Изобретение преимущественно касается непрерывно осуществляемого процесса, предназначенного для промышленного получения водных щелочных или щелочноземельных растворов метионинатов, которые могут быть использованы непосредственно как, например, кормовые добавки, а также введены для выделения аминокислот или их солей, применяемых в качестве кормовых добавок.
Способ предназначен, в частности, для получения раствора метионината. Метионин, а также водные растворы солей метионина, в частности метионината натрия (DE 31 05 009 C), в том числе такие заменители, как гидроксианалог метионина (МНА), используют во всем мире как кормовые добавки при разведении домашней птицы, свиней и других домашних животных и применяют, главным образом, для получения животного белка.
Принимая во внимание рост населения в мире и возрастание проблемы питания, метионин и его различные формы приобретают особое значение ввиду их недорогостоящего получения, а также в первую очередь как одна из самых необходимых аминокислот для процесса роста животных. При необходимости можно применять как твердое вещество, так жидкие формы.
Промышленно поставляемый раствор метионината натрия имеет концентрацию 40 мас. % метионина и по своей биологической валентности соответствует, в отличие от заменителя МНА, твердому метионину при сравнении с эквимолярной основой. Для получения таких растворов метионината натрия применяют, главным образом, три способа.
1. Простое растворение выделенного метионина.
Несмотря на то, что благодаря этому способу продукт получают в самой чистой форме, он представляет собой более дорогостоящую дополнительную рабочую стадию по сравнению с получением твердого вещества и, к тому же, этот способ получения метионина является малорентабельным.
2. Щелочной гидролиз 5- β -метилмеркаптоэтил)гидантоина NaOH или смесью NaOH/Ca(OH)2), при котором в целях устранения нежелательного образования побочных продуктов необходимо вводить около 2 - 3 эквивалентов гидроксида.
Хотя при применении Ca(ОН)2 может быть отделен избыток омыляющих веществ (DE 31 05 006 С) в форме карбоната кальция, однако вряд ли пригодная отработанная соль может быть удалена или преобразована в восстановимую форму гидроксида путем таких дорогостоящих дополнительных стадий, как сгорание и гашение.
При применении одного раствора едкого натра, напротив, избыток должен удаляться либо в форме полученного карбоната натрия (DE 31 04 997 A) или после нейтрализации карбоната натрия с, серной кислотой - в форме полученного сульфата натрия (ЕР 253 740). Недостатками этих способов являются образование соли, а также необходимость дорогостоящего отделения соли и, кроме того, опасность наличия нежелательного остатка соли в растворе продукта.
3. Щелочной гидролиз метионинамида.
Как известно, например, из ЕР 0 228 938, щелочной гидролиз метионинамида можно проводить примерно со стехиометрическим количеством гидроксида без образования большего количества побочных продуктов. В этом заключается существенное преимущество по сравнению со способом 2.
Получение метионинамида можно осуществлять согласно известному способу гидролиза метиониннитрила, который проводят путем прямого синтеза из обычных конечных веществ метилмеркаптопропиональдегида (ММР), синильной кислоты или цианида аммония и аммиака.
Гидролиз нитрила, катализируемый кислотой, исключают из-за неизбежного образования нейтральной соли, в то время как щелочной гидролиз нитрила проводят преимущественно при добавлении каталитически эффективных карбонильных соединений, в частности кетонов (Houben-Weyl: Methoden der Organischen Chemie, Erweiterungs- und Folgebande zur 4. Aufl. 1985, Bd. E5, S. 535ff. и далее цитируемая литература).
Поскольку способ выделения чистого метионинамида в целях его кристаллизации, заключающийся в его гидролизе с NaOH до NaMet (патент ЕР 228 938), является довольно дорогостоящим и приводит к большим потерям, то такой способ является неэкономичным.
Согласно другому примеру из данного патента в резервуаре давления объемом 500 мл из ММР и HCN вначале обычным способом получают ММР-цианогидрин, который превращают в метиониннитрил путем добавления избытка конденсированного NH3. После отделения и охлаждения с 60oC до 10oC на него (см. Houben-Weyl) действуют водным раствором ацетона и NaOH, затем удаляют остаточный аммиак и ацетон путем помещения в вакуум, прежде чем полученный метионинамид будет гидролизован после добавления раствора едкого натра к метионинату натрия при температуре выше 180oC. Последующее отделение и удаление остаточного аммиака в вакууме приводит к образованию раствора метионината натрия, содержащего наряду с метионином в незначительном количестве еще 10 других побочных продуктов различной концентрации, которой нельзя пренебречь. Эта лабораторно-технически полученная форма непригодна для применения при обычном промышленном производстве, где каждая установка производит 10000 jato метионина. В данном патенте далее не приводится никакого упоминания об остатках ацетона и соответственно о рециклизации, а также о рециклизации введенного большого количества избытка NH3. Полная рекуперация введенного в избытке аммиака, а также необходимого катализатора кетона в рециклированной форме и их последующее повторное использование, наряду с оптимальным осуществлением избранного пути синтеза относительно выхода и избирательности, являются предпосылкой создания экономичного способа, который должен проводиться непрерывно в соответствующем масштабе.
До сих пор еще не был описан непрерывный процесс гомогенного щелочного гидролиза аминонитрила, катализируемого кетоном. Причина этого может заключаться в том, что до сих пор не использовался эффективный и непрерывный способ технической очистки смеси, получаемой из кетона, аммиака, воды, а также при необходимости - из побочных продуктов.
Проблема состоит в том, что в качестве заменителя кетона разработаны каталитически активные, несущие карбонильную группу, смолы полимера (ЕР 84 470, DD 208 349), которые можно легко и непрерывно выделять из реакционной среды фильтрацией или получать в форме неподвижного слоя раствора субстрата.
Однако этот на первый взгляд очень привлекательный вариант имеет недостаток, заключающийся в том, что такие катализаторы до сих пор не поступают в продажу и к тому же могли бы быть очень дорогими при возможном изготовлении необходимых количеств для широкого производства в отличие от имеющихся в продаже недорогих кетонов. Кроме того, описанные смолы полимера быстро становятся токсичными из-за побочных реакций в карбонильных группах и их восстановление является дорогостоящим.
В варианте этого способа (ЕР 168 282, US 4,677,224) используют продление стойкости катализатора путем сильного обратного разбавления введенного для гидролиза раствора аминонитрила от концентрации около 1 M до 0,1 M на следующей ступени омыления раствора полученного продукта.
Однако это является также невыгодным для крупного производства. Во-первых, используют соответственно большие колонны, наполненные полимерными катализаторами, количество которых должно быть как минимум двукратным, причем наблюдают частичное восстановление катализатора. Во-вторых, около 90% горячего раствора продукта с температурой около 80oC требуется постоянно охлаждать вновь до необходимой для предварительной стадии температуры, равной 30oC, чтобы снова нагревать его до первоначальной температуры омыления после прохождения стадии.
При таком процессе необходимы также устранение и соответственно минимизация подаваемого потока, а также оптимальное проведение процесса как с экономической, так и с современной экологической точки зрения.
Целью изобретения является разработка способа получения метионина или его соли или такой их предварительной стадии, как метиониннитрил или метионинамид, который основан на непосредственном введении уже упомянутых основных химикалий и может быть осуществлен промышленным образом без выделения промежуточных стадий. Способ должен быть рентабельным и, в частности, непрерывным, причем лишний аммиак, а также введенный кетон должны быть, по возможности, возвращены и восстановлены без потерь.
Настоящий способ решает эту задачу в соответствии с формулой изобретения, причем, в частности, возможна комбинация пунктов формулы для получения настоящих преимуществ.
Способы, осуществляемые преимущественно непрерывно или частично непрерывно, а также с перерывами, содержат всего 5 или 6 стадий осуществления главного способа.
1). Получение сырого ММР-циангидрина (CH) из ММР и HCN (предварительная стадия)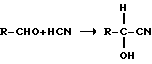
2). Получение сырого ММР-аминонитрила из сырого - ММР-СН и NH3 или смеси NH3 и воды или из ММР, HCN и NH3 и соответственно из смеси NH3 и воды (предварительная стадия)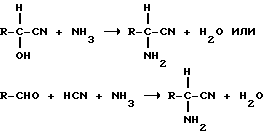
3). Щелочной гидролиз, катализируемый кетоном, при котором из сырого ММР-аминонитрила получают сырой ММР-амид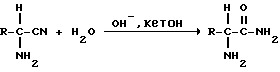
4). Щелочное омыление метионинамида (Met-АН), содержащего смесь сырого продукта, полученную 3), при необходимости путем одновременного частичного или полного выделения кетона, NH3 и при необходимости других летучих компонентов.
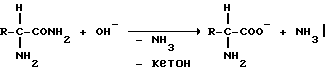
5). При необходимости последующая обработка полученного согласно 4) раствора сырого метионината, включающая, например, последующую реакцию, стадию очистки и соответственно выпаривание до желаемой конечной концентрации.
6). Очистка смеси аммиака и кетона и рециклизация
R=(CH2)2SCH3
Касается 1) и 2).
Известно получение аминонитрилов как предварительной стадии образования α-аминокислоты из альдегидов с синильной кислотой и аммиака (прямой синтез) или из первично полученных циангидринов и аммиака (вариант по Tiemann), что исследовано в многочисленных примерах (Houben-Weyl: Methoden der Organischen Chemie, 4. Aufl., 1952, Bd. 8, S. 274ff и S. 279ff и далее в следующем томе 4. Aufl. E5 S. 1425ff). Обе реакции протекают при соответствующем выборе условий реакции с выходом > 90%.
В данном случае вариант no Tiemann имеет преимущество, заключающееся в том, что может сохраняться относительно большая теплота реакции образования циангидрина. Это возможно, в частности, при непрерывном режиме и более точной установке температуры реакции последующего экзотермического получения аминонитрила.
Получение ММР-цианогидрина.
Получение ММР-цианогидрина осуществляется спонтанно из ММР и жидкой или газообразной синильной кислоты при pH 5 -9 и температуре до 50oC, предпочтительно при pH 5,5 - 7,5 и температуре от 20 до 30oC.
При необходимости значение pH остается постоянным благодаря добавке незначительного количества основания в заданном диапазоне. Применяют также такие органические основания, как аминосоединения, например триэтиламин или пиридин, а также неорганические основания как, например, щелочи или гидроксиды щелочноземельных металлов, и соответственно такие цианиды, как, например, NaOH или NaCN, преимущественно в форме водных растворов.
Реакционные вещества необходимо вводить примерно в эквимолярных количествах. По возможности следует избегать избытка альдегидов, так как на следующих стадиях реакции он приводит к образованию побочных продуктов и продуктов распада в конечном продукте.
Необходимо также удалять большой избыток HCN, так как синильная кислота должна быть либо удалена на соответствующей стадии, либо разрушена, для чего потребуется дополнительная энергия и соответственно возникнут технологические затраты. В частности, наличие незначительного избытка HCN - до 5 мол. % может быть выгодным, так как при этом можно достигнуть количественного превращения ММР с избирательностью практически до 99,9%.
Получение ММР-СН можно проводить из ММР и газообразной или жидкой синильной кислоты как периодически, например, в котле с мешалкой, так и преимущественно непрерывно в трубе или петлевом реакторе или в комбинации обоих вышеописанных способов. Хорошее перемешивание реагентов при постоянном контроле pH и температуры является предпосылкой оптимального проведения этого этапа способа.
С помощью обычных методов анализа, например ВЭЖХ (высокоэффективная жидкостная хроматография) или ТХ (тонкослойная хроматография), подтверждают выход 99,9%.
Получение аминонитрила.
Полученный сырой ММР-циангидрин может также состоять примерно в эквимолярных количествах из ММР и HCN или любой смеси из сырого ММР-циангидрина и ММР/HCN как, например, при не полностью полученном ММР-СН в стадии циангидрина. Путем проведения реакции с аммиаком или смесью аммиака и воды сырой ММР-циангидрин может быть превращен в сырой ММР-аминонитрил.
В опубликованных ранее патентах прежде всего описаны превращения жидкого NH3 и MMP/HCN и соответственно ММР-СН, причем подчеркнуто предпочтительное применение водного аммиака при получении аминонитрила.
В патенте DE-OS 16 43 535 описано применение избытка NH3 выше 4,6 - 55 экв. относительно концентраций ММР и NH3 > 50 мас.% при избыточном давлении при максимальной температуре до 60oC, преимущественно при 15 - 40oC. Длительность обработки для непрерывного способа, в частности, составляет от 4,5 до 5,5 часов. Кроме того, при описанном способе возникает слишком много побочных продуктов (DN и DNMA), которые не приемлемы для непрерывного крупного производства. Выход метионина после последующего кислого гидролиза аминонитрила будет составлять от 97 до 99% относительно данного ММР. В патенте DE-OS 20 06 979 описан одновременно протекающий в данных условиях способ, который проводят непрерывно в котле с мешалкой, причем суть этого изобретения состоит в частичном возвращении в котел реакции водной фазы, полученной после отделения фазы сырого аминонитрила, богатой NH3 и содержащей AN. Вследствие этого должен уменьшаться выход соли при последующем кислом омылении. Частичное возвращение сразу полученного продукта ограничивает рентабельность этого способа, в частности, по причине кислого омыления.
В патенте DE 26 45 544 описан предпочтительный способ применения водного аммиака с концентрацией NH3 от 24 - 48,6 мас.%, в частности от 32 до 42 мас. %, и избытке от 4 - 7 экв. NH3 относительно введенного заранее полученного циангидрина при температуре от 50 до 100oC и давлении от 1 до 10 бар. Получение аминонитрила осуществляют непрерывно и в трубообразном реакторе менее 30 мин однако в лучшем случае конечный выход аминонитрила достигает только 96% относительно введенного ММР-цианогидрина. Промежуточный выход ниже 100% не является достаточным для многоступенчатого способа как с экономической, так и с экологической точек зрения, так как непреобразованные сточные продукты или возникающие из него побочные продукты приходится отделять и удалять дорогостоящим путем. Это, в частности, неосуществимо в больших масштабах, что является обычным при производстве метионина.
Во всех цитируемых патентах выход составляет едва 90% и, к тому же, не приводится никакого упоминания по поводу пробела, встречающегося в балансе.
Согласно изобретению было обнаружено, что имеются главные побочные органические компоненты в сыром аминонитриле, полученном из соответствующего замещенного иминодинитрила (DN) (DN = 2,2'-бис-(2-метилмеркаптоэтил)иминодиацетонитрил), его производного динитрилмоноамида (DNMA) (DNNA = 2,2-бис-(2-метилмеркаптоэтил)иминодиацетонитрил-моноамид), а также из полученного здесь метионинамида (AM).
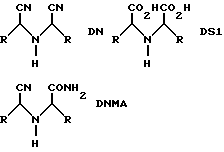
где R имеет вышеупомянутое значение.
До сих пор еще не описанные в литературе соединения иминодинитрила DN и DNHA полностью или частично гидролизуют в следующих стадиях до соответствующей иминодикислоты (DS1 = 2,2-бис(2-метилмеркаптоэтил)иминодиуксусная кислота), содержание которой, как чужеродного компонента в продукте, должно быть по возможности низким. Следовательно, в первую очередь становится выгодной оптимально полученная форма стадии аминонитрила для того, чтобы при практическом количественном конечном выходе аминонитрила получить возможно минимальное образование побочных продуктов, в частности, при учете важных аспектов рентабельного способа.
Было обнаружено, что DN + доля DNMA имеют небольшое допустимое значение от < 2 мол.% введенного циангидрина при выходе AN > 98 мол.% относительно преобразованного альдегида, если работают при молярном соотношении NH3/CH и соответственно соотношение NH3/альдегид от по меньшей мере 4:1, в частности 5: 1, до 15:1 и введенной концентрации NH3 > 50 мас.%, предпочтительно 55 - 85 мас. %, в частности, при молярном соотношении NH3/H2O от 1,5, при температуре выше 40oC, однако не выше 80oC и при времени реакции от минимум 5 до максимум 60 мин. В частности, высокие температуры или снижающаяся концентрация NH3 и соответственно избыток NH3, а также большая длительность обработки позволяют повысить долю динитрила, одновременно низкое содержание воды увеличивает необходимое время реакции. В основном, реакция завершается позднее всего тогда, если получают 2% сухого продукта (мол.% относительно ММР + CH) DN + DNMA, иначе доля DS в конечном продукте будет высокой. В целом реакцию проводят таким образом, что возможна минимальная доля DN + DNMA.
Предпочтительная форма пути осуществления способа заключается в применении около 5-8 экв. NH3 c концентрацией от 55 до 85 мас.%, в частности 60-80 мас.%, в температурном диапазоне от 50 до 70oC при времени реакции от 10 до 50 мин, и установленном при этом давлении системы. В этой области можно понижать долю DN + DNMA даже на < 1 мол.%.
Выбранные условия способствуют непрерывному характеру движения с выходом выше 96%, что необходимо для крупного производства. Для осуществления способа устанавливают чистый трубчатый реактор или комбинацию петельного и трубчатого реакторов, причем в трубчатом реакторе продолжительность реакции должна быть в несколько раз больше, чем в петлевом реакторе, что позволит снизить образование динитрила. Описанный способ отличается тем, что вместо свежей смеси аммиака и воды можно использовать аммиак, полученный на более поздних стадиях рециклизации, а также соответствующую смесь аммиака и воды.
Полученную таким образом смесь сырого аминонитрила, воды и аммиака вводят либо непосредственно для проведения следующего гидролиза, либо после релаксации при нормальном давлении или давлении, установленном на стадии (получения) аминонитрила между основным давлением в системе и нормальным давлением. При релаксации на стадии аминонитрила здесь может быть возвращена часть избыточного NH3.
Гидролиз аминонитрила = получение метионинамида.
В патентах DE 26 37 204 и DE 27 53 828 для получения амидов аминокислот путем гидролиза соответствующего аминонитрила, катализируемого кетоном, предложенные алифатический и циклоалифатический кетон обладают четкими различиями в их применимости для гидролиза аминонитрила.
Самой высокой скорости реакции достигают при участии ацетона. Этот способ является недорогим и представляет наименьшую опасность для здоровья, однако преимущественно для промышленных целей вводят также кетон.
Гидролиз описанного полученного аминонитрила с разбавленным раствором едкого натра в присутствии 0,2 - 2,0 экв. ацетона при температуре от 10 до 30oC приводит к получению смеси продукта, состоящей главным образом из метионинамида, метионината натрия, соединений имидазолидинона LM1 и IМ2, а также ММР и других компонентов, встречающихся в меньших концентрациях.
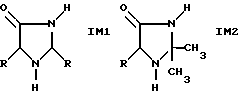
где R имеет вышеупомянутое значение.
Имидазолидинон также превращают в метионин в последующем омылении при более высокой температуре, однако наличие долей IM1 и ММР приводит к потерям в конечном выходе и возникновению проблемы очистки. Согласно изобретению восстановление альдегида можно значительно снизить, а получение IM1 при пониженной температуре и содержание щелочи как следствие повышает избирательность общей последовательности реакции. В патенте DE 27 53 829 описан способ получения α-аминокислот, при котором к смеси сырого аминонитрила добавляют необходимое для омыления аминонитрила количество щелочи, в частности 1 экв. гидроксида, и проводят омыление в шаге, приводящем в данном случае к достаточно плохим результатам. Кроме того, при повышении температуры выше 40oC может наблюдаться повышенное содержание альдегида - до 7 мол.%. Доля альдегида может быть снижена на менее 2 мол.% при температуре ниже 30oC. Так как известно, что скорость реакции гидролиза аминонитрила значительно повышается вместе с концентрацией кетона (Commeyras et al., Tetrahedron 1978, 34, 2275ff на примере от ацетона и α-аланиннитрила), то это свойство можно использовать для управления реакцией. Не представляет интереса большой, например, неоднократный молярный избыток кетона по причинам рентабельности и чистоты продукта. Было обнаружено, что в диапазоне избытка H2O при замедленном введении ацетона и преимущественно низком количестве гидроксида можно достичь высоких скоростей реакций и при этом достаточно хорошей избирательности в стадии гидролиза. В данном случае можно следующим образом ограничить оптимальный диапазон (относительно нитрила): 15-50 экв., предпочтительно 20-30 экв. H2O; 0,2-2,0 экв., предпочтительно 0,3-1,0 экв. кетона: 0,1-1,1 экв. , преимущественно 0,15-0,4 экв. и, в частности, 0,15-0,3 экв. OH- и 1-7 экв., предпочтительно 2-5 экв. NH3 при температуре от 10 до 30oC, преимущественно от 15-28oC, и время реакции от 10 до 90 мин, предпочтительно от 20 до 60 мин. Описанный здесь способ отличается тем, что вместо кетона и воды в следующей поздней стадии рециклизации применяют полученный водный раствор кетона, в частности раствор ацетона, включающий содержащиеся там побочные компоненты.
Так же, как и при предыдущих шагах способа, гидролиз сырого аминонитрила можно осуществлять как прерывисто, так и предпочтительно непрерывно, во-первых, например, в реакционном сосуде при перемешивании. Во-вторых, можно использовать простую реакционную трубу, предпочтительно петлевой реактор, в частности, комбинацию петлевого реактора и реакционной трубы. Давление на этой стадии является некритическим и может быть свободно выбрано в зависимости от способа. При данном способе применяют преимущественно установленное давление в системе.
Гидролиз амида аминокислоты = получение метионата.
Полученный таким образом раствор можно вводить непосредственно на стадии последующего омыления щелочью и соответственно гидроксидом щелочноземельного металла. Кроме того, в предыдущей стадии гидролиза содержание введенного гидроксида повышают путем добавки другого эквивалента гидроксида, так что результирующее содержание гидроксида соответствует соотношению от 0,95 до 1,1 экв., предпочтительно 1,0-1,05 экв., относительно введенного ММР. Температура омыления должна составлять как минимум 85oC, преимущественно как минимум 90oC, как правило выше 100oC, в частности 110-140oC. Как правило, температура не превышает 200oC. Концентрация амида составляет предпочтительно 10-35 мас.%, преимущественно < 30 мас.%, в частности < 25 мас.%, причем при температуре омыления > 160oC предпочтительна концентрация 25 мас.%. При выбранной температуре устанавливают давление в системе от 1 до нескольких бар и при необходимости проводят одновременное выпаривание реакционного раствора. Время реакции составляет минимум 10, максимум - 90 мин, преимущественно 20-40 мин, в случае гидроксида щелочноземельного металла и соответственно до 180 мин в случае гидроксида щелочноземельного металла.
Способ согласно изобретению отличается тем, что в течение и/или после омыления амида аммиак, кетон и воду перегоняют при температуре > 85oC и/или помещают в вакуум. Аммиак освобождают преимущественно от кетона и проводят синтез нитрила (см. выше). Способ согласно изобретению может осуществляться при всех синтезах метионина, при которых метиониннитрил вначале омыляется в амид в присутствии кетона при температуре ниже 60oC.
Согласно изобретению установлено, что нет необходимости заранее отделять кетон, полученный в смеси сырого метионинамида, как это осуществляют, например, согласно EP-B 228 938 путем помещения в вакуум полученного раствора метионинамида, а также предлагают в DE 26 37 204 C2 (ввиду низкого pH). Как установлено согласно изобретению, часть кетона (2-10% введенного количества) в отсутствие имидазолидинона (IM2 в случае ацетона) не существует в химически связанной форме и освобождается только в стадии омыления. При выборе метода, исходя из экономических и экологических причин, учитывают необходимость рециркуляции и общей рециркуляции свободного и освобожденного кетона в течение и соответственно после омыления амида. Амид кетона предпочтительно не вводят перед шагом омыления, исключая таким образом второй парциальный поток с кетоном, содержащим вторичный пар, и, вместе с тем, дополнительную эксплуатацию аппаратуры. Стадию омыления могут проводить как прерывисто в реакторе, работающем под давлением, и при перемешивании, лучше непрерывно, например, в колонне омыления. При непрерывном характере движения особенно предпочтительно удалять остаточный и образующийся в ходе реакции NH3 вместе с еще имеющимся кетоном, а также водяной пар, полученный при температуре омыления in situ, или дополнительно впрыскиваемый нагревающий пар. Эту смесь, удаленную в верхней части, возвращают и вводят для рециклизации аммиака и кетона. При этом в конце колонны образуется раствор, содержащий аммиак и не включающий кетоны. В зависимости от температуры выпаривания получают светло-желтый раствор соли аминокислоты различной концентрации с содержанием аминокислоты от 10 до 45 мас.%.
Поскольку в растворах сырой соли аминокислоты еще может встречаться остаток цианида в концентрации от нескольких сотен млн.-1, то его необходимо разрушать. Согласно изобретению это осуществляют путем термической обработки, преимущественно непосредственно полученным раствором соли аминокислоты при температуре выше 150oC, предпочтительно до 200oC, и длительности обработки до 40 мин, в частности как минимум 10 мин. В основном это осуществляют: при ≥ 160oC - минимум 15 мин, при ≥ 180oC - минимум 7 мин и при ≥ 190oC минимум - 4 мин.
Из DE-OS 33 34 328 известно, что остаточные концентрации цианида являются нестойкими вследствие повышения температуры. В DE-OS 33 34 328 также в щелочном растворе описано уничтожение связанной синильной кислоты и акрилнитрила в сыром ацетонитриле, причем в отличие от превращения аминокислот учитывали изменение значения температуры и значения pH.
Полученные таким образом растворы имеют содержание цианида от < 10 млн. -1 относительно содержания аминокислоты 40 мас.%. Это обозначает четкое снижение допустимого предела. Последующую термическую обработку можно успешно проводить при каждой полученной концентрации аминокислоты от 10-45 мас.%, однако преимущественно при 15-30 мас.%. Обработку можно проводить как в подключенном шаге, например в дополнительной реакционной трубе, так и во время собственно омыления, например путем соответствующего повышения температуры в нижней части колонны омыления. Омыление цианида приводит к появлению в растворе продукта незначительной доли щелочи, иногда - формиата щелочного металла, которые не являются побочными, так как формиат, в частности формиат кальция используют как кормовую аминокислотную добавку.
B основном, при конечных концентрациях от < 40 мас.% требуется дополнительное выпаривание аминокислоты. Его проводят предпочтительно при нормальном или пониженном давлении и при необходимости - при температуре кипения при возможно лучших условиях, например с применением пленочного испарителя для осаждения. Полученный вторичный пар могут снова вводить в процесс в другом месте.
Экономически выгодной является также фильтрация раствора продукта, предпочтительно через активированный уголь. Фильтрацию можно проводить, в частности, при непрерывном характере движения, предпочтительно в колонне. После устранения даже незначительного помутнения, а также небольшой доли побочных продуктов происходит осветление раствора. Этот шаг можно проводить не только перед, но и после выпаривания. Общий выход метионина при представленном здесь способе составляет по меньшей мере 95% относительно введенного альдегида. Содержание выявленных в растворе нетоксичных компонентов иминодиуксусной кислоты DS1 и OS2 (N-(1-карбокси-1-метилэтил)-метионин)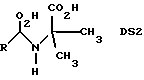
где R имеет вышеупомянутое значение, составляет при непрерывном омылении в сумме ниже 1 мас.% относительно метионина. Качество растворов продукта соответствует качеству, установленному для кормовых средств.
Очистка смеси аммиака и кетона и рециклизация.
Для рентабельности этого способа является важным повторно использовать избыточный аммиак, в частности возвращенный в область образования аминонитрила. Так как в ходе омыления аминонитрила вновь образуется эквивалент NH3, то при успешной рециклизации возможно даже проведение всего процесса без применения NH3 В конечном счете из синильной кислоты образуется азот, помещенный в молекулу аминокислоты.
Также по причинам рентабельности необходима по возможности полная рециклизация кетона, вводимого в качестве катализатора.
Согласно изобретению показано, что введенный для получения аминонитрила аммиак не должен содержать кетоны, так как небольшие доли приводят к пониженному конечному выходу аминонитрила и четко измеряемой доли альдегида в сыром аминонитриле и, вместе с тем, к убыткам при выходе и ухудшению качества конечного продукта. Рециклированный аммиак должен содержать преимущественно менее чем 25 моль/эквивалент и, в частности, менее чем 10 моль/эквивалент кетона относительно циангидрина и/или альдегида. Следовательно, это означает, что выпаренная смесь NH3, кетона и воды не может быть непосредственно рециклированна, причем несущественным является то, когда проводят выпаривание - перед, во время или после омыления амида. Обе последние альтернативы предпочтительны в зависимости от способа их выполнения. Прежде всего необходимо добиться по возможности количественного выхода в NH3, не содержащем кетоны, в то время как рециклируемый кетон может содержать как воду, так и NH3.
Эту задачу по разделению можно решить путем дистилляции. Образующийся в результате омыления вторичный пар вводят прежде всего в воду в стадии адсорбции, и этот раствор помещают между верхней и нижней частями, преимущественно между средней и нижней областями дистилляционной колонны. Если кетоном является, например, ацетон, то в колонне происходит разделение на ацетон и безводный аммиак, который при соответствующем соотношении оборотного продукта конденсируется частично или полностью в верхней части колонны и непрерывно отводится. Также в нижнюю часть колонны непрерывно отводят водный раствор, содержащий 5-30 мас.% ацетона и 0-5 мас.% NH3. Колонна функционирует при температуре верхней части 100- 200oC и давлении 1-20 бар. Температура верхней части может находиться соответственно 20 и 55oC.
Давление преимущественно устанавливают 5-18 бар, в частности 9-15 бар, температура верхней части составляет соответственно 140-190oC, в частности 150-180oC. После разбавления с водой до заданной концентрации от > 50 мас.% NH3 продукт верхней части может быть возвращен непосредственно в стадию получения аминонитрила. При этом способе введенный ацетон может также оказаться в концентрации выше 95% на конце нижней части. Этот раствор при необходимости можно использовать непосредственно или по желанию - после фильтрации как адсорбционное средство, как, например, активированный уголь, вместо свежего ацетона для гидролиза аминонитрила. При этом, если это необходимо, целесообразно путем разбавления водой или восполнения незначительных потерь добиться тех концентраций ацетона, которые вместе со смесью сырого аминонитрила определяют соответствующую желаемую заданную концентрацию ацетона и воды в стадии гидролиза аминонитрила. Необходимый эквивалент гидроксида могут вводить либо непосредственно путем добавки к смеси полученного водного раствора кетона, либо также путем сепаратной дозировки соответствующего раствора гидроксида щелочного металла и соответственно суспензии гидроксидов щелочноземельных металлов на стадии гидролиза. Возрастающее содержание побочных продуктов при более длительной рециклизации, состоящих из различных пиридиновых соединений, практически не имеют негативного влияния на выход гидролиза аминонитрила согласно способу изобретения. Уровень побочного продукта можно устанавливать постоянно на желаемом уровне путем непрерывного извлечения меньших долей, например от 1-10 мас.% жидкого раствора при перегонке под давлением, и добавлением соответствующих долей в свежий раствор. Удаленные доли могут быть либо просто удалены, либо повторно переработаны, в частности очищены от побочных продуктов и затем восстановлены.
Как показано на чертеже, разделенном на одиннадцать этапов, осуществляемый при вышеописанных условиях способ может быть выполнен технически, преимущественно абсолютно непрерывно, но также и избирательно частично непрерывно или прерывно. В реакторе для получения циангидрина 1 из ММР и HCN образуется катализируемый основанием (КАТ.) ММР-цианогидрин, который в реакторе для получения аминонитрила 2 вместе с водным аммиаком концентрации от > 50 мас.% подвергается превращению до стадии абсорбции и образования смеси 11. Реактор для получения циангидрина 1 и реактор для получения аминонитрила 2 могут быть сведены также к одной стадии. Полученную смесь сырого метиониннитрила, воды и аммиака либо вносят в стадии релаксации 3 при более низком давлении, причем освобождающуюся при этом долю NH3 сепаратно отводят по пути (а) или b)) или для следующей стадии гидролиза 4 сырую смесь непосредственно вводят в максимальном количестве в стадии релаксации. Здесь проводят гидролиз аминонитрила с помощью одновременно поданного водного раствора гидроксида щелочного металла или суспензии гидроксидов щелочноземельных металлов (MOH) и водного раствора кетона, преимущественно в форме смесей 10, полученных путем дистилляции продуктов, уходящих из низа колонны, и при необходимости - путем добавки свежего кетона (Δ > C = 0) и соответственно воды (Δ) . Полученный таким образом раствор кетона, при необходимости - NH3 и вышеупомянутых продуктов реакции в воде, вносят одновременно с соответствующим водным раствором гидроксида щелочного металла или суспензии гидроксидов щелочноземельных металлов (MOH) в реактор для омыления 5 и подвергают превращениям. Полученную таким образом щелочь или раствор метионината щелочноземельных металлов после перехода зоны термической последующей обработки 6, которая также может быть интегрирована в реактор для омыления 5, пропускают через фильтр 7 и при необходимости далее концентрируют в стадии выпаривания 8. После зоны последующей обработки 6 альтернативно или дополнительно может быть также установлена стадия выпаривания 8а. При этом фильтр 7 имеет минимальную пропускную способность жидкости. К фильтру подводят новое адсорбционное средство (C = активированный уголь и/или вспомогательное средство фильтра) и/или при необходимости - вспомогательное средство фильтрации и изымают израсходованное средство (C-отходы).
Вторичный пар, выведенный из реактора для омыления 5 и содержащий аммиак, кетон, воду и долю побочных продуктов, при необходимости вместе с NH3, образующимся в стадии релаксации 3 (а), и вторичным паром стадии выпаривания 8 и соответственно 8а, в степени адсорбции 9, поглощает такое количество воды, что полученная концентрация NH3 по меньшей мере составляет 20 мас.%. Затем этот раствор поступает в дистилляционную установку 10. Полученный раствор, содержащий кетоны, в виде дистиллята нижней части после фильтрации (7а) проходит через адсорбционное и/или вспомогательное средство фильтра или при необходимости после извлечения небольших долей (отходов) в стадию гидролиза 4. NH3, полученный как продукт верхней части, при необходимости поступает вместе с полученным на стадии релаксации 3 парциальным током NH3 (b) в стадию абсорбции и смешения 11, где путем добавки воды устанавливают желаемую концентрацию NH3 и при необходимости компенсируют незначительные потери NH3 (Δ-NH).
Полученный согласно этому способу раствор метионата можно использовать непосредственно как состав жидкого метионина в качестве кормовых средств согласно DE-OS 31 05 009 или при желании - для выделения метионина или производных метионина согласно известным из литературы методам.
Пример 1. 2-Гидрокси-4-метилмеркаптобутиронитрил (ММР-циангидрин).
(Поз. 1 на чертеже) В 400-литровый котел с анкерной мешалкой, pH-электродом, термометром, дифлегматором, дозировочной установкой и подключенным испарителем синильной кислоты помещали 250,0 кг (2400 моль) свежего дистиллированного 3-(метилмеркапто)пропиональдегида (ММР) и 110 мл триэтиламина при 20oC и pH 7,0. При перемешивании в течение 7 часов вводили 66,0 кг (2442 моль) свежей выпаренной газообразной синильной кислоты таким образом, чтобы температура в котле не превышала 30oC. Путем одновременной добавки 130 мл триэтиламина pH поддерживали между 5,5 и 7,5. Согласно ВЭЖХ-анализу ММР-оборот составлял 100%. Выход ММР-цианогидрина составлял 316 кг с содержанием циангидрина 99,6 мас.%. Этот циангидрин вводили в последующих примерах.
Пример 2.
(Поз. 1) В 250-литровом котле с анкерной мешалкой, pH-электродом, термометром, дифлегматором, дозировочной установкой и присоединенным запасным контейнером с жидкой HCN в течение 4 часов при условиях, аналогичных примеру 1, проводили реакцию 133,9 кг (1285 моль) ММР и 400 мл (0,29 кг) триэтиламина с 35,8 кг (1324 моль) жидкой синильной кислоты. Выход ММР-циангидрина составлял 170,0 кг с содержанием циангидрина 99,2 мас.%.
Пример 3. 2-Амино-4-метилмеркаптомасляной кислоты нитрил (D,L-метиониннитрил).
(Поз. 2) В 250-миллиметровый стальной автоклав с мешалкой, манометром, внутренним термометром, подающей линией, насосом, сборником и нагревательной баней помещали 51,0 г (2,40 моль) 80%-ного водного аммиака при 44oC. Затем в течение 2 мин, при перемешивании добавляли 51,5 г (0,391 моль) ММР-цианогидрина. При этом температура повышалась до 57oC, давление составляло 13 бар. Реакционную смесь перемешивали еще 20 мин при такой же температуре, затем охлаждали в ледяной бане и подвергали релаксации до нормального давления. ВЭЖХ-анализ реакционной смеси дал следующие результаты: 99,1% сухого продукта метиониннитрила, 0,0% сухого продукта метионинамида (AM), 0,8% сухого продукта динитрила (DN) и 0,0% динитрилмоноамида (DNMA).
Пример 4. (Поз. 2) приборы как в примере 3.
В автоклав при 50oC помещали 68,0 г (2,40 моль) 60%-ного водного аммиака. Затем в течение 2 мин при перемешивании добавляли 53,6 г (0,407 моль) ММР-цианогидрина. При этом температура возрастала до 58oC, давление составляло 7 бар. Реакционную смесь перемешивали еще 10 мин при той же температуре, затем охлаждали в ледяной бане и подвергали релаксации до нормального давления. ВЭЖХ-анализ реакционной смеси дал следующие результаты: 99,0% сухого продукта метиониннитрила, 0,0% сухого продукта AM, 0,9% сухого продукта динитрила (DN) и 0,0% сухого продукта DNMA.
Пример 5 (сравнительный). Аналогично примеру 3 проводили реакцию 102,0 г (2,40 моль) 40%-ного водного аммиака с 55,2 г (0,419 моль) ММР-цианогидрина. ВЭЖХ-анализ дал следующие результаты: 98,0% сухого продукта метиониннитрила, 0,3% сухого продукта метионинамида, 1,4% сухого продукта DN и 0,3% сухого продукта динитрилмоноамида (DNMA).
Пример 6 (сравнительный). Аналогично примеру 3 проводили реакцию 122,4 г (1,80 моль) 25%-ного водного аммиака с 41,1 г (0,312 моль) ММР-цианогидрина. HHLC-анализ дал следующие результаты: 95,8% сухого продукта метиониннитрила, 1,6% сухого продукта метионинамида, 1,6% сухого продукта DN и 1,0% сухого продукта DNMA.
Пример 7 (сравнительный). (Поз. 2 без поз. 1). В приборы, описанные в примере 3, в частности в автоклав, помещали 122,4 г (1,80 моль) 25%-ного водного аммиака при 42oC. Затем в течение 4 мин при перемешивании добавляли раствор 8,5 г (0,314 моль) HCN в 31,7 г (0,304 моль) ММР. При этом температура повышалась до 53oC, давление составляло 1 бар. Реакционную смесь перемешивали еще 10 мин при такой же температуре, затем охлаждали в ледяной бане и подвергали релаксации до нормального давления. ВЭЖХ-анализ реакционной смеси дал следующие результаты 91,9% d Th. метиониннитрила, 1,8% сухого продукта метионинамида, 1,7% сухого продукта DN, 1,2% сухого продукта DNMA.
Пример 8. (Поз. 2 без поз. 1). Аналогично примеру 7 в автоклав при 32oC помещали 51,0 г (1,80 моль) 60%-ного водного аммиака. Затем в течение 0,7 мин при перемешивании добавляли раствор 8,5 г (0,314 моль) HCN в 32,2 г (0,309 моль) ММР. При этом температура возрастала до 54oC, давление составляло 5 бар. Реакционную смесь перемешивали еще 10 мин при той же температуре, затем охлаждали в ледяной бане и подвергали релаксации до нормального давления. ВЭЖХ-анализ реакционной смеси дал следующие результаты: 97,6% сухого продукта метиониннитрила, 0% сухого продукта метионинамида, 1,5% сухого продукта DN, 0,5% DNMA, 0,2% сухого продукта ММР и 0,2% сухого продукта CH.
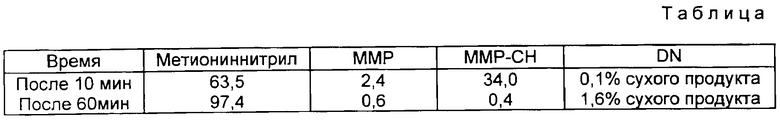
Примеру 9. (Поз. 2). В 250-миллиметровый стальной автоклав с мешалкой, манометром, внутренним термометром, подающей линией, установкой для отбора пробы, насосом, сборником и нагревательной баней помещали 68,0 г (2,40 моль) 60%-ного водного аммиака при 31oC. Затем в течение 2 мин при перемешивании добавляли 52,9 г (0,402 моль) ММР-цианогидрина. При этом температура возрастала до 39oC. Реакционную смесь перемешивали еще 60 мин при 40oC и после 10 и соответственно 60 мин проводили ВЭЖХ-анализ пробы. При этом получали следующие показатели выхода (см. таблицу).
Пример 10 (сравнительный). (Поз. 2). Аналогично примеру 3 в автоклав при 68oC помещали 116,3 г (2,60 моль) 38%-ного водного аммиака. Затем в течение 1,6 мин при перемешивании добавляли 54,3 г (0,412 моль) ММР-цианогидрина. При этом температура возрастала до 80oC, давление составляло 5 бар. Реакционную смесь перемешивали еще 10 мин при такой же температуре, затем охлаждали в ледяной бане и подвергали релаксации до нормального давления. ВЭЖХ-анализ реакционной смеси дал следующие результаты: 93,0% сухого продукта метиониннитрил, 0,45% сухого продукта метионинамида, 1,35% сухого продукта ММР, 4,60% сухого продукта DN, 0,55% сухого продукта DNMA.
Пример 11 (сравнительный). (Поз. 2). Аналогично примеру 3 в автоклав при 44oC помещали 68,0 г (2,40 моль) 60%-ного водного аммиака и 4,7 г (0,08 моль) ацетона. Затем в течение 2 мин при перемешивании добавляли 53,7 г (0,408 моль) MMP-цианогидрина. При этом температура возрастала до 56oC, давление составляло 6 бар. Реакционную смесь перемешивали еще 10 мин при такой же температуре, затем охлаждали в ледяной бане и подвергали релаксации до нормального давления. ВЭЖХ-анализ реакционной смеси дал следующие результаты: 91,6% сухого продукта метиониннитрила и 8,4% сухого продукта ММР.
Пример 12. (Поз. 2). При непрерывном способе в нагреваемую реакционную трубу с подключенным смесительным штреком, измерением внутренней температуры, установкой для отбора пробы и присоединенной подачей ММР-СН со сборником и подачей NH3 со сборником и промежуточно включенным теплообменником в течение часа по дозам вводили одновременно 2,55 кг (89,8 моль) подогретого до 40oC водного 60%-ного аммиака и 1,923 кг (14,60 моль) ММР-циангидрина. Установленная внутренняя температура на входе трубы составляла 55oC, на выходе трубы - 56oC, давление составляло 7,8 бар, продолжительность реакции 17,3 мин. ВЭЖХ-анализ проб, полученных на выходе трубы, дал следующие результаты: 97,2% сухого продукта метиониннитрила, 0,3% сухого продукта метионинамида, 0,5% сухого продукта ММР и 1,8% сухого продукта DN.
Пример 13. (Поз. 2). Аналогично примеру 12 в течение часа проводили реакцию 2,45 кг (86,3 моль) 60%-ного водного аммиака с 2,371 кг (18,0 моль) ММР-цианогидрина при продолжительности реакции 21,8 мин. ВЭЖХ-анализ дал следующие результаты: 95,2% сухого продукта метиониннитрила, 0,0% сухого продукта метионинамида, 1,7 сухого продукта DN, 0,7% сухого продукта ММР-СН, 0,4% сухого продукта ММР.
Пример 14. (Поз. 2). Аналогично примеру 12 в течение часа по дозам одновременно вводили 2,07 кг (97,2 моль) подогретого до 53oC водного 80%-ного аммиака и 1,917 кг (14,56 моль) ММР-цианогидрина. Установленная внутренняя температура на входе трубы составляла 57oC, на выходе трубы - 56oC, давление составляло 14,4 бар, продолжительность реакции 24,5 мин. ВЭЖХ-анализ проб, полученных на выходе трубы, дал следующие результаты: 98,2% сухого продукта метиониннитрила, 0,0% сухого продукта метионинамида, 1,3% сухого продукта DN и 0,4% сухого продукта ММР.
Пример 15. Раствор D,L-метионинамида.
(Поз. 2, 3 и 4). В 250-миллилитровый стальной автоклав с мешалкой, манометром, внутренним термометром, подающей линией, насосом, сборником и нагревательной баней помещали 50,0 г (1,76 моль) 60%-ного водного аммиака при 45oC. Затем в течение 1,5 мин при перемешивании добавляли 37,25 г (0,283 моль) ММР-цианогидрина. При этом температура возрастала до 54oC, давление составляло 7 бар. Реакционную смесь перемешивали еще 10 мин при такой же температуре и затем одновременно охлаждали и осуществляли релаксацию до нормального давления.
В теплую смесь при температуре 15oC при одновременном охлаждении воды вводили раствор 2,5 г (0,063 моль) NaOH и 8,8 г (0,152 моль) ацетона в 110 г (6,11 моль) воды, причем температура возрастала до 27oC. Реакционную смесь перемешивали еще 30 мин, при 25oC и тогда сразу анализировали. Прозрачный бледно-желтый раствор (201,1 г) имел следующий состав:
19,93 мас.% метионинамида, - 95,60% сухого продукта
0,38 мас.% метионина, - 1,80% сухого продукта
0,06 мас.% IM1, - 0,35% сухого продукта
0,46 мас.% IM2, - 1,70% сухого продукта
0,05 мас.% ММР, - 0,35% сухого продукта
Пример 16. (Поз. 2, 3 и 4). Аналогично примеру 15 в автоклав помещали 51,0 г (1,80 моль) 60%-ного водного аммиака при 45oC. Затем в течение 1,5 мин при перемешивании добавляли 42,0 г (0,319 моль) ММР-цианогидрина. При этом температура возрастала до 54oC, давление составляло 7 бар. Реакционную смесь перемешивали еще 10 мин при такой же температуре и затем подвергали релаксации до нормального давления при одновременном охлаждении.
В теплую смесь с температурой 15oC при одновременном охлаждении воды добавляли раствор 12,2 г (0,31 моль) NaOH и 8,8 г (0,152 моль) ацетона в 113 г (6,27 моль) воды, причем температура возрастала до 27oC. Реакционную смесь перемешивали еще 30 мин при 25oC и сразу анализировали. Мутный бледно-желтый раствор (208,3 г) имел следующий состав:
18,8 мас.% метионинамида, - 82,8% сухого продукта
2,8 мас.% метионина, - 12,2% сухого продукта
0,0 мас.% IM1, - 0% сухого продукта
0,77 мас.% IM2, - 2,7% сухого продукта
0,17 мас.% MMP, - 1,1% сухого продукта
Пример 17. В нагреваемой гидравлической трубе с измеряемой внутренней температурой (поз. 2) и с подключенным смесительным штреком расположена подающая линия ММР-СН, связанная со сборником, и присоединена подающая линия аммиака, которая через промежуточно включенный теплообменник связана со стадией абсорбции и смешения (поз. 11). Гидравлическая труба связана через автоматический вентиль для поддержания давления с вращающимся испарительным сосудом (поз. 3), газовая камера которого связана проводкой со стадией адсорбции (поз. 9), а нижний сток которого через насос подведен к стадии гидролиза (поз. 4). Стадия гидролиза является термостатической и состоит из петлевого реактора с циркуляционным насосом и подключенной гидравлической трубы. Петлевой реактор с одной стороны связан со сборником водного раствора ацетона и NaOH через транспортер и насос, а гидравлическая труба связана с приемником через регулятор давления. Стадия адсорбции (поз. 9) является системой адсорбции NH3, которая состоит из резервуара нижней части с циркуляционным насосом, тушильной установки и присоединенной ректификационной колонны.
Верхняя часть колонны снабжается свежей водой из резервуара с помощью транспортера и насоса. Конец нижней части расположен в приемнике.
В гидравлическую трубу (поз. 2) в течение часа по дозам подавали приблизительно 1,925 кг (14,61 моль) ММР-циангидрина, а также 2,55 кг (89,8 моль) подогретого до 40oC 60%-ного аммиака из поз. 11 с отрегулированным потоком. Установленная внутренняя температура составляла 55oC на входе трубы и 56oC на выходе трубы, давление составляло 7,8 бар, продолжительность реакции 23 мин. Давление в испарительном сосуде (поз. 3) составляло 1,2 бар, температура 35oC. Уровень сырого аминонитрила в испарительном сосуде постоянно поддерживали при постоянном количестве транспортируемого для стадии гидролиза материала (поз. 4).
В петлевой реактор стадии гидролиза (поз. 4) наряду со смесью сырого аминонитрила из стадии релаксации (поз. 3) вводили из запасного резервуара в течение часа 6,40 кг водного раствора 7,25 мас.% (0,464 кг, 7,99 моль) рециклизированного ацетона, 2,0 мас.% (0,128 кг, 3,2 моль) NaOH и 3,3 мас.% NH3 (0,211 кг, 12,4 моль). Температура в петле составляла 27oC, в реакционной трубе 26oC, вся продолжительность реакции составляла 50 мин.
Температура при абсорбции NH3 (поз. 9) составляла 22oC при давлении 1,2 бар. По окончании стадии гидролиза (поз. 4) получили в среднем 10,58 кг продукта в виде окрашенного бледно-желтого водного раствора следующего состава:
19,19 мас.% метионинамид, - 93,8% сухого продукта
0,31 мас.% метионина, - 1,5% сухого продукта
0,38 мас.% IM2, - 1,5% сухого продукта
0,26 мас.% ММР, - 1,8% сухого продукта
Пример 18. Раствор D,L-метионината натрия
(Поз. 2, 3, 4, 5 и 6). К 250-миллилитровому стальному автоклаву с мешалкой, манометром, внутренним термометром и нагревательной баней присоединяли подающую линию с насосом и сборником.
В автоклав помещали 38,3 г (1,80 моль) 80%-ного водного аммиака при 42oC. Затем при перемешивании в течение 1,5 мин добавляли 37,80 г (0,287 моль) ММР-цианогидрина. Реакционную смесь перемешивали еще 20 мин, при 56oC и при одновременном охлаждении подвергали релаксации от давления 11 бар до нормального давления. При этом получали 14,4 г (0,84 моль) NH3. В теплую смесь с температурой 20oC при одновременном охлаждении воды добавляли раствор 2,5 r (0,063 моль) NaOH и 8,8 г (0,152 моль) ацетона в 122 г (6,77 моль) воды, причем внутренняя температура возрастала до 25oC. Перемешивание продолжали еще 35 мин, при такой же температуре. Затем добавляли 18,6 г (0,233 моль) 50%-ного раствора едкого натра и перемешивали при нормальном давлении 30 мин при 100-105oC. Затем перемешивали еще 30 мин, при 160oC и 15 бар. Полученный светло-желтый раствор фильтровали и концентрировали при нормальном давлении. Получали 102,3 г желтого раствора следующего состава:
40, 15 мас.% метионина, - 95,9% сухого продукта
Следы DS1
0,42 мас.% DS2, - 0,64% сухого продукта
< 10 млн.-1 цианида
Пример 19, Аналогично примеру 18 в автоклав помещали 51,0 г (1,80 моль) 60%-ного водного аммиака при 44oC. Затем при перемешивании в течение 1,5 мин добавляли 38,5 г (0, 292 моль) ММР-цианогидрина. Реакционную смесь перемешивали еще 11 мин при 55oC и при одновременном охлаждении подвергали релаксации от давления 6 бар до нормального давления. При этом получали 12,3 г NH3. В теплую смесь температурой 20oC при одновременном охлаждении воды добавляли раствор 2,5 г (0,063 моль) NaOH и 8,8 г (0,152 моль) ацетона в 115 г (6,38 моль) воды, причем внутренняя температура возрастала до 25oC. При такой же температуре продолжали перемешивание еще 32 мин. Затем добавляли 18,6 г (0,233 моль) 50%-ного раствора едкого натра и перемешивали еще 45 мин при 160oC и 21 бар. Полученный светло-желтый раствор фильтровали и концентрировали при нормальном давлении. После охлаждения до комнатной температуры получали 100,8 г желтого раствора следующего состава:
41,7 мас.% метионина, - 96,5% сухого продукта
< 0,04 мас.% DS1, - < 0,01% сухого продукта
0,42 мас.% DS2, - 0,6% сухого продукта
< 10 млн.-1 цианида.
Пример 20. Раствор метионината натрия получали аналогично примеру 19 из 50,0 г (1,76 моль) 60%-ного аммиака, 39,3 г (0,298 моль) ММР-цианогидрина, раствора 2,5 г (0,063 моль) NaOH и 3,6 г (0,062 моль) ацетона в 79,6 г (4,42 моль) воды и 18,8 г (0,235 моль) 50%-ного раствора едкого натра. Получали 101,8 г светло-коричневого раствора следующего состава:
41,2 мас.% метионина, - 94,3% сухого продукта
0,20 мас.% DS1, - 0,5% сухого продукта
0,66 мас.% DS2, - 1,0% сухого продукта
Пример 21 (сравнительный). Аналогично примеру 18 в автоклав при 50oC помещали 122,2 г (1,80 моль) 25%-ного водного аммиака. Затем при перемешивании добавляли в течение 1,5 мин 39,4 г (0,299 моль) ММР-цианогидрина. Реакционную смесь перемешивали еще 10 мин при 55oC и 2 бар и затем охлаждали. В теплую смесь с температурой 20oC, не оказывающую давления, добавляли 6,2 г (0,062 моль) 40%-ного раствора едкого натра и 3,6 г (0,062 моль) ацетона, причем внутренняя температура возрастала до 36oC. Затем смесь перемешивали еще 60 мин при 33oC. Далее добавляли 19,1 г (0,239 моль) 50%-ного раствора едкого натра и перемешивали еще 45 мин при 160oC и 21 бар. Полученный раствор фильтровали и концентрировали при нормальном давлении. После охлаждения до комнатной температуры получали 104,2 г красновато-коричневого раствора следующего состава:
39,65 мас.% метионина, - 92,6% сухого продукта
0,59 мас.% DS1, - 1,5% сухого продукта
0,56 мас.% DS2, - 0,8% сухого продукта
Пример 22. К нагреваемой гидравлической трубе с измерением внутренней температуры (поз. 2) через подключенный смесительный канал присоединяли подающую линию ММР-СН, связанную со сборником, и подающую линию аммиака, которая через промежуточно включенный теплообменник связана со стадией абсорбции и смешения (поз. 11). Гидравлическая труба (поз. 2) непосредственно связана со стадией гидролиза (поз. 4), которая является термостатической и состоит из петлевого реактора с циркуляционным насосом и подключенной гидравлической трубой. Петлевой реактор со стороны входа связан через транспортер и насос со сборником для водного раствора ацетона и NaOH и со стороны выхода через гидравлическую трубу и регулятор давления связан с реактором для омыления (поз. 5). Реактор для омыления состоит из ректификационной колонны, верхнее основание которой связано через подачу питания и насос с запасным сосудом раствора едкого натра и верхней частью через регулятор давления со стадией адсорбции (поз. 9). Нижняя часть состоит из циркуляционного выпарного аппарата. Конец нижней части реактора для омыления с регулируемым уровнем связан через промежуточный резервуар и насос с зоной последующей обработки (поз. 6). Зона последующей обработки состоит из прибора предварительного нагрева и подключенного объекта управления (штрека) продолжительностью реакции и непосредственно связан через термостатическую линию и регулятор давления при помощи фильтра (поз. 7). Фильтр состоит из тонкого фильтра, конец которого ведет в стадию выпаривания (поз. 8). Стадия выпаривания представляет собой систему выпаривания, состоящую из пленочного испарителя для осаждения, подключенного конденсатора и приемника продукта. Стадия адсорбции (поз. 9) функционирует как система адсорбции NH3, которая состоит из контейнера нижней части с циркуляционным насосом, тушильной установки и присоединенной ректификационной колонны. Верхняя часть колонны снабжена резервуаром со свежей водой посредством транспортера и насоса. Подающая линия вторичного пара в тушильной установке примыкает к верхней части колонны омыления (поз. 5) и подающей линии конденсата стадии выпаривания (поз. 8). Конец нижней части стадии адсорбции (поз. 9) связан через насос с дистилляционной установкой (поз. 10), который функционирует как система перегонки под давлением для непрерывного выделения NH3 из питательной смеси и выполнена в виде ректификационной колонны. Подача питания из стадии адсорбции (поз. 9) осуществляется в середине колонны.
Нижняя часть состоит из циркуляционного выпарного аппарата, который имеет охлаждаемый сток с регулируемым уровнем, через который он соединен с приемником. К циркуляционному выпарному аппарату присоединен запасной резервуар для водного раствора ацетона и NaOH.
Верхняя часть состоит из конденсатора с примыкающим приемником, который посредством регулятора потока связан со стадией абсорбции и смешения (поз. 11). Регулятор потока состоит из котла с мешалкой с постоянным давлением. Котел дополнительно снабжен жидким аммиаком с возможностью подачи свежей воды.
В гидравлическую трубу (поз. 2) в течение часа по дозам вводили в среднем 1,774 кг (13,47 моль) ММР-цианогидрина, а также 2,55 кг (89,8 моль) подогретого до 40oC 60%-ного аммиака из поз. 11 с регулируемым потоком. Установленная внутренняя температура составляла 55oC на входе трубы и 50oC на выходе трубы, давление составляло 7,8 бар, продолжительность реакции 23 мин. В петлевой реактор (поз. 4) в течение часа добавляли 6,0 кг водного раствора с 7,25 мас. % (7,50 моль) ацетона и 2,0 мас.% (3,00 моль) NaOH из запасного резервуара. Температура в петле составляла 29oC, в трубе реакции 25oC, общая продолжительность реакции составляла 50 мин. В реактор для омыления (поз. 5) добавляли в течение часа 0,86 кг (10,75 моль) 50%-ного раствора едкого натра. Температура составляла 132oC в нижней части и 118oC в верхней части колонны, давление - 1,7 бар, общая продолжительность реакции - 26 мин.
Путем охлаждения и добавления в течение часа в среднем 1 л воды при адсорбции пара (поз. 9) температуру поддерживали на уровне 19-22oC, а давление - на уровне 0,3-1,0 бар. В течение часа 5,5-6,5 л раствора подводили в колонну для перегонки под давлением. Температура в нижней части составляла 161-163oC и 28-29oC в верхней части, давление - 10 бар, установленное соотношение рециркуляции 0,5-0,6. В приемник нижней части собирали в течение часа в среднем 4,0 кг водного раствора, содержащего 11-12 мас.% ацетона и 2,0-2,7 мас. % NH3. В этот приемник добавляли определенное количество воды, ацетона и раствора едкого натра до тех пор, пока полученная общая концентрация не соответствовала упомянутой заданной концентрации. Затем этим раствором наполняли запасной резервуар для стадии гидролиза (поз. 4). В приемнике верхней части дистилляционной колонны в течение часа смешивали конденсированный аммиак (Поз. 11) вместе с 1 л свежей воды. Время от времени незначительные потери возмещали добавлением свежего NH3 и свежей воды. Температура в дополнительном реакторе (поз. 6) составляла 180oC, давление - 12 бар и продолжительность реакции - 20 мин. После релаксации до нормального давления и охлаждения до 90-95oC раствор фильтровали (поз. 7) и затем конденсировали в пленочном испарителе для осаждения (поз. 8) при 135-140oC и 0,5 бар. По окончании стадии выпаривания (поз. 8) получали приблизительно 4,69 кг водного окрашенного желтого раствора метионината натрия следующего состава:
40,7 мас.% метионина, - 95,0% сухого продукта
0,15 мас.% DS1, - 0,4% сухого продукта
0,18 мас.% DS2, - 0,3% сухого продукта
6,74 мас.% натрия
< 5 млн.-1 цианида
Пример 23. Способ осуществляли как в примере 22, причем между гидравлической трубой (поз. 2) и стадией гидролиза (поз. 4) была промежуточно включена стадия разрядки (поз. 3).
Стадия разрядки (поз. 3) представляет собой испарительный сосуд с перемешиванием, газовая камера которого связана со стадией адсорбции (поз. 9) и стоком нижней части через насос со стадией гидролиза (поз. 4).
Фильтр (поз. 7) состоит из двух соединенных в ряд фильтрующих слоев активированного угля, которые проходят снизу наверх таким образом, что конец второй колонны ведет в стадию выпаривания (поз. 8).
В гидравлическую трубу (поз. 2) по дозам вводили в течение часа примерно 1,931 кг (14,66 моль) ММР-цианогидрина, а также 2,61 кг (92,0 моль) подогретого до 40oC 60%-ного аммиака (из поз. 11) с отрегулированным потоком. Установленная внутренняя температура составляла 55oC на входе трубы и 56oC на выходе трубы, давление 7,8 бар, продолжительность реакции 23 мин. Реакционную смесь подвергали релаксации в соответствующей стадии поз. 3 до 0,9-1,2 бар при 30oC и помещали в петлевой реактор (поз. 4), в который одновременно в течение часа добавляли из запасного резервуара 6,0 кг водного раствора, содержащего 7,25 мас. % (7,5 моль) ацетона и 2,0 мас.% (3,0 моль) NaOH. Температура в петле составляла 26oC, в реакционной трубе 25oC, общая продолжительность реакции 50 мин.
В реактор для омыления (поз. 5) добавляли в течение часа 0,92 кг (11,50 моль) 50%-ного раствора едкого натра. Температура в нижней части составляла 135oC и в верхней части колонны 122oC, давление - 1,7 бар, общая продолжительность реакции - 26 мин.
Посредством охлаждения и добавления приблизительно 1,8 л воды при адсорбции пара (поз. 9) температуру поддерживали на уровне 21-27oC и давление - на уровне 0,9-1,2 бар. Из поз. 9 в колонну для перегонки под давлением подводили в течение часа 5,5-6,5 л раствора. Температура в нижней части составляла 160-161oC и в верхней части 28-29oC, давление - 10 бар, установленное соотношение рециркуляции - 0,5-0,6. В приемнике стока нижней части собирали в течение часа примерно 4,0 кг водного раствора, содержащего 10-12 мас. % ацетона и 2,0-3,0 мас.% NH3. Его смешивали с определенным количеством воды, ацетона и раствора едкого натра до тех пор, пока полученная общая концентрация не соответствовала вышеупомянутой заданной концентрации. Затем этим раствором наполняли запасной резервуар для стадии гидролиза (поз. 4). В приемнике верхней части дистилляционной колонны в течение часа смешивали 2,5 л конденсированного аммиака в поз. 11 с 1 л свежей водой. Время от времени незначительные потери возмещали добавлением свежего NH3 и свежей воды. Температура в дополнительном реакторе (поз. 6) составляла 180oC, давление - 12 бар и продолжительность реакции - 20 мин. После релаксации до нормального давления и охлаждения до 80oC раствор при этой температуре пропускали через 13 кг активированного угля (поз. 7) и затем концентрировали в пленочном испарителе для осаждения (поз. 8) при 135-140oC и 0,5 бар. В результате получали в поз. 8 приблизительно 4,945 кг водного окрашенного светло-желтого раствора метионината натрия следующего состава:
42,16 мас.% метионина, - 95,3% сухого продукта
0,18 мас.% DS1, - 0,4% сухого продукта
0,20 мас.% DS2, - 0,3% сухого продукта
6,74 мас.% натрия
< 2 млн.-1 цианида
Пример 24. Проводили синтез как в примере 23.
В гидравлическую трубу (поз. 2) в течение часа по дозам вводили приблизительно 1,978 кг (15,02 моль) ММР-цианогидрина, а также 2,01 кг (94,4 моль) подогретого до 40oC 80%-ного аммиака (из поз. 11) с отрегулированным потоком. Установленная внутренняя температура составляла 58oC на входе трубы и 56oC на выходе трубы, давление - 14,2 бар, продолжительность реакции - 24,6 мин. Реакционную смесь подвергали релаксации в соответствующей стадии (поз. 3) до 0,9-1,0 бар при 30oC и добавляли в петлевой реактор (поз. 4). Одновременно в течение часа из запасного резервуара добавляли 6,7 кг водного раствора, содержащего 6,55 мас.% (7,56 моль) ацетона и 1,8 мас.% (3,02 моль) NaOH. Температура в петле составляла 26oC, в реакционной трубе 25oC, общая продолжительность реакции - 50 мин.
В реактор для омыления (поз. 5) вводили в течение часа 0,96 кг (11,98 моль) 50%-ного раствора едкого натра. Температура составляла в нижней части 133oC и в верхней части колонны 118oC, давление - 1,7 бар, общая продолжительность реакции - 26 мин.
Посредством охлаждения и добавления приблизительно 2,4 л воды в час при адсорбции пара (поз. 9) поддерживали температуру на уровне 19-22oC и давление - на уровне 0,9-1,0 бар. Из поз. 9 в колонну для перегонки под давлением за час подводили 5,5-6,5 л раствора. Температура в нижней части составляла 159-161oC и в верхней части 28-29oC, давление - 10 бар, установленное соотношение рециркуляции 0,4-0,5. В приемник стока нижней части собирали за час приблизительно 4,0 кг водного раствора, содержащего 10-12 мас.% ацетона и 2,0-3,0 мас.% NH3. В этот раствор добавляли определенное количество воды, ацетона и раствора едкого натра до тех пор, пока полученная общая концентрация не соответствовала вышеупомянутой заданной концентрации. Затем раствором наполняли запасной резервуар для стадии гидролиза (поз. 4). В приемнике верхней части дистилляционной колонны смешивали 2,5 л в час конденсированного аммиака в поз. 11 с 0,4 л свежей воды в час. Время от времени незначительные потери восполняли добавлением свежего NH3 и свежей воды. Температура в дополнительном реакторе (поз. 6) составляла 180oC, давление - 12 бар и продолжительность реакции - 20 мин. После релаксации до нормального давления и охлаждения до 80oС раствор при этой температуре пропускали через 13 кг активированного угля (поз. 7). Затем раствор концентрировали в пленочном испарителе для осаждения (поз. 8) при 135-140oC и 0,5 бар. В результате в поз. 8 получали примерно 4,87 кг водного окрашенного светло-желтого раствора метионината натрия следующего состава:
44,25 мас.% метионина, - 96,2% сухого продукта
0,14 мас.% DS1, - 0,3% сухого продукта
0,18 мас.% DS2, - 0,2% сухого продукта
7,08 мас.% натрия
< 5 млн.-1 цианида
Пример 25. Раствор метионината кальция.
(Поз. 5). В 350-миллилитровый стальной автоклав с мешалкой, монометром, внутренним термометром и нагревательной баней вводили при перемешивании раствор 50,0 г (0,330 моль) метионинамида (97,8%-ный) в 223,2 г воды и 13,1 г (0,170 моль) Ca(ОН)2 (96%-ный). После нагревания до 130oC смесь перемешивали при этой температуре и давлении 2 бар в течение 120 мин. Реакционную смесь подвергали релаксации при одновременном охлаждении до нормального давления. Получали 286,2 г желтоватого фильтрата, содержащего 17,06 мас.% метионина, 99,2% сухого продукта и 0,08 мас.% метионинамида, 0,5% сухого продукта.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ МЕТИОНИНА | 2001 |
|
RU2265593C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВОДНЫХ РАСТВОРОВ МЕТИОНИНАТА НАТРИЯ И ПРИМЕНЕНИЕ ЭТИХ РАСТВОРОВ ДЛЯ ПОЛУЧЕНИЯ ГРАНУЛЯТОВ | 1999 |
|
RU2222526C2 |
| СПОСОБ ПОЛУЧЕНИЯ СОЛИ МЕТИОНИНА | 2012 |
|
RU2618042C2 |
| СПОСОБ ПРЕВРАЩЕНИЯ МЕТИЛМЕРКАПТОПРОПИОНОВОГО АЛЬДЕГИДА, ПОЛУЧАЕМОГО ИЗ СЫРЫХ АКРОЛЕИНА И МЕТИЛМЕРКАПТАНА | 2012 |
|
RU2615734C2 |
| СПОСОБ ПОЛУЧЕНИЯ АММОНИЕВОЙ СОЛИ 2-ГИДРОКСИ-4-МЕТИЛТИОМАСЛЯНОЙ КИСЛОТЫ | 2004 |
|
RU2355678C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2-ГИДРОКСИ-4-МЕТИЛТИОМАСЛЯНОЙ КИСЛОТЫ (ГИДРОКСИАНАЛОГАМЕТИОНИНА, ГАМ), ГАМ | 1995 |
|
RU2130925C1 |
| СПОСОБ ПОЛУЧЕНИЯ МЕТИОНИНА | 2016 |
|
RU2708258C2 |
| СПОСОБ ОЧИСТКИ СОДЕРЖАЩИХ ДИОКСИД УГЛЕРОДА ГАЗОВЫХ ПОТОКОВ | 2005 |
|
RU2388521C2 |
| СПОСОБ ПОЛУЧЕНИЯ D,L-МЕТИОНИНА ИЛИ ЕГО СОЛИ (ВАРИАНТЫ) | 1996 |
|
RU2176240C2 |
| Способ получения 30-60%-ного раствора метионината натрия | 1986 |
|
SU1598867A3 |
Изобретение относится к способу получения метионина, который применяют в качестве кормовой добавки, в частности, в виде водного раствора. Метионин получают прямым омылением метиониннитрилов до получения метионинамида в присутствии кетона и последующего омыления амида основанием, при этом во время и/или после омыления амида смесь аммиака, кетона и воды удаляют при температуре ≥ 85oC и/или вакууме. Используют метиониннитрил, полученный взаимодействием метилмеркаптопропиональдегида (MMP) с синильной кислотой и аммиаком или взаимодействием соответствующего циангидрина (CH) с аммиаком, или взаимодействием любой смеси MMP с синильной кислотой и соответствующего циангидрина с аммиаком. Данный способ осуществляют без выделения промежуточных продуктов, непрерывно, при минимальных потерях. Используемый кетон может быть рекуперирован с высоким выходом после омыления амида, и он не содержит побочных продуктов реакции, отделяют аммиак от кетона, причем получаемую при реакции смесь аммиака, кетона и воды разделяют в колонне под давлением. Если амид омыляют при ≥ 160oC, то предпочтительной является концентрация амида ≤ 25 мас.%. Метиониннитрил получают путем обработки до крайней мере 4 эквивалентом ≥ 50 мас.% аммиака при температуре от 40 до 80oC в течение от 5 до 60 мин. Остаток цианида разрушают нагреванием до температуры выше 150oC. 16 з.п. ф-лы 1 табл., 1 ил.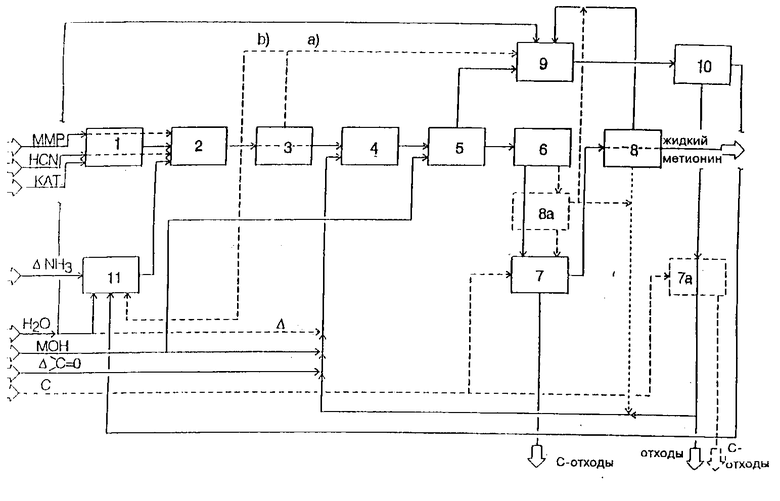
| EP 0228938, C 07 C 148/00, 1987. |

