Изобретение относится к новым производным эритромицина, к методу их получения и к их использованию в качестве медикаментов.
Предметом настоящего изобретения являются продукты формулы (l):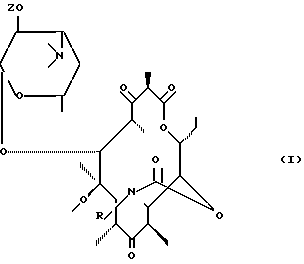
где
R - радикал 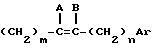 , в котором m и n, одинаковые или различные, имеют целочисленное значение от 0 до 6;
, в котором m и n, одинаковые или различные, имеют целочисленное значение от 0 до 6;
и или A и B, одинаковые или различные, представляют собой атом водорода, галогенный атом или алкильный радикал, включающий до 8 атомов углерода, при этом геометрией двойной связи является E или Z или смесь E + Z,
или A и B образуют третью связь между атомами углерода, с которыми они связаны,
и Ar представляет собой :
либо карбоциклический арильный радикал, включающий до 18 атомов углерода с возможностью замещения одним или несколькими радикалами, выбранными из группы, состоящей из карбоксильных радикалов, свободных, превращенных в соль, в сложный эфир или аминированных, гидроксильного радикала, галогенных атомов, радикалов NO2, C ≡ N радикалов, таких как алкильный, алкенильный и алкинильный, O-алкильный, O-алкенильный и O-алкинильный, S-алкильный, S-алкенильный и S-алкинильный, N-алкильный, N-алкенильный и N-алкинильный, линейных, разветвленных или циклических, включающих до 12 атомов углерода, с возможностью замещения одним или несколькими галогенными-атомами, радикал 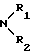 , в котором R1 и R2, одинаковые или различные, представляют собой атом водорода или алкильный радикал, включающий до 12 атомов углерода, арильный, O-арильный и S-арильный карбоциклические радикалы и арильный, O-арильный и S-арильный гетероциклические радикалы, включающие один или несколько гетероатомов; при этом имеется возможность замещения одним или несколькими заменителями из числа вышеперечисленных;
, в котором R1 и R2, одинаковые или различные, представляют собой атом водорода или алкильный радикал, включающий до 12 атомов углерода, арильный, O-арильный и S-арильный карбоциклические радикалы и арильный, O-арильный и S-арильный гетероциклические радикалы, включающие один или несколько гетероатомов; при этом имеется возможность замещения одним или несколькими заменителями из числа вышеперечисленных;
либо Ar представляет собой гетероциклический арильный радикал, включающий один или несколько гетероатомов, с возможностью замещения одним или несколькими заменителями из числа вышеперечисленных, а Z представляет собой атом водорода или остаток карбоциклической кислоты, включающей до 18 атомов углерода, а также добавляемые соли с кислотами соединений формулы (I).
В определении заменителей:
карбоциклическим арильным радикалом является, в первую очередь фенильный или нафтильный радикал;
Под гетероциклическим арильным радикалом понимают либо моноциклический гетероарильный радикал с 5 или 6 атомами, включающий один или несколько гетероатомов, либо конденсированную полициклическую систему, каждый цикл которой содержит 5 или 6 атомов и, возможно, один или несколько гетероатомов:
гетероциклический арильный радикал включает один или несколько гетероатомов, выбранных прежде всего из кислорода, серы и азота;
моноциклическим гетероарильным радикалом с 5 атомами является, в первую очередь, тиенильный, фурильный, пиролильный, тиазолильный, пиридильный, пиримидинильный, пиридазинильный или пиразинильный радикал;
конденсированным полициклическим гетероарильным радикалом может быть, например, индолильный, бензофурильный, бензотиенильный или хинолинильный радикал или остаток пуринового основания, такого как аденин;
алкильным, алкенильным или алкинильным радикалом является, в первую очередь, радикал, такой как метильный, этильный, пропильный, изопропильный, n-бутильный, изобутильный, трет-бутильный, децильный или додецильный, винильный, аллильный, этинильный, пропинильный, пропаргильный, циклобутильный, циклопентильный или циклогексильный:
галогеном является, в первую очередь, фтор, хлор или бром;
алкильным радикалом, замещенным галогенным атомом, является прежде всего такой радикал как CHCL2, CHBr2, CHF2, CCl3, CBr3, CF3, CH2CF3, CH2CH2Cl3,
CH2CH2CF3;
карбоциклическим кислотным остатком является, в первую очередь, ацетильный, пропионильный, бутирильный, изобутирильный, n-валерильный, изовалерильный, трет-валерильный и пивалильный.
Предметом изобретения, в частности, являются :
соединения формулы (I), в которых Z представляет собой атом водорода;
соединения формулы (I), в которых A и B представляют собой атом водорода или образуют третью связь между атомами углерода, с которыми они связаны;
соединения формулы (I), в которых m представляет собой число 1 или 2;
соединения формулы (I), в которых n представляет собой число 0 или 1;
соединения формулы (I), в которых Ar представляет собой карбоциклический арильный радикал с возможностью замещения одним из вышеперечисленных заменителей, например фенильным радикалом с возможностью замещения атомом фтора, радикалом CF3, фенильным радикалом и в первую очередь те, в которых Ar представляет собой незамещенный фенильный радикал.
Предметом настоящего изобретения, в первую очередь, является соединение Примера 1 и его соли с кислотами.
Продукты общей формулы (I) обладают очень высокой антибиотической активностью в отношении грамположительных бактерий, таких как стафилококки, стрептококки, пневмококки.
Поэтому соединения, являющиеся предметом настоящего изобретения, могут использоваться в качестве медикаментов при лечении инфекций с чувствительными микробами, в частности при лечении стафилококковых инфекций, таких как стафилококковые септицемии, лицевые или кожные злокачественные стафилококковые инфекции, пиодермии, септические или гнойные раны, фурункулы, карбункулы, флегмоны, рожа и угри, такие стафилококковые инфекции, как первичные или пост-гриппозные острые ангины, бронхопневмонии, легочные нагноения, такие стрептококковые инфекции, как острые ангины, отиты, синуситы, скарлатины, такие пневмококковые инфекции, как пневмонии, бронхиты; бруцеллез, дифтерия, гонококковое заболевание.
Продукты, являющиеся предметом настоящего изобретения, обладают также активностью против инфекций, вызванных такими микробами, как Haemophilus Influenzae. Rickettsies, Mycoplasma pneumoniae, Chlamydia, Legionella, Ureaplasma, Toxoplasma, или микробами типа микобактерий, листерий, менингококков и кампилобактерий.
Таким образом, предметом настоящего изобретения на медикаментозном уровне, в частности в качестве антибиотических средств, также являются продукты формулы (I), как указано выше, а также их добавляемые соли с допустимыми с фармацевтической точки зрения неорганическими или органическими кислотами.
Предметом настоящего изобретения на медикаментозном уровне, в частности в качестве антибиотических средств, также является продукт Примера 1 и его допустимые с фармацевтической точки зрения соли.
Предметом настоящего изобретения также являются фармацевтические соединения, включающие, в качестве действующего начала, как минимум один из указанных выше медикаментов.
Указанные составы могут применяться внутрь, ректальным путем, парентеральным путем или локально нанесением на кожу или на слизистые оболочки, однако их предпочтительно применять внутрь.
Указанные составы могут быть твердыми или жидкими и иметь любую фармацевтическую форму, широко применяемую при лечении человека, как, например, простые или дражевидные таблетки, капсулы, гранулы, суппозитории, препараты для инъекций, мази, кремы, гели; они производятся обычными методами. Действующее начало (действующие начала) вводится в основы, обычно используемые при изготовлении фармацевтических составов, такие как тальк, аравийская камедь, лактоза, амиден, стеарат магния, какао-масло, водные или безводные связующие, жиры животного или растительного происхождения, производные парафина, гликоли, различные увлажняющие, диспергирующие или эмульсионные агенты, консерванты.
Указанные составы могут также иметь форму порошка, предназначенного для растворения перед приемом в соответствующем связующем, например в стерильной апирогенной воде.
Обычно применяемые дозы зависят от заболевания, которое предстоит лечить, от особенностей больного, от способа применения и от самого продукта. В случае продукта Примера 1 они могут составлять, например, от 50 до 300 мг в день для взрослых при приеме внутрь.
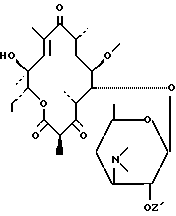
Предметом настоящего изобретения также является метод получения соединений формулы (I), отличающийся тем, что соединение формулы (II):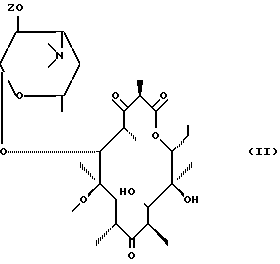
в котором Z' представляет собой остаток карбоновой кислоты, включающий до 18 атомов углерода, подвергают воздействию агента, способного выборочно активировать гидроксил в положении 11 для получения соединения формулы (III):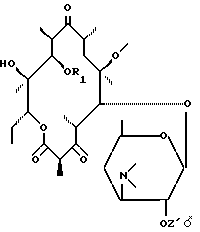
в которой R1 представляет собой остаток активирующей группы, который подвергают воздействию основания для получения соединения формулы (IV):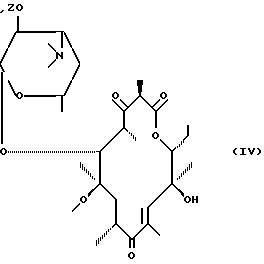
после чего соединение формулы (IV) подвергают:
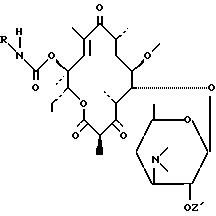
либо воздействию соединения формулы (V):
R-N=C=O
в которой R имеет вышеуказанное значение, для получения соединения формулы (VI):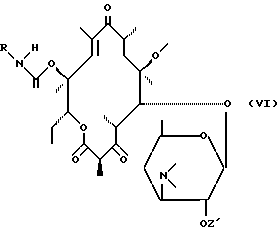
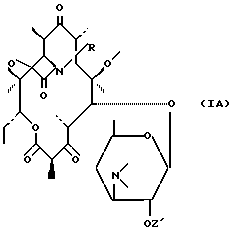
которое циклизируется либо самопроизвольно путем нагревания, либо подвергается воздействию циклизирующего средства для получения соединения формулы (IA):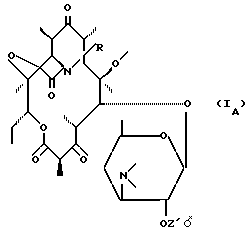
соответствующего продукту формулы (I), в которой Z не представляет собой атом водорода;
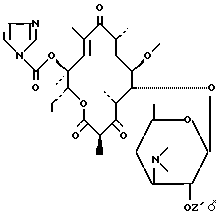
либо воздействию карбонилдиимидазола для получения соединения формулы (VII):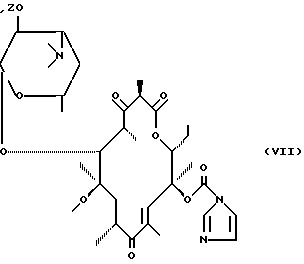
затем воздействию соединения формулы (VIII):
RNH2
в которой R имеет вышеуказанное значение, для получения вышеуказанного соединения формулы (VI), которое самопроизвольно циклизируется путем нагревания, либо подвергается воздействию циклизирующего средства для получения соответствующего соединения формулы (IA), после чего, при необходимости, указанное соединения формулы (IA) подвергается воздействию средства выделения гидроксильной функциональной группы в положении 2' и/или, при необходимости, воздействию кислоты для получения соли.
В предпочтительных условиях применения метода, являющегося предметом настоящего изобретения:
веществом, способным выборочно активировать гидроксил в положении 11, является сульфоновый ангидрид, такой как метансульфоновый, паратолуолсульфоновый или трифторметансульфоновый ангидрид;
основанием, способным создать двойную связь 10(11), является диазабициклоундецилен, например ДБУ (1,8-диазабицикло [5-4-0] ундецил-7-ен), диазабициклононен, 2,6-лутидин, 2,4,6-коллидин или тетраметилгуанидин;
реакция между соединением формулы (IV) и соединением формулы (V) производится в присутствии основания, такого как пиридин, триэтиламин, морфолин, N-метилморфолин, причем циклизация соединения формулы (VI) происходит либо самопроизвольно, либо путем нагревания при температуре от 50oC до 100oC;
реакция между соединением формулы (IV) и карбонилдиимидазолом протекает в присутствии основания, такого как гидрид натрия, триэтиламин, карбонат, бикарбонат натрия или калия, или в отсутствие основания в растворителе, таком как метиленхлорид, тетрагидрофуран или диметилформамид;
реакция между соединением формулы (VII) и соединением RNH2 протекает в среде растворителя, такого как, например, ацетонитрил, диметилформамид или тетрагидрофуран, диметоксиэтан или диметилсульфоксид, при этом циклизация соединения формулы (VI) производится, как правило, в ходе реакции или же за счет воздействия на выделенное соединение формулы (VI) таким основанием, как тетрабутилат калия, в среде растворителя, такого как тетрагидрофуран;
гидролиз сложноэфирной функциональной группы в положении 2' производится с помощью метанола или водного раствора хлористоводородной кислоты;
солеобразование осуществляется обычными методами с использованием кислот.
Соединениями формулы (II), используемыми в качестве исходных продуктов, являются общеизвестные продукты, которые могут быть получены как описано в патентной заявке ЕЭС 0.487.411.
Соединениями формул RN=C=0 и RNH2 также являются общеизвестные продукты.
Соединения формулы RNH2 могут быть получены, например методом, описанным в Ann. Chem. 690-98-114.
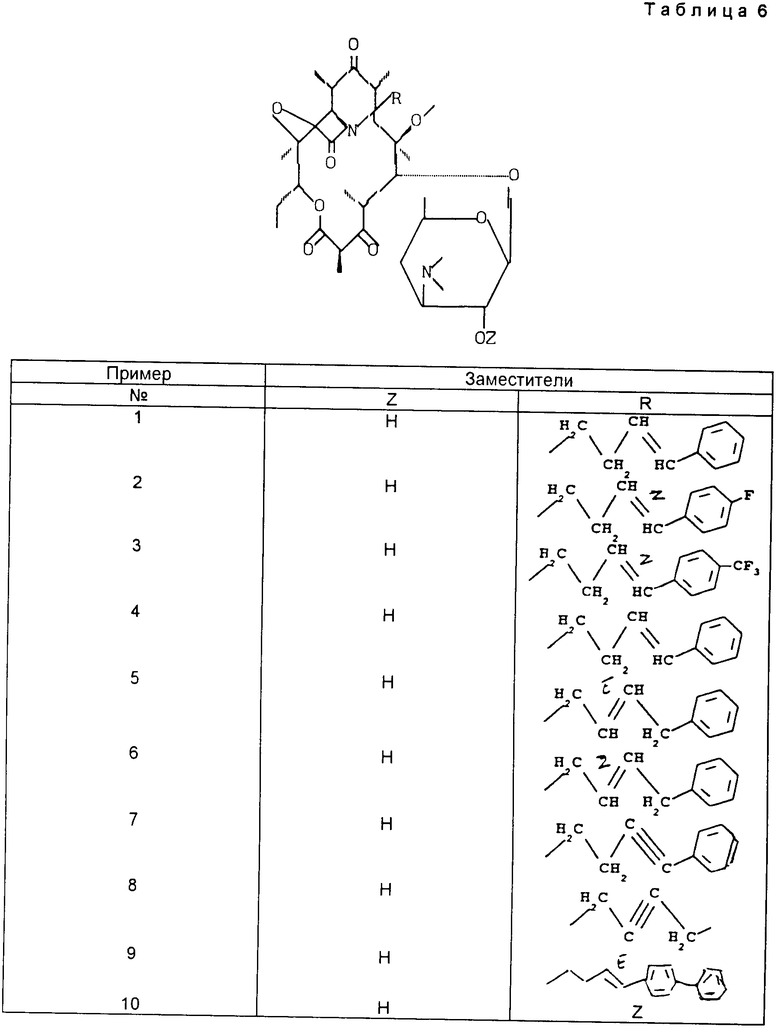
Пример 1. 11,12-дидеокси 3-де ((2,6-дидеокси 3-C-метил 3-О-метил α -L-рибогексопиранозил) окси) 6-0-метил 3-оксо 12,11-(оксикарбонил((4-фенил 3-бутенил) имино)) эритромицин.
Этап А: 2'-ацетат 3-де ((2,6-дидеокси 3-С-метил 3-О- метил α -L-рибогексопиранозил)окси) 6-О-метил 11-O-(метилсульфонил) 3-оксо эритромицина.
В среде азота при взбалтывании в 17 г 2'-ацетата 3-де ((2,6-дидеокси 3-C-метил 3-О-метил α -L-рибогексопиранозил)окси) 6-O-метил 3-оксоэритромицина добавляют 100 мл пиридина. Затем полученную смесь охлаждают до +10oC и добавляют 11,9 r метансульфонового ангидрида. После этого температуре дают подняться до уровня комнатной и выдерживают смесь, взбалтывая, в течение 5 ч. Полученный осадок фильтруют. Затем производят концентрирование, обрабатывают водой и экстрагируют с использованием этилацетата. Органические фазы промывают водой, высушивают, фильтруют и концентрируют. Таким образом получают 20,9 г сырого искомого продукта, которые подвергают очистке путем солеобразования с использованием щавелевой кислоты и выделения основания с помощью аммиака. В результате получают 15, 16 г искомого продукта (tпл 210 - 212oC).
Этап Б: 2'-ацетат 11-деокси 10,11-дидегидро 3-де (2,6- дидеокси 3-C-метил 3-O-метил α -L-рибогексопиранозил) окси) 6-O- метил 3-оксо эритромицина.
При взбалтывании вводят 8,26 г продукта, полученного на Этапе А, в 35 мл ацетона. После этого по капле добавляют 2,19 мл ДБУ. Взбалтывание при комнатной температуре продолжают еще в течение 20 ч. Затем реакционную смесь обрабатывают метиленхлоридом. Органические фазы промывают водой, высушивают на сульфате натрия, фильтруют и концентрируют. Таким образом получают 10 г продукта, который обрабатывают эфиром. Затем выполняются центрифугирование и промывка этиловым эфиром. В результате получают 6,33 г искомого продукта (tпл 230 - 232oC).
Этап В: 2'-ацетат11-деокси] 10,11-дидегидро 3-де(2,6- дидеокси 3-C-метил 3-O-метил α -L-рибогексопиранозил) окси) 12-О-((1Н- имидазол-1-ил)карбонил) 6-O-метил 3-оксо эритромицина.
96 мг 50-процентного раствора гидрида натрия в масле вводят в 15 мл тетрагидрофурана. Полученную суспензию охлаждают до 0oC и по капле вводят раствор 611 мг продукта, полученного на предыдущем этапе, в 17 мл тетрагидрофурана. Затем при температуре 0oC вводят раствор 486 мг карбонилдиимидазола в 15 мл тетрагидрофурана. Взбалтывание продолжают в течение 4 ч 30 мин. После этого температуре реакционной смеси дают подняться до уровня комнатной и выполняют фильтрацию и концентрирование. Затем обрабатывают этилацетатом промывают дигидрогенофосфатом натрия, экстрагируют с использованием этилацетата, высушивают, фильтруют и концентрируют. В результате получают 852 мг искомого продукта.
Этап Г: 2'-ацетат 11,12-дидеокси 3-де ((2,6-дидеокси 3-C-метил 3-O-метил α -L-рибогексопиранозил) окси) 6-O-метил 3-оксо 12,11- (оксикарбонил ((4-фенил 3-бутенил) имино)) эритромицина;
В течение 5 ч 30 мин при температуре +55oC взбалтывают смесь, состоящую из 0,9 г 2'-ацетат 11-деокси 10,11-дидегидро 3-де ((2,6 - дидеокси 3-C-метил 3-O-метил α -L-рибогексопиранозил)окси) 12-O-((1Н-имидазол-1-ил)карбонил) 6-O-метил 3-оксо эритромицина, 3 мл ацетонитрила и 0,6 г 4-фенил 3-бутениламина. Затем реакционную смесь вливают в водный раствор дигидрогенофосфата натрия (молярная концентрация = 0,5 моль/л), экстрагируют с использованием этилацетата, промывают водой, высушивают, фильтруют и концентрируют. Таким образом получают 0,9 г вязкой жидкости, которую подвергают хроматографии на двуокиси кремния (элюант : этилацетат/триэтиламин (96 : 4)). Гомогенные фазы собирают в CCM* и концентрируют (CCM - тонкослойная хроматография). В результате получают 0,32 г продукта, который сгущают гексаном, центрифугируют и высушивают при температуре +70oC. Таким образом выделяют 0,215 r искомого продукта (tпл 201-203oC).
Этап Д: 11,12-дидеокси 3-де((2,6-дидеокси 3-C-метил 3-O-метил α -L-рибогексопиранозил)окси) 6-О-метил 3-оксо 12, 11- (оксикарбонил ((4- фенил 3-бутенил) имина)) эритромицина.
Раствор, включающий 0,194 г продукта, полученного на Этапе А, и 6 мг метанола взбалтывают в течение 15 ч при комнатной температуре. Затем метанол выпаривают с помощью аппарата "Ротовапор". Полученный продукт подвергают хроматографии на двуокиси кремния (элюант : метиленхлорид/метанол/ аммоний (95 : 5 : 0,2)). Гомогенные фазы собирают, концентрируют, высушивают, фильтруют и концентрируют. Таким образом получают 0,155 г продукта; который подвергают центрифугированию и высушивают при температуре +70oC. В результате получают 0,124 г целевого продукта (tпл 257-259oC).
При выполнении действий, указанных в Примере 1, с использованием соответствующих аминов были получены следующие продукты:
Пример 2. ((Z) 11,12-дидеокси 3-де ((2,6-дидеокси 3-C-метил 3-О-метил α -L-рибогексопиранозил)окси) 6-0-метил 3-оксо 12,11-(оксикарбонил ((4-(4-фторфенил 3-бутенил)имино)) эритромицин
tпл 222oC.
Пример 3. (Z) 11,12-дидеокси 3-де ((2,6-дидеокси 3-C-метил 3-О- метил α -L-рибогексопиранозил)окси) 6-O-метил 3-оксо 12,11- (оксикарбонил((4-трифторметил 3-бутенил)имино))эритромицин
tпл 230oC
Пример 4. (цис) 11,12-дидеокси 3-де ((2,6-дидеокси 3-С-метил 3-О-метил α -L-рибогексопиранозил)окси) 6-O-метил 3-оксо 12,11-(оксикарбонил ((4- фенил 3-бутенил)имино))эритромицин
tпл 220 - 225oC
[α]D = + 10,5°C (c = 0,9 % CHCl3)
Пример 5. (E) 11,12- дидеокси 3-де ((2,6-дидеокси 3-C-метил 3-О-метил α -L-рибогексопиранозил) окси) 6-O-метил 3-оксо 12,11-(оксикарбонил((4-фенил 2-бутенил)имино)) эритромицин
tпл ≈ 78oC
Пример 6. (Z) 11,12-дидеокси 3-де ((2,6-дидеокси 3-C-метил 3-О-метил α -L-рибогексопираноэил)окси) 6-O-метил 3-оксо 12,11-(оксикарбонил((4- фенил 3-бутенил)имино))эритромицин
tпл 220oC
[α]D = + 16°C (c = 1 % CHCl3)
Пример 7. 11,12-дидеокси 3-де ((2,6-дидеокси 3-C-метил 3-О-метил α -L-рибогексопиранозил)окси) 6-O-метил 3-оксо 12,11-(оксикарбонил ((4-фенил 3-бутенил)имино)) эритромицин
tпл 252oC
Пример 8. 11,12-дидеокси 3-де ((2,6-дидеокси 3-C-метил 3-О-метил α -L-рибогексопиранозил)окси) 6-O-метил 3-оксо 12,11- (оксикарбонил ((4-фенил 2-бутенил)имино)) эритромицин
tпл ≈ 98oC
Пример 9. (Е) 11,12-дидеокси 3-де((2,6-дидеокси 3-C-метил 3-O-метил α - -L-рибогексопиранозил) окси) 6-O-метил 3-оксо 12,11-(оксикарбонил ((4-(1,2'-бифенил 4-ил)3-бутенил)имино))эритромицин
tпл 215 - 217oC
[α]D = - 46,5°C (c = 1% CHCl3)
Пример 10. (Z) 11,12-дидеокси 3-де((2,6-дидеокси 3-C-метил 3-O-метил α -L-рибогексопиранозил)окси) 6-О-метил 3-оксо 12,11-(оксикарбонил((4- (1,2'-6ифенил 4-ил)3-бутенил)имино))эритромицин
tпл 133-137oC
[α]D = - 2,5°C (c = 1 % CHCl3)
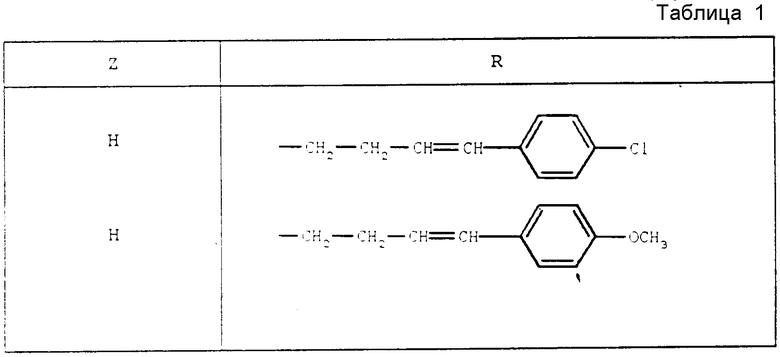
Действуя идентичным образом, были получены следующие продукты, приведенные в табл. 1.
Приготовление 1: [4-фторфенил-4-ил] 3-бутениламин.
Этап А : N-[[4-фторфенил-4-ил] 3-бутенил]фталимид.
Суспензию, включающую 150 мл тетрагидрофурана, 6 г 4-фторбензальдегида и 25,6 г бромида трифенилфосфония N-(3-бромпропил)фталимида, охлаждают до -40oC. Затем вводят 5,42 г трет-бутилата калия. Температуре дают подняться до -15oC и выдерживают при взбалтывании в течение 1 ч.
Затем смесь выливают на лед, экстрагируют с использованием этилацетата, промывают водой, высушивают органические фазы на Na2SO4, фильтруют и концентрируют. Таким образом получают 26,7 г продукта, который растворяют в метиленхлориде и подвергают хроматографии на двуокиси кремния (элюант : этилацетат/гексан (1 : 9)). Затем выполняют концентрацию при пониженном давлении и выделяют 5,2 г искомого продукта (tпл 94oC).
Микроанализ.
Рассчитано, %: C 73,40; H 4,77; N 4,74; F 4,43
Получено, %: C 73,2; H 4,8; N 4,6; F 6,6
Этап Б: [4-фторфенил-4-ил] 3-бутениламин.
В течение 16 ч при температуре 0oC взбалтывают смесь, включающую 200 мл этанола, 5,5 г продукта, полученного на Этапе А, и 1,5 мл гидразингидрата. После этого температуре дают подняться до уровня комнатной, растворитель выпаривают, остаток забирают в эфире, подкисляют до pH = 1 c помощью 2 н. раствора хлористоводородной кислоты, растворитель удаляют, забирают в воде, добавляют раствор карбоната натрия, экстрагируют с использованием эфира и выпаривают растворители. После хроматографии осадка на двуокиси кремния (элюант : метиленхлорид/метанол/аммоний (9 : 1 : 0,5)) получают 2,2 г целевого продукта.
Приготовление 2: [4-трифторметилфенил-4-ил] 3-бутениламин.
Этап А: N-[[4-трифторметил-4-ил]3-бутенил]фталимид.
Операции выполняются как на Этапе А Приготовления 1 с использованием, на начальном этапе, 6 г 4-(трифторметил) бензальдегида. В результате получают 4 г искомого продукта (tпл 88oC).
Этап Б: [4-трифторметилфенил-4-ил] 3-бутениламин.
Операции выполняются как на Этапе Б Приготовления 1 с использованием, на начальном этапе, 2,13 г продукта, полученного на Этапе А, и 0,84 см3 гидразингидрата. В результате получают 0,850 г целевого амина.
Приготовление 3: 4-[(1,1'-6ифенил)4-ил] 3-бутениламин.
Этап А:N-[4[(1,1'-бифенил)4-ил] 3-бутенил]фталимид.
Суспензию, включающую 150 мл тетрагидрофурана, 5,46 г 4-фенилбензальдегида и 15,9 бромидтрифенилфосфония N-(3-бромпропил) фталимида, охлаждают до температуры -40oC. Затем вводят 3,37 г тетрабутилата калия, дают температуре дают подняться до -15oC и выдерживают в течение 1 ч при взбалтывании при температуре -15oC.
Затем смесь выливают на лед, экстрагируют с использованием этилацетата, промывают водой, высушивают органические фазы на Na2SO4, фильтруют и концентрируют. Таким образом получают 19 г продукта, который растворяют в метиленхлориде и подвергают хроматографии на двуокиси кремния (элюант : этилацетат/гексан (3 : 7)). Затем выполняют концентрацию, сгущают гексаном, центрифугируют и высушивают при пониженном давлении и выделяют 8,5 г искомого продукта (tпл 112≈114oC).
Микроанализ.
Рассчитано, %: C 81,56; H 5,42; N 3,96.
Получено, %: C 81,4; H 5,3; N 3,8.
Этап Б: 4-[(1,1'бифенил)-4-ил] 3-бутениламин.
Смесь, включающую 280 мл этанола, 7,9 г продукта, полученного на Этапе А, и 1,3 мл гидразингидрата, доводят до температуры кипения. Температуре дают опуститься до уровня комнатной, фильтруют полученный осадок и промывают этанолом. После этого производят концентрирование, вливают в 2 н. раствор хлористоводородной кислоты и экстрагируют с использованием этилацетата. Затем органические фазы промывают водой, высушивают, фильтруют и концентрируют при пониженном давлении. В результате получают 2,89 г целевого продукта (tпл 188≈194oC).
Пример фармацевтического соединения. Были приготовлены таблетки, отвечающие следующей формуле:
Продукт примера 1 - 150 мг
Основа для готовой таблетки - 1 г
Деталировка основы - крахмал, тальк, стеарат магния.
Фармакологические исследования продуктов изобретения. Метод разведения в жидкой среде.
Подготавливают серию пробирок, по которым распределяется одинаковое количество стерильной питательной среды. Затем в каждую пробирку вливают возрастающее количество исследуемого продукта, после чего каждая пробирка засеивается бактериальным штаммом.
После инкубации в сушильном шкафу в течение 24 ч при температуре +37oC ингибирование роста оценивается путем просвечивания того, что дает возможность определить минимальную ингибирующую концентрацию (M.И.К.), выражаемую в микрограммах на см3.
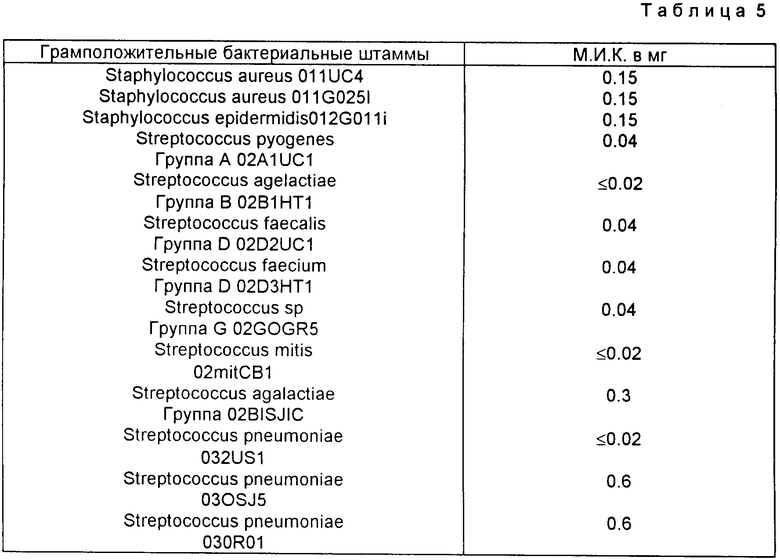
Для продукта Примера 1 были получены нижеследующие результаты (считывание результатов производилось через 24 ч):
Фармакологическое исследование. Метод разведения в жидкой фазе.
Готовят серию пробирок, по которым распределяют одинаковое количество стерильной питательной среды и вводят возрастающее количество исследуемого продукта в каждую пробирку. Затем, каждая пробирка засевается бактериальным штаммом и после инкубации в течение 24 ч в сушильном шкафу при температуре 37oC, ингибирование роста оценивают путем просвечивания, что позволяет определить минимальную ингибирующую концентрацию (M.И.К.), выраженную в микрограмках на миллилитр.
Для соединений примеров 5, 7 и 10 были получены результаты (считывание происходит после 24 ч), приведенные ниже.
Приложение; результаты испытаний соединений примеров 5, 7 и 10, приведенных в табл. 4 - 6. Химическая структура соединений согласно примерам описания приведена в табл. 4.


Описывается производные эритромицина формула 1, где 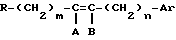 , m = 1,2, n = 1,2, Ar-фенил, возможно замещенный атомами галогена, алкил - или алкоксигруппой, CF3-группой или фенилом, A=B=H или A и B вместе образуют углерод- углеродную связь, Z = H или остаток карбоновой кислоты C1-C18, обладающие антибиотической активностью, в отношении грамположительных бактерий. Соединения + получают способом, включающим стадии ацилирования, аминирования и циклизации. Фармацевтическая композиция в качестве активного начала содержит соединение 1 в эффективном количестве, поэтому может быть использована в качестве медикаментов при лечении инфекций с чувствительными микробами. 3 с. и 11 з.п. ф-лы, 6 табл.
, m = 1,2, n = 1,2, Ar-фенил, возможно замещенный атомами галогена, алкил - или алкоксигруппой, CF3-группой или фенилом, A=B=H или A и B вместе образуют углерод- углеродную связь, Z = H или остаток карбоновой кислоты C1-C18, обладающие антибиотической активностью, в отношении грамположительных бактерий. Соединения + получают способом, включающим стадии ацилирования, аминирования и циклизации. Фармацевтическая композиция в качестве активного начала содержит соединение 1 в эффективном количестве, поэтому может быть использована в качестве медикаментов при лечении инфекций с чувствительными микробами. 3 с. и 11 з.п. ф-лы, 6 табл. 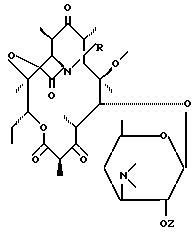 \
\
Производные эритромицина общей формулы I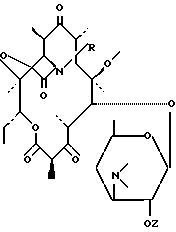
где R - радикал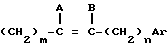
где m = 1 или 2;
n = 1 или 2;
Ar - фенил, возможно замещенный одним или несколькими атомами, алкил- или алкоксигруппой, CF3-группой или фенилом;
A = B - H или A и B вместе образуют третью углерод-углеродную связь;
Z - H или остаток карбоновой кислоты, включающей до 18 атомов углерода.
| Ann.Chem | |||
| Регистратор для дел | 1925 |
|
SU690A1 |
| EP, 0487411, C 07 H 17/08, 1990. | |||

