Изобретение относится к аналогам дидезоксикарбоциклических нуклеозидов. Более конкретно, настоящее изобретение касается аналогов карбоциклических 2', 3'-дидезокси-2', 3'-дидегидропуриннуклеозидов и их использования в терапии, в частности, в качестве противовирусных агентов.
В свете сходства между вирусными и хозяин-клеточными функциями трудно селективно поражать вирус и одновременно оставлять клетку хозяина неповрежденной. Таким образом существуют всего несколько агентов, эффективных против вирусов per se и очень трудно найти противовирусные агенты, имеющие приемлемый терапевтический индекс, то есть агенты, которые имеют многозначительный противовирусный эффект при уровне дозировки, при котором агент имеет приемлемую токсичность или побочное действие, профиль.
Одной группой вирусов, которая недавно получила основное значение, являются ретровирусы, ответственные за синдром приобретенного иммунодефицита (СПИД) человека. Такие вирусы ранее имели различную терминологию, однако теперь в основном упоминаются как вирусы человеческого иммунодефицита (HIY'S) ; два таких вируса HIY-I и HIY-II репродуктивно выделены из пациентов, страдающих СПИДом и родственными состояниями, такими как СПИД-связанный комплекс (ARC) и устойчивая распространенная лимфаденопатия.
Хотя целый ряд нуклеозидов рассматривают как полезные в лечении состояний, ассоциируемых с заражениями HIY, только зидовудин (AZ1, Petrovir) получил постоянное одобрение для лечения таких состояний. Однако известно, что зидовудин имеет крайне нежелательные побочные эффекты, вызывающие угнетение костного мозга, что ведет к снижению количества лейкоцитов с последующей выраженной анемией, и существует необходимость в эффективных агентах, являющихся менее цитотоксичными.
Заявитель обнаружил новый класс нуклеозидных аналогов, имеющих противовирусную активность. Поэтому предлагается первый вариант соединения формулы (I):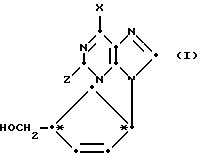
где
X обозначает водород, NRR1, SR, OR или галоген, Z обозначает водород, OR2 или NRR1, R, R1 и R2 могут быть одинаковыми или различными, и их выбирают из водорода, C1-C4-алкила и арила, а также его фармацевтически приемлемых производных.
Специалист поймет, что соединения формулы (I) представляют собой цис-соединения, а циклипентен - кольцо соединений формулы (I) содержит два хиральных центра (показаны в формуле (I) под обозначением *) и их смеси, включая рацемические смеси. Все такие изомеры и их смеси, включая рацемические смеси, включены в объем настоящего изобретения. Так, в соединениях формулы (I) либо хиральный центр, к которому присоединено основание, находится в R-конфигурации, а хиральный центр, к которому присоединена часть CH2OH, находится в S-конфигурации (далее D-изомер), либо хиральный центр, к которому присоединено основание, находится в S-конфигурации, а хиральный центр, к которому присоединена часть CH2OH, находится в R-конфигурации (далее L-изомер). Соединения могут быть в виде либо рацемической смеси, либо в основном как чистый D-изомер.
D-изомеры могут быть представлены формулой (Ia):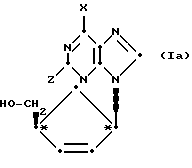
где
X и Z имеют определенные выше значения. В дальнейшем ссылка на соединения формулы (I) включает соединения формулы (Ia).
Следует также понять, что некоторые соединения формулы (I) могут существовать как ряд таутомерных форм, и все такие таутомеры включены в объем настоящего изобретения.
Как он используется здесь, термин галоген касается фтора, хлора, брома, и йода; когда X обозначает галоген, он предпочтительно является хлором.
C1-C4-алкил относится здесь к прямоцепочечной или разветвленной алкиловой группе, например метил, этил, н-пропил, изопропил, н-бутил, втор-бутил и трет-бутил. Предпочтительно, C1-C4-алкил обозначает метил.
Арил здесь относится к любой моно- или полициклической ароматической части и включает незамещенный и замещенный арил (такой, как фенил, толил, ксилил, анизил) и незамещенный и замещенный аралкил, включая ар (C1-C4)алкил, такой как фен(C1-C4)алкил, например бензил или фенетил.
В соединениях формулы (I) Z предпочтительно обозначает амино.
В одном предпочтительном классе соединений формулы (I) X обозначает OR, в частности OH.
В другом предпочтительном классе соединений формулы (I) X обозначает NRR1, в частности NH2, или водород.
Особенно предпочтительны соединения формулы (I), в которых Z обозначает NH2 и X обозначает H, NH2 или, особенно, OH. Такие соединения имеют особенно желательные терапевтические индексы в качестве противовирусных агентов.
Под "фармацевтически приемлемым производным" подразумевают любую фармацевтически приемлемую соль, сложный эфир или соль такого сложного эфира соединения формулы (I) или любого другого соединения, которое при введении реципиенту способно приводить к получению (непосредственно или опосредованно) соединения формулы (I) или его противовирусно активного метаболита или остатка.
Предпочтительные сложные эфиры соединения формулы (I) включают сложные эфиры карбоновой кислоты, в которых некарбонильную часть группировки сложного эфира выбирают из водорода, прямоцепочечного или разветвленного алкила (например, метила, этила, н-пропила, трет-бутила, н-бутила), алкоксиалкила (например, метоксиметила), аралкила (например, бензила), арилоксиалкила (например, феноксиметила), арила (например, фенила, по выбору замещенного галогеном, C1-C4-алкилом или C1-C4-алкокси); сложные эфиры сульфокислоты, такие как алкил или аралкилсульфонил (например, метансульфонил), аминокислотные сложные эфиры (например, L-валил или L-изолейцил) и сложные эфиры моно-, ди- или три-фосфорной кислоты.
Что касается вышеупомянутых сложных эфиров, то в том случае, если не указано что-либо иное, любая присутствующая алкильная часть преимущественно содержит от 1 до 18 атомов углерода, в частности, от 1 до 4 атомов углерода. Любая арильная часть, присутствующая в таких сложных эфирах, преимущественно содержит фенильную группу.
Фармацевтически приемлемые соли соединений формулы (I) включают те, которые произошли от фармацевтически приемлемых неорганических и органических кислот и оснований. Примеры пригодных кислот включают хлористоводородную кислоту, бромистоводородную кислоту, серную кислоту, азотную кислоту, перхлорную кислоту, фумаровую кислоту, малеиновую кислоту, фосфорную кислоту, гликолевую кислоту, молочную кислоту, салициловую кислоту, янтарную кислоту, толуол-н-сульфокислоту, винную кислоту, уксусную кислоту, лимонную кислоту, метансульфокислоту, муравьиную кислоту, бензойную кислоту, малоновую кислоту, нафталин-2-сульфокислоту и бензолсульфокислоту. Другие кислоты, такие как щавелевая кислота, хотя и не являются фармацевтически приемлемыми сами по себе, могут быть использованы в получении солей, пригодных в качестве промежуточных продуктов при получении соединений настоящего изобретения и их фармацевтически приемлемых кислых аддитивных солей.
Соли, происходящие от пригодных оснований, включают соли щелочных металлов (например, натрия), соли щелочно-земельных металлов (например, магния), аммониевые соли и соли NR
В дальнейшем ссылки на соединение в соответствии с изобретением включают как соединения формулы (I), так и их фармацевтически приемлемые производные.
Специфические соединения формулы (I) включают: / 1α,4α /-4-/6-хлор-9H-пурин-9-ил/-2-циклопентенил-карбинол,
/ 1α,4α /-4-/6-гидрокси-9H-пурин-9-ил-/2-циклопентенил-карбинол,
/ 1α,4α /-4-/6-амино-9H-пурин-9-ил-/2-циклопентенил-карбинол,
/ 1α,4α /-4-/6-меркапто-9H-пурин-9-ил-/2-циклопентенил-карбинол,
/ 1α,4α /-4-/2-амино-6-хлор-9H-пурин-9-ил-/2-циклопентенил-карбинол,
/ 1α,4α /-4-/2-амино-6-гидрокси-9H-пурин-9-ил-/2-циклопентенил-карбинол,
/ 1α,4α /-4-/2,6-диамино-9H-пурин-9-ил-/2-циклопентенил-карбинол
в форме рацемической смеси или простого энантиомера.
Соединения настоящего изобретения либо сами по себе обладают противовирусной активностью, либо они метаболизируются в такие соединения. В частности, эти соединения эффективны при ингибировании репликации ретровирусов, включая человеческие ретровирусы, такие как вирусы иммунодефицита человека (HIY'S) возбудители СПИДа.
Некоторые соединения настоящего изобретения обладают противораковой активностью, в частности соединения, в которых обозначает водород.
Таким образом, предлагается другой вариант настоящего изобретения относительно соединения формулы (I) или его фармацевтически приемлемого производного для использования в качестве противовирусного агента, например, при лечении ретровирусных заражений, или в качестве противоракового агента.
В другом или альтернативном варианте настоящего изобретения предлагается способ лечения вирусной инфекции, в частности инфекции, вызванной ретровирусом, таким как HIY, у млекопитающих, включая человека, при котором вводят эффективное количество противовирусного агента в виде соединения формулы (I) или его фармацевтически приемлемого производного.
Также предлагается в качестве дополнительного или альтернативного варианта изобретения использование соединения формулы (I) или его фармацевтически приемлемого производного для производства лекарственного препарата для лечения вирусной инфекции или использования в качестве противоракового средства.
Соединения настоящего изобретения, имеющие противовирусную активность, также пригодны при лечении СПИД-связанных состояний, таких как СПИД-связанный комплекс (ARC), прогрессивная распространенная ламфаденопатия (PGL), СПИД-связанные неврологические состояния (такие как слабоумие или тропический парапарез), анти-HIY антитело-положительные и HIY-положительные состояния, идиопатическая множественная геморрагическая саркома (ангиоматоз Капощи) и тромбоцитопеническая пурпура.
Противовирусные соединения настоящего изобретения также пригодны в предотвращении развития клинического заболевания пациентов, которые являются анти-HIY антитело- или HIY-антиген-положительными, и при профилактике вслед за экспозицией HIY.
Противовирусные соединения формулы (I) или их фармацевтически приемлемые производные могут быть также пригодны для предотвращения вирусного заражения физиологических жидкостей, таких как кровь или семенная жидкость, in vitro.
Некоторые соединения формулы (I) также пригодны в качестве промежуточных соединений при получении других соединений настоящего изобретения.
Специалисту станет ясно, что ссылка здесь на лечение предполагает профилактику и лечение установленных заражений или симптомов.
Ясно, что количество соединения настоящего изобретения, необходимое для использования при лечении, будет варьироваться не только в зависимости от типа выбранного соединения, но также и в зависимости от пути введения лекарственного средства, природы состояния, подлежащего лечению, возраста и состояния пациента и будет полностью в компетенции врача или ветеринара. Вообще пригодная доза будет составлять диапазон приблизительно от 1 приблизительно до 750 мг/кг, например, приблизительно от 1 до 750 мг/кг веса тела в день или приблизительно от 3 до 120 мг/кг веса тела реципиента в день, предпочтительно в диапазоне от 6 до 90 мг/кг, наиболее предпочтительно в диапазоне от 15 до 60 мг/кг/день.
Желаемая доза может присутствовать в виде однократной дозы или в виде раздельных дозировок, вводимых при соответствующих интервалах, например, в виде двух, трех, четырех или более поддозировок в день.
Соединение удобно вводить в форме унифицированной дозы, например, содержащей от 10 до 1500 мг, удобнее от 20 до 1000 мг, наиболее удобно от 50 до 700 мг активного компонента на лекарственную стандартную дозу.
Идеально активный ингредиент должен вводиться с тем, чтобы достичь пиковой концентрации активного соединения в плазме, равной приблизительно от 1 до 75 мкМ, предпочтительно около 2-50 мкМ, наиболее предпочтительно около 3-30 мкМ. Это может быть достигнуто, например, путем внутривенного введения 0,1-5%-ного раствора компонента по выбору в физиологическом растворе или путем перорального введения в виде болюса, содержащего приблизительно от 1 до 100 мг/кг активного компонента. Желаемые уровни крови могут быть поддержаны путем постоянного вливания с получением приблизительно от 0,01 до 5,0 мг/кг/ч или путем прерывистых вливаний, содержащих приблизительно от 0,4 до 15 мг/кг активного компонента.
Хотя для использования в терапии можно вводить соединение настоящего изобретения в виде сырого химиката, предпочтительно, чтобы активный компонент присутствовал в виде фармацевтического состава.
Настоящее изобретение также предлагает фармацевтический состав, содержащий соединение формулы (I) или его фармацевтически пригодное производное вместе с одним или более фармацевтически приемлемыми носителями и, необязательно, другими терапевтическими и/или профилактическими компонентами. Носитель (носители) должен быть "приемлемым" в смысле совместимости с другими компонентами состава и не должен быть вредным для реципиента.
Фармацевтические составы включают те, которые пригодны для перорального, ректального, назального, локального (включая трансбуккальный и подъязычный), вагинального и парентерального (включая внутримышечный и внутривенный) введения или в форме, пригодной для введения путем ингаляции или инсуффляции. Составы, где это подходит, могут присутствовать в виде дискретных дозированных единиц и могут быть получены любыми методами, хорошо известными в области фармацевтического дела. Все методы включают стадию связывания активного соединения с жидкими носителями или мелкодисперсными твердыми носителями, или обоими и, если необходимо, стадию формирования продукта в желаемую конфигурацию.
Фармацевтические составы, пригодные для перорального введения, могут присутствовать в виде дискретных единиц, таких как капсулы, крахмальные облатки или таблетки, при этом каждая содержит установленное количество активного компонента в виде порошка или гранул, в виде раствора, суспензии или эмульсии. Активный компонент может также присутствовать в виде болюса, электуария (лекарственной кашки) или пасты. Таблетки и капсулы для перорального введения могут содержать традиционные наполнители, такие как связывающие вещества, замасливатели, смачивающие вещества или дезинтеграторы. Таблетки могут покрываться в соответствии с методами, хорошо известными в данной области техники. Жидкие препараты для перорального введения могут существовать в форме, например, водных или маслянистых суспензий, растворов, эмульсий, сиропов или эликсиров, или они могут присутствовать в виде сухого продукта для составления вместе с водой или другим пригодным носителем перед использованием. Такие жидкие препараты могут содержать традиционные присадки, такие как суспендирующие вещества: эмульгаторы, неводные носители (которые могут включать съедобные масла) или консервирующие вещества.
Соединения в соответствии с настоящим изобретением могут также составляться для парентерального введения (например, путем инъекции, например, введением шарика или непрерывным вливанием) и могут присутствовать в форме унифицированной дозировки в ампулах, предварительно наполненных шприцах или в упаковке лекарственных средств для многократного приема с добавлением консервирующего вещества. Композиции могут принимать такие формы, как суспензии, растворы или эмульсии в маслянистых или водных носителях и могут содержать суспендирующие вещества, стабилизаторы и/или диспергаторы. Альтернативно активный компонент может быть в форме порошка, полученной асептическим выделением стерильного твердого вещества или лиофилизацией из раствора для составления вместе с пригодным наполнителем, например стерилизованной апирогенной водой, перед использованием.
Для местного применения к эпидермису соединения в соответствии с настоящим изобретением могут быть составлены в виде мазей, кремов или примочек или же в виде трансдермального пластыря. Мази и кремы могут, например, составляться вместе с водным или маслянистым основанием и могут также содержать один или более эмульгаторов, стабилизаторов, диспергаторов, суспендирующих веществ, загустителей или окрашивающих веществ.
Составы, пригодные для местного применения во рту, включают таблетки, содержащие активный компонент в корригентной основе (обычно сахарозе и аравийской камеди) или трагаканте; пастилки, содержащие активный компонент в инертном основании, таком как желатина и глицерина или сахароза и аравийская камедь; а также жидкости для полоскания рта, содержащие активный компонент в пригодном жидком носителе.
Фармацевтические составы, пригодные для ректального введения, причем носителем является твердое вещество, наиболее предпочтительно представлены в виде суппозиториев однократного применения. Пригодные носители включают кокосовое масло и другие вещества, обычно используемые в данной области техники, и суппозитории могут быть удобно составлены путем смешивания активного соединения с размягченным или расплавленным носителем (носителями) с последующим охлаждением и формованием в пресс-формах.
Составы, пригодные для вагинального введения, могут быть в виде вагинальных суппозиториев, тампонов, кремов, гелей, паст, пены или аэрозолей, содержащих, кроме активного компонента, такие носители, которые известны в данной области.
Для внутриносового введения соединения настоящего изобретения могут быть использованы в качестве жидкого состава для разбрызгивания или в форме капель.
Капли могут составляться вместе с водным или неводным основанием, также содержащим один или более диспергаторов, солюбилизаторов или суспнедирующих веществ. Жидкие составы для разбрызгивания удобно заключены в герметизированные пакеты.
Для введения ингаляцией соединения в соответствии с настоящим изобретением удобно доставляются из инсуффлятора, распылителя или герметизированного пакета либо другими пригодными средствами доставки аэрозолей. Герметизированные пакеты могут содержать пригодный диспергатор, такой как дихлордифторметан, трихлорфторметан, дихлортетрафторэтан, двуокись углерода или другой пригодный газ. В случае герметизированной аэрозоли дозировку можно определить путем снабжения клапаном с тем, чтобы доставить измеренное количество соединения.
Альтернативно для введения ингаляцией или инсуффляцией соединения в соответствии с настоящим изобретением могут принимать форму композиции сухого порошка, например порошковой смеси соединения и пригодного порошкового основания, такого как лактоза или крахмал. Порошковая композиция может быть представлена в лекарственной форме, например в капсулах или гильзах, или, например, в желатиновых или вытяжных прозрачных упаковках, из которых порошок может быть введен с помощью ингалятора или инсуффлятора.
При желании вышеописанные составы могут использоваться с получением пролонгированного высвобождения активного компонента.
Фармацевтические композиции в соответствии с настоящим изобретением также содержат другие активные компоненты, такие как противомикробные средства или консервирующие вещества.
Соединения настоящего изобретения могут также использоваться в комбинации с другими терапевтическими средствами, например другими противоинфекционными веществами. В частности, соединения настоящего изобретения могут быть использованы вместе с известными противовирусными веществами.
Таким образом, настоящее изобретение в дополнительном своем варианте предусматривает комбинацию, содержащую соединение формулы (I) или его физиологически приемлемое производное вместе с другим терапевтически активным веществом, в частности противовирусным веществом.
Упомянутые выше комбинации могут быть представлены для использования в форме фармацевтической прописи, и такие фармацевтические составы, содержащие определенную выше комбинацию, вместе с фармацевтически приемлемым носителем составляют дополнительный вариант настоящего изобретения.
Пригодные терапевтические агенты для использования в таких комбинациях включают ациклические нуклеозиды, такие как ацикловир, интерфероны, такие как интерферон, ренальные ингибиторы экскреции, такие как пробеницид, ингибиторы нуклеозидного транспорта, такие как дипиридамол, 2',3'-дидезоксинуклеозиды, такие как 2',3'-дидезоксицитидин, 2',3'-дидезоксиаденозин, 2',3'-дидезоксиинозин, 2', 3'-дидезокситимидин и 2',3'-дидезокси-2',3'-дидегидротимидин, а также иммуномодуляторы, такие как интерлейкин II (1L2) и гранулоцитарномакрофагальный колониестимулирующий фактор (GM-CSF), эритропоэтин и амплиген.
Отдельные компоненты таких комбинаций могут быть введены либо последовательно, либо одновременно в отдельных или совместных фармацевтических составах.
Когда соединение формулы (I) или его фармацевтически приемлемое производное используют в сочетании с вторым терапевтическим агентом, активным против того же самого вируса, доза каждого соединения может отличаться от той дозы, когда соединение используют отдельно. Подходящие дозы станут понятны специалисту.
Соединения формулы (I) и их фармацевтически приемлемые производные могут быть получены любым известным в данной области способом получения соединений аналогичной структуры.
Пригодными способами получения соединений формулы (I) и их фармацевтически приемлемых производных являются описываемые ниже; группы X и Z имеют вышеприведенные значения за исключением тех случаев, когда указаны иные значения. Следует понять, что следующие реакции могут потребовать использования или удобно могут быть приспособлены к исходным материалам, имеющим защищенные функциональные группы, и освобождение от защиты поэтому может потребоваться в качестве промежуточной или конечной стадии для получения желаемого соединения. Защиту и освобождение от защиты функциональных групп можно осуществлять с использованием традиционных способов. Так, например, аминогруппы могут быть защищены группой, выбранной из аралкила (например, бензила), ацила или арила (например, 2,4-динитрофенила); последующее отщепление защитной группы осуществляют при необходимости гидролизом или гидрогенолизом с использованием стандартных условий. Гидроксильные группы могут быть защищены с использованием любой традиционной гидроксильной защитной группы, например, как описано в работе "Защитные группы в органической химии", Ред. T.F.W. Mс Omie (Plenum Press, 1973) или работе "Защитные группы при органическом синтезе" Theodora W. Greene/Tohu Wiley and Sons, 1981). Примеры пригодных гидроксильных защитных групп включают группы, выбранные из алкила (например, метил, трет-бутил или метоксиметил), аралкила (например, бензил, дифенилметил или трифенилметил), гетероциклические группы, такие как тетрагидропиранил, ацильные (например, ацетил или бензоил) и силильные группы, такие как триалкилсилил (например, трет-бутилдиметилсилил). Гидроксильные защитные группы могут быть отщеплены традиционными методами. Так, например, алкильные, силильные, ацильные и гетероциклические группы могут быть отщеплены сольволизом, например гидролизом, в кислотных или основных условиях. Аралкильные группы, такие как трифенилметил, могут быть аналогично отщеплены сольволизом, например гидролизом, в кислотных условиях. Аралкильные группы, такие как бензил, могут быть отщеплены гидрогенолизом в присутствии катализатора благородного металла, такого как палладированный уголь. Силильные группы могут быть также отщеплены с использованием источника фторид-ионов, таких как тетра-н-бутиламмонийфторид.
В первом процессе (А) соединения формулы (I) и их фармацевтически приемлемые производные могут быть получены путем взаимодействия соединения формулы (II)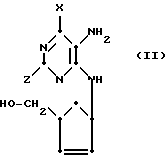
где
X и Z обозначают заместители, имеющие значение формулы (I), или обозначают их защитные формы, а гидроксильная группа в группировке циклопентенилкарбинола может быть в защитной форме или его фармацевтически приемлемого производного с реагентом, выбранным из муравьиной кислоты или ее реакционноспособных производных, с последующим, где необходимо, отщеплением нежелательных групп, вводимых таким реагентом, и/или отщеплением любых защитных групп.
Примеры пригодных производных муравьиной кислоты, которые могут быть использованы в процессе (A), включают ортоформиаты (например, триэтилортоформиат), диалкоксиметилацетаты (например, диэтоксиметилацетат), дитиомуравьиную кислоту, формамид, триазин или формамидинацетат.
Нежелательные группы, вводимые муравьиной кислотой или ее реакционноспособным производным, могут быть удобно отщеплены гидролизом в мягких условиях, например, с использованием неорганической кислоты, такой как водная хлористоводородная кислота.
Когда используют триалкилортоформиат, такой как триэтилортоформиат, он также представляет собой растворитель для реакции. Другие растворители, которые могут быть использованы, включают амиды (например, диметилформамид или диметилацетамид), хлорированные углеводороды (например, дихлорметан), простые эфиры (например, тетрагидрофуран) или нитрилы (например, ацетонитрил).
В некоторых случаях (например, когда используют триалкилортоформиат, такой как триэтилортоформиат) реакцию предпочтительно осуществлять в присутствии катализатора, такого как сильная кислота (например, концентрированная хлористоводородная кислота, азотная кислота или серная кислота). Реакцию можно осуществлять при температуре от -25 до 150oC, например, от 0 до 100oC, и удобно при температуре окружающей среды.
В другом процессе (B) соединения формулы (I) и их фармацевтически приемлемые производные или их защищенную форму подвергают реакции взаимопревращения, при которой заместитель X, присутствующий изначально, замещают другим заместителем X, и/или группу Z, присутствующую изначально, замещают другой группой Z и, если необходимо, затем отщепляют любую присутствующую защитную группу.
В одном варианте процесса (B) соединения формулы (I), в которых X обозначает группу RR1 (где R и R1 имеют вышеприведенные значения), могут быть получены аминированием соответствующего соединения формулы (I), в котором X обозначает атом галогена (например, хлора). Аминирование можно осуществлять путем взаимодействия с реагентом HNRR1 (где R и R1 имеют вышеприведенные значения) в растворителе, таком как спирт (например, метанол). Реакцию можно осуществлять при любой пригодной температуре и удобно при повышенной температуре, такой как температура флегмы, или, когда используют жидкий аммиак в запаянной трубке при температуре около 50 - 80oC. Пригодные условия для превращения галогенидов во вторичные и третичные амины также описаны Harrison et. al., Compendium of Organic Sinthetic Methods, Wiley-Jntesscience, Нью-Йорк (1971) на с. 250 - 252.
В другом варианте процесса (B) соединения формулы (I), в которых X обозначает группу OR (где R имеет вышеприведенное значение), могут быть получены замещением атома галогена (например, хлора) соответствующим анионом RO-. Когда R обозначает атом водорода, реакцию замещения можно осуществлять в воде или в смеси воды и смешивающегося с водой растворителя, такого как спирт (например, метанол или этанол), простой эфир (например, диоксан или тетрагидрофуран), кетон (например, ацетон), амид (например, диметилформамид) или сульфоксид (например, диметилсульфоксид), удобно в присутствии кислоты или основания. Пригодные кислоты включают органические кислоты, такие как п-толуолсульфокислота, и неорганические кислоты, такие как хлористоводородная кислота, азотная кислота или серная кислота. Пригодные основания включают неорганические основания, такие как гидроокиси или карбонаты щелочных металлов (например, гидроокись или карбонат натрия или калия). В качестве реакционного растворителя также можно использовать водную кислоту или основание. Гидролиз можно удобно осуществлять при температуре от -10 до +150oC, например, при температуре флегмы. Когда R обозначает C1-C4-алкил или арил, анион RO- образуют из соответствующего спирта ROH с использованием неорганического основания, такого как щелочной металл (например, натрий) или гидрид щелочного металла (например, гидрид натрия). Реакцию с образованием in situ анионом можно удобно осуществлять при температуре окружающей среды.
В другом варианте осуществления процесса (B) соединения формулы (I), в которых X обозначает группу SH, могут быть получены путем взаимодействия гало-соединения формулы (I) с тиомочевиной в пригодном растворителе, таком как спирт (например, н-пропанол), при повышенной температуре (например, температуре флегмы) с последующим основным гидролизом. Пригодные основания, которые могут быть использованы в данном случае, включают гидроокиси щелочных металлов (например, гидроокись натрия). Реакцию можно осуществлять в соответствии с методом G.G. Urquart и др. Org. Syn. Coll., том. 3, с. 363, 1953, например, нагреванием с обратным холодильником промежуточного продукта, используя водный раствор NaOH, в течение приблизительно 0,25 - 5 ч.
В дополнительном варианте осуществления процесса (B) соединения формулы (I), в которых X обозначает атом водорода, могут быть получены восстановлением гало-соединения формулы (I), используя восстановительную систему, которая не оказывает воздействия на покой молекулы. Пригодными восстановителями, которые могут быть использованы для осуществления требуемой реакции дегалогенирования, являются цинк/вода. Восстановление данного типа описано T. R. Marshall и др. J. Chem. Soc., 1004 (1951). Альтернативно реакцию можно проводить фотолизом в пригодном растворителе, таком как тетрагидрофуран, содержащий 10% триэтиламина, и удобно в реакторе фотохимическом Rayonet (2537A) в соответствии с методом V. Nair и др., J. Org. Chem., 52, 1344 (1987).
В еще одном варианте осуществления процесса (B) соединения формулы (I), в которых X обозначает атом галогена, могут быть получены из другого гало-соединения формулы (I) традиционными методами галогенид-галогенидного обмена. Альтернативно, когда X обозначает хлор, данный заместитель может быть замещен другими атомами галогена с использованием различных п-/гало/бензол-диазонийхлоридов в соответствии с хорошо известными методиками.
Соединения формулы (I), в которых X обозначает группу SR, где R обозначает группу C1-C4-алкила или арила, могут быть получены из соответствующих тиолов с использованием стандартных методик алкилирования или арилирования, например, как описано в патенте США N 438114.
Соединения формулы (I), в которых Z обозначает гидроксильную группу, могут быть удобно получены из соответствующего соединения формулы (I), в которых Z обозначает NH2, путем взаимодействия с азотистой кислотой, например, с использованием метода, описанного J. Davoll in J. Amer. Chem. Soc., 73, 3174 (1951).
Многие реакции, описанные выше, широко изложены в контексте пурин-нуклеозидного синтеза, например, в работе Nucleoside Analogs-Chemistry, Biology and Medical Applications, R. T. Walker и др., Ред. Пленум Пресс, Нью-Йорк (1979) на с. 193 - 223, описание которой введено сюда в качестве ссылки.
Фармацевтически приемлемые соли соединений настоящего изобретения могут быть получены, как описано в патенте США N 4383114, описание которого введено сюда в качестве отсылки. Так, например, когда необходимо получить кислую аддитивную соль соединения формулы (I), продукт любой из вышеуказанных методик может быть превращен в соль путем обработки полученного свободного основания пригодной кислотой с использованием традиционных методов. Фармацевтически приемлемые кислые аддитивные соли могут быть получены путем взаимодействия свободного основания с подходящей кислотой по выбору - в присутствии пригодного растворителя, такого как сложный эфир (например, этилацетат) или спирт (например, метанол, этанол или изопропанол). Неорганические основные соли могут быть получены путем взаимодействия свободного основания с пригодным основанием, таким как алкоксид (например, метоксид натрия), по выбору в присутствии растворителя, такого как спирт (например, метанол). Фармацевтически приемлемые соли могут быть также получены из других солей, включая другие фармацевтически приемлемые соли, соединений формулы (I) с использованием традиционных способов.
Соединение формулы (I) может быть превращено в фармацевтически приемлемый фосфат или другой сложный эфир путем взаимодействия с фосфорилирующим веществом, таким как POCl3, или пригодным эстерифицирующим веществом, таким как галоидангидрид или ангидрид кислоты. Сложный эфир или соль соединения формулы (I) можно превратить в родственное соединение, например, путем гидролиза.
Соединения формулы (II) и их соли представляют собой новые соединения, и они составляют дополнительный отличительный признак настоящего изобретения.
Соединения формулы (II), в которых Z обозначает водород или гидроксил, могут быть получены непосредственно из соединения 2a.
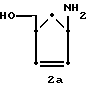
путем взаимодействия с избытком пиримидина формулы (III):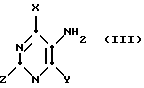
(где
Y обозначает атом галогена, например хлора, и Z обозначает водород или гидроксильную группу) в присутствии аминового основания, такого как триэтиламин, и в спиртовом растворителе (например, н-бутаноле), удобно при температуре флегмы.
Соединения формулы (II), в которых Z обозначает NH2, могут быть получены с использованием соединения формулы 2a путем взаимодействия с избытком пиримидина формулы (IV):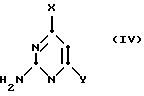
(где
Y обозначает то же, что и в формуле (III) выше) при таких же самых условиях, которые только что описаны для получения соединений формулы (II), в которых Z обозначает водород или гидроксильную группу, с получением соединения формулы (V):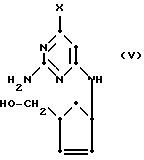
которое может быть диазотировано с использованием диазониевой соли ArN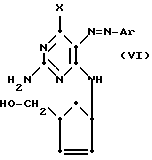
(где
Ar имеет вышеприведенное значение), которое можно превратить в желаемое соединение формулы (II) восстановлением с использованием, например, восстановительного металла, такого как цинк, в присутствии кислоты, например уксусной кислоты. Следует понять, что выбор восстановителя зависит от природы группы X.
Соединение 2a может быть получено из универсального предшественника, 1α -ацетиламино- 3α -ацетокси-метилциклопент-2-ена (1a), гидролизом в присутствии слабого основания, такого как гидроокись щелочноземельного металла.
Чрезвычайно удобный синтез соединений формулы (I) посредством 6-хлоросоединений формулы (II) приведен ниже.
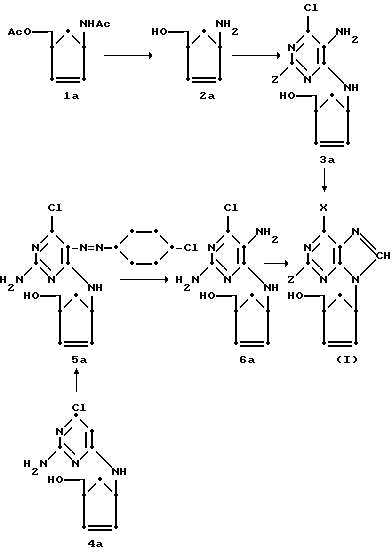
Соединение 2a и соединения формул (V) и (VI) являются новыми промежуточными соединениями и образуют дополнительные признаки настоящего изобретения.
Соединение 1a является известным соединением, описанным в патенте США N 4138562.
Когда соединение формулы (I) желательно иметь в виде простого изомера, оно может быть получено либо пептизацией конечного продукта, либо стереоспецифическим синтезом из изомерически чистого исходного вещества или любого подходящего промежуточного соединения.
Пептизацию конечного продукта или промежуточного, или исходного продукта можно осуществлять любым пригодным методом, известным в данной области знания: см., например, "Стереохимия углеродных соединений", E.L. Eliel (McGraw Hill, 1962) и "Таблицы пептизирующих агентов", написанные S.H.Wilen.
Одним удобным способом получения хирально чистых соединений формулы (I) является ферментативное превращение рацемической смеси соединения или его предшественника. При помощи такого способа (+) и (-) соединения формулы (I) могут быть получены в оптически чистой форме. Пригодные ферменты включают дезаминазы, такие как аденозиндезаминаза.
Настоящее изобретение далее описывается со ссылкой на следующие подробные примеры, в которых элементарный анализ был осуществлен при участии M-H-W Laboratories, Phoenix, AZ. Точки плавления были определены на аппарате Mel - Temp и откорректированы. Спектры ядерного магнитного резонанса были получены на спектрометрах Yed FX 90QFT или Nicollet NT300 и были записаны в DMCO-d6. Химические сдвиги выражены в млн. долях из Me4Si. ИК-спектры были определены в качестве гранул KBr спектрометром ИК Nicollet 50XC FT и УФ-спектры были определены на спектрофотометре Beckmann DU-8. Масс-спектры были получены масс-спектрометром AEI Scientific Apparatus Limited MS-30. Тонкослойную хроматографию (ТСХ) проводили на 0,25 мм слоях силикагеля Мерка (230-400 меш). Все химические вещества и растворители имеют чистоту реактива, если не указано что-либо иное. Термин "активный ингредиент", как он используется в примерах, означает соединение формулы (I) или его фармацевтически приемлемое производное.
Пример 1. (±)-( 1α,4α )-4-/5-Амино-6-хлор-4-пиримидинил) амино-2-циклопентенилкарбинол (3a)
Смесь 1α -ацетиламино- 3α -ацетоксиметилциклопент-2-ена (1a) (3,0 г, 15 ммоль) и водного раствора гидроокиси бария (0,5 н, 300 мл) нагревают с обратным холодильником в течение ночи. После охлаждения смесь нейтрализуют сухим льдом. Осадок отфильтровывают и водный раствор концентрируют до сухости. Остаток экстрагируют абсолютным этанолом и концентрируют вновь с получением соединения 2a в виде бесцветного сиропа (1,6 г, 14 ммоль).
К этому сиропу прибавляют 5-амино-4,6-дихлорпиримидин (4,59 г, 28 ммоль), триэтиламин (4,2 г, 42 ммоль) и н-бутанол (50 мл) и смесь нагревают с обратным холодильником в течение 24 ч. Летучие растворители удаляют, остаток абсорбируют на силикагеле (7 г), укладывая во флеш-колонну (4,0 • 12 см) и элюируют CHCl3-MeOH (20:1) с получением 2,69 г (74%) соединения 3a; температура плавления 130-132oC. Аналитический образец получают перекристаллизацией из этилацетата (EtOAc), температура плавления 134-135oC, масс-спектр (MC) (30 эВ, 200oC), m/e 240 и 242 (M+ и M++2), 209 (M+-31), 144 (B+),
ИК: 3600-2600 (OH), 1620, 1580 (C=C, C=N), Анал. (C10H13ClN4O) C, H, N.
Пример 2
(±)-( 1α,4α )-4-(/2-Амино-6-хлор-4-пиримидинил)/амино/-2-циклопентенилкарбинол (4a)
К 14 ммоль сырого соединения 2a (пример 1) прибавляют 2-амино-4,6-дихлорпиримидин (3,74 г, 22,8 ммоль), триэтиламин (15 мл) и н-бутанол (75 мл) и смесь нагревают с обратным холодильником в течении 48 ч. Летучие растворители удаляют, остаток обрабатывают метанолом с тем, чтобы отделить нерастворенный побочный продукт (двойной пиримидин-нуклеозид). Метаноловый раствор абсорбируют на силикагеле (8 г), упаковывают в колонку (4,0 • 14 см) и элюируют CHCl3-MeOH (40:1) с получением 1,52 г (42%) сырого соединения 4a. Продукт перекристаллизовывают из этилацетата с получением соединения 4a; температура плавления 132-134oC, MC (30 эВ, 200oC), m/e 240 и 242 (M+ и M++2), 209 (M+-31), 144 (B+),
ИК: 3600-3000 (NH2, OH), 1620, 1580 (C=C, C=N). Анал. (C10H13Cl-N4) C, H, N.
Пример 3.
(±)-( 1α,4α )-4-[/2-Амино-6-хлор-5-/4-хлорфенил/азо-4-пиримидинил-амино] -2-циклопентенилкарбинол (5a)
Холодный раствор диазопиевой соли получают из п-хлоранилина (1,47 г, 11,5 ммоль) в 3н. растворе HCI (25 мл) и нитрила натрия (870 мг, 12,5 ммоль) в воде (10 мл). Этот раствор прибавляют в смесь соединения 4a (2,40 г, 10 ммоль), уксусной кислоты (50 мл), воды (50 мл) и тригидрата ацетата натрия (20 г). Реакционную смесь перемешивают в течение ночи при комнатной температуре. Осадок желтого цвета фильтруют и промывают холодной водой до тех пор, пока не станет нейтральным, затем сушат на воздухе с получением 3,60 г (94%) соединения 5a; температура плавления 229oC (разложение). Аналитический образец получают из смеси ацетон-метанол (1:2) температура плавления 241-243oC (разложение). MC (30 эВ, 260oC), m/e 378 и 380 (M+ и M++2), 282 (B+),
ИК: 3600-3000 (NH2, OH), 1620, 1580 (C=C, C=N). Анал. (C16H16Cl2N6O) C, H, N.
Пример 4.
(±)-( 1α,4α )-4-[/2,5-Диамино-6-хлор-4-пиримидинил/амино] -2-циклопентенилкарбинол (6a)
Смесь соединения 5a (379 мг, 1 ммоль), цинкового порошка (0,65 г, 10 ммоль) уксусной кислоты (0,32 мл), воды (15 мл) и этанола (15 мл) нагревают с обратным холодильником под азотом в течение 3 ч. Цинк удаляют и растворители упаривают. Остаток абсорбируют на силикагеле (2 г), укладывают в колонну (2,0 • 18 см) и элюируют CHCl3-MeOH (15:1). Получают желтовато-зеленый сироп. Дальнейшая очистка из метанол-простого эфира приводит к получению соединения 6a в виде кристаллов желтовато-зеленого цвета, 170 мг (66%), температура плавления 168-170oC, MC (30 эВ, 220oC), m/e 225 и 257 (M+ и M++2), 224 (M+-31), 159 (B+),
ИК: 3600-3000 (NH2OH) 1620, 1580 (C=C, C=N). Анал. (C10H14ClN5) C, H, N.
Пример 5
(±)-( 1α,4α )-4-/6-Хлор-8H-пурин-9-ил/-2-циклопентатенилкарбинол (7a)
Смесь соединения 3a (1,30 г, 5,4 ммоль), триэтилортоформиата (30 мл) и хлористоводородной кислоты (12 н, 0,50 мл) перемешивают в течение ночи при комнатной температуре. Растворитель упаривают при температуре 35oC в вакууме. К остатку прибавляют 0,5 н. раствор (водный) хлористоводородной кислоты (30 мл) и смесь перемешивают в течение 1 ч, затем смесь нейтрализуют до pH 7-8 1 н. раствором гидроокиси натрия и абсорбируют на силикагеле (8 г), укладывают в колонну (4,0 • 8 см) и элюируют CHCl3-MeOH (20:1) с получением кристаллов белого цвета соединения 7a, 1,12 г (82%). Сырой продукт перекристаллизовывают из этилацетата с получением соединения 7a, температура плавления 108-110oC, MC (30 эВ, 200oC), m/e 250 и 252 (M+ и M++2), 219 (M+-31), 154 (B+),
ИК: 3600-2800 (OH), 1600 (C=C, C=N). Анал. (C11H11ClN4O) C, H, N.
Пример 6.
(±)-( 1α,4α )-4-/6-Гидрокси-9H-пурин-9ил/-2-циклопентенилкарбинол (8a)
Смесь соединения (7a) (251 мг, 1 ммоль) и водного раствора гидроокиси натрия (0,2 н, 10 мл) нагревают с обратным холодильником в течение 3 ч. После охлаждения реакционную смесь доводят до pH 5-6 с использованием уксусной кислоты. Реакционную смесь абсорбируют на силикагеле (2 г), укладывают в колонну (2,0 • 11 см) и элюируют CHCl3-MeOH (10:1) с получением 105 мг (45%) соединения 8a. Сырой продукт белого цвета перекристаллизовывают из вода-метанол (3: 1) с получением соединения 8a, температура плавления 248-250oC (разложение), MC (30 эВ, 300oC), m/e 232 (M+), 214 (М+-18), 136 (B+),
ИК: 3600-2600 (OH), 1680, 1600 (C=O, C=C, C=N). Анал. (C11H12N4O2) C, H, N.
Пример 7
(±)-/ 1α,4α - /-4-/-6-Амино-8H-пурин-9-ил/-2-циклопентенилкарбинол (9а)
Жидкий аммиак пропускают в автоклав, содержащий раствор соединения 7a (250 мг, 1 ммоль) в метаноле ( 5 мл) при температуре -80oC. Автоклав герметизируют и нагревают при температуре 60oC в течение 24 ч. Аммиак и метанол упаривают и остаток перекристаллизовывают из воды с получением не совсем белых кристаллов соединения 9a, 187 мг (81%), температура плавления 198-200oC. МС (30 эВ, 210oC), m/e 231 (M+), 213(M + -18), 135 (B+).
ИК: 3600-2600 (NH2, OH), 1700, 1600 (C=C, C=N). Анал. (C11H13N5O) C, H, N.
Пример 8
(±)-( 1α,4α ) -4-(6-Меркапто-9H-пурин-9-ил)-2-циклопентенилкарбинол (10a)
Смесь соединения 7a (125 мг, 0,5 ммоль), тиомочевины (40 мг, 0,64 ммоль) и н-пропанола (5 мл) нагревают с обратным холодильником в течение 2 ч. После охлаждения осадок выделяют фильтрацией, промывают н-пропанолом и растворяют в гидроокиси натрия (1 н. раствор, 5 мл). Раствор доводят до pH 5 с использованием для этой цели уксусной кислоты. Сырое соединение 10a (90 мг, 73%) выделяют вновь, температура плавления 260-262oC (разложение), и перекристаллизовывают из N,N-диметилформамида с получением соединения 10a, температура плавления 263-265oC (разложение). МС (30 эВ, 290oC): m/e 248 (M+), 230 (M+-18), 152 (B+),
ИК: 3600-3200 (OH), 3100, 2400 (SH), 1600 (C=C, C=N). Анал. (C11H12N4OS), C, H, N.
Пример 9.
(±)-( 1α,4α ) -4-(2-Амино-6-хлор-9H-пурин-9-ил)-2-циклопентенилкарбинол (13a).
Смесь соединения 6a (1,41 г, 5,5 ммоль), триэтилортоформиата (30 мл) и хлористоводородной кислоты (12 н, 1,40 мл) перемешивают в течение ночи. Суспензию сушат в вакууме. Прибавляют разведенную хлористоводородную кислоту (0,5 н, 40 мл) и смесь подвергают взаимодействию при комнатной температуре в течение 1 ч. Смесь нейтрализуют до pH 8 с использованием 1 н. раствора гидроокиси натрия и абсорбируют на силикагеле (7,5 г), укладывают в колонну (4,0•10 см) и элюируют CHCl3-MeOH (20: 1) с получением не совсем белых кристаллов соединения 13a, 1,18 г (80%). Сырой продукт перекристаллизуют из этанола с получением соединения 13a, температура плавления 145-147oC. MC (30 эВ, 220oC): m/e 265 и 267 (M+ и M+ +2), 235 (M+- 30), 169 (B+),
ИК: 3600-2600 (NH2, OH), 1620, 1580 (C=C, C=N). Анал. (C11H12-N5OCl•3/4 H2O)C, H, N.
Пример 10.
(±)-( 1α,4α ) -4-(2-Амино-6-гидрокси-9H-пурин-9-ил)-2-циклопентенилкарбинол (14a).
Смесь соединения 13a (266 мг, 1 ммоль) и водного раствора гидроокиси натрия (0,33 н.) нагревают с обратным холодильником в течение 5 ч, абсорбируют на силикагеле (2 г), укладывают в колонне (2,0•7,5 см) и элюируют CHCl3-MEOH (5:1). Сырой продукт перекристаллизовывают из смеси метанол-вода (1: 4) с получением белых кристаллов соединения 14a, 152 мг (61%), температура плавления 254-256oC (разложение). MC (30 эВ, 200oC): m/e 247 (M+), 217 (M+ -30), 151 (B+),
ИК: 3600-2600 (NH2, OH), 1700, 1600 (C=O, C=C, C=N), Анал. (C11H13N5O2•3/4 H2O) C, H, N.
Пример 11
(±)-( 1α,4α ) -4-(2,6-Диамино-9H-пурин-9-ил)-2-циклопентилкарбинол (15а).
Жидкий аммиак пропускают в раствор соединения 13a (265 мг, 1ммоль) в метаноле (10 мл) при температуре -80oC в автоклаве. Автоклав герметизируют и нагревают при температуре 75oC в течение 48 ч. Аммиак и метанол упаривают. Остаток абсорбируют на силикагеле (2 г), укладывают в колонну (2,0•10 см) и элюируют CHCl3-MeOH (15:1). Сырой продукт перекристаллизовывают из этанола с получением 196 мг (80%) соединения 15а, температура плавления 152-155oC. MC (30 эВ, 200oC), m/e 246 (M+), 229 (M+ -17), 216 (M+ -30), 150 (B+),
ИК: 3600-3000 (NH2, OH) 1700, 1650, 1600 (C= O, C=C, C=N). Анал. (C11H14N6O) C, H, N.
Пример 12.
(1S, 4R)-4-(2,6-Диамино-9H-пурин-9-ил/-2-циклопентенилкарбинол[/1S, 4R/-4-/2,6-Диамино-9H-пурин-9-ил/-2-циколопентенметанол]
(а) Промежуточное соединение I: /1R, 2S, 3R, 5R/-3-[6-Амино-9H-пурин-9-ил/]-5-[/1,1-диметилэтил/- диметилсилилокси/метил]-1,2-циклопентандиол
(-) Аристеромицин1 (12,505 г), трет-бутилдиметилсилилхлорид (7,8 г) и имидазол (12,96 г) в сухом диметилформамиде (85 мл) перемешивали при температуре окружающей среды в течение 2,5 ч. Полученный раствор разбавляют этилацетатом (500 мл) затем промывают водой (3•100 мл) и солевым раствором (50 мл), что приводит к выкристаллизовыванию белого твердого тела. Данное тело собирают фильтрацией, промывают этилацетатом, затем сушат в вакууме с получением указанного в заголовке соединения (3,92 г).
1H ЯМР (DMCO-d6) 8,15 (8,09 1H), 7,19 (2H), 5,0 (1H), 4,72 (1H), 4,69 (1H), 4,36 (1H), 3,85 (1H), 3,67 (2H), 2,23 (1H), 2,09 (1H), 1,79 (1H), 0,89 (9H), 0,07 (6H).
1. J. Am. Chem. Society, 1983, том 105, с. 4049-4055.
(b) Промежуточное соединение 2: (4R, 3a, 6R, 6aR)-4-[6-Амино-9H-пурин-9-ил] -6-[/1,1-диметилэтил/- диметилсилилокси/метил]-3a,5,6,6a-тетрагидро-4H-циклопента-1,3- диоксол-2-тион.
Перемешанную суспензию промежуточного соединения 1 (3,45 г) в сухом диметилформамиде (56 мл) обрабатывают 1,1'-тиокарбонилдиимидазолом (3,3 г), получая раствор желтого цвета. Через 15,5 ч при температуре окружающей среды полученный раствор объединяют с раствором, оставшимся от ранее проведенного эксперимента (6% шкала), и растворитель удаляют упариванием. Остаточное масло разбавляют этилацетатом (100 мл), затем промывают водой (2•20 мл) и солевым раствором (2•20 мл), сушат при сульфате магния и упаривают до твердого вещества желтого цвета, которое промывают простым диэтиловым эфиром (25 мл), затем собирают фильтрацией, далее промывают простым эфиром (25 мл) и сушат в вакууме с получением указанного в заголовке соединения в виде светло-кремового твердого вещества (3,61 г).
λmax (этанол) 240,0 нм ( E
(c) Промежуточное соединение 3:/I'R, 4'6/-9-[4-///1,1- Диметилсилилокси/метил/-2-циклопентен-1-ил]-9H-пурин-6-амин
Раствор промежуточного соединения 2 (3,57 г) в сухом тетрагидрофуране (25 мл) обрабатывают раствором 1,3-диметил-2-фенил-1,3,2-диазафосфолидина (4,94 г) в сухом тетрагидрофуране (10 мл), затем перемешивают при температуре окружающей среды в течение 8,25 ч. Растворитель удаляют упариванием. Остаточное масло объединяют с маслом, полученным в результате предыдущего эксперимента (40% шкала), затем подвергают колоночной хроматографии на кремнеземе (200 г, МЕРК 7734), элюируют хлороформом, затем смесью хлороформ-этанол, получая твердое вещество белого цвета. Данное вещество промывают простым диэтиловым эфиром (10 мл), сушат в вакууме с получением указанного в заголовке соединения (1,47 г).
λmax (этанол) 261,4 нм E
1H ЯМР (DMCO-d6), 8,14 (1H), 8,00 (1H), 7,20 (2H), 6,12 (1H), 5,95 (1H), 5,60 (1H), 3,66 (2H), 2,96 (1H), 2,69 (1H), 1,65 (1H), 0,74 (9H), 0,02 (6H).
(d) Промежуточное соединение 4: /1', R, 4' S/-9[4-///1, 1-Диметилэтил/диметилсилилокси/метил/-2-циклопентен-1-ил]-9H-пурин- 6-амин, 1-оксид
Раствор промежуточного соединения 3 (1,37 г) в хлороформе (30 мл) обрабатывают 80-90%-ной м-хлорпероксибензойной кислотой (1,29), затем перемешивают при температуре окружающей среды в течение 3 ч. Растворитель удаляют упариванием и остаточную камедь растворяют в этилацетате (10 мл). Выкристаллизовывается твердое тело белого цвета, которое (а вместе с ним и вещество), восстановленное упариванием фильтрата, растворяют в хлороформе (100 мл), затем промывают насыщенным водным раствором бикарбоната натрия (3 • 10 мл) и соленым раствором ( 2• 10 мл). Водные промывки подвергают обратной экстракции хлороформом (50 мл). Объединенные органические растворы сушат в присутствии сульфата магния, затем упаривают до твердого тела, которое промывают простым диэтиловым эфиром (25 мл), после чего собирают фильтрацией. Белое твердое вещество далее промывают простым эфиром (10 мл), сушат в вакууме, получая в результате этого указанное в заголовке соединение (1,16 г).
λmax (этанол) 235,4 нм E
1H -ЯМР(CDCl3) 8,72 (1H), 8,02 (1H), 7,16 (2H), 6,21 (1H), 5,87 (1H), 5,72 (1H), 3,68 (2H), 3,04 (1H), 2,82 (1H), 1,74 (1H) 0,89 (9H), 0,06 (6H).
(e) Промежуточное соединение 5: (I', R, 4' S )-7-[4-,///1,1-диметилэтил/диметилсилилокси/метил/-2-циклопентен-1-ил] -2-имино-1,2-дигидро [1,2,4] оксадиазоло [3,2-i]-9H-пурин-бромгидрат
Перемешанную, охлажденную на льду суспензию промежуточного соединения 4 (1,08 г) в метаноле (20 мл) обрабатывают раствором цианогенбромида (0,34 г) в метаноле (20 мл), добавленным в течение 5 мин. Через 15 мин суспензию нагревают до температуры окружающей среды, получая раствор. Через 90 мин растворитель удаляют упариванием. Остаток промывают простым диэтиловым эфиром (25 мл), собирают фильтрацией. Твердое тело промывают затем простым эфиром (25 мл) и сушат в вакууме с получением указанного в заголовке соединения (1,37),
λmax (этанол) 228,2 нм ( E
1H -ЯМР (CDCl3) 10,20 (1H), 10,02 (1H), 8,37 (1H), 6,25 (1H), 6,01 (1H), 5,90 (1H), 3,69 (2H), 3,05 (1H), 2,86 (1H), 1,73 (1H), 0,86 (9H), 0,03 (6H).
(f) Промежуточное соединение 6: (I', R, 4'S)-9-[4-///1,1-диметилэтил/диметилсилилокси/метил/-2-циклопентен-1-ил] -6-цианоимино-1,6-дигидро-1-метокси-9H-пурин
Раствор промежуточного соединения 5 (1,36 г) в диметилформамиде (10 мл) перемешивают при температуре окружающей среды, затем обрабатывают триэтиламино (1,2 мл). Через 40 мин прибавляют в иодометан (0,54 мл), что приводит к образованию раствора желтого цвета. Через 3 ч 45 мин растворитель удаляют упариванием. Остаток распределяют между этилацетатом (100 мл) и водой (20 мл). Органический раствор промывают водой (2 • 20 мл) и солевым раствором (20 мл), сушат в присутствии сульфата магния и упаривают до твердого тела. Это твердое тело промывают простым диэтиловым эфиром (25 мл), после чего собирают фильтрацией. Затем это твердое тело белого цвета промывают простым эфиром (10 мл), сушат в вакууме с получением указанного в заголовке соединения (0,865 г),
λmax (этанол) 227,2 нм ( E
1H-ЯМР 8,23 (1H), 7,96 (1H), 6,24 (1H), 5,85 (1H), 5,65 (1H), 4,21 (3H), 3,66 (2H), 3,04 (1H), 2,77 (1H), 1,68 (1H), 0,88 (9H), 0,05 (6H).
(g) Промежуточное соединение 7: (1', R, 4'S)-9-[4-///1, 1-диметилэтил/диметилсилилокси/метил/-2-цклопентен-1-ил-6-метокси-амино-9H-пурин-2-амин.
Раствор промежуточного соединения 6 (802 мг) и 1,8-диазабицикло [5,4,0] ундец-7-ена (0,45 мл) в этаноле (80 мл) перемешивают и нагревают с обратным холодильником. Нагревание прекращают через 9 ч и раствор оставляют при температуре окружающей среды на ночь. Растворитель удаляют упариванием. Остаточное масло объединяют с тем, которое получено в результате предыдущего эксперимента (4% шкала), затем подвергают колоночной хроматографии на кремнеземе (40 г, Мерк 9385), элюируя хлороформом, затем смесью хлороформ-этанол с получением пены. Эту пену порошкуют простым диэтиловым эфиром (10 мл), и полученное твердое тело собирают фильтрацией. Твердое тело затем промывают простым эфиром (5 мл), сушат в вакууме, что приводит к получению указанного в заголовке соединения (594 мг),
λmax (этанол) 282,2 нм ( E
1H-ЯМР (DMCO-d6) 9,76 (1H),7,32,(1H), 6,53 (2H), 6,08 (1H), 5,26 (1H), 3,72 (3H), 3,61 (2H), 2,90 (1H), 2,50 (1H) 1,52 (1H), 0,83 (9H), 0,02(6H).
(h) Промежуточное соединение 8: /1S, 4 R/-4-[2-амино-6-метоксиамино-9H-пурин-9-ил]-2-циклопентен-метанол
Раствор промежуточного соединения 7 (356 мг) в тетрагидрофуране (35 мл) перемешивают при температуре окружающей среды, затем обрабатывают тетрабутиламмонийфторидом (1,0 М раствор в тетрагидрофуране, 1,4 мл). Через 90 мин реакционную смесь охлаждают водой (1 мл), затем растворители удаляют упариванием. Остаточное масло подвергают колоночной хроматографии на кремнеземе (20 г, Мерк 7734), элюируют хлороформом, затем смесью хлороформ-этанол с получением указанного в заголовке продукта в виде твердого тела (243 мг),
λmax (pH 6 буфер) 250,2 нм ( E
1H-ЯМР (DMCO-d6) 9,75 (1H), 7,39 (1H), 6,52 (2H), 6,10 (1H), 5,84 (1H), 5,27 (1H), 4,73 (1H), 3,40 (2H), 2,83 (1H), 2,55 (1H), 1,52 (1H).
/1S, 4P/-4-[2,6-Диамино-9H-пурин-9-ил]-2-циклопентенкарбинол.
Перемешанный, охлажденный на льду раствор промежуточного соединения 6 (210 мг) в воде (10 мл) и тетрагидрофуране (50 мл) обрабатывают амальгамой (из алюминия (237 мг) и 0,5%-ного водного раствора хлористой ртути), прибавляемой маленькими порциями за 15 мин. Через 40 мин перемешанную смесь нагревают до температуры окружающей среды. Через 15 мин полученную смесь фильтруют через кизельгур с тем, чтобы удалить нерастворимые вещества. Их промывают смесью вода : тетрагидрофуран (1:5, 60 мл). Объединенные фильтраты упаривают. Остаток подвергают колоночной хроматографии на кремнеземе (10 г, Мерк 9385), элюируют смесью хлороформ-этанол с получением указанного в заголовке продукта в виде пены (159 мг),
[α]D -81o (с, 1,04, метанол);
λmax (pH 6 буфер) 255,0 нм ( E
1H-ЯМР (DMCO-d6) 7,61 (1H), 6,66 (2H), 6,10 (1H), 5,87 (1H), 5,76 (2H), 5,38 (1H), 4,76 (1H), 3,45 (2H), 2,87 (1H), 2,60 (1H), 1,60 (1H).
Пример 13
/1S, 4R/-4-/2-Амино-6-гидрокси-9H-пурин-9-ил-/- 2-циклопентенилкарбинол
/1', R, 4'S/-2-Амино-1,9-дигидро-9-[4-гидроксиметил-2-циклопентен-1-ил]- 6H-пурин-6-он
Мутный раствор указанного в заголовке примера 12 соединения (144 мг) в 0,1 M, pH 6 буфере (10 мл) (из 28,4 г динатрийортофосфата в 2 л воды, доведенных ортофосфорной кислотой) обрабатывают раствором аденозин-дезаминазы (0,5 мл, 778 единиц) в 50%-ной смеси глицерин - 0,01 M фосфат калия, pH 6,0, затем перемешивают и нагревают до температуры 37oC. Через 18,5 ч полученную суспензию охлаждают. Собранное твердое тело перекристаллизовывают из воды с получением указанного в заголовке продукта в виде твердого тела белого цвета (86 мг),
[α]D -49o (с, 0,5, диметилсульфоксид),
λmax (pH 6 буфер) 252,6 нм E
1H-ЯМР (DMCO-d6) 10,60 (1H), 7,60 (1H), 6,47 (2H), 6,10 (1H), 5,86 (1H), 5,33 (1H), 4,72 (1H), 3,45 (2H), 2,59 (1H), 1,58 (1H).
Пример 14
Получение энантиомеров ( 1α,4α )-4-(2-Амино-6-гидрокси-9H-пурин- 9-ил/-2-циклопентенкарбинола
(а) /1S, 4R/-4-/2-Амино-6-гидрокси-9H-пурин-9-ил/-2-циклопентенил-карбинол
Диаминоаналог (100 г) (пример 11) растворяют в 3 мл, 0,05 M K2PO4 буфера (pH 7,4) с нагреванием (50oC). Раствор охлаждают до комнатной температуры и 40 единиц аденозин-дезаминазы (Сигма, Тип VI, телячья кишечная слизистая оболочка) прибавляют в раствор, который инкубируют в течение трех дней при комнатной температуре, после чего образованный осадок удаляют фильтрацией, получая при этом 18,2 мг. Фильтрат концентрируют до 1,5 мл и охлаждают в течение 2 дн. При фильтрации получают дополнительное количество (26,8 мг) твердого тела. Две фракции твердого тела перекристаллизовывают из воды с получением чистого продукта, указанного в заголовке, температура плавления 269-272oC, [α]
(b) /1R, 4S/-4-/2-Амино-6-гидрокси-9H-пурин-9-ил/-2-циклопентенил-карбинол
Фильтраты в результате получения 1S,4R-изомера (пример 14а) объединяют и упаривают до сухости. Неизменный диаминоисходный материал разделяют на флэш-колонне с силикагелем, используя 10%-ную смесь метанол/хлороформ. Диамино-соединение растворяют в 0,05M K2PO4 буфере, pH 7,4 (15 мл), и 800 единиц аденозин-дезаминазы прибавляют в раствор, который инкубируют при температуре 37oC в течение 96 ч. Тонкослойная хроматография отмечает наличие некоторого количества непрореагировавшего продукта. Раствор нагревают в кипящей воде в течение 3 мин и фильтруют с тем, чтобы удалить денатурированный белок. Затем прибавляют еще 800 ед. аденозин-дезаминазы и процесс повторяют. Депротеинированный раствор упаривают до сухости и продукт кристаллизуют из воды. Указанное в заголовке соединение в виде твердого тела белого цвета собирают фильтрацией из воды, температура плавления 265-270oC.
[α]
Пример 15
/±/-/ 1α,4α /-4-/2-Амино-6-гидрокси-9H-пурин-9-ил/-2-циклопентенилацетоксикарбинол
К суспензии продукта примера 10 (130 мг, 0,50 ммоль) и 4-диметиламинопиридина (5 мг, 0,04 ммоль) в смеси ацетонитрила (6 мл) и триэтиламина (0,09 мл, 0,66 ммоль) прибавляют ангидрид уксусной кислоты (0,06 мл, 0,6 ммоль). Смесь перемешивают при комнатной температуре в течение 3 ч. Метанол (1 мл) прибавляют с тем, чтобы охладить реакционную смесь. Раствор концентрируют и абсорбируют на силикагеле (1,5 мг), укладывают в колонну (2,0 • 12 см), элюируют CHCl3-MeOH (20 : 1). Фракции продукта собирают и концентрируют с получением твердого тела белого цвета. Твердый продукт промывают смесью MeOH-AcOEt: выход 123 мл (85%). Дальнейшая очистка из метанола приводит к получению указанного в заголовке соединения в виде игольчатых кристаллов, температура плавления 237 - 239oC. Анал. (C13H15N5O3) C, H, N.
Пример 16
/1S,4R/-4-[2-Амино-9H-пурин-9-ил]-2-циклопентенилкарбинол
Перемешанный, охлажденный на льду раствор (1S,4R/-4-[2-амино-6-метоксиамино-9H-пурин-9-ил] -2-циклопентен-метанола (промежуточное соединение 8, пример 12) (1,202 г) в тетрагидрофуране (250 мл) и воде (50 мл) обрабатывают амальгамой алюминия (из алюминия (1,761 г) и 0,5%-ного водного раствора хлористой ртути), прибавляемой маленькими порциями за 1 ч 47 мин. Через 35 мин перемешанную смесь нагревают до температуры окружающей среды. Через 16 ч 50 мин прибавляют еще амальгаму алюминия (из 235 мг алюминия) в течение 14 мин. Через 4 ч 10 мин полученную смесь фильтруют через кизельгур с тем, чтобы удалить нерастворимые тела, которые промывают смесью тетрагидрофуран : вода (5: 1, 300 мл). Объединенные фильтраты упаривают с получением желтой пены. Пену подвергают колоночной хроматографии на кремнеземе (33,8 г, Мерк 7734) в хлороформе и элюируют смесью хлороформ-этанол с получением нескольких фракций (578 мг, 420 мг и 40 мг). Две наибольшие фракции отдельно кристаллизуют из изопропанола. Фильтраты объединяют с наиболее мелкой колоночной фракцией и подвергают препаративной тонкослойной хроматографии (Мерк 5717), проявляя три раза в смеси хлороформ : метанол (10 : 1). Пластинки элюируют смесью этилацетатэтанол (1 : 1) с получением твердого тела коричневого цвета (45 мг). Твердое тело подвергают колоночной хроматографии на кремнеземе (2,7, Мерк 7734) в хлороформе и элюируют смесями хлороформ-метанол-триэтиламин с получением камеди (17 мг). Вслед за неудавшейся кристаллизацией из изопропанола и обработкой древесным углем в метаноле водный раствор восстановленного вещества сушат вымораживанием с получением указанного в заголовке соединения - 15 мг.
1H-ЯМР (DMCO-d6) 1,62 (1H), 2,63 (1H), 2,89 (1H), 3,45 (2H): 4,73 (1H), 5,48 (1H), 5,91 (1H), 6,14 (1H), 6,50 (2H), 7,98 (1H), 8,57 (1H).
Масс-спектр: [MH]+ 232.
Пример 17
Таблетированные препараты
A. Следующий препарат получен мокрым гранулированием ингредиентов и раствора повидона в воде, сушкой и просеиванием с последующим добавлением стеарата магния и прессованием (мг-таблетка):
(а) Активный ингредиент - 250
(б) Лактоза B.P. (British Pharmacopеia - британская фармакопея) - 210
(в) Повидон B.P. - 15
(г) Натриевая соль карбоксиметилкрахмала - 20
(д) Стеарат магния - 5 - 500
B. Следующий препарат получен прямым прессованием, используют тип лактозы для прямого прессования (мг/таблетка):
(а) Активный ингредиент - 250
(б) Лактоза - 145
(в) Авицел - 100
(д) Стеарат магния - 5 - 500
C. (Препарат с контролируемым выделением). Препарат получен мокрым гранулированием указанных ниже ингредиентов с раствором повидона в воде, сушкой и просеиванием, с последующим добавлением стеарата магния и прессованием (мг/таблетка):
(а) Активный ингредиент - 500
(б) Гидроксипропилметилцеллюлоза (Methocel k4m Premium) - 112
(в) Лактоза B.P. - 53
(г) Повидон B.P. - 28
(д) Стеарат магния - 7 - 700
Пример 18
Капсулированный препарат
Капсулированный препарат получают смешением указанных ниже ингредиентов и заполнением твердых желатиновых капсул, состоящих из двух частей, (мг/таблетка):
Активный ингредиент - 125
Лактоза - 72,5
Авицел - 50
Стеарат магния - 2,5 - 500
Пример 19
Препарат для инъекций
Активный ингредиент - 0,200 г
0,1 M Раствор гидроксида натрия - до pH около 11
Стерильная вода - до 10 мл
Активный ингредиент суспендируют в воде, которая может быть теплой, и с помощью гидроксида натрия доводят pH приблизительно до 11. Порцию затем доводят до требуемого объема и фильтруют через стерильный мембранный фильтр в стерильный стеклянный пузырек объемом 10 мл, герметично закрывают стерильной пробкой и поверх - металлической крышкой.
Пример 20
Свечи (мг):
Активный ингредиент (63 мкм) - 250
Твердый жир B.P. - 1770 - 2020
Одну пятую часть твердого жира растапливают в бане, снабженной паронагреваемой рубашкой, при температуре не выше 45oC. Активный ингредиент просеивают через сито (200 мкм), добавляют к расплавленной основе и перемешивают до получения мягкой дисперсии с использованием высокоэффективной мешалки. Оставляют смесь при 45oC, добавляют к суспензии оставшуюся часть твердого жира и перемешивают до получения гомогенной смеси. Продолжая перемешивание, всю суспензию пропускают через сито из нержавеющей стали (250 мкм) и дают охладиться до 40oC. При температуре смеси от 38 до 40oC подходящие пластиковые формы объемом в 2 мл заполняют 2,02 г смеси и дают охладиться до комнатной температуры.
Пример 21
Антивирусная активность
(А) Анализ на энти-HIY
Соединения формулы (I) подвергали скринингу на энти-HIY активность в Национальном раковом институте на исследовательской аппаратуре изучения раковых заболеваний Фредерика, Фредерика Мэрилэнд (FCPF). Далее описывают современные операционные методы скрининга, используемые в FCPF. Протокол состоит из трех разделов:
(I) получение инфицированных клеток и распределение их на тестовых пластинках;
(II) приготовление пластинок с разбавленным лекарством и распределение на тестовых пластинках; и
(III) процедура XTT анализа.
См. работу Д.А. Скудьеро с сотр. "Новый упрощенный анализ из тетрозолий для роста клеток и определения чувствительности к лекарствам в культуре". Cancer Res. 48 3827 (1988).
I. Инфицирование и распределение ATH8 клеток на микротитрометрических лотках
Клетки, подлежащие инфицированию (нормальная лимфобластоидная линия, которая экспрессирует СД4), помещали в 50 мл конические пробирки центрифуги и в течение 1 ч обрабатывали 1-2 мкг/мл полибрена при 37oC. Затем клетки в течение 8 мин центрифугировали при скорости вращения 1200 об/мин. Вирус HIY, разбавленный в соотношении 1:10 средой (RMRI-1640, 10% человеческой сыворотки или 15% сыворотки телячьего плода - FCS), в присутствии 1α-2- и антибиотиков) добавляли в систему для обеспечения MOI порядка 0,001. К контрольным клеткам, не содержащим вируса, добавляли только среду. Полагая титр инфицирующего вируса равным 10-4, значение MOI, равное 0,001, представляет собой 8 инфицирующих вирусных частиц на 10000 клеток. Примерно 500000 кл. /пробирку экспонировали 400 мкл вирусного разбавителя. Полученную в результате смесь инкубировали в течение 1 ч при 37oC в атмосфере: воздух - CO2. Инфицированные или неинфицированные клетки разбавляли до значения 1 • 10-4 (человеческой сыворотки) или 2 • 10-4 (сывороткой телячьего плода) кл. /100 мкл.
Инфицированные или неинфицированные клетки (100 мкл) распределяли по соответствующим углублениям микротитрометрической пластинки с V-образным днищем, содержащей 96 углублений. Каждое разбавление соединения тестировали инфицированными клетками, причем опыты дублировали. Неинфицированные клетки исследовали на чувствительность к лекарствам в единичном углублении для каждого разбавления соединения. Опыты с контрольными клетками, как инфицированными, так и неинфицированными, повторяли три раза. Углубления B2-G2 служили контролями на реагент, в них помещали только среду. Пластинки инкубировали при 37oC в атмосфере: воздух - CO2 перед давлением лекарства.
II. Разбавление и добавление лекарства
Пластинки для разбавления (микротитрометрические пластинки с плоским дном, содержащие 96 углублений) в течение ночи обрабатывали фосфатным буфферным раствором (PBS) или средой, содержащей по крайней мере 1% FCS или 1% человеческой сыворотки (в зависимости от среды, используемой в испытании), начиная со дня, предшествующего анализу. Использовали методику "блокирования" с целью ограничения адсорбции лекарства на микротитрометрической пластине в ходе процесса разбавления. Углубления полностью заполняли блокирующим раствором и выстаивали при комнатной температуре в увлажненной камере под колпаком.
Процесс разбавления начинали путем первого разбавления испытуемого соединения до соотношения 1:20. Заблокированные разбавительные пластинки готовили путем вытряхивания блокирующего раствора и промокания досуха стерильной марлей. Затем все углубления в каждой пластине заполняли 225 мкл соответствующей среды с использованием системы Cetus для обращения с жидкостями. Затем по 25 мкл каждого из соединений, разбавленных в соотношении 1:20, вручную добавляли в ряд А заблокированной и заполненной разбавительной пластинки. На разбавительную пластинку добавляли по четыре соединения, что достаточно для двух испытательных пластинок. Затем эти четыре соединения подвергли 10-кратному серийному разбавлению от ряда A до H с использованием системы Цетуса. В этот момент исходное разбавление каждого соединения в ряду A составило 1:200. Пластинки с разбавлениями хранили на льду до использования.
С использованием многоканальной пипетки с 6 микронаконечниками 100 мкл каждого разбавителя лекарства переносили на испытательную пластинку, которая уже содержала 100 мкл среды и клетки. Конечное разбавление на испытательной пластинке начиналось со значения 1:400 (углубления B4-G4). Такое разбавление (до 0,25% DMCO) предотвращает отрицательное влияние DMCO на рост клеток. К несодержащим лекарства инфицированным и неинфицированным клеткам (углубления B3-G3) и контрольным реагентам (B2-G2) добавляли только среду. Два оставшихся соединения затем переносили на углубления H7-H12 на вторую испытательную пластинку с использованием аналогичной методики. Пластинки инкубировали при 37oC в атмосфере воздуха - CO2 в течение 7-14 дн или до лизирования вирусных контролей согласно макроскопическим определениям.
III. Количественная оценка вирусной цитопатогенности и активности лекарства
A. Материалы
1. Раствор 2,3-бис/2-метокси-4-нитро-5-сульфофенил/-5-/(фениламино)карбонил/-2H-тетразолий гидрокси, (ХТТ) - раствор с концентрацией 1 мг/мл в среде, не содержащей FCS. Раствор хранили при 4oC. Готовили еженедельно.
2. Основной раствор метосульфоната феназина (PMS). Этот раствор может быть приготовлен заранее и он хранится в замороженном состоянии до использования при -20oC. Концентрация Pb в таком растворе может составлять 15,5 мг/мл.
B. Анализ на тетразолий в микрокультуре (МГА)
1. Приготовление: раствор XTT-PMS-XTT-PMS готовили непосредственно перед его добавлением в углубления пластины с культурой.
Основной раствор PMS разбавляли до значения 1:100 (0,153 мг/мл). Разбавленный PMS добавляли к каждому мл XTT до конечной концентрации PMS 0,02 мМ. 50 мкл ликвоты смеси XTT-PMS добавляли в каждое из соответствующих углублений и пластинку инкубировали в течение 4 ч при 37oC. С пластинок снимали колпаки и заменяли их на липкие закупоривающие пластины (Dynatech cat 001-010-3501). Закрытые таким образом пластинки встряхивали на миксере для пластинок с микрокультурой и определяли поглощение при длине волны 450 нм.
IV. Результаты
Полученные данные представлены в виде графика зависимости процентного количества испытуемых клеток по отношению к неинфицированным клеткам (%) как для инфицированных, так и для неинфицированных клеток, от увеличивающейся концентрации испытуемого соединения. Такие графики позволяют рассчитать эффективную концентрацию (EC50) по отношению к инфицированным клеткам, ингибиторную концентрацию (IC50)) по отношению к нормальным клеткам и терапевтический индекс (TI50).
Ингибиторные концентрации против HIY, определенные в соответствии с описанным выше для соединений примеров 7, 9, 10, 11 и 14b, представлены в табл. 1 (в мкг/мл) и в табл. 2.
Терапевтический индекс рассчитывали как частное от деления IC50 на EC50.
В этом скрининге соединения примеров 5 и 8 проявляют также антивирусную активность.
Более ранний анализ, проведенный с соединением примера 10 в южном исследовательском институте, дал значения TI50, равное 200, при культурировании клеток МТ-2 в присутствии H9/HIIV-IIIB.
(B) Активность против вируса лейками Фелина
Антивирусный скрининг на активность против FeIV-JAIDS проводили на пластинках с 96 углублениями (корнинг) с использованием индикаторных клеток 81C в среде Дулбекко, модифицированной Исковым, которая была дополнена 10% термодезактивированной сыворотки телячьего плода (FBS). За двадцать часов до анализа пластинки засевали клетками 81C с концентрацией 5 • 103 кл. /углубление. В день анализа клетки предварительно обрабатывали в течение 30 мин при 37oC DEAE-декстраном (25 мкг/мл) в 0,1 мл сбалансированного солевого раствора Ханкса. Раствор удаляли и затем в каждое углубление добавляли по 0,1 мл растительной среды, содержащей 32 TCID50FIY-FAIDS, или 0,1 мл только растительной среды. Вирусу давали адсорбироваться в течение часа и затем добавляли 0,1 мл испытуемого или положительного контрольного соединения (2', 3'-дидеоксицитидин; ddc), либо растительную среду. Пластинки инкубировали при 37oC. На 4 день после инфицирования клетки кормили свежей растительной средой, содержащей соединение. Культурную среду полностью обменивали и заменяли свежей средой, содержащей соединение на 7-ой день после инфицирования. На 10-й день после инфицирования клетки фиксировали формалином, окрашивали 0,1%-ным раствором красителя Космаши Бриллиантовый голубой R-25o и наблюдали под микроскопом на CPE и лекарственную цитотоксичность.
Соединение примера 14a имело значение ED50 порядка 1,9 мкг/мл.
(C) Активность против AIDS Мурин
Пластинки Фалкон с 6 углублениями для ткановой культуры засевали 1,75 • 105 кл. на углубление в общем объеме 2,5 мл EMEM, содержащей 5% термодезактивированной FBS. Через двадцать часов после высеивания клеток среду сливали и в каждое углубление добавляли по 2,5 мл DEAEдекстрана (25 мкг/мл в растворе фосфатного буфера). Культуры инкубировали в течение 1 ч при 37oC, после чего DEAE-декстрановый раствор сливали и клеточные слои сразу ополаскивали 2,5 мл среды (без вируса или лекарства). Контрольные культуры на лекарство получали 2,5 мл среды, содержащей лекарство без вируса. Инфицированные вирусом контрольные культуры получали 0,5 мл соответствующего разбавления маточного CAS-BR-M с целью продуцирования подсчитываемых бляшек плюс 2,0 мл среды. К испытуемым образцам добавляли 0,5 мл соответствующего вирусного разбавления и 2,0 мл среды лекарственного разбавления. Испытывали шесть концентраций испытуемого соединения, разбавленного серийными полулогарифмическими разбавлениями. Испытывали три концентрации положительного контрольного лекарства ddc. В каждый анализ включали по три углубления для каждой концентрации испытуемого соединения, а также 6 вирусов и 6 клеточных контрольных культур. На 3 день послевирусную инокуляционную токсичность лекарства по отношению к клеткам C-1 определяли микроскопическим исследованием окрашенных дублированных клеточных культур и культур с контрольным лекарством. Оставшиеся испытуемые и контрольные культуры облучали светом от УФ-лампы в течение 20 с и к каждой культуре добавляли клетки XC (5 • 105 кл. /углубление в 2,5 EMEM, содержащей 10% термодезективированной FBS). На 3 день после облучения культуры фиксировали формалином и окрашивали кристаллическим фиолетовым. Бляшки подсчитывали с помощью аналитического микроскопа.
Антивирусную активность в отношении уменьшения числа CAS-BR-M бляшек выражали уменьшением среднего числа бляшек, подсчитанных в обработанных лекарством, инфицированных вирусом культурах по сравнению со средним числом бляшек, подсчитанных в необработанных зараженных вирусом культурах (процент контроля). Соединение примера 14a имело значение ED50 порядка 1,1 мкг/мл.
(D) Активность против ретровируса SAIDS (SRY-2). Симиана
Антивирусный скрининг против вируса SAIDS (Д/Вашингтон) осуществляли анализом на ингибирование синцития на клетках Реджи. Лекарство разбавляли в полной среде Искова и затем по 100 мкл каждого разбавления добавляли в соответствующие углубления в пластинке с 96 углублениями. Затем в каждое углубление добавляли активно растущие клетки Реджи, 5 • 103 кл. в 50 мл полной среде Искова. Затем добавляли 50 мкл осветленного верхнего слоя из SRY-2/клеточной сокультуры Реджи. В этом анализе в качестве положительного контрольного лекарства использовали ДДС. Пластинки инкубировали при 37oC в увлажненной атмосфере, содержащей 5% CO2. На 7 день после инфицирования подсчитывали синцитий. Токсичность лекарства устанавливали путем сравнения числа живых клеток в неинфицированном, обработанном лекарством образце с жизнеспособностью неинфицированного, необработанного контроля. Соединение примера 14a имеет значение ED50 2,8 мкг/мл.
(E) Активность против вируса Вишна Маэди
Антивирусную активность против штамма вируса Вишна Маэди (VMV) WL C-1 определяли путем измерения уменьшения степени вирус-специфичного, иммуногистохимического окрашивания. Монослои клеток хороидного оплетения овцы инфицировали VMV и нагружали серийными разбавлениями испытуемых соединений. После инкубирования в течение пяти дней монослои дополнительно инкубировали в присутствии вирус-специфичной антисыворотки, конъюгированной с пероксидазой ложевицы приморской (HRP). Далее проводили инкубацию монослоев в присутствии хромогенного субстрата HRP, штаммовых площадей вирусной репликации. Подсчитывали такие дискретные очаги и рассчитывали концентрацию испытуемого соединения, требуемую для уменьшения числа до 50% от соответствующего числа в контрольных образцах, необработанных лекарством. Соединение примера (+14)a имело значение ED50, равное 0,2 мкг/мл.
Пример 22. Цитотоксическая активность
Соединения примеров 7a, 9a и 10a, определенные в примере 17, проявили цитотоксическую активность при испытании против клеточной культуры лейкемии мышей P388 согласно анализу, описанному Р.С. Алоквистом и Р. Винсом в J. Med. Chem. 16, 1396 (1973). Полученные значения ED50 (мкг/мл) составили:
Соединение 5 - 12
Соединение 7 - 40
Соединение 8 - 3
Дополнительные примеры
Пример 23
(22R)- 16α,17α -бутилидендиокси-21-каприлилокси- 6α,9α -дифтор-11 β -гидрооксипрегн-4-ен-3,20-дион
В раствор (22R)- 16α,17α -бутилидендиокси- 6α,9α -дифтор-β , 21-дигидроксипрегн-4-ен-3,20-диона (60 мг) в 4 мл метиленхлорида добавили каприлилхлорид (35 мг) и 4-диметиламинопиридин (30 мг). Реакционную смесь перемешивали при комнатной температуре в течение 45 мин. После выпаривания остаток очищали хроматографией на колонке с Merk Keselgel 60, используя смесь гептан: этилацетат 1: 1 в качестве подвижной фазы. Полученный продукт очищали хроматографией на колонке Sephadex LH-20 (внутренний диаметр - 85•2,5 см), используя в качестве подвижной фазы хлороформ. Фракцию 280-315 мл собирали и выпаривали, остаток осаждали из смеси метиленхлорид:петролейный эфир, получая 28 мг (22R)- 16α,17α -бутилидендиокси-21-каприлилокси-6α,9α-дифтор-11β-гидроксипрегн-4-3,20-диона. Температура плавления 158-165 градусов Цельсия.
[α]
Пример 24. (22R)-16α,17α-бутилидендиокси-21-каприлокси-6α,9α-дифтор-11β-гидрокипрегн-4-ен-3,20-дион.
Раствор каприлхлорида (75 мг) в 2 мл диоксана по капле добавили в раствор (22R)- 16α,17α -бутилидендиокси- 6α,9α -дифтор- 11β , 21-дигидрооксипрегн-4-ген-3,20-диона (60 мг) в 4 мл пиридина. Реакционную смесь перемешивали при комнатной температуре в течение ночи и обрабатывали как в примере 1. Неочищенный продукт очищали хроматографией на колонке Sephadex LH-20 (внутренний диаметр 89•2,5 см), используя смесь гептан:хлороформ:этанол 20: 20: 1 в качестве подвижной фазы. Фракцию 245-315 мл собирали и выпаривали, остаток осаждали из смеси метиленхлорид: петролейный эфир, получая 58 мг (22R)- 16α,17α -бутилендиокси-21-каприлокси- 6α,9α -дифтор- 11β -гидркосипрегн-4-ен-3,20-диона. Температура плавления 168-171oC.
[α]
Пример 25.
(22R)- 16α,17α -бутилидендиокси-21-каприлокси- 6α,9α -дифтор- 11β -гидроксипрегн-4-ен-3,20-дион
В раствор (22R)- 16α,17α -бутилидендиокси- 6α,9α -дифтор- 11β , 21-дигидроксипрегн-4-ен-3,20-диоена (60 мг) в 4 мл метиленхлорида добавили каприлилхлорид (25 мг) и 4-диметиламинопиридин (30 мг). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин и обрабатывали как в примере 28. После хроматографии на колонке с Merk Keselgel 60 продукт очищали хроматографией на колонке Sephadex LH-20 (внутренний диаметр - 85•2,5 см), используя в качестве подвижной фазы хлороформ. Фракцию 300-345 мл собирали и выпаривали, остаток осаждали из смеси метиленхлорид:петролейный эфир, получая 20 мг (22R)- 16α,17α -бутилидендиокси-21-каприлокси- 6α,9α -дифтор- 11β -гидроксипрегн-4-ен-3,20-диона. Температура плавления 148-160oС.
[α]
Пример 26.
(22R)- 16α,17α -бутилидендиокси-21-бутирилокси- 6α,9α -дифтор- 11β -гидроксипрегн-4-ен-3,20-дион
Раствор бутирилхлорида (170 мг) в 5 мл диоксана по капле добавили в раствор (22R)- 16α,17α -бутилидендиокси- 6α,9α -дифтор- 11β , 21-дигидроксипрегн-4-ен-3,20-диона (150 мг) в 10 мл пиридина. Реакционную смесь перемешивали при комнатной температуре в течение ночи и обрабатывали как в примере 1. Неочищенный продукт очищали хроматографией на колонке Sephadex LH-20 (внутренний диаметр 70•6,3 см), используя хлороформ в качестве подвижной фазы. Фракцию 1440-1740 мл собирали и выпаривали, далее очищали на колонке Sephadex LH-20 (внутренний диаметр 70•6,3 см), используя смесь гептан:хлороформ:этанол 20:20:1 в качестве подвижной фазы. Фракцию 2010-2230 мл собирали и выпаривали. Остаток осаждали из смеси метиленхлорид:петролейный эфир, получая 118 мг (22R)- 16α,17α -бутилидендиокси-21-бутирилокси- 6α,9α -дифтор- 11β -гидроксипрегн-4-ен-3,20-диона. Температура плавления 208-216oC.
[α]
Пример 27 (по схемам I и II).
Синтез (гидроксиметил) циклопентил-производных формул ба-11а и (гидроксиметил) циклопентил-производных формул 6b-11b на прекурсора цис-(4-ацетамидоциклопент-2-енил)метилацетата (1а) выполняют в соответствии со схемами I и II (см. в конце текста). Соединение 1а гидролизуют водным гидроксидом бария для получения аминоспирта 2а. Конденсация 2а с 2-амино-4,6-дихлорпиримидином приводит к получению соответствующего пиридиниламинопроизводного 3а. 5-[(Пара-хлорфенил)азо]пиримидин 4а получают с помощью хлорида пара-хлорбензолдиазония. Shealy and Clayton, J.Pharm. Sci., 62, 1482 (1973). Восстановление 4а цинком и уксусной кислотой приводит к получению пиримидина 5а, который затем превращают в 9-замещенный 2-амино-6-хлорпурин 6а посредством образования циклического соединения с триэтил-орто-формиатом. 2-Амино-6-хлорпурин 6а превращают в карбоциклический 2', 3'-дидегидро-2',3'-дидеоксигуанозиновый аналог 7а при кипячении с обратным холодильником в 0,8 N гидроксида натрия, в то время как обработка 6а жидким аммиаком приводит к получению 2,6-диаминопуринового аналога 8а.
Образование циклического соединения (реакция циклизации) 5а с нитритом натрия и уксусной кислотой приводит к получению (+)-(цис)-4-(5-амино-7-хлор-3H-1,2,3-триазоло[4,5-d] пиримидин-3-ил] -2-циклопентилкарбинола (9а) с хорошим выходом. Основной гидролиз 9а гидроксидом натрия приводит к получению 8-аза-аналога 10а - карбоциклического 2',3'-дидегидро-2',3'-дидеоксигуанозина, тогда как обработка жидким аммиаком приводит к получению соответствующего 2,6-диаминопурин-аналога 11а.
2', 3'-насыщенные производные получают в соответствии со схемой II. Каталитическое восстановление 1а приводит к получению соответственно замещенного циклопентанового прекурсора 1b. Остальные стадии синтеза соединений ряда b аналогичны стадиям синтеза соединений ряда а.
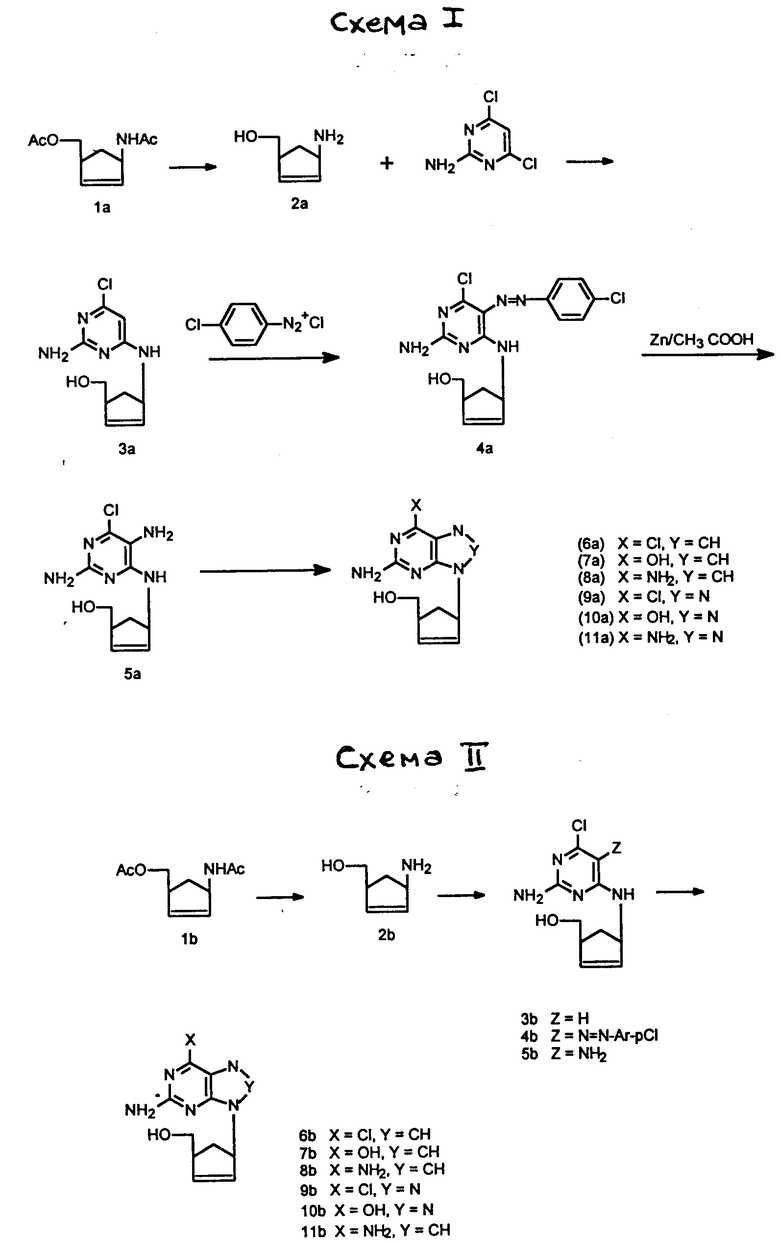
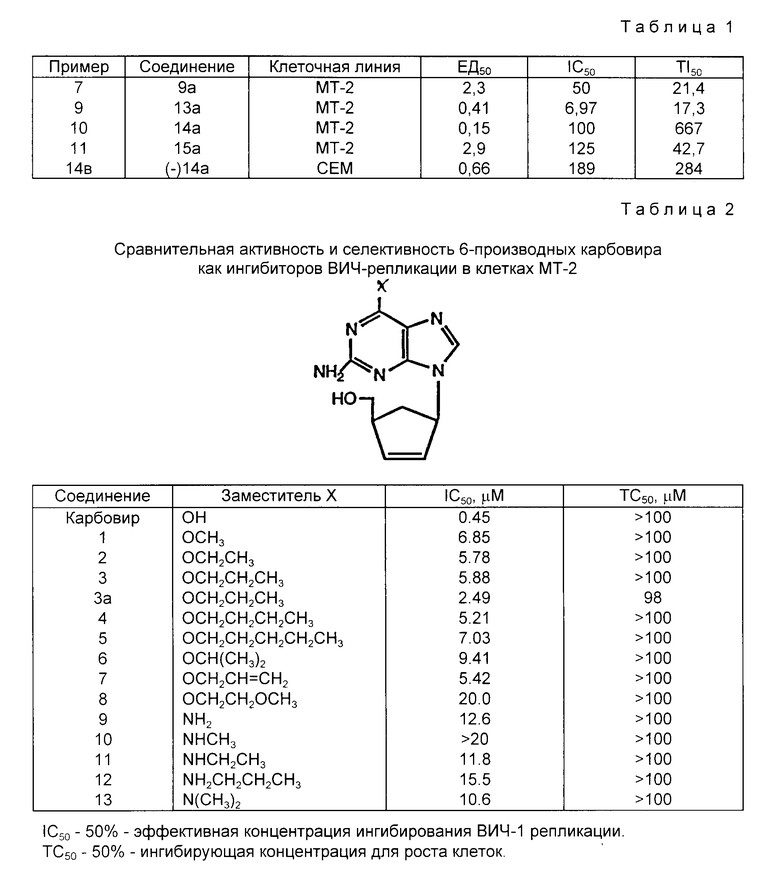
Предложены соединения формулы :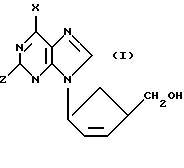
где X представляет собой H, NRR1, SR, OR или галоген; Z представляет собой H, OR2 или NRR1; R, R1 и R2 представляют собой H, C1-C4-алкил или арил и их фармацевтически приемлемые соли. Предложено также применение этих соединений в качестве антивирусных и антиопухолевых агентов, фармкомпозиции на их основе и методы получения исходных и промежуточных соединений. 3 с. и 13 з.п. 2 табл.
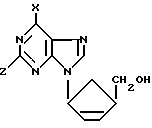
где X - NRR1, SR, OR2, H или атом галогена;
Z - атом водорода или NH2;
R, R1, R2, одинаковые или различные, - атом водорода, C1 - C4-алкил,
или их фармацевтически приемлемые производные.
(1а,4а)-4-(6-хлор-9Н-пурин-9-ил)-2-циклопентенилкарбинола;
(1а,4а)-4-(6-гидрокси-9Н-пурин-9-ил)-2-циклопентенилкарбинола;
(1а,4а)-4-(6-амино-9Н-пурин-9-ил)-2-циклопентенилкарбинола;
(1а,4а)-4-(6-меркапто-9Н-пурин-9-ил)-2-циклопентенилкарбинола;
(1а,4а)-4-(2,6-диамино-9Н-пурин-9-ил)-2-циклопентенилкарбинола;
(1а,4а)-4-(2-амино-6-хлор-9Н-пурин-9-ил)-2-циклопентенилкарбинола;
(1а,4а)-4-(2-амино-9Н-пурин-9-ил)-2-циклопентенилкарбинола;
9. (1а,4а)-4-(2-амино-6-гидрокси-9Н-пурин-9-ил)-2-циклопентенилкарбинол.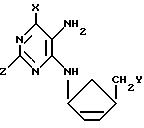
где X - NRR1, SR, OR2, атом галогена; где R, R1, R2 имеют значения, указанные в п.1;
Y - OH-группа, возможно защищенная;
Z - NH2,
или их фармацевтически приемлемые производные.
Приоритеты по пунктам:
20.01.88 - по пп.3, 4, 8 и 9;
07.09.88 - по пп.1 - 16;
05.12.88 - по пп.1 - 16.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Chemistry, biology and Medical Applications, R.T.Walker Plenum-Press, n-y, 1979, p.193 | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| EP, A, 0072027, C 07 D 473/16, 1983 | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| FR, A, 22 8289 2, A 61 K 31/52, 1976 | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| US, A, 4362729, C 07 D 239/70, 1982. | |||
