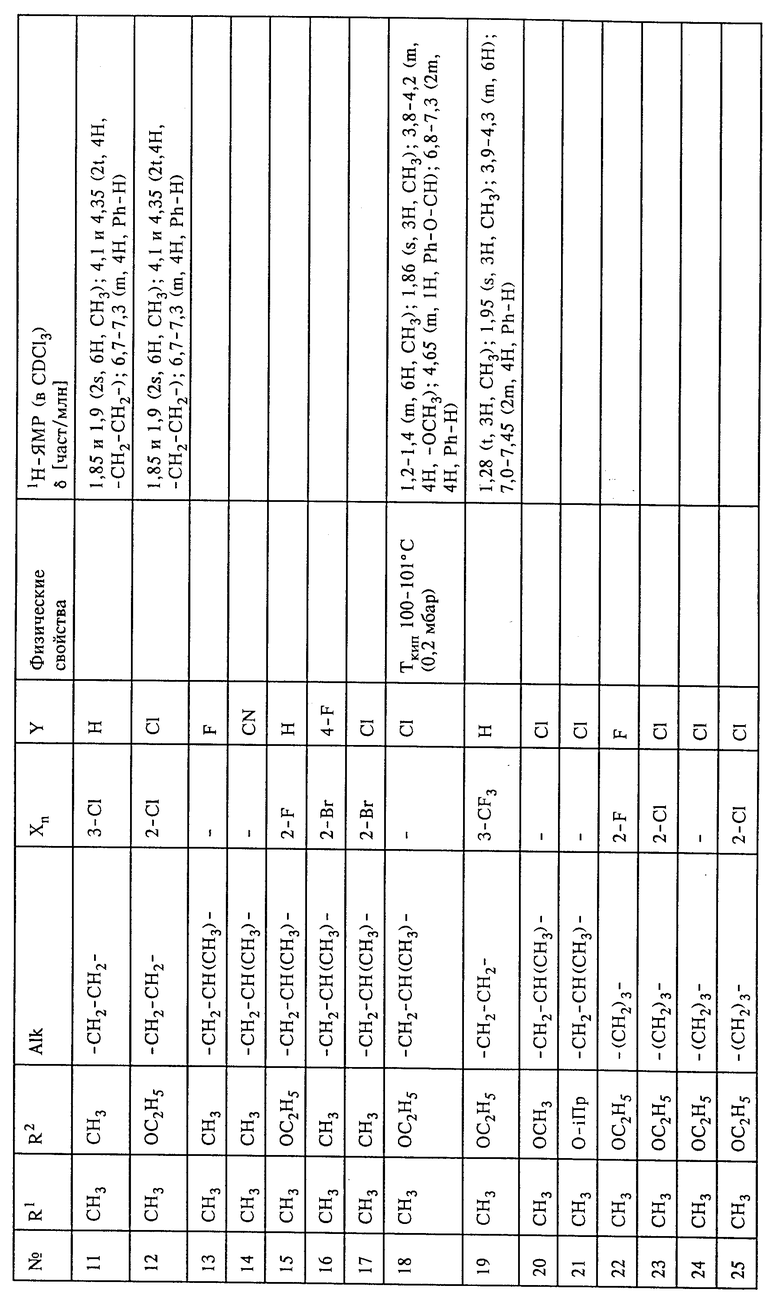
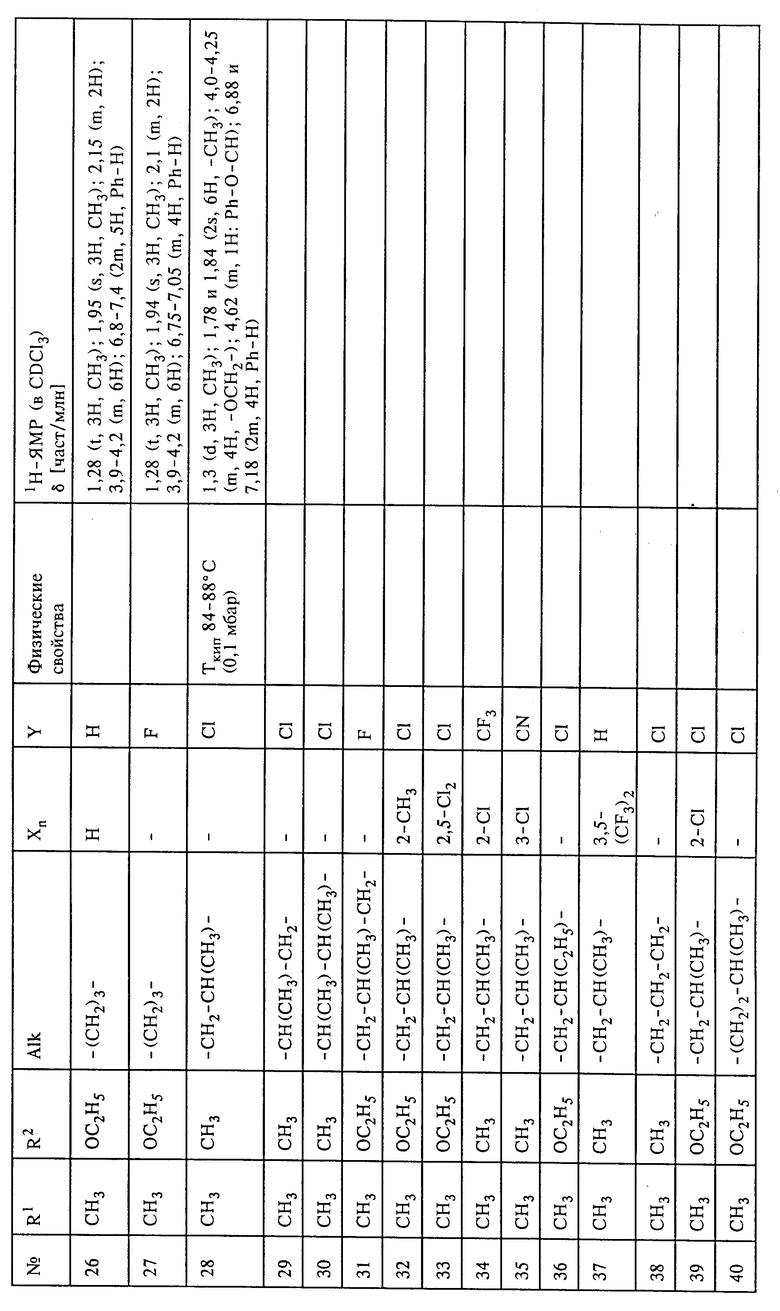
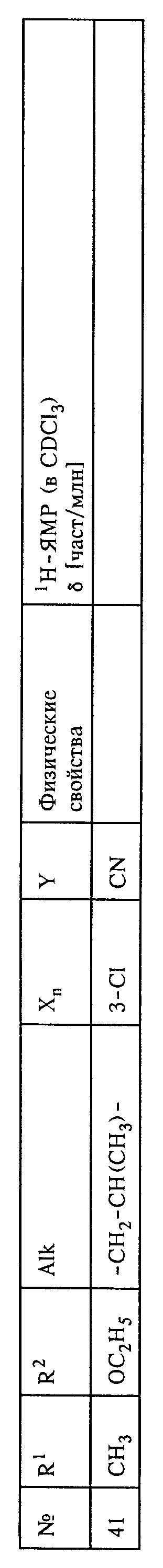
Настоящее изобретение относится к способу и промежуточным продуктам для получения простых гидроксиламиновых эфиров формулы I,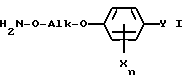
в которой переменные имеют следующие значения:
X обозначает нитро, циано, галоген, C1-C4-алкил и C1-C4-галогеналкил;
Y обозначает водород, нитро, циано, галоген, C1-C4-алкил, C1-C4-галогеналкил;
n обозначает 0-2 либо 1-4 в случае, если Y и все X обозначают галоген;
Alk обозначает C2- либо C3-алкиленовую цепь, которая при необходимости может нести от одной до трех C1-C3-алкильных групп, а также к их солям, получаемым с помощью минеральных кислот или сильных органических кислот.
Настоящее изобретение относится, кроме того, к новым оксиминопроизводным формулы IVa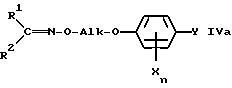
в которой переменные имеют следующие значения:
R1 обозначает C1-C4-алкил;
R2 обозначает C1-C4-алкил либо C1-C6-алкокси или
R1 и R2 оба вместе обозначают C4-C6-алкилен;
X обозначает нитро, циано, галоген, C1-C4-алкил и C1-C4-галогеналкил;
Ya обозначает водород, нитро, циано либо галоген;
m обозначает 0-2, если Y представляет собой нитро, циано либо галоген; обозначает 1, если Y представляет собой водород, а X нитро, циано либо C1-C4-алкокси и обозначает 2 или 3, если Y представляет собой водород;
Alk обозначает C2- либо C3-алкиленовую цепь, которая при необходимости может нести от одной до трех C1-C3-алкильных групп, причем, если R1 и R2 оба вместе обозначают метил, Alk обозначает -(CH2)3- и Xn представляет собой 2,5-Cl2, Y не может обозначать хлор.
Получение соединений формулы I не может осуществляться непосредственно O-алкилированием гидроксиламина, а предполагает применение защитных групп. Такие методы синтеза простых гидроксиламиновых эфиров типа соединений I описываются, например, в Houben-Weyl, Methoden der orfanischen Chemie, том E16a, 1990, стр.214 и далее. Из этой публикации известен также так называемый "N-гидроксифталимидный метод", с помощью которого согласно европейской заявке EP-A 456112, заявкам Германии DE-A 4204203 и DE-A 4204206 до настоящего времени получали простые гидроксиламиновые эфиры типа соединений I. Однако при промышленном применении этот метод характеризуется существенными недостатками. Так, при отщеплении защитной группы наряду с желаемыми O-замещенными гидроксиламинами образуются, как правило, непригодные для использования продукты сочетания, например гидразид фталевой кислоты, образующийся при отщеплении гидразином. Возвращение использованной защитной группы обычно также невозможно.
Из Journ. Agric. Food. Chem. 38, 514 (1990) известен способ получения ацетон-[O-3-(4-феноксифенокси)пропил] -оксима в присутствии трет.-бутилата калия в диоксане в качестве растворителя. Однако описанный способ непригоден для получения оксиминопроизводных IV промышленным путем, так как для этой цели требуются большие количества растворителей. Осуществление соответствующего химического процесса с меньшими количествами растворителей невозможно в силу того, что в этом случае получают вязкие эмульсии.
В другой публикации (Journ. Am. Chem. Soc., 74, 3956 (1952)) описывается конденсация, в частности 2-феноксиэтилбромида, с помощью натриевой соли ацетоноксима (при этом, однако, не приводится никаких данных об условиях осуществления способа) и отщепление полученного оксимового эфира с помощью соляной кислоты.
В заявках Германии DE-A 2651083 и DE-A 2651085 и в патенте Японии JP-A91/258757 при алкилировании гидроксииминопроизводными типа соединений формулы II используют относительно дорогие, связанные с технологическими трудностями основания, как гидриды щелочных металлов, например гидрид натрия, амиды щелочных металлов, например амид натрия, или металлорганические соединения, например бутиллитий. Необходимое условие при этом - работать с безводными соединениями, что связано с большими техническими затратами.
Некоторые из оксиминопроизводных формулы IV уже известны из патента США US 4647698 (ср. формулу B в патенте), а именно, применяемые в качестве пестицидов. Для их получения из гидроксииминосоединений формулы II и алкилирующих средств типа соединений формулы III рекомендуется присутствие гидрида натрия в качестве основания. Недостатком указанного способа являются значительные технические трудности, обусловленные работой в атмосфере инертного газа.
Согласно Bioorg. Khim 12, 1662 (1986) 1-(1-этоксиэтилиденаминоокси)-2-феноксиэтан может быть получен взаимодействием 1-(1-этоксиэтилиденаминоокси)-2-бромэтана с фенолятом натрия в метаноле. Недостаток данного способа заключается при этом в низком выходе как названного первым компонента реакции, составляющем всего лишь 28%, так и самого конечного продукта, составляющем лишь 40%.
Далее, в европейской заявке EP-A 023560 описан способ, по которому определенные кетоксимы с помощью (цикло)алкил- либо арилалкилгалогенидов трансформируют в O-замещенные кетоксимы. Применение эфиров сульфокислот типа соединений формулы III в качестве алкилирующих средств в названной публикации не упоминается.
Касательно гидролиза окисминопроизводные формулы IV, в которых R2 обозначает C1-C4-алкил, в имеющихся публикациях можно найти лишь небольшое число аналогичных примеров. Так, Bajwa et. al., Heterocycles 20, 839 (1983) при гидролизе соединения формулы IV, где R1 и R2 обозначают метил, Alk обозначает 1,3-пропилиден и Y обозначает хлор, а X2 обозначает 2,5-дихлор, осуществляемом в смеси из соляной кислоты, этанола и воды, получали соответствующий простой гидроксиламиновый эфир формулы I в виде гидрохлорида.
В основу настоящего изобретения была положена задача создать улучшенные возможности для получения простых гидроксиламиновых эфиров формулы I.
В соответствии с этой задачей был найден способ получения простых гидроксиламиновых эфиров формулы I, а также их солей с помощью минеральных кислот или сильных органических кислот, отличающийся тем, что либо гидроксииминосоединение формулы II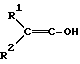
в которой
R1 обозначает C1-C4-алкил, R2 обозначает C1-C4-алкил либо C1-C6-алкокси или R1 и R2 оба вместе представляют собой C4-C6-алкиленовую цепь, в присутствии гидрокисда щелочного металла, алкоголята щелочного металла, гидрокарбоната щелочного металла или карбоната щелочного металла в качестве основания, либо соответствующий анион гидроксииминосоединения II с помощью алкилирующего средства формулы III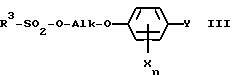
в которой
R3 обозначает C1-C4-алкил, C1-C4-галогеналкил или при определенных условиях замещенный фенил, трансформируют в оксиминопроизводное формулы IV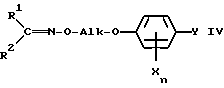
и что затем оксиминопроизводное IV с помощью минеральной кислоты или сильной органической кислоты расщепляют в соответствующую соль гидроксиламинового эфира I и эту последнюю при необходимости с помощью основания переводят в свободное соединение I.
Кроме того, были найдены новые оксиминопроизводные формулы IVa.
Радикалы R1, R2, X, Y и Alk имеют каждый в отдельности следующие значения:
R1 обозначает C1-C4-алкил, как метил, этил, n-пропил, 1-метилэтил, n-бутил, 1-метилпропил, 2-метилпропил и 1,1-диметилэтил, предпочтительно метил, этил, n-пропил, n-бутил и 1-метилэтил, прежде всего метил и этил;
R2 обозначает метил, этил, n-пропил, 1-метилэтил, n-бутил, 1-метилпропил, 2-метилпропил, 1,1-диметилэтил либо метокси, этокси, n-пропокси, 1-метилэтокси, n-бутокси, 1-мектилпропокси, 2-метилпропокси, 1,1-диметилэтокси, n-пентокси, 1-метилбутокси, 2-метилбутокси, 3-метилбутокси, 2,2-диметилпропокси, 1-этилпропокси, n-гексокси, 1,1-диметилпропокси, 1,2-диметилпропокси, 1-метилпентокси, 2-метилпентокси, 3-метилпентокси, 4-метилпентокси, 1,1-диметилбутокси, 1,2-диметилбутокси, 1,3-диметилбутокси, 2,2-диметилбутокси, 2,3-диметилбутокси, 3,3-диметилбутокси, 1-этилбутокси, 2-этилбутокси, 1,1,2-триметилпропокси, 1,2,2, -триметилпропокси, 1-этил-1-метилпропокси, 1-этил-2-метилпропокси, предпочтительно метил, этил, n-пропил, n-бутил и метокси, этокси, n-пропокси и n-бутокси, прежде всего метил, этил, метокси и этокси;
или
R1 и R2 оба вместе представляют собой алкиленовую цепь, как -CH2CH2CH2CH2-, -CH2CH2CH2CH2CH2- и -CH2CH2CH2CH2CH2CH2-; X обозначает нитро, циано; фтор, хлор, бром и иод, прежде всего фтор и хлор; C1-C4-алкил, как метил, этил, n-пропил, 1-метилэтил, n-бутил, 1-метилпропил, 2-метилпропил, 1,1-диметилэтил, предпочтительно метил, этил, n-пропил либо n-бутил, прежде всего метил либо этил; C1-C4-галогеналкил, прежде всего фторметил, дифторметил, трифторметил, хлорфторметил, дихлорфторметил, хлордифторметил, 1-фторэтил, 2-фторэтил, 2,2-дифторэтил, 2,2,2-трифторэтил, 2-хлор-2-фторэтил, 2-хлор-2,2-дифторэтил, 2,2-дихлор-2-фторэтил, 2,2,2-трихлорэтил и пентафторэтил, предпочтительно трифторметил, дифторметил и фторметил;
Y обозначает водород, нитро, циано; фтор, хлор, бром и иод, предпочтительно фтор и хлор; C1-C4-алкил, как и для X, предпочтительно метил, этил, n-пропил или n-бутил, прежде всего метил либо этил; C1-C4-галогеналкил, прежде всего C1-C2-галогеналкил, как и для X, предпочтительно трифторметил, дифторметил и фторметил;
Alk обозначает 1,2-этилен либо 1,3-пропилен, которые могут быть оба незамещенными или могут нести от одной до трех C1-C3-алкильных групп, как метил, этил, n-пропил и 1-метилэтил, предпочтительно метил и этил, прежде всего метил.
Особенно предпочтительным значением R1 и R2 является соответственно C1-C4-алкил.
Среди новых оксиминопроизводных IVa особенно предпочтительными являются те из них, в которых заместители имеют следующие значения:
R1 обозначает C1-C4-алкил, предпочтительно метил, этил, n-пропил либо изопропил, прежде всего метил либо этил;
R2 обозначает C1-C4-алкил, предпочтительно метил, этил, n-пропил либо изопропил, прежде всего метил либо этил, или C1-C4-алкокси, предпочтительно метокси, этокси, n-пропокси либо изопропокси, прежде всего метокси либо этокси;
X обозначает галоген, прежде всего фтор либо хлор;
Ya обозначает водород либо галоген, предпочтительно фтор либо хлор, прежде всего хлор;
m обозначает 0 либо 1, если Y обозначает галоген, или 2, если Y обозначает водород, прежде всего 0;
Alk представляет собой 1,2-этиленовую цепь, либо 1,3-пропиленовую цепь, причем эта цепь несет одну либо две метиловые и/или этиловые группы.
Гидроксииминовые соединения II могут быть частично приобретены коммерческим путем либо их можно получать по методам, известным из публикаций (ср. , например, патент США US 4743701).
Алкилирующие средства III также частично известны или их можно получать с помощью методов, известных из соответствующих публикаций.
На примере двух приведенных ниже уравнений схематически представлен один из возможных путей синтеза, используемого для получения соединений III, исходя из феноксиалкановых кислот или их эфиров. Взаимодействием с соответствующими восстановителями, например аллюмогидридом лития либо борогидридом натрия, в инертном растворителе, как тетрагидрофуран, получают феноксиалканолы формулы V [ср., например, Journ. Pharmacol. Chemother. 7, 197 (1952)]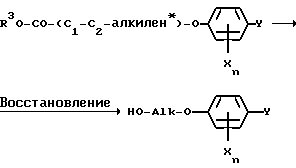
* при определенных условиях замещенный на C1-C3-алкильные группы;
R3 обозначает водород либо низший алкил.
Феноксиалканолы V могут затем взаимодействием с галогенидами неорганических либо органических кислот переводиться в алкилирующие средства III. Так, например, обмен гидроксильной группы на CH3-SO2-O- осуществляют с помощью хлорида метансульфокислоты в присутствии третичного амина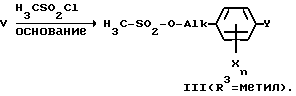
Если гидроксильную группу феноксиалканолов V заменить на хлор, бром (например, с помощью трибромида фосфора в присутствии третичного амина) или иод, то получают галогенированные алкилирующие средства формулы VI, которые могут использоваться вместо алкилирующих средств III.
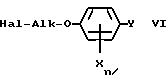
Hal обозначает хлор, бром, иод.
Взаимодействие гидроксииминосоединений формулы II с алкилирующими средствами формулы III осуществляют следующим образом: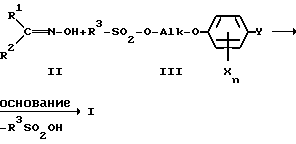
Температура реакции лежит обычно в диапазоне от 20 до 150oC, предпочтительно от 40 до 120oC, прежде всего от 60 до 100oC.
В качестве оснований могут использоваться, например, гидроксиды щелочных металлов, как гидроксид натрия либо гидроксид калия, алкоголяты щелочных металлов, как метилат лития, метилат натрия, метилат калия, этилат лития, этилат натрия, этилат калия, трет.-бутилат натрия и трет.-бутилат калия, карбонаты щелочных металлов, как карбонат натрия и карбонат калия, гидрокарбонаты щелочных металлов, как гидрокарбонат натрия и гидрокарбонат калия.
Особенно предпочтительны среди перечисленных выше натриевые соединения, прежде всего гидроксид натрия и метилат натрия.
Основание целесообразно применять в эквимолярном количестве по отношению к соединению формулы II.
Касательно определения диполярно-апротонных растворителей можно сослаться на Chr. Recichardt, Loesungsmitel-Effekte in der organischen Chemie, изд-во Verlag Chemie, 1969 г. Под диполярно-апротонными растворителями понимаются, в частности, такие растворители, которые не являются донорами протона, образующими водородную связь, а имеют ярко выраженный дипольный момент (μ больше 2,5 единиц Дебая), а также высокую диэлектрическую константу (Σ больше 16).
Пригодными для указанной выше цели диполярно-апротонными растворителями являются, например, сульфоксиды, как диметилсульфоксид, диэтилсульфоксид, диметилсульфон, диэтилсульфон, метилэтилсульфон, тетраметиленсульфон; нитрилы, как ацетонитрил, бензонитрил, бутиронитрил, изобутиронитрил, m-хлорбензонитрил; N,N-двузамещенные амиды карбоновой кислоты, как диметилформамид, N, N-диметилбензамид, N,N-диметилацетамид, N,N-диметилфенилацетамид, амид N, N-диметилциклогексанкарбоновой кислоты, амид N,N-диметилпропионовой кислоты и гомологенный пиперидид карбоновой кислоты, морфолид карбоновой кислоты, пирролидид карбоновой кислоты; соответствующие амиды N,N-диэтиловой, N,N-ди-n-пропиловой, N,N-диизопропиловой, N,N-диизобутиловой, N,N-дибензиловой, N-метил-N-фенильной, N-циклогексил-N-метиловой карбоновой кислоты, N-метилформанилид; N-алкиллактамы, как N-этилпирролидон, N-октилпирролидон, N-циклогексилпирролидон, N-метилпирролидон, N-бутилпирролидон; четырехзамещенные циклические и ациклические мочевины, как тетраметилмочевина, тетрабутилмочевина, 1,3-диметил-2-имидазолинон, 1,3-диметил-3,4,5,6-тетрагидро-2-(1H)-пиримидон и смеси названных растворителей. Предпочтительными являются замещенные N, N-диалкилом амиды карбоновых кислот, как диметилформамид и диметилацетамид, или замещенные N-алкилом лактамы, как N-метилпирролидон.
Растворитель, соответственно смесь растворителей применяют, как правило, в количестве от 0,3 до 1,0 л, предпочтительно от 0,4 до 0,8 л, прежде всего от 0,5 до 0,7 л из расчета на 1 моль гидроксииминосоединения формулы II.
Эдукты формул II и III применяют, как правило, в эквимолярных количествах, однако для оптимизации выхода может оказаться целесообразным использовать эдукты II с избытком порядка 0,1-0,5 мол.экв., предпочтительно 0,2-0,4 мол.экв., прежде всего 0,2-0,3 мол.экв. по отношению к эдуктам III.
После окончания реакции отгонкой при пониженном давлении большая часть использованного растворителя может возвращаться обратно. После смешивания остатка при комнатной температуре с водой продукты IV способа можно отделять, при необходимости посредством экстракции с помощью, например, углеводородов, как толуол и циклогексан. Если оксиминопроизводные IV требуется представить в чистом виде, то может осуществляться очистка сырых продуктов по известной методике, например, путем кристаллизации либо фракционной отгонки при пониженном давлении.
Саму обменную реакцию целесообразно проводить следующим образом. Сначала приготавливают раствор гидроксииминосоединения II, в который добавляют основание, затем эту смесь доводят до требуемой температуры, некоторое время для обеспечения солеобразования перемешивают, после чего производят добавку алкилирующего средства III при необходимости в виде раствора.
Перед добавкой алкилирующего средства III может оказаться целесообразным отделить высвобожденный в результате солеобразования спирт, соответственно высвобожденную воду путем отгонки при пониженном давлении. Это можно порекомендовать особенно в тех случаях, когда вместо алкилирующего средства III применяют алкилирующее средство VI.
Гидроксииминосоединение формулы II может быть переведено на предварительной стадии также в его соль щелочного металла и при необходимости как таковая выделена. Затем эту соль смешивают с выбранным для проведения реакции растворителем и без использования каких-либо оснований осуществляют реакцию с помощью алкилирующего средства III. Для этой цели пригодны предпочтительно уже упоминавшиеся выше карбонаты и гидрокарбонаты щелочных металлов, гидроксиды и алкоголяты щелочных металлов. Эти вещества в обычных для таких случаев растворителях, например спиртах, либо в воде подвергают взаимодействию в стехиометрических количествах с соответствующим гидроксииминосоединением II в диапазоне температур от 0 до 50oC. Особенно хорошо зарекомендовала себя при этом работа с раствором металла натрия, при определенных условиях с добавками углеводородов, как толуол. Целесообразно при этом после непродолжительного перемешивания (в течение 10-60 мин) удалять легколетучие компоненты, что обычно проводят при пониженном давлении. Полученный остаток содержит соль щелочного металла соединения II.
Хотя получение оксиминопроизводных формулы IV в принципе возможно также из соединений II или из соответствующего аниона соединения II и алкилирующего средства VI, предпочесть следует особенно хорошо зарекомендовавшие себя алкилирующие средства III. R3 обозначает при этом предпочтительно C1-C4-алкил, C1-C4-галогеналкил, фенил либо фенил, однократно- трехкратно замещенный на галоген и/или C1-C4-алкил. Особенно предпочтительны CH3-SO2-O-, C6H5-SO2-O-, (4-H3-C6H4)-SO2-O- либо [2,4,6-(CH3)3-C6H2]-SO2-O-. Гидроксииминосоединения II, в которых R2 обозначает C1-C6-алкоксигруппу, позволяют получить особое преимущество, заключающееся в том, что при использовании алкоголятов щелочных металлов в качестве оснований спирт, высвобожденный в результате солеобразования в начале или во время реакции, может оставаться в реакционной смеси. Благодаря этому такие нежелательные побочные реакции, как элиминирование R3-SO2-OH или образование простого эфира при реакции между спиртом и алкилирующим средством III, удаляется подавить в очень сильной степени.
Что же касается гидроксииминосоединений II, в которых R2 обозначает C1-C6-алкил, то применение гидроксидов щелочных металлов в качестве оснований и N-алкилпирролидонов, предпочтительно N-метилпирролоидона, в качестве растворителей дает особое преимущество, поскольку в этих случаях вода, высвобожденная в результате солеобразования в начале или во время реакции, может оставаться в реакционной смеси. При таком осуществлении способа в очень сильной степени удается подавить нежелательные побочные реакции, как элиминирование R3-SO2-OH или омыление алкилирующего средства III.
Из соединений IV и IVa путем кислотного гидролиза может быть высвобожден соответствующий простой гидроксиламиновый эфир формулы I. Последний образуется при этом сначала в виде соли используемой кислоты, а затем может быть выделен либо как таковая, либо, после добавки основания, в виде свободного гидроксиламинового эфира I.
Пригодными для осуществления реакции расщепления зарекомендовали себя минеральные кислоты, предпочтительно соляная кислота и фосфорная кислота, и сильные органические кислоты, как трихлоруксусная кислота и трифторуксусная кислота. Те оксиминопроизводные IV и IVa, в которых R2 обозначает C1-C4-алкил, могут расщепляться особенно эффективно с помощью минеральных кислот.
Среди минеральных кислот особенно предпочтительна соляная кислота, в которую при необходимости может добавляться сорастворитель. В качестве сорастворителей могут использоваться, например, спирты.
Обычно расщепление проводят в диапазоне температур от 50 до 120oC, причем реакция протекает с достаточной быстротой.
Количество кислоты не является критическим. Для осуществления полного гидролиза требуется по крайней мере эквивалентное количество кислоты по отношению к соединениям IV или IVa. Как правило, достаточные количества составляет 1-10 моль кислоты на 1 моль соединения IV или, если соединение IV не выделяют, на 1 моль соединения II или III. Применение большего количества кислоты также допустимо, однако обычно это не дает никаких преимуществ.
Оксиминопроизводные формулы IV, в которых R1 обозначает метил, а R2 обозначает этокси, можно гидролизовать также аналогично методу, описанному в заявке Германии DE-A 2651083.
Все названные выше стадии способа могут осуществляться, как правило, при атмосферном давлении либо при собственном давлении соответствующей системы.
Согласно предлагаемому по изобретению способу простые гидроксиламиновые эфиры I можно получать технические простым путем. Существенное преимущество при этом состоит в том, что при расщеплении оксиминопроизводных IV наряду с гидроксиламиновыми эфирами I получают также другие ценные продукты, а именно, производные (дочерние) продукты фрагментов, содержащих защитные группы, т.е. кетоны или сложные эфиры. Во многих случаях существует даже возможность возвращения защитной группы и ее повторного использования для получения гидроксииминосоединений II. Так, например, при значениях R1 и R2, соответствующих метилу, возможен возврат образующегося при гидролизе ацетона для повторного использования при получении ацетоноксима II (R1, R2 обозначает метил).
Простые гидроксиламиновые эфиры формулы I представляют собой важные промежуточные продукты для средств защиты растений и фармацевтических препаратов. В качестве свободных оснований или солей их можно, например, конденсировать по известной методике с помощью циклогексантрионов либо пиронов VII с целью получения соответствующих простых оксимовых эфиров VIII, применяемых предпочтительно в качестве гербицидов для защиты растений (ср., например, европейские заявки EP-A 136702, EP-A 142741 и EP-A 456112).
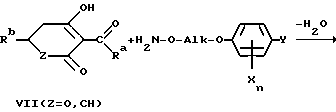
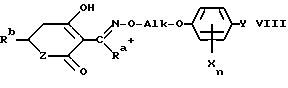
Ra обозначает предпочтительно C1-C4-алкил и Rb представляет собой, например, алкоксиалкил, алкилтиоалкил, при определенных условиях замещенную циклоалкильную либо циклоалкенильную группу, при определенных условиях замещенный 5-звенный гетероцикл или гетероароматический углеводород, при определенных условиях замещенный 6- либо 7-звенный гетероцикл или при определенных условиях замещенное фенильное либо пиридиловое кольцо.
Примеры получения.
Пример 1. 2-(4-хлорфенокси)-1-(1-этоксиэтилиденаминоокси)- пропан {= этиловый эфир O-[2-(4-хлорфенокси)пропил] ацетгидроксимовой кислоты} (таблица, соединение N 18)
а) Алкилирование метилатом калия в качестве основания.
К 155 г (1,5 моль) этилового эфира ацетгидроксимовой кислоты в 1500 мл абсолютного диметилформамида добавляли 105,2 г (1,5 моль) метилата калия (слегка экзотермическая реакция). После перемешивания в течение 45 мин при температуре 25-30oC получали прозрачный раствор, который в течение 2,5 ч добавляли в нагретый до 50oC раствор из 265 г (1 моль) 2-(4-хлорфенокси)пропилметансульфоната в 600 мл диметилформамида. После окончания добавки продолжали перемешивать еще в течение 4 ч при температуре 50oC, после чего еще в течение 1 ч нагревали до 100oC и затем охлаждали. Далее удаляли диметилформамид в водоструйном вакууме при температуре ванны макс. 100oC. После охлаждения остаток растворяли в 1 л толуола и 1 л 1 мас.%-ного едкого натра. Водную фазу отделяли и экстрагировали один раз с помощью 200 мл толуола. Соединенные органические фазы промывали дважды порциями по 200 мл 1 мас. %-ного едкого натра и один раз водой, после чего сушили и концентрировали. Полученный сырой продукт со степенью чистоты 94,4% (ГХ - процент по площади) можно было непосредственно использовать для проведения следующей стадии (получение простого гидроксиламинового эфира).
При необходимости продукт можно очищать путем фракционной перегонки при пониженном давлении.
Выход: 89%; Ткип 100-101oC (при 0,2 мбар).
б) Алкилирование метилатом натрия в качестве основания.
К 30,9 г (0,3 моль) этилового эфира ацетгидроксимовой кислоты в 450 мл диметилформамида при температуре 50oC по каплям добавляли 30 мас.%-ный раствор метилата натрия в метаноле (0,3 молья метилата натрия). Полученную смесь перемешивали еще в течение 30 мин, после чего при пониженном давлении при внутренней температуре макс. 50oC из реакционной смеси отгоняли 180 мл жидкости. В концентрат в течение 40 мин при той же температуре добавляли по каплям 52,9 г (0,2 моль) 2-(4-хлорфенокси)пропилметансульфоната, растворенного в 70 мл диметилформамида. После дальнейшего перемешивания в течение 18 ч при температуре 50oC работали аналогично тому, как это описано выше в а).
Таким путем получали указанное в заготовке соединение с выходом 79%.
Пример 2. 2-(4-хлорфенокси)-1-изопропилиденаминооксипропан {=ацетон-[O-2-(4-хлорфенокси)пропил]-оксим} (таблица, соединение N 28).
а) Алкилирование гидроксидом натрия в качестве основания
К 68,3 г (0,94 моль) ацетоноксима и 306 мл N-метилпирролидона при перемешивании добавляли 37,4 г (0,94 моля) гидроксида натрия. Затем смесь нагревали до внутренней температуры 100oC и в течение 45 мин по каплям добавляли 24,5 г (0,85 моль) 2-(4-хлорфенокси)пропилметансульфоната, растворенного в 155 мл N-метилпирролидона. Через 2 ч реакционную смесь охлаждали до 30oC, после чего при пониженном давлении при температуре кипения 46oC (2 мбар) отгоняли 415 г N-метилпирролидона, который может быть использован при дальнейшем осуществлении способа. Затем давали остыть, добавляли 500 мл воды, перемешивали в течение 45 мин и экстрагировали пять раз соответственно порциями по 250 мл циклогексана. После сушки и концентрирования сырой продукт очищали путем фракционной перегонки.
В результате получали указанное в заголовке соединение с выходом 80%. Ткип 83-87oC (при 0,1 мбар).
б) Алкилирование натриевой солью ацетоноксима.
30 мас.%-ный раствор метилата натрия в метаноле разбавляли в трехкратном объеме толуола, после чего добавляли эквивалентное количество ацетоноксима. Затем при пониженном давлении удаляли низкокипящие компоненты.
Затем к 142,6 г (1,5 моль) натриевой соли ацетоноксима, помещенного в 490 мл N-метилпирролидона, при температуре 100oC добавляли по каплям 264,1 г (1 моль) 2-(4-хлорфенокси)пропилметансульфоната, растворенного в 180 мл N-метилпирролидона. После этого реакцию проводили еще в течение 1 ч и далее работали аналогично тому, как это описано выше.
Таким образом повторно получали 590 мл N-метилпирролидона. Выход соединения, указанного в заголовке, составлял 81% (степень чистоты согласно ГХ 96%).
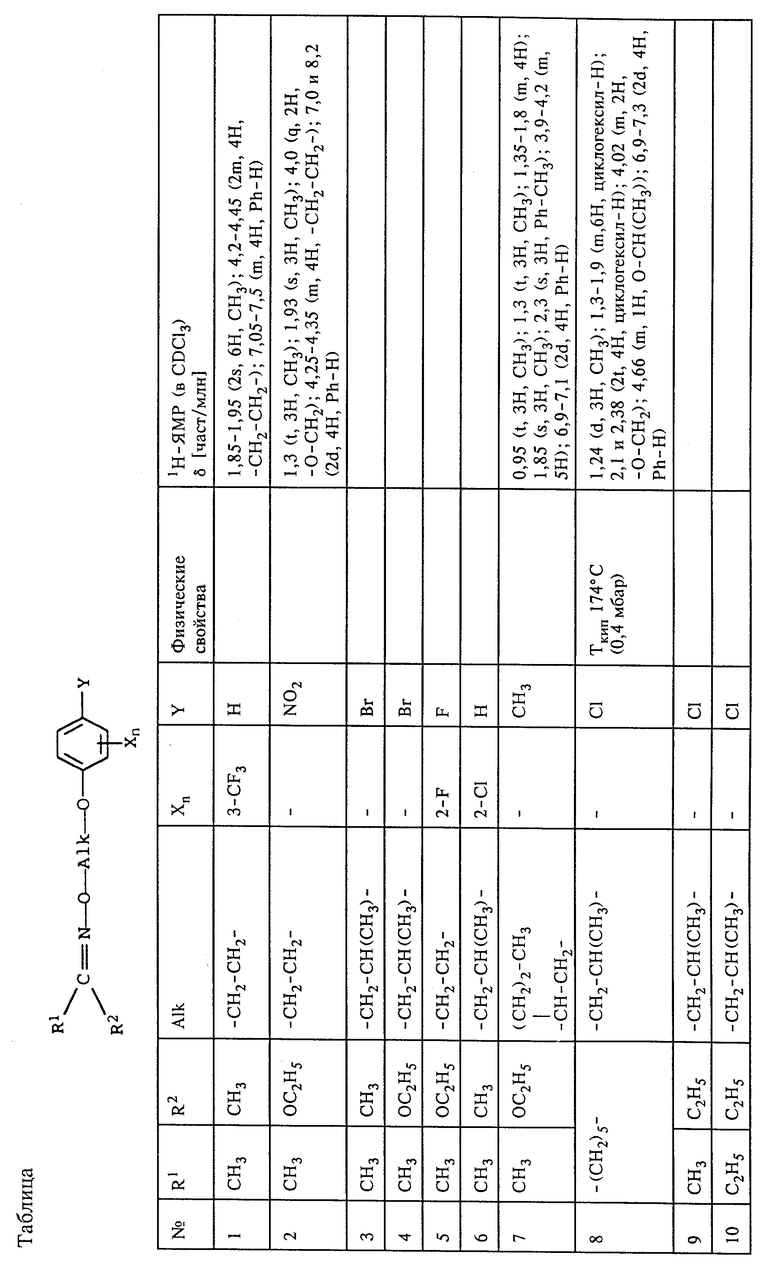
В таблице представлены другие оксиминопроизводные формул IV и IVa, которые получали по аналогичной методике или которые могут быть получены таким путем.
Пример 3. 1-аминоокси-2-(4-хлорфенокси)пропан { = 2-(4-хлорфенокси)пропоксиамин}.
а) Гидролиз этилового эфира O-[2-(4-хлорфенокси)пропил]ацетгидроксимовой кислоты.
К 1,7 л (3,4 моль) 2H соляной кислоты при температуре 20-25oC по каплям добавляли в течение 60 мин 485 г (1,7 моль) этилового эфира O-[2-(4-хлорфенокси)пропил] ацетгидроксимовой кислоты (таблица, соединение N 18; степень чистоты 95% согласно ГХ), после чего смесь в течение 30 мин нагревали с обратным холодильником. Затем смесь охлаждали, при охлаждении ледяной ванной с помощью 280 мл 50 мас%-ного едкого натра устанавливали на pH 10 и трижды экстрагировали соответственно порциями по 400 мл дихлорметана. Соединенные органические фазы промывали водой и затем сушили и концентрировали.
Таким путем получали указанное в заголовке соединение с выходом 97% (степень чистоты 96,9% согласно анализу ГХ).
При необходимости полученное соединение можно очистить путем перегонки; Ткип 102-104oC (0,4 мбар).
250 МГц, 1H-ЯМР (в CDCl3): δ [част/млн]=1,25 (d, 3H, CH3); 3,6-3,9 (m, 2H; -O-CH2); 4,64 (m, 1H; Ph-O-CH(CH3); 5,5 (широкий s, 2H; NH2); 6,9 и 7,2 (2d, 4H; Ph-H).
б) Гидролиз ацетон-[O-2-(4-хлорфенокси)пропил] оксима трихлоруксусной кислотой.
В смесительном устройстве с насадкой в виде 30-сантиметровой колонки при температуре 78oC и пониженном давлении 450 мбар в течение 10 ч нагревали смесь из 10 г (4,13 ммоль) ацетон-[O-2-(4-хлорфенокси)пропил] -оксима (соединение N 28 в таблице) и 44 г 30 мас%-ного водного раствора трихлоруксусной кислоты, причем в реакционную смесь непрерывно добавляли по каплям 110 г воды, а нагрев проводили при непрерывной отгонке образующейся смеси воды и ацетона. Для дальнейшей переработки реакционный продукт подщелачивали 10 мас%-ным едким натром и затем экстрагировали с помощью толуола. Таким путем выделяли 6 г 2-(4-хлорфенокси)пропоксиамина. Выход составлял 72%.
в) Гидролиз ацетон-[O-2(4-хлорфенокси)пропил] оксима трифторуксусной кислотой.
Аналогично эксперименту 3б) 10 г (4,13 ммоль) ацетон-[O-2-(4-хлорфенокси)пропил] -оксима и 31 г 30%-ного водного раствора трифторуксусной кислоты нагревали при пониженном давлении 430 мбар в течение 8 3/4 ч до 80oC, причем в реакционную смесь непрерывно добавляли по каплям 100 г воды, а нагрев проводили при непрерывной отгонке смеси воды и ацетона. После обработки, аналогично описанной в 3б), получали 6,4 г 2-(4-хлорфенокси)пропоксиамина. Выход составлял 76%.
г) Гидролиз ацетон-[O-2-(4-хлорфенокси)пропил]оксима соляной кислотой.
В смесительном устройстве 10 г (4,13 ммоль) ацетон-[O-2-(4-хлорфенокси)пропил] оксима растворяли в смеси из 250 г n-пропанола, 38 г концентрированной соляной кислоты (38 мас%-ной) и 60 г воды. Этот раствор нагревали в течение 6 ч до 80oC, после чего для дальнейшей обработки отгоняли n-пропанол и воду. После перекристаллизации остатка из 20 мас%-ной соляной кислоты получали 7,7 г гидрохлорида 2-(4-хлорфенокси)-пропоксиамина. Выход составлял 78%.
Экспериментальные исследования.
Пример А. Алкилирование ацетоноксима 2-(4-хлорфнокси)пропил-метансульфонатом с карбонатом калия в качестве основания в N-метилпирролидоне.
В 65 мл N-метилпирролидона помещали 14,6 г (0,2 моль) ацетоноксима и 27,64 г (0,2 моль) карбоната калия. Затем при перемешивании нагревали до внутренней температуры 100oC в течение 22 мин по каплям добавляли раствор из 54,08 г (0,18 моль) 2-(4-хлорфенокси)пропилметансульфоната в 33 мл N-метилпирролидона. Перемешивание продолжали еще в течение 14 ч и 40 мин при этой температуре, после чего при пониженном давлении практически полностью удаляли растворитель, остаток растворяли в 400 мл воды и трижды экстрагировали порциями по 200 мл циклогексана соответственно. Объединенные органические фазы сушили над сульфатом магния и концентрировали. ГХ-анализ показал наличие в остатке ацетон-[O-2-(4-хлорфенокси)пропил]оксима.
Пример Б. Алкилирование ацетоноксима 2-(4-хлорфенокси)пропилметансульфонатом с карбонатом калия в качестве основания в N-метилпирролидоне.
В 65 мл N-метилпирролидона помещали 14,6 г (0,2 моль) ацетоноксима и 55,28 г (0,4 моль) карбоната калия. Затем при перемешивании нагревали до внутренней температуры 100oC и в течение 17 мин по каплям добавляли раствор из 54,08 г (0,18 моль) 2-(4-хлорфенокси)пропилметансульфоната в 33 мл N-метилпирролидона. Перемешивание продолжали еще в течение 32 ч и 18 мин при этой температуре, после чего при пониженном давлении практически полностью удаляли растворитель, остаток растворяли в 400 мл воды и трижды экстрагировали порциями по 200 мл циклогексана соответственно. Объединенные органические фазы сушили над сульфатом магния и концентрировали. ГХ-анализ показал наличие в остатке ацетон-[O-2-(4-хлорфенокси)пропил]оксима 32,6%.
Пример В. Алкилирование ацетоноксима 2-(4-хлорфенокси)пропилметансульфонатом с гидрокарбонатом калия в качестве основания в N-метилпирролидоне.
В 65 мл N-метилпирролидона помещали 14,6 г (0,2 моль) ацетоноксима и 40,05 г (0,4 моль) гидрокарбоната калия. Затем при перемешивании нагревали до внутренней температуры 100oC и в течение 25 мин по каплям добавляли раствор из 54,08 г (0,18 моль) 2-(4-хлорфенокси)пропилметансульфоната в 33 мл N-метилпирролидона. Перемешивание продолжали еще в течение 14 ч и 15 мин при этой температуре, после чего при пониженном давлении практически полностью удаляли растворитель, остаток растворяли в 400 мл воды и трижды экстрагировали порциями по 200 мл циклогексана соответственно. Объединенные органические фазы сушили над сульфатом магния и концентрировали. ГХ-анализ показал наличие в остатке ацетон-[O-2-(4-хлорфенокси)пропил]оксима.
Пример Г. Алкилирование ацетоноксима 2-(4-хлорфенокси)пропилметансульфонатом с карбонатом натрия в качестве основания в N-метилпирролидоне.
В 65 мл N-метилпирролидона помещали 14,6 г (0,2 моль) ацетоноксима и 42,4 г (0,4 моль) карбоната натрия. Затем при перемешивании нагревали до внутренней температуры 100oC и в течение 22 мин по каплям добавляли раствор из 54,08 г (0,18 моль) 2-(4-хлорфенокси)пропилметансульфоната в 33 мл N-метилпирролидона. Перемешивание продолжали еще в течение 13 ч и 34 мин при этой температуре, после чего при пониженном давлении практически полностью удаляли растворитель, остаток растворяли в 400 мл воды и трижды экстрагировали порциями по 200 мл циклогексана соответственно. Объединенные органические фазы сушили над сульфатом магния и концентрировали. ГХ-анализ показал наличие в остатке ацетон-[O-2-(4-хлорфенокси)пропил]оксима.
Пример Д. Алкилирование ацетоноксима 2-(4-хлорфенокси)пропилметансульфонатом с гидрокарбонатом натрия в качестве основания в N-метилпирролидоне.
В 65 мл N-метилпирролидона помещали 14,6 г (0,2 моль) ацетоноксима и 33,6 г (0,4 моль) гидрокарбоната натрия. Затем при перемешивании нагревали до внутренней температуры 100oC и в течение 25 мин по каплям добавляли раствор из 54,08 г (0,18 моль) 2-(4-хлорфенокси)пропилметансульфоната в 33 мл N-метилпирролидона. Перемешивание продолжали еще в течение 14 ч и 15 мин при этой температуре, после чего при пониженном давлении практически полностью удаляли растворитель, остаток растворяли в 400 мл воды и трижды экстрагировали порциями по 200 мл циклогексана соответственно. Объединенные органические фазы сушили над сульфатом магния и концентрировали. ГХ-анализ показал наличие в остатке ацетон-[O-2-(4-хлорфенокси)пропил]оксима.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 3-ГАЛОГЕН-3-ГЕТАРИЛКАРБОНОВОЙ КИСЛОТЫ, ГЕРБИЦИДНОЕ СРЕДСТВО | 1994 |
|
RU2146255C1 |
| ПРОСТЫЕ ЦИКЛОГЕКСЕНОНОКСИМОВЫЕ ЭФИРЫ И ГЕРБИЦИДНАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2110513C1 |
| ОРТОЗАМЕЩЕННЫЕ АМИДЫ 2-МЕТОКСИИМИНОФЕНИЛУКСУСНОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, СРЕДСТВО ДЛЯ БОРЬБЫ С ВРЕДОНОСНЫМИ ГРИБАМИ И ИНСЕКТОАКАРИЦИДНОЕ СРЕДСТВО | 1994 |
|
RU2130924C1 |
| СПОСОБ ПОЛУЧЕНИЯ АСИММЕТРИЧНО ЗАМЕЩЕННЫХ ТРИАЗИНОВ | 1994 |
|
RU2125995C1 |
| ПРОИЗВОДНЫЕ 3-АРИЛОКСИКАРБОНОВОЙ КИСЛОТЫ, ГЕРБИЦИДНЫЙ ПРЕПАРАТ И СРЕДСТВО ДЛЯ ПОДАВЛЕНИЯ РОСТА РАСТЕНИЙ | 1994 |
|
RU2135479C1 |
| ПРОИЗВОДНЫЕ 3-(ГЕТ)АРИЛКАРБОНОВОЙ КИСЛОТЫ, ГЕРБИЦИДНЫЙ ПРЕПАРАТ И СРЕДСТВО ДЛЯ ПОДАВЛЕНИЯ РОСТА | 1994 |
|
RU2140413C1 |
| ПРОИЗВОДНЫЕ ГИДРОКСИМЕТИЛФУРАЗАНКАРБОНОВОЙ КИСЛОТЫ И ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ | 1994 |
|
RU2134687C1 |
| ЦИКЛИЧЕСКИЕ И ГЕТЕРОЦИКЛИЧЕСКИЕ N-ЗАМЕЩЕННЫЕ α-ИМИНОГИДРОКСАМОВЫЕ И КАРБОНОВЫЕ КИСЛОТЫ | 1996 |
|
RU2164914C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 3-ПИРРОЛИН-2-КАРБОНОВОЙ КИСЛОТЫ | 1997 |
|
RU2199529C2 |
| АМИДЫ КАРБАМОИЛКАРБОНОВОЙ КИСЛОТЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, СРЕДСТВО И СПОСОБЫ ДЛЯ БОРЬБЫ С ВРЕДОНОСНЫМИ ГРИБАМИ | 1995 |
|
RU2145956C1 |
Простые гидроксиламиновые эфиры формулы I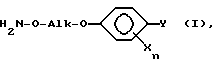
являются промежуточными продуктами для средства защиты растений и фармацевтических препаратов. Эфиры I, где X - галоген, C1-С4-алкил и C1-C4-галогеналкил; Y - H, NO2, галоген, C1-C4-алкил; n - 0-2, Alk - C2-или C3-алкиленовая цепь, которая может нести от одной до трех C1-C3-алкильных групп, получают либо взаимодействием гидроксииминосоединения II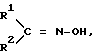
где R1 = алкил, R2 = алкил, алкокси или R1+R2 = алкиленовая цепь, с алкилирующим агентом, в присутствии основания, либо соответствующий анион соединения II непосредственно алкилирующим средством III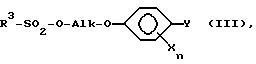
переводят в оксиминопроизводное IV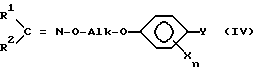
затем IY с помощью минеральной кислоты или сильной органической кислоты расщепляют в соль соединения I. Согласно предлагаемому способу простые гидроксиламиновые эфиры I получают технически простым путем. Все стадии способа могут осуществляться, как правило, при атмосферном давлении либо при собственном давлении соответствующей системы. 2 с. и 7 з.п.ф-лы, 1 табл.
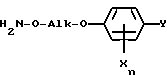
в которой X обозначает галоген, C1-C4-алкил и C1-C4-галогеналкил;
Y обозначает водород, нитро, галоген, C1-C4-алкил;
n обозначает 0 - 2, либо 1 - 4 в случае, если Y и все X означают галоген;
Alk обозначает C2- либо C3-алкиленовую цепь, которая при необходимости может нести от одной до трех C1-C3-алкильных групп,
а также их солей с минеральными кислотами или сильными органическими кислотами, отличающийся тем, что либо гидроксииминосоединение формулы II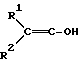
в которой R1 обозначает C1-C4-алкил, R2 обозначает C1-C4-алкил или C1-C6-алкоксил, или R1 и R2 вместе представляют собой C4-C6-алкиленовую цепь, в присутствии гидроксида щелочного металла, алкоголята щелочного металла, гидрокарбоната щелочного металла или карбоната щелочного металла в качестве основания, либо соответствующий анион гидроксииминосоединения II подвергают взаимодействию с алкилирующим средством формулы III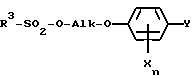
в которой R3 обозначает C1-C4-алкил, с получением оксиминопроизводного формулы IV, в котором R1, R2, Alk, X, Y и n имеют вышеуказанные значения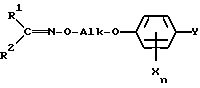
которое затем с помощью минеральной кислоты или сильной органической кислоты расщепляют с образованием соответствующей соли гидроксиламинового эфира I и эту последнюю, при необходимости, с помощью основания переводят в свободное соединение I.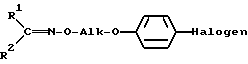
в которой R1 обозначает C1-C4-алкил;
R2 обозначает C1-C4-алкил либо C1-C6-алкоксил или R1 и R2 вместе обозначают C4-C6-алкилен;
Alk обозначает C2 либо С3-алкиленовую цепь, которая при необходимости может нести от одной до трех C1-C3-алкильных групп.
| ЕР, 23560, 1981 | |||
| EP, 456112, 1991 | |||
| EP, 440582, 1991. |
