Изобретение касается ряда новых гексагидронафталиновых производных, относящихся к классу соединений, известному как класс "ML-236B", которые обладают способностью ингибировать синтез холестерина и могут использоваться для лечения и профилактики гиперхолестеринемии и различных сердечных расстройств. Изобретение также касается методов и композиций на основе этих соединений, а также способов их получения.
Чрезмерные уровни содержания холестерина в крови оказываются причиной возникновения многих жизненно угрожающих расстройств и, следовательно, ощущается потребность в лекарственных препаратах, которые воздействуют понижающим образом на уровни содержания холестерина в крови. Один возможный путь достижения этого посредством использования лекарственного препарата сводится к торможению биосинтеза холестерина.
Известен ряд соединений, которые в общем случае могут быть описаны как 7-[(замещенные)-1,2,3,5,6,7,8,8а-октагидро-1-нафтил] -3,5-дигидрооксигептаноаты, и такие соединения раскрыты в Европейской патентной публикации N 314435, в которой также более подробно описывают разработка и предшественники этих типов соединений. Полагают, однако, что ближайшими соединениями к соединениям настоящего изобретения являются соединения, раскрытые в описании изобретения Великобритании N 2077264 и в Японской патентной заявке N 59-175450, которые могут быть представлены соответственно формулами (A) и (B):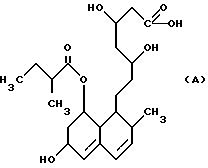
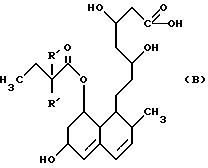
Эти известные соединения, как и соединения настоящего изобретения, обладают способностью ингибировать биосинтез холестерина и тем самым могут быть использованы для лечения и профилактики различных заболеваний, вызванных гиперхолестеринемией, таких как атеросклероз и различные сердечные расстройства.
Цель настоящего изобретения состоит в предоставлении ряда новых гексагидронафталиновых производных.
Далее и точнее цель настоящего изобретения состоит в предоставлении соединений, которые обладают способностью ингибировать биосинтез холестерина.
Другие цели и преимущества настоящего изобретения станут очевидными из последующего описания.
Таким образом, настоящее изобретение касается соединений формулы (I)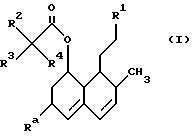
в которой R1 представляет собой группу формулы (II) или (III)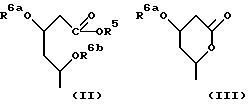
R2 представляет собой алкильную группу с числом атомов углерода от 1 до 6, алкенильную группу с числом атомов углерода от 2 до 6 или алкинильную группу с числом атомов углерода от 2 до 6;
R3 и R4 независимо выбираются из группы, в которую входят атом водорода, алкильные группы с числом атомов углерода от 1 до 6, алкенильные группы с числом атомов углерода от 2 до 6 и алкинильные группы с числом атомов углерода от 2 до 6;
R5 представляет собой атом водорода или карбоксизащищающую группу;
Ra представляет собой атом водорода или группу формулы -OR6
R6, R6a и R6b независимо выбираются из группы, в которую входят атомы водорода, гидроксизащищающие группы, алкильные группы с числом атомов углерода от 1 до 6, алкансульфонильные группы с числом атомов углерода от 1 до 6, галоидированные алкансульфонильные группы с числом атомов углерода от 1 до 6 и арилсульфонильные группы, у которых арильная часть представляет собой ароматическое углеводородное кольцо, которое содержит от 6 до 14 атомов углерода и является незамещенным или замещенным, по крайней мере, одним заместителем, выбранным из группы, в которую входят заместителя α, определенные ниже;
упомянутые заместители α выбраны из группы, в которую входят атомы галогена, алкильные группы с числом атомов углерода от 1 до 6, алкоксигруппы с числом атомов углерода от 1 до 6, карбоксигруппы, нитрогруппы, цианогруппы, алкилендиоксигруппы с числом атомов углерода от 1 до 4, ациламиногруппы, алкоксикарбонильные группы с числом атомов углерода от 2 до 7 и арильные группы;
При условии, что, когда R2 представляет собой этильную группу и R3 представляет собой атом водорода, R4 не является метильной группой, и, когда R2 представляет собой этильную группу и R3 представляет собой алкильную группу, R4 также не является алкильной группой;
и их фармацевтически приемлемых солей и сложных эфиров.
Изобретение также представляет фармацевтическую композицию (состав), включающую агент, ингибирующий биосинтез холестерина в смеси с фармацевтически приемлемым носителем или разбавителем, при этом упомянутый агент выбирается из группы, в которую входят соединения формулы (I), определенные выше, и их фармацевтически приемлемые соли и сложные эфиры.
Изобретение также касается способа лечения млекопитающего, страдающего расстройством, обусловленным дисбалансом холестерина в крови, который включает введение упомянутому млекопитающему эффективно действующего количества вещества, ингибирующего биосинтез холестерина и выбранного из группы, в которую входят соединения формулы (I), определенные выше, и их фармацевтически приемлемые соли и сложные эфиры.
Изобретение также касается способов получения соединений формулы (I) и их фармацевтически приемлемых солей и сложных эфиров, которые более подробно описаны ниже.
К соединениям настоящего изобретения относятся соединения формул (Ia) и (Ib) следующего вида:
в которых группы R1, R2, R3, R4 и R6 имеют значения, определенные ранее. Для устранения всяких сомнений в приведенных выше двух формулах показана также система частичной нумерации гексагидронафталиновых колец, как она используется в этом описании.
В случае соединений настоящего изобретения, в которых R2, R3, R4, R6, R6a или R6b представляет собой алкильную группу, эта группа может представлять собой алкильную группу с прямой или разветвленной цепью, которая содержит от 1 до 6 атомов углерода; желательно, чтобы число атомов углерода составляло от 1 до 4. Примерами таких групп являются метильная, этильная, пропильная, изопропильная, бутильная, изобутильная, втор-бутильная, трет-бутильная, пентильная, изопентильная, 2-метилбутильная, неопентильная, 1-этилпропильная, гексильная, 4-метилпентильная, 3-метилпентильная, 2-метилпентильная, 1-метилпентильная, 3,3-диметилбутильная, 2,2-диметилбутильная, 1,1-диметилбутильная, 1,2-диметилбутильная, 1,3-диметилбутильная, 2,3-диметилбутильная, и 2-этилбутильная группы, из которых предпочтение следует отдавать метильной, этильной, пропильной, изопропильной, бутильной и трет-бутильной группам. В случае группы R2 более предпочтительными являются метильная и этильная группы, причем этильная группа является самой предпочтительной. В случае группы R3 более предпочтительными группами являются метильная, этильная пропильная и изопропильная группы, причем самыми предпочтительными группами являются этильная и изопропильная группы. В случае группы R4 более предпочтительными группами являются этильная, пропильная, изопропильная, бутильная и трет-бутильная группы, причем самыми предпочтительными являются этильная и изопропильная группы.
Когда R2, R3 или R4 представляет собой алкенильную группу, то она может представлять собой алкенильную группу с прямой или разветвленной цепью, содержащей от 2 до 6 атомов углерода; желательно, чтобы она содержала от 2 до 4 атомов углерода. Примерами таких групп являются винильная, 1-пропенильная, аллильная (т.е. 2-пропенильная), 1-метил-2-пропенильная, 2-метил-1-пропенильная, 2-метил-2-пропенильная, 2-этил-2-пропенильная, 1-бутенильная, 2-бутенильная, 1-метил-2-бутенильная, 2-метил-2-бутенильная, 3-метил-2-бутенильная, 1-этил-2-бутенильная, 3-бутенильная, 1-метил-3-бутенильная, 2-метил-3-бутенильная, 1-этил-3-бутенильная, 1-пентильная, 2-пентенильная, 1-метил-2-пентенильная, 2-метил-2-пентенильная, 3-пентенильная, 1-метил-3-пентенильная, 2-метил-3-пентенильная, 4-пентенильная, 1-метил-4-пентенильная, 2-метил-4-пентенильная, 1-гексенильная, 2-гексенильная, 3-гексенильная, 4-гексенильная и 5-гексенильная группы, из которых предпочтительными являются винильная, аллильная и 3-бутенильная группы, причем самой предпочтительной является аллильная группа.
Если группы R2, R3 или R4 представляют собой алкинильную группу, то тогда она может представлять собой алкинильную группу с прямой или разветвленной цепью, содержащей от 2 до 6 атомов углерода, причем желательно, чтобы она содержала от 2 до 4 атомов углерода. Примерами таких групп являются этильная, 2-пропильная, 1-метил-2-пропинильная, 2-метил-2-пропинильная, 2-этил-2-пропинильная, 2-бутилильная, 1-метил-2-бутинильная, 2-метил-2-бутинильная, 1-этил-2-бутинильная, 3-бутинильная, 1-метил-3-бутинильная, 2-метил-3-бутинильная, 1-этил-3-бутинильная, 2-пентинильная, 1-метил-2-пентинильная, 3-пентенильная, 1-метил-3-пентинильная, 2-метил-3-пентинильная, 4-пентинильная, 1-метил-4-пентинильная, 2-метил-4-пентинильная, 2-гексинильная, 3-гексинильная, 4-гексинильная и 5-гексинильная группы, из которых предпочтительной является 2-пропинильная группа.
Термин "карбоксизащищающая группа", как он используется в определении группы R5, распространяется на защищающую группу, которая может быть удалена химическими способами (такими как гидрогенолиз, гидролиз, электролиз или фотолиз) с образованием свободной карбоксигруппы, или распространяется на защищающую группу, которая может быть удалена ин виво биологическими способами, такими как гидролиз.
Примерами карбоксизащищающих групп, которые могут быть удалены химическими способами, являются сложноэфирные и другие группы, такие как
алкильные группы с числом атомов углерода от 1 до 20, причем желательно, чтобы число атомов углерода составляло от 1 до 6, такие как группы, приведенные в качестве примеров выше в связи с рассмотрением группы R2, и т.д., и высшие алкильные группы, такие как группы, хорошо известные в этой области техники, например гептильная, октильная, нонильная, децильная, додецильная, тридецильная, пентадецильная, октадецильная, нонадецильная и икосильная группы, но при этом наиболее предпочтительными являются метильная, этильная и трет-бутильная группы;
галоидированные алкильные группы с числом атомов углерода от 1 до 6, желательно, чтобы число атомов углерода было от 1 до 4, в которых алкильная часть является такой, как определена и проиллюстрирована примерами в связи с проведением выше рассмотрения алкильных групп, и в которых атомом галогена является хлор, фтор, бром или иод, такие как 2,2,2-трихлорэтильная, 2-галоэтильная (например, 2-хлорэтильная, 2-фторэтильная, 2-бромэтильная или 2-иодэтильная группы), 2,2-дибромэтильная и 2,2,2-трибромэтильная группы;
циклоалкильные группы с числом атомов углерода от 3 до 7, например циклопропильная, циклобутильная, циклопентильная, циклогексильная и циклогептильная группы;
аралкильные группы, в которых алкильная часть содержит от 1 до 3 атомов углерода, и арильная или арильная часть представляет собой карбоциклическую ароматическую группу с числом атомов углерода от 6 до 14, которая может быть замещенной или незамещенной и, если она является замещенной, содержит, по крайней мере, один из заместителей α, определенных и проиллюстрированных примерами ниже; на алкильной группе может находиться 1, 2 или 3 таких арильных заместителя; примерами таких аралкильных групп являются бензильная, фенэтильная, 1-фенилэтильная, 3-фенилпропильная, 2-фенилпропильная, α-нафтилметильная, β-нафтилметильная, 2-(α-нафтил)этильная, 2-(2-нафтил)этильная, бензгидрильная (т.е. дифенилметильная), трифенилметильная (т. е. тритильная), α-нафтилдифенилметильная, 4-метилбензильная, 2,4,6-триметилбензильная, 3,4,5-триметилбензильная, 4-метоксибензильная, 4-метоксифенилдифенилметильная, 2-нитробензильная, 4-нитробензильная, 3-нитробензильная, 4-хлорбензильная, 4-бромбензильная, 4-цианобензильная, 4-цианофенилдифенилметильная, бис-(о-нитрофенил)метильная, 9-антрилметильная и пиперонильная группы;
алкенильные группы с числом атомов углерода от 2 до 6, такие как винильная, аллильная, 2-метилаллильная, 1-пропенильная, изопропенильная, 1-бутенильная, 2-бутенильная, 3-бутенильная, 1-пентенильная, 2-пентенильная, 3-пентенильная, 4-пентенильная, 1-гексенильная, 2-гексенильная, 3-гексенильная, 4-гексенильная, и 5-гексенильная группы, из которых предпочтительными являются винильная, аллильная, 2-метилаллильная, 1-пропенильная, изопропенильная и бутенильная группы, причем наиболее предпочтительными являются аллильная и 2-метилаллильная группы;
замещенные силилалкильные группы, в которых алкильная часть имеет значения, определенные и проиллюстрированные примерами ранее, и силильная группа содержит до трех заместителей, выбранных из алкильных групп с числом атомов углерода от 1 до 6 и фенильных групп, которые являются незамещенными или содержат, по крайней мере, один заместитель, выбранный из заместителей α , определенных и проиллюстрированных примерами ниже, например 2-триметилсилилэтильная группа;
арильные группы с числом атомов углерода от 6 до 14 и необязательно замещенные одним или несколькими заместителями α, определенными и проиллюстрированными примерами ниже, например фенильная, α-нафтильная, β-нафтильная, инданильная и антренильная группы, желательно, чтобы ими были фенильная или инданильная группа, и более желательно, чтобы ею была фенильная группа; любые из этих арильных групп могут быть незамещенными или замещенными и, если они являются замещенными, то желательно, чтобы присутствовала, по крайней мере, одна алкильная группа с числом атомов углерода от 1 до 4 или ациламиногруппа; примерами замещенных групп являются толильная и бензамидофенильная группы;
фенацильные группы, которые могут быть незамещенными или могут содержать один из заместителей α, определенных и проиллюстрированных примерами ниже, например сама фенацильная группа или п-бромфенацильная группа; и
циклические и ациклические терпенильные группы, например геранильная, нерильная, линалильная, фитильная, ментильная (особенно м- и п-ментильная), туйильная, карильная, пинанильная, борнильная, норкарильная, норпинанильная, норборнильная, ментенильная, камфенильная и норборненильная группы.
Примерами карбоксизащищающих групп, которые могут быть удалены ин виво биологическими способами, такими как гидролиз, являются сложноэфирные и другие группы, такие как
алкоксиалкильные группы, у которых алкокси и алкильная части каждая содержит от 1 до 5 атомов углерода, причем желательно, чтобы содержалось от 1 до 4 атомов углерода, особенно алкоксиметильные группы и такие группы, которые содержат, по крайней мере, один заместитель, желательно от одного до пяти заместителей, а лучше - от одного до трех заместителей, но самое лучшее - один заместитель, выбранный, желательно, из низших алкоксиметильных групп и иных алкоксиалкильных групп (таких как метоксиметильная, этоксиметильная, пропоксиметильная, изопропоксиметильная, бутоксиметильная и трет-бутоксиметильная групп); низших алкоксизамещенных низших алкоксиметильных групп (таких как 2-метоксиэтоксиметильная группа), галогенированных низших алкоксиметильных групп (таких как 2,2,2-трихлорэтоксиметильная и бис-(2-хлорэтокси)метильная группы) и низших алкоксизамещенных этильных и высших алкильных групп (таких как 1-этоксиэтильная, 1-метил-1-метоксиэтильная и 1-изопропоксиэтильная группы);
другие замещенные этильные группы, желательно выбранные из галоидированных этильных групп (таких как 2,2,2-трихлорэтильная группа), и арилселенилзамещенные этильные группы, у которых арильная составляющая является такой, какой она определена ранее, желательно, чтобы ею была фенильная группа (такая как 2-(фенилселенил)этильная группа);
алифатические ацилоксиалкильные группы, у которых ацильная группа преимущественно является алканоильной группой (которая может быть замещенной или может иметь, по крайней мере, один заместитель, выбранный из группы, в которую входят аминогруппы, алкиламиногруппы и диалкиламиногруппы) и, что более желательно, алканоильной группой с числом атомов углерода от 2 до 6 и у которых алкильная составляющая характеризуется наличием от 1 до 6 атомов углерода, желательно от 1 до 4 атомов углерода, такие как ацетооксиметильная, диметиламиноацетооксиметильная, пропионилоксиметильная, бутирилоксиметильная, изобутирилоксиметильная, пивалоилоксиметильная, 1-пивалоилоксиэтильная, 1-ацетоксиэтильная, 1-изобутирилоксиэтильная, 1-пивалоилоксипропильная, 2-метил-1-пивалоилоксипропильная, 2-пивалоилоксипропильная, 1-изобутирилоксиэтильная, 1-изобутирилоксипропильная, 1-ацетоксипропильная, 1-ацетокси-2-метилпропильная, 1-пропионилоксиэтильная, 1-пропионилоксипропильная, 2-ацетоксипропильная и 1-бутирилоксиэтильная группы;
алкоксикарбонилоксиалкильные группы, особенно 1-(алкоксикарбонилокси)этильные группы, у которых алкоксисоставляющая содержит от 1 до 10, желательно от 1 до 6 и, еще лучше от 1 до 4 атомов углерода и у которых алкильная составляющая содержит от 1 до 6, желательно от 1 до 4, атомов углерода, такие как метоксикарбонилоксиметильная, этоксикарбонилоксиметильная, пропоксикарбонилоксиметильная, изопропоксикарбонилоксиметильная, бутоксикарбонилоксиметильная, изобутоксикарбонилоксиметильная, 1-метоксикарбонилоксиэтильная, 1-этоксикарбонилоксиэтильная, 1-пропоксикарбонилоксиэтильная, 1-изопропоксикарбонилоксиэтильная, 1-бутоксикарбонилоксиэтильная, 1-изобутоксикарбонилоксиэтильная, 1-втор-бутоксикарбонилоксиэтильная, 1-трет-бутоксикарбонилоксиэтильная, 1-(1-этилпропоксикарбонилокси)этильная и 1-(1,1-дипропилбутоксикарбонилокси)этильная группы, и другие алкоксикарбонилалкильные группы, у которых как алкоксигруппа, так и алкильная группа содержат от 1 до 6, желательно от 1 до 4, атомов углерода, такие как 2-метил-1-(изопропоксикарбонилокси)пропильная, 2-(изопропоксикарбонилокси)пропильная, изопропоксикарбонилоксиметильная, трет-бутоксикарбонилоксиметильная, метоксикарбонилоксиметильная и этоксикарбонилоксиметильная группы;
циклоалкилкарбонилоксиалкильная и циклоалкилоксикарбонилоксиалкильная группы, у которых циклоалкильная группа содержит от 3 до 10, желательно от 3 до 7, атомов углерода, является моно- или полициклической и является произвольно замещенной, по крайней мере, одним (а желательно только одним) заместителем в виде алкильной группы с числом атомов углерода от 1 до 4 (например, группа, выбранная из тех алкильных групп, которые в качестве примера назывались ранее) и у которых алкильная составляющая содержит от 1 до 6, но более желательно от 1 до 4, атомов углерода (например, группа, выбранная из тех алкильных групп, которые в качестве примера назывались ранее) и, что наиболее желательно, представляла собой метильную, этильную или пропильную группу, например, ими могут быть циклогексилоксикарбонилоксиметильная, 1-метилциклогексилкарбонилоксиметильная, 1-метилциклогексилоксикарбонилоксиметильная, циклопентилоксикарбонилоксиметильная, циклопентилкарбонилоксиметильная, 1-циклогексилоксикарбонилоксиэтильная, 1-циклогексилкарбонилоксиэтильная, 1-циклопентилоксикарбонилоксиэтильная, 1-циклопентилкарбонилоксиэтильная, 1-циклогептилоксикарбонилоксиэтильная, 1-циклогептилкарбонилоксиэтильная, 1-метилциклопентилкарбонилоксиметильная, 1-метилциклопентилоксикарбонилоксиметильная, 2-метил-1-(1-метилциклогексилкарбонилокси)пропильная, 1-(1-метилциклогексилкарбонилокси)пропильная, 2-(1-метилциклогексилкарбонилокси)пропильная, 1-(циклогексилкарбонилокси)пропильная, 2-(циклогексилкарбонилокси)пропильная, 2-метил-1-(1-метилциклопентилкарбонилокси)пропильная, 1-(1-метилциклопентилкарбонилокси)пропильная, 2-(1-метилциклопентилкарбонилокси)пропильная, 1-(циклопентилкарбонилокси)-пропильная, 2-(циклопентилкарбонилокси)пропильная, 1-(1-метилциклопентилкарбонилокси)этильная, 1-(1-метилциклопентилкарбонилокси)пропильная, адамантилоксикарбонилоксиметильная, адамантилкарбонилоксиметильная, 1-адамантилоксикарбонилоксиэтильная, 1-адамантилкарбонилоксиэтильная и циклогексилоксикарбонилокси)циклогексил)метильная группы;
циклоалкилзамещенные алифатические ацилоксиалкильные группы, у которых ацильная группа предпочтительно являет алканоильной группой и, что более желательно, алканоильной группой с числом атомов углерода от 2 до 6, циклоалкильный заместитель содержит от 3 до 7 атомов углерода и алкильная составляющая содержит от 1 до 6, желательно от 1 до 4, атомов углерода, такие как (циклогексилацетокси)метильная, 1-(циклогексилацетокси)этильная, 1-(циклогексилацетокси)пропильная, 2-метил-1-(циклогексилацетокси)пропильная, (циклопентилацетокси)метильная, 1-(циклопентилацетокси)этильная, 1-(циклопентилацетокси)пропильная и 2-метил-1-(циклопентилацетокси)пропильная группы;
циклоалкилалкоксикарбонилоксиалкильные группы, у которых алкоксигруппа содержит единственный циклоалкильный заместитель, причем циклоалкильный заместитель содержит от 3 до 10, желательно от 3 до 7, атомов углерода и является моно- или полициклическим, например, ими могут быть циклопропилметоксикарбонилоксиметильная, циклобутилметоксикарбонилоксиметильная, циклопентилметоксикарбонилоксиметильная, циклогексилметоксикарбонилоксиметильная, 1-(циклопропилметоксикарбонилокси)этильная, 1-(циклобутилметоксикарбонилокси)этильная, 1-(циклопентилметоксикарбонилокси)этильная и 1-(циклогексилметоксикарбонилокси)этильная группы;
терпенилкарбонилоксиалкильная и терпенилоксикарбонилоксиалкильная группы, у которых терпенильная группа является такой, какие в качестве примера приводили ранее, и желательно, чтобы ею являлась циклическая терпенильная группа, например, такая, как 1-(метилкоксикарбонилокси)этильная, 1-(метилкарбонилокси)этильная, ментилоксикарбонилоксиметильная, ментилкарбонилоксиметильная, 1-(3-пинанилоксикарбонилокси)этильная, 1-(3-пинанилкарбонилокси)этильная, 3-пинанилоксикарбонилоксиметильная и 3-пинанилкарбонилоксиметильная группы;
5-алкильная и 5-фенильная (которые могут быть замещенными, по крайней мере, одним заместителем α, определенным и проиллюстрированным примерами ниже) (2-оксо-1-,3-диоксолен-4-ил)алкильная группы, у которых каждая алкильная группа (которые могут быть одинаковыми или различными) обладают от 1 до 6, желательно от 1 до 4, атомов углерода, например, такие, как (5-метил-2-оксо-1,3-диоксолен-4-ил)метильная, (5-фенил-2-оксо-1,3-диоксолен-4-ил)метильная, (5-изопропил-2-оксо-1,3-диоксолен-4-ил)метильная, (5-трет-бутил-2-оксо-1,3-диоксолен-4-ил)метильная и 1-(5-метил-2-оксо-1,3-диоксолен-4-ил)этильная группы; и
фталидильные группы, которые могут быть незамещенными или могут быть замещенными, по крайней мере, одним заместителем, выбранным из группы, в которую входят заместители α, определенные и проиллюстрированные примерами ниже, причем желательно, чтобы ими была алкильная группа или алкоксигруппа, например, такая, как фталидильная, диметилфталидильная и диметоксифталидильная группы;
любая из алкильных групп, проиллюстрированных примерами ранее;
карбоксиалкильные группы с числом атомов углерода от 2 до 7, такие как карбоксиметильная группа; и
амидообразующие остатки аминокислот, такие как фенилаланин.
Примерами заместителей α, о которых говорилось выше, являются
атомы галогена, такие как атомы фтора, хлора, брома и иода;
алкильные группы с числом атомов углерода от 1 до 6, такие как группы, приведенные в качестве примера ранее, особенно метильная, этильная и трет-бутильная группы;
алкоксигруппы с числом атомов углерода от 1 до 6, такие как метокси-, этокси-, пропокси-, изопропокси-, бутокси-, изобутокси-, втор-бутокси-, трет-бутокси-, пентилокси-, изопентилокси-, неопентилокси-, гексилокси- и изогексилоксигруппы, которые содержат от 1 до 4 атомов углерода, причем желательно, чтобы ими были метокси-, этокси-, пропокси-, изопропокси-, бутокси- и изобутоксигруппы, и наиболее предпочтительной является метоксигруппа;
карбоксигруппы, нитрогруппы и цианогруппы;
алкилендиоксигруппы с числом атомов углерода от 1 до 4, такие как метилендиоксигруппа;
ациламиногруппы, содержащие ациламиногруппы, соответствующие алифатическим и ароматическим ацильным группам, примеры которых будут приведены ниже в связи с рассмотрением гидроксизащищающих групп, причем желательно, чтобы ими была ацетамидогруппа или бензамидогруппа;
алкоксикарбонильные группы с числом атомов углерода от 2 до 7, желательно от 2 до 5, такие как метоксикарбонильная, этоксикарбонильная, пропоксикарбонильная, изопропоксикарбонильная, бутоксикарбонильная, изобутоксикарбонильная и трет-бутоксикарбонильная группы; и
арильные группы, такие как приведенные в качестве примера ранее, с условием, что любая такая арильная группа, которая содержится в заместителях α, не является дополнительно замещенной арильной группой.
Для выявления возможности отщепления защищающей группы биологическими способами соединение, содержащее такую группу или его фармацевтически приемлемую соль, вводят посредством внутривенного впрыскивания тестируемому животному, такому как крыса или мышь, и продукты метаболизма, извлеченные впоследствии из жидкостей организма использованного животного, анализируют на наличие отщепленной группы. Из описанных выше защищающих групп предпочтение следует отдавать тем группам, которые могут быть отщеплены ин виво биологическими способами, такими как гидролиз. Следует, разумеется, понимать, что, по крайней мере, некоторые из этих групп, которые способны отщепляться ин виво биологическими способами, могут быть также отщеплены и химическими способами.
Термин "гидроксизащищающая группа", как он используется в определениях групп R6, R6a и R6b, распространяется на защищающую группу, способную отщепляться при воздействии химическими методами (такими как гидрогенолиз, гидролиз, электролиз или фотолиз) с образованием свободной гидроксигруппы, или на защищающую группу, способную отщепляться ин виво при воздействии биологическими методами, такими как гидролиз.
Примерами гидроксизащищающих групп, которые могут быть отщеплены химическими способами, являются
алифатические ацильные группы, предпочтительно алканоильные группы с числом атомов углерода от 1 до 25, более предпочтительно, чтобы они были с числом атомов углерода от 1 до 20, еще более предпочтительно, чтобы они были с числом атомов углерода от 1 до 6, и лучше всего, чтобы они были с числом атомов углерода от 1 до 4 (такие как формильная, ацетильная, пропионильная, бутирильная, изобутирильная, пивалоильная, валерильная, изовалерильная, гексаноильная, гептаноильная, октаноильная, лауроильная, миристоильная, тридеканоильная, пальмитоильная, и стеароильная группы, из которых ацетильная группа является наиболее предпочтительной); галогенированные алканоильные группы с числом атомов углерода от 2 до 6, особенно галогенированные ацетильные группы (такие как хлорацетильная, дихлорацетильная, трихлорацетильная и трифторацетильная группы); низшие алкоксиалканоильные группы, в которых алкоксичасть содержит от 1 до 6, желательно от 1 до 3, атомов углерода и алканоильная часть содержит от 2 до 6 атомов углерода и, желательно, представляет собой ацетильную группу (такую как метоксиацетильная группа); и ненасыщенные аналоги таких групп, особенно алкеноильные или алкиноильные группы с числом атомов углерода от 3 до 6 (такие как акрилоильная, метакрилоильная, пропиолоильная, кротоноильная, изокротоноильная и (E)-2-метил-2-бутеноильная группы);
ароматические ацильные группы, желательно арилкарбонильные группы, у которых арильная составляющая содержит от 6 до 14, более желательно от 6 до 10, еще более желательно 6 или 10 и наиболее желательно 6, кольцевых атомов углерода и представляет собой карбоциклическую группу, которая является незамещенной или содержит от 1 до 5, желательно от 1 до 3, заместителей, выбранных из группы, состоящей из заместителей α, определенных и проиллюстрированных примерами ранее, например такие, как незамещенные группы (такие как бензоильная, α-нафтоильная и β-нафтоильная группы); галоидированные арилкарбонильные группы (такие как 2-бромбензоильная и 4-хлорбензоильная группы); низшие алкилзамещенные арилкарбонильные группы, в которых алкильный или каждый алкильный заместитель содержит от 1 до 6, желательно от 1 до 4, атомов углерода (такие как 2,4,6-триметилбензоильная и 4-толуоильная группы); низшие алкоксизамещенные арилкарбонильные группы, в которых алкокси- или каждый алкоксизаместитель преимущественно содержит от 1 до 6, а еще лучше от 1 до 4, атомов углерода (такие как 4-анизоильная группа); карбоксизамещенные арилкарбонильные группы (такие как 2-карбоксибензоильная, 3-карбоксибензоильная и 4-карбоксибензоильная группы); нитрозамещенные арилкарбонильные группы (такие как 4-нитробензоильная и 2-нитробензоильная группы); низшие алкоксикарбонилзамещенные арилкарбонильные группы, в которых алкоксикарбонильный или каждый алкоксикарбонильный заместитель преимущественно содержит от 2 до 6 атомов углерода (такая как 2-(метоксикарбонил)бензоильная группа); и арилзамещенные арилкарбонильные группы, в которых арильный заместитель является таким, каким он был определен ранее, исключая лишь то, что, если он является замещенным дополнительной арильной группой, то такая арильная группа не является сама замещенной арильной группой (такая как 4-фенилбензоильная группа);
гетероциклические группы с числом кольцевых атомов 5 или 6, из которых 1 или 2, атома представляют собой гетероатомы, выбранные из группы, состоящей из кислорода, серы и азота, желательно из атомов кислорода или серы, причем такие группы могут быть незамещенными или могут содержать, по крайней мере, один заместитель, выбранный из группы, в которую входят заместители α и атомы кислорода, желательно атомы халькогена и алкоксигруппы; примерами являются тетрагидропиранильные группы, которые могут быть замещенными или незамещенными, такие как тетрагидропиран-2-ильная, 3-бромтетрагидропиран-2-ильная и 4-метокситетрагидропиран-4-ильная группы; тетрагидротиопиранильные группы, которые могут быть замещенными или незамещенными, такие как тетрагидротиопиран-2-ильная и 4-метокситетрагидротиопиран-4-ильная группы; тетрагидрофуранильные группы, которые могут быть замещенными или незамещенными, такие как тетрагидрофуран-2-ильная группа; и тетрагидротиенильная группы, которые могут быть замещенными или незамещенными, такие как тетрагидротиен-2-ильная группа;
тризамещенные силильные группы, в которых все три или два, или один из заместителей представляют собой алкильные группы с числом атомов углерода от 1 до 5, предпочтительно от 1 до 4, и ни один или один, или два из заместителей являются арильными группами, определенными выше, предпочтительно фенильная или замещенная фенильная группы, предпочтительно три(низший алкил) силильные группы, такие как триметилсилильная, триэтилсилильная, изопропилдиметилсилильная, трет-бутилдиметилсилильная, метилдиизопропилсилильная, метил-ди-трет-бутилсилильная и триизопропилсилильная группы; и три(низший алкил)силильные группы, в которых одна или две из алкильных групп замещены арильными группами, такие как дифенилметилсилильная, дифенилбутилсилильная, дифенил-трет-бутилсилильная, дифенилизопропилсилильная и фенилдиизопропилсилильная группы;
алкоксиалкильные группы, в которых алкокси и алкильная части содержат каждая от 1 до 6, предпочтительно от 1 до 4, атомов углерода, особенно алкоксиметильные группы и такие группы, которые содержат, по крайней мере, один, предпочтительно от 1 до 6, еще предпочтительнее от 1 до 3 и наиболее предпочтительно 1, заместитель, предпочтительно низшие алкоксиметильные группы и иные алкоксиалкильные группы (такие как метоксиметильная, этоксиметильная, пропоксиметильная, изопропоксиметильная, бутоксиметильная и трет-бутоксиметильная группы); низшие алкоксизамещенные низшие алкоксиметильные группы (такие как 2-метоксиэтоксиметильная группа); галоидированные низшие алкоксиметильные группы (такие как 2,2,2-трихлорэтоксиметильная и бис-(2-хлорэтокси)метильная группы) и низшие алкоксизамещенные этильные группы (такие как 1-этоксиэтильная, 1-метил-1-метоксиэтильная и 1-изопропоксиэтильная группы);
другие замещенные этильные группы, предпочтительно галоидированные этильные группы (такие как 2,2,2-трихлорэтильная группа); и арилселенилзамещенные этильные группы, в которых арильная часть является такой, какой определена ранее (такая как 2-(фенилселенил)этильная группа);
аралкильные группы, предпочтительно алкильные группы, содержащие от 1 до 4, более предпочтительно от 1 до 3 и наиболее предпочтительно 1 или 2, атомов углерода, которые являются замещенными одной-тремя арильными группами, определенными и проиллюстрированными примерами ранее, которые могут быть незамещенными (такими как бензильная, фенэтильная, 1-фенилэтильная, 3-фенилпропильная, α-нафтилметильная, β-нафтилметильная, дифенилметильная, трифенилметильная, α-нафтилдифенилметильная и 9-антрилметильная группы) или замещенными в арильной части низшей алкильной группой, низшей алкоксигруппой, нитрогруппой, атомом галогена, цианогруппой или алкилендиоксигруппой с числом атомов углерода от 1 до 3, предпочтительно метилендиоксигруппой, такой как 4-метилбензильная, 2,4,6-триметилбензильная, 3,4,5-триметилбензильная, 4-метоксибензильная, 4-метоксифенилдифенилметильная, 2-нитробензильная, 4-нитробензильная, 4-хлорбензоильная, 4-бромбензильная, 4-цианобензильная, 4-цианобензилдифенилметильная, бис-(2-нитрофенил)метильная и пиперонильная группы;
алкоксикарбонильные группы, особенно такие группы, которые содержат от 2 до 7, предпочтительно от 2 до 5, атомов углерода и которые могут быть незамещенными (такими как метоксикарбонильная, этоксикарбонильная, трет-бутоксикарбонильная и изобутоксикарбонильная группы) или замещенными атомом галогена или тризамещенной силильной группой, например, три(низшей алкилсилильной) группой (такой как 2,2,2-трихлорэтоксикарбонильная и 2-триметилсилилэтоксикарбонильная группы);
алкенилоксикарбонильные группы, в которых алкенильная часть содержит от 2 до 6, желательно от 2 до 4, атомов углерода (такие как винилоксикарбонильная и аллилоксикарбонильная группы);
сульфогруппы и
аралкилоксикарбонильные группы, в которых аралкильная часть является такой, какой определена и проиллюстрирована примерами ранее, и в которых арильное кольцо, если оно является замещенным, замещено, по крайней мере, одним заместителем, выбранным из группы, состоящей из заместителей α, определенных и проиллюстрированных примерами ранее, одним или двумя низшими алкокси- или нитрозаместителями, такие как бензилоксикарбонильная, 4-метоксибензилоксикарбонильная, 3,4-диметоксибензилоксикарбонильная, 2-нитробензилоксикарбонильная и 4-нитробензилоксикарбонильная группы.
Примерами гидроксизащищающих групп, которые обладают способностью отщепляться ин виво при воздействии биологическими методами, такими как гидролиз, являются
диоксоленилалкильные группы, алифатические ацильные группы и ароматические ацильные группы, такие как группы, приведенные в качестве примеров выше в связи с рассмотрением карбоксизащищающих групп;
остаток, который образует соль полуэфира дикарбоновой кислоты, такой как янтарная кислота;
остаток, которые образует соль в виде фосфата;
остаток сложного эфира аминокислоты и
карбонилоксиалкилоксикарбонильные группы, такие как пивалоилоксиметоксикарбонильная группа. \\\ В том случае, кода группа R1 представляет собой группу формулы (II), две группы, представляемые группами R6a и R6b, могут вместе образовывать одну из следующих бидентатных защищающих групп:
низшая алкилиденовая группа с числом атомов углерода от 1 до 46 такая как метилиденовая, этилиденовая или изопропилиденовая группа;
аралкилиденовая группа, в которой арильная часть может быть такой, как определена выше, и алкилиденовая часть содержит от 1 до 4 атомов углерода, такая как бензилиденовая группа;
алкоксиэтилиденовая группа, в которой алкоксичасть содержит от 1 до 6, желательно от 1 до 4, атомов углерода, такая как метоксиэтилиденовая или этоксиэтилиденовая группа;
оксометиленовая группа и
триоксометиленовая группа.
Способность защищающих групп, описанных выше, отщепляться при воздействии биологических методов может определяться таким же образом, как описанная выше в отношении карбоксизащищающих групп.
Из этих гидроксизащищающих групп изобретатели предпочтение отдают силильной группе и защищающим группам, способным отщепляться ин виво с помощью биологических методов.
Когда группы R6, R6a или R6b представляют собой алкильную группу, ею может быть любая из алкильных групп, проиллюстрированных примерами в отношении группы R2 и др.
Когда группы R6, R6a или R6b представляют собой алкансульфонилоксигруппу, ею может быть группа с прямой или разветвленной цепью, содержащая от 1 до 6 атомов углерода, например, такая, как метансульфонилоксигруппа, этансульфонилоксигруппа и пропансульфонилоксигруппа.
Когда группы R6, R6a или R6b представляют собой галоидированную алкансульфонилоксигруппу, ею может быть любая из незамещенных алкансульфонилоксигрупп, перечисленных выше, и предпочтительно ею является фторированная алкансульфонилоксигруппа, такая как трифторметансульфонилоксигруппа или пентафторэтансульфонилоксигруппа.
Когда группы R6, R6a или R6b представляют собой арилсульфонилоксигруппу, арильная часть может быть такой, как определена и проиллюстрирована примерами выше, и примерами таких групп являются бензолсульфонилоксигруппа и п-толуолсульфонилоксигруппа.
Из этих групп изобретатели отдают предпочтение алкильным группам.
Те соединения, отвечающие настоящему изобретению, которые содержат свободную карбоксигруппу, например, те соединения, в которых группа R1 представляет собой группу формулы (II) и группа R5 представляет собой атом водорода, могут образовывать соли. Примерами таких солей являются соли со щелочным металлом, таким как натрий, калий или литий, соли со щелочноземельным металлом, таким как барий или кальций, соли с еще одним металлом, таким как магний, алюминий, железо, цинк, медь, никель или кобальт, аммониевые соли, соли органического основания, в частности соли с органическими аминами, такие как соль с триэтиламином, диизопропиламином, циклогексиламином, трет-октиламином, дибензиламином, морфолином, глюкозамином, фенилглициналкильными сложными эфирами, этилендиамином, N-метилглюкамином, гуанидином, диэтиламином, триэтиламином, дициклогексиламином, N,N'-дибензилэтилендиамином, хлорпрокаином, прокаином, диэтаноамином, N-бензилфенэтиламином, пиперазином, тетраметиламмонием или трис(гидроксиметил)аминометаном, и соли с основной аминокислотой, такой как гистидин, α,β-диаминомасляная кислота, лизин, аргинин, орнитин, глутаминовая кислота или аспарагиновая кислота.
Кроме того, когда соединение настоящего изобретения содержит основную группу в своей молекуле, оно может образовывать аддитивные соли кислот. Примерами таких солей являются соли с минеральными кислотами, особенно галоидоводородными кислотами (такими как фтористоводородная кислота, бромистоводородная кислота, иодистоводородная кислота или хлористоводородная кислота), азотной кислотой, угольной кислотой, серной кислотой или фосфорной кислотой, соли с низшими алкилсульфоновыми кислотами, такими как метансульфоновая кислота, трифторметансульфоновая кислота или этансульфоновая кислота, соли с арилсульфоновыми кислотами, такими как бензолсульфоновая кислота или п-толуолсульфоновая кислота, соли с органическими карбоновыми кислотами, такими как уксусная кислота, фумаровая кислота, винная кислота, щавелевая кислота, малеиновая кислота, яблочная кислота, янтарная кислота, бензойная кислота, миндальная кислота, аскорбиновая кислота, молочная кислота, глюконовая кислота или лимонная кислота, и соли с аминокислотами, такими как глутаминовая кислота или аспарагиновая кислота.
Соединения настоящего изобретения могут содержать один или несколько асимметричных атомов углерода в своих молекулах и, таким образом, могут образовывать оптические изомеры. Хотя все эти соединения представлены в настоящем описании единственной молекулярной формулой, настоящее изобретение распространяется как на индивидуальные, выделенные изомеры, так и на их смеси, включая их рацематы. Если проводится стереоспецифический синтез или в качестве исходных веществ применяются оптически активные соединения, могут быть непосредственно получены индивидуальные изомеры; с другой стороны, если получают смесь изомеров, то индивидуальные изомеры могут быть получены обычными способами выделения.
Предпочтительными классами соединений настоящего изобретения являются соединения формул (I), (Ia) или (Ib) и их фармацевтически приемлемые соли и сложные эфиры, в которых
(A) R2 представляет собой алкильную группу с числом атомов углерода от 1 до 4, алкенильную группу с числом атомов углерода от 2 до 4 или алкинильную группу с числом атомов углерода от 2 до 4 и
R3 и R4 являются одинаковыми или разными и каждый представляет собой атом водорода, алкильную группу с числом атомов углерода от 1 до 4, алкенильную группу с числом атомов углерода от 2 до 4 или алкинильную группу с числом атомов углерода от 2 до 4;
или
(B) R2 представляет собой алкильную группу с числом атомов углерода от 1 до 4, алкенильную группу с числом атомов углерода от 2 до 4 или алкинильную группу с числом атомов углерода от 2 до 4;
R3 представляет собой алкильную группу с числом атомов углерода от 1 до 4, алкенильную группу с числом атомов углерода от 2 до 4 или алкинильную группу с числом атомов углерода от 2 до 4 и
R4 представляет собой атом водорода, алкильную группу с числом атомов углерода от 1 до 4, алкенильную группу с числом атомов углерода от 2 до 4 или алкинильную группу с числом атомов углерода от 2 до 4;
(C) R2 представляет собой алкильную группу с числом атомов углерода от 1 до 4 или алкенильную группу с числом атомов углерода от 2 до 4;
R3 представляет собой алкильную группу с числом атомов углерода от 1 до 4 или алкенильную группу с числом атомов углерода от 2 до 4 и
R4 представляет собой атом водорода, алкильную группу с числом атомов углерода от 1 до 4 или алкенильную группу с числом атомов углерода от, 2 до 4;
(D) R2 представляет собой этильную группу;
R3 представляет собой алкильную группу с числом атомов углерода от 1 до 4 и
R4 представляет собой алкильную группу с числом атомов углерода от 2 до 4;
(E) R2 представляет собой этильную группу;
R3 представляет собой алкильную группу с числом атомов углерода от 1 до 3 и
R4 представляет собой алкильную группу с числом атомов углерода 2 или 3;
(F) R1 представляет собой группу с формулой (II) и, что более желательно, R2, R3 и R4 имеют значения, определенные в одном из приведенных выше пунктов (A) - (E);
(G) R1 представляет собой группу с формулой (II) и
R6, R6a и R6b представляют собой атом водорода и более предпочтительно R2, R3 и R4 имеют значения, определенные выше по одному из пунктов (A) - (E);
(H) фармацевтически приемлемые соли соединений, определенных выше по п. (G);
(I) R5 представляет собой атом водорода или защищающую группу, способную отщепляться ин виво с помощью биологических методов;
(J) R6, R6a и R6b каждый представляет атом водорода или защищающую группу, способную отщепляться ин виво с помощью биологических методов, таких как гидролиз; и
(K) R5 представляет собой атом водорода.
Конкретные примеры индивидуальных соединений настоящего изобретения даются следующими формулами (I-1), (I-1a), (I-2) и (I-2a), в которых различные использованные символы имеют значения определенные в соответствующей одной из табл. 1 и 2, т.е. табл.1 относится к формулам (I-1) и (I-1a), а табл.2 относится к формулам (I-2) и (I-2a). В этих таблицах для некоторых групп использованы следующие сокращения:
All - аллильная группа,
Bu - бутильная группа,
iBu - изобутильная группа,
tBu - трет-бутильная группа,
Me - метильная группа,
Pr - пропильная группа,
iPr - изопропильная группа,
Et - этильная группа.
Из соединений, перечисленных выше, предпочтительными соединениями являются соединения NN: 1-4, 1-5, 1-6, 1-7, 1-9, 1-10, 1-13, 1-19, 1-20, 1-21, 1-22, 1-23, 1-25, 1-29, 1-30, 1-32, 1-33, 1-38, 1-39, 1-41, 1-45, 1-48, 1-49, 1-52, 1-54, 1-56, 1-57, 1-60, 1-63, 1-65, 1-66, 1-70, 1-71, 1-73, 1-74, 1-75, 1-79, 1-82, 1-86, 1-87, 1-90, 1-93, 1-94, 1-96, 1-97, 1-99, 1-100, 1-101, 1-102, 1-103, 1-105, 1-106, 1-108, 1-109, 1-112, 1-115, 1-116, 1-118, 1-120, 1-123, 1-125, 1-128, 1-129, 1-130, 1-135, 1-137, 1-139, 1-140, 1-141, 1-142, 1-146, 1-147, 1-151, 1-154, 1-155, 1-157, 2-4, 2-5, 2-6, 2-7, 2-9, 2-10, 2-13, 2-19, 2-20, 2-21, 2-22, 2-23, 2-25, 2-29, 2-30, 2-32, 2-33, 2-38, 2-39, 2-41, 2-45, 2-48, 2-49, 2-52, 2-54, 2-56, 2-57, 2-60, 2-63, 2-65, 2-66, 2-70, 2-71, 2-73, 2-74, 2-75, 2-79, 2-82, 2-86, 2-87, 2-90, 2-93, 2-94, 2-96, 2-97, 2-99, 2-100, 2-101, 2-102, 2-103, 2-105, 2-106, 2-108, 2-109, 2-112, 2-115, 2-116, 2-118, 2-120, 2-123, 2-125, 2-128, 2-129, 2-130, 2-135, 2-137, 2-139, 2-140, 2-141, 2-142, 2-146, 2-147, 2-151, 2-154, 2-155 и 2-157.
Более предпочтительными соединениями являются соединения NN: 1-5, 1-7, 1-13, 1-19, 1-22, 1-23, 1-25, 1-29, 1-32, 1-33, 1-38, 1-41, 1-45, 1-49, 1-52, 1-54, 1-56, 1-57, 1-60, 1-63, 1-65, 1-66, 1-70, 1-73, 1-74, 1-75, 1-79, 1-82, 1-87, 1-90, 1-93, 1-94, 1-96, 1-99, 1-100, 1-101, 1-102, 1-103, 1-105, 1-109, 1-112, 1-120, 1-155, 2-4, 2-5, 2-7, 2-13, 2-19, 2-22, 2-23, 2-25, 2-29, 2-32, 2-33, 2-38, 2-41, 2-45, 2-49, 2-52, 2-54, 2-56, 2-57, 2-60, 2-63, 2-65, 2-66, 2-70, 2-73, 2-74, 2-75, 2-79, 2-82, 2-87, 2-90, 2-93, 2-94, 2-96, 2-99, 2-100, 2-101, 2-102, 2-103, 2-105, 2-109, 2-112, 2-120 и 2-155.
Наиболее предпочтительными соединениями являются соединения со следующими номерами.
1-4. 3,5-Дигидрокси-7-(6-гидрокси-2-метил-8-изовалерилокси- 1,2,6,8,8а-гексагидро-1-нафтил)гептановая кислота.
1-5. 3,5-Дигидрокси-7-(6-гидрокси-2-метил-8-гексаноилокси- 1,2,6,7,8,8а-гексагидро-1-нафтил)гептановая кислота.
1-7. 3,5-Дигидрокси-7-[6-гидрокси-2-метил-8-(3,3-диметилбутирилокси)- 1,2,6,7,8,8а-гексагидро-1-нафтил]гептановая кислота.
1-13. 3,5-Дигидрокси-7-[6-гидрокси-2-метил-8-(2-метил-пентаноилокси)- 1,2,6,7,8,8а-гексагидро-1-нафтил]гептановая кислота.
1-22 3,5-Дигидрокси-7-[6-гидрокси-2-метил-8-(2-этилбутирилокси)- 1,2,6,7,8,8а-гексагидро-1-нафтил]гептановая кислота.
1-32. 3,5-Дигидрокси-7-[6-гидрокси-2-метил-8-(2-пропил-пентаноилокси)- 1,2,6,7,8,8а-гексагидро-1-нафтил]гептановая кислота.
1-33. 3,5-Дигидрокси-7-[(6-гидрокси-2-метил-8-(2-изопропил-3- метибутирилокси)-1,2,6,7,8,8а-гексагидро-1-нафтил]гептановая кислота.
1-41. 3,5-Дигидрокси-7-[6-гидркоси-2-метил-8-(2-бутилгексаноилокси)- 1,2,6,7,8,8а-гексагидро-1-нафтил]гептановая кислота.
1-49. 3,5-Дигидрокси-7-[6-гидркоси-2-метил-8(2-аллил-4-пентеноилокси)- 1,2,6,7,8,8а-гексагидро-1-нафтил]гептановая кислота.
1-52. 3,5-Дигидрокси-7-(6-гидрокси-2-метил-8-пивалоилокси- 1,2,6,7,8,8а-гексагидро-1-нафтил)гептановая кислота.
1-54. 3,5-Дигидрокси-7-[6-гидрокси-2-метил-8-(2,2-диметилпентаноилокси)- 1,2,6,7,8,8а-гексагидро-1-нафтил]гептановая кислота.
1-56. 3,5-Дигидрокси-7-[6-гидрокси-2-метил-8-(2,2, -диметилгексаноилокси)- 1,2,6,7,8,8а-гексагидро-1-нафтил]гептановая кислота.
1-60. 3,5-Дигидрокси-7-[6-гидрокси-2-метил-8-(2,2-диметил- 4-пентеноилокси)-1,2,6,7,8,8а-гексагидро-1нафтил]гептановая кислота.
1-63. 3,5-Дигидрокси-7-[6-гидрокси-2-метил-8-(2-этил-2- метилпентаноилокси)-1,2,6,7,8,8а-гексагидро-1-нафтил]гептановая кислота.
1-65. 3,5-Дигидрокси-7-[6-гидрокси-2-метил-8-(2-этил-2-метилбутирилокси)- 1,2,6,7,8,8а-гексагидро-1-нафтил]гептановая кислота.
1-73. 3,5-Дигидрокси-7-[6-гидрокси-2-метил-8-(2-метил-2- пропилпентаноилокси)-1,2,6,7,8,8а-гексагидро-1-нафтил]гептановая кислота.
1-90. 3,5-Дигидрокси-7-[6-гидрокси-2-метил-8-(2-аллил-2-метил-4- пентеноилокси)-1,2,6,7,8,8а-гексагидро-1-нафтил]гептановая кислота.
1-93. 3,5-Дигидрокси-7-[6-гидрокси-2-метил-8-(2,2-диэтилбутирилокси)- 1,2,6,7,8,8а-гексагидро-1-нафтил]гептановая кислота.
1-94. 3,5-Дигидрокси-7-[6-гидрокси-2-метитл-8-(2,2-диэтилпентаноил)- 1,2,6,7,8,8а-гексагидро-1-нафтил]гептановая кислота.
1-100. 3,5-Дигидрокси-7-[6-гидрокси-2-метил-8-(2,2-диэтил-4- пентеноилокси)-1,2,6,7,8,8а-гексагидро-1-нафтил]гептановая кислота.
1-120. 3,5-Дигидрокси-7-[6-гидрокси-2-метил-8-(2-аллил-2-этил-4- пентеноилокси)-1,2,6,7,8,8а-гексагидро-1-нафтил]гептановая кислота.
1-155. 3,5-Дигидрокси-7-[6-гидрокси-2-метил-8-(2,2-диаллил-4- пентеноилокси)-1,2,6,7,8,8а-гексагидро-1-нафтил]гептановая кислота.
2-4. 3,5-Дигидрокси-7-(2-метил-8-изовалирелокси-1,2,6,7,8-8а-гексагидро- 1-нафтил)гептановая кислота.
2-5. 3,5-Дигидрокси-7-(2-метил-8-гексаноилокси-1,2,6,7,8,8а-гексагидро- 1-нафтил)гептановая кислота.
2-7. 3,5-Дигидрокси-7-[2-метил-8-(3,3-диметилбутирилокси)-1,2,6,7,8,8а- гексагидро-1-нафтил]гептановая кислота.
2-13. 3,5-Дигидрокси-7-[2-метил-8-(2-метилпентаноилокси)-1,2,6,7,8,8а- гексагидро-1-нафтил]гептановая кислота.
2-22. 3,5-Дигидрокси-7-[2-метил-8-(2-этилбутирилокси)-1,2,6,7,8,8а- гексагидро-1-нафтил]гептановая кислота.
2-32. 3,5-Дигидрокси-7-[2-метил-8-(2-пропилпентаноилокси)-1,2,6,7,8,8а- гексагидро-1-нафтил]гептановая кислота.
2-33. 3,5-Дигидрокси-7-[2-метил-8-(2-изопропил-3-метибултирилокси)- 1,2,6,7,8,8а-гексагидро-1-нафтил]гептановая кислота.
2-41. 3,5-Дигидрокси-7-[2-метил-8-(2-бутилгексаноилокси)-1,2,6,7,8,8а- гексагидро-1-нафтил]гептановая кислота.
2-49. 3,5-Дигидрокси-7-[2-метил-8-(2-аллил-4-пентеноилокси)- 1,2,6,7,8,7а-гексагидро-1-нафтил]гептановая кислота.
2-52. 3,5-Дигидркоси-7-(2-метил-8-пивалоилокси-1,2,6,7,8,8а- гексагидро-1-нафтил)гептановая кислота.
2-54. 3,5-Дигидрокси-7-[2-метил-8-(2,2-диметилпентаноилокси)- 1,2,6,7,8,8а-гексагидро-1-нафтил]гептановая кислота.
2-56. 3,5-Дигидрокси-7-[2-метил-8-(2,2-диметилгексаноилокси)- 1,2,6,7,8,8а-гексагидро-1-нафтил]гептановая кислота.
2-60. 3,5-Дигидрокси-7-[2-метил-8-(2,2-диметил-4-пентеноилокси)- 1,2,6,7,8,8а-гексагидро-1-нафтил]гептановая кислота.
2-63. 3,5-Дигидрокси-7-[2-метил-8-(2-этил-2-метилпентаноилокси)- 1,2,6,7,8,8а-гексагидро-1-нафтил]гептановая кислота.
2-65. 3,5-Дигидрокси-7-[2-метил-8-(2-этил-2-метилбутирилокси)- 1,2,6,7,8,8а-гексагидро-1-нафтил]гептановая кислота.
2-73. 3,5-Дигидрокси-7-[2-метил-8-(2-метил-2-пропилпентаноилокси)- 1,2,6,7,8,8а-гексагидро-1-нафтил]гептановая кислота.
2-90. 3,5-Дигидрокси-7-[2-метил-8-(2-аллил-2-метил-4-пентеноилокси)- 1,2,667,8,8а-гексагидро-1-нафтил]гептановая кислота.
2-93. 3,5-Дигидрокси-7-[2-метил-8-(2,2-диэтилбутирилокси)- 1,2,6,7,8,8а-гексагидро-1-нафтил]гептановая кислота.
2-94. 3,5-Дигидрокси-7-[2-метил-8-(2,2-диэтилпентаноилокси)- 1,2,6,7,8,8а-гексагидро-1-нафтил]гептановая кислота.
2-100. 3,5-Дигидрокси-7-[2-метил-8-(2,2, -диэтил-4-пентеноилокси)- 1,2,6,7,8,8а-гексагидро-1-нафтил]гептановая кислота.
2-120. 3,5-Дигидрокси-7-[2-метил-8-(2-аллкил-2-этил-4-пентеноилокси)- 1,2,6,7,8,8а-гексагидро-1-нафтил]гептановая кислота.
2-155. 3,5-Дигидрокси-7-[2-метил-8-(2,2-диалкил-4-пентеноилокси)- 1,2,6,7,8,8а-гексагидро-1-нафтил]гептановая кислота;
и лактоны с замкнутым кольцом, соответствующие гидроксикислотам, перечисленным выше;
и их фармацевтически приемлемые соли и сложные эфиры.
Соединения настоящего изобретения могут быть получены самыми разными способами, хорошо известными при получении соединений этого типа. Например, в общем случае они могут быть получены взаимодействием соединения формулы (IV)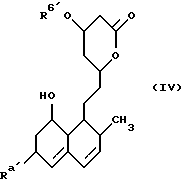
(в которой Ra′ представляет собой атом водорода или группу с формулой R6′ O- и символы R6′ каждый представляет собой любую из групп, представленных R6, но не может быть атомом водорода) с реакционноспособным соединением, содержащим группу R6′, предпочтительно с ацилирующим агентом с получением соединения формулы (V)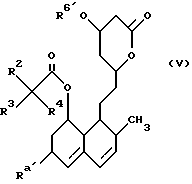
в которой группы R2, R3, R4 и R6′ имеют значения, определенные ранее, и, при необходимости, удалением защищающих групп, и, при необходимости, подвержением соединения формулы (V) гидролизу или сольволизу с раскрытием кольца и, если желательно, когда Ra представляет атом водорода, с введением группы формулы R6O- вместо группы Ra.
Более подробно соединения настоящего изобретения могут быть получены как это проиллюстрировано следующими реакционными схемами A, B, C и D.
Реакционная схема A
Соединения формулы (Ia) могут быть получены как проиллюстрировано следующей реакционной схемой A (табл.3).
В этом способе исходным веществом формулы (VI) может быть известное соединение правастатин, в котором гидроксигруппа в положении 6 находится в β-конфигурации. По всей реакционной схеме соответствующие группы, находящиеся в 6-положении, стереохимически считаются находящимися в β-конфигурации. Альтернативно, в качестве исходного вещества на стадии AI можно использовать эпимерный изомер в 6-положении правастатина, в случае чего можно получить требуемые соединения формул (X), (XI) и (XII), в которых заместители в 6-положении находятся в α-конфигурации. Хотя стереохимия для 6-положения и других положений не отображается в последующих формулах, в настоящем изобретении предполагается использование как индивидуальных выделенных изомеров, например правастатина или его эпимеров, так и смесей этих изомеров.
В приведенных формулах (табл.3):
R5′ представляет атом водорода, карбоксизащищающую группу, определенную для R5, или катионную часть соли;
R6′ представляет гидроксизащищающую группу, алкильную группу, алкансульфонильную группу, галоидированную алкансульфонильную группу или арилсульфонильную группу, все из которых определены и проиллюстрированы примерами в отношении R6 и т.д.;
R7 представляет собой группу формулы
-CO-C(R2)(R3)(R4)
в которой R2, R3 и R4 имеют тот же смысл, что и ранее и
M представляет собой атом водорода или катионную часть соли.
Если R5' или M представляет собой катионную часть соли, ею может быть любой из катионов, приведенных в качестве примера ранее в связи с фармацевтически приемлемыми солями.
Стадия AI
На стадии AI этой реакционной схемы соединение формулы (VII) получают гидролизом соединения формулы (VI) или его фармацевтически приемлемой соли. Гидролиз может проводиться обычными способами, например использованием основания в растворителе для превращения сложноэфирной боковой цепи, в положении 8 в гидроксигруппу.
Реакцию обычно и предпочтительно проводят в присутствии растворителя. Отсутствуют какие-либо особые ограничения, касающиеся природы используемого растворителя, при условии, что он не оказывает отрицательного воздействия на ход реакции или на реактивы, участвующие в ней, и что он способен растворять реактивы, по крайней мере, в некоторой степени. Примерами приемлемых растворителей являются вода и органические растворители, такие как простые эфиры, например такие, как тетрагидрофуран, диоксан, диметоксиэтан и диэтиленгликоль и диметиловый простой эфир; спирты, например, такие как метанол, этанол, пропанол, изопропанол, бутанол, изобутанол, трет-бутанол, изоамиловый спирт, диэтиленгликоль или этиленгликолевый монометиловый простой эфир; и смеси воды с одним или несколькими этими органическими растворителями.
Отсутствуют какие-либо особые ограничения, касающиеся природы используемого основания, и любое основание, обычно используемое в качестве основания в обычных реакциях, может быть в одинаковой мере использовано и здесь. Примерами предпочтительных оснований являются неорганические основания, такие как карбонаты щелочных металлов (например, карбонат натрия, карбонат калия или карбонат лития), водородсодержащие карбонаты щелочных металлов (например, бикарбонат натрия, бикарбонат калия или бикарбонат лития), гидроксиды щелочных металлов (например, гидроксид натрия, гидроксид калия, гидроксид бария или гидроксид лития) и алкоксиды щелочных металлов (например, такие как метоксид натрия, этоксид натрия, метоксид калия, этоксид калия, трет-бутоксид калия и метоксид лития).
При использовании в качестве основания карбоната щелочного металла, бикарбоната щелочного металла или гидроксида щелочного металла, реакцию желательно вести, используя один или несколько эквивалентов основания в расчете на моль соединения с формулой (VI). При использовании в качестве основания алкоксида щелочного металла реакцию ведут при использовании такого количества основания, которое превышает каталитически необходимое количество основания.
Реакция может протекать в широком диапазоне температур, и точная величина температуры не является критической величиной в изобретении. В общем случае изобретателями установлено, что реакцию удобно вести либо при температуре, находящейся в области от -20oC до 150oC, более предпочтительно от 80oC до 120oC, либо при температуре кипения используемого растворителя. Время, необходимое для протекания реакции, может также изменяться в широких пределах, что зависит от многих факторов, а именно от температуры проведения реакции и природы реагирующих веществ, основания и растворителя, используемых при проведении реакции. Однако, если реакцию ведут при предпочтительных условиях, оговоренных выше, обычно достаточен период от 3 до 100 ч, более предпочтительно от 24 до 60 ч.
По завершении реакции требуемый продукт формулы (VII) может быть выделен из реакционной смеси обычными способами. Например, в случае одной из приемлемых методик извлечения реакционную смесь адекватным образом нейтрализуют, при наличии нерастворимых веществ их удаляют фильтрованием, к реакционной смеси или к фильтрату добавляют воду и несмешиваемый с водой органический растворитель, такой как этилацетат, и конечный продукт экстрагируют растворителем, экстракт промывают водой и сушат, например, над безводным сульфатом магния, и затем растворитель отгоняют, получая в виде остатка требуемый продукт.
Соединение формулы (VII), полученное таким способом, представляет собой соль гидроксикислоты, и, если необходимо, она может быть очищена обычным способом, например перекристаллизацией, переосаждением или различными хроматографическими способами. Примерами хроматографических методик являются распределительная хроматография с использованием синтетических абсорбентов, таких как сефадексТМ LH-20 (торговое название вещества, фирмы "Фармация, инк. "), амберлитТМ XAD-11 (торговое название материала, фирма "Ром энд Хаас ко.") или диаионТМ HP-20 (торговое название материала, фирма "Мицубиси касеи корпорейшн"); колонная хроматография с пропусканием вещества через регулярно-фазовую или обращенно-фазовую колонку с насадкой из силикагеля или алкилированного силикагеля (желательно использовать высокопроизводительную жидкостную хроматографию) или комбинация из этих способов с последующим элюированием при использовании подходящего элюирующего растворителя.
Стадия A2
На этой стадии лактоновое соединение формулы (VIII) получают взаимодействием соли гидроксикислотного соединения формулы (VII) с одним или несколькими эквивалентами кислоты с получением свободной карбоновой кислоты, и затем подвергают продукт реакции циклизации.
Реакцию обычно и предпочтительно ведут в присутствии растворителя. Отсутствуют какие-либо особые ограничения, касающиеся природы растворителя, при условии, что он не оказывает отрицательного воздействия на ход реакции или на реагирующие вещества, участвующие в ней, и что он может растворять реагирующие вещества, по крайней мере, в некоторой степени. Примерами подходящих растворителей являются вода и органические растворители, такие как простые эфиры (например, такие как тетрагидрофуран, диоксан, диметоксиэтан и диэтиленгликолевый диметиловый простой эфир); спирты (например, такие как метанол, этанол, пропанол, изопропанол, бутанол, изобутанол, трет-бутанол, диэтиленгликоль и циклогексанол) и смеси воды с одним или несколькими этими органическими растворителями.
Отсутствуют также какие-либо особые ограничения, касающиеся природы кислоты, используемой в первой части этой стадии; и здесь может быть использован любой катализатор, обычно применяемый при проведении реакции этого типа. Примерами предпочтительных кислот являются неорганические кислоты, такие как хлористоводородная кислота, бромистоводородная кислота, серная кислота, перхлорная кислота или фосфорная кислота.
Реакция может протекать в широком интервале температур, и точная температура взаимодействия не является критической для изобретения. В общем случае установлено, что удобно вести реакцию при температуре в интервале -20oC и до 50oC, более предпочтительно в интервале от 0oC и примерно до комнатной температуры. Время, необходимое для протекания реакции, также может меняться в широких пределах в зависимости от многих факторов, а именно от температуры взаимодействия и природы реагентов кислоты и растворителя, применяемых в реакции. Если реакцию ведут в предпочтительных условиях, оговоренных ранее, то тогда она может завершаться сразу же после добавления кислоты; или же реакцию ведут в течение промежутка времени до 2 ч, более предпочтительно в течение периода до 30 мин.
После завершения реакции требуемый продукт этой реакции, может быть извлечен из реакционной смеси обычными способами. Например, в случае одного из приемлемых способов извлечения реакционную смесь адекватным образом нейтрализуют, при наличии нерастворимых веществ их удаляют фильтрованием, к реакционной смеси или фильтрату добавляют воду и несмешиваемый с водой органический растворитель, такой как этилацетат, и продукт экстрагируют растворителем, экстракт промывают водой и сушат, например, над безводным сульфатом магния и растворитель отгоняют, получая в остатке требуемый продукт. Альтернативно после завершения реакции требуемое соединение может быть извлечено отгонкой растворителя из реакционной смеси, смешиванием остатка с органическим растворителем, отфильтровыванием нерастворимых твердых веществ и отгонкой растворителя. Примерами органических растворителей, которые могут быть использованы в этой методике извлечения, являются алифатические углеводороды, такие как гексан, гептан, лигроин или петролейный эфир; ароматические углеводороды, такие как бензол толуол и ксилол; галогенированные углеводороды, такие как хлористый метилен, хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол и дихлорбензол; сложные эфиры, такие как этилформиат, этилацетат, пропилацетат, бутилацетат и диэтилкарбонат; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диэтиленгликолевый диметиловый простой эфир; спирты, такие как метанол, этанол, пропанол, изопропанол, бутанол, изобутанол, трет-бутанол, диэтиленгликоль или циклогексанол; и кетоны, такие как ацетон и метилэтиловый кетон.
Требуемое соединение, полученное таким способом, может быть, если это необходимо, подвергнуто очистке обычными способами, например перекристаллизацией, переосаждением или хроматографическими способами. Примерами приемлемых хроматографических методик являются распределительная хроматография с пропусканием очищаемого вещества через синтетический абсорбент, такой как сефадексТМ LH-20 (торговое название материала, фирма "Фармация, инк."), амберлитТМ XAD-11 (торговое название материала, фирма "Ром энд Хаас ко.") или диаионТМ HP-20 (Мицубиси касеи корпорейшн"); колонная хроматография с пропусканием очищаемого вещества через регулярно-фазовую или обращенно-фазовую колонку с насадкой из силикагеля или алкилированного силикагеля (желательно использовать высокопроизводительную жидкостную хроматографию) или комбинацию из этих способов с последующим элюированием при использовании подходящего элюирующего растворителя.
Проведением циклизующей лактонизации во второй части стадии гидроксикислоту превращают в соединение с лактоновым циклом. Реакция может быть проведена разными способами, например, следующими методами:
метод 1 включает простое нагревание соответствующей гидроксикислоты в растворителе;
метод 2 включает обработку соответствующей гидроксикислоты сложноэтерифицирующим агентом в растворителе.
Метод 1.
Взаимодействие проводят в присутствии растворителя. Отсутствуют какие-либо особые ограничения, касающиеся природы применяемого растворителя, при условии, что он не оказывает отрицательного воздействия на ход реакции или на реагенты, участвующие в ней, и что он способен растворять реагенты, по крайней мере, в некоторой степени. Примерами подходящих растворителей являются алифатические углеводороды, такие как бензол, толуол или ксилол; галоидированные углеводороды, такие как хлористый метилен, хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол или дихлорбензол; сложные эфиры, такие как этилформиат, этилацетат, пропилацетат, бутилацетат или диэтилкарбонат; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан или диэтиленгликолевый диметиловый эфир; кетоны, такие как ацетон, метилэтиловый кетон, метилизобутиловый кетон, изофорон или циклогексанон; и нитрилы, такие как ацетонитрил или изобутиронитрил.
Взаимодействие может протекать в широком диапазоне температур, и точная температура не является критической. В общем установлено, что реакцию удобно вести при температуре в области от 0oC до температуры дефлегмации используемого растворителя, более предпочтительно в интервале примерно от комнатной до 100oC. Время, необходимое для взаимодействия, может также изменяться в широких пределах, в зависимости от многих факторов, особенно от температуры взаимодействия и природы реагентов и растворителя, применяемых в реакции. Если же реакцию ведут при предпочтительных условиях, оговоренных выше, обычно достаточен период от 10 мин до 6 ч, более предпочтительно от 30 мин до 3 ч.
Реакция может быть ускорена использованием кислоты в качестве катализатора. Отсутствует какое-либо особое ограничение, касающееся природы используемой кислоты, и здесь в равной мере может быть использована любая кислота, которая является приемлемой для использования в качестве кислотного катализатора в обычных реакциях. Примерами таких кислот являются органические кислоты, такие как уксусная кислота, муравьиная кислота, щавелевая кислота, метансульфоновая кислота, п-толуолсульфоновая кислота, трифторуксусная кислота или трифторметансульфоновая кислота; и кислоты Льюиса, такие как треххлористый бор, трехфтористый бор и трехбромистый бор. Из этих соединений предпочтение следует отдавать органическим кислотам, причем более желательно, чтобы ими были сильные органические кислоты.
Метод 2.
Взаимодействие по методу 2 обычно следует и желательно вести в присутствии растворителя. Отсутствует какое-либо особое ограничение, касающееся природы растворителя, который должен быть применен при условии, что он не оказывает отрицательного воздействия на ход реакции или на реагенты, участвующие в ней, и что он может растворять реагенты, по крайней мере, в некоторой степени. Растворитель должен быть безводным. Примерами подходящих растворителей являются алифатические углеводороды, такие как гексан или гептан; ароматические углеводороды, такие как бензол, толуол или ксилол; галоидированные углеводороды, такие как хлористый метилен, хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол или дихлорбензол; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан, и диэтиленгликолевый диметиловый эфир; кетоны, такие как ацетон, метилэтилкетон, метилизобутилкетон, изофорон или циклогексанон; нитрилы, такие как ацетонитрил или изобутиронитрил; и амиды, такие как формамид, диметилформамид, диметилацетамид, N-метил-2-пиридон, N-метилпирролидинон или гексаметилфосфорный триамид.
Примерами этерифицирующего агента, который может быть использован в методе 2, являются конденсирующие вещества, примеры которых приведены ниже; алкилгалоидформиаты, такие как метилхлорформиат или этилхлорформиат; и сложные диэфиры цианофосфорной кислоты, такие как диэтилцианофосфонат. Примерами конденсирующих веществ являются N-гидроксипроизводные, такие как N-гидроксисукцинимид, 1-гидроксибензотриазол и N-гидрокси-5-норборнен-2,3-дикарбоксимид; дисульфидные соединения, такие как 2,2'-дипиридилдисульфид; соединения янтарной кислоты, такие как N,N'-дисукцинимидилкарбонат; фосфиновые хлористые соединения, такие как N,N'-бис-(2-оксо-3-оксазолидинил)фосфинхлорид; оксалатные производные, такие как N,N'-дисукцинимидилоксалат, N,N'-дифталимидоксалат, N,N'-бис-(норборненилсукцинимид)оксалат, 1,1'-бис-(бензотриазолил)оксалат, 1,1'-бис-(6-хлорбензотриазолил)оксалат или 1,1'-бис-(6-трифторметилбензотриазолил)оксалат; триарилфосфины, такие как трифенилфосфин; комбинации ди(низший алкил)азодикарбоксилата и триарилфосфина, такие как комбинация диэтилазодикарбоната и трифенилфосфина; N-(низший алкил)-5-арилизоксазоиум-3'-сульфонаты, такие как N-этил-5-фенилизоксазолиум-3'-сульфонат; карбодиимидные производные, включающие N',N'-дициклоалкилкарбодиимиды, такие как N',N'-дициклогексилкарбодиимид или 1-этил-3-(3-диметиламинопропил)карбодиимид; дигетероарильные диселениды, такие как ди-2-пиридилдиселенид; арилсульфонильные триазолидины, такие как п-нитробензолсульфонилтриазолидин; 2-галоид-1-(низший алкил)пиридиниевые галогениды, такие как 2-хлор-1-метилпиридиниевый иодид; диарилфосфорилазиды, такие как дифенилфосфорилазид; имидазольные производные, такие как 1,1'-оксалилдиимидазол или N, N'-карбонилдиимидазол; бензотриальные производные, такие как 1-гидроксибензотриазол; и дикарбоксимидные производные, такие как N-гидрокси-5-норборнер-2,3-дикарбоксимид. Из этих соединений изобретатели предпочтение отдают диарилфосфорильным азидам.
Взаимодействие может проходить в широком интервале температур, и точная температура не является критической для изобретения. В общем случае установлено, что удобно вести реакцию при температуре в области от -20oC до 100oC, более желательно в области от 0oC и примерно до комнатной температуры. Время, необходимое для взаимодействия, также может меняться в широких пределах в зависимости от многих факторов, особенно от температуры взаимодействия и природы применяемых реагентов и растворителя. Если же реакцию ведут при предпочтительных условиях, оговоренных выше, обычно достаточен период от 10 мин до 8 ч, предпочтительно от 30 мин до 4 ч.
После завершения реакции требуемое соединение с формулой (VIII) может быть извлечено из реакционной смеси обычными способами. Например, в случае одного приемлемого способа извлечения реакционную смесь подвергают нейтрализации, при наличии нерастворимых веществ их удаляют фильтрованием, к фильтрату или к нейтрализованной реакционной смеси добавляют воду и несмешиваемый с водой органический растворитель, такой как этилацетат, и продукт экстрагируют растворителем, экстракт промывают водой и сушат, например, над безводным сульфатом магния, и затем растворитель отгоняют, получая в остатке требуемый продукт.
Требуемый продукт, полученный таким способом, может быть, если это необходимо, подвергнут дальнейшей очистке обычными средствами, например перекристаллизацией, переосаждением или различными хроматографическими способами. Примерами подходящих хроматографических методик являются абсорбционная хроматография с пропусканием очищаемого вещества через носитель, такой как силикагель, оксид алюминия или флорисил (торговое название материала, представляющего собой силикагель, содержащий магний); распределительная хроматография с пропусканием очищаемого вещества через синтетический абсорбент, такой как сефадексТМ LH-20 (торговое название материала, фирма "Фармация, инк. "), амберлитТМ XAD-11 (торговое название материала, фирма "Ром энд Хаас ко.") или диаионТМ HP-20 (торговое название материала, фирма "Мицубиси касеи корпорейшн"); колонная хроматография с пропусканием очищаемого вещества через регулярно-фазовую или обращенно-фазовую колонку с насадкой из силикагеля или алкилированного силикагеля (желательно использовать высокопроизводительную жидкостную хроматографию); или комбинация из этих способов с последующим элюированием при использовании подходящего растворителя.
Стадия A3
На этой стадии соединение формулы (IX) получают с помощью селективной защиты двух гидроксигрупп, иных, чем гидроксигруппа, в положении 8, соединении формулы (VIII) группой R6′.
Защита может быть осуществлена различными способами, что, частично, зависит от природы выбранной защищающей группы, как это, например, проиллюстрировано следующими методами 1 - 3.
Метод 1.
В этом случае проводится взаимодействие соединения формулы (VIII) с подходящим количеством, например с 1 - 4 эквивалентами (более желательно использовать от двух до трех эквивалентов), соединения формулы R6′-X или соединения формулы 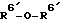 (в которых R6′ имеет значения, определенные ранее, предпочтительно она представляет ацильную группу, и X представляет собой отщепляемую группу) в растворителе в присутствии или в отсутствии основания. В приведенных выше формулах R6′ имеет значения, определенные ранее, и предпочтительно представляет собой гидроксизащищающую группу, более предпочтительно силильную группу, и наиболее предпочтительно трет-бутилдиметилсилильную группу.
(в которых R6′ имеет значения, определенные ранее, предпочтительно она представляет ацильную группу, и X представляет собой отщепляемую группу) в растворителе в присутствии или в отсутствии основания. В приведенных выше формулах R6′ имеет значения, определенные ранее, и предпочтительно представляет собой гидроксизащищающую группу, более предпочтительно силильную группу, и наиболее предпочтительно трет-бутилдиметилсилильную группу.
Отсутствует какое-либо особое ограничение, касающееся природы отщепляемой группы, при условии, что она представляет собой группу, способную отщепляться в виде нуклеофильного остатка, такого как те, которые являются хорошо известными в этой области техники. Примерами предпочтительных отщепляемых групп являются атомы галогена, такие как атомы хлора, бром и иода; низшие алкоксикарбонилоксигруппы, такие как метоксикарбонилоксигруппа и этоксикарбонилоксигруппа; галогенированные алкилкарбонилоксигруппы, такие как хлорацетоксигруппа, дихлорацетоксигруппа, трихлорацетоксигруппа и трифторацетоксигруппа; низшие алкансульфонилоксигруппы, такие как метансульфонилоксигруппа и этансульфонилоксигруппа; низшие галоидалкансульфонилоксигруппы, такие как трифторметансульфонилоксигруппа и пентафторэтансульфонилоксигруппа; и арилсульфонилоксигруппы, такие как бензолсульфонилоксигруппа, п-толуолсульфонилоксигруппа и п-нитробензолсульфонилоксигруппа. Из этих групп изобретатели отдают предпочтение атомам галогена, низшим галоалкансульфонилоксигруппам и арилсульфонилоксигруппам.
Реакцию обычно и предпочтительно проводят в присутствии растворителя. Отсутствует какое-либо ограничение, касающееся природы растворителя, который должен быть применен, при условии, что он не оказывает отрицательного воздействия на ход реакции или на реагенты, участвующие в ней, и что он может растворять их, по крайней мере, в некоторой степени. Примерами приемлемых растворителей являются алифатические углеводороды, такие как бензол, толуол и ксилол; галоидированные углеводороды, такие как хлористый метилен, хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол и дихлорбензол; сложные эфиры, такие как этилформиат, этилацетат, пропилацетат, бутилацетат и диэтилкарбонат; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диэтиленгликольдиметиловый простой эфир; нитрилы, такие как ацетонитрил и изобутиронитрил; и амиды, такие как формамид, диметилформамид, диметилацетамид, N-метил-2-пирролидон, N-метилпирролидинон и гексаметилфосфорный триамид.
Отсутствует какое-либо особе ограничение, касающееся природы основания, используемого в методе 1, и любое основание, которое может быть использовано в общеизвестных реакциях этого типа, может быть в одинаковой мере использовано и здесь. Примерами предпочтительных оснований являются органические основания, такие как N-метилморфолин, триэтиламин, трибутиламин, дициклогексиламин, N-метилпиперидин, пиридин, 4-(1-пирролидинил)пиридин, пиколин, 4-(N, N-диметиламино)пиридин, 2,6-ди-трет-бутил-4-метилпиридин, хинолин, N, N-диметиланилин и N,N-диэтиланилин. При желании можно использовать каталитические количества 9-(N,N-диметиламино)пиридина, 4-(1-пирролидинил)пиридина или комбинацию из других оснований. Чтобы содействовать более эффективному протеканию реакции в реакционную систему могут быть добавлены четвертичная аммониевая соль (такая как хлористый бензилтриэтиламмоний или хлористый тетрабутиламмоний) или краун-эфиры (такие как дибензо-18-краун-6).
Взаимодействие может происходить в широком интервале температур, и точная температура не является в изобретении критической. В общем случае установлено, что реакцию удобно вести при температуре в области от -20oC и до температуры дефлегмации используемого растворителя, более предпочтительно в области от 0oC до температуры дефлегмации используемого растворителя. Время, необходимое для взаимодействия, также может изменяться в широких пределах в зависимости от многих факторов, а именно от температуры взаимодействия и природы применяемых реагентов, основания и растворителя. Если же реакцию ведут в предпочтительных условиях, оговоренных выше, то тогда обычно достаточна продолжительность от 10 мин до 3 дней, более предпочтительно от 1 до 6 ч.
Метод 2.
Этот метод включает взаимодействие соединения формулы (VIII) с соединением формулы R6′-OH (в которой R6′ имеет значения, определенные ранее, и, предпочтительно, представляет ацильную группу) в растворителе в присутствии этерифицирующего агента, такого как те, что проиллюстрированы примерами ранее в методе 2 на стадии A2, и каталитического количества основания.
Реакцию обычно и предпочтительно проводят в присутствии растворителя. Отсутствует какое-либо особое ограничение, касающееся природы используемого растворителя при условии, что он не оказывает отрицательного воздействия на ход реакции или на реагенты, участвующие в реакции, и что он может растворять реагенты, по крайней мере, в некоторой степени. Примерами подходящих растворителей являются алифатические углеводороды, такие как гексан и гептан; ароматические углеводороды, такие как бензол, толуол и ксилол; галоидированные углеводороды, такие как хлористый метилен, хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол и дихлорбензол; сложные эфиры, такие как этилформиат, этилацетат, пропилацетат, бутилацетат и диэтилкарбонат; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диэтиленгликольдиметиловый эфир; нитрилы, такие как ацетонитрил и изобутиронитрил; и амиды, такие как формамид, диметилформамид, диметилацетамид, N-метил-2-пирролидон, N-метилпирролидинон и гексаметилфосфорный триамид.
Примерами оснований, которые могут быть использованы в методе 2, являются те же основания, что описаны в предшествующем методе 1.
Взаимодействие может протекать в широком интервале температур, и точная температура не является в изобретении критической. В общем случае установлено, что реакцию удобно вести при температуре в области от -20 и до 80oC, более предпочтительно от 0oC примерно до комнатной температуры. Время, необходимое для протекания взаимодействия, может также изменяться в широких пределах в зависимости от многих факторов, а именно от температуры взаимодействия и природы применяемых реагентов и растворителя. Если же реакцию ведут в предпочтительных условиях, оговоренных ранее, обычно достаточна продолжительность от 10 мин до 3 дней, более предпочтительно от 30 мин до одного дня.
Метод 3.
Этот метод включает взаимодействие соединения формулы (VIII) с соединением формулы R6′-OH (в которой R6′ является таким, как определен ранее, и, предпочтительно, представляет ацильную группу) в растворителе в присутствии галоидированного диалкильного сложного эфира фосфорной кислоты, такого как диэтилхлорфосфат, и основания.
Взаимодействие обычно и предпочтительно проводят в присутствии растворителя. Отсутствует какое-либо особое ограничение, касающееся природы применяемого растворителя, при условии, что он не оказывает отрицательного воздействия на ход реакции или на реагенты участвующие в реакции и что он может растворять реагенты, по крайней мере, в некоторой степени. Примерами подходящих растворителей являются алифатические углеводороды, такие как гексан и гептан; ароматические углеводороды, такие как бензол, толуол и ксилол; галоидированные углеводороды, такие как хлористый метилен, хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол и дихлорбензол; сложные эфиры, такие как этилформиат, этилацетат, пропилацетат, бутилацетат и диэтилкарбонат; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диэтиленгликольдиметиловый простой эфир; нитрилы, такие как ацетонитрил и изобутиронитрил; и амиды, такие как формамид, диметилформамид, диметилацетамид, N-метил-2-пирролидон, N-метилпирролидинон и гексаметилфосфорный триамид.
Примерами оснований, которые могут быть использованы в методе 3, являются те же, что описаны для использования в предшествующем методе 1.
Взаимодействие может протекать в широком интервале температур, и точная температура не является в изобретении критической. В общем случае установлено, что реакцию удобно вести при температуре в области от 0oC и до температуры дефлегмации используемого растворителя, более предпочтительно примерно от комнатной температуры до 50oC. Время, необходимое для протекания взаимодействия, также может меняться в широких пределах в зависимости от многих факторов, особенно от температуры взаимодействия и природы применяемых реагентов и растворителя. Если же реакцию ведут в предпочтительных условиях, оговоренных выше, то тогда продолжительность взаимодействия обычно составляет от 10 мин и до трех дней, предпочтительно от 30 мин до одного дня.
Если группа R6′ представляет собой низшую алкильную группу, она может быть введена в соединение формулы (VIII) обычными способами, например взаимодействием соединения формулы (VIII) с диалкилсульфатом, таким как диметилсульфат или диэтилсульфат. Использованием защищающих реагентов с разной реакционной способностью можно получить соединение с двумя гидроксигруппами, защищенными различными группами R6′.
После завершения реакции требуемое соединение формулы (IX) может быть извлечено из реакционной смеси обычными способами. Например, в случае одной приемлемой методики извлечения реакционную смесь подвергают нейтрализации, при наличии нерастворимых веществ их удаляют фильтрованием, к реакционной смеси или к нейтрализованной реакционной смеси добавляют воду и несмешиваемый с водой растворитель, такой как этилацетат, и продукт экстрагируют растворителем, экстракт промывают водой и сушат, например, над безводным сульфатом магния, и затем растворитель отгоняют, получая требуемый продукт.
Соединение, полученное таким способом, может быть, при необходимости, очищено общепринятыми способами, например перекристаллизацией, переосаждением или различными хроматографическими способами. Примерами приемлемых хроматографических методик являются абсорбционная колонная хроматография с пропусканием очищаемого вещества через носитель, такой как силикагель, оксид алюминия или флорисил (силикагель, содержащий магний); распределительная колонная хроматография с пропусканием очищаемого вещества через синтетический абсорбент, такой как сефадексТМ LH-20 (торговое название материала производства фирмы "Фармация, инк.") амберлитТМ XAD-11 (торговое название материала производства фирмы "Ром энд Хаас ко. ") или диаионТМ HP-20 (торговое название материала производства фирмы "Мицубиси касеи корпорейшн"); колонная хроматография с пропусканием очищаемого вещества через регулярно-фазовую или обращенно-фазовую колонку с насадкой из силикагеля или из алкилированного силикагеля (желательно использовать высокопроизводительную жидкостную хроматографию); или комбинация из этих способов с последующим элюированием надлежащим элюирующим растворителем.
Стадия A4
На этой стадии сложноэфирное соединение формулы (X) получают ацилированием гидроксигруппы, в положении 8 в соединении формулы (IX), группой R7. Реакцию проводят по методике, описанной при рассмотрении стадии A3, используя любой из методов, описанных ниже.
Метод 1.
Этот метод включает взаимодействие соединения формулы (IX) с надлежащим количеством, например с 1 - 4 эквивалентами (более желательно, с двумя-тремя эквивалентами) соединения формулы R7-X или R7-O-R7 (в которой R7 и X являются такими, как определены ранее) в растворителе в присутствии или в отсутствии основания.
Метод 2.
Этот метод включает взаимодействие соединения формулы (IX) с соединением формулы R7-OH (в которой R7 является такой, как определена ранее) в растворителе в присутствии этерифицирующего вещества, такого как проиллюстрированные примерами ранее в методе 2 на стадии A2, и каталитического количества основания.
Метод 3.
Этот метод включает взаимодействие соединения формулы (IX) с соединением формулы R7-OH (в которой группа R7 является такой, как определена ранее) в растворителе в присутствии галоидированного диэтилового сложного эфира фосфорной кислоты, такого как диэтилхлорфосфат, и основания.
Стадия A5
На этой стадии соединение формулы (XI) получают удалением гидроксизащищающей группы, представленной группой R6′, из соединения формулы (X) и затем, при необходимости, блокированием некоторых или всех образовавшихся свободных гидроксигрупп при использовании одной и той же или различных защищающих групп, желательно таких, которые могут быть отщеплены ин виво биологическими методами, такими как гидролиз.
Реакционные условия, применяемые при удалении гидроксизащищающей группы, представленной группой R6′, меняются в широких пределах в зависимости от природы защищающей группы; но при этом реакцию обычно проводят способами, хорошо известными в этой области техники, такими, например, как описанные ниже.
Удаление с использованием фторидного аниона или органической кислоты. В тех случаях, когда гидроксизащищающая группа представляет собой силильную группу, она обычно может быть удалена обработкой защищенного соединения соединением, способным давать фторидный анион, таким как фтористый тетрабутиламмоний или фтористоводородная кислота, или обработкой его органической кислотой, такой как метансульфоновая кислота, п-толуолсульфоновая кислота, трифторуксусная кислота или трифторметансульфоновая кислота. В случае использования фторидного аниона в качестве деблокирующего вещества реакцию иногда можно ускорить добавлением органической кислоты, такой как муравьиная кислота, уксусная кислота или пропионовая кислота. Эта реакция удаления характеризуется тем преимуществом, что при ее проведении подавляются побочные реакции.
Реакцию обычно и предпочтительно проводят в присутствии растворителя. Отсутствует какое-либо особое ограничение, касающееся природы растворителя, который должен быть применен, при условии, что он не оказывает отрицательного воздействия на ход реакции или на реагенты, участвующие в реакции, и что он может растворять реагенты, по крайней мере, в некоторой степени. Примерами подходящих растворителей являются простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диэтиленгликолдиметиловый эфир; и нитрилы, такие как ацетонитрил и изобутиронитрил.
Взаимодействие может происходить в широком интервале температур, и точная температура не является в изобретении критической. В общем случае установлено, что реакцию удобно вести при температуре, в области от 0oC до 50oC, более предпочтительно при температуре, близкой к комнатной. Время, необходимое для обеспечения взаимодействия, может также изменяться в широких пределах в зависимости от многих факторов, особенно от температуры взаимодействия и природы применяемых реагентов и растворителя. Если же реакцию ведут в предпочтительных условиях, оговоренных выше, то тогда продолжительность взаимодействия обычно составляет от 2 до 24 ч.
Удаление посредством восстановления или окисления. В том случае, когда гидроксизащищающая группа представляет собой аралкильную или аралкилоксикарбонильную группу, она в предпочтительном варианте может быть удалена контактированием защищенного соединения с восстанавливающими веществами (желательно проведением каталитического восстановления с применением водорода в присутствии катализатора, например, проведением взаимодействия при примерно комнатной температуре) в растворителе или использованием окисляющего вещества.
Реакцию восстановления обычно и предпочтительно проводят в присутствии растворителя. Отсутствует какое-либо особое ограничение, касающееся природы растворителя, который должен быть применен, при условии, что он не оказывает отрицательного воздействия на ход реакции или на реагенты участвующие в реакции и что он может растворять их, по крайней мере, в некоторой степени. Примерами подходящих растворителей являются спирты, такие как этанол и изопропанол, простые эфиры, такие как диэтиловый эфир, тетрагидрофуран и диоксан; ароматические углеводороды, такие как толуол, бензол и ксилол; алифатические углеводороды, такие как гексан и циклогексан; сложные эфиры, такие как этилацетат и пропилацетат; амиды, такие как формамид, диметилформамид, диметилацетамид, N-метил-2-пиридин и гексаметилфосфорный триамид; алифатические кислоты, такие как муравьиная кислота и уксусная кислота; или вода. Может быть использован какой-то один из этих растворителей или может быть использована смесь, состоящая из двух или нескольких растворителей. Из этих соединений изобретатели предпочтение отдают спиртам, алифатическим кислотам, смеси спирта с простым эфиром, смеси спирта с водой или смеси алифатической кислоты с водой.
Отсутствует какое-либо особое ограничение, касающееся природы используемого катализатора, и любой катализатор, обычно используемый при проведении каталитического восстановления, в равной мере может быть использован и здесь. Примерами предпочтительных катализаторов являются палладий, нанесенный на активированный уголь, палладиевая чернь, никель Ренея, оксид платины, платиновая чернь, родий на оксиде алюминия, комбинация из трифенилфосфина и хлорида родия и палладий на сульфате бария.
Давление водорода, используемое при проведении реакции, не является критической величиной; однако реакцию обычно ведут при давлении, в области от давления окружающей среды до 10 ат.
Взаимодействие может протекать в широком интервале температур, и точная температура не является в изобретении критической, хотя предпочтительная температура может меняться в зависимости от таких факторов, как природа реагентов и катализатора. В общем случае установлено, что реакцию удобно вести при температуре в области от 0oC до 100oC, более желательно от 20oC до 70oC. Время, необходимое для протекания взаимодействия, может также изменяться в широких пределах в зависимости от многих факторов, а именно от температуры взаимодействия и природы применяемых реагентов и растворителя. Если же реакцию ведут в предпочтительных условиях, оговоренных выше, продолжительность взаимодействия обычно составляет от 5 мин до 48 ч, более предпочтительно от 1 до 24 ч.
В случае проведения реакции окисления реакцию обычно предпочтительно проводят в присутствии растворителя. Здесь также отсутствует какое-либо особое ограничение, касающееся природы растворителя, который должен быть использован при условии, что он не оказывает отрицательного воздействия на ход реакции или на реагенты, участвующие в реакции, и что он может растворять их, по крайней мере, в некоторой степени. Примерами приемлемых растворителей являются водные органические растворители. Примерами таких органических растворителей являются кетоны, такие как ацетон; галоидированные углеводороды, такие как хлористый метилен, хлороформ и четыреххлористый углерод; нитрилы, такие как ацетонитрил; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран и диоксан; амиды, такие как диметилформамид, диметилацетамид и гексаметилфосфорный триамид; и сульфоксиды, такие как диметилсульфоксид.
Отсутствует какое-либо особое ограничение, касающееся природы используемого окисляющего вещества, и любое окисляющее вещество, обычно используемое при проведении реакций окисления этого типа, одинаково пригодно для использования и здесь. Примерами предпочтительных окисляющих веществ являются персульфат калия, персульфат натрия, аммониевый нитрат церия и 2,3-дихлор-5,6-дициано-п-бензохинон.
Взаимодействие может протекать в широком интервале температур, и точная температура не является в изобретении критической. В общем случае установлено, что реакцию удобно вести при температуре в области от 0oC и до 150oC. Время, необходимое для протекания взаимодействия, также может изменяться в широких пределах в зависимости от многих факторов, а именно от температуры взаимодействия и природы применяемых реагентов и растворителя. Если же реакцию ведут в предпочтительных условиях, оговоренных выше, то тогда продолжительность взаимодействия обычно составляет от 10 мин до 24 ч.
Удаление посредством обработки щелочным металлом. Защищающая группа может быть удалена обработкой щелочным металлом, таким как литий или натрий, в жидком аммиаке или в спирте, таком как метанол или этанол, при надлежащей температуре, например в области от -78oC до -20oC.
Удаление посредством обработки хлоридом алюминия. Можно также удалить защищающую группу контактированием защищенного соединения со смесью хлорида алюминия с иодидом натрия или с алкилсилильным галогенидом, таким как иодистый триметилсилил.
Реакцию обычно и предпочтительно проводят в присутствии растворителя. Отсутствует какое-либо особое ограничение, касающееся природы растворителя, который должен быть применен, при условии, что он не оказывает отрицательного воздействия на ход реакции или на реагенты, участвующие в ней, и что он может растворять их, по крайней мере, в некоторой степени. Примерами подходящих растворителей являются нитрилы, такие как ацетонитрил; и галоидированные углеводороды, такие как хлористый метилен и хлороформ. Может быть использован какой-то один из этих растворителей или может быть использована смесь из двух или более растворителей.
Взаимодействие может протекать в широком интервале температур, и точная температура не является в изобретении критической. В общем случае установлено, что реакцию удобно вести при температуре в области от 0oC до 50oC. Время, необходимое для протекания взаимодействия, также может меняться в широких пределах в зависимости от многих факторов, а именно от температуры взаимодействия и природы применения реагентов и растворителя. Однако, если реакцию ведут в предпочтительных условиях, оговоренных выше, продолжительность взаимодействия обычно составляет в области от 5 мин и до трех дней.
Если реакционный субстрат содержит атом серы, предпочтительно использовать смесь хлорида алюминия с иодидом натрия.
Удаление посредством обработки основанием. В том случае, когда гидроксизащищающая группа представляет собой алифатическую ацильную группу, ароматическую ацильную группу или алкоксикарбонильную группу, защищающая группа может быть удалена обработкой защищенного соединения основанием в растворителе.
Отсутствует какое-либо особое ограничение, касающееся природы используемого основания при условии, что другие части соединения не подвергаются воздействию при удалении защищающей группы. Примерами предпочтительных оснований являются алкоксиды металлов, такие как метоксид натрия; карбонаты щелочных металлов, такие как карбонат натрия, карбонат калия и карбонат лития; гидроксиды щелочных металлов, такие как гидроксид натрия, гидроксид калия, гидроксид лития и гидроксид бария; и аммиак, например, в виде водного раствора аммиака или в виде смеси концентрированного аммиака с метанолом.
Реакцию обычно и предпочтительно проводят в присутствии растворителя. Отсутствует какое-либо особое ограничение, касающееся природы растворителя, который должен быть применен, при условии, что он не оказывает отрицательного воздействия на ход реакции или на реагенты, участвующие в реакции, и что он может растворять реагенты, по крайней мере, в какой-то степени. Примерами подходящих растворителей являются вода; органические растворители, например спирты, такие как этанол и пропанол; простые эфиры, такие как тетрагидрофуран и диоксан; или смесь воды с каким-либо одним или с несколькими этими органическими растворителями.
Взаимодействие может протекать в широком интервале температур, и точная температура не является критической для изобретения. В общем случае установлено, что реакцию удобно вести при температуре от 0oC и до 150oC. Время, необходимое для протекания взаимодействия, может также изменяться в широких пределах, в зависимости от многих факторов, особенно от температуры взаимодействия и природы применяемых реагентов и растворителя. Если же реакцию ведут в предпочтительных условиях, оговоренных выше, то тогда продолжительность взаимодействия обычно составляет от 1 до 10 ч.
В том случае, когда гидроксизащищающей группой является алкенилоксикарбонильная группа, деблокирование может быть также осуществлено посредством обработки основанием, и реакционные условия являются таким же, что и тогда, когда гидроксизащищающей группой является алифатическая ацильная, ароматическая ацильная или алкоксикарбонильная группа.
Удаление посредством обработки кислотой. В тех случаях, когда гидроксизащищающая группа представляет собой алкоксиметильную, тетрагидропиранильную, тетрагидротиопиранильную, тетрагидрофуранильную, тетрагидротиенильную или замещенную этильную группу, она может быть обычно удалена обработкой защищенного соединения кислотой.
Отсутствует какое-либо особое ограничение, касающееся природы кислоты, и любые кислоты, обычно используемые для этой цели, включая кислоты Бренстеда и кислоты Льюиса, могут быть здесь в равной мере использованы. Примерами предпочтительных кислот являются неорганические кислоты, такие как хлористый водород, хлористоводородная кислота, серная кислота и азотная кислота; кислоты Бренстеда, включая органические кислоты, такие как уксусная кислота, трифторуксусная кислота, метансульфоновая кислота или п-толуолсульфоновая кислота; кислоты Льюиса, такие как треххлористый бор; и сильно кислотные катионообменные смолы, такие как дауэкс-50WТМ (торговое название материала).
Реакцию обычно и предпочтительно проводят в присутствии растворителя. Отсутствует какое-либо особое ограничение, касающееся природы применяемого растворителя, при условии, что он не оказывает отрицательного воздействия на ход реакции на реагенты, участвующие в ней, и что он может растворять реагенты, по крайней мере, в некоторой степени. Примерами подходящих растворителей являются алифатические углеводороды, такие как гексан и гептан; ароматические углеводороды, такие как бензол, толуол и ксилол; галоидированные углеводороды, такие как хлористый метилен; хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол и дихлорбензол; сложные эфиры, такие как этилформиат, этилацетат, пропилацетата, бутилацетат и диэтилкарбонат; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диэтиленгликолдиметиловый эфир; спирты, такие как этанол, пропанол, изопропанол, бутанол, изобутанол, трет-бутанол, изоамиловый спирт, диэтиленгликоль и циклогексанол; кетоны, такие как ацетон, метилэтиловый кетон, метилизобутиловый кетон, изофорон и циклогексанон; или вода. Может быть использован любой один из этих растворителей или смесь двух или большего числа растворителей. Из указанных соединений изобретатели предпочтение отдают галоидированным углеводородам, сложным эфирам и простым эфирам.
Взаимодействие может происходить в широком интервале температур, и точная температура не является критической для изобретения. В общем случае установлено, что реакцию удобно вести при температуре от -10oC и до 100oC, более предпочтительно от -5oC до 50oC. Время, необходимое для протекания взаимодействия, также может изменяться в широких пределах в зависимости от многих факторов, а именно от температуры взаимодействия и природы применяемых реагентов и растворителя. Если же реакцию ведут в предпочтительных условиях, оговоренных выше, то тогда продолжительность взаимодействия обычно составляет от 5 мин до 48 часов, более предпочтительно от 30 мин до 10 часов.
Удаление воздействием палладия и трифенилфосфина или тетракарбонила никеля. В том случае, кода гидроксизащищающая группа представляет собой арилоксикарбонильную группу, она может быть просто удалена использованием комбинации из палладия и трифенилфосфина или тетракарбонильного никеля, что имеет преимущество, поскольку подавляются побочные реакции.
Введение гидроксизащищающей группы. При желании, полученная в результате свободная гидроксигруппа может быть впоследствии защищена защищающей группой, особенно защищающей группой, способной отщепляться ин виво биологическими методами, такими как гидролиз. Сказанное может быть осуществлено использованием соответствующего реагента, содержащего требуемую защищающую группу, что проводится по методике, описанной при рассмотрении стадии A3.
В том случае, когда число защищаемых гидроксигрупп оказывается большим, чем одна группа, они могут быть защищены одной и той же защищающей группой или различными защищающими группами, как это, например, проиллюстрировано ниже:
1) когда две гидроксигруппы защищаются различными защищающими группами, представленными R6′, каждая из этих групп может быть удалена избирательно и получающаяся в результате свободная гидроксигруппа может быть затем защищена соответствующими защищающими реагентами, с получением соединения с гидроксигруппами, защищенными различными группами R6, или
2) две гидроксигруппы защищаются различными защищающими группами, представленными R6, с использованием различия в реакционных способностях защищающих реагентов, как хорошо известно в данной области техники.
После завершения взаимодействия требуемое соединение формулы (XI) может быть извлечено из реакционной смеси обычными методами. Например, в случае одной из приемлемых методик извлечения реакционную смесь подвергают нейтрализации, при наличии нерастворимых веществ их удаляют фильтрованием, к фильтрату или к нейтрализованной реакционной смеси добавляют воду и несмешиваемый с водой растворитель, такой как этилацетат, и продукт экстрагируют растворителем, экстракт промывают водой и сушат, например, над безводным сульфатом магния; и затем растворитель отгоняют из экстракта, получая в остатке требуемый продукт.
Требуемое соединение, полученное таким способом, может быть, если это необходимо, очищено обычными способами, например перекристаллизацией, переосаждением или различными хроматографическими способами. Примерами подходящими хроматографических способов являются абсорбционная колонная хроматография с пропусканием очищаемого вещества через носитель, такой как силикагель, оксид алюминия или флорисил, содержащий магний и силикагель; распределительная колонная хроматография с пропусканием очищаемого вещества через абсорбент, такой как сефадексТМ LH-20 (фирма "Фармация, инк."), амберлитТМ XAD-11 (Ром энд Хаас ко.") или диаионТМ ("Мицубиси Касеи Корпорейшн"); жидкостная хроматография с пропусканием очищаемого вещества через регулярно-фазовую или обращенно-фазовую колонку с насадкой из силикагеля или из алкилированного силикагеля (желательно использовать высокопроизводительную жидкостную хроматографию); или комбинация из этих способов с последующим элюированием при использовании надлежащего элюирующего растворителя.
Стадия A6
На этой стадии соединение формулы (XII), которое является соединением настоящего изобретения, получают, проводят гидролиз или сольволиз лактонового кольца соединения формулы (XI), получая соль карбоновой кислоты или сложный эфир карбоновой кислоты. Эта реакция может быть, при желании, проведена посредством
1) получения свободной карбоновой кислоты;
2) защиты некоторых или всех свободных гидроксигрупп одной и той же или различными защищающими группами, желательно способными отщепляться ин виво биологическими методами, такими как гидролиз;
3) защиты получающейся в результате карбоксигруппы защищающей группой, желательно способной отщепляться ин виво биологическими методами, такими как гидролиз, или получения еще одной соли карбоновой кислоты и/или,
4) при желании, проведения циклизации соединения карбоновой кислоты с получением снова лактонового соединения.
Получение соли карбоновой кислоты можно осуществить проведением обычной реакции гидролиза с использованием основания, предпочтительно от 1 до 2 моль основания.
Реакцию обычно и предпочтительно проводят в присутствии растворителя. Отсутствует какое-либо особое ограничение, касающееся природы применяемого растворителя, при условии, что он не оказывает отрицательного воздействия на ход реакции или на реагенты, участвующие в ней, и что он может растворять реагенты, по крайней мере, в некоторой степени. Примерами приемлемых растворителей являются вода или смесь воды с одним или несколькими органическими растворителями, такими как простые эфиры, такие как тетрагидрофуран, диоксан или диэтиленгликолдиметиловый простой эфир; спирты, такие как этанол, пропанол, изопропанол, бутанол или изобутанол; кетоны, такие как ацетон или метилэтиловый кетон; нитрилы, такие как ацетонитрил или изобутиронитрил; и амиды, такие как формамид, диметилформамид, диметилацетамид, N-метил-2-пирролидон, N-метилпирролидинон или гексаметилфосфорный триамид).
Отсутствует также какое-либо определенное ограничение, касающееся природы используемого основания, и любое основание, обычно используемое в общепринятых реакциях, может быть с одинаковым успехом использовано и здесь. Примерами предпочтительных оснований являются карбонаты щелочных металлов, такие как карбонат натрия, карбонат калия или карбонат лития; водородсодержащие карбонаты щелочных металлов, такие как бикарбонат натрия, бикарбонат калия или бикарбонат лития; оксиды щелочных металлов, такие как гидроксид натрия, гидроксид калия, гидроксид кальция, гидроксид бария или гидроксид лития; и алкоксиды щелочных металлов, такие как метоксид натрия, этоксид натрия, метоксид калия, этоксид калия, трет-бутоксид калия или метоксид лития.
Взаимодействие происходит в широком интервале температур, и точная температура не является критической. В общем установлено, что реакцию удобно вести при температуре от -10oC и до 100oC, более предпочтительно от 0oC и примерно до комнатной температуры. Время, необходимое для протекания взаимодействия, также может меняться в широких пределах в зависимости от многих факторов, а именно от температуры взаимодействия, используемого основания и природы реагентов. Однако в большинстве случаев продолжительность взаимодействия составляет от 30 мин до 10 ч, предпочтительно от 1 до 5 ч.
Реакция получения сложного эфира карбоновой кислоты может осуществляться проведением сольволиза в присутствии кислотного катализатора и растворителя, содержащего спирт.
Реакцию обычно и предпочтительно проводят в присутствии растворителя. Отсутствует какое-либо особое ограничение, касающееся природы применяемого растворителя, при условии, что он не оказывает отрицательного воздействия на ход реакции или на реагенты, участвующие в ней, и что он может растворять реагенты, по крайней мере, в некоторой степени. Примерами приемлемых растворителей являются алифатические углеводороды, такие как гексан или гептан; ароматические углеводороды, такие как бензол, толуол или ксилол; галоидированные углеводороды, такие как хлористый метилен, хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол или дихлорбензол; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан или диэтиленгликолдиметиловый эфир; кетоны, такие как ацетон, метилэтиловый кетон, метилизобутиловый кетон, изофорон или циклогексанон; нитрилы, такие как ацетонитрил или изобутиронитрил; и амиды, такие как формамид, диметилформамид, диметилацетамид, N-метил-2-пирролидон, N-метилпирролидинон или гексаметилфосфорный триамид. Однако изобретатели предпочитают использовать в качестве растворителя спирт, который соответствует сложноэфирному остатку, который необходимо ввести.
Отсутствует также какое-либо особое ограничение, касающееся природы используемого кислотного катализатора, и любая кислота, обычно используемая в качестве катализатора при проведении общеизвестных реакций, может быть в равной мере использована и здесь. Примерами предпочтительных кислотных катализаторов являются неорганические кислоты, такие как хлористоводородная кислота, бромистоводородная кислота, серная кислота, перхлорная кислота или фосфорная кислота; кислоты Бренстеда, например, такие как органические кислоты, включая карбоновые кислоты (такие как уксусная кислота, щавелевая кислота, муравьиная кислота и трифторуксусная кислота) и сульфоновые кислоты (такие как метансульфоновая кислота, п-толуолсульфоновая кислота и трифторметансульфоновая кислота); кислоты Льюиса, такие как треххлористый бор, трехфтористый бор или трехбромистый бор; и кислотные ионообменные смолы. Из них изобретатели отдают предпочтение органическим кислотам и, более предпочтительно, сильным органическим кислотам.
Взаимодействие проходит в широком интервале температур, и точная температура не является критической. Обычно реакцию удобно вести при температуре от 0oC и до температуры кипения используемого растворителя, более предпочтительно от 50oC и до температуры кипения используемого растворителя. Время, необходимое для протекания взаимодействия, также может меняться в широких пределах в зависимости от многих факторов, а именно от температуры взаимодействия и природы используемых реагентов и растворителя. Однако в большинстве случаев продолжительность обычно составляет от 10 мин и до 6 дней, более предпочтительно от 30 мин и до 3 дней.
По завершении реакции требуемое соединение может быть извлечено из реакционной смеси обычными способами. Например, если реакцию проводят с использованием кислотной ионообменной смолы в качестве кислотного катализатора, то тогда приемлемая методика извлечения включает фильтрование реакционной смеси и удаление отгонкой растворителя из фильтра с получением в остатке требуемого продукта. Если реакцию проводят с использованием еще одной кислоты в качестве кислотного катализатора, то тогда приемлемая методика извлечения включает нейтрализацию реакционной смеси, при наличии нерастворимых веществ их удаление фильтрованием, добавление воды и несмешиваемого с водой растворителя, такого как этилацетат, к нейтрализованной реакционной смеси или к фильтрату и экстракцию продукта растворителем, промывку экстракта водой и сушку его, например, над безводным сульфатом магния, и затем удаление растворителя отгонкой с получением в остатке требуемого продукта.
Требуемый продукт, полученный таким способом, подвергают, если это необходимо, очистке, используя обычные способы, например перекристаллизацию, переосаждение или различные хроматографические методики. Примерами таких хроматографических методик являются распределительная колонная хроматография с пропусканием очищаемого вещества через синтетический абсорбент, такой как сефадексТМ LH-20 ("Фармация, инк."), амберлитТМ XAD-11 ("Ром энд Хаас ко".) или диаионТМ HP-20 ("Мицубиси касеи корпорейшн"); жидкостная хроматография с пропусканием очищаемого вещества через регулярно-фазовую или обращенно-фазовую колонку с насадкой из силикагеля или из алкилированного силикагеля (желательно использовать высокопроизводительную жидкостную хроматографию), или надлежащее сочетание этих методик с последующим элюированием с использованием надлежащего элюирующего растворителя.
Свободная карбоновая кислота предпочтительно получается путем доведения pH фильтрата, содержащего соль карбоновой кислоты, полученной выше, до значения менее 5, предпочтительно до pH от 3 до 4, добавлением подходящей кислоты.
Отсутствует какое-либо особое ограничение, касающееся типа используемой кислоты, и любая органическая или минеральная кислота может быть использована при условии, что она не оказывает отрицательного воздействия на требуемое соединение. Примерами предпочтительных кислот являются неорганические кислоты, такие как хлористоводородная кислота, бромистоводородная кислота, серная кислота, перхлорная кислота или фосфорная кислота; кислоты Бренстеда, включая органические кислоты, такие как уксусная кислота, муравьиная кислота, щавелевая кислота, метансульфоновая кислот, п-толуолсульфоновая кислота, трифторуксусная кислота или трифторметансульфоновая кислот; и кислотные ионообменные смолы.
Соединение в виде свободной карбоновой кислоты, полученное таким способом, может быть извлечено и очищено обычными способами, например экстракцией, промывкой, сушкой и им подобными способами, и затем оно может быть использовано в последующих взаимодействиях.
Гидроксигруппа у получающегося соединения (которое содержит в своей молекуле солевую группу карбоновой кислоты, карбоновокислотную сложноэфирную группу или свободную карбоновокислотную группу) может защищаться предпочтительно защитной группой, способной отщепляться ин виво биологическими методами, такими как гидролиз. Реакционные условия, применяемые при введении этой защитной группы, аналогичны тем, которые применяются на стадии A5.
Если продукт представляет собой соединение формулы (II), содержащей две свободные гидроксигруппы, гидроксигруппы могут быть защищены одновременно диол-защищающей группой, такой как изопропилиденовая, бензилиденовая или этилиденовая группа, путем взаимодействия соединения с подходящим реагентом в присутствии кислотного катализатора.
Отсутствует какое-либо особое ограничение, касающееся природы реагента, используемого для введения диолзащищающей группы, и любой агент, обычно используемый для защиты диольной группы, в равной мере может быть использован и здесь. Примерами предпочтительных реагентов являются альдегидные производные, такие как бензальдегид; кетоновые производные, такие как ацетон; и диметоксисоединения, такие как 2,2-диметоксипропан или диметоксибензил.
Реакцию обычно и предпочтительно проводят в присутствии растворителя. Отсутствует какое-либо особое ограничение, касающееся природы растворителя, при условии, что он не оказывает отрицательного воздействия на ход реакции или на реагенты, участвующие в ней, и что он может растворять реагенты, по крайней мере, в некоторой степени. Примерами приемлемых растворителей являются галоидированные углеводороды, такие как хлористый метилен или хлороформ; простые эфиры, такие как диоксан или тетрагидрофуран; углеводороды, такие как гексан или пентан; ароматические углеводороды, такие как бензол или толуол; сложные эфиры, такие как этилацетат; и полярные растворители, такие как диметилформамид или ацетон.
Отсутствует какое-либо особое ограничение, касающееся природы используемого кислотного катализатора, и любая кислота, обычно используемая в качестве катализатора при проведении реакций этого типа, может быть в равной мере использована. Примерами предпочтительных кислотных катализаторов являются органические кислоты, такие как п-толуолсульфоновая кислота, камфорсульфоновая кислота и п-толуолсульфонат пиридиния; и неорганические кислоты, такие как хлористоводородная кислота.
Взаимодействие происходит в широком интервале температур, и точная температура не является критической, хотя предпочтительная температура меняется в зависимости от природы использованных кислотного катализатора и исходного соединения. Однако обычно реакцию удобно вести при температуре от 0oC и до 100oC. Время, необходимое для протекания взаимодействия, может также изменяться в широких пределах в зависимости от многих факторов, особенно от температуры и природы реагентов. Однако в большинстве случаев продолжительность реакции обычно составляет от 0,1 до 24 ч.
Если защищающая группа, способная отщепляться ин виво биологическими методами, используемая в виде карбоксизащищающей группы, представляет собой алкильную группу или аналогичную, соединение, содержащее карбоновокислотную солевую группу или свободную карбоновокислотную группу, может быть защищено следующими методами.
Метод 1.
В этом методе соединение, подлежащее защите, вводят во взаимодействие с соединением формулы R5″-X' (в которой R5″ представляет собой защищающую группу, способную отщепляться ин виво биологическими методами, включенную в определение R5, и X' представляет собой группу или атом, способные отщепляться в виде нуклеофильного остатка). Примерами групп и атомов, способных отщепляться в виде нуклеофильного остатка, является атомы галогена, также как атомы хлора, брома и иода; низшие алкансульфонилоксигруппы, такие как метансульфонилоксигруппа и этансульфонилоксигруппа; галоалкансульфонилоксигруппы, такие как трифторметансульфонилоксигруппа и пентафторэтансульфонилоксигруппа; и арилсульфонилоксигруппы, такие как бензолсульфонилоксигруппа, п-толуолсульфонилоксигруппа и п-нитробензолсульфонилоксигруппа. Примерами таких соединений являются алифатические ацилоксиметилгалогениды, такие как ацетоксиметилхлорид, пивалоилоксиметилбромид и пивалоилоксиметилхлорид; низшие алкоксикарбонилоксиалкильные галогениды, такие как этоксикарбонилоксиметилхлорид, изопропоксикарбонилоксиметилхлорид, 1-(этокси-карбонилокси)этилхлорид и 1-(этоксикарбонилокси)этилиодид; фталидилгалогениды и (5-метил-2-оксо-5-метил-1,3-диоксолен-4-ил)метилгалогениды.
Реакцию обычно предпочтительно ведут в присутствии растворителя. Отсутствует какое-либо особое ограничение, касающееся природы применяемого растворителя, при условии, что он не оказывает отрицательного воздействия на реакцию и на реагенты, участвующие в ней, и что он может растворять реагенты, по крайней мере, в некоторой степени. Примерами подходящих растворителей являются алифатические углеводороды, такие как гексан или гептан; ароматические углеводороды, такие как хлористый метилен, хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол и дихлорбензол; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и диэтиленгликолдиметиловый эфир; кетоны, такие как ацетон, метилэтиловый кетон, метилизобутиловый кетон, изофорон и циклогексанон; нитрилы, такие как ацетонитрил и изобутиронитрил; и амиды, такие как формамид, диметилформамид, диметилацетамид, N-метил-2-пирролидон, N-метилпирролидинон и гексаметилфосфорный триамид.
Реакция также идет в присутствии основания. Нет каких-либо особых ограничений, касающихся природы используемого основания, и любое основание, обычно используемое при проведении реакций этого типа, может быть использовано. Примерами предпочтительных оснований являются карбонаты щелочных металлов, такие как карбонат натрия, карбонат калия и карбонат лития; водородсодержащие карбонаты щелочных металлов, такие как бикарбонат натрия, бикарбонат калия и бикарбонат лития; гидриды щелочных металлов, такие как гидрид лития, гидрид натрия и гидрид калия; гидроксиды щелочных металлов, такие как гидроксид натрия, гидроксид калия, гидроксид бария и гидроксид лития; фториды щелочных металлов, такие как фторид натрия и фторид калия; алкоксиды щелочных металлов, такие как метоксид натрия, этоксид натрия, метоксид калия, этоксид калия, трет-бутоксид калия и метоксид лития; алкилтиолаты щелочных металлов, такие как метилтиолат натрия и этилтиолат натрия; органические основания, такие как N-метилморфолин, триэтиламин, трибутиламин, диизопропилэтиламин, дициклогексиламин, N-метилпиперидин, пиридин, 4-пирролидинопиридин, пиколин, 4-(N,N-диметиламино)пиридин, 2,6-ди(трет-бутил)-4-метилпиридин, хинолин, N,N-диметиланилин, N,N-диэтиланилин, 1,5-диазабицикло[4.3.0] нона-5-ен, 1,4-диазабицикло[2.2.2]октан и 1,8-диазабицикло[5.4.0]ундек-7-ен; металлоорганические основания, такие как бутиллитий, диизопропиламид лития и бис(триметилсилил)амид лития.
Взаимодействие проходит в широкой области температур, и точная температура не является критической. Обычно реакцию удобно вести при температуре от -20oC и до 120oC, более предпочтительно от 0oC до 80oC. Время, необходимое для взаимодействия, также может меняться в широких пределах в зависимости от многих факторов, а именно от температуры и природы реагентов. Однако в большинстве случаев продолжительность взаимодействия обычно составляет от 0,5 до 10 час.
Метод 2.
Этот метод включает взаимодействие незащищенного соединения формулы R5′-OH (в которой R5′ является такой, как определена ранее) в растворителе в присутствии этерифицирующего агента и каталитического количества основания. Реакцию проводят по методике, описанной при рассмотрении стадии A3 метода 2.
Метод 3.
Этот метод включает взаимодействие незащищенного соединения R5'-OH (в которой R5′ является такой, как определена ранее) в растворителе в присутствии галоидированного диэтилового эфира фосфорной кислоты, такого как диэтилхлорфосфат, и основания. Реакцию проводит по методике, описанной при рассмотрении стадии A3 метода 3.
Метод 4.
Этот метод может быть использован, когда защищающая группа представляет собой алкильную группу, и включает взаимодействие незащищенного соединения с соответствующим спиртом, используемым в качестве реагента, такого как метанол, этанол, пропанол и бутанол, в растворителе. Нет каких-либо особых ограничений, касающихся природы используемого растворителя, при условии, что он не оказывает отрицательного воздействия на реакцию и что он может растворять исходное вещество, по крайней мере, в некоторой степени. Примерами предпочтительных растворителей являются те же спирты, что и используемые в качестве реагентов; алифатические углеводороды, такие гексан и гептан; ароматические углеводороды, такие как бензол, толуол и ксилол; галоидированные углеводороды, такие как хлористый метилен, хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол и дихлорбензол; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметилоксиэтан и диэтиленгликолдиметиловый простой эфир; кетоны, такие как ацетон, метилэтиловый кетон, метилизобутилкетон, изофорон и циклогексанон; нитрилы, такие как ацетонитрил и изобутиронитрил; и амиды, такие как формамид, диметилформамид, диметилацетамид, N-метил-2-пирролидон, N-метилпирролидинон и гексаметилфосфорный триамид. Из этих веществ изобретатели предпочитают те же спирты, что и используемые в качестве реагентов. Реакцию ведут в присутствии кислотного катализатора. Нет каких-либо особых ограничений, касающихся природы используемого кислотного катализатора, и всякая кислота, обычно используемая в качестве катализатора при проведении обычных реакций этого типа, может быть в равной мере использована. Примерами предпочтительного кислотного катализатора являются неорганические кислоты, такие как хлористоводородная кислота, бромистоводородная кислота, серная кислота, перхлорная кислота и фосфорная кислота; кислоты Бренстеда, включая органические кислоты, такие как уксусная кислота, муравьиная кислота, щавелевая кислот, метансульфоновая кислота, п-толуолсульфоновая кислота, трифторуксусная кислота и трифторметансульфоновая кислота; кислоты Льюиса, такие как треххлористый бор, трехфтористый бор и трехбромистый бор; и кислотные ионообменные смолы.
Взаимодействие проходит в широком интервале температур, и точная температура реакции не является критической. Обычно реакцию удобно вести при температуре от 0o до 100oC, более предпочтительно от 20oC до 60oC. Время, необходимое для протекания взаимодействия, также может меняться в широких пределах в зависимости от многих факторов, а именно от температуры взаимодействия и природы реагентов. Однако, в большинстве случаев, продолжительность взаимодействия обычно составляет от 1 до 24 часов.
Метод 5.
Этот метод включает взаимодействие незащищенного карбоновокислотного соединения либо с
I) галоидирующим агентом, например пентахлоридом фосфора, хлористым тионилом или хлористым оксалилом, при подходящей температуре, например при комнатной температуре, в течение промежутка времени, например, от 30 мин и до 5 ч с получением соответствующего галоидангидрида кислоты, либо с
II) хлорформиатом, таким как метилхлорформиат или этилхлорформиат, в присутствии органического амина (такого как триэтиламин), что может быть осуществлено при той же температуре и в течение того же промежутка времени, что и в случае 1, выше, с получением соответствующего ангидрида кислоты; с последующей обработкой получающегося ангидрида кислоты или галоидангидрида кислоты подходящим спиртом или алкоксидом щелочного металла с получением требуемого сложного эфира. Для получения трет-бутилового эфира предпочтительно использовать трет-бутоксид калия.
Реакцию обычно и предпочтительно ведут в присутствии растворителя. Нет каких-либо особых ограничений, касающихся природы используемого растворителя, при условии, что он не оказывает отрицательного воздействия на ход реакции или на реагенты, участвующие в ней, и что он может растворять реагенты, по крайней мере, в некоторой степени. Примерами подходящих растворителей являются ароматические углеводороды, такие как бензол, толуол и ксилол; галоидированные углеводороды, такие как хлористый метилен и хлороформ; сложные эфиры, такие как этилацетат и пропилацетат; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан и диметоксиэтан; и нитрилы, такие как ацетонитрил. Реакцию можно также вести в присутствии основания, природа которого не играет критической роли, например в присутствии триэтиламина. Взаимодействие может протекать в широком интервале температур, и точная температура не является критической. Обычно реакцию удобно вести при температуре от -10oC и до 150oC, более предпочтительно примерно при комнатной температуре. Время, необходимое для взаимодействия, может также меняться в широких пределах в зависимости от многих факторов, а именно от температуры взаимодействия и от природы применяемых реагентов и растворителя. Однако, если реакцию ведут в предпочтительных условиях, оговоренных выше, тогда для взаимодействия обычно достаточно от 10 мин до 15 ч, предпочтительно от 30 мин до 10 ч.
Метод 6.
Этот метод включает взаимодействие незащищенного свободного карбоновокислотного соединения с диазоалканом, таким как диазометан или диазоэтан (обычно в эфирном растворе диазоалкана) при подходяще температуре, например приблизительно при комнатной температуре; однако, при необходимости, реакцию ведут при нагревании.
Или же в качестве исходного соединения может быть использован эфир карбоновой кислоты, при этом требуемое соединение может быть получено обычными способами, т. е. проведением переэтерификации соединением формулы R5′-OH, в которой R5′ является таким, как определен выше.
Если карбоксизащищающая группа, способная отщепляться ин виво биологическими методами, представляет собой группу амидного типа, реакция защиты может быть проведена по следующему методу.
Метод 7.
Этот метод включает превращение соли карбоновой кислоты или свободной карбоновой кислоты, которая может быть получена по способу, описанному выше, в галоидангидрид кислоты или в ангидрид кислоты по способу, описанному в методе 5, и затем включает взаимодействие галоидангидрида кислоты или ангидрида кислоты с соответствующим основанием, например с газообразным аммиаком или с диметиламином.
Метод 8.
Этот метод включает подвержение эфира карбоновой кислоты, который может быть получен по описанным выше методам 1 - 6, обычной эфирамидной обменной реакции.
Получение солей.
Реакции, которые ведут к получению соли карбоновой кислоты, могут быть проведены описанными ниже способами.
1. Металлические соли карбоновых кислот. Требуемая соль может быть получена контактированием свободной карбоновой кислоты с надлежащим соединением металла, например с гидроксидом металла или карбонатом металла, в водном растворителе.
Примерами предпочтительных водных растворителей являются сама вода и смесь воды с органическим растворителем, таким как спирт, например с метанолом или этанолом; или кетон, например ацетон. Особое предпочтение изобретатели отдают смеси воды с гидрофильным органическим растворителем.
Обычно реакцию предпочтительно вести примерно при комнатной температуре или, при необходимости, ее можно вести при нагревании.
2. Аминовые соли карбоновых кислот. Требуемая соль может быть получена контактированием свободной карбоновой кислоты с подходящим амином в присутствии водного растворителя.
Примерами предпочтительных водных растворителей являются сама вода или смесь воды с органическим растворителем, таким как спирт, например метанол или этанол; простой эфир, например тетрагидрофуран; или нитрил, например ацетонитрил. Из этих веществ изобретатели предпочитают водный раствор ацетона.
Обычно реакцию предпочтительно проводят при pH от 7,0 до 8,5 и при температуре ниже комнатной, в частности при температуре от 5oC и до 10oC. Реакция идет сразу же до завершения.
Или же требуемая соль может быть получена проведением соль-аминовой взаимообменной реакции, т. е. растворением металлической соли карбоновой кислоты, которая может быть получена по описанному выше п.1, в водном растворителе и затем добавлением соли минеральной кислоты требуемого амина (например, соли галогеноводородной кислоты, такой как хлористоводородная кислота). Реакция может быть проведена при тех же условиях, что и описанные выше.
3. Аминокислотные соли карбоновых кислот. Требуемая соль может быть получена контактированием свободной карбоновой кислоты с требуемой аминокислотной в водном растворителе.
Примерами предпочтительных водных растворителей являются сама вода и смесь воды с органическим растворителем, таким как спирт, например метанол или этанол; или простой эфир, такой как тетрагидрофуран.
Реакцию обычно ведут при нагревании, предпочтительно при температуре от 50oC до 60oC.
Получение лактона.
Требуемое лактоновое соединение может быть получено контактированием соединения карбоновой кислоты, полученного как описано выше, с каталитическим количеством кислоты.
Реакцию обычно предпочтительно проводят в присутствии растворителя. Нет каких-либо особых ограничений, касающихся природы применяемого растворителя, при условии, что он не оказывает отрицательного воздействия на ход реакции и на реагенты, участвующие в ней, и может растворять реагенты, по крайне мере, в некоторой степени. Примерами подходящих растворителей являются вода, простые эфиры, такие как тетрагидрофуран, диоксан, диметоксиэтан и диэтиленгликолдиметиловый простой эфир; кетоны, такие как ацетон и метилэтиловый кетон; нитрилы, такие как ацетонитрил и изобутиронитрил; амиды, такие как формамид, диметилформамид, диметилацетамид, N-метил-2-пирролидон и гексаметилфосфорный триамид; сульфоксиды, такие как диметилсульфоксид и сульфонан; или смеси одного или нескольких таких растворителей с водой.
Нет каких-либо ограничений, касающихся природы используемого кислотного катализатора, и любой кислотный катализатор, обычно используемый при проведении обычных реакций этого типа, может быть использован. Примерами предпочтительных кислотных катализаторов являются неорганические кислоты, такие как хлористоводородная кислота, бромистоводородная кислота, серная кислота, перхлорная кислота и фосфорная кислота; кислоты Бренстеда, включая органические кислоты, такие как уксусная кислота, муравьиная кислота, щавелевая кислота, метансульфоновая кислота, п-толуолсульфоновая кислота, трифторуксусная кислота и трифторметансульфоновая кислота; кислоты Льюиса, такие как хлористый цинк, тетрахлорид олова, треххлористый бор, трехфтористый бор и трехбромистый бор; и кислотные ионообменные смолы. Из этих веществ изобретатели предпочтение отдают неорганическим кислотам.
Реакция может протекать в широком интервале температур, и точная температура не является критической. Обычно реакцию ведут при температуре от -20oC до 170oC, предпочтительно от 0oC до 50oC. Время, необходимое для протекания взаимодействия, также может меняться в широких пределах в зависимости от многих факторов, а именно от температуры и природы применяемых реагентов и растворителя. Однако, если реакцию ведут в предпочтительных условиях, оговоренных выше, тогда продолжительность взаимодействия обычно составляет от 10 мин до одного дня.
По завершении реакции получающееся соединение формулы (XII) может быть извлечено и очищено с помощью любого подходящего сочетания различных видов способов извлечения и очистки, таких как описанные и проиллюстрированные примерами выше, особенно различных хроматографических методик. Примерами таких методик являются распределительная колонная хроматография с пропусканием очищаемого вещества через синтетический абсорбент, такой как сефадексТМ LH-20 ("Фармация, инк."), амберлитТМ XAD-11 ("Ром энд Хаас ко.") или диаионТМ HP-20 ("Мицубиси касеи корпорейшн"); ионообменная хроматография; гель-фильтрация с пропусканием очищаемого вещества через колонку с сефадексом; жидкостная хроматография с пропусканием очищаемого вещества через регулярно-фазовую или обращенно-фазовую колонку с насадкой из силикагеля или алкилированного силикагеля (желательно использовать высокопроизводительную жидкостную хроматографию); или любое сочетание этих хроматографических методик; требуемое соединение может быть затем элюировано использованием надлежащего элюирующего растворителя. В ином варианте продукт может быть эффективно экстрагирован органическим растворителем, таким как диэтиловый простой эфир, этилацетат или хлороформ.
Если требуемое соединение, полученное проведением описанных выше стадий, образуется в виде смеси стереоизомеров и необходимо провести выделение индивидуальных изомеров, то тогда каждый из изомеров может быть выделен и очищен обычными способами, описанным выше, в конце каждой реакции или в любое приемлемое время после завершения каждой реакции.
Реакционная схема B.
Другой возможный способ получения соединения, отвечающего настоящему изобретению, показан на реакционной схеме B (табл.4).
В приведенных выше формулах R5′, R6, R6a, R6b, R6′ и R7, являются такими, как определены ранее.
Реакционная схема B дает способ получения соединений формул (XVIII) и (XIX) настоящего изобретения, и альтернативный способ получения соединений формул (XI) и (XII), которые также являются соединениями настоящего изобретения.
Стадия B1.
На этой стадии соединение формулы (XIV) получают гидролизом сложноэфирной боковой цепи, находящейся в положении 8 в исходном соединении формулы (XIII), для чего используют основание в растворителе. Эта реакция по существу является такой же, что и реакция, описанная при рассмотрении стадии A1 реакционной схемы A, и она может быть проведена с использованием тех же реагентов и в тех же реакционных условиях.
Стадия B2.
На этой стадии лактоновое соединение формулы (XV) получают нейтрализацией соли гидроксикислоты формулы (XIV), предпочтительно в растворителе, содержащем один или два эквивалента кислоты, и затем циклизацией получающейся свободной кислоты. Эта реакция по существу является такой же, что описанная при рассмотрении стадии A2 реакционной схемы A, и может быть проведена с использованием тех же реагентов и в тех же реакционных условиях.
Стадия B3.
На этой стадии соединение с формулой (XVI) получают путем избирательной защиты гидроксигруппы, отличной от гидроксигруппы в положении 8 в соединении формулы (XV), группой R6′. Эта реакция по существу является такой же, что описанная при рассмотрении стадии A3 реакционной схемы A, и может быть проведена с использованием тех же реагентов и в тех же реакционных условиях.
Стадия B4.
На этой стадии соединение формулы (XVII) получают ацилированием гидроксигруппы в положении 8 в соединении формулы (XVI) группой R7. Реакция по существу является такой же, что описанная при рассмотрении стадии A4 реакционной схемы A, и может проводиться с использованием тех же реагентов и в тех же реакционных условиях.
Стадия B5.
На этой стадии соединение формулы (XVIII), которое представляет собой соединение настоящего изобретения, получают удалением гидроксизащищающей группы, представленной R6′, в соединения формулы (XVII) и затем, при необходимости, защитой получающейся гидроксигруппы еще одной защищающей группой, желательно способной отщепляться ин виво биологическими методами, такими как гидролиз. Эта реакция по существу является такой же, что описанная при рассмотрении стадии A5 реакционной схемы A, и может быть проведена с использованием тех же реагентов и в тех же реакционных условиях.
Стадия B6.
На этой стадии соединение формулы (XIX) получают гидролизом или сольволизом лактонового кольца соединения формулы (XVIII) с получением соли карбоновой кислоты или эфира карбоновой кислоты, и затем, при необходимости, продукт взаимодействия подвергают одной из следующих реакций:
1) получения свободной карбоновой кислоты;
2) защиты некоторых или всех свободных гидроксигрупп защищающими группами, желательно группами, способными отщепляться ин виво биологическими методами, такими как гидролиз;
3) защиты получающейся карбоксигруппы защищающей группой, желательно группой, способной отщепляться ин виво биологическими методами, такими как гидролиз, или получения других солей карбоновой кислоты и/или
4) при необходимости получения сплава лактонового соединения замыканием кольца. Реакцию проводят, следуя методике, описанной при рассмотрении стадии 6.
Стадии B7, B8 и B9.
На этих стадиях соединения формул (XI) и (XII) получают введением стереоспецифически гидроксигруппы в положении 6 карбоновокислотного соединения формулы (XIX), его фармацевтически приемлемой соли или сложного эфира, или лактонового соединения формулы (XVIII) ферментным гидролизом. Это может осуществляться с использованием процедуры, описанной ниже в разделе "Получение биологическими методами". После этого, при необходимости, могут проводиться следующие реакции:
1) гидролиза или сольволиза;
2) получения свободной карбоновой кислоты;
3) защиты некоторых или всех свободных гидроксигрупп защищающими группами, желательно группами, способными отщепляться ин виво биологическими методами, такими как гидролиз, группы могут быть одними и теми же или отличными друг от друга.
4) защиты получающейся карбоксигруппы защищающей группой, которая предпочтительно способна отщепляться ин виво биологическими методами, такими как гидролиз, или получение иных солей карбоновой кислоты и/или
5) замыкания кольца с получением состава лактонового соединения.
Эти реакции по существу являются такими же, что и реакции, описанные при рассмотрении стадии A6 реакционной схемы A, и могут проводиться с использованием тех же реагентов и в тех же реакционных условиях.
Реакционная схема C.
Эта схема дает альтернативный способ получения соединения формулы (XI), используемого в качестве промежуточного соединения по реакционной схеме A, и соединения формулы (XVIII), используемого в качестве промежуточного соединения по реакционной схеме B (табл.5).
В приведенных формулах (табл. 5) R6, R6a и R7 являются такими же, что определены ранее.
Соединение формул (XI) и (XVIII), используемые в качестве промежуточных соединений, могут быть получены ацилированием всех гидроксигрупп соединения формулы (VIII) или (XV) группой R7 с получением соединения формулы (XX) или (XXI). Эта реакция по существу является такой же, что описанная при рассмотрении стадии A4 реакционной схемы A, и может быть проведена с использованием тех же реагентов и в тех же реакционных условиях. Одну или две защищающие группы, отличных от ацилированной гидроксигруппы в положении 8 затем селективно удаляют, следуя методике, изложенной в описании патента Великобритании N 2255974 A, после чего, при необходимости, любую или обе деблокированные группы защищают защищающей группой, желательно группой, способной отщепляться ин виво биологическими методами, такими как гидролиз, причем группы могут быть одними и теми же или отличными друг от друга. Эта реакция по существу является такой же, что и описанная при рассмотрении стадии A5 реакционной схемы A, и может быть проведена с использованием тех же реагентов в тех реакционных условиях.
Реакционная схема D.
Эта схема дает альтернативный метод получения соединений формулы (I') и (XXII) ферментацией (табл.6)
В приведенных формулах (табл.6) R5, R6, R6a и R6b являются такими же, что определены выше.
На стадии D1 соединение формулы (I'), являющиеся соединением настоящего изобретения, получают инокулированием микроорганизма, способного продуцировать упомянутое соединение, который принадлежит к роду Penicillium. Это может осуществляться с использованием методики, описанной ниже в разделе "Получение биологическими методами".
После чего, при желании, проводят одну или несколько следующих реакций:
1) гидролиза или сольволиза;
2) получения свободной карбоновой кислоты;
3) защиты некоторых или всех гидроксигрупп защищающими группами, желательно группами, способными отщепляться ин виво биологическими методами, такими как гидролиз, которые могут быть одними и теми же или отличными друг от друга;
4) защиты получающейся гидроксигруппы защищающей группой, которая, желательно, способна отщепляться ин виво биологическими методами, такими как гидролиз, или получения иных солей карбоновой кислоты и/или
5) при необходимости, снова замыкания кольца с образованием лактонового соединения.
Соединение формулы (XIII), используемое в качестве исходного вещества по схеме B, может быть получено химически по следующей методике, описанной в любом из приведенных ниже литературных источников:
(1) D.J. Clive et al., J. Am. Chem. Soc., 112, 3018 (1990);
(2) C.T. Hsu et al., J. Am. Chem. Soc., 105, 593 (1983);
(3) n. n. Jirotra et al., Tetrahedron Lett., 23 5501 (1982); ibid., 24, 3687 (1983) and ibid., 25 5371 (1983);
(4) M. Hirama te al., J. Am. Chem. Soc., 104, 4252 (1982);
(5) P.A. Jrieco et al., J. Am. Chem. Soc., 108, 5908 (1986);
(6) T. Rosen et al., J. Am. Chem. Soc., 107, 3731 (1985);
(7) J.E. Keck et al., J. Am. Chem. 51, 2487 (1986);
(8) A.P. Kozikowski et al., J. Am. Chem., 52, 3541 (1987).
(9) S.J. Danishefsky et al., J. Am. Chem. Soc., 111, 2599 (1989).
По методикам, описанным в Японской патентной заявке N Sho 56-12114 и в Японской патентной заявке Sho 51-136885, исходные соединения формул (XII) и (XV), применяемые в реакционных схемах B и C, могут быть получены микробиологически. На стадии D1 реакционной схемы D оба соединения могут быть приготовлены одновременно.
Правастатин, который может использоваться в качестве исходного вещества, может быть получен ферментативно проведением стереоизбирательного гидроксилирования соединения формулы (XIII) в положении 6 с получением соединения с 6 β-гидроксигруппой по методике, раскрытой в Японской патентной публикации N 61-13699 или в стадиях B7, B8 и B9.
Эпимер в положении 6 правастатина, т.е. соединение с 6-гидроксигруппой в α-конфигурации, может быть также использовано в качестве исходного вещества на стадии A1. Это исходное соединение может быть получено с помощью стереоизбирательного гидроксилирования в положении 6 соединения формулы (XIII), аналогично синтезу правастатина, следуя методике, раскрытой в Японской патентной публикации N Sho 61-13699 или при рассмотрении стадий B7, B8 и B9.
Карбоновая кислота формулы R7-OH, которую используют в качестве исходного вещества в способе настоящего изобретения, может быть легко получена известными методами, например методом, описанным автором P.E. Pfeffer, J. Org. Chem., 37,451 (1972)).
Получение биологическими методами.
Некоторые соединения настоящего изобретения могут быть также получены биологическими методами, как это более подробно описано ниже.
Получение соединений формулы (I')
Например, те соединения формулы (IV), которые имеют 2-метилпентаноилоксигруппу в положении 8, т.е. соединения формулы (I')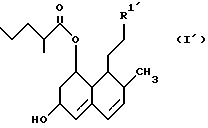
где R1′ представляет собой группу формулы (II') или (III')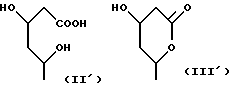
могут быть получены культивированием микроорганизма рода Penicillium в питательной для него среде и отделением упомянутого соединения формулы (I') от питательной среды. Этот способ также составляет часть настоящего изобретения.
Нет каких-либо особых ограничений, касающихся вида микроорганизма, используемого для получения соединения формулы (I'), при условии, что он принадлежит к роду Penicillium и обладает способностью продуцировать соединение формулы (I'). Примером штамма микроорганизма, способного продуцировать соединение формулы (I'), является штамм Thom Sank 13380 вида Penicillium citrinum, который принадлежит к роду Penicillium и который депонирован по Будапештскому договору в Научно-исследовательский институт ферментации, Агентство промышленной науки и технологии, Министерство внешней торговли и промышленности, Токио, Япония, под инвентарным номером FERM BP-4129, дата депонирования 22 декабря 1992 г.
Микробиологические свойства штамма SANK 13380 являются следующими.
Колонии, выращенные на среде Чапека (Czapek) из дрожжевого автолизатного агара, составляли в диаметре 1,8 см после выращивания в течение 7 дней при 25oC. Цвета поверхности были от белого (1 A 1) до светло-желтого (2 A 4), и поверхность была покрыта белыми хлопьевидными воздушными гифами. Обратная сторона окрашена от белого цвета (1 A 1) до светло-желтого цвета (2 A 4), и наблюдались радиальные складки. Не обнаруживались ни экссудаты, ни растворимые пигменты.
Колонии, выращенные на среде из ангара с солодовым экстрактом, составляли в диаметре 1,3 см (после выращивания при 25oC в течение 7 дней). Поверхность окрашена в бледно-желтый цвет (2 A 3), и внешний вид поверхности изменялся от бархатистого до порошкообразного. Обратная сторона окрашена в коричневато-оранжевый цвет (7 C 7).
Колонии, выращенные на среде из 25%-ного (по весу в расчете на объем) глицеринового нитратного агара, составляли в диаметре 1,6 см (после выращивания при 25oC в течение 7 дней). Окраска поверхности изменялась от белой (1 A 1) до желтовато-белой (1 A 2), и поверхность покрыта хлопьевидноопушенной гифой. Обратная сторона окрашена в бледно-желтый цвет (2 A 3).
Не наблюдали никакого роста ни на одной из этих сред при 5oC или 37oC.
Поверхности у конидиофор были гладкими и бимутовчатыми. Метулы цилиндрические с небольшой пузырчатостью и имеют размер 9 - 15 на 3 - 4 мкм. Фиалиды ампулообразные и имеют размер 8 - 10 на 3 - 4 мкм. Конидии шариковидные и их поверхности гладкие с проявлением небольшой шероховатости при значениях диаметра от 2,5 до 4 мкм.
Из сравнения этих свойств со свойствами, наблюдаемыми у известных видов следует, что свойства этого штамма соответствуют свойствам штамма Thom вида Penicillium citrinum, описанного Питтом (J.I.Pitt in "The genus Penicillium and its teleomorpholic states, Eupenicillium and Falaromyces", p.634, Academic Press (1979)). В соответствии со сказанным этот штамм идентифицирован как Penicillium citrinum Thom.
При описании цветовых тонов следовали рекомендации (A.Kornerup) и Wansher "Methuen Handbook of Colour" 3-е изд. (1978) опубликовано Eyre Methuen (Лондон).
Следует заметить, что штамм SANK 13380 или любой другой штамм, способный продуцировать соединение формулы (I'), может быть субкультивирован или биотехнологически изменен или модифицирован с образованием микроорганизма с иными характеристиками. Единственное требование состоит в том, чтобы получающийся микроорганизм обладал способностью продуцировать требуемое соединение. Изменения могут происходить естественным или искусственным образом под воздействием, например, ультрафиолетового излучения, высокочастотных волн, радиации при химическом воздействии с образованием мутагенов.
Такие изменения и модификации могут принимать любую желаемую форму, и они могут быть следствием действия таких факторов как, например, условия выращивания. Штаммы могут быть модифицированы выращиванием и подвергнуты селекции с проявлением таких характеристик, как усиленный рост или рост при пониженных/повышенных температурах.
Биотехнологические модифицирования обычно носят целенаправленный характер и ими могут быть приданы заданные характеристики, такие как биостатическая резистентность или восприимчивость или сочетание таковых характеристик, чем обеспечивается поддержание чистоты культуры или достигается очистка культур, особенно затравочных культур, время от времени.
Другие характеристики, которые могут быть приданы генетическим манипулированием, представляют собой любые характеристики, допустимые у видов рода Penicillium. Например, могут быть удалены любые плазмиды естественного происхождения. К предпочтительным плазмидам относятся те из них, которые придают ауксотрофию. Плазмиды могут быть получены из любого подходящего источника или они могут быть сконструированы выделением естественно встречающейся плазмиды рода Penicillium и встраиванием требуемого гена или генов, взятых из еще одного источника. Природные плазмиды могут быть также модифицированы любым другим способом, который может представляться желательным.
Любой такой модифицированный штамм может быть применен в способе настоящего изобретения, при условии, что штамм обладает способностью продуцировать соединение формулы (VI), что может быть легко установлено проведением простого и обычного эксперимента.
Для получения соединения формулы (VI) из культуры надлежащего микроорганизма такой микроорганизм следует подвергнуть ферментации в надлежащей среде. Такие среды обычно являются хорошо известными в этой области техники и часто они относятся к типу, широко используемому при продуцировании других продуктов ферментации.
В типичном случае необходимо, чтобы среда включала сочетание источника углерода, источника азота и одной или нескольких неорганических солей, приемлемых для надлежащего микроорганизма. Минимальное требование к среде состоит в том, что она должна содержать такие ингредиенты, которые являются существенными для роста микроорганизма.
Приемлемыми источниками углерода являются всякие углеродсодержащие вещества, которые могут быть усвоены микроорганизмом, например карбогидраты (углеводы), такие как глюкоза, фруктоза, мальтоза, лактоза, сахароза, крахмал, маннит, декстрин, глицерин, густая солодовая патока, меласса, черная патока, овсяная мука, ржаная мука, кукурузный крахмал, картофельный крахмал, кукурузная мука, соевая мука или солодовый экстракт; масла или жиры, такие как соевое масло, хлопковое масло, оливковое масло, рыбий жир или свиное сало; органические кислоты, такие как лимонная кислота, аскорбат натрия, яблочная кислота, уксусная кислота, фумаровая кислота, винная кислота, янтарная кислота или глюконовая кислота; спирты, такие как метанол, этанол, пропанол, изопропанол, бутанол, изобутанол или трет-бутанол; и аминокислоты, такие как глутаминовая кислота. Эти вещества могут быть использованы индивидуально или может быть использована смесь, приготовленная из любых двух или большего числа веществ. Типичные количества находятся в диапазоне примерно от 1 до 10% (по весу в расчете на объем) от количества среды, хотя количество может изменяться по желанию и в соответствии с требуемым результатом.
Приемлемыми источниками азота являются любые азотсодержащие вещества, которые могут быть усвоены микроорганизмом, например вещества, содержащие белок, или иной легко усваиваемый источник азота. Представительными примерами источника азота являются органические источники азота животного и растительного происхождения и ими могут быть экстракты, полученные из таких природных источников, как соевая мука, пшеничные отруби, пшеничные проростки, арахисовая мука, мука из хлопковых семян, хлопковое масло, выделенный соевый белок, казаминокислота (казеиновая аминокислота), казеиновый гидролизат, фермамин, рыбная мука, кукурузный экстракт, пептон, мясной экстракт, дрожжи, дрожжевой автолизат, дрожжевой экстракт, солодовый экстракт и мочевина; аминокислоты, такая как аспарагиновая кислота, глутамин, цистин или аланин; аммониевые соли, такие как сульфат аммония, нитрат аммония, хлорид аммония или фосфат аммония; и неорганические азотные соединения, такие как нитрат натрия или нитрат калия. Как и в случае источника углерода, они могут применяться индивидуально или в любом сочетании. В типичных случаях приемлемые количества находятся в диапазоне примерно от 0,2 до 6% (по весу в расчете на объем) от количества среды.
Приемлемыми питательными неорганическими солями являются соли, которые дают следовые элементы, а также основную составляющую соли. Желательно, чтобы соль была источником таких ионов, как натрий, калий, магний, аммоний, кальций, фосфат, сульфат, хлорид или карбонат, находящиеся в усваиваемой форме, и желательно, чтобы в следовых количествах содержались такие металлы, как молибден, бор, медь, кобальт, марганец и железо. Примерами подходящих соединений являются хлорид натрия, хлорид марганца, хлорид кобальта, хлорид калия, хлорид кальция, карбонат кальция, алюминиевокалиевый сульфат, сульфат марганца, сульфат меди (2), сульфат кобальта, сульфат цинка, сульфат железа (2), сульфат магния, первичный кислый фосфат калия, вторичный кислый фосфат калия, вторичный кислый фосфат натрия или молибдат алюминия. Кроме того, в любой приемлемой комбинации могут быть использованы всякие иные добавки, необходимые для роста микроорганизма и способствующие образованию соединения с формулой (I').
Добавление соединения, серы, усваиваемой микроорганизмом из среды, иногда может усиливать продуцирование требуемого соединения. Подходящими соединениями серы являются неорганические соединения серы, к которым относятся сульфаты, такие как сульфат цинка, сульфат меди (2), сульфат железа (2) или сульфат аммония; тиосульфаты, такие как тиосульфат аммония; и сульфиты, такие как сульфит аммония; или органические серные соединения, к которым относятся серусодержащие аминокислоты, такие как цистин, цистеин или L-тиазолин-4-карбоновая кислота; сульфатные соединения тяжелых металлов, такие как сульфат железа (2) или сульфат меди (2); витамины, такие как витамин В1 или биотин; и факторы, содействующие росту бактерий, такие как тиамин.
К среде может быть добавлено пеногасящее вещество, такое как силиконовое масло, полиалкиленгликолевый эфир, растительное масло, или надлежащее поверхностно-активное вещество. Такая добавка может оказаться особенно уместной, когда микроорганизм подвергают ферментированию в виде жидкой культуры.
Желательно, чтобы величина pH среды с культурой, предназначенной для культивирования штамма Penicillium citrinum Thom SANK 13380, когда его используют для продуцирования соединения формулы (I'), поддерживалась в области от 5,0 до 8,0, причем более желательно, чтобы величина pH находилась в области от 6,0 до 7,0, хотя единственное требование сводится к тому, что величина pH не должна препятствовать росту микроорганизма или отрицательно и необратимо воздействовать на качество конечного продукта.
В общем случае штамм Penicillium citrinum Thom SANK 13380 выращивают при температурах, находящихся от 15oC до 35oC, и рост идет хорошо при температурах от 22oC до 30oC. Другие температуры, не попадающие в пределы этих диапазонов, могут оказаться применимыми при разработке штамма, который может расти при пониженных или повышенных температурах, или для других специальных целей, как хорошо известно в данной области техники. Для продуцирования соединения формулы (I') предпочтительная температура составляет от 15oC до 35oC, более предпочтительно от 22oC до 26oC и лучше всего примерно 24oC.
Отсутствует какое-либо особое ограничение, касающееся методики культивирования, используемой для получения соединения формулы (I'), и здесь может быть использован любой метод работы с культурой, обычно используемый для проведения бактериального роста. Однако соединение формулы (I') идеально получается при аэробном выращивании и могут быть применены всякие приемлемые способы аэробного выращивания, такие как способ твердофазного выращивания, способ с перемешиваемой культурой, способ со стационарной культурой, выращивание со встряхиванием или выращивание с перемешиванием за счет аэрации.
Если выращивание проводят в небольшом масштабе, то тогда предпочтение обычно отдают способу ферментирования со встряхиванием культуры, проводя выращивание при температурах от 20oC до 30oC в течение нескольких дней, причем более желательно выращивание проводить примерно при 24oC.
Для начала ферментативного выращивания в предпочтительном варианте применяют исходный инокулюм, приготовленный в одну или две стадии, например, в колбе Эрленмейера, которая предпочтительно снабжена отбойными перегородками (стекой, направляющей поток воды). Для культуральной среды источник углерода и источник азота могут быть использованы в сочетании. Колбу с затравкой встряхивают в термостатированном инкубаторе при подходящей температуре, например, от 20 до 30oC, более предпочтительно от 22oC до 26oC, наиболее предпочтительно примерно 24oC, в течение подходящего промежутка времени, обычно от 2 до 7 дней или до тех пор, пока не замечается достаточный рост, предпочтительно от 3 до 5 дней. Получающаяся затравочная культура может быть затем использована для инокуляции второй затравочной культуры или производственной культуры. Если проводят вторую затравку, то это может быть сделано аналогичным образом и она может быть частично использована для инокулирования продуцирующей среды. Колбу, в которой инокулируется затравочная культура, встряхивают в течение надлежащего промежутка времени, например от 2 до 7 дней или до тех пор, пока не достигается максимальное продуцирование, при подходящей температуре, например при 24oC. После завершения инкубации содержимое колбы может быть собрано центрифугированием или фильтрованием.
Если культура должна быть получена в крупном масштабе, то тогда культивирование желательно вести в ферментере с перемешиванием при аэрации. В этом варианте питательная среда может быть приготовлена в ферментере. Среду сначала подвергают стерилизации при приемлемой высокой температуре, например примерно при 120oC, после чего ее охлаждают и затравливают инокулюмом, выращенным ранее в среде, подвергнуто стерилизации. Выращивание желательно вести при температуре от 20oC до 26oC, наиболее предпочтительно от 22oC до 24oC, при перемешивании и аэрации. Эта методика является пригодной для получения большого количества соединения.
Количество соединения формулы (I'), продуцируемое культурой по мере прохождения времени, может контролироваться взятием пробы и оценкой содержания соединения формулы (I') посредством, например, высокопроизводительной жидкостной хроматографии. Соединение формулы (I') может существовать как в лактоновой форме, таки в гидроксиформе, и оно обычно продуцируется в виде смеси этих форм. Можно определить количества каждой из форм, образовавшихся за один и тот же промежуток времени. В общем случае продуцируемое количество соединения формулы (I') достигает максимума по истечении промежутка времени между 72 и 300 часами.
Соединение формулы (I'), продуцированное культурой, находится как в фильтрате культуры, так и в бактериальных клетках. Оно может существовать либо в гидроксикислотной форме, либо в лактоновой форме, каждая из которых может переходить из одной в другую. Кроме того, гидроксикислотная форма может образовывать соответствующую соль, будет устойчивой.
Следовательно, соединение формулы (I') может быть экстрагировано и собрано прямым использованием этого свойства в сочетании с другими свойствами, например, как проиллюстрировано ниже.
Метод 1.
Бактериальные клетки и другие твердые материалы, находящиеся в среде, отделяют центрифугированием или фильтрованием с использованием фильтрующего материала, такого как диатомовая земля, с разделением на надосадочную жидкость и бактериальные клетки.
1. Надосадочная жидкость. Лактоновое кольцо в лактоновой форме соединения формулы (I'), существующего в надосадочной жидкости, подвергают гидролизу в щелочных условиях (предпочтительно при pH 12 и более), в результате чего происходит дециклизация, и все соединения формулы (I') переходит в гидроксикислотную форму. Соль затем превращается в соответствующую свободную гидроксикислоту путем осторожного подкисления; затем соединение формулы (I') получают из этой смеси в виде свободной гидроксикислоты, экстракцией несмешиваемым с водой органическим растворителем, например алифатическим углеводородом, таким как гексан или гептан; ароматическим углеводородом, таким как бензол, толуол или ксилол; галоидированным углеводородом, таким как хлористый метилен, хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол или дихлорбензол; простым эфиром, таким как диэтиловый эфир или диизопропиловый эфир; или сложным эфиром, таким как этилформиат, этилацетат, пропилацетат, бутилацетат или диэтилкарбонат. Эти растворители могут использоваться индивидуально или в виде смеси двух или более растворителей.
2. Бактериальные клетки. Бактериальные клетки смешивают со смешиваемым с водой органическим растворителем, например спиртом, таким как метанол или этанол; кетоном, таким как ацетон; нитрилом, таким как ацетонитрил или диизобутиронитрил; амидом, таким как диметилформамид, диметилацетамид, N-метил-2-пирролидон, N-метилпирролидинон или гексаметилфосфорный триамид. Конечная концентрация бактериальных клеток в получающейся смеси составляет предпочтительно от 50% до 90%. Полученную смесь предпочтительно затем обрабатывают по способу, описанному выше применительно к надосадочной жидкости, получая свободную гидроксикислоту.
Метод 2.
Среду с культурой обрабатывают в щелочных условиях (предпочтительно при pH 12 или более) при нагревании или при комнатной температуре, для разрушения клеток и гидролизуют с раскрытием лактонового кольца молекулы. В таком случае все соединение формулы (I') переходит в форму гидроксикислотной соли. Соединение формулы (I'), в свободной гидроксикислотной форме, получают после превращения солевой формы в его соответствующую свободную гидроксикислотную форму, с помощью обработки, аналогичной описанной выше при рассмотрении надосадочной жидкости в методе 1.
Полученная свободная гидроксикислотная форма может растворяться в виде соли в водном растворе основания щелочного металла, например гидроксида щелочного металла, такого как гидроксид натрия. Кроме того, свободная гидроксикислотная форма может быть превращена в солевую форму, которая является легко получаемой и наиболее устойчивой.
Или же получающаяся свободная гидроксикислотная форма может быть превращена в лактоновую форму дегидратацией при нагревании или циклизацией в органическом растворителе.
Выделение и очистка свободной гидроксикислотной формы, гидроксикислотной солевой формы и лактоновой формы, полученных таким способом, могут быть осуществлены обычными способами, широко используемыми для выделения и очистки органических соединений. Примерами таких методов являются метод с использованием синтетического адсорбента, такой как распределительная хроматография с использованием носителя, например, такого, как сефадекс LH-20 (торговая марка материала фирмы "Фармация"), амберлит XAD-11 (торговая марка материала "Ром энд Хаас") или диаион HP-20 (торговая марка материала "Мицубиси кемикал индастри"). Альтернативно они могут выделяться и очищаться с использованием обычно-фазовой или обращенно-фазовой колонной хроматографии с применением силикагеля или алкилированного силикагеля (предпочтительно высокопроизводительной жидкостной хроматографии, после чего проводят элюирование подходящим растворителем.
Лактоновая форма может быть также очищена применением адсорбционной колонной хроматографии с использованием носителя, такого как силикагель, оксид алюминия или флорисил (торговая марка носителя магнийсиликагельного типа).
Примерами растворителей, которые могут применяться в качестве элюента, являются алифатические углеводороды, такие как гексан, гептан, лигроин или петролейный эфир; ароматические углеводороды, такие как бензол, толуол или ксилол; галоидированные углеводороды, такие как хлористый метилен, хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол или дихлорбензол; сложные эфиры, такие как этилформиат, этилацетат, пропилацетат, бутилацетат или диэтилкарбонат; и простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан или диэтиленгликолдиметиловый простой эфир.
Или же соединения могут быть получены пропусканием экстрагированного раствора через колонку с адсорбентом для удаления примесей или адсорбцией свободной гидроксикислотной формы на такой колонке с последующим элюированием водным спиртом, таким как водный метанол, водный этанол, водный бутанол или водный изопропанол; или водным кетоном, таким как водный ацетон. Приемлемым адсорбирующим веществом, которое может быть применено, является активированный уголь или адсорбирующая смола, такая как амберлит XASD-2 или XAD-4 (торговое название продукта фирмы "Ром энд Хаас") или диаион HP-10; HP-20, CHP-20, HP-50 (торговая марка продукта "Мицубиси кемикал индастри").
Свободная гидроксикислота и соль гидроксикислоты могут быть превращены друг в друга обычными способами и подвергнуты очистке в любой требуемой форме.
Гидроксилирование соединения формулы (Ib) в соединение формулы (Ia)
Соединение формулы (Ib)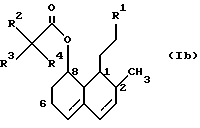
в которой R1 является такой же, как определен ранее, или соответствующее соединение, в котором реакционноспособные группы защищены, может быть превращено в соединение формулы (Ia)
в которой R1 является такой же, что и определенная ранее, или в соответствующее соединение, в котором реакционноспособные группы защищены с помощью гидролизирующего фермента.
Гидролизирующий фермент может быть получен с использованием микроорганизма рода, выбранного из группы, состоящей из Amycolata, Nocardia, Syncephalastrum, Mucor, Rhizopus, Zygorynchus, Circinella, Actinomucor, Jongronella, Phycomyces, Absidia, Cunninghamella, Mortierella, Pychpoporus (прежнее родовое название: Trametes), Streptomyces и Rhizoctonio.
Этот гидролиз может быть проведен любым следующим методом:
метод 1 - включает добавление соединения формулы (Ib) к жидкой среде в ходе культивирования конвертирующих микроорганизмов и затем продолжение культивирования;
метод 2 - включает контактирование соединения формулы (Ib) с выращенными клетками, собранными из жидкой среды с культурой упомянутого микроорганизма; или
метод 2 - включает контактирование соединения формулы (Ib) со свободным от клеток экстрактом, полученным из упомянутого микроорганизма.
В любом из этих методов микроорганизм культивируют в условиях, пригодных для достижения максимального продуцирования и максимальной эффективности действия фермента в подходящей среде выращивания. Состав среды может быть таким же, что и описанный выше в связи с культивированием микроорганизмов рода Penicillium.
Нет каких-либо особых ограничений, касающихся вида используемого микроорганизма, при условии, что он способен вводить гидроксигруппу в положение 6 соединения формулы (Ib). Примерами таких микроорганизмов являются:
грибы класса Zygomycetes: родов Syncephalastrum, Mucor, Rhezopus, Zygorynchus, Circinella, Actinomucor, Jongronella, Phycomyces, Absidia, Cunninghamella и Mortierella;
грибы других классов, отличных от Zygomycetes: родов Pychnoporus (прежнее родовое название: Frametes) и Rhizoctonia;
актиномицеты: родов Amycolata, Nocardia и Streptomyces; желательно
штаммы, принадлежащие к роду Syncephalastrum, включая:
Cyncephalastrum racemosum (Cohn) Schroeter SANK 91872 (FERM BP-4107); Syncephalastrum nigricans Vuillemin SANK 42372 IFO 4814 (FERM BP-4106); Syncephalastrum nigricans SANK 42172 (FERM P-6041); Syncephalastrum nigricans SANK 42272 (FERM P-6042); и Syncephalastrum racemosum IFO 4828;
штаммы, принадлежащие к роду Mucor, включая:
Mucor hiemalis Wehmer SANK 36372, IFO 5834 (FERM BP-4108); Mucor hiemalis f. hiemalis IFO 5303; Mucor hiemalis f. hiemalis IFO 8567; Mucor hiemalis f. hiemalis IFO 8449; Mucor hiemalis f. hiemalis IFO 8448; Mucor hiemalis f. hiemalis IFO 8565; Mucor hiemalis f. hiemalis CBS 117.08; Mucor hiemalis f. hiemalisCBS 109.19; Mucor hiemalis f. hiemalis CBS 200.28; Mucor hiemalis f. hiemalis CBS 242.35; Mucor hiemalis f. hiemalis CBS 110.19; Mucor hiemalis f. hiemalis CBS 201.65; Mucor bacilliformis NRRL 2346; Mucor circinelloides f. circenelloides IFO 4554; Mucor circinelloides f. circenelloides IFO 5775; Mucor hiemalis f. corticolus SANK 34572 (FERM P-5913); Mucor dimorphosporus IFO 4556; Mucor fragillis CBS 23635; Mucor genevesis IFO 4584; Mucor globosus SANK 35472 (FERM P-5915); и Mucor circinelloides f. griseocyanus IFO 4563;
штаммы, принадлежащие к роду Rhizopus, включая: Rhizopus chinensis IFO 4772; Rhizopus circinans ATCC 1225; и Rhizopus arrhizus ATCC 11145;
штаммы, принадлежащие к роду Zygorynchus, включая: Zygorynchus moelleri IFO 4833;
штаммы, принадлежащие к роду Circinella, включая: Circinella muscae IFO 4457; Circinella umbellata IFO 4452; и Circinella umbellata IFO 5842;
штаммы, принадлежащие к роду Actinomucor, включая: Actinomucor elegans ATCC 6476;
штаммы, принадлежащие к роду Jongronella, включая: Jongronella butleri IFO 8080;
штаммы, принадлежащие к роду Phycomyces, включая: Phycomyces blakesleeanus SANK 45172 (FERM P-5914);
штаммы, принадлежащие к роду Absidia, включая Absidia coerulea IFO 4423; и Absidia glauca var.paradoxa IFO 4431;
штаммы, принадлежащие к роду Cunninghamella, включая Cunninghamella echinulata IFO 4445; Cunninghamella echinulata IFO 4444; и Cunninghamella echinulata ATCC 9244;
штаммы, принадлежащие к роду Mortierella, включая: Mortierella isabellina IFO 6739;
штаммы, принадлежащие к роду Amycolata, включая: Amycolata autotropica SANK 62981 (FERM BP-4105); Amycolata autotropica SANS 62781 (FERM P-6181); Amycolata autotropica subsp. canberrica subsp. nov. SANK 62881 (FERM P-6182); и Amycolata autotropica IFO 12743;
штаммы, принадлежащие к роду Nocardia, включая: Nocardia asteroides IFO 3424; Nocardia farcinica ATCC 3318; и Nocardia coeliaca ATCC 17040;
штаммы, принадлежащие к роду Pychnoporus, включая: Pychnoporus coccineus SANK 11280 (FERM P-5916);
штаммы, принадлежащие к роду Streptomyces, включая: Streptomyces carbophilus SANK 62585 (FERM BP-4128); Streptomyces roseochromogenus IFO 3363; Streptomyces roseochromogenus IFO 3411; и Streptomyces halstedii IFO 3199;
штаммы, принадлежащие к роду Rhizoctonia, включая: Rhizoctonia solani SANK 22972 (FERM P-5917).
Из них наиболее предпочтительными микроорганизмами являются:
Amycolata autotrophica SANK 62981 (FERM BP-41-5);
Syncephalastrum racemosum (Cohn) Schroeter SANK 41872 (FERM BP-4107);
Syncephalastrum nigricans Vuillemin SANK 42372 (FERM BP-4106);
Mucor hiemalis wehmer SANK 36372 (FERM BP-4108) и
Streptomyces carbophilus SANK 62585 (FERM BP-4128).
Микроорганизмы, описанные выше, депонированы в коллекции культур Научно-исследовательского института ферментации, Агентства промышленной науки и технологии, Министерства внешней торговли и промышленности, или они могут быть получены из официальных агентств (IFO, CBS, NRRL и ATCC (Американская коллекция типовых культур микроорганизмов)) без ограничений доступности. Следующие примеры, в которых использованы вышеприведенные грибы, приведены для того, чтобы настоящее изобретение могло быть лучше понято.
Следует заметить, что штаммы, упомянутые выше, или любые другие штаммы, способные проявлять аналогичную активность, могут быть субкультивированы, или биотехнологически изменены или модифицированы с получением организма с иными характеристиками. Единственное требование состоит в том, чтобы получающийся микроорганизм был способен продуцировать требуемое соединение. Могут происходить изменения естественным или искусственным способом.
Такие изменения и модификации могут принимать любую требуемую форму или же они могут основываться на таких соображениях, как, например, условия выращивания. Штаммы могут быть модифицированы, выращиванием и подвергнуты селекции, которая способствует проявлению таких характеристик, как усиленный рост или рост при пониженных/повышенных температурах.
Биотехнологические модифицирования обычно носят целенаправленный характер и ими могут вводиться характеристики избирательности, такие как бактериостатическая резистентность или восприимчивость, или комбинация таковых для чистоты культуры, или для очистки культур, особенно затравочных культур время от времени.
Другими характеристиками, которые могут быть введены генетическим манипулированием, являются всякие характеристики, которые оказываются допустимыми для вида, к которому относятся указанные выше штаммы. Например, могут быть встроены плазмиды, котирующие резистентность, или могут быть удалены плазмиды естественного происхождения. Предпочтительными плазмидами являются те плазмиды, которые проявляют ауксотрофию. Плазмиды могут быть получены из любого подходящего источника или же они могут быть сконструированы выделением природно встречающихся плазмид и встраиванием в требуемый ген или гены, взятые из другого источника. Природные плазмиды могут быть также модифицированы любым иным образом, который считается желательным.
Любой такой модифицированный штамм может быть применен в способе настоящего изобретения, если штамм способен проявлять требуемую активность, что может легко оцениваться проведением простого и обычного эксперимента.
Микологические свойства этих штаммов приведены ниже.
Микологические свойства штамма.
Amycolata autotrophica SANK 62981
Согласно методам Shirling и Jottlieb (International Journal of Systematic Bacteriology, 16. 313-340 (1968) и S.A.Waksman (The Actinomyces) наблюдение за штаммом вели в течение 14 дней.
1. Микробиологические характеристики.
Форма верха воздушных гифов - Rectus-flexibilis
Вид гифального ветвления - Простое ветвление
Гифальное деление - Можно наблюдать
Поверхностная структура артроспор - Гладкая
Другие органы - Отсутствуют
2. Свойства, получаемые на средах различных видов, для классификации.
Штамм хорошо растет на любой тестированной среде.
Штамм SANK 62981 растет, обнаруживая окраску от светло-коричневато-белой до бледно-желтовато-оранжевой. По мере длительности культивирования наблюдается появление пятен от светло-коричневого до фиолетового.
На других средах, отличных от дрожжевого экстракта, таких как агаровая среда с солодовым экстрактом, наблюдается образование светло-коричневато-серых воздушных гифов.
Никакое образование растворимого пигмента не наблюдается.
В таблице 7 обозначения G, AM, R и SP означают рост, воздушный мицелий, обратная сторона и растворимый пигмент соответственно.
Цветовые тона указаны в приведенной выше таблице согласно номерам цветов окраски (Color Tip Numbers), приведенным в руководстве (Standard Color Table), опубликованном японским издательством (Nihon Shikisai kenkyujo).
3. Физиологические свойства.
Уменьшение содержания нитрата - Положительный результат
Гидролиз крахмала - Отрицательный результат
Образование меланоидного пигмента - Отрицательный результат
Определения проводили на следующих трех средах:
среда 1: триптон-жидкая среда с дрожжевым экстрактом (ISP I)
среда 2: пептон-дрожжевой экстракт-агар с железом (ISP 6)
среда 3: тирозиновый агар (ISP 7).
4. Усваиваемость различных видов источников углерода.
Использованием агаровой среды Pridham-Jottlieb (ISP 9) изучали усваиваемость различных источников углерода и о ней судили после проведения выращивания в течение 14 дней при 28oC.
В следующей таблице обозначения означают:
+ усваивание,
± слабое усваивание и
- отсутствие усваивания.
D-Глюкоза - +
L-Арабиноза - +
D-Ксилоза - +
D-Фруктоза - +
L-Рамноза - ±
Инозит - +
Сахароза - -
Рафиноза - -
D-Маннит - +
Контрольный опыт - -
5. Внутриклеточные компоненты. Согласно методам B.Becker и др. (Applied Microbiology 12, 236 (1965)) и (M.P. Lechevalier и др. (The Actinomycetales by H. Prauser p. 311 91970)) кислотные гидролизаты клеток этих штаммов анализировали методом бумажной хроматографии. В клеточных стенках была обнаружена мезо-2,6-диаминопимелиновая кислота и были замечены арабиноза и галактоза как сахарные компоненты бактериальных клеток, что подтверждало, что бактериальные компоненты относятся к типу IV-A.
Миколовая кислота не обнаружена.
На основании этих результатов штамм SANK 62981 был определен принадлежащим к виду Amycolata autotrophica.
Однако так как вегетативный рост штамма SANK 62981 обнаруживает цветовой тон, подобный аметисту, сделан вывод, что вид представляет подвид вида Amycolata autotrophica.
Этот штамм депонирован на условиях Будапештского договора в постоянной коллекции культур Научно-исследовательского института ферментации, Агентство промышленной науки и технологии. Министерство внешней торговли и промышленности, Япония, под номером FERM-BP-4105.
Этот штамм идентифицирован согласно стандарту Международного стрептомицинового проекта (International Streptomyces Project) (Bergey's Manual of Determinative Bacteriology, 8th Ed., the Actinomycetes, Vol.2 by S.A.Waksman) и последним сообщениям, касающимся актиномицетов. Род Amycolata был до этого классифицирован как часть рода Nocardia. Однако из-за различий в компонентах бактериальных клеток теперь полагают, что род Amycolata является отличным от рода Nocardia и каждый из них образует новый род (International Journal of Systematic 36, 29 (1986)).
Микологические свойства штамма Syncephalastrum racemosum (Cohn) Schroeter SANK 41872
Этот штамм был получен пересевом штамма, депонированного в IFO под номером IFO 4814. Он повторно депонирован в Научно-исследовательском институте ферментации, Агентство промышленной науки и технологии, Министерство внешней торговли и промышленности, и ему присвоен номер FERM BP-4197.
Микологические свойства штамма Syncephalastrum nigricans Vuillemin SANK 42372.
Вегетативные гифы развиваются хорошо и растут быстро. Спорангиефоры идут вертикально от гифов, характеризуются бледно-коричневой окраской при наличии ризоидных и направальных ветвей и образуют перегородки.
Боковые ветви иногда сильно искривлены.
На верхушках основной оси и боковых ветвях образуются пузырьки. Пузырьки характеризуются субсферической или овальной формой, иногда эллиптической формой, и те пузырьки, которые образуются на верхушке основной оси, имеют диаметр от 28 до 50 мкм, а пузырьки, которые образуются на верхушке боковых ветвей, имеют диаметр от 15 до 25 мкм.
Многие мероспорангии образуются по всей поверхности. Спорангиефоры являются одностержневыми или пальцеобразными по форме и часто образуется от 5 до 10 спор в линию.
Споры почти бесцветные с гладкими поверхностями, одноклеточные и характеризуются формой от субсферической до овальной с диаметром от 3,5 мкм до 6,5 мкм.
Зигоспоры не наблюдаются.
Из сравнения этих свойств со свойствами известных штаммов следует, что свойства этого штамма хорошо согласуются со свойствами вида Syncephalastrum nigricans описанного Vuillemin ("An Illustrated Book of Funji" Ed. by Keisuke Tsubaki and Shun ichi Udagawa, Kodansha, стр.303-304 (1978)).
Этот штамм депонирован на условиях Будапештского договора в Научно-исследовательском институте ферментации, Агентства промышленной науки и технологии, Министерство внешней торговли и промышленности, под инвентарным номером FERM BP-4106.
Микологические свойства штамма Mucolhiemalis Wehmer SANK 36372.
Этот штамм был получен пересевом штамма, депонированного в IFO под инвентарным номером IFO 5834. Он повторно депонирован в Научно-исследовательском институте ферментации, Агентства промышленной науки и технологии, Министерства внешней торговли и промышленности под номером FERM BP-4108.
Микологические свойства штамма Streptomyces carbophilus SANK 62585.
1. Морфологические характеристики. Морфологию штамма наблюдали под микроскопом после 14 дней культивирования при 28oC в среде, предписанной требованиями Международного стрептомицинового проекта (International Streptomyces Project (ISP).
Субстратные гифы довольно удлиненные и разветвленные, воздушные мицелии просто разветвлены. Спорангиеносцы являются или прямыми или искривленными, или иногда образуют спирали, и поверхность спор является гладкой.
Не наблюдали каких-либо специальных органов, таких как завитки, склероции, фрагментация субстратных гифов или спорангий.
2. Свойства, получаемые на различных видах сред для классификации. Свойства штамма SANK 62585 определяли на различных средах после 14 дней инкубирования при 28oC. Результаты приведены в табл.8.
В приведенной таблице 8 использованы те же сокращения, что и в табл.7.
Цветовые тона, указанные в приведенной выше таблице, соответствуют номерам цветов окраски (Color Tip Numbers), приведенным в руководстве (Standard Color Table), опубликованном японским издательством (Nihon Shikisai Kenkyujo).
Физиологические свойства.
Гидролиз крахмала - Положительный
Разжижение желатины - Отрицательное
Понижение содержания нитрата - Положительное
Коагуляция молока - Положительная
Пентонизация молока - Положительная
Температурный интервал роста
(среда 1) - 4-45oC
Температурный интервал оптимального роста
(среда 1) - 15-35oC
Продуцирование меланоидных пигментов
(среда 2) - Отрицательное
(среда 3) - Псевдоположительное.
(Меланоидный пигмент иногда продуцируется в последний период инкубации.)
(среда 4) - Отрицательное
Среды, использованные в приведенных выше тестах, были следующими:
среда 1 - агар с содержанием дрожжей и солода (ISP 2);
среда 2 - жидкая среда с триптон-дрожжевым экстрактом (ISP 1);
среда 3 - агар с пептон-дрожжевым экстрактом и железом (ISP 6);
среда 4 - тирозиновый агар (ISP 7).
4. Усваиваемость углеродных источников. Усваиваемость источника углерода, который был использован в агаровой базисной (ISP 9) среде (Pridham-Jottlieb), изучали добавлением D-глюкозы, L-арабинозы, D-ксилозы, инозита, D-маннита, D-фруктозы, L-рамнозы, сахарозы, рафинозы, целлобиозы или трегалозы. Ферментацию с применением этого микроорганизма проводили при температуре 28oC в течение 14 дней. Поскольку штамм хорошо растет в контрольной среде и без добавления какого-либо источника углерода, усваиваемость источников углерода еще должна быть определена. Однако вегетативный рост этого штамма в средах, содержащих D-глюкозу, D-ксилозу, инозит, рафинозу, целлобиозу или трегалозу, гораздо лучше, чем в контрольной среде.
5. Внутриклеточные компоненты. Компоненты клеточной стенки штамма SANK 62585 анализировали, следуя методу, описанному (B.Becker и др. Applied Microbiology, 12, 421-423 (1964)). Были обнаружены L,L-диаминопимелиновая кислота и глицин. Таким образом подтверждалось, что стенки клеток этого штамма являются клеточной стенкой первого типа. Компоненты сахара всей клетки анализировали, следуя методу, описанному M.B.Lechevalier и др. (Journal of Laboratory and Clinical Medicine, 71, 934 (1968)), но не обнаружили характерных особенностей.
На основании приведенных выше данных очевидно, что штамм SANK 62585 принадлежит к роду Streptomyces, одному из родов актиномицетов.
Идентификацию штамма SANK проводили согласно стандарту ISP (The International Streptomyces Project (международный стрептомициновый проект); Bergey's Manual of Determinative Bacteriology (the 8th edition), S.A.Waksman: The Actinomycetes and recent literature on Actinomycetes).
Из тщательного сопоставления приведенных выше данных с опубликованными описаниями известных микроорганизмов вытекает значительное различие, из чего следует, что штамм SANK 62585 должен быть классифицирован как новый вид, принадлежащий к роду Streptomyces. На основании сказанного он обозначен как Streptomyces carbophilus. Этот штамм депонирован в постоянной коллекции культур Научно-исследовательского института ферментации. Агентство промышленной науки и технологии, Министерство внешней торговли и промышленности, и ему присвоен инвентарный номер FERM-BP 4128.
Отсутствует какое-либо особое ограничение, касающееся способа культивирования, применяемого для выращивания конвертирующего микроорганизма, и любой метод, обычно используемый для культивирования микроорганизмов, может быть с одинаковым успехом использован и здесь. Примерами таких методов являются твердофазное выращивание, выращивание в стационарных условиях, выращивание со встряхиванием, выращивание при перемешивании и выращивание с аэрацией. Из этих методов предпочтение отдается методу выращивания с аэрацией, т. е. из способа с перемешиванием, со встряхиванием или с аэрацией, более предпочтительным является способ со встряхиванием.
Ферментацию на промышленном уровне желательно вести в условиях перемешивания культуры с принудительной аэрацией.
Величина pH питательной среды, используемой для выращивания конвертирующего микроорганизма, обычно находится в диапазоне от 5,0 до 8,0, предпочтительно от 6,0 до 7,0.
Ферментацию с использованием конвертирующего микроорганизма предпочтительно ведут при температуре в диапазоне от 15 до 35oC, более предпочтительно от 26 до 30oC и наиболее предпочтительно при 28oC.
Метод 1.
Этот способ ферментативного гидролиза проводится инкубированием штамма конвертирующего микроорганизма и добавлением соединения формулы (Ib) в ходе проведения ферментации.
Время, в которое добавляют соединение, может меняться в зависимости от условий оптимального культивирования для применяемого конвертирующего микроорганизма, в частности, от типа аппаратуры, применяемой для выращивания, состава среды, температуры выращивания и других условий; предпочитается добавлять соединение формулы (Ib), когда гидроксилирующая способность конвертирующего микроорганизма начинает возрастать. В общем, предпочтение следует отдавать времени, составляющему от 1 до 3 дней от начала инкубирования конвертирующего микроорганизма.
Количество соединения формулы (Ib), которое следует добавить, обычно находится в диапазоне от 0,01 до 5,0%, более предпочтительно в диапазоне от 0,025 до 2,0% из расчета объема среды.
Время, необходимое для инкубирования, может меняться в широких пределах в зависимости от многих факторов, включая условия культивирования и природу микроорганизма, но обычно достаточен промежуток времени от 3 до 5 дней после добавления соединения формулы (Ib).
Метод 2.
Этот метод проводится при инкубировании конвертирующего микроорганизма в присутствии небольшого количества субстрата, по методике метода 1 до тех пор, пока гидроксилирование с помощью микроорганизма, не достигнет максимальной продуктивности.
Гидроксилирующая способность изменяется в зависимости от типа среды выращивания, температуры ферментации и других условий, но обычно она достигает максимальной величины по прошествии 4 - 5 дней после начала выращивания. Выращивание обычно завершают в этот момент времени.
Клетки затем собирают, подвергая культуральный бульон центрифугированию, фильтрованию или им подобным воздействиям. Желательно, чтобы собранные таким образом клетки промывались перед использованием физиологическим солевым раствором или соответствующим буферным раствором.
Соединение формулы (Ib) обычно контактирует с клетками, полученными таким способом, в водном растворителе, например в фосфатном буфере с pH, от 5 до 9.
Реакцию гидролиза предпочтительно проводят при температуре от 20o до 45oC, более предпочтительно от 25o до 35oC.
Концентрация соединения формулы (Ib) предпочтительно находится в диапазоне от 0,01 до 5,0% в расчете на объем среды.
Время, необходимое для взаимодействия, меняется в зависимости от многих факторов, таких как концентрация соединения формулы (Ib), температура взаимодействия и другие условия, однако взаимодействие обычно завершается за период от 1 до 5 дней.
Метод 3.
В этом методе свободный от клеток экстракт получают разрушением клеток, что может быть достигнуто физическими или химическими методами, например измельчением или ультразвуковой обработкой, в результате чего получают суспензию, содержащую клеточные компоненты, включая фермент. Или же это может быть достигнуто обработкой клеток органическим растворителем, поверхностно-активным веществом или ферментом, способным давать свободный от клеток экстракт. Клетки могут быть получены так, как это описано в методе 2. Экстракт затем приводят в контакт с соединением формулы (Ib).
Условия, применяемые при контактировании свободного от клеток экстракта с соединением формулы (Ib), являются аналогичным условием, описанным в методе 2.
Согласно методам, описанным выше, подходящий субстрат (гидроксикислота или лактоновое соединение) вводят во взаимодействие с конвертирующим микроорганизмом или с его свободным от клеток и содержащим фермент экстрактом для стереоизбирательного введения гидроксигруппы в положение 6 субстрата. Требуемое соединение с 6 β-гидроксигруппой, могут быть избирательно получены при использовании подходящих сочетаний, например:
1) лактоновое соединение и штамм Mucor hiemalis Wehmer;
2) гидроксикислотное соединение и штамм Streptomyces carbophilus; или
3) гидроксикислотное соединение и штамм Amycolata autotrophica.
Требуемые соединения с 6 α-гидроксигруппой, могут быть получены использованием подходящего сочетания, например:
1) лактоновое соединение и штамм Syncephalastrum nigricans Vuillemin или
2) лактоновое соединение и штамм Syncephalastrum racemosum (Cohn) Schroete.
Продукты, полученные с помощью описанных выше методов настоящего изобретения, обнаруживаются в фильтрате, полученном из жидкой среды, и в мицелиях в конце периода ферментации. Соединение настоящего изобретения существует в форме либо гидроксикислоты, либо лактона, и эти формы могут быть превращены одна в другую. Важное преимущество гидроксикислотного соединения состоит в том, что оно может образовывать устойчивую соль.
Соответственно экстракция и извлечение требуемого продукта из всей ферментационной жидкой среды могут быть осуществлены, например, следуя методу 1 или методу 2.
Метод 1.
Всю ферментационную жидкую среду подвергают центрифугированию или фильтрованию на фильтре, таком как диатомовая земля, для отделения надосадочной жидкости от мицелия и других твердых материалов. Они затем обрабатываются следующим образом.
1. Надосадочная жидкость. Если надосадочная жидкость содержит лактоновое соединение, то его подвергают гидролизу в щелочных условиях (предпочтительно pH 12 или более) для раскрытия лактонового кольца. Гидролизат затем подкисляют осторожно для получения свободной гидроксикислоты. Этот подкисленный гидролизат или надосадочную жидкость, содержащую свободную гидроксикислоту, затем экстрагируют несмешиваемым с водой органическим растворителем, и растворитель удаляют из экстракта, например, отгонкой при пониженном давлении. Примерами подходящих несмешиваемых с водой органических растворителей являются алифатические углеводороды, такие как гексан и гептан; ароматические углеводороды, такие как бензол, толуол или ксилол; галоидированные углеводороды, такие как хлористый метилен, хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол или дихлорбензол; простые эфиры, такие как диэтиловый эфир или диизопропиловый эфир; сложные эфиры, такие как этилформиат, этилацетат, пропилацетат, бутилацетат или диэтилкарбонат; и смеси любых двух или большего числа приведенных выше растворителей.
2. Мицелий. Лепешку из мицелия смешивают с несмешиваемым с водой органическим растворителем так, чтобы конечная концентрация у лепешки составляла от 50 до 90%, считая на объем смеси. Полученную смесь затем обрабатывают аналогичным образом, как и выше, в случае обработки надосадочной жидкости. Примерами подходящих несмешиваемых с водой органических растворителей являются спирты, такие как метанол или этанол; кетоны, такие как ацетон; нитрилы, такие как ацетонитрил или изобутиронитрил; и амиды, такие как формамид, диметилформамид, диметилацетамид, N-метил-2-пирролидон, N-метилпирролидинон или гексаметилфосфорный триамид.
Метод 2.
Ферментационную жидкую среду подвергают гидролизу в щелочных условиях (предпочтительно при pH 12 или более) либо при нагревании, либо при комнатной температуре, для раскрытия лактонового кольца, в то же самое время, когда разрушается мицелий. Все активные соединения, находящиеся в жидкой среде, превращают в соль гидроксикислотного соединения, и требуемая свободная гидроксикислота может быть извлечена из смеси аналогичной обработкой, что и обработка, описанная выше применительно к надосадочной жидкости.
Свободное гидроксикислотное соединение, полученное таким способом, может быть, если необходимо, растворено в водном растворе соли щелочного металла или гидроксида щелочного металла, такого как гидроксид натрия, с образованием соответствующей соли по методике, описанной при рассмотрении стадии 6. Гидроксикислота может быть затем извлечена удобным образом в форме ее наиболее устойчивой соли.
Альтернативно для извлечения требуемого соединения свободное гидроксикислотное соединение, полученное таким способом, подвергают дегидратации, нагревая в органическом растворителе с получением соединениям с лактоновым кольцом, по методике, описанной при рассмотрении стадии 6.
Смесь, состоящая из соединений, включающих свободную гидроксикислоту, одну или несколько солей гидроксикислоты и лактоновое соединение, может быть разделена обычными способами, используемыми в органической химии. Например, они могут быть разделены и извлечены различными хроматографическими способами, включающими в себя распределительную колонную хроматографию с пропусканием вещества через синтетический абсорбент, такой как сефадекс LH-20 ("Фармация, инк. "), амберлитТМ XAD-11 ("Ром энд Хаас ко.") или диаионТМ HP-20 ("Мицубиси касеи корпорейшн"); жидкостная хроматография с пропусканием вещества через регулярно-фазовую или обращенно-фазовую колонку с насадкой из силикагеля или из алкилированного силикагеля (предпочтительно высокоскоростная жидкостная хроматография; или с помощью подходящего сочетания приемов; после чего соединение может быть получено элюированием подходящим элюирующим растворителем.
Лактоновое соединение может быть также очищено абсорбционной колонной хроматографией с пропусканием вещества через носитель, такой как силикагель, оксид алюминия или флорисил (содержащий силикагель и магний).
Примерами предпочтительных растворителей, используемых для элюирования, являются алифатические углеводороды, такие как гексан, гептан, лигроин или петролейные эфиры; ароматические углеводороды, такие как бензол, толуол, или ксилол; галоидированные углеводороды, такие как хлористый метилен, хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол или дихлорбензол; сложные эфиры, такие как этилформиат, этилацетат, пропилацетат, бутилацетат или диэтилкарбонат; и простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан или диэтиленгликолдиметиловый простой эфир.
Или же экстракт может быть очищен абсорбционной колонной хроматографией, использованной для удаления примесей. Требуемое гидроксикислотное соединение может быть получено абсорбцией его в абсорбционной колонке и затем элюированием его элюирующим растворителем, например водным спиртом, таким как водный метанол, водный этанол, водный пропанол или водный изопропанол; или водным кетоном, таким как водный ацетон. Примерами таких абсорбентов являются активированный уголь или абсорбционная смола, такая как амберлитТМ XAD-2 или XAD-4 ("Ром энд Хаас ко.") или диаионТМ HP-10, HP-20, CHP-20 или HP-50 ("Мицубиси Касеи корпорейшн").
Для очистки требуемое соединение может быть использовано в форме либо свободной гидроксикислоты, либо соли гидроксикислоты, поскольку обе формы характеризуются взаимопревращаемостью, по методике, описанной при рассмотрении стадии 6.
Биологическая активность.
Соединения настоящего изобретения обладают заметной способностью понимать уровни содержания сывороточного холестерина. В частности, соединения ингибируют биосинтез холестерина в ферментной системе или в системе клеточной культуры, отделенной от экспериментального животного ингибированием 3-гидрокси-3-метилглутарил-CoA-редуктазы (HMGGCo'A) фермента, лимитирующего скорость стерольного биосинтеза, за счет конкуренции с HMG-CoA. Из сказанного следует, что соединения проявляют сильный эффект в отношении понижения содержания сывороточного холестерина, когда их применяют при лечении людей и других животных.
Эксперимент 1.
Определение ингибирующей активности 3-гидрокси-3-метилглутарил-KoA-редуктазы.
Способность предпочтительных тестируемых соединений ингибировать активность 3-гидрокси-3-метилглутарил-KoA-редуктазы определяли, следуя методу (Koga) и др. (Eur. J. Biochem. 209, 315-319 (1992)), улучшенной методике (Kuroda) и др. (Biochem. Biophys. Acta, 485, 70-81 (1977)), которая является модификацией метода Shapiro и др. (Anal. Biochem. 31, 383-390,(1969)).
Раствор из 5 мл предпочтительно тестируемого соединения, растворенного в дистиллированной воде, добавляли к 45 мкл реакционной смеси, содержащей 100 мМ фосфата калия в виде буферного раствора (с величиной pH 7,4), 0,2 мМ (14C)3-гидрокси-3-метилглутарил-KoA-редуктазы, 10 мМ двунатриевой соли этилендиаминтетрауксусной кислоты, 10 мМ дитиотреитола, 10 мМ восстановленного никотинамидадениндинуклеотидфосфата и раствор фермента (микросомальная фракция из печени крысы). Содержание веществ указаны в расчете на конечный объем анализируемой смеси величиной в 50 мкм. Полученную смесь инкубировали в течение 15 мин при 37oC. Реакцию затем завершали, добавляя 10 мкл 2 н. раствора хлористоводородной кислоты для лактонизации образовавшегося (14C)мевалоната. После 15 мин инкубирования добавляли 1 мл 1:1 по объему водной суспензии биорекса-5, и ампулы энергично перемешивали, используя смеситель "Вортекс". Смесь затем центрифугировали при 3000 оборотов в 1 мин в течение 10 мин при 4oC. Надосадочную жидкость (400 мки) смешивали с 4,5 мл вещества оптифтоуТМ в сцинцилляционных пробирках, и активность (14C)мевалонолактона определяли жидкостным сцинциляционным счетчиком.
Полученные результаты приведены в табл.9.
Эксперимент 2.
Определение ингибирующей активности в отношении подавления стерольного синтеза в мышиной печени.
Стерольный синтез в печени мышей оценивали по методу Koga и др. (Biochem. Biophys. Acta, 1045, 115-120,(1990)).
Каждой мыши интраперитонеально вводили 15 мкл (14C)ацетата. По прошествии одного часа животное умерщвляли обезглавливанием и вырезали печень. Стерольный синтез в печени оценивали введением активного по углероду-14 препарата в осаждаемые дигитонином стеролы. Предпочтительные тестируемые соединения, растворенные в 1 %-ном твине 80, вводили мышам орально за 2 часа до инъекции (14C)ацетата.
Активность в отношении стерольного синтеза у контрольного животного, получившего только 1%-ный раствор твина 80, принимали за 100%. Определяли относительное ингибирование стерольного синтеза в печени у мышей, которые получили различные дозы тестируемого соединения, и рассчитывали величины ED50 (мг/кг, доза, необходимая для торможения стерольного синтеза в печени на 50%).
Полученные результаты приведены в табл.9.
Применяемое известное соединение имеет формулу (XXIII) и представляет собой соединение примера 4, описанного в Японской патентной публикации N Hei 3-33696.
Как ясно видно из результатов испытания, приведенных выше, соединения настоящего изобретения конкурируют с 3-гидрокси-3-метилглутарил-CoA, который ответственен за протекание лимитирующей скорость стадии биосинтеза холестерина в ферментной системе, отделенной от лабораторного животного или в печени мыши. Соответственно активность 3-гидрокси-3-метилглутарил-CoA-редуктазы ингибируется и биосинтез холестерина предотвращается.
Соединения настоящего изобретения обнаруживают сильную активность в отношении понижения содержания холестерина в сыворотке крови животных. Кроме того, их токсичность является очень низкой. Следовательно, они применимы в качестве медицинского препарата в терапии гиперлипемии и для предупреждения атериосклероза, а также в качестве противогрибковых и противоопухолевых средств.
С этой целью соединения формул (I) и (IV) могут назначаться орально в форме таблеток, капсул, гранул, порошков или сиропов, или парентерально посредством внутривенной инъекции, суппозиториев и им подобными способами. Эти фармацевтические рецептуры могут быть приготовлены смешением соединении настоящего изобретения, с одним или несколькими адъювантами, такими как наполнители (например, органические наполнители, включая производные сахаров, такие как лактоза, сахароза, глюкоза, маннит, или сорбит; производные крахмала, такие как кукурузный крахмал, картофельное пюре, α-крахмал, декстрин или карбоксиметиловый крахмал; производные целлюлозы, такие как кристаллическая целлюлоза, низкомолекулярная гидроксипропилзамещенная целлюлоза, гидроксипропилметилцеллюлоза, карбоксиметилцеллюлоза, кальциевая карбоксиметильная целлюлоза или внутренне сшитая натриевая карбоксиметильная целлюлоза; гуммиарабик; декстран и пуллулан (фирменное название материала); неорганические наполнители, включая силикаты, такие как светлый ангидрид кремниевой кислоты, синтетический алюмосиликат или магниевый мета-кремниевокислотный алюминат; фосфаты, такие как фосфат кальция; карбонаты, такие как карбонат кальция; и сульфаты, такие как сульфат кальция); смазочные агенты (например, стеараты металлов, такие как стеариновая кислота, стеарат кальция или стеарат магния; тальк; коллоидный кремнезем; воски, такие как пчелиный воск или спермацет; борная кислота; адипиновая кислота; сульфаты, такие как сульфат натрия; гликоль; фумаровая кислота; бензоат натрия; DL-лейцин; натриевые соли алифатических кислот; лаурилсульфаты, такие как лаурилсульфат натрия или лаурилсульфат магния; силикаты, такие как ангидрид кремниевой кислоты или гидрат кремниевой кислоты; и вышеназванные производные крахмала), связывающие вещества (например, поливинилпиридон, макрогол (фирменное название материала) и соединения, аналогичные описанным выше наполнителям); разрыхляющие вещества (например, соединения, аналогичные описанным выше наполнителям; и химически модифицированные крахмал-целлюлозы, такие как кросскармелозный натрий, натриевый карбоксиметильный крахмал или мостиковый поливинилпирролидон); стабилизаторы (например, п-гидроксибензоаты, такие как метилпарабен или пропилпарабен; спирты, такие как хлорбутанол, бензиловый спирт или фенилэтиловый спирт; хлористый бензалконий; фенолы, такие как фенол или креазол; тимеросал; дигидроуксусная кислота и сорбиновая кислота); корригенты (например, подслащиватели, уксус или отдушки, такие как обычно используемые); разбавители и аналогичные.
Доза может быть различной в зависимости от состояния и возраста пациента и от пути и характера введения; например, соединения настоящего изобретения, могут быть введены орально с дневной дозой от 0,01 до 1000 мг на 1 кг веса тела (предпочтительно от 0,05 до 200 мг на 1 кг веса тела), в виде одной или раздельных доз.
Получение некоторых из соединений настоящего изобретения проиллюстрировано далее нижеследующими примерами. Последующие синтезы, а также примеры A и B иллюстрируют получение некоторых исходных веществ, используемых в этих примерах.
Эти примеры включают получение представительных соединений настоящего изобретения прямым выделением из микроорганизмов. Способы, описанные в этих примерах, носят чисто иллюстративный характер и они могут модифицироваться на основе, например, свойств требуемого соединения.
Пример A. (4R, 6R)-6-{ 2-[(1S, 2S, 6S,8S,8aR)-1,2,6,7,8,8a-Гексагидро- 6,8-дигидрокси-2-метил-1-нафтил]этил}тетрагидро-4-гидрокси-2Н-пиран-2-он
A-(1) Натриевый (3R, 5R)-3,5-дигидрокси-7-[(1S, 2S,6S,8S,8aR)-6,8- дигидрокси-2-метил-1,2,6,7,8,8a-гексагидро-1-нафтил]гептаноат.
Раствор метоксида натрия с концентрацией 28% (по весу в расчете на объем) в количестве 50 мл (0,24 моль), добавляли к раствору 100 г (0,31 моль) (3R, 5R)-3,5-дигидрокси-7-[(1S, 2S,6S,8S,8aR)-6-гидрокси-2-метил-8- [(S)-2-метилбутирилокси] -1,2,6,7,8,8a-гексагидро-1-нафтил]гептаноата (правастатин; приготовлен по способу, описанному в патенте США N 4 346 227) в 900 мл метанола, и полученную смесь нагревали в сосуде с обратным холодильником в течение 60 ч. В конце этого времени смесь охлаждали до комнатной температуры, и метанол затем удаляли из реакционной смеси дистилляцией при пониженном давлении. Полученный остаток промывали 200 мл гексана и затем сушили в вакууме, в результате чего получали 120 г названного соединения.
A-(2) (3R, 5R)-3,5-Дигидрокси-7-[(1S, 2S,8S,8S,8aR)-6,8-дигидрокси- 2-метил-1,2,6,7,8,8a-гексагидро-1-нафтил]гептановая кислота.
Весь натриевый (3R,5R)-3,5-дигидрокси-7-[(1S,2S,6S,8S,8aR)- 6,8-дигидрокси-2-метил-1,2,6,7,8,8a-гексагидро-1-нафтил)гептаноат, полученный в стадии 1 выше, растворяли без проведения дополнительной очистки в 300 мл воды. Величину pH у раствора поддерживали на уровне 4,0, добавляя 35%-ный (по весу в расчете на объем) раствор хлористого водорода. Воду затем удаляли из смеси дистилляцией при пониженном давлении. Остаток сушили в вакууме, после чего высушенный остаток растворяли в 300 мл этанола. Хлорид натрия, образовавшийся при взаимодействии, удаляли затем фильтрованием, после чего полученный фильтрат концентрировали упариванием при пониженном давлении. Полученный остаток сушили, получая 94 г названного соединения.
A-(3) (4R, 6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6,8- дигидрокси-2-метил-1-нафтил]этил}тетрагидро-4-гидрокси-2Н-пиран-2-он.
Всю сырую (3R, 5R)-3,5-дигидрокси-7-[(1S,2S,6S,3S,8aR)-6,8-дигидрокси- 2-метил-1,2,6,7,8,8a-гексагидро-1-нафтил] гептановую кислоту, полученную в стадии 2 выше, смешивали с 1000 мл тетрагидрофурана. 3атем к смеси добавляли 38 мл (9,27 моль) триэтиламина, после чего добавляли 38 мл (0,25 моль) диэтилцианофосфоната при охлаждении льдом и перемешивании. Полученную смесь затем перемешивали при комнатной температуре в течение 1,5 ч. В конце этого времени тетрагидрофуран удаляли из реакционной смеси дистилляцией при пониженном давлении, и остаток растирали в смеси простого диэтилового эфира с этанолом, чем стимулировали кристаллизацию. Полученные кристаллы собирали фильтрованием, получая 47,7 г названного соединения. Это соединение затем перекристаллизовывали из смеси этилацетата с этанолом, получая бесцветные пластинчатые кристаллы с температурой плавления от 161 до 163oC.
Спектр ядерного магнитного резонанса (270 МГц, гексадейтерированный диметилсульфоксид) δ, млн. дол.
0,82 (3Н, дублет, J = 6,3 Гц);
4,07-4,15 (2Н, мультиплет);
4,29 (1Н, дублет, J = 4,4 Гц, взаимозаменяемый с D2O);
4,23-4,35 (1Н, мультиплет);
4,52 (1Н, дублет, J = 6,4 Гц, взаимозаменяемый с D2O);
4,51-4,62 (1Н, мультиплет);
5,15 (1Н, дублет, J = 2,9 Гц, взаимозаменяемый с D2O);
5,40 (1Н, широкий синглет);
5,84 (1Н, дублет из дублетов, J = 6,2 и 9,3 Гц);
5,90 (1Н, дублет, J = 9,8 Гц).
Элементный анализ для C18H26O5:
Вычислено: C - 67,06%, H - 8,13%
Найдено: C - 66,31%, H - 3,37%.
Инфракрасный абсорбционный спектр (KBr) νax(см-1) - 3436, 3339, 3222, 1730, 1260, 1217, 1042.
Масс-спектр (m/e): 322 (M+), 304, 286, 268.
[α]
Пример B. (4R, 6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-гидрокси-2-метил-1-нафтил] этил}тетрагидро-4- трет-бутилдиметилсилилокси-2Н-пиран-2-он.
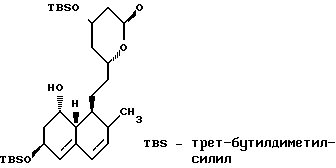
Раствор 9,04 г (60,0 ммоль) трет-бутилдиметилсилилхлорида в 35 мл диметилформамида, добавляли по каплям к раствору 9,65 г (30,0 ммоль) (4R, 6R)-6-{ 2-[(1S, 2S, 6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6,8-дигидрокси- 2-метил-1-нафтил]этил}тетрагидро-4-гидрокси-2Н-пиран-2-она (полученного, как описано выше в примере A) и 6,12 г (90,0 ммоль) имидазола в 45 мл диметилформамида при охлаждении льдом и перемешивании. Полученную смесь затем перемешивали при комнатной температуре в течение 5 ч, после чего растворитель удаляли дистилляцией при пониженном давлении. Полученный остаток растворяли в 500 мл этилацетата, и раствор затем промывали сначала водой, а затем насыщенным водным раствором хлорида натрия. Раствор затем сушили над безводным сульфатом магния, после чего фильтровали. Полученный фильтрат затем концентрировали упариванием при пониженном давлении. Концентрат очищали флэш (мгновенной) хроматографией, на колонке, пропуская через силикагель с использованием метода градиентного элюирования смесью гексана с этилацетатом, как элюента, с объемным соотношением от 2:1 до 1:1, получая 13,3 г названного соединения в виде бесцветного твердого вещества. Это соединение затем перекристаллизовывали из простого диизопропилового эфира, получая бесцветные игольчатые кристаллы с температурой плавления от 132 до 134oC.
Элементный анализ для C30H54O5Si2:
Вычислено: C - 65,40%, H - 9,88%,
Найдено:: C - 65,29% H - 9,96%.
Спектр ядерного магнитного резонанса (270 МГц, гексадейтерированный диметилсульфоксид) δ, в миллионных долях,
0,79 - 0,92 (21Н, мультиплет);
4,07-4,15 (1Н, мультиплет);
4,27-4,34 (1Н, мультиплет);
4,38 (1Н, дублет, J = 3,9 Гц, взаимозаменяемый с D2O);
4,48-4,60 (2Н, мультиплет);
5,33 (1Н, широкий синглет);
5,82 (1Н, дублет из дублетов, J = 6,2 и 9,8 Гц);
5,92 (1Н, дублет, J = 9,8 Гц)
νmax(см-1) инфракрасного абсорбционного спектра (KBr) 3497, 2956, 2929, 2857, 1736, 1711, 1361, 1257, 1071, 837.
Масс-спектр (m/e): 550 (M+), 532, 493, 475, 343, 275.
[α]
В последующих примерах 1 - 23 описано поучение соединений формулы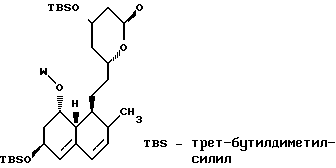
т. е. соединений формулы (I), в которой R1 представляет группу формулы (III) и R6 представляет собой трет-бутилдиметилсилильную группу. Каждая группа W, как она определена в последующих примерах, присоединена к формуле, показанной выше, через связь, обозначенную Z.
Пример 1. (4R, 6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-(3,3-диметилбутирилокси)-2-метил- 1-нафтил] этил}тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-он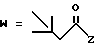
3,3-Диметилмасляную кислоту 0,46 мл (3,6 ммоль), 741 мг (3,6 ммоль) дициклогексилкарбодиимида и 13 мг (0,09 ммоль) 4-(1-пирролидинил)пиридина добавляли к раствору, 1,00 г (1,8 ммоль) (4R,6R)-6-{2-[1S,2S,6S,8S,8aR)-1,2,6,7,3,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-гидрокси-2-метил-1-нафтил] -этил} тетрагидро- 4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного как описано выше в примере B) в 15 мл хлористого метилена при охлаждении льдом. Полученную смесь перемешивали при той же температуре в течение 30 мин и затем перемешивали при комнатной температуре еще в течение 19 ч. В конце этого времени растворитель отгоняли дистилляцией при пониженном давлении, и получающийся остаток смешивали с 20 мл простого диэтилового эфира. Нерастворимые вещества удаляли фильтрованием, и затем дважды промывали простым диэтиловым эфиром используя на каждую промывку по 5 мл. Фильтрат и промывные растворы затем соединяли, и растворитель удаляли дистилляцией при пониженном давлении. Полученный остаток очищали флэш-хроматографией, пропуская через силикагель при использовании в качестве элюента смеси гексана и этилацетата 4:1, по объему получая 555 мг (выход 47%) названного соединения.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ, в миллионных долях,
2,20 (2Н, синглет);
4,24-4,32 (1Н, мультиплет);
4,39-4,46 (1Н, мультиплет);
4,53-4,65 (1Н, мультиплет);
5,37 (1Н, широкий синглет);
5,45 (1Н, широкий синглет);
5,84 (1Н, дублет из дублетов, J = 9,8 и 5,8 Гц);
5,99 (1Н, дублет, J = 9,6 Гц).
νmax(см-1) инфракрасного абсорбционного спектра (CHCl3): 2950, 1800, 1250, 1080, 840.
Масс-спектр (m/e): 648 (M+), 633, 591, 532, 475.
[α]
Пример 2. (4R, 6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдимитилсилилокси-8-(2-этилбутирилокси)-2-метил-1-нафтил] этил} тетрагидро- 4-трет-бутилдиметилсилилокси-2Н-пиран-2-он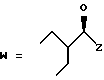
1,26 мл (9,1 ммоль триэтиламина, 15 мг (0,1 ммоль) 4-(1-пирролидинил)пиридина и 0,63 мл (2,73 ммоль) ангидрида 2-этилмасляной кислоты добавляли к раствору, 1,00 г (1,8 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2-6,7,8,8a-гексагидро-6-трет-бутилдиметилсилилокси-8-гидрокси-2-метил-1-нафтил]этил}тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного как описано выше в примере B) в 10 мл хлористого метилена при охлаждении льдом. Полученную смесь перемешивали при комнатной температуре в течение трех дней. В конце этого времени реакционную смесь разбавляли 50 мл этилацетата, и разбавленную смесь промывали 20 мл воды, 10%-ным (по весу в расчете на объем) водным раствором лимонной кислоты, 20 мл насыщенного водного раствора бикарбоната натрия и 20 мл насыщенного водного раствора хлорида натрия, в указанном порядке. Промытую смесь затем сушили над безводным сульфатом магния, после чего смесь фильтровали. Фильтрат, полученный при этом, концентрировали упариванием при пониженном давлении, и полученный концентрат очищали флэш-хроматографией, пропуская через силикагель при использовании в качестве элюента смеси гексана с этилацетатом в объемном соотношении 5:1, получая 1,04 г (выход 88%) названного соединения.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ, в миллионных долях,
4,24-4,32 (1Н, мультиплет);
4,37-4,49 (1Н, мультиплет);
4,51-4,62 (1Н, мультиплет);
5,42 (1Н, широкий синглет);
5,47 (1Н, широкий синглет);
5,84 (1Н, дублет из дублетов, J = 9,8 и 5,9 Гц);
5,98 (1Н, дублет, J = 9,9 Гц).
νmax(см-1) инфракрасного абсорбционного спектра (CHCl3): 2950, 1720, 1250, 1080, 840.
Масс-спектр (m/e):
648 (M+), 633, 591, 532, 475.
[α]
Пример 3. (4R, 6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8а-гексагидро-6-трет- бутилдиметилсилилокси-8-[(S)-2-метилвалерилокси] -2-метил-1-нафтил] этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-он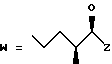
690 мг (6,8 ммоль) триэтиламина и 713 мг (4,1 ммоль) диэтилхлорфосфата добавляли к раствору 400 мг (3,4 ммоль) (S)-2-метилвалериановой кислоты в 15 мл сухого бензола, и полученную смесь перемешивали при комнатной температуре в течение часа. К смеси затем добавляли 1,58 г (2,9 ммоль) (4R,6R)-6-{2- [(1S, 2S, 6S, 8S, 8aR)-1,2,6,7,8,8a-гексагидро-6-трет-бутилдиметилсилилокси- 8-гидрокси-2-метил-1-нафтил] этил} тетрагидро-4-трет-бутилдиметилсилилокси- 2Н-пиран-2-она (полученного, как описано выше в примере B) и 250 мг (1,7 ммоль) 4-(1-пирролидинил)пиридина. Смесь затем перемешивали при комнатной температуре в течение 24 ч, после чего смесь разбавляли 20 мл бензола. Разбавленную смесь затем промывали 20 мл воды, 20 мл 10%-ного (по весу в расчете на объем) водного раствора лимонной кислоты, насыщенным водным раствором бикарбоната натрия и насыщенным водным раствором хлорида натрия, в указанной последовательности. Органический слой сушили над безводным сульфатом магния, и растворитель отгоняли дистилляцией при пониженном давлении. Полученный остаток очищали колонной флэш-хроматографией, пропуская через силикагель с использованием в качестве элюента смеси гексана с этилацетатом в объемном соотношении 6:1, получая 1,36 г (выход 74%) названного соединения.
Спектр ядерного магнитного резонанса: (400 МГц, CDCl3) δ, в миллионных долях,
1,11 (3Н, дублет, J = 7,1 Гц);
4,27-4,30 (1Н, мультиплет);
4,40-4,44 (1Н, мультиплет);
4,55-4,61 (1Н, мультиплет);
5,36 (1Н, широкий синглет);
5,48 (1Н, широкий синглет);
5,85 (1Н, дублет из дублетов, J = 9,7 и 5,9 Гц);
5,99 (1Н, дублет, J = 9,7 Гц),
νmax(см-1) инфракрасного абсорбционного спектра (CHCl3): 2950, 1720, 1250, 1080, 840.
Масс-спектр (m/e): 648 (M+), 591, 532, 475.
[α]
Пример 4. (4R, 6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-(2-пропилвалерилокси)-2-метил- 1-нафтил]этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-он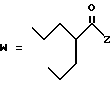
Вещество 4-(1-пирролидинил)пиридин (15 мг, 0,01 ммоль) и 592 мг (3,6 ммоль) хлористого 2-пропилвалерила добавляли к раствору 1,0 г (1,8 ммоль) (4R, 6R)-6{ 2-[1S, 2S, 6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-гидрокси-2-метил-1-нафтил] этил} -тетрагидро- 4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного как описано выше в примере B) в 5 мл сухого пиридина при охлаждении льдом, и полученную смесь перемешивали при 70oC в течение трех часов. В конце этого времени реакционную смесь разбавляли 100 мл этилацетата, и разбавленную смесь затем промывали 100 мл воды, 100 мл 10%-ного (по весу в расчете на объем) водного раствора хлористого водорода, насыщенным водным раствором бикарбоната натрия и насыщенным водным раствором хлорида натрия, в указанной последовательности. Органический слой затем сушили над безводным сульфатом магния, после чего этот слой удаляли фильтрованием. Фильтрат концентрировали упариванием при пониженном давлении, и полученный остаток очищали колонной флэш хроматографией, через силикагель при использовании в качестве элюента смеси гексана с этилацетатом в объемном соотношении 5:1, получая 1,15 г (выход 93%) названного соединения.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
4,25-4,31 (1Н, мультиплет);
4,36-4,48 (1Н, мультиплет);
4,51-4,62 (1Н, мультиплет);
5,40 (1Н, широкий синглет);
5,47 (1Н, широкий синглет);
5,85 (1Н, дублет из дублетов, J = 9,3 и 5,9 Гц);
5,99 (1Н, дублет, J = 9,8 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 295, 1720, 1250, 840.
Масс-спектр (m/e):
676 (M+), 621, 549, 532, 475.
[α]
Пример 5. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-(2-этил-2-метилбутилтирилокси)-2-метил-1-нафтил] этил}- тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-он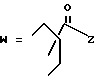
Триэтиламин (0,76 мл, 5,4 ммоль), 807 мг (5,4 ммоль) 4-(1-пирролидинил)пиридина и 674 мг (4,5 ммоль) 2-этил-2-метилбутирилхлорида добавляли к раствору 500 мг (0,91 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-гидрокси-2-метил-1-нафтил]-этил} тетрагидро-4-трет- бутилдиметилсилилокси-2Н-пиран-2-она (полученного как описано выше в примере B) в 10 мл бензола, и смесь нагревали с обратным холодильником в течение 5 ч. В конце этого времени реакционную смесь разбавляли 50 мл этилацетата. Разбавленную смесь затем промывали 30 мл воды, 30 мл 10%-ного (по весу в расчете на объем) водного раствора лимонной кислоты, насыщенным водным раствором бикарбоната натрия и насыщенным водным раствором хлорида натрия в указанном порядке. Органическую фазу затем сушили над безводным сульфатом магния и отфильтровывали. Фильтрат концентрировали упариванием при пониженном давлении, и концентрат очищали испарительной колонной хроматографией пропуская через силикагель при использовании в качестве элюента смеси гексана с этилацетатом, в объемном соотношении 5:1, получая 601 мг (выход 100%) названного соединения.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
1,07 (3Н, синглет);
4,23-4,32 (1Н, мультиплет);
4,37-4,48 (1Н, мультиплет);
4,51-4,64 (1Н, мультиплет);
5,35 (1Н, широкий синглет);
5,46 (1Н, широкий синглет);
5,84 (1Н, дублет из дублетов, J = 9,8 и 5,9 Гц);
5,98 (1Н, дублет, J = 9,3 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 2950, 1720, 1250, 1180, 840.
Масс-спектр (m/e): 662 (M+), 647, 605, 549, 532.
[α]
Пример 6. (4R, 6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,3,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-(2,2-диэтилбутирилокси)-2-метил-1-нафтил]этил} тетрагидро-4-трет-бутилдиметилсилилокси-2-Н-пиран-2-он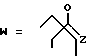
2,2-диэтилбутирилхлорид (1,48 г, 9,1 ммоль) добавляли к раствору 1,0 г (1,8 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-гидрокси-2-метил-1-нафтил] этил} тетрагидро-4-трет- бутилдиметилсилилокси-2Н-пиран-2-она (полученного как описано выше в примере B), 1,67 г (11,3 ммоль) 4-(1-пирролидинил)пиридина и 1,0 мл (7,1 ммоль) триэтиламина в 10 мл толуола, и смесь нагревали с обратным холодильником в течение 10 ч. В конце этого времени реакционную смесь обрабатывали по методике, аналогичной описанной выше в примере 5, получая 1,09 г (выход 89%) названного соединения.
Спектр ядерного магнитного резонанса: (360 МГц, CDCl3) δ , в миллионных долях,
0,96 (9Н, триплет, J = 7,7 Гц);
1,22-1,29 (1Н, мультиплет);
1,42-1,47 (2Н, мультиплет);
4,26-4,29 (1Н, мультиплет);
4,42-4,45 (1Н, мультиплет);
4,53-4,60 (1Н, мультиплет);
5,39 (1Н, широкий синглет);
5,46 (1Н, широкий синглет);
5,35 (1Н, дублет из дублетов, J = 9,7 и 5,9 Гц);
5,99 (1Н, дублет, J = 9,7 Гц),
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 2950, 1715, 1260, 340.
Масс-спектр (m/e): 676 (M+), 661, 619, 532, 475, 400.
[α]
Пример 7. (4R, 6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-3-(2,2-диметил-4-пентеноилокси)-2-метил-1-нафтил]этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-он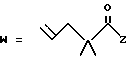
Следовали методике, аналогичной описанной выше в примере 3, но при этом использовали 1,0 г (1,8 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет-бутил- диметилсилилокси-8-гидрокси-2-метил-1-нафтил] этил} тетрагидро-4-трет- бутилдиметилсилилокси-2Н-пиран-2-она (полученного как описано выше в примере B) и 466 мг (3,6 ммоль) 2,2-диметил-4-пентеновой кислоты, получая 231 мг названного соединения.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
1,41 (6Н, синглет);
2,26 (2Н, дублет, J = 7,3 Гц);
4,25-4,33 (1Н, мультиплет);
4,38-4,47 (1Н, мультиплет);
5,00-5,10 (2Н, мультиплет);
5,34 (1Н, широкий синглет):
5,45 (1Н, широкий синглет);
5,60-5,76 (1Н, мультиплет);
5,83 (1Н, дублет из дублетов, J = 9,8 и 5,9 Гц);
5,97 (1Н, дублет, J = 9,8 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 2950, 1720, 1250, 1180, 840.
Масс-спектр (m/e): 645 (М+-15), 603, 535, 517, 475.
[α]
Пример 8. (4R, 6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-(2-аллил-4-пентеноилокси)-2-метил-1-нафтил] этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-он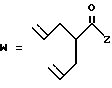
Следовали методике, аналогичной описанной выше в примере 3, но при этом использовали 1,10 г (2,0 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-гидрокси-2-метил-1-нафтил] этил} тетрагидро-4- трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного как описано выше в примере B) и 560 мг (4,0 ммоль) 2-аллил-4-пентеновой кислоты, получая 1,02 г названного соединения.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
4,26-4,31 (1Н, мультиплет);
4,40-4,48 (1Н, мультиплет);
4,52-4,62 (1Н, мультиплет);
4,98-5,11 (4Н, мультиплет);
5,40 (1Н, широкий синглет);
5,47 (1Н, широкий синглет);
5,63-5,80 (2Н, мультиплет);
5,85 (1Н, дублет из дублетов, J = 9,8 и 5,9 Гц);
5,99 (1Н, дублет, J = 9,8 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 2950, 1720, 1250, 1080, 840.
Масс-спектр (m/e): 672 (M+), 615, 532, 475.
[α]
Пример 9. (4R, 6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-(2-бутилгексаноилокси)-2-метил-1-нафтил] этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-он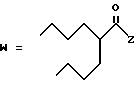
Следовали методике, аналогичной описанной выше в примере 3, но при этом использовали 1,0 г (1,3 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-гидрокси-2-метил-1-нафтил] этил} тетрагидро-4- трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного как описано выше в примере B) и 627 мг (3,6 ммоль) 2-бутилгексановой кислоты, получив 797 мг названного соединения.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
4,25-4,26 (1Н, мультиплет);
4,39-4,48 (1Н, мультиплет);
4,52-4,63 (1Н, мультиплет);
5,42 (1Н, широкий синглет);
5,48 (1Н, широкий синглет);
5,86 (1Н, дублет из дублетов, J = 9,8 и 5,9 Гц);
6,00 (1Н, дублет, J = 9,8 Гц).
νmax(см-1) инфракрасного абсорбционного спектра (CHCl3): 2950, 1850, 1720, 1460, 1250.
Масс-спектр (m/e): 689 (M+-15), 647, 549, 532.
[α]
Пример 10. (4R, 6R)-6-{2-[1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-Гексагидро-6-трет- бутилдиметилсилилокси-8-гексаноилокси-2-метил-1-нафтил] этил} тетрагидро-4- трет-бутилдиметилсилилокси-2Н-пиран-2-он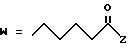
Следовали методике, аналогичной описанной выше в примере 3, но использовали 1,0 г (1,8 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-гидрокси-2-метил-1-нафтил] этил} тетрагидро-4-трет- бутилдиметилсилилокси-2Н-пиран-2-она (полученного как описано выше в примере B) и 423 мг (3,6 ммоль) гексановой кислоты, получали 364 мг названного соединения.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
4,25-4,32 (1Н, мультиплет);
4,39-4,46 (1Н, мультиплет);
4,55-4,65 (1Н, мультиплет);
5,38 (1Н, широкий синглет);
5,48 (1Н, широкий синглет);
5,85 (1Н, дублет из дублетов, J =9,8, и 5,9 Гц);
6,00 (1Н, дублет, J = 9,8 Гц),
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 2950, 1720, 1250, 1180, 840.
Масс-спектр (m/e): 591 (M+-57), 532, 517, 475.
[α]
Пример 11. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-изовалерилокси-2-метил-1-нафтил] -этил}тетрагидро-4- трет-бутилдиметилсилилокси-2Н-пиран-2-он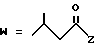
Следовали методике, аналогичной описанной в примере 4, но использовали 1,10 г (2,0 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-гидрокси-2-метил-1-нафтил] этил} тетрагидро-4-трет- бутилдиметилсилилокси-2Н-пиран-2-она (полученного как описано выше в примере B) и 361 мг (3,0 ммоль) изовалерилхлорида, получая 1,14 г названного соединения.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях, составляла
0,94 (6Н, дублет, J = 6,4 Гц);
4,27-4,29 (1Н, мультиплет);
4,40-4,50 (1Н, мультиплет);
4,55-4,65 (1Н, мультиплет);
5,39 (1Н, широкий синглет);
5,48 (1Н, широкий синглет);
5,65 (1Н, дублет из дублетов, J = 9,8 и 5,9 Гц);
5,98 (1Н, дублет, J = 9,8 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 2875, 1725, 1225, 1080, 840.
Масс-спектр (m/e): 634 (M+), 577, 532, 475.
[α]
Пример 12. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-пивалоилокси-2-метил-1-нафтил] -этил} тетрагидро-4- трет-бутилдиметилсилилокси-2Н-пиран-2-он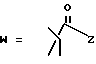
Следовали методике, аналогичной описанной в примере 4, но использовали 1,10 г (2,0 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8a-гексагидро-6-трет-бутил- диметилсилилокси-8-гидрокси-2-метил-1-нафтил] этил} тетрагидро-4-трет- бутилдиметилсилилокси-2Н-пиран-2-она (полученного как описано выше в примере B) и 486 мг (4,0 ммоль) хлористого пивалоила, получая 594 мг названного соединения.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
1,17 (9Н, синглет);
4,27-4,31 (1Н, мультиплет);
4,40-4,44 (1Н, мультиплет);
4,56-4,63 (1Н, мультиплет);
5,32 (1Н, широкий синглет);
5,48 (1Н, широкий синглет);
5,84 (1H, дублет из дублетов, J = 9,7 и 5,3 Гц);
5,98 (1Н, дублет, J = 9,7 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 2950, 1720, 1255, 1080, 340.
Масс-спектр (m/e): 634 (M+), 577, 532, 475, 343.
[α]
Пример 13. (4R,6R)-6-{2-[(1S,2S,6S,6S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-(2,2-диметилвалерилокси)-2-метил-1-нафтил] этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-он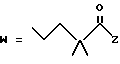
Следовали методике, аналогичной описанной в примере 4, но использовали 2,0 г (3,6 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,3S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-гидрокси-2-метил-1-нафтил]этил}тетрагидро-4-трет- бутилдиметилсилилокси-2Н-пиран-2-она (полученного как описано выше в примере B) и 2,16 г (14,5 ммоль) 2,2-диметилпентаноилхлорида, получая 1,31 г названного соединения.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
1,13 (6Н, синглет);
4,25-4,32 (1Н, мультиплет);
4,36-4,46 (1Н, мультиплет);
4,52-4,64 (1Н, мультиплет);
5,32 (1Н, широкий синглет);
5,45 (1Н, широкий синглет);
5,83 (1Н, дублет из дублетов, J = 9,8 и 5,9 Гц);
5,98 (1Н, дублет, J = 9,8 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 2950, 1720, 1250, 1080, 840.
Масс-спектр (m/e) 662, 647, 605, 532, 475.
[α]
Пример 14. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексаридро-6-трет- бутилдиметилсилилокси-8-(2-аллил-2-метил-4-пентеноилокси)-2-метил-1- нафтил]этил}тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-он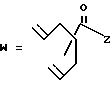
Следовали методике, аналогичной описанной в примере 4, но использовали 2,0 г (3,6 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-гидрокси-2-метил-1-нафтил]этил}тетрагидро-4-трет- бутилдиметилсилилокси-2Н-пиран-2-она (полученного как описано выше в примере B) и 1,26 г (7,3 ммоль) хлористого 2-аллил-2-метил-4-пентеноила, получая 2,13 г названного соединения.
Спектр ядерного магнитного резонанса: (270МГц, CDCl3) δ , в миллионных долях,
1,08 (3Н, синглет);
4,28-4,31 (1Н, мультиплет);
4,41-4,45 (1Н, мультиплет);
4,56-4,60 (1Н, мультиплет);
5,04-5,03 (4Н, мультиплет);
5,38 (1Н, широкий синглет);
5,46 (1Н, широкий синглет);
5,62-5,72 (2Н, мультиплет);
5,85 (1Н, дублет из дублетов, J = 9,7 и 5,9 Гц);
5,98 (1Н, дублет, J = 9,7 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 2950, 1720, 1250, 1080, 835.
Масс-спектр (m/e):
686 (M+), 629, 532, 475.
[α]
Пример 15. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-(2-метил-2-пропилвалерилокси)-2-метил-1-нафтил]этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-он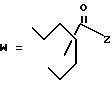
Следовали методике, аналогичной описанной в примере 4, но использовали 2,0 г (3,3 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро- 6-трет-бутилдиметилсилилокси-8-гидрокси-2-метил-1-нафтил] этил}тетрагидро-4- трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного как описано выше в примере B) и 1,92 г (10,9 ммоль) 2-метил-2-пропилвалерилхлорида, получая 1,05 г названного соединения.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
1,08 (3Н, синглет);
4,26-4,32 (1Н, мультиплет);
4,38-4,45 (1Н, мультиплет);
4,53-4,60 (1Н, мультиплет);
5,45 (1Н, широкий синглет);
5,47 (1Н, широкий синглет);
5,85 (1Н, дублет из дублетов, J = 9,7 и 5,9 Гц);
5,98 (1Н, J = 9,7 Гц),
νmax (см-1 инфракрасного абсорбционного спектра (CHCl3); 2950, 1720, 1250, 1180, 840.
Масс-спектр (m/e): 690 (M+), 675, 633, 549, 532.
[α]
Пример 16. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-(2,2-диэтилвалерилокси)-2-метил-1-нафтил]этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-он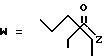
Следовали методике, аналогичной описанной в примере 4, но использовали 2,0 г (3,6 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6- трет-бутил-диметилсилилокси-8-гидрокси-2-метил-1-нафтил] этил} тетрагидро-4- трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного как описано выше в примере B) и 1,29 г (7,3 ммоль) хлористого 2,2-диэтилвалерила, получая 188 мг названного соединения.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
4,20-4,25 (1Н, мультиплет);
4,33-4,37 (1Н, мультиплет);
4,46-4,53 (1Н, мультиплет);
5,32 (1Н, широкий синглет);
5,39 (1Н, широкий синглет);
5,79 (1Н, дублет из дублетов, J = 9,7 и 5,8 Гц);
5,92 (1Н, дублет, J = 9,7 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 2950, 1720, 1250, 1080.
Масс-спектр (m/e): 690 (M+), 675, 633, 568, 532.
[α]
Пример 17. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-(2-изопропил-3-метилбутирилокси)-2-метил-1-нафтил] этил}тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-он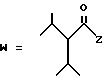
Следовали методике, аналогичной описанной в примере 4, но использовали 1,0 г (1,8 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилокси-8-гидрокси-2-метил-1-нафтил]этил}тетрагидро-4-трет- бутилдиметилсилилокси-2Н-пиран-2-она (полученного как описано выше в примере B) и 888 мг (5,5 ммоль) хлористого 2-изопропил-3-метилбутирила, получая 198 мг названного соединения.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
4,26-4,32 (1Н, мультиплет);
4,45-4,60 (2Н, мультиплет);
5,45-(2Н, широкий синглет);
5,85 (1Н, дублет из дублетов, J = 9,7 и 6,0 Гц);
5,99 (1Н, дублет, J = 9,7 Гц).
νmax (см-1) спектра инфракрасного абсорбционного поглощения (CHCl3): 2950, 1720, 1250, 1180, 840.
Масс-спектр (m/e): 676 (M+), 661, 619, 568, 532.
[α]
Пример 18. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-(2,2-диэтил-4-пентеноилокси)-2-метил-1-нафтил] этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-он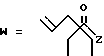
Следовали методике, аналогичной описанной в примере 6, но использовали 2,0 г (3,6 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-гидрокси-2-метил-1-нафтил]этил}тетрагидро-4-трет- бутилдиметилсилилокси-2Н-пиран-2-она (полученного как описано выше в примере B) и 3,17 г (18,1 ммоль) хлористого 2,2-диэтил-4-пентаноила, получая 1,95 г названного соединения.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
4,22-4,23 (1Н, мультиплет);
4,38-4,47 (1Н, мультиплет);
4,52-4,62 (1Н, мультиплет);
5,00-5,14 (2Н, мультиплет);
5,41 (1Н, широкий синглет);
5,46 (1Н, широкий синглет);
5,54-5,72 (1Н, мультиплет);
5,85 (1Н, дублет из дублетов, J = 9,7 и 5,9 Гц);
5,99 (1Н, дублет, J = 9,7 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 2950, 1720, 1250, 1080, 840.
Масс-спектр (m/e): 688(M+), 631, 623, 568, 532.
[α]
Пример 19. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-(2-алкил-2-этил-4-пентеноилокси)-2-метил-1-нафтил] этил}тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-он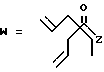
Следовали методике, аналогичной описанной в примере 6, но использовали 1,65 г (3,0 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-гидрокси-2-метил-1-нафтил] этил} тетрагидро-4-трет- бутилдиметилсилилокси-2Н-пиран-2-она (полученного как описано выше в примере B) и 2,30 г (15,0 ммоль) хлористого 2-аллил-2-этил-4-пентеноила, получая 1,63 г названного соединения.
Спектр ядерное магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
0,81 (3Н, триплет, J = 7,4 Гц),
4,17-4,29 (1Н, мультиплет);
4,41 - 4,45 (1Н, мультиплет);
4,54-4,60 (1Н, мультиплет);
5,04-5,12 (4Н, мультиплет);
5,42-(1Н, широкий синглет);
5,46 (1Н, широкий синглет);
5,60-5,69-(2Н, мультиплет);
5,85 (1Н, дублет из дублетов, J = 9,7 и 5,9);
5,98 (1Н, дублет, J = 9,7 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 2950, 1720, 1255, 1030, 840.
Масс-спектр (m/e): 700 (M+), 643, 532, 475, 400.
[α]
Пример 20. (4R,6R)-6-2{(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-Гексагидро-6-трет- бутилдиметилсилилокси-8-[(2,2-диалиил-4-пентеноилокси)-2-метил-1-нафтил] этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-он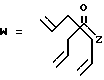
Следовали методике, аналогичной описанной в примере 6, но использовали 1,10 г (2,0 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет-бутил- диметилсилилокси-8-гидрокси-2-метил-1-нафтил] этил} тетрагидро-4-трет- бутилдиметилсилилокси-2Н-пиран-2-она (полученного как описано выше в примере B) и 584 мг (2,9 ммоль хлористого 2,2-диаллил-4-пентеноила, получая 585 мг названного соединения.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
2,29 (3Н, дублет, J = 7,2 Гц);
3,38 (3Н, дублет, J = 7,2 Гц);
4,28-4,30 (1Н, мультиплет);
4,41-4,45 (1Н, мультиплет);
4,54-4,61 (1Н, мультиплет);
5,03-5,16 (6Н, мультиплет);
5,43 (1Н, широкий синглет);
5,45 (1Н, широкий синглет);
5,53-5,80 (3Н, мультиплет);
5,85 (1Н, дублет из дублетов, J = 9,7 и 5,9 Гц);
5,98 (1Н, дублет, J = 9,7 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 2950, 1720, 1250, 1080, 835.
Масс-спектр (m/e): 712 (M+), 555, 532, 475, 343.
Пример 21. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилокси-8-(2-этил-2-метилвалерилокси)-2-метил-1-нафтил] этил} тетрагидро-4-трет-бутилдиметилсилилокси-2H-пиран-2-он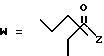
Следовали методике, аналогичной описанной в примере 6, но использовали 1,0 г (1,8 ммоль) (4R,6R)-6{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-гидрокси-2-метил-1-нафтил]этил}тетрагидро-4-трет- бутилдиметилсилилокси-2Н-пиран-2-она (полученного как описано выше в примере B) и 1,18 г (7,3 ммоль) хлористого 2-этил-2-метилвалерина, получая 956 мг названного соединения.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
4,27-4,30 (1Н, мультиплет);
4,40-4,44 (1Н, мультиплет);
4,54 - 4,53 (1Н, мультиплет);
5,36 (1Н, широкий синглет);
5,46 (1Н, широкий синглет);
5,85 (1Н, дублет из дублетов, J = 9,7 и 5,9 Гц);
5,98 (1Н, дублет, J = 9,7 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 2950, 1720, 1250, 1080, 840.
Масс-спектр (m/e): 676 (M+), 619, 591, 532, 475.
[α]
Использованием стереоспецифических исходных веществ, т.е. использованием (2S)- или (2S)-2-этил-2-метилвалерилхлорида, могут быть получены соответствующие стереоизомеры названного соединения, как, например, показано в примере 22. Любой из этих двух стереоизомеров, полученных этим способом, может быть затем использован в качестве исходного вещества в примере 43.
Пример 22. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-[(2S)-2-этил-2-метилвалерилокси] -2-метил-1-нафтил] этил}тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-он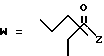
Хлористый тионил (121 мкл (1,66 ммоль) добавляли к 60 мг (0,42 ммоль) (-)-(2S)-2-этил-2-метилпентановой кислоты (полученной, как описано в синтезе 16), и смесь нагревали при 100oC в течение одного часа. В конце этого времени смесь концентрировали упариванием при пониженном давлении. Весь полученный хлористый (-)-(2S)-2-этил-2-метилвалерил добавляли без очистки к раствору 453 мг (0,83 ммоль) (4R,6R)-6- {2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет-бутилдиметилсилилокси- 8-гидрокси-2-метил-1-нафтил] этил} тетрагидро-4-трет-бутилдиметилсилилокси- 2H-пиран-2-она (полученного как описано в примере B), 203 мг (1,66 ммоль) 4-(N,N-диметиламино)пиридина, каталитического количества (20 мг) 4-диметиламинопиридина и 232 мкл триэтиламина в 2,5 мл толуола, и полученную смесь нагревали с обратным холодильником в течение 24 ч. В конце этого времени реакционную смесь охлаждали до комнатной температуры и затем смешивали с 10 мл 10%-ного (по весу в расчете на объем) водного раствора хлористого водорода. Водную смесь трижды экстрагировали, используя каждый раз 20 мл этилацетата. Соединенные экстракты затем промывали насыщенным водным раствором хлорида натрия, после чего промытый раствор сушили над безводным сульфатом натрия. Растворитель затем удаляли дистилляцией при пониженном давлении, и полученный бледно-желтый маслянистый остаток очищали колонной флэш-хроматографией, пропуская через силикагель с использованием в качестве элюента смеси гексана с этилацетатом, в объемном соотношении 5:1, получая 88 мг (выход 31%) названного соединения в виде пенообразного вещества.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
4,27-4,30 (1Н, мультиплет);
4,40-4,44 (1Н, мультиплет);
4,54-4,58 (1Н, мультиплет);
5,36 (1Н, широкий синглет);
5,46 (1Н, широкий синглет);
5,85 (1Н, дублет из дублетов, J = 9,7 и 5,9 Гц);
5,98 (1Н, дублет, J = 9,7 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 2950, 1720, 1250, 1080, 840.
Масс-спектр (m/e): 676 (M+).
[α]
Пример 23. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-(2,2-диметилгексаноилокси)-2-метил-1-нафтил] этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-он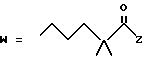
Следовали методике, аналогичной описанной в примере 6, но использовали 1,10 г (2,0 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-8-трет- бутилдиметилсилилокси-8-гидрокси-2-метил-1-нафтил] этил} тетрагидро-4-трет- бутилдиметилсилилокси-2Н-пиран-2-она, (полученного как описано выше в примере B) и 1,33 г (10,0 ммоль) 2,2-диметилгексаноилхлорида, получая 1,22 г названного соединения.
Спектр ядерного магнитного резонанса: (400 МГц, CDCl3) δ , в миллионных долях,
1,20 (6Н, синглет);
4,27-4,30 (1Н, мультиплет);
4,40-4,44 (1Н, мультиплет);
4,55-4,61 (1Н, мультиплет);
5,34 (1Н, широкий синглет);
5,47 (1Н, широкий синглет);
5,84 (1Н, дублет из дублетов, J = 9,6 и 5,9 Гц);
5,98 (1Н, дублет, J = 9,6 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 2950, 1720, 1250, 1080, 835.
Масс-спектр (m/e): 676 (M+), 619, 532, 475, 343.
[α]
В каждом из нижеследующих примеров с 24 до 46 описано получение соединений следующий формулы: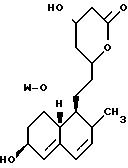
т. е. получение соединении формулы (I), в которой R1 представляет собой группу формулы (III) и R6 представляет собой атом водорода. Каждая группа W, как она определена в последующих примерах, присоединяется к формуле, показанной выше, через связь, обозначенную Z.
Пример 24. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-8- (2-этил-2-метилбутирилокси)-2-метил-1-нафтил)этил} тетрагидро-4-гидрокси- 2Н-пиран-2-он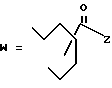
Раствор 600 мг (0,9 ммоль) (4R,6R)-6-{2-[1S,2S,6S,8S,8aR)-1,2,6,7,8,8a- гексагидро-6-трет-бутилдиметилсилилокси-8-(2-этил-2-метилбутирилокси)-2- метил-1-нафтил] этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного как описано выше в примере 5) в 2 мл тетрагидрофурана, добавляли к смеси 12,7 мл 1 М раствора фтористого тетрагидробутиламмония в тетрагидрофуране и 1,27 мл уксусной кислоты, и смесь перемешивали при комнатной температуре в течение 15 ч. В конце этого времени тетрагидрофуран удаляли из реакционной смеси отгонкой при пониженном давлении. Остаток затем разбавляли 50 мл этилацетата, и разбавленный раствор дважды промывали водой по 60 мл, трижды промывали насыщенным водным раствором бикарбоната натрия, каждый раз по 30 мл, и один раз промывали насыщенным водным раствором хлорида натрия, в указанной последовательности. Органический слой затем сушили над безводным сульфатом магния и удаляли из смеси фильтрованием, после чего растворитель удаляли дистилляцией при пониженном давлении. Остаток очищали испарительной колонной хроматографией, пропуская через силикагель при использовании в качестве элюента этилацетата, получая 387 мг (выход 98%) названного соединения в виде бесцветного твердого вещества. Это соединение перекристаллизовывали из смеси гексана с этилацетатом, получая названное соединение в виде бесцветных призматических кристаллов, плавящихся при температурах от 152 до 154oC.
Элементный анализ для C25H38O6:
Вычислено: C - 69,10%, H - 8,81%;
Найдено: C - 68,83%, H - 8,70%
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
0,81 (3Н, триплет, J = 7,3 Гц);
0,82 (3Н, триплет, J = 7,3 Гц);
0,90 (3Н, дублет, J = 7,3 Гц);
1,03 (3Н, синглет);
4,33-4,44 (2Н, мультиплет);
4,54-4,85 (1Н, мультиплет);
5,04 (1Н, широкий синглет);
5,37 (1Н, широкий синглет);
5,89 (1Н, дублет из дублетов, J = 5,9 и 9,8 Гц);
6,00 (1Н, дублет, J = 9,8 Гц).
νmax (см-1 инфракрасного абсорбционного спектра (CHCl3): 3450, 2950, 1720, 1150.
Масс-спектр (m/e): 434 (M+), 416, 304, 286.
[α]
Пример 25. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси- 8-(2-этилбутирилокси)-2-метил-1-нафтил] этил}тетрагидро-4-гидрокси-2Н-пиран- 2-он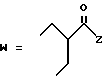
Следовали методике, аналогичной описанной в примере 24, но использовали 1,0 г (1,6 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,6S,8aR)-1,2,6,7,8,6а-гексагидро- 6-трет-бутилдиметилсилилокси-8-(2-этилбутирилокси)-2-метил-1-нафтил] - этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного, как описано выше в примере 2), получая 649 мг названного соединения, плавящегося при 158oC.
Элементный анализ для C24H36O6:
Вычислено: C - 68,55%, H - 8,63%;
Найдено: C - 68,33%, H - 8,71%,
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
4,32-4,46 (2Н, мультиплет);
4,54 - 4,66 (1Н, мультиплет);
5,45 (1Н, широкий синглет);
5,58 (1Н, широкий синглет);
5,90 (1Н, дублет из дублетов, J = 9,8 и 5,9 Гц);
6,02 (1Н, дублет, J = 9,8 Гц).
Значения νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 3450, 2950, 1720.
Масс-спектр (m/e): 420 (M+), 403, 321, 304, 286.
[α]
Пример 26. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси- 8-[(S)-2-метилвалерилокси] -2-метил-1-нафтил]этил}-тетрагидро-4-гидрокси- 2Н-пиран-2-он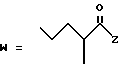
Следовали методике, аналогичной описанной в примере 24, но использовали 1,38 г (2,1 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро- 6-трет-бутилдиметилсилилокси-8-[(S)-2-метилвалерилокси] -2-метил-1-нафтил] - этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного, как описано выше в примере 3), получая 674 мг названного соединения, плавящегося при 134oC.
Элементный анализ для C24H36O6:
Вычислено: C - 68,55%, H - 8,63%,
Найдено: C - 68,36%, H - 8,77%.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
0,89 (3Н, триплет, J = 7,3 Гц);
0,91 (3Н, дублет, J = 7,3 Гц);
1,11 (3Н, дублет, J = 7,3 Гц);
2,32 (1Н, широкий синглет, взаимозаменяемый с D2O);
2,73 (1Н, дублет из дублетов, J = 17,7 и 5,1 Гц);
4,33-4,43 (2Н, мультиплет);
4,57-4,64 (1Н, мультиплет);
5,41 (1Н, синглет);
5,57 (1Н, синглет);
5,90 (1Н, дублет из дублетов, J = 9,5 и 5,9 Гц);
6,00 (1Н, дублет, J = 9,5 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 3501, 3453, 2964, 1724, 1699, 1132, 1044, 861.
Масс-спектр (m/e): 420 (M+), 403, 304.
[α]
Пример 27. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси- 8-(2-пропилвалерилокси)-2-метил-1-нафтил] этил} тетрагидро-4-гидрокси-2Н- пиран-2-он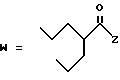
Следовали методике, аналогичной описанной в примере 24, но использовали 1,13 г (1,7 ммоль) (4R,6R))-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро- 6-трет-бутилдиметилсилилокси-8-(2-пропилвалерилокси)-2-метил-1-нафтил] - этил}тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного как описано выше в примере 4), получая 668 г названного соединения, плавящегося при температурах от 165 до 166oC.
Элементный анализ для C26H40O6:
Вычислено: C - 69,61%, H - 8,99%:
Найдено: C - 69,67%, H - 6,95%.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
4,33-4,45 (2Н, мультиплет);
4,54-4,65 (1Н, мультиплет);
5,43 (1Н, широкий синглет);
5,56 (1Н, широкий синглет);
5,90 (1Н, дублет из дублетов, J = 9,3 и 5,9 Гц);
6,01 (1Н, дублет, J = 9,8 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 3450, 2950, 1720.
Масс-спектр (m/e): 448 (M+), 430, 304, 286.
[α]
Пример 28. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси- 8-(3,3-диметилбутирилокси)-2-метил-1-нафтил] этил}-тетрагидро-4-гидрокси- 2Н-пиран-2-он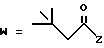
Следовали методике, аналогичной описанной в примере 24, но использовали 1,45 г (1,8 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро- 6-трет-бутилдиметилсилилокси-8-(3,3-диметилбутирилокси)-2-метил-1-нафтил] -этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного как описано выше в примере 1), получая 640 мг названного соединения, плавящегося при 155oC.
Элементный анализ для C24H36O6:
Вычислено: C - 68,55%, H - 8,63%;
Найдено: C - 68,32%, H - 8,81%,
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
0,80 (3Н, дублет, J = 6,8 Гц);
1,02 (9Н, синглет);
2,05 (1Н, мультиплет, взаимозаменяемый с D2O);
2,20 (2Н, синглет);
4,32-4,48 (2Н, мультиплет);
4,56-4,67 (1Н, мультиплет);
5,40 (1Н, широкий синглет);
5,55 (1Н, широкий синглет);
5,88 (1Н, дублет из дублетов, J = 9,8 и 5,9 Гц);
6,00 (1Н, дублет, J = 9,8 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 3400, 2950, 1720.
Масс-спектр (m/e): 420 (M+), 402, 384, 346, 321.
[α]
Пример 29. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси- 8-(2,2-диэтилбутирилокси)-2-метил-1-нафтил] этил} -тетрагидро-4-гидрокси- 2Н-пиран-2-он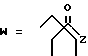
Следовали методике, аналогичной описанной в примере 24, но использовали 2,52 г (3,8 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8а-гексагидро- 6-трет-бутилдиметилсилилокси-3-(2,2-диэтилбутирилокси)-2-метил-1-нафтил] - этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного, как описано выше в примере 6), получая 1,05 г названного соединения, плавящегося при температурах от 146 до 148oC с разложением.
Элементный анализ для C26H40O6:
Вычислено: C - 69,61%, H - 8,99%;
Найдено: C - 69,53%, H - 9,10%.
Спектр ядерного магнитного резонанса: (360 МГц, CDCl3) δ , в миллионных долях,
0,76 (9Н, триплет, J = 7,5 Гц);
0,91 (3Н, дублет, J = 7,0 Гц);
4,35-4,41 (2Н, мультиплет);
4,56-4,64 (1Н, мультиплет);
5,45 (1Н, широкий синглет);
5,57 (1Н, широкий синглет);
5,90 (1Н, дублет из дублетов, J = 9,7 и 5,9 Гц);
6,01 (1Н, дублет, J = 9,7 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (KBr): 3428, 2967, 1717, 1255, 1142, 1041.
Масс-спектр (m/e): 448 (M+), 430, 304, 286.
[α]
Пример 30. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси- 8-(2,2-диметил-4-пентаноилокси)-2-метил-1-нафтил] -этил}тетрагидро-4- гидрокси-2Н-пиран-2-он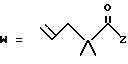
Следовали методике, аналогичной описанной в примере 24, но использовали 227 мг (0,3 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-(2,2-диметил-4-пентеноилокси)-2-метил-1-нафтил] этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного, как описано выше в примере 7), получая 127 мг названного соединения, плавящегося при температурах от 141 до 142oC.
Элементный анализ для C25H36O6;
Вычислено: C - 69,42%, H - 8,39%;
Найдено:: C - 69,15%, H - 8,34%.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
0,90 (3Н, дублет, J = 7,3 Гц);
1,14 (6Н, синглет);
2,25 (2Н, дублет, J = 7,3 Гц);
4,33-4,45 (2Н, мультиплет);
4,55-4,36 (1Н, мультиплет);
5,01-5,10 (2Н, мультиплет);
5,37 (1Н, широкий синглет);
5,57 (1Н, широкий синглет);
5,61-5,76 (1Н, мультиплет);
5,79 (1Н, дублет из дублетов, J = 9,8 и 5,9 Гц);
6,00 (1Н, дублет, J = 9,8 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 3450, 2950, 1720, 1250.
Масс-спектр (m/e): 432 (M+), 415, 345, 304, 286.
[α]
Пример 31. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси-8- (2-аллил-4-пентеноилокси)-2-метил-1-нафтил]этил}-тетрагидро-4-гидрокси- 2Н-пиран-2-он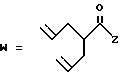
Следовали методике, аналогичной описанной в примере 24, но использовали 966 мг (1,4 ммоль) (4R,6R)-6{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,6а-гексагидро-6-трет- бутилдиметилсилилокси-8-(2-аллил-4-пентеноилокси)-2-метил-1-нафтил] -этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного, как описано выше в примере 8), получая 555 мг названного соединения, плавящегося при температурах от 159 до 160oC.
Элементный анализ для C26H36O6•1/2H2O:
Вычислено: C - 66,85%, H - 8,22%;
Найдено: C - 68,85%, H - 8,10%.
Спектр ядерного магнитного резонанса: (270 МГц, гексадейтерированный диметилсульфоксид) δ, в миллионных долях,
0,84 (3Н, дублет, J = 6,8 Гц);
4,08-4,25 (2Н, мультиплет);
4,41-4,52 (1Н, мультиплет);
4,76 (1Н, дублет, J = 5,9 Гц, взаимозаменяемый с D2O);
4,99-5,07 (4Н, мультиплет);
5,17 (1Н, дублет, J = 2,9 Гц, взаимозаменяемый с D2O);
5,26 (1Н, широкий синглет);
5,49 (1Н, широкий синглет);
5,61-5,78 (2Н, мультиплет);
5,84 (1Н, дублет из дублетов, J = 9,8 и 5,9 Гц);
5,96 (1Н, дублет, J = 9,8 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 3400, 2950, 1720, 1240.
Масс-спектр (m/e): 444 (M+), 427, 304, 161.
[α]
Пример 32. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси-8- (2-бутилгексаноилокси)-2-метил-1-нафтил)этил} -тетрагидро-4-гидрокси-2Н- пиран-2-он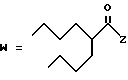
Следовали методике, аналогичной описанной в примере 24, но использовали 785 мг (1,1 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-(2-бутилгексаноилокси)-2-метил-1-нафтил] -этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного, как описано выше в примере 9), получая 520 мг названного соединения, плавящегося при температурах от 143 до 145oC.
Элементный анализ для C28H44O6:
Вычислено: C - 70,56%, H - 9,30%;
Найдено: C - 70,27%, H - 9,36%.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
4,34 - 4,45 (2Н, мультиплет);
4,55 - 4,65 (1Н, мультиплет);
5,47 (1Н, широкий синглет);
5,59 (1Н, широкий синглет);
5,89 (1Н, дублет из дублетов, J = 9,8 и 5,9 Гц);
6,01 (1Н, дублет, J = 9,8 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 3450, 2950, 1720.
Масс-спектр (m/e): 476 (M+), 459, 356, 321.
[α]
Пример 33. (4R, 6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-Гексагидро-6-гидрокси-8- гексаноилокси-2-метил-1-нафтил)этил} тетрагидро-4-гидрокси-2Н-пиран-2-он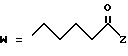
Следовали методике, аналогичной описанной в примере 24, но использовали 338 мг (0,5 ммоль) (4R,6R)-6{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,6а-Гексагидро- 6-трет-бутилдиметилсилилокси-8-гексаноилокси-2-метил-1-нафтил] этил} тетрагидро- 4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного как описано выше в примере 10), получая названное соединение, плавящееся при температуре от 138 до 139oC.
Элементный анализ для C24H36O6:
Вычислено: C - 68,55%, H - 8,63%;
Найдено: C - 68,34%, H - 8,67%.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
4,35 - 4,46 (2Н, мультиплет);
4,58 - 4,68 (1Н, мультиплет);
5,42 (1Н, широкий синглет);
5,57 (1Н, широкий синглет);
5,90 (1Н, дублет из дублетов, J = 9,8 и 5,9 Гц);
6,00 (1Н, дублет, J = 9,8 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 3450, 2950, 1720, 1250.
Масс-спектр (m/e): 420 (M+), 403, 321, 304.
[α]
Пример 34. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси- 8-изовалерилокси-2-метил-1-нафтил]этил}тетрагидро-4-гидрокси-2Н-пиран-2-он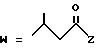
Следовали методике, аналогичной описанной в примере 24, но использовали 1,1 г (1,7 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-изовалерилокси-2-метил-1-нафтил] этил}-тетрагидро- 4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного, как описано выше в примере 11), получая 488 мг названного соединения, плавящегося при температурах от 153 до 155oC.
Элементный анализ для C23H34O6•1/2H2O:
Вычислено: C - 67,96%, H - 8,43%,
Найдено: C - 67,31%, H - 8,30%.
Спектр ядерного магнитного резонанса: (270 МГц, гексадейтерированный диметилсульфоксид) δ , в миллионных долях,
0,84 (3Н, дублет, J = 6,9 Гц);
0,88 (6Н, дублет, J = 6,3 Гц);
4,04-4,10 (1Н, мультиплет);
4,10-4,16 (1Н, мультиплет);
4,43-4,50 (1Н, мультиплет);
4,77 (1Н, дублет, J = 6,3 Гц, взаимозаменяемый с D2O);
5,16 (1Н, дублет, J = 2,9 Гц, взаимозаменяемый с D2O);
5,23 (1Н, широкий синглет);
5,49 (1Н, широкий синглет);
5,84 (1Н, дублет из дублетов, J = 9,3 и 5,9 Гц);
5,96 (1Н, дублет, J = 9,8 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 3350, 2880, 1725, 1250.
Масс-спектр (m/e): 406 (M+), 322, 304.
[α]
Пример 35. (4R,6R))-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси-3- пивалоилокси-2-метил-1-нафтил] этил} тетрагидро-4-гидрокси-2Н-пиран-2-он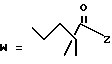
Следовали методике, аналогичной описанной в примере 24, но использовали 571 мг (0,0 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-пивалоилокси-2-метил-1-нафтил] этил}тетрагидро- 4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного, как описано выше в примере 12), получая 354 мг названного соединения, плавящегося при температурах от 132 до 133oC.
Элементный анализ для C23H34O6:
Вычислено: C - 67,96%, H - 8,43%;
Найдено: C - 37,87%, H - 8,53%.
Спектр ядерного магнитного резонанса: (270 МГц, гексадейтерированный диметилсульфоксид) δ , в миллионных долях,
0,85 (3Н, дублет, J = 7,0 Гц);
1,10 (9Н, синглет);
4,08-4,15 (2Н, мультиплет);
4,46-4,50 (1Н, мультиплет);
4,78 (1Н, дублет, J = 6,3 Гц, взаимозаменяемый с D2O);
5,17 (1Н, широкий синглет);
5,17 (1Н, дублет, J = 3,3 Гц, взаимозаменяемый с D2O);
5,51 (1Н, широкий синглет);
5,84 (1Н, дублет из дублетов, J = 9,7 и 5,8 Гц);
5,97 (1Н, дублет, J = 9,7 Гц).
νmax(см-1) инфракрасного абсорбционного спектра (CHCl3): 3450, 2950, 1720, 1160.
Масс-спектр (m/e): 406 (M+), 321, 304, 286.
[α]
Пример 36. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси- 8-(2,2-диметилвалерилокси)-2-метил-1-нафтил] этил}-тетрагидро-4-гидрокси-2Н-пиран-2-он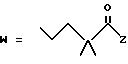
Следовали методике, аналогичной описанной в примере 24, но использовали 1,29 г (1,9 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,65,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-(2,2-диметилвалерилокси)-2-метил-1-нафтил]-этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного, как описано выше в примере 13), получая 817 мг названного соединения, плавящегося при температурах от 143 до 144oC.
Элементный анализ для C25H38O6:
Вычислено: C - 69,10%, H - 8,81%;
Найдено: C - 68,86%, H - 8,91%.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
1,13 (6Н, синглет);
4,32-4,43 (2Н, мультиплет);
4,54-4,66 (1Н, мультиплет);
5,35 (1Н, широкий синглет);
5,56 (1Н, широкий синглет);
5,90 (1Н, дублет из дублетов, J = 9,8 и 5,9 Гц);
6,01 (1Н, дублет, J = 9,8 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 3450, 2950, 1720, 1160.
Масс-спектр (m/e): 434 (M+) 321, 304, 286.
[α]
Пример 37. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси-8- (2-аллил-2-метил-4-пентеноилокси)-2-метил-1-нафтил] этил}тетрагидро-4-гидрокси- 2Н-пиран-2-он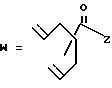
Следовали методике, аналогичной описанной в примере 24, но использовали 2,07 г (3,0 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-(2-аллил-2-метил-4-пентеноилокси)-2-метил-1-нафтил] этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного, как описано выше в примере 14), получая 1,29 г названного соединения, плавящегося при температурах от 115 до 116oC.
Элементный анализ для C27H38O6:
Вычислено: C - 70,72%, H - 8,35%;
Найдено: C - 70,48%, H - 8,46%.
Спектр ядерного магнитного резонанса: (270 МГц, гексадейтерированный диметилсульфоксид) δ , в миллионных долях,
0,84 (3Н, дублет, J = 6,9 Гц);
1,01 (3Н, синглет);
4,09-4,11 (1Н, мультиплет);
4,15-4,18 (1Н, мультиплет);
4,45-4,50 (1Н, мультиплет);
4,79 (1Н, дублет, J = 6,0 Гц, взаимозаменяемый: с D2O);
5,04-5,08 (4Н, мультиплет);
5,19 (1Н, дублет, J = 3,2 Гц, взаимозаменяемый с D2O);
5,25 (1Н, широкий синглет);
5,50 (1Н, широкий синглет);
5,59-5,70 (2Н, мультиплет);
5,84 (1Н, дублет из дублетов, J = 9,5 и 5,9 Гц);
5,97 (1Н, дублет, J = 9,7 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 3450, 2950, 1720, 1250.
Масс-спектр (m/e): 458 (M+), 422, 304, 286.
[α]
Пример 38. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,9а-гексагидро-6-гидрокси-8-(2- метил-3-пропилвалерилокси)-2-метил-1-нафтил] -этил}тетрагидро-4-гидрокси- 2Н-пиран-2-он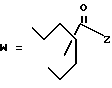
Следовали методике, аналогичной описанной в примере 24, но использовали 956 мг (1,4 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-(2-метил-2-пропилвалерилокси)-2-метил-1-нафтил] этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного, как описано выше в примере 15), получая 550 мг названного соединения, плавящегося при температурах от 109 до 111oC.
Элементный анализ для C27H42O6•H2O
Вычислено: C - 67,61%, H - 8,83%;
Найдено: C - 67,65%, H - 8,79%.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях;
4,35-4,40 (2Н, мультиплет);
4,57-4,63 (1Н, мультиплет);
5,40 (1Н, широкий синглет);
5,58 (1Н, широкий синглет);
5,90(1Н, дублет из дублетов, J = 9,7 и 5,9 Гц);
6,02 (1Н, дублет, J = 9,7 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 3450, 2950, 1720, 1150.
Масс-спектр (m/e): 462 (M+), 444, 321, 304.
[α]
Пример 39. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси- 8-(2,2-диэтилвалерилокси)-2-метил-1-нафтил] этил} -тетрагидро-4-гидрокси- 2Н-пиран-2-он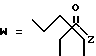
Следовали методике, аналогичной описанной в примере 24, но использовали 184 мг (0,3 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро- 6-трет-бутилдиметилсилилокси-8-(2,2-диэтилвалерилокси)-2-метил-1-нафтил] - этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного, как описано выше в примере 16), получая 97 мг названного соединения, плавящегося при температуре от 130 до 131oC.
Элементный анализ для C27H42O6•CH3COOC2H5:
Вычислено: C - 67,60%, H - 9,15%,
Найдено: C - 67,32%, H - 9,10%.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
0,76 (3Н, триплет, J = 7,6 Гц);
2,74 (1Н, дублет из дублетов, J = 17,6 и 5,1 Гц);
4,35-4,42 (2Н, мультиплет);
4,56-4,63 (1Н, мультиплет);
5,44 (1Н, широкий синглет);
5,57 (1Н, широкий синглет);
5,89 (1Н, дублет из дублетов, J = 9,7 и 5,9 Гц);
6,01 (1Н, дублет, J = 9,7 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (KBr): 3350, 2950, 1720, 1700.
Масс-спектр (m/e): 462 (M+), 444, 321, 304.
[α]
Пример 40. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8а-гексагидро-6-гидрокси-8- (2-изопропил-3-метилбутирилокси)-2-метил-1-нафтил] -этил}тетрагидро-4-гидрокси- 2Н-пиран-2-он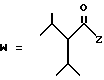
Следовали методике, аналогичной в примере 24, но использовали 190 мг (0,3 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8а-Гексагидро-6-трет- бутилдиметилсилилокси-8-(2-изопропил-3-метилбутирилокси)-2-метил-1-нафтил] этил}тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного как описано выше в примере 17), получая 100 мг названного соединения, плавящегося при температурах от 210 до 211oC.
Элементный анализ для C26H40O6:
Вычислено: C - 69,61%, H - 8,99%;
Найдено: C - 69,35%, H - 9,04%.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
2,32-2,44 (2Н, мультиплет);
2,56-2,66 (2Н, мультиплет);
2,75 (1Н, дублет из дублетов, J = 17,6 и 5,1 Гц);
4,34-4,40 (1Н, мультиплет);
4,43-4,50 (1Н, мультиплет);
4,56 - 4,64 (1Н, мультиплет);
5,50 (1Н, широкий синглет);
5,57 (1Н, широкий синглет);
5,90 (1Н, дублет из дублетов, J = 9,8 и 6,0 Гц);
6,01 (1Н, дублет, J = 9,8 Гц);
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 3450, 2950, 1720.
Масс-спектр (m/e): 448 (M+), 418, 321, 304.
[α]
Пример 41. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси-8- (2,2-диэтил-4-пентеноилокси)-2-метил-1-нафтил] -этил} тетрагидро-4-гидрокси-2Н- пиран-2-он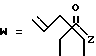
Следовали методике, аналогичной описанной в примера 24, но использовали 1,95 г (2,8 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-(2,2-диэтил-4-пентеноилокси)-2-метил-1-нафтил] этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного, как описано выше в примере 18), получая 1,04 г названного соединения, плавящегося при температурах от 107 до 108oC.
Элементный анализ для C27H40O6•CH2Cl2:
Вычислено: C - 61,64%, H - 7,76%;
Найдено: C - 61,63%, H - 7,95%;
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных частях,
0,90 (3Н, дублет, J = 7,1 Гц);
2,30 (2Н, дублет J = 7,3 Гц);
2,75 (1Н, дублет из дублетов, J = 17,6 и 5,1 Гц);
4,35-4,45 (2Н, мультиплет);
4,55-4,64 (H, мультиплет);
5,03-5,12 (2Н, мультиплет);
5,45 (1Н, широкий синглет);
5,57 (1Н, широкий синглет);
5,57-5,69 (1Н, мультиплет);
5,90 (1Н, дублет из дублетов, J = 9,7 и 5,9 Гц);
6,01 (1Н, дублет, J = 9,7 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (KBr): 3340, 2970, 1720, 1690.
Масс-спектр (m/e): 460 (M+), 442, 321, 304.
[α]
Пример 42. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси-8- (2-аллил-2-этил-4-пентеноилокси)-2-метил-1-нафтил] этил} тетрагидро-4-гидрокси- 2Н-пиран-2-он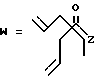
Следовали методике, аналогичной описанной в примере 24, но использовали 1,63 г (2,3 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-(2-аллил-2-этил-4-пентеноилокси)-2-метил-1-нафтил] этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного, как описано выше в примере 19), получая 1,10 г названного соединения, плавящегося при температурах от 99 до 100oC.
Элементный анализ для C28H40O6•1/2H2O:
Вычислено: C - 69,82%, H - 8,58%;
Найдено: C - 69,33%, H - 8,62%.
Спектр ядерного магнитного резонанса: (270 МГц, гексадейтерированный диметилсульфоксид)δ, в миллионных долях,
0,75 (3Н, триплет, J = 7,4 Гц);
0,84 (3Н, дублет, J = 6,8 Гц);
4,09-4,10 (1Н, мультиплет);
4,14-4,17 (1Н, мультиплет);
4,44-4,48 (1Н, мультиплет);
4,81 (1Н, дублет, J = 6,2 Гц, взаимозаменяемый с D2O);
5,06-5,10 (4Н, мультиплет);
5,19 (1Н, дублет, J = 3,1 Гц, взаимозаменяемый с D2O);
5,29 (1Н, широкий синглет);
5,50 (1Н, широкий синглет);
5,56-5,66 (2Н, мультиплет);
5,84 (1Н, дублет из дублетов, J = 9,7 и 5,8 Гц);
5,98 (1Н, дублет, J = 9,7 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (KBr): 3350, 2950, 1710, 1255, 1040.
Массовый спектр (m/e):
472 (M+), 321, 304, 286.
[α]
Пример 43. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-8- (2,2-диаллил-4-пентеноилокси)-2-метил-1-нафтил] этил}-тетрагидро-4-гидрокси- 2Н-пиран-2-он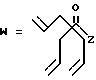
Следовали методике, аналогичной описанной в примере 24, но использовали 267 мг (0,4 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR))-1,2,6,7,8,8a-Гексагидро-6-трет- бутилдиметилсилилокси-8-(2,2-диаллил-4-пентеноилокси)-2-метил-1-нафтил] этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного, как описано выше в примере 20), получая 180 мг названного соединения, плавящегося при температурах от 118 до 119oC.
Элементный анализ для C29H40O6:
Вычислено: C - 71,87%, H - 8,32%;
Найдено: C - 71,84%, H - 8,29%.
Спектр ядерного магнитного резонанса: (270 МГц, гексадейтерированный диметилсульфоксид) δ , в миллионных долях,
0,84 (3Н, дублет, J = 7,0 Гц);
2,21 (6Н, дублет, J = 7,3 Гц);
4,08-4,12 (1Н, мультиплет);
4,16-4,19 (1Н, мультиплет);
4,45-4,49 (1Н, мультиплет);
4,80 (1Н, дублет, J = 6,3 Гц, взаимозаменяемый с D2O);
5,06-5,10 (6Н, мультиплет);
5,20 (1Н, дублет, J = 3,3 Гц, взаимозаменяемый D2O);
5,30 (1Н, широкий синглет);
5,51 (1Н, широкий синглет);
5,59-5,71 (3Н, мультиплет);
5,84 (1Н, дублет из дублетов, J = 9,7 и 5,8 Гц);
5,98 (1Н, дублет, J = 9,7 Гц),
νmax (см-1 инфракрасного абсорбционного спектра (CHCl3): 3450, 2950, 1720, 1220.
Масс-спектр (m/e): 484 (M+), 438, 304, 286.
[α]
Пример 44. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси-8- (2-этил-2-метилвалерилокси)-2-метил-1-нафтил] -этил} -тетрагидро-4-гидрокси- 2Н-пиран-2-он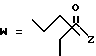
Следовали методике, аналогичной описанной в примере 24, но использовали 899 мг (2,0 ммоль) (4R,6R)-8-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро- 6-трет-бутилдиметилсилилокси-8-(2-этил-2-метилвалерилокси)-2-метил-1- нафтил]этил}тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного, как описано выше в примере 21), получая 279 мг названного соединения, плавящегося при температурах от 126 до 128oC.
Элементный анализ для C26H40O6:
Вычислено: C - 69,61%, H - 8,99%;
Найдено: C - 69,33%, H - 9,22%.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
1,07 (3Н, синглет);
4,37-4,39 (2Н, мультиплет);
4,57-4,63 (1Н, мультиплет);
5,41 (1Н, широкий синглет);
5,57 (1Н, широкий синглет);
5,89 (1Н, дублет из дублетов, J = 9,7 и 5,9 Гц);
6,00 (1Н, дублет, J = 9,7 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3):
3400, 2950, 1720, 1150.
Масс-спектр (m/e):
448 (M+), 304, 286, 268,
[α]
Методика примера 44, может быть применена с использованием одного из стереоизомеров, полученных в примере 21, в качестве исходного вещества для получения соответствующего стереоизомера соединения, примера 44, как, например, показано в примере 45.
Пример 45
(4R, 6R)-6-{ 2-[(1S, 2S,6S,8S,8aR)-1,2,6,7,8,8a-Гексагидро-6-гидрокси-8- [(2S)-2-этил-2-метилвалерилокси-2-метил-1-нафтил] -этил} тетрагидро-4- гидрокси-2Н-пиран-2-он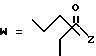
Следовали методике, аналогичной описанной в примере 28, но использовали 80 мг (0,12 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8а-гексагидро-6-трет- бутилдиметилсилилокси-8-[(2S)-2-этил-2-метилвалерилокси-2-метил-1-нафтил] этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного как описано выше в примере 22), получая 50 мг названного соединения, плавящегося при 127oC.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
1,07 (3Н, синглет);
4,37-4,39 (2Н, мультиплет);
4,57-4,63 (1Н, мультиплет);
5,41 (1Н, широкий синглет);
5,57 (1Н, широкий синглет);
5,89 (1Н, дублет из дублетов, J = 9,7 и 5,9 Гц);
6,00 (1Н, дублет, J = 9,7 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 3400, 2950, 1720, 1150.
Масс-спектр (m/e): 448 (M+).
[α]
Пример 46. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси-8- (2,2-диметилгексаноилокси)-2-метил-1-нафтил]-этил}-тетрагидро-4- гидрокси-2Н-пиран-2-он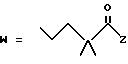
Следовали методике, аналогичной описанной в примере 24, но использовали 1,16 г (1,72 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8а-гексагидро-6-трет- бутилдиметилсилилокси-8-(2,2-диметилгексаноилокси)-2-метил-1-нафтил] этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного, как описано выше в примере 23), получая 660 мг названного соединения.
Элементный анализ для C26H40O6•1/4H2O:
Вычислено: C - 68,91%, H - 9,01%;
Найдено: C - 69,05%, H - 8,96%.
Спектр ядерного магнитного резонанса: (400 МГц, гексадейтерированный диметилсульфоксид) δ , в миллионных долях,
0,84 (3Н, дублет, J = 7,0 Гц);
0,85 (3Н, триплет, J = 7,0 Гц);
1,06 (6Н, синглет);
4,08-4,15 (2Н, мультиплет);
4,45-4,49 (1Н, мультиплет);
4,79 (1Н, дублет, J = 6,0 Гц);
5,18 (1Н, дублет, J = 3,6 Гц);
5,20 (1Н, широкий синглет);
5,50 (1Н, широкий синглет);
5,84 (1Н, дублет из дублетов, J = 9,7 и 6,0 Гц);
5,97 (1Н, дублет, J = 9,7 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 3450, 2950, 1720, 1160.
Масс-спектр (m/e): 448 (M+), 304, 286, 268.
[α]
В каждом из последующих примеров 47 - 69 описано получение соединений нижеприведенной формулы: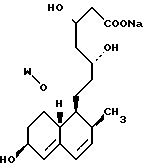
т. е. получение соединений формулы (I), в которой R1 представляет собой группу формулы (III), R5 представляет собой атом натрия и R6 представляет собой атом водорода. Каждая группа W, как она определена в последующих примерах, присоединяется к формуле, показанной выше, через связь, обозначенную буквой Z.
Пример 47. Натриевый (3R, 5R)-3,5-дигидрокси-7-[(1S,2S,6S,8S,8aR)-6-гидрокси- 2-метил-8-(3,3-диметилбутирилокси)-1,2,6,7,8,8а-гексагидро-1-нафтил]гептаноат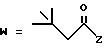
Воду (0,5 мл) добавляли к раствору 32 мг (0,076 ммоль) (4R,6R)-6-{2-[(1S, 2S, 6S,8S,8aR)-1,2,6,7,8,8а-гексагидро-6-гидрокси-8- (3,3-диметилбутирилокси)-2-метил-1-нафтил] этил} тетрагидро-4- гидрокси-2Н-пиран-2-она (полученному, как описано выше в примере 28) в 1 мл диоксана, после чего к смеси добавляли 0,8 мл (0,08 ммоль) 0,1 н. водного раствора гидроксида натрия. Полученную смесь перемешивали при комнатной температуре в течение 30 мин. В конце этого времени реакционную смесь подвергали лиофилизации, получая 35 мл названного соединения в виде бесцветного порошка.
Пример 48. Натриевый (3R, 5R)-3,5-дигидрокси-7-{(1S,2S,6S,8S,8aR)-6-гидрокси- 2-метил-8-(2-этилбутирилокси)-1,2,6,7,8,8а-гексагидро-1-нафтил] гептаноат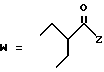
Следовали методике, аналогичной описанной в примере 47, но использовали 31 мг (0,074 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси- 8-(2-этилбутирилокси)-2-метил-1-нафтил]этил}тетрагидро-4-гидрокси-2Н- пиран-2-она (полученного как описано выше в примере 25), получая 36 мл названного соединения в виде бесцветного порошка.
Пример 49. Натриевый (3R, 5R)-3,5-дигидрокси-7-[(1S,2S,6S,8S,8aR)-6-гидрокси- 2-метил-8-[(S)-2-метилвалерилокси] -1,2,6,7,8,8a-гексагидро-1-нафтил]гептаноат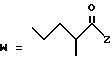
Следовали методике, аналогичной описанной в примере 47, но использовали 537 мг (1,28 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро- 6-гидрокси-8[(S)-2-метилвалерилокси-2-метил-1-нафтил] этил} тетрагидро-4- гидрокси-2Н-пиран-2-она (полученного, как описано выше в примере 26), получая 587 г названного соединения в виде бесцветного порошка.
Пример 50. Натриевый (3R, 5R)-3,5-дигидрокси-7-[(1S,2S,6S,8S,8aR)-6-гидрокси- 2-метил-8-(2-пропилвалерилокси)-1,2,6,7,8,8a-гексагидро-1-нафтил] гептаноат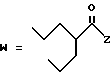
Следовали методике, аналогичной описанной в примере 47, но использовали 23 мг (0,051 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8а-гексагидро-6-гидрокси-8- {2-пропилвалерилокси)-2-метил-1-нафтил]этил}тетрагидро-4-гидрокси-2Н- пиран-2-она (полученного, как описано в примере 27), получая 25 мг названного соединения в виде бесцветного порошка.
Пример 51. Натриевый (3R, 5R)-3,5-дигидрокси-7-[(1S,2S,6S,8S,8aR)-6-гидрокси- 2-метил-8-(2-этил-2-метилбутирилокси)-1,2,6,7,8,8a-гексагидро- 1-нафтил]гептаноат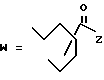
Следовала методике, аналогичной описанной в примере 47, но использовали 22 мг (0,051 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси- 8-(2-этил-2-метилбутирилокси)-2-метил-1-нафтил)этил} тетрагидро-4- гидрокси-2Н-пиран-2-она (полученного, как описано в примере 24), получая 24 мг названного соединения в виде бесцветного порошка.
Пример 52. Натриевый (3R, 5R)-3,5-дигидрокси-7-[(1S,2S,6S,8S,8aR)-6-гидрокси-2-метил-3-(2,2- диэтилбутирилокси)-1,2,6,7,8,8а-гексагидро-1-нафтил]гептаноат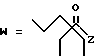
Следовали методике, аналогичной описанной в примере 47, но использовали 215 мг (0,46 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси-8- (2,2-диэтилбутирилокси)-2-метил-1-нафтил]этил}тетрагидро-4-гидрокси- 2Н-пиран-2-она (полученного, как, описано в примере 29), получая 234 мг названного соединения в виде бесцветного порошка.
Пример 53. Натриевый (3R, 5R)-3,5-дигидрокси-7-[(1S,2S,6S,8S,8aR)-6-гидрокси-2-метил-8-(2,2- диметил-4-пентеноилокси)-1,2,6,7,8,8a-гексагидро-1-нафтил]гептаноат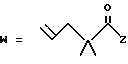
Следовали методике, аналогичной описанной в примере 47, но использовали 23 мг (0,053 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси-8- (2,2, -диметил-4-пентеноилокси)-2-метил-1-нафтил)этил}тетрагидро-4- гидрокси-2Н-пиран-2-она (полученного, как описано в примере 30), получая 26 мг названного соединения в виде бесцветного порошка.
Пример 54. Натриевый (3R, 5R)-3,5-дигидрокси-7-[(1S,2S,6S,8S,8aR)-6-гидрокси-2- метил-8-[2-аллил-4-пентеноилокси] -1,2,6,7,8,8a-гексагидро-1-нафтил]гептаноат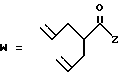
Следовали методике, аналогичной описанной в примере 47, но использовали 27 мг (0,061 ммоль)(4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8а-гексагидро-6-гидрокси- 8-(2-аллил-4-пентеноилокси)-2-метил-1-нафтил] этил}тетрагидро-4- гидрокси-2Н-пиран-2-она (полученного, как, описано в примере 31), получая 29 мг названного соединения в виде бесцветного порошка.
Пример 55. Натриевый (3R, 5R)-3,5-дигидрокси-7-[(1S,2S,6S,85,8aR)-7-гидрокси-2-метил-3-(2- бутилгексаноилокси)-1,2,6,7,8,8a-гексагидро-1-нафтил] гептаноат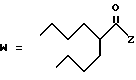
Следовали методике, аналогичной описанной в примере 47, но использовали 22 мг (0,046 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси-8-(2- бутилгексаноилокси)-2-метил-1-нафтил] этил}тетрагидро-4-гидрокси-2Н-пиран-2-она (полученного, как описано в примере 32), получая 24 мг названного соединения в виде бесцветного порошка.
Пример 56. Натриевый (3S, 5S)-3,5-дигидрокси-7-[(1S,2S,6S,8S,8aR)-6-гидрокси-2- метил-8-гексаноилокси-1,2,6,7,8,8a-гексагидро-1-нафтил]гептаноат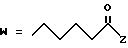
Следовали методике, аналогичной описанной в примере 47, но использовали 21 мг (0,50 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси-8- гексаноилокси-2-метил-1-нафтил] этил} тетрагидро-4-гидрокси-2Н-пиран-2-она (полученного, как описано в примере 33), получая 23 мг названного соединения в виде бесцветного порошка.
Пример 57. Натриевый (3R, 5R)-3,5-дигидрокси-7-[(1S,2S,6S,8S,8aR)-6-гидрокси-2-метил-3- изовалерилокси-1,2,6,7,8,8a-гексагидро-1-нафтил]гептаноат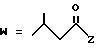
Следовали методике, аналогичной описанной в примере 47, но использовали 26 мг (0,064 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси-8- изовалерилокси-2-метил-1-нафтил] этил}тетрагидро-4-гидрокси-пиран-2-она (полученного, как описано в примере 34) и получали 29 мг названного соединения в виде бесцветного порошка.
Пример 58. Натриевый (3R,5R)-3,5-дигидрокси-7-[(1S,2S, 6S,8S,8aR)-6-гидрокси-2-метил-8-пивалоилокси-1,2,6,7,8,8a-гексагидро-1- нафтил]гептаноат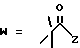
Следовали методике, аналогичной описанной в примере 47, но использовали 24 мг (0,060 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси-8- пивалоилокси-2-метил-1-нафтил]этил}тетрагидро-4-гидрокси-2Н-пиран-2-она (полученного как описано в примере 35), получая 29 мг названного соединения в виде бесцветного порошка.
Пример 59. Натриевый (3R, 5R)-3,5-дигидрокси-7-[(1S,2S,6S,8S,8aR)-6-гидрокси-2-метил-8- (2,2-диметилвалерилокси)-1,2,6,7,8,8a-гексагидро-1-нафтил]гептаноат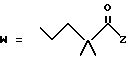
Следовали методике, аналогичной описанной в примере 47, но использовали 27 мг (0,062 ммоль) (4R,6R))-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси-8- (2,2-диметилвалерилокси)-2-метил-1-нафтил] этил} тетрагидро-4-гидрокси-2Н- пиран-2-она (полученного, как описано в примере 36), получали 29 мг названного соединения в виде бесцветного порошка.
Пример 60. Натриевый (3R, 5R)-3,5-дигидрокси-7-[(1S,2S,6S,8S,8aR)-6-гидрокси-2-метил-8-(2- аллил-2-метил-4-пентеноилокси)-1,2,6,7,8,8a-гексагидро-1-нафтил]гептаноат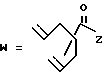
Следовали методике, аналогичной описанной выше в примере 47, но использовали 27 мг (0,059 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси-8- (2-аллил-2-метил-4-пентеноилокси)-2-метил-1-нафтил] этил} -тетрагидро-4- гидрокси-2Н-пиран-2-она (полученного и, как описано в примере 37), получили 30 мг названного соединения в виде бесцветного порошка.
Пример 61. Натриевый (3R,5R)-3,5-дигидрокси-7-[(1S,2S,6S,8S,8aR)-6-гидрокси-2-метил-8- (2-метил-2-пропилвалерилокси)-1,2,6,7,8,8a-гексагидро-1-нафтил гептаноат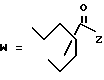
Следовали методике, аналогичной описанной в примере 47, но использовали 22 мг (0,048 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси-8- (2-метил-2-пропилвалерилокси)-2-метил-1-нафтил] этил} тетрагидро-4-гидрокси- 2Н-пиран-2-она (полученного, как описано в примере 38), получали 24 мг названного соединения в виде бесцветного порошка.
Пример 62. Натриевый (3R, 5R)-3,5-дигидрокси-7-[(1S,2S,6S,8S,8aR)-6-гидрокси-2-метил-8- (2,2-диэтилвалерилокси)-1,2,6,7,8,8a-гексагидро-1-нафтил]гептаноат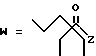
Следовали методике, аналогичной описанной в примере 47, но использовали 19 мг (0,041 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси-8- (2,2-диэтилвалерилокси)-2-метил-1-нафтил]этил}тетрагидро-4- гидрокси-2Н-пиран-2-она (полученного, как описано в примере 39), получали 21 мг названного соединения в виде бесцветного порошка.
Пример 63. Натриевый (3R, 5R)-3,5-дигидрокси-7-[(1S,2S,6S,8S,8aR)-6-гидрокси- 2-метил-8-(2-изопропил-3-метилбутирилокси)-1,2,6,7,8,8a-гексагидро-1- нафтил]гептаноат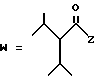
Следовали методике, аналогичной описанной в примере 47, но использовали 17 мг (0,038 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси-8- (2-изопропил-3-метилбутирилокси)-2-метил-1-нафтил] этил}-тетрагидро-4- гидрокси-2Н-пиран-2-она (полученного, как описано в примере 40), получали 19 мг названного соединения в виде бесцветного порошка.
Пример 64. Натриевый (3R, 5R)-3,5-дигидрокси-7-[(1S,2S,6S,8S,8aR)-6-гидрокси-2-метил-8- (2,2-диэтил-4-пентаноилокси)-1,2,6,7,8,8a-гексагипро-1-нафтил]гептаноат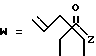
Следовали методике, аналогичной описанной в примере 47, но использовали 12 мг (0,026 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси-8- (2,2-диэтил-4-пентеноилокси)-2-метил-1-нафтил} этил}тетрагидро-4- гидрокси-2Н-пиран-2-она (полученного, как описано в примере 41), получали 13 мг названного соединения в виде бесцветного порошка.
Пример 65. Натриевый (3R, 5R)-3,5-дигидрокси-7-[(1S,2S,6S,8S,8aR)-6-гидрокси-2- метил-8-(2-аллил-2-этил-4-пентеноилокси)-1,2,6,7,8,8a-гексагидро-1- нафтил]гептаноат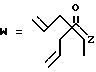
Следовали методике, аналогичной описанной в примере 47, но использовали 24 мг (0,051 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси-8- (2-аллил-2-этил-4-пентеноилокси)-2-метил-1-нафтил] этил} тетрагидро-4- гидрокси-2Н-пиран-2-она (полученного, как описано в примере 42), получали 25 мг названного соединения в виде бесцветного порошка.
Пример 66. Натриевый (3R, 5R)-3,5-дигидрокси-7-[(1S,2S,6S,8S,8aR)-6-гидрокси-2-метил-8- (2,2-диаллил-4-пентеноилокси)-1,2,6,7,8,8a-гексагидро-1-нафтил]гептаноат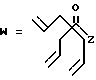
Следовали методике, аналогичной описанной в примере 47, но использовали 22 мг (0,045 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси-8- (2,2-диаллил-4-пентеноилокси)-2-метил-1-нафтил] этил} тетрагидро-4- гидрокси-2Н-пиран-2-она (полученного, как описано в примере 43), получали 25 мг названного соединения в виде бесцветного порошка.
Пример 67. Натриевый (3R, 5R)-3,5-дигидрокси-7-[(1S,2S,6S,8S,8aR)-6-гидрокси-2-метил-8- (2-этил-2-метилвалерилокси)-1,2,6,7,8,8a-гексагидро-1-нафтил]гептаноат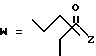
Следовали методике, аналогичной описанной в примере 47, но использовали 18 мг (0,040 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси-8- (2-этил-2-метилвалерилокси)-2-метил-1-нафтил] этил} тетрагидро-4- гидрокси-2Н-пиран-2-она (полученного, как описано в примере 44), получали 20 мг названного соединения в виде бесцветного порошка.
Методика примера 67 может быть применена с использованием одного из стереоизомеров, полученных в примере 44, в качестве исходного вещества, для получения соответствующего стереоизомера соединения примера 67, как проиллюстрировано в примере 68.
Пример 68. Натриевый (3R, 5R)-3,5-дигидрокси-7-[(1S,2S,6S,8S,8aR)-6-гидрокси-2-метил-8- [(2S)-2-этил-2-метилвалерилокси-1,2,6,7,8,8a-гексагидро-1-нафтил]гептаноат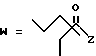
Следовали методике, аналогичной описанной в примере 47, но использовали 5,8 мг (0,012 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси-8- [(2S)-2-этил-2-метилвалерилокси]-2-метил-1-нафтил}этил} -тетрагидро-4- гидрокси-2Н-пиран-2-она (полученного, как описано в примере 45), получали 6,2 мг названного соединения в виде бесцветного порошка.
Пример 69. Натриевый (3R, 5R)-3,5-дигидрокси-7-[(1S,2S,6S,8S,8aR)-6-гидрокси-2- метил-8-[2,2-диметилгексаноилокси)-1,2,6,7,8,8a-гексагидро-1-нафтил] гептаноат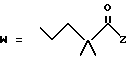
Следовали методике, аналогичной описанной в примере 47, но использовали 28 мг (0,062 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-гидрокси-3- (2,2-диметилгексаноилокси)-2-метил-1-нафтил] этил} тетрагидро-4-гидрокси- 2Н-пиран-2-она (полученного, как описано в примере 46), получали 32 мг названного соединения в виде бесцветного порошка.
В каждом из следующих примеров 70 - 74 описано получение соединения формулы: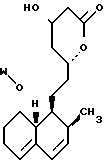
т. е. получение соединения формулы (IV), в которой R1 представляет собой группу формулы (III) и R6 представляет собой атом водорода. Каждая группа W, как она определена в последующих примерах, присоединяется к формуле, показанной выше, через связь, обозначенную буквой Z.
Пример 70. (4R, 6R)-6-{ 2-[(1S, 2S,8S,8aR)-1,2,6,7,8,8a-Гексагидро-8-[(S)-2- метилвалерилокси] -2-метил-1-нафтил]этил}-тетрагидро-4-гидрокси-2Н- пиран-2-он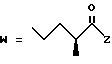
70-(1) (4R, 6R)-6-{2-(1S,2S,8S,8aR)-1,2,6,7,8,8a-Гексагидро-8- [(S)-2-метилвалерилокси] -2-метил-1-нафтил] этил} -тетрагидро-4-трет- бутилдиметилсилилокси-2Н-пиран-2-он.
Следовали методике, аналогичной описанной в примере 4, но использовали 12,6 г (30,0 ммоль) (4R,6R)-6-{2-[(1S,2S,8S,8aR)-1,2,6,7,8,8a-гексагидро-8-гидрокси-2-метил- 1-нафтил]этил}тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного, как описано в Японской патентной заявке N Sho 59-175450) и 4,0 г (29,7 ммоль) хлористого (S)-2-метилвалерила, и получали 12,2 г названного соединения.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
1,12 (3Н, дублет, J = 7,3 Гц);
4,27-4,30 (1Н, мультиплет);
4,54-4,64 (1Н, мультиплет);
5,32 (1Н, широкий синглет);
5,56 (1Н, широкий синглет);
5,75 (1Н, дублет из дублетов, J = 9,2 и 5,9 Гц);
5,98 (1Н, дублет, J = 9,2 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 2950, 1720, 1250, 1080.
Масс-спектр (m/e): 519 (М++1), 477, 435, 387.
[α]
70-(2) (4R, 6R)-6-{ 2-[(1S,2S,8S,8aR)-1,2,6,7,8,8a-Гексагидро-8-[(S)-2- метилвалерилокси]-2-метил-1-нафтил)этил}-тетрагидро-4-гидрокси-2Н-пиран-3-он
Следовали методике, аналогичной описанной в примере 28, но использовали 12,2 г (23,5 ммоль) (4R,6R)-6-{2-[(1S,2S,8S,8aR)-1,2,6,7,8,8a-гексагидро- 8-[(S)-2-метил-валерилокси] -2-метил-1-нафтил] этил(тетрагидро-4-трет- бутилдиметилсилилокси-2Н-пиран-2-она (полученного, как описано на стадии 1), и получали 5,5 г названного соединения, плавящегося при температурах от 110 до 111,5oC.
Элементный анализ для C24H36O5:
Вычислено: C - 71,26%, H - 8,97%;
Найдено: C - 71,00%, H - 8,82%.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях;
1,12 (3Н, дублет, J = 6,8 Гц);
4,35-4,40 (1Н, мультиплет);
4,56-4,66 (1Н, мультиплет);
5,33 (1Н, широкий синглет);
5,55 (1Н, широкий синглет);
5,74 (1Н, дублет из дублетов, J = 9,3 и 5,9 Гц);
5,98 (1Н, дублет, J = 9,3 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 3450, 2950, 1720, 1250, 1080.
Масс-спектр (m/e): 404 (M+), 270, 255, 229.
[α]
Пример 71. (4R,6R)-6-{2-[(1S,2S,8S,8aR)-1,2,6,7,8,8a-гексагидро-8-(2-этил- 2-метилбутирилокси)-2-метил-1-нафтил]этил}тетрагидро-4-гидрокси- 2Н-пиран-2-он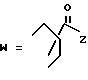
71- (1) (4R, 6R)-6-{ 2-[(1S,2S,8S,8aR)-1,2,6,7,8,8a-Гексагидро-8-(2-этил-2- метилбутирилокси)-2-метил-1-нафтил] этил} -тетрагидро-4-трет- бутилдиметилсилилокси-2Н-пиран-2-он.
Следовали методике, аналогичной описанной в примере 4, но использовали 1,0 г (2,4 ммоль) (4R,6R)-6-{2-[(1S,2S,8S,8aR)-1,2,6,7,8,8a-гексагидро-8-гидрокси-2- метил-1-нафтил] этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного, как описано в Японской патентной заявке N Sho 59-175450) и 1,4 г (9,4 ммоль) хлористого 2-этил-2-метилбутирила, получали 951 мг названного соединения.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
4,26-4,29 (1Н, мультиплет);
4,54-4,61 (1Н, мультиплет);
5,31 (1Н, широкий синглет);
5,54 (1Н, широкий синглет);
5,73 (1Н, дублет из дублетов, J = 9,7 и 6,0 Гц);
5,98 (1Н, дублет, J = 9,7 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 2950, 1720, 1250, 1150, 1080, 840.
Масс-спектр (m/e): 532 (M+), 402, 345, 327.
[α]
71- (2) (4R, 6R)-6-{2-[(1S,2S,8S,8aR)-1,2,6,7,8,8a-Гексагидро-8- (2-этил-2-метилбутирилокси)-2-метил-1-нафтил]этил}-тетрагидро-4-гидрокси- 2Н-пиран-2-он
Следовали методике, аналогичной описанной в примере 28, но использовали 951 мг (1,9 ммоль) (4R,6R)-6-{2-[(1S,2S,8S,8aR)-1,2,6,7,8,8a-гексагидро-8-(2-этил-2- метилбутирилокси)-2-метил-1-нафтил] этил} тетрагидро-4- третбутилдиметилсилилокси-2Н-пиран-2-она (полученного как описано на стадии 1), получали 581 мг, названного соединения, плавящегося при температурах от 61 до 64oC.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
0,81 (3Н, триплет, J = 7,4 Гц);
0,83 (3Н, триплет, J = 7,4 Гц);
0,90 (3Н, дублет, J = 7,1 Гц);
1,06 (3Н, синглет);
4,37 (1Н, широкий синглет);
4,57-4,64 (1Н, мультиплет);
5,34 (1Н, широкий синглет);
5,55 (1Н, широкий синглет);
5,74 (1Н, дублет из дублетов, J = 9,7 и 6,0 Гц);
5,99 (1Н, дублет, J = 9,7 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 3450, 2950, 1720, 1250, 1150, 1080, 840.
Масс-спектр (m/e): 418 (M+), 400, 369, 288.
Элементный анализ для C25H38O5:
Вычислено: C - 71,74%, H - 9,15%;
Найдено: C - 71,19%, H - 9,29%.
[α]
Пример 72. (4R, 6R)-6-{2-[(1S,2S,8S,8aR)-1,2,6,7,8,8a-Гексагидро-8-(2- пропилвалерилокси)-2-метил-1-нафтил] этил} тетрагидро-4-гидрокси- 2Н-пиран-2-он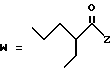
72- (1) (4R, 6R)-6-{ 2-[(1S, 2S, 8S,8aR)-1,2,6,7,8,8a-гексагидро-8-(2- пропилвалерилокси)-2-метил-1-нафтил] этил} тетрагидро-4-трет- бутилдиметилсилилокси-2Н-пиран-2-он.
Следовали методике, аналогичной описанной в примере 3, но использовали 1,0 Г (2,4 ммоль) (4R,6R)-6-{2-[(1S,2S,8S,8aR)-1,2,6,7,8,8a-гексагидро-8-гидрокси-2- метил-1-нафтил] этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного, как описано в Японской патентной заявке N Sho 59-175450) и 686 мг (4,8 ммоль) 2-пропилвалериановой кислоты, получали 1,3 г названного соединения.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
4,25-4,31 (1Н, мультиплет);
4,53-4,61 (1Н, мультиплет);
5,35 (1Н, широкий синглет);
5,55 (1Н, широкий синглет);
5,74 (1Н, дублет из дублетов, J = 9,8 и 5,9 Гц);
5,96 (1Н, дублет, J = 9,8 Гц).
νmax(см-1) инфракрасного абсорбционного спектра (CHCl3): 2950, 1720, 1250, 1080, 838.
Масс-спектр (m/e): 546 (M+), 402, 345, 327.
[α]
72- (2) (4R,6R)-6-{2-[(1S,2S,8S,8aR)-1,2,6,7,8,8a-гексагидро-8-(2- пропилвалерилокси)-2-метил-1-нафтил]этил}тетрагидро-4-гидрокси-2Н-пиран-2-он
Следовали методике, аналогичной описанной в примере 28, но использовали 1,20 г (2,2 ммоль) (4R,6R)-6-{2-[(1S,2S,8S,8aR)-1,2,6,7,8,8a-гексагидро-8-(2- пропилвалерилокси)-2-метил-1-нафтил] этил}тетрагидро-4-трет- бутилдиметилсилилокси-2Н-пиран-2-она (полученного, как описано на стадии 1), получали 650 мг названного соединения, плавящегося при температурах от 92 до 94oC.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
4,37 (1Н, широкий синглет);
4,57-4,64 (1Н, мультиплет);
5,38 (1Н, широкий синглет);
5,56 (1Н, широкий синглет);
5,75 (1Н, дублет из дублетов, J = 9,6 и 6,0 Гц);
5,97 (1Н, дублет, J = 9,6 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (KBr): 3450, 2950, 1720.
Масс-спектр (m/e): 432 (M+), 414, 368, 357.
Элементный анализ для C26H40O5•1/2H2O:
Вычислено: C - 70,72%, H - 9,36%;
Найдено: C - 70,80%, H - 9,31%.
[α]
Пример 73. (4R,6R)-6-{2-[(1S,2S,8S,8aR)-1,2,6,7,8,8a-Гексагидро-8-(2,2- диэтилбутирилокси)-2-метил-1-нафтил]этил}тетрагидро-4-гидрокси-2Н-пиран-2-он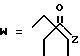
73- (1) (4R, 6R)-6-{ 2-[(1S,2S,8S,8aR)-1,2,6,7,8,8a-Гексагидро-8-(2,2- диэтилбутирилокси)-2-метил-1-нафтил] этил} тетрагидро-4-трет- бутилдиметилсилилокси-2Н-пиран-2-он
Следовали методике, аналогичной в примере 8, но использовали 1,26 г (3,0 ммоль) (4R, 6R)-6-{ 2-[(1S,2S,8S,8aR)-1,2,6-7,8,8a-гексагидро-8-гидрокси-2- метил-1-нафтил] этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного, как описано в Японской патентной заявке N Sho 59-175450) и 2,22 г (13,6 ммоль) хлористого 2,2-диэтилбутирила, получили 800 мг названного соединения.
Спектр ядерного магнитного резонанса: (400 МГц, CDCl3) δ , в миллионных долях,
0,75 (9Н, триплет, J = 7,5 Гц);
1,56 (6Н, квартет, J = 7,5 Гц);
4,26-4,30 (1Н, мультиплет);
4,54-4,61 (1Н, мультиплет);
5,34 (1Н, широкий синглет);
5,55 (1Н, широкий синглет);
5,74 (1Н, дублет, из дублетов, J = 9,6 и 6,0 Гц);
5,98 (1Н, дублет, J = 9,6 Гц).
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 2875, 1715, 1255, 1080, 840.
Масс-спектр (m/e): 546 (M+), 489, 387, 345, 327.
[α]
73- (2) (4R, 6R)-6-{ 2-[(1S,2S,8S,8aR)-1,2,6,7,8,8a-Гексагидро-8-(2,2- диэтилбутирилокси)-2-метил-1-нафтил]этил}тетрагидро-4-гидрокси-2Н-пиран-2-он
Следовали методике, аналогичной описанной в примере 28, но использовали 830 мг (1,5 ммоль) (4R,6R)-6-{2-[(1S,2S,8S,8aR)-1,2,6,7,8,8a-гексагидро-8-(2,2- диэтилбутирилокси)-2-метил-1-нафтил)этил} тетрагидро-4-трет- бутилдиметилсилилокси-2Н-пиран-2-она (полученного, как описано на стадии 1), получали 575 мг названного соединения, плавящегося при температурах от 65 до 68oC.
Элементный анализ для C26H40O5:
Вычислено: C - 72,19%, H - 9,32%;
Найдено: C - 72,00%, H - 9,56%.
Спектр ядерного магнитного резонанса: (400 МГц, гексадейтерированный диметилсульфоксид) δ , в миллионных долях,
0,71 (9Н, триплет, J = 7,4 Гц);
0,84 (3Н, дублет, J = 6,9 Гц);
1,48 (6Н, квартет, J = 7,4 Гц);
4,08-4,10 (1Н, мультиплет);
4,42-4,48 (1Н, мультиплет);
5,17 (1Н, дублет, J = 3,2 Гц, взаимозаменяемый с D2O);
5,23 (1Н, широкий синглет);
5,53 (1Н, широкий синглет);
5,74 (1Н, дублет из дублетов, J = 9,6 и 6,0 Гц);
5,95 (1Н, дублет, J = 9,6 Гц);
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 3350, 2880, 1710, 1220.
Масс-спектр (m/e): 432 (M+), 353, 288, 270, 210.
[α]
Пример 74. (4R, 6R)-6-{ 2-[(1S,2S,8S,8aR)-1,2,6,7,8,8a-гексагидро-8-(2,2-диэтил- 4-пентеноилокси)-2-метил-1-нафтил]этил}тетрагидро-4-гидрокси-2Н-пиран-2-он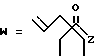
74- (1) (4R,6R)-6-{2-[(1S,2S,8S,8aR)-1,2,6,7,8,8a-Гексагидро-8- (2,2-диэтил-4-пентеноилокси)-2-метил-1-нафтил] этил} -тетрагидро-4-трет- бутилдиметилсилилокси-2Н-пиран-2-он.
Следовали методике, аналогичной описанной в примере 6, но использовали 1,26 г (3,0 ммоль) (4R,6R)-6-{2-[(1S,2S,8S,8aR)-1,2,6,7,8,8a-гексагидро-8-гидрокси-2-метил- 1-нафтил] этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного, как описано в Японской патентной заявке N Sho 59-175450) и 2,09 г хлористого 2,2-диэтил-4-пентеноила, получали 1,30 г названного соединения.
Спектр ядерного магнитного резонанса: (400 МГц, CDCl3) δ , в миллионных долях,
0,778 (3Н, триплет, J = 7,4 Гц);
0,784 (3Н, триплет, J = 7,4 Гц);
2,31 (2Н, дублет, J = 7,2 Гц);
4,27-4,30 (1Н, мультиплет);
4,55-4,61 (1Н, мультиплет);
5,01 - 5,09 (2Н, мультиплет);
5,36 (1Н, широкий синглет);
5,54 (1Н, широкий синглет);
5,59-5,69 (1Н, мультиплет);
5,74 (1Н, дублет из дублетов, J = 9,7 и 6,0 Гц);
5,98 (1Н, дублет, J = 9,7 Гц),
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 2875, 1715, 1255, 1080, 840.
Масс-спектр (m/e): 558 (M+), 501, 387, 345, 327.
[α]
74- (2) (4R,6R)-6-{2-[(1S,2S,8S,8aR)-1,2,6,7,8,8a-гексагидро-8- (2,2-диэтил-4-пентеноилокси)-2 метил-1-нафтил]этил}-тетрагидро-4-гидрокси- 2Н-пиран-2-он
Следовали методике, аналогичной описанной в примере 28, но использовали 1,15 г (2,1 ммоль) (4R,6R)-6-{2-[(1S,2S,8S,8aR)-1,2,6,7,8,8a-гексагидро-8-(2,2-диэтил-4- пентеноилокси)-2-метил-1-нафтил] этил}тетрагидро-4-трет- бутилдиметилсилилокси-2Н-пиран-2-она (полученного, как описано на стадии 1), получали 770 мг названного соединения.
Элементный анализ для C27H40O5:
Вычислено: C - 72,94%, H - 9,07%;
Найдено: C - 72,54%, H - 9,33%.
Спектр ядерного магнитного резонанса: (400 МГц, гексадейтерированный диметилсульфоксид) δ , в миллионных долях
0,74 (6Н, триплет, J = 7,3 Гц);
0,84 (3Н, дублет, J = 7,0 Гц);
2,23 (2Н, дублет, J = 7,3 Гц);
4,08-4,12 (1Н, мультиплет);
4,43-4,49 (1Н, мультиплет);
5,04-5,11 (2Н, мультиплет);
5,18 (1Н, дублет, J = 3,4 Гц, взаимозаменяемый с D2O);
5,24 (1Н, широкий синглет);
5,54 (1Н, широкий синглет);
5,55-5,65 (1Н, мультиплет);
5,73 (1Н, дублет из дублетов, J = 9,6 и 6,0 Гц);
5,95 (1Н, дублет, J = 9,6 Гц),
νmax (см-1) инфракрасного абсорбционного спектра (CHCl3): 3350, 2880, 1710, 1220.
Масс-спектр (m/e): 445 (M+), 427, 288, 270, 210.
[α]
В каждом из следующих примеров 75 - 79 описано получение соединений следующей формулы: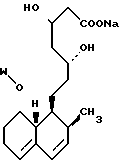
т. е. соединений формулы (IV), в которой R1 представляет собой группу формулы (II), R5 представляет собой атом натрия и R6 представляет собой атом водорода. Каждая группа W как она определена в последующих примерах, присоединяется к формуле, показанной выше, через связь, обозначенную буквой Z.
Пример 75. Натриевый (3R, 5R)-3,5-дигидрокси-7-[(1S,2S,8S,8aR,)-2-метил-8-[(S)-2- метилвалерилокси]-1,2,6,7,8,8a-гексагидро-1-нафтил]гептаноат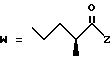
Следовали методике, аналогичной описанной в примере 47, но использовали 1,01 г (2,5 ммоль) (4R,6R)-6-{2-[(1S,2S,8S,8aR)-1,2,6,7,8,8a-гексагидро- 8-[(S)-2-метилвалерилокси] -2-метил-1-нафтил] этил} тетрагидро-4-гидрокси- 2Н-пиран-2-она (полученного, как описано в примере 70), получали 1,12 г названного соединения в виде бесцветного порошка.
Пример 76. Натриевый (3R,5R)-3,5-дигидрокси-7-[(1S,2S,8S,8aR)-2-метил-8-(2-этил-2- метилбутирилокси)-1,2,6,7,8,8a-гексагидро-1-нафтил]гептаноат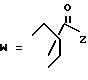
Следовали методике, аналогичной описанной в примере 47, но использовали 210 мг (0,50 ммоль) (4R,6R)-6-{2-[(1S,2S,8S,8aR)-1,2,6,7,8,8a-гексагидро-8-(2-этил-2- метилбутирилокси)-2-метил-1-нафтил] этил} тетрагидро-4-гидрокси-2Н-пиран-2-она (полученного, как описано в примере 71), получали 227 мг названного соединения в виде бесцветного порошка.
Пример 77. Натриевый (3R,5R)-3,5-дигидрокси-7-[(1S,2S,8S,8aR)-2-метил-8-(2- пропилвалерилокси)-1,2,6,7,8,8a-гексагидро-1-нафтил]-гептаноат.
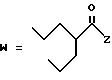
Следовали методике, аналогичной описанной в примере 47, но использовали 200 мг (0,45 ммоль) (4R,6R)-6-{2-[(1S,2S,8S,8aR)-1,2,6,7,8,8a-гексагидро-8-(2- пропилвалерилокси)-2-метил-1-нафтил] этил}тетрагидро-4-гидрокси- 2Н-пиран-2-она (полученного, как описано в примере 72), получали 223 мг названного соединения в виде бесцветного порошка.
Пример 78. Натриевый (3R,5R)-3,5-дигидрокси-7-[(1S,2S,8S,8aR)-2-метил-8-(2,2-диэтилбутирилокси)- 1,2,6,7,8,8a-гексагидро-1-нафтил]-гептаноат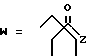
Следовали методике, аналогичной описанной в примере 47, но использовали 20 мг (0,047 ммоль) (4R,6R)-6-{2-[(1S,2S,8S,8aR)-1,2,6,7,8,8a-гексагидро-8-(2,2- диэтилбутирилокси)-2-метил-1-нафтил]этил}тетрагипро-4-гидрокси-2Н-пиран- 2-она (полученного, как описано в примере 73) и получали 22 мг названного соединения в виде бесцветного порошка.
Пример 79. Натриевый (3R,5R)-3,5-дигидрокси-7-[(1S,2S,8S,8aR)-2-метил-8-(2,2-диэтил-4- пентеноилокси)-1,2,6,7,8,8a-гексагидро-1-нафтил]гептаноат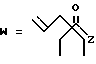
Следовали методике, аналогичной описанной в примере 47, но использовали 22 мг (0,043 ммоль) (4R,6R)-6-{2-[(1S,2S,8S,8aR)-1,2,6,7,8,8a-гексагидро-8-(2,2-диэтил-4- пентеноилокси)-2-метил-1-нафтил] этил}тетрагидро-4-гидрокси-2Н-пиран-2-она (полученного, как описано в примере 74), получали 23 мг названного соединения в виде бесцветного порошка.
В каждом из следующих примеров 80 - 84 описано получение соединений следующей формулы: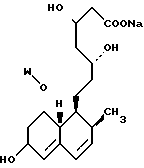
т. е. соединений формулы (I), в которой R1 представляет собой группу формулы (II), R5 представляет собой атом натрия и R6 представляет собой атом водорода. Каждая группа W, как она определена в последующих примерах, присоединяется к формуле, показанной выше, через связь, обозначенную буквой Z.
Пример 80. Натриевый (3R, 5R)-3,5-дигидрокси-7-[(1S,2S,6S,8S,8aR)-6-гидрокси-2-метил-8-[(S)- 2-метилвалерилокси] -1,2,6,7,8,8a-гексагидро-1-нафтил]гептаноат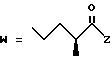
Метод 1. Каждую из двадцати колб Эрленмейера на 500 мл, содержащих 100 мл в расчете на колбу среды TS-C указанного ниже состава, инокулировали инокулюмом Mucorhiemalis Wehmer SANK 36372 (FERM BP-4108) одной платиновой петлей. Инокулированные колбы инкубировали в течение трех дней при 26oC на ротационной мешалке, вращающейся со скоростью 200 об/мин.
Среда для выращивания TS-C, % (вес на объем):
Глюкоза - 1
Полипептон (фирма "Дейго нутришн кемикалз ко.") - 0,2
Мясной экстракт - 0,1
Дрожжевой экстракт (Дифко) - 0,1
Водопроводная вода - до 100
Величину pH не регулировали.
В конце этого времени 0,1 мл раствора (4R,6R)-6-{2-[(1S,2S,8S,8aR)- 1,2,6,7,8,8a-гексагидро-8-[(S)-2-метилвалерилокси] -2-метил-1-нафтил] этил} тетрагидро-4-гидрокси-2Н-пиран-2-она (полученного, как описано в примере 70) и диметилсульфоксиде добавляли в каждую колбу, в результате чего конечная концентрация соединения в среде для выращивания становилась 0,01% (вес на объем). Культивирование затем продолжали еще в течение 3 дней в указанных выше условиях.
В конце этого дополнительного периода культивирования ферментационную жидкую среду фильтровали, и фильтрат адсорбировали на колонке, содержащей 200 мл смолы диаион HP-20 ("Мицубиси касеи корпорейшн"). Смолу затем промывали 500 мл дистиллированной воды, после чего фракции, содержащие названное соединение, элюировали с колонки 600 мл 50%-ного (по объему) водного раствора ацетона. Элюаты соединяли, и получающийся раствор концентрировали до достижения сухого состояния упариванием при пониженном давлении. Полученный остаток очищали хроматографией, пропускав через препаративную ODS-колонку (ODS-H-5251 - Это торговое название материала, фирма "Сенсю сайентифик ко. , лтд.") с использованием в качестве элюента смеси ацетонитрила, воды и уксусной кислоты в объемном соотношении 450:550:1. Хроматографию контролировали проведением ультрафиолетовой абсорбции при длине волны 237 нм. Величину pH элюата доводили до 8,0 водным раствором гидроксида натрия, и полученную смесь концентрировали до сухого выпаривания при пониженном давлении. Остаток затем растворяли в 20 мл воды, и раствор адсорбировали на колонке, содержащей 20 мл диаиона HP-20ТМ. Смолу промывали 50 мл воды, после чего смолу элюировали 60 мл 50%-ного (по объему) водного раствора ацетона, получая 8 мг по существу чистого названного соединения.
Метод 2. Одну платиновую петлю инокулята Streptomyces carbophilus SANK 62585 (FERM BP-4128) вводили в колбу Эрленмейера на 500 мл, содержащую 100 мл среды SC, состав которой указан ниже. Инокулированную колбу затем инкубировали при 28oC на роторной мешалке, вращающейся со скоростью 200 об/мин.
Среда SC, %(вес на объем):
Дрожжевой экстракт (фирма "Дифко") - 0,1
Полипептон (фирма "Дейго нутришн кемикалз ко.") - 1,0
Глюкоза - 2,0
Водопроводная вода - до 100
Величина pH составляла 7,0 (перед стерилизацией).
После инкубирования инокулированной среды в течение трех дней порцию такой среды переносили в каждую из двадцати колб Эрленмейера на 500 мл, содержащих 100 мл среды SC в каждой колбе, в результате чего концентрация инокулированной затравочной среды в свежей среде составляла 5,0% (по весу на объем). Эту свежеинокулированную среду затем инкубировали еще в течение трех дней в условиях оговоренных выше.
В конце периода инкубирования некоторое количество водного раствора натриевого (3R, 5R)-3,5-дигидрокси-7-[(1S, 2S,8S,8aR)-2-метил-8-[(S)-2- метилвалерилокси] -1,2,6,7,8,8a-гексагидро-1-нафтил] гептаноата (полученного, как описано в примере 75) добавляли к среде до конечной концентрации указанного соединения в растворе 0,01% (по весу на объем). Культивирование затем еще продолжали в течение трех дней в условиях, оговоренных выше.
В конце периода культивирования ферментационную жидкую среду фильтровали, и фильтрат адсорбировали на колонке, содержащей 200 мл смолы диаион HP-20ТМ. Смолу промывали 300 мл дистиллированной воды, после чего фракции, содержащие названное соединение, элюировали 400 мл 50%-ного (по объему) водного раствора ацетона. Полученные фракции соединяли, и элюат концентрировали до сухого испарением при пониженном давлении. Остаток затем очищали хроматографией, пропуская вещество через препаративную ODS-колонку (ODS-H-5251 "Сенсю сайентифик ко. , лтд") при использовании в качестве элюента смеси, ацетонитрила, воды и уксусной кислоты, в объемном соотношении 450: 550: 1. Хроматографию контролировали проведением ультрафиолетовой абсорбции при 237 мм.
Величину pH объединенных фракций, содержащих очищенное соединение, доводили затем до 8,0 добавлением водного раствора гидроксида натрия, и полученную смесь концентрировали до сухого упариванием при пониженном давлении. Полученный остаток растворяли в 20 мл воды, после чего раствор адсорбировали на колонке, содержащей 20 мл диаиона HP-20. Смолу промывали 30 мл воды и затем элюировали 100 мл 50%-ного (по объему) водного раствора ацетона, получая 10 мг по существу чистого названного соединения.
Было показано, что физико-химические свойства соединения, полученного таким способом, идентичны таковым соединения, полученного в приведенном выше примере 49.
Пример 81. Натриевый (3R, 5R)-3,5-дигидрокси-7-[(1S,2S,6S,8S,8aR)-6-гидрокси-2- метил-8-(2-этил-2-метилбутирилокси)-1,2,6,7,8,8a-гексагидро- 1-нафтил]гептаноат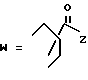
Одну платиновую петлю инокулята Amycolata autotrophica SANK 62981 (FERM BP-4105) инокулировали в колбу Эрленмейера на 500 мл, содержащую 100 мл дрожжевой среды МУ, состав которой приведен ниже. Инокулированную колбу инкубировали при 28oC на ротационной мешалке, вращающуейся со скоростью 200 об/мин.
Дрожжевая среда МУ, %(вес на объем):
Дрожжевой экстракт (фирма "Дифко") - 0,3
Солодовый экстракт (фирма "Дифко") - 0,3
Полипептон (фирма "Дейго нутришн кемикалж ко.") - 0,5
Глюкоза - 1,0
Водопроводная вода - до 100
Величину pH у среды не регулировали.
После инкубирования инокулированной затравочной среды в течение трех дней затравочную среду делили и переносили в двадцать колб Эрленмейера на 500 мл, содержащих 100 мл дрожжевой среды МУ в каждой колбе, в результате чего концентрация затравочной среды в свежей дрожжевой среде МУ составляла 0,5% (по весу в расчете на объем). Колбы затем инкубировали еще в течение двух дней в условиях, оговоренных выше.
В конце инкубационного периода некоторое количество водного раствора натриевого (3R, 5R)-3,5-дигидрокси-7-[(1S, 2S, 8S,8aR)-2-метил-8-(2-этил-2- метилбутирилокси)-1,2,6,7,8,8a-гексагидро-1-нафтил] гептаноата (полученного, как описано в примере 76) добавляли к жидкой среде выращивания, чтобы конечная концентрация указанного соединения в растворе составляла 0,01% (по весу на объем). Культивирование затем продолжали еще в течение пяти дней в условиях, оговоренных выше.
В конце периода культивирования ферментационную жидкую среду фильтровали, и фильтрат подвергали адсорбции на колонке, содержащей 200 мл диаиона HP-20ТМ. Смолу промывали 300 мл дистиллированной воды, после чего фракции, содержащие названное соединение, элюировали 400 мл 50%-ного (по объему) водного раствора ацетона.
Элюат концентрировали до сухого упариванием при пониженном давлении, и остаток затем очищали хроматографией, пропуская вещество через препаративную ODS-колонку (ODS-H-5251 "Сенсю сайентифик ко., лтд") при использовании в качестве элюента смеси ацетонитрила, воды и уксусной кислоты, в объемном соотношении 450: 550:1. Хроматографию контролировали проведением ультрафиолетовой адсорбции при 237 нм. Величину pH элюата, полученного после проведения хроматографии, доводили затем до 8,0 водным раствором гидроксида натрия, после чего раствор концентрировали до сухого испарением при пониженном давлении. Концентрат растворяли в 20 мл воды, и раствор затем подвергали адсорбции на колонке, содержащей 20 мл диаиона НР-20ТМ. Смолу промывали 30 мл воды и затем элюировали 100 мл 50%-ного (по объему) водного раствора ацетона, получая 5,1 мг по существу чистого названного соединения.
Метод 2. Одну платиновую петлю инокулята Mucor hiemalis Wehmer SANK 36372 (FERM BP-4108) инокулировали в каждую из двадцати колб Эрленмейера на 500 мл, содержащих в каждой колбе по 100 мл среды TS-C, и инокулированные колбы инкубировали при 26oC на ротационной мешалке со скоростью 200 об/мин.
По истечении трех дней инкубирования при этих условиях 0,1 мл раствора (4R, 6R)-6-{ 2-[(1S, 2S,8S,8aR)-1,2,6,7,8,8a-гексагидро-8-[(S)-2- метилвалерилокси] -2-метил-1-нафтил] этил} тетрагидро-4-гидрокси-2Н-пиран-2-она (полученного, как описано в примере 70) в диметилсульфоксиде добавляли к среде, в результате чего конечная концентрация указанного соединения в среде становилась 0,01% (по весу на объем). Инкубирование затем продолжали еще в течение трех дней в условиях, оговоренных выше.
В конце инкубационного периода ферментационную жидкую среду фильтровали, и фильтрат подвергали адсорбции в колонке, содержащей 200 мл диаиона HP-20ТМ. Смолу промывали 300 мл дистиллированной воды, после чего фракции, содержащие названое соединение, элюировали 400 мл 50%-ного (по объему) водного раствора ацетона. Элюат затем концентрировали до сухого упариванием при пониженном давлении, и концентрат очищали хроматографией, пропуская вещество через препаративную ODS-колонку (ODS-H-5251ТМ "Сенсю сайентифик ко. , "лтд") с использованием в качестве элюента смеси ацетонитрила, воды и уксусной кислоты, в объемном соотношении 450:550:1. Хроматографию контролировали проведением ультрафиолетовой абсорбции при 237 нм. Величину pH элюата затем доводили до 3,0 добавлением водного раствора гидроксида натрия, после чего раствор концентрировали до сухого упариванием при пониженном давлении. Концентрат затем растворяли в 20 мл воды, и раствор подвергали адсорбции на колонке, содержащей 20 мл диаиона НР-20ТМ. Смолу промывали 30 мл воды и затем элюировали 100 мл 50%-ного (по объему) водного раствора ацетона, получая 48 мг по существу чистого названного соединения.
Физико-химические свойства названного соединения, полученного таким способом, были идентичными таковым соединения, полученного в примере 51.
Метод 3. Одну платиновую петлю инокулята Syncephalastrum nigricans SANK 42372 (FERM BP-4106) использовали для инокулирования 100 мл среды TS-C в колбе Эрленмейера на 500 мл. Инокулированную среду затем инкубировали при 26oC на ротационной мешалке, вращающейся со скоростью 200 об/мин.
После инкубирования при этих условиях в течение периода три дня 0,1 мл раствора (4R, 6R)-6-{2-[(1S,2S,8S,8aR)-1,2,6,7,8,8а-гексагидро-8-(2-этил-2- метилбутирилокси)-2-метил-1-нафтил]этил}тетрагидро-4-гидрокси-2Н- пиран-2-она (полученного, как описано в примере 71) в диметилсульфоксиде добавляли к среде, в результате чего конечная концентрация указанного соединения в среде становилась равной 0,01% (по весу на объем). Культивирование продолжали еще в течение 9 дней в условиях, оговоренных выше.
В конце этого периода культивирования ферментационную жидкую среду фильтровали, и фильтрат подвергали адсорбции на колонке, содержащей 200 мл диаиона HP-20ТМ. Смолу затем промывали 300 мл дистиллированной воды, после чего фракции, содержащие названное соединение, элюировали 600 мл 50%-ного (по объему) водного раствора ацетона. Элюаты соединяли и затем концентрировали до сухого упариванием при пониженном давлении. Полученный остаток очищали хроматографией, пропуская вещество через препаративную ODS-колонку (ODS-H-5251ТМ "Сенсю сайентифик ко. , лтд") с использованием в качестве элюента смеси ацетонитрила, воды и уксусной кислоты в объемном соотношении 450: 550: 1. Хроматографию контролировали проведением ультрафиолетовой абсорбции при 237 нм. Элюат, полученный после проведения хроматографии, затем нейтрализовали, смешивая элюат, без дополнительной очистки, с 0,1 М водным раствором (pH 8,0) бикарбоната натрия и гидроксида натрия. Величину pH фракций, содержащих названное соединение, затем доводили до 8,0 и смесь концентрировали до сухого упариванием при пониженном давлении. Полученный остаток растворяли в 20 мл воды, после чего раствор подвергали адсорбции на колонке, содержащей 20 мл диаиона HP-20ТМ. Смолу затем промывали 30 мл дистиллированной воды и элюировали 100 мл 50%-ного (по объему) водного раствора ацетона, получая 24,1 мг по существу чистого названного соединения.
Спектр ядерного магнитного резонанса: (360 МГц, CD3OD) δ , в миллионных долях,
0,35-0,92 (6Н, мультиплет);
0,92 (3Н, дублет, J = 7,1 Гц);
1,15-1,74 (14Н, мультиплет);
1,76 (1Н, мультиплет);
1,92 (1Н, двойной дублет из дублетов, J = 15,4; 5,9 и 2,1 Гц);
2,15-2,50 (6Н, мультиплет);
3,69 (1Н, мультиплет);
4,10 (1Н, мультиплет);
4,25 (1Н, мультиплет);
5,33 (1Н, мультиплет);
5,65 (1Н мультиплет);
5,95 (1Н, дублет из дублетов, J = 9,7 и 6,1 Гц);
6,02 (1Н, дублет, 1 = 9,7 Гц).
Молекулярный вес 438 (по данным масс-спектрометрии высокого разрешения с бомбардировкой быстрыми атомами определено как C26H41O7Na).
[α]
Пример 82.1 Натриевый (3R,5R)-3,5-дигидрокси-7- [(1S,2S,6S,8S,8aR)-6-гидрокси-2-метил-3-(2-пропилвалерилокси)- 1,2,6,7,8,8a-гексагидро-1-нафтил] гептаноат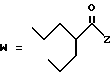
Метод 1. Одну платиновую петлю инокулята Mucor hiemalis Wehmer SANK 36372 (FERM BP-4103) использовали для инокулирования 100 мл среды TS-C состава, указанного в примере 30, в каждой из двадцати колб Эрленмейера на 500 мл. Инокулированные колбы затем инкубировали при 26oC на ротационной мешалке со скоростью 200 об/мин.
В конце третьего дня инкубирования 0,1 мл раствора (4R,6R)-6-{2-[(1S,2S, 8S, 8aR)-1,2,6,7,8,8a-гексагидро-8-(2- пропилвалерилокси)-2-метил-1-нафтил)этил} тетрагидро-4-гидрокси- 2Н-пиран-2-она (полученного, как описано в примере 72) в диметилсульфоксиде добавляли к среде до конечной концентрации указанного соединения в среде 0,01% (по весу на объем). Культивирование затем продолжали еще в течение трех дней в условиях, оговоренных выше.
В конце этого дополнительного периода культивирования ферментационную жидкую среду фильтровали, и фильтрат подвергали адсорбции на колонке, содержащей 200 мл диаиона HP-20ТМ. Смолу промывали 500 мл дистиллированной воды, после чего фракции, содержащие название соединение, элюировали 600 мл 50%-ного водного раствора ацетона (указана объемная концентрация). Требуемые фракции затем соединяли, после чего объединенный элюат концентрировали до сухого упариванием при пониженном давлении. Полученный остаток затем очищали хроматографией, пропуская вещество через препаративную ODS-колонку (ODS-H-5251ТМ "Сенсю сайентифик ко. , лтд") при использовании в качестве элюата смеси ацетонитрила, воды и уксусной кислоты в соотношении 450:550:1. За ходом хроматографии следили по ультрафиолетовой адсорбции при 237 мм. Величину pH элюата затем доводили до 8,0 добавлением водного раствора гидроксида натрия, после чего смесь концентрировали досуха упариванием при пониженном давлении. Остаток растворяли в 20 мл воды, и полученный раствор подвергали адсорбции на колонке, содержащей 20 мл диаиона HP-20ТМ. Смолу промывали 50 мл воды, после чего смолу элюировали 60 мл 50%-ного (по объему) водного раствора ацетона, получая 48 мг по существу чистого названного соединения.
Физико-химические свойства соединения, полученного таким способом, идентичны свойствам соединения, синтезированного в примере 50.
Метод 2. Одну платиновую петлю инокулята Syncephalastrum racemosum (Cohn) Schroeter SANK 41872 (FERM BP-4107) использовали для инокулирования 100 мл среды TS-C (состав которой указан выше в примере 80) в каждой из двадцати колб Эрленмейера на 500 мл. Инокулированные колбы затем инкубировали при 26oC на ротационной мешалке со скоростью 200 об/мин.
По истечении трех дней инкубирования 0,1 мл раствора (4R,6R)-6-{2- [(1S, 2S, 8S, 8aR)-1,2,6,7,8,8a-гексагидро-8-(2-пропилвалерилокси)-2- метил-1-нафтил] этил} тетрагидро-4-гидрокси-2Н-пиран-2-она (полученного как описано в примере 72) в диметилсульфоксиде добавляли к среде до концентраций указанного соединения в среде 0,01% (по весу на объем). Культивирование затем продолжали еще в течение 7 дней при 26oC на ротационной мешалке, вращающейся со скоростью 200 об/мин.
В конце этого дополнительного периода культивирования ферментационную жидкую среду фильтровали, и фильтрат подвергали адсорбции на колонке, содержащей 200 мл диаиона HP-20ТМ. Смолу промывали 300 мл дистиллированной воды, после чего фракции, содержащие названное соединение, элюировали 600 мл 50%-ного (по объему) водного раствора ацетона. Полученные элюаты соединяли и затем концентрировали досуха упариванием при пониженном давлении. Полученный остаток очищали хроматографией, пропуская вещество через колонку препаративного назначения с набивкой из ODS (ODS-H-5251ТМ "Сенсю сайентифик ко., лтд"), при использовании в качестве элюента смеси ацетонитрила, воды и уксусной кислоты в соотношении 450:550:1. Требуемые фракции элюировали с одновременным проведением ультрафиолетовой абсорбции при 237 нм. Полученный элюат нейтрализовали смешением без дополнительной очистки с 0,1 М водным раствором вторичного кислого фосфата натрия (pH 8) и гидроксида натрия. Величину pH фракций, содержащих названное соединение, доводили по 8,0, после чего смесь концентрировали досуха упариванием при пониженном давлении. Полученный остаток растворяли в 20 мл воды, и раствор затем подвергали адсорбции на колонке, содержащей 20 мл диаиона HP-20ТМ. Смолу промывали 50 мл воды и затем элюировали 100 мл 50%-ного (по объему) водного ацетона, получая 33 мг по существу чистого соединения.
Спектр ядерного магнитного резонанса: (360 МГц, CD3OD) δ , в миллионных долях,
0,83 (6Н, триплет, J = 7,4 Гц);
0,91 (3Н, дублет, J = 7,2 Гц):
1,07 (3Н, синглет);
1,2-1,9 (11Н, мультиплет);
1,92 (1Н, удвоенный дублет из дублетов, J = 15,4, 6,1 и 2,1 Гц).
2,2-2,5 (5Н, мультиплет):
3,69 (1Н, мультиплет);
4,10 (1Н, мультиплет);
4,28 (1Н, мультиплет);
5,27 (1Н, мультиплет);
5,64 (1Н, мультиплет);
5,94 (1Н, дублет из дублетов, J = 9,7 и 6,1 Гц);
6,02 (1Н, дублет, J = 9,7 Гц).
Молекулярный вес 474 (C25H39O7Na по данным масс-спектрометрии высокого разрешения с бомбардировкой быстрыми атомами).
[α]
Пример 83. Натриевый (3R,5R)-3,5-дигидрокси-7-[(1S,2S,6S,8S,8aR)-6-гидрокси- 2-метил-8-(2,2-диэтилбутирилокси)-1,2,6,7,8,8a-гексагидро-1-нафтил] гептаноат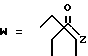
Метод 1. Одну платиновую петлю инокулята Mucor hiemalis Wehmer SANK 36372 (FERM BP-4108) использовали для инокулирования 100 мл среды TS-C (состав которой приведен выше в примере 80), в каждой из двадцати колб Эрленмейера на 500 мл. Инокулированные колбы инкубировали при 26oC на ротационной мешалке со скоростью 200 об/мин.
В конце 3-дневного инкубационного периода 0,1 мл раствора (4R,6R)-6-{ 2-[(1S, 2S, 8S,8aR)-1,2,6,7,8,8a-гексагидро-8-(2,2- диэтилбутирилокси)-2-метил-1-нафтил] этил} тетрагидро-4-гидрокси- 2Н-пиран-2-она (полученного, как описано в примере 73) в диметилсульфоксиде добавляли к среде до конечной концентрации указанного соединения 0,01% (по весу на объем). Культивирование затем продолжали еще в течение трех дней при 26oC на ротационной мешалке со скоростью 200 об/мин.
В конце этого времени ферментационную жидкую среду фильтровали, и фильтрат подвергали адсорбции на колонке, содержащей 200 мл диаиона HP-20ТМ. Смолу промывали 500 мл дистиллированной воды, и фракции, содержащие названное соединение, элюировали 800 мл 50%-ного (по объему) водного раствора ацетона. Требуемые фракции соединяли, и полученный раствор концентрировали досуха упариванием при пониженном давлении. Остаток очищали хроматографией, пропуская вещество через препаративную ODS-колонку (ODS-H-5251ТМ "Сенсю сайентифик ко., лтд.") при использовании в качестве элюента смеси ацетонитрила, воды и уксусной кислоты в объемном соотношении 450:550:1. За ходом хроматографии следили по ультрафиолетовой абсорбции при 237 нм. Величину pH элюата доводили затем до 8,0 добавлением водного раствора гидроксида натрия, и смесь затем концентрировали досуха упариванием при пониженном давлении. Остаток растворяли в 20 мл воды, и раствор подвергали адсорбции на колонке, содержащей 20 мл диаиона НР-20ТМ, после чего смолу промывали 80 мл воды и затем элюировали 100 мл 50%-ного (по объему) водного раствора ацетона, получая 78 мг по существу чистого названного соединения.
Было показано, что физико-химические свойства продукта являются идентичными таковым соединения, синтезированного в примере 52.
Метод 2. Одну платиновую петлю инокулята Syncephalastrum nigricans Vuillemin SANK 42372 (FERM BP-4106) использовали для инокулирования 100 мл среды TS-C (состав которой указан в примере 80), в каждой из двадцати колб Эрленмейера на 500 мл. Инокулированные колбы затем инкубировали при 26oC на ротационной мешалке, вращающейся со скоростью 200 об/мин.
После трех дней инкубирования при этих условиях 0,1 мл раствора (4R, 6R)-6-{ 2-[(1S, 2S, 8S,8aR)-1,2,6,7,8,8a-гексагидро-8-(2,2- диэтилбутирилокси)-2-метил-1-нафтил]этил}тетрагидро-4-гидрокси-2Н- пиран-2-она (полученного, как описано в примере 73) в диметилсульфоксиде добавляли к среде так, чтобы конечная концентрация указанного соединения в среде была 0,01% (по весу на объем). Культивирование в тех же условиях продолжали еще в течение пяти дней.
В конце этого дополнительного периода культивирования ферментационную жидкую среду анализировали, используя высокоскоростную жидкостную хроматографию с пропусканием вещества через патрон C18 с набивкой из нова-пакТМ ("Уотерзз инк", патрон размером 8 на 100 мм). Колонку элюировали, используя смесь ацетонитрила и 0,1%-ного (вес на объем) триэтиламина (водным раствором фосфорной кислоты pH доводили до 3,2), со скоростью потока 1,5 мл/мин. Названное соединение элюировали в виде фракции со временем удерживания 6,38 мин. (То же самое соединение, полученное по методу 1, описанному выше, элюированное с колонки в виде фракции со временем удерживания 5,13 мин).
Ферментационный бульон фильтровали, и фильтрат подвергали адсорбции на колонке, содержащей 200 мл диаиона HP-20ТМ. Смолу промывали 300 мл дистиллированной воды, и фракции, содержащие названное соединение, элюировали 800 мл 50%-ного (по объему) водного раствора ацетона. Элюат концентрировали досуха упариванием при пониженном давлении, и концентрат очищали хроматографией с пропусканием вещества через ODS-колонку (ODS-H-5251ТМ "Сенсю сайентифик ко., лтд. ") при использовании в качестве элюента смеси ацетонитрила, воды и уксусной кислоты в объемном соотношении 450:550:1. За ходом хроматографии следили с помощью ультрафиолетовой абсорбции при 237 нм. Элюат нейтрализовали, непосредственно смешивая с 0,1 М водным раствором вторичного кислого фосфата натрия и гидроксида натрия (pH 8,0). Величину pH фракций, содержащих названное соединение, затем доводили до 8,0, после чего фракции концентрировали досуха, упариванием при пониженном давлении. Полученный концентрат затем растворяли в 20 мл воды, и раствор подвергали адсорбции на колонке, содержащей 20 мл диаиона HP-20ТМ. Смолу промывали 30 мл воды и затем элюировали 100 мл 50%-ного (по объему) водного раствора ацетона, получая 68 мг по существу чистой формы очищенного полевого соединения.
Спектр ядерного магнитного резонанса: (270 МГц, CD3OD) δ , в миллионных долях,
0,78 (9Н, триплет, J = 7,4 Гц);
0,91 (3Н, дублет, J = 7,0 Гц);
1,2-1,9 (13Н, мультиплет);
1,94 (1Н, удвоенный дублет из дублетов, J = 15,5, 6,2 и 2,0 Гц);
2,2-2,5 (5Н, мультиплет);
3,68 (1Н, мультиплет);
4,07 (1Н, мультиплет);
4,28 (1Н, мультиплет);
5,30 (1Н, мультиплет);
5,63 (1Н, мультиплет);
5,94 (1Н, дублет из дублетов, J = 9,7 и 6,1 Гц);
6,02 (1Н, дублет, J = 9,7 Гц).
Молекулярный вес 488 (C27H41O7Na, установлено методом масс-спектрометрии высокого разрешения с бомбардировкой быстрыми атомами).
[α]
Пример 84. Натриевый (3R,5R)-3,5-дигидрокси-7-[(1S,2S,6S,8S,8aR)-6-гидрокси- 2-метил-8-(2,2-диэтил-4-пентеноилокси)-1,2,6,7,8,8а-гексагидро-1- нафтил]гептаноат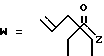
Одну платиновую петлю инокулята Amycolata autotrophica SANK 62981 (FERM BP-4105) использовали для инокулирования 100 мл дрожжевой среды МУ (состав которой указан выше в примере 81) в колбе Эрленмейера на 500 мл. Инокулированную колбу затем инкубировали при 28oC на ротационной мешалке со скоростью 200 об/мин.
После трех дней инкубирования при этих условиях часть инокулированной затравочной среды переносили в каждую из двадцати колб Эрленмейера на 500 мл, содержащих свежую дрожжевую среду МУ, так, чтобы конечная концентрация затравочной среды в свежей среде составляла 0,5% (по весу на объем). Колбы затем дополнительно инкубировали при 28oC на ротационной мешалке, вращающейся со скоростью 200 об/мин.
После двух дней инкубирования при этих условиях к среде добавляли водный раствор натриевого (3R, 5R)-3,5-дигидрокси-7-[(1S, 2S, 8S,8aR)-2-метил-8-(2,2-диэтил-4- пентеноилокси)-1,2,6,7,8,8a-гексагидро-1-нафтил] гептаноата (полученного как описано в примере 79) так, чтобы конечная концентрация указанного соединения в среде составляла 0,01% (по весу на объем). Культивирование затем продолжали еще в течение 5 дней в условиях, оговоренных ранее.
В конце этого периода культивирования ферментационную жидкую среду фильтровали, и фильтрат подвергали адсорбции на колонке, содержащей 200 мл диаиона HP-20ТМ. Смолу промывали 300 мл дистиллированной воды, после чего фракции, содержащие названное соединение, элюировали 800 мл 50%-ного (по объему) водного раствора ацетона.
Требуемые фракции соединяли, и объединенный элюат концентрировали, досуха упариванием при пониженном давлении. Полученный остаток очищали хроматографией, пропуская вещество через препаративную ODS-колонку (ODS-H-5251ТМ "Сенсю сайентифик ко., лтд.") при использовании в качестве элюента смеси из ацетонитрила, воды и уксусной кислоты в объемном соотношении 450:550:1. За ходом хроматографии следили по ультрафиолетовой адсорбции при 237 нм. Величину pH элюата доводили до 8,0, добавляли водный раствор гидроксида натрия, и полученную смесь концентрировали досуха упариванием при пониженном давлении. Полученный остаток растворяли в 50 мл воды, и раствор затем подвергали адсорбции на колонке, содержащей 20 мл диаиона НР-20ТМ. Смолу промывали 100 мл дистиллированной воды и затем элюировали 300 мл 50%-ного (по объему) водного раствора ацетона, получая 28 мг по существу чистого названного соединения.
Было показано, что физико-химические свойства соединения, полученного этим способом, являются идентичными таковым соединения, синтезированного в примере 64.
Пример 85. (4R, 6R)-6-{ 2-[(1S, 2S,8S,8aR)-1,2,6,7,8,8a-гексагидро-8-[(S)-2- метилвалерилокси] -2-метил-1-нафтил]этил}тетрагидро-4-гидрокси-2Н-пиран-2-он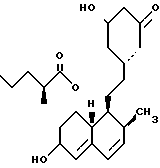
Ацетонитрил удаляли из фракций со временем удерживания от 40 до 50 мин (полученных проведением высокопроизводительной жидкостной хроматографии на стадии 2 синтеза 1, приведенного ниже) выпариванием при пониженном давлении в условиях использования роторного испарителя. Полученный концентрат экстрагировали дважды, используя каждый этилацетат в количестве половина объема концентрата. Экстракты соединяли и затем концентрировали упариванием при пониженном давлении, получая 5,2 г маслянистого вещества. Это вещество затем обрабатывали одним из двух нижеследующих способов.
1. Маслянистое вещество, полученное этим способом, растворяли в 20 мл ацетонитрила. Полученный раствор в количестве 2 мл затем впрыскивали в ODS-колонку (с внутренним диаметром 30 мм и длиной 300 мм; фирмы "Уай-Эм-Эс инк. ") с набивкой на УМС-пакS-346-15 S-15ТМ. Колонку затем подвергали элюированию, и элюирование производили в виде подвижной фазы при использовании 70%-ного (по весу в расчете на объем) водного раствора ацетонитрила при скорости потока 10 мл/мин, причем за ходом элюирования следили, используя рефрактометр. Элюаты со временем удерживания в пределах от 51 до 54 мин собирали.
Часть элюата, полученного выше, очищали жидкостной хроматографией высокой производительности, пропуская вещество через патрон с радиал-пакТМ (8 NVC 184, внутренний диаметр 8 мм на 10 см; фирмы "Уотерз ко.") с использованием в качестве подвижной фазы 70%-ного (по объему) водного раствора метанола при скорости потока 2,0 мл/мин. Требуемые фракции характеризовались ультрафиолетовой абсорбцией при 236 нм. Требуемая фракция имела время удерживания 4,7 мин.
При условиях, оговоренных выше, время удерживания соединения, полученного в синтезе 1, составляло 3,6 мин.
Описанный выше процесс хроматографической очистки затем повторяли еще десять раз до получения фракций с временем удерживания в 3,6 мин. Элюаты, полученные в ходе проведения этой дополнительной очистки, соединяли, и смесь затем концентрировали досуха упариванием при пониженном давлении с использованием роторного испарителя, получая 30 мг названного соединения в виде сырого продукта.
II. По альтернативному метолу маслянистое вещество, полученное ранее, растворяли в 1,5 мл ацетонитрила, и раствор затем впрыскивали в препаративную колонку (ODS-5251-SТМ, внутренний диаметр 20 мм на 250 мм, "Сенсю сайентифик ко., лтд"). Колонку элюировали с использованием в качестве подвижной фазы 70%-ного (по весу на объем) водного раствора ацетонитрила при скорости потока 5 мл/мин. Элюаты со временем удерживания от 33 до 37 мин собирали, причем за ходом элюирования следили по рефрактометру.
Полученные элюаты собирали и обеспечивали добавлением 15 мг порошка активированного угля, после чего смесь перемешивали при комнатной температуре в течение 10 мин. Смесь затем фильтровали через фильтровальную бумагу, и обесцвеченный фильтрат концентрировали досуха упариванием при пониженном давлении при использовании роторного испарителя, получая 13 мг по существу чистого названного соединения.
Масс-спектр (m/e) 404 (M+).
Молекулярная формула: C24H36O5.
λmax (нм) (Е1% 1см) ультрафиолетового спектра (этанол): 236,5 (576).
Спектр ядерного (углерод-13) магнитного резонанса (90 МГц, CDCl3) δ , в миллионных долях (в качестве внутреннего стандарта использовали тетраметилсилан; сигнал от дейтерированного хлороформа обнаруживался при δ 70,0 млн. долей): 170,3; 176,9; 132,6; 133,6; 128,1; 123,6; 76,2; 67,6; 62,61; 20,90; 38,61; 36,20; 39,94; 37,51; 36,89; 26,19; 33,03; 35,92; 30,9; 24,0; 20,6; 17,4; 13,9; 13,9.
В спектре ядерного магнитного резонанса на ядрах углеводорода-13 наблюдали сигналы 24 атомов углерода в соответствии с данными масс-спектрального анализа.
Спектр ядерного (водород-1) магнитного резонанса (360 МГц, CDCl3) δ , в миллионных долях: 5,88; 5,98; 5,51; 3,68; 5,36; 4,10; 4,29; 2,34; 2,24; 1,53; 1,57; 2,45; 2,37; 1,67; 1,59; 2,48; 1,35; 1,54; 1,22; 1,55; 2,42; 1,32 (каждый, 1Н); 1,12; 0,92; 0,91 (каждый, 3Н).
νmax (см-1 инфракрасного спектра (KBr): 3513, 1741, 1700, 1234, 1180.
[α]
Из приведенных спектральных данных следует, что соединение является идентичным соединению, полученному в примере 70.
Пример 86. Натриевый (3R,5R)-3,5-дигидрокси-7-[(1S,2S,8S,8aR)-2-метил-8- [(S)-2-метилвалерилокси]-1,2,6,7,8,8a-гексагидро-1-нафтил]гептаноат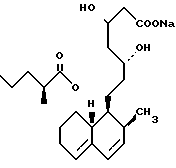
0,1 м 0,1 н. водного раствора гидроксида натрия добавляли к раствору 10 мг (4R, 6R)-6-{2-[(1S,2S,8S,8aR)-1,2,6,7,8,8a-гексагидро-8-[(S)-2- метилвалерилокси] -2-метил-1-нафтил] этил} тетрагидро-4-гидрокси-2Н- пиран-2-она (полученного, как описано в примере 85) в 0,2 мл 1,4-диоксана, и смесь нагревали при 60oC в течение 30 мин. В конце этого времени добавляли к смеси 10 мл воды и pH смеси доводили до 8,5 добавлением 0,1 н. водного раствора хлорида натрия. Полученную смесь затем адсорбировали на колонке, содержащей 5 мл диаиона НР-20ТМ. Смолу промывали 20 мл воды и затем элюировали 60%-ным (по весу на объем) водным раствором ацетона. Полученный элюат концентрировали упариванием при пониженном давлении с использованием роторного испарителя и концентрат подвергали лиофилизации, получая 9,8 мг названного соединения.
Молекулярный вес: 444 (по данным масс-спектрометрии с бомбардировкой быстрыми атомами).
Молекулярная формула: C24H37O6. Na (установлена методом масс-спектрометрии высокого разрешения с бомбардировкой быстрыми атомами).
λmax (нм) ультрафиолетового спектра (H2O): 237,4.
Спектр ядерного (углерод-13) магнитного резонанса (90 МГц, CD3OD) δ , в миллионных долях (в качестве внутреннего стандарта использовали тетраметилсилан; сигнал дейтерированного метанола появлялся при 49,0 млн. долей; сигналы наблюдали от 24 атомов углерода, что согласуется с молекулярной формулой), 180,5; 178,5; 135,4; 133,9; 129,3; 124,1; 71,8; 69,4; 69,4; 45,4; 45,2; 41,2; 38,8; 38,5; 37,2; 35,8; 32,1; 27,1; 25,6; 21,9; 21,6; 17,9; 14,4; 14,1.
Спектр ядерного (водород-1) магнитного резонанса (360 МГц, CD3OD) δ , в миллионных долях, 5,9; 5,7; 5,5; 5,3; 4,1; 3,7; 3,3 (каждая, 1Н); 1,1 (3Н); 0,9 (6Н).
νmax (см-1 инфракрасного спектра (KBr): 3385, 2936, 1728, 1578, 1409, 1085, 836.
[α]
Показано, что физико-химические данные соединения, полученного этим способом, идентичны таковым соединения, полученного в примере 75.
Пример 87. Натриевый (3R,5R)-3,5-дигидрокси-7-[(1S,2S,6S,8S,8aR)-8-гидрокси- 2-метил-8-[(S)-2-метилвалерилокси]-1,2,6,7,8,8a-гексагидро-1-нафтил] гептаноат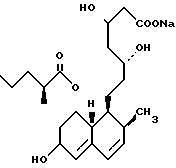
Одну платиновую петлю инокулята Streptomyces carbophilus SANK 62585 (FERM BP-4128) использовали для инокулирования 100 мл среды SC (состав которой указан в вышеприведенном примере 80) в колбе Эрленмейера на 500 мл, и инокулированную колбу инкубировали при 28oC на ротационной мешалке, вращающейся со скоростью 200 об/мин.
После трехдневного инкубационного периода часть инокулированной затравочной среды переносили в каждую из пяти колб Эрленмейера на 500 мл, содержащих по 100 мл свежей среды SC, так, чтобы концентрация затравочной среды в свежей среде составляла 5,0% (по весу в расчете на объем). Колбы затем инкубировали при 28oC на ротационной мешалке, вращающейся со скоростью 200 об/мин.
После еще трех дней инкубирования водный раствор 100 мг натриевого (3R, 5R)-3,5-дигидрокси-7-[(1S, 2S, 8S, 8aR)-2-метил-8-[(S)-2-метилвалерилокси] - 1,2,6,7,8,8a-гексагидро-1-нафтил]гептаноата (полученного, как описано в примере 10), добавляли к среде выращивания, так, чтобы конечная концентрация указанного соединения в среде составляла 0,02% (по весу в расчете на объем). Культивирование затем продолжали еще в течение трех дней в условиях, оговоренных выше.
В конце этого дополнительного периода культивирования ферментационную жидкую среду подвергали центрифугированию в течение 10 мин при скорости 3000 об/мин для разделения смеси на мицелий и надосадочную жидкость. 400 мл надосадочной жидкости удаляли, и pH жидкости доводили до 6 добавлением соответствующего количества 2 н. водного раствора гидроксида натрия. Смесь подвергали адсорбции на колонке, содержащей 20 мл диаиона НР-20ТМ ("Мицубиси касеи корпорейшн"). Смолу промывали 200 мл дистиллированной воды и затем элюировали 20 мл 20%-ного (по объему) водного раствора метанола, 20 мл 40%-ного (по объему) водного раствора метанола, и 40 мл 80%-ного (по объему) водного раствора метанола в указанном порядке.
Фракции, которые элюировались 40%-ным (по объему) водным раствором метанола и 60%-ным (по объему) водным раствором метанола соединяли и затем концентрировали досуха упариванием при пониженном давлении с использованием роторного испарителя, получая 50 мг названного соединения в виде сырого продукта.
Сырой продукт очищали хроматографией, пропуская вещество через колонку с микробонда-ПАКТМ (ODS; 8 мм x 30 см; "Уотерз инк.") в виде подвижной фазы при использовании в качестве элюента смеси метанола, воды и уксусной кислоты в объемном соотношении 550:450:1 при скорости подачи элюента 3 мл/мин. За ходом элюирования следили, используя дифференциальный рефрактометр. Фракции со временим удерживания 13 мин собирали.
pH собранных фракций доводили до 9 добавлением соответствующего 2 н. водного раствора гидроксида натрия, и метанол удаляли из смеси дистилляцией при пониженном давлении с использованием роторного испарителя. Величину pH остатка доводили до 8, и смесь подвергали адсорбции на колонке, содержащей 2 мл диаиона HP-20ТМ. Смолу промывали 10 мл дистиллированной воды и затем элюировали 20 мл 60%-ного (по весу на объем) водного раствора метанола.
Полученный элюат концентрировали упариванием при пониженном давлении, и затем подвергали лиофилизации, получая 3,4 мг по существу чистого названного соединения.
Молекулярный вес по данным масс-спектрометрии с бомбардировкой быстрыми атомами) (M+H)+:
Найдено: 461,2524;
Вычислено: 461,2515.
Молекулярная формула: C24H37O7•Na (определена методом масс-спектрометрии с бомбардировкой быстрыми атомами).
λmax (нм) (Е1% 1см) ультрафиолетового спектра (H2O): 238,7 (629).
Спектр ядерного (углерод-13) магнитного резонанса (90 МГц, CD3OD) δ , в миллионных долях (в качестве внутреннего стандарта использовали тетраметилсилан; сигнал от дейтерированного метанола появлялся при δ 49,0 миллионных долей): 179,8; 178,1; 136,8, 136,5; 128,6; 127,4; 71,5; 71,0; 69,2; 65,4; 45,1; 45,1; 41,2; 38,9; 38,3; 37,1; 37,1; 35,7; 32,3; 21,6; 17,8; 14,4; 13,9.
Спектр ядерного (водород-1) магнитного резонанса (360 МГц, CD3OD) δ , в миллионных долях: 5,88; 5,98; 5,51; 3,68; 5,36; 4,10; 4,29; 2,34; 2,24; 1,53; 1,57; 2,45; 2,37; 1,67; 1,58; 2,48; 1,35; 1,54; 1,22; 1,55; 2,42 (каждая, 1Н); 1,32 (2Н); 1,12; 0,92; 0,91 (каждая, 3Н).
νmax (см-1) инфракрасного спектра (KBr): 3391, 2960, 2935; 1728, 1400, 1181, 1043, 855.
[α]
Показано, что физико-химические данные соединения, полученного этим способом, идентичны таковым соединения из примера 49.
Синтез 1. Получение ML-236B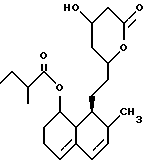
1. Выращивание
Среда для затравочной культуры:
Глицерин - 30 г
Глюкоза - 30 г
Соевая мука - 20 г
Микуни-пептон - 8 г ("Микуни кемикал индастриз ко., лтд.")
Нитрат натрия - 2 г
Сульфат магния - 1 г
Водопроводная вода (pH 6,0 - 6,5) - до 1000 мл
Среду для выращивания затравочной культуры в количестве 50 мл (состав приведен выше) загружали в колбу Эрленмейера на 500 мл и выдерживали в автоклаве при 120oC в течение 30 мин перед инокулированием микроорганизма. Одну платиновую петлю исходной культуры Penicillium citrinum Thom SANK 13380 (FERM BP-4129) на скошенном агаре асептически переносили в колбу, содержащую среду. Инокулированную колбу инкубировали при 24oC в течение трех дней на ротационной мешалке, вращающейся со скоростью 210 об/мин.
Колбу Эрленмейера на 2000 мл, содержащую 700 мл среды для выращивания затравочной культуры, помещали затем в автоклав и выдерживали при 120oC в течение 30 мин, после чего инокулировали все количество (примерно 50 мл) ферментационной жидкой среды, полученной как описано выше. Колбу инкубировали в течение двух дней при 24oC на ротационной мешалке, вращающейся со скоростью 210 об/мин, получая вторую генерацию культуры.
Следующие среды использовали при последующем продуцировании названного соединения.
Продуцирующая среда с культурой (I).
Достаточное количество водопроводной воды добавляли к 150 г глицерина и 600 г жидкого Sanmalt "Санва корнстач индастри, лтд") с доведением суммарного объема раствора до 5 л. Среду (I) затем стерилизовали в автоклаве в течение 30 мин при 120oC.
Продуцирующая среда с культурой (2). Смешивали нижеприведенные компоненты.
Соевая мука - 300 г
Микуни-пептон (фирма "Микуни кемикл индастриз ко., лтд") - 150 г
Онен CSальфа (фирма "Онен корпорейшн") - 300 г
Клейковина (фирма "Нихон сокухин какао ко., лтд") - 150 г
Сульфат магния - 15 г
pH устанавливали от 6,0 до 6,5 добавлением 10%-ного (по весу на объем) водного раствора гидроксида натрия, и затем общий объем доводили до 10 л добавлением водопроводной воды. Среду 2 затем стерилизовали в автоклаве в течение 30 мин при 120oC.
Подпитывающая среда А.
Водопроводную воду добавляли к смеси 1600 г глицерина и 6400 г санмолта S (фирма "Санва корнстач индастри, лтд.") и затем смесь нагревали до температуры выше 90oC. После полного растворения санмолта S к раствору добавляли водопроводную воду, доводя объем до 10 л. Раствор затем обрабатывали в автоклаве при 120oC в течение 30 мин.
Подпитывающая среда В.
Среду санникс PP 2000 ("Сане кемикал индастриз лтд.") в количестве 600 мл обрабатывали в автоклаве при 120oC в течение 30 мин.
5 л среды и 10 л среды 2 обрабатывали в автоклаве и затем загружали в цилиндрический ферментер из нержавеющей стали на 30 л для получения культуры второй генерации.
Все содержимое колбы Эрленмейера (примерно 700 мл), содержащей культуру второй генерации, полученную как описано выше, использовали затем для инокулирования обработанной в автоклаве среды, предназначенной для использования с продуцирующей культурой и находящейся в цилиндрическом ферментере. Ферментер инкубировали при 24oC в условиях перемешивания с автоматическим поддержанием скорости вращения в пределах от 260 до 500 об/мин; при этом аэрацию производили потоком воздуха с расходом 7,5 л/мин и давление поддерживали на уровне 0,5 кг/см2 для обеспечения концентрации растворенного кислорода от 3 до 5 частей на миллион.
В период с третьего по шестой день после совершения инкубирования 150 мл подпитывающей среды B добавляли к среде выращивания один раз в день (всего добавляли 4 раза). После того как концентрация сахара составляла не более 1%, начинали непрерывно добавлять подпитывающую среду A для поддержания величины pH жидкой среды примерно 4.
По прошествии 14 дней производили сбор клеток полученной жидкой среды.
2. Выделение.
Величину pH жидкой среды выращивали (40 л), доводили до 12, добавляли 800 мл 6 н. водного раствора гидроксида натрия, и полученную смесь перемешивали в течение 60 мин при комнатной температуре. В конце этого времени жидкую среду смешивали с 1,5 кг целитового фильтрующего материала (Целит N 545; торговое название продукта фирмы "Джонз-Манвилл продактс корп."), и смесь перемешивали. Полученную смесь фильтровали, пропуская через фильтр-пресс, получая фильтрат.
850 мл 6 н. водной соляной кислоты осторожно добавляли к фильтрату, и pH смеси доводили до 5,0. 80 л этилацетата добавляли к полученному раствору, и смесь перемешивали для экстракции требуемого продукта. Органический слой отделяли, и водный слой обрабатывали 40 л этилацетата и перемешивали, экстрагируя требуемый продукт. Объединенные этилацетатные экстракты затем экстрагировали 10 л 3%-ного (по весу на объем) водного раствора бикарбоната натрия. Водный слой отделяли, и органический слой снова экстрагировали 3%-ным (по весу на объем) водным раствором бикарбоната натрия.
1600 мл н. водной соляной кислоты осторожно добавляли к объединенным водным экстрактам, и pH смеси доводили до 5,0. Этилацетат в количестве 20 л добавляли к полученной смеси, и смесь перемешивали экстрагируя требуемый продукт. Органический слой отделяли и водный слой обрабатывали 10 л этилацетата и перемешивали для экстракции требуемого продукта. Объединенные этилацетатные экстракты промывали 15 л 10%-ного (по весу на объем) водного раствора хлорида натрия. Экстракт затем сушили над 3000 г безводного сульфата натрия, и растворитель удаляли упариванием досуха при пониженном давлении с использованием роторного испарителя, при этом получали маслянистый остаток.
Этот маслянистый остаток растворяли в 1000 мл этилацетата. К раствору добавляли 0,5 мл трифторуксусной кислоты, и смесь нагревали в условиях дефлегмации в течение 30 мин с обратным холодильником. Содержимое охлаждали до 10oC и затем дважды промывали 500 мл порциями 3%-ного (по весу на объем) водного раствора бикарбоната натрия, и один раз промывали 800 мл 10%-ного (по весу на объем) водного раствора хлорида натрия в указанной последовательности. Органический слой сушили над 100 г безводного сульфата натрия и фильтровали. Фильтрат освобождали от растворителя выпариванием досуха при пониженном давлении с использованием роторного испарителя, получая 50 г маслянистого остатка.
Весь маслянистый остаток растворяли в 500 мл ацетонитрила и полученный раствор делили на пять частей. Каждую часть очищали хроматографией, пропуская вещество через ODS-обращенно-фазовую колонку (ODS-1050-20SR; 10 см внутренний диаметр) на 50 см; 15-30 мкм (размер частиц); ("Курита когио ко., лтд. "). Колонку элюировали 70%-ным (по объему) водным раствором ацетонитрила, используемого в качестве подвижной фазы, при скорости потока 200 мл/мин. Фракции, извлеченные из колонки, контролировали по ультрафиолетовой абсорбции, и на основе обнаруженных пиков собирали фракции, время удерживания которых было между 30 и 36 мин.
Чистоту этих фракций оценивали проведением жидкостной хроматографии высокого разрешения, пропуская вещество через колонку (ODS-262; "Сенсю сайентифик ко., лтд.") при использовании в качестве подвижной фазы 70%-ного (по объему) водного раствора метанола при скорости потока 1,0 мл/мин; за фракциями следили по ультрафиолетовой абсорбции при 236 нм. Фракция со временем удерживания 11 мин характеризовалась наличием единственного пика ультрафиолетовой абсорбции.
Элюаты со временем удерживания от 40 до 50 мин хранили, и затем их использовали для извлечения соединения, синтезированного в примере 85.
Фракции, время удерживания которых при хроматографии с обращенно-фазовой колонкой находилось в области от 30 до 36 мин, подвергали концентрированию посредством дистилляции при пониженном давлении с использованием роторного испарителя для отгонки ацетонитрила. Концентрат дважды экстрагировали половинным объемом (от объема концентрата) этилацетата. Этилацетатные экстракты соединяли и концентрировали упариванием досуха при пониженном давлении, получая 30 г маслянистого остатка.
Маслянистое вещество растирали со смесью этанола с водой для инициирования кристаллизации. Получали 17 г названного соединения в виде бесцветных кристаллов.
Физико-химические свойства этого соединения известны и идентичны свойствам, описанным в Японской патентной публикации N Sho-56-12114 (патент Великобритании N 1453425) и в другой литературе.
Синтез 2. Получение натриевой соли правастатина.
Колбу Эрленмейера на 500 мл, содержащую 100 мл дрожжевой среды МУ, предназначенной для выращивания, состав которой приведен в примере 61, инокулировали платиновой петлей исходной культуры Amycolota autotrophica SANK 62981 (FERM BP-4105) со скошенного агара. Колбу инкубировали при 28oC на ротационной мешалке, вращающейся со скоростью 200 об/мин.
Через три дня двадцать колб Эрленмейера на 500 мл, содержащих по 100 мл дрожжевой среды МУ для выращивания культуры, состав которой указан в примере 81, инокулировали посевной культурой, в количестве 0,5% содержимого колбы. Культуры затем инкубировали при 28oC на ротационной мешалке со скоростью 200 об/мин. По прошествии двух дней водный раствор натриевой соли соединения ML-236B добавляли до конечной концентрации в 0,1% по натриевой соли, и смесь инкубировали при 28oC на ротационной мешалке, вращающейся со скоростью 200 об/мин в течение 5 дней.
В конце этого времени ферментационную жидкую среду фильтровали, и фильтрат подвергали абсорбции на 200 мл неионной смолы диаион НР-20. Смолу промывали 300 мл дистиллированной воды, и фракции, содержащие названное соединение, элюировали 800 мл 50%-ного (по объему) водного раствора ацетона.
Элюат концентрировали упариванием досуха при пониженном давлении, и концентрат очищали хроматографией, пропуская вещество через препаративную ODS-колонку (ODS-H-5251) при использовании в качестве элюента смеси ацетонитрила, воды и уксусной кислоты в объемном соотношении 460:520:1; фракции при этом контролировали измерением ультрафиолетовой абсорбции при 237 нм. Требуемые фракции собирали, и pH доводили до 3,0 добавлением водного раствора гидроксида натрия. Смесь затем концентрировали упариванием при пониженном давлении. Концентрат растворяли в 50 мл воды, и полученный водный раствор обрабатывали 50 мл диаиона НР-20. Смолу промывали 100 мл дистиллированной воды и затем элюировали 200 мл 50%-ного (по объему) водного раствора ацетона, получая 618 мг названного соединения.
Физико-химические свойства известны и идентичны свойствам, описанным в Японской патентной публикации N Sho 61-13699 (патент Великобритании N 2077264) и в другой литературе.
Синтез 3. (4R, 6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-Гексагидро-6-трет- бутилдиметилсилилокси-8-(2,2,3,3-тетраметилциклопропанкарбонилокси)- 2-метил-1-нафтил]этил}тетрагидро-4-третдиметилсилилокси-2Н-пиран-2-он.
Следовали методике, аналогичной описанной в примере 4, но использовали 1,0 г (1,8 ммоль) (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-6-трет- бутилдиметилсилилокси-8-гидрокси-2-метил-1-нафтил] этил}тетрагидро- 4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного как описано в примере B) и 1,17 г хлористого 2,2,3,3,-тетраметилциклопропанкарбонила, получая 833 мг названного соединения.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
4,24-4,29 (1Н, мультиплет);
4,30-4,49 (1Н, мультиплет);
4,56-4,63 (1Н, мультиплет);
5,41 (1Н, широкий синглет);
5,84 (1Н, дублет из дублетов, J = 9,3 и 5,9 Гц);
5,99 (1Н, дублет, J = 9,3 Гц).
νmax (см-1) инфракрасного спектра (CHCl3): 2950, 1720, 1250, 1080, 840.
Масс-спектр (m/e): 674 (M+), 659, 617, 532.
[α]
Синтез 4. (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-Гексагидро-6-гидрокси-8- (2,2,3,3, -тетраметилциклопропанкарбонилокси)-2-метил-1-нафтил] этил} тетрагидро-2Н-пиран-2-он
Следовали методике, аналогичной описанной в примере 24, но использовали 312 мл (4R,6R)-6-{2-[(1S,2S,6S,8S,8aR)-1,2,6,7,8,8a-гексагидро-8-трет- бутилдиметилсилилокси-8-(2,2,3,3-тетра-метилциклопропанкарбонилокси)-2- метил-1-нафтил] этил} тетрагидро-4-трет-бутилдиметилсилилокси-2Н-пиран-2-она (полученного как описано в синтезе 3) и получали 480 мг названного соединения, плавящегося при температурах от 124 до 126oC.
Элементный анализ для C26H38O6•1/2H2O:
Вычислено: C - 68,54%, H - 8,41%;
Найдено: C - 68,65%, H - 8,60%.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
0,91 (3Н, дублет, J = 7,3 Гц);
1,15 (3Н, синглет);
1,17 (3Н, синглет);
1,22 (3Н, синглет);
1,24 (3Н, синглет);
2,93-3,03 (1Н, мультиплет);
4,35-4,50 (1Н, мультиплет);
4,56-4,68 (1Н, мультиплет);
5,39 (1Н, широкий синглет);
5,59 (1Н, широкий синглет);
5,90 (1Н, дублет из дублетов, J = 9,8 и 5,9 Гц);
6,01 (1Н, , дублет, J = 9,3 Гц).
νmax (см-1) инфракрасного спектра (CHCl3): 3450, 2950, 1720, 1180.
Масс-спектр (m/e): 446 (M+), 423, 321, 304.
[α]
Синтез 5. Получение соединения формулы (XIV).
Величину pH 4 л среды с затравочной культурой, приготовленной так, как описано на стадии 1 в синтезе 1, доводили до 12, добавляя 80 мл 6 н. водного раствора гидроксида натрия. Смесь затем перемешивали при комнатной температуре в течение 60 мин.
В конце этого времени 0,1 кг целита (целит N 545 "Джонз-Мэнвилл продактс корп. ") смешивали с жидкой средой в качестве средства для фильтрования, и жидкую среду затем фильтровали. Величину pH фильтрата доводили до 5,0 осторожным добавлением 85 мл 6 н. водного раствора хлористого водорода, после чего смесь экстрагировали 8 л этилацетата.
Органический слой затем отделяли, и водный слой снова экстрагировали 4 л этилацетата. Экстракты соединяли и затем экстрагировали дважды, используя каждый раз по 1 л 3%-ного (по весу на объем) водного раствора бикарбоната натрия. Полученные водные экстракты соединяли и pH доводили до 5,0 осторожным добавлением 160 мл 6 н. водного раствора хлористого водорода. Смесь экстрагировали 2 л этилацетата, после чего водный слой отделяли и экстрагировали еще раз 1 л этилацетата. Экстракты соединяли, промывали 1,5 л насыщенного водного раствора хлорида натрия и затем сушили над 300 г безводного сульфата натрия. Растворитель удаляли дистилляцией при пониженном давлении, используя роторный испаритель и получали маслянистый остаток. Трифторуксусную кислоту (0,1 мл) добавляли к раствору, полученному при растворении остатка в 100 мл этилацетата, и смесь нагревали в условиях дефлегмации в течение 30 мин в колбе, снабженной обратным холодильником.
В конце этого времени смесь охлаждали до 10oC, и полученную смесь дважды промывали 50 мл (каждый раз) 3%-ного (по весу на объем) водного раствора бикарбоната натрия и затем дважды промывали 50 мл (каждый раз) 10%-ного (по весу на объем) водного раствора хлорида натрия. Органический слой затем сушили над 10 г безводного сульфата натрия, после чего этот слой фильтровали и концентрировали упариванием при пониженном давлении с использованием роторного испарителя, получая 5 г маслянистого вещества.
Весь полученный маслянистый материал растворяли в 100 мл ацетонитрила и раствор очищали хроматографией, пропуская через обращенно-фазовую ODS-колонку (ODS-1050-20-SRТМ внутренний диаметр 10 см х 50 см; 15-30 мкм (размер части); "Курита уотер индастриз лтд.") при использовании в качестве подвижной фазы 40%-ного (по объему) водного раствора ацетонитрила, подаваемого со скоростью 200 мл/мин. За ходом хроматографии следили по ультрафиолетовой абсорбции при 236 нм. Собирали фракции со временем удерживания от 33 до 39 мин. Ацетонитрил затем удаляли из фракций отгонкой при пониженном давлении в условиях использования роторного испарителя, получая маслянистое вещество.
Все полученное маслянистое вещество растворяли в 5 мл ацетонитрила и затем снова очищали хроматографией, пропуская через препаративную ODS-колонку (ODS-H-5251ТМ "Сенсю сайентифик ко., лтд".) при использовании в качестве подвижной фазы смеси ацетонитрила и воды в объемном соотношении 35:65. За ходом хроматографии следили по показателю преломления, полученному на дифференциальном рефрактометре. Фракции со временем удерживания от 30 до 35 мин собирали, и ацетонитрил удаляли отгонкой при пониженном давлении в условиях использования роторного испарителя. Остаток затем дважды экстрагировали, порциями этилацетата, составляющими половину объема остатка. Экстракты объединяли и затем концентрировали досуха упариванием при пониженном давлении 100 мг названного соединения.
Физико-химические свойства соединения известны и идентичны свойствам, описанным для соединения в Японской патентной заявке N Sho 51-136885.
Масс-спектр (m/e): 306 (M+), 270, 210, 145.
Спектр ядерного (водород-1) магнитного резонанса (270 МГц, CDCl3) δ , в миллионных долях,
5,9 (1Н, дублет);
5,75 (1Н, дублет из дублетов);
5,55 (1Н, широкий синглет);
4,7 (1Н, мультиплет);
4,35 (1Н, мультиплет);
4,25 (1Н, мультиплет);
0,9 (2Н, дублет).
Спектр ядерного (углерод-13) магнитного резонанса (90 МГц, CDCl3) δ , в миллионных долях: 171,3; 133,4; 128,4; 123,7; 76,4; 64,4; 62,5; 38,8; 38,5; 36,4; 36,1; 32,7; 30,8; 29,2; 23,8; 20,4; 13,9.
В следующих синтезах 6 - 18 описано получение различных стереоизомерных соединений, подходящих для использования в качестве исходных веществ в предшествующих примерах согласно следующей реакционной схеме (табл.10).
Синтез 6. (+)-(2S)-1,2-Диметил-2-фенил-1-циклопентанол (соединение 2).
Каталитическое количество иода добавляли к суспензии 5,24 г (216 ммоль) магния в 30 мл сухого диэтилового простого эфира при перемешивании в токе азота. Затем по каплям в течение часа к смеси добавляли раствор 13,4 мл (216 ммоль) иодистого метила в 180 мл диэтилового эфира. Смесь затем перемешивали в течение 20 мин. В конце этого времени к смеси по каплям добавляли раствор 3,76 г (21,6 ммоль) (+)-(2S)-2-метил-2-фенилциклопентана (соединение 1) (оптическая энантиомерная чистота составляла 95%), синтезированного по методике Кога (Koga) и др. (Chemical and Pharmaceutical Bulletin (Япония) 27, 2760 (1979)), в 30 мл диэтилового эфира в течение 10 мин. Полученную смесь затем нагревали с обратным холодильником в течение 2 ч. В конце этого времени реакционную смесь охлаждали льдом, после чего к смеси на протяжении 20 мин по каплям добавляли 250 мл насыщенного водного раствора хлорида аммония. Полученную смесь затем разбавляли 100 мл воды и образовавшуюся водную смесь экстрагировали дважды порциями по 100 мл этилацетата. Экстракты соединяли, промывали насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия. Растворитель затем удаляли отгонкой при пониженном давлении и получали названное соединение в виде бледно-желтой маслянистой жидкости в форме двух диастереоизомеров. Продукт, состоящий из двух диастереоизомеров, может использоваться непосредственно в последующей реакции, т. е. без разделения стереоизомеров. Бледно-желтую маслянистую жидкость очищали колонной флэш-хроматографией с пропусканием через силикагель при использовании в качестве элюента смеси гексана и этилацетата в объемном соотношении 5:1 и получали 863 мг (выход 21%) продукта в виде бледно-желтого масла из менее полярных фракций.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
1,26 (3Н, синглет);
1,32 (3Н, синглет);
1,60-2,10 (6Н, мультиплет; 1Н способен обмениваться с D2O);
2,69-2,80 (1Н, мультиплет);
7,22-7,53 (5Н, мультиплет).
νmax (см-1) инфракрасного спектра (CHCl3): 3350, 2950, 1730, 1500, 1440, 1380, 1140, 700.
Масс-спектр (m/e): 190 (M+).
[α]
Использование приведенной выше методики давало 2,43 г (выход 59%) названного соединения в виде бледно-желтого масла из более полярных фракций.
Спектр ядерного магнитного резонанса (270 МГц, CDCl3) δ , в миллионных долях,
0,93 (3Н, синглет);
1,38 (3Н, синглет);
1,70-1,97 (6Н, мультиплет, 1Н способен обмениваться с D2O);
2,25-2,36 (1Н, мультиплет);
7,17-7,46 (5Н, мультиплет);
νmax (см-1) инфракрасного спектра (CHCl3): 3350, 2950, 1730, 1600, 1500, 1370, 1100, 1050, 700.
Масс-спектр (m/e): 190 (M+).
[α]
Синтез 7. (+)-(1S)-1,2-Диметил-1-фенил-2-циклопентен (соединение 3).
Хлороксид фосфора (38,6 мл) на протяжении 30 мин добавляли по каплям к раствору 7,47 г (39,2 ммоль) (+)-(2S)-1,2-диметил-2-фенил-1-циклопентанола (полученного, как описано в синтезе 6) в 77 мл сухого пиридина при охлаждении льдом в токе азота. Полученную смесь затем перемешивали сначала при комнатной температуре в течение 16 ч, а затем при 70oC в течение 2 ч. В конце этого времени реакционную смесь охлаждали льдом и выливали понемногу в 700 мл смеси воды со льдом. Полученную водную смесь дважды экстрагировали, каждый раз по 400 мл этилацетата. Экстракты соединяли, промывали сначала насыщенным водным раствором бикарбоната натрия и затем насыщенным водным раствором хлорида натрия, и затем смесь сушили над безводным сульфатом натрия. Растворитель удаляли дистилляцией при пониженном давлении, получая бледно-желтый маслянистый остаток в количестве 6,70 г.
Бледно-желтый маслянистый остаток (6,70 г) растворяли в 500 мл диоксана, и затем к раствору добавляли 6,70 г (38,9 ммоль) п-толуолсульфоновой кислоты. Полученную смесь нагревали с обратным холодильником в течение 18 ч. В конце этого времени реакционную смесь концентрировали упариванием при пониженном давлении, и концентрат разбавляли 300 мл воды и затем дважды экстрагировали, каждый раз по 400 мл этилацетата. Экстракты соединяли, промывали сначала насыщенным водным раствором бикарбоната натрия, а затем насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом натрия. Растворитель удаляли дистилляцией при пониженном давлении и получали бледно-желтый остаток. Остаток очищали колонной флэш-хроматографией, пропуская через силикагель с использованием в качестве элюента гексана, получая 4,84 г (выход 72%) названного соединения в виде бледно-желтого масла.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
1,47 (3Н, синглет);
1,50 (3Н, синглет);
1,95-2,16 (2Н, мультиплет);
2,30-2,39 (2Н, мультиплет);
5,53 (1Н, синглет);
7,16-7,34 (5Н, мультиплет);
νmax (см-1) инфракрасного спектра (CHCl3): 2950, 1600, 1490, 1440, 1370, 1020, 700.
Масс-спектр (m/e): 172 (M+).
[α]
Синтез 8. (+)-(2S)-6,6-Диметокси-3-фенил-3-метил-2-гексанон (соединение 4).
Поток воздуха, содержащий 10 г/м3 озона, барботировали через раствор 764 мг (4,43 ммоль) (+)-(1S)-1,2-диметил-1-фенил-2-циклопентена (полученного, как описано в синтезе 7) в 15 мл метанола в течение 2,5 ч при охлаждении льдом. В конце этого времени реакционную смесь охлаждали до -78oC, после чего к реакционной смеси добавляли 0,65 мл диметилсульфида. Температуре реакционной смеси давали подняться до комнатной, и реакционную смесь затем перемешивали в течение 15 ч. Смесь затем концентрировали упариванием при пониженном давлении. Полученный концентрат разбавляли 50 мл воды, и разбавленный раствор дважды экстрагировали, каждый раз по 50 мл этилацетата. Экстракты соединяли, промывали насыщенным водным раствором хлорида натрия, и затем сушили над безводным сульфатом натрия. Растворители затем удаляли дистилляцией при пониженном давлении, и получали бесцветный маслянистый остаток. Этот остаток очищали колонной флэш хроматографией, пропуская через силикагель при использовании в качестве элюента смеси гексана с этилацетатом, в объемном соотношении 5:1, получая 971 мг (выход 88%) названного соединения в виде бесцветного масла.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
1,26-1,47 (2Н, мультиплет);
1,50 (3Н, синглет);
1,92 (3Н, синглет);
1,92-2,00 (2Н, мультиплет);
3,24 (3Н, синглет);
3,30 (3Н, синглет);
4,32 (1Н, триплет, J = 5,9 Гц);
7,20-7,39 (5Н, мультиплет).
νmax (см-1) инфракрасного спектра (CHCl3): 2950, 1700, 1440, 1350, 1120.
Масс-спектр (m/e): 249 (М+-1).
[α]
Синтез 9. (+)-(4S)-5-Оксо-4-фенил-4-метилгексанал (соединение 5).
6 мл воды и затем 6,0 мл трифторуксусной кислоты добавляли к раствору 953 мг (3,81 ммоль) (+)-(2S)-6,6-диметокси-3-фенил-3-метил-2-гексанона (полученного, как описало в синтезе 3) в 12 мл хлороформа при охлаждении льдом и перемешивании, и полученную смесь энергично перемешивали при комнатной температуре в течение 3 ч. В конце этого времени реакционную смесь разбавляли 50 мл воды, и разбавленный раствор дважды экстрагировали, каждый раз по 100 мл хлористого метилена. Экстракты соединяли, затем промывали сначала насыщенным водным раствором бикарбоната натрия и затем насыщенным водным раствором хлорида натрия и сушили над безводным сульфатом натрия. Растворитель удаляли дистилляцией при пониженном давлении, получая бесцветный маслянистый остаток. Этот остаток очищали колонной флэш-хроматографией, пропуская через силикагель при использовании в качестве элюента смеси гексана с этилацетатом в объемном соотношении 3:1, получая 699 мг (выход 90%) названного соединения в виде бесцветного масла.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
1,51 (3Н, синглет);
1,93 (3Н, синглет);
2,15-2,33 (4Н, мультиплет);
7,20-7,41 (5Н, мультиплет);
9,66 (1Н, синглет).
νmax (см-1) инфракрасного спектра (CHCl3): 1720, 1600, 1350.
Масс-спектр (m/e): 203 (M+-1).
[α]
Синтез 10. (-)-(3S)-5-Гидрокси-3-фенил-3-метил-2-гексанол (соединение 6).
Боргидрид натрия (259 мг, 6,84 ммоль) добавляли понемногу к раствору 699 мг (3,42 ммоль) (+)-(4S)-5-оксо-4-фенил-4-метил-гексанола (полученного, как описано в синтезе 9) в 14 мл этанола, и полученную смесь перемешивали при комнатной температуре в течение часа. В конце этого времени 4,0 мл ацетона добавляли к смеси, и смесь перемешивали в течение 20 мин. Реакционную смесь затем концентрировали упариванием при пониженном давлении, и концентрат смешивали с 30 мл воды. Смесь дважды экстрагировали, каждый раз по 30 мл этилацетата, экстракты соединяли, промывали насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия. Растворитель затем удаляли дистилляцией при пониженном давлении и получали названное соединение, состоящее из двух диастереоизомеров в виде бесцветного масла. Смесь, состоящая из двух диастереоизомеров, может быть использована непосредственно в следующей реакции, т.е. без дальнейшего разделения.
Полученный маслянистый продукт очищали колонной флэш-хроматографией, пропуская через силикагель при использовании в качестве элюента смеси гексана с этилацетатом в объемном соотношении 1:1, получая 208 мг (выход 29%) первого изомера названного соединения в виде бесцветных порошкообразных кристаллов из менее полярных фракций, и 286 мг (выход 54%) второго изомера в виде бесцветных порошкообразных кристаллов из более полярных фракций.
Соединение, которое элюировалось первым, имело следующие характеристики.
Температура плавления: 100oC (после перекристаллизации из смеси хлористого метилена с гексаном).
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
1,08-1,19 (1Н, мультиплет);
1,12 (3Н, дублет, J = 6,5 Гц);
1,31 (3Н, синглет);
1,34-1,49 (1Н, мультиплет);
1,54-1,62 (3Н, мультиплет; 2Н способный обмениваться с D2O);
1,90-1,99 (1Н, мультиплет);
3,56 (2Н, триплет, J = 6,5 Гц);
3,87 (1Н, квартет,, J = 6,5 Гц);
7,21-7,39 (5Н, мультиплет).
νmax (см-1) инфракрасного спектра (CHCl3): 3600, 2950, 1380, 1260, 1150, 1130, 1100, 700.
Масс-спектр (m/e): 209 (M+ + 1).
Элементный анализ для C13H20O2:
Вычислено: C - 74,96%, H - 9,68%;
Найдено: C - 74,75%, H - 9,65%.
[α]
Соединение, которое элюировало позже, имело следующие характеристики.
Соединение плавится между 105 и 106oC (после перекристаллизации из смеси хлористого метилена с гексаном).
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
0,96 (3Н, дублет, J = 6,4 Гц);
1,16-1,27 (1Н, мультиплет);
1,32 (3Н, синглет);
1,38-1,55 (3Н, мультиплет; 2Н могут обмениваться с D2O);
1,71 - 1,79 (1Н, мультиплет);
1,82-2,02 (1Н, мультиплет);
3,58 (2Н, триплет, J = 6,5 Гц);
3,87 (1Н, квартет, J = 6,4 Гц);
7,19-7,38 (5Н, мультиплет).
νmax (см-1) инфракрасного спектра (CHCl3): 3650, 3450, 2950, 1380, 1150, 700.
Масс-спектр (m/e) 209 (M+ + 1).
Элементный анализ для C13H20O2:
Вычислено: C - 74,96%, H - 9,66%.
Найдено: C - 74,70%, H - 9,63%.
[α]
Синтез 11. (-)-(3S)-6-Бензоилокси-3-фенил-3-метил-2-гексанол (соединение 7).
Каталитическое количество (20 мг) 4-диметиламинопиридина добавляли в токе азота к раствору 4,04 г (19,4 ммоль) смеси двух диастереоизомеров (-)-(8S)-6-гидрокси-3-фенил-3-метил-2-гексанола (полученного, как описано в синтезе 10,) в 100 мл сухого пиридина, после чего к смеси при перемешивании и охлаждении льдом по каплям в течение 15 мин добавляли 2,36 мл (20,4 ммоль) хлористого бензоила. Температуре смеси давали подняться от температуры льда до комнатной температуры, и затем реакционную смесь перемешивали в течение 16 ч. В конце этого времени смесь концентрировали упариванием при пониженном давлении. Концентрат разбавляли 300 мл воды, и разбавленный раствор дважды экстрагировали, каждый раз по 200 мл этилацетата. Экстракты соединяли, затем промывали 5%-ным (по весу в расчете на объем) водным раствором хлористого водорода, насыщенным водным раствором бикарбоната натрия и насыщенным водным раствором хлорида натрия, в указанном порядке, и затем сушили над безводным сульфатом натрия. Растворитель затем удаляли дистилляцией при пониженном давлении и получали бесцветное масло. Это масло представляет собой смесь двух диастереоизомеров, получившихся из диастереоизомерного исходного вещества. Продукт очищали колонной флэш-хроматографией, пропуская через силикагель с использованием в качестве элюента смеси гексана с этилацетатом в объемном соотношении 2:1, 5,54 г (выход 91%) названного соединения, состоящего из двух диастереоизомеров, в виде бесцветного масла.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
0,97 (1,2H, дублет, J = 6,6 Гц);
1,12 (1,8H, дуплет, J = 6,6 Гц);
1,30-1,52 (1H, мультиплет);
1,34 (1,6 H, синглет);
1,35 (1,2H, синглет);
1,54-1,75 (2H, мультиплет; 1H способен обмениваться с D2O);
1,80-1,91 (1H, мультиплет);
2,03-2,12 (1H, мультиплет);
3,82-3,91 (1H, мультиплет);
4,21-4,27 (2H, мультиплет);
7,22-7,59 (8H, мультиплет);
8,02 (2H, дублет, J = 7,9 Гц).
νmax (см-1) инфракрасного спектра (CHCl3): 3600, 2950, 1710, 1280, 1120.
Масс-спектр (m/e): 313 (M+ + 1).
Синтез 12. (-)-(4S)-5-трет-Бутилдиметилсилилокси-4-фенил-4-метил-1-гексанол (соединение 8).
Имидазол (4,88 г (70,8 ммоль), а затем 8,02 г (53,1 ммоль) хлористого трет-бутилдиметилсилила добавляли в токе азота к раствору 5,54 г (17,7 ммоль) смеси двух диастереоизомеров соединения (-)-(8S)-6-бензилокси-3-фенил-3-метил-2-гексанола (полученного, как описано в синтезе 11) в 20 мл диметилформамида, и полученную смесь перемешивали при комнатной температуре в течение 15 ч. В конце этого времени реакционную смесь концентрировали упариванием при пониженном давлении, и концентрат разбавляли 400 мл воды. Разбавленный раствор дважды экстрагировали, каждый раз по 300 мл этилацетата. Экстракты соединяли, промывали насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия. Растворитель затем удаляли дистилляцией при пониженном давлении и получали бесцветное масло. Это вещество состоит из двух диастереоизомеров, происходящих из исходного соединения. 53 мл 1 н. водного раствора гидроксида натрия добавляли к раствору 3,03 г указанной выше диастереоизомерной смеси в 250 мл этанола, и полученную смесь перемешивали при 60oC в течение 2,5 ч. В конце этого времени реакционную смесь концентрировали упариванием при пониженном давлении, и концентрат разбавляли 400 мл воды. Разбавленный раствор дважды экстрагировали, каждый раз по 300 мл этилацетата, экстракты соединяли, промывали насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия. Растворитель удаляли дистилляцией при пониженном давлении, получая бесцветный маслянистый остаток. Этот остаток очищали мгновенной колонной хроматографией, пропуская через силикагель при использовании в качестве элюента смеси гексана с этилацетатам, в соотношении 4:1 и получали 5,37 г (выход 94%) названного соединения, состоящего из двух диастереоизомеров в виде бесцветного масла.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
0,00-0,10 (6H, мультиплет);
0,80 (1,8H, дублет, J = 6,4 Гц);
0,90 (9H, синглет);
0,97 (1,2 H, дублет, J = 6,5 Гц);
1,03-1,14 (1H, мультиплет);
1,26 (1,2H, синглет);
1,28 (1,8H, синглет);
1,32-1,55 (2H, мультиплет; 1H способен обмениваться с D2O);
1,72-1,84 (2H, мультиплет);
3,49-3,57 (2H, мультиплет);
3,79 (0,4H квартет, J = 6,4 Гц);
3,90 (0,6H, квартет, J = 6,4 Гц);
7,14-7,33 (5H, мультиплет).
νmax (см-1) инфракрасного спектра (CHCl3): 3650, 2950, 1250, 1150, 1090, 640.
Масс-спектр (m/e) был следующим: 321 (M+-1).
Синтез 13. (-)-(3S)-2-трет-бутилдиметилсилилокси-3-фенил-3-метил-6-иодгексан (соединение 9).
Диэтилазодикарбоксилат (5,24 мл, 33,2 ммоль), а затем 3,11 мл (49,8 ммоль) иодистого метила добавляли к раствору 5,37 г (16,6 ммоль) смеси двух диастереоизомеров (-)-(4S)-5-трет-бутилдиметилсилилокси-4-фенил-4-метил-1-гексанола (полученного, как описано выше в синтезе 12) и 8,73 г (33,2 ммоль) трифенилфосфина в 70 мл сухого тетрагидрофурана при охлаждении льдом в токе азота, и полученную смесь перемешивали при комнатной температуре в течение одного часа. В конце этого времени 2,18 г (8,3 ммоль) трифенилфосфина добавляли к смеси, и смесь охлаждали льдом. Диэтилазодикарбоксилат (2,62 мл, 16,6 ммоль) и 1,55 мл (43,8 ммоль) иодистого метила добавляли затем к смеси, и полученную смесь перемешивали при комнатной температуре в течение 30 мин. В конце этого времени реакционную смесь концентрировали упариванием при пониженном давлении, и концентрат очищали мгновенной хроматографией, пропуская через силикагель с использованием в качестве элюента гексана, получая 6,29 г (выход 67%) названного соединения, состоящего из двух диастереоизомеров, в виде бесцветного масла.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
-0,22 (1,8 H, синглет);
-0,04 (1,3 H, синглет);
0,04 (1,2 H, синглет);
0,05 (1,2 H, синглет);
0,79 (1,2 H, дублет, J = 5,9 Гц);
0,82 (5,4 H, синглет);
0,92 (3,6 H, синглет);
0,95 (1,8 H, дублет, J = 5,9 Гц);
1,25 (1,2 H, синглет);
1,28 (1,8 H, синглет);
1,28-1,40 (1H, мультиплет);
1,54 - 1,65 (1Н, мультиплет);
1,74 - 2,10 (2Н, мультиплет);
3,02-3,14 (2Н, мультиплет);
3,77 (0,6 H, квартет, J = 6,6 Гц);
3,90 (0,4, квартет, J = 6,6 Гц);
7,16 - 7,32 (5Н, мультиплет).
νmax (см-1) инфракрасного спектра (CHCl3): 2950, 1250, 1100, 980, 840, 700.
Масс-спектр (m/e): 431 (M+-1).
Синтез 14. (-)-(3S)-2-Гидрокси-3-фенил-3-метилгексан (соединение 10).
Гидрид трибутилолова (19,2 мл, 71,0 ммоль) и 3,51 г (21,3 ммоль) азобисизобутиронитрила добавляли в токе азота к раствору 6,16 г (14,2 ммоль) смеси двух диастереоизомеров (-)-(35)-2-трет-бутилдиметилсилилокси-3-фенил-3-метил-6-иодгексана (полученного, как описано в синтезе 13) в 80 мл толуола, и полученную смесь перемешивали при 80oC в течение одного часа. В конце этого времени реакционную смесь концентрировали упариванием при пониженном давлении, и концентрат очищали мгновенной колонной хроматографией, пропуская через силикагель с использованием в качестве элюента гексана. Продукт, полученный таким способом, растворяли в 200 мл ацетонитрила, после чего 20 мл 46%-ного (по весу на объем) водного раствора фтористого водорода добавляли к смеси, которую затем перемешивали при комнатной температуре в течение 4 ч. В конце этого времени реакционную смесь концентрировали упариванием при пониженном давлении, и концентрат смешивали с 300 мл воды. Водную смесь затем дважды экстрагировали, каждый раз по 200 мл этилацетата. Экстракты соединяли, промывали насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия, растворитель удаляли дистилляцией при пониженном давлении, и бесцветный маслянистый остаток очищали мгновенной колонной хроматографией, пропуская через силикагель при использовании в качестве элюента смеси гексана с этилацетатом при объемном соотношении 4: 1, получая 2,84 г (выход 65%) названного соединения (состоящего из двух диастереоизомеров) в виде бесцветного масла.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
0,82-0,97 (6Н, мультиплет);
1,10-1,21 (1Н, мультиплет);
1,29 (1,6H, синглет);
1,31 (1,2H, синглет);
1,35-1,93 (4Н, мультиплет; 1Н может обмениваться с D2O);
3,83-3,92 (1Н, мультиплет);
7,22-7,41 (5Н, мультиплет).
νmax (см-1) инфракрасного спектра (CHCl3):
3600, 2950, 1100, 700.
Масс-спектр (m/e) 177 (M+-15).
Синтез 15. (+)-(3S)-3-фенил-3-метил-2-гексанон (соединение 11).
Раствор 1,86 мл (26,2 ммоль) диметилсульфоксида в 5 мл хлористого метилена добавляли по каплям на протяжении 5 мин в токе азота и при температуре -78oC к раствору 1,43 мл (16,4 ммоль) хлористого оксалила в 25 мл сухого хлористого метилена, и полученную смесь перемешивали при -78oC в течение 10 мин. В конце этого времени раствор 2,10 г (10,9 ммоль) смеси двух диастереоизомеров (-)-(3S)-2-гидрокси-3-фенил-3-метилгексана (полученного, как описано в синтезе 14) в 10 мл сухого хлористого метилена добавляли к смеси по каплям на протяжении 5 мин. Смесь, полученную таким способом, перемешивали при -78oC в течение 15 мин, после чего к смеси по каплям на протяжении 5 мин добавляли 7,0 мл (50 ммоль) триэтиламина. Смесь затем перемешивали при -78oC в течение 20 мин, после чего смесь перемешивали при 0oC в течение одного часа и затем смешивали с 50 мл воды. Водную смесь экстрагировали трижды, каждый раз по 100 мл этилацетата. Экстракты соединяли, промывали сначала насыщенным водным раствором бикарбоната натрия, потом насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия. Растворитель удаляли дистилляцией при пониженном давлении, и полученный бесцветный маслянистый остаток очищали мгновенной колонной хроматографией, пропуская через силикагель при использовании в качестве элюента смеси гексана с этилацетатом, в объемном соотношении 20:1 и получали 1,91 г (выход 92%) названного соединения в виде бесцветного масла.
Спектр ядерного магнитного резонанса:
(270 МГц, CDCl3) δ , в миллионных долях,
0,91 (3Н, триплет, J = 7,3 Гц);
1,03-1,93 (2Н, мультиплет);
1,46 (3Н, синглет);
1,87-1,93 (2Н, мультиплет);
1,89 (3Н, синглет);
7,22-7,34 (5Н, мультиплет).
νmax (см-1) инфракрасного спектра (CHCl3): 2950, 1700, 1350, 1130, 700.
Масс-спектр (m/e): 190 (M+).
[α]
Синтез 16. 2-Метил-2-[(-)-(2S)-1-метил-1-фенилбутил] -1,3-дитиан (соединение 12).
1,2-Этандиол (0,99 мл (9689 ммоль)) и 0,17 мл диэтилового эфирата трехфтористого бора добавляли к раствору 1,25 г (6,59 ммоль) (+)-(3S)-3-фенил-3-метил-2-гексанона (полученного, как описано в синтезе 15) в 25 мл сухого хлористого метилена, и полученную смесь перемешивали при комнатной температуре в течение 16 ч. В конце этого времени 0,33 мл диэтилового эфирата трехфтористого бора добавляли к смеси, и смесь перемешивали еще в течение 16 ч. К смеси добавляли 30 мл 5%-ного (по весу на объем) водного раствора гидроксида натрия, и полученную смесь дважды экстрагировали, каждый раз по 100 мл этилацетата. Экстракты соединяли, промывали насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия. Растворитель затем удаляли дистилляцией при пониженном давлении, и образовавшийся бесцветный маслянистый остаток очищали мгновенной колонной хроматографией, пропуская через силикагель при использовании в качестве элюента смеси гексана с этилацетатом при объемном соотношении 30:1, получая 1,20 г (выход 65%) названного соединения в виде бесцветного масла.
Элюат от приведенной выше процедуры хроматографии давал также 454 мл непрореагировавшего исходного вещества (+)-(3S)-3-фенил-3-метил-2-гексанона.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
0,88-0,94 (4Н, мультиплет);
1,12-1,28 (1Н, мультиплет);
1,66 (3Н, синглет);
1,74 (3Н, синглет);
1,76-1,82 (1Н, мультиплет);
1,95-2,06 (1Н, мультиплет);
2,41-2,50 (1Н, мультиплет);
2,58-2,67 (2Н, мультиплет);
2,86-3,00 (2Н, мультиплет);
7,23-7,33 (3Н, мультиплет);
7,46 (2Н, дублет, J = 7,3 Гц).
νmax (см-1) инфракрасного спектра (CHCl3): 2950, 1450, 1270, 700.
Масс-спектр (m/e): 280 (M+).
[α]
Синтез 17. (-)-(S)-3-фенил-3-метилгексан (соединение 13).
Никель Ренея (W-2) (20 г) добавляли к раствору 1,20 г (4,26 ммоль) 2-метил-2[(-)-(2S)-1-метил-1-фенилбутил]-1,3-дитиана (полученного, как описано в синтезе 16 в 50 мл абсолютного спирта, и смесь нагревали до дефлегмации в течение 4,5 ч. В конце этого времени реакционную смесь охлаждали до комнатной температуры и затем фильтровали при использовании целитаТМ. Любой никель Ренея, оставшийся на фильтре, тщательно смывали 1 л этанола. Фильтрат и промывные воды соединяли и затем концентрировали упариванием при пониженном давлении. Полученный концентрат очищали мгновенной колонной хроматографией, пропуская через силикагель с использованием в качестве элюента смеси гексана с этилацетатом в объемном соотношении 5:1, получая 536 мг (выход 71%) названного соединения в виде бледно-желтого масла.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
0,66 (3Н, триплет, J = 7,3 Гц);
0,81 (3Н, триплет, J = 7,3 Гц);
0,85-1,05 (2Н, мультиплет);
1,09-1,22 (1Н, мультиплет);
1,26 (3Н, синглет);
1,43-1,78 (3Н, мультиплет);
7,15-7,18 (1Н, мультиплет);
7,28-7,33 (4Н, мультиплет),
νmax (см-1 инфракрасного спектра (CHCl3): 2950, 1600, 1500, 1460, 1380, 700.
Масс-спектр (m/e): 176 (M+).
[α]
Синтез 18. (-)-(2S)-2-этил-2-метилвалериановая кислота (соединение 14).
Струя воздуха, содержащая 10 г/м3 озона, барботировалась через раствор 423 мг (2,4 ммоль) (-)-(S)-3-фенил-3-метилгексана (полученного, как описано в синтезе 17) в 25 мл уксусной кислоты при комнатной температуре в течение 8 ч. Затем к реакционной смеси добавляли 8,4 мл 30%-ного (по весу на объем) водного раствора перекиси водорода, и полученную смесь перемешивали при комнатной температуре в течение 14 ч. В конце этого времени к смеси добавляли 20 мг платины, и смесь перемешивали в течение 4 ч. Смесь затем фильтровали через целитТМ, фильтрат концентрировали упариванием при пониженном давлении, и концентрат смешивали с 10%-ным (по весу на объем) водным раствором гидроксида натрия и 50 мл этилацетата. Водный слой удаляли и pH слоя доводили до 2 добавлением соответствующего количества концентрированной хлористоводородной кислоты. Смесь затем трижды экстрагировали, каждый раз по 50 мл этилацетата. Экстракты соединяли, промывали насыщенным водным раствором хлорида натрия и затем сушили над безводным сульфатом натрия. Растворитель затем удаляли дистилляцией при пониженном давлении, и бледно-желтый маслянистый остаток, который образовывался при этом, очищали дистилляцией (130oC при 2 мм рт.ст.) и получали 70 мг названного соединения в виде бесцветного масла. Оптическая чистота продукта составляет 93% избытка энантиомера по данным жидкостной хроматографии высокой производительности с пропусканием через оптически активную колонку.
Спектр ядерного магнитного резонанса: (270 МГц, CDCl3) δ , в миллионных долях,
0,87 - (3Н, триплет, J = 7,3 Гц);
0,91 (3Н, триплет, J = 7,3 Гц);
1,12 (3Н, синглет);
1,16-1,78 (6Н, мультиплет);
3,40-4,20 (1Н, мультиплет, способен обмениваться с D2O).
νmax (см-1) инфракрасного спектра (CHCl3): 3000, 2950, 1700, 1440, 1120.
Масс-спектр (m/e): 145 (M++1).
[α]
Приведенная выше методика может быть также использована для получения (+)-(2R)-2-этил-2-метилвалериановой кислоты при использовании в качестве исходного вещества (-)-(2R)-2-метил-2-фенилциклопентана.
Рецептура 1. Твердые капсулы
Следующие ингредиенты вводили в стандартные твердые желатиновые капсулы, состоящие из двух частей, получая единую капсулу, которую затем промывали и сушили.
Соединение из примера 50 - 5 мг
Гидроксипропилцеллюлоза (низкого замещения) - 10 мг
Гидроксипропилцеллюлоза - 3 мг
Стеарат магния - 1 мг
Лактоза - 81 мг
Всего - 100 мг
Рецептура 2. Порошковая рецептура.
Порошковую рецептуру, содержащую ингредиенты, перечисленные ниже, готовили, используя обычные способы.
Соединение из примера 64 - 5 мг
Гидроксипропилцеллюлоза (низкого замещения - 20 мг
Гидроксипропилцеллюлоза - 40 мг
Стеарат магния - 5 мг
Лактоза - 930 мг
Всего - 1000 мг
Рецептура 3. Таблеточная рецептура.
Таблетки, содержащие ингредиенты, перечисленные ниже, готовили, используя обычные способы.
Соединение из примера 65 - 5 мг
Гидроксипропилцеллюлоза (низкого замещения) - 10 мг
Гидроксипропилцеллюлоза - 3 мг
Стеарат магния - 1 мг
Лактоза - 81 мг
Всего - 100 мг
На таблетки, при желании, может быть нанесено покрытие. Способы нанесения покрытия и составляющие компоненты покрытий хорошо известны в этой области техники.
Соединение примера 52.
Спектр ЯМР (400 МГц, D2O) δ, млн.дол.:
0,76 (9Н, т., J = 7,4 Гц),
0,91 (3Н, д., J = 6,9 Гц),
1,23 ≈ 1,35 (2Н, м.)
1,37 ≈ 1,49 (1Н, м.),
4,07 ≈ 4,14 (1Н, м.),
4,49 ≈ 4,43 (1Н, м.),
5,45 (1Н, широкий синглет),
5,59 (1Н, широкий синглет),
6,04 (1Н, дублет дублетов, J = 9,7 Гц и 5,7 Гц),
6,10 (1Н, д., J = 9,7 Гц).
| название | год | авторы | номер документа |
|---|---|---|---|
| ЭФИРНЫЕ ПРОИЗВОДНЫЕ ГЕКСАГИДРОНАФТАЛИНА, ИХ ПОЛУЧЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1994 |
|
RU2114101C1 |
| КРИСТАЛЛИЧЕСКИЙ ПИВАЛОИЛОКСИМЕТИЛ (IR, 5S, 6S)-[(4R)-2-ОКСО-4-ПИРРОЛИДИНИЛТИО]-6-[(IR)-1-ГИДРОКСИЭТИЛ]-1-МЕТИЛ-1-КАРБАПЕН-2-ЕМ-3-КАРБОКСИЛАТ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1993 |
|
RU2090567C1 |
| ПРОИЗВОДНЫЕ АЗЕТИДИНОНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2047602C1 |
| ПРОИЗВОДНЫЕ КАРБАПЕНЕМА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2097383C1 |
| α,ω ДИАРИЛАЛКАНЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2105752C1 |
| ПРОИЗВОДНЫЕ ДИАРИЛАЛКАНОВ, СОДЕРЖАЩИЕ АЛИЦИКЛИЧЕСКУЮ ГРУППУ, ПРОЯВЛЯЮЩИЕ АНТАГОНИЗМ В ОТНОШЕНИИ СЕРОТОНИН-2-РЕЦЕПТОРОВ И/ИЛИ ИНГИБИРУЮЩУЮ АКТИВНОСТЬ В ОТНОШЕНИИ СКВАЛЕН-СИНТАЗЫ, КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ СЕРДЕЧНО-СОСУДИСТЫХ ЗАБОЛЕВАНИЙ | 1997 |
|
RU2139277C1 |
| ПРОИЗВОДНЫЕ ЦИКЛОГЕКСАНА ИЛИ ТЕТРАГИДРОПИРАНА, ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ИЛИ ГИДРАТЫ, ИЛИ СОЛЬВАТЫ ЭТИХ СОЕДИНЕНИЙ, ИЛИ ИХ СОЛЕЙ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ И ФУНГИЦИДНОЕ СРЕДСТВО | 1992 |
|
RU2084439C1 |
| ФУНГИЦИДНЫЕ ПРОИЗВОДНЫЕ ОКСЕТАНА И ИХ СОЛИ | 1992 |
|
RU2044736C1 |
| ПЕПТИДНОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1993 |
|
RU2106357C1 |
| АЗАСТЕРОИДНЫЕ СОЕДИНЕНИЯ | 1991 |
|
RU2070204C1 |
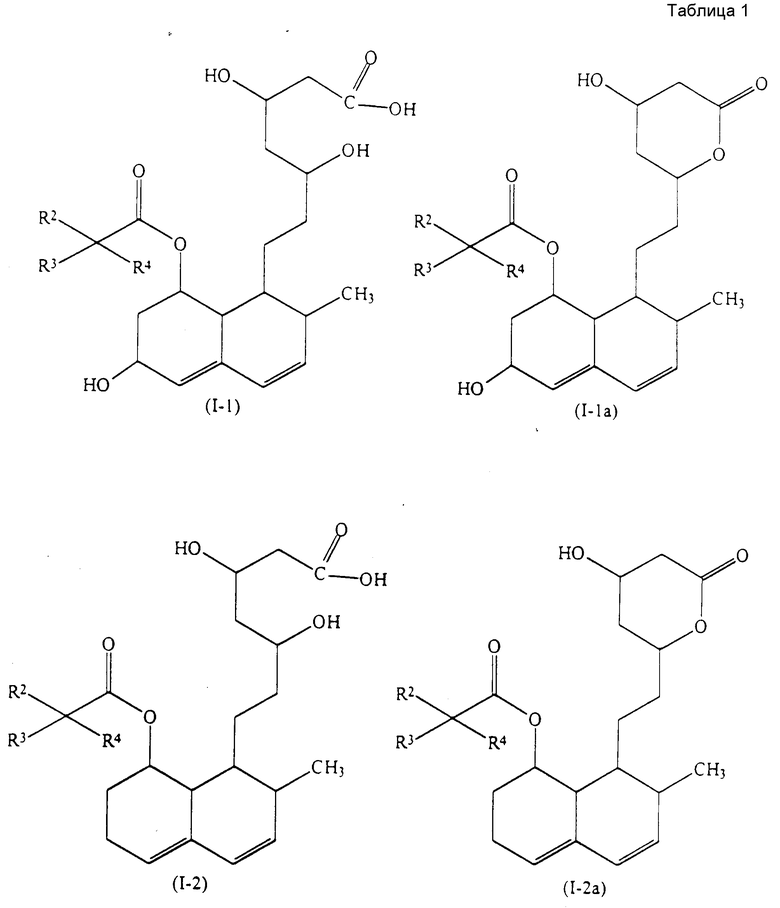
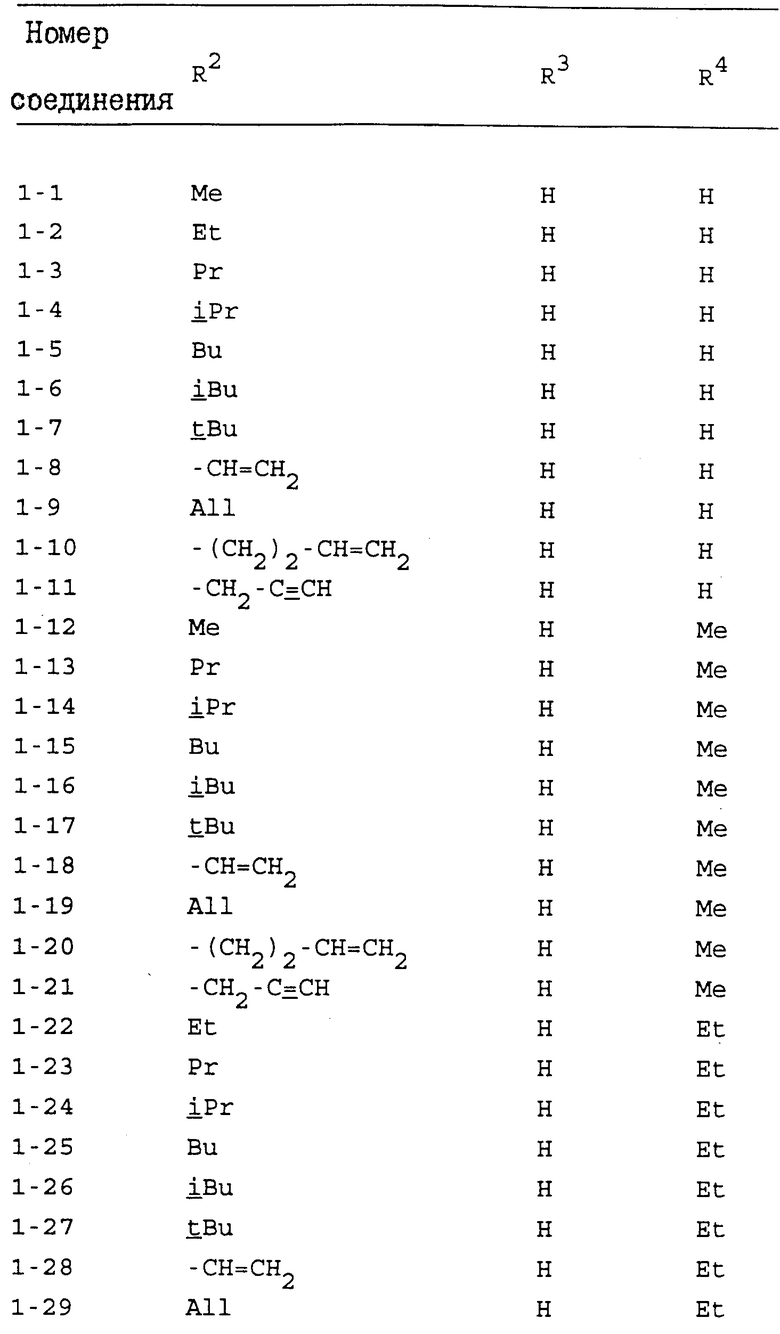


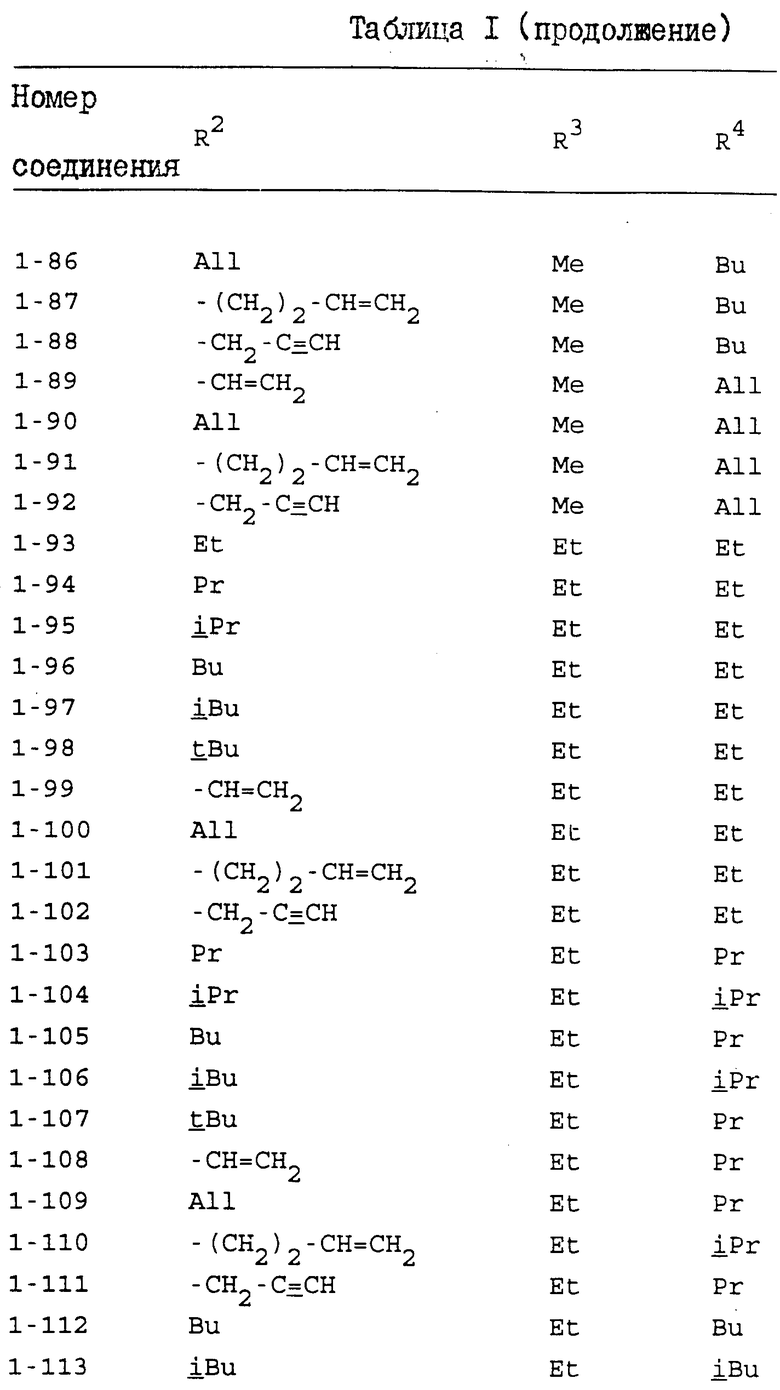

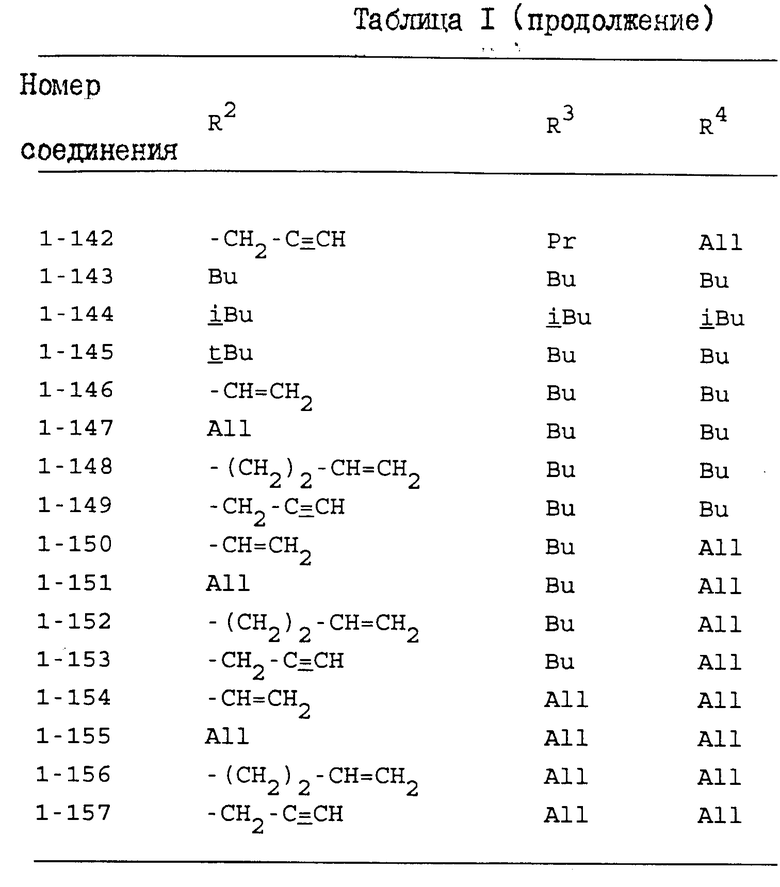

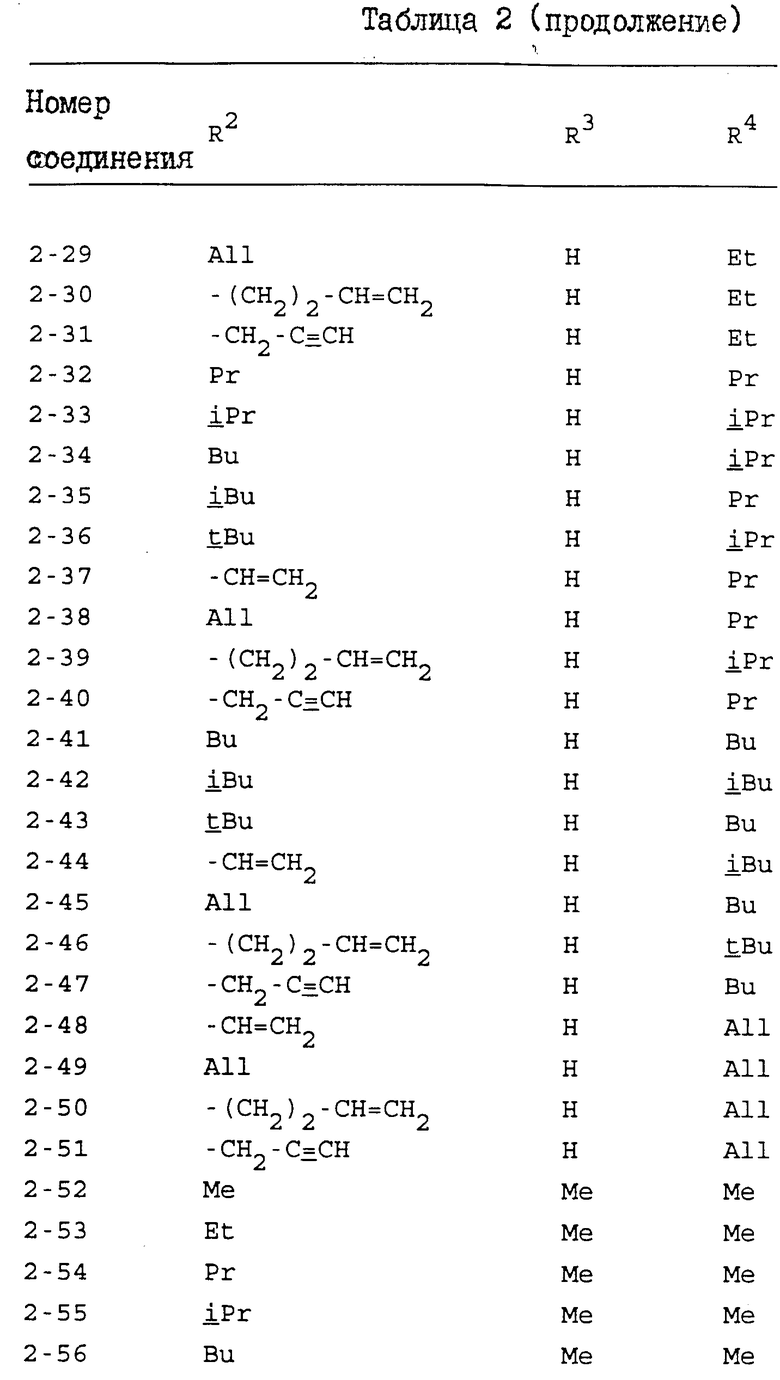
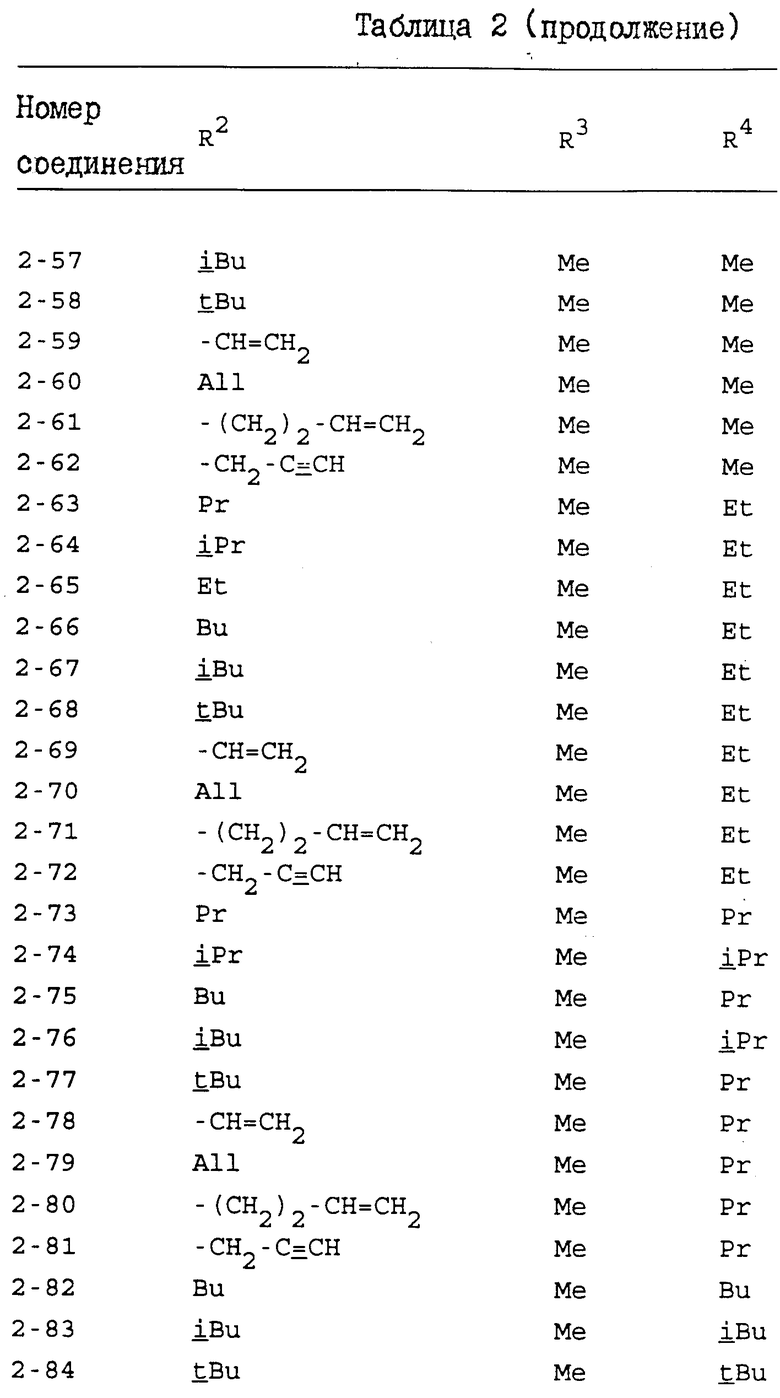
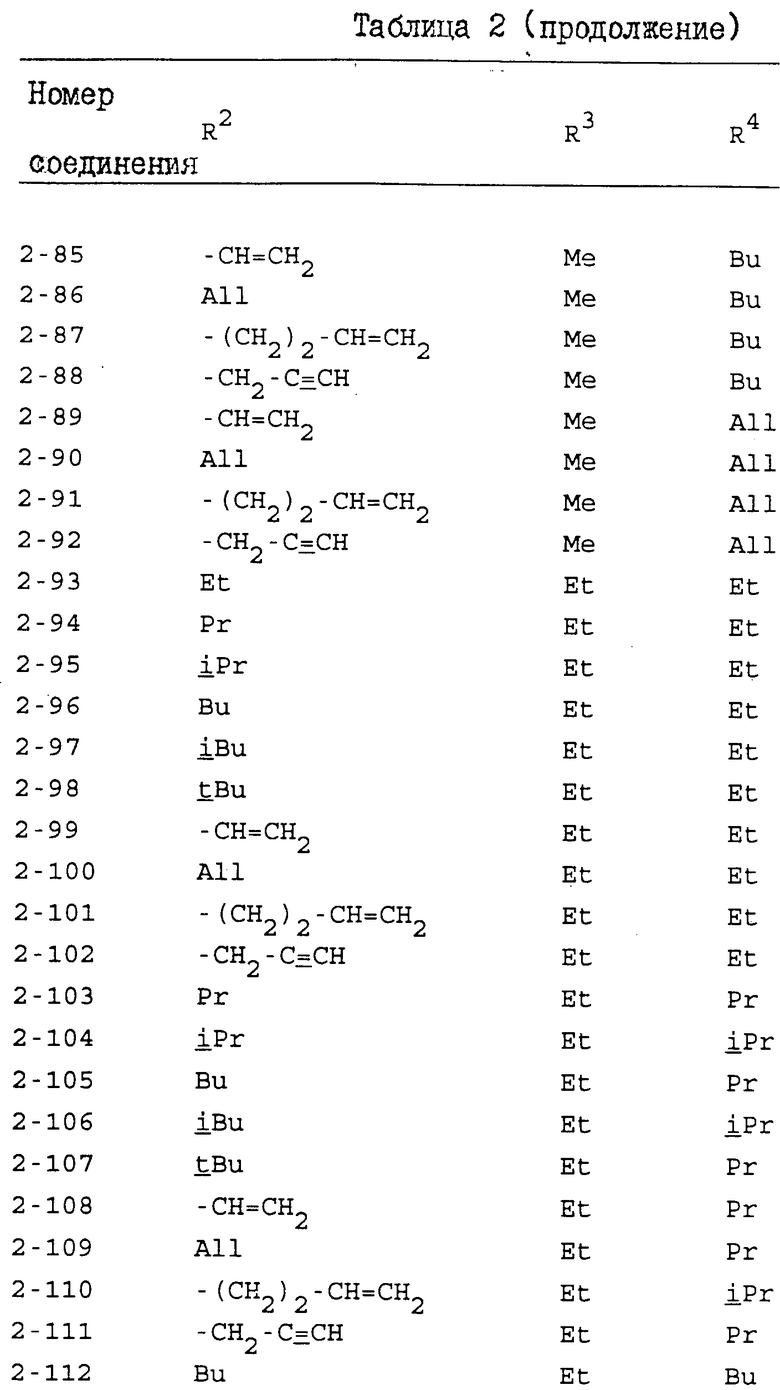

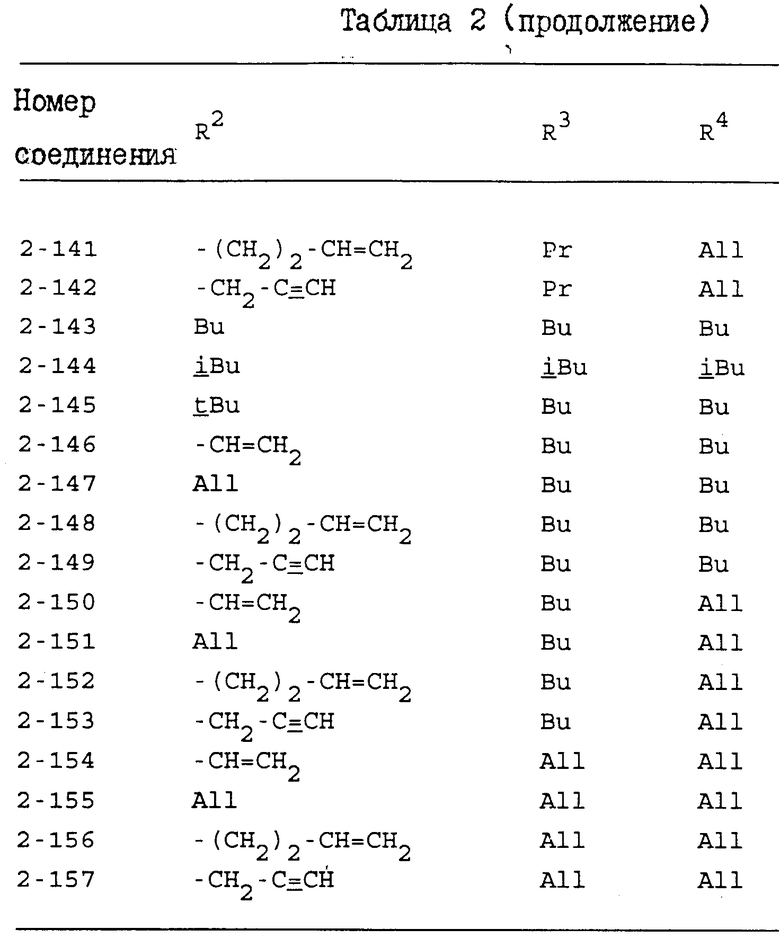
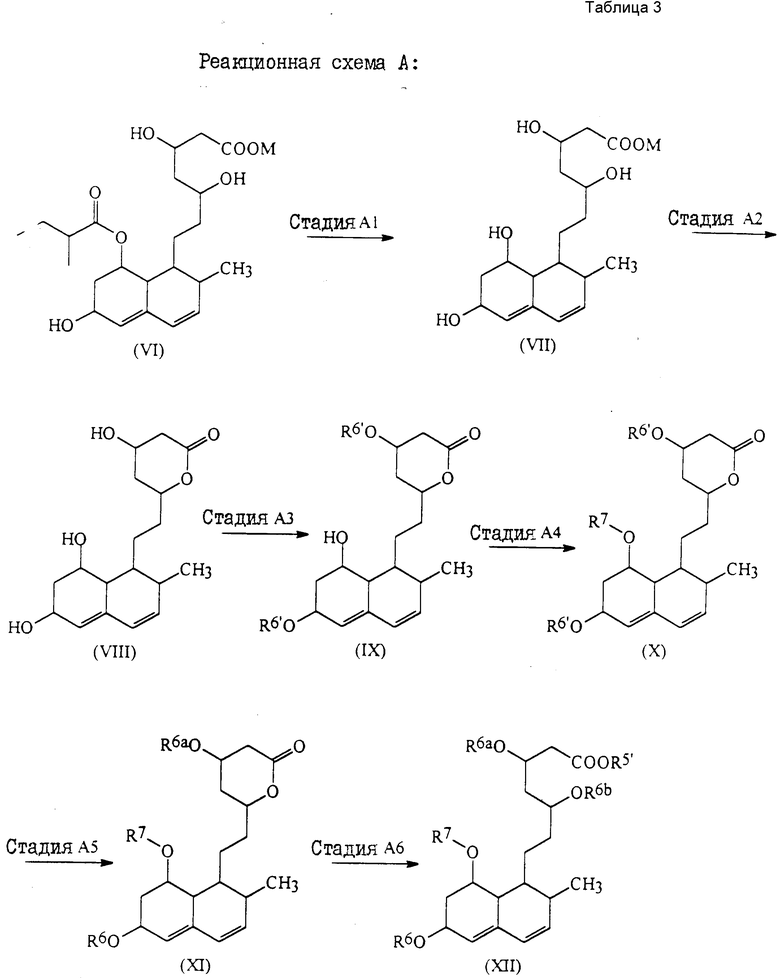
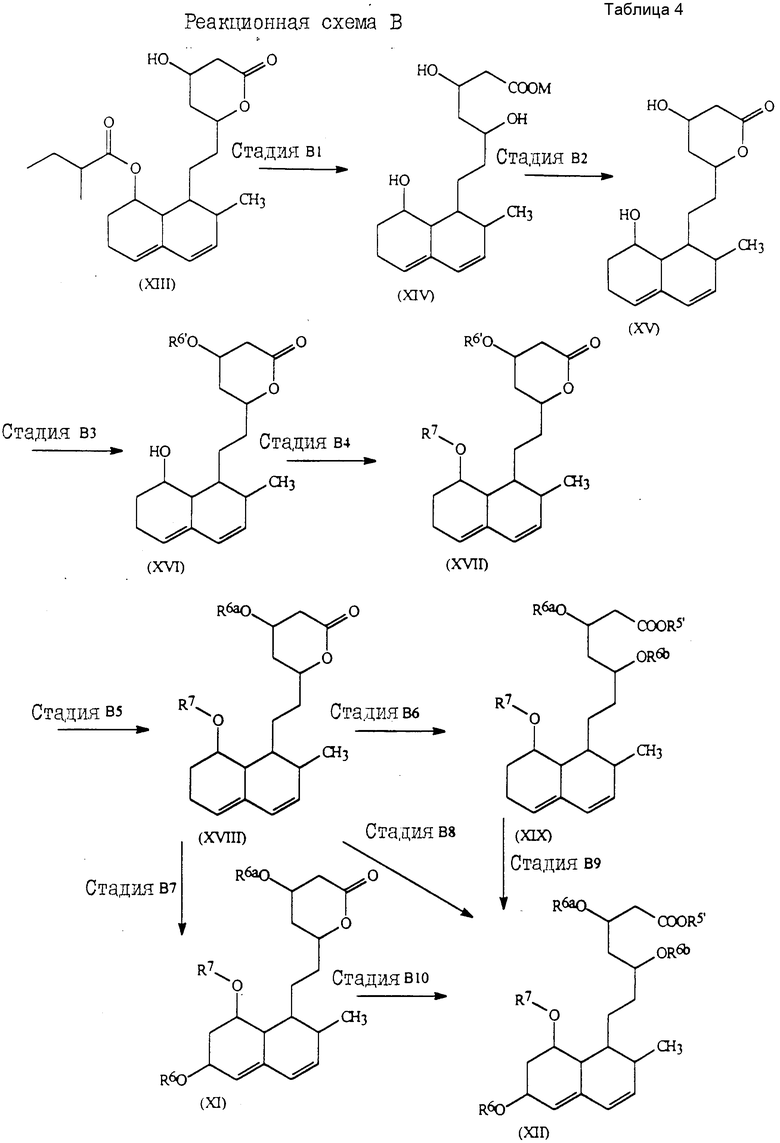
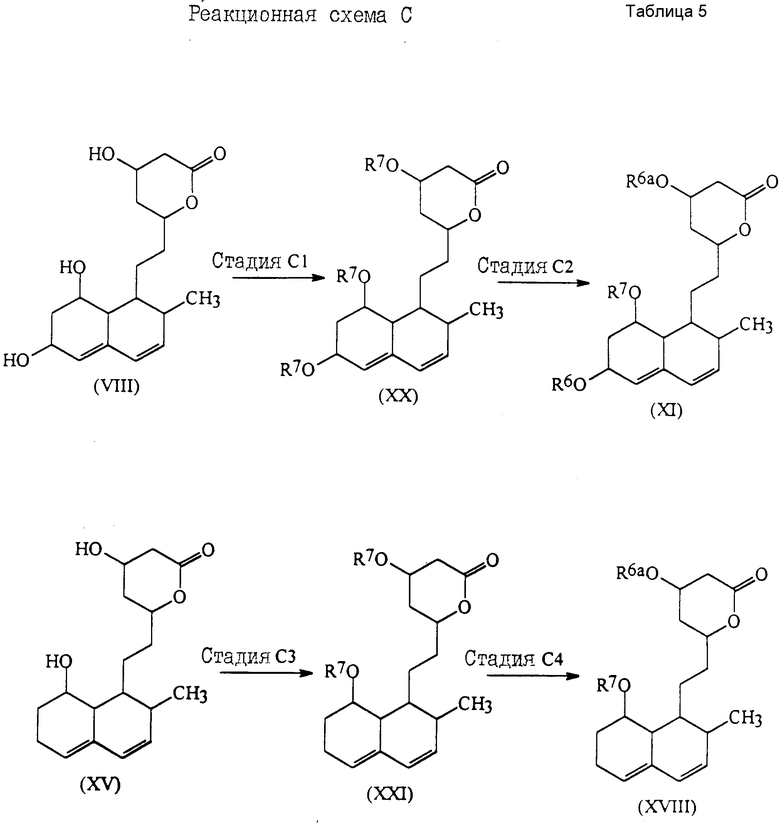
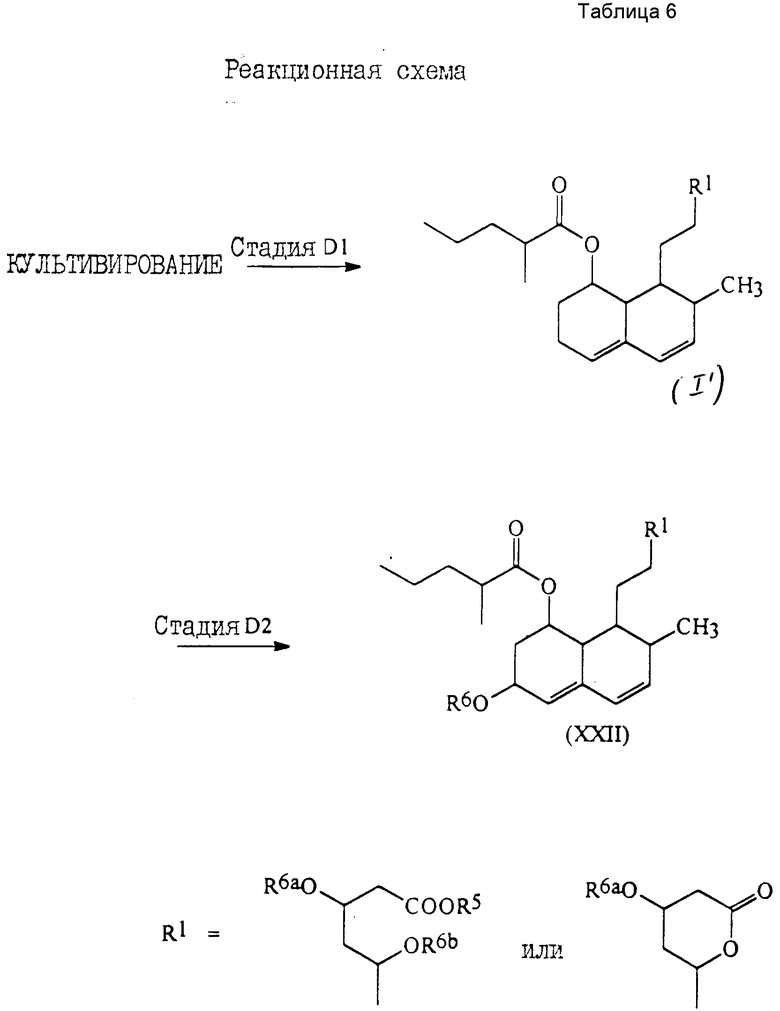
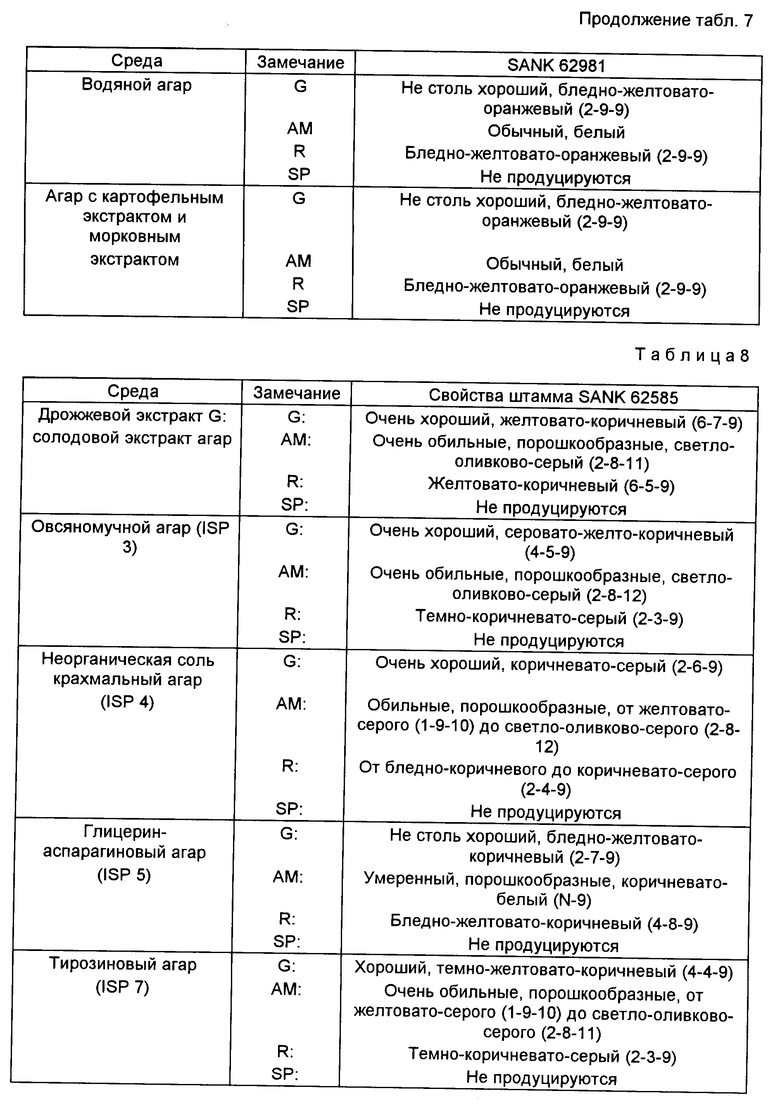
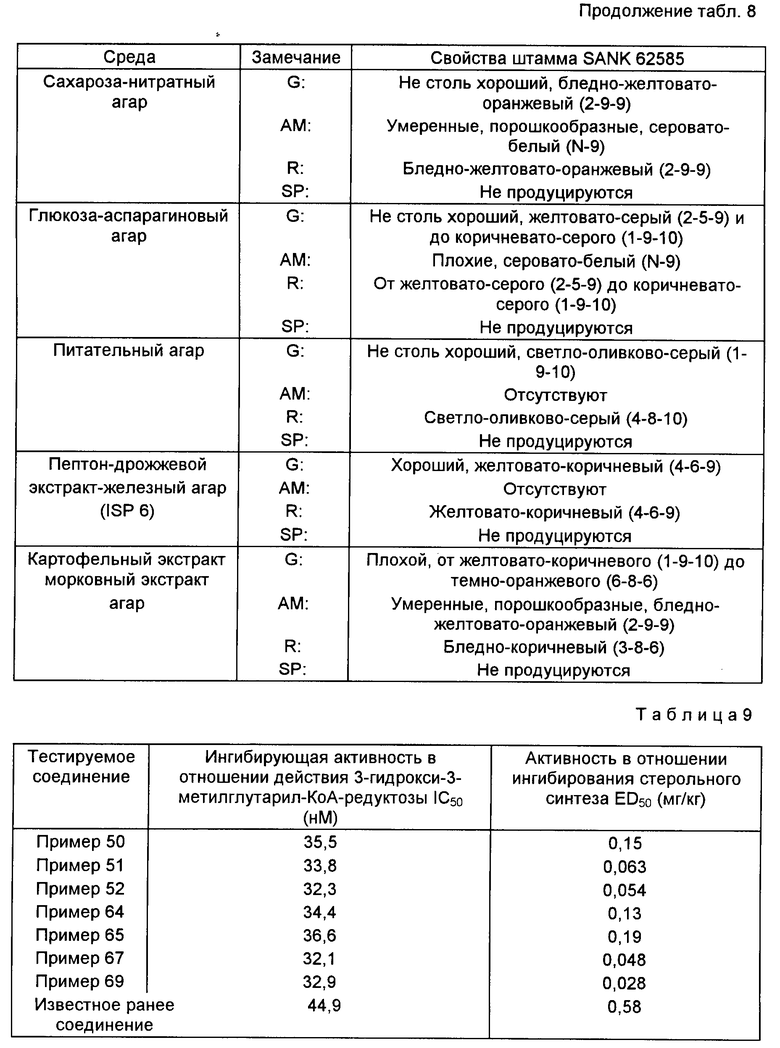
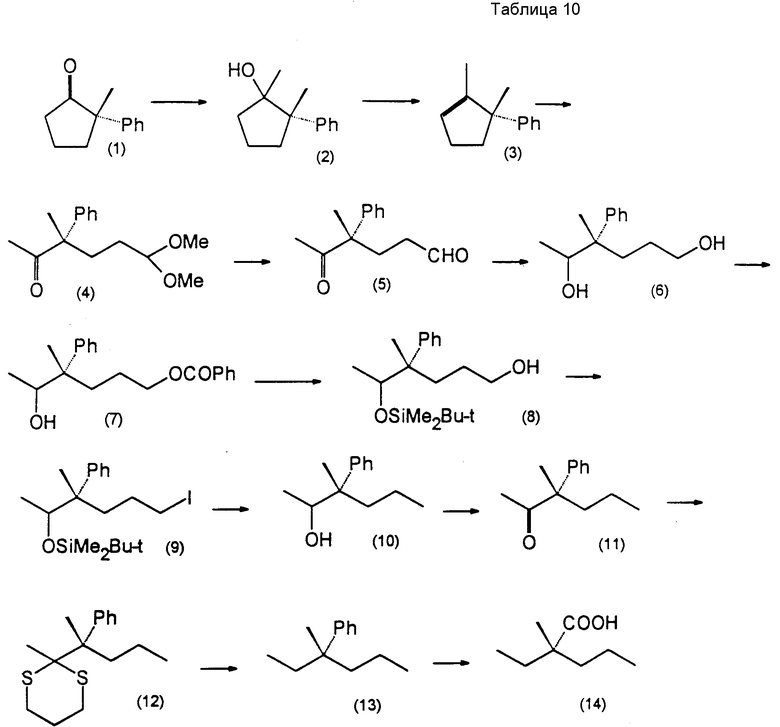
Предложены гексагидронафталиновые сложноэфирные соединения формулы (I), в которой R1 представляет группу формулы (II) (III), R5 представляет атом водорода; Ra представляет гидроксигруппу; R6a представляет атом водорода или гидроксизащитную группу; и или: (a) R2 представляет этильную группу, R3 представляет алкильную группу, имеющую от 1 до 4 атомов углерода, или алкенильную группу, имеющую от 2 до 6 атомов углерода, и R4 представляет алкильную группу, имеющую от 2 до 4 атомов углерода, или алкенильную группу, имеющую от 2 до 6 атомов углерода; или (b) R2 представляет пропильную группу, R3 представляет атом водорода и R4 представляет пропильную группу; или (c) R2 представляет бутильную группу, R3 представляет метильную группу и R4 представляет метильную группу; и его фармацевтически приемлемые соли. Предложена фармацевтическая композиция, ингибирующая биосинтез холестерина, включающая в качестве активного ингредиента соединения формулы (I) и фармацевтически приемлемый носитель. 2 с. и 7 з.п.ф-лы, 10 табл.
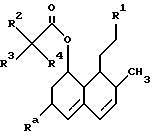
в которой R1 группа формулы II или III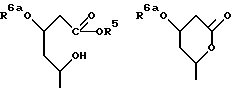
R5 атом водорода;
Ra гидроксигруппа;
R6 a атом водорода или гидроксизащитная группа
и/или
(а) R2 этильная группа;
R3 алкильная группа, имеющая от 1 до 4 атомов углерода, или алкенильная группа, имеющая от 2 до 6 атомов углерода;
R4 алкильная группа, имеющая от 2 до 4 атомов углерода;
(b) R2 алкенильная группа, имеющая от 2 до 6 атомов углерода, или
R1 пропильная группа;
R3 атом водорода;
R4 пропильная группа, или
(с) R2 бутильная группа;
R3 метильная группа;
R4 метильная группа,
и его фармацевтически приемлемые соли.
R2 этильная группа;
R3 алкильная группа, имеющая от 1 до 4 атомов углерода, или аллильная группа;
R4 алкильная группа, имеющая от 2 до 4 атомов углерода, или аллильная группа.
R2 этильная группа;
R3 алкильная группа, имеющая от 1 до 3 атомов углерода;
R4 алкильная группа, имеющая 2 или 3 атома углерода.
(3R, 5R)-3,5-дигидрокси-7-[(1S, 2S, 6S, 8S, 8aR)- 6-гидрокси-2-метил-8-(2-пропилпентаноилокси)-1,2,6,7,8,8а-гексагидро-1-нафтил] гептановой кислоты;
(3R, 5R)-3,5-дигидрокси-7-[(1S, 2S, 6S, 8S, 8aR)-6-гидрокси-2-метил-8-(2,2-диметилгексаноилокси)-1,2,6,7,8,8а-гексагидро-1-нафтил] гептановой кислоты;
(3R, 5R)-3,5-дигидрокси-7-[(1S, 2S, 6S, 8S, 8aR)-6-гидрокси-2-метил-8-(2-этил-2-метилпентаноилокси)- 1,2,6,7,8,8а-гексагидро-1-нафтил] гептановой кислоты;
(3R, 5R)-3,5-дигидрокси-7-[(1S, 2S, 6S, 8S, 8aR)-6-гидрокси-2-метил-8- (2-этил-2-метилбутирилокси)-1,2,6,7,8а-гексагидро-1-нафтил] гептановой кислоты;
(3R, 5R)-3,5-дигидрокси-7-[(1S, 2S, 6S, 8S, 8aR)-6-гидрокси-2-метил-8-(2,2-диэтилбутирилокси)- 1,2,6,7,8,8а-гексагидро-1-нафтил] гептановой кислоты;
(3R, 5R)-3,5-дигидрокси-7-[(1S, 2S, 6S, 8S, 8aR)-6-гидрокси-2-метил-8- (2,2-диэтил-4-пентеноилокси)-1,2,6,7,8,8а-гексагидро-1-нафтил] гептановой кислоты и
(3R, 5R)-3,5-дигидрокси-7[(1S, 2S, 6S, 8S, 8aR)-6-гидрокси-2-метил-8-(2-аллил-2-этил-4-пентеноилокси)-1,2,6,7,8,8а-гексагидро-1-нафтил]гептановой кислоты,
и его фармацевтически приемлемые соли.
| GB, патент 2077264, C 07 C 69/03, 1981 | |||
| EP, патент 314435, C 07 D 309/30, 1988. |

