Изобретение относится к аналогам пуринового нуклеозида, содержащим вместо остатка сахара карбоциклическое кольцо, их фармацевтически приемлемым производным, и их применению в медицинской терапии, в особенности для лечения определенных вирусных инфекций.
Вирус гепатита B (ВГВ) представляет собой вирус, содержащий небольшую ДНК, который инфицирует людей. Он является представителем класса близкородственных вирусов, известных как гепа-ДНК-вирусы, каждый из которых селективно инфицирует организмы либо млекопитающих, либо птиц, таких как североамериканские лесные сурки или утки.
В мировом масштабе ВГВ является вирусным возбудителем заболеваний первостепенного значения. Он наиболее распространен в странах Азии и преобладает в субсахарной Африке. Этот вирус этиологически связан с первичным печеночно-клеточным раком и полагают, что он вызывает 80% случаев заболеваний рака печени в мире. В Соединенных Штатах каждый год госпитализируют более десяти тысяч человек с заболеванием, обусловленным ВГВ, в среднем 250 больных умирают от внезапного и быстро развивающегося заболевания.
По оценкам, в Соединенных Штатах в настоящее время содержится пул из 500000 - 1 млн. переносчиков инфекции. У более 25% носителей разовьется хронический активный гепатит, и он часто прогрессирует в цирроз. По оценкам каждый год в США 5000 людей умирают от связанного с ВГВ цирроза, и возможно 1000 человек умирают от связанного с ВГВ рака печени. Даже если имеется универсальная вакцина против ВГВ, будет сохраняться потребность в эффективных соединениях против ВГВ. Большая масса постоянно инфицированных носителей, которая в мире оценивается как 220 млн. человек, не получит никакой пользы от вакцинации и будет продолжать существовать с высоким риском заболеваний печени, индуцированных ВГВ. Эта популяция носителей служит источником инфекции для восприимчивых индивидуумов, навсегда сохраняющих факт заболевания, особенно в эндемических районах или в группах высокого риска, таких как токсикоманы, использующие внутривенное введение и гомосексуалисты. Таким образом, существует большая необходимость в эффективных антивирусных агентах как для контроля хронической инфекции, так и для уменьшения прогрессии к печеночно-клеточному раку.
Клинические эффекты инфекции ВГВ простираются от головной боли, лихорадки, недомогания, тошноты, рвоты, анорексии и болей в животе. Репликация вируса обычно контролируется иммунной реакцией, при этом протекание выздоровления у людей длится неделями или месяцами, однако инфекция может быть более серьезной и приводить к указанному выше устойчивому хроническому заболеванию печени.
В книге "Viral Infections of Humans" (second edition, Ed. Evans, A.S. (1982 Plenum Press Publishing Corporation, New. York) в главе 12 детально описана этиология герпеса является источником многих обычных вирусных заболеваний человека. Эта группа включает вирус цитомегалии (ВЦМ), вирус Эпштейна/Барра (ВЭБ), вирус ветряной оспы (ВВО), вирус герпеса (ВГ) и вирус герпеса человека 6 (ВГЧ6).
Как и в случае других вирусов герпеса, инфекция ВЦМ приводит к связыванию вируса и организма хозяина на всю жизнь и после первичной инфекции вирус может скрываться несколько лет. Клинические эффекты простираются от смерти и серьезных болезней (микроцефалия, гепатоспленомегалия, желтуха, олигофрения), через неблагоприятное состояние организма, восприимчивость к инфекциям в грудной клетке и уже до отсутствия какого-либо явного эффекта заболевания. Для заболевших СПИДом инфекция ВЦМ является доминирующей причиной смертности, в то время как у 40 - 80% взрослого населения она присутствует в скрытой форме и может быть повторно активирована у пациентов с недостатком иммунитета.
ВЭБ вызывает инфекционный мононуклеоз, и также предполагают, что он является возбудителем рака носоглотки, иммунобластической лимфомы, лимфомы Беркитта и лейкоплакии волос.
ВВО вызывает ветряную оспу и опоясывающий герпес. Ветряная оспа является первичным заболеванием, продуцируемым в организме без иммунитета. У малых детей это обычно легкое заболевание, характеризуемое везикулярной сыпью и лихорадкой. Опоясывающий герпес является рецидивирующей формой заболевания взрослых, которые ранее были инфицированы ветряной оспой. Клинические проявления опоясывающего герпеса включают невралгию и везикулярную сыпь на коже с односторонним и дерматомным распределением. Распространение воспаления может приводить к параличу или судорогам, а если будет затронут мозг - к коме. У пациентов с ослабленным иммунитетом ВВО может диссеминировать, вызывая серьезные или даже смертельные заболевания.
ВГ 1 и ВГ 2 являются одними из наиболее распространенных инфицирующих агентов человека. Большинство этих вирусов способно постоянно жить в нервных клетках организма хозяина. Будучи однажды инфицированными индивидуумы имеют риск рецидивного клинического проявления инфекции, что может приводить как к физическому, так и психологическому дистрессу. Инфекция ВГ часто характеризуется обширными патологическими изменениями кожи, рта и/или половых органов. Первичные инфекции могут быть бессимптомными, хотя они имеют склонность быть более серьезными, чем инфекции в индивидуумах, предварительно подвергнувшихся воздействию вируса. Глазные инфекции ВГ могут приводить к кератиту или катарактам. Инфекции у новорожденных, пациентов с ослабленным иммунитетом или проникновение инфекции в центральную нервную систему могут оказаться смертельными. ВГЧ6 является возбудителем болезни roseola infantum (exanthum subitum) у детей, которая характеризуется лихорадкой и появлением сыпи после снижения жара. ВГЧ6 также вовлечен в синдромы лихорадки и/или сыпи, а также пневмонии или гепатита у пациентов с ослабленным иммунитетом.
Обнаружено, что определенные замещенные бензимидазольные соединения, указанные ниже, могут быть использованы для лечения или профилактики определенных вирусных инфекций. Согласно первому аспекту данного изобретения предлагаются новые соединения формул (I) и (I-1):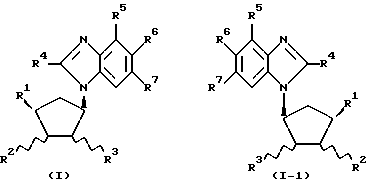
где R1 - H, CH3 или CH2OH;
R2 - H или OH;
R4 - H или OH;
или R2 и R3 вместе образуют связь;
R4 - H, Cl, Br, I, C1-4-алкил, C3-7-циклоалкил, C3-7-циклоалкил C1-4-алкил, C1-4-перфторалкил (например, трифторметил), NH2, C1-4-алкиламино-, C1-4-диалкиламино, C3-7-циклоалкиламино-, ди-C3-7-циклоалкиламино-, N-C1-4-алкил-N-C3-7-циклоалкиламино-, N-C1-4-алкил-N-C3-7-циклоалкил-C1-4-алкиламино-, ди-C3-7-циклоалкил-C1-4-алкиламино-, C3-7-циклоалкил-C1-4-алкиламино-, N-C3-7-циклоалкил-N-C3-7-циклоалкил-C1-4- алкиламиногруппы, SH, C1-4-алкилтио-, C6-10-арил-C1-4-алкилтиогруппы, OH, C1-4-алкокси-, C6-10-арил-C1-4-алкокси-группы или C6-10-арил-C1-4-алкил;
R5, R6 и R7 независимо выбраны из числа H, F, Cl, Br, I, CF3 и CH3 при условии, что по меньшей мере один из R1, R2 и R3 является OH или содержит эту группу;
а также их фармацевтически приемлемые производные.
Предпочтительные соединения с формулами (I) и (I-1) изображены формулами (IA) и (IA-1).
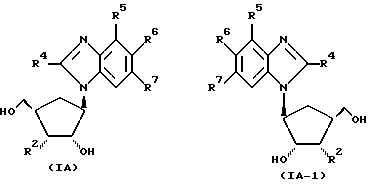
где R2 - H или OH.
Наиболее предпочтительны соединения с формулами (I), (I-1), (IA) и (IA-1), в которых R4 - CH3, Cl или Br; R5 - H, а каждый из R6 и R7 представляет Cl.
Следует понять, что настоящее изобретение охватывает особые энантиомеры, изображенные в формулах (I) и (I-1), включая таутомеры пурина как таковые или в комбинации с их зеркальными энантиомерами. Энантиомеры, изображенные формулой (I), являются предпочтительными, и предпочтительно чтобы они были в основном свободны от соответствующего энантиомера в такой степени, чтобы в смеси в основном содержалось менее 10% (в/в), предпочтительно менее 5% (в/в), более предпочтительно менее 2% (в/в) и наиболее предпочтительно менее 1% (в/в) соответствующего энантиомера исходя из общего веса смеси. Энантиомеры, изображенные формулой (I-1), наиболее предпочтительны, и предпочтительно чтобы они были в основном свободны от соответствующего энантиомера в такой степени, чтобы в смеси в основном содержалось менее 10% (в/в), предпочтительно менее 5% (в/в), более предпочтительно менее 2% (в/в) и наиболее предпочтительно менее 1% (в/в) соответствующего энантиомера исходя из общего веса смеси.
Особенно предпочтительными примерами соединений с формулой (I) являются:
(1R,2S,3S,5S)-5-(2-бром-5,6-дихлор-1H-бензимидазол-1-ил)-3-(гидроксиметил)-1,2-циклопентандиол;
(1S,2R,3R,5R)-5-(2-бром-5,6-дихлор-1H-бензимидазол-1-ил)-3-(гидроксиметил)-1,2-циклопентандиол;
(+/-)-(1R*, 2S*, 3S*, 5S*)-5-(2,5,6-трихлор-1H-бензимидазол-1-ил)-3-(гидроксиметил)-1,2-циклопентандиол;
(+/-)-(1R*, 2S*, 3R*)-3-(2-бром-5,6-дихлор-1H-бензимидазол-1-ил)-1,2-циклопентандиол и
(+/-)-(1R*, 2S*, 3S*,5S*)-5-(5,6- дихлор-2-метил-1H-бензимидазол-1-ил)-3-(гидроксиметил)-1,2- циклопентандиол
и их фармацевтически приемлемые соли.
В качестве соединений по изобретению здесь обозначаются соединения с формулами (I) и (I-1) и их фармацевтически приемлемые производные.
Дальнейшим аспектом изобретения является применение соединений по изобретению в медицинской терапии, в особенности для лечения или профилактики вирусных инфекций, таких как вирусные герпесные инфекции. В настоящее время показано, что соединения по изобретению проявляют активность против инфекций вирусом гепатита B (ВГВ) и вирусом цитомегалии (ВЦМ), хотя предварительные результаты предполагают, что изобретение также может быть использовано против других герпесных вирусных инфекций, таких как ВЭГ, ВВО, ВГI и II и ВГЧ6.
Иные вирусные состояния, которые можно лечить и в соответствии с изобретением, обсуждены в приведенном выше введении.
В следующем аспекте изобретения предлагаются:
а) метод лечения или профилактики гепа-ДНК-вирусной инфекции, такой как гепатит B, или герпесной вирусной инфекции, такой как ВЦМ, который включает применение к субъекту терапевтически эффективного количества соединения по изобретению.
б) Применение соединения по изобретению в производстве медикамента для лечения или профилактики любого из вышеупомянутых инфекций или состояний.
Под "фармацевтически приемлемым производным" подразумевается любая фармацевтически или фармакологически приемлемая соль, сложный эфир или соль такого эфира соединения по изобретению, или любое соединение, которое способно при применению к реципиенту давать (прямо или косвенно) соединение по изобретению, или обладающий антивирусной активностью метаболит, или его остаток.
Предпочтительные сложные эфиры соединений по изобретению включают эфиры карбоновых кислот, в которых не содержащая карбонил часть сложноэфирной группы выбрана из числа линейного или разветвленного алкила, например, н-пропила, трет-бутила, н-бутила, алкоксиалкила (например, метоксиметила), арилалкила (например, бензила), арилоксиалкила (например, феноксиметила, арила (например, фенила, необязательно замещенного галогеном, C1-4-алкилом или C1-4-алкокси- или аминогруппой); эфиры сульфокислот, такие как алкильные или арилалкилсульфонильные (например, метансульфонильные) эфиры аминокислот (например, L-валильные или L-изолейцильные); а также моно-, ди- и триэфиры фосфорной кислоты. Эфиры фосфорной кислоты могут далее быть этерифицированы, например, C1-20-спиртом, или его реакционноспособным производным, или 2,3-ди(C6-24)ацилглицерином.
Для вышеописанных сложных эфиров, если не указано иначе, любой присутствующий алкильный фрагмент предпочтительно содержит от 1 до 18 атомов углерода, особенно от 3 до 6 атомов углерода, так, как в случае пентаноата. Любой арильный фрагмент, присутствующий в этих эфирах, предпочтительно включает фенильную группу.
Все упоминания о любом из вышеназванных соединений также включают их фармацевтически приемлемые соли.
Физиологически приемлемые соли включают соли органических карбоновых кислот, таких как уксусная, молочная, винная, яблочная, изэтионовая, лактобионовая, п-аминобензойная и янтарная кислота; соли органических сульфокислот, таких как метансульфо-, этансульфо-, бензолсульфо- и п-толуолсульфокислота; а также неорганических кислот, таких как соляная, серная, фосфорная и сульфаминовая кислота.
Вышеуказанные соединения по изобретению и их фармацевтически приемлемые производные могут применяться вместе с иными терапевтическими агентами для лечения упомянутых выше инфекций или состояний. В число примеров таких дополнительных терапевтических агентов входят агенты, эффективные для лечения вирусных инфекций или связанных с ними состояний, такие как ациклические нуклеозиды (например, ацикловир), иммуномодулирующие агенты, такие как тимозин, ингибиторы рибонуклеотидной редуктазы, такие как 2-ацетилпиридин-5-[(2-хлоранилино)тиокарбонил] тиокарбогидразон, интерфероны, такие как α-интерферон, 1-  - D-арабинофуранозил-5-(1-пропинил)урацил, 3'-азидо-3'-дезокситимидин, рибавирин и фосфономуравьиная кислота. Соединения, являющиеся компонентами такой составной терапии, могут применяться одновременно, в виде либо раздельных, либо объединенных композиций, или в разное время, то есть последовательно, так, чтобы достигался объединенный эффект.
- D-арабинофуранозил-5-(1-пропинил)урацил, 3'-азидо-3'-дезокситимидин, рибавирин и фосфономуравьиная кислота. Соединения, являющиеся компонентами такой составной терапии, могут применяться одновременно, в виде либо раздельных, либо объединенных композиций, или в разное время, то есть последовательно, так, чтобы достигался объединенный эффект.
Соединения по изобретению, называемые здесь также активными ингредиентами, могут применяться в терапии любым подходящим способом, включая пероральный, ректальный, носовой, местный (включая сквозькожный, ротовой и подъязычный), вагинальный и парентеральный (включая подкожный, внутримышечный, внутривенный и внутрикожный). Следует понять, что предпочтительный способ применения может изменяться в зависимости от состояния и возраста реципиента, природы инфекции и выбранного активного ингредиента.
Как правило, подходящая доза для любого из вышеупомянутых состояний будет в диапазоне от 0,01 до 250 мг на килограмм веса тела реципиента (например, человека) в день, предпочтительно в диапазоне от 1,0 до 20 мг на килограмм веса тела в день. (Если не указано иное, все веса активных ингредиентов рассчитаны на исходное соединение с формулой (I); для их солей или сложных эфиров веса нужно пропорционально увеличить). Желаемая доза предпочтительно представляется двумя, тремя, четырьмя, пятью, шестью или большим количеством субдоз, применяемых через надлежащие интервалы времени в течение дня. Эти субдозы могут применяться в виде форм единичных доз, содержащих, например, 10 - 1000 мг, предпочтительно 20 - 500 мг, наиболее предпочтительно 100 - 400 мг активного ингредиента в форме единичной дозы.
В идеальном случае активный ингредиент должен применяться до достижения максимальной концентрации активного соединения в плазме от около 0,025 до около 100 мкмоль, предпочтительно от около 0,1 до 70 мкмоль, наиболее предпочтительно около 0,25 - 50 мкмоль. Это может быть достигнуто, например, путем внутривенной инъекции 0,1 - 5% раствора активного ингредиента, необязательно в физиологическом растворе, или пероральном применением в виде болюса, содержащего от около 0,1 до около 250 мг/кг активного ингредиента. Желательные уровни содержания вещества в крови могут поддерживаться непрерывным вливанием (около 0,01 - 5,0 мг/кг/ч) или путем прерывистых вливаний по приблизительно 0,4 - 15 мг/кг активного ингредиента.
Хотя активный ингредиент можно применять сам по себе, предпочтительно использовать его в виде фармацевтической композиции. Композиция по настоящему изобретению включают по меньшей мере один активный ингредиент, определенный выше, вместе с одним или более приемлемым носителем и необязательно с иными терапевтическими агентами. Каждый носитель должен быть "приемлем" в том смысле, что он совместим с другими ингредиентами композиции и не приносит вреда пациенту. В число композиций входят пригодные для перорального, ректального, носового, местного (включая сквозькожное, ротовое и подъязычное), вагинального или парентерального (включая подкожное, внутримышечное, внутривенное и внутрикожное) применения. Композиции могут быть удобно представлены в форме единичных доз и могут быть приготовлены любыми известными в фармацевтике методами. Эти методы включают стадию связывания активного ингредиента с носителем, который состоит из одного или более вспомогательных ингредиентов. Как правило, композиции получают путем равномерного и плотного связывания активного ингредиента с жидкими носителями, или с мелкодисперсными твердыми носителями, или с обоими носителями вместе, после чего при необходимости продукту придают форму.
Композиции, пригодные для сквозькожного применения, могут быть выполнены в виде отдельных пластырей, приспособленных для нахождения в тесном контакте с эпидермисом реципиента в течение длительного времени. Удобно, чтобы такие пластыри содержали активное соединение 1) в необязательно буферном водном растворе; 2) растворенное и/или диспергированное в клее или 3) диспергированное в полимере. Подходящая концентрация активного соединения составляет около 1 - 25%, предпочтительно около 3 - 15%. В особом возможном случае применения активное соединение может выделяться из пластыря методами электротранспорта или ионтофореза, как описано в общем виде в Pharmaceutigal Research, 3, (6), 318 (1986).
Композиции по настоящему изобретению, пригодные для перорального применения, могут быть представлены в виде дискретных форм, таких как капсулы, каше или таблетки, каждая из которых содержит предварительно определенное количество активного ингредиента; в виде порошка или гранул; в виде раствора или суспензии в водной или неводной жидкой среде; или в виде жидких эмульсий типа масло/вода или вода/масло. Активный ингредиент также может быть представлен в виде болюса, электруария или пасты.
Таблетка может быть изготовлена прессованием или формованием, необязательно с одним или более вспомогательным ингредиентом. Прессованные таблетки могут быть изготовлены прессованием на подходящем оборудовании активного ингредиента в свободно текучей форме, такой как порошок или гранулы, необязательно смешанного со связующим веществом (например, с повидоном, желатиной или гидроксипропилметилцеллюлозой), смазкой, инертным разбавителем, консервантом, дезинтегрантом (например, натриевым гликолятом крахмала, сшитым повидоном, сшитой натриевой солью карбоксиметилцеллюлозы), поверхностно-активным или диспергирующим агентом. Формованные таблетки могут быть изготовлены путем формования на подходящем оборудовании смеси превращенного в порошок соединения, увлажненного инертным жидким разбавителем. Таблетки могут быть необязательно покрыты защитным покрытием или содержать насечки, а также могут быть изготовлены таким образом, чтобы осуществлять медленное или контролируемое выделение используемого в них активного ингредиента, используя, например, гидроксипропилметилцеллюлозу в различных пропорциях для получения желательного профиля выделения. Таблетки необязательно могут иметь энтеросолюбильное покрытие для обеспечения выделения в частях внутренностей, отличных от желудка.
Композиции, пригодные для местного применения в полости рта, включают лепешки, содержащие активный ингредиент в корригентной основе, обычно сахарозе и акации или тарганте; пастилки, содержащие активный ингредиент в инертной основе, такой как желатина с глицерином или сахароза с акацией; а также жидкости для полоскания рта, содержащие активный ингредиент в подходящем жидком носителе.
Композиции для ректального применения могут быть представлены в виде суппозитория с подходящей основой, включающей, например, масло какао или салицилат.
Композиции, пригодные для вагинального применения, могут быть представлены в виде пессариев, тампонов, кремов, гелей, паст, пен или распыляемых композиций, содержащих в дополнение к активному ингредиенту известные пригодные носители.
Композиции, пригодные для парентерального применения, включают водные и неводные изотонические стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферы, бактериостатические факторы и растворенные вещества, которые делают композицию изотоничной с кровью предлагаемого реципиента; а также водные и неводные стерильные суспензии, которые могут содержать суспендирующие агенты и загустители. Композиции могут быть представлены в герметичных упаковках, содержащих единичную дозу или несколько доз, например, в ампулах и пузырьках, а также могут храниться в высушенном при замораживании (лиофилизированном) состоянии, требующем только добавления стерильного жидкого носителя, например, воды для инъекций, непосредственно перед применением. Импровизированные растворы и суспензии для инъекций могут быть приготовлены из стерильных порошков, гранул и таблеток описанного ранее вида.
Предпочтительными композициями с единичной дозой являются композиции, содержащие дневную дозу или единицу, дневную поддозу, как описано выше, или надлежащую часть активного ингредиента.
Следует понять, что кроме ингредиентов, особо отмеченных выше, композиции по изобретению могут включать другие обычные агенты, имеющие отношение к типу рассматриваемой композиции; например, композиции для перорального применения могут включать такие дополнительные агенты, как подсластители, загустители и ароматизаторы.
Кроме того, в изобретение включен следующий схематично изображенный способ получения соединений с формулой (I) и их производных, либо самих по себе, либо в комбинации с их соответствующими энантиомерами.
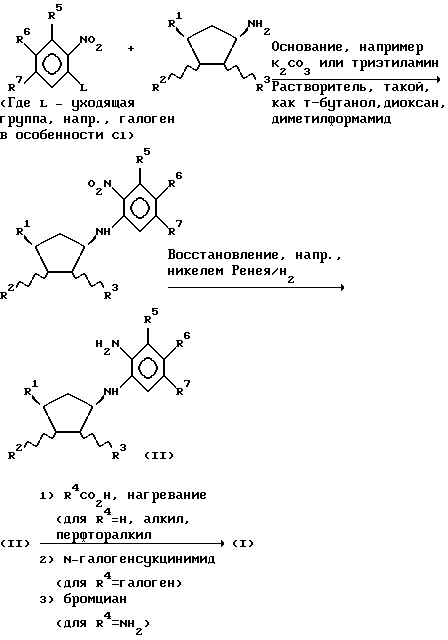
Соединения с формулами (I) и (I-1), где R4 - галоген, например, хлор могут быть превращены в соединения с формулами (I) и (I-1), где R4 - SH (или S - алкил), OH (или алкокси) хорошо известными методами, например, реакцией со спиртовым раствором NaH (или NaS-алкил) или с водным раствором NaOH (или щелочным раствором алкоксида натрия) соответственно.
Таким образом, в соответствии с дальнейшим содержанием настоящего изобретения мы предлагаем способ получения соединений с формулами (I) и (I-1) как таковых или в комбинации с их зеркальными энантиомерами и их фармацевтически приемлемых производных, который включает:
(A) реакцию соединения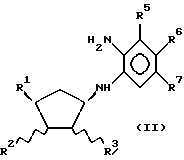
или его зеркального энантиомера
а) либо с соединением с формулой R4CO2H, где R4 - H, C1-4-алкил или C1-4-перфторалкил, предпочтительно при повышенной температуре, либо с соединением с формулой R4C(OR)3, где R4 - H, C1-4-алкил или C1-4-перфторалкил и R - C1-4-алкил, предпочтительно при комнатной температуре и в кислой среде, с образованием соединения с формулой (I) и (I-1), где R4 - H, C1-4-алкил или C1-4-перфторалкил; или
б) с бромцианом с образованием соединения с формулой (I) или (I-1), где R4 - NH2; или
в) с 1,1'-карбонил-диимидазолом с образованием соединения с формулой (I) или (I-1), где R4 - OH; или
г) с 1,1'-тиокарбонил-диимидазолом или тиомочевиной с образованием соединения с формулой (I) или (I-1), где R4 - SH; или (Б)
а) превращение соединения с формулой (I) или (I-1), где R4 - водород, в дальнейшее соединение с формулой (I) или (I-1), где R4 - различная группа; например, путем обработки N-(Cl, Br или I) - сукцинимидом с образованием соединения, где R4 - Cl, Br или I; или
б) превращение соединения с формулой (I) или (I-1), где R4 - Cl, Br или I в дальнейшее соединение с формулой (I) или (I-1), где R4 - различная группа, определенная выше; например, путем обработки спиртовым NaSH или соединением NaS - C1-4-алкил, например, в спиртовом растворе, с образованием соединений, в которых R4 - SH или C1-4-алкилтиогруппа соответственно; путем обработки, например, водным NaOH или спиртовым C1-4-алкоксидом натрия с образованием соединений, в которых R4 - OH или C1-4-алкоксигруппа; или путем обработки C1-4-алкиламином или ди-C1-4-алкиламином с образованием соединений, в которых R4 - C1-4-алкиламино- или ди-C1-4-алкиламиногруппа; или (В) реакцию соединения с формулой
(где R4 - водород, а R5, R6 и R7 определены ранее) или его функционального эквивалента с соединением с формулой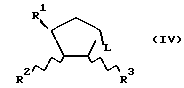
где R1, R2 и R3 определены выше, а L - уходящая группа, например, органическая сульфонилоксигруппа (например, п-толуолсульфонилокси- или метансульфонилоксигруппа), галоген или трифлатная (OSO2CF3) группа, например, в присутствии основания, такого как карбонат натрия или гидрид натрия, в растворителе, таком как диметилформамид, благоприятно при повышенной температуре, например, при 80 - 100oC, с образованием соединения с формулой (I) или (I-1), где R4 - водород; с необязательным превращением соединения с формулой (I) или (I-1) в его фармацевтически приемлемое производное.
В альтернативном варианте вышеуказанного способа (С) соединение с формулой (IV) можно заменить соединением, в котором группы I и R3 заменены циклической сульфатной группой.
Предполагается, что все изображенные выше структуры представляют показанные энантиомеры, их зеркальные изомеры и их смеси. Таким образом, предполагается, что настоящее изобретение охватывает и рацематы, и чистые энантиомеры, в основном свободные от их зеркальных изомеров.
Соединение с формулой (I) или (I-1) может быть превращено в фармацевтически пригодный сложный эфир путем реакции с надлежащим этерифицирующим агентом, например, с галогенангидридом или ангидридом кислоты. Соединение с формулой (I) или (I-1), включая его сложные эфиры, можно превратить в их фармацевтически приемлемые соли обычным способом, например, обработкой нужной кислотой. Сложный эфир или его соль с формулой (I) или (I-1) можно превратить в исходное соединение, например, гидролизом.
Следующие примеры предназначены только для иллюстрации и никоим образом не для ограничения объема изобретения. Термин "активный ингредиент", использованный в примерах, означает соединение с формулой (I) или (I-1) или его фармацевтически приемлемое производное.
Пример А:
Композиции для таблеток
Следующие композиции А и Б были получены влажным гранулированием ингредиентов с использованием раствора повидона с последующим добавлением стеарата магния и прессованием (см. табл. 1 и 2).
Следующие композиции Г и Д были получены прямым прессованием смешанных ингредиентов. В композиции Д использовали лактозу для прямого прессования (Dairy Crest "Zeparox").
Композиция Г - мг/таблетку
Активный ингредиент - 250
Прежелатинизированный крахмал NF15 - 150 - 400
Композиция Д - мг/таблетку
Активный ингредиент - 250
Лактоза - 150
Авицел - 100 - 500
Композиция Е (с контролируемым выделением)
Эта композиция была получена влажным гранулированием ингредиентов (см. ниже) с использованием раствора повидона с последующим добавлением стеарата магния и прессованием. - мг/таблетку
(а) Активный ингредиент - 500
(б) Гидроксипропилметилцеллюлоза (Methocel K4M Premium) - 112
(в) Лактоза В.Р. - 53
(г) Повидон В.Р.С. - 28
(д) Стеарат магния - 7 - 700
Пример Б
Капсульные композиции:
Композиции А
Капсульная композиция была получена смешиванием ингредиентов композиции Г из примера 1 и заполнением жесткой желатиновой капсулы, состоящей из двух частей. Аналогично была получена композиция Б (инфра).
Композиция Б - мг/капсулу
(а) Активный ингредиент - 250
(б) Лактоза В.Р. - 143
(в) Натриевый гликолят крахмала - 25
(г) Стеарат магния - 2 - 420
Композиция В - мг/капсулу
(а) Активный ингредиент - 250
(б) Макрогол 4000 В.Р. - 350 - 600
Капсулы получали расплавлением макрогола 4000 В.Р., диспергированием активного ингредиента в расплаве и заполнением расплавом жесткой желатиновой капсулы, состоящей из двух частей.
Композиция Г - мг/капсулу
Активный ингредиент - 250
Лецитин - 100
Арахисовое масло - 100 - 450
Капсулы получали диспергированием активного ингредиента в лецитине и арахисовом масле и заполнением дисперсией мягких эластичных желатиновых капсул.
Композиция Д (капсулы с регулируемым выделением)
Следующая композиция для капсул с регулируемым выделением была получена путем выдавливания ингредиентов а, б и в с помощью экструдера с последующим приданием экструдату сферической формы и высушиванием. Высушенные гранулы затем покрывали мембраной (г), контролирующей выделение, и помещали в жесткие желатиновые капсулы, состоящие из двух частей. - мг/капсулу
(а) Активный ингредиент - 250
(б) Микрокристаллическая целлюлоза - 125
Лактоза В.Р. - 125
(г) Этилцеллюлоза - 13 - 513
Пример В:
Композиция для инъекций
Композиция А
Активный ингредиент - 0,200 г
0,1 М раствор соляной кислоты - до pH 4,0 - 7,0
0,1 М раствор гидроксида натрия - до pH 4,0 - 7,0
Стерильная вода - до 10 мл
Активный ингредиент был растворен в большей части воды (35 - 40oC) и pH раствора доводили до величины в диапазоне от 4,0 до 7,0 соляной кислотой или гидроксидом натрия в зависимости от условий. Порцию доводили до объема водой и фильтровали через стерильный микропористый фильтр в стерильный стеклянный пузырек из темного стекла объемом 10 мл (тип 1) и затем закрывали стерильными пробками и крышками.
Композиция Б
Активный ингредиент - 0,125 г
Стерильный апирогенный фосфатный буфер pH 7 - до 25 мл
Пример Г:
Внутримышечная инъекция
Активный ингредиент - 0,20 г
Бензиловый спирт - 0,10 г
Гликофурол - 1,45 г
Вода для инъекций - до 3,00 мл
Активный ингредиент растворяли в гликофуроле. Затем добавляли и растворяли бензиловый спирт и затем воду до 3 мл. После этого смесь фильтровали через стерильный микропористый фильтр и запаивали в стерильные стеклянные ампулы (тип 1) из темного стекла объемом 3 мл.
Пример Д:
Сироп
Активный ингредиент - 0,2500 г
Раствор сорбита - 1,5000 г
Глицерин - 2,0000 г
Бензоат натрия - 0,0050 г
Ароматизатор Peach 17.42.3169 - 0,0125 мл
Очищенная вода - до 5,0000 мл
Активный ингредиент растворяли в смеси глицерина и большей части очищенной воды. После этого к раствору добавляли водный раствор бензоата натрия, затем раствор сорбита и, наконец, ароматизатор. Доводили до конечного объема очищенной водой и хорошо перемешивали.
Пример Е:
Суппозиторий - мг/суппозиторий
Активный ингредиент (631 m)* - 250
Твердый жир В.Р. (Witepsol H15 - Dynamit Nobel) - 1770 - 2020
* Активный ингредиент использовался в виде порошка, в котором по меньшей мере 90% частиц имели диаметр 631 m или менее.
Одну пятую часть продукта Witepsol H15 расплавляли в сосуде с паровой рубашкой при максимальной температуре 45oC. Активный ингредиент просеивали через сито 2001 m и при перемешивании добавляли к расплавленной основе, используя сильверсон, оборудованный рассекающей головкой до достижения состояния тонкой дисперсии. Поддерживая температуру смеси 45oC, к суспензии добавляли оставшееся количество Witepsol H15 и перемешивали до образования гомогенной смеси. Всю суспензию пропускали через сито из нержавеющей стали (2501 m) и при непрерывном перемешивании оставляли охлаждаться до 40oC. При температуре от 38oC до 40oC 2.02 г смеси помещали в подходящие пластмассовые формы объемом 2 мл. Суппозитории оставляли охлаждаться до комнатной температуры.
Пример Е:
Пессарии - мг/пессарий
Активный ингредиент (631 m) - 250
Безводная декстроза - 380
Картофельный крахмал - 363
Стеарат магния - 7 - 1000
Указанные ингредиенты непосредственно смешивали и изготавливали пессарии путем непосредственного прессования полученной смеси.
Антивирусные испытания
1. Анти-ВЦМЧ
Вирус цитомегалии человека (ВЦМЧ) испытывают в монослоях клеток MRC5 (из легких эмбриона человека) в многоячеечных кюветах. Активность соединений определяют испытанием на уменьшение розеток, в котором монослой клеток инфицируют суспензией ВЦМЧ. В накладке из карбоксиметилцеллюлозы создают диапазон концентраций испытуемого соединения (известной молярности). Число розеток при каждой концентрации выражают в процентах от контрольного испытания и строят кривую зависимости доза-эффект. Из этой кривой оценивают концентрацию с 50% ингибированием (IC50).
Анти-ВЦМЧ активность
Соединение - IC50 (мкМ)
Пример 4 - 1,9
Пример 13 - 1,0
2. Анти-ВГВ
а. Общее описание
Анти-ВГВ активность соединений формул (I) и (I-1) определяли высокопроизводительным испытанием на эффективность ассессии. Супернатанты из растущих клеток, продуцирующих ВГВ (HepG2 2.2.15, линия клеток P5A) в планшетах с 96 лунками накладывали на лунки планшета для микротитрования, покрытые специфичным моноклональным антителом по отношению к поверхностному антителу ВГВ (HBsAg). Присутствующие в супернатантах частицы вируса связываются с антителом и остаются иммобилизованными, в то время как другие инородные вещества удаляют промыванием. После этого данные частицы вируса денатурируют с выделением нитей ДНК ВГВ, которые далее увеличивают в цепной реакции полимеразы (polymerase chain reacti on PCR) и регистрируют колориметрическим анализом на захват гибрида. Количественное охарактеризование достигается путем подгонки стандартной кривой к разбавлениям клеточного супернатанта с известным содержанием ДНК ВГВ. Степень анти-ВГВ активности получается путем сравнения содержания ДНК ВГВ в контрольных супернатантах необработанных клеток и в супернатантах, содержащих соединение формулы (I) или (I-1).
6. Иммуноаффинный захват ВГВ
Продуцирующие ВГВ клетки (2500 клеток/лунку) засевали в 96-луночные лотки для культур в смесь RPM1/10% эмбриональная бычья сыворотка/2 мМ глутамин (RPM1/10/2). На 1, 3, 5 и 7 день среды пополняли разбавлениями соединений с формулой (I) или (I-1) в смеси RPM1-10/2 до конечного объема 150 мкл. В каждую лунку круглодонного планшета для микротитрования добавляли пятьдесят мкл моноклонального антитела мыши anti-HBsAG (10 мкг/мл в фосфатно-солевом буферном растворе ФСБР, PBS). После инкубирования в течение ночи при 4oC растворы отсасывали и заменяли 100 мкл 0,1% раствора БСА в ФСБР. Образцы инкубировали 2 ч при 37oC и три раза промывали раствором ФСБР/0/0,01% Tween-20 (ФСБР/Т), используя промыватель Nипс. После этого во все лунки добавляли микропипеткой 10 мкл 0,035% раствора Tween 20 в ФСБР. Микропипеткой в лунки переносили клеточные супернатанты (25 мкл), содержащие внеклеточный вирион ДНК; конечная концентрация Tween 20 составляла 0,01%. В два ряда лунок, которые служили в качестве кривой внутреннего стандарта для количественного охарактеризования, вносили по 25 мкл стандартных разбавлений ВГВ в смеси RPM1/10/2; планшеты герметизировали и инкубировали в течение ночи при 4oC. Образцы 5 раз промывали смесь ФСБР/Т и два раза ФСБР, последнюю промывку отсасывали. После этого в каждую лунку микропипеткой добавляли 25 мкл раствора 0,09 н. NaOH/0,01% NP40, лунки с образцами герметизировали и инкубировали 60 мин при 37oC. Затем образцы нейтрализовали 25 мкл раствора 0,09 н. NaOH/100 ТРИС (pH 8,3).
В. Цепная реакция полимеразы (PCR):
Цепную реакцию полимеразы (Saiki, P. K. et al., Science, 239 (4839) 487-91 (1988)) проводили на образцах 5 мкл, используя приспособление для PCR фирмы Perkin Elmer. PCR выполняют в трубках "MicroAmp tubes" в конечном объеме 25 мкл. Праймеры выбирали из сохранившихся областей генома ВГВ, что было определено путем соединения нескольких последовательностей. Один праймер биотинилировали на конце в положении 5 с целью облегчения обнаружения продуктов PCR методом захвата гибридов. Все праймеры получены от Synthecell Corp., Rockville, MD 20850.
г. Обнаружение продуктов PCR методом захвата гибридов.
Продукты PCR обнаруживали олигонуклеотидными зондами, меченными пероксидазой хрена (Synthecell Corp., Rockville, MD 20850), которые образовывали гибриды с биотинилированными нитями денатурированных продуктов PCR прямо в лунках планшета для микротитрования, покрытых стрептавидином; использовался в основном метод Holodiniy, M. et Аl., BioTecnigues, 12 (1), 37 - 39 (1992). Модификации включали использование реакционных объемов PCR 25 λ и проведение денатурации гидроксидом натрия, а не теплом. Одновременное связывание биотинового фрагмента со связанным с планшетом стрептавидином во время гибридизации служит для "захвата" гибридов. Несвязанные меченые зонды отмывались перед колориметрическим определением связанной (гибридизованной) пероксидазы хрена. Количества ДНК ВГВ, присутствующие в исходных образцах, рассчитывали путем сравнения со стандартами. С целью определения эффективности анти-ВГВ активности эти величины затем сравнивались с полученными для необработанных клеточных культур.
IC50 (концентрация половины ингибирования) представляет количество соединения, приводящее к 50% уменьшению ДНК ВГВ. Приблизительные величины IC50 соединений из примеров 4, 13 и 69 приведены ниже.
Анти-ВЦМЧ активность
Соединение - IC50 (мкм)
Пример 4 - 0,74, 2,5
Пример 13 - 5,0
Пример 69 - 0,72, 1,3
д. Селективное ингибирование роста T-клеток
Соединения по изобретению были испытаны на ингибирование роста T-клеток (Molt 4) и B-клеток (IM9) методом Averett, D., Journal of Virological Methods, 23, (1989), 263 - 276 (см. табл. 3).
Пример 1
(+/-)-(1R*, 2S*, 3S*,5S*)-3-(ацетоксиметил) -5-(4,5-дихлор-2-нитроанилино)-1,2-циклопентандиилдиацетат
(+/-)-(1R*,2S*,3R*,4R*)-трет-бутил-N- [2,3-дигидрокси-4-(гидроксиметил)-1-циклопентил] карбамат (6,27 г, 25,1 ммоль) и 1 н. соляную кислоту (50 мл) перемешивали в течение ночи. Полученный прозрачный раствор концентрировали в вакууме и высушивали путем выпаривания метанола и этанола; получили гидрохлорид (+/-)-(1S*,2R*,3S*,5R*)-3-амино-5-(гидроксиметил)-1,2-циклопентандиол в виде твердого пенного вещества. Эту твердую пену энергично кипятили с обратным холодильником в течение 24 ч с триэтиламином (7,5 г, 75 ммоль), 1,2,4-трихлор-5-нитробензолом (5,84 г, 25,0 ммоль, 97%, Aldrich) и 2-метоксиэтанолом (75 мл). Образовавшуюся смесь черного цвета выпаривали досуха, и остаток хроматографировали на силикагеле, продукт элюировали смесью метанол : хлороформ (1:10) и получали в виде темно-оранжевого стекловидного вещества (6,9 г). Кристаллизация из смеси этанол-вода давала оранжевый порошок (3,00 г), который в течение ночи перемешивали в смеси уксусного ангидрида (3,0 мл) и пиридина (20 мл) при комнатной температуре. Выпаривание летучих веществ с последующей кристаллизацией их смеси этилацетат-гексаны приводило к получению указанного в заглавии соединения в виде оранжевых игл (2,82 г, 24%), т. пл. 153 - 156oC;
1H ЯМР (ДМСО-d6) δ: 8,25 и 7,51 (оба с., каждый 1, C6H2), 8,07 (д, J=7,8 Гц, 1, NH), 5,23 и 5,09 (оба м, 2, 2CHO), 4,3 (м, 1, CHN), 4,2-4,0 (м, 2, CH2O), 2,5-2,35 (м, 2, 2CH), 2,04, 2,03, 2,02 (все с., 9, 3CH3CO), 1,5-1,4 (м, 1, CH).
Анализ C15H20N2O5Cl2:
Рассчитано: C 46,67; H 4,35; N 6,05; Cl 15,31.
Найдено: C 46,66; H 4,37; N 6,02; Cl 15,38.
Пример 2
(+/-)-(1R*, 2S*, 3S*, 5S*)-3-(ацетоксиметил)-5-(5,6-дихлор-1H-бензимидазол-1-ил)-1,2- циклопентандиилдиацетат
(+/-)-(1R*, 2S*, 3S*, 5S*)-3-(ацетоксиметил)-5-(4,5-дихлор-2-нитроанилино)-1,2- циклопентадиилдиацетат (2,75 г, 5,93 ммоль) и никель Ренея (водная суспензия, Aldrich, 300 мг влажного) в изопропаноле (250 мл) трясли в атмосфере водорода (40 psi) в аппарате для встряхивания Парра в течение 2,25 ч. Катализатор отфильтровывали на целите, фильтрат подкисляли 98% муравьиной кислотой (5 мл) и концентрировали до получения оранжевого масла. Масло разбавляли дополнительным количеством 98% муравьиной кислоты (45 мл) и полученный оранжевый раствор кипятили с обратным холодильником в течение 40 мин. Летучие вещества удаляли, и оставшееся темное масло растворяли в хлороформе (100 мл). Раствор в хлороформе промывали насыщенным водным раствором бикарбоната натрия (3 х 10 мл), осушали (сульфат натрия) и выпаривали до получения пены, которую хроматографировали на силикагеле. Соединение, указанное в заглавии, элюировали смесью метанол-хлороформ (3:97) и получали в виде белой пены из этилацетата (2,26 г, 86%); 1H ЯМР (ДМСО-d6) δ: 8,57, 8,17, 7,97 (все с., каждый 1, 3 CH бензимидазола), 5,6 (м, 1, CHO), 5,3-5,1 (м, 2, CHO и CHN), 4,35-4,15 (м, 2, CH2O), 2,6-2,4 (м, перекрывающийся с растворителем, 2CH), 2,10, 2,06, 1,92 (все с.), перекрывающиеся с 2,0 (м, общее количество 10, 3CH3CO и CH).
Анализ для C19H20N2O6Cl2:
Рассчитано: С 51,49; H 4,55; N 6,32; Cl 16,00.
Найдено: C 51,39; H 4,58; N 6,22; Cl 16,07.
Пример 3
(+/-)-(1R*, 2S*, 3S*,5S*)-3-(ацетоксиметил)-5-(2-бром-5,6-дихлор-1H-бензимидазол-1-ил)- 1,2-циклопентандиилдиацетат
(+/-)-(1R*,2S*,3S*, 5S*)-3-(ацетоксиметил)-5-(5,6-дихлор-1H-)бензимидазол-1-ил)-1,2- циклопентандиилдиацетат (1,32 г, 2,98 ммоль) в сухом N, N-диметилформамиде (6 мл) нагревали при 60oC. В течение 5 ч добавляли N-бромсукцинимид (1,59 г, 8,93 ммоль) порциями (около 1 ммоль каждая). Нагревание продолжали в течение дополнительных 4 ч. Летучие вещества удаляли в вакууме и остаток хроматографировали на силикагеле. Указанное в заголовке соединение элюировали смесью гексан-этилацетат (1: 1) в виде коричневого порошка (1,1 г, 69%), при этом 1H ЯМР характеризовался спектром, идентичным со спектром перекристаллизованного образца. Такой образец перекристаллизовывали из смеси этанол-вода и получали белый порошок.
Т. пл. 156-159oC; 1H-ЯМР (ДМСО-d6) δ: 8,34, 7,97 (оба C, 1 каждый 2, бензимидазольных CH), 5,6 (М, 1, OCH), 5,3 (М, 1, OCH), 5,2-5,0 (М, 1, NCH), 4,4-4,2 (М, 2, OCH2), 2,7-2,5 (М, 1, CH), 2,4-2,0 (М) перекрывается при 2,1 и 2,07 (оба C, всего 8, CH2 и 2CH3CO), 1,92 (с, 3, CH3CO); масс-спектр (ХИ): 527 (6,6) 525 (45), 523 (100), 521 (65, М+1), 257 (48, М-В).
Анализ для C19H19N2O6BrCl2:
Рассчитано: C 43,71; H 3,67; N 5,37; общий галоген в виде Br 45.91.
Найдено: C 43.64; H 3,63; N 5,30; общий галоген в виде Br 45,77.
Пример 4
(+/-)-(1R*, 2S*,3S*, 5S*)-5-(2-бром-5,6-дихлор-1H-бензимидазол-1-ил)-3-(гидроксиметил)-1,2-циклопентандиол
(+/-)-(1R*, 2S*, 3S*, 5S*)-3-(ацетоксиметил)-5-(2-бром-5,6-дихлор-1H-бензимидазол-1-ил)-1,2- циклопентандиилдиацетат (600 мг, 1,15 ммоль) добавки к перемешиваемой смеси карбоната натрия (122 мг) в растворителе: вода (2 мл) - этанол (10 мл) - метанол (10 мл). Через два с половиной часа при комнатной температуре pH доводили до 7 ледяной уксусной кислотой. Летучие вещества удаляли в вакууме, остаток растирали с водой (5 мл), фильтровали и получали белое твердое вещество. Перекристаллизация твердого вещества из смеси этанол-метанол (1: 1) приводила к указанному в заголовке веществу в виде белого порошка (282 мг, 62%) - далее Т.пл. 208-211oC; 1H-ЯМР (ДМСО-d6) δ: 8,23, (C, 1, бензимидазольный H7), 7,95 (C, бензимидазольный H4), 5,13 (T, J=4,1 Гц, 1, CH2OH), 5,03 (Д, J = 6,2 Гц, 1, OH), 5,0 - 4,85 (М, 1, H5), 4,71 (Д, J= 3,5 Гц, 1, OH), 4,55-4,45 (М, 1, H1), 3,85-3,80 (М, 1, H2), 3,7-3,6 и 3,55-3,45 (оба М, 1 каждый, OCH2), 2,2-1,95 (М, 3, H3 и 2H4); масс-спектр (ХИ): 395 (М+1).
Анализ для C13H13N2O3Cl2Br:
Рассчитано: C 39,43; H 3,31; N 7,07; общий галоген в виде Br 60,52.
Найдено: C 39,50; H 3,33; N 7,02; общий галоген в виде Br 60,71.
Пример 5
(±)-(1R*, 2S*, 3S*, 5S*)-3-(ацетоксиметил)-5-(-2,5,6-трихлор-1H-бензимидазол-1-ил)- 1,2-циклопентандиилдиацетат
(±)-(1R*,2S*,3S*, 5S*)-3-(ацетоксиметил)-5-(5,6-дихлор-1H-бензимидазол-1-ил)-1,2- циклопентандиилдиацетат (850 мг, 1,92 ммоль) в сухом N,N-диметилформамиде (5 мл) выдерживали при 95oC в течение 3 ч, при этом порциями добавляли N-хлорсукцинимид (760 мг). Нагревание продолжали в общей сложности 6 ч. Летучие вещества удаляли в вакууме и остаток хроматографировали на силикагеле. Соединение, указанное в заголовке, элюировали смесью этилацетат : гексаны (3:7) как желтое твердое вещество (160 мг, 17%); спектр 1H ЯМР был в согласии со структурой и почти идентичен спектру соединения, указанного в заголовке примера 3.
Пример 6
(±)-(1R*, 2S*, 3S*, 5S*)-5-(2,5,6-трихлор-1H-бензимидазол-1-ил)- 3-гидроксиметил-(1,2-циклопентандиол)
(±)-(1R*, 2S*, 3S*, 5S*)-3-(ацетоксиметил)-5-(2,5,6-трихлор-1H-бензимидазол-1-ил)- 1,2-циклопентандиилдиацетат (160 мг, 3,40 ммоль) был деоцетилирован, как в примере 4: после элюирования с силикагеля, получено указанное вещество в виде белого порошка (36 мг, 31%) Т.пл. 206-210oC; 1H-ЯМР (ДМСО-d6) δ: 8,24 (C, 1, бензимидазольный H7), 7,98 (C, 1, бензимидазольный H4), 5,15-5,0 (М, 2, CH2OH и OH), 5,0-4,8 (М, 1, H5), 4,74 (g, J=3,5 Гц, 1, OH), 4,55-4,40 (М, 1, H1), 3,9-3,75 (M, 1, H2), 3,7-3,4 (М, 2, OCH2), 2,2-1,9 (М, 3, H3 и 2H4).
Анализ для C13H13N2O3Cl3:
Рассчитано: C 44.41; H 3,73, N 7,97; Cl 30.25.
Найдено: C 44.20; H 3,81; N 7,94; Cl 30.08
Пример 7
(1 α, 3 β, 4 β -(3,4-дигидрокси-1-циклопентил)метилбензоат
К перемешиваемому охлажденному (0oC) раствору 4-гидроксиметилциклопентена (37,0 г, 276 ммоль) в 450 мл пиридина (J. P. Depres and A.E.Greeh, J. Org, Chem. ) 1984, 49, 928-93 (и ссылки в этой работе) в течение 30 мин добавляли бензоилхлорид (32,1 мл, 276 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 1,25 ч. Добавляли воду (50 мл), и летучие вещества удаляли в вакууме. Оставщееся масло растворяли в хлороформе и проводили экстракцию из раствора водой, затем его осушали над сульфатом натрия. Выпариванием растворителя получен (3-циклопентен-1-ил)метилбензоат в виде желтого масла (53,94 г, 91%), достаточно чистого для применения 1H-ЯМР (ДМСО-d6) δ: 7,98, 7,67, 7,56 (М, 5, C6H5), 5,72 (C, 2, CH=CH), 4,19 (М, 2, OCH2), 2,71 (М, 1, CH), 2,56-2,77 М, перекрыт с растворителем 2CH), 2,21-2,14 (М, 2, 2CH).
(3-циклопентен-1-ил)метилбензоат (37,6 г, 0,161 ммоль) в 200 мл ацетона по каплям добавляли в течение 2 ч к перемешиваемому раствору N-метилморфолин-N-оксида (33,1 г, 60%-ный раствор в воде, 0,169 моль), тетраоксида осмия (2,5%-ный раствор в трет-бутаноле, Aldrich, 3,0 мл) и ацетона (200 мл) при комнатной температуре. Перемешивание дополнительно продолжали 16 ч. Добавляли 500 мл хлороформа и 150 мл воды. Органический слой отделяли, промывали холодным раствором 1 н. соляной кислоты (2х150 мл) и затем насыщенным раствором бикарбоната натрия в воде (100 мл), после чего высушивали (MgSO4). Летучие соединения удаляли и оставшееся твердое вещество кристаллизовали из толуола (200 мл); получили указанное в заголовке вещество в виде белых кристаллов (26,9 г, 73%), далее Т.пл. 92-94oC; 1H-ЯМР (ДМСО-d6) δ: 7,96, 7,65, 7,56 (М, 5, C6H5), 4,38 (g, J=4,1 Гц, 2, 2OH), 4,14 (g, J=6,6 Гц, 2, CH2O), 3,90 (М, 2, 2 OCH), 2,58 (М, перекрывается с растворителем CH), 1,75 (М, 2, 2CH), 1,55 (М, 2, 2CH).
Анализ для C13H16O4:
Рассчитано: C 66,09; H 6,83.
Найдено: C 66,19; H 6,86.
Концентрирование маточных растворов давало 10,33 г белого твердого вещества, содержащего дополнительное количество указанного в заголовке вещества, загрязненного (+/-)-(1_, 3_,4_)-3,4-дигидрокси-1-циклопентил)метилбензоатом в приблизительном соотношении 2:3 по данным 1H ЯМР.
Пример 8
(3a- α , 5 α , 6a- α ) -(тетрагидро-4H-циклопента-1,3,2-диоксатиол-5-ил)метилбензоат-S-оксид
К раствору (1 β , 3 α , 4 α )-(3,4-дигидрокси-1-циклопентил)метилбензоата (10,0 г, 42,3 ммоль) в 150 мл четыреххлористого углерода добавляли тионилхлорид (6,04 г, 50,8 ммоль). Раствор кипятили с обратным холодильником в течение 1,5 ч. Растворитель выпаривали и получали указанное в заголовке вещество в виде густого масла, достаточной для использования чистоты (см. следующий пример). Этот образец кристаллизовался из толуола в виде воскообразного твердого вещества Т.пл. 48-57oC; 1H-ЯМР (ДМСО-d6) δ: 7,96, 7,66, 7,52 (М, 5, C6H5), 5,46 и 5,32 (оба М, 1,2 OCH, смесь изомерных S-оксидов N 1: 1), 4,28 (М, 2, OCH2), 2,90 и 2,43 (оба M, 1, CH двух изомерных S-оксидов, 2,10 и 1,74 (оба М, 4, 4CH).
Анализ для C13H14O5:
Рассчитано: C 55,31; H 5,00; S 11,36.
Найдено: C 55,41; H 5,04; S 11,30.
Пример 9
(3a - α , 5 α , 6a - α )-(тетрагидро-4H-циклопента-1,3,2-диоксатиол-5-ил)-метилбензоат-S,S-диоксид
(3a- α , 5 α , 6a- α )-(тетрагидро-4H-циклопента-1,3,2-диоксатиол-5-ил)метилбензоат-S-оксид (предыдущий пример, 42,3 ммоль) перемешивали в смеси четыреххлористый углерод (40 мл) - ацетонитрил (40 мл) вода (60 мл) при добавлении метапериодата натрия (8,98 г, 42,3 мэкв) и трихлорида рутения (44 мг, 0,21 мэкв). Через 30 мин добавляли дополнительное количество метапериодата натрия (179 мг) с целью доведения реакции до конца, о чем судили путем ТСХ (силикагель, смесь метанол-хлороформ 1:19, проявление в парах иода). Спустя в общей сложности 1 ч, добавляли 300 мл хлористого метилена. Органический слой отделяли и проводили экстракцию из водного слоя дополнительным количеством хлористого метилена (300 мл). Объединенные органические слои промывали насыщенным водным раствором бикарбоната натрия (100 мл), затем насыщенным водным раствором хлорида натрия (100 мл), осушали (MgSO4) и концентрировали в вакууме; получали указанное в заголовке соединение в виде белого порошка (12,37 г, 98%), Т.пл. 114-119oC, 1H-ЯМР (ДМСО-d6) δ: 8,02, 7,70, 7,55 (все М, 5, C6H5), 5,62 (М, 2OCH), 4,34 (д, J = 5,8 Гц, 2, OCH2), 2,79 - 2,64 (М, 1, CH), 2,32 - 2,21 и 1,97 - 1,79 (М, 4, 2 CH2)
Анализ для C13H14SO6:
Рассчитано: C 52,35; H 4,73; S 10,75.
Найдено: C 52,32; H 4,73; S 10,69.
Пример 10
(+)-(1R*,2R*,4S*)-2- (5,6-дихлор-1H-бензимидазол-1-ил)-4-(гидроксиметил) циклопентанол
К раствору 5,6-дихлорбензимидазола (L. B. Townsend and G.R.Rewankar, Chem. Rev. , 1970, 70, 389 и ссылки из этой работы) (1,50 г 8,00 ммоль) в сухом N,N-диметилформамиде (35 мл) добавляли гидрид натрия (416 мг, 10,4 мэкв, в виде 60% масляной дисперсии). Смесь перемешивали при 25oC в течение 45 мин. В течение 5 ч порциями добавляли (3a- α , 5 α , 6a - α )-(тетрагидро-4H-циклопента-1,3,2-диоксатиол-5-ил)метилбензоат- S,S-диоксид (3,05 г, 10,2 ммоль). Перемешивание продолжали в течение ночи при комнатной температуре. Летучие вещества удаляли в вакууме и оставшееся масло растворяли в 1,4-диоксане (130 мл) в смеси водой (10 мл) при кипячении с обратным холодильником в присутствии 4 M серной кислоты (2,3 мл). После 10 мин кипячения раствор подщелачивали 5 н. раствором гидроксида натрия, дополнительно нагревали один час при 50oC, а затем нейтрализовали дополнительным количеством кислоты. Выпаривание летучих веществ в вакууме приводило к остаточным твердым веществам, которые подвергали экстракции хлороформом с целью удаления непрореагировавшего 5,6-дихлорбензимидазола и далее перекристаллизовывали из смеси этанол-вода; получено указанное в заголовке вещество в виде белого порошка (2,09 г, 87%). Перекристаллизация такого образца из смеси этанол-вода приводила к указанному в заголовке веществу в виде белых гранул. Т.пл. 244-245oC; 1H-ЯМР (ДМСО-d6) δ: 8,47, 8,05, 7,93 (все C, 3, арильный CH, 5,19 (д, J = 5,3 Гц, 1, CHOH), 4,71 (T, J = 5,3 Гц, 1, CH2OH), 4,6 - 4,5 (М, 1, NCH), 4,37 - 4,25 (М, 1, OCH), 3,41 (М, 2, OCH2), 2,4 - 2,2 и 1,95 - 1,62 (М, 5, 5CH).
Анализ для C13H14N2Cl2 • 0,02 C2H5OH :
Рассчитано: C 51,85; H 4,71; N 9,27; Cl 23,47.
Найдено: C 51,87; H 4,74; N 9,28; Cl 23,60.
Пример 11
(±)-(1R*, 2R*, 4S*)-4-(ацетоксиметил)- 2-(5,6-дихлор-1H-бензимидазол-1-ил)циклопентилацетат
(±)-(1R*,2R*, 4S*)-2-(5,6-дихлор-1H-бензимидазол-1-ил)-4-(гидроксиметил) циклопентанол (7,80 г, 25,8 ммоль) растворяли в смеси пиридин (50 мл) - уксусный ангидрид (50 мл) и раствор перемешивали в течение ночи. Летучие вещества удаляли в вакууме и оставшееся масло подвергали распределению между хлористым метиленом (150 мл) и насыщенным водным раствором бикарбоната натрия (100 мл). Органический слой осушали (сульфат натрия) и выпаривали до получения стеклообразного вещества (9,91 г, 99%); 1H-ЯМР (ДМСО-d6) δ:: 8,58, 8,08, 7,96 (C, 3, арильный CH), 5,39 - 5,32 (М, 1, OCH), 5,09 - 5,04 (М, 1, NCH), 4,11 (д, J = 6,6 Гц, 2, OCH2), 2,59 - 2,50 (М, перекрывается с растворителем, CH), 2,41 - 2,35 (М, 1, CH), 2,17 - 1,86 (М, перекрывается с 2,06 и 1,94, оба C, всего 9, 3CH и 2CH3CO).
Анализ для C17H18N2O2Cl2 • 0,1 CH2Cl2:
Рассчитано: C 52,96; H 4,70; N 7,26; Cl 18.55.
Найдено: C 52,86; H 4,74; N 7,25; Cl 18,50.
Пример 12.
(±)-(1R*, 2R*, 4S*)-4-(ацетоксиметил)-2-(2-бром-5,6-дихлор-1H-бензимидазол-1-ил)циклопентилацетат
К раствору (±)-(1R*, 2R*,4S*)-4- (ацетоксиметил)-2-(5,6-дихлор-1H-бензимидазол-1-ил) циклопентилацетата (8,95 г, 23,2 ммоль) в 46 мл сухого N, N-диметилформамида добавляли N-бромсукцинимид (4,54 г, 25,5 ммоль). Раствор держали при приблизительно 70oC (масляная баня) в течение 5 ч. Летучие вещества удаляли в вакууме и оставшийся оранжевый сироп хроматографировали на силикагеле. Соединение, указанное в заголовке, было элюировано хлороформом как бледно-желтое твердое вещество (5,14 г, 48%), Т.пл. 122-125oC; 1H-ЯМР (ДМСО-d6) δ:: 8,16 (C, 1, бензимидазольный H7), 7,95 (C, 1, бензимидазольный H4), 5,60 - 5,55 (М, 1, OCH), 5,12 - 5,03 (М, 1, NCH), 4,15 (д, J = 6,3 Гц, 2, OCH2), 2,66 - 2,60 (М, 1, CHCH2), 2,29 - 2,14 (М, 3, CH), 2,06 (C, 3, CH3CO), 1,93 (C, перекрытый M, 4, CH3CO + CH); масс-спектр (ХИ): 469 (5,8), 467 (37,5), 465 (95), 463 (54, M+1), 199 (100, M-B).
Анализ для C17H17N2Cl2BrO4:
Рассчитано: C 43,99; H 3,69; N 6,04; общий галоген в виде Br 51,65.
Найдено: C 44,06; H 3,70; N 5,97; общий галоген в виде Br 51,74.
Пример 13
(±)-(1R*, 2S*, 4S*)-2-(2-бром-5,6-дихлор-1H-бензимидазол-1-ил)-4-(гидроксиметил) циклопентанол
(±)-(1R*, 2R*, 4S*)-4-(ацетоксиметил)-2-(2-бром-5,6-дихлор-1H-бензимидазол-1-ил) циклопентилацетат (2,75 г, 5,92 ммоль) добавляли в перемешиваемую смесь карбонат натрия (0,63 г) - вода (11 мл) - этанол (55 мл) метанол (55 мл). Через 2 ч при комнатной температуре pH доводили до 7 ледяной уксусной кислотой. Летучие вещества удаляли в вакууме, остаток растирали с водой (30 мл), фильтровали и получали белое твердое вещество. Перекристаллизацией этого твердого вещества из смеси этанол-метанол (1:1), получено указанное в заголовке соединение в виде белого порошка. Т.пл. 218-220oC; 1H-ЯМР (ДМСО-d6) δ:: 8,18 (C, бензимидазольный H7), 7,97 (C, 1, бензимидазольный H4), 5,20 (М, 1, OH), 4,95 (М, 1, OH), 4,80 - 4,60 (M, 2, OCH и NCH), 3,50 - 3,40 (М, 2, OCH2), 2,45 - 2,20 (М, 1, CH), 2,20 - 1,60 (М, 4, 4CH); масс-спектр (XИ): 379 (M+1).
Анализ для C13H13N2O2Cl2Br:
Рассчитано: C 41,08; H 3,45; N 7,37; общий галоген в виде Br 63,07.
Найдено: C 41,27; H 3,49; N 7,28; общий галоген в виде Br 62,88.
Пример 14
(±)-(1R*,2S*,3S*)- 3-азидо-1,2-циклопентандиол
Смесь (±)-(1R*, 2R*, 3S*)-2,3-эпокси-1-циклопентанола (R.Steyn and H.Z. Sabbe, Tet 1969, 25, 3579) (36,2 г, 0,362 моль), азида натрия (47,1 г, 0,724 экв), сульфата аммония (23,9 г, 0,181 экв), диоксана (200 мл) и воды (180 мл) медленно нагревали до кипячения с обратным холодильником. После того, как первоначальное выделение тепла кончилось, раствор кипятили в мягких условиях в течение 18 ч. Летучие вещества удаляли и проводили экстракцию оставшегося материала абсолютированным этанолом (250 мл). Этанол выпаривали и оставшееся масло хроматографировали на силикагеле. Элюировали указанное в заголовке вещество в виде бледно-желтого масла (41,0 г, 79%). 1H-ЯМР (ДМСО-d6) δ:: 4,96 (д, J = 6,4 Гц, 1, OH), 4,55 (д, J = 3,9 Гц, 1, OH), 3,85 - 3,55 (M, 3, 2 OCH NCH), 2,1 - 1,75 и 1,55 - 1,2 (М, 4, 2CH2).
Анализ для C5H9N3O2:
Рассчитано: C 41,95; H 6,34; N 29,35.
Найдено: C 41,71; H 6,36; N 29,15.
Продолженное элюирование колонки хлористым метиленом привело к получению 5,32 г масла, состоящего из дополнительного количества указанного в заголовке соединения, загрязненного примерно 50% (±)-(1 α , 2 β , 3 α )-2-азидо-1,3-циклопентандиола.
Пример 15
(±)-(1R*,2S*, 3R*)-3-амино-1,2-циклопентандиол
Смесь (±)-(1R*, 2S*, 3R*)-3-азидо-1,2-циклопентандиола (2,10 г, 14,7 моль), 5% палладия на угле (250 мг) и абсолютированного этанола (150 мл) трясли на аппарате для встряхивания Парра в атмосфере водорода (50 psi) в течение 3 ч. Катализатор отфильтровывали на целите, выпаривали растворитель в вакууме и получали указанное в заголовке соединение в виде белого твердого вещества (1,65 г, 96%). Т.пл. 74-76oC.
Анализ для C5H11NO2:
Рассчитано: C 51,26; H 9,46; N 11,96.
Найдено: C 51,16; H 9,52; N 11,91.
Пример 16
(±)-(1R*,2S*, 3R*)-3-(4,5-дихлор-2-нитроанилино)-1,2-циклопентандиол
(±)-(1R*, 2S*, 3R*)-3-амино-1,2-циклопентандиол (7,10 г, 50,0 ммоль), 1,2,4-трихлор-5-нитробензол (11,67 г, 50,0 ммоль в виде 97%-го, Aldrich), триэтиламин (10 мл) и трет-бутанол (50 мл) кипятили с обратным холодильником в атмосфере азота в течение 7 ч. Летучие вещества удаляли в вакууме, и черный остаток хроматографировали на силикагеле. Элюировали указанное в заголовке соединение смесью метанол-хлороформ (1: 24) в виде оранжевого твердого вещества (6,45 г, 42%). Кристаллизацией данного образца из смеси этанол-вода получено указанное в заголовке соединение в виде желтых иголок. Т. пл. 137-140oC; 1H-ЯМР (ДМСО-d6) δ:: 8,26 и 7,49 (оба, 1 каждый, 2 ароматических CH), 8,06 (д, J = 7,0 Гц, 1, NH), 5,04 (д, J = 6,1 Гц, 1, OH), 4,65 (д, J = 4,1 Гц, 1, OH), 4,0 - 3,7 (М, 3, 2 OCH и NCH), 2,2 - 2,1, 2,0 - 1,8, 1,7 - 1,3 (все М, 4, 4 CH).
Анализ для C11H12N2O4Cl2:
Рассчитано: C 43,02; H 3,94; N 9,12; Cl 23,09.
Найдено: C 43,01; H 3,92; N 9,04; Cl 23,15.
Пример 17.
(±)-(1R*, 2S*, 3R*)-3-(2-амино-5,6-дихлор-1H-бензимидазол-1-ил)-1,2-циклопентандиол
(±)-(1R*, 2S*, 3R*)-3-(4,5-дихлор-2-нитроанилино)-1,2-циклопентандиол (2,00 г, 6,51 ммоль) в этаноле (100 мл) трясли в аппарате для встряхивания Парра с никелем Ренея (Aldrich, предварительно промыт водой до нейтральной реакции, примерно 1 tsp) в атмосфере водорода (50 psi) в течение 1.5 ч, пока не прекратилось поглощение водорода. ТСХ (силикагель, смесь метанол-хлороформ 1: 10) обнаруживает одно пятно с Rf меньшим, чем у исходного материала. Катализатор отфильтровывали (целит), летучие вещества выпаривали и получали стекловидный продукт (2.0 г), который быстро темнел на воздухе. Его немедленно растворяли в ацетонитриле (20 мл), добавляли бромциан (1 М раствор в ацетонитриле, Aldrich, 1.4 мл, 7.0 ммоль), и раствор в течение ночи перемешивали при комнатной температуре. Образовавшийся темный осадок растворяли при добавлении 20 мл воды. Раствор пурпурного цвета нейтрализовали 1 н. раствором гидроксида натрия. Полученный осадок отфильтровывали, промывали водой и кристаллизовали из 95% этанола; (75 мл); получили серо-белый порошок (1,14 г, 58%). Т.пл. >250oC разл.; 1H-ЯМР (ДМСО-d6) δ:: 7,37 и 7,32 (оба с, 2,2 бензимидазольных CH), 6,6 (м.с, 2, NH2), 4,81 (д, J = 7,3 Гц, 1, OH), 4,73 (д, J = 3,2 Гц, 1, OH), 4,7-4,5 (М, 1, NCH), 4,4 (М, OCH), 2,4-1,6 (М, 4,4 CH).
Анализ для C12H13N3O2Cl2:
Рассчитано: C 47,70; H 4,34; N 13,91; Cl 23,47.
Найдено: C 47,80; H 4,35; N 13,83; Cl 23,45.
Пример 18
(±)-(1R*, 2S*, 3R*)-3-(5,6-дихлор-1H- бензимидазол-1-ил)-1,2-циклопентандиилдиацетат
(±)-(1R*, 2S*, 3R*)-3-(4,5-дихлор-2-нитроанилино)-1,2-циклопентандиол (2,00 г, 6,51 ммоль) в течение ночи при комнатной температуре перемешивали в смеси уксусный ангидрид (1,8 мл) - пиридин (15 мл). Летучие вещества удаляли в вакууме. Оставшееся масло разделяли между насыщенным водным раствором бикарбоната натрия и H2O. Слой CHCl3 высушивали (Na2SO4) и растворитель выпаривали; получили желтое твердое вещество (2,56 мг). Этот диацетат восстанавливали никелем Ренея в атмосфере водорода согласно примеру 17. Катализатор отфильтровывали и к этанольному фильтрату добавляли 5 мл 98% муравьиной кислоты. Летучие вещества удаляли в вакууме и остаток в течение 30 мин кипятили с обратным холодильником в 35 мл 98% муравьиной кислоты. Муравьиную кислоту выпаривали и оставшееся масло распределяли между хлороформом и избытком насыщенного водного раствора бикарбоната натрия. Слой хлороформа осушали (Na2SO4) и концентрировали до получения коричневого масла. Это масло хроматографировали на силикагеле. Соединение, указанное в заголовке, элюировали 6-10% смесью метанол-хлороформ. Кристаллизацией из смеси этанол-вода получено указанное в заголовке вещество в виде белого порошка (1,77 г, 72%). Т. пл. 95-97oC; 1H-ЯМР (ДМСО-d6) δ:: 8,56, 8,16, 7,96 (все C, 1 каждый, 3 бензимидазольных CH), 5,6-5,5 (М, 1, OCH), 5,35-5,30 (М, 1, OCH), 5,15-5,05 (М, 1, NCH), 2,55-2,25 (М, перекрывается с растворителем, 2 CH), 2,1-2,0 (М, перекрывается с при 2,10, 4, CH и CH3CO), 2,0-1,8 (М, перекрывающийся с при 1,88, 4, CH и CH3CO).
Анализ для C16H16N2O4Cl2•0,45H2O:
Рассчитано: C 50,66; H 4,49; N 7,39; Cl 18,69.
Найдено: C 50,73; H 4,49; N 7,36; Cl 18,70.
Пример 19
(±)-(1R*, 2S*, 3R*)-3-(2-бром-5,6- дихлор-1H-бензимидазол-1-ил)-1,2-циклопентандиилдиацетат
(±)-(1R*, 2S*, 3R*)-3-(дихлор-1H- бензимидазол-1-ил)-1,2-циклопентандиилдиацетат (800 мг, 2,16 ммоль) бромировали согласно примеру 3 и указанное в заголовке вещество элюировали из колонки с силикагелем смесью этилацетат-гексаны (3:7) в виде белого порошка (450 мл, 46%).
Этот образец переосаждали из смеси EtOAc-гексаны и получили указанное в заголовке соединение в виде белого порошка, Т.пл. 140-146oC; 1H-ЯМР (ДМСО-d6) δ:: 8,24 и 7,96 (оба с, 1 каждый, H7 и H4), 5,8-5,7 (М, 1, OCH), 5,4-5,3 (М, 1, OCH), 5,2-5,1 (М, 1, NCH), 2,6-2,4 (М, перекрывается с растворителем CH), 2,4-2,15 (М, 2,2 CH), 2,11 (С, 3, CH3CO), 1,95-1,80 (М, перекрывается с с при 1,88, 4, CH и CH3CO); масс-спектр (ХИ): 455 (1,3), 453 (29), 451 (68), 449 (45, M+1).
Анализ для C16H15N2O4Cl2Br:
Рассчитано: C 42,70; H 3,36; N 6,22; общий галоген в виде Cl 23,63.
Найдено: C 42,77; H 3,41; N 6,16; общий галоген в виде Cl 23,68.
Пример 20
(±)-(1R*, 2S*, 3R*)-3-(2-бром-5,6- дихлор-1H-бензимидазол-1-ил)-1,2-циклопентандиол
Диацетат из примера 19 (315 мг, 0,700 ммоль) деацетилировали как описано в примере 4 с хроматографированием сырого продукта на силикагеле. Указанное в заголовке соединение элюировали смесью метанол-хлороформ (1:9) в виде белого порошка (180 мг, 74%). Т.пл. 169-175oC; 1H-ЯМР (ДМСО-d6) δ:: 7,99 и 7,95 (оба с, 2, 2 бензимидазольных CH), 5,08 (д, J = 6,3 Гц, 1, OH), 5,0-4,85 (М, 1, NCH), 4,76 (д, J = 3,0 Гц, 1, OH), 4,55-4,45 (М, 1, OCH), 4,05-3,95 (М, 1, OCH), 2,35-2,0 (М, 3, 3CH), 1,7-1,6 (М, 1, CH).
Анализ для C12H11N2Cl2O2Br:
Рассчитано: C 39,20; H 3,06; N 7,62; общий галоген в виде Cl 28,93.
Найдено: C 38,99; H 3,07; N 7,49; общий галоген в виде Cl 29,16.
Пример 21.
Метансульфонат (-)-(1S,4R)-4-амино-2-циклопентен-1-карбоновой кислоты
Раствор (-)-2-азабицикло[2.2.1] гепт-5-ен-3-она (97,45 г, 0,8929 ммоль, Enzymatix Ltd) в 500 мл тетрагидрофурана фильтровали и нагревали до 35oC. В течение 1,5 ч добавляли раствор метансульфокислоты (63,7 мл, 0,9817 ммоль) в воде (24,1 мл, 1,34 ммоль) так, чтобы последующее выделение тепла не приводило к температуре выше 45oC. Полученную суспензию в течение 3 ч нагревали при 60oC, а затем оставляли ее остывать до комнатной температуры в течение 15 ч. Суспензию фильтровали и отжатый осадок дважды промывали 200 мл безводного тетрагидрофурана. Отбирали образец отжатого осадка для анализа, высушивали его и получали указанное в заголовке соединение в виде белого твердого вещества (1,264 г). Т.пл. 167-169,2oC; 1H-ЯМР (ДМСО-d6) δ:: 12,6 (мс, 1H, CO2H), 8,04 (мс, 3H, 11H3 +), 6,10 (дт, J = 5,6, 2,0, 2,0 Гц, 1H, винил, 5,85 (дт, J = 5,3, 2,3, 2,3 Гц, 1H, винил), 4,19 (мс, w1/2 = 20 Гц, 1H, аллильный H), 3,61 (м, w1/2 = 22 Гц, 1H, аллильный H), 2,53 (квинтет, J = 5,3 Гц (перекрывается с сигналом ДМСО), 1/2 CH2), 2,39 (с, 3H, CH3O3H), 1,93 (дт, J = 6,7, 6,7, 13,7 Гц, 1H, 1/2 CH2); [α]
Анализ для C7H13NO5S:
Рассчитано: С 37,66; H 5,87; N 6,27; S 14,36.
Найдено: C 37,65; H 5,88; N 6,30; S 14,44.
Оставшийся влажный отжатый осадок непосредственно использовали в последующем примере.
Пример 22
(-)-(1S,4R)-4-амино-2-циклопентен-1-метанол.
Влажный от тетрагидрофурана отжатый осадок метилсульфоната (-)-(1S, 4R)-4-амино-2-циклопентен-1-карбоновой кислоты, полученный в примере 21, суспендировали в 400 мл сухого тетрагидрофурана и переносили через трубку в быстроперемешиваемый раствор литийалюминийгидрида в тетрагидрофуране (1,0 М, 1600 мл, 1,6 ммоль, Aldrich), охлаждаемый в бане со смесью лед/ацетон. Скорость переноса ограничивали для контроля скорости газовыделения и для поддержания температуры между 0oC и 10oC (общее время добавления 1,5 ч). Полученную смесь в течение двух часов нагревали до начала кипения, а затем кипятили с обратным холодильником в течение 16 ч.
Путем перегонки удаляли примерно 1,6 л растворителя, оставшуюся суспензию охлаждали в бане со смесью лед/ацетон, и затем обрабатывали диэтиловым эфиром (сухой, 1 л) и фторидом натрия (403,3 г, 9,605 ммоль, Aldrich). Медленно добавляли воду (86 мл, 4,8 ммоль) с такой скоростью (3 ч, чтобы температура была ниже 5oC и выделение водорода было умеренным. Полученную взвесь фильтровали и сжатый осадок промывали 200 мл тетрагидрофурана, затем 7% смесью вода-тетрагидрофуран (500 мл). Количественный анализ ЖХВД (см. ниже пример 23) фильтрата показал, что он содержит 60,04 г указанного в заголовке соединения. Отжатый осадок повторно суспендировали в 7% смеси вода-тетрагидрофуран (1 л) в течение получаса, фильтровали и промывали 400 мл 7% смеси вода-тетрагидрофуран, а затем 300 мл 10% смеси вода-тетрагидрофуран. Количественный анализ ЖХВД (см. ниже пример 23) показал, что фильтрат содержит 26,70 г указанного в заголовке соединения. Отжатый осадок повторно суспендировали в 1 л метанола в течение 16 ч, фильтровали и промывали 500 мл метанола. Количественный анализ ЖХВД (см. ниже пример 23) фильтрата показал, что он содержит 4,09 г указанного в заголовке соединения. Общий выход указанного в заголовке соединения, таким образом составил 90,83 г, 0,8027 ммоль или 90,5% от теоретического выхода с поправкой на взятие образца для анализа.
Пример 23.
Анализ (-)-(1S, 4R)-4-амино-2-циклопентен-1-метанола и его энантиомера (+)-(1R,4S)-4-амино-2-циклопентен-1-метанола
Образцы указанного в заголовке соединения были охарактеризованы методом Брюкнера Г. , Виттнера Р. и Годеля Г. - Автоматическое разделение энантиомеров аминокислот путем получения их производных с о-фталевым диальдегидом и N-ацилированными цистеинами (J. Chrom, 476 (1989), 73-82). В качестве реагентов для образования производных использовали о-фталевый диальдегид и N-ацетил-L-цистеин. При хроматографическом разделении использовали Optima II ODS 100 x 4,5 мм, трехмикронную колонку (III Supplies Co., Meriden, CT), и градиентное элюирование со скоростью 0,9 мл/мин, начиная со 100% натрийацетатного буфера (40 мм, рH 6,5) с линейным переходом к 18% ацетонитрилу в течение 15 мин и последующей поддержкой 18% ацетонитрила в течение 15 мин. Детектирование осуществлялось при 338 нм. Образцы растворяли в 0,1 М боратном буфере, pH 10,4. Идентичность и чистоту образцов устанавливали путем сравнения с аутентичными стандартами (см. EP 434450 (26 июня 1991 г.)). Время удерживания (1S, RS)-изомера составляло около 21 мин. Время удерживания (1R,4S)-изомера было около 22 мин.
Пример 24
(-)-(1R,4S)-трет-бутил-N-[4-гидроксиметил-2-циклопентен-1-ил]карбамат
Первый фильтрат из примера 22, содержащий (-)-(1S,4R)-4-амино-2-циклопентен-1-метанол, охлаждали в бане со смесью лед/ацетон и обрабатывали дитретбутилдикарбонатом (199,42 г, 0,9265 ммоль, Aldrich). Смесь концентрировали в вакууме до объема 300 мл и добавляли к второму фильтрату из примера 22, который тем временем охлаждали в бане со смесью лед/ацетон. Смесь оставляли при перемешивании нагреваться до комнатной температуры в течение 18 ч; в это время имело место выделение газа и образование прозрачного раствора. Раствор объединяли с последним фильтратом из примера 22, и который выпаривали в вакууме, получая смесь масла и твердых веществ. Полученный раствор выпаривали в вакууме и получали масло. Масло распределяли между 300 мл этилацетата и фосфатным буфером (100 мл 1,5 М раствора калийдигидрофосфата с pH, доведенным до 7,0 50% смесью гидроксида натрия и воды. Фазы разделяли, водную фазу дважды реэкстрагировали этилацетатом (200 мл). Органические фазы сушили над сульфатом натрия и фильтровали через силикагель (50 г). Растворитель удаляли в вакууме и получили масло (220,78 г), которое помещали в гексаны (300 мл). С целью растворения масла добавляли минимальное количество этилацетата (около 50 мл), и раствор оставляли кристаллизоваться в течение трех дней. Кристаллы отфильтровывали, промывали 20% смесью этилацетат/гексаны и осушали путем отсасывания до достижения постоянного веса (156,1 г, 0,732 ммоль, 82,6% от теоретического) указанного в заголовке соединения; (c = 5,07, метанол; ХИ-МС (CH4) 214 (M+1); ТСХ (окись кремния, 10% смеси метанол-хлороформ, проявление иодом Rf = 0,51.
Анализ для C11H19O3N:
Рассчитано: C 61,95; H 8,98; N 6,57.
Найдено: C 61,87; H 8,96; N 6,59.
Дополнительные 10,14 г кристаллического материала было получено из маточного раствора путем кристаллизации и хроматографии, доводя общий выход до 166,24 г (0,780 ммоль, 87,9% от теории, исходя из лактама из примера 21).
Также было обнаружено, что указанное в заголовке соединение можно удобно получить непосредственно из 2-азабицикло[2.2.1]гепт-5-ен-3-она, как в рацемической форме, так и в виде (-) энантиомера по следующей процедуре. (-)-2-азабицикло[2.2.1] гепт-5-ен-3-он (6,00 г, 55,0 ммоль) в безводном тетрагидрофуране (30 мл) нагревали до 34oC и перемешивали; в течение 10 мин по каплям добавляли метансульфокислоту (3,6 мл, 55 ммоль) и воду (0,99 мл, 55 ммоль). В течение 5 мин наблюдался подъем температуры на 10oC и начало выпадать кристаллическое вещество. Смесь кипятили с обратным холодильником (масляная баня при 74oC) в течение 2,5 ч. Затем смесь охлаждали до -10oC и добавляли 100 мл 1,0 М раствора литийалюминийгидрида в тетрагидрофуране. Первые 15 мл добавляли в течение 10 мин и наблюдали разогрев на 7oC. Оставшиеся 85 мл добавляли быстро, и дальнейшего разогрева замечено не было. Смесь доводили до кипения в течение 30 мин и продолжали кипячение с обратным холодильником 18 ч. После этого смесь охлаждали до 25oC, добавляли фторид натрия (25,2 г, 0,600 ммоль), и после 30-минутного перемешивания к охлажденной (0oC) смеси добавляли в течение 10 мин по каплям 5,3 мл воды. Смесь перемешивали 30 мин при 25oC и добавляли дитретбутилдикарбонат (12,6 мл, 55,0 ммоль). Данную смесь перемешивали 16 ч, фильтровали, и корку растирали с этилацетатом (2 х 50 мл). Объединенную смесь фильтрата с промывкой промывали водой (20 мл), осушали (Na2SO4), выпаривали и получившийся сироп кристаллизовали из смеси этилацетат-гексаны 1:2 (30 мл); получили указанное в заголовке соединение в виде белых кристаллов (10,32 г, 88%), по свойствам идентичное вышеописанному образцу.
Пример 25
(-)-(1R, 2S, 3R, 4R)-трет-бутил-N-[2,3-дигидрокси-4-(гидроксиметил)- 1-циклопентил]карбамат
К смеси N-метилморфолин-N-оксида (146,2 г, 60%-ный раствор в воде, 0,749 моль) и тетраоксида осмия (9,75 г, 2,5%-ный раствор в трет-бутаноле, 0,959 ммоль) в ацетоне (1 л), перемешиваемой при -8oC в бане со смесью лед/ацетон, одной порцией добавили (-)-(1R,4S)-трет-бутил-N-[4-гидроксиметил)-2-циклопентен-1-ил] карбамат (152,10 г, 0,7132 ммоль, из примера 24). Полученную смесь оставляли нагреваться до комнатной температуры в течение 16 ч; в это время смесь стала гомогенной. Добавили дополнительное количество тетраоксида осмия (2,602 г, 0,256 ммоль) и раствор перемешивали при 20oC четыре часа, затем два часа при 40o, при этом о завершении реакции судили по ТСХ (оксид кремния, 10% смесь метанол-хлороформ, проявление иодом с ванилиновым углем, исходный материал: Rf = 0,51, продукты: Rf = 0,22 (2S,3R)-изомер и Rf = 0,36 (2R, 3S)-изомер. По данным ЯМР 1H и ТСХ отношение изомеров (2S,3R)/(2R,3S) составляло около 73: 27. Добавляли воду (75 мл) и затем хлороформ (2 л). Полученную двухфазную смесь охлаждали в ледяной бане и при очень мягком перемешивании (для предотвращения смешивания фаз) несколькими порциями добавляли безводный сульфат меди (457,8 г, Alfa). Полученную взвесь оставляли при перемешивании при комнатной температуре на примерно 16 ч, затем ее отфильтровали на фильтрах Целит 545 и 512. Отжатый осадок промывали 6 л тетрагидрофурана до прекращения элюирования продукта. Фильтрат выпаривали в вакууме до темного масла, в котором в основном отсутствовал N-метилморфолин. Масло фильтровали через силикагель (300 г) и элюировали 3 л тетрагидрофурана до полного вымывания продукта. Элюат концентрировали до 200 мл и добавляли гексаны (около 300 мл). Кристаллизация начиналась спонтанно и ее продолжали при -5o в течение 16 ч. Кристаллы получали фильтрованием, их обильно промывали 50% смесью этилацетат-гексаны и высушивали при отсасывании до постоянного веса (105,78 г, 0,428 ммоль, 60% от теоретического выхода). Перекристаллизацией из кипящего этилацетата (200 мл) получено указанное в заголовке соединение в виде белых кристаллов (93,85 г, 0,3795 ммоль, 53,2% от теоретического выхода); Т.пл. 115,8-117o; 1H-ЯМР (ДМСО-d6) δ:: 6,71 (мд, J = 7,4 Гц, 1H, 11H), 4,52 (Т, J = 5,2 Гц, 1H, CH2OH), 4,43 (д, J = 5,1 Гц, 1H, CHOH), 4,31 (д, J = 4,9 Гц, 1H, CHOH), 3,54-3,41 перекрывает мультиплет и 3H, CHN и CHOH), 3,34 (М, перекрывается с НОД, w1/2 = 20 Гц, CH2OH), 1,99 (дт, J = 12,5, 6,8, 6,8 Гц, 1H, HOCH2CH), 1,85 (м.М, w1/2 = 30 Гц, 1H, 1/2 CH2), 1,39 (с, 9H, C(CH3)3), 0,98 (дт, J = 12,4, 7,8, 7,8 Гц, 1H, 1/2 CH2); [α]
Анализ для C11H21O5N:
Рассчитано: C 53,43; H 8,56; N 5,66.
Найдено: C 53,45; H 8,58; N 5,69.
Образец (-)-(2R,3S)-изомера (25,60 г) был получен из маточных растворов путем дробной кристаллизации из этилацетата: Т. пл. 106-107,2oC; 1H-ЯМР (ДМСО-d6) δ:: 5,93 (шд, J = 7,6 Гц, 1H, NH), 4,77 (д, J = 4,9 Гц, 1H, CHOH), 4,58 (д, J = 4,1 Гц, 1H, CHOH), 4,35 (шт, w1/2=15 Гц, 1H, CH2OH), 3,89 (шс, w1/2= 10 Гц, 1H, OCH), 3,73 (шс, 2H, OCH, NCH), 3,50 (шм, w1/220 Гц, 1H, 1/2 OCH2), 3,38 (шм, заслонен НОД, 1/2 OCH2), 1,90 (м, w1/2=24 Гц, 2H, OCH2CH, 1/2 CH2), 1,38 (с, 9H, C(CH3)3), 1,27 (М, 1H, 1/2 CH2); [α]
Анализ для C11H21O5N, 0,05 H2O:
Рассчитано: C 53,23; H 8,57; N, 5,64.
Найдено: C 53,20; H 8,55; N 5,61.
Таким же образом рацемат из примера 28 был превращен в рацемат указанного в заголовке соединения: т.пл. 134-136oC (из этилацетата), 51%. 1H ЯМР идентичен (-)-энантиомеру.
Пример 26.
4-Толуолсульфонат (+)-цис-4-амино-2-циклопентен-1-карбоновой кислоты
В трехгорлую колбу объемом 500 мл с вертикальными шлифами помещали (±)-2-азабицикло[2.2.1] гепт-5-ен-3-он (48,66 г, 0,4459 ммоль, Cambridge); колбу оборудовали механической мешалкой, термометром с адаптером для ввода газа, присоединенным к источнику азота, а также воронкой для сыпучих веществ. Добавляли 200 мл тетрагидрофурана (reagent grade) и начинали перемешивание для растворения твердого вещества. Замечено охлаждение до 13oC. Из воронки для сыпучих веществ через адаптер для ввода газа подавали слабый поток азота и добавляли гидрат-4-толуолсульфокислоты (93,52 г, 0,416 ммоль, 1,1 экв) вместе с небольшим количеством указанного в заголовке соединения в качестве затравки. Воронку для сыпучих веществ заменяли обратным холодильником и колбу помещали в масляную баню с предварительно уравновешенной температурой 35oC. Через 10 мин началась кристаллизация с последующим разогревом до 60oC через последующие 15 мин. После достижения максимального разогрева температуру в бане установили равной 60-65oC и реакционную смесь нагревали 2 ч при температуре 60-65oC (внутри) до тех пор, пока ТСХ супернатанта (оксид кремния, элюент этилацетат, проявление иодом) не стала свидетельствовать об отсутствии исходного лактама в сравнении с аутентичным пятном. После этого смесь охлаждали в ледяной бане до 5oC. Стеклянную трубку с пористым фильтром на конце через гибкий шланг присоединяли к колбе для фильтрования, которая, в свою очередь, была связана с источником вакуума. Из колбы со взвесью удаляли холодильник, выключали мешалку, и при потоке азота через газовый вход конец трубки с пористым фильтром вводили ко дну колбы под перемешиватель. До полного удаления жидкости включали вакуум, твердый материал повторно суспендировали в 100 мл сухого тетрагидрофурана и повторяли операцию фильтрования. Оставшееся белое твердое вещество повторно суспендировали в сухом тетрагидрофуране (200 мл) и открытую горловину накрывали перегородкой. Полученную взвесь указанного в заголовке соединения непосредственно использовали в следующем примере; аналогично приготовляли образец для анализа, за исключением того, что его высушивали сперва отсасыванием, а затем с использованием вакуума: Т. пл. 191-193oC; 1H-ЯМР (ДМСО-d6), δ:: 12,62 (мс, 1H, CO2H), 7,93 (шс, 3H, NH3+), 7,47 и 7,11 (дд, 8,0 Гц, 2H каждый, Ar-H), 6,11 (дт, J = 5,7, 1,9 Гц, 1H, винил), 5,82 (дт, J = 5,7, 2,8, 2,8 Гц, 1H винильный), 4,20 (шм, w1/2= 21 Гц, 1H, алкильный H), 3,61 (штт?, w1/2=21 Гц, 1H, алкильный), 2,29 (с, 3H, CH3), 2,50 (дт?, J = 5,8, 5,8, 11,5 Гц перекрывается с пиком ДМСО), 1/2 CH2), 1.92 (дт, J = 6,7, 6,7, 13,4 Гц, 1H, 1/2CH2).
Анализ для C13H17O5N5:
Рассчитано: C 52,16; H 5,72; N 4,68; S 10,71.
Найдено: C 52,16; H 5,76; N 4,66; S 10,62.
Пример 27
(±)-цис-4-амино-2-циклопентен-1-метанол
Сухая трехгорлая колба объемом 2 л была оборудована механической мешалкой, термометром с адаптером для ввода газа, соединенным с источником азота и перегородкой. Колбу продували азотом, погружали в баню со смесью лед-ацетон и через трубку добавляли раствор литийалюминийгидрида в тетрагидрофуране (1 М, 800 мл, 0,80 ммоль, Aldrich). Для смывки раствора литийалюминийгидрида использовали сухой тетрагидрофуран (2 х 15 мл). Когда раствор охладился до 0oC, была введена через трубку взвесь 4-толуолсульфонатной соли (±)-цис-4-амино-2-циклопентен-1-карбоновой кислоты в тетрагидрофуране, полученная в предыдущем примере, при хорошем перемешивании с такой скоростью, чтобы температура была менее 10oC и имело место умеренное выделение водорода (около одного часа). Колбу промывали сухим тетрагидрофураном (2 х 15 мл) и вместо перегородки устанавливали обратный холодильник. Полученный прозрачный со слегка янтарным цветом раствор медленно подогревали в течение двух часов до слабого кипения, при котором он становился мутным. После кипячения с обратным холодильником в течение ночи (16 ч), нагревающую баню опускали, добавляли фторид натрия (136,3 г, 3,25 ммоль, порошок квалификации reagent grade) и холодильник переставляли для нисходящей перегонки. Смесь дистиллировали в тонкую взвесь (собрано 700 мл дистиллята) и затем охлаждали в ледяной бане. Добавляли 500 мл сухого диэтилового эфира и холодильник заменяли капельной воронкой с водой (43 мл, 2,4 моль). Воду добавляли очень медленно (два часа) при внимательном контроле за скоростью выделения водорода и поддерживании температуры 10±5oC. Тем временем к вышеполученному дистилляту добавляли воду (54 мл) и достаточное дополнительное количество тетрагидрофурана до доведения общего объема до 900 мл (6% воды). Реакционную смесь фильтровали при отсасывании и отжатый осадок промывали 100 мл тетрагидрофурана. Часть 6% водно-тетрагидрофуранового раствора (300 мл) использовали для взвешивания и промывания отжатого осадка, который затем возвращали в реакционную колбу. Отжатый осадок растирали в течение 25 мин со смесью вода - тетрагидрофуран (6%, 400 мл), фильтровали и удаляли с промыванием 200 мл 6% смеси вода-тетрагидрофуран. Соединенные фильтры концентрировали до бледно-желтого масла в вакууме (44,07 г, 67,8% по ЖХВД, см. пример 3). Это масло, содержащее чистое вещество, указанное в заголовке, воду и следы тозилатной соли, быстро темнеет в нормальных условиях. Его быстро вводили в реакцию для образования производного N-BOC, стабильного, кристаллического вещества (см. последующий пример). Отжатые осадки с фильтра возвращали в колбу и растирали в метаноле (800 мл) в течение 48 ч. Полученную взвесь фильтровали под резиновой перемычкой и отжатый осадок промывали 200 мл метанола. Фильтрат концентрировали в вакууме и получили желтое твердое вещество (56,80 г, 20,9% выход согласно ЖХВД, общий суммарный выход 88,7%). Этот экстракт также использовали для N-BOC-производных (см. следующий пример).
Пример 28
(±)-цис-трет-бутил-N-[4-(гидроксиметил)-2-циклопентен-1-ил]карбамат
Первый экстракт из предыдущего примера, содержащий (±)-цис-4-амино-2-циклопентен-1-метанол (0,459 ммоль) был растворен в 1,2 л смеси 1,4-диоксан - вода (2: 1). Добавляли бикарбонат натрия (48,69 г, 0,580 ммоль), смесь охлаждали в бане со смесью лед-вода, и при быстром перемешивании одной порцией добавляли дитретбутилкарбонат (110,25 г, 0,490 ммоль, Aldrich, 97%). Полученную смесь в течение одного часа нагревали до комнатной температуры, затем концентрировали в вакууме до объема около 400 мл. Взвесь помещали в 300 мл хлороформа, фазы разделяли, и из водной (верхней) фазы проводили вторичную экстракцию хлороформом (пять порций по 300 мл каждая) до тех пор, пока в экстракте не обнаруживался продукт ТСХ: оксид кремния, 10% смесь метанол-хлороформ, проявление иодом, Rf = 0,51). Соединенные органические фазы осушали над сульфатом натрия, фильтровали и концентрировали в вакууме; получали указанный в заголовке продукт в виде масла. Последний экстракт из предыдущего примера аналогичным образом вводили в реакцию и полученное сырое вещество, указанное в заголовке, объединяли с вышеуказанной порцией; объединенный продукт помещали в гексаны и выпаривали в вакууме с целью удаления остатка хлороформа. После этого масло спонтанно кристаллизовалось. Его растирали в холодных гексанах, фильтровали, и получали сырое указанное в заголовке соединение в виде кристаллического твердого вещества, которое осушали при отсасывании до постоянного веса (79,98 г, 0,3750 ммоль). Перекристаллизацией из кипящих этилацетата (70 мл) и гексанов (300 мл) получено указанное в заголовке вещество в виде нечисто белого кристаллического вещества (73,43 г, 0,3443 ммоль); Т.пл. 54-55,5oC; 1H-ЯМР (ДМСО-d6) δ:: 6,72 (д, J = 7,9 Гц, 1H, NH), 5,80 и 5,60 (два М, 2H, CH=CH), 4,59 (Т, J = 5,2 Гц, 1H, OH), 4,45 (М, 1H, CHN), 3,35 (М, перекрывается с H2O CH2O), 2,60 (М, 1H, CH), 2,30 (М, 1H, 1/2CH2), 1,40 (с, 9H, C(CH3)3), 1,2 (М, 2H, 1/2CH2).
Анализ для C11H19NO3:
Рассчитано: C 61,94; H 8,98; N 6,57.
Найдено: C 62,00; H 8,99; N 6,55.
Маточные растворы объединяли, хроматографировали на силикагеле (700 г, 30% смесь этилацетат-гексаны и 5% смесь метанол-хлороформ) и кристаллизовали, как указано выше; получили вторую порцию указанного в заглавии соединения (10,49 г, 0,0492 ммоль). Таким образом, общий выход составил 0,3935 ммоль или 88,9% от теоретического, исходя из начального (±)-2-азабицикло[2.2.1] гепт-5-ен-3-она (с поправкой на взятые аликвоты).
Пример 29
(±)-цис-4-амино-2-циклопентен-1-метанол
С использованием метода, описанного в примерах 26 и 27, но в двойном масштабе (97,40 г, 0,8924 ммоль (±)-2-азабицикло[2.2.1]гепт-5-ен-3-она) получено указанное в заголовке соединение в виде содержащих его экстрактов (0,7926 ммоль, 88,8% от теоретического выхода с учетом удаленных аликвот; определенно по методу из примера 23.
Пример 30
(±)-цис-трет-бутил-N-[4-(гидроксиметил)-2-циклопентен-1-ил]-карбамат.
Объединенные тетрагидрофурановые экстракты из предыдущего примера концентрировали в вакууме до 1031 г, охлаждали в бане со смесью лед-вода и добавляли смесь бикарбоната натрия (97,46 г, 1,16 ммоль) с водой (500 мл). После этого добавляли дитретбутилкарбонат (204,5 г, 0,9501 ммоль). Смесь перемешивали при 5oC в течение двух дней. Метанольные экстракты из предыдущего примера выпаривали до маслянистого твердого вещества (136,64 г), которое добавляли в смесь. После нагревания до комнатной температуры органические растворители выпаривали в вакууме, а полученную взвесь экстрагировали гексанами, тремя порциями хлористого метилена и затем опять гексанами (по 200 мл каждая). Органические экстракты выпаривали до масла, которое кристаллизовали из гексанов (около 300 мл) и получали указанное в заголовке соединение (154,15 г, 0,7229 ммоль), идентичное продукту из примера 28. Дополнительное количество продукта было получено хроматографированием маточных растворов (10,5 г, 0,0491 ммоль, 86,6% от теоретического выхода, исходя из начального лактам, с учетом удаленных аликвот).
Пример 31
(±)-цис-4-амино-2-циклопентен-1-карбоновая кислота, метансульфонат.
Исходя из (±)-2-азабицикло[2.2.1]гепт-5-ен-3-она (5,111 г, 46,83 ммоль, Cambridge) по методу из примера 31 получено указанное в заголовке соединение (10,268 г, 45,99 ммоль, 98,2%); Т.пл. 137-139oC; 1H-ЯМР (ДМСО-d6) δ:: 12,6 (шс, 1H, CO2H), 8,04 (шс, 3H, NH3 +), 6,10 (дт, J = 5,6, 2,0, 2,0 Гц, 1H, винил), 5,85 (дт, J = 5,3, 2,3, 2,3 Гц, 1H, винил), 4,19 (шс, w1/2 = 20 Гц, 1H, аллильный H), 3,61 (М, w1/2 = 22 Гц, 1H, аллильный H), 2,53 (квинтет) J = 5,3 Гц перекрывается с пиком ДМСО), 1/2 CH2), 2,39 (С, 3H, CH3SO3H), 1,93 (дт, J = 6,7, 6,7, 13,7 Гц, 1H, 1/2CH2); ХИ-МС (CH4): 128 (M+1); ЭИ-МС: 127 (M).
Анализ для C7H13NO5S:
Рассчитано: C 37,66; H 5,87; N 6,27; S 14,36
Найдено: C 37,60; H 5,85; N 6,25; S 14,30
Пример 32
(±)-цис-4-амино-2-циклопентен-1-карбоновая кислота, 4-толуолсульфонат
К раствору, содержащему каталитическое количество 4-толуолсульфокислоты (10 мг) в 30%-ной водной перекиси водорода (0,30 мл, 2,7 ммоль) добавляли 3-тозил-2-азабицикло[2.2.1] гепта-2,5-диен (369 мг, 1,49 ммоль), полученный по методике J.C.Jagd and A.M. van Lensen, J, Org. Chem.) 1974, 39, 564-566, порциями при быстром перемешивании. Было отмечено большое тепловыделение, стабилизировавшееся при 75oC в течение второй половины периода добавления. После перемешивания при 70oC в течение 40 мин смесь несколько раз разбавляли водой (всего 6 мл) и фильтровали до получения прозрачного раствора. Раствор упаривали до кристаллизующегося масла (349 мг). Последнее растирали в тетрагидрофуране, фильтровали и высушивали в вакууме; получили указанное в заголовке соединение (202 мг, 45,2% от теоретического выхода). Спектр 1H ЯМР был идентичен спектру продукта из примера 26.
Пример 33
(±)-цис-[4-(4,5-дихлор-2-нитроанилино)-2-циклопентен-1-ил]метанол
(±)-цис-трет-бутил-N-[4-(гидроксиметил)-2-циклопентен-1-ил] карбамат (50,0 г, 0,230 ммоль) перемешивали в 25%-ном растворе трифторуксусной кислоты в хлористом метилене (1,5 л) при 0oC в течение 1,0 ч. После выпаривания летучих веществ оставалась трифторуксусная соль амина, описанного в примере 27 в виде темного масла. К этому маслу добавляли трет-бутанол (350 мл), карбонат калия (65 г) и 1,2,4-трихлор-5-нитробензол (Aldrich, 54,7 г, 0,230 ммоль, 97%). Полученную смесь 3 дня кипятили с обратным холодильником при энергичном перемешивании. Летучие вещества удаляли в вакууме и остаток растирали с метанолом. Растворимый в метаноле материал хроматографировали на силикагеле. Сырой продукт элюировали 2% смесью метанол-хлороформ и получали оранжевое твердое вещество (38,0 г). Кристаллизация из смеси этилацетат-гексаны давала указанное в заголовке соединение в виде оранжевых кристаллов (34,0 г, 49%), т.пл. 96-98oC; спектры 1H ЯМР (ДМСО-d6) и масс-спектры (ХИ) были в согласии со структурой и идентичны со спектрами образцов хиральных энантиомеров, описанных в примерах 53 и 64.
Анализ для C12H12N2Cl2O3:
Рассчитано: C 47,55; H 3,99; N 9,24; Cl 23,39.
Найдено: C 47,75; H 4,10; N 9,20; Cl 23,52.
Продолженное элюирование из колонки давало последующие фракции, содержащие указанное в заголовке соединение с малым количеством примесей характеризующихся низкими значениями Rf. Эти фракции объединяли с маточным раствором после вышеупомянутой кристаллизации и перекристаллизовывали из смеси этилацетат-гексаны; получили дополнительное количество оранжевого твердого вещества (16,7 г) с идентичным спектром 1H ЯМР; общий выход доходил до 73%.
Пример 34
(±)-цис-[4-(5,6-дихлор-1H-бензимидазол-1-ил)-2-циклопентен- 1-ил] метанол.
(±)-цис-[4-(4,5-дихлор-2-нитроанилино)-2-циклопентен-1-ил] метанол (5,00 г, 16,5 ммоль) в смеси этанол (100 мл) - вода (35 мл) нагревали, при этом добавляли порошкообразное железо (325 меш, 99,9%, Aldrich, 9,2 г, 0,165 экв) и гептагидрат сульфата двухвалентного железа (Aldrich, 98+%, 2,3 г, 8,2 мэкв). Смесь кипятили с обратным холодильником в течение 1,75 ч. Твердые вещества отфильтровывали и этанольный фильтрат-промывку концентрировали до состояния масла. К маслу добавляли триэтилортоформиат (75 мл) и метансульфокислоту (0,05 мл); полученный раствор перемешивали при комнатной температуре в течение 18 ч. Концентрированием в вакууме получено масло, которое повторно растворяли в смеси 1 н. соляная кислота (25 мл) - диоксан (5 мл). Через 3 ч pH доводили до 7 1 н гидроксидом натрия, и проводили экстракцию из раствора хлороформом (3 х 50 мл), Содержимое осушенного (сульфат натрия) раствора в хлороформе хроматографировали на силикагеле. Указанное в заголовке соединение элюировали 5% смесью метанол-хлороформ и кристаллизовали из смеси этилацетат-гексаны; получили белые кристаллы (3,85 г, 82%), т.пл. 166-169oC, спектры 1H ЯМР (ДМСО-d6) и масс-спектры (ХИ) соответствовали структуре.
Анализ для C13H12N2Cl2O:
Рассчитано: C 55,14; H 4,27; N 9,89; Cl 25,04.
Найдено: C 55,20; H 4,32; N 9,84; Cl 24,94.
Пример 35
(±)-цис-[4-(2-бром-5,6-дихлор-1H-бензимидазол-1-ил)-2- циклопентен-1-ил] метанол.
(±)-цис-[4-(5,6-дихлор-1H-бензимидазол-1-ил)-2- циклопентен-1-ил]метанол (1,55 г, 5,47 ммоль) ацетилировали в пиридине (17 мл) уксусным ангидридом (0,6 мл) при комнатной температуре в течение 2 дней. Летучие вещества удаляли в вакууме и оставшееся масло распределяли между хлороформом и водным раствором бикарбоната натрия. Раствор в хлороформе осушали (сульфат натрия) и концентрировали досуха. Остаток растворяли в сухом диоксане (10 мл) и раствор доводили до кипения. Через 4 мин кипячения темный раствор охлаждали. Летучие вещества выпаривали в вакууме и остаток хроматографировали на силикагеле. Элюирование 2 - 5% смесью метанол-хлороформ давало фракции, содержащие исходный материал (1,05 г) и последующие фракции, содержащие указанное в заголовке вещество в виде темного масла (0,28 г). Выделенный исходный материал снова обрабатывали N-бромсукцинимидом и сырой продукт хроматографировали. Все содержащие продукт фракции объединяли и подвергали повторному хроматографированию на силикагеле с элюированием смесью гексаны-этилацетат; получили ацетат указанного в заглавии вещества в виде желтого масла (0,62 г). Деацетилирование осуществляли, как в примере 4. Летучие продукты выпаривали в вакууме из нейтрализованного раствора и остаток хроматографировали на силикагеле. Элюирование 2% смесью метанол-хлороформ давало (±)-цис-[4-(2-бром-5,6-дихлор-1H-бензимидазол-1-ил)-2-циклопентен-1-ил] метанол в виде порошка не чисто белого цвета (293 мг, 15%) после переосаждения из смеси гексаны-этилацетат, Т. пл. 126 - 128,5oC; 1H-ЯМР (ДМСО-d6) δ:: 8,09 и 7,95 (оба с каждой, ароматический CH), 6,24 - 6,20 и 5,98 - 5,94 (оба M, 2, CH= CH), 5,81 - 5,76 (2M, 1 каждый, CH2O), 4,96 (T, J = 4,9 Гц, 1, OH), 3,02 - 2,95 (M, 1, CH), 2,67 - 2,51 (M, перекрывается с растворителем, 1/2 CH2), 2,0 - 1,8 (M, 1, 1/2 CH2); Масс-спектр (ХИ) в согласии со структурой.
Анализ для C13H11N2BrCl2O:
Рассчитано: C 43,13; H 3,06; N 7,74; общий галоген в виде Cl 29,38.
Найдено: C 43,20; H 3,07; N 7,71; общий галоген в виде Cl 29,35.
Пример 36
(+/-)-цис-3-(5,6-дихлор-1H-бензимидазол-1-ил)-1-циклопентанметанол/
Смесь (+/-)-цис-4-(4,5-дихлор-2-нитроанилино)-2-циклопентен-1-ил метанола (5,00 г, 16,5 ммоль) и никеля Ренея (Aldrich, взвесь в воде 500 мг сырого веса) в н-пропаноле (250 мл) трясли в атмосфере водорода (50 psi) в аппарате для встряхивания Парра в течение 2 ч. Катализатор отфильтровывали, растворитель выпаривали в вакууме и остаток растворяли в системе триэтилортоформиат (300 мл) - метансульфокислота (200 мл). Через 18 ч раствор концентрировали до сиропа, который растворяли в 1 н. соляной кислоте (40 мл) и перемешивали в течение ночи при комнатной температуре. pH доводили до 7 1 н. раствором гидроксида натрия и проводили экстракцию из раствора хлороформом (3 х 50 мл). Содержимое высушенного над сульфатом натрия раствора в хлороформе хроматографировали на силикагеле. Указанное в заголовке соединение элюировали 5% смесью метанол-хлороформ в виде бесцветного масла, которое отверждалось из смеси этилацетат-гексаны с образованием белого порошка (3,98 г, 85%), т. пл. 142-145oC, 1H ЯМР (ДМСО-d6) и масс-спектр (ХИ) были в согласии со структурой.
Анализ для C13H14N2Cl2O:
Рассчитано: C 54,75; H 4,95; N 9,82; Cl 24,86.
Найдено: C 54,88; H 4,99; N 9,70; Cl 24,74.
Пример 37
(+/-)-цис-3-(2-бром-5,6-дихлор-1H-бензимидазол-1-ил)-1- циклопентанметанол.
(+/-)-цис-3-(5,6-дихлор-1H-бензимидазол-1-ил)-1-циклопентанметанол (3,65 г, 12,8 ммоль) ацетилировали в 40 мл пиридина уксусным ангидридом (2 мл) при комнатной температуре в течение ночи. Летучие продукты выпаривали и остаток распределяли между хлороформом и насыщенным водным раствором карбоната натрия. Слой хлороформа осушали (сульфат натрия) и выпаривали до стеклообразного продукта, который направляли в реакцию с N-бромсукцинидом, как описано в примере 3. Сырой продукт бромирования хроматографировали на силикагеле, и ацетат указанного в заголовке соединения элюировали 5% смесью метанол-хлороформ; получили порошок не число белого цвета (2,8 г, 55%); 1H ЯМР и масс-спектр (ХИ) были в согласии со структурой. Этот порошок (2,00 г, 4,92 ммоль) деацетилировали, как в примере 4, и после элюирования из колонки с силикагелем 2-3% смесью метанол-хлороформ получили бесцветное твердое пенообразное вещество. Кристаллизацией из смеси этилацетат-гексаны получен (+/-)-цис-3-(2-бром-5,6-дихлор-1H-бензимидазол-1-ил)-1-циклопентан метанол в виде кристаллов не число белого цвета (1,08 г, 60%). Т.пл. 111-112oC; 1H-ЯМР (ДМСО-d6) δ:: 8,08 и 7,96 (оба с, 1 каждый, 2 ароматический CH), 5,06 - 5,01 (M, 1, NCH), 4,79 (T, J = 5,1 Гц, 1, OH), 3,56 - 3,51 (M, 2, CH2OH), 2,28 - 2,05 (M, 5, 2 CH2 и CH); Масс-спектр (ХИ): 367 (54), 365 (100), 363 (71, M+1).
Анализ для C13H13N2BrCl2O:
Рассчитано: C 42,89; H 3,60: N 7,69; общий галоген в виде Br 65,84, в виде Cl 29,21.
Найдено: C 42,94; H 3,63; N 7,62; общий галоген в виде Br 65,75, в виде Cl 29,17.
Пример 38
(+/-)-(1R*, 2S*, 3S*, 5S*)-3-(ацетоксиметил)-5-(4,5-дихлор-2-нитроанилино)-1,2 -циклопентандиилдиацетат и (+/-)-(1R*,2S*, 3R*,5R*)-3-(ацетоксиметил)-5-(4,5-дихлор-2-нитроанилино)- 1,2-циклопентандиилдиацетат.
К раствору (+/-)-цис-[4-(4,5-дихлор-2-анилино)-2-циклопентен-1-ил] метанола (20,0 г, 66,0 ммоль) и N-метил-морфолин-N-оксида (Aldrich, 60%-ный водный раствор, 12,0 мл, 69 ммоль), в 280 мл ацетона добавляли тетраоксид осмия (2,5%-ный раствор в трет-бутиловом спирте, Aldrich, 1,24 мл). После перемешивания при комнатной температуре в течение 18 ч летучие вещества удаляли в вакууме и остаток дополнительно в течение 18 ч перемешивали со смесью пиридин (200 мл) - уксусной ангидрид (40 мл)
Раствор концентрировали до густого красного масла, которое распределяли между насыщенным водным раствором карбоната натрия и хлороформом. Слой хлороформа осушали (сульфат натрия) и затем концентрировали в вакууме до масла. Смесь изомерных соединений, указанных в заголовке, элюировали из колонки с силикагелем 20 смесью метанол-хлороформ и кристаллизовали из смеси этилацетат-гексаны с введением затравки кристалло (1R*,2S*)-изомера, полученного методом из примера 1; получили (+/-)-(1R*, 2S*, 3S*,5S*)-3-(ацетоксиметил)-5-(4,5-дихлор-2-нитроанилино)-1,2 -циклопентандиилдиацетат в виде оранжевых кристаллов (17,4 г, 57%), т.пл. 154-156oC, спектр 1H ЯМР (ДМСО-d6) был идентичен спектру образца, описанного в примере 1.
Продолженной кристаллизацией содержимого маточного раствора из смеси этилацетат-гексаны получен (+/-)-(1R*, 2S*, 3R*,5R*)-3-(ацетоксиметил)-5-(4,5-дихлор-2-нитроанилино)-1,2-циклопентандиилдиацетат в виде оранжевых кристаллов (8,82 г, 29%), т.пл. 105 - 107oC. Спектр 1H ЯМР (ДМСО-d6) был идентичен спектру образца, описанного в примере 6.
Анализ для C18H20N2Cl2O8:
Рассчитано: C 46,67; H 4,35; N 6,05; Cl 15,31.
Найдено: C 46,50; H 4,33; N 5,96; Cl 15,23.
Пример 39
(+/-)-(1R*, 2S*, 3R*, 5R*)-3-(ацетоксиметил)-5-(2-бром-5,6-дихлор-1H-бензимидазол-1-ил)-1,2 -циклопентандиилдиацетат
(+/-)-(1R*, 2S*, 3R*, 5R*)-3-(ацетоксиметил)-5-(4,5-дихлор-2-нитроанилино)-1,2 -циклопентандиилдиацетат (5,00 г, 10,8 ммоль) перемешивали в течение 18 ч при комнатной температуре в растворе аммиак/метанол (около 2 н. 100 мл). После выпаривания летучих веществ в вакууме оставалось оранжевое твердое вещество (+/-)-(1R*, 2S*, 3R*,5R*)-5-(4,5-дихлор-2-нитроанилино)-3-(гидроксиметил)-1,2 -циклопентандиол, который имел величину Rf на силикагелевых пластинках для ТСХ, идентичную с величиной для хирального образца, описанного в примере 54. Это твердое вещество восстанавливали никелем Ренея в атмосфере водорода (45 psi) в 200 мл изопропанола. Катализатор отфильтровывали на целите. Фильтрат с промывкой досуха упаривали в вакууме. Остаток кипятили с обратным холодильником с 50 мл 96% муравьиной кислоты в течение одного часа, как описано в примере 2. Масло, оставшееся после выпаривания муравьиной кислоты, растворяли в метаноле. pH доводили до 13 водным 5 и раствором гидроксида натрия и раствор перемешивали при комнатной температуре в течение одного часа с целью гидролиза эфиров муравьиной кислоты pH доводили до 7 1 н. соляной кислотой и летучие вещества выпаривали в вакууме. К остатку добавляли 100 мл пиридина и 4 мл уксусного ангидрида и смесь перемешивали при комнатной температуре в течение ночи. После выпаривания летучих веществ в вакууме с последующей хроматографией на силикагеле с использованием 1% смеси метанол-хлороформ получен (+/-)-(1R*,2S*, 3R*,5R*)-3-(ацетоксиметил)-5-(5,6-дихлор-1H-бензимидазол- 1-ил)-1,2-циклопентандиилдиацетат в виде белых кристаллов (из смеси этанол-вода) (2,6 г, 53%), 1H ЯМР (ДМСО-d6) был в согласии со структурой.
(+/-)-(1R*, 2S*, 3R*, 5R*)-3-(ацетоксиметил)-5-(5,6-дихлор-1H-бензимидазол-1-ил)-1,2-циклопентандиилдиацетат (2,5 г, 5,7 ммоль) растворяли в сухом диоксане (15 мл) и раствор кипятили с обратным холодильником, при этом одной порцией добавляли свежеперекристаллизованный N-бромсукцинимид (2,10 г, 11,5 ммоль). После 5 мин кипячения красно-коричневый раствор выпаривали в вакууме до получения красного масла. Раствор этого масла в хлороформе промывали водой и затем высушивали над сульфатом натрия. Раствор в хлороформе концентрировали до состояния масла, которое хроматографировали на силикагеле. Фракции, содержащие продукт, элюировали 2-4% смесью метанол-хлороформ. Кристаллизацией из смеси этилацетат-гексаны получено твердое вещество не чисто белого цвета (1,5 г, 50%); спектр 1H ЯМР (ДМСО-d6) был в согласии со структурой указанного в заголовке соединения. Этот образец повторно хроматографировали на силикагеле с элюированием хлороформом и получили (+/-)-(1R*, 2S*, 3R*,5R*)-3-(ацетоксиметил)-5-(2-бром-5,6-дихлор-1H-бензимидазол- 1-ил)-1,2-циклопентадиилдиацетат в виде белых кристаллов после кристаллизации из смеси этилацетат-гексаны. Т.пл. 166-167oC; 1H-ЯМР (ДМСО-d6) δ: 8,14 и 7,96 (оба с, 1 каждый, 2 ароматических CH), 5,6 - 5,35 (M, 3,2 OCH и NCH), 4,4 - 4,1 (M, 2, OCH2), 2,8 - 2,4 (M, перекрывается с растворителем, 2CH), 2,4 - 2,1 (M, перекрывается с при 2,25, всего 4, CH и CH3), 2,04 (с, 3, CH3), 1,37 (с, 1, CH3); масс-спектр (ХИ); 525 (53), 523 (100), 521 (54, M+1).
Анализ для C19H19N2BrCl2O6:
Рассчитано: C 43,70; H 3,67; N 5,37; общий галоген в виде Cl 20,37.
Найдено: C 43,65; H 3,68; N 5,35; общий галоген в виде Cl 20,32.
Пример 40
(+/-)-(1R*, 2S*, 3S*, 5S*)-3(ацетоксиметил)-5-(2-бром-5,6-дихлор-1H-бензимидазол-1-ил)-1,2- циклопентандиилдиацетат
(+/-)-(1R*, 2S*, 3S*, 5S*)-3-(ацетоксиметил)-5-(5,6-дихлор-1H-бензимидазол-1-ил)-1,2 -циклопентандиилдиацетат (4,20 г, 9,48 ммоль) кипятили с обратным холодильником в 25 мл сухого диоксана; при этом одной порцией добавили свежеперекристаллизованный N-бромсукцинимид (3,37 г, 18,9 ммоль). После 5 мин перемешивания летучие вещества удаляли в вакууме. Остаток распределяли между водой и хлороформом. Слой хлороформа осушали над сульфатом натрия и выпаривали до получения масла, которое хроматографировали на силикагеле при элюировании смесью гексаны-этилацетат (1: 1). Переосаждение из смеси этанол-вода давало указанное в заголовке соединение в виде белого порошка (3,10 г, 63%), т. пл. 157-158,5oC; спектр 1H ЯМР (ДМСО-d6) идентичен спектру образца, описанного в примере 3.
Анализ для C19H19N2BrCl2O6:
Рассчитано: C 43,71; H 3,67; N 5,37; общий галоген в виде Cl 20,37.
Найдено: C 43,66; H 3,72; N 5,34; общий галоген в виде Cl 20,32.
Пример 41
(+/-)-(1R*, 2S*, 3S*,5S*)-3-(ацетоксиметил)- 5-(2-амино-4,5-дихлоранилино)-1,2-циклопентандиилдиацетат
(+/-)-(1R*, 2S*, 3S*,5S*)-3-(ацетоксиметил)- 5-(4,5-дихлор-2-нитроанилино)-1,2-циклопентандиилдиацетат (4,30 г, 9,28 ммоль) в 250 мл изопропанола трясли с никелем Ренея (Aldrich, взвесь в воде, 400 мг сырого веса) в атмосфере водорода (50 psi) в аппарате для встряхивания Парра в течение 2,75 ч. Катализатор отфильтровывали на целите и фильтрат с промывкой (350 мл) хранили при -5oC. Медленно формировались желтые кристаллы указанного в заголовке соединения (2,35 г, 58%), Т.пл. 124-125oC; 1H-ЯМР (ДМСО-d6) δ: 6,70 и 6,57 (оба с, 1 каждый, 2 ароматических CH), 5,15 - 4,9 (M, 5, NH2, NH, и 2 OCH), 4,2 - 4,0 (M, 2, CH2O), 3,9 - 3,75 (M, 1, NCH), 2,5 - 2,4 (M, перекрывается с растворителем CH2), 2,07, 2,04 и 2,01 (все с, 9, 3 CH3), 1,4 - 1,2 (М, 1, CH); масс-спектр (ХИ): 433 (М+1).
Анализ для C18H22N2Cl2O6:
Рассчитано: C 49,90; H 5,12; N 6,47; Cl 16,37.
Найдено: C 50,00; H 5,13; N 6,38; Cl 16,28.
Концентрированием маточного раствора получено дополнительное количество указанного в заголовке соединения в виде желтого порошка (1,00 г, 25%) с достаточной для использования чистотой (пример 44).
Пример 42
(+/-)-(1R*, 2S*,3S*,5S*)-5-(5,6-дихлор-2-метил-1H-бензимидазол-1-ил)-3-(гидроксиметил)-1,2-циклопентандиол
(+/-)-(1R*, 2S*, 3S*,5S*)-3-(ацетоксиметил)- 5-(4,5-дихлор-2-нитроанилино)-1,2-циклопентандиилдиацетат (1,00 г, 2,16 ммоль), никель Ренея (Aldrich, взвесь в воде, 100 мг сырого веса) и изопропанол (200 мл) трясли в атмосфере водорода (50 psi) в течение 1,25 ч. Катализатор отфильтровывали на целите и фильтрат с промывкой выпаривали досуха. Оставшееся желтое масло растворяли в 20 мл триэтилортоацетата с одной каплей метансульфокислоты и раствор перемешивали 18 ч при комнатной температуре. Летучие вещества удаляли в вакууме и остаток перемешивали в смеси диоксана (20 мл и 1 н. гидроксида натрия (10 мл) в течение 5 ч при комнатной температуре. После этого pH доводили до 1 концентрированной соляной кислоты и продолжали перемешивание 15 мин, после чего ТСХ (силикагель, обработанный 20% смесью метанол-хлороформ) показывала одно пятно, поглощающее УФ-излучение, с Rf 0,25. Раствор нейтрализовали гидроксидом натрия и летучие вещества выпаривали в вакууме. Остаток хроматографировали на колонке с силикагелем и элюировали указанное в заголовке соединение 10 - 15% смесью метанол-хлороформ. Кристаллизацией из смеси этанол-вода получен (+/-)-(1R*,2S*,3S*,5S*)-5-(5,6-дихлор-2-метил- 1H-бензимидазол-1-ил)-3-(гидроксиметил)-1,2-циклопентандиол в виде мелких белых кристаллов (0,42 г, 67%). Т.пл. 222-226oC; 1H-ЯМР (ДМСО-d6) δ: 8,30 и 7,72 (оба с, 2,2 ароматических CH), 5,0 - 4,8 (M, 3,2 OH и NCH), 4,56 (Т, J = 5,1 Гц, 1, CH2OH), 4,2 - 4,1 (M, 2,2 OCH), 3,8 - 3,5 (M, 2, CH2O), 2,58 (c, 3, CH3), 2,4-2,2 (M, 1, CH), 2,2-1,9, CH2); масс-спектр (ХИ): 332 (M+1).
Анализ для C14H16N2Cl3O3:
Рассчитано: C 50,78; H 4,87; N 8,46; Cl 21,24.
Найдено: C 50,72; H 4,91; N 8,54; Cl 21,13.
Пример 43
(+/-)-(1R*, 2S*,3S*,5S*)-5-(5,6-дихлор- 2-этил-1H-бензимидазол-1-ил)-3-гидроксиметил)-1,2-циклопентандиол.
Таким же образом, как описано в примере 42, (+/-)-(1R*,2S*,3S*,5S*)-3-(ацетоксиметил)-5-(4,5-дихлор-2-нитроанилино)-1,2-циклопентандиилдиацетат (1,00 г, 2,16 ммоль) был превращен в указанное в заголовке соединение с использованием триэтилортопропионата (Aldrich, 97%, 22 мл) для формирования бензимидазола. Осаждение из смеси этанол-вода давало (+/-)-(1R*,2S*,3S*, 5S*)-5-(5,6-дихлор- 2-этил-1H-бензимидазол-1-ил)-3-(гидроксиметил)-1,2-циклопентандиол в виде белого порошка (0,463 г, 63%). Т.пл. 191 - 193oC; 1H-ЯМР (ДМСО-d6) δ: 8,10 и 7,86 (оба с, 2, 2 ароматических CH), 5,10 (т, J = 4,7 Гц, 1, OH), 4,94 (д, J = 6,9 Гц, 1 OH), 4,8 - 4,6 (M, перекрывается с при 4,71, J = 3,9 Гц, всего 2, NCH и OH), 4,5 - 4,3 (M, 1, OCH), 3,9 - 3,8 (M, 1, OCH), 3,75 - 3,45 (M, 2, OCH2), 2,93 квадруплет, J = 7,4 Гц, 2, CH2CH3), 2,2 - 2,0 (M, 3, CH2 и CH), 1,34 (Т, J = 7,4 Гц, 3, CH3); масс-спектр (ХИ): 345 (М+1).
Анализ для C15H18N2Cl2O3 • 0,25H2O:
Рассчитано: C 51,52; H 5,33; N 8,01; Cl 20,27.
Найдено: C 51,59; H 5,31; N 8,05; Cl 20,19.
Пример 44
(+/-)-(1R*, 2S*,3S*,5S*-3-(ацетоксиметил)-5- (5,6-дихлор-2,3-дигидро-2-тиоксо-1H-бензимидазол-1-ил)- 1,2-циклопентандиилдиацетат
(+/-)-(1R*, 2S*, 3S*, 5S*-3-(ацетоксиметил)-5- (2-амино-4,5-дихлоранилино)-1,2-циклопентандиилдиацетат (2,25 г, 5,19 ммоль) 1,1-тиокарбонилдиимидазол (Aldrich, 1,03 г, 5,19 ммоль при 90%) и толуола (125 мл) кипятили с обратным холодильником 30 мин. Добавили дополнительное количество 1,1-тиокарбонилдиимидазола (0,51 г) и продолжали кипячение еще 15 мин. Летучие вещества выпаривали в вакууме и оставшееся желтое масло растворяли в хлороформе (75 мл) и промывали водой (2 х 25 мл). Слой хлороформа осушали над сульфатом натрия и концентрировали в вакууме до желтого масла. Кристаллизация из смеси этилацетат-гексаны давала рыжевато-коричневые кристаллы (2,30 г, 93%) со спектром 1H ЯМР, идентичным описанному выше. Этот образец затем очищали элюированием из колонки с силикагелем 4% смесью метанолхлороформ перед кристаллизацией из смеси этилацетат-гексаны; получен (+/-)-(1R*,2S*,3S*, 5S*)-3-(ацетоксиметил)-5-(5,6-дихлор-2,3-дигидро-2-тиоксо-1H-бензимидазол-1-ил)-1,2-циклопентандиилдиацетат в виде кристаллов не чисто белого цвета. Т. пл. 208-209oC; 1H-ЯМР (ДМСО-d6) δ: 13,2 (шс, 1, NH), 8,05 и 7,39 (оба с, 2, 2 ароматических CH), 6,0-5,8 (M, 1, OCH), 5,6-5,3 (M, 2, OCH и NCH), 4,40 - 4,15 (M, 2, OCH2), 2,6 - 2,5 (M, перекрывается с растворителем CH), 2,4 - 2,0 (M, перекрывается с двумя с при 2,09 и 2,06, всего 8, CH2 и 2 CH3), 1,93 (с, 3, CH3); масс-спектр (ХИ): 475 (М+1).
Анализ для C19H20N2Cl2O6S:
Рассчитано: C 48,01; H 4,24; N 5,89; Cl 14,92; S 6,75.
Найдено: C 48,11; H 4,22; N 5,90: Cl 14,85; S 6,74.
Пример 45
(+/-)-(1R*, 2S*, 3S*,5S*)-5-[2- (бензилтио)-5,6-дихлор-1H-бензимидазол-1-ил)]-3-(гидроксиметил)-1,2- циклопентандиол
Смесь (+/-)-(1R*, 2S*,3S*,5S*)-3- (ацетоксиметил)-5-(5,6-дихлор-2,3-дигидро-2-тиоксо-1H-бензимидазол-1-ил)- 1,2-циклопентандиилдиацетата (0,50 г, 1,05 ммоль), бромистого бензила (0,3 мл, 2,5 ммоль) и безводного карбоната калия (0,145 г, 1,05 мэкв в виде 98% продукта) в диоксане (5 мл) энергично перемешивали при комнатной температуре в течение 2,5 дней. Летучие вещества выпаривали в вакууме и остаток распределяли между хлороформом и водой. Слой хлороформа осушали над сульфатом натрия и концентрировали до масла, которое перемешивали в метанольном растворе аммиака (40 мл наполовину насыщенного раствора) в течение 18 ч. Летучие вещества выпаривали и остаток переосаждали из смеси метанол-вода (3:1); получили указанное в заголовке вещество в виде белого порошка (300 мг, 65%). Т.пл. 160-162oC; 1H-ЯМР (ДМСО-d6) δ: 8,04 и 7,89 (оба с, 1 каждый, 2 ароматических CH), 7,5 - 7,2 (M, 5, C6H5), 5,06 (Т, J = 4,7 Гц, 1, OH), (д, J = 6,2 Гц, 1, OH), 4,75 - 4,55 М, перекрывается с д при 4,68, J= 3,7 Гц, и с при 4,63, всего 4, NCH, OH и SCH2); масс-спектр (ХИ): 439 (M+1).
Анализ для C20H20N2Cl2O3S:
Рассчитано: C 54,68; H 4,59; N 6,38; общий галоген в виде Cl 16,14; S 7,30
Найдено: C 54,75; H 4,62; N 6,38; общий галоген в виде Cl 16,21; S, 7,27.
Пример 46
(+/-)-(1R*, 2S*, 3S*, 5S*)-5- (5,6-дихлор-2-метокси-1H-бензимидазол-1-ил)-3-(гидроксиметил)- 1,2-циклопентандиол.
В сухой метанол (15 мл) добавляли натрий (шарики, 0,1 г, 4,8 мэкв). К раствору метоксида натрия в метаноле добавляли (+/-)-(1R*,2S*,3S*, 5S*)-3-(ацетоксиметил)-5-(2-бром-5,6-дихлор-1H-бензимидазол-1- ил)-1,2-циклопентандиилдиацетат (0,560 г, 0,958 ммоль) и раствор перемешивали при комнатной температуре в атмосфере азота в течение 5 ч. Затем раствор нейтрализовали 1 н соляной кислотой и летучие вещества удаляли в вакууме. Оставшееся твердое вещество хроматографировали на силикагеле (+/-)-(1R*,2S*, 3S*,5S*)-5-(5,6-дихлор-2-метокси-1H-бензимидазол- 1-ил)-3-(гидроксиметил)-1,2-циклопентандиол после растирания с диэтиловым эфиром элюировали 10% смесью метанол-хлороформ в виде белого порошка (0,211 г, 64%). Т.пл. 204-206oC разл.; 1H-ЯМР (ДМСО-d6) δ: 7,82 и 7,67 (оба с, 2, ароматические CH), 4,9 (M, 2, 2 OH), 4,7-4,6 (M, перекрывается с д при 4,61, J = 4,1 Гц, всего 2, NCH и OH), 4,4-4,3 (M, 1, OCH), 4,11 (с, 3, OCH3), 3,8 (M, 1, OCH), 3,6-3,4 (M, 2, OCH2), 2,1-2,0 (M, 2, CH2), 1,9-1,8 (M, 1, CH); масс-спектр (ХИ): 347 (M+1).
Анализ для C14H16N2Cl2O4:
Рассчитано: C 48,43; H 4,65; N 8,07; Cl 20,42.
Найдено: C 48,23; H 4,71; N 7,98; Cl 20,51.
Пример 47
(+/-)-(1R*, 2S*,3S*, 5S*)-5-(5,6-дихлор-2-фенокси-1H-бензимидазол-1-ил)- 3-(гидроксиметил)-1,2-циклопентандион
Фенол (72 мг, 0,77 ммоль) и безводный карбонат калия (106 мг, 0,77 ммоль) перемешивали в сухом N,N-диметилформамиде (5 мл) в атмосфере азота в течение 1,0 ч. Добавляли (+/-)-(1R*,2S*, 3S*,5S*)-3-(ацетоксиметил)-5-(2-бром-5,6-дихлор-1H-бензимидазол- 1-ил)-1,2-циклопентандиилдиацетат (400 мг, 0,770 ммоль) и смесь перемешивали при 80oC (масляная баня) в течение 18 ч. Летучие вещества удаляли в вакууме и остаток распределяли между хлороформом и водой. Слой хлороформа осушали над сульфатом натрия и летучие вещества выпаривали. Оставшееся масло перемешивали в 4 н. растворе аммиака в метаноле (30 мл) при комнатной температуре в течение 18 ч. Выпариванием и хроматографированием остатка на силикагеле с элюированием 2% смесью метанол-хлороформ получен (+/-)-(1R*, 2S*,3S*,5S*)-5-(5,6-дихлор-2-фенокси- 1H-бензимидазол-1-ил)-3-(гидроксиметил)-1,2-циклопентандиол в виде белого твердого вещества (из этилацетата) (215 мг, 68%). 1H-ЯМР (ДМСО-d6) δ: 7,98 и 7,71 (оба с, 2, ароматические CH), 7,6-7,3 (M, 5, C6H5), 5,07 (д, J=6,4 Гц, 1, OH), 5,0-4,75 (M, перекрывается с T при 4,95, J=3,1 Гц, всего 2, NCH и OH), 4,71 (д, J=3,9 Гц, 1, OH), 4,5-4,35 (M, 1, OCH), 3,9-3,8 (M, 1, OCH), 3,7-3,4 (M, 2, OCH2), 2,3-1,9 (M, 3, CH2 и CH); масс-спектр (ХИ): 409 (M+1).
Анализ для C19H18N2Cl2O4:
Рассчитано: C 55,15; H 4,51; N 6,77; Cl 17,14.
Найдено: C 55,14; H 4,52; N 6,72; Cl 17,07.
Пример 48
(+/-)-(1R*, 2S*, 3S*, 5S*)-5-[(5,6-дихлор-2-(циклопропиламино)-1H-бензимидазол-1-ил)]-3-(гидроксиметил)- 1,2-циклопентандиол
(+/-)-(1R*, 2S*, 3S*, 5S*)-3-ацетоксиметил)-5-(2-бром-5,6- дихлор-1H-бензимидазол-1-ил)-1,2-циклопентандиилдиацетат (500 мг, 0,958 ммоль) растворяли в абсолютированном этаноле (5 мл) и добавляли циклопропиламин (0,66 г, 9,6 ммоль). Раствор кипятили с обратным холодильником в атмосфере азота в течение 2 ч. Добавляли дополнительное количество циклопропиламина (0,66 мл) и кипячение продолжали еще 18 ч. Раствор охлаждали и добавляли метанол, насыщенный при 0oC аммиаком (5 мл). Через два дня стояния смеси при комнатной температуре летучие вещества удаляли в вакууме и остаток хроматографировали на силикагеле. (+/-)-(1R*, 2S*, 3S*, 5S*)- 5-[(5,6-дихлор-2-(циклопропиламино)-1H-бензимидазол-1- ил)] -3-(гидроксиметил)-1,2-циклопентандиол элюировали 7% смесью метонол-хлороформ и высаживали из смеси этилацетат-гексаны в виде твердого вещества (210 мг, 59%). Т.пл. 223-224oC; 1H-ЯМР (ДМСО-d6) δ: 7,64 и 7,46 (оба с, 2, ароматический CH), 7,11 (M, 1, NH), 5,11 (Т, J=4,3 Гц, 1, OH), 4,77 (д, J=7,0 Гц, 1, OH), 4,67 (д, J=3,7 Гц, 1, OH), 4,65-4,30 (М, 2, OCH и NCH), 3,85-3,75 (М, 1, OCH), 3,7-3,4 (М, 2, OCH2), 2,85-2,70 (М, 1, NCH) циклопропила, 2,15-1,80 (М, 3, CH2 и CH циклопентана), 0,80-0,50 (М, 4, 2 CH2) циклопропила; масс-спектр (ХИ): 372 (M+1).
Анализ для C16H19N3Cl2O3:
Рассчитано: C 51,63; H 5,15; N 11,29; Cl 19,05.
Найдено: C 51,41; H 5,20; N 11,19; Cl 19,16.
Пример 49
(+/-)-(1R*, 2S*, 3S*,5S*)-5-[(5,6- дихлор-2-(диметиламино)-1H-бензимидазол-1-ил)]-3-(гидроксиметил)-1,2- циклопентандиол
(+/-)-(1R*, 2S*, 3S*, 5S*)-3-(ацетоксиметил)-5-(2-бром-5,6-дихлор-1H-бензимидазол-1-ил) -1,2-циклопентандиилдиацетат (500 мг, 0,958 ммоль) и 40%-ный водный раствор N,N-диметиламина (6,0 мл) кипятили с обратным холодильником 2,5 ч. Летучие вещества удаляли в вакууме и остаток осаждали из смеси этанол-вода (1:1); получили (+/-)-(1R*,2S*,3S*,5S*)-5-[(5,6- дихлор-2-(диметиламино)-1H-бензимидазол-1-ил)]-3-(гидроксиметил)- 1,2-циклопентандиол в виде белого порошка (263 мг, 76%). Т.пл. 190-193oC; 1H-ЯМР (ДМСО-d6) δ: 7,89 и 7,66 (оба с, 2, ароматический CH), 5,07 (Т, J=4,5 Гц, 1, OH), 4,97 (д, J=5,9 Гц, 1, OH), 4,8-4,6 (м, перекрывается с д при 4,62, J=3,5 Гц всего 2, NCH и OH), 4,55-4,4 (М, 1, OCH), 3,9-3,75 (М, 1, OCH), 3,7-3,4 (М, 2, OCH2), 2,92 (с, 6, 2 CH3), 2,2-1,9 (М, 3, CH2 CH); масс-спектр (ХИ): 360 (М+1).
Анализ для C15H19N3Cl2O3 • 0,65 H2O:
Рассчитано: C 48,44; H 5,50; N 11,30; Cl 19,06.
Найдено: C 48,39; H 5,31; N 10,88, Cl 19,49.
Пример 50
(+/-)-(1R*, 2S*, 3S*,5S*)-5-[(5,6- дихлор-2-(циклопентиламино)-1H-бензимидазол-1-ил)]-3-(гидроксиметил)- 1,2-циклопентандиол
(+/-)-(1R*, 2S*, 3S*,5S*)-3- ацетоксиметил)-5-(2-бром-5,6-дихлор-1H-бензимидазол-1-ил)-1,2- циклопентандиилдиацетат (500 мг, 0,958 ммоль) и циклопентиламин (0,95 мг, 9,6 ммоль) кипятили с обратным холодильником в 5 мл абсолютированного этанола в течение 3,5 ч. Добавляли раствор аммиака в метаноле (25 мл, насыщенный при 0oC) и продолжали перемешивание еще 18 ч. Летучие вещества удаляли и в вакууме и остаток хроматографировали на силикагеле. Элюированием 6-8% смесью метанол-хлороформ с последующей кристаллизацией из смеси этилацетат-гексаны получен (+/-)-(1R*,2S*,3S*, 5S*)-5-[(5,6-дихлор-2-(циклопентиламино)-1H-бензимидазол-1-ил] - 3-(гидроксиметил)-1,2-циклопентандиол в виде белого порошка (155 мг, 41%). Т.пл. 270-271oC разл. 1H-ЯМР (ДМСО-d6) δ: 7,58 и 7,37 (оба с, 2, 2 ароматических CH), 6,67 (д, J=6,8 Гц, 1, NH), 5,11 (Т, J=4,5 Гц, 1, OH), 4,79 (д, J=3,7 Гц, 1, OH), 4,6-4,5 (М, 1, NCH), 4,4-4,3 (М, 1, OCH) 4,2-4,1 (M, 1, NCH) циклопентиламиногруппы, 3,79-3,77 (M, 1, OCH), 3,75-3,5 (M, 2, OCH2), 2,1-1,8 (M, 5, 2CH2 и CH), 1,7-1,3 (M, 6,3 CH2); масс-спектр (ХИ): 400 (M+1).
Анализ для C18H23N3Cl2O3 • 0,10 EtOAc:
Рассчитано: C 54,02; H 5,86; N 10,27; Cl 17,33.
Найдено: C 53,77; H 5,83; N 10,30; Cl 17,52.
Пример 51
(+/-)-(1R*, 2S*, 3S*,5S*)-3- (ацетоксиметил)-5-(5,6-дихлор-2-иод-1H-бензимидазол-1-ил)-1,2-циклопентандиилдиацетат
(+/-)-(1R*), 2S*, 3S*,5S*)-3-(ацетоксиметил)-5-(5,6-дихлор-1H-бензимидазол-1-ил)-1,2-циклопентандиилдиацетат (500 мг, 1,13 ммоль) растворяли в 4 мл сухого N, N-диметилформамида и нагревали до 95-105oC. В течение 5,5 ч порциями добавляли N-иодсукцинимид (534 мг, 2,3 ммоль в виде 95% продукта). Летучие вещества удаляли в вакууме и остаток хроматографировали на силикагеле. Элюированием 10% смесью этилацетат-гексаны с последующим осаждением из смеси этанол-вода получен (+/-)-(1R*, 2S*,3S*,5S*)-3- (ацетоксиметил)-5-(5,6-дихлор-2-иод-1H-бензимидазол-1-ил)-1,2- циклопентандиилдиацетат в виде белого порошка (255 мг, 40%). Т.пл. 152,5-153oC; 1H-ЯМР (ДМСО-d6) δ: 8,32 и 7,94 (оба с, 2, 2 ароматических CH), 5,8-5,7 (M, 1, OCH), 5,4-5,3 (M, 1, OCH), 5,2-5,0 (M, 1, NCH), 4,35-4,2 (M, 2, OCH2), 2,7-2,6 (M, 1, CH), 2,35-2,25 (M, 2, CH2), 2,11, 2,08 и 1,93 (все с, 3 каждый, 3 CH3); масс-спектр (ХИ) 569 (M+1).
Анализ для C19H19N2Cl2IO6:
Рассчитано: C 40,10; H 3,37; N, 4,92; общий галоген в виде Cl 18,69.
Найдено: C 40,27; H 3,39; N 4,88; общий галоген в виде Cl 18,63.
Пример 52
(+/-)-(1R*, 2S*, 3S*,5S*)-5- (5,6-дихлор-2-иод-1H-бензимидазол-1-ил)-3-(гидроксиметил)-1,2- циклопентандиол.
В воде (0,7 мл) растворяли карбонат натрия (40 мг, 0,37 ммоль) и добавляли 3 мл метанола и 3 мл этанола. К этой перемешиваемой смеси добавляли (+/-)-(1R*,2S*, 3S*,5S*)-3-(ацетоксиметил)-5-(5,6-дихлор-2-иод-1H- бензимидазол-1-ил)-1,2-циклопентандиилдиацетат (215 мг, 0,37 ммоль). После выдерживания в течение 2 ч при комнатной температуре добавляли уксусную кислоту до pH 7 и летучие вещества выпаривали в вакууме. Переосаждением оставшегося твердого вещества из смеси этанол-вода (3:1) получен (+/-)-(1R*,2S*,3S*, 5S*)-5-(5,6-дихлор-2-иод-1H-бензимидазол-1-ил)-3-(гидроксиметил)-1,2-циклопентандиол в виде белого порошка (153 мг, 94%). Т.пл. 209-210oC разл. 1H-ЯМР (ДМСО-d6) δ: 8,22 и 7,91 (оба с, 2,2 ароматических CH), 5,12 (Т, J = 4,5 Гц, 1, OH), 4,95 (д, J = 6,2 Гц, 1, OH), 4,9-4,8 (M, 1, NCH), 4,70 (Т, J = 3,5 Гц, 1, OH), 4,6-4,5 (M, 1, OCH), 3,9-3,8 (M, 1, OCH), 3,7-3,6 и 3,55-3,45 (оба M, 1 каждый, OCH2), 2,2-2,0 (M, 3, CH2 и CH); масс-спектр (ХИ): 443 (M+1).
Анализ для C13H13N2Cl2IO2:
Рассчитано: C 35,24; H 2,96, N 6,32; общий галоген в виде Cl 24,01.
Найдено: C 35,30; H 3,01; N 6,23; общий галоген в виде Cl 23,95.
Пример 53
(1S,4R)-[4-(4,5-дихлор-2-нитроанилино)-2-циклопентен-1-ил]- метанол
(-)-(1R, 4S)-трет-бутил-N-[4-(гидроксиметил)-2-циклопентен-1-ил] - карбамат (15,00 г, 70,3 ммоль) был превращен методом, описанным в примере 33, в (1S, 4R)-[4-(4,5-дихлор-2-нитроанилино)-2-циклопентен-1-ил] метанол, который был выделен в виде желтого порошка после элюирования из колонки с силикагелем смесью гексаны-хлороформ (1:1 и переосаждения из смеси этилацетат-гексаны (9,97 г, 47%). Т.пл. 94,5-96,5oC; 1H-ЯМР (ДМСО-d6) δ: 8,24 (с, 1, бензимидазольный CH), 8,09 (д, J = 8,1 Гц, 1, NH), 7,51 (с, 1, бензимидазольный CH), 5,95 и 5,85 (оба M, 2, CH=CH), 4,9-4,7 (M, перекрывается с T при 4,78, J = 5,1 Гц, всего 2, CHN и OH), 3,4 (M, 2, CH2O), 2,80 (M, 1, CH), 2,6-2,4 (M, перекрывается с растворителем CH), 1,5-1,4 (M, 1, CH); масс-спектр (ХИ): 303 (M+1); [α]
Анализ для 12H12N2Cl2O3 • 0,18 C6H14:
Рассчитано: C 49,30; H 4,59; N 8,79, Cl 22,25.
Найдено: C 49,64; H 4,64; N 8,68; Cl 22,10.
Пример 54
(1S, 2R, 3R, 5R)-5-(4,5-дихлор-2-нитроанилино)-3- (гидроксиметил)-1,2-циклопентандиол и (1R,2S,3R,5R)-5-(4,5- дихлор-2-нитроанилино)-3-(гидроксиметил)-1,2-циклопентандиол
К раствору (1S, 4R)-[4-(4,5-дихлор-2-нитроанилино)-2-циклопентен-1-ил] метанола (8,60 г, 27,6 ммоль) и N-метилморфолин-N-оксида (Aldrich, 60%-ный водный раствор, 5,02 мл, 29,0 ммоль) в 90 мл ацетона добавляли тетраоксид осмия (Aldrich, 25%-ный раствор в трет-бутиловом спирте, 0,51 мл). После перемешивания при комнатной температуре в течение 18 ч добавляли дополнительно 0,25 мл 60%-ного водного раствора N-метилморфолин-N-оксида и раствор перемешивали еще 5 ч. Летучие вещества выпаривали в вакууме и остаток дважды перекристаллизовывали из 95% этанола; получили (1S,2R,3R,5R)-5-(4,5-дихлор-2-нитроанилино)-3-(гидроксиметил)-1,2- циклопентандиол в виде желтого порошка (1,78 г, 19%). Т.пл. 197-199oC; 1H-ЯМР (ДМСО-d6) δ: 8,23 (с, 1, CH бензимидазола), 8,1 (д, J = 7,0 Гц, 1, NH), 7,50 (с, 1, CH бензимидазола), 5,02 (д, J = 4,9 Гц, 1, OH), 4,74 (Т, J = 5,1 Гц, 1, CH2OH 4,58 (д, J = 5,1 Гц, 1, OH), 4,0 - 3,8 (М, 1, NCH), 3,8 - 3,7 (M, 2, 2 OCH), 3,5 - 3,4 (М, 2, CH2O), 2,45 - 2,25 (М, 1, CH), 2,1 - 1,9 (М, 1, CH), 1,4 - 1,2 (М, 1, CH); масс-спектр (ХИ): 337 (М+1); [α]
Анализ для C12H14N2Cl2O5:
Рассчитано: C 42,75; H 4,19; N 8,31; Cl 21,03.
Найдено: C 42,84; H 4,21; N 8,24; Cl 21,09.
Хроматографированием содержимого маточного раствора на силикагеле при элюировании 7-8% смесью метанол-хлороформ получен (1R,2S)-изомер; после двух перекристаллизацией из 90% этанола получен (1R,2S,3R,5R)-5-(4,5-дихлор-2-нитроанилино)-3-(гидроксиметил)-1,2- циклопентандиол в виде желтого порошка (1,57 г, 17%), Т.пл. 179-181oC; 1H-ЯМР (ДМСО-d6) δ: 8,70 (д, J = 7,1 Гц, 1, NH), 8,22 и 7,32 (оба с, 1 каждый, 2 бензимидазольных CH), 5,28 (д, J = 5,6 Гц, 1, OH), 4,77 (д, J = 3,9 Гц, 1 OH), 4,45 (Т, J = 4,9 Гц, 1, CH2OH), 4,1 - 3,9 (М, 3, 2 OCH и NCH), 3,6 - 3,5 и 3,45 - 3,35 оба M частично перекрываются с H2O, 2, CH2O), 2,45 - 2,25 (М, 1, CH), 2,1- 3,9 (М, 1, CH), 1,35 - 1,25 (М, 1, CH); масс-спектр (ХИ): 337 (М+1); [α]
Анализ для C12H14N2Cl2O5:
Рассчитано: C 42,75; H 4,19; N 8,31; Cl 21,03.
Найдено: C 42,87; H 4,15; N 8,30, Cl 21,14.
Элюированием 8-10% смесью метанол-хлороформ получено белое твердое вещество (2,9 г); согласно 1H ЯМР оно представляет смесь двух изомеров в приблизительном соотношении 1:1.
Продолженное элюирование из колонки 10-20% смесью метанол-хлороформ давало фракции, содержащие дополнительные количества (1S,2R,3R,5R)-5-(4,5-дихлор-2-нитроанилино)-3-(гидроксиметил)- 1,2-циклопентандиола, который осаждали из 90% этанола в виде белого порошка (2,23 г), доводя таким образом общий выход данного изомера до 43%.
Пример 55
(1S, 2R, 3R, 5R)-3-(ацетоксиметил)-5-(4,5-дихлор-2-нитроанилино)-1,2- циклопентандиилдиацетат
(1S, 4R)-[4-(4,5-дихлор-2-нитроанилино)-2- циклопентен-1-ил] -метанол (3,75 г, 11,1 ммоль) ацетилировали в смеси пиридин-уксусный ангидрид как описано в примере 38. Сырой продукт элюировали из колонки с силикагелем 2% смесью метанол-хлороформ и осаждали из этилацетата; получили (1S,2R,3R, 5R)-3-(ацетоксиметил)-5-(4,5- дихлор-2-нитроанилино)-1,2-циклопентандиилдиацетат в виде желтого порошка (5,13 г, 100%) со спектром ЯМР, идентичным с приведенным в примере 1. Этот образец кристаллизовался из смеси этилацетат-гексаны в указанное в заголовке соединение в виде желтого порошка, т.пл. 128-130oC; спектры 1H-ЯМР (ДМСО-d6) и масс-спектры (ХИ) были идентичны со спектрами из примера 1, [α]
Анализ для C18H20N2Cl2O8:
Рассчитано: C 46,67; H 4,35; N 6,05; Cl 15,31.
Найдено: C 46,74; H 4,36; N 5,96; Cl 15,38.
Пример 56
(1S, 2R, 3R,5R)-3-(ацетоксиметил)-5-(5,6-дихлор-1H- бензимидазол-1-ил)-1,2-циклопентандиилдиацетат
(1S, 2R,3R,5R)-3-(ацетоксиметил)-5-(4,5-дихлор-2-нитроанилино)-1,2- циклопентандиилдиацетат (4,42 г, 9,97 ммоль) был превращен в указанное в заголовке соединение тем же способом, что и рацемический образец из примера 3. Сырой продукт хроматографировали на силикагеле при элюировании 5% смесью метанол-хлороформ и растворители выпаривали; получен (1S, 2R, 3R, 5R)-3-(ацетоксиметил)-5-(5,6- дихлор-1H-бензимидазол-1-ил)-1,2-циклопентандиилдиацетат в виде твердого пенистого вещества не чисто белого цвета (из этанола) (4,0 г, 90%); спектры 1H ЯМР (ДМСО-d6) и масс-спектры (ХИ) были идентичны со спектрами рацематов, описанных в примере 2; [α]
Анализ для C19H20N2Cl2O6:
Рассчитано: C 51,49; H 4,55; N 6,32; Cl 16,00.
Найдено: C 51,33; H 4,58; N 16,27; Cl 15,90.
Пример 57
(1S, 2R, 3R,5R)-5-(5,6-дихлор-1H-бензимидазол-1-ил)-3- (гидроксиметил)-1,2-циклопентандиол
(1S, 2R, 3R, 5R)-3-(ацетоксиметил)-5-(5,6-дихлор-1H-бензимидазол-1-ил)- 1,2-циклопентандиилдиацетат (0,96 г, 2,17 ммоль) и карбонат натрия (0,230 г, 2,17 ммоль) перемешивали в смеси вода (3 мл)-этанол (15 мл) - метанол (15 мл) 24 ч при комнатной температуре. pH доводили до 7 уксусной кислотой и летучие вещества удаляли в вакууме. Оставшееся твердое вещество суспендировали в воде (25 мл) и фильтровали. Переосаждением из смеси этанол-метанол (2:1) получен (1S,2R,3R,5R)-5-(5,6-дихлор-1H-бензимидазол-1-ил)-3-(гидроксиметил)-1,2-циклопентандиол в виде белого порошка (408 мг, 60%). Т. пл. 222-225oC; 1H-ЯМР (ДМСО-d6) δ: 8,49, 8,09 и 7,96 (все с, 1 каждый, 3 бензимидазольных CH), 5,04 (д, J = 7,0 Гц, 1, OH), 4,87 (T, J = 5,1 Гц, 1, CH2OH), 4,8 - 4,6 (M перекрывается с д при 4,76, J = 4,3 Гц, 2, NCH и OH), 4,25 - 4,10 (М, 1, OCH), 3,9 - 3,8 (М, 1, OCH), 3,6 - 3,45 (M, 2, CH2O) 2,45 - 2,25 (М, 1, CH), 2,2 - 2,0 (М, 1, CH), 1,85 - 1,65 (М, 1, CH); масс-спектр (ХИ): 317 (М+1); [α]
Анализ для C13H14N2Cl2O3:
Рассчитано: C 49,23; H 4,45; N 8,83; Cl 22,36.
Найдено: C 49,25; H 4,47; N 8,83; Cl 22,46.
Пример 58
(1S,2R,3R,5R)-5-(2-бром-5,6-дихлор-1H-бензимидазол-1-ил)-3- (гидроксиметил)-1,2-циклопентандиол
(1S, 2R, 3R,5R)-3-(ацетоксиметил)-5-(5,6-дихлор-1H- бензимидазол-1-ил)-1,2-циклопентандиилдиацетат (2,00 г, 4,51 ммоль) растворяли в сухом N,N-диметилформамиде (9 мл) и нагревали до 90oC. В течение 5 ч 4 порциями добавляли N-бромсукцинимид (1,62 г, 9,02 ммоль). Летучие вещества выпаривали в вакууме. Остаток хроматографировали на силикагеле, продукт получен элюированием 30-50% смесью этилацетат-гексаны в виде желтого стеклообразного вещества (1,00 г, 43%); спектр 1H ЯМР (ДМСО-d6) были в согласии со структурой. Образец деацетилировали карбонатом натрия (203 мг, 1,9 ммоль) в смеси вода (3 мл) - этанол (15 мл) - метанол (15 мл) при комнатной температуре в течение 5 ч. pH доводили до 7 уксусной кислотой. Раствор выпаривали в вакууме досуха, остаток растирали с водой и получали белый порошок, который хроматографировали. Элюированием из колонки с силикагелем 10-12% смесью метанол-хлороформ получен (1S, 2R, 3R, 5R)-5-(2-бром-5,6-дихлор-1H-бензимидазол-1-ил)-3- (гидроксиметил)-1,2-циклопентандиол в виде белого порошка после высаживания из смеси этанол-метанол (1:1), (410 мг, 54%), т.пл. 212-215oC; спектры 1H ЯМР (ДМСО-d6) и масс-спектры были идентичны спектрам рацемата, описанного в примере 4; [α]
Анализ для C13H13N2ArCl2O3:
Рассчитано: C 39,43; H 3,31; N 7,07; общий галоген в виде Cl 26,86.
Найдено: C 39,62; H 3,37; N 7,02; общий галоген в виде Cl 26,75.
Пример 59
(1S,2R,3R,5R)-5-(5,6-дихлор-2-метил-1H-бензимидазол-1-ил)-3- (гидроксиметил)-1,2-циклопентандиол.
Указанное в заголовке примера 55 вещество (514 мг, 1,11 ммоль) восстанавливали никелем Ренея и водородом, как описано в примере 56. Катализатор отфильтровывали, этанол выпаривали в вакууме; муравьиную кислоту заменяли ледяной уксусной кислотой (5 мл). Раствор в уксусной кислоте кипятили с обратным холодильником несколько часов, добавляли уксусный ангидрид (5 мл) и продолжали кипячение в течение 0,5 ч. Летучие вещества выпаривали в вакууме и оставшееся темное масло хроматографировали на силикагеле. (Элюирование 4% смесью метанол-хлороформ приводило к фракциям, содержащим триацетат указанного в заголовке соединения в виде желтого масла (0,46 г); спектр 1H ЯМР был в согласии со структурой. Деацетилирование проводили с применением карбоната натрия, как описано в примере 57. Твердое вещество, полученное при растирании с водой, перекристаллизовывали из смеси метанол-этанол (1:1) с несколькими каплями воды; получен (1S,2R,3R,5R)-5-(5,6-дихлор-метил-1H-бензимидазол-1-ил)-3- (гидроксиметил)-1,2-циклопентандиол в виде белого порошка (220 мг, 75%); Т.пл. 222-224oC; 1H-ЯМР (ДМСО-d6) δ: 8,09 и 7,82 (оба с, 1 каждый, 2 бензимидазольных CH), 5,09 (Т, J = 4,5 Гц, 1 CH2OH), 4,95 (д, J = 7,2 Гц, 1, OH), 4,8 - 4,6 (M, перекрывается с д при 4,75, J = 3,9 Гц, 2, NCH и OH), 4,5 - 4,3 (M, 1, OCH), 3,9 - 3,8 (M, 1, OCH), 3,7 - 3,45 (M, 2, CH2O), 2,59 (c, перекрывается с растворителем CH3), 2,3 - 1,9 (M, 3, CH2 и CH); масс-спектр (ХИ): 33 (M+1); [α]
Анализ для C14H16N2Cl2O3:
Рассчитано: C 50,78; H 4,87; N 8,46; Cl 21,41.
Найдено: C 50,85; H 4,88; N 8,45; Cl 21,32.
Пример 60
(1S, 2S, 3R, 4R)-5,6-дихлор-1-[2,3-дигидрокси-4-(гидроксиметил)- циклопентил]-1H-бензимидазол-2-(3H)-он.
Указанное в заголовке примера 55 соединение (500 мг, 1,08 ммоль) восстанавливали никелем Ренея и водородом, как описано в примере 56. Катализатор отфильтровывали, этанол выпаривали в вакууме; оставшееся масло растворяли в 50 мл хлороформа и доводили до кипения. Во время кипячения раствора с обратным холодильником (3,5 ч) добавляли порциями 1,1'-карбонилдиимидазол (845 мг, 4,40 ммоль). Раствор охлаждали и затем проводили экстракцию водой (2 х 20 мл), осушали над сульфатом натрия и концентрировали до темного масла, которое хроматографировали на силикагеле. Элюированием 5% смесью метанол-хлороформ получен триацетат указанного в заголовке соединения в виде белого порошка (400 мг, 81%), спектр 1H ЯМР был в согласии со структурой. Деацетилирование проводили в диоксане (10 мл) 1 н. раствором гидроксида натрия в количестве, достаточном до доведения pH до 14. Через 1 ч (комнатная температура) pH доводили до 7 1 н. раствором соляной кислоты. Летучие вещества выпаривали в вакууме и оставшееся твердое вещество хроматографировали на силикагеле. Продукт элюировали 10-15% смесью метанол-хлороформ и перекристаллизовывали из смеси этанол-вода, затем из смеси метанол-вода, получен (1R, 2S, 3R, 4R)-5,6-дихлор-1-[2,3-дигидрокси-4-(гидроксиметил) циклопентил-1H-бензимидазол-2-(3H)-он в виде белых игольчатых кристаллов (130 мг, 45%). Т. пл. 219-220oC; 1H-ЯМР (ДМСО-d6) δ: 11,2 (ш.м, 1, имидазольный NH, 7,56 и 7,19 (оба с, 1 каждый, 2 бензимидазольных CH), 4,9 - 4,7 (M, 2, 2OH), 4,7 - 4,45 (M, 2, OH и OCH), 4,45 - 4,3 (M, 1, NCH), 3,80 (M, 1, OCH), 3,50 (M, 2, OCH2), 2,1 - 1,8 (M, 3, CH2 и CH); масс-спектр (ХИ): 333 (M+1); [α]
Анализ для C13H14N2Cl2O4:
Рассчитано: C 46,87; H 4,24; N 8,41; Cl 21,28.
Найдено: C 46,98; H 4,26; N 8,37; Cl 21,30.
Пример 61
(1R, 2S,3R,5R)-3-(ацетоксиметил)-5-(4,5-дихлор-2-нитроанилино)-1,2- циклопентандиилдиацетат
(1R, 2S, 3R, 5R)-5-(4,5-дихлор-2-нитроанилино)-3- (гидроксиметил)-1,2-циклопентандиол (1,30 г, 3,86 ммоль) в течение 2 дней при комнатной температуре перемешивали в смеси пиридин (10 мл) - уксусный ангидрид (2,2 мл). Летучие вещества выпаривали в вакууме и остаток распределяли между хлороформом и насыщенным водным раствором бикарбоната натрия. Слой хлороформа осушали над сульфатом натрия и концентрировали до масла, которое хроматографировали на силикагеле. Элюированием 2% смесью метанол-хлороформ получен (1R,2S,3R, 5R)-3-(ацетоксиметил)-5-(4,5-дихлор-2-нитроанилино)-1,2- циклопентандиилацетат в виде желтых игольчатых кристаллов (1,60 г, 89%) после кристаллизации из смеси этилацетат-гексаны, т. пл. 125-130oC: спектры 1H ЯМР (ДМСО-d6) и масс-спектры (XИ) были в согласии со структурой; [α]
Анализ для C18H20N2Cl2O8:
Рассчитано: C 46,67; H 4,35: N 6,05; Cl 15,31.
Найдено: C 46,76; H 4,35; N 6,08; Cl 15,23.
Пример 62
(1R,2S,3R,5R)-5-(5,6-дихлор-2-метил-1H-бензимидазол-1-ил)-3- (гидроксиметил)-1,2-циклопентандиол
(1R,2S,3R,5R)-3-(ацетоксиметил)-5-(4,5-дихлор-2-нитроанилино)-1,2- циклопентандиилдиацетат (1,29 г, 2,78 ммоль) в н-пропаноле (100 мл) трясли с никелем Ренея (Aldrich, взвесь в воде, 200 мг сырого веса) в атмосфере водорода (40 psi) в аппарате для встряхивания Парра 4 ч. Катализатор отфильтровывали на целите и летучие вещества выпаривали в вакууме. Оставшееся масло 5 ч кипятили с обратным холодильником в 96% муравьиной кислоте (45 мл). Добавляли 5 мл концентрированной соляной кислоты и продолжали кипячение еще 4 ч. Летучие вещества выпаривали в вакууме и остаток растворяли в 10 мл 1 н. раствора гидроксида натрия. Раствор перемешивали при комнатной температуре 18 ч, нейтрализовали уксусной кислотой и концентрировали в вакууме. Остаток хроматографировали на силикагеле. Элюированием 15% смесью метанол-хлороформ получен (1R, 2S, 3R, 5R)-5-(5,6-дихлор-2-метил-1H-бензимидазол-1-ил)-3-(гидроксиметил)- 1,2-циклопентандиол в виде белого порошка (236 мг, 26%). Т. пл. 198-200oC; 1H-ЯМР (ДМСО-d6) δ: 8,30 и 7,72 (оба с, 1 каждый, 2 ароматических CH), 5,0-4,8 (М, 3,2 OH и NCH), 4,56 (Т, J = 5,1 Гц, 1, CH2OH), 4,2-4,1 (М, 2, 2 OCH), 3,8-3,5 (М, 2: CH2O), 2,58 (с, 3, CH3), 2,4-2,2 (M, 1, CH), 2,2-1,9 (М, 2, CH2); масс-спектр (ХИ): 331 (М+1); [α]
Анализ для C14H16N2Cl2O3:
Расчитано: C 50,78; H 4,87; N 8,46; Cl 21,41.
Найдено: C 50,68; H 4,88; N 8,39; Cl 21,31.
Пример 63
(1R,4S)-4-амино-2-циклопентен-1-метанол
Смесь (-)-(1S, 4R)-4-амино-2-циклопентен-1-карбоновой кислоты (Chiros Itd, Cambridge, England; 40,00 г, 0,315 ммоль) в сухом тетрагидрофуране (300 мл) перемешивали на бане со льдом 1,5 ч, при этом добавляли 1 М раствор литийалюминийгидрида в тетрагидрофуране (Aldrich, 485 мл). При этом перемешивании не допускали повышения температуры выше 0oC. Температуру смеси доводили до комнатной и затем до температуры кипения в течение одного часа, после чего поддерживали кипячение с обратным холодильником в течение 2,5 ч. Смесь оставляли охлаждаться до комнатной температуры, добавляли 89,6 г фторида натрия и перемешивали еще 0,5 ч. Смесь охлаждали в бане со льдом и медленно добавляли 23 мл воды. Перемешивание продолжали еще 0,5 ч. Осадок фильтровали и проводили экстракцию 40% смесью метанол-тетрагидрофуран (2х300 мл). Фильтрат с промывкой концентрировали в вакууме до бесцветного масла, которое быстро темнело на воздухе и на свету и было сразу же использовано (пример 64). Этот образец осушали при комнатной температуре (0,2 мм рт. ст.) и получили бледно-желтое масло; спектр 1H ЯМР (ДМСО-d6) идентичен спектру энантиомера, описанного в примере 22, δ: 5,67 (М, 2, CH=CH) 3,8-3,7 (М, 1, CHN), 3,32 (д, J=6,0 Гц, перекрывается с широким, обменивающимся с D2О, с центром при 3,18, CH2O, OH, NH2 и H2O в растворителе), 2,68-2,56 (М, 1, H-1), 2,28-2,18 (М, 1, 1/2 CH2), 1,08-0,98 (М, 1, 1/2 CH2); масс-спектр (ХИ): 114 (М+1); [α]
Анализ для C6H11NO•0.31 H2O:
Рассчитано: C 60,69; H 9,86; N 11,80.
Найдено: С 61,12; H 9,79; N, 11,38.
Пример 64
(1R,4S)-[4-(4,5-дихлор-2-нитроанилино)-2-циклопентен-1-ил]метанол.
Фильтрат с промывкой из примера 63 концентрировали и к оставшемуся маслу добавляли 400 мл трет-бутанола. Этот раствор использовали для проведения конденсации с 1,2,4-трихлор-5-нитробензолом (Aldrich, 71,3 г, 0,315 моль в виде 97% продукта) по методу из примера 33. После выпаривания летучих веществ в вакууме реакционную смесь хроматографировали на колонке с силикагелем при элюировании смесью гексаны-этилацетат (1:1) и этилацетатом. Повторное хроматографирование сырого продукта на силикагеле осуществляли при элюировании 4-6% смесью метанол-хлороформ. Из объединенных фракций, содержащих продукт, после выпаривания растворителей получено 58 г твердого вещества красноватого цвета. Это вещество переосаждали из смеси этилацетат-гексаны и получили (1R,4S)-[4-(4,5-дихлор-2-нитроанилино)- 2-циклопентен-1-ил]метанол в виде желтого порошка (34,5 г, 36% в расчете на (-)-(1S,4R)-4-амино-2-циклопентен-1-карбоновую кислоту); т.пл. 95-97oC; спектр 1H ЯМР (ДМСО-d6) и масс-спектр (ХИ) были идентичны спектрам энантиомера, описанного в примере 53; [α]
Анализ для C12H12N2Cl2O3:
Рассчитано: C 47,55; H 3,99; N 9,24; Cl 23,39.
Найдено: C 47,56; H 4,01; N 9,25; Cl 23,30.
Дальнейшее элюирование из колонки (см выше) давало дополнительное количество желтого порошка (18,0 г, 19%), который по данным 1H ЯМР является дополнительным количеством указанного в заголовке соединения, загрязненным примерно 15% (1R,4S)-[4-(2,5-дихлор-4-нитроанилино)-2-циклопентен-1-ил]метанола.
Пример 65
(1R, 2S, 3S, 5S)-3-(ацетоксиметил)-5-(4,5-дихлор-2-нитроанилино)-1,2- циклопентандиилдиацетат и (1S,2R,3S,5S)-3-(ацетоксиметил)- 5-(4,5-дихлор-2-нитроанилино)-1,2-циклопентандиилдиацетат
(1R, 4S)-[4-(4,5-дихлор-2-нитроанилино)-2-циклопентен-1-ил] -метанол (17,00 г, 56,1 ммоль) гидроксилировали и смесь триолов ацетилировали, как описано в примере 3. Выделенный после ацетилирования сырой продукт в виде красного масла хроматографировали на силикагеле; смесь указанных в заголовке соединений элюировали 2% смесью метанол-хлороформ. Дробной кристаллизацией из смеси этилацетат-гексаны в два захода получен (1R,2S,3S,5S)-3-(ацетоксиметил)-5-(4,5-дихлор-2-нитроанилино)-1,2-циклопентандиилдиацетат в виде желтых игольчатых кристаллов (12,78 г, 49%), т.пл. 127-128oC, спектр 1H-ЯМР (ДМСО-d6) и масс-спектр (ХИ) были идентичны спектрам рацемического образца, описанного в примере 1, и энантиомера, описанного в примере 55; [α]
Анализ для C18H20N2Cl2O8:
Рассчитано: C 46,67; H 4,35; N 6,05; Cl 15,31.
Найдено: C 46,74; H 4,40; N 6,09; Cl 15,22.
Дальнейшей дробной кристаллизацией содержимого маточного раствора из смеси этилацетат-гексаны получен (1S, 2R,3S,5S)-3-(ацетоксиметил)-5-(4,5- дихлор-2-нитроанилино)-1,2-циклопентандиилдиацетат в виде оранжевых кристаллов (2,45 г, 10%), т.пл. 122-124oC, спектр 1H ЯМР (ДМСО-d6) был идентичен со спектром хирального образца, описанного в примере 61.
Выпаривание объединенных маточных растворов приводило к получению дополнительного количества (9,50 г, 40%) смеси указанных в заголовке соединений приблизительного состава 1:1 (по данным 1H ЯМР).
Пример 66
(1R, 2S, 3S,5S)-3-(ацетоксиметил)-5-(5,6-дихлор-1H- бензимидазол-1-ил)-1,2-циклопентандиилдиацетат
(1R, 2S,3S,5S)-3-(ацетоксиметил)-5-(4,5-дихлор-2-нитроанилино)- 1,2-циклопентандиилдиацетат был превращен в указанное в заголовке соединение согласно примеру 2. После обработки муравьиной кислотой сырой продукт хроматографировали на силикагеле при элюировании 10% смесью этилацетат-гексаны. После выпаривания фракций, содержащих продукт, оставался (1R, 2S, 3S, 5S)-3-(ацетоксиметил)-5-(5,6-дихлор- 1H-бензимидазол)-1-ил-1,2-циклопентандиилдиацетат в виде твердого пенистого вещества (из этилацетата) (1,85 г, 95%); спектр 1H-ЯМР (ДМСО-d6) и масс-спектр (ХИ) были идентичны спектрам рацемата, описанного в примере 2, и энантиомера, описанного в примере 56; [α]
Анализ для C19H20N2Cl2O6 • 0,1 EtOAc:
Рассчитано: C 51,54; H 4,64; N 6,20; Cl 15,68.
Найдено: C 51,29; H 4,69; N 6,19; Cl 15,91.
Пример 67
(1S, 2R, 3S, 5S)-5-(5,6-дихлор-1H-бензимидазол-1-ил)-3-(гидроксиметил)-1,2-циклопентандиол и (1R,2S,3S,5S)-5-(5,6-дихлор-1H-бензимидазол-1-ил)-3-(гидроксиметил)- 1,2-циклопентандиол
Смесь (1R, 2S, 3S, 5S)-3-(ацетоксиметил)-5-(4,5-дихлор-2-нитроанилино)- 1,2-циклопентандиилдиацетата и (1S,2R,3S,5S)-3-(ацетоксиметил)-5-(4,5-дихлор-2-нитроанилино)-1,2- циклопентандиилдиацетата приблизительного состава 1: 1 (4,30 г, 9,28 ммоль) деацетилировали карбонатом натрия (97 мг) в смеси вода-этанол-метанол (1: 1:1, 100 мл) при комнатной температуре в течение 24 ч, pH доводили до 7 уксусной кислотой и летучие вещества удаляли в вакууме. Оставшееся твердое вещество экстрагировали метанолом. Метанольный фильтрат досуха выпаривали в вакууме. Оставшееся твердое вещество растворяли в смеси этанол (55 мл) - вода (20 мл), pH доводили до 5-6 серной кислотой и смесь кипятили с обратным холодильником с порошком железа (325 меш, 99,9%, Aldrich, 5,18 г, 93 мэкв) и гептагидратом сульфата двухвалентного железа (Aldrich, 98+%, 1,30 г, 4,58 мэкв) в течение 4 ч. Твердые продукты отфильтровывали и этанольный фильтрат с промывкой концентрировали до состояния масла. К маслу добавляли 55 мл триэтилортоформиата и 0,05 мл метансульфокислоты; полученный раствор перемешивали 18 ч при комнатной температуре. После концентрирования в вакууме получено масло, которое повторно растворяли в смеси 1 н. соляная кислота (50 мл) - диоксан (5 мл). Через 2,5 ч pH доводили до 7 1 н раствором гидроксида натрия и летучие вещества выпаривали в вакууме. Оставшееся твердое вещество хроматографировали на силикагеле. Элюирование 10-12% смесью метанол-хлороформ давало фракции, содержащие (1S,2R,3S, 5S)-5-(5,6-дихлор-1H-бензимидазол-1-ил)-3- (гидроксиметил)-1,2-циклопентандиол, который выделяли в виде белых кристаллов (540 мг, 18%) кристаллизацией из смеси этилацетат-гексаны. Т.пл. 201-202oC; 1H-ЯМР (ДМСО-d6) δ: 8,42, 8,07 и 7,92 (все с, 1 каждый, 3 бензимидазольных CH), 5,1-4,8 M, перекрывающийся с д при 5,02, J = 5,7 Гц и д при 4,93, J = 3,9 Гц, всего 3, NCH и 2 OH), 4,54 (T, J = 4,8 Гц, 1, OH), 4,2 - 4,0 (M, 2, 2OCH), 3,75 - 3,45 (M, 2, OCH2), 2,4 - 1,9 (M, 3, CH2 и CH); масс-спектр (ХИ): 317 (M+1); [α]
Анализ для C13H14N2Cl2O3:
Рассчитано: C 49,23; H 4,45; N 8,83; Cl 22,36.
Найдено: C 49,20; H 4,45; N 8,78; Cl 22,37.
Последующее элюирование из колонки 15-20% смесью метанол-хлороформ давало фракции, содержащие смесь указанных в заголовке соединений, за которыми шли фракции, содержащие только (1R,2S,3S,5S)-5-(5,6-дихлор-1H-бензимидазол-1-ил)-3- (гидроксиметил)-1,2-циклопентандиол, который изолировали в виде белых кристаллов (605 мг, 21%) при кристаллизации из 10% смеси метанол-этилацетат, т. пл. 221-222oC; спектр 1H ЯМР (ДМСО-d6) и масс-спектр (ХИ) были идентичны спектру энантиомера, описанного в примере 57; [α]
Анализ для C13H14N2Cl2O3:
Рассчитано: C 49,23; H 4,45; N 8,83; Cl 22,36.
Найдено: C 49,29; H 4,46; N 8,87; Cl 22,26.
Пример 68
(1R, 2S, 3S, 5S, )-3-(ацетоксиметил)-5-(2-бром-5,6-дихлор-1H- бензимидазол-1-ил)-1,2-циклопентандиилдиацетат
(1R, 2S, 3S, 5S)-3-(ацетоксиметил)-5-(5,6-дихлор-1H-бензимидазол-1-ил)- 1,2-циклопентандиилдиацетат (1,40 г, 2,94 ммоль) бромировали, как описано в примере 3. Летучие вещества удаляли в вакууме и остаток хроматографировали на силикагеле. Элюированием 20 - 30% смесью гексан-этилацетат получен сырой продукт в виде бесцветного масла. Раствор этого масла в хлороформе промывали водой с целью удаления примесей сукцинимида. Раствор в хлороформе осушали над сульфатом натрия и выпаривали в вакууме досуха; получили указанное в заголовке соединение в виде твердого пенистого вещества (из этанола) - (760 мг, 50%); спектр 1H ЯМР (ДМСО-d6 ) и масс-спектр (ХИ) были идентичны спектрам рацемата, описанного в примере 3; [α]
Анализ для C19H19N2BrCl2O6 • 0,05 EtOH:
Рассчитано: C 43,74; H 3,71; N 5,34; общий галоген в виде Cl 20,28.
Найдено: C 43,74; H 3,69; N 5,35; общий галоген в виде Cl 20,41.
Пример 69
(1R,2S,3S,5S)-5-(2-бром-5,6-дихлор-1H-бензимидазол-1-ил)-3- (гидроксиметил)-1,2-циклопентандиол
(1R, 2S, 3S,5S)-3-(ацетоксиметил)-5-(5,6-дихлор-1H-бензимидазол-1-ил) -1,2-циклопентандиилдиацетат (660 мг, 1,26 ммоль) деацетилировали, как описано в примере 4, и получили указанное в заголовке соединение в виде белого порошка после осаждения из смеси этанол-метанол (1:1) - (415 мг, 83%), т.пл. 213-216oC, спектр 1H ЯМР (ДМСО-d6) и масс-спектр (ХИ) были идентичны спектрам рацемата, описанного в примере 4; [α]
Анализ для C13H13N2BrCl2O3:
Рассчитано: C 39,43; H 3,31; N 7,07; общий галоген в виде Cl 26,86.
Найдено: C 39,48; H 3,29; N 7,00; общий галоген в виде Cl 26,90.
Пример 70
(+/-)-4,5-дихлор-N-(2-циклопентен-1-ил)-2-нитроаналин
Гидрохлорид 3-аминоциклопентена (R. Vince and S.Daluge, J. Med. Chem., 1974, 17, 578) (5,10 г, 42,8 ммоль), 1,2,4-трихлор-5-нитробензол (Aldrich, 10,0 г, 42,8 ммоль) и карбонат калия (98%, Aldrich, 15,00 г, 107 ммоль) кипятили с обратным холодильником в трет-бутиловом спирте (100 мл) в атмосфере азота в течение 24 ч. Летучие вещества выпаривали в вакууме и остаток хроматографировали на силикагеле. Указанное в заголовке соединение элюировали 20-30% смесью хлороформ-гексаны в виде оранжевого порошка (5,14 г, 44%), т.пл. 85-86oC; 1H-ЯМР (ДМСО-d6) δ: 8,27 (с, 1, ароматический CH), 7,88 (д, J = 7,3 Гц, 1, NH), 7,48 (с, 1, ароматический CH) 6,2 - 6,0 (M, 1, =CH), 6,0 - 5,8 (M, 1, =CH), 5,0 - 4,7 (M, 1, NCH), 2,6 - 2,2 (M, перекрывается с растворителем, 3 CH), 1,8 - 1,5 (M, 1, 1/2 CH2); масс-спектр (ХИ): 273 (M+1)
Анализ для C11H10N2Cl2O2:
Рассчитано: C 48,37; H 3,69; N 10,26; Cl 25,96.
Найдено: С 48,39; H 3,72; N 10,28; Cl 25,87.
Пример 71
(+/-)-(1R*,2S*, 3R*)-3-(4,5-дихлор-2-нитроанилино)-1,2-циклопентандиол и (+/-)-(1S*,2R*, 3R*)-3-(4,5-дихлор-2-нитроанилино)-1,2-циклопентандиол
(±)-4,5-дихлор-N-(циклопентен-1-ил)-2-нитроанилин (6,30 г, 23,1 ммоль) гидроксилировали согласно примеру 38. Летучие вещества выпаривали в вакууме и остаток хроматографировали на силикагеле. Смесь указанных в заголовке соединений элюировали 10% смесью метанол-хлороформ; получено 7,00 г оранжевого вещества. Кристаллизацией из смеси этанол-вода получен (+/-)-(1S*, 2R*,3R*)-3-(4,5-дихлор-2-нитроанилино)-1,2-циклопентандиол в виде оранжевого порошка (4,03 г, 57%) - Т.пл. 134-136oC; 1H-ЯМР (ДМСО-d6) δ: 8,72 (д, J = 7,6 Гц, 1, NH), 8,25 и 7,36 (оба с, 1 каждый, 2 ароматических CH), 5,25 (д, J = 4,5 Гц, 1, OH) 4,80 (д, J = 5,1 Гц, 1, OH), 4,14 - 3,86 (M, 3,2 OCH, и 1 NCH), 2,2 - 2,1 (M, 1, CH), 1,8 - 1,4 (M, 3, CH и CH2); масс-спектр (ХИ): 307 (M+1).
Анализ для C11H12N2Cl2O4:
Рассчитано: C 43,02; H 3,94; N 9,12; Cl 23,09.
Найдено: C 43,09; H 3,99; N 9,03; Cl 23,03.
Концентрирование маточного раствора давало дополнительное количество оранжевого твердого вещества (2,86 г), которое согласно 1H ЯМР является смесью (+/-)-(1R*, 2S*, 3R*)-3-(4,5-дихлор-2-нитроанилино)-1,2-циклопентандиола (спектр ЯМР описан в примере 16) и (+/-)-(1S*, 2R*, 3R*)-3-(4,5-дихлор-2-нитроаналино)-1,2-циклопентандиола в приблизительном соотношении 1:1.
Пример 72
(+/-)-(1S*, 2R*, 3R*)-3-(5,6-дихлор-1H-бензимидазол-1-ил)-1,2-циклопентандиол
(+/-)-(1S*, 2R*, 3R*)-3-(4,5-дихлор-2-нитроанилино)-1,2-циклопентандиол (3,77 г, 12,3 ммоль) в 250 мл изопропанола трясли в аппарате для встряхивания Парра с никелем Ренея (Aldrich, предварительно промыт водой до нейтральной реакции, около 1 tsp) в атмосфере водорода (50 psi) в течение 2 ч, пока не прекратилось поглощение водорода. ТСХ (силикагель, смесь метанол-хлороформ 1: 10) показывала одно пятно с Rf меньшим, чем у исходного материала. Катализатор отфильтровывали на целите, летучие вещества выпаривали и получали стеклообразное вещество, которое 40 мин кипятили с обратным холодильником в 65 мл муравьиной кислоты. Муравьиную кислоту выпаривали и оставшееся масло растворяли в 50 мл этанола, pH доводили до 13 5 н. раствором гидроксида натрия. После перемешивания смеси при комнатной температуре в течение 18 ч pH доводили до 7 соляной кислотой и летучие вещества удаляли выпариванием в вакууме. Остаток растворяли с метанолом. Раствор в метаноле досуха выпаривали и получали белое твердое вещество, которое содержало продукт и соли (7,41 г). Порцию твердого вещества (1,05 г) хроматографировали на силикагеле и продукт элюировали 6-8% смесью метанол-хлороформ; после высаживания из этанола получили 350 мг белого порошка. Т.пл. 180 - 182oC; 1H-ЯМР (ДМСО-d6) δ: 8,46, 8,08 и 7,93 (все с, 1 каждый, 3 ароматических CH) 4,96 (д, J = 4,7 Гц, 1, OH), 4,92 - 4,85 (M, 1, NCH), 4,81 (д, J = 5,9 Гц, 1, OH), 4,19 - 4,11 (M, 1, OCH), 4,08 - 3,93 (M, 1, OCH), 2,38 - 2,04 (M, 2, CH2), 2,03 - 1,72 (M, 2, CH2); масс-спектр (ХИ): 287 (M+1).
Анализ для C12H12N2Cl2O2:
Рассчитано: C 50,19; H 4,21; N 9,76; Cl 24,69.
Найдено: C 50,18; H 4,24; N 9,74; Cl 24,60.
Пример 73
(+/-)-(1S*, 2R*, 3R*)-3-(2-бром-5,6-дихлор-1H-бензимидазол-1-ил)-1,2 -циклопентандиилдиацетат
(+/-)-(1S*, 2R*, 3R*)-3-(5,6-дихлор-1H-бензимидазол-1-ил)-1,2 циклопентандиол (3,00 г, 10,5 ммоль) ацетилировали и бромировали как описано в примере 39; указанное в заголовке соединение элюировали из колонки с силикагелем 1% смесью метанол-хлороформ и получили продукт в виде рыжевато-коричневого твердого пенистого вещества (2,86 г, 66%). Дальнейшая очистка этого образца хроматографией на силикагеле при элюировании 10% смесью гексаны-хлорофором с последующим переосаждением из метанола дала (+/-)-(1S*,2R*, 3R*)-3-(2-бром-5,6-дихлор-1H-бензимидазол-1-ил)-1,2 -циклопентандиилдиацетат в виде желтого порошка. Т.пл. 203 - 205oC; 1H-ЯМР (ДМСО-d6) δ: 8,03 и 7,93 (оба с, 1 каждый, 2 ароматических CH) 5,42 - 5,33 (M, 3,2 OCH и NCH), 2,69 - 2,62 (M, 1, 1/2CH2), 2,28 - 2,09 (М, 6, CH2, CH и CH3), 1,54 - 1,51 (M, 3, CH3); масс-спектр (ХИ): 455 (7,1), 453 (47), 451 (100), 449 (455, M+1).
Анализ для C16H15N2Cl2BrO4:
Рассчитано: C 42,69; H 3,36; N 6,22; общий галоген в виде Cl 23,63.
Найдено: C 42,77; H 3,39; N 6,17; общий галоген в виде Cl 23,62.



Производные бензимидазола формул I и I', где R1 - Н, СH2OH; R2 и R3 - H или OH или вместе образуют связь; R4 - H, Cl, Br, J, алкил, циклоалкил, NH2, алкиламино, диалкиламино, циклоалкиламино, алкокси, C6-10-арил, C6-10-арилокси, алкил тио и SH; R5, R6, R7 - Н, Сl, при условии что по меньшей мере один из R1, R2, R3 представляет или содержит ОН, или их фармацевтически приемлемые производные обладают антивирусным действием. 2 с. и 5 з.п.ф-лы.
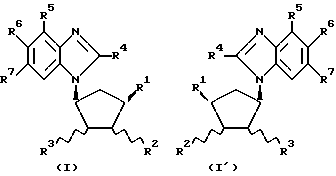
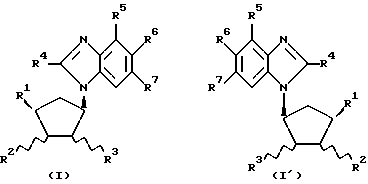
где R1 - H или CH2OH;
R2 - H или OH;
R3 - H или OH; R2 и R3 вместе образуют образуют связь;
R4 - H, Cl, Br, J, C1-4-алкил, C3-7-циклоалкил, NH2, C1-4-алкиламино, C1-4-диалкиламино, C3-7-циклоалкиламино, C1-4-алкокси, C6-10-арил, C6-10-арилокси, C1-4-алкилтио и SH; R5, R6 и R7 независимо, выбраны из H или Cl, при условии, что по меньшей мере один из R1, R2 и R3 представляет или содержит OH,
или их фармацевтически приемлемые производные.
или его зеркальный энантиомер подвергают взаимодействию с соединением формулы R4CO2H, в которой R4 представляет H, C1-4-алкил или C1-4-перфторалкил, или с соединением формулы R4C(OR)3, где R4 представляет H, C1-4-алкил или C1-4-перфторалкил и R представляет C1-4-алкил, с образованием соединения формулы I и I', где R4 - H, C1-4- или C1-4-перфторалкил или с цианбромидом с образованием соединения формулы I или I', где R4 означает NH2, с выделением целевого продукта или с превращением в его фармацевтически приемлемое производное.
в которой R1 - R3 и R5 - R7 имеют значения, определенные выше,
или его энантиомер зеркального отражения подвергают реакции с 1,1-тиокарбонилдиимидазолом или тиомочевиной с образованием соединения формулы I и I', в которой R4 представляет SH, с последующим выделением целевого продукта или с превращением в его фармацевтически приемлемое производное.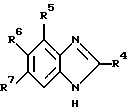
где R4 представляет водород и R5 - R7 имеют вышеуказанные значения,
подвергают реакции с соединением формулы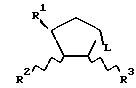
где R1 - R3 имеют вышеуказанные значения и L представляет уходящую группу,
с образованием соединения формулы I и I', в которой R4 представляет водород, с выделением целевого продукта или с превращением в его фармацевтически приемлемое производное.
| ТЕПЛООБМЕННАЯ ТРУБА | 1997 |
|
RU2127408C1 |
| Устройство для управления электроприводами механизмов перемещения грузоподъемного средства | 1987 |
|
SU1421674A1 |
| Способ получения основнозамещенных производных 4-оксибензимидазола или их солей | 1975 |
|
SU580836A3 |
| US 4410385, 1984 | |||
| Способ обработки метастабильных аустенитных сталей методом интенсивной пластической деформации | 2015 |
|
RU2610196C1 |
| US 4677210, 1987. | |||
