Изобретение относится к новым производным азетидинона общей формулы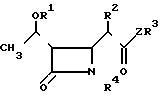 в которой R1 представляет собой гидроксизащитную группу;
в которой R1 представляет собой гидроксизащитную группу;
R2 представляет собой алкильную группу, имеющую от 1 до 3 углеродных атомов;
R3 представляет собой пиридильную группу, которая не замещена или замещена С1-С3-алкильной группой;
незамещенную хинолильную группу или фенильную группу, которая имеет заместитель формулы -СYNR5R6 и/или С1-С3-алкильную группу, где Y представляет собой атом кислорода или серы, а R5 и R6 одинаковые или различные и каждый представляет собой алкильную группу, имеющую от 1 до 6 углеродных атомов, или фенильную группу;
или R5 и R6 вместе образуют пирролидинил, пиперидил, морфолинил, или азепинил;
R4 представляет собой атом водорода;
Z представляет собой атом серы или атом кислорода.
Целью изобретения является разработка способа, позволяющего получать новые производные азетидинона, которые могут быть использованы как промежуточные продукты при получении некоторых антибиотиков карбапенема.
Пространственная конфигурация соединений I является важной поэтому предпочтительными соединениями являются те, чья конфигурация показана в формуле Ia
CH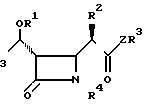
Соединения согласно изобретению неизбежно содержат несколько асимметрических атомов углерода и поэтому могут образовать оптические изомеры, включая, например, соединения, представленные формулой (Ia). В дополнение в зависимости от природы различных заместителей, возможны другие оптические и геометрические изомеры. Хотя все такие изомеры показаны здесь единственной формулой, но настоящее изобретение включает все такие изомеры, а также их смеси. Если получена смесь соединений согласно изобретению, то она может быть разделена традиционными методами расщепления. Альтернативно, в некоторых случаях может быть использована смесь изомеров. Однако, следует иметь в виду, что преимуществом настоящего изобретения является возможность получения желаемого 1 β-изомера быстро и с хорошим выходом.
П р и м е р 1. (3S,4S)-3-/(1R)-1-трет-бутилдиметилсилилоксиэтил/-4-(1R)-(2-пири-дилтиокарбо нил )-этилазетидин-2-он (соединение I-I)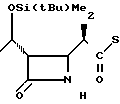
1(a) Z(0)-2-(1-трет-бутилдиметилсилилокси-1-пропенилтио)-пиридин
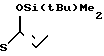 22,5 мл раствора, содержащего 1,2 эквивалента бутиллития в гексане, добавляли при комнатной температуре к раствору 7,5 мл гексаметилдисилазана в 50 мл тетрагидрофурана, полученную смесь перемешивали 30 мин, чтобы получить 1,2 эквивалента гексаметилдисилазана лития. Затем реакционную смесь охлаждали до -78оС и добавляли в следующем порядке 6,25 мл (1,2 эквивалента) триамида гексаметилфосфора, 8,37 мл (2,0 эквивалента) триэтиламина и 9,60 г (2,0 эквивалента) хлористого трет-бутилдиметилсилила с последующим добавлением раствора 5,0 г 2-пропионилтиопиридина (полученного как описано в получении I) в 10 мл тетрагидрофурана. Затем реакционную смесь перемешивали 10 мин, после чего ее разбавляли этилацетатом. Органический слой отделяли, два раза промывали водой и затем сушили над безводным сульфатом магния. Затем растворитель удаляли дистилляцией под пониженным давлением, остаток подвергали дробной перегонке и получали 8,4 г (выход 88%) соединения, указанного в названии, кипящего при 130оС и 0,1 мм ртутного столба (13,3 Па).
22,5 мл раствора, содержащего 1,2 эквивалента бутиллития в гексане, добавляли при комнатной температуре к раствору 7,5 мл гексаметилдисилазана в 50 мл тетрагидрофурана, полученную смесь перемешивали 30 мин, чтобы получить 1,2 эквивалента гексаметилдисилазана лития. Затем реакционную смесь охлаждали до -78оС и добавляли в следующем порядке 6,25 мл (1,2 эквивалента) триамида гексаметилфосфора, 8,37 мл (2,0 эквивалента) триэтиламина и 9,60 г (2,0 эквивалента) хлористого трет-бутилдиметилсилила с последующим добавлением раствора 5,0 г 2-пропионилтиопиридина (полученного как описано в получении I) в 10 мл тетрагидрофурана. Затем реакционную смесь перемешивали 10 мин, после чего ее разбавляли этилацетатом. Органический слой отделяли, два раза промывали водой и затем сушили над безводным сульфатом магния. Затем растворитель удаляли дистилляцией под пониженным давлением, остаток подвергали дробной перегонке и получали 8,4 г (выход 88%) соединения, указанного в названии, кипящего при 130оС и 0,1 мм ртутного столба (13,3 Па).
Спектр ЯМР на 1Н (СDСl3, 270 МГц), δ, млн. доли: 0,09 (6H, синглет), 0,88 (9Н, синглет), 1,73 (3Н, дублет, J=6,6 Гц), 5,45 (1Н, квартет, J=6,6 Гц), 6,97-7,02 (1Н, мультиплет), 7,32 (1Н, дублет, J=8,6 Гц), 7,51-7,75 (1Н, мультиплет), 8,42 (1Н, дублет, J=4 Гц).
Этот процесс повторяли за исключением того, что гексаметил фосфорный триамид заменяли добавками, показанными ниже. Во всех случаях количество литиевой соли гексаметилдисилазана составляло 1,2 эквивалента и по два эквивалента каждого из триэтиламина и хлористого трет-бутилдиметилсилила, применяемого в реакции. Температура реакции равна -78оС. Используя 1,3-диметил-3,4,5,6-тетрагидро-2(1Н)-пири- мидинон в качестве добавки и проводя реакцию в течение 10 мин, целевое соединение получали с выходом 88% Используя в качестве добавки N,N-диметилформамид и проводя реакцию в течение 10 мин, целевое соединение получали с 80% выходом. Используя в качестве N,N-диметилацетамид и проводя реакцию в течение 1 ч, целевое соединение получали с выходом 46%
1(b) (3S, 4S)-3-/(1R)-1-трет-бутилдиметилсилилоксиэтил/-4-/(1R)-(2-пиридилтио-карб онил)-этил/-азетидин-2-он.
163 мг (2,0 эквивалента) безводного хлористого цинка (свеже расплавленного) добавляли к раствору 171 мг Z(0)-(3S,4R)- 3-/(1R)-1-трет-бутилдиметилсилилоксиэтил/- 4-ацетоксиазетедин-2-она и 337 мг (два эквивалента) 2-(1-трет-бутилдиметилсилилокси-1-пропенилтио)-пиридина (полученного как описано на стадии (а) выше) в 15 мл хлористого метилена и смесь перемешивали при температуре бани 12оС в течение 15 ч. По окончании этого времени реакционную смесь смешивали с хлористым метиленом и органический слой три раза промывали водой. Затем ее сушили над безводным сульфатом магния и растворитель удаляли отгонкой при пониженном давлении. Остаток очищали колоночной хроматографией через силикагель, используя смесь циклогексана и этилацетата в отношении 1:1 по объему в качестве элюента, и получали целевое соединение, имеющее значение Rf 0,2 и плавящееся при 109оС.
Спектр поглощения в ИК-области (КВr), λмакс cм-1: 1757, 1718, 1696, 1564, 3181, 3099.
Спектр ЯМР на 1Н (CDCl3, 270 МГц), δ млн.доли: 0,07 (6H, синглет), 0,87 (9Н, синглет), 1,19 (3Н, дублет, J=5,9 Гц), 1,35 (3Н, дублет, J=7,2 Гц), 3,0-3,05 (2Н, мультиплет), 3,99 (1Н, двойной дублет, J=1,98 и 5,28 Гц), 4,19-4,23 (1Н, мультиплет), 5,90 (1Н, синглет), 7,3-7,32 (1Н, мультиплет), 7,60 (1Н, дублет, J=7,9 Гц), 7,73-7,93 (1Н, мультиплет), 8,63 (1Н, дублет), J=3,9 Гц).
1(с) (3S, 4S)-3-/(1R)-1-трет-бутилдиметилсилилоксиэтил/-4-/(1R)-(2-пиридилтиокарбо нил)-этил-азетидин-2-он.
Здесь предложен альтернативный метод получения того же соединения, которое получено на стадии (b) выше.
200 мг (5,7 ммоля) Z(0)-(3S,4S)-3-/(1R)-1-трет-бутилдиметилсилилоксиэтил/-4-фенил- сульфинилазетидин-2-она (получен способом, аналогичным описанному в получении 3) добавляли к раствору 160 мг (5,7 ммоля) 2-(1-трет-бутилдиметилсилилокси-1-пропе- нилтио)-пиридина (полученного как описано на стадии (а) выше) в 18 мл хлористого метилена, и полученную смесь нагревали при 15оС 4 ч. По истечении этого времени реакционную смесь смешивали с хлористым метиленом, органический слой отделяли и затем промывали водой и насыщенным водным раствором хлористого натрия в такой же последовательности. Затем раствор сушили над безводным сульфатом магния и растворитель удаляли отгонкой при пониженном давлении. Полученный остаток очищали колоночной хроматографией, используя в качестве элюента смесь циклогексан-этилацетат в отношении 1: 1 по объему и получали 60 мг (выход 28,7%) смеси целевого соединения и его изомера в отношении 18: 1, в котором (изомере) метильная группа, образующая часть этильной группы в 4-м положении азетидинонового кольца, находится в α-конфигурации, вместо β-конфигурации.
П р и м е р 2. (3S,4S)-3-/(1R)-1-трет-бутилдиметилсилилоксиэтил/-4-/(1R)-(2-хино- линтиокарбонил)-этил/-азетидин-2-он (соединение N 1-5)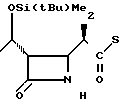
2(а) Z(0)-2-(1-трет-бутилдиметилсилилокси-1-пропенилтио)-хинолин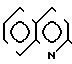
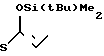
0,9 мл раствора, содержащего 1,2 эквивалента бутиллития в гексане, добавляли при комнатной температуре к раствору 0,3 мл гексаметилдисилазана в 10 мл безводного тетрагидрофурана, и полученную смесь перемешивали 30 мин. Затем реакци- онную смесь охлаждали до -78оС и добавляли в следующем порядке 0,25 мл (1,2 эквивалента) гексаметилфосфорного триамида, 0,33 мл (два эквивалента) триэтиламина и 360 мг (два эквивалента) хлористого трет-бутилдиметилсилила с последующим добавлением 300 мг пропионилтиохинолина (полученного как описано в получении 2). Затем реакционную смесь перемешивали 10 мин, после чего ее смешивали с этилацетатом, органический слой отделяли и промывали водой. Растворитель удаляли отгонкой при пониженном давлении и полученный остаток очищали колоночной хроматографией, используя смесь цикло- гексан-этилацетат в отношении 10:1 по объему в качестве элюента, и получали 320 мг (выход 80%) целевого соединения, имеющего значение Rf 0,8.
Cпектр ядерного магнитного резонанса на 1Н (CDCl3, 270 МГц), δ млн.доли: 0,10 (6H, синглет), 0,89 (9Н, синглет), 1,73 (3Н, дублет, J=6,9 Гц), 5,53 (1Н, квартет, J=6,9 Гц), 7,43-7,50 (2Н, мультиплет), 7,66-7,73 (1Н, мультиплет), 7,74 (1Н, дублет, J=8 Гц), 7,96 (1Н, дублет, J=8,6 Гц), 8,00 (1Н, дублет, J=8,6 Гц).
2(b) (3S,4S)-/3-/(1R)-1-трет-бутилдиметилсилилоксиэтил/-4-/(1R)-(2-хинолинтиокарб онил)-этил/-азетидин-2-он.
120 мг (два эквивалента) безводного хлористого цинка добавляли к раствору 126 мг Z(0)-(3S,4S)-3-/(1R)-1-трет-бутилдиметилсилилоксиэтил/-4-ацетоксиазетидин-2- она и 320 мг (два эквивалента) 2-(1-трет-бутилдиметилсилилокси-1-пропенилтио)-хинолина (полученного как описано на стадии (а) выше) в 15 мл безводного хлористого метилена, и полученную смесь перемешивали при температуре бани 28-30оС 3 ч. По истечении этого времени реакционную смесь смешивали с хлористым метиленом. Затем органический слой отделяли, промывали водой и сушили над сульфатом магния. Затем растворитель удаляли отгонкой при пониженном давлении. Полученный остаток очищали колоночной хроматографией, используя в качестве элюента смесь циклогексан-этилацетат в отношении 1:1 по объему, и получали 64 мг (выход 32%) целевого соединения, указанного в названии стадии, имеющего значение Rf, равное 0,2.
Cпектр ядерного магнитного резонанса на 1Н (CDCl3, 270 МГц), δ мл.доли: 0,01 (6Н, синглет), 0,88 (9Н, синглет), 1,22 (3Н, дублет, J=6 Гц), 1,40 (3Н, дублет, J=7,2 Гц), 3,07-3,11 (2Н, мультиплет), 4,04 (1Н, дублет дублетов, J= 2,0 и 5,28 Гц), 4,22-4,26 (1Н, мультиплет), 5,91 (1Н, синглет), 7,63-7,68 (2Н, мультиплет), 7,7-7,8 (1Н, мультиплет), 7,87 (1Н, дублет, J=8,5 Гц), 8,11 (1Н, дублет, J=8,5 Гц), 8,22 (1Н, дублет, J=8,58 Гц).
П р и м е р 3. (3S,4S)-3-/(1R)-1-трет-бутилдиметилсилилоксиэтил/-4-{ (1R)-1-/3/- метил-2-пиридилтиокарбонил/-этил} /азетидин-2-он.
(Соединение 1-2)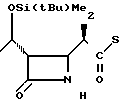
3(а) Z(0)-2-(1-трет-бутилдиметилсилилокси-1-пропенилтио)-3-метилпиридин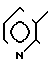 S
S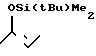
15,17 мл (24,3 ммоля) 1,6 м раствора бутиллития в гексане по каплям добавляли при -78оС к раствору 5,59 мл (26,5 ммоля) гексаметилдиосилазана в смеси с 40 мл тетрагидрофурана и 4,61 мл (26,5 ммоля) триамидо гексаметилфосфора и затем к полученной смеси добавляли 6,65 г (44,1 ммоля) хлористого трет-бутилдиметилсилила и 9,25 мл (66,3 ммоля) триэтиламина. Полученную смесь затем продолжали перемешивать еще 10 мин, после чего к реакционной смеси добавляли по каплям раствор 4,00 г (22,1 ммоля) 2-пропионилтио-3-метилпиридина в 10 мл безводного тетрагидрофурана. Затем смесь перемешивали при -78оС в течение 10 мин, и температуру реакционной смеси снижали до комнатной температуры. К смеси примешивали насыщенный водный раствор бикарбоната натрия и полученную смесь три раза экстрагировали каждый раз по 60 мл пентана. Экстракт сушили над безводным сульфатом магния, и затем растворитель удаляли отгонкой при пониженном давлении. Полученный остаток очищали колоночной хроматографией через глинозем, используя в качестве элюента смесь гексан-этилацетат в отношении 5:1, и получали 3,38 г (выход 52%) целевого соединения в виде бесцветного масла.
Спектр ядерного магнитного резонанса на 1Н (CDСl3), δ млн. доли: 0,04 (6Н, синглет), 0,85 (9Н, синглет), 1,74 (3Н, дублет, J=7 Гц), 2,25 (3Н, синглет), 5,33 (3Н, квартет, J=7 Гц), 6,95 (1Н, дублет дублетов, J=7 и 5 Гц), 7,33 (1Н, двойной дублет, J=7 и 1 Гц), 8,37 (1Н, двойной дублет, J=5 и 1 Гц).
3(b) (3S,4S)-3-/(1R)-1-трет-бутилдиметилсилилоксиэтил/-4-{(1R)-1-/3-метил-2-пи- ридилтиокарбонил/-этил}-азетидин-2-он.
190 мг (1,39 ммоля) хлористого цинка добавляли к раствору 200 мг (0,70 ммоля) Z(0)-(3S, 4S)-3-/(1R)-1-трет-бутилдиметилси- лилоксиэтил/-4-ацетоксиазетидин-2-она в 5 мл хлористого метилена и к нему добавляли затем раствор 411 мг 2-(1-трет-бутилдиметилсилилокси-1-пропенилтио)-3-метилпи-ридина (полученного как описано на стадии (а), в 2,0 мл хлористого метилена при охлаждении льдом и в атмосфере азота). Полученную смесь перемешивали при комнатной температуре 8 ч, после чего добавляли 50 мл хлористого метилена. Органический слой промывали водой со льдом и сушили над безводным сульфатом магния, растворитель удаляли отгонкой при пониженном давлении. Полученный остаток очищали колоночной хроматографией среднего давления через силикагель, используя в качестве элюента смесь гексан-этилацетат в отношении 1:2 по объему, и получали 246 мг (выход 87%) целевого соединения в виде бесцветных кристаллов. Образцы для анализа перекристаллизовывали из диизопропилового эфира и определена точка плавления 120-122оС.
Cпектр ядерного магнитного резонанса на 1Н (CDCl3) δ мл.д. 0,07 (6H, синглет), 0,87 (9Н, синглет), 1,20 (3Н, дублет, J=6 Гц), 1,35 (3Н, дублет, J=7 Гц), 2,36 (3Н, синглет), 3,02-3,13 (2Н, мультиплет), 4,00 (1Н, дублет дублетов, J= 4 и 2 Гц), 4,20 (1Н, мультиплет), 5,89 (1Н, широкий синглет) 7,28 (1Н, дублет дублетов, J=8 и 5 Гц), 7,64 (1Н, дублет дублетов, J=8 и 1 Гц), 8,50 (1Н, двойной дублет, J=5 и 1 Гц).
П р и м е р 4. (3S,4S)-3-/(1R)-1-трет-бутилдиметилсилилоксиэтил/-4-{ (1R)-1-/4-ме- тил-2-пиридилтиокарбонил/-этил}-азети- дин-2-он
Соединение 1-3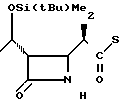
4(a) Z(0)-2-(1-трет-бутилдиметилсилилокси-1-пропенилтио)-4-метилпиридин.
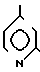
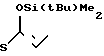
Выполняя процедуру, аналогичную описанной в примере 3(а), но используя 1,51 г (8,33 ммоля) 2-пропионилтио-4-метилпиридина, получали 1,25 г (выход 51%) целевого соединения в виде бесцветного масла.
Спектр ядерного магнитного резонанса на 1H (CDCl3), δ м.д. 0,09 (6Н, синглет), 0,89 (9Н, синглет), 1,74 (3Н, дублет, J=7 Гц), 2,30 (3Н, синглет), 5,43 (1Н, квартет, J=7 Гц), 6,82 (1Н, дублет, J=5 Гц), 7,14 (1Н, синглет), 8,27 (1Н, дублет, J=5 Гц).
4(b) (3S, 4S)-3-/(1R)-1-трет-бутилметилсилилоксиэтил/-4-{(1R)-1-/4-метил-2-пири-ди лтиокарбонил/-этил}-азетидин-2-он.
Следуя процессу, аналогичному описанному в примере 3(b), но используя 500 мг (1,74 ммоля) Z(0)-(3S, 4S)-3-/(1R)-1-трет-бутилдиметилсилилоксиэтил/-4-ацетоксиазе- тидин-2-он, 1,03 г (3,49 ммоля) 2-(1-трет-бутилдиметилсилилокси-1-пропенилтио)-4- метилпиридина (полученного как описано на стадии (а) выше) и 474 мг (3,48 ммоля) хлористого цинка, получали 502 мг (выход 71% ) целевого соединения в виде бесцветных кристаллов. Образцы для анализа перекристаллизовывали из диизопропилового эфира и определяли точку плавления 123-125оС.
Спектр ядерного магнитного резонанса (СDCl3), δ илн.доли: 0,07 (6H, синглет), 0,87 (9Н, синглет), 1,19 (3Н, дублет, J=6 Гц), 1,35 (3Н, дублет, J= 7 Гц), 2,40 (3Н, синглет), 2,95-3,10 (2Н, мультиплет), 3,98 (1Н, двойной дублет, J=5 и 2 Гц), 4,21 (1Н, мультиплет), 7,13 (1Н, двойной дублет, J=5 и 1 Гц), 7,42 (1Н, дублет, J=1 Гц), 8,48 (1Н, дублет, J=5 Гц).
П р и м е р 5. (3S,4S)-3-/(1R)-1-трет-бутилдиметилсилилоксиэтил/-4-{ (1R)-1-/5-ме- тил-2-пиридилтиокарбонил/-этил}-азети- дин-2-он.
(Соединение 1-4)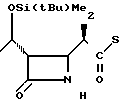
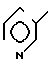
5(a) Z(0)-2-(1-трет-бутилдиметилсилилокси-1-пропенилтио)-5-метилпиридин
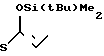
Выполняя процесс, аналогичный описанному в примере 3(а), но используя 1,80 г (9,93 ммоля) 2-пропионилтио-5-метилпиридина, получая 1,50 г (выход 51%) целевого соединения в виде бесцветного масла.
Спектр ядерного магнитного резонанса на 1Н (CDCl3), δ мл.д. 0,09 (6Н, синглет), 0,88 (9Н, синглет), 1,72 (3Н, дублет, J=7 Гц), 2,27 (3Н, синглет), 5,42 (1Н, квартет, J= 7 Гц), 7,22 (1Н, дублет, J=8 Гц), 7,36 (1Н, дублет дублетов, J=8 и 2 Гц), 8,26 (1Н, дублет, J=2 Гц).
5(b) (3S, 4S)-3-/(1R)-1-трет-бутилдиметилсилилоксиэтил/{ (1R)-1-/5-метил-2-пири- дилтиокарбонил/-этил}-ацетидин-2-он.
Следуя процедуре, аналогичной описанной в примере 3(b), но используя 500 мг (1,74 ммоля) Z(0)-(3S,4S)-3-/(1R)-1-трет-бутилдиметилсилилоксиэтил/-4-ацетоксиазе- тидин-2-она, 1,03 г (3,49 ммоля) 2-(1-трет-бутилдиметилсилилокси-1-пропенилтио)-5- метилпиридина (полученного как описано на стадии (а) выше), и 474 мг (3,48 ммоля) хлористого цинка, получали 583 мг (выход 82%) целевого соединения в виде бесцветных кристаллов. Образцы для анализа перекристаллизовывали из диизопропилового эфира и расплавляли при 86-88оС.
Спектр ядерного магнитного резонанса на 1Н (CDCl3), δ млн.д. 0,07 (6Н, синглет), 0,87 (9Н, синглет), 1,19 (3Н, дублет, J=6 Гц), 1,34 (3Н, дублет, J= 7 Гц), 2,37 (3Н, синглет), 2,95-3,08 (2Н, мультиплет), 3,98 (1Н, дублет дублетов, J= 5 и 2 Гц), 4,21 (1Н, мультиплет), 5,90 (1Н, широкий синглет), 7,46 (1Н, дублет, J=8 Гц), 7,56 (1Н, двойной дублет, J=8 и 2 Гц), 8,47 (1Н, дублет, J=2 Гц).
П р и м е р 6. S-2-диэтилкарбамоилфениловый эфир 2(R)-{(3S,4S)-3-/1-(R)-(трет-бутилдиметилсилилокси)-этил/-2-оксо-4-азе-тидин ил}-тиопропионовой кислоты и его 2(S)-изомер. (Соединение 2-2).
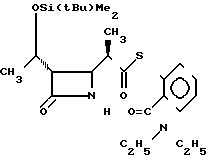
330 г (2,42 ммоля) безводного хлористого цинка добавляли к раствору 911 мг (2,40 ммоля) 1-трет-бутилдиметилсилилокси-1-(2-диэтилкарбамоил)-фенилтио-1-пропена (полученного как описано в получении 19 ниже, и 347 мг (1,21 ммоля) (3R,4R)-3-/(1R)-(трет-бутилдиметилсилилокси)-этил/-4-аце- токси-2-азетидинона в 12 мл хлористого метилена, и полученную смесь перешивали при комнатной температуре два часа. По истечении этого времени реакционную смесь разбавляли этилацетатом, смесь промывали водой и сушили над безводным сульфатом магния. Затем растворитель удаляли отгонкой при пониженном давлении. Полученный остаток очищали хроматографией через колонку Лобара, используя в качестве элюента смесь гексана-этилацетат в отношении 1:1 по объему, и получали 61 мг (выход 10%) 2S-изомера целевого соединения, плавящегося при 125-126,5оС (после перекристаллизации из смеси гексан-этилацетат), и 487 мг (выход 82% ) 2R-изомера целевого соединения, плавящегося при 130,5-132оС (после перекристаллизации из диизопропилового эфира).
ИК-спектр поглощения (КВr), λмакс см-1 (2S-изомер): 3182, 1765, 1711, 1629, 965, 829;
(2R-изомер): 3086, 1762, 1700, 1637, 965, 829.
Спектр ядерного магнитного резонанса на 1Н (CDCl3, 270 МГц),
(2S-изомер): δ млн. доли: 0,06 (3H, синглет), 0,07 (3Н, синглет), 0,87 (9Н, синглет), 1,05 (3Н, триплет, J=7 Гц), 1,26 (3Н, дублет, J=6 Гц), 1,26 (3Н, триплет, J=7 Гц), 1,28 (3Н, дублет, J=7 Гц), 2,71-2,83 (2Н, мультиплет); 3,00-3,21 (2Н, нонетоподобный, J=7 Гц), 3,35-3,80 (2Н, широкий), 3,63 (1Н, дублет, J= 9 Гц), 4,14 (1Н, квинтет, J=6 Гц), 7,00-7,30 (1Н, широкий синглет), 7,30-7,35 (1Н, мультиплет), 7,24-7,53 (3Н, мультиплет);
(2R-изомер): 0,08 (6H, синглет), 0,87 (9Н, cинглет), 1,03 (3Н, триплет, J= 7 Гц), 1,21 (3Н, дублет, J=6 Гц), 1,25 (3Н, триплет, J=7 Гц), 1,29 (3Н, дублет, J= 7 Гц), 2,96-3,15 (4Н, мультиплет), 3,20-3,85 (2Н, широкий), 3,96 (1Н, двойной дублет, J=2 и 4 Гц), 4,19 (1Н, квинтет, J=6 Гц), 5,90-6,10 (1Н, широкий синглет); 7,30-7,35 (1Н, мультиплет), 7,41-7,51 (3Н, мультиплет).
Масс-спектр (m/z): (2R- и 2S-изомеры): 492 (M+, C25H40N2O4SSi).
Вычислено, C 60,94; H 8,18; N 5,69; S 6,51.
C25H40N2O4SSi.
Найдено, 2S-изомер: C 60,72; H 8,01; N 5,70; S 6,51.
Найдено, 2R-изомер: C 60,85; H 8,10; N 5,62; S 6,50.
П р и м е р ы 7-17. Следуя процедуре, аналогичной описанной в примере 6, синтезировали следующие соединения.
П р и м е р 7. 2-Диметилкарбамоилфениловый эфир 2(R)-{(3S,4S)-3-/1(R)-трет-бутилдиметилсилилокси)-этил/-2-оксо-4-азе-тидинил -тиопропионовой кислоты (соединение 2-1)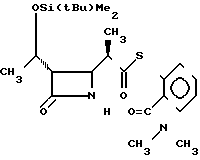
Выход 2R-изомера составил 79% и отношение выходов 2R-изомера к 2S-изомеру равнялось 4,9:1,0.
Cпектр ядерного магнитного резонанса (СDCl3), δ млн.доли: 0,08 (6H, cинглет), 0,88 (9Н, синглет), 1,21 (3Н, дублет, J=6 Гц), 1,29 (3Н, дублет, J=7 Гц), 2,79 (3Н, синглет), 2,96-3,08 (2Н, мультиплет), 3,10 (3Н, синглет), 3,94 (1Н, дублет дублетов, J=2 и 5 Гц), 4,19 (1Н, двойной квартет, J=5 и 6 Гц), 6,10-6,20 (1Н, широкий синглет), 7,31-7,36 (1Н, мультиплет), 7,40-7,70 (3Н, мультиплет).
2R-изомер был получен в форме игольчатых кристаллов с температурой плавления 99-101оС (после перекристаллизации из смеси этилацетат-гексан).
П р и м е р 8. S-2-дипропилкарбамоилфениловый эфир 2(R)-{(3S,4S)-3-/1(R)-(трет-бутилдиметилсилилокси)-этил/-2-оксо- 4-азетидинил} -тиопропионовой кислоты (соединение 2-3).
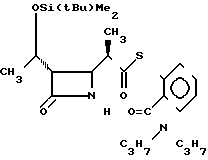
Выход 2R-изомера составил 74% и отношение выходов 2R-изомера к 2S-изомеру равнялось 3,5:1,0.
Спектр ядерного магнитного резонанса (СDCl3), δ млн.доли: 0,08 (6Н, синглет), 0,72 (3Н, триплет, J=7 Гц), 0,88 (9Н, синглет), 1,00 (3Н, триплет, J=7 Гц), 1,22 (3Н, дублет, J=6 Гц), 1,20-1,40 (3Н, широкий), 1,46 (2Н, секстет, J= 7 Гц), 1,70 (2Н, секстет, J=7 Гц), 2,91-3,06 (2Н, мультиплет), 3,10-3,80 (2Н, широкий), 3,96 (2Н, двойной дублет, J=2 и 4 Гц), 4,19 (1Н, двойной квартет, J= 5 и 6 Гц), 5,90-6,20 (1Н, широкий синглет), 7,92-7,35 (1Н, мультиплет), 7,40-7,52 (3Н, мультиплет).
2R-изомер имел форму игольчатых кристаллов, плавящихся при 112-113оС (после перекристаллизации из смеси этилацетатгексан).
П р и м е р 9. S-2-Диизобутилкарбамоилфениловый эфир 2(R)-{(3S,4S)-1(R)-(трет-бутилдиметилсилилокси)-этил/-2-оксо-4- азетидинил}-тиопропионовой кислоты (соединение 2-6)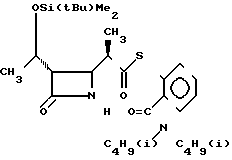
Выход 2R-изомера составил 70% и отношение выходов 2R-изомера к 2S-изомеру равнялось 4,6:1,0.
Спектр ядерного магнитного резонанса (CDCl3), δ млн.доли: 0,08 (6Н, синглет), 0,74 (6Н, дублет, J=7 Гц), 0,87 (9Н, синглет), 1,02 (6Н, дублет, J=7 Гц), 1,21 (3Н, дублет, J=6 Гц), 1,20-1,40 (3Н, широкий), 1,81 (1Н, септет, J= 7 Гц), 2,12 (1Н, септет, J=7 Гц), 2,80-3,06 (4Н, мультиплет), 3,20-3,57 (2Н, широкий), 3,92-4,05 (1Н, широкий синглет), 4,13-4,28 (1Н, широкий), 5,95-6,15 (1Н, широкий), 7,29-7,35 (1Н, мультиплет), 7,42-7,50 (3Н, мультиплет).
2R-изомер получен в форме игольчатых кристаллов, плавящихся при 144-146оС (после перекристаллизации из смеси этилацетат-гексан).
П р и м е р 10. S-2-(N-метил-N-фенилкарбамоил)-фениловый эфир 2(R)-{(3S, 4S)-3-/1(R)-(трет-бутилдиметилсилилокси)-этил/2-оксо-4-азетидин} -т иопропионовой кислоты (соединение 2-16)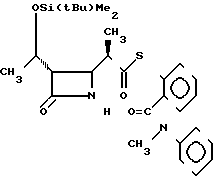
Выход 2R-изомера составил 64% и отношение выходов 2R-изомера к 2S-изомеру было как 2,6:1,0.
Спектр ядерного магнитного резонанса (CDCl3), δ млн.доли: 0,08 и 0,09 (вместе 6Н, каждый синглет), 0,88 (9Н, синглет), 1,23 (3Н, дублет, J=6 Гц), 1,34 (3Н, дублет, J=7 Гц), 3,01-3,12 (2Н, мультиплет), 3,49 (3Н, синглет), 4,00-4,08 (1Н, широкий синглет), 4,20 (1Н, двойной квартет, J=6 и 6 Гц), 6,05-6,20 (1Н, широкий синглет), 6,95-7,63 (3Н, мультиплет).
2R-изомер имел форму игольчатых кристаллов, плавящихся при 158-159оС (после перекристаллизации из смеси этилацетат-гексан).
П р и м е р 11. S-2-(1-пирролидинилкарбонил)-фениловый эфир 2(R)-{(3S, 4S)-3-/1(R)-(трет-бутилдиметилсилилокси)-этил/-2-оксо-4-азетидинил тиопропионовой кислоты (соединение 3-3)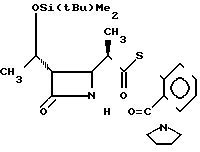
Выход изомера 2R составил 85% и отношение выходов 2R-изомера к 2S-изомеру равно 7,1:1,0.
Cпектр ядерного магнитного резонанса (CDCl3), млн.доли: 0,08 (6Н, синглет), 0,88 (9Н, синглет), 1,21 (3Н, дублет, и J=6 Гц), 1,29 (3Н, дублет, J=7 Гц), 1,75-2,00 (4Н, мультиплет), 2,95-3,06 (2Н, мультиплет), 3,18 (2Н, триплет, J=7 Гц), 3,60 (2Н, триплет, J=7 Гц), 3,96 (1Н, двойной дублет, J=2 и 4 Гц), 4,20 (1Н, двойной квартет, J=5 и 6 Гц), 6,10-6,25 (1Н, широкий синглет), 7,37-7,53 (4Н, мультиплет).
2R-изомер имел форму пеноподобного вещества.
П р и м е р 12. S-2-(1-пиперидилкарбонил)-фениловый эфир 2(R)-{(3S, 4S)-3-/1(R)-(трет-бутилдиметилсилилокси)-этил/-2-ок-со-4-азетидини л}-тиопропионовой кислоты (соединение 3-4)
CH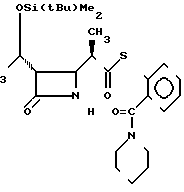
Выход 2R-изомера составил 75% и отношение выходов 2R-изомера к 2S-изомеру было 4,8:1,0.
Спектр ядерного магнитного резонанса (СDCl3), δ млн.доли: 0,07 и 0,08 (вместе 6Н, два синглета), 0,87 и 0,88 (вместе 9Н, два синглета), 1,16-1,25 (3Н, мультиплет), 1,28 и 1,33 (вместе 3Н, два дублета, J=7 и 7 Гц), 1,37-1,52 (2Н, широкий), 1,54-1,77 (4Н, широкий), 2,95-3,26 (4Н, мультиплет), 3,47-3,60 (1Н, широкий), 3,80-3,95 (1Н, широкий), 3,97 (1Н, двойной дублет, J= 2 и 4 Гц), 4,12-4,26 (1Н, широкий), 6,00-6,16 (1Н, широкий), 7,26-7,52 (4Н, мультиплет).
2R-изомер получен в форме стекловидного тела.
П р и м е р 13. S-2-морфолинокарбонилфениловый эфир 2(R)-{(3S,4S)-3-/1(R)-(трет-бутилдиметилсилилокси)-этил/-2-оксо-4- азетидинил} -тиопропионовой кислоты (соединение 3-8)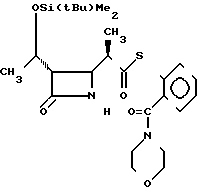
Выход 2R-изомера составил 83% и отношение выхода 2R-изомера в 2S-изомеру составило 7,9:1,0.
Спектр ядерного магнитного резонанса (СDCl3), δ млн.доли: 0,08 (6Н, синглет), 0,88 (9Н, синглет), 1,21 (3Н, дублет, J=7 Гц), 1,22-1,38 (3Н, мультиплет), 2,97-3,08 (2Н, мультиплет), 3,12-3,32 (2Н, мультиплет), 3,50-3,60 (2Н, мультиплет), 3,70-3,84 (4Н, широкий), 3,93-4,01 (1Н, широкий синглет), 4,19 (1Н, двойной квартет, J=5 и 5 Гц), 5,90-6,10 (1Н, широкий), 7,20-7,38 (1Н, мультиплет), 7,42-7,55 (3Н, мультиплет).
2R-изомер получен в форме стекловидного вещества.
П р и м е р ы 14. S-2-(1-Азепинилкарбонил)-фениловый эфир 2(R)-{(3S, 4S)-3-/1(R)-(трет-бутилдиметилсилилокси)-этил/-2-ок-со-4-азетидини л} -тиопропионовой кислоты (соединение 3-5)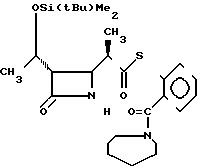
Выход 2R-изомера составил 87% и отношение выходов 2R-изомера к 2S-изомеру было 9,5:1,0.
Cпектр ядерного магнитного резонанса (CDCl3), δ млн.доли: 0,08 (6Н, синглет), 0,88 (9Н, синглет), 1,22 (3Н, дублет, J=6 Гц), 1,20-1,40 (3Н, широкий), 1,50-1,96 (8Н, широкий), 2,97-3,10 (2Н, мультиплет), 3,06-3,32 (2Н, широкий), 3,40-3,90 (2Н, широкий), 3,96 (1Н, двойной дублет, J=2 и 4 Гц), 4,20 (1Н, двойной квартет, J=6 и 6 Гц), 6,05-6,25 (1Н, широкий), 7,30-7,37 (1Н, мультиплет), 7,42-7,52 (3Н, мультиплет).
2R-изомер получен в форме стекловидного вещества.
П р и м е р 15. S-2-диэтилкарбамоил-6-метилфениловый эфир 2(R)-{(3S, 4S)-3-/1(R)-(трет-бутилдиметилсилилокси)-этил/-2-ок-со-4-азетидини л}-тиопропионовой кислоты (соединение 2-40)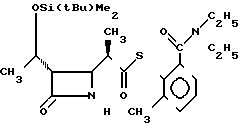
Выход 2R-изомера составил 88% и отношение выходов 2R-изомера к 2S-изомеру составило 12,3:1,0.
Cпектр ядерного магнитного резонанса (CDCl3), δ млн.доли: 0,06 (3Н, синглет), 0,86 (9/2H, синглет), 0,89 (9/2H, синглет), 1,02 (3Н, триплет, J=7 Гц), 1,19-1,29 (7,5Н, мультиплет), 1,35 (1,5Н, дублет, J=7 Гц), 2,35 (3Н, синглет), 2,92-3,16 (4Н, мультиплет), 3,28-3,41 (1Н, мультиплет), 3,74 (1Н, двойной квартет, J=14 и 7 Гц), 3,95-4,00 (1Н, мультиплет), 4,17 (1Н, квинтет, J=6 Гц), 6,00-6,30 (1Н, широкий синглет), 7,14 (1Н, двойной дублет, J=3 и 6 Гц), 7,34-7,41 (2Н, мультиплет).
2R-изомер получен в форме игольчатых кристаллов, плавящихся при 150-150,5оС (после перекристаллизации из диизопропилового эфира).
П р и м е р 16. S-2-/Диэтил-(тиокарбамоил)/-6-метилфениловый эфир 2(R)-{ (3S, 4S)-3-/1(R)-(трет-бутилдиметилсилилок- си)-этил/-2-оксо-4-азетидин}-тиопропионо- вой кислоты (соединение 2-63)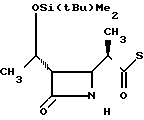
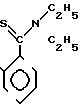
Выход 2R-изомера составил 81% и отношение выходов 2R-изомера к 2S-изомеру равнялось 4,8:1,0.
Спектр ядерного магнитного резонанса (СDCl3), δ млн.доли: 0,09 (6H, синглет), 0,89 (9Н, синглет), 1,09 (3Н, триплет, J=7 Гц), 1,22 (3Н, дублет, J= 6 Гц), 1,26 (3Н, дублет, J=7 Гц), 1,38 (3Н, триплет, J=7 Гц), 2,96-3,06 (2Н, мультиплет), 3,20 (1Н, двойной квартет, J=14 и 7 Гц), 3,36 (1Н, двойной квартет, J= 14 и 7 Гц), 3,76 (1Н, двойной квартет, J=14 и 7 Гц), 3,97 (1Н, двойной дублет, т J=2 и 5 Гц), 4,20 (1Н, двойной квартет, J=5 и 6 Гц), 4,46 (1Н, двойной квартет, J=14 и 7 Гц), 7,22-7,26 (1Н, мультиплет), 7,33-7,46 (3Н, мультиплет).
2R-изомер получен в форме игольчатых кристаллов, плавящихся при 163-165оС (после перекристаллизации из смеси этилацетат-гексан).
П р и м е р 17. S-2-Диэтилкарбамоилфениловый эфир 2(R)-{(3S,4S)-3-/1(R)-(трет-бутилдиметилсилилокси)-этил/-2-оксо-4-азе- тидинил}-пропионовой кислоты (соединение 2-56).
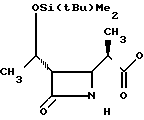
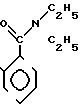
Выход 2R-изомера составил 61% и отношение выходов 2R-изомера к 2S-изомеру равнялось 6,8:1,0.
Спектр ядерного магнитного резонанса (CDCl3), δ млн.доли: 0,09 (6Н, синглет), 0,88 (9Н, синглет), 1,10 (3Н, триплет, J=7 Гц), 1,22 (3Н, триплет, J= 7 Гц), 1,25 (3Н, дублет, J=6 Гц), 1,32 (3Н, дублет, J=7 Гц), 2,95 (1Н, двойной квартет, J=4 и 7 Гц), 3,00 (1Н, дублет, J=6 Гц), 3,22 (2Н, квартет, J= 7 Гц), 3,42-3,63 (1Н, мультиплет), 4,08-4,16 (1Н, мультиплет), 4,18 (1Н, квинтет, J= 6 Гц), 6,45 (1Н, широкий синглет), 7,17 (1Н, дублет), J=8 Гц), 7,26-7,28 (2Н, мультиплет), 7,38-7,43 (1Н, мультиплет).
2R-изомер был получен в форме стекловидного вещества.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЧЕТЫРЕХКООРДИНАЦИОННЫЕ КОМПЛЕКСЫ ДВУХВАЛЕНТНОЙ ПЛАТИНЫ | 1992 |
|
RU2039064C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОТИВООПУХОЛЕВОГО КОМПЛЕКСА ПЛАТИНЫ | 1988 |
|
RU2007413C1 |
| ПРОИЗВОДНЫЕ КАРБАПЕНЕМА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2097383C1 |
| ФУНГИЦИДНЫЕ ПРОИЗВОДНЫЕ ОКСЕТАНА И ИХ СОЛИ | 1992 |
|
RU2044736C1 |
| ГЕКСАГИДРОНАФТАЛИНОВЫЕ СЛОЖНОЭФИРНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2104997C1 |
| α,ω ДИАРИЛАЛКАНЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2105752C1 |
| АЗАСТЕРОИДНЫЕ СОЕДИНЕНИЯ | 1991 |
|
RU2070204C1 |
| КРЕМНИЙСОДЕРЖАЩИЕ СОЕДИНЕНИЯ ИЛИ ИХ СОЛИ, ОБЛАДАЮЩИЕ ФУНГИЦИДНОЙ АКТИВНОСТЬЮ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2069213C1 |
| КРИСТАЛЛИЧЕСКИЙ ПИВАЛОИЛОКСИМЕТИЛ (IR, 5S, 6S)-[(4R)-2-ОКСО-4-ПИРРОЛИДИНИЛТИО]-6-[(IR)-1-ГИДРОКСИЭТИЛ]-1-МЕТИЛ-1-КАРБАПЕН-2-ЕМ-3-КАРБОКСИЛАТ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1993 |
|
RU2090567C1 |
| Способ получения производных пергидротиазепина или их кислотно-аддитивных фармацевтически приемлемых солей | 1985 |
|
SU1435151A3 |
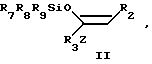
Использование: в качестве промежуточных продуктов в синтезе антибиотиков карбапенема. Сущность изобретения: продукт производные азетидиона ф-лы I, где R1 гидроксизащитная группа, R2 C1-C3 -алкил, R3 незамещенный или замещенный C1-C3 -алкилом пиридил, незамещенный хинолил, фенил, замещенный C1-C3 -алкилом или группой CYNR5-R6, где Y кислород или сера, а R5 и R6 одинаковые или различные и означают C1-C6 -алкил или фенил, или R5 и R6 вместе образуют пирролидинил, пиперидил, морфолинил или азепинил: R4 водород, Z атом серы или атом кислорода. Реагент 1: соединение ф-лы II, где R2 R3 и Z указано выше R7, R8 и R9 одинаковые или различные и означают C1-C4 -алкил или фенил. Реагент 2: соединение ф-лы III, где R1 и R4 указано выше, R10 ацилокси-, алкилсульфонил-, арилсульфонил, алкилсульфинил- или арилсульфинилгруппа. Структура ф-лы I, II, III: 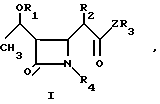
 2 с. и 2 з. п. ф-лы.
2 с. и 2 з. п. ф-лы.
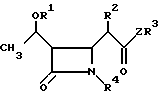
где R1 гидроксизащитная группа;
R2 C1 C3-алкильная группа;
R3 пиридильная группа, которая не замещена или замещена C1 C3-алкильной группой, незамещенная хилолиловая или фенильная группа, которая имеет заместитель общей формулы CYNR5R6, и/или C1 C3-алкильная группа, где Y кислород или сера, R5 и R6, одинаковые или различные, и каждый C1 C6-алкильная группа или фенильная группа, или R5 и R6 вместе образуют пирролидинил, пиперидил, морфолинил или азепинил;
R4 водород;
Z сера или кислород.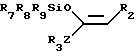
где R2,R3 и Z имеют указанные значения;
R7, R8 и R9, одинаковые или различные, и каждый - C1 C4-алкильная группа или фенильная группа,
подвергают взаимодействию с соединением общей формулы III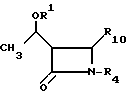
где R1 и R4 имеют указанные значения;
R10 ацилокси-, алкилсульфонильная, арилсульфонильная, алкилсульфинильная или арилсульфинильная группа.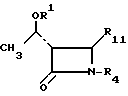
для получения соединения общей формулы Iа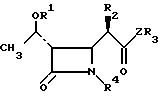
где R1, R2, R3, R4 и Z имеют указанные значения
Приоритет по признакам:
31.05.91 R1 R6 и Z имеют значения, указанные в п. 1 формуле изобретения;
R3 незамещенная или замещенная C1 C3 -алкилом пиридильная группа или незамещенная хинолильная группа.
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
