Изобретение относится к неизвестному доныне классу соединений, которые демонстрируют противовоспалительную и иммуномодуляторную активности, а также обладают высокой активностью по индуцированию дифференциации и ингибированию нежелательной пролиферации определенных клеток, включая раковые клетки и клетки кожи, к фармацевтическим препаратам, содержащим эти соединения, к единицам доз таких препаратов и к их использованию при лечении и профилактике гиперпаратиреоза, в частности вторичного гиперпаратиреоза, связанного с почечной недостаточностью, для промотирования остеогенеза и лечения остеопороза и ряда болезненных состояний, включая диабетическое воспаление щек (diabetes mellitus), гипертензию, акне, алопецию, старение кожи, разбаланс иммунной системы, такие воспалительные заболевания, как ревматоидный артрит и астма, а также заболеваний, характеризующихся ненормальной клеточной дифференциацией и/или клеточной пролиферацией, например, псориаза и рака.
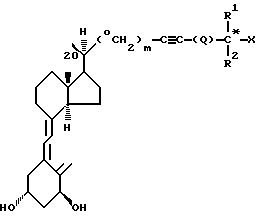
Соединения настоящего изобретения составляют новый класс аналогов витамина D и представлены общей формулой I:
где Х представляет гидрокси;
R1 и R2, которые могут быть одинаковы или различны, представляют метил, этил или трифторметил;
Q представляет простую связь или Q=(CH2)n, где n = 0, 1, 2, 3, 4;
m представляет 0 или 1,
а также диастереоизомеры этого соединения в чистом виде или смесь диастереоизомеров этого соединения.
Наиболее предпочтительными являются соединения:
а) 1(S), 3(R)-дигидрокси-20(R)-(6'-этил-6'-гидрокси-окт-1'-ин-1' -ил)-9,10-секо-прегна-5(Z),7(Е),10(19)-триен,
b) 1(S), 3(R)-дигидрокси-20(R)-(7'-этил-7'-гидрокси-нон-1'-ин-1' -ил)-9,10-секо-прегна-5(Z),7(Е),10(19)-триен.
Как можно видеть из формулы I, в зависимости от значений R1, R2, Q и Х, соединения настоящего изобретения могут включать несколько диастереоизомерных форм (например, R или S конфигурации у помеченного звездочкой атома углерода). Настоящее изобретение охватывает все эти диастереоизомеры как в чистом виде, так и в виде их смесей. Кроме того, производные 1, в которых одна или более из гидроксильных групп помечены как группы, которые можно реконвертировать в гидроксильные группы in vivo, также включены в объем настоящего изобретения ("биопревращаемые производные или предшественники 1").
Термин "биопревращаемые производные или предшественники 1" включают (но не ограничиваются ими) производные соединений формулы I, в которых одна или более из гидроксильных групп была трансформирована в -О-ацил или -О-гликозил, или группы фосфатного сложного эфира, причем такие замаскированные группы могут гидролизоваться in vivo.
Соединения формулы I, в которых Х является водородом, могут также действовать как предшественники, так как эти соединения относительно неактивны in vitro, но могут быть превращены в активные соединения формулы I за счет ферментативного гидроксилирования после приема пациентом.
Недавно было показано, что 1 α , 25-дигидрокси-витамин D3 (1,25/ОН/2 D3) влияет на действие и/или продуцирование интерлейкинов (Immunol Lett. 17, 361-366 (1988)), что говорит о возможности применения этого соединения при лечении заболеваний, характеризуемых дисфункцией иммунной системы, например, аутоиммунных заболеваний, СПИДа, реакций на прививки и отторжения трансплантатов, или других состояний, характеризующихся ненормальным продуцированием интерлейкина-1, например, таких воспалительных заболеваний, как ревматоидный артрит и астма.
Было также показано, что 1,25/ОН/2D3 способен стимулировать дифференциацию клеток и ингибировать избыточную клеточную пролиферацию (Abe, E. et. al. , Proc. Natl. Acad. Sci. USA, 78, 4990-4944 (1981)), и было высказано предположение о том, что это соединение может быть полезно при лечении заболеваний, характеризующихся ненормальной клеточной пролиферацией и/или клеточной дифференциацией, таких как лейкемия, миелофиброзы и псориазы.
Кроме того, было предложено использование 1,25/ОН/2D3 или его предшественника 1 α -OH-D3 для лечения гипертензии (Lind. L. et. al. Acta, Med. Scand. 222, 423-427 (1987)) и при диабете (Inomata, S. et. al. Bone Mineral. 1, 187-192 (1986)). Другое показание для 1,25/ОН/2D3 предложено в результате недавнего наблюдения связи между врожденной сопротивляемостью витамину D и алопецией: лечение 1,25/ОН/2 D3 может промотировать рост волос (Lanut, March. 4, 1989, p. 478). Кроме того, тот факт, что поверхностное нанесение 1,25/ОН/2D3 снижает размеры сальных желез в ушах самцов сирийского хомячка, дает возможность предположить, что это соединение может быть полезным при лечении акне. (Malloy, V.L. et al. Tricontinental Muting for Investigative Dermatology, Washington, 1989).
Однако терапевтические возможности таких показаний 1,25/ОН/2D3 существенно ограничены хорошо известным потенциальным действием этого гормона на метаболизм кальция, что приводит к повышению его концентрации в крови и быстрому возникновению гиперкальциемии. Так, это соединение и его потенциальные синтетические аналоги не полностью удовлетворительны для использования в качестве лекарственных препаратов при лечении, например, псориаза, лейкемии или иммунных заболеваний, которые могут потребовать регулярного приема препарата в относительно высоких дозах.
Недавно был описан целый ряд аналогов витамина D, которые демонстрируют некоторую степень селективности в отношении стимулирования клеточной дифференциации и ингибирования клеточной пролиферации по сравнению с влиянием на метаболизм кальция.
Так, аналог витамина D3, МС 903, содержащий 22, 23 двойную связь, 24-гидроксигруппу, и в котором 25, 26 и 27 атомы углерода включены в трехчленные циклы, является потенциальным стимулятором клеточной дифференциации и ингибитором клеточной пролиферации, который демонстрирует лишь умеренную активность по отношению к метаболизму кальция in vivo (Binderap, L. and Bramm, E. Biochemical Pharmacology, 37, 889-895 (1988)). Однако эта селективность не подтверждается исследованиями in vitro, в которых показано, что МС 903 также хорошо, как 1,25/ОН/2D3, связывается с интестипальными рецепторами витамина D. Поэтому может оказаться, что низкая активность in vivo МС 903 по отношению к метаболизму кальция связана с быстрым метаболизмом соединения, что лимитирует возможность использования этого соединения для систематического применения.
Было заявлено, что 24-гомо-1,25-дигидроксивитамин D3 и 26-гомо-1,25-дигидроксивитамин D3 (вместе с их 22, 23-дидегидроаналогами) (Ostrem. V.K., Tanaka, J. , Prahl. J. Deluca, H.F. and Jkekawa, N., Proc. Natl. Acad. Sci. USA, 84, 2410-14 (1987)) обладают таким же связывающим средством, что и 1,25/ОН/2D3 по отношению к желудочным рецепторам как крысы, так и цыпленка, и к рецепторам линии клеток миелоидной лейкемии человека (HL-60), и в 10 раз более эффективны в стимулировании дифференциации HL-60 клеток in vitro, нежели 1,25/ОН/2D3. In vivo эти соединения "значительно менее эффективны" и "более эффективны" нежели 1,25/ОН/2D3 при оценке метаболизма кальция.
26,27-диметил-1 α , 25-дигидроксивитамин D3 был синтезирован, но опубликованная информация относительно его биологической активности противоречива (Sai, H., Takasuto, S., Hara, N., and Jkekawa, N., Chem. Pharm. Bull. 33, 878-881 (1985) и Jkekawa, N., Eguchi, T., Hara, N., Takatsuto, S., Honda, A. , Mori. J. and Atomo, S., Chem. Pharm. Bull., 35, 4362-4365 (1987)). Эти авторы также сообщают о близко родственном 26,27-диэтил-1 α ,25-дигидроксивитамин D3, как о "почти не имеющем активности витамина D" (то есть действия на метаболизм кальция), хотя и являющемся в 10 раз более эффективным, нежели 1,25/ОН/2D3 в стимулировании клеточной дифференциации.
В патенте США 4804502 раскрыты соединения, содержащие тройную связь в боковой цепи витамина D, и эти соединения, как заявлено, пригодны для лечения болезненных состояний, характеризующихся недостаточным метаболизмом кальция.
Тот факт, что существует лишь небольшая структурная разница между вышеуказанными соединениями, указывает на то, что существующий уровень знаний не позволяет предсказать структуру аналогов витамина D, которые будут демонстрировать благоприятную степень селективности, что отражается на активности по клеточной дифференциации in vitro по сравнению со сродством по отношению к интестинальным рецепторам витамина D in vitro. Более того, проблема осложняется из-за наблюдения, что рецепторное сродство in vitro не всегда соответствует исследованиям in vivo, что вероятно отражает фармакокинетические различия между соединениями.
Соединения настоящего изобретения структурно отличаются от вышеуказанных аналогов витамина D, некоторые из которых, как было сообщено, обладают потенциальным действием на клеточную дифференциацию/пролиферацию в конфигурации метильных групп при углероде-20. Такая "неестественная" конфигурация имеется в соединении 1 и, как было неожиданно обнаружено, обладает серьезным и выгодным биологическим значением. Так, конкретное соединение формулы I, при сравнении с соответствующим соединением, включающим "естественную" конфигурацию С-20 (изменены положения метильного и водородного радикалов), как наблюдают, демонстрирует одно или более из следующих преимуществ:
(а) более эффективно по отношению клеточной дифференциации/пролиферации,
(b) обладают более высокой селективностью в плане потенциального влияния на клеточную дифференциацию/пролиферацию по сравнению с влиянием на метаболизм кальция,
(с) оказывают большее воздействие на продуцирование и действие интерлейкинов,
(d) обладают большей селективностью в плане воздействия на продуцирование и действие интерлейкина по сравнению с влиянием на метаболизм кальция.
Поэтому соединения настоящего изобретения наиболее пригодны как для локального, так и систематического лечения и профилактики заболеваний человека и ветеринарных расстройств, которые характеризуются 1) ненормальной клеточной пролиферацией и/или клеточной дифференциацией, как, например, при некоторых кожных заболеваниях, включая псориаз и некоторые формы рака, 2) расстройством иммунной системы, например, при аутоиммунных заболеваниях, включая сахарный диабет, реакции пациента на прививки и отторжение трансплантатов; и кроме того, для лечения воспалительных заболеваний, таких как ревматоидный артрит и астма. Акне, алопеция и гипертензия являются другими состояниями, которые могут подвергаться лечению соединениями настоящего изобретения. И наконец, так как утолщение кожи наблюдается после местного лечения соединениями настоящего изобретения, эти соединения могут быть полезны для лечения или предотвращения старения кожи, включая фотостарение.
Благодаря невысокой тенденции соединений к провоцированию гиперкальциемии при продолжительном приеме ожидается, что они могут быть ценны при длительном лечении гиперпаратириоза (особенно вторичного гиперпаратириоза, связанного с почечной недостаточностью), и для промотирования остеогенеза и лечения остеопорозов. При таких показаниях описываемые соединения обладают более высокой терапевтической ценностью, нежели соединения, известные ранее (см. патенты США 4948789 и EP 0385446 A2).
Соединения настоящего изобретения можно использовать в сочетании с другими лекарственными препаратами. Для предотвращения отторжения трансплантатов и реакций пациента на прививки лечение соединениями настоящего изобретения можно с успехом сочетать, например, с лечением циклоспоринами.
Еще одним аспектом изобретения является способ получения соединения формулы I: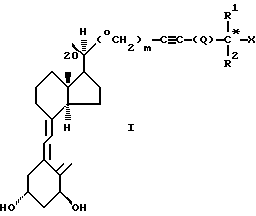
где Х представляет гидрокси,
R1 и R2, которые могут быть одинаковы или различны, представляют метил, этил или трифторметил;
Q представляет простую связь или Q=(CH2)n, где n = 0, 1, 2, 3, 4, а
m равно 0 или 1, в котором
а) 1(S), 3(R)-бис-(гидрокси-защищенный)-20(R)-формил-9,10-секо-прегна-5(Е), 7(Е), 10(19)-триен обрабатывают вначале литий бис(этилендиоксиборил) метилидом, а затем водным перборатом натрия до получения соединения формулы II, где m = 1.

где -О-Prot представляет защищенную гидроксигруппу,
b) соединение формулы II подвергают реакции типа реакции Виттинга до получения соединения формулы III: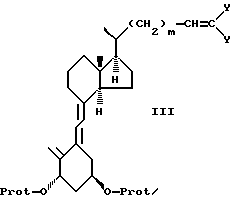
где m и -O-Prot имеют указанные ранее значения, а Y, будучи одинаковыми или различными, представляют Cl или Br,
с) соединение формулы III подвергают взаимодействию с подходящим основанием до получения промежуточного ацетилида, который без промежуточного выделения подвергают взаимодействию в присутствии катализатора с электрофильным реагентом формулы R-Z, где Z представляет отщепляемую группу, а R представляет -Q-C(R1)(R2)X, или радикал, который можно в него превратить на более поздней стадии (стадиях), или с соединением, одной из следующих формул: R1COR2 или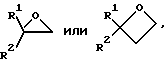
где R1, R2, Q и Х имеют указанные ранее значения,
до получения соединения формулы IV:
где R, m и -O-Prot имеют указанные ранее значения;
d) соединение формулы IV подвергают триплет-сенсибилизированной фотоизомеризации и необязательной модификации функциональных групп в боковой цепи R до получения соединения формулы V: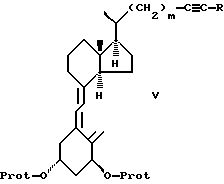
где R, m и -O-Prot имеют указанные ранее значения,
е) соединение формулы V подвергают реакции удаления защитных групп и необязательной модификации функциональной группы в боковой цепи R с образованием целевого соединения формулы I.
Соединение настоящего изобретения предназначено для использования в фармацевтических композициях, которые полезны при лечении заболеваний человека и животных, как было описано ранее.
Нужное количество соединения формулы I (здесь и далее именуемого активным ингредиентом) для терапевтического воздействия будет, естественно, зависеть как от конкретного соединения, так и от способа приема и пациента. Соединения настоящего изобретения можно вводить парэнтерально, интраартикулярно, энтерально или поверхностным нанесением (местно). Они все хорошо абсорбируются при энтеральном введении, и это предпочтительный способ введения при лечении системных расстройств. При лечении дерматологических расстройств, таких как псориаз или глазные болезни, предпочтительным является поверхностное или энтериальное введение.
При лечении респираторных заболеваний, таких как астма, предпочтительны аэрозоли.
Хотя активный ингредиент можно вводить отдельно, в виде отдельного химического препарата, предпочтительно вводить его в виде фармацевтической композиции. Обычно активный ингредиент содержит от 0,0001 до 0,1 вес.% композиции.
Под термином "единичная доза" понимают единичную дозу, которую можно ввести пациенту и которую можно легко приготовить и упаковать, причем она остается физически и химически стабильной единичной дозой, содержащей либо активный материал отдельно, либо в смеси его с твердым или жидким фармацевтическим разбавителем или носителем.
Композиции, как для медицинского, так и для ветеринарного использования, настоящего изобретения содержат активный ингредиент в сочетании с фармацевтически приемлемым носителем, и необязательно с другим (другими) терапевтическими ингредиентами. Носитель (носители) должен быть "приемлемым" в том смысле, что он должен быть совместимым с другими ингредиентами композиции и не оказывать вредного воздействия на ее реципиента.
Композиции включают, например, такие формы, пригодные для орального, ректального, парэнтерального (включая подкожное, внутримышечное и внутривенное), интра-артикулярное и поверхностное введение.
Композиции могут быть представлены в единичных дозированных формах и могут быть получены любым известным в фармации способом. Все способы включают стадию перемешивания активного ингредиента с носителем, который может состоять из одного или нескольких вспомогательных ингредиентов. Вообще, композиции приготавливают путем равномерного и тщательного перемешивания активного ингредиента с жидким или тонко измельченным твердым носителем или с тем и с другим, а затем, при необходимости, оформляют полученный продукт в целевые формы.
Композиции настоящего изобретения, пригодные для орального приема, могут быть в форме дискретных единиц, например капсул, саше, таблеток, облаток, каждая из которых содержит определенное количество активного ингредиента; в форме порошка или гранул; в форме раствора или суспензии в водной или неводной жидкости; или в форме эмульсии типа масло в воде или вода в масле.
Композиции могут также содержать другие терапевтически активные соединения, обычно используемые при лечении вышеуказанных патологических состояний.
Настоящее изобретение относится также к способу подавления вышеуказанных заболеваний или состояний, который состоит во введении пациенту, нуждающемуся в лечении, эффективного количества одного или более из соединений формулы I, одного или в сочетании с одним или более из других терапевтически активных соединений, обычно используемых при лечении указанных патологических состояний. Лечение соединениями настоящего изобретения и/или другими терапевтически активными соединениями может проводиться одновременно или с интервалами.
При лечении системных заболеваний эффективные дозы соединения формулы I могут составлять от 0,1 до 100 мкг в день, предпочтительно от 0,2 до 25 мкг. При поверхностном лечении кожных заболеваний используют мази, кремы или лосьоны, содержащие от 0,1 до 500 мкг/г и предпочтительно от 0,1 до 100 мкг/г соединения I. Для поверхностного нанесения в офтальмологические мази, капли или гели вводят от 0,1 до 500 мкг/г и предпочтительно от 0,1 до 100 мкг/г соединения формулы I. Композиции для орального приема изготавливают предпочтительно в форме таблеток, капсул или капель, содержащих от 0,05 до 50 мкг, предпочтительно от 0,1 до 25 мкг соединения формулы I на единичную дозу.
Далее изобретение иллюстрируется следующими нелимитирующими примерами получения и примерами:
Примеры получения, примеры, биологические тесты
Общие условия
Примеры соединений формулы I приведены далее в таблице 1.
В спектрах ядерного магнитного резонанса (300 МГц) значения химических сдвигов (δ) указаны для растворов в дейтерохлороформе относительно внутреннего стандарта тетраметилсилана (δ = 0) или хлороформа (δ = 7,25), обозначения мультиплетов: либо указаны - дублет (д), триплет (т), квартет (кв) или не указаны (м) и даются приблизительно по значению середины сигнала, если не указан интервал (с - синглет, шир. - широкий). Константы взаимодействия (J) приведены в Герцах и иногда округлены до ближайших значений.
Эфир является диэтиловым эфиром, высушенным над натрием. Тетрагидрофуран сушат над натрий-бензофеноном. Петролейный эфир относится к пентановой фракции. Реакции ведут при комнатной температуре, если нет других указаний. Упоминающаяся процедура обработки сводится к разбавлению специфическим растворителем (отличающимся от растворителя реакции), экстрагированию водой, а затем рассолом (раствором соли), высушиванию над безводным сульфатом магния и концентрированию в вакууме до получения остатка. Хроматографию осуществляют на силикагеле.
Пример получения 1. 1(S),3(R)-бис-трет.-бутилдиметил-силилокси- 20(R)-2,2-дихлорвинил-9,10-втор-прегна-5(Е),7(Е),10(19)-триен (соединение 1)
К раствору альдегида общей формулы II (м = 0), (1,15 г) и бромтрихлорметана (0,44 г) в сухом дихлорметане (6 мл), охлажденному до -20oC и перемешиваемому в атмосфере аргона, прикапывают в течение 40 минут раствор трис/диметил-амино/фосфина (0,72 г) в сухом дихлорметане (4 мл). Полученную смесь перемешивают еще 30 минут при -20oC, а затем в течение 20 часов при 20oC. Полученную смесь разделяют между разбавленным рассолом (75 мл) и дихлорметаном (75 мл), затем органический слой промывают водой и сушат. После концентрирования в вакууме получают остаток, который затем очищают хроматографически (1% эфир в петролейном эфире в качестве элюента), а затем кристаллизуют из метанола до получения указанного в заглавии соединения.
Т. плавления 120-123oC.
ЯМР: δ = 0,06 (м, 12H), 0,51 (с, 3H), 0,86 (с, 9H), 0,89 (с, 9H), 0,96 (д, 3H), 1,00-2,10 (м, 13H), 2,30 (шир.д., 1H), 2,43 (м, 1H), 2,55 (д д, 1H), 2,88 (дд, 1H), 4,21 (м, 1H), 4,53 (м, 1H), 4,94 (м, 1H), 4,98 (м, 1H), 5,71 (д, 1H), 5,82 (д, 1H), 6,45 (д, 1H).
Пример получения 2. 1(S),3(R)-бис-трет.-бутилдиметил-силилокси- 20(R)-(6'-этил-6'-триметилсилилокси-окт-1'-ин-1'-ил)-9,10-втор-прегна-5(Е), 7(Е), 10(19)-триен (соединение 2)
К раствору соединения 1 (0,28 г) в сухом ТГФ (16 мл), охлажденному до -70oC и перемешиваемому в атмосфере аргона, прикапывают в течение 5 минут раствор бутиллития (1,6 М в гексане, 0,7 мл). Перемешивание продолжают при -70oC в течение часа, а затем при 20oC в течение двух часов. Полученную смесь охлаждают до -10oC, а затем добавляют 0,38 г 1-бром-4-этил-4-триметилсилилоксигексана в течение 3 минут, затем ГМФА (2 мл) в течение 4 минут. Перемешивание продолжают при -10oC в течение 7 минут, а затем 20oC в течение трех часов. Реакционную смесь выливают в 25 мл полунасыщенного рассола и экстрагируют эфиром (2 • 50 мл). Объединенные органические фазы экстрагируют (быстро) 1 H соляной кислотой (25 мл), 1% водным бикарбонатом натрия (25 мл), водой (25 мл) и рассолом (25 мл). После сушки над сульфатом магния и концентрируют в вакууме, в результате чего получают масло, которое дважды очищают хроматографически (сначала 2% эфиром в петролейном эфире, затем 0,2% этилацетатом в гексане в качестве элюентов) до получения указанного в заглавии соединения.
ЯМР: δ = 0,05 (м, 12H), 0,09 (с, 9H), 0,57 (с, 3H), 0,81 (т, 6H), 0,85 (с, 9H), 0,89 (с, 9H), 1,11 (д, 3H), 1,45 (кв, 4H), 1,00-2,08 (м, 16H), 2,12 (м, 2H), 2,22 (м, 1H), 2,31 (шир. д, 1H), 2,41 (шир.д., 1H), 2,55 (дд, 1H), 2,88 (шир. д. 1H), 4,21 (шир.д., 1H), 4,53 (м, 1H), 4,93 (м, 1Н), 4,98 (м, 1H), 5,81 (д, 1H), 6,46 (д, 1H).
Пример получения 3. 1(S),3(R)-бис-трет.-бутилдиметил-силилокси- 20(R)-(6'-этил-6'-триметилсилилокси-окт-1'-ин-1'-ил)-9,10-втор-прегна-5(Z), 7(Е), 10(19)-триен (соединение 3)
Раствор соединения 2 (20 мг), антрацена (10 мг) и триэтиламина (0,005 мл) в дихлорметане (4 мл) в атмосфере аргона в пирексной колбе облучают светом ультрафиолетовой лампы высокого давления, тип TQ 760Z2 (Hanau) при температуре около 10oC в течение 20 минут при перемешивании. Реакционную смесь концентрируют в вакууме и обрабатывают петролейным эфиром (1 мл), содержащим 0,25% триэтиламина. После фильтрования полученный фильтрат концентрируют в вакууме и очищают хроматографически (1% эфир в петролейном эфире в качестве элюента) до получения указанного в заглавии соединения.
ЯМР: δ = 0,05 (м, 12H), 0,09 (с, 9H), 0,56 (с, 3H), 0,81 (т, 6H), 0,86 (с, 9H), 0,87 (с, 9H), 1,11 (д, 3H), 1,44 (кв, 4H), 0,85-2,75 (м, 22H), 2,83 (шир. д, 1H), 4,18 (м, 1H), 4,36 (м, 1H), 4,85 (м, 1H), 5,17 (м, 1H), 6,00 (д, 1H), 6,24 (д, 1H).
Пример получения 4. 1(S),3(R)-бис-трет.-бутилдиметил-силилокси- 20(R)-(7'-этил-7'-триметилсилилокси-нон-1'-ин-1'- ил)-9,10-втор-прегна-5(Е),7(Е), 10(19)-триен (соединение 4)
По способу примера получения 2, но заменяя 1-бром-5-этил-5-триметилсилилоксигептаном 1-бром-4-этил-4-триметилсилилоксигексан, получают указанное в заглавии соединение.
ЯМР: δ = 0,05 (м, 12H), 0,09 (с, 9H), 0,57 (с, 3H), 0,80 (т, 6H), 0,86 (с, 9H), 0,89 (с, 9H), 1,11 (д, 3H), 1,15-2,5 (м, 23H), 1,43 (кв, 4H), 2,55 (м, 1H), 2,88 (м, 1H), 4,22 (м, 1H), 4,53 (м, 1H), 4,93 (м, 1H), 4,98 (м, 1H), 5,81 (д, 1H), 6,46 (д, 1H).
Пример получения 5. 1(S),3(R)-бис-трет.-бутилдиметил-силилокси- 20(R)-(7'-этил-7'-триметилсилилокси-нон-1'-ин-1'-ил)-9,10-втор-прегна-5(Z), 7(Е), 10(19)-триен (соединение 5)
По способу примера 3, но заменяя соединение 2 на соединение 4, получают указанное в заглавии соединение.
ЯМР: δ = 0,05 (м, 12H), 0,10 (с, 9H), 0,56 (с, 3H), 0,80 (т, 6H), 0,86 (с, 18H), 1,10 (д, 3H), 1,05-1,93 (м, 22H), 1,98 (шир.т. 1H), 2,15 (м, 2H), 2,21 (дд, 1H), 2,38 (шир. д, 1H), 2,44 (дд, 1H), 2,83 (дд, 1H), 4,18 (м, 1H), 4,36 (м, 1H), 4,86 (м, 1H), 5,17 (м, 1н), 6,00 (д, 1H), 6,24 (д, 1H).
Пример 1
1(S), 3(R)-дигидрокси-20(R)-(6'-этил-6'-гидрокси-окт-1'-ин-1'- ил)-9,10-втор-прегна-5(Z),7(Е),10(19)-триен (соединение 103)
Раствор соединения 3 (12 мг) и тетра-н-бутил-аммоний-фторидтригидрата (31 мг) в ТГФ (1, 5 мл) нагревают при 60oC в атмосфере аргона при перемешивании в течение одного часа. После охлаждения реакционный раствор разделяют между этилацетатом (8 мл) и 2% раствором бикарбоната натрия, содержащим 4% натрийхлорида (8 мл). Органический слой промывают водой и рассолом, сушат и концентрируют в вакууме. Остаток очищают хроматографически 20% петролейным эфиром в этилацетате в качестве элюента до получения соединения 103.
УФ: λмакс (EtOH): 264 нм.
ЯМР: δ = 0,58 (с, 3H), 0,87 (т, 6H), 1,12 (д, 3H), 1,46 (кв, 4H), 1,10-2,25 (м, 22H), 2,31 (дд, 1H), 2,40 (шир. д., 1H), 2,60 (дд, 1H), 2,84 (дд, 1H), 4,23 (м, 1H), 4,43 (м, 1H), 5,00 (м, 1H), 5,32 (м, 1H), 6,01 (д, 1H), 6,39 (д, 1H).
Пример 2
1(S), 3(R)-дигидрокси-20(R)-(7'-этил-7'-гидрокси-нон-1'-ин-1'- ил)-9,10-втор-прегна-5(Z),7(Е),10(19)-триен (соединение 131)
По способу примера 1, но заменяя соединение 3 на соединение 5, получают соединение 131.
УФ: λмакс (EtOH): 263 нм.
ЯМР: δ = 0,58 (с, 3H), 0,86 (т, 6H), 1,11 (д, 3H), 1,30-2,10 (м, 25H), 2,17 (м, 3H), 2,32 (дд, 1H), 2,40 (м, 1H), 2,60 (дд, 1H), 2,85 (дд, 1H), 4,23 (м, 1H), 4,43 (м, 1H), 5,00 (м, 1H), 5,32 (м, 1H), 6,01 (д, 1H), 6,38 (д, 1H).
Другие полученные по описанным методикам и примерам соединения по изобретению приведены в таблице 1.
Пример 3
Капсулы, содержащие соединение 103
Соединение 103 растворяют в арахисовом масле до окончательной концентрации 1 мкг/мл масла. 10 вес. частей желатина, 5 вес. частей глицерина, 0,08 вес. частей сорбита калия и 14 вес. частей дистиллированной воды смешивают вместе при нагревании и формуют в мягкие желатиновые капсулы. Их заполняют 100 мкл соединения 103 в масляном растворе в каждую капсулу, так что в каждой из них содержится по 0,1 мкг указанного соединения.
Пример 4
Дерматологический крем, содержащий соединение 103
В 1 г миндального масла растворяют 0,05 мг соединения 103. К этому раствору добавляют 40 г минерального масла и 20 г самоэмульгирующегося пчелиного воска. Полученную смесь нагревают до ожижения. После добавления 40 мл горячей воды смесь хорошо перемешивают. Полученный крем содержит приблизительно 0,5 мкг активного соединения (103) на грамм крема.
Биологическое тестирование
Влияние на раковые клетки in vitro
Исследование клеточного роста осуществляли с использованием опухолевой клеточной линии человеческой гистиоцитемической лимфомы U 937 (Американская коллекция типовых культур - American Type Culture Collection) Rockville, MD). Клетки U 937 доводили до 1 • 105 клеток/мл в RPMI 1640 (Gibco, США). В среду добавляли 2 мМ глутамина, 100 М.Е./мл пенициллина, 100 мкг/мл стрептомицина и 10% фетальной телячьей сыворотки (FCS/Gibco). Клетки (5 мл) инкубировали в увлажненной атмосфере в течение 96 часов при 37oC в присутствии 1,25 (ОН)2D3 или аналогов витамина D (от 10-12 до 10-7 М). К контрольным культурам добавляли 0,2% растворителя. В конце инкубирования клетки были сосчитаны и концентрация тестируемого соединения, приводящая к 50% ингибированию (IC50) роста клеток, рассчитывалась из кривой доза-ответ. Типичное значение IC50 для 1,25 (ОН)2D3 составляло 5 • 10-8 М. Каждый эксперимент включал контрольный эксперимент в присутствии и в отсутствие 1,25 (ОН)2D3.
Влияние на гиперпролиферацию клеток кожи in vitro
Самопроизвольно иммортализованная клеточная линия незлокачественного человеческого псориаза НаСаТ была любезно предоставлена доктором N.E.Fusenig, Немецкий Научно-исследовательский центр рака (German Cancer Research Center) (Гейдельберг, Германия) и банком опухолей (Tumorbank), DKFZ (Гейдельберг). Клетки растили в среде феноловый красный - свободная среда Дульбекко, модифицированная по способу Игла, с добавлением 2 мМ глутамина, 100 М.Е./мл пенициллина, 100 мкг/мл стрептомицина и 5% фетальной телячьей сыворотки (FCS/Gibco). Клетки высеивали при плотности 1 • 104 клеток на лунку на мультипланшеты. Тестируемые соединения (1,25 (ОН)2D3 или аналоги витамина D (от 10-11 до 10-7 М) добавляли спустя 2 часа после посева. После инкубации в увлажненной атмосфере в течение 120 часов при 37oC, синтез ДНК определяли посредством внедрения тимидина: 1мкСи/мл Н3 - меченого тимидина добавляли к клеткам и кластеры инкубировали в течение 4 часов. Затем клетки промывали трижды NaCl+25 мг/л немеченного тимидина, солюбилизировали в течение 5-10 минут в 1 мл 0,5 М раствора NaOH. Внедренный [Н3] тимидин измеряли счетчиком бета-излучения. Рассчитывали концентрацию, приводящую к 50% максимального ответа (IC50). Типичное значение IC50 для 1,25 (ОН)2D3 составляло 5 • 10-8 М. Каждый эксперимент включал контрольное определение в присутствии и в отсутствие 1,25 (ОН)2D3, и каждый образец тестировался четырехкратно.
Значения, полученные для U 937 и НаСаТ, приведенные в таблице 2, относятся к величине для 1,25 (ОН)2D3, то есть представляют собой соотношение между IC50 для 1,25 (ОН)2D3 и IC50 для аналога витамина D (соединение по изобретению). Табличные значения таким образом указывают, что при исследовании соединение было более активным и сильнодействующим, чем 1,25 (ОН)2D3.
Из таблицы видно, что большинство из 10 соединений - примеров являются либо более, либо гораздо более эффективными, нежели 1,25(ОН)2D3 в U 937-анализе (модель рака) и в НаСаТ-анализе (модель псориаза).

Изобретение относится к соединениям формулы (1), где Х -гидрокси; R1 и R2 могут быть одинаковы или различны, представляют метил, этил или трифторметил, Q представляет простую связь или Q = (CH2)n, где n = 0,1-4, a m - 0 или 1. Соединения обладают противовоспалительной и иммуномодуляторной активностью, а также высокой способностью вызывать дифференциацию и ингибирование нежелательной пролиферации некоторых клеток. Описывается их способ получения, промежуточное соединение для получения соединения формулы I, а также фармацевтическая композиция, проявляющая антипролиферативную активность и способ подавления болезненных состояний, характеризующихся патологической клеточной дифференциацией или пролиферацией, таких как рак и псориаз, с использованием соединения формулы I. 5 с. и 6 з.п. ф-лы, 2 табл.

где X представляет гидрокси;
R1 и R2, которые могут быть одинаковы или различны, представляют метил, этил, или трифторметил;
Q представляет простую связь или Q = (CH2)n, где n = 0, 1, 2, 3, 4;
m = 0 или 1.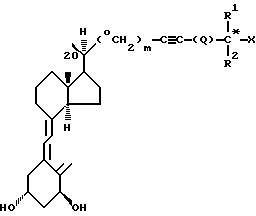
где X представляет гидрокси;
R1 и R2, которые могут быть одинаковы или различны, представляют метил, этил, или трифторметил;
Q представляет простую связь или Q = (CH2)n, где n = 0, 1, 2, 3, 4;
m = 0 или 1,
отличающийся тем, что а) 1(S), 3(R)-бис-(гидрокси-защищенный)-20(R)формил-9, 10-секо-прегна-5(E), 7(E), 10(19)-триен обрабатывают вначале литий бис(этилендиоксиборид)метилидом, а затем водным перборатом натрия до получения соединения формулы II, где m = 1,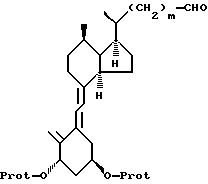
a -O-Prot представляет защищенную гидроксигруппу,
b) которое затем подвергают реакции типа реакции Виттига с получением соединения формулы III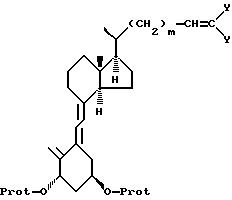
где m и -O-Prot имеют указанные выше значения;
Y одинаковы или различны и представляют CL или Br,
c) которое далее подвергают взаимодействию с подходящим основанием для получения промежуточного ацетилида, который без промежуточного выделения подвергают взаимодействию в присутствии катализатора с электрофильным реагентом формулы
R-Z,
где Z представляет уходящую группу;
R представляет -(Q)-C(R1)(R2)X,
либо соединение III подвергают взаимодействию с соединением одной из следующих формул:
R1COR2,
или 
или 
где R1, R2, Q и X имеют указанные ранее значения,
с получением соединения формулы 4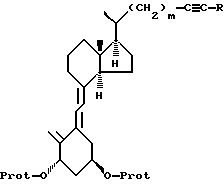
где R, m и -O-Prot имеют указанные выше значения,
d) которое затем подвергают триплет-сенсибилизированной фотоизомеризации и необязательной модификации функциональных групп в боковой цепи R с получением соединения формулы V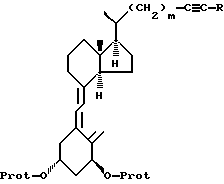
где R, m и -O-Prot имеют указанные выше значения,
e) которое подвергают реакции удаления защитных групп и необязательной модификации функциональной группы в боковой цепи R с получением целевого соединения формулы I.
| ВОГ-СОЮЗПАП^ 'Г":^^'Г>&Г' V>&\''^'','''^'~-l!l l..:i^^tii.^~i,fiili\ 'il^ lil-БИБЛИОТЕКААНТИФРИЗ | 0 |
|
SU296800A1 |
| СБОРНАЯ ЧЕРВЯЧНАЯ ФРЕЗА | 0 |
|
SU184112A1 |
| EP 326875 A1, 1989 | |||
| РАБОЧИЙ ОРГАН ВЯЗАЛЬНОЙ МАШИНЫ | 0 |
|
SU398217A1 |
| Тринус Ф.П | |||
| Фармако-терапевтический справочник | |||
| - Киев: Здоровья, 1989, с.288. | |||

