виде жидкостей или эмульсий.
Фармацевтические композиции можно получить способами, известными специалистам. Так например, композиции соединений можно получить с обычными наполнителями, разбавителями или носителями, и приготовить в виде таблеток, капсул, суспензий, порошков и т.п.
Примеры наполнителей, разбавителей и носителей, которые пригодны для таких композиций, включают следующие: такие наполнители и агенты, увеличивающие объем, как крахмал, сахар, маннит и производные кремния; такие связывающие агенты, как карбоксиметилцеллюлоза и другие производные целлюлозы, альгинаты, желатин и поливинилпирролидон; такие увлажнители, как глицерин; такие разрыхлители, как карбонат кальция и бикарбонат натрия; такие агенты, замедляющие растворение, как парафин; такие ускорители ресорбции, как соединения четвертичного аммония; такие поверхностно-активные агенты, как цетиловый спирт, моностеарат глицерина; такие адсорбтивные носители, как каолин и бентонит; и такие скользящие, как тальк, стеарат кальция и магния, и твердые полиэтиленгликоли.
Соединения также можно приготовить в композициях в виде эликсиров или растворов для удобства перорального приема, или в виде растворов для парэнтерального введения, например, внутримышечного, подкожного или внутривенного. Кроме того, соединения пригодны для приготовления композиций в виде лекарственных форм для замедленного выделения и т.п. Композиции могут быть также приготовлены таким образом, чтобы они выделяли активный ингредиент только (или предпочтительно) в определенном участке кишечника и возможно в течение определенного промежутка времени. Можно использовать покрытия, облатки и защитные матрицы, например, из полимерных материалов или восков.
Дозы согласно настоящего изобретения составляет от около 55 до около 150 мг/день и предпочтительно от 60 до 150 мг/день, и наиболее предпочтительно от 60 до 100 мг/день. Конкретными дозами в рамках настоящего изобретения являются 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 105, 110, 115, 120, 125, 130, 135, 140, 145 и 150 мг/день.
Композиции, предпочтительно, создают в виде стандартных лекарственных форм, причем каждая доза содержит от около 55 до около 150 мг и более предпочтительно указанные выше количества. Термин "стандартная лекарственная форма" относится к физически дискретной единице, например, к таблеткам или капсулам, пригодным для одноразового приема, особенно для одноразового ежедневного приема, для людей и других млекопитающих, причем каждая доза содержит заранее определенное количество активного материала, рассчитанное для достижения желательного терапевтического эффекта, вместе с подходящим фармацевтическим наполнителем.
Длительность приема людьми дозы от около 55 до около 150 мг/день будет зависеть от тяжести состояния, здоровья пациента и сопутствующих факторов, которые должен оценить лечащий врач. Предполагается, что курс лечения должен составлять, по крайней мере, шесть месяцев, обычно, по крайней мере, один год и предпочтительно не должен прерываться.
Примеры композиций в дозированном интервале следующие:
Композиции
Рецептура 1: желатиновые капсулы
Твердые желатиновые капсулы получают, используя следующие ингредиенты, мг/капсулу:
Ралоксифен - 55-150
Крахмал, NF - 0-650
Крахмал в виде пересыпающегося порошка - 0-650
Силиконовая жидкость, 350 - 0-15
Ингридиенты смешивают, пропускают через сито 45 меш США и заполняют в твердые желатиновые капсулы.
Примеры композиций капсул включают приводимые далее:
Рецептура 2: капсулы ралоксифена, мг/капсулу:
Ралоксифен - 60
Крахмал, NF - 112
Свободно пересыпающийся порошок крахмала - 225,3
Силиконовая жидкость, 350 сСт - 1,7
Рецептура 3: ралоксифена, мг/капсулу:
Ралоксифен - 75
Крахмал, NF - 108
Свободно пересыпающийся порошок крахмала - 225,3
Силиконовая жидкость, 350 сСт - 1,7
Рецептура 4: капсулы ралоксифена, мг/капсулу:
Ралоксифен - 100
Крахмал, NF - 103
Свободно пересыпающийся порошок крахмала - 225,3
Силиконовая жидкость, 350 сСт - 1,7
Рецептура 5: ралоксифеновые капсулы, мг/капсулу:
Ралоксифен - 125
Крахмал, NF - 150
Свободно пересыпающийся порошок крахмала - 397
Силиконовая жидкость, 350 сСт - 3
Рецептура 6: ралоксифеновые капсулы, мг/капсулу:
Ралоксифен - 150
Крахмал, NF - 150
Свободно пересыпающийся порошок крахмала - 397
Силиконовая жидкость, 350 сСт - 3
Указанные выше конкретные рецептуры можно изменить в соответствии с разумными приложенными вариантами.
Рецептуры таблеток получают, используя приведенные далее ингредиенты.
Рецептура 7: таблетки, мг/таблетку:
Ралоксифен - 60
Микрокристаллическая целлюлоза - 0-650
Мелкодисперсная двуокись кремния - 0-650
Стеариновая кислота - 0-15
Рецептура 8: таблетки, мг/таблетку:
Ралоксифен - 75
Микрокристаллическая целлюлоза - 0-650
Мелкодисперсная двуокись кремния - 0-650
Стеариновая кислота - 0-15
Рецептура 9: таблетки, мг/таблетку:
Ралоксифен - 100
Микрокристаллическая целлюлоза - 0-650
Мелкодисперсная двуокись кремния - 0-650
Стеариновая кислота - 0-15
Рецептура 10: таблетки, мг/таблетку:
Ралоксифен - 125
Микрокристаллическая целлюлоза - 0-650
Мелкодисперсная двуокись кремния - 0-650
Стеариновая кислота - 0-15
Рецептура 11: таблетки, мг/таблетку:
Ралоксифен - 150
Микрокристаллическая целлюлоза - 0-650
Мелкодисперсная двуокись кремния - 0-650
Стеариновая кислота - 0-15
Эти компоненты смешивают и прессуют в таблетки.
В другом варианте можно получить таблетки, каждая из которых содержит от 55 до 150 мг активного ингредиента, следующим образом:
Рецептура 12: таблетки мг/таблетки:
Ралоксифен - 60
Крахмал - 45
Микрокристаллическая целлюлоза - 35
Поливинилпирролидон (в виде 10% раствора в воде) - 4
Натрийкарбоксиметилцеллюлоза - 4,5
Стеарат магния - 0,5
Тальк - 1
Рецептура 13: таблетки, мг/таблетку:
Ралоксифен - 75
Крахмал - 45
Микрокристаллическая целлюлоза - 35
Поливинилпирролидон (в виде 10% раствора в воде) - 4
Натрийкарбоксиметилцеллюлоза - 4,5
Стеарат магния - 0,5
Тальк - 1
Рецептура 14: таблетки мг/таблетку:
Ралоксифен - 100
Крахмал - 45
Микрокристаллическая целлюлоза - 35
Поливинилпирролидон (в виде 10% раствора в воде) - 4
Натрийкарбоксиметилцеллюлоза - 4,5
Стеарат магния - 0,5
Тальк - 1
Рецептура 15: таблетки, мг/таблетку:
Ралоксифен - 125
Крахмал - 45
Микрокристаллическая целлюлоза - 35
Поливинилпирролидон (в виде 10% раствора в воде) - 4
Натрийкарбоксиметилцеллюлоза - 4,5
Стеарат магния - 0,5
Тальк 1 - 1
Рецептура 16: таблетки, мг/таблетку:
Ралоксифен - 150
Крахмал - 45
Микрокристаллическая целлюлоза - 35
Поливинилпирролидон (в виде 10% раствора в воде) - 4
Натрийкарбоксиметилцеллюлоза - 4,5
Стеарат магния - 0,5
Тальк - 1
Активный ингредиент, крахмал и целлюлозу пропускают через сито N 45 меш США и тщательно смешивают. С полученным порошком смешивают раствор поливинилпирролидона, и все это пропускают через сито N 14 меш США. Полученные таким образом гранулы сушат при 50-60oC и пропускают через сито N 18 меш США. Затем к гранулам добавляют предварительно пропущенные через сито N 60 меш США натрийкарбоксиметилцеллюлозу, стеарат магния и тальк, и все это после перемешивания прессуют в устройстве для изготовления таблеток.
Суспензии, каждая из которых содержит 55-150 мг медикамента на 5 мл дозу, приготавливают следующим образом:
Рецептура 17: суспензии, мг/5 мл:
Ралоксифен - 60
Натрийкарбоксиметилцеллюлоза - 50
Сироп - 1,25
Раствор бензойной кислоты - 0,10
Корригент - 0..
Краситель - 0..
Очищенная вода, мл - До 5
Рецептура 18: суспензии, мг/5 мл:
Ралоксифен - 75
Натрийкарбоксиметилцеллюлоза - 50
Сироп - 1,25
Раствор бензойной кислоты, мл - 0,10
Корригент - 0..
Краситель - 0..
Очищенная вода, мл - До 5
Рецептура 19: суспензии, мг/5 мл:
Ралоксифен - 100
Натрийкарбоксиметилцеллюлоза - 50
Сироп - 1,25
Раствор бензойной кислоты, мл - 0,10
Корригент - 0..
Краситель - 0..
Очищенная вода, мл - До 5
Рецептура 20: суспензии, мг/5 мл:
Ралоксифен - 125
Натрийкарбоксиметилцеллюлоза - 50
Сироп - 1,25
Раствор бензойной кислоты, мл - 0,10
Корригент - 0..
Краситель - 0..
Очищенная вода, мл - До 5
Рецептура 21: суспензии, мг/5 мл:
Ралоксифен - 150
Натрийкарбоксиметилцеллюлоза - 50
Сироп - 1,25
Раствор бензойной кислоты, мл - 0,10
Корригент - 0..
Краситель - 0..
Очищенная вода, мл - До 5
Медикаменты пропускают через сито N 45 меш США и смешивают с натрийкарбоксиметилцеллюлозой и сиропом до получения однородной пасты. Раствор бензойной кислоты, вкусовой агент и краситель разбавляют небольшим количеством воды и добавляют при перемешивании. Затем добавляют остальную воду до нужного объема. Нижеследующие примеры иллюстрируют получение соединений, использованных в настоящем изобретении.
Пример 1
6-гидрокси-2-(4-гидроксифенил)-3-[4-(2-пиперидиноэтокси) бензоил] бензо [b]тиофен
Порцию 4 г гидрохлорида 6-метансульфонилокси-2-(4-метансульфонилоксифенил)-3-[4-(2- пиперидиноэтокси) бензоил]бензо[b]тиофена соединяют с 10 мл денатурированного спирта и 10 мл 5н гидроксида натрия, и перемешивают при кипячении с обратным холодильником в течение 1,5 часов в атмосфере азота. Затем реакционную смесь выпаривают досуха в вакууме, а остаток растворяют в 200 мл воды и промывают 300 мл диэтилового эфира. Водный слой дегазируют в вакууме, а затем через него барботируют азот для полного удаления следов эфира. Затем полученную смесь подкисляют 1н соляной кислотой, затем подщелачивают избытком бикарбоната натрия. Осадок собирают фильтрованием и промывают холодной водой до получения 2,4 г неочищенного продукта. Его очищают на колонке с силикагелем (2х30 см), элюируя вначале 700 мл 55% метанолом в хлороформе, а затем 1 литром 10% метанола в хлороформе. Вначале выходят примеси, а содержащие продукт фракции объединяют и выпаривают в вакууме до получения 1,78 г масла желтого цвета. Это масло растворяют в 6 мл ацетона, вводят затравку и охлаждают в холодильнике до получения 1,2 г очищенного продукта, т.пл. 143-147oC. Идентичность продукта подтверждают следующим образом:
Спектр ЯМР (100 МГц, в DMCO-d6) δ: 1,20-1,65 (6H, м, N(CH2CH2)2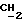 ); 2,30-2,45 (4H, м, N(
); 2,30-2,45 (4H, м, N(  CH2)2CH2); 2,60 (2H, т, J=6 Гц, OCH2
CH2)2CH2); 2,60 (2H, т, J=6 Гц, OCH2 N); 4,06 (2H, т, J= 6 Гц, O
N); 4,06 (2H, т, J= 6 Гц, O 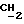 CH2N); 6,68 (2H, д, J7=9 Гц, ароматич. до ОН); 6,85 (1H, JH4-H5= 9 Гц, JH5-H7=2H, H5 бензотиофенового кольца); 6,90 (2H, д, J=9 Гц, ароматич. кольцо до OCH2CH2); 7,18 (2H, д, J=9 Гц, ароматик. мет до ОН); 7,25 (1H, д, J= 9 Гц, H4 бензотиофенового кольца); 7,66 (2H, д, J=9 Гц, ароматич. орто до СО); 9,72 (2H, шир. с., ОН).
CH2N); 6,68 (2H, д, J7=9 Гц, ароматич. до ОН); 6,85 (1H, JH4-H5= 9 Гц, JH5-H7=2H, H5 бензотиофенового кольца); 6,90 (2H, д, J=9 Гц, ароматич. кольцо до OCH2CH2); 7,18 (2H, д, J=9 Гц, ароматик. мет до ОН); 7,25 (1H, д, J= 9 Гц, H4 бензотиофенового кольца); 7,66 (2H, д, J=9 Гц, ароматич. орто до СО); 9,72 (2H, шир. с., ОН).
Ультрафиолетовый спектр (в этаноле); νмакс (E): 290 нм (34000).
Масс-спектр (бомбардировка электронами) МТ при м/e 473.
Пример 2
6-Гидрокси-2-(4-гидроксифенил)-3[4-(2- пиперидиноэтоксибензоил] бензо[b] тиофен
Порцию в 3,6 г 6- метансульфонилокси-2-(4-метансульфонилоксифенил)-3-[4-(2-пиперидиноэтокси) бензоил] бензо[b]тиофена растворяют в 100 мл тетрагидрофурана и 40 мл метанола и добавляют 10 мл 5н гидроксида натрия. Полученную смесь перемешивают в течение 16 часов при комнатной температуре, а затем обрабатывают по способу примера 1 до получения 3,5 г твердого желтого продукта. Этот продукт очищают на хроматографической колонке с силикагелем, элюируя (с градиентом) растворителем от 5% метанола в хлороформе до 30% метанола в хлороформе. Фракции, содержащие продукт, выпаривают до получения 1,85 г маслянистого продукта, который перекристаллизовывают из ацетона и получают 1,25 г очищенного продукта. Т.пл. 141-144oC.
Пример 3
Гидрохлорид 6-гидрокси-2- (4-гидроксифенил) -3[4- (2- пиперидиноэтокси) бензоил] бензо[b]тиофена
В атмосфере азота смесь 3 г гидрохлорида 4-(2-пиперидиноэтокси) бензойной кислоты, 2 капель диметилформамида, 2,5 мл тионилхлорида и 40 мл хлорбензола нагревают при 70-75oC в течение около одного часа. Затем отгоняют избыток тионилхлорида и 15-20 мл растворителя. Оставшуюся суспензию охлаждают до комнатной температуры, и к этому добавляют 100 мл дихлорметана, 2,7 г 6-метокси-2-(4-метоксифенил) бензо[b]тиофена и 10 г алюминийхлорида. Полученный раствор перемешивают около одного часа, добавляют 7,5 мл этантиола, и полученную смесь перемешивают в течение 45 мин или более. Затем добавляют 40 мл тетрагидрофурана, затем 15 мл 20%- ной соляной кислоты с экзотермом до рефлюкса. Добавляют 50 мл воды и 25 мл насыщенного водного натрийхлорида. Полученную смесь перемешивают и оставляют охлаждаться до комнатной температуры. Осадок собирают фильтрованием и промывают последовательно 30 мл воды, 40 мл 25%-ного водного тетрагидрофурана и 35 мл воды. Затем твердую часть сушат при 40oC в вакууме до получения 5,05 г продукта, который идентифицируют по данным ЯМР:
δ: 1,7 (6H, м, N(CH2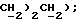 2,6-3,1 (2H, м, NCH2); 3,5-4,1 (4H, м, NCH2); 4,4 (2H, м, OCH2); 6,6-7,4 (9H, м, ароматика); 7,7 (2H, д, ароматика орто до СО); 9,8 (2H, м, ОН).
2,6-3,1 (2H, м, NCH2); 3,5-4,1 (4H, м, NCH2); 4,4 (2H, м, OCH2); 6,6-7,4 (9H, м, ароматика); 7,7 (2H, д, ароматика орто до СО); 9,8 (2H, м, ОН).
Результаты тестов
Провели 8-недельные параллельные, двойные слепые, плацебо исследования на 160 здоровых женщинах в постклимактерический период. Использованные дозы ралоксифена в этом исследовании составили 10, 50 и 200 мг. Доза 10 мг не давала заметной активности ни с одним костным маркером. (см. таблицу 1). На основании наблюдений во времени для многих костных маркеров, по-видимому, можно считать дозу ралоксифена 50 мг/день полностью активной при оценке в результате исследований большой длительности.
Сокращения: n - наибольшее число тестированных для каждого маркера; SEM - стандартное отклонение от среднего, * - статистически значимое (p < 0,051), отличное от плацебо (двойное сравнение).
На уровни липидов сыворотки оказывают влияние дозы ралоксифена 50 и 200 мг (таблица II). Снижение ЛНП холестерина наблюдается для обработанных ралоксифеном объектов при значении 50 мг и сопоставимое значение снижения для доз 200 мг. У получивших ралоксифен изменения уровней ЛВП.
Статистически значимые снижения ЛВП: ЛНП и уровней полного сывороточного холестерина наблюдаются для получавших ралоксифен как в дозах 50, так и 200 мг.
Сокращения:
ЛНП-Х - холестерин липопротеинов низкой плотности;
ЛВП-Х - холестерин липопротеинов высокой плотности;
n - наибольшее число тестированных для каждого маркера;
SEM - средняя стандартная ошибка;
# - статистическая значимость (p < 0,050) больше, чем все остальные обработки (двойное сравнение);
* - статистическая значимость (p < 0,050), отличная от плацебо (двойное сравнение).


Предложены новый способ снижения холестерина в сыворотке, включающий введение нуждающемуся в лечении пациенту соединения формулы I
или его фармацевтически приемлемой соли, и фармацевтическая композиция того же назначения. Изобретение отличает неожиданно низкая доза этого соединения - 55 -150 мг/день, обеспечивающая получение эквивалентного известному ранее результата. 2 с. и 5 з.п. ф-лы, 2 табл.

или его фармацевтически приемлемой соли в количестве 55 - 150 мг/день.
или его фармацевтически приемлемой соли, и наполнитель, разбавитель или носитель.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Прибор для охлаждения жидкостей в зимнее время | 1921 |
|
SU1994A1 |
| Black L.J | |||
| et al | |||
| ПРЕОБРАЗОВАТЕЛЬ УГЛОВОГО ПОЛОЖЕНИЯ ВАЛА В КОД | 0 |
|
SU139481A1 |
| J.-Clin | |||
| - Invest | |||
| Прибор для охлаждения жидкостей в зимнее время | 1921 |
|
SU1994A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| WO 9310113 A1, 27.05.93 | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| ИНДУКТИВНЫЙ ДАТЧИК ДАВЛЕНИЯ МОДЕЛИ ДЛЯ АЭРОДИНАМИЧЕСКИХ ИССЛЕДОВАНИЙ | 0 |
|
SU307290A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Сергеев П.В | |||
| и др | |||
| Антиэстрогены | |||
| Молекулярные механизмы действия | |||
| Химико-фармацевтический журнал | |||
| - М.: Медицина, 1990, N 5, с.4-8. | |||

