Настоящее изобретение относится к новому способу получения производных пирролин-2-карбоновой кислоты.
Замена пролина на 3,4-дегидропролин в биологически активных пептидах или пептидомиметиках редко приводит к потере активности (А.М. Felix et al. Int. J. Pept. Prot. Res. 10, 299 (1977); С.R. Botos et al. J. Med. Chem. 22, 926 (1979); G. H. Fisher, W. Ryan, FEBS Lett. 107, 273 (1979)), но в некоторых случаях даже усиливает действие при одновременном уменьшении токсичности (G. H. Fisher, W. Ryan, FEBS Lett. 107. 273 (1979); S. Natarajan et al., в: Peptide, Structure and Biological Function, редакторы: E. Gross, J. Meienhofer, Pierce Chemical Company, 1979, с. 463).
Синтез N-защищенных 3,4-дегидропролинов в промышленном масштабе, осуществляемый в соответствии с известными из литературы способами, требует очень больших затрат, как показывает, например, термическое цис-удаление S-метилксантогената из гидроксипролина по Чугаеву. Недостатки этого способа состоят в том, что приходится работать с большими количествами метилиодида, а также в том, что образуются метилмеркаптан и оксисульфид углерода (J.-R. Dormay et al. , Angew. Chem. 92, 761 (1980); Houben-Weil, Methoden der Organischen Chemie, том 5/1b, 126 (1972)).
Также проблематично восстановление пиррол-2-карбоновой кислоты иодидом фосфония в дымящей иодистоводородной кислоте из-за необходимости работать с большим избытком газообразного йодистого водорода, а также по причине заметного снижения выхода и начинающейся полимеризации при больших степенях конверсии (J. W. Scott et al., Synth. Commun. 10 (7), 529 (1980)). Напротив, удаление Бок-защищенного метилового эфира 4-фенилселенинилпролина протекает при значительно более мягких условиях (J. -R. Dormay, Synthesis 9, 753 (1982). Удаление происходит при комнатной температуре и приводит с высокой селективностью к получению Δ3-олефина. Термические цис-удаления сопровождаются образованием значительных количеств изомерного Δ4-олефина. Кроме того, недостатком является как удаление оксидов селена из-за образующихся токсичных селенсодержащих отходов, обезвреживание которых как раз при получении в полупромышленном масштабе является дорогостоящей проблемой, так и присоединение ранее отщепленной селенистой кислоты по двойным связям, особенно если речь идет о лекарственных активных веществах, в которых уже минимальные количества селенсодержащих соединений приводят к появлению токсичных свойств.
Из N-замещенного 3-метилсульфонилокси-пирролидина были получены небольшие количества 3-пирролина (Т. Uno et al., J. Heterocycl. Chem. 24, 1025 (1987). Отщепление сульфонатов, таких как, например, метилсульфонат, для получения производных 3-пирролин-2-карбоновой кислоты до сих пор описано не было.
Названные литературные способы получения производных 3-пирролин-2-карбоновой кислоты не пригодны для промышленных синтезов.
Настоящее изобретение относится к способу получения производных 3-пирролин-2-карбоновой кислоты формулы I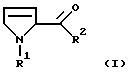
в которой R1 обозначает водород, алкил с 1-6 атомами углерода, бензил, замещенный в фенильном кольце бензил, аллилоксикарбонил, C1-С6-алкилоксикарбонил, бензилоксикарбонил, в котором бензильный остаток может быть замещен остатками метокси, или С1-С4-алкилкарбонил или
R1 обозначает связанный через С-конец остаток аминокислоты, которая может быть алкилирована или ацилирована у азота,
и
R2 обозначает гидрокси, алкокси с 1-4 атомами углерода, бензилокси или группу NR3R4,
где R3 и R4 независимо друг от друга обозначают водород, алкил с 1-4 атомами углерода, бензил, фенил или пиридил, причем ароматические соединения в R3 и R4 могут иметь до трех одинаковых или различных заместителей, выбранных из группы, состоящей из метила, метокси, гидрокси, циано и галогена,
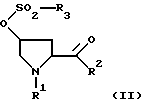
который состоит в том, что из сульфоната формулы II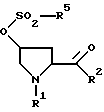
где R1 и R2 имеют вышеуказанное значение, a R5 обозначает алкил с 1-6 атомами углерода, бензил, трифторметил, нафтил или фенил, который может быть при необходимости замещен остатками из группы метил, нитро, галоген, отщепляют с помощью основания сульфокислотный остаток.
В качестве остатка R1 предпочтительны С1-С4-алкилкарбонил, бензил, замещенный в фенильное кольцо бензил, C1-С6-алкоксикарбонил и бензилоксикарбонил. Если бензилоксикарбонильный остаток замещен группой метокси, то метоксигруппа находится предпочтительно в пара-положении. Особенно предпочтительным является C1-С6-алкилоксикарбонильный остаток.
Предпочтительными остатками R2 являются гидрокси и алкокси с 1-4 атомами углерода.
Предпочтительными остатками R5 являются алкил с 1-6 атомами углерода и бензил, в частности алкил с 1-4 атомами углерода.
Соединения I содержат один асимметричный С-атом, соединения II содержат два асимметричных С-атома в 5-членном кольце. Соединения формулы II могут применяться в виде рацематов, смесей диастереомеров, чисто диастереомерных и также чисто энантиомерных соединений. Поэтому соединения I в зависимости от стереохимической структуры применяемых в качестве аддуктов соединений II и условий реакции могут быть получены в виде рацематов или в оптически активной форме.
Соединения формулы II могут быть получены известными из литературы методами (см., например, D.J. Abraham, M. Mokotoff, L. Sheh, J.E. Simmons, J. Med. Chem. 26(4), 549 (1983)).
Отщепление сульфокислотного остатка, т.е. группы -O-SO2-R5, из оптически активных соединений формулы II протекает с рацемизацией, если R2 является алкокси с 1-4 атомами углерода или бензилокси. Таким образом получают рацемические эфиры 3,4-дегидропролина, из которых с помощью последующего ферментативного расщепления рацемата могут быть получены производные как D-, так и L-3,4-дегидропролина (способ А).
Способ А:
(R5=СН3, R2=ОСН3)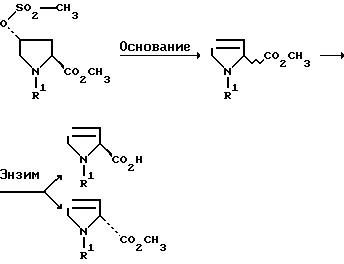
Особенно предпочтительный вариант осуществления способа состоит в том, что при применении соединений формулы II, в которой R2 обозначает гидрокси и установлена абсолютная конфигурация остатка карбоновой кислоты, т.е. эта конфигурация соответствует либо R- либо S-конфигурации, соответствующие карбоновые кислоты формулы I могут быть получены без рацемизации (способ Б).
Способ Б:
(R5=СН3, R2=OH)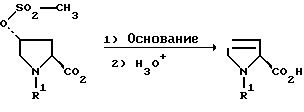
Для реакций отщепления А и Б пригодны непротонные растворители, в частности диметилформамид, диоксан, тетрагидрофуран, диметоксиэтан, диметилсульфоксид, CH3CN, причем растворитель может при необходимости содержать небольшие количества воды или спирта.
В зависимости от способа основание применяется в количестве от 1,0 до 1,5 эквивалентов (способ А), соответственно в количестве от 2,0 до 3,0 эквивалентов (способ Б), причем в качестве оснований могут рассматриваться гидриды, амиды или алкоголяты лития, натрия, калия, рубидия, цезия, кальция или магния, предпочтительно, однако, гидриды, амиды или алкоголяты натрия и калия. Алкоголяты натрия предпочтительно используются в виде оснований, в которых алкоголятными остатками являются первичные, вторичные и третичные спирты. Могут применяться также диолы, триолы, эфироспирты типа моноэфира три-, ди- или моноэтиленгликоля или аминоспирты. Предпочтительно следует назвать: монометиловый эфир триэтиленгликоля, монометиловый эфир диэтиленгликоля или монометиловый эфир этиленгликоля, диметиламиноэтанол или 2-[2-(диметиламино)-этокси]-этанол.
Отщепление сульфонатной группы происходит уже при температуре -20oС. В принципе реакция может быть проведена при температурах от -20oС до +100oС. Предпочтительно она проводится при температурах от -10oС до +60oС. Отщепление сульфонатной группы из соответствующих сложных эфиров по способу А происходит у α-углеродного атома образующегося при этом эфира 3,4-дегидропролина уже при -20oС с рацемизацией.
Неожиданно оказалось, что способ согласно особенно предпочтительному варианту Б может проводиться с аддуктами формулы II, где R2 обозначает гидрокси и остаток карбоновой кислоты имеет либо R-, либо S-конфигурацию, почти без рацемизации. В качестве оснований в этом варианте предпочтительны гидриды, первичные алкоголяты, первичные эфироспирты или первичные аминоалкоголяты. Особенно предпочтительно применяются 2-метоксиэтанолят, 2-(2-метоксиэтокси)-этанолят или 2-[2-(диметиламино)-этокси]-этанолят. Предпочтительно на один эквивалент аддукта расходуется от 2,0 до 2,5 эквивалентов основания. Предпочтительный температурный интервал для реакции по способу Б лежит в пределах от -10oС до +25oС.
Основание, применяемое для отщепления, может вводиться в реакционную смесь в твердой форме, но его можно также получать перед реакцией in situ. Если основанием, применяемым для отщепления, является 2-метоксиэтанолят натрия, то это основание можно предпочтительно получать in situ, добавляя по каплям соответствующий алкоголят к раствору, соответственно к суспензии натриевой соли более сильного основания, такого как, например, гидрид натрия, трет-бутилат натрия или бис-(триметилсилил)-амид натрия.
Реакция может проводиться в полунепрерывном режиме либо с подачей раствора основания к растворенному аддукту формулы II, либо предпочтительно с подачей раствора аддукта II к загруженному в реактор раствору, соответственно суспензии основания.
Обработка реакционной смеси может осуществляться путем дистилляции, экстракции, кристаллизации, хроматографии или комбинации этих способов.
Целевой энантиомер может быть выделен из рацемического 3,4-дегидропролина с помощью либо (+)-, либо (-)-винной кислоты (см. J. W. Scott et al., Synthetic Communications 10, 529 (1980), соответственно US 4111951) или же расщепление рацемата может быть проведено с помощью оптически активного 1-(4-нитрофенил)-этиламина после получения Бок-защищенной аминокислоты (патент США US 4066658, J.U. Kahl, T. Wieland, Liebigs Ann. Chem. 8, 1445 (1981)).
N-защищенные 3,4-дегидропролины, полученные в соответствии с предпочтительным способом Б без рацемизации, могут быть целесообразным образом очищены путем их кристаллизации в виде солей аммония с ахиральными аминами. В частности, можно получать в чистой форме L-Бок-3,4-дегидропролин в качестве диэтиламмониевой соли.
Предметом изобретения являются поэтому соединения формулы IV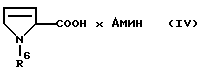
где R6 является аминозащитной группой, а амин представляет собой моно-, ди- или триалкиламин, в котором алкильные остатки содержат 1-4 С-атома и могут быть заменены циклоалкильными остатками с 5-7 атомами углерода и их оптически активными D- и L-формами. R6 предпочтительно обозначает Бок-защитную группу, а "амин" - предпочтительно диэтиламин или дицикло-гексиламин.
Полученные по способу А сложные эфиры могут быть легко расщеплены частично с помощью ферментов, таких как липазы, эстеразы и протеазы, причем образуется один антипод свободной кислоты, в то время как другой антипод остается в форме сложного эфира.
В качестве гидролаз в названном способе может быть использовано большое количество ферментов. Предпочтительно используются протеазы, эстеразы и особенно липазы. В качестве липаз весьма пригодны прежде всего бактериальные липазы, которые, например, могут быть выделены из дрожжей или бактерий. Другими особенно хорошо пригодными гидролазами являются реализуемые на коммерческой основе фирмой Novo Nordisk (Enzyme Toolbox) ферменты, в частности липазы SP 523, SP 524; SP 525, SP 526 и ®Novozym 435.
Кроме того, в предлагаемом способе с успехом могут быть использованы липазы "Chirazyme L1-L8", которые можно приобрести на коммерческих началах у фирмы Boehringer Mannheim.
Могут быть использованы также эстеразы (такие как эстераза, полученная из свиной печени).
Ферменты могут применяться в нативной или иммобилизованной форме.
Расщепление сложных эфиров производится в буфере при рН 6-8 и предпочтительно при комнатной температуре.
Новый способ обеспечивает простое получение соединений I. Это особенно важно для производных дегидропролина, которые до сих пор можно было получать лишь с трудом и отчасти с плохим выходом.
Особенно благоприятно то, что новый способ позволяет получать оптически активные N-защищенные производные 3-пирролин-2-карбоновой кислоты в виде свободной кислоты или в виде сложного эфира, из которого свободную кислоту можно выделить, предпочтительно, ферментативно.
Если оптически активную кислоту получают из ее эфира ферментативно, то антипод эфира, как правило, остается в неизменной форме. Его можно рацемизировать, например, основаниями и снова подвергнуть ферментативному расщеплению.
Преимущество настоящего изобретения состоит в том, что оно впервые обеспечивает при мягких и одновременно экологически безвредных условиях реакции простой доступ к получению производных 3,4-дегидропролина в стерически чистой форме также и промышленном масштабе. Поразительным является тот факт, что отщепление сульфокислотного остатка может быть осуществлено уже при низких температурах.
Полученные новым способом вещества представляют большой интерес. Они являются, например, ценными промежуточными продуктами для получения тромбин-ингибирующих низкомолекулярных производных пептида (ср. между народная заявка на патент WO 94/29336), у которых пролиновый остаток заменен на дегидропролиновый остаток. Далее было установлено, что 3,4-дегидропролин представляет собой пригодный агент для подавления синтеза коллагена (патент США US 4066658).
Что касается дальнейшей переработки продукта, то особое преимущество способа согласно изобретению состоит в том, что полученный этим способом сырой продукт может быть использован без дополнительной очистки в процессе получения последующих промежуточных продуктов для синтеза производных пептида, которые в свою очередь очень легко могут быть очищены. Эти промежуточные продукты имеют формулу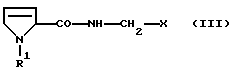
где R1 имеет вышеуказанное значение и Х обозначает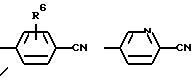
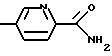
(R6=водород, метил, метокси, гидрокси или галоген)
Они могут быть получены из соединений I тем, что соединения I активируют в присутствии основания, такого как триэтиламин или диизопропилэтиламин, и конденсирующего агента, такого как ангидрид пропилфосфоновой кислоты, пивалоилхлорид или смесь дициклогексилкарбодиимида с гидроксисукцинимидом, и затем связывают с H2N-CH2-X, получая в результате соединения формулы III. Это взаимодействие целесообразно проводить в растворителе, таком как дихлорметан, тетрагидрофуран, диоксан, трет-бутил-метиловый эфир, диметоксиэтан или ацетонитрил, и при температуре от -20oС до +30oС.
Примеры
В примерах использованы следующие сокращения:
Бнс = бензилсульфонил,
Бок = трет-бутилоксикарбонил,
ДИПЭА = диизопропилэтиламин,
ДМЭ = диметоксиэтан,
ДМФ = диметилформамид,
КОтБ = трет-бутилат калия,
Мс = метилсульфонил,
АПФК = ангидрид пропилфосфоновой кислоты,
Pro = пролин,
Руr = 3,4-дегидропролин,
КТ = комнатная температура,
ТГФ = тетрагидрофуран,
ЖХВР = жидкостная хроматография высокого разрешения,
ДМСО = диметилсульфоксид.
А. Получение исходных веществ
а) Бок-(L)-(4-МсО)-Рrо-ОСН3 и Бок-(L)-(4-БнсО)-Рrо-ОСН3:
Метиловый эфир (4R)-N-Бок-4-гидрокси-(L)-пролина подвергают взаимодействию с метилсульфонилхлоридом, получая метиловый эфир (4R)-N-Бок-4-метилсульфонилокси-(L)-пролина (D. J. Abraham, M. Mokotoff, L. Sheh, J. Е. Simmons, J. Med. Chem. 26(4), 549 (1983)). Аналогично взаимодействию с метилсульфонилхлоридом при взаимодействии с бензилсульфонилхлоридом получают метиловый эфир (4R)-N-Бок-4-бензилсульфонилокси-(L)-пролина после кристаллизации из этанола с выходом 76%.
1Н-ЯМР (CDCl3, δ, частей на миллион): 7,40 и 7,28 (с, 5Н, ароматическое соединение), 4,95 и 4,85 (м, 1Н, O-СН), 4,40 (с, 2Н, SO2-CH2), 4,4-4,25 (1H, N-CN), 3,72 и 3,71 (с, 3Н, CO2C□3), 3,7-3,50 (2Н, N-CH2), 2,53-1,95 (2Н, CH2), 1,45 и 1,42 (с, 9Н, Бок); (2 ротамера).
б) (4R)-N-Бoк-(4-McO)-Pro-OH:
186 г (575 ммоль) метилового эфира Бок-(L)-(4-МсО)-Рrо-ОСН3 гидролизуют при 0oС в 500 мл диоксана и 1150 мл 1н. NaOH в течение 2,5 часов. После экстракции эфиром доводят рН водной фазы с помощью 2 н. соляной кислоты до значения 3 и продукт экстрагируют уксусным эфиром. После сушки над Na2SO4 и полного удаления растворителя получают 163 г (92%) желтоватого масла с 94%-ной чистотой. Продукт медленно застывает, превращаясь в твердое вещество.
[α] D 22= -50,5o (с=1,01; МеОН); после кристаллизации из диизопропилового эфира
1Н-ЯМР (CDCl3, δ, частей на миллион): около 9-8 (СООН), 5,35-5,20 (м, 1Н, O-СН), 4,60-4,40 (1Н, N-CH), 3,95-3,65 (2Н, N-CH2), 3,08 (с, 3Н, SO2СН3), 2,85-2,25 (2Н, CH2), 1,50 и 1,40 (с, 9Н, Бок); (2 ротамера).
Б. Получение конечных продуктов
Пример 1
Получение Бок-(D/L)-Руr-ОСН3:
а) 100 г (309 ммоль) Бок-(L)-(4-МсО)-Рrо-ОСН3 растворяют в 600 мл сухого ДМФ. В полученный раствор при температуре от 0 до 5oС добавляют по каплям в течение 1 часа раствор 36,45 г (325 ммоль) КОтБ в 300 мл сухого ДМФ и перемешивают в течение 30 мин при 0-5oС и затем еще в течение 2 часов при КТ. Затем смесь выливают на ледяную воду, трижды экстрагируют смесью эфира с уксусным эфиром (5:1) и органическую фазу еще раз промывают водой. После сушки над Na2SО4 растворитель полностью удаляют при 35oС. Получают 68 г сырого сложного эфира, который перегоняют при 1,7 мбар и 100-102oС. Полученное таким образом бесцветное масло застывает, превращаясь в твердое вещество (выход: 56%).
1Н-ЯМР (CDCl3, δ, частей на миллион): 6,05-5,95 (м, 1Н, 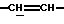 ), 5,80-5,67 (м, 1Н,
), 5,80-5,67 (м, 1Н, 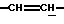 ), 5,05 и 4,98 (м, 1Н, N-CH), 4,35-4,15 (м, 2Н, N-CH2), 3,75 и 3,74 (с, 3Н, СО2СН3), 1,47 и 1,43 (с, 9Н, Бок); (2 ротамера).
), 5,05 и 4,98 (м, 1Н, N-CH), 4,35-4,15 (м, 2Н, N-CH2), 3,75 и 3,74 (с, 3Н, СО2СН3), 1,47 и 1,43 (с, 9Н, Бок); (2 ротамера).
б) То же соединение (Бок-(D/L)-Руr-ОСН3) получают также из бензилсульфоната Бок-(L)-(4-БнсО)-Рrо-ОСН3. Из 5 г (12,5 ммоль) названного бензилсульфоната в 50 мл сухого ДМФ, который добавляют по каплям при -10oС к суспензии 0,5 г (12,5 ммоль) NaH в 10 мл сухого ДМФ и затем в течение ночи перемешивают при КТ, получают после обработки аналогично отщеплению метилата согласно а) 2,2 г сырого сложного эфира. Продукт можно было очищать хроматографически на колонке (подвижная среда: смесь уксусного эфира с гексаном в соотношении 2: 3). Однако из-за слабого хромофора предпочтение следует отдать дистилляции.
Пример 2
Получение Бок-(L)-Руr-ОН:
а) К 14,5 г 55-65%-ного NaH (около 364 ммоль) в 400 мл ДМЭ прибавляют при КТ в течение 45 мин 50,0 г (161,6 ммоль) Бок-(L)-(4-МсО)-Рrо-ОН, растворенного в 650 мл ДМЭ (в который добавляют 25 ммоль воды), причем температуру повышают без дополнительного охлаждения приблизительно до 30oС. Смесь перемешивают в течение 15 часов при КТ и затем еще 1 час при 50oС, после чего выливают на ледяную воду и трижды промывают смесью эфира с уксусным эфиром (2: 1). Водную фазу подкисляют 2 н. соляной кислотой до рН 2 и экстрагируют продукт уксусным эфиром. После сушки над Na2SO4 и полного удаления растворителя получают 36 г сырого вещества в виде желтоватого масла, которое содержит 70% продукта. В результате детектирования установлено, что соотношение продукт/аддукт= 97: 3 (ЖХВР: смесь воды с ацетонитрилом (8:2)+0,1% ТФА; Merck ®Purospher RP-18e; детектирование при 210,4 нм), а соотношение энантиомеров (L): (D) - =90:10. Определение долей энантиомеров производят после связывания кислотной группы с производным 3-пиколиламина в качестве соответствующего Бок-3,4-дегидропролил-(3-пиколил)-амида на хиральной хроматографической колонке высокого разрешения. Предшествующие исследования показывают, что само связывание протекает почти без рацемизации.
После проведения аналогичной реакции, но с 3 эквивалентами NaH и времени перемешивания 4 часа при КТ получают 65%-ный продукт (продукт : аддукт = 94: 6; (L):(D)=96:4).
1H-ЯРМ (CDCl3, δ, частей на миллион): 10,5-9,5 (СООН), 6,10-5,90 (1H, 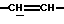 ), 5,88-5,70 (1H,
), 5,88-5,70 (1H, 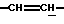 ), 5,12-4,95 (1H, N-CH), 4,30-4,15 (2Н, N-CH2), 1,55-1,35 (9Н, Бок); (2 ротамера).
), 5,12-4,95 (1H, N-CH), 4,30-4,15 (2Н, N-CH2), 1,55-1,35 (9Н, Бок); (2 ротамера).
Методами классической рацемизации производят кристаллизацию Бок-3,4-дегидропролина и (+)-дегидроабиетиламина из ацетона в виде соответствующей соли аммония и затем без дальнейшей перекристаллизации после отщепления амина выделяют Бок-(L)-3,4-дегидропролин с чистотой 85% и соотношением энантиомеров (L):(D)=96:4.
б) 46,2 г трет-амилата натрия (398,5 ммоль) вносят в 150 мл ТГФ. Затем при 10oС к смеси прибавляют 32,9 г метоксиэтанола (429,5 ммоль). После этого прибавляют по каплям раствор 50 г Бок-(L)-(4-МсО)-Рrо-ОН (159,4 ммоль) в 100 мл ТГФ так, чтобы внутренняя температура не превышает 8-10oС. По окончании добавления продолжают перемешивать еще в течение 20 часов при 10oС. После добавления 300 мл ледяной воды при 5-10oС однократно экстрагируют с помощью 50 мл метил-трет-бутилового эфира и затем подкисляют соляной кислотой до рН 2. Сырой продукт экстрагируют метиленхлоридом и после выпаривания растворителя выделяют в виде желтоватого масла.
Получают 40,9 г сырого продукта, в том числе 18 г Бок-(L)-3,4-дегидропролина, определенного с помощью аналитической ЖХВР-техники, калиброванной по внешнему стандарту (начальный градиент вода (0,1% Н3РO4)/ацетонитрил 70: 30; колонка: Prodigy (ODS3) 100А; детектирование при 210 нм). Соотношение энантиомеров (L):(D)=99:1 определяют также с помощью ЖХВР-техники (гексан/изопропанол 8,75:1,25, 0,1%
НСООН; колонка: Chiracel OD; детектирование при 230 нм).
В одном опыте, проведенном при аналогичных условиях, взаимодействию подвергают 70 г Бок-(L)-(4-МсО)-Рrо-ОН (224 ммоль). После экстракции метиленхлоридом сырой продукт переводят путем дистиллятивного обмена растворителей в 220 мл метил-трет-бутилового эфира и затем осаждают содержащийся в нем Бок-(L)-3,4-дегидропролин путем добавления 16,5 г диэтиламина (224 ммоль) в виде диэтиламмониевой соли.
Получают 23,8 г этой соли. В осажденном таким образом продукте вышеуказанными аналитическими ЖХВР-методами (D)-энантиомер не обнаруживают.
1Н-ЯМР (ДМСО, δ, частей на миллион): 5,86-5,67 (2Н, -СН=СН-), 4,6-4,5 (N-CH), 4,1-3,9 (N-CH2), 2,88-2,7 (4Н кв, 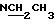 ), 1,45-1,25 (9Н, Бок, 2 ротамера), 1,2-1,05 (6Н,
), 1,45-1,25 (9Н, Бок, 2 ротамера), 1,2-1,05 (6Н, 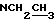 )
)
[α]D 22=-240,1o (с=1,08, МеОН)
Точка плавления: 130-133oС.
в) 10,52 г бис-(триметилсилил)-натрийамида (57,4 ммоль) помещают в 25 мл ТГФ, добавляют по каплям в течение 15 мин при охлаждении 8,25 г 2-[2-(диметиламино)-этокси] -этанола (62 ммоль) в 15 мл ТГФ и перемешивают в течение 30 мин при КТ. Затем добавляют по каплям при -5oС в течение 20 мин 7,1 г Бoк-(L)-(4-McO)-Pro-OH (23,0 ммоль), растворенного в 15 мл ТГФ, перемешивают в течение 1 часа при -5oС, в течение 2 часов при 0oС и в течение ночи при КТ. Затем смесь выливают на 125 г ледяной воды, четырежды экстрагируют метил-трет-бутиловым эфиром, водную фазу подкисляют до рН 2,2 с помощью 60 мл 10%-ной лимонной кислоты и оставляют на ночь перемешиваться при КТ. После трехкратной экстракции реакционного раствора метил-трет-бутиловым эфиром собранные органические фазы последовательно промывают водой, насыщенным раствором поваренной соли и водой, сушат над сульфатом магния и концентрируют в вакууме. Получают 4,1 г Бок-(L)-3,4-дегидропролина в качестве сырого продукта, который затем растворяют в 20 мл метил-трет-бутилового эфира и смешивают по каплям с раствором 1,35 г диэтиламина (18,52 ммоль) в 10 мл метил-трет-бутилового эфира. Для полного осаждения соли добавляют петролейный эфир. После отсасывания и сушки продукта получают 4,0 г Бок-(L)-3,4-дегидропролина. Из маточного раствора дополнительно получают вторичный кристаллизат в количестве 0,3 г, что доводит общий выход целевого продукта до 66%.
Пример 3
Получение Бок-(D,L)-Pyr-OH:
К 8 г 60%-ного NaH (200 ммоль) в 150 мл ДМЭ при охлаждении прибавляют по каплям 13 г изопропанола (215 ммоль). После прекращения образования Н2 добавляют раствор 25 г Бок-(L)-(4-МсО)-Рrо-ОН (80 ммоль) в 100 мл ДМЭ. После выдержки в течение 1 часа при 0oС нагревают в течение 20 часов до 20oС и затем добавляют 150 мл воды. После однократной экстракции метил-трет-бутиловым эфиром подкисляют соляной кислотой до рН 2 и экстрагируют метиленхлоридом. Получают 17 г продукта. Соотношение энантиомеров (L):(D)=57:43 определяют с помощью ЖХВР-анализа (гек-сан/изопропанол 8,75: 1,25, 0,1% НСООН; колонка: Chiracel OD; детектирование при 230 нм).
Пример 4
Ферментативное расщепление Бок-(D/L)-Руr-ОСН3 до Бок-(L)-Руr-ОН
5,68 г (25 ммоль) Бок-(D/L)-Руr-ОСН3 встряхивают в 100 мл фосфатного буфера (рН 7,0) и 15 мл ТГФ с 3,12 г ®Novozym 435 в течение 24 часов при КТ. При этом рН доводят до первоначального значения добавлением 1 н. NaOH. За протеканием реакции следят по расходу раствора едкого натра. Твердое вещество отфильтровывают и рН фильтрата доводят до значения 10 с помощью 1 н. NaOH. Непрореагировавший антипод Бок-(D)-Руr-ОСН3 экстрагируют смесью уксусного эфира и простого эфира (1:1). Значение рН водной фазы доводят 1 н. соляной кислотой до 1 и продукт Бок-(L)-Pyr-OH трижды экстрагируют уксусным эфиром. Получают 2,14 г продукта, который кристаллизуют из смеси толуола с гексаном или смеси эфира с гексаном. После кристаллизации продукт имеет угол поворота плоскости поляризации [α]D 22=-273,8o (с=1,03; метанол). (Согласно литературным данным [α]D 25=-272o (с=1,0; метанол) J.U. Kahl. Т. Wieland, Liebigs Ann. Chem. 8, 1445 (1981)).
Пример применения 1
ПолучениеН-(L)-Руr-(6-карбоксамидо)-3-пиколиламиддигидрохлорида
К 1,5 г сырого Бок-Руr-ОН из примера 2 в 30 мл дихлорметана прибавляют по каплям при -10oС 5,3 мл (30,3 ммоль) ДИПЭА, через 5 мин добавляют 1,58 г (7,0 ммоль) (6-карбоксамидо)-3-пиколиламин-дигидрохлорида и затем еще через 5 мин 5,7 мл (7,9 ммоль) АПФК (50%-ный раствор в уксусном эфире) в 5 мл дихлорметана. Реакционной смеси дают в течение 1 часа нагреться с -10oС до 0oС, затем ее разбавляют дихлорметаном и последовательно промывают насыщенным раствором NaHCO3, 5%-ной лимонной кислотой и насыщенным раствором поваренной соли. После сушки органической фазы над Na2SO4 и полного удаления растворителя получают 1,8 г сырого Бок-(L)-Руr-(6-карбоксамидо)-3-пиколиламида, который перемешивают в течение 50 мин при 50oС в 30 мл 0,9-молярного изопропанольного раствора НСl. Выпавший при этом осадок отделяют на нутч-фильтре, растворяют в небольшом количестве метанола, осаждают изопропанолом и снова отделяют. После сушки при 45oС в вакууме получают 2,0 г Н-(L)-Руr-(6-карбоксамидо)-3-пиколиламиддигидрохлорида в виде белого порошка (чистота: 95%); (L):(D)>99:1.
1Н-ЯМР (ДМСО-d6, δ, частей на миллион): 10,9 и 8,9 (по одному 1Н, -NH2-⊕), 9,77 (т, 1Н, CO-NH), 8,60, 8,10 и 8,00 (по одному 1Н, ароматическое соединение-Н), 8,25 и 7,75 (по одному 1Н, CO-NH2), 6,03 (с, 2Н, -СН=СН-), 5,10 (1Н, N-CH-CO-), 4,47 (□, 2Н, CH2), 4,00 (2Н, СН2).
Пример применения 2
Получение Н-(L)-Руr-(4-СN)-бензиламид-гидрохлорида:
10,0 г сырого Бок-Руr-ОН аналогично примеру применения 2 подвергают взаимодействию с 6,2 г п-циано-бензиламина. После обработки получают 14,7 г сырого Бок-(L)-Руr-(4-СN)-бензиламида, который перемешивают в течение 2 часов при 50oС в 230 мл 1-молярного изопропанольного раствора НСl. После того как раствор остывает до КТ, вещество начинает выпадать в осадок. Твердое вещество отделяют на нутч-фильтре. Получают 3,7 г H-(L)-Руr-(4-СN)-бензиламид-гидрохлорида в виде белого порошка (чистота: 96%); (L): (D)>99:1.
1Н-ЯМР (ДМСО-d6, δ, частей на миллион): 10,9 и 8,9 (по одному 1Н, -NH2-⊕), 7,82 и 7,47 (по одному 2Н, ароматическое соединение-Н), 6,02 (с, 2Н, -СН=СН-), 5,10 (1Н, N-CH-CO-), 4,45 (д, 2Н, СН2), 4,02 (2Н, СН2).
Пример 5
10,6 г (50 ммоль) бок-3,4-дегидропролина, растворенного в 80 мл тетрагидрофурана, вместе с 5,7 г гидроксисукцинимида и 10,2 г дициклогексилкарбодиимида в хлористом метилене перемешивают при температуре 0oС в течение 30 минут. Затем каплями добавляют при 0oС 8,0 г (50 ммоль) 4-аминометил-3-метокси-бензолнитрила, растворенного в 50 мл тетрагидрофурана, и полученную смесь перемешивают при комнатной температуре в течение 20 часов. Получаемое твердое вещество фильтруют, фильтрат смешивают с одинаковым объемом этилацетата и промывают холодным раствором 5%-ного гидрогенсульфата натрия и насыщенным раствором хлористого натрия. Получают 11,5 г (65%) продукта.
Если вместо 4-аминометил-3-метокси-бензолнитрила применять другие соединения формулы V, у которых Х означает бензолнитрил, замещенный метилом, гидроксилом или хлором в положении 3 или 5, или цианозамещенный пиридин, то с аналогичным выходом получают соответствующие производные 3,4-дегидропролина формулы III.
Пример 6
11,4 г (31,7 ммоль) продукта примера 5 растворяют в 130 мл хлористого метилена и насыщают HCl при температуре 0-5oС. Через два часа аминозащитная группа Бок полностью отщеплена. Растворитель удаляют в вакууме и продукт выделяют без очистки.
1H-ЯМР (ДМСО-d6; млн. дол.): 10,55 и 8,80 (2Н, 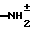 ); 9,65 (1Н, СО-NН-); 7,55 (1Н, аром. ); 7,47 (1Н, аром.); 7,43 (1Н, аром.); 5,91 (2Н, -СН=СН-); 4,95 (1Н, N-СН-СО); 4,30 (2Н, -СН2-); 3,98 (2Н, -CH2-); 3,90 (3Н, O-СН3).
); 9,65 (1Н, СО-NН-); 7,55 (1Н, аром. ); 7,47 (1Н, аром.); 7,43 (1Н, аром.); 5,91 (2Н, -СН=СН-); 4,95 (1Н, N-СН-СО); 4,30 (2Н, -СН2-); 3,98 (2Н, -CH2-); 3,90 (3Н, O-СН3).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ АМИДИНЫ, ОБЛАДАЮЩИЕ СВОЙСТВАМИ ИНГИБИТОРОВ ТРОМБИНА, СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ПРОИЗВОДНЫЕ ПЯТИЧЛЕННОГО ГЕТЕРОЦИКЛИЧЕСКОГО АМИДИНА, В КАЧЕСТВЕ СОСТАВНОЙ ЧАСТИ ИНГИБИТОРОВ СЕРИНПРОТЕАЗЫ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1997 |
|
RU2175328C2 |
| ЦИКЛОАЛКИЛЬНЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1997 |
|
RU2197458C2 |
| ПРОТИВООПУХОЛЕВЫЕ ПЕПТИДЫ | 1996 |
|
RU2182911C2 |
| НОВЫЕ БЕНЗАМИДОАЛЬДЕГИДЫ | 1997 |
|
RU2189973C2 |
| ЗАМЕЩЕННЫЕ ДИАМИНОКАРБОНОВЫЕ КИСЛОТЫ | 1998 |
|
RU2196768C2 |
| ПРОИЗВОДНЫЕ ДИПЕПТИДНЫХ П-АМИДИНО-БЕНЗИЛАМИДОВ С N-КОНЦЕВЫМИ СУЛЬФОНИЛЬНЫМИ ОСТАТКАМИ И ИХ СОЛИ С ФИЗИОЛОГИЧЕСКИ ПРИЕМЛЕМЫМИ КИСЛОТАМИ | 1995 |
|
RU2152953C1 |
| ИНГИБИТОРЫ ТРОМБИНА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ | 1995 |
|
RU2172741C2 |
| ПРОИЗВОДНЫЕ ДОЛАСТАТИНА 15 | 1998 |
|
RU2195462C2 |
| ПЕРФТОРАЛКИЛСОДЕРЖАЩИЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) И ФАРМАЦЕВТИЧЕСКОЕ СРЕДСТВО, СОДЕРЖАЩЕЕ УКАЗАННЫЕ СОЕДИНЕНИЯ | 1997 |
|
RU2242477C2 |
| ЗАМЕЩЕННЫЕ 6- И 7-АМИНОТЕТРАГИДРОИЗОХИНОЛИН-КАРБОНОВЫЕ КИСЛОТЫ | 1998 |
|
RU2212405C2 |
Изобретение относится к новому способу получения производных 3-пирролин-2-карбоновой кислоты формулы I, где R1 - C1-С6-алкилоксикарбонил; R2 - гидроксил, С1-С4-алкокси или их аммониевых солей, отщеплением основанием сульфокислотного остатка от соединения формулы II, где R1 и R2, как указано выше; R3 - C1-С6-алкил, бензил, трифторметил, нафтил, фенил, который может быть замещен остатком, включающий СН3, NO2, галоген. Основание выбирают из группы, включающей гидриды, бисалкилсилиламиды, алкоголяты натрия и калия, в среде непротонного растворителя с последующим, при необходимости, переведением полученного рацемата при R2=С1-С4-алкокси в оптически активные свободные кислоты и выделением целевого продукта в свободном виде или в виде аммониевой соли. Описан способ получения производных 3,4-дегидропролина формулы III, где R1 указан выше; Х означает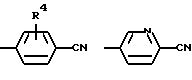
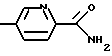
взаимодействием соединения I с соединением V: H2N-CH2-X, где Х указан выше. Описываются аммонийные соли соединения I. 3 с. и 5 з.п. ф-лы.
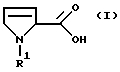
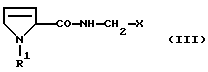
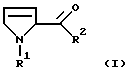
в которой R1 - C1-C6-алкилоксикарбонил;
R2 - гидроксил, алкокси с 1-4 атомами углерода;
или их аммониевых солей, отличающийся тем, что от соединения формулы II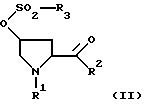
где R1 и R2 имеют вышеуказанное значение;
R3 означает алкил с 1-6 атомами углерода, бензил, трифторметил, нафтил или фенил, который может быть замещен остатками из группы, включающей метил, нитро, галоген,
отщепляют сульфокислотный остаток путем обработки основанием, выбранным из группы, включающей гидриды, бисалкилсилиламиды и алкоголяты натрия и калия, в среде непротонного растворителя с последующим, при необходимости, переведением полученного рацемата при R2=C1-C4-алкокси в оптически активные свободные кислоты и выделением целевого продукта в свободном виде или в виде аммониевой соли.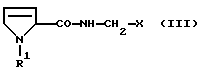
где R1 означает водород или C1-C6-алкилоксикарбонил и Х означает группы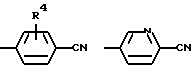
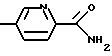
где R4 означает водород, метил, метокси, гидроксил или галоид,
отличающийся тем, что соединение формулы I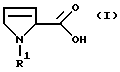
где R1 имеет вышеуказанное значение,
подвергают взаимодействию с соединением формулы V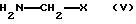
где Х имеет вышеуказанное значение,
в присутствии основания и агента конденсации и, при необходимости, в среде органического растворителя, при температуре от -20 до +30oС.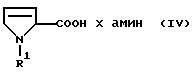
где R1 означает С1-С6-алкилоксикарбонил,
амин представляет собой моно-, ди- или триалкиламин, в котором алкильные остатки содержат 1-4 С-атома, которые могут быть заменены циклоалкильными остатками с 5-7 атомами углерода.
| US 4066658 А, 03.01.1978 | |||
| US 4111951 А, 05.09.1978 | |||
| US 4501901 А, 26.02.1985 | |||
| RU 94028194 А1, 20.04.1996. |

