Изобретение относится к неизвестному до настоящего времени классу соединений, который проявляет как противовоспалительное и усиливающее иммунную систему действие, так и высокую активность в индуцировании дифференциации и ингибирования нежелательной пролиферации некоторых клеток, включая раковые клетки и клетки кожи, а также относится к фармацевтическим препаратам, содержащим эти соединения, к единичным дозировкам этих препаратов и к их использованию при лечении и профилактике увеличенной щитовидной железы, особенно при вторичном увеличении щитовидной железы, связанном с почечной недостаточностью; ряда болезненных состояний, включая сахарный диабет, повышенное кровяное давление, воспаление сальных желез, облысение, старение кожи, дисбаланс иммунной системы; воспалительных заболеваний, таких, как ревматоидный артрит или астма; заболеваний, характеризуемых аномальной дифференциацией клеток, таких, например, как псориаз и рак, для предотвращения и/или лечения атрофии кожи, вызванной стероидами, и для способствования остеогенезу и лечения остеопороза.
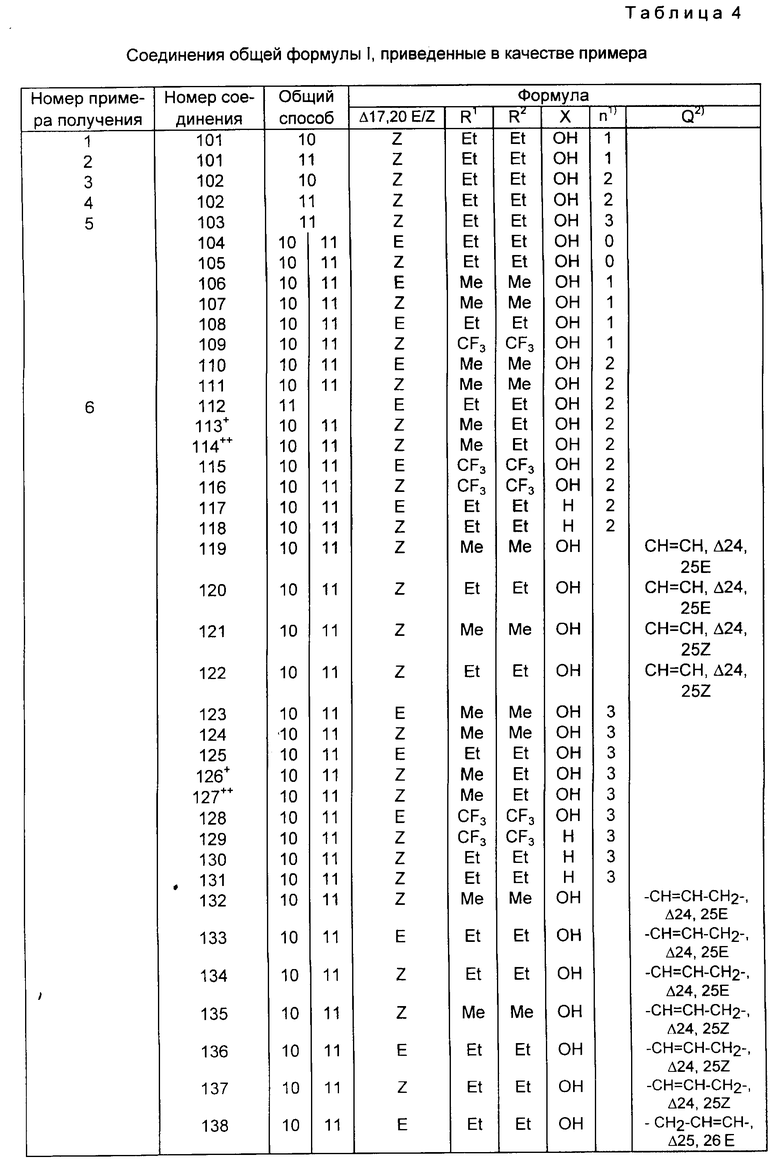
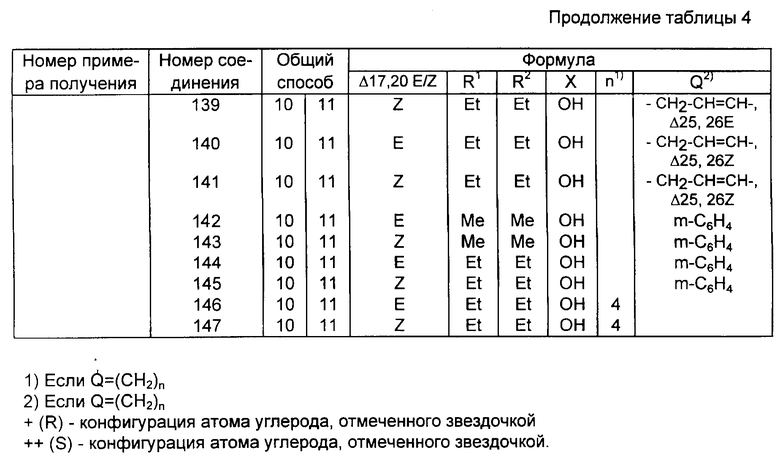
Соединения изобретения содержат новый класс аналогов витамина D общей формулы I: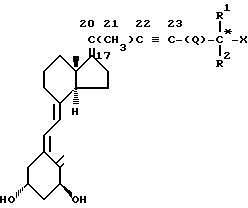 ,
,
в которой
X представляет собой кислород или гидрокси группу; R1 и R2, которые могут быть одинаковыми или различными, обозначают водород или C1-C6 -гидрокарбильный радикал; или R1 и R2, взятые вместе с атомом углерода (отмечен звездочкой в формуле I), несущих группу X, могут образовывать C3-C8 - карбоциклическое кольцо, Q представляет собой одинарную связь или C1-C8 - гидрокарбиленовый бирадикал, R1 и R2 и или Q необязательно могут быть замещены одним или более атомами дейтерия или фтора.
В контексте данного изобретения выражение гидрокарбиловый радикал (гидрокарбиленовый бирадикал) означает остаток после удаления 1(2) атома(ов) углерода из прямого, разветвленного или циклического углеводорода.
R1 и R2, взятые отдельно, могут включать (отдельно от водорода), но не ограничиваться ими, метил, трифторметил, этил, винил, пропил, изопропил, циклопропил и 1-метилвинил.
R1 и R2 вместе, могут быть ди-, три-, тетра- и пентаметилен.
Примеры Q включают одинарную связь, метилен, ди-, три- и тетраметилен -CH2-CH= CH-, -CH2-C=C-, -CH=CH-CH2-, -C=C-CH2-, фенил Ен (C6H4; орто, мета, пара), -CH2-(C6H4)-, и -(C6H4)-CH2-.
Конфигурация вокруг двойной связи 17, 20 может быть либо E-, либо Z-типа. Как можно увидеть из формулы I, в зависимости от значения R1, R2, Q и X, соединения изобретения могут включать несколько диастереомерных форм (например, R или S конфигурации отмеченного звездочкой атома углерода). Изобретение включает все эти диастереомеры в чистой форме, также как и в форме смеси диастереомеров.
В частности, включают оба диастереомера, имеющих две возможные конфигурации вокруг двойной связи 17, 20.
Кроме того, про-лекарства I, в которых одна или более гидроксигрупп представляют собой производные гидрокси группы, которые могут быть обратно преобразованы в гидроксигруппы in vivo, также находятся в объеме изобретения.
Соединения формулы I, в которых X является водородом также могут быть про-лекарством, поскольку эти соединения являются относительно неактивными in vitro, но преобразуются в активные соединения формулы I путем ферментативного гидроксилирования после приема пациентом.
Известно, что Iα,25-дигидроксивитамин D3 (1,25(OH)2D3) влияет на действие и/или образование интерлейкинов (Muller, K. et al., Immunol. Lett. 17, 361 - 366 (1988)), указывая на возможное использование этого соединения при лечении заболеваний, характеризуемых дисфункцией иммунной системы, например аутоиммунные заболевания, СПИД, общая реакция на прививки и отторжение трансплантатов или другие состояния, характеризуемые ненормальным образованием интерлейкина-1, например воспалительные заболевания, такие, как ревматоидный артрит и астма.
Известно также, что 1,25(OH)2D3 способно стимулировать дифференциацию клеток и ингибировать избыточную пролиферацию клеток (Abe, E. et al., Proc. Natl. Acad. Sci, USA 78, 4990-4994 (1981)) и сделано предложение, что это вещество может быть полезным при лечении заболеваний, характеризуемых ненормальной пролиферацией клеток и/или дифференциацией клеток, таких, как лейкемия, миелофиброз и псориаз.
Также, предлагают использовать 1,25(OH)2D3 или его про-лекарства Iα-OH-D3 для лечения повышенного кровяного давления (Lind. L. et al., Asta Med. Scand 222, 423-427 (1987) и сахарного диабета (Inomata, S. et al., Bone Mineral 1, 187-192 (1986)). Другое показание для 1,25 (OH)2D3 связано с недавним наблюдением взаимосвязи между наследственной устойчивостью к витамину D и облысением: лечение с помощью 1,25(OH)2D3 может способствовать росту волос (Editorial, Lancet, March 4, 1989, p.478). Также тот факт, что наружное нанесение 1,25(OH)2D3 вызывает уменьшение размера сальных желез в ушах самцов сирийского хомяка дает возможность предположить, что соединение может быть пригодным для использования при лечении воспаления сальных желез (Malloy, V. L. et al., the Tricontinental Muting for investigative Dermotology, Washington, 1989)
Однако терапевтические возможности 1,25(OH)2D3 при таких показаниях строго ограничены из-за хорошо известного сильного влияния этого гормона на метаболизм кальция: его повышенная концентрация в крови немедленно вызывает гиперкальцемию. Таким образом, это соединение и его сильнодействующие синтетические аналоги полностью не удовлетворяют требованиям, предъявляемым к лекарствам при лечении, например, псориаза, лейкемии или иммунных болезней, которые могут требовать непрерывного приема лекарства в относительно высоких дозах.
Известно несколько аналогов витамина D, которые проявляют некоторую степень селективности в отношении активности индукции дифференциации клеток /ингибирования пролиферации клеток, если сравнивать с влиянием на метаболизм кальция.
Таким образом, аналог витамина D3: кальципотриол, содержащий 22, 23 - двойную связь, 24 - гидроксигруппу и в котором атомы углерода 25, 26 и 27 являются включенными в трехчленное кольцо, является сильным индуктором дифференциации клеток и ингибитором пролиферации клеток, который проявляет только умеренную активность в метаболизме кальция in vivo (Binderup, L. and Bramm, E., Biochem. Pharmacol. 37, 889 - 895 (1988)).
Однако эта селективность не подтверждается при исследованиях in vitro, которые показывают, что кальципотриол связывается также хорошо, как и 1,25(OH)2D3 с кишечным рецептором витамина D. Возможно низкая активность кальципотриоле in vivo в кальциевом метаболизме, вызвана быстрым метаболизмом соединения, ограничивающим таким образом потенциал этого соединения для системного использования.
24-Гомо-1,25-дигидроксивитамин D3 и 26-гомо-1,25-дигидрокси-витамин D3 (вместе с их 22,23-дидегидроаналогами) (Ostrem, V Jamaka, Y, Prahl, J.; Deluca, H.F.; and Ikekawa, N.; Proc Natl. Acad. Sci. USA 84, 2610-14 (1987)), имеют ту же самую афинность при связывании, как и в 1,25(OH)2D3, как с кишечными рецепторами мышей и кур, так и с рецепторами линии клеток (HL-60) миелоидной лейкемии человека, и еще являются в 10 раз более сильнодействующим, чем 1,25(OH)2D3, при индуцировании дифференциации клеток HL-60 in vitro. In vivo эти соединения являются соответственно "значительно менее сильнодействующими" и "более сильнодействующими", чем 1,25(OH)2D3 в отношении метаболизма кальция.
26,27-Диметил- Iα, 25-дигидроксивитамин D3 синтезирован, однако информация, относящаяся к его биологической активности, противоречива (Sai, H.; Takatsuto; S; Hara, N; and Ikekawa, N.; Chem. Pharm. Bull. 33 878-881 (1985) and Ikekawa, N; Eguchi, T., Hara, N; Takatsuto, S; Honda, A; Mori, V.; and Otomo, S.; Chem. Pharm. Bull. 35, 4362-4365 (1987)).
Известно, что близкий к нему 26,27-диэтил -Iα,25-дигидроксивитамин D3, почти "не имеет активности витамина D" (т.е. влияния на метаболизм кальция), он в то же время является в 10 раз более сильнодействующим, чем 1,25(OH)2D3 при индуцировании дифференциации клеток.
Патент США 4804502 описывает соединения, содержащие тройную связь в боковой цепи витамина D, и эти соединения являются пригодными для использования при лечении болезненных состояний, характеризуемых недостаточностью метаболизма кальция.
Поскольку существуют только малые структурные отличия между известными из литературы соединениями это означает, что современное состояние знаний не позволяет предсказать структуру аналогов витамина D, которые проявят наиболее благоприятный уровень селективности, что отражается в более высокой активности при дифференциации клеток in vitro по сравнению с афинностью при связывании с кишечным рецептором витамина D in vitro. Причем афинность при связывании с рецептором in vitro не всегда подтверждается в исследованиях in vivo, вероятно отражая фармакокинетическое различие между соединениями.
Известно, что соединения, которые структурно отличаются от указанных аналогов витамина D конфигурацией метильной группы углерода-20, как имеющие сильное влияние на дифференциацию/пролиферацию клеток. Эта "неестественная" конфигурация, представленная в заявках PCT N 90 00156, WO N 91 00271, PCT N 91 00200, WO 92 03414, заявке на международный патент PCT 93 00105 заявке на патент Великобритании номер 92202720, заявке на патент Великобритании номер 92204395, заявке на патент Великобритании номер 9220614 и заявке на патент Великобритании номер 92268770, как неожиданно обнаружено, имеет глубокое и многообещающее биологическое значение.
Соединения данного изобретения отличаются от обоих эпимерных типов с C-20 ранее известных аналогов витамина D присутствием двойной связи в положении 17, 20, которая дает измененные стереохимические условия, и проявляют одно или более следующих преимуществ по сравнению с соединениями, известными из литературы:
(a) более сильное влияние на клеточную дифференциацию/пролиферацию;
(b) более высокую селективность в предпочтении сильного влияния на клеточную дифференциацию/пролиферацию по сравнению с влиянием на метаболизм кальция;
(c) более сильное влияние на образование и действие интерлейкинов;
(d) более высокая селективность в отношении влияния на образование и действие и интерлейкина по сравнению с влиянием на метаболизм кальция.
Следовательно, соединения согласно изобретению являются особенно пригодными как для локального, так и для системного лечения и профилактики расстройств человека и животных, которые характеризуются 1) ненормальной клеточной пролиферацией и/или клеточной дифференциацией, такие, как некоторые дерматологические расстройства, включая псориаз и определенные формы рака, 2) дисбалансом иммунной системы, например, при автоиммунных болезнях, включая сахарный диабет, общую реакцию на прививку и отторжение трансплантатов, и кроме того, при лечении воспалительных болезней, таких, как ревматоидный артрит и астма. Воспаление сальных желез, облысение и повышенное кровяное давление являются другими состояниями, которые могут лечиться соединениями изобретения. Наконец, поскольку после наружного употребления соединений изобретения наблюдается уплотнение кожи, эти соединения могут быть пригодными для использования при лечении или предотвращении старения кожи, включая старение под действием света.
Поскольку существует низкая тенденция соединений к образованию гиперкальцемии при непрерывном употреблении, ожидается, что они будут иметь ценность при долговременном лечении увеличенной щитовидной железы (особенно, вторичного увеличения щитовидной железы, связанного с почечной недостаточностью), и для способствования остеогенезу, и при лечении остеопороза. Для этих показаний описываемые здесь соединения имеют более высокое терапевтическое отношение, чем соединения, известные по литературе (см. патент США N 4948 789 и EP 0385 446 A2).
Данные соединения могут быть использованы с другими медикаментами. При предотвращении отторжения прививки или общей реакции на прививку, лечение с помощью данных соединений может проводиться совместно с лечением, например, с помощью циклоопорина.
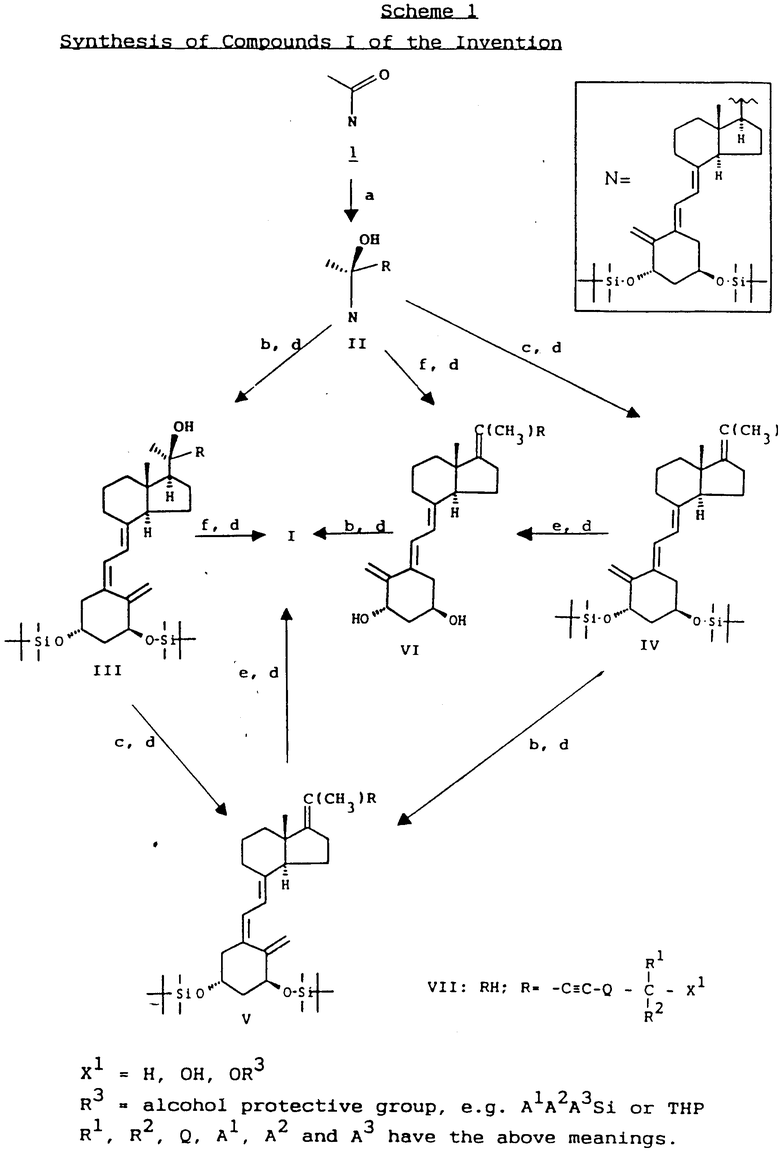
Соединения I могут быть получены из производного витамина D кетонового соединения I (Схема I), синтез которого известен [Hansen K., Calverley M.J. and Binderup L.: Syntesis and Biological Activity of 22-Oxa Vitamin D analogues. In: Vitamin D, Proc. Eighth Werkshop on Vitamin D, Paris, July 5-10, 1991, p. 161; Walter de Gruyter, Berlin 1991], например, путем, представленным на схеме I, приведенной в конце описания.
В описании используются следующие стандартные сокращения Me = метил; Et = этил; Bu = n-бутил; THF = тетрагидро-4H-пиран-2-ил; TMS = триметилсилил; DMAP = 4-диметиламинопиридин; пет.эфир = петролейный эфир; THF - тетрагидрофуран; TBAF = тетра(n-бутил)-аммоний фторил; т.к. = температура кипения; ПТСХ = препаративная тонкослойная хроматография; Tf = трифторметансульфонил; DMF = N, N - диметилформамид; "HF" = 5% фторид водорода в ацетонитрил:вода (7: 1); TBDMS - трет-бутилдиметилсилил; HCl = соляная кислота; "NaHCO3" = насыщенный водный раствор бикарбоната натрия; A1A2A3SiZ: силилирующий агент, где A1, A2 и A3, которые могут быть одинаковыми или различными, представляют собой C1-C6-гидрокарбонил, C1-C6-гидрокарболокси или арил, и представляет собой хорошо полимеризующуюся группу, такую, как -Cl, -Br или OTf (трифторметансульфонат или трифлат); NOE = ядерное усиление Оверхаузера; PPTS = пиридиний-p-толуолсульфонат.
Примечания к схеме I.
a) (i) Соединение I взаимодействует с анионом R-, производимым от блока, образующего боковую цепь, RH, общей формулы VII, с соответствующим основанием.
(ii) Основной продукт, (предполагаемое) R-соединение из двух возможных C-20 эпимеров изолируют с помощью хроматографии.
b) Изомеризация соединений II, IV или VI в соответствующее соединение III, V или I посредством ультрафиолетового света в присутствии триплетного сенсибилизатора, например антрацена.
(c) (i) Дегидрирование соединений II или III для соответствующих соединений IV или V производят путем обработки безводной кислотой при соответствующих условиях, например с фосфорной кислотой (0,2 М, в ацетонитриле при 50oC в течение 1-2 ч). Кислотно-чувствительные блокирующие группы, такие, как THP, могут быть удалены на этой стадии.
(ii) Если формируют оба 17, 20-еновые изомера, они могут быть при этом разделены или альтернативно это может быть проделано на более поздней стадии. Разделение и очистку предпочтительно осуществляют с помощью хроматографии.
d) Необязательное модифицирование функциональной группы в боковой цепи.
e) Деблокировка соединения IV или V в соединении VI или I соответственно например, посредством "HF" или TBAF.
f) Дегидратирование, сопровождающееся деблокировкой, соединений II или III в соединение VI или I соответственно например, посредством "HF".
Блоки, образующие боковую цепь, RH, общей формулы VII либо являются известными соединениями, либо могут быть получены с помощью стандартных способов, известных специалистам. В особенности это относится к блокам, образующим боковую цепь, необходимым для получения используемых в качестве примеров соединений (101-147). В тех случаях, когда не дается специальных указаний действия в соответствии со схемой I могут производиться аналогично конкретным процессам и примерам, рассматриваемым впоследствии.
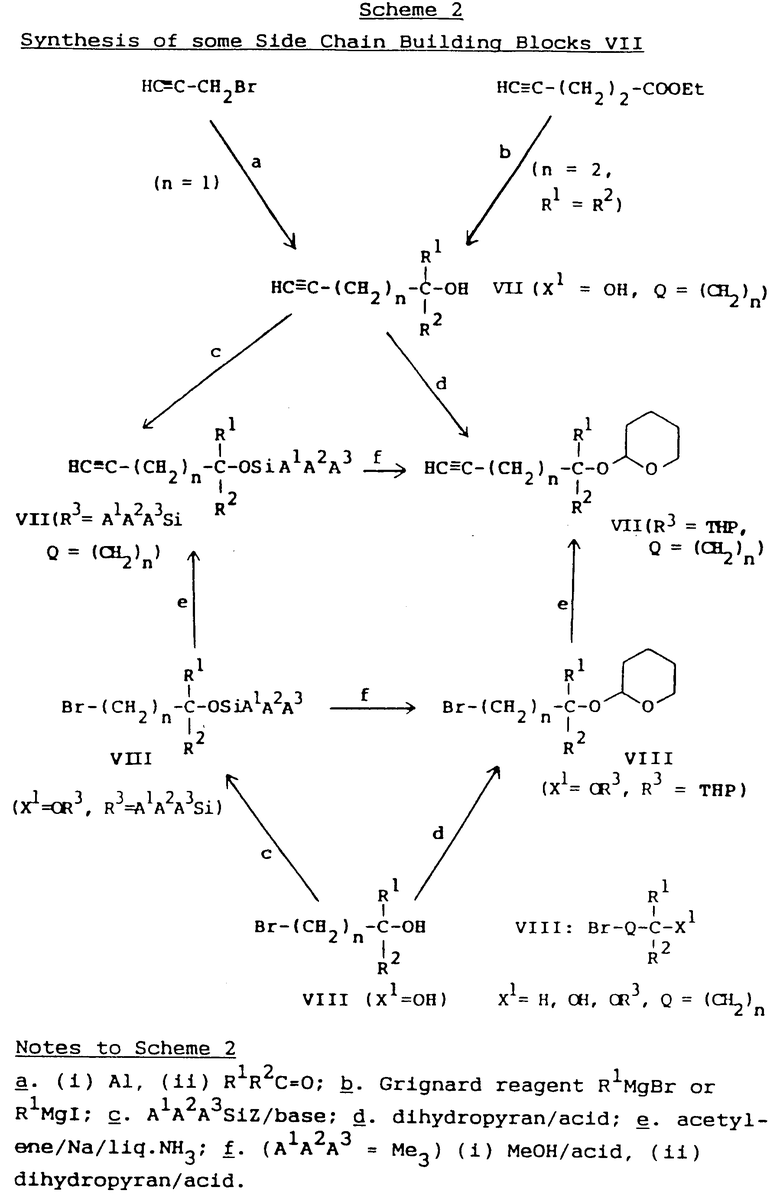
В качестве неограничивающей иллюстрации, получение некоторых соединений общей формулы VII, где Q = (CH2)n(n=0-3), X1=OR3 и R3= - Si A1A2A3 или THF, представлено в общих чертах на схеме 2, но подобные соединения формулы VII с другими Q и/или X могут быть получены с помощью аналогичных способов. Некоторые конкретные блоки, образующие боковые цепи RH, представлены в табл. 1 и способы их синтеза описываются в примерах получения (схема 2), приведенная в конце текста описания).
Некоторые блоки, образующие боковую цепь, RH, общей формулы VII (O= (CH2)n, n=0 - 3, R1=R2=CH3 или C2H5; R3=TMS, TBDMS или THP
Промежуточные соединения для получения блоков, образующих боковую цепь, RH, в табл. 1 являются либо известными соединениями, либо могут, например, быть получены из соединений, перечисленных в табл. 2. Процессы синтеза этих соединений описаны в примерах получения.
Реакция кетона, соединения I с блоками, образующими боковую цепь RH=H-C= C-O-C(R1) (R2)X1, может быть проведена с помощью стандартных способов нуклеофильного добавления ацетиленовых анионов (R-) к карбонильным соединениям, т. е. путем обработки RH соответствующим основанием, таким, как n-Buli, в соответствующем неводном растворителе, таком, как THF, затем добавляя соединение I, получая соединение II после обычного водного обогащения (которое обычно производят во всех реакциях по схемам I или II).
Обычно продукт реакции II является смесью двух возможных эпимеров C-20. Обычно является предпочтительным разделение этих эпимеров, которое удобно может быть проделано с помощью хроматографии.
Один из эпимеров образуется с гораздо более высоким выходом, чем второй и является менее полярным хроматографически. Этот основной эпимер по аналогии с подобными реакциями, как предполагается, является формой 20-R, и это тот эпимер, который используют в реакциях, представленных на схеме I, приводящим к образованию соединений I изобретения. Табл. 3 содержит неограничивающие иллюстрации таких соединений формулы II и далее другие такие промежуточные соединения типов III, IV и V на схеме I также перечислены в табл. 3.
Дегидрирование соединения II или III, в которых OH группа C-20 устраняется вместе с C-17, предпочтительно производят путем умеренно кислотного удаления, которое удобно может быть достигнуто путем обработки безводной фосфорной кислотой в ацетонитриле, как описано в общем способе 9.
Альтернативно возможно проводить дегидратацию 17, 20- соединения II или III одновременно с удалением достаточно лабильных блокирующих групп путем обработки "HF", как описано в общем способе 10.
Оба процесса иллюстрированы на схеме I.
Путем дегидратации, ведущей к образованию соединений, содержащих двойную связь 17, 20, является возможным получение двух различных изомеров: E- и Z-форм. Как правило, одна из форм образуется в качестве доминирующей, и она после очистки используется в качестве промежуточного вещества, ведущего в конце концов к образованию соединения I. Это и есть случай соединений 13, 15 и 17. Иногда, как в случае изомерных соединений 13 и 14, является возможным изолировать также и другой меньший 17, 20-еновый изомер. Путем сравнения химического сдвига ЯМР 1H 21-метиловых групп соединения 13 ( δ 1, 72) и соединения 14 ( δ 1,84) с величинами, описанными в литературе для стероидов с подобной 17, 20-еновой, 22, 23-иновой структурой (Chaudhuri, N.K., et al., JACK 87, 3737, (1965)) можно увидеть, что соединение 13 является Z-формой, а соединение 14 является E-формой, так что разумно предположить, что другие (главные) изомеры соединения 15 и 17, являются также Z-формами. Далее соединение 14 проявляет NOE-эффект между H18 и H21, а также между H18 и экваториально H12, как можно ожидать для E-конфигурации, и соединение 13 проявляет NOE между H16 и H21, в согласии с Z-формой. Хотя и несколько эмпирически, следующие соединения перечисляются в качестве Z-формы: соединения 13, 15, 16, 17, 101, 103 и 103. В качестве Е-формы перечисляются соединения 14, 22 и 112.
Приведенные в качестве примеров соединения I изобретения перечислены в табл. 4, перечисленные примеры содержат упоминание иллюстрированных способов синтеза вместе со спектроскопическими данными для тех же самых соединений примеров.
Необходимо отметить, что примеры получения и примеры схем 1 и 2 являются только иллюстративными, поскольку конкретный синтез каждой стадии и порядок, в котором производится каждая стадия может сильно изменяться. Далее радикал R: -C ≡ C-Q-C(R1χR2)(X1) может быть таким, как описано, или быть радикалом, который может быть преобразован в указанный радикал на любой удобной поздней стадии (или на нескольких стадиях). Таким образом, R в соединениях II, III. IV и I не должен обязательно иметь одно и то же значение, на протяжении конкретной последовательности синтеза. Преобразование R в -C ≡ C -Q-C(R1)(R2)(X1) вполне может включать несколько стадий и возможно включать временную блокировку чувствительной триеновой системы молекулы. Не считая любой необходимой модификации в боковой цепи (R), превращение I в II включает стадию фотоизомеризации и стадию деблокирования (если до этого не произведена одновременно со стадией 17, 20-дегидратации, как рассмотрено выше), аналогично стадиям, использованным на последних ступенях синтеза других аналогов витамина D (см. EP N 0227836).
Данные соединения предполагается использовать в фармацевтических композициях, которые являются пригодными для использования при лечении расстройств у людей и животных, как описано выше.
Требуемое количество соединения формулы I (далее упоминаемого как активный ингредиент) для терапевтического воздействия, является, конечно, функцией как конкретного соединения, так и способа приема и млекопитающего, подвергающегося лечению. Соединения изобретения могут приниматься путем парентерального, внутрисуставного, энтерального введения или наружного употребления. Они хорошо поглощаются при энтеральном введении, что является предпочтительным способом употребления при лечении системных расстройств. При лечении дерматологических расстройств, подобных псориазу или глазных болезней, наружная или энтеральная формы являются предпочтительными.
При лечении респираторных заболеваний, подобных астме, предпочтительным является аэрозоль.
Активный ингредиент может быть введен сам по себе, но предпочтительно его использовать в виде фармацевтического препарата. Как правило, активный ингредиент составляет от 0,1 млн.ч. до 0,1% по массе от всего препарата.
Под термином "единичная дозировка" подразумевается унитарная, т.е. единичная доза, которую можно назначать пациенту, и с которой просто обращаться, которая может быть легко упакована, оставаясь в качестве физически и химически стабильной единичной дозы, включая либо активный материал как таковой, либо смесь его с твердыми или жидкими фармацевтическими разбавителями и носителями.
Препараты, используемые как для лечения человека, так и животных, данного изобретения, содержат активный ингредиент в соединении с фармацевтически приемлемым носителем, следовательно, и необязательно другой(ие) терапевтический(е) ингредиент(ы). Носитель(и) должен(ны) быть "приемлемым(и)", т.е. совместимым(и) с другими ингредиентами препаратов и не быть вредным(и) для того, кто его(их) принимает.
Препараты включают, например, последние в форме, пригодной для орального, ректального, парентерального (включая подкожное, внутримышечное и внутривенное), внутрисуставное введение и наружное употребление.
Препараты удобно могут быть представлены в форме единичной дозировки и могут быть приготовлены с помощью любого из способов, хорошо известных из литературы по фармацеи. Все способы включают стадию приведения активного ингредиента в соприкосновение с носителем, который содержит один или более дополнительных ингредиентов. Как правило, препараты готовят путем одновременного и тесного приведения активного ингредиента в соприкосновение с жидким носителем или тонко измельченным твердым носителем, или с обоими, и затем, если это необходимо, формированием продукта в желаемый препарат.
Препараты данного изобретения, пригодные для орального приема, могут быть в форме отдельных единиц, в виде капсул, саше, таблеток или лепешек, каждая из которых содержит заданное количество активного ингредиента; в форме порошка или гранул; в форме раствора или суспензии в водной жидкости или в неводной жидкости; или в форме эмульсии "масло в воде", или эмульсии "вода в масле". Активированный ингредиент может также назначаться в форме шариков, кашки или пасты.
Таблетка может быть получена путем прессования или формования активного ингредиента, необязательно с одним или более вспомогательными ингредиентами. Прессованные таблетки могут быть получены путем прессования в соответствующей машине активного ингредиента в свободно поступающей форме, такой, как порошок или гранулы, необязательно смешанные со связующим, смазывающим веществом инертным разбавителем, поверхностно-активным или диспергирующим веществом. Формованные таблетки могут быть получены путем формования в соответствующей машине смеси порошкообразного активного ингредиента и соответствующего носителя, смоченного инертным жидким разбавителем.
Препараты для ректального введения могут быть в форме суппозитариев, включающих активный ингредиент и носитель, такой, как масло какао, или в форме клизмы.
Препараты, пригодные для парентерального введения, обычно включают стерильный водный или масляный препарат активного ингредиента, который предпочтительно является изотоническим с кровью пациента.
Препараты, пригодные для внутрисуставного введения, могут быть в форме стерильного водного препарата активного ингредиента, который может быть в микрокристаллической форме, например в форме водной суспензии микрокристаллов. Липосомальные препараты или бидеградируемые полимерные системы могут также быть использованы для представления активного ингредиента как для внутрисуставного, так и для офтальмологического назначения.
Препараты, пригодные для наружного употребления, включая лечение глаз, включают жидкие или полужидкие препараты, такие, как линименты, примочки, гели, аппликации, эмульсии "масло в воде" или "вода в масле", такие, как кремы, примочки или пасты, или растворы или суспензии, такие, как капли.
При лечении астмы может быть использована ингаляция порошка, самораспыляющихся или распыляющихся под давлением препаратов, распределяемых в аэрозольном баллончике, распылителях различных видов. Препараты после диспергирования, предпочтительно, имеют размеры частиц в пределах 10 - 100 мкм.
Такие препараты являются наиболее предпочтительными в форме тонко измельченного порошка для пульмонарного введения из приспособления для порошковых ингаляций или самораспыляющихся порошковых препаратов. В случае самораспыляющегося раствора или препарата в аэрозольном баллончике эффект может быть достигнут либо подбором клапана, имеющего желаемые характеристики распыления (т.е. являющегося пригодным для получения капель желаемого размера), либо путем включения активного ингредиента в виде суспендированного порошка с контролируемым размером частиц. Эти самораспыляющиеся препараты могут быть либо препаратами с распаляющимся порошком, либо препаратом, распространяющим активный ингредиент в виде капель раствора или суспензии.
Самораспыляющиеся, распространяющие порошок препараты предпочтительно содержат дисперсные частицы твердого активного ингредиента и жидкий пропеллент, имеющий температуру кипения ниже 18oC при атмосферном давлении. Жидкий пропеллент может быть любым пропеллентом, о котором известно, что он пригоден для медицинского применения и может включать один или более C1-C6-алкильных углеводородов или галогенированных C1-C6-алкильных углеводородов, или их смеси; хлорированные и фторированные C1-C6-алкильные углеводороды являются особенно предпочтительными. В целом пропеллент составляет от 45 до 99,9 мас.% препарата, в то время как активный ингредиент составляет от 0,1 млн.ч. до 0,1 мас.% препарата.
В дополнение к рассмотренным ингредиентам препараты данного изобретения могут включать один или более дополнительных ингредиентов, таких, как разбавители, буферы, ароматизаторы, связующие, поверхностно-активные вещества, сгущающие средства, смазывающие вещества, консерванты, например метилгидроксибензоат (включая антиоксиданты), эмульгирующие агенты и им подобные.
Композиции могут содержать другие терапевтически активные соединения, обычно применяемые при лечении рассмотренных патологических состояний.
Данное изобретение касается способа лечения пациентов, страдающих от одного из указанных патологических состояний, и который включает введение пациенту, нуждающемуся в лечении, эффективного количества одного или более компонентов формулы I, как таковое или в сочетании с одним или более терапевтическими активными соединениями, обычно применяемыми при лечении указанных патологических состояний. Лечение с помощью данных соединений и/или других терапевтически активными соединениями может проводиться одновременно или с интервалами.
При лечении системных расстройств назначаются дневные доза соединения формулы I 0,1 - 100 мкг, предпочтительно 0,2 - 25 мкг. При наружном лечении дерматологических расстройств назначаются мази, кремы или примочки, содержащие 0,1 - 50 мкг/г, предпочтительно 0,1 - 100 мкг/г соединения формулы I. Для наружного употребления в офтальмологических мазях, каплях или гелях назначают 0,1 - 500 мкг/г, предпочтительно 0,1 - 100 мкг/г соединения формулы I. Композиции для орального приема готовятся предпочтительно в виде таблеток, капсул или капель, содержащих 0,05 - 50 мкг, предпочтительно 0,1 - 25 мкг соединения формулы I в единичной дозировке.
Изобретение описывается в последующих неограничивающих общих способах, примерах получения и примерах.
Представленные в качестве примеров соединения I перечислены в табл. 4.
Для спектров ядерного магнитного резонанса (300 Мгц) значения химического сдвига (δ) выражены в единицах сдвига для дейтерий-хлороформных растворов относительно внутреннего тетраметилсилана (δ = 0) или хлороформа (δ = 7,25). Значение для мультиплета либо определено (дублет (d), триплет (t), квартет (q)), либо нет - (m) дано при примерно средней точке, за исключением случаев, когда оценены пределы изменения (S=синглет, b=уширенная линия). Константы связи (J) даны в Гц и иногда округлены до ближайшей единицы.
Эфир является диэтиловым эфиром и его высушивают над натрием. ТГР высушивают над натрий бензофенном. Петролейный эфир относится к пентановой фракции. Реакции проходят при комнатной температуре, если не указано иного. Процедура обогащения включает разбавление специальным растворителем (иным, чем органический реакционный раствор), экстракцию водой, а затем рассолом и концентрирование в вакууме для получения остатка. Хроматографию проводят на силикагеле.
Общий способ 1. Взаимодействие кетонов R1R2C=O с металлоорганическим реагентом, полученным из пропаргил бромида и алюминия, для получения соответствующего третичного спирта VII (схема 2, табл. 2, пример получения 1).
Смесь алюминиевых чешуек (3,6 г), хлорида ртути (0,1 г) и безводного ТГФ (20 мл) перемешивают при 20oC в течение 20 мин в атмосфере аргона. Раствор пропаргил бромида (23,8 г) в безводном ТГФ (20 мл) добавляют при перемешивании в течение 40 мин, поддерживая температуру 25 - 30oC путем перемежающегося охлаждения. Реакционную смесь перемешивают при 40 - 45oC, при необходимости подогревая в течение 30 мин. После охлаждения примерно до 25oC добавляют в течение 1 ч при перемешивании раствор соответствующего кетона R1R2C=O (0,2 моль) в безводном эфире (25 мл), охлаждая слегка, чтобы поддерживать температуру около 25oC. Перемешивание продолжают в течение следующего получаса при 30 - 35oC, после чего реакционную смесь обогащают (эфир). Остаток очищают путем дистилляции in vacuo через колонку Rodbielniak длиной 50 см, получая указанное в заглавии соединение в виде масла.
Общий способ 2. Блокировка третичных спиртов VII или VIII для получения соответствующих 2-тетрагидропропираниловых соединений VII или VIII (схема 2, табл. 1, примеры получения 2, 17 и 19).
Смесь соответствующего соединения VII или VIII (0,01 моль), 3,4-дигидро-2H-пирана (1,26 г), PPTS (0,25 г) и безводного дихлорметана (25 мл) перемешивают в атмосфере аргона в течение 4 ч при 20oC. К реакционной смеси добавляют 100 мл эфира и 50 мл полунасыщенного водного раствора хлорида натрия. Органическую фазу отделяют, сушат и выпаривают in vacuo, получая исходный продукт, который очищают с помощью хроматографии (смесь эфира и пет. эфира в качестве элюента), получая указанное в заглавии примера соединение.
Общий способ 3. Реакция этилового эфира 4-пентиновой кислоты с реагентами Гриньяра R1MgX2 для получения соответствующего третичного спирта VII (схема 2, табл. 2, примеры получения 3 и 18). (X2 = Cl, Br, I)
К 1,1 г магниевой стружки (качество по Гриньяру) в сухой колбе добавляют по каплям при перемешивании раствор соответствующего алкил галогенида R1X2 (0,045 мл) в безводном эфире (20 мл). Реакция проходит в атмосфере аргона при перемешивании и нагревании с обратным холодильником и длится 20 мин. Перемешивание и нагревание с обратным холодильником продолжают в течение следующих 10 мин.
Этот реагент Гриньяра переносят в дополнительную воронку в атмосфере аргона и добавляют по каплям при перемешивании и охлаждении примерно до -20oC и к раствору этилового эфира 4-пентиновой кислоты (1,9 г) в безводном эфире (20 мл). Эквимолярное количество соответствующего другого низкомолекулярного алкилового эфира, например метилового или пропилового эфира, может быть использовано вместо этилового эфира.
Добавление продолжается 15 мин и после этого перемешивание продолжают в течение 20 мин при -20oC и в течение 1 ч при 30oC.
Реакционную смесь выливают в 100 г смеси лед/вода и 4н. соляной кислоты (15 мл) при перемешивании. После добавления водного раствора бикарбоната натрия для установления pH около 5, смесь экстрагируют дважды эфиром (25 мл каждый раз). Объединенные органические фазы промывают водой и насыщенным водным раствором хлорида натрия, высушивают in vacuo, получая исходный продукт. Последний очищают либо путем дистилляции in vacuo, либо с помощью хроматографии (смесь эфира и петролитного эфира в качестве элюента), получая указанное в заглавии примера получения соединение.
Общий способ 4. Блокировка третичных спиртов VII или VIII для получения соответствующего A1A2A3 силилового соединения VII или VIII (схема 2, табл. 1, примеры получения 4 и 16).
К раствору соответствующего соединения VII или VIII (14 ммоль) в соответствующем безводном растворителе, например в дихлорметане или DMF, добавляют одно или более соответствующих оснований, например триэтиламин, DMAP или имидазол, в атмосфере аргона и при перемешивании, и охлаждении на ледяной бане. Соответствующий силилирующий агент A1A2A3 SiZ, например TMSCl, TBDMSOTf, триэтилсилилтрифталат или дифенилметилсилил хлорид, добавляют по каплям при перемешивании в течение 20 мин при 0oC. Перемешивание продолжают в течение достаточного времени (как правило, в течение 0,5 - 24 ч) при соответствующей температуре (как правило при 25 - 50oC). После соответственного обогащения исходный продукт очищают с помощью хроматографии, получая указанное в заглавии примера получения соединение.
Общий способ 5. Преобразование TMS-блокированных спиртов типа VII или VIII в соответствующее THP-блокированное соединение типа VII или VIII (схема 2, табл. 2, пример получения 5).
К раствору соответствующего TMS-блокированного третичного спирта VII или VIII (0,02 моль) в метаноле (25 мл) добавляют 5 капель 6 М хлористого водорода в метаноле и смесь перемешивают в течение 15 мин при 20oC. Реакционную смесь выпаривают до тех пор, пока не будет удален метанол и остаток растворяют вновь в дихлорметане (40 мл). К этому раствору добавляют 3,4-дигидро-2-H-пиран (3,3 г) и PPTS (0,16 г) по частям при перемешивании и охлаждении на ледяной бане. После чего смесь перемешивают при 20oC в течение 3 ч и затем разбавляют эфиром (200 мл). Эфирную фазу экстрагируют насыщенным водным раствором бикарбоната натрия, водой и насыщенным раствором хлорида натрия, высушивают и выпаривают in vacuo, получая исходный продукт. Последний очищают с помощью хроматографии (смесь эфира и пет. эфира в качестве элюента), получая указанное в заглавии примера получения соединение в виде масла.
Общий способ 6. Преобразование соединений VIII с концевым атомом брома в соответствующее соединение VII с концевой этиловой группой (схема 2, табл. 1, пример получения 6).
Через безводный жидкий аммиак (около 75 мл) пропускают безводный ацетилен с объемной скоростью примерно 200 мл в минуту при перемешивании. В это же время добавляют натрий (0,5 г) малыми порциями в течение 5 мин. Примерно через 5 мин поток ацетилена прерывают и добавляют соответствующее бромистое соединение VIII (3 ммоль) в течение 5 мин, перемешивание при комнатной температуре продолжают до тех пор, пока не испарится весь аммиак (2 - 4 ч). Петр. эфир (100 мл) и смесь лед/вода (100 г) добавляют при перемешивании. Органическую фазу отделяют, промывают несколько раз водой до тех пор, пока она не станет нейтральной, высушивают и выпаривают in vacuo, получая исходный продукт. Последний очищают с помощью хроматографии (дихлорметан или смесь дихлорметана и петр. эфира в качестве элюентов), получая указанное в заглавии примера получения соединение.
Общий способ 7. Реакция соединения I с блоками VII (RH), образующими боковую цепь, для получения соединения II (схема 1, табл. 3, примеры получения 7 - 9).
К раствору соответствующего соединения VII (1,5 ммоль) в безводном ТГФ (5 мл), охлажденному до -70oC и перемешиваемому в атмосфере аргона, добавляют по каплям раствор n-бутиллития (1,6 мм в гексане; 0,65 мл). Перемешивание продолжают при -70oC в течение 10 мин и затем при 20oC в течение 1 ч. Смесь повторно охлаждают до -70oC и добавляют по каплям раствор кетона, соединение I (0,28 г; 0,5 моль) в безводном ТГФ (5 мл) в течение 4 мин, после чего продолжают перемешивание при -70oC в течение 30 мин. Реакционную смесь обогащают (эфир), получая исходный продукт, который очищают посредством хроматографии (смесь эфира и петр. эфира в качестве элюента), получая указанное в заглавии примера получения соединение.
Общий способ 8. Изомеризация соединений II, IV или VI в соответствующее соединение III, V или I (схема 1, табл. 3, примеры получения 10, 11 и 14).
Раствор соответствующего соединения II, IV или VI (0,3 ммоль), антрацена (100 мг) и триэтиламина (0,05 мл) в дихлорметане (20 мл) в атмосфере аргона в колбе из стекла Pirex облучают ультрафиолетовым светом от ультрафиолетовой лампы высокого давления типа TQ 760Z2 (Hanau) примерно при 10oC в течение 20 мин при перемешивании. Реакционную смесь концентрируют в вакууме и обрабатывают петр. эфиром (2•5 мл). После фильтрования фильтрат концентрируют в вакууме и очищают с помощью хроматографии (смесь эфира и петр. эфира в качестве элюента), получая указанное в заглавии примера соединение.
Общий способ 9. Дегидратирование третичных спиртов II или III с помощью фосфорной кислоты в ацетонитриле для получения соответствующих 17,20-еновых соединений IV или V (схема 1, табл. 3, примеры получения 12 - 13 и 15).
К раствору соответствующего соединения II или III (1 ммоль) в смеси 1:1 ацетонитрила и этилацетата (25 мл) добавляют 0,2 М раствор безводной фосфорной кислоты в ацетонитриле (10 мл) в атмосфере аргона при перемешивании при 50oC. Перемешивание продолжают в течение 1 ч при 50oC. Реакционную смесь обогащают (этилацетат с дополнительной экстракцией NaHCO3). Остаток очищают в соответствующем элюенте, если это необходимо, то с помощью повторной хроматографии, обычно в установке Waters Prep - 500R или с помощью ПТСХ).
Общий способ 10. Деблокирование соединений IV или V или альтернативно дегидратирование, сопровождающееся деблокировкой, соединений II или III в соответствующее соединение VI или I, с помощью обработки "HF" (схема 1, табл. 4, примеры 1 и 3).
К раствору соответствующего соединения II, III, IV или V (0,07 ммоль) в этилацетате (0,2 мл) добавляют ацетонитрил (2 мл) с последующим добавлением 5%-ного раствора плавиковой кислоты в смеси ацетонитрил-вода 7:1 (1,2 мл) в атмосфере аргона при перемешивании. Перемешивание продолжают в течение 10 - 60 мин при 20 - 60oC. Добавляют насыщенный водный раствор бикарбоната натрия (10 мл) и обогащают реакционную смесь (этилацетат). Остаток очищают с помощью хроматографии (этилацетат или смесь этилацетата и гексана или пентана в качестве элюента), получая указанное в заголовке примера соединение.
Общий способ 11. Деблокирование соединений IV или V в соответствующее соединение VI или I путем обработки TBAF (схема I, табл. 4, примеры 2, 4, 5 и 6).
К раствору соответствующего соединения IV или V (0,07 ммоль) в THF (6 мл) добавляют TBAF (120 мг), растворенный в ТГФ (4 мл) в атмосфере аргона и при перемешивании. Перемешивание продолжают в течение 1-30 ч при 60-150oC (в закрытом сосуде, рассчитанном на высокие давления, если требуется). Добавляют насыщенный водный раствор бикарбоната натрия (10 мл) и обогащают реакционную смесь (этилацетат). Остаток очищают с помощью хроматографии (этилацетат или смесь этилацетата и гексана или пентана в качестве элюента), получая указанное в заглавии примера соединение.
Пример получения 1: соединение 2.
Способ: общий способ 1.
Исходный материал: диэтилкетон.
Хроматографический элюент: 10% эфира в петр.эфире.
Т.кип.соединения 2: 71-72oC, 30 мбар.
ЯМР: δ = 0,90 (t, 6H), 1,60 (m, 4H), 1,75 (s, 1H), 2,05 (t, 1H), 2,35 (m, 2H).
Пример получения 2: соединение 3
Способ: общий способ 2.
Исходный материал VII: соединение 2.
Хроматографический элюент: от 0 до 5% эфира в петр.эфире.
ЯМР: δ = 0,90 (m, 6H), 1,45-1,92 (m, 10H), 1,96 (t, 1H), 2,46 (d, 2H), 3,47 (m, 1H), 3,98 (m, 1H), 4,81 (m, 1H).
Пример получения 3: соединение 4.
Способ: общий способ 3.
Исходный материал: этилмагний бромид.
Хроматографический элюент: 25% эфира в петр.эфире.
ЯМР: δ = 0,87 (t, 6H), 1,48 (m, 4H), 1,71 (m, 2H), 1,97 (t, 2H), 2,26 (m, 2H).
Пример получения 4: соединение 5.
Способ: общий способ 4.
Исходный материал VII: соединение 4.
Растворитель: дихлорметан (30 мл).
Основание: 2,6-лютидин (6,8 мл).
Силилирующий агент: TBDMSOTf (9,6 мл).
Температура реакции: 25oC
Время реакции: 0,5 ч.
Обогащение: эфир, с дополнительными экстракциями 1н. HCl с последующей обработкой "NaHCO3".
Хроматографический элюент: от 0 до 10% эфира в петр.эфире.
ЯМР: δ = 0,07 (s, 6H), 0,83 (t, 6H), 0,87 (s, 9H), 1,46 (m, 4H), 1,70 (m, 2H), 1,91 (t, 1H), 2,19 (m, 2H).
Пример получения 5: соединение 6.
Способ: общий способ 5.
Исходный материал VIII: 1-бром-4-этил-4-триметилсилилоксигексан.
Хроматографический элюент: 10% эфира в петр.эфире
ЯМР: δ = 0,83 (m, 6H), 1,45-2,05 (m, 14H), 3,43 (t, 2H), 3,45 (m, 1H), 3,94 (m, 1H), 4,68 (m, 1H).
Пример получения 6: соединение 7.
Способ: общий способ 6.
Исходный материал: VIII: соединение 6.
Хроматографический элюент: дихлорметан.
ЯМР: δ = 0,83 (t, 6H), 1,54 (q, 4H), 1,45-1,90 (m, 10H), 1,95 (t, 1H), 2,17 (m, 2H), 3,44 (m, 1H), 3,95 (m, 1H), 4,69 (m, 1H).
Пример получения 7: соединение 8.
Способ: общий способ 7.
Исходный материал VII: соединение 3.
Хроматографический элюент: от 15 до 25% эфира в петр.эфире.
ЯМР: δ 0,05 (m, 12H), 0,81 (s, 3H), 0,86 (s, 9H), 0,89 (s, 9H), 0,83-0,90 (m, 6H), 1,46 (bs, 3H), 1,27-2,07 (m, 23H), 2,15 (bd, 1H), 2,31 (bd, 1H), 2,45 (bs, 2H), 2,55 (dd, 1H), 2,86 (m, 1H), 3,44 (m, 1H), 3,95 (m, 1H), 4,21 (m, 1H), 4,53 (m, 1H), 4,79 (m, 1H), 4,93 (m, 1H), 4,98 (m, H), 5,80 (d, 1H), 6,45 (d, 1H).
Пример получения 8: соединение 9.
Способ: общий способ 7.
Исходный материал VII: соединение 5
Хроматографический элюент: 5% эфира в петр.эфире.
ЯМР: δ = 0,05 (s, 9H), 0,06 (s, 9H), 0,80 (t, 6H), 0,81 (s, 3H), 0,85 (s, 9H), 0,86 (s, 9H), 0,89 (s, 9H), 1,46 (s, 3H), 1,25-2,10 (m, 19H), 2,10-2,25 (m, 3H), 2,33 (bd, 1H), 2,53 (dd, 1H), 2,86 (m, 1H), 4,22 (m, 1H), 4,52 (m, 1H), 4,93 (m, 1H), 4,98 (m, 1H), 5,82 (d, 1H), 6,45 (d, 1H).
Пример получения 9: соединение 10.
Способ: общий способ 7.
Исходный материал VII: соединение 7
Хроматографический элюент: от 15 до 20% эфира в петр.эфире.
ЯМР: δ = 0,05 (m, 12H), 0,80 (bs, 3H), 0,82 (t, 6H), 0,86 (s, 9H), 0,88 (s, 9H), 1,45 (bs, 3H), 1,10-2,07 (m, 27H), 2,15 (m, 3H), 2,37 (bd, 1H), 2,48 (dd, 1H), 2,85 (bd, 1H), 3,44 (m, 1H), 3,93 (m, 1H), 4,20 (m, 1H), 4,50 (m, 1H), 4,70 (m, 1H), 4,91 (m, 1H), 4,98 (m, 1H), 5,81 (d, 1H), 6,44 (d, 1H).
Пример получения 10: соединение 11.
Способ: общий способ 8.
Исходный материал II: соединение 8
Хроматографический элюент: от 15 до 20% эфира в петр.эфире.
ЯМР: δ = 0,06 (m, 12H), 0,80 (s, 3H), 0,86 (s, 9H), 0,87 (s, 9H), 0,85-0,92 (m, 6H), 1,45 (bs, 3H), 1,25-2,05 (m, 23H), 2,07-2,27 (m, 2H), 2,43 (m, 1H), 2,44 (s, 2H), 2,81 (m, 1H), 3,45 (m, 1H), 3,94 (m, 1H), 4,18 (m, 1H), 4,37 (m, 1H), 4,79 (m, 1H), 4,86 (m, 1H), 5,18 (m, 1H), 6,00 (d, 1H), 6,22 (d, 1H).
Пример получения 11: соединение 12.
Способ: общий способ 8.
Исходный материал II: соединение 10
Хроматографический элюент: от 12,5% эфира в петр.эфире.
ЯМР: δ = 0,06 (m, 12H), 0,79 (s, 3H), 0,81 (t, 6H), 0,86 (s, 9H), 0,87 (s, 9H), 1,45 (bs, 3H), 1,25-2,25 (m, 3H), 2,42 (dd, 1H), 2,81 (m, 1H), 3,43 (m, 1H), 3,93 (m, 1H), 4,17 (m, 1H), 4,38 (t, 1H), 4,70 (m, 1H), 4,86 (m, 1H), 5,19 (m, 1H), 6,00 (d, 1H), 6,21 (d, 1H).
Пример получения 12: соединение 13 и 14.
Способ: общий способ 9.
Исходный материал II: соединение 9.
Хроматографический элюент: от 1 до 10% дихлорметана в гексане.
ЯМР 13: δ = 0,06 (m, 18H), 0,76 (s, 3H), 0,82 (t, 6H), 0,8 (s, 9H), 0,87 (s, 9H), 0,90 (s, 9H), 1,72 (bs, 3H), 1,20-1,87 (m, 13H), 1,92 (m, 1H), 2,16 (dd, 1H), 2,20-2,47 (m, 5H), 2,57 (dd, 1H), 2,75 (bd, 1H), 2,86 (m, 1H), 4,22 (m, 1H), 4,53 (m, 1H), 4,94 (m, 1H), 4,99 (m, 1H), 5,85 (d, 1H), 6,45 (d, 1H).
ЯМР 14: δ = 0,06 (m, 18H), 0,75 (s, 3H), 0,83 (t, 6H), 0,85 (s, 9H), 0,86 (s, 9H), 0,90 (s, 9H), 1,84 (bs, 3H), 1,35-2,65 (m, 22H), 2,84 (bd, 1H), 4,22 (m, 1H), 4,52 (m, 1H), 4,94 (m, 1H), 4,98 (m, 1H), 5,87 (d, 1H), 6,43 (d, 1H).
Пример получения 13: соединение 15.
Способ: общий способ 9.
Исходный материал III: соединение 11.
Хроматографический элюент: 33% эфира в петр.эфире (ПТСХ).
ЯМР: δ = 0,05 (m, 12H), 0,73 (s, 3H), 0,86 (s, 9H), 0,87 (s, 9H), 0,89 (t, 6H), 1,73 (bs, 3H), 2,47 (s, 2H), 1,35-2,55 (m, 18H), 2,69 (bd, 1H), 2,80 (m, 1H), 4,17 (m, 1H), 4,37 (m, 1H), 4,86 (m, 1H), 5,18 (m, 1H), 6,03 (d, 1H), 6,22 (d, 1H).
Пример получения 14: соединение 16.
Способ: общий способ 8.
Исходный материал IV: соединение 13
Хроматографический элюент: от 0 до 1% эфира в петр.эфире.
ЯМР: δ = 0,06 (m, 18H), 0,75 (s, 3H), 0,83 (t, 6H), 0,86 (s, 9H), 0,86 (s, 9H), 0,87 (s, 9H), 1,72 (bs, 3H), 1,35-2,50 (m, 21H), 2,78 (m, 2H), 4,19 (m, 1H), 4,37 (m, 1H), 4,87 (m, 1H), 5,18 (m, 1H), 6,04 (d, 1H), 6,23 (d, 1H).
Пример получения 15: соединение 17.
Способ: общий способ 9.
Исходный материал: соединение 12
Хроматографический элюент: 10% эфира в петр.эфире.
ЯМР: δ = 0,06 (m, 12H), 0,74 (s, 3H), 0,86 (t, 6H), 0,86 (s, 9H), 0,87 (s, 9H), 1,45 (q, 4H), 1,72 (bs, 3H), 1,35-2,55 (m, 20H), 2,77 (m, 2H), 4,18 (m, 1H), 4,37 (m, 1H), 4,86 (m, 1H), 5,18 (m, 1H), 6,03 (d, 1H), 6,22 (d, 1H).
Пример получения 16: соединение 18.
Способ: общий способ 4.
Исходный материал VII: 3-этил-1-пентин-3-ол.
Растворитель: дихлорметан (20 мл).
Основание: N-этил-диизопропиламин (2,0 г).
Силилирующий агент: хлорметил-силан (1,7 г).
Температура реакции: 20oC.
Время реакции: 1 ч.
Обогащение: дополнительная экстракция фосфатным буфером (pH 6,5, 0,07 М, 60 мл).
ЯМР: δ = 0,17 (s, 9H), 0,95 (t, 6H), 1,63 (q, 4H), 2,42 (s, 1H).
Пример получения 17: соединение 19.
Способ: общий способ 2.
Исходный материал VII: 2-метил-4-пентин-2-ол.
Хроматографический элюент: 5% эфира в петр.эфире.
ЯМР: δ = 1,34 (s, 3H), 1,35 (s, 3H), 1,51 (m, 4H), 1,67 (m, 1H), 1,84 (m, 1H), 2,00 (t, 1H), 2,44 (m, 2H), 3,45 (m, 1H), 3,97 (m, 1H), 4,81 (m, 1H).
Пример получения 18: соединение 20.
Способ: общий способ 3.
Исходный материал: метилмагний иодид.
Очистка путем дистилляции в вакууме.
Т.кип. соединения 20: 58-59oC 12 мм Hg.
ЯМР: δ = 1,24 (s, 6H), 1,69 (s, 1H), 1,75 (t, 2H), 1,98 (t, 1H), 2,31 (m, 2H).
Пример получения 19: соединение 21.
Способ: общий способ 2.
Исходный материал VII: соединение 20.
Хроматографический элюент: от 0 до 5% эфира в петр.эфире.
ЯМР: δ = 1,21 (s, 3H), 1,23 (s, 3H), 1,51 (m, 4H), 1,64 (m, 1H), 1,78 (t, 2H), 1,83 (m, 1H), 1,92 (t, 1H), 2,29 (m, 2H), 3,45 (m, 1H), 3,93 (m, 1H), 4,73 (m, 1H).
Пример получения 20: соединение 22.
Способ: общий способ 8.
Исходный материал IV: соединение 14.
Хроматографический элюент: 1% эфира в петр.эфире.
ЯМР: δ = 0,05 (m, 18H), 0,74 (s, 3H), 0,83 (t, 6H), 0,86 (s, 18H), 0,87 (s, 9H), 1,84 (bs, 3H), 1,35-2,62 (m, 22H), 2,79 (bd, 1H), 4,18 (m, 1H), 4,37 (m, 1H), 4,87 (m, 1H), 5,18 (m, 1H), 6,05 (d, 1H), 6,21 (d, 1H).
Пример 1. 1(S), 3(R)-Дигидрокси-20-(4-этил-4-гидрокси-1-гексин-1-ил)- 9,10-секо-прегна-5(Z), 7(E), 10(19),17(20) (Z)-тетраен (Соединение 101).
Способ: общий способ 10.
Исходный материал III: соединение 11.
Температура реакции: 25oC.
Время реакции: 45 мин.
Хроматографический элюент: от 50 до 0% петр.эфира этилацетата..
ЯМР: δ = 0,76 (s, 3H), 0,90 (t, 6H), 1,75 (bs, 3H), 1,40-2,43 (m, 19H), 2,49 (bs, 2H), 2,60 (dd, 1H), 2,71 (m, 1H), 2,82 (m, 1H), 4,24 (m, 1H), 4,44 (m, 1H), 5,01 (m, 1H), 5,34 (m, 1H), 6,04 (d, 1H), 6,37 (d, 1H).
Пример 2. 1(S), 3(R)-Дигидрокси-20-(4-этил-4-гидрокси-1-гексин-1-ил)- 9,10-секо-прегна-5(Z), 7(E), 10(19), 17(20) (Z)-тетраен (Соединение 101).
Способ: общий способ 11.
Исходный материал V: соединение 15.
Температура реакции: 60oC.
Время реакции: 1 ч.
Хроматографический элюент: от 50 до 0% петр.эфира в этилацетате.
ЯМР: δ = 0,76 (s, 3H), 0,90 (t, 6H), 1,75 (bs, 3H), 1,40-2,43 (m, 19H), 2,49 (bs, 2H), 2,60 (d, 1H), 2,71 (m, 1H), 2,82 (m, 1H), 4,24 (m, 1H), 4,44 (m, 1H), 5,01 (m, 1H), 5,34 (m, 1H), 6,04 (d, 1H), 6,37 (d, 1H).
Пример 3. 1(S), 3(R)-Дигидрокси-20-(5-этил-5-гидрокси-1-гептин-1-ил)- 9,10-секо-прегна-5(Z), 7(E), 10(19), 17(20) (Z)-тетраен (Соединение 102).
Способ: общий способ 10.
Исходный материал V: соединение 16.
Температура реакции: 50oC.
Время реакции: 10 мин.
Хроматографический элюент: от 50 до 0% петр.эфира в этилацетате.
ЯМР: δ = 0,75 (s, 3H), 0,87 (t, 6H), 1,72 (bs, 3H), 1,35-2,50 (m, 23H), 2,61 (dd, 1H), 2,67-2,90 (m, 2H), 4,24 (m, 1H), 4,44 (m, 1H), 5,01 (m, 1H), 5,34 (m, 1H), 6,04 (d, 1H), 6,38 (d, 1H).
Пример 4. 1(S), 3(R)-Дигидрокси-20-(5-этил-5-гидрокси-1-гептин-1-ил)- 9,10-секо-прегна-5(Z), 7(E), 10(19), 17(20) (Z)-тетраен (Соединение 102).
Способ: общий способ 11.
Исходный материал V: соединение 16.
Температура реакции: 100oC.
Время реакции: 17 ч.
Хроматографический элюент: 40% петр.эфира в этилацетате.
ЯМР: δ = 0,75 (s, 3H), 0,87 (t, 6H), 1,72 (bs, 3H), 1,35-2,50 (m, 23H), 2,61 (dd, 1H), 2,67-2,90 (m, 2H), 4,24 (m, 1H), 4,44 (m, 1H), 5,01 (m, 1H), 5,34 (m, 1H), 6,04 (d, 1H), 6,38 (d, 1H).
Пример 5. 1(S), 3(R)-Дигидрокси-20-(6-этил-6-гидрокси-1-октин-1-ил)- 9,10-секо-прегна-5(Z), 7(E), 10(19), 17(20) (Z)-тетраен (Соединение103).
Способ: общий способ 11.
Исходный материал V: соединение 17.
Температура реакции: 60oC.
Время реакции: 90 мин.
Хроматографический элюент: от 50 до 0% петр.эфира в этилацетате.
ЯМР: δ = 0,76 (s, 3H), 0,87 (t, 6H), 1,47 (q, 4H), 1,73 (bs, 3H), 1,40-2,50 (m, 21H), 2,60 (dd, 1H), 2,78 (m, 2H), 4,24 (m, 1H), 4,44 (m, 1H), 5,01 (m, 1H), 5,34 (m, 1H), 6,04 (d, 1H), 6,38 (d, 1H).
Пример 6. 1(S), 3(R)-Дигидрокси-20-(5-этил-5-гидрокси-1-гептин-1-ил)- 9,10-секо-прегна-5(Z), 7(E), 10(19), 17(20) (E)-тетраен (Соединение 112).
Способ: общий способ 11.
Исходный материал V: соединение 22.
Температура реакции: 100oC.
Время реакции: 20 ч.
Хроматографический элюент: от 50 до 0% петр.эфира в этилацетате.
ЯМР: δ = 0,74 (s, 3H), 0,86 (t, 6H), 1,49 (q, 4H), 1,83 (s, 3H), 2,41 (bs, 2H), 1,15-2,65 (t, 19H), 2,79 (m, 1H), 4,22 (m, 1H), 4,43 (m, 1H), 5,00 (m, 1H), 5,33 (m, 1H), 6,04 (d, 1H), 6,35 (d, 1H).
Соединение 102 растворяют в арахисовом масле до достижения конечной концентрации 1 мг соединения/102 мл масла, 10 мас.ч. желатина, 5 мас.ч. глицерина, 0,08 мас. ч. сорбата калия и 14 мас.ч. дистиллированной воды смешивают вместе при нагревании и формуют в мягкие желатиновые капсулы. Последние затем наполняют 100 мкл раствора соединения 102 в масле, так что каждая капсула содержит 0,1 мкг соединения 102.
Пример 8. Дерматологический крем, содержащий соединение 102.
В 1 г миндального масла растворяют 0,05 мг соединения 102. К этому раствору добавляют 40 г минерального масла и 20 г самоэмульгирующегося пчелиного воска. Смесь нагревают до расплавления. После добавления 40 мл горячей воды микстуру как следует перемешивают. Получающийся в результате крем содержит примерно 0,5 мкг соединения 102 нг крема.
Изобретение относится к производным витамина D общей формулы 1, где X - H, OH, R1 и R2 одинаковые или различные-C1-C6-алкил, возможно замещенный атомами фтора, Q-одинарная связь или группа (CH2)n, где n = 0, 1, 2 или 3, причем имеющиеся OH-группы могут быть защищены таким образом, что обратное их превращение в OH группу происходит in vivo, или их диастереомеры. Соединения проявляют противовоспалительное и иммуномодулирующее действие, также как и сильную активностью в индуцировании дифференциации и ингибировании пролиферации некоторых клеток. 4 з.п. ф-лы, 4 табл., 2 ил. 
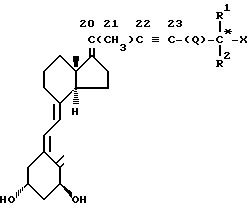
где X - водород или гидроксигруппа;
R1 и R2 одинаковые или различные - алкил C1 - C6, возможно замещенный атомами фтора;
Q - одинарная связь или группа (CH2)n, где n = 1, 2 или 3,
причем имеющиеся гидроксигруппы могут быть защищены таким образом, что они могут быть преобразованы в гидроксигруппы in vivo или их диастереомеры.
| WO, 9203414 A, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| US, 4804502 A, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

