Изобретение относится к новому препарату вещества, которое представляет собой специфическую кристаллическую форму пивалоилоксиметилового эфира производного карбапенема, известного как (1R,5S,6S)-2-[(4R)-2-оксо-4-пирролидинилтио] -6-[(1R)-1-гидроксиэтил] -1-метил-1-карбапен-2-ем-3-карбоновая кислота.
Соединение имеет формулу:
Производные карбапенема этого типа обладают хорошей активностью в качестве антибиотика и могут использоваться как оральные фармацевтические препараты.
В патенте США N 5104867 и европейском патенте N 337637 (в примере 39) раскрывается получение производного карбапанема, которое относится к соединениям формулы (I). По методике этих патентов производное карбопенема первоначально получали только в виде аморфного порошка. Однако наши исследования показали, что соединение формулы (I), которое получается в виде аморфного порошка при использовании методов, описанных в работах предшествующего уровня, трудно переводить в оральные фармацевтические препараты, и, кроме того, они немного нестабильны, особенно после длительного хранения и, следовательно, имеют ограниченное применение в качестве лекарства.
Мы неожиданно обнаружили, что первоначально не реализуемая кристаллическая форма этого соединения обладает значительной и в целом неожиданной стабильностью, что делает это производное карбапенема более простым при работе с ним и при приготовлении из него лекарств и, следовательно, имеет намного большее значение в качестве фармацевтического средства.
Объектом настоящего изобретения является получение соединения формулы (I) в кристаллической форме, предпочтительно в такой форме, которая стабильна и с которой легко работать и использовать при получении лекарств.
Еще одним объектом настоящего изобретения является получение соединения формулы (I) в форме кристаллов, и эти соединения обладают приемлемой антибактериальной активностью.
Таким образом, настоящее изобретение предлагает кристаллическую форму пивалоилоксиметила (1R,5S,6S)-2-[(4R)-2-оксо-4-пирролидинилтио]-6-[(1R)-1-гидроксиэтил]-1-метил-1-карбапен-2-ем-3-карбоксилата формулы (I):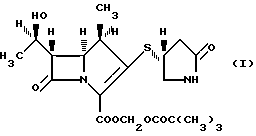
Изобретение также предлагает фармацевтический препарат, содержащий антибиотик в смеси с фармацевтически приемлемым носителем или разбавителем, в котором антибиотик представляет собой соединение формулы (I) в кристаллической форме, описанной выше.
Изобретение дополнительно предлагает способ лечения или профилактики бактериальной инфекции у млекопитающих, например у человека, который включает назначение эффективного количества антибиотика вышеуказанному млекопитающему, причем вышеуказанный антибиотик представляет собой соединение формулы (I) в кристаллической форме, описанной выше.
Изобретение также предлагает способ получения кристаллической форму соединения формулы (I), которая более подробно описана ниже.
На чертеже представлена дифракционная рентгенограмма кристаллов настоящего изобретения на рентгеновских лучах при порошковом методе с использованием Kα-лучей (медь),  Вертикальная ось представляет собой силу дифракции в числах в секунду (cps), горизонтальная ось показывает величину при дифракционном угле 20o.
Вертикальная ось представляет собой силу дифракции в числах в секунду (cps), горизонтальная ось показывает величину при дифракционном угле 20o.
Кристаллическая форма соединения формулы (I) может быть охарактеризована различными физическими параметрами, в том числе температурой плавления кристаллов и рентгенограммой, получаемой при дифракции рентгеновских лучей. Предпочтительные кристаллы настоящего изобретения имеют температуру плавления 189oC. Эти кристаллы также обычно имеют основные пики при межплоскостных расстояниях 18,41; 9,21; 6,24; 5,28; 5,04 и 4,72 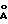 , определенных с помощью дифракции рентгеновских лучей при порошковом методе с использованием Kα-лучей (медь),
, определенных с помощью дифракции рентгеновских лучей при порошковом методе с использованием Kα-лучей (медь), 
Кристаллическая форма соединения формулы (I) может быть получена при добавлении растворителя и пивалоилоксиметиловому эфиру (1R, 5S, 6S)-2-[(4R)-2-оксо-4-пирролидинилтио] -6-[(1R)-1-гидроксиэтил] -1-метил-1-карбапен-2-ем-3-карбоновой кислоты с последующим активным или пассивным удалением растворителя и промывкой, сушкой и выделением получаемых кристаллов.
Более конкретно, кристаллы могут быть получены с помощью следующих стадий.
1. Реакция пивалоилоксиметилйодида с натровой солью (1R,5S,6S)-2-[(4R)-2-оксо-4-пирролидинилтио] -6-[(1R)-1-гидроксиэтил] -1-метил-1-карбапен-2-ем-3-карбоновой кислоты с последующим разбавлением смеси этилацетатом. Разбавленная смесь может быть затем промыта водой и сконцентрирована, например, путем упаривания в вакууме.
2(а). Полученный таким образом остаток может быть затем растворен в сильном растворителе. Примерами растворителей, которые могут быть использованы на этой стадии, являются: галогенсодержащие углеводороды, особенно галогенированные алифатические углеводороды, такие как метиленхлорид, 1,2-дихлорэтан или хлороформ, предпочтительно метиленхлорид; диалкилсульфоксиды, такие как диметилсульфоксид; амиды, такие как N,N-диметилформамид или N,N-диметилацетамид; кетоны, такие как ацетон или 2-бутанон, предпочтительно ацетон; спирты, такие как метанол. Затем к раствору добавляют более слабый растворитель или нерастворитель. Примерами более слабых растворителей и нерастворителей, которые могут быть использованы на этой стадии, являются: спирты, такие как этанол; эфиры, такие как диэтиловый эфир, тетрагидрофуран или диоксан, предпочтительно диэтиловый эфир; эфиры, такие как этилацетат; циклические и ароматические углеводороды, такие как циклогексан, толуол или бензол, предпочтительно циклогексан или толуол; вода. Кристаллы могут быть затем получены после стояния смеси и упаривания растворителя естественным путем или после упаривания части растворителя при пониженном давлении и затем при стоянии смеси для дальнейшего упаривания растворителя.
2(б). По другому способу остаток, полученный при концентрировании на стадии 1, может быть растворен в смеси растворителей или в смеси растворителя и слабого растворителя или нерастворителя, предпочтительно в смеси сильного растворителя и слабого растворителя или нерастворителя, например в смеси метиленхлорида и этанола. Количество растворителя и слабого или нерастворителя, используемой на этой стадии, и соотношение между растворителем и слабым растворителем или нерастворителем не являются критическими для настоящего изобретения, поскольку растворитель и слабый растворитель или нерастворитель присутствуют в количестве и соотношении, достаточных для того, чтобы имела место кристаллизация. Мы установили, что отношение метиленхлорида к этанолу приблизительно от 4:1 до 1:4 (по объему), предпочтительно приблизительно от 1:1 до 1:4 (по объему), достаточно для этих целей. Однако соотношение от 1: 1 до 1:2 (по объему) наиболее предпочтительно. Кристаллы могут быть затем получены после стояния смеси и упаривания растворителя естественным путем или после упаривания части растворителя при пониженном давлении и затем при стоянии смеси для дальнейшего упаривания растворителя.
Кристаллы, полученные на стадиях, описанных выше, могут быть затем промыты, высушены и выделены с использованием стандартных методик. Обычно кристаллы промывают, например, этанолом и сушат при пониженном давлении при температуре приблизительно от 20 до 25oC.
Кристаллы настоящего изобретения, полученные в соответствии со стадиями, описанными выше, плавятся приблизительно при температуре 189oC и имеют основные пики при межплоскостных расстояниях 18,41; 9,21; 6,24; 5,28; 5,04 и 4,72  , определенных с помощью дифракции рентгеновских лучей при порошковом методе с использованием Kα-лучей (медь)
, определенных с помощью дифракции рентгеновских лучей при порошковом методе с использованием Kα-лучей (медь) 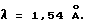
Кристаллы настоящего изобретения обладают прекрасной антибактериальной активностью против широкого спектра грамположительных, грамотрицательных и цефалоспоринпродуцирующих бактерий, сравнимой с активностью соединения формулы (I) в форме порошка, особенно при оральном применении.
После культивирования при 37oC в течение 1 часа в сыворотке лошади антибактериальная активность кристаллов настоящего изобретения оценивалась с помощью метода разбавления агаровых пластин. В этом случае кристаллы настоящего изобретения показали активность против широкого спектра патогенных микроорганизмов, в том числе против грамположительных бактерий, таких как Staphilococcus aureus или Enterococcus faecalis, а также грамотрицательных бактерий, таких как Escherichia coli, вид Shigella, Klebsiella pneumoniae, вид Proteus, вид Serratia, вид Enterobacter и Pseudomonas aeruginosa, а также против анаэробных бактерий, таких как Bacteroides fragilis, и, таким образом, являются весьма полезными при лечении болезней, вызываемых такими микроорганизмами у человека и животных.
После орального введения кристаллов настоящего изобретения мышам, полностью инфицированным по всему телу или Staphilococcus aureus или Escherichia coli, конечные терапевтические эффекты были прекрасными. Более того, большие количества кислотной формы соединения формулы (I) были обнаружены в моче мышей, после орального введения соединения формулы (I) в кристаллической форме.
Кристаллы соединения формулы (I) также обладают низкой токсичностью, причем эта токсичность ниже, чем токсичность соединения формулы (I) в аморфной форме, вероятно, из-за отсутствия в кристаллах продуктов разложения. Следовательно, кристаллы соединения формулы (I) имеют большое значение в качестве терапевтических средств для лечения бактериальных инфекций.
Кроме того, кристаллы настоящего изобретения не разлагаются после стояния в течение нескольких недель при температуре около 60oC, что указывает на то, что кристаллическая форма отличается более высокой стабильностью, чем аморфная порошкообразная форма, как это показано в примерах.
Кристаллы настоящего изобретения могут применяться орально для лечения заболеваний человека и животных, вызываемых патогенными микроорганизмами. Кристаллы могут быть приготовлены в любой, обычной для применения форме. Например, для орального применения кристаллы могут быть смешаны с подходящим фармацевтически приемлемым наполнителем, включая, например: крахмал; сахар, такой как лактоза или сахароза; карбонат щелочного металла, такой как карбонат кальция, карбонат калия или карбонат магния, предпочтительно карбонат кальция; фосфат щелочного металла, такой как фосфат кальция, фосфат магния или фосфат калия, предпочтительно фосфат кальция; эфиры, такие как полиэтиленгликоль; связующее средство, например гумми-арабик, карбоксиметилцеллюлозу, гидроксипропилцеллюлозу, желатин или поливинилпирролидон; смазывающее вещество, такое как стеарат магния, тальк, лаурилсульфат натрия или полиэтиленгликоль; диспергирующее средство, такое как альгиновая кислота, соли альгиновой кислоты или кальциевая соль карбоксиметилцеллюлозы; окрашивающее средство; отдушки; подслащивающее средство. Подходящими формами для орального применения кристаллов настоящего изобретения являются таблетки, гранулы, капсулы, порошки и сиропы.
Доза кристаллов настоящего изобретения будет изменяться в зависимости от природы заболевания, которое должно быть подвергнуто лечению, симптомов, возраста, веса тела и состояния пациента, а также в зависимости от формы и числа приемов лекарства. Однако в общем случае дневная доза для взрослого, как ожидается, должна быть от 50 мг до 2 г кристаллов, которые могут быть введены в одну порцию или два-четыре раза в день.
Стабильность соединения формулы I в кристаллической форме
Кристаллы, полученные в примере 1, и аморфная порошкообразная форма соединения формулы (I), полученная в соответствии с методикой, описанной в примере 39 европейского патента N 337637 (или примера 16 японской заявки N Hei 2-49783), хранили в десикаторе с силикагелем при температуре 60oC. Стабильность двух соединений через 7 дней и окончательно через 28 дней оценивалась путем определения количества оставшегося соединения (см. табл. 1). Исследование проводили с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ) при следующих условиях:
Колонка: Intersil ODS-2(4,6 мм диаметр 150 мм);
Подвижная фаза: 20 мМ 3-(N-морфолино)пропансульфокислотного буфера (pH 7) CH3CN 70 30 (об.);
Детектор: ультрафиолетовый, поглощение при 322 нм.
В приведенных ниже примерах и методиках, если не указано специально, спектры ядерного магнитного резонанса получены с использованием тетраметилсилана в качестве внешнего и внутреннего стандарта.
Пример 1. В 35 мл N,N-диметилацетамида растворяли 4,76 г натровой соли (1R,5S,6S)-2-[(4R)-2-оксо-4-пирролидинилтио]-6-[(1R)-1-гидроксиэтил]-1-метил-1-карбапен-2-ем-3-карбоновой кислоты (получена в соответствии с методикой 6). Затем к полученному раствору при охлаждении льдом добавляли 3,60 г пивалоилоксиметилйодида, и полученную смесь перемешивали в течение 30 мин. Далее смесь разбавляли этилацетатом, промывали водой и насыщенным водным раствором хлористого натрия. Этилацетатный слой затем сушили безводным сульфатом натрия и упаривали в вакууме. Полученный остаток в форме аморфного порошка (4,54 г) растворяли в смеси этанола и метиленхлорида с объемным соотношением 1:1, после чего кристаллы формировались при упаривании метиленхлорида при пониженном давлении. Кристаллы выделяли фильтрованием от оставшегося этанола, и после сушки получали 3,68 г бесцветных кристаллов; т.пл. 189oC.
Спектр инфракрасного поглощения (KBr) νmax см-1:
3336, 1764, 1751, 1691, 1542, 1347, 1213, 1160, 1114, 9957.
Спектр ультрафиолетового поглощения (CH3CN) λmax нм: 324.
Спектр ядерного магнитного резонанса:
(400 мГц, гексадейтерированный диметилсульфоксид, внутренний стандарт - тетраметилсилан) δ ppm:
1,10-1,80 (15Н, мультиплет);
2,11 (1Н, дублет дублетов, J 17,0 и 4,3 Гц);
2,87 (1Н, дублет дублетов, J 17,0 и 7,7 Гц);
3,09 (1Н, дублет дублетов, J 10,9 и 3,9 Гц);
3,25 (1Н, дублет дублетов, J 6,2 и 2,5 Гц);
4,44-3,48 (1Н, мультиплет);
3,71 (1Н, дублет дублетов, J 10,9 и 7,6 Гц);
3,94-4,00 (1Н, мультиплет);
9,04-4,09 (1Н, мультиплет);
4,23 (1Н, дублет дублетов, J 9,5 и 2,5 Гц);
5,08 (1Н, дублет, J 5,1 Гц);
5,73 (1Н, дублет, J 5,9 Гц);
5,88 (1Н, дублет, J 5,9 Гц);
7,84 (1Н, синглет).
Рентгенограмма кристаллов, полученная с использованием порошкового метода, Kα-лучи (медь),  , приведена на чертеже.
, приведена на чертеже.
Методика 1
(4R)-4-Ацетилтио-2-оксопирролидин
1-(1) (4S)-4-Метансульфонилокси-2-оксопирролидин
В 100 мл пиридина растворял 1,90 г (4S)-4-гидрокси-2-оксопирролидина, затем по каплям добавляли 2,26 г метансульфонилхлорида, перемешивая и охлаждая льдом. Полученную смесь затем перемешивали при комнатной температуре в течение 1,5 ч, после чего смесь концентрировали в вакууме. Добавляли 9 мл насыщенного водного раствора кислого карбоната натрия, и смесь упаривали досуха в вакууме. К полученному остатку добавляли смесь этилацетата и метанола с объемным соотношением 1:1, нерастворившуюся часть отфильтровывали, и полученный раствор упаривали в вакууме. Полученный остаток после упаривания раствора хроматографировали на колонке (Merck 9385, 150 мл), с силикагелем, с использованием метода градиентного элюирования с использованием в качестве элюента смесей этилацетата и метанола с объемным соотношением от 9:1 до 4:1. Фракции, содержащие соединения настоящего изобретения, объединяли и концентрировали в вакууме, а требуемое соединение перекристаллизовывали из смеси этилацетата и метанола. Получали 2,44 г названного соединения в виде кристаллов, имеющих т.пл. 137,5-139oC.
Оптическое вращение [α]
Спектр инфракрасного поглощения (KBr) νmax см-1:
1719, 1697, 1659, 1305, 1177, 1171, 1159, 963.
Спектр ядерного магнитного резонанса:
(400 мГц, гексадейтерированный диметилсульфоксид) 8 ppm:
2,28 (1Н, дублет дублетов, J 17,6 и 1,8 Гц);
2,71 (1Н, дублет дублетов, J 17,6 и 6,3 Гц);
3,24 (3Н, синглет);
3,37 (1Н, дублет, J 11,9 Гц);
3,66 (1Н, дублет дублетов, J 11,9 и 5,3 Гц);
5,31-5,34 (1Н, мультиплет);
7,85 (1Н, уширенный синглет);
1-(2) (4R)-4-Ацетилтио-2-оксопирролидин
В 90 мл безводного ацетонитрила растворяли 896 мг соединения, полученного на стадии 1, описанной выше, и затем добавляли 857 мг тиоацетата калия. Раствор нагревали до кипения на масляной бане при температуре 85oC и выдерживали при этой температуре в течение 2 ч. Затем отфильтровывали нерастворившийся остаток и фильтрат упаривали в вакууме. К остатку добавляли этилацетат, и снова отфильтровывали нерастворившийся остаток. Раствор подвергали хроматографированию на колонке с силикагелем с помощью градиентного метода, с использованием в качестве элюента смесей этилацетата и метанола с объемным соотношением от 98: 2 до 98:4 до 94:6. Фракции, содержащие продукт, объединяли и концентрировали в вакууме. Получали 593 мг кристаллического остатка, который перекристаллизовывали из смеси этилацетата с циклогексаном. Получали 455 мг названного соединения в виде кристаллов, т.пл. 59-60oC.
Оптическое вращение [α]
Спектр инфракрасного поглощения (KBr) νmax см-1:
1689, 1125.
Спектр ядерного магнитного резонанса:
(400 мГц, CDCl3) δ ppm:
2,30 (1Н, дублет дублетов, J 17,4 и 6,0 Гц);
2,35 (3Н, синглет);
2,80 (1Н, дублет дублетов, J 17,4 и 9,1 Гц);
3,31 (1Н, дублет дублетов, J 10,2 и 5,1 Гц);
3,80 (1Н, дублет дублетов, J 10,2 и 7,2 Гц);
4,15-4,23 (1Н, мультиплет);
7,27 (1Н, уширенный синглет).
Методика 2
(4R)-4-Ацетилтио-2-оксиопирролидин
В 21 мл безводного тетрагидрофурана суспендировали 380 мг (4S)-4-гидрокси-2-оксопирролидина, и при комнатной температуре к суспензии добавляли 1,18 г трифенилфосфина. Затем к полученному раствору по каплям добавляли 783 мг диэтилазодикаоксилата, поддерживая температуру около -30oC. Затем температуру смеси постепенно поднимали до 4oC, после чего смесь перемешивали при этой температуре в течение 30 мин до получения гомогенной смеси. Далее реакционную смесь охлаждали до -20oC и к охлажденной смеси по каплям добавляли 320 мкл тиоуксусной кислоты. Температуру полученной смеси постепенно повышали до температуры ледяной бани, и смесь перемешивали при этой температуре в течение 1,5 ч. Затем реакционную массу концентрировали в вакууме и хроматографировали на колонке с силикагелем (Merck 9385, 150 мл), методом градиентного элюирования с использованием в качестве элюента смесей бензола и ацетонитрила с объемным соотношением от 2:1 до 1:1. Фракции, содержащие продукт, объединяли, концентрировали в вакууме, получали 69 г кристаллического остатка, который перекристаллизовывали из смеси этилацетата и циклогексана. Получали 54 г названного соединения в виде кристаллов.
Температура плавления, угол оптического вращения, спектр инфракрасного поглощения и спектр ядерного магнитного резонанса соединения, полученного этим способом, идентичны соответствующим значениям для соединения, полученного на стадии 2 методики 1.
Методика 3
(4R)-4-Ацетилтио-2-оксопирролидин
3-(1) (4S)-4-[(1S)-10-Камфорсульфонилокси]-2-оксопирролидин
В 50 мл пиридина растворяли 1911 мг 4-гидрокси-2-оксопирролидина, и при охлаждении на ледяной бане добавляли к раствору 2,63 мг хлорангидрида (1S)-10-камфорсульфокислоты. Смесь перемешивали при этой температуре 10 мин и затем перемешивали при комнатной температуре в течение 30 мин, после чего реакционную массу концентрировали в вакууме. Полученный остаток растворяли в этилацетате, промывали насыщенным водным раствором хлористого натрия. Этилацетатный слой концентрировали в вакууме, и полученный остаток хроматографировали на колонке с силикагелем (Merk 9385, 100 мл) методом градиентного элюирования с использованием в качестве элюента либо этилацетата, либо смеси этилацетата и метанола с объемным соотношением от 95:5 до 9:1. Фракции, содержащие продукт, объединяли и концентрировали в вакууме. Полученный остаток растворяли в 50 мл этилацетата. При стоянии из полученного раствора выпадали кристаллы, которые отфильтровывали и сушили. Получали 262 мг названного соединения, т.пл. 114-116oC.
Спектр ядерного магнитного резонанса:
(270 мГц, CDCl3) d ppm:
0,89 (3Н, синглет);
1,10 (3Н, синглет);
1,47 (1Н, дублет дублета дублетов, J 12,5, 9,2 и 3,3 Гц);
1,70 (1Н, дублет дублета дублетов, J 13,8, 9,2 и 4,6 Гц);
1,97 (1Н, дублет, J 17,5 Гц);
2,02-2,17 (2Н, мультиплет);
2,35-2,49 (2Н, мультиплет);
2,63 (1Н, дублет дублетов, J 17,8 и 2,6 Гц);
2,79 (1Н, дублет дублетов, J 17,8 и 6,6 Гц);
3,05 (1Н, дублет, J 15,0 Гц);
3,61 (1Н, дублет, J 15,0 Гц);
3,66 (1Н, дублет, J 11,9 Гц);
3,82 (1Н, дублет дублетов, J 11,9 и 6,0 Гц);
5,43-5,48 (1Н, мультиплет);
6,01 (1Н, уширенный синглет).
3-(2) (4R)-4-Ацетилтио-2-оксопирролидин
В 16 мл сухого ацетонитрила растворяли 160 г соединения, полученного на стадии 1, и к полученному раствору добавляли 90 мг тиоацетата калия. Смесь кипятили на масляной бане с температурой 98oC в течение 5 ч. Затем по методике, аналогичной методике, описанной на стадии 2 методики 1, получали 61 мг названного соединения в виде кристаллов.
Температура плавления, угол оптического вращения, спектр инфракрасного поглощения и спектр ядерного магнитного резонанса соединения, полученного этим способом, идентичны соответствующим значениям для соединения, полученного по методике.
Методика 4
(4R)-4-Меркапто-2-оксопирролидин
В 5 мл метанола растворяли 375 мг (4R)-4-ацетилтио-2-оксопирролидина (получен по любой из методик 1-3), и к полученному раствору при охлаждении льдом добавляли 3,35 мл 1 н. раствора метилата натрия в метаноле. Полученную смесь перемешивали при этой температуре в течение 20 мин. Затем к реакционной смеси добавляли 2,35 мл 1 н. водного раствора хлористого водорода, после чего смесь упаривали досуха в вакууме. К полученному остатку добавляли этилацетат и фильтровали. Фильтрат упаривали в вакууме, и получали 275 мг названного соединения в виде кристаллов. Т.пл. 69,5-70oC.
Оптическое вращение [α]
Спектр инфракрасного поглощения (KBr) νmax см-1:
2539, 1699, 1683.
Спектр ядерного магнитного резонанса:
(400 мГц, CDCl3) δ ppm:
1,96 (1Н, дублет J 7,2 Гц);
2,32 (1Н, дублет дублетов, J 17,2 и 6,8 Гц);
2,80 (1Н, дублет дублетов, J 17,2 и 8,2 Гц);
3,32 (1Н, дублет дублетов, J 10,2 и 5,6 Гц);
3,62-3,70 (1Н, мультиплет);
3,81 (1Н, дублет дублетов, J 10,2 и 7,3 Гц);
7,27 (1Н, уширенный синглет).
Методика 5
4-Нитробензил (1R,5S,6S)-2-[(4R)-2-оксо-4-пирролидинил-тио]-6-[(1R)-1-гидроксиэтил]-1-метил-1-карбапен-2-ем-3-карбоксилат
В 10 мл ацетонитрила растворяли 1000 мг 4-нитробензил-(1R,5S,6S)-2-дифенилфосфонилокси-6-[(1R)-1-гидроксиэтил] -1-метил-1-карбапен-2-ем-3-карбоксилата (получен по методике японской заявки N Hei 4-330085). К полученному раствору при охлаждении льдом добавляли раствор 200 мг (4R)-4-меркапто-2-оксопирролидина (получен по методике 4) в 3 мл ацетонитрила и 296 л ) диизопропилэтиламина. Полученную смесь перемешивали при этой температуре 1 ч и затем выдерживали в течение ночи при 4oC. Из реакционной смеси за это время выпадал кристаллический осадок, который отфильтровывали и сушили. Получали 672 мг названного соединения. Т.пл. 219-221oC.
Спектр ядерного магнитного резонанса:
(270 мГц, гексадейтерированный диметилсульфоксид) d ppm:
1,16 (3Н, дублет, J 6,3 Гц);
1,17 (3Н, дублет, J 7,3 Гц);
2,13 (1Н, дублет дублетов, J 17,1 и 4,4 Гц);
2,79 (1Н, дублет дублетов, J 17,1 и 7,8 Гц);
3,10 (1Н, дублет дублетов, J 108 и 3,4 Гц);
3,16-3,35 (1Н, мультиплет);
3,40-3,51 (1Н, мультиплет);
3,70 (1Н, дублет дублетов, J 10,7 и 7,3 Гц);
3,95-4,12 (2Н, мультиплет);
4,25 (1Н, дублет дублетов, J 9,3 и 2,5 Гц);
5,07 (1Н, дублет, J 5,4 Гц);
5,30, 5,46 (2Н, АВ, J 14,2 Гц);
7,71 (2Н, дублет, J 8,8 Гц);
8,23 (2Н, дублет, J 8.8 Гц).
Методика 6
(1R, 5S, 6S)-2-[(4R)-2-Оксо-4-пирролидинилтио]-6-[(1R)-1-гидроксиэтил]-1-метил-1-карбапен-2-ем-3-карбоксилат натрия
В смеси 19 мл тетрагидрофурана и 18 мл 0,1 М водного раствора фосфатного буфера растворяли 390 мг соединения, полученного по методике 5. К раствору добавляли 300 мг 10-ного (мас./мас.) палладия на угле, и полученную смесь интенсивно перемешивали в атмосфере водорода в течение 2,5 ч. Затем катализатор отфильтровывали и фильтрат дважды промывали диэтиловым эфиром. Водный слой упаривали в вакууме, и полученный остаток подвергали хроматографированию на MCI GEL CHP-20P (торговая марка "Мицубиси Касеи Корп.", 75-150 мкм, 50 мл), элюент вода. Фракции, содержащие продукт, упаривали в вакууме, после чего остаток сушили при температуре ниже 0oC, и получали 225 мг названного соединения в виде бесцветного порошка.
Спектр ядерного магнитного резонанса:
(270 мГц, дейтерированная вода) d ppm:
1,02 (3Н, дублет, J 7,3 Гц);
1,10 (3Н, дублет, J 6,6 Гц);
2,22 (1Н, дублет дублетов, J 17,6 и 4,4 Гц);
2,77 (1Н, дублет дублетов, J 17,6 и 8,4 Гц);
3,08-3,25 (2Н, мультиплет);
3,25 (1Н, дублет дублетов, J 5,9 и 2,6 Гц);
3,68 (1Н, дублет дублетов, J 11,4 и 6,4 Гц);
3,84-3,96 (1Н, мультиплет);
4,00-4,12 (2Н, мультиплет).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ КАРБАПЕНЕМА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2097383C1 |
| КРИСТАЛЛИЧЕСКИЕ ПРОИЗВОДНЫЕ 1-МЕТИЛКАРБАПЕНЕМА | 2000 |
|
RU2214411C2 |
| ЧЕТЫРЕХКООРДИНАЦИОННЫЕ КОМПЛЕКСЫ ДВУХВАЛЕНТНОЙ ПЛАТИНЫ | 1992 |
|
RU2039064C1 |
| ПРИМЕНЕНИЕ ПРОИЗВОДНЫХ 1-МЕТИЛКАРБОПЕНЕМА, АНТИБАКТЕРИАЛЬНАЯ В ОТНОШЕНИИ HELICOBACTER PYLORI КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА 1-МЕТИЛКАРБОПЕНЕМОВЫЕ ПРОИЗВОДНЫЕ И СПОСОБ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ ИНФЕКЦИОННЫХ ЗАБОЛЕВАНИЙ | 1997 |
|
RU2173147C2 |
| 1-МЕТИЛКАРБАПЕНЕМ ИЛИ ЕГО ФАРМАКОЛОГИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, КОМПОЗИЦИЯ, СПОСОБ ПРЕДУПРЕЖДЕНИЯ ИЛИ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 1997 |
|
RU2162088C2 |
| ГЕКСАГИДРОНАФТАЛИНОВЫЕ СЛОЖНОЭФИРНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2104997C1 |
| ПРОИЗВОДНЫЕ АЗЕТИДИНОНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2047602C1 |
| ПРОИЗВОДНЫЕ 1-МЕТИЛКАРБАПЕНЕМА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ АНТИБАКТЕРИАЛЬНОГО ВОЗДЕЙСТВИЯ | 1996 |
|
RU2160737C2 |
| α,ω ДИАРИЛАЛКАНЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2105752C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОТИВООПУХОЛЕВОГО КОМПЛЕКСА ПЛАТИНЫ | 1988 |
|
RU2007413C1 |
Сущность изобретения: предлагается стабильная при хранении кристаллическая форма пивалоилоксиметилового эфира производного карбапенема, известного как (1R,5S,6S)-2-[(4R)-2-оксо-4-пирролидинилтио]-6-[(1R)-1-гидроксиэтил] -1-метил-1-карбапен-2-ем-3-карбоновая кислота. 2 с. и 2 з.п. ф-лы, 1 табл., 1 ил.

имеющий основные пики при межплоскостных расстояниях 18.41, 9.21, 6.24, 5.28, 5.04, 4.72 , определенные методом рентгеноструктурного анализа при облучении порошковой пробы Kα - излучением меди с длиной волны
, определенные методом рентгеноструктурного анализа при облучении порошковой пробы Kα - излучением меди с длиной волны 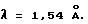
2. Соединение по п.1, имеющее температуру плавления 189oС.
| ЦЕНТРОИСКАТЕЛЬ | 0 |
|
SU337637A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

