Ноотропные соединения (усилители умственных возможностей) представляют собой перспективную группу лекарственных веществ. Известные ноотропные соединения включают пирацетам (N-карбамидо-метилпирролидон-2), который был широко введен в медицинскую практику в начале 80-х годов. В дальнейшем были синтезированы N-замещенные-2-пирролидоны (например, этирацетам, оксирацетам, анирацетам, прамирацетам, ролцирацетам и др.).
В патенте США N 4743616 Танаки и др. описаны N-ацилпирролидиновые соединения, имеющие эндопептидазную ингибиторную активность и оказывающие антиамнестическое действие. В отличие от производных пирацетама, соединения, описанные Танакой и др., содержат пролиновую группу.
Т. Гудашева и Р. Островская описали (Chem.Pharmac. J., 1985, N 11, pp. 1322-1329) биологически активные N-концевые производные пироглутаминовой кислоты, имеющие формулу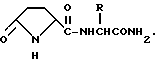
В другом источнике (Т.А.Гудашева и др. Chem.Pharmac. J., 1988, N 3, pp. 271-2759) описано биологически активное соединения N-ацилпролина, имеющее формулу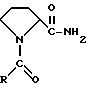
Необходимость в высокоактивных нетоксичных ноотропных препаратах, которые могут быть использованы для лечения ухудшений умственных способностей, вызванных различными деструктивными факторами, существует до сих пор. Настоящее изобретение направлено на удовлетворение этой потребности.
Настоящее изобретение относится к новой группе соединений, которые обладают различными типами психотропной активности, в частности антиамнестическим, антигипоксическим и анорексигенным действием.
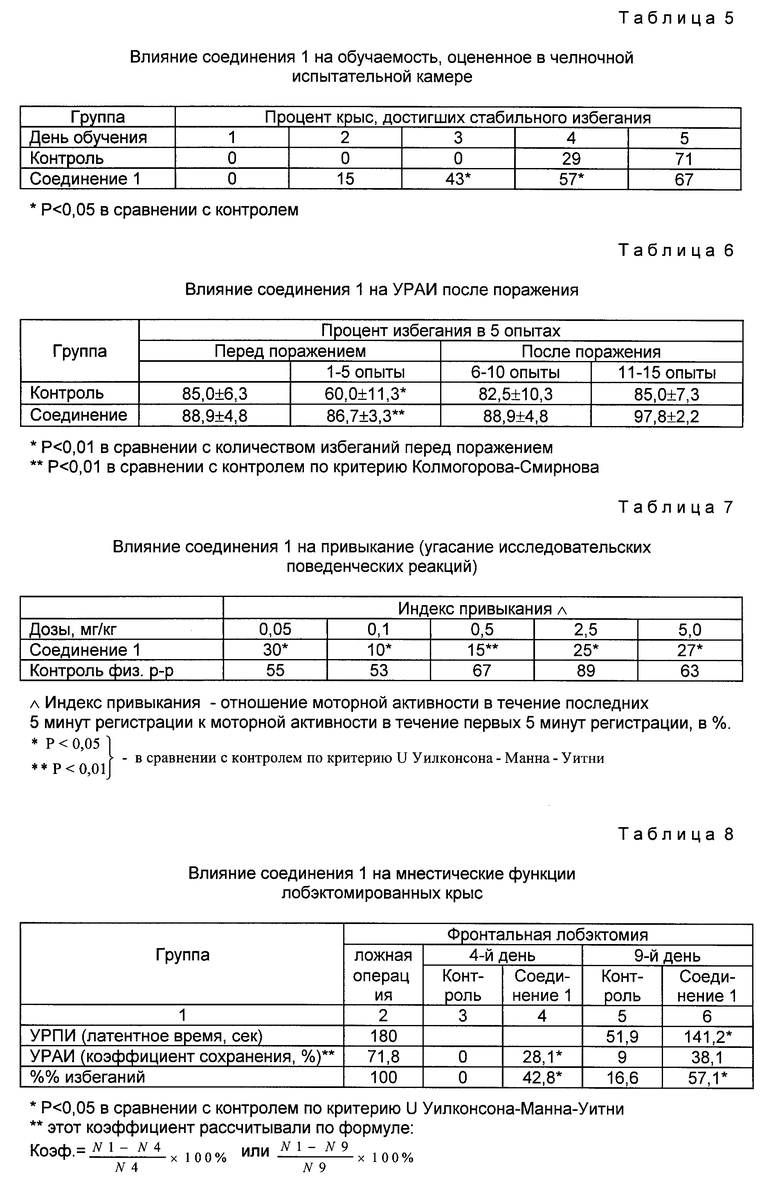
Согласно настоящему изобретению предложены производные N-ацилпролилдипептидов общей формулы I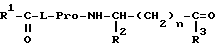
где
R1=(C4-C5)алкил, циклоалкил, аралкил или арил;
R2 = H, (C1-C4)алкил, карбамидоалкил или карбалкоксиалкил;
R3 = гидрокси, алкокси, амино, алкиламино или диалкиламино;
n = 0-2;
при этом
а) когда R1=фенил, R2=водород или изопропил, а n=0, R3 не метокси,
б) когда R1=трет-бутил, R2=водород, а n=0 или 1, R3 = не метиламино,
в) когда R1=трет-бутил, R2= изобутил, метил, бензил или карбамидобутил, а n=0, R3 не метиламино,
г) когда R1= трет-бутил, R2= карбометоксиметил, n=0, R3 не изопропиламино.
Производные N-ацилпролилдипептидов могут представлять собой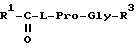
где
R1 = изобутил, пентил, 1-адамантил, фенил, фенилметил или фенилпропил
R3 = OH, OC2H5, NH2, NHCH3, N(CH3)2; или
где
R1 = фенилметил или фенил;
R3 = OC2H5 или NH2; или же
где
R1 = фенилметил или фенил.
Производное N-ацилпролилдипептида согласно изобретению может быть выбрано из следующих соединений:

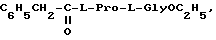


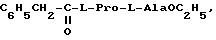
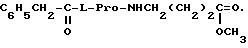
Соединения формулы I отличаются от известных ноотропных препаратов тем, что они содержат в своей структуре остаток природной аминокислоты L-пролина (вместо пирролидона), совместно с остатком второй природной аминокислоты. Эти N-ацил-L-пролиновые дипептиды имеют предельно низкую токсичность и высокоактивны.
Примерами осуществления изобретения могут служить следующие соединения:
I N-фенацетил-L-пролилглицинэтиловый эфир
II N-фенацетил-L-пролилглицинамид
III N-фенацетил-L-пролил -β- аланинэтиловый эфир
IV N-фенилацетил-L-пролил -β- аланинамид
V N-фенилацетил-L-пролил-L-аспарагиновой кислоты диэтиловый эфир
VI N-фенилацетил-L-пролил-L-аспарагинамид
VII N-бензоил-L-пролилглицинэтиловый эфир
VIII N-изовалерил-L-пролилглицинэтиловый эфир
IX N-фенилацетил-L-пролил-L-валинэтиловый эфир
X N-бензоил-L-пролил-L-валинэтиловый эфир
XI N-бензоил-L-пролил -β- аланинэтиловый эфир
XII N-бензоил-L-пролил -β- аланинамид
XIII N-бензоил-L-пролилглицинамид
XIV N-фенилацетил-L-пролилглицин-N-метиламид
XV N-фенилацетил-L-пролилглициндиметиламид
XVI N-фенилацетил-L-пролил-L-глутаминовой кислоты диэтиловый эфир
XVII N-фенилацетил-L-пролил-L-лейцинамид
XVIII N-фенилацетил-L-пролилглицин
XIX N-фенилацетил-L-пролил-ГАМК-метиловый эфир
XX N-фенилацетил-L-пролил-L-аланинэтиловый эфир
XXI N-капроил-L-пролилглицинэтиловый эфир
XXII N-(1-адамантоил)-L-пролилглицинэтиловый эфир
XXIII N-фенилбутирил-L-пролилглициновый эфир
Настоящее изобретение направлено также на получение фармацевтических составов и способы лечения, которые предусматривают использование в качестве активного вещества фармацевтически эффективных количеств N-ацилпролилдипептида формулы 1, как определено выше, предпочтительно соединение общей формулы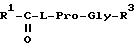
где
R1 выбирают предпочтительно из группы, состоящей из изобутила, пентила, L-адамантила, фенила, фенилметила и фенилпропила; R3 выбирают предпочтительно из группы, состоящей из NH2, NH CH3, N(CH3)2, OH и OC2H5. Более предпочтительно в фармацевтический состав и методы лечения включать согласно изобретению эффективную дозу соединения формулы
Здесь "фармацевтически эффективное количество" или "эффективное количество" обозначает количество, способное обеспечить эффективное лечение синдрома, из-за которого пациент страдает.
Соединения согласно изобретению обладают психотропной активностью, включая и антиамнестическую активность, и улучшают умственные функции, нарушенные вследствие мозговой травмы, интоксикации, старения и гипоксии. Эти соединения также проявляют анорексигенный эффект, антиалкогольную активность и уменьшают психические расстройства пренатально алкоголизированного потомства.
Настоящее изобретение относится также к различным способам лечения больных людей путем введения эффективной дозы соединения согласно изобретению. Такие способы предусматривают лечение людей, страдающих снижением умственных способностей вызванных мозговой травмой, старением, старческим слабоумием, и умственно отсталых детей; ожирения; нарушения центральной нервной системы (ЦНС) в результате химической интоксикации, преимущественно вызванной сатурнизмом; серповидной клеточной анемии; синдрома отмены бензодиазепина, который проявляется в виде агрессивности, тревожности и припадков; а также алкогольной зависимости.
Подробное описание изобретения
Приведенные выше соединения формулы 1 были получены хорошо известными способами синтеза пептидов. Обычный процесс получения рассматриваемых соединений состоит в смешивании и конденсации требуемых аминокислот, как правило, в гомогенной фазе.
Конденсация в гомогенной фазе может быть выполнена следующим образом:
а) конденсация аминокислоты, имеющей свободную карбоксильную группу и защищенную другую реакционноспособную группу, с аминокислотой, которая имеет свободную аминогруппу и защищенные другие реакционноспособные группы, в присутствии конденсирующего агента, такого как карбодиимид;
б) конденсация аминокислоты, имеющей активированную карбоксильную группу и защищенную другую реакционноспособную группу, с аминокислотой, которая имеет свободную аминогруппу и защищенные другие реакционноспособные группы;
в) конденсация аминокислоты, имеющей свободную карбоксильную группу и защищенную другую реакционноспособную группу, с аминокислотой, имеющей активированную аминогруппу и защищенные другие реакционноспособные группы.
Карбоксильная группа может быть активирована превращением ее в хлорангидридную, азидную, ангидридную группу или активированный эфир, такой как N-оксисукцинимидный, N-ок-сибензтриазольный, пентахлорфениловый или паранитрофениловый эфир. Аминогруппа может быть активирована превращением ее в фосфитамид или "фосфоразным" методом.
Наиболее общими для рассмотренных выше реакций конденсации являются: карбодиимидный метод; азидный метод; метод смешанных ангидридов; метод активированных эфиров. Эти методы описаны в "The Peptides". Vol.1. 1965 (Academic Press), E. Schroeder, K.Lubke, или в "The Peptides", Vol.1, 1979 (Academic Press) E.Cross, L.Meinhofen.
Предпочтительными методами конденсации при получении пептидов формулы 1 являются метод смешанных ангидридов или карбодиимидный метод. Реакцию конденсации, осуществляемую по методу смешанных ангидридов, предпочтительно проводят с соблюдением "Андерсоновских" условий (G.W.Anderson et al. J.Am. Chem. Soc, 89, 50-12-5017 (1967)). Карбоксильную составляющую (для образования смешанного ангидрида N-ацилпролина предпочтительно активируют изобутилхлорформиатом. Могут быть использованы также этилхлороформиат и метилхлороформиат.
Лучшими растворителями являются смесь этилацетата с диметилформамидом, чистый диметилформамид и хлороформ. Лучшими третичными основаниями являются N-метилфорфолин, N-этилморфолин и триэтиламин.
Реакцию конденсации, осуществляемую карбодиимидным методом, предпочтительно проводят в присутствии оксибензотриазола (W.Konig, R.Geiger, Chem. Ber., 103, 788-789 (1979)). Конденсация по карбодиимидному методу может быть также проведена в присутствии других добавок, таких как паранитрофенол, пентахлорфенол или N-оксисукцинимид.
Реакционноспособные группы, которые не должны участвовать в конденсации, могут быть защищены группами, которые легко удаляются, например, гидролизом или восстановлением. Так, карбоксильная группа может быть защищена этерфикацией этанолом, метанолом, трет-бутанолом, бензиловым спиртом.
Аминогруппу обычно эффективно защищают кислотными группами, например остатками алифатических, ароматических, гетероциклических карбоновых кислот, такими как ацетил, бензоил, пиридинкарбоксил, кислотными группами, производными от угольной кислоты, такими как этоксикарбонил, бензилоксикарбонил, третбутилоксикарбонил; или кислотными группами, производными от сульфокислоты, такой как пара-толуосульфониловая.
Термином "функциональные производные пептидов формулы 1" обозначены:
1) N-ацильные производные алифатической или ароматической кислот;
2) эфиры, производные от низших алифатических спиртов;
3) амиды, или моноалкил-, или диалкилзамещенные амиды, в которых алкильные группы имеют один или два C-атомов.
При синтезе заявляемых пролилдипептидов N-ацильные производные предпочтительно получали, используя пролин, который предварительно ацетилировали подходящей ацильной группой. Эта ацильная группа функционировала также в качестве защитной группы в дальнейшем ходе синтеза. Можно также ввести желательную аминогруппу после синтеза пептидов, ацилируя дипептиды обычными способами. Предпочтительными N-ацильными группами являются N-фенилацетил и N-бензоил.
Эфиры пептидов согласно формуле 1 получают предпочтительно путем использования аминокислоты в форме желаемого эфира. Они могут быть получены также соответствующей этерификацией полученного пептида. Предпочтительно, чтобы эфиры были производными метанола или этанола.
Амиды пептидов формулы 1 получают аммонолизом (т.е. реакцией с NH3) алкилэфира соответствующего дипептида или реакцией с аминокислотой в виде желательного амида. Амиды дипептидов также могут быть получены введением амидной группы в соответствующий дипептид каким-либо подходящим способом, например обработкой амином в присутствии конденсирующего агента. Более предпочтительны незамещенные амиды, монометил- и диметиламиды.
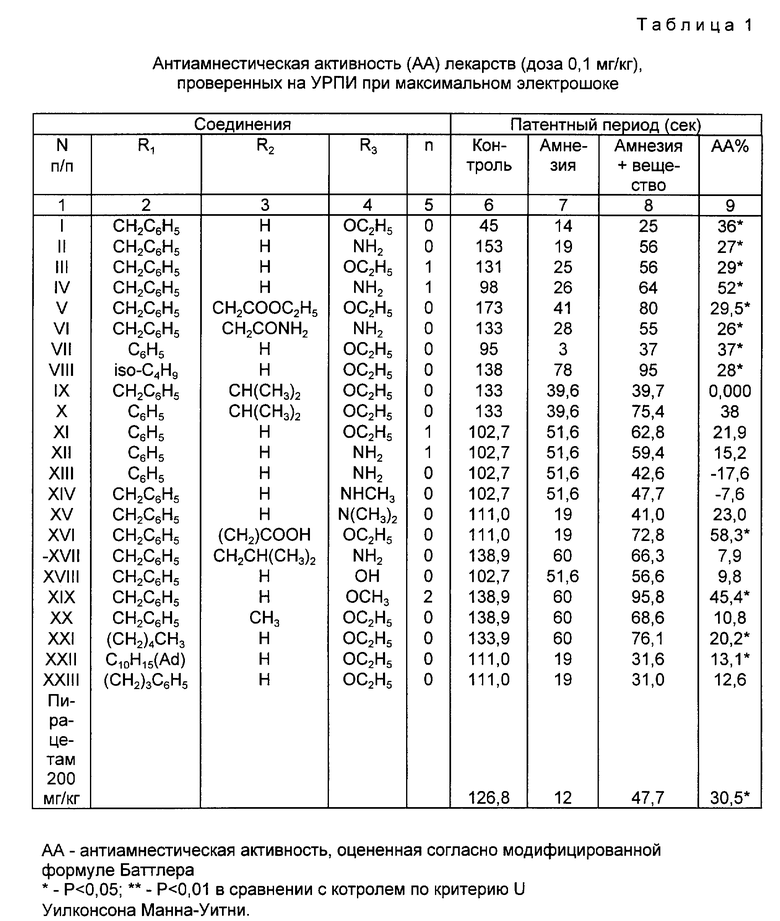
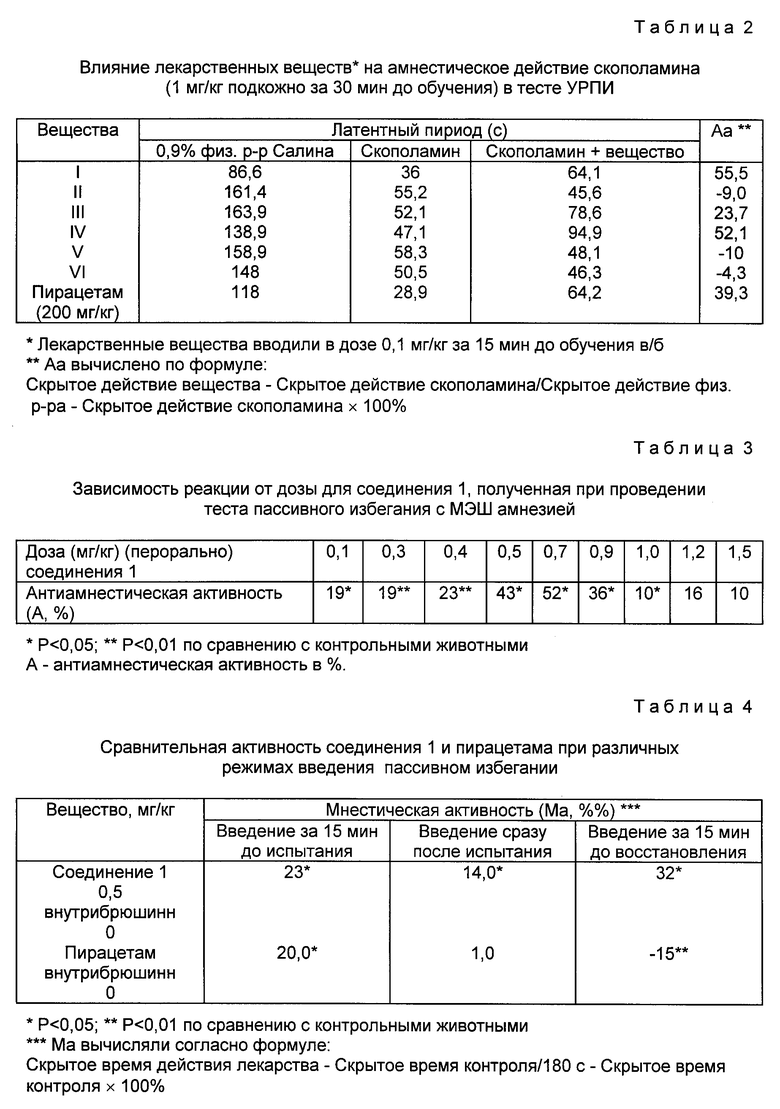
Была изучена способность соединений согласно изобретению препятствовать нарушению памяти, вызываемому максимальным электрошоком или приемом скополамина. В отличие от N-ацилпирролидиновых соединений, ранее описанных Танака и др. , соединения согласно изобретению не рассчитаны на подавлением активности пролилэндопептидазы. Как описано в примерах, испытанные соединения проявляют антиамнестическую активность (пример 12). Было показано, что предпочтительное соединение, имеющее формулу 1 (см. табл. 1), способно облегчить разные фазы формирования памяти: получение информации, сохранение и восстановление ее в памяти в различных процедурах пассивного и активного избегания (примеры 12, 13). Показано, что это соединение способно повышать скорость адаптации (пример 14). Соединение 1 предотвращает снижение умственных способностей, вызванное фронтальной лобэктомией (пример 15), пренатальной алкоголизацией и пренатальной гипоксией (пример 16), старением (пример 17). Оно проявляет также антигипоксическую активность (пример 18).
При наблюдении за тем, как пирацетам взывал некоторые из вышеописанных эффектов, установлено, что уровень активной дозы для него составляет 200-800 мг/кг. В отличие от этого заявляемого соединения вызывает такие же эффекты при дозах приблизительно 0,1-0,5 мг/кг.
Описанное здесь соединение 1 имеет дополнительное полезное свойство - анорексигенное действие. В отличие от известных анорексигенных лекарств соединение 1 не вызывает адренергической стимуляции (возбуждения, увеличения кровяного давления и др.). Соединение 1, кроме того, нетоксично (пример 20) и действует при пероральном приеме (пример 12). Показано, что соединение 1 способно ослаблять абстинентный синдром при отмене бензодиазепина, уменьшая тревожность, агрессивность и выраженность пентилентетразолового киндлинга (пример 21).
Вещества согласно изобретению могут быть использованы в любой форме, которая пригодна для перорального приема, такой как пилюли, таблетки, драже. Описанные здесь вещества могут быть также использованы парентерально путем инъекций или вливаний в форме фармацевтических препаратов, содержащих активный ингредиент в сочетании с фармацевтически допустимым носителем. Вещества согласно изобретению могут быть использованы для лечения больных со снижением умственных способностей, вызванным мозговой травмой, интоксикациями, процессами старения, синдром Корсакова, болезнью Альцгеймера, органическим мозговым синдромом, алкоголизмом, включая пренатальные алкогольные повреждения, гипоксией , задержкой умственного развития детей, ожирением, инсультом, мозговыми нарушениями вследствие врожденных дисфункций или генетических отклонений, нарушением ЦНС в результате химической интоксикации, включая сатурнизм для лечения от токсикомании, включая поддержание абстиненции и отмены, и некоторыми гематологическими отклонениями, включая серповидноклеточную анемию. Предпочтительно эти вещества могут быть использованы в дозах 0,5-5,0 мг в день.
Далее изобретение подробно описано с учетом различных специфических и предпочтительных применений и в дальнейшем будет описываться со ссылками на последующие примеры. Очевидно, однако, что имеется множество вариантов расширения, вариаций и модификаций, выходящих за рамки приведенных примеров и подробного описания, но соответствующих общему изобретательскому замыслу и объему притязаний.
Примеры.
В дальнейшем используются следующие сокращения:
Pro - пролил
Asp - аспартил
Аsn - аспарагинил
Val - валил
Ala - аланил
β-Ala -β-аланил
Leu - лейцил
Gly - глицил
Glu - глутамил
EtOH - этанол
MeOH - метанол
DMF - диметилформамид
DCC - дициклогексилкарбодимид
DCU - дициклогексилмочевина
EtAC - этилацетат
BZ - бензоил
Ad - 1-адамантил
OHBt - 1-оксибензтриазол
Et3N - триэтиламин
Phac - фенилацетил
TLC - тонкослойная хроматография
В примерах была использована следующая аппаратура:
точки плавления определяли по серно-кислотному прибору в открытых капиллярах без корректировки;
удельное вращение плоскости поляризации света регистрировали на автоматическом поляриметре Perkin-Elmer-241;
спектры ядерного магнитного резонанса (ЯМР) были получены на спектрометре Bruker AC-250;
Химические сдвиги выражали в млн-1 относительно Me4Si.
Для обозначения резонансных сигналов использовали следующие сокращения: s - синглет, d - дуплет, t - триплет, g - квадруплет, m - мультиплет.
Константы спин-спинового взаимодействия даны в Гц.
Тонкослойную хроматографию (TLC) выполняли на силикагелевых пластинах 60F254 Merck с проявлением в иодной камере или ультрафиолетовых лучах.
В экспериментах на живых организмах использовали самцов беспородных крыс-альбиносов массой приблизительно 180-220 г и/или самцов беспородных мышей-альбиносов массой около 18-22 г.
В алкогольных тестах на живых организмах использовали потомство алкоголизированных матерей подобного вида. Самцов крыс-альбиносов линии Вистар в возрасте 24-26 месяцев использовали в экспериментах, связанных со старением.
Пример 1.
Синтез этилового эфира из N-фенилацетил-L-пропил-глицина
N-Phac-L-Pro-Gly-O Et (1)
a) N-фенилацетил-L-пропил, N-Phac-L-Pro-OH
К 5,75 г (0,05 моль) L-пролина в 25 мл 2N NaOH добавляли по каплям с перемешиванием при температуре ниже 10oC 12,5 мл 4N NaOH и 6,6 мл (0,05 моль) N-фенилацетилхлорида (tкип 89 - 90oC/10 Torr). Реакционную смесь перемешивали 15 мин, экстрагировали EtAc для удаления хлорида, затем подкисляли 2N HCl до pH 3, экстрагировали хлороформом, высушивали сульфатом натрия и выпаривали. Получали 6,2 г N-Phac-L-Pro-OH в виде белых кристаллов c: tпл 150 - 152oC, [α]
1H-ЯМР-спектр в CDCl1 δ/ (млн-1): 1,77 - 2,29 (m, CβH2-CγH2 , Pro, 4H); 3,40 - 3,63 (m, CδH2 , Pro, 2H); 3,63 и 3,73 (каждый s, CH2-C6H5, 2H); 4,56 и 4,38 (каждый dd, CαH Pro, 1H); 7,18 - 7,39 (m, CH2C6H5, 5H); 11,38 (ушир. s, COOH, 1H).
Рассчитано для C13H15NO3 : C = 66,93; H = 6,49; N = 6,00.
Найдено: C = 66,65; H = 6,40; N = 5,86.
б) Этиловый эфир N-фенилацетил-L-пролилглицина
В раствор 2,33 г (0,01 моль) N-Phac-L-Pro-OH (tпл 150 - 151oC) в 50 мл DMF добавляли с перемешиванием при -10oC 1,39 мл (0,01 моль) Et3N и затем 1,34 мл (0,01 моль) изобутилхлорформиата. Через 2 мин к реакционной смеси добавляли по каплям в течение 25 мин, 1,4 г (0,01 моль) гидрохлорида глицинэтилового эфира (tпл 140 - 142oC) и раствор 1,39 мл (0,01 моль) Et3N в 25 мл DMF, чтобы избежать повышения температуры. Затем перемешивание продолжали при охлаждении в течение 30 мин и при комнатной температуре в течение 1,5 ч. Осадок отфильтровывали и фильтрат выпаривали в вакууме, осадок растворяли в CHCl3, раствор промывали 5% NaHCO3, водой, 1N HCl и вновь водой, высушивали сульфатом натрия и выпаривали. Образованные 1,66 г (54%) масла смешивали с эфиром до получения белых кристаллов: tпл 96 - 97oC; [α]
1H-ЯМР спектр в (CD3)2SO, δ (млн-1): 1,18 (t, CH3CH2O, 55% 5H); 1,17 (t, CH3CH2O, 45% 3H); 1,65 - 2,35 (m, CβH2-CγH2Pro , 4H); 3,2 - 3,4 (m, CδH2 , Pro, 2H); 3,40 (s, CH2-С6H5, 45% H); 3,67 (s, CH2-C6H5, 55% 2H); 3,80 (d, CαH2Gly , J = 5,9, 55% 2H); 3,86 (d, J = 5,9, CαH2Gly , 45% 2H); 4,08 (q, CH3CH2-O, 55% 2H); 4,09 (q, CH3CH2-O, 45% 2H); 4,32 (dd, CαH , Pro, 55% 1H); 4,48 (dd, CαH , Pro, 45% 1H); 7,1 - 7,6 (m, CH2C6H5, 5H); 8,29 (t, J = 5,9, NH Gly, 55% 1H); 8,63 (t, J = 5,9 NH Gly, 45% 1H).
Рассчитано для C17H22N4O4: C = 64,15; H=6,92; N=8,80.
Найдено: C=63,93; H=6,81; N=9,07.
Пример 2.
Синтез N-фенилацетил-L-пролилглицинамида Phac-L-Pro-Gly- NH2 (II)
N-Phac-L-Pro-Gly-OEt (0,53 г; tпл 96 - 97oC [α]
1H-ЯМР спектр в CHCl3, δ (млн-1) : 1,8 - 2,3 (m, CβH2-CγH2 , Pro, 4H); 3,3 - 3,5 (m, CδH2 , Pro, 2H); 3,55 - 3,75 (m, AB-часть ABX-системы, CαH2Gly , 2H); 3,66 (s, CH2C6H5, 2H): 4,07 (dd, CαH , Pro, 1H); 4,37 (t, NHGly, 1H); 5,63 и 7,86 (каждый s, NH2); 7,2 - 7,4 (m, C6H5, 5H).
Рассчитано для C15H19N3O3 : C=62,53; H=6,48; N=14,61.
Найдено: C=62,28; H=6,54; N=14,53.
Пример 3.
Синтез этилового эфира N-фенил-L-пролил -β- аланина
N-Phac-L-Pro -β- Ala-OEt (III)
К раствору 0,61 г (2,6 ммоль) N-фенилацетил-L-пролина (tпл 151oC, [α]
1H-ЯМР спектр с (CD3)2SO, δ (млн-1): 1,17 (t, CH3CH2-O, 68% 3H); 1,13 (t, CH3CH2O, 32% 3H); 1,7-2,2 (m, CβH2-CγH2, Pro, 4H); 2,42 (m, CαH2β-Ala, 2H); 3,2-3,3 (m, CδH2, Pro, 2H); -3,40 (s, CH2-C6H5, 32% 2H) при HDO-сигнале; 3,66 (m, CH2-C6H5, 68% 2H); 4,01 (q, CH3CH2O, 32% 2H); 4,04 (q, CH3-CH2O, 68% 2H); 4,41 (m, CβH2β-Ala, 2H); 4,21 (dd, CαH, Pro, 1H); 7,1-7,36 (m, CH2-C6H5, 5H); 7,93 (t, NH, 1H).
Рассчитано для C18H24N2O4: C=65,03; H=7,29; N=8,42.
Найдено: C=65,37; H=7,77; N=8,38.
Пример 4.
Синтез N-фенилацетил-L-пролил -β аланинового амида N-Phac-L-Pro -β- Ala-NH2 (IV)
Для сатурации этанольного раствора (15 мл) 0,36 г N-фенилацетил-L-пролил -β- аланин-этилового эфира, полученного в примере 3, барботировали газообразный аммиак. Отстоявшийся в течение ночи раствор выпаривали, а остаток очищали в хроматографической колонке (силикагель), используя CHCl3 в качестве элюента. Амид IV был получен в виде масла: выход 0,22 г (61%); Rf= 0,28 (силикагель, CHCl3 - метанол, 9:1); [α]
1H-ЯМР (Me2SO-d6), δ/ (млн-1); 1,69-2,2 (m, CβH2-CγH2 Pro, 4H); 2,16-2,31 (m, CαH2β-Ala 2H); 3,1-3,3 (m, CβH2 , β-Ala , 2H); 3,3-3,45 (m, CδH2 Pro, 2H); 3,66 (s, CH2-C6H5, 2H); 4,22 и 4,41 (каждый dd, CαH Pro, 1H); 6,84, 7,36 и 6,86, 7,38 (каждый шир. s, NH2, 2H); 7,12-7,35 (m, CH2C6H5, 5H); 7,89 и 8,22 (каждый t, NH β-Ala , 1H).
Рассчитано для C16H21N3O3: C=63,34; H=6,99; N=13,85.
Найдено: C=63,81; H=7,03; N=14,01.
Пример 5.
Синтез диэтилового эфира N-фенилацетил-L-пролил-L-аспарагиновой кислоты N-Phac-L-Pro-L-Asp (OEt)2, (V)
К хорошо перемешанному раствору 1 г (4,3 ммоль) N-фенилацетил-L-пролина в 40 мл абсолютного EtAc добавили 0,48 мл (4,3 ммоль) N-метилморфолина при -10oC. После этого последовательно добавляли 0,57 мл (4,3 ммоль) изо-BuOC(O)Cl, через 2-3 мин - смесь 0,91 г (4,3 ммоль) L-Asp(OEt)2HCl; 0,48 мл (4,3 ммол) N-метилморфолина и 15 мл EtAc. Перемешивание продолжали в течение 1 ч при -10oC. После того как смесь отстоялась в течение 1 часа, остаток отфильтровывали, раствор выпаривали и осадок растворяли в смеси этанола и эфира. Полученный в результате твердый остаток отделяли, маточный раствор выпаривали и осадок (1,65 г) очищали в хроматографический колонке (силикагель) с использованием CHCl3 и смеси CHCl3-EtOH в качестве элюентов. Диэтилэфир N-фенилацетил-L-пролил-L-аспарагиновой кислоты был получен в виде масла: Rf=0,87 (силикагель, CHCl3-EtOH, 9:3); [α]
1H-ЯМР (Me2SO-d6), δ (млн-1): 1,23 (t, J=7,16 Hz, CH3CH2O, 90% 3H); 1,24 (t, J=7,16, CH3CH2O, 10% 3H); 4,10 (q, CH3CH2-O, 90% 2H); 4,12 (q, CH3CH2-O, 10% 2H); 1,25 (t, J=7,14, CH3CH2O, 90% 3H); 1,26 (t, J=7,14, CH3CH2O, 10% 3H); 4,19 (q, CH3CH2-O, 90% 2H); 4,21 (q, CH3CH2O, 10% 2H); 1,75-2,40 (m, CβH2-CγH2 Pro, 4H); 3,45-3,65 (m, CδH2 , Pro, 2H); 4,58 (dd, J=8,00; J=2,59; CαH Pro, 1H); 2,78; 2,95 (dd, AB-часть ABX-системы, JAB=17,04; JAX=4,88; JBX = 4,88 CβH2 Asp, 90% 2H); 2,80; 3,00 (dd CβH2 Asp, 10% 2H); 4,81 (dt, J=CαH, NH 8,50; CαH Asp 1H); 7,5 (d, J=8,50; NH Asp, 90% 1H); 7,03 (d, J=8,40; NHAsp, 10% 1H); 3,70 (s, CH2C6H5, 2H); 7,20-7,36 (m, C6H5, 5H).
Рассчитано для C21H28N2O6: C=62,35; H=6,99; N=6,92.
Найдено: C=62,63; H=7,01; N=6,74.
Пример 6.
Синтез амида N-фенилацетил-L-пролил-L-аспарагина
N-Phac-L-Pro-L-Asn-NH2 (VI)
Для сатурации метанольного раствора (25 мл) 0,5 г N-Phac-L-Pro-L-Asp(OEt)2, полученного так же, как в примере 5, через него барботировали газообразный аммиак при 0oC. После отстаивания в течение ночи раствор выпаривали, осадок растворяли в нагретой смеси этанола с хлороформом и к остатку добавляли пентан. Полученный в результате остаток собирали и высушивали; выход составлял 0,45 г амида N-фенилацетил-L-пролил- L-аспарагина: tпл 170 - 172oC; Rf = 0,24 (силикагель, CHCl3-EtOH, 9:3), [α]
1H-ЯМР (Мe2SO-d6), δ (млн-1): 1,60 - 2,30 (m, CβH2-CγH2 , Pro, 4H); 2,35 - 2,50 (m, CβH2 , Asn, 2H) при сигнале растворителя; 3,63 (s, CH2=C6H5, 2H), 4,36 (m, CαH Pro, 1H); 4,4 - 4,6 (m, CαH Asn, 1H); 6,80 - 7,60 (m, C6H5, 5H); 6,8 - 7,1 (s, NH2, 4H); 8,15; 8,25; 8,35 (каждый d, NH Asn, 1H).
Рассчитано для C17H22N4O4: C = 58,94; H = 6,41; N = 16,16.
Найдено: C = 59,23; H = 6,66; N = 6,04.
Пример 7.
Синтез этилового эфира N-бензоил-L-пролилглицина, N-BZ-L-Pro-Gly-OEt (VII)
а) N-бензоил-L-пролин
К хорошо перемешиваемому раствору L-пролина (5,75 г, 0,05 моль) в 2N NaOH (25 мл) добавляли капельно из разных капельных воронок 4N NaOH (12,5 мл) и бензоилхлорид (5,8 мл, 0,05 моль), поддерживая температуру около 0 - 4oC. Смесь через 15 мин подкисляли 1N HCl. Выделившееся масло экстрагировали CHCl3, смешанный органический экстракт высушивали (MgSO4) и растворитель удаляли. К осадку добавляли эфир и смесь отстаивали в течение ночи при 0oC. Отделяли кристаллы N-бензоил-L-пролина. Выход составлял 3,42 г (60%), tкип 152 - 154oC, [α]
Рассчитано для C12H13NO3: C = 65,73; H = 5,99; N = 6,39.
Найдено: C = 65,64; H = 6,03; N 6,54.
б) Этиловый эфир N-бензоил-L-пролилглицина
К хорошо перемешиваемому раствору N-бензоил-L-пролина (2,19 г, 0,01 моль) в смеси абсолютного EtAc (50 мл) и DMF (10 мл) добавляли капельно при -10oC N-метилморфин (1,12 мл, 0,01 моль) и изо-BuOC(O)Cl (1,34 мл, 0,01 моль). Через 2 мин добавляли смесь NH2CH2COOEt • HCl (1,4 г, 0,01 моль), N-метилморфолина (1,12 мл, 0,01 моль), и DMF (20 мл). После 30-минутного перемешивания с охлаждением и полуторачасового при комнатной температуре растворитель удаляли в вакууме. Остаток растворяли в хлороформе, промывали 5%-ным водным раствором NaHCO3, 1N HCl, водой и затем высушивали с помощью Na2SO4. После фильтрации растворитель выпаривали, к осадку добавляли эфир и отделяли полученные кристаллы эфира VII. Выход 1,67 г (76%); tкип 63 - 65oC [α]
1H-ЯМР (Me2SO-d6) δ (млн-1): 1,18 (t, J = 7,04, CH3CH2O, 75% 3H); 1,09 (t, J = 7,04, CH3CH2O, 25% 3H); 1,73 - 2,28 (m, CβH2-CγH2 , Pro, 4H); 3,3 - 3,4 (m, CγH2 Pro, 2H); 3,60 и 3,74 (dd, J = 5,87 и J = 8,22; CαH2Gly , 25% 2H); 3,85 (d, J = 5,87, CαH2Gly , 75%, 2H); 4,10 (q, CH3 CH2O), 75 % 2H); 4,13 (q, CH3CH2O, 25% 2H); 4,47 (dd, CαH Pro, 25% 1H); 4,48 (dd, CαH Pro, 75% 1H), 7,33 - 7,62 (m, C6H5, 5H); 8,36 (t, J = 5,87; NH, 75% 1H); 8,40 (t, J = 5,87; NH, 25% 1H).
Рассчитано для C16H20N2O4: C = 63,14; H = 6,16; N = 9,20.
Найдено: C = 63,56; H = 6,87; N = 9,48.
Пример 8.
Синтез этилового эфира N-изовалерил-L-пролилглицина
Изо-C4H9C(O)-LPro-Gly-OЕt (VIII)
а) N-изовалерил-L-пролин
К хорошо перемешиваемому раствору L-пролина (1,15 г; 0,01 моль) в 2N- водн. NaOH (5 мл) добавляли одновременно по каналам при температуре около 0 - 4oC 4N-водн. NaOH (2,5 мл) и изовалерил хлорид (1,4 мл; 0,012 моль). Через 15 мин смесь подкисляли (pH 2 - 3) 1N HCl и экстрагировали с помощью CHCl3. Смешанный органический экстракт высушивали, используя MgSO4 и растворитель удаляли. N-изовалерил-L-пролин был получен в виде масла. Выход составил 0,85 г; Rf = 0,66 (силикагель, n-C4H9OH-AcOH-H2O, 5:1:2); [α]
1H-ЯМР (CDCl3), δ/ (млн-1): 0,99 (d, -CH(CH3)2, 6H); 2,25 (m, CH(CH3)2, 1H); 2,3 (m, CH2CH(CH3)2, 2H); 1,9 - 2,65 (m, CβH2-CγH2, 4H); 3,45 - 3,70 (m, CδH2 Pro, 2H); 8,85 (шир. s, COOH, 1H).
Рассчитано для C10H17NO3: C = 60,27; H = 8,62; N = 7,02.
Найдено: C = 60,34; H = 8,74; N = 7,23.
б) Этиловый эфир N-изовалерил-L-пролилглицина (VIII)
К хорошо перемешиваемому раствору N-изовалерил-L-пролина (0,74 г; 0,0037 моль) в abs.EtAc (15 мл) добавляли N-этилморфолин (0,45 мл; 0,0037 моль) и изо-BuOC(O)Cl (0,5 мл; 0,0037 моль). Через 2 - 3 мин капельно добавляли при температуре от -10 до -5oC смесь NH2CH2COOEt • HCl (0,52 г; 0,0037 моль), N-этилморфолина (0,47 мл, 0,0037 моль) и DMF (10 мл). Перемешивание продолжали в течение 30 мин с охлаждением и 1,5 ч при комнатной температуре. Остаток отделяли фильтрацией и маточную жидкость выпаривали. Осадок растворяли в хлороформе, промываемом 5%-ным водным раствором NaHCO3, водой, IN водным раствором HCl, водой и затем высушивают посредством MgSO4. После фильтрации растворитель выпаривают, а остаток (0,6 г) очищают в хроматографической колонке (силикагель), используя CHCl3 в качестве элюента. Получают 0,49 г (49%) эфира VIII в виде прозрачного масла, Rf = 0,55 (силикагель, CHCl3-MeOH 9:1), [α]
Найдено, % : C 58,78; H 8,74; N 9,87 C14H24H2O4
Вычислено, % : C 59,12; H 8,52; N 9,85.
Пример 9.
Этиловый эфир N-фенилацетил - L - пролил- L -валина,
N - Phac - L - Pro - L - Val - OC2H5 (IX)
Получен в условиях примера 1 из 2,33 г (0,01 м) N - фенилацетил- L -пролина и 1,8 г (0,01 моль) хлоргидрата этилового эфира L -валина с выходом 72% в виде масла, Rf = 0,64 (силикагель, диоксан-вода 9:1), [α]
ПМР спектры CDCl3, δ м.д.): 0,83 и 0,86 (каждый д, J = 6,9, CβH(CH3)2Val 90% 6H); 0,89 и 0,95 (каждый д, J=6,9, CH(CH3)2 Val, 10% 6H); 1,27 (т, CH3-CH2-O, 90% 3H), 1,28 (т, CH3-CH2-O, 10% 3H); 1,7-2,5 (м,CβH2-CγH2Pro,4H) 2,25 (м,CβH Val,1H); 3,4-3,7 (м,CβH2Pro,2H); 3,7 (с, CH2Ar, 2H); 4,18 (кв, CH3CH2-O, 2H); 4,38 (д.д.,CαHVal,J=8,4, J= 4,9, 90% 1H); 4,54 (д.д., CαH Val,10% 1H);4,68(д.д.CαHPro1H); 7,28 (м, C6H5, 5H); 7,44 (д., J=8,4; NH 90% 1H); 6,48 (д, NH, 10% 1H).
Найдено, %: C 66,58; H 7,74; N 7,85 C20H28N2O4
Вычислено, %: C 66,63; H 7,84; N 7,76.
Пример 10.
Этиловый эфир N-бензоил- L -пролил- L -валина,
N-Bz-L-Pro-L-Val-OEt (X)
Получен в условиях примера 1 из 2,2 г (0,01 моль) N-бензоил-L-пролина и 1,8 г (0,01 моль) хлоргидрата этилового эфира L-валина. Выход 2,6 г (72,3%), т. пл. 96-97oC (из эфира); Rf=0,61 (силикагель, диоксан-вода 9:1), Rf 0,86 (силикагель, CHCl3-C2H5OH 3:1); [α]
ПМР-спектр в CDCl3, δ (м. д. ) : 0,92 и 0,96 (каждый д, J=6,9 Гц, CβH(CH3)2Val6H); 1,28 (т, CH3CH2O, 3H); 1,75-2,7 (м,CβH2-CγH2Pro4H), 2,21 (м,CβH Val,1H), 3,36-3,70 (м, Cδ H2 Pro, 2H), 4,21 (кв, CH2-CH2-O, 2H), 4,51 (д. д. J=8,6, J = 6,7CαHVal, 1H), 4,82 (д.д.,CαHPro,1H), 6,40 (уш.с., NE 5% 1H), 7,3-7,6 (м, C6H5 и NH 5H + 95% 1H).
Найдено, %: C 65,54; H 7,47; N 8,21, C19H26N2O4
Вычислено,% : C 65,86; H 7,58; N 8,08.
Пример 11.
N-бензоил- L -пропил -β- аланинэтилового эфира.
N-BZ-L-PrO-β-Ala-Oεt (XI)
К раствору в ДМФА (25 мл), 0,9 г (6 ммоль) β-Ala-Oεt•HCl, 1,3 г (6 ммоль) N-BZ-L-Pro-OH и 0,87 г (6,4 ммоль) N-гидроксибензтриазола добавляли 0,83 мл (6,01 ммоль) триэтиламина. Смесь была охлаждена до 3oC, и в нее добавили 1,54 г (7,5 ммоль) DCC. Смесь отстаивалась при 3oC всю ночь и при комнатной температуре 1,5 часа, а затем ее охладили до -15oC. Полученный DCU удалили фильтрацией. Для фильтрации добавили водный калий бикарбонат и получили остаток. Он был профильтрован, промыт водой и выкристаллизован из смеси этанола с водой. Твердая часть профильтрована, промыта и высушена. Получено 1,5 г (78,9%) продукта: tпл 88-98oC, [α]
H-ЯМР спектры DMSO-d6, δ (млн-1 : 1,03 и 1,07 (каждый t, CH3-CH2O, 3H); 1,6-2,3 (м,CβH2-CγH2Pro4H); 2,45 (t,CαH2β-Ala,2H); 4,04 (g, CH3CHO, 2H); 3,14 и 3,40 (каждый m, Cβ H2Ala, 2H); 3,3-3,6 (m, Cγ H2 Pro, 2H); 4,37 и 4,14 (каждый dd CαH Pro, 1H); 7,33-7,62 (m, C6H5, 5H); 8,02 и 8,00 (каждый, t, NH, 1H).
Рассчитано для C17H22N2O6 : C = 64,15; H = 6,92; N 8,80
Найдено: C=64,23; H=7,03; N=8,91
Пример 12.
Амид N-бензоил-L-пролил -β- аланина,
N-BZ-L-Pro -β- Ala-Nh2 (XII).
получен в условиях примера 2 из этилового эфира N-бензоил-L-пролил -β-/ аланина с выходом 72%, т. пл. 135-137oC (из эфира); Rf=0,21 (силикагель, CHCl3-C2H5OH 9:1); [α]
Найдено, %: C 62,31; H 6,70; N 14,58 C15H19N3O3
Вычислено, %: C 62,28, H 6,57, N 14,53.
Пример 13.
Амид N-бензоил-Т-пропилилицина, N-Bz-L-Pro-Gly-NH2(XIII)
Получен в условиях примера 4 из этилового эфира N-бензоил-L-пролилицина с выходом 75%, т.пл. 64-74oC (гигр.), Rf=0,54 (силикагель, CHCl3-C2H5OH, 3: 1), Rf=0,3 (силикагель, CHCl3-C2H5OH, 9:1), Rf=-17,9o (с = 0,45, CHCl3).
ПМР спектр в (CD3)2SO, δ (м.д.): 1,68-2,0 и 2,05-2,30 (м,CβH2-CγH2Pro,4H), 3,3-3,45 (м, CH2 под НДО, 2H), 3,53-3,75 (м,CβH2-Gly,2H), 4,20 и 4,40 (каждый, м CαHPro 1H и 90% 1H), 6,95-7,65 (м, C6H5 и NH2, 7H), 8,05 и 8,41 (каждый т, NH, 10% 1H + 90% 1H),
Найдено, %: C 61,32, H 6,31, N15,21
C14H17N3O3
Вычислено,%: C 61,09 H 6,20, N 15,27
Пример 14.
N-фенилацетил-L-пролилглицинметиламида
N-Phac-L-Pro-Gly-NHMe (XIV)
Раствор 1,6 г (5 моль) N-Phac-L-Pro-Gly-OEt в 50 мл этанола охладили до 0oC, затем в течение 30 мин через раствор барботировали монометиламином (высушенным пропусканием через колонку с NaOH). Раствор оставили на 5 ч при комнатной температуре. Этанол выпаривали в вакууме. К осадку был добавлен эфир, и твердая часть была отфильтрована и высушена в вакууме при 25oC. Получены 1,6 г (99%) продукта: t
1H-ЯМР спектр в DMSO-d6, δ (млн-1): 1,66-2,24 (м,CβH2CγH2Pro,4H); 2,49 (d, NHCH3, 85% 3H); 2,60 (d, NHCH3, 15% 3H); 3,61 и 3,63 (каждый m, Cα H2Gly, 85% 2H); 3,52 и 3,62 (каждый м,CαH2Gly 15% 2H); 3,40 - 3,60 (м,CδH2Pro,2H); 3,70 (s, CH7Ar, 85% 2H); 3,68 (s, CH2Ar, 15% 2H); 4,23 (dd, CαHPro, 85% 1H); 4,44 (dd,CαHPro 15% 1H); 7,16-7,36 (m, C6H5, 5H); 7,58 (q, NH-CH3, 85% 1H); 7,84 (q, NHCH3, 15% 1H); 8,38 (t, NHGly, 85% 1H); 8,36 (t, NHGly, 15% 1H).
Найдено: C 63,61; H 6,75; N 14,01.
C16H21N3O3.
Вычислено: 63,34; 6,93; N 13,86.
Последующие продукты были приготовлены способом, соответствующим примеру 1.
Пример 15.
Диметиламид N-фенилацетил-L-пролилглицина
Дипептид формулы N-Phac-L-Pro-Gly-NMe2 (XV), выход 78%; масло; Rf = 0,68; силикагель, диоксан: вода 9:1);
[α]
H-ЯМР спектр в DMSO-d6δ(млн-1):1,71-2,06(m,CβH2-CγH2Pro,4H) 2,83, 2,93 и 2,84 2,96 (каждый s, N(CH3)2, 6H); 3,3-3,6 (m, Cδ H2Pro, 2H); 3,67 (s, CH2C6H5 2H); 3,89 и 3,95 (каждый d, Cα H2Gly 2H); 4,37 и 4,52 (dd, каждый Cα H Pro, 1Н); 7,15-7,34 (m, C6H5, 5H); 7,88 и 8,24 (каждый t , NH, 1H).
Найдено: C = 64,53; H = 7,48; N = 13,01.
С17H23H3O3
Вычислено: C = 64,32; H = 7,32; N = 13,23.
Пример 16.
Диэтиловый эфир N-фенилацетил-L-пролил-L-глутаминовой кислоты.
Дипептид формулы: N-Phac-L-Pro-L-Glu-(OEt)2 (XVI) выход 69%, масло; Rf = 0,9 (силикагель, диоксан: вода, 9:1); Rf = 0,7 (силикагель, CHCl3 : EtOH, 3: 1); [α]
1H-ЯМР спектр в CDCl3 δ (млн-1; 1,25 и 1,27 (каждый t, 2 CH3-CH2-O, 6H); 1,76-2,49 (m, CβH2CγH2,Pro,CβH2-CγH2 Gly, 8H); 3,39-3,92 (m,CδH2Pro,2H); 3,71 (s, CH2-C6H5, 2H); 4,13 и 4,19 (каждый q, 2 CH3-CH2-O, 4H); 4,35 и 4,49 (каждый m, Cα HGly, 1H); 4,49 и 4,61 (каждый dd, 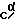 HPro, 1H); 7,15-7,38 (m, C6H5, 5H); 7,30 и 7,43 (каждый d, NH Clu 1H);
HPro, 1H); 7,15-7,38 (m, C6H5, 5H); 7,30 и 7,43 (каждый d, NH Clu 1H);
Найдено: C=63,24; H=7,11; N=6,81.
C22H30N2O6
Вычислено: C=63,13; H= 7,24; N=6,69.
Пример 17.
N-фенилацетил-L-пролил-L-лейциламид
Дипептид формулы N-Phac-L-Pro-L-Leu-NH2 (XVII), выход 83%; tпл 174-175oC, Rf = 0,5 (силикагель, CHCl3: EtOH, 9:1); [α]
1H-ЯМР спектр в DMSO-d6, δ (млн-1; 0,82 и 0,88 (каждый d, Cβ H (CH3)2Leu, 6H); 1,50 (m, Cβ H Leu, 1H); 1,31-1,93 и 1,7-2,40 (каждый m, CβH2-CγH2Pro 4H); 3,43-3,67 (m, CβH2Pro 2H); 3,69 (s, CH2-C6H5, 2H); 4,17 и 4,34 (каждый m, CαH Leu 1H); 4,28 и 4,58 (каждый dd CαHPro, 1H), 7,0 и 7,13 (каждый s, NH2); 7,15-7,35 (m, C6H5, 5H); 7,10-7,45 (m, два s, C6H5 и NH2); 7,86 и 8,27 (каждый d, NHLeu, 1H).
C19H27H3O3•0,5H2O
Вычислено: C=64,37; H=7,98; N=11,85.
Найдено: C=64,13; H=7,70; N=11,95.
Пример 18.
N-фенилацетил-L-пролилглицина
N-Phac-L-Pro-Gly-OH (XVIII)
Суспензию 1,06 г (3,5 ммоль) этилового эфира N-фенилацетил-L-пролоиглицин (пример 1) в 5 мл 1N NaOH перемешивали при комнатной температуре в течение 3 ч до получения раствора. Затем его подкислили 2N HCl до pH 3. Раствор был выпарен в вакууме до получения масла, которое растворили в 15 мл хлороформа. Нерастворившуюся часть отфильтровали, фильтрат испарили. К осадку добавили эфир, твердую часть отфильтровали и высушили в вакууме при комнатной температуре: получили, 0,9 г (89,9%) продукта: tпл 159-160oC (субл.); Rf = 0,54 (силикагель, диоксан-вода, 9:1); [α]
1H-ЯМР спектр в DMSO-d6, δ (млн-1): 1,80-2,25 (m, CβH2CγH2, Pro, 4H); 3,36-3,63 (m, Cγ H2 Pro, 2H); 3,64 и 3,68 (каждый s, CH2-C6H5, 2H); 3,86; 4,00 и 3,83; 4,02 (каждый dd, CαH2 Gly, 2H); 4,57 и 4,44 (каждый dd CαH Pro, H); 7,11-7,38 (m, C6H5, 5H); 7,52 и 7,32 (каждый t, NH, 1H) 12,06 (шир. s, COOH, 1H).
Найдено: C=62,11; H=6,26; N=10,09.
C15H18N2O4
Вычислено: C=62,04; H=6,22; N= 9,64.
Пример 19.
Метиловый эфир N-фенилацетил-L-пролил-j-аминомасляной кислоты
N-Phac-L-Pro-GABA-OMe (XIX)
Получен в условиях, приведенных в примере 1. Выход 86%, масло, Rf 0,65 (силикагель, CHCl3 - EtOH 9:1), [α]
1H-ЯМР спектр в DMSO - d6 δ (млн-1); 1,63 (m, Cβ H GABA 2H); 1,65-2,15 (m, CβH2-CγH2 Pro 4H); 2,29 (m, Cα H2 GABA, 2H), 3,05 (m, CγH2 GABA, 2H), 3,2-3,4 (m, CβH2 Pro при HDO 2H); 3,58 (s, OCH3 3H); 3,66 (s, CH2-C6H5, 2H); 4,20 и 4,40 (каждый dd, Cα H Pro, 1H); 7,02-7,37 (m, C6H5 5H); 7,85 и 8,20 (каждый t, NH, 1H).
Найдено: C=65,37; H=7,41; N=8,28.
C18H24N2O4
Вычислено: C=65,03; H=7,29; N=8,42.
Пример 20.
Этиловый эфир N-фенилацетил-L-пролил-L-аланина
N-Phac-L-Pro-L-Ala-OEt (XX)
Получен в условиях, приведенных в примере 1, выход 78%, т.пл. 58-51oC, Rf 0,75 (силикагель, диоксан-вода 10:1) [α]
1H-ЯМР спектр в DMSO-d6, δ (млн-1): 1,16 (t, CH3-CH2-O, 3H) 1,27 и 1,31 (каждый d, CH3 Alа, 3H); 1,68-2,27 (m, CβH2-CγH2 Pro, 4H); 3,46-3,61 (m, Cδ H2 Pro, 2H); 3,65 (s, CH2-C6H5, 2H); 3,98-4,14 (q, CH3-CH2-O, 2H); 4,19 и 4,29 (каждый dq, Cα H Ala, 1H); 4,34 и 4,48 (каждый dd, CαHPro, 1H); 7,12-7,36 (m, C6H5, 5H); 8,28 и 8,60 (каждый d, NH Ala, 1H).
Найдено: C=65,07; H=7,32; N=8,45.
C18H24N2O4
Вычислено: C=65,03; H=7,29; N=8,42.
Пример 21.
Этиловый эфир N-капроил-L-пролилглицина
Получен в условиях, приведенных в примере 1. Выход 54%, масло, Rf 0,8 (силикагель, диоксан-вода 9:1), [α]
1H-ЯМР спектр в DMSO-d6,δ (млн-1): 0,90 и 0,91 (каждый CH3-(CH2)4, 3H); 1,19 (t, CH3-CH2-O, 3H); 1,27, 1,50 и 2,25 ( два m и t, CH3-(CH2)4, 8 H), 1,70 - 2,20 (m, CβH2-CγH2 Pro, 4H); 3,35 - 3,50 (m, Cδ H2 Pro, 2H); 3,78 и 3,82 (каждый d, CαH2/ Gly, 2H); 4,08 (I, CH3-CH2-O, 2H); 4,30 и 4,36 (каждый dd, CαH Pro, 1H); 8,15 и 8,36 (каждый t, NH Gly, 1H).
C15H26N2O4
Найдено: C = 60,35; H = 8,84; N 9,31.
Вычислено: C = 60,39; H = 8,78; N = 9,38.
Пример 22.
Этиловый эфир N-(1-адамантоил)-L-пролил-глицина
N-Ad-C(O)-L-Pro-Gly-OEt (XXII), выход 81%, tпл 177 - 179oC, Rf = 0,93 (силикагель, CHCl3 EtOH, 2:3); [α]
1H-ЯМР спектр в DMSO-d6 δ (млн-1): 1,18 (t, CH3-CH2-O, 3H); 1,66, 1,88 и 1,96 (m, Ad); 1,6 - 2,0 (m, CβH2-CγH2 Pro при Ad); 2,23 - 3,37 (m, Cδ H2 Pro, 2H); 3,72 и 3,84 (каждый dd, CαH2, Gly, J = 16,5, 2H); 4,08 (q, CH3CH2-O, 2H); 4,39 (mрасшир. CαH Pro, 1Р); 8,07 (tрасшир. NH Gly, 1H).
Найдено: C = 66,49; H = 8,36; N = 8,13.
C20H30N2O4
Вычислено: C = 66,28; H = 8,33; N = 7,73.
Пример 23.
Этиловый эфир N-фенилбутирил-L-пролилглицина N-C2H5(CH2)3C(O)-t-Pro-Gly-OEt (XXIII), получен в условиях примера 1, выход 84%, масло; Rf = 0,87 (силикагель, диоксан: вода, 9:1); Rf = 0,75 (силикагель, CHCl3; EtOH, 9:1) [α]
1H-ЯМР-спектр в DMSO-d6, δ (млн-1): 1,18 (t, CH3-CH2-O-, 3H); 1,64 - 2,23 (m, CβH2-CγH2 Pro, 4H); 1,79, 2,28 и 2,59 (m, два t, -(CH3)3, 6H); 3,2 - 3,6 (m, CδH2 Pro, 2H); 3,78 и 3,81 (каждый d, CαH2 Gly, 2H); 4,07 и 4,09 (каждый q, CH3-CH2 - O, 2H); 4,33 и 4,36 (каждый dd, CαH Pro, 1 H); 7,04 - 7,35 (m, C6H5, 5H); 8,18 и 8,47 (каждый t, NH Gly, 1H).
Рассчитано для C19H26N2O3: C = 69,05; H = 7,95; N = 8,47
Найдено: C = 69,21; H = 7,99; N = 8,52.
Пример 24.
У заявляемых соединений, включая вещества, описанные в примерах 1 - 23, была оценена способность предотвращать нарушение памяти, вызываемые максимальным электрошоком (МЭШ) или скополамином в тесте условного рефлекса пассивного избегания (УРПИ) с помощью примера (установка Laffaette Co, США). Эксперименты, описываемые здесь (примеры 12, 13, 15 и 19) были проведены на самцах беспородных крыс-альбиносов массой 180 - 220 г. Каждое вещество вводили внутрибрюшинно в дозе 0,1 мг/кг за 15 мин до выработки УРПИ. Амнезию вызывали транскорнеальным МЭШ сразу же после обучения или скополамином (1 мг/кг, подкожно), вводимым за 30 мин до обучения. Сохранность рефлекса определяли через 24 ч путем измерения латентного периода перехода в темную камеру.
Степень антиамнестической активности оценивали по модифицированной формуле Баттлера: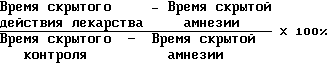
Чем выше этот показатель, тем больше антиамнестическая активность.
В контрольной группе животных МЭШ так же, как и скополамин, вызывал амнезию: значительное уменьшение латентного периода при переходе в темную камеру.
Заявляемые соединения вызывали значительный антиамнестический эффект: увеличение латентного периода и индекса антиамнестической активности (АА) (табл. 1,2).
Учитывая прекрасное антиамнестическое действие соединения 1 в обоих видах амнезии, его и выбрали для дальнейшего исследования.
Было показано, что его эффективность сохраняется после перорального введения; в экспериментах с максимальной электрошоковой амнезией была продемонстрирована перевернутая U-образная зависимость реакции от дозы (табл. 3).
Положительный амнестический эффект был выявлен также в условиях снижения интенсивности тренировки на выработку рефлекса пассивного избегания. Облегчающий эффект соединения 1 был продемонстрирован в случае его введения за 15 мин. до испытания, сразу же после него или за 15 мин до испытания восстановления памяти (табл. 4).
Соединение I может облегчить все основные фазы формирования памяти:
введение информации, закрепление и восстановление.
Пирацетам не облегчал восстановление в этих экспериментах.
Пример 25.
Влияние соединения I на обучение активному избеганию было исследовано в челночной испытательной камере (Уго Базиль, Италия). Условный рефлекс активного избегания (УРАИ) вырабатывали в течение 5 дней с 50 ежедневными пробами для каждой крысы. Учебное задание заключалось в том, чтобы крысы избегали электрошока, вызываемого через пол клетки, непосредственно после звукового сигнала. Крысы могли избежать шока, перемещаясь в другой отсек клетки, но только в то время, когда звучит сигнал (3c).
Соединение I (0,1 мг/кг, ежедневно, внутрибрюшинно), вводимое в течение 14 дней до обучения и ежедневно во время обучения, продемонстрировало свою способность повысить обучаемость (табл. 5).
В других экспериментах реакция стабильного активного избегания была нарушена одновременным включением звукового сигнала и электрошока (в конечностях) в пяти опытах. Соединение I предотвращало ухудшение избегания (табл. 6).
Пример 26.
Влияние соединения I на острое угасание локомоторной активности было оценено по уменьшению передвижений в течение 30 мин. Эксперименты выполнены на самцах беспородных мышей-альбиносов (18 - 22 г), которых помещали группами по 10 животных в регистрационную клетку (Optovarimex, Colomb., США).
Введение за 15 мин до начала регистрации дозами размером от 0,05 до 5,0 мг/кг соединения 1 продемонстрировало увеличение степени привыкания ("усвоение негативного навыка") без изменения начальной локомоторной активности (табл. 7).
Пример 27.
Ухудшение обучения и памяти достигали также фронтальной лобэктомией, которую выполняли после обучения пассивному или активному избеганию. Введение соединения I начинали с первого послеоперационного дня и заканчивали на 9-й лень. Тестирование проводили на 4-й и 9-й день. В контрольной группе (ложная операция) обученность пассивному и активному избеганию не изменялась. Следствием фронтальной лобэктомии явилось сокращение латентного периода перехода в темный отсек; соединение I восстановило реакцию пассивного избегания (табл. 8).
Лобэктомированные крысы проявляли полное отсутствие рефлекса активного избегания на 4-й день и слабую тенденцию к его восстановлению на 9-й день. Введение соединения I увеличило процент животных, проявляющих активную реакцию избегания и коэффициент сохранения (табл. 8).
Пример 28.
Ухудшение обучаемости потомства крыс вызывали пренатальной алкоголизацией или пренатальной гипоксией. Пренатальная алкоголизация достигалась пероральным введением 5 г/кг день (25%-ный раствор) этанола поросным самкам крыс в течение всего периода поросности. Для достижения пренатальной гипоксии крыс на 15-й день поросности крыс помещали в барометрическую камеру с большим разрежением ("подъем" на высоту 8500 м; выдержка 2 ч).
Лечение молодняка производили 8-го по 20-й день. Соединение I вводили подкожно в дозе 0,1 мг/кг/день. Тестирование выполняли на двухмесячных крысах.
Установлено, что обе разновидности поражения (алкоголизация, гипоксия) вызывали нарушение обучаемости в тесте активного избегания (УРАИ). Ранее постнатальное введение соединения I восстанавливало способность к обучению (таблица 9).
Было показано, что пренатальная алкоголизация уменьшает степень привыкания в открытом пространстве. Постоянное постнатальное введение соединения I восстанавливало нормальный ход привыкания (таблица 10).
Пример 29.
Влияние соединения 1 на старческое снижение умственных способностей изучали в экспериментах на 24-месячных крысах линии Вистар. Вещество вводили постоянно в дозе 0,1 мг/кг/день в течение 24 дней внутрибрюшинно перед опытом и 14 дней в течение опыта.
Регистрация локомоторной активности в многоканальном анализаторе Optovarimex показала, что соединение 1 повышало, при постоянном введении, начальную горизонтальную активность. Тестирование активности в течение 5 мин выявило снижение подвижности вследствие острого привыкания ("усвоения негативного навыка"). Это испытание пригодно для изучения ноотропов. В контрольной группе коэффициент привыкания составлял 0,29. У животных, предварительно получивших соединение 1, выявлено более выраженное привыкание с коэффициентом 0,03 (табл. 11).
Постоянное введение соединения 1 облегчало сохранение памяти по истечении 24 ч и особенно 14 дней после участия в тесте пассивного избегания (табл. 11). Из этих экспериментов очевидно, что соединение 1 способно улучшать мнестическую функцию у старых крыс.
Пример 30.
Противогипоксическое действие соединения 1 оценивали с использованием барометрической камеры посредством "подъема" беспородистых мышей-альбиносов (18-22 г) на высоту 11000 м со скоростью 1000 м/мин. Через час выдержки количество предварительно обработанных 0,9%-ным физ. р-ром выживших мышей составило 15%. Введение соединения 1 (0,5 мг/кг за 15 мин перед гипоксией) увеличило это значение до 43,7%.
Пример 31.
Влияние соединения 1 на вес и потребление пищи изучали в экспериментах на беспородистых крысах. Вещество, вводимое внутрибрюшинно в течение 3 недель в дозе от 0,1 мг/кг/день, замедляло динамику увеличения веса по сравнению с контролем: увеличение массы (в % к начальному) составляло в обработанной группе 12% и в контрольной 24%. Количество потребляемой еды, проверенное между 19-м и 21-м днями, было меньше у обработанных животных (39,5 г/день/животное), чем у контрольных (68,6 г/день/животное). Подученные данные подтверждают анорексигенное действие соединения 1.
Пример 32.
Были изучены влияние соединения 1 на общее (свободное) поведение и его острая токсичность. В опытах на беспородных самцах мышей-альбиносов (18-22 г) соединение 1 при дозировке в пределах 1 - 10 мг/кг (внутрибрюшинно) не увеличивало и не уменьшало спонтанной двигательной активности, а также не усиливало стимулирующий эффект амфитамина на нее. В этих дозах соединение 1 также не изменяло ректальную температуру. При дозе 25 мг/кг соединение 1 снижало стимулирующий эффект амфитамина.
Увеличение дозы до 500 мг/кг не изменяло двигательной координации (тест вращающегося стержня) и не разрушало общего состояния (свободного поведения) животных.
При дозе 500 - 3000 мг/кг (внутрибрюшинно) соединение 1 не вызывало смерти животных в течение 24 ч. При дозе 100 мг/кг оно вызывало возбуждение у 50% животных. При дозе 5000 мг/кг (внутрибрюшинно) соединение вызывало смерть 50% животных. Соединения по изобретению могут быть отнесены к малотоксичным.
Пример 33.
Влияние соединения 1 на синдром отмены, вызванный прекращением постоянного введения бензодиазепинового транквилизатора феназепама (Т.А. Воронина и др. , Феназепам; А.В.Богатский, издательство "Наукова думка", Киев, 1982; с. 67-169) было изучено в экспериментах на самцах крыс-альбиносов массой 190 - 200 г в начале эксперимента и 300 - 350 г в конце эксперимента.
Феназепам (2 мг/кг внутрибрюшинно) или физ. р-р (контрольная группа крыс) вводили животным ежедневно в течение 57 дней. Синдром проявлялся на 24-72 ч после последней инъекции. Основными проявлениями синдрома отмены были: "анксиогенно-подобное" состояние, агрессивность и ускоренное развитие киндлинга, вызванного пентилэнтетразолом. Соединение 1 вводили крысам, у которых была произведена отмена, в дозах 0,5 мг/кг внутрибрюшинно за 15 мин до тестирования.
Для оценки уровня "тревожности" крыс использовали тест конфликтной ситуации (JR. Vogel et.al., Psychopharmаcologia, 21 1-7. (1971)). Этот эксперимент заключался в предварительной дрессировке крыс, которые были ограничены в приеме жидкости, пить воду из поилки. На следующий день через поилку пропускали электрический ток 0,5 мА. Конфликтную ситуацию создавали путем столкновения двух различных рефлексов (питьевого и самозащиты).
Через 24 ч после прекращения постоянного испытания крыс, которым отменили феназепам, у них отмечено уменьшение частоты питья по сравнению с контрольной группой. Такое поведение в тесте конфликтной ситуации можно рассматривать как "анксиогенно-подобное". Соединение 1 противодействовало "анксиогенно-подобной" реакции на отмену бензодиазепина и увеличивало частоту питья в конфликтной ситуации (табл. 12).
В эксперименте на крысах определяли порог агрессивности поведения (R. Tedeshi et. al. J. pharmacol. Exp. Ther. 125 28 - 34 (1959)). Пару крыс помещали на настил, подключенный к электрической цепи; регистрировали силу тока, которая провоцировала драку.
У крыс с отмененным феназепамом наблюдалось уменьшение этого порока. Соединение 1 при введении через 2 ч после отмены феназепама увеличивало порог агрессивной реакции (табл. 13).
Исследовали судорожные реакции крыс с использованием химического киндлинга (Mason C. R. et. al Epilepsia, 13, 663-674 (1972); R.M.Post et.al., Handbook of Biological Psychiatry, Part IV, N 7, Mapcel -Dekker (1981), p.p. 609-651), вызываемого малыми дозами пентилен-тетразола (35 мг/кг, внутрибрюшинно), вводимого через 24, 48 и 72 ч после отмены феназепама. Уровень судорог оценивали по 4-бальной системе: дрожание (1 балл); конвульсии (2 балла); клонические судороги (3 балла); тонические припадки и смерть (4 балла).
Наши эксперименты выявили, что судороги, вызываемы пентиден-тетразолом, были более выраженными у крыс, которым отменили феназепам, чем у контрольных. Соединение 1 уменьшает степень киндлинга, вызываемого пентилен-тетразолом (табл. 14).
Из этих экспериментов очевидно, что соединение 1 способно уменьшить синдром отмены бензодиазепина: оно снижает тревожность, агрессивность и киндлинг, вызываемый пентилентетразолом.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ ПРОЛИЛТИРОЗИНЫ, ОБЛАДАЮЩИЕ ПСИХОТРОПНОЙ АКТИВНОСТЬЮ | 1995 |
|
RU2091390C1 |
| Новые глипролины с ноотропной, антигипоксической, нейропротективной и анксиолитической активностью | 2016 |
|
RU2646604C2 |
| Дипептидные лиганды TSPO, обладающие нейропсихотропной активностью | 2018 |
|
RU2756772C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИНДИВИДУАЛЬНЫХ СТЕРЕОИЗОМЕРОВ 4-ЗАМЕЩЕННЫХ ТИОПРОИЗВОДНЫХ ГЛУТАМИНОВОЙ КИСЛОТЫ | 1994 |
|
RU2083560C1 |
| Малые молекулы с NGF-подобной активностью, обладающие антидиабетическими свойствами | 2013 |
|
RU2613314C2 |
| ЗАМЕЩЕННЫЙ БИСДИПЕПТИД С НЕЙРОПРОТЕКТИВНЫМ И АНТИДЕПРЕССИВНЫМ ЭФФЕКТОМ | 2014 |
|
RU2559880C1 |
| L-ПИРОГЛУТАМИЛ-L-АСПАРАГИН И ЕГО ПРОИЗВОДНЫЕ, ОБЛАДАЮЩИЕ СПОСОБНОСТЬЮ РЕГУЛИРОВАТЬ ПРОЦЕССЫ ОБУЧЕНИЯ И ПАМЯТИ | 1988 |
|
RU1619684C |
| ЗАМЕЩЕННЫЕ ДИПЕПТИДЫ С НЕЙРОПСИХОТРОПНОЙ АКТИВНОСТЬЮ | 2014 |
|
RU2573823C2 |
| ПРОИЗВОДНЫЕ АЗА-АНТРАЦИКЛИНОНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2159245C2 |
| ЛЕКАРСТВЕННОЕ СРЕДСТВО С АНТИАРИТМИЧЕСКИМ И АНТИФИБРИЛЛЯТОРНЫМ ДЕЙСТВИЕМ | 2010 |
|
RU2477144C2 |
Использование: в медицине как соединения, обладающие антиамнестическим, антигипоксическим и анорексигенным действием, проявляющие низкую токсичность, способствующие улучшению способности к обучению и памяти. Сущность изобретения: производные N-ацил-пролилдипептидов общей формулы I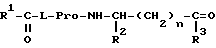
где R1 = (C1 - C4)алкил, циклоалкил, аралкил или арил; R2 = Н, (C4 - C5)алкил, карбамидоалкил или карбалкоксиалкил; R3 = гидрокси, алкокси, амино, алкиламино или диалкиламино; n = 0 - 3, при этом а) когда R1 = фенил, R2 = водород или изопропил, а n = 0, R3 = не метокси; б) когда R1 = третбутил, R2 = водород, а n = 0 или 1, R3 = не метиламино; в) когда R1 = трет-бутил, R2 = изобутил, метил, бензил или карбамидометил, а n = 0, R3 = не метиламино, г) когда R1 = трет-бутил, R2 = карбометоксиметил, n = 0, R3 = не изопропиламино. Соединения малотоксичны. Описываемые соединения получают путем конденсации требуемых кислот в гомогенной фазе при блокировании не вступающих в реакцию функциональных групп с использованием конденсирующих агентов, таких как, например, карбодиимид. 10 з.п. ф-лы, 14 табл.
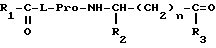
где R1 = (C4-C5)алкил, циклоалкил, аралкил или арил;
R2 = H, (C1-C4)алкил, карбамидоалкил или карбалкоксиалкил;
R3 = гидрокси, алкокси, амино, алкиламино или диалкиламино;
n = 0-3,
при этом:
а) когда R1 = фенил, R2 = водород или изопропил, а n = 0, R3 = не метокси;
б) когда R1 = трет-бутил, R2 = водород, а n = 0 или 1, R3 = не метиламино;
в) когда R1 = трет-бутил, R2 = изобутил, метил, бензил или карбамидометил, а n = 0, R3 не метиламино;
г) когда R1 = трет-бутил, R2 = карбометоксиметил, n = 0, R3 не изопропиламино.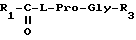
где R1 = изобутил, пентил, 1-адамантил, фенил, фенилметил или фенилпропил;
R3 = OH или OC2H5, NH2, NHCH3, N(CH3)2
3. Производные N-ацилпролилдипептидов общей формулы I по п.1, представляющие собой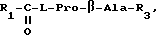
где R1 = фенилметил или фенил, R3 = OC2H5 или NH2.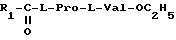
где R1 = фенилметил или фенил.
6. Производное N-ацилпролилдипептидов по п.1, представляющее собой
7. Производное N-ацилпролилдипептидов по п.1, представляющее собой
8. Производное N-ацилпролилдипептида по п.1, представляющее собой
9. Производное N-ацилпролилдипептида по п.1, представляющее собой
10. Производное N-ацилпролилдипептида по п.1, представляющее собой
11. Производное N-ацилпролилдипептида по п.1, представляющее собой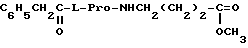
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Гудашева Т., Островская Р | |||
| Chem | |||
| Pharmac | |||
| J, 1985, N 11, pp.1322 - 1329 | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Гудашева Т.А | |||
| и др | |||
| Chem Pharm | |||
| J, 1988, N 3, pp.271 - 2759 | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Шредер Э., Любке К | |||
| Пептиды | |||
| Мир, ч.1, 1967, с.116. | |||

