Настоящее изобретение касается новых производных эритромицина, способа их получения и применения в качестве медикаментов.
Объектом изобретения являются соединения формулы (I):
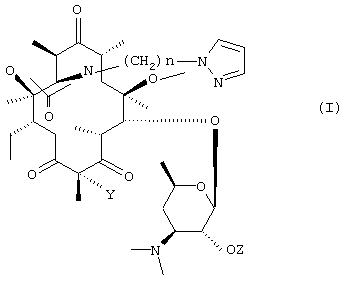
в которой:
Y обозначает атом водорода или фтора;
n обозначает целое число от 1 до 8;
Z обозначает атом водорода или остаток карбоновой кислоты, в которой пиразольный цикл замещен гетероарилом, содержащим один атом азота, а также их солевые аддукты с кислотами.
В качестве примера солевых аддуктов настоящих производных с минеральными или органическими кислотами могут быть названы соли, образованные со следующими кислотами: уксусная, пропионовая, трифторуксусная, малеиновая, винная, метансульфоновая, бензолсульфоновая, п-толуолсульфоновая, хлористоводородная, бромистоводородная, иодистоводородная, серная, фосфорная и особенно применимы стеариновая, этилкоричная или лаурилсульфоновая.
Более конкретно, настоящее изобретение относится к соединениям формулы 1, в которой Z является атомом водорода, к соединениям I, где n равно 4 и к соединениям I, у которых радикал
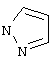
замещен радикалом
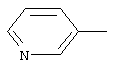
Еще более конкретно, настоящее изобретение относится к соединениям формулы I, в которой Z является атомом водорода.
Из предпочтительных соединений следует особо назвать соединения, получение которых приведено ниже в экспериментальной части, и из последних следует особо выделить соединение, полученное в примере 1.
Соединения общей формулы I обладают очень высокой антибиотической активностью на грамположительных бактериальных штаммах стафилококков, стрептококков и пневмококков.
Таким образом, соединения настоящего изобретения могут быть использованы в качестве лекарственных средств при лечении инфекций, носители которых восприимчивы к лекарственным средствам, а именно при лечении стафилококковых инфекций таких как стафилококковый сепсис, злокачественных стафилококковых инфекций лица и кожи, пиодермитов, септических или гнойных ран, фурункулов, карбункулов, флегмон, рож и угрей, таких стафилококковых инфекций как первичные или послегрипповые острые ангины, бронхопневмонии, легочные нагноения, таких стрептококковых инфекций как острые ангины, отиты, синуситы, скарлатина, таких пневмококковых инфекций как пневмонии, бронхиты, а также бруцеллез, дифтерия и гонококковые инфекции.
Соединения настоящего изобретения обладают также активностью в отношении инфекций, вызываемых такими бактериями как Haemophilus influenzae, Rickettsies, Mycoplasma pneumoniae, Chlamydia, Legionella, Ureaplasma, Toxoplasma или бактериями семейства Mycobacterium.
Объектом изобретения является также использование в качестве лекарственных средств и, более конкретно, в качестве антибиотических лекарственных средств, определенных выше соединений формулы I, а также их фармацевтически приемлемых солевых аддуктов с минеральными и органическими кислотами.
Более конкретно, изобретение предлагает использовать в качестве лекарственного средства, а именно антибиотического лекарственного средства, соединение примера 1 и его фармацевтически приемлемые соли.
Изобретение относится к также фармацевтической композиции, содержащей в качестве активного начала по меньшей мере одно из названных выше лекарственных средств.
Эти композиции могут применяться перорально, ректально, парентерально или местно путем нанесения на кожу или слизистые ткани, однако, предпочтительным способом применения является пероральный.
Названные композиции могут быть твердыми или жидкими и выпускаться в обычно применяемых в медицине лекарственных формах, таких, например, как приготовленные обычными способами простые или дражевидные таблетки, желатиновые капсулы, гранулы, свечи, препараты для инъекций, мази, кремы, гели. Активное начало или активные начала могут вводиться в наполнители, обычно применяемые в фармацевтических композициях, такие как тальк, гуммиарабик, лактоза, крахмал, стеарат магния, масло какао, водные и неводные разбавители, животные и растительные жиры, парафиновые производные, гликоли, различные увлажняющие агенты, диспергаторы или эмульгаторы, консерванты.
Композиции могут также находиться в виде порошка, предназначенного для разбавления в момент применения в подходящем разбавителе, например в апирогенной стерильной воде.
Применяемая доза зависит от характера заболевания, конкретного больного, способа применения и применяемого препарата. Эта доза может, например, составлять от 50 до 3000 мг в сутки для взрослого при пероральном применении продукта примера 1.
Изобретение относится также к способу получения соединения формулы I, заключающемуся в том, что соединение формулы II:
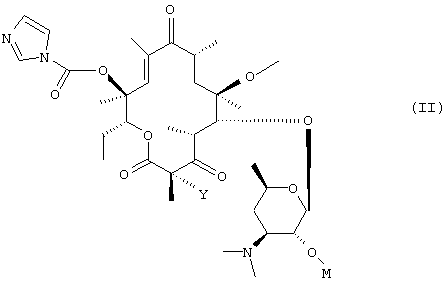
в которой Y имеет указанное выше значение и М обозначает остаток кислоты обрабатывают соединением формулы III:
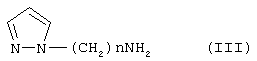
в которой гетероциклический радикал может быть замещен, как указано выше, с получением соединения формулы 1, в которой Z представляет собой остаток кислоты и затем, при необходимости, соединение 1 подвергают действию агента высвобождения гидроксила в положении 2' агента с целью получения соответствующего соединения формулы I, в котором Z является атомом водорода с последующим, при желании, добавлением кислоты для получения соли.
При этом реакцию соединения формулы II с соединением формулы III проводят в растворителе таком, например, как ацетонитрил, диметилформамид или тетрагидрофуран, диметоксиэтан или диметилсульфоксид;
гидролиз сложноэфирной функции в положении 2' осуществляют с помощью метанола или водной соляной кислоты;
образование солей осуществляют с помощью кислот, применяя традиционные способы.
Соединения формулы II, у которых Y является атомом водорода и которые используются в качестве исходных соединений, описаны и заявлены в Европейской патентной заявке 0596902.
Соединения формулы II, у которых Y является атомом фтора, могут быть получены в соответствии со способом, приведенном ниже в экспериментальной части.
Изобретение относится также к новым химическим продуктам. представляющим собой соединения формулы III и, в частности, соединения формулы III, получение которых приведено в экспериментальной части.
Пример 1
11,12-дидезокси-3-де[(2,6-дидезокси-3-С-метил-3-0-метил-альфа-L-рибогексопиранозил)окси]-6-0-метил-3-оксо-12,11-[оксикарбонил[[4-[3-(3-пиридинил)-1H-пиразол-1-ил]бутил]имино]]-эритромицин.
Смесь 26 см3 ацетонитрила, 2,5 см3 воды, 5,13 г полученного ниже (приготовление 1) амина и 6,20 г 2'-ацетата и 12-(1Н-имидазол-1-илкарбоксилата)10,11-дидегидро-11-дезокси-3-де[(2,6-дидезокси-3-С-метил-3-0-метил-альфа-L-рибогексопиранозил)окси]-6-0-метил-3-оксо-эритромицина нагревают в течение 21 ч при 55°С, выливают реакционную смесь в воду, экстрагируют этилацетатом и сушат. Получают 8,02 г продукта, который вводят в 70 см3 метанола. Полученную реакционную смесь нагревают при температуре кипения с обратным холодильником в течение 1,5 ч. Полученный продукт хроматографируют на силикагеле, элюируя смесью хлористого метилена, метанола и гидроксида аммония в соотношении 95:5:0.5. Получают 3,59 г продукта с т.пл. 143-145°С.
Результаты полученного приготовления приведены в табл. 1.

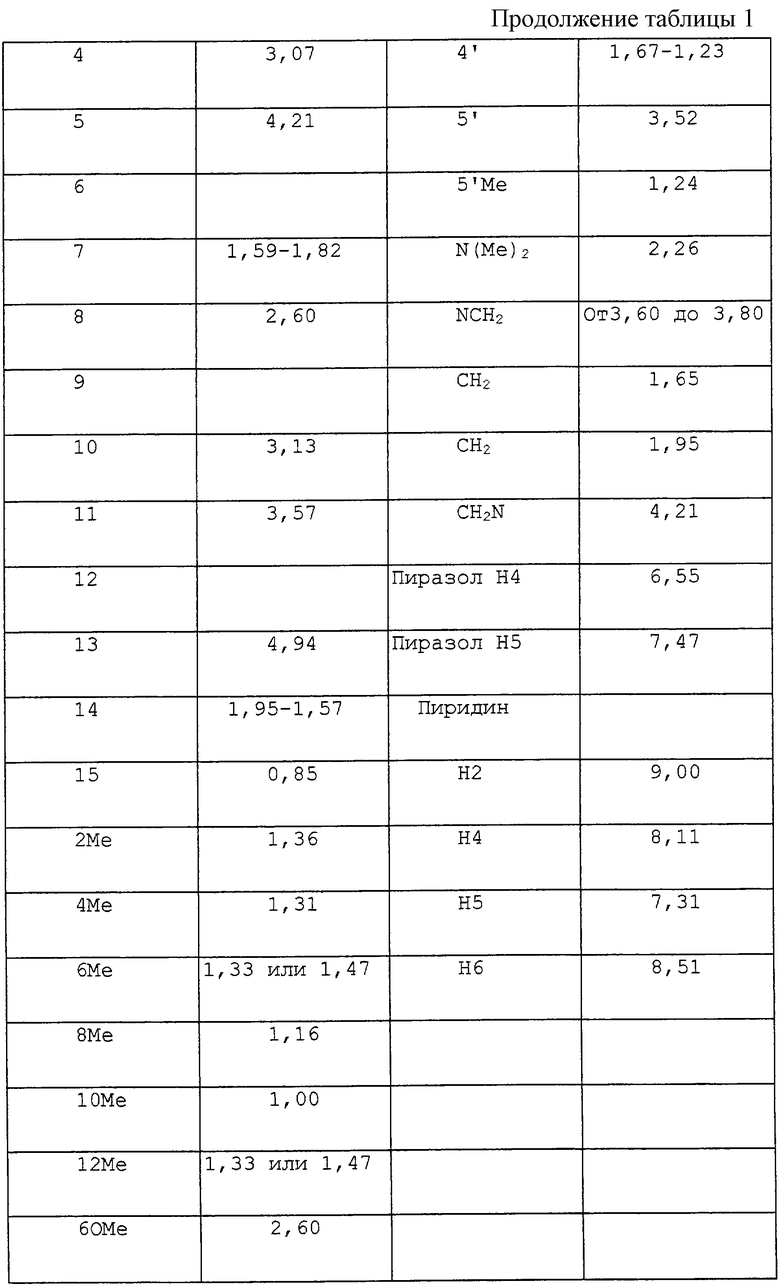
Приготовление 1:
3-(3-пиридинил)-1Н-пиразол-1-бутанамин
Стадия А:
2-[4-[3-пиридинил]-1Н-пиразол-1-ил]бутил]-1H-изоиндол-1,3(2Н)-дион.
15,45 г 3-(1H-пиразол-3-ил)пиридина, полученного по методу, описанному в СА 68р 95812д (1968), прибавляют по каплям в течение 1 ч к смеси 20 мл диметилформамида (ДМФ) и 6.13 г гидрида натрия, поддерживая температуру ниже 30°С или равную 30°С. Затем прибавляют по каплям раствор 29,90 г 2-(4-бромбутил)-1Н-изоиндол-1,3(2Н)-диона и 110 мл ДМФ, перемешивают 30 мин при комнатной температуре, концентрируют, вливают в 300 мл охлажденной до 10°С воды, экстрагируют этилацетатом, промывают водой, высушивают, фильтруют и концентрируют. Поглощают метиленхлоридом, сушат, фильтруют и концентрируют. Получают 35,87 г продукта, который кристаллизуют из диэтилового эфира, отжимают, промывают водой и сушат, получая 22,93 г целевого продукта.
Стадия В:
3-(3-пиридинил)-1H-пиразол-1-бутанамин
7 мл гидразин-гидрата добавляют к суспензии, содержащей 450 мл этанола и 22,33 г продукта стадии А и кипятят с обратным холодильником в течение 15 ч. После этого выпаривают этанол, перемешивают реакционную смесь с 200 мл этилацетата, промывают подсоленной водой, высушивают, фильтруют и концентрируют. Получают 9,60 г целевого продукта.
Пример 2
11,12-дидезокси-3-де[(2,6-дидезокси-3-С-метил-3-0-метил- альфа-L-рибогексопиранозил)окси}-2-фтор-6-0-метил-3-оксо-12,11-[оксикарбонил[[4-[3-(3-пиридинил)-1Н-пиразол-1-ил]бутил]имино]]-эритромицин.
Повторяя описанные выше операции, но исходя из соответствующего производного формулы II, содержащего фтор в положении 2, полученного как описано ниже, получают целевой продукт с т.пл.117-121°С.
Получение соединения формулы (II), в которой У означает атом фтора 2'-ацетокси-2-α-фтор-12-оксикарбонилимидазол)-11-дезокси-10,11-дидегидро-3-де[[2,6-дидезокси-3-С-метил-3-0-метил-альфа-L-рибогексопиранозил)окси]-6-0-метил-3-оксо-эритромицин.
Стадия А:
11-дезокси-10,11-дидегидро-3-де[(2,6-дидезокси-3-0-метил-альфа-L-рибогексопиранозил)окси]-6-0-метил-3-оксо-эритромицин
Смесь 8,722 г 2'-ацетата 11-дезокси-10,11-дидегидро-3-де[(2,6-дидезокси-3-0-метил-альфа-L-рибогексопиранозил)-окси]-6-0-метил-3-оксо-эритромицина и 350 мл безводного метанола (ЕР 596802) перемешивают 44 ч, получая 8,794 г целевого продукта.
Стадия В:
2'-триметилсилилокси-11-дезокси-10,11-дидегидро-3-де[(2,6-дидезокси-3-0-метил-альфа-L-рибогексопиранозил)-окси]-6-0-метил-3-оксо-эритромицин.
Смесь 3,08 г продукта, полученного на предыдущей стадии, 340 мг имидазола, 32 мл безводного тетрагидрофурана (ТГФ) и 1,06 мл гексаметилдисилилазана перемешивают в течение 4 суток при комнатной температуре, досуха упаривают и поглощают смесью 60 мл хлористого метилена и 60 мл 0,5 М кислого фосфата натрия. После этого реакционную смесь перемешивают 15 мин, декантируют, экстрагируют метиленхлоридом, высушивают и досуха упаривают, получая 3,345 г целевого продукта.
Стадия С:
2'-триметилсилилокси-2α-фтор-11-дезокси-10,11-дидегидро-3-де[(2,6-дидезокси-3-0-метил-альфа-L-рибогексопиранозил)окси]-6-0-метил-3-оксо-эритромицин.
1,24 мл 0,97 М раствора трет-бутилата калия в ТГФ добавляют при температуре -12°С в атмосфере аргона к раствору 668 мг 2'-триметилсилилокси-де-11-дезокси-10,11-дидегидро-3-де[(2,6-дидезокси-3-0-метил-альфа-L-рибогексопиранозил)-окси]-6-0-метил-3-оксо-эритромицина в 6,7 мл безводного ТГФ. Перемешивают 5 мин и добавляют 378 мг N-фтор-дибензолсульфонамида. Перемешивают 10 мин при -12°С и в течение 1,5 ч поднимают температуру до комнатной. После этого выделяют и очищают целевой продукт. Выход 695 мг.
Стадия D:
2α-фтор-11-дезокси-10,11-дидегидро-3-де[(2,6-дидезокси-3-0-метил-3-С-метил-альфа-L-рибогексопиранозил)окси]-6-0-метил-3-оксо-эритромицин.
Смесь 5,476 г продукта примера 2,50 мл ТГФ и 11,2 мл 1 М раствора тетрабутиламмонийфторида в ТГФ перемешивают в течение 3,5 ч, отгоняют растворитель, добавляют 37 мл этилацетата, 37 мл воды и 7,5 мл 20%-ного раствора аммиака, перемешивают еще 10 мин, декантируют и экстрагируют этилацетатом. Экстрактный раствор высушивают, фильтруют и досуха упаривают. Полученный продукт хроматографируют на силикагеле, элюируя смесью хлористого метилена и аммиачного метанола в соотношении 99:1, затем 98:2, 97:3, 96:4, 95:5. Получают 2,452 г целевого продукта.
Стадия Е:
2'-ацетокси-2-α-фтор-11-деэокси-10,11-дидегидро-3-де[(2,6-дидезокси-3-0-метил-альфа-L-рибогексопиранозил)-окси]-6-0-метил-3-оксо-аритромицин.
1,02 г продукта стадии А, 10 мл хлористого метилена и 241 μл уксусного ангидрида выдерживают при перемешивании в течение 3 час, упаривают и добавляют 10 мл воды и 10 мл этилацетата. После этого температуру реакционной смеси поднимают до комнатной в течение 1 часа при перемешивании, декантируют, высушивают и упаривают. Получают 1,01 г целевого продукта.
Стадия F:
2'-ацетокси-2-α-фтор-12-(оксикарбонилимидазол)-11-дезокси-10,11-дидегидро-3-де[(2,6-дидезокси-3-С-метил-3-0-метил-альфа-L-рибогексопиранозил)окси]-6-0-метил-3-оксо-эритромицин.
0,388 г карбонилдиинидазола и 24 μл 1,8-диазабицикло[5.4.0]ундец-7-ена добавляют при 0°С к раствору 1,01 г продукта предыдущей стадии в 10 мл безводного ТГФ, отгоняют ТГФ и добавляют 10 мл воды и 10 мл этилацетата. Реакционную смесь после этого перемешивают 10 мин, экстрагируют, высушивают и упаривают, получая 0,902 г неочищенного целевого продукта. Последний хроматографируют, элюируя смесью этилацетат-триэтиламин (96:4), получая 0,573 целевого продукта.
Пример 3
Повторяя операции примера I и используя 4-(3-пиридинил)-1Н-пиразол-1-аминобутан получают 11,12-дидезокси-3-де[(2,6-дидезокси-3-С-метил-3-0-метил-альфа-L-рибогексопиранозил)окси]-6-0-метил-3-оксо-12,11-[оксикарбонил[[4-[4-(3-пиридинил)-1Н-пиразол-1-ил]бутил]имино]]-эритромицин.
Масс-спектр МН+=812+
Спектр ЯМР (300 МГц, СDСl3)
Н2: 3,84 мд; Н4: 3006 мд; Н5: 4,22 мд; Н7:1,58,1,83 мд; Н8: 2,58 мд; Н10: 3,12 мд; Н11: 3,56 мд; Н13: 4,92 мд; Н14: 1,55,1,94 мд; Н15: 0,81 мд; 2Ме: 1,35 мд; 4Ме: 1,29 мд; 6Ме: 1,32 или 1,46 мд; 8Ме: 1,16 мд; 10Ме: 1,01 мд; 12Ме: 1,32 или 1,46; 60Ме: 2,6 мд; 1': 4,27 мд; 2': 3,17 мд; 3': 2,44 мд; 4': 1,67 и 1,24 мд; 5': 3,55 мд; Nme2: 2,26 мд; NCH3: 3,69 мд; СН2: 1,64,1,94 мд; CH2N: 4,19 мд; пиразол: 7,77 мд; пиридин: 8,76, 7,75, 7,27, 8,44 мд.
Масс-спектр
812+:MH+
850+:MK+
Исходный амин был получен в соответствии с Приготовлением 1 исходя из продукта, полученного по следующей схеме:
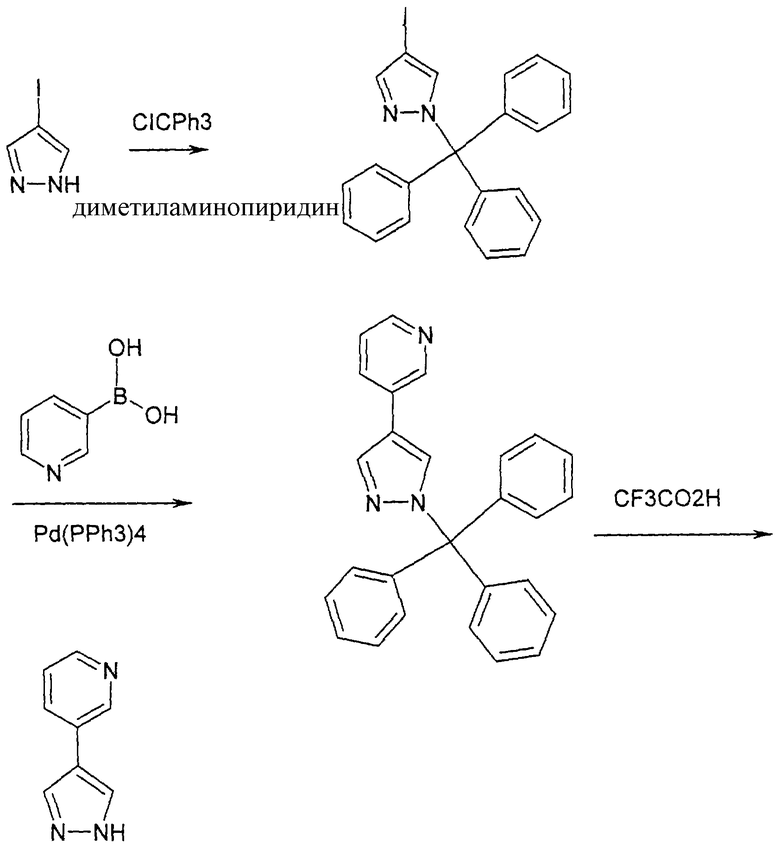
Пример 4
11,12-дидезокси-3-де[(2,6-дидезокси-3-С-метил-3-0-метил-альфа-L-рибогексопиранозил)окси]-2-фтор-6-0-метил-3-оксо-12,11-[оксикарбонил[[4-[4-(3-пиридинил)-1Н-пиразол-1-ил]бутил]имино]]-эритромицин
Спектр ЯМР (300 МГц, СDСl3)
Н4: 3,53 мд; Н5: 4,05 мд; Н7: 1,51,1,88 мд; Н8: 2,61 мд; Н10: 3,10 мд; Н11: 3,42 мд; Н13: 4,86 мд; Н14: 1,63,1,95 мд; Н15: 0,85 мд; 2Ме: 1,75 мд; 4Ме: 1,29 мд; 6Ме: 1,31 или 1,49 мд; 8Ме: 1,17 мд; 10Ме: 1,00 мд; 12Ме: 1,31 или 1,49; 60Ме: 2,51 мд; 1': 4,30 мд; 2': 3,19 мд; 3': 2,48 мд; 4': 1,68 и 1,26 мд; 5': 3,53 мд; 5Ме: 1,24; Nme2: 2,28 мд; NCH2: от 3,55 до 3,80 мд; CH2: 1,61,1,93 мд; CH2N: 4,19 мд; пиразол: 7,75, 7,78 мд; пиридин: 8,77, 7,77, 7,27, 8,44 мд.
Масс-спектр
830+: МН+
158+: дезаминир.
673+: 830+-158+H
Пример фармацевтической композиции
Приготовлены таблетки, содержащие:
продукт примера 1 150 мг
экципиент в достаточном количестве до 1 г
Составные части наполнителя: крахмал, тальк, стеарат магния
Фармакологическое исследование продуктов изобретения
А - Метод разбавлении жидкой средой
Приготовляют ряд пробирок, в которые помещают одинаковые количества стерильной питательной среды. Затем в пробирки помещают возрастающие количества исследуемого продукта, после чего каждую пробирку осеменяют бактериальным штаммом. После инкубирования в течение 24 час в термостате при 37°С оценивают ингибирование роста путем просвечивания, что позволяет установить минимальные концентрации ингибирования, выраженные в μг/см3. Для продукта примера 1 получены следующие результаты (после выдержки в течение 24 час).
Использованные бактериальные штаммы были грамположительными. Результаты их использования приведены в таблице 2.
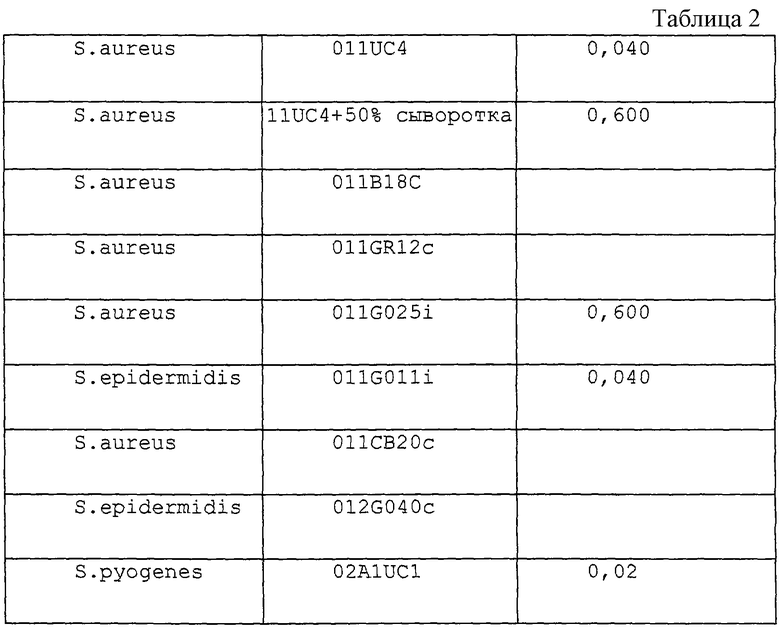
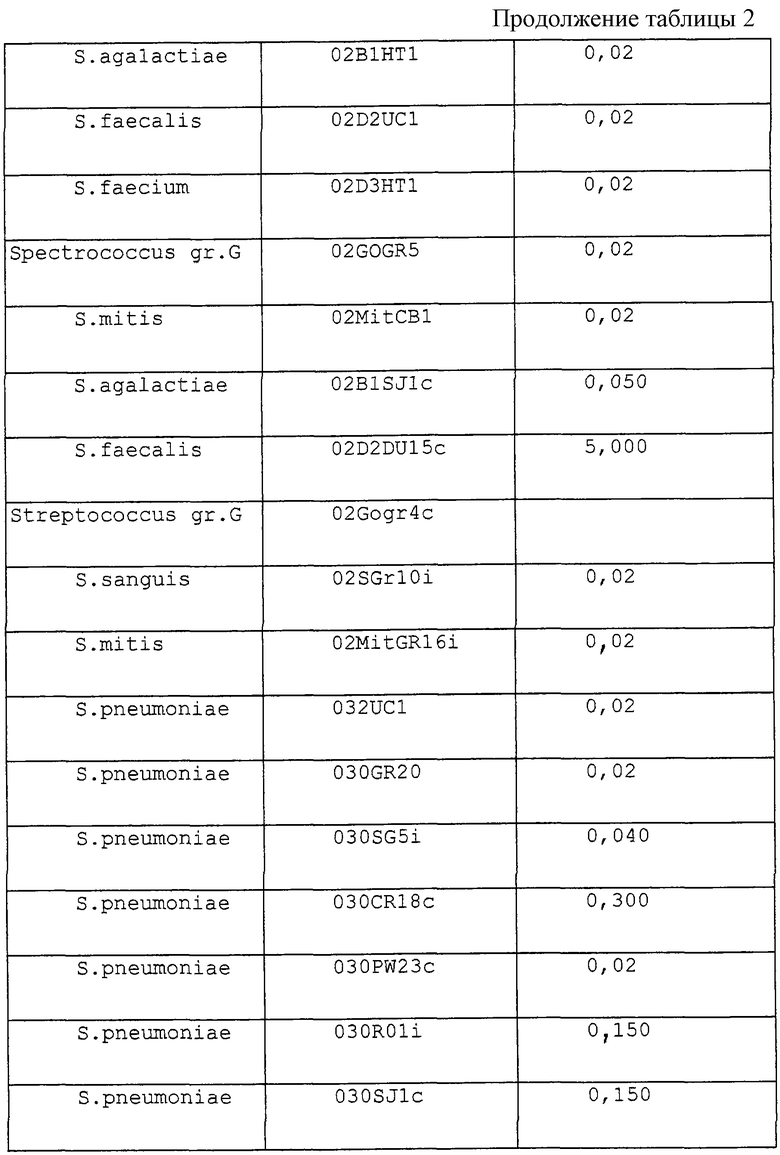
Наряду с этим, продукт примера 1 обладает, в частности, значительной активностью в отношении грамотрицательных бактериальных штаммов Haemophilus Influenzae 351 НТЗ, 35В12, 35СА1 и 351GR6.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ЭРИТРОМИЦИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ЭРИТРОМИЦИНА | 1993 |
|
RU2114859C1 |
| ПРОИЗВОДНЫЕ ЭРИТРОМИЦИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1993 |
|
RU2126416C1 |
| ПРОИЗВОДНЫЕ ЭРИТРОМИЦИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1995 |
|
RU2141965C1 |
| ПРОИЗВОДНЫЕ ЭРИТРОМИЦИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1995 |
|
RU2144036C1 |
| ПРОИЗВОДНЫЕ ЭРИТРОМИЦИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНТИБИОТИЧЕСКОЙ АКТИВНОСТЬЮ | 1991 |
|
RU2100367C1 |
| ФОСФОЛИПИДНЫЕ ПРОИЗВОДНЫЕ НУКЛЕОЗИДОВ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2104282C1 |
| КЕТОЛИДНЫЕ СОЕДИНЕНИЯ | 2011 |
|
RU2601550C2 |
| КЕТОЛИДНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ АНТИБАКТЕРИАЛЬНЫХ АГЕНТОВ | 2005 |
|
RU2397987C2 |
| ПРОИЗВОДНЫЕ 10,13,15-ТРИОКСАТРИЦИКЛО (9.2.1.1.)-ПЕНТАДЕКАНОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЕ ЭТИ СОЕДИНЕНИЯ ЛЕКАРСТВЕННЫЕ СРЕДСТВА | 1997 |
|
RU2181727C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ УЛУЧШЕННЫМ ПРОТИВООПУХОЛЕВЫМ ДЕЙСТВИЕМ И/ИЛИ СНИЖЕННЫМИ ПОБОЧНЫМИ ЭФФЕКТАМИ, СОДЕРЖАЩАЯ ПРОТИВООПУХОЛЕВЫЙ АГЕНТ И ПРОИЗВОДНОЕ ГИДРОКСАМОВОЙ КИСЛОТЫ | 1998 |
|
RU2254129C2 |
Изобретение относится к производным эритромицина формулы (I)
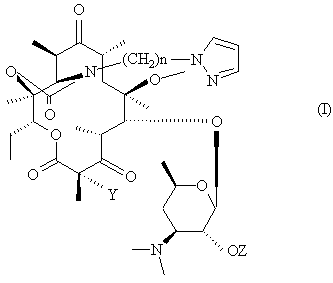
в которой Y обозначает атом водорода или фтора; n обозначает целое число от 1 до 8; Z обозначает атом водорода или остаток карбоновой кислоты, и в которой пиразольный цикл замещен гетероарильным радикалом, содержащим один атом азота; а также их солевым аддуктам с килотами. Соединения формулы (I) обладают антибиотическими свойствами. Также изобретение относится к способу получения соединений формулы (I), содержащей их фармацевтической композиции и промежуточным соединениям для их получения. 4 н. и 8 з.п. ф-лы, 2 табл.
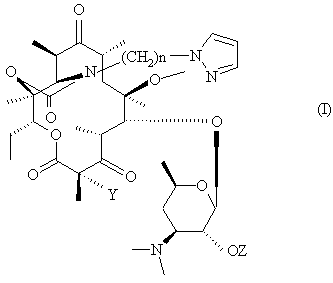
в которой Y обозначает атом водорода или фтора;
n обозначает целое число от 1 до 8;
Z обозначает атом водорода или остаток карбоновой кислоты,
и в которой пиразольный цикл замещен гетероарильным радикалом, содержащим один атом азота,
а также их солевые аддукты с кислотами.
замещен радикалом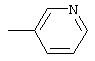
11,12-дидезокси-3-де[(2,6-дидезокси-3-С-метил-3-O-метил-.альфа.-L-рибогексопиранозил)окси]-6-О-метил-3-оксо-12,11-[оксикарбонил[[4-[3-(3-пиридинил)-1Н-пиразол-1-ил]бутил]-имино]]-эритромицин.
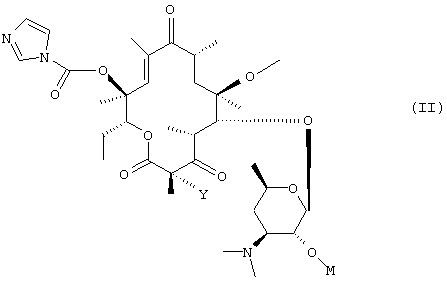
в которой Y имеет указанное в п.1 значение;
М обозначает остаток кислоты, обрабатывают соединением формулы (III)
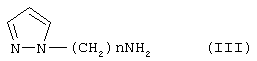
в которой пиразольный цикл замещен как указано в п.1, с получением соединения формулы (I), в которой Z обозначает остаток кислоты, и затем, при желании, полученное соединение формулы (I) подвергают действию агента высвобождения гидроксила в положении 2’ для получения соответствующего соединения формулы (I), в которой Z является атомом водорода, с последующим, при желании, добавлением кислоты для получения соли.
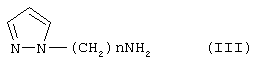
в которой n обозначает целое число от 1 до 8, а пиразольный цикл замещен гетероарильным радикалом, содержащим один атом азота.
| Короб шлаковика мартеновской печи | 1976 |
|
SU596802A1 |
| СПОСОБ ПОВЫШЕНИЯ БАКТЕРИЦИДНОЙ АКТИВНОСТИ ПЕРОКСИДА ВОДОРОДА, ПРИМЕНЯЕМОГО ДЛЯ ОБЕЗЗАРАЖИВАНИЯ ПИТЬЕВОЙ ВОДЫ | 2005 |
|
RU2288174C1 |
| Ferroni R | |||
| et al | |||
| Arzneimittel-Forshung | |||
| Способ приготовления консистентных мазей | 1919 |
|
SU1990A1 |
| Асратян Г.В | |||
| и др | |||
| Журнал прикладной химии | |||
| Пневматический водоподъемный аппарат-двигатель | 1917 |
|
SU1986A1 |
| LIX, № 6, с | |||
| Предохранительная фрикционная муфта | 1924 |
|
SU1296A1 |
| ПРОИЗВОДНЫЕ ЭРИТРОМИЦИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ЭРИТРОМИЦИНА | 1993 |
|
RU2114859C1 |

