Изобретение относится к гиполипидемическим и антиатеросклеротическим соединениям, фармацевтическим композициям на их основе и применению названных соединений.
С самого начала истории медицины атеросклероз описывается как серьезное заболевание. Однако только в двадцатом веке было установлено, что инфаркт миокарда всегда сопровождается коронарным атеросклерозом и тромбозом. Эпидемиологические исследования, проведенные в семидесятые годы, показали, что около пятидесяти процентов всех смертей в западных индустриально развитых странах обусловлены сердечно-сосудистыми заболеваниями.
Известны наиболее важные факторы возникновения и развития атеросклероза. В самом начале исследований было установлено значение повышенных концентраций холестерина и триглицеридов. Холестерин и триглицериды переносятся в форме так называемых липопротеинов, фракции которых с низкой плотностью (ЛНП - липиды с низкой плотностью), обогащенные холестерином, и фракции с очень низкой плотностью (ЛОНП - липиды с очень низкой плотностью), обогащенные триглицеридами, считаются атерогенными, если их концентрация в крови повышена.
Однако только недавно было установлено, что кроме их количества решающее значение также имеет "качество" ЛПН. Так, в опытах с животными можно было показать, что одинаково высокие концентрации ЛПД-холестерина являются более или менее атерогенными в зависимости от свойств лекарства, используемого для снижения повышенного уровня ЛНП-холестерина. Причина такого явления также показана авторами исследования, то есть лекарства, которые также обладают способностью уменьшать окисление ЛНП, наиболее эффективны при лечении атеросклероза (Carew et al., Proc. Nat. Acad. Sci., USA, 84, 7725-7729, ноябрь 1987). Этот опыт однозначно показывает значение "качества" ЛНП. В организме ЛНП и ЛОНП подвергаются непрерывному окислению, которое физиологически удерживается в равновесии с помощью естественных так называемых антиоксидантов, таких как токоферол или аскорбиновая кислота. Однако при повышенных величинах для ЛНП (холестерин) и ЛОНП (триглицериды) этот гомеостаз нарушается и сдвигается в сторону повышения окисления.
Таким образом, опасность развития и проявления атеросклероза повышается при наличии двух факторов: повышенная концентрация так называемых атерогенных липопротеинов (ЛНП, ЛОНП) и содержание в них продуктов окисления (например, окисленных липидов).
Таким образом, при лечении необходимо воздействовать на оба фактора.
Помимо гуморальных факторов, то есть липопротеинов, клеточные составляющие стенок сосудов также играют важную роль при атерогенезисе. Следовательно, в случае химического или механического повреждения внутреннего покрытия сосудов - эндотеция - возникают условия для быстрого размножения клеток, что стимулирует подслойный слой клеток мягких тканей. Этот процесс особенно интенсивен после эндотелиального повреждения в результате пластических операций на сосудах. За относительно короткий промежуток времени спровоцированная профелерация клеток может закрыть сосуд до такой степени, что возможно возникновение инфаркта. Кроме механических эндотелиальных повреждений также важное значение имеют окисленные ЛНП.
Таким образом, кроме двух возможных процессов, указанных выше, эффективное противодействие атеросклерозу может быть осуществлено при сохранении эндотеция и предотвращении избыточного быстрого размножения клеток гладких мышц.
Соединения, которые удовлетворяют указанным выше требованиям, должны содержать два фрагмента, обладающие различным фармакологическим действием, например никотиновую кислоту или ее приемлемое производное в качестве липидопонижающего элемента; антиоксидант в качестве антиатеросклеротического элемента.
Никотиновая кислота представляет собой лекарство, которое действует не только на липиды, но и сама по себе обладает антисклеротической активностью.
Кроме того, для использования в медицинских целях необходимо решить проблему побочных эффектов, возникающих при применении никотиновой кислоты, например, прилив крови к лицу из-за быстрого увеличения концентрации никотиновой кислоты в крови. Действительно, было установлено, что быстрое увеличение и высокий уровень никотиновой кислоты не являются необходимыми условиями для достижения липидопонижающего эффекта. Следовательно, низкие концентрации никотиновой кислоты и достаточное снижение липидов должны быть основными исходными требованиями к соединениям, тип которых описывался выше.
В DE 2716125 в качестве предпочтительного соединения раскрывается эфирное производное БГТ (бутилированного гидрокситолуола) (обозначаемого здесь как Mrz 3/156), которое имеет формулу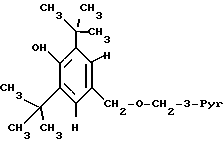
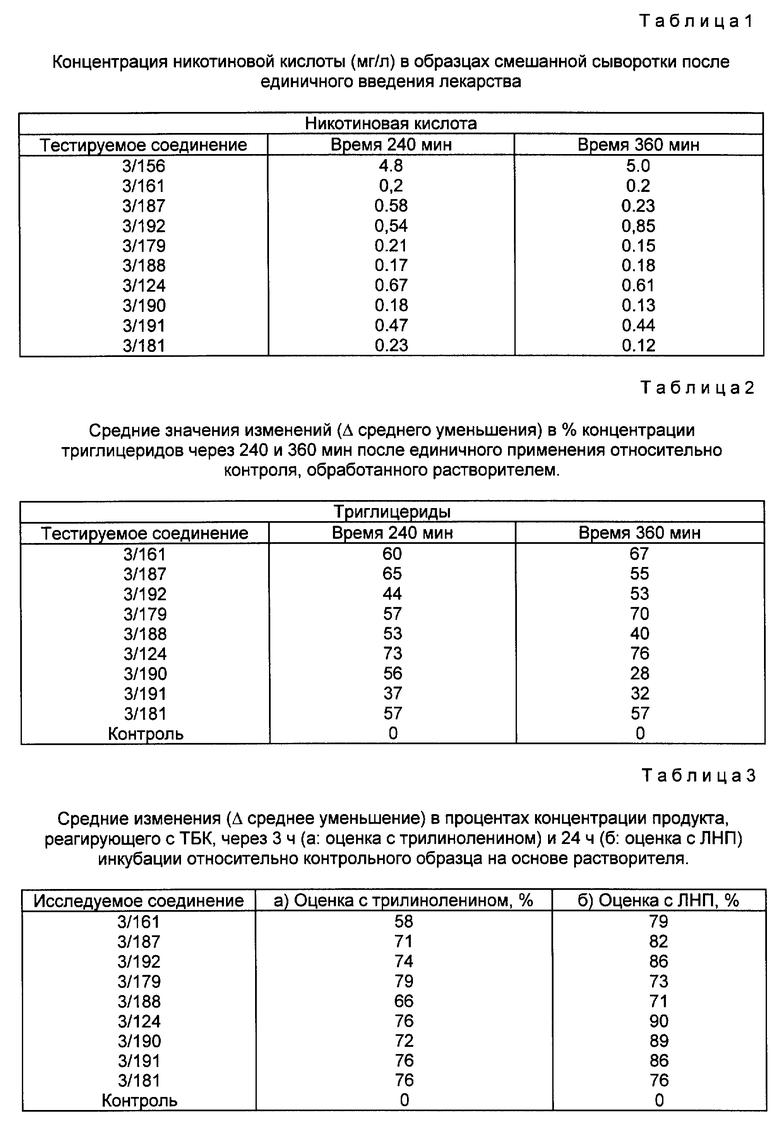
При оценке требуемых свойств наблюдались исключительно высокие уровни никотиновой кислоты (см. табл. 1), что показывает, что это соединение не соответствует установленным требованиям.
В результате интенсивных и кропотливых синтетических и фармакологических исследований установлено, что существует ряд структурных фрагментов, наличие которых необходимо для соблюдения всех критериев.
Сделан вывод, что только соединения приведенной ниже общей формулы представляют интерес.
Можно увидеть, что соединения в соответствии с настоящим изобретением эффективно понижают липиды без появления избыточно-высокого содержания никотиновой кислоты; эффективно препятствуют окислению липидов и липопротеинов; эффективно уменьшают эндотелиальное повреждение, обусловленное окисленными ЛНП, и эффективно снижают избыточный рост клеток гладких мышц на стенках сосудов.
Целью настоящего изобретения является создание новых и более эффективных гиполипидемических и антиатеросклеротических соединений, фармацевтических композиций на их основе и способ лечения гиперлипемии и атеросклероза. Еще одной целью настоящего изобретения является создание таких новых соединений, композиций и способа, которые удовлетворяют перечисленным выше теоретическим требованиям. Другие цели настоящего изобретения будут понятными из представленного описания и будут очевидны для квалифицированного в данной области специалиста.
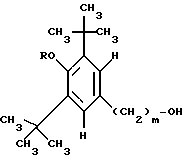
Данное изобретение относится к простым эфирам производных БГТ (производных бутилированного гидрокситолуола) и ω- -пиридилалкил-, алкенил- или алкинил-спиртам в качестве активных гиполипидемических и антиатеросклеротических соединений. Более конкретно оно относится к простым эфирам формулы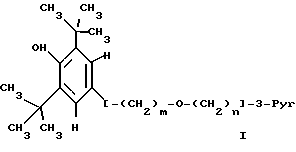
где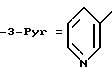
m = 1 и n = 4-7 или m = 3, а n = 1-7 и
где фрагмент (CH2)n может необязательно включать двойную или тройную связь, сопряженную с положением 3 пиридинового кольца, то есть связь между двумя атомами углерода фрагмента (CH2)n, соседнего с пиридиновым кольцом, может быть простой, двойной или тройной, и к их фармацевтически приемлемым кислотно-аддитивным солям, а также к фармацевтической композиции, содержащей заявляемое соединение в качестве активного ингредиента, которые могут быть использованы в качестве антилипидемических и антиатеросклеротических агентов, а также к способу лечения гиперлипемии и атеросклероза с их помощью.
Фармакология
а) Кинетика никотиновой кислоты.
Кинетика никотиновой кислоты, выделяющейся из исследуемых соединений, была изучена на крысах. Единичную дозу (0,2 ммоль/кг) тестируемого соединения вводят некормленным нормолипидемическим крысам через желудочный зонд. Через 240 и 360 мин отбирают образцы крови и определяют концентрацию никотиновой кислоты в сыворотке крови с помощью хромато-масс-спектрометрии (n = 5 крыс в единицу времени).
Полученные данные приведены в табл. 1.
б) Доказательство липидопонижающих свойств.
Липидопонижащие свойства соединений настоящего изобретения исследуют на крысах. По этому методу единичную дозу (0,2 ммоль/кг) тестируемого соединения вводят орально некормленным нормолипидемическим крысам. Через 240 и 360 мин после введения отбирают образцы крови и определяют концентрацию триглицеридов по стандартной методике (а именно GPO-PAP-методом, E. Merck-system, Darnistadt, Germany) (n = 5 крыс в измеряемое время).
В табл. 2 приведены средние значения изменений (в %) в сравнении с растворителем-контролем (кремофор, 30% в дистиллированной воде).
в) Доказательство антиоксидантных свойств.
Антиоксидантные свойства соединений изобретения изучают в условиях in vitro. В этих опытах вещество выдерживают в субстрате, который должен быть окислен (синтетический триглицерид с ненасыщенными жирными кислотами, например трилиноленин или человеческий ЛНП). По окончании выдерживания степень окисления определяют с использованием так называемого метода тиобарбитуровой кислоты (ТБК). Тиобарбитуровая кислота реагирует с малондиальдегидом (МДА), который представляет собой один из основных продуктов окисления. Количество продукта реакции оценивают фотометрически.
в1) Оценка с использованием трилиноленина.
В течение 3 ч при 37oC 400 мкл трилиноленина выдерживают с 2 мл среды HAM F 10 с растворителем (например, с этанолом, конечная концентрация 0,5%) или с исследуемым соединением (концентрация 5•10-5 M) (окисление протекает эффективно без использования специального катализатора).
Общее количество материала, способного реагировать с ТБК, определяют в 1 мл инкубата при добавлении 1.5 мл 0,67%-ного раствора ТБК к 0.05 н. раствору NaOH и 1.5 мл к 20%-ному раствору трихлоруксусной кислоты. Смесь выдерживают в течение 60 мин на кипящей бане, после охлаждения определяют поглощение при 532 нм.
Для одного исследуемого соединения проводилось по три опыта.
в2) Оценка с использованием ЛНП.
ЛНП (d = 1.019 - 1.063) получают из человеческой плазмы при ультрацентрифугировании с добавлением ЭДТУ.
В течение 32 ч при 37oC 100 мкл ЛНП выдерживают с 0.5 мл среды НАМ F 10 и с растворителем, например, с эталоном, конечная концентрация 0,5%) или с исследуемым соединением (концентрация 5•10-5 М) в присутствии 10 мкл Cu2+.
Общее количество продукта, реагирующего с ТБК, определяют в соответствии с методикой, приведенной для оценки с использованием трилиноленина.
Для каждого исследуемого соединения проводят три опыта.
В табл. 3 представлены средние значения изменений в процентах в сравнении с растворителем-контролем.
в) Доказательство эндотелиальной защиты относительно цитотоксических эффектов окисленных ЛНП.
Человеческие эндотелиальные клетки умбиликальной вены выделяют после коллагеновой обработки сосудов и культивирования.
Для проведения оценки клетки диспергируют с плотностью 50000 на чашку и выращивают в течение 4 дней в среде Ham's F 12/DMEM (соотношение 4:1) при добавлении 15% фетальной телячьей сыворотки, 5% лошадиной сыворотки и 10 нг/мл гепарина ECGF.
Через 4 дня клетки промывают средой, не содержащей сыворотку, и используют при проведении цитотоксического анализа.
Для этого анализа клетки культивируют в течение 24 ч при 37oC в среде Ham's F 10(+гепарин ECGF) с ЛНП (200 мкг/мл). При культивировании добавляют либо растворитель (то есть этанол, конечная концентрация 0,5%) - контрольный образец) или исследуемое соединение (2•10-5 М). Параллельно один образец культивируют без ЛНП и без другого соединения и с растворителем (0,5% этанола) для оценки максимальной скорости роста клеток (контрольный образец 2).
Через 24 ч в среде определяют полученное количество продукта, реагирующего с ТБК, или подсчитывают количество клеток.
Для каждого соединения исследуют три образца.
В табл. 4а представлены значения средних изменений в процентах относительно контрольного образца 1.
В табл. 4б показано число клеток в процентах относительно соответствующего образца 2.
e) Доказательство антипролиферационного эффекта в опытах in vitro на гладких мышцах.
Клетки гладких мышц, полученные после выделения из аорты крыс, катетеризированных баллонным катетером, культивируют в среде Игла, модифицированной по способу Дульбекко (DMED), добавлением 10% фетальной телячьей сыворотки и подвергают пересеву.
Для проведения оценки 10000 клеток одного пересева культивируют в чашке и выдерживают в течение 3 дней или с растворителем (то есть с DMCO, конечная концентрация 0,5%) или с испытуемым соединением (5•10-5 М). Через 3 дня клетки подсчитывают. Для каждого испытуемого соединения готовят шесть образцов.
В таблице 5 показана величина средних значений изменения в процентах относительно контроля с растворителем.
Синтез соединений изобретения
Синтез соединений настоящего изобретения осуществляют, исходя из ω- пиридилалкил-, алкенил- или -алкинил-спиртов, которые реагируют с 3,5-дитрет. -бутил-4-гидроксибензиловым спиртом в виде его ацетата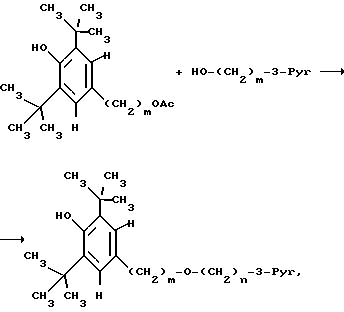
где фрагменты и переменные принимают описанные выше значения.
ω- Пиридилалкиловый спирт получают по реакции Виттига из пиридил-3-альдегида и фосфониевой соли, синтезируемой из соответствующего галогеналкилового спирта. Полученный ненасыщенный 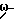 пиридилалкиловый спирт непосредственно, или после гидрирования, превращают в заявляемый эфир.
пиридилалкиловый спирт непосредственно, или после гидрирования, превращают в заявляемый эфир.
THP = тетрагидропиранил.
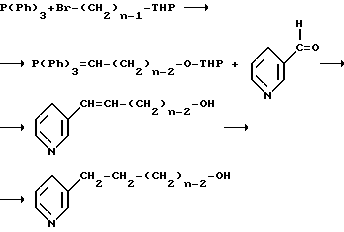
Альтернативно исходный ω- пиридилалкиловый спирт может быть получен реакцией 3-бромпиридина с соответствующим ω- алкиниловым спиртом. Полученный ω- пиридилалкиниловый спирт может быть непосредственно, или после гидрирования до ω- пиридилалкенилового спирта или ω- пиридилалкилового спирта, превращен в заявляемое соединение формулы I.
Изобретение также включает фармацевтически приемлемые кислотно-аддитивные соли указанных выше соединений.
1. 2,6-Ди-трет.-бутил-4-[8-(3-пиридил)-2-оксаоктил]фенол Mrz 3/161).
2. 2,6-Ди-трет.-бутил-4-[6-(3-пиридил)-2-оксагексил]фенол (Mrz 3/187).
3. 2,6-Ди-трет.-бутил-4-[7(3-пиридил)-2-оксагептил]фенол (Mrz 3/192).
4. (Z)-2,6-Ди-трет. -бутил-4-[8-(3-пиридил)-2-оксаокт-7-енил] фенол (Mrz 3/181).
5. 2,6-Ди-трет.-бутил-4-[9(3-пиридил)-2-оксанонил]фенол (Mrz 3/188).
6. 2,6-Ди-трет.-бутил-4-[5-(3-пиридил)-4-оксапентил]фенол (Mrz 3/124).
7. 2,6-Ди-трет.-бутил-4-[7-(3-пиридил)-4-оксагептил]фенол (Mrz 3/190).
8. 2,6-Ди-трет.-бутил-4-[9-(3-пиридил)-4-оксанонил]фенол (Mrz 3/191)э
9. 2,6-Ди-трет. -бутил-4-[8-(3-пиридил)-2-оксаокт-7-инил] фенол (Mrz 3/179),
из которых предпочтительными являются 2,6-ди-трет.бутил-4-[8-(3-пиридил)-2-оксаоктил] фенол и 2,6-ди-трет.-бутил-4-[5-(3-пиридил)-4-оксапентил] фенол.
Приведенные ниже примеры даны для иллюстрации получения соединений настоящего изобретения, но не должны рассматриваться как примеры, ограничивающие его объем.
Пример 1. Синтез 2,6-ди-трет.-бутил-4-[8-(3-пиридил)-2-оксаоктил]фенола.
Стадия 1. 3,5-Ди-трет.бутил-4-гидроксибензилацетат.
К раствору 118 г (0,5 моля) 3,5-ди-трет.-бутил-4-гидроксибензилового спирта в 990 мл пиридина при 0 oC при перемешивании медленно добавляют 10 - 2,1 г (1 моль) уксусного ангидрида. Через 15 мин охлаждение снимают и выдерживают еще 2 часа при комнатной температуре. Затем при интенсивном перемешивании смесь выливают в воду со льдом, через 15 мин выделившийся осадок отделяют фильтрацией, промывают водой, сушат и перекристализовывают из гексана, получают 110 г (80%) 3,5-ди-трет.-бутил-4-гидроксибензилацетата в виде желтоватых кристаллов, т. пл. 108oC,
(C17H26O3; молекулярный вес (М. в. 278,2).
Rf:0,57 (SiO2 60; н-гексан-диэтиловый эфир, 3:1).
Стадия 2. 6-(3-Пиридил)гексанол.
Раствор 45 мл (0,47 моль) 3-бромпиридина и 52 г (0,53 моль) 5-гексин-1-ола в 150 мл триэтиламина и 500 мл метиленхлорида продувают аргоном в течение 15 мин и добавляют 3 г (4,3 моль) бис-(трифенилфосфин)палладий(II)хлорида и 450 мг моноиодистой меди. Смесь кипятят в течение 3 ч., охлаждают, разбавляют 1 л метиленхлорида, промывают водой, рассолом, сушат (K2CO3), упаривают и перегоняют "из колбы в колбу" (т. кип. 120 - 130o/0,05 мбар), получают 6-(3-пиридил)гекс-5-инол в виде желтого масла. Полученный продукт растворяют в 300 мл изопропанола и гидрируют в течение 16 ч с 6 г 10%-ного палладия на угле в приборе Парра. Сырой продукт перегоняют (т. кип. 123 - 130oC/0,05 мбар), получают 61,8 г (65%) 6-(3-пиридил)гексанола в виде слабо-желтой вязкой жидкости.
(C11H17NO; М.в. 179,3).
Стадия 3. 2,6-Ди-трет.-бутил-4-[8-(3-пиридил)-2-оксаоктил]фенол.
Раствор 1,53 г (5,5 ммоль) 3,5-ди-трет.-бутил-4-гидроксибензилацетата, 895 мг (5 ммоль) 6-(3-пиридил)гексанола и 30 мл тетракис(трифенилфосфин)палладия (0) в 60 мл обескислороженного сухого ацетонитрила перемешивают при комнатной температуре в атмосфере азота в течение 5 дней. Растворитель упаривают, остаток распределяют между эфиром и насыщенным водным раствором бикарбоната. Высушенный экстракт упаривают и разделяют колоночной хроматографией на силикагеле, получают 59% 2,6-ди-трет.бутил-4-[8-(3-пиридил)-2-оксаоктил] фенола в виде желтоватого вязкого масла, которое со временем кристаллизуется, т. пл. 63oC.
(C26H39NO2; М.в. 397,6).
Rf: 0,6 (SiO2 60; н-гексан-этилацетат, 3:1).
Пример 2. Синтез 2,6-ди-трет.бутил-4-[6-(3-пиридил)-2-оксагексил]фенола.
Стадия 1. 2-(3-Бромпропокси)тетрагидропиран.
К раствору 35 г (0,251 моль) 3-бромпропинола и 1 г n-толуол-сульфокислоты в 500 мл диэтилового эфира при охлаждении водой со льдом добавляют по каплям 32,5 мл (0,357 моль) 3,4-дигидро-2H-пирана. Раствор перемешивают при комнатной температуре в течение 2 ч, нейтрализуют насыщенным водным раствором бикарбоната, промывают рассолом, сушат, упаривают, получают 56 г 2-(3-бромпропокси)тетрагидропиран с практически количественным выходом в виде вязкой жидкости, которая может быть далее использована без дополнительной очистки.
(C8H15BrO2; М.в. 223,1).
Rf: 0,9 (SiO2 60; н-гексан-этилацетат, 1:1).
Стадия 2. 3-(Тетрагидропиранилокси)пропил- трифенилфосфонийбромид.
Смесь 66,5 г (0,298 моль) 2-(3-бромпропокси)тетрагидропирана, 82 г (0,312 моль) трифенилфосфина и 1 г тетрабутиламмонийиодида в 500 мл ацетонитрила кипятят в течение 24 ч и упаривают растворитель. Остаток несколько раз растирают с кипящим диэтиловым эфиром, получают 144 г (100%) 3-(тетрагидропиранилокси)пропилтрифенилфосфонийбромида в виде тяжелого масла,
(C26H30Br NO2P; М.в.485,4).
Стадия 3. 2-[4-(3-Пиридил)бут-3-енокси]тетрагидропиран.
К раствору 33,6 мл (0,24 моль) диизопропиламина в 300 мл сухого тетрагидрофурана при -78oC в атмосфере азота по каплям добавляют 149 мл (0,244 моль) 15%-ного раствора бутиллития в гексане. Смесь перемешивают в течение 15 мин, медленно добавляют раствор 115 г (0,237 моль) 3-(тетрагидропиранилокси)пропил-трифенилфосфонийбромида в 400 мл сухого тетрагидрофурана и перемешивают еще в течение 30 мин. К смеси добавляют раствор 19 мл (0,199 моль) свежеперегнанного пиридин-3-альдегида, перемешивают 3 часа при -78oC и 16 ч при комнатной температуре. Обычной обработкой выделяют сырой продукт, из которого хроматографированием выделяют 87 г (79%) 2-[4-(3-пиридил)бут-3-енокси]тетрагидропирана в виде вязкой жидкости.
(C14H19NO2; М.в. 233,3).
Rf : 0,2 (SiO2 60; н-гексан-диэтиловый эфир, 1:1).
Стадия 4. 4-(3-Пиридил)бутанол.
Раствор 10 г (43 ммоль) 2-[4-(3-пиридил)-бут-3-енокси]- тетрагидропирана в 200 мл смеси метанол-вода (1:1 об.) подкисляют 2 н. соляной кислотой и гидрируют в течение 16 ч на 2 г палладия на углероде. Реакционную массу фильтруют, упаривают, остаток нейтрализуют насыщенным водным раствором бикарбоната и экстрагируют несколькими порциями эфира. Сырой продукт очищают колоночной хроматографией на силикагеле, продукт 15,7 г (87%) 4-(3-пиридил)-бутанола в виде бесцветного вязкого масла.
(C9H13NO; М.в. 151,2).
Rf: 0,2 (SiO2 60; н-гексан-этилацетат, 1:1).
Стадия 5. 2,6-Ди-трет.-бутил-4-[6-(3-пиридил)-2- оксагексил]фенол.
Синтез осуществляют по методике стадии 3 примера 1. Получают 2,6-ди-трет. -бутил-4-[6-(3-пиридил)-2-оксагексил] фенол в виде янтарного масла. Выход на данной стадии составляет 62%.
(C24H35NO2; М.в. 369,6).
Rf: 0,42 (SiO2 60; н-гексан-этилацетат, 3:2).
Пример 3. Синтез 2,6-ди-трет.-бутил-4-[7-(3-пиридил)-2- оксагептил]фенола осуществляют по методике стадии 2 примера 1 (исходный продукт 4-пентил-1-ол; E.R.H. Jones et al., Org. Synthesis Coll. Vol. 4,755 (1963)) и стадии 3. Получают 2,6-ди-трет. -бутил-4-[7-(3-пиридил)-2-оксагептил]фенол в виде тяжелого желтого сиропа с выходом 59% на последней стадии образования простого эфира.
(C29H37NO2; М.в. 383,6).
Rf: 0,49 (SiO2 60; н-гексан-этилацетат, 3:2).
Пример 4. Синтез (Z)-2,6-ди-трет.-бутил-4-[8-(3-пиридил)-2- оксаокт-7-енил]фенола.
Стадия 1. (Z)-6-(3-Пиридил)гекс-5-енол.
Через раствор 45 мл (0,47 моль) 3-бромпиридина и 52 г (0,53 моль) 5-гексин-1-ола в 150 мл триэтиламина и 500 мл метиленхлорида пропускают ток аргона в течение 15 мин и добавляют 3 г (4,3 ммоль) бис(трифенилфосфин)палладий (II)хлорида и 450 мл моноиодистой меди. Смесь кипятят в течение 3 ч, по охлаждении разбавляют 1 л метиленхлорида, промывают водой, рассолом, сушат (K2CO3), упаривают и перегоняют "из колбы в колбу" (т. кип. 120 - 130oC/0,05 мбар), получают 63 г 6-(3-пиридил)гекс-5-инола в виде желтого масла. Полученный продукт растворяют в 500 мл этилацетата, добавляют 15 мл хинолина и гидрируют в течение 5 ч на катализаторе Линдлара (отравленный свинцом палладиевый катализатор) в аппарате Парра. Сырой продукт перегоняют (т. кип. 120 - 130oC/0.05 мбар), получают 61 г (73%) (Z)-6-(3-пиридил)гекс-5-енола в виде бесцветной вязкой жидкости.
(C11H15NO; М.в. 177,3).
Rf: 0,21 (SiO2 60; CH2Cl2 - MeOH, 97:3).
Стадия 2. (Z)-2,6-Ди-трет.-бутил-4-[8-(3-пиридил)-2- оксаокт-7-енил]-фенол получают в соответствии с методикой стадии 3 примера 1, используя в качестве исходного (Z)-6-(3-пиридил)гекс-5-енол как спиртовой компонент реакции образования простого эфира. Выход (Z)-2,6-ди-трет.-бутил-4-[8- (3-пиридил)-2- оксаокт-7-енил]фенола составляет 49%, продукт представляет собой желтоватое масло, которое кристаллизуется, т. пл. 44oC.
(C26H39NO2; М.в. 397,6).
Rf: 0,4 (SiO2 60; н-гексан-этилацетат, 2:1).
Пример 5. Синтез 2,6-ди-трет.бутил-4-[9-(3-пиридил)-2- оксанонил]фенола.
Исходный 7-(3-пиридил)гептанол получают реакцией Виттига из 6-бромгексанола и никотинового альдегида, аналогично тому, как это описано в примере 2, стадии 1 - 4. По методике стадии 3 примера 1 получают 2,6-ди-трет.-бутил-4-[9-(3-пиридил)-2-оксанонил] фенол (43%) в виде вязкой жидкости янтарного цвета.
(C27H41NO2; М.в. 411,6).
Rf: 0.48 (SiO2 60; н-гексан-этилацетат, 3:2).
Пример 6. Синтез 2,6-ди-трет.-бутил-4-[5-(3-пиридил)-4- оксапентил]фенола.
Стадия 1. В трехгорлой колбе на 1 л, снабженной механической мешалкой, обратным холодильником с приспособлением для ввода азота и капельной воронкой с противодавлением на 500 мл, готовят раствор 57,76 г (0,2 моль; 70%-ного раствора в толуоле) бис(2-метокси-этокси)алюмогидрида натрия. При охлаждении на ледяной бане в атмосфере азота медленно в течение 30 мин добавляют раствор 29,24 г (0,1 моль) метилового эфира 3-(3,5-ди-трет.бутил-4- гидроксифенил)пропионовой кислоты (Ionox 520; Shell Chemie GmbH) в 300 мл безводного толуола. По окончании добавления смесь перемешивают в течение 30 мин при комнатной температуре и кипятят в течение 80 мин. Смесь охлаждают (при этом выделяется вязкий стеклообразный осадок) и осторожно промывают 1 л 3 М соляной кислоты. После обычной обработки выделяют маслообразный сырой продукт, который перегоняют "из колбы в колбу" при 145 - 150oC (1 Торр), получают 25,7 г (97%) 3-(3,5-ди-трет.бутил-4-гидроксифенил)пропанола в виде бесцветного твердого порошка, после перекристаллизации из н-гексана т.пл. 69 - 70oC.
(C17H28O2; М.в. 264,4).
Rf: 0,49 (SiO2 60; толуол-изопропанол, 9:1).
Стадия 2. 3-(3,5-Ди-трет.-бутил-4-гидроксифенил)-1- (тетрагидропиран-2-илокси)пропан.
К раствору 31,06 г (0,117 моль) 3-(3,5-ди-трет.бутил-4- гидроксифенил)пропанола и 500 мл пиридиний п-толуолсульфоната (ППТС) в 120 мл безводного метиленхлорида при охлаждении на ледяной бане по каплям в течение 15 мин добавляют 11,91 г (0,142 моль) 3,4-дигидро-2H-пирана, перемешивают 60 мин при охлаждении льдом и в течение ночи при комнатной температуре. Реакционную смесь промывают насыщенным водным раствором бикарбоната, рассолом, сушат Na2SO4 и упаривают в вакууме, получают 40,74 г (практически 100%) 3-(3,5-ди-трет. -бутил-4-гидроксифенил)-1-(тетрагидропиран-2- илокси)пропана в виде желтого масла, которое далее используют без дополнительной очистки.
(C22H36O3; М.в. 348,5).
Rf : 0,57 (SiO2 60; толуол-изопропанол, 9:1).
Стадия 3. 3-(4-Ацетокси-3,5-ди-трет. -бутилфенил)-1-(тетрагидропиран- 2-илокси)пропан.
К смеси 40,74 г (0,117 моль) 3-(3,5-ди-трет.-бутил-4-гидроксифенил)-1-(тетрагидропиран-2-илокси)пропана, 5,85 г (16,7 ммоль)тетрабутиламмонийгидросульфата, 250 мл метиленхлорида и 250 мл 50%-ного водного раствора гидроксида натрия при интенсивном перемешивании при 0oC в атмосфере азота добавляют по каплям раствор 14,32 г (0,140 моль) уксусного ангидрида в 100 мл метиленхлорида. По окончании добавления смесь перемешивают в течение 4 ч при комнатной температуре и осуществляют экстракцию обычным способом, получают 42,69 г (93%) 3-(4-ацетокси-3,5-ди-трет.бутилфенил)-1-(тетрагидропиран-2-илокси)пропана в виде коричневой вязкой жидкости, которую используют на следующей стадии снятия защиты без дополнительной очистки.
(C24H38O4; М.в. 390,6).
Rf:0,52 (SiO2 60; толуол-изопропанол, 9:1)
Стадия 4. 3-(4-Ацетокси-3,5-ди-трет.бутилфенил)пропанол.
Раствор 14,6 г (37,38 ммоль) 3-(4-ацетокси-3,5-ди-трет.- бутилфенил)-1-(тетрагидропиран-2-илокси)пропана в 80 мл безводного метанола перемешивают вместе с 1 г амберлиста 15 (H+-форма) при 45oC в течение 2 ч и оставляют на ночь при комнатной температуре. Суспензию разбавляют 100 мл метанола, катализатор отделяют фильтрованием, фильтрат упаривают в вакууме, получают 11,53 г сырого продукта, который очищают быстрой хроматографией на силикагеле (элюент гексан-этилацетат, 3: 1). Из фракций, содержащих продукт, выделяют 6,69 г (61%) 3-(4-ацетокси-3,5-ди-трет.- бутилфенил)пропанола в виде желтоватого масла, которое со временем кристаллизуется, т.пл. 93-95oC.
(C19H30O3; M.в. 306,4).
Rf : 0,47 (SiO2 60; толуол-изопропанол, 9:1).
Стадия 5. 2,3-Ди-трет.-бутил-4-[5-(3-пиридил)-4-оксапентил]фенола.
К раствору 19,74 г (64,42 ммоль) 3-(4-ацетокси-3,5-ди-трет.бутилфенил)пропанола, 12,68 г (77, 3 ммоль) хлоргидрата-3-пиколилхлорида и 5 г тетрабутиламмоний гидросульфата в 250 м толуола при интенсивном перемешивании (не менее 400 об/мин) и охлаждении водой в атмосфере азота добавляют по каплям 250 мл 50%-ного водного раствора гидроксида натрия. Темную реакционную смесь перемешивают при комнатной температуре в течение 5 ч и после обычной процедуры экстракции получают сырой продукт (26,3 г), который очищают быстрой хроматографией на силикагеле (элюент этилацетат-гексан, 2:1), выделяют 16,9 г (66%) 2,6-ди-трет.-бутил-4-[5-(3-пиридил)-4-оксапентил]фенилацетата в виде бесцветного масла. Полученный продукт растворяют в 100 мл сухого тетрагидрофурана и по каплям при -78oC (баня сухой лед-ацетон) в атмосфере инертного газа при перемешивании добавляют к суспензии 1,77 г (46,76 ммоль) алюмогидрида лития в 100 мл безводного тетрагидрофурана. Реакционную массу перемешивают в течение ночи в постепенно нагревающейся охлаждающей бане, выдерживают 2 ч при 40oC и при интенсивном перемешивании и охлаждении водой со льдом останавливают реакцию, осторожно добавляя последовательно 1,77 г воды, 1,77 г 15%-ного водного раствора гидроксида натрия и затем 5,31 г воды. Смесь выдерживают в течение 1 ч, разбавляют 200 мл ТГФ, сушат 20 г Na2SO4, выделившийся осадок алюмината отделяют фильтрацией через пористый стеклянный фильтр, фильтрат упаривают в вакууме, получают 12,9 г сырого 2,6-ди-трет.-бутил-4-[5-(3-пиридил)-4-оксапентил] фенола в виде твердого вещества желтого цвета. После перекристаллизации из смеси н-гексан-этилацетат выделяют 11,49 г (76% по стадии снятия защиты) чистого продукта, т.пл. 105oC.
C23H33NO2; M.в. 355.5).
Rf : 0,47 (SiO2 60; толуол-изопропанол, 9:1).
Пример 7. Синтез 2,6-ди-трет.-бутил-4-[7-(3-пиридил)-4-оксагептил]фенола.
Стадия 1. 3-(4-Ацетокси-3,5-ди-трет.-бутилфенил)-1-(тозилокси)пропан.
К раствору 8,41 г (27,44 ммоль) 3-(4-ацетокси-3,5-ди-трет.-бутилфенил)пропанола (см. пример 6, стадии 3 и 4) в 125 мл безводного пиридина при 0oC добавляют 5,49 г (28,80 ммоля) п-толуолсульфохлорида. Смесь перемешивают при той же температуре в течение 1,5 ч, выдерживают в течение ночи в холодильнике и проводят экстракцию обычным способом. Сырой 3-(4-ацетокси-3,5-ди-трет. -бутилфенил)-1-(тозилокси)пропан очищают быстрой хроматографией на силикагеле (элюент н-гексанэтилацетат, 4:1), получают 8,28 г чистого тозилата в виде бесцветной вязкой жидкости.
(C26H36SO5; M.в. 460,6).
Rf: 0,42 (SiO2 60; толуол-изопропанол, 9:1).
Стадия 2. 2,6-Ди-трет.-бутил-4-[7-(3-пиридил)-4-оксагептил]фенол.
К суспензии 2,56 г (53,33 ммоль) 60%-ной суспензии NaH в масле в 35 мл безводного ТГФ в атмосфере инертного газа при перемешивании при 0oC через шприц добавляют раствор 3,66 г (26,68 ммоль) 3-(3-пиридил)пропанола в 30 мл безводного ТГФ и 3,5 мл гексаметилфосфортриамида (ГМФТА) и кипятят в течение 2 ч. Полученный раствор алкоголята охлаждают до 0oC, добавляют раствор 10,68 г (23,19 ммоль) 3-(4-ацетокси-3,5-дитрет.-бутилфенил)-1-(тозилокси)пропана в 35 мл безводного ТГФ, кипятят в течение 2 ч и перемешивают при комнатной температуре в течение ночи. После обычной обработки сырой продукт (9,88 г; коричневое масло) очищают быстрой хроматографией на силикагеле (элюент н-гексан-этилацетат, 3:1), получают 2,88 г (62%) 2,6-ди-трет.-бутил-4-[7-(3-пиридил)-4-оксагептил]фенола в виде янтарного масла.
(C25H37NO2; M.в. 383.6).
Rf: 0,44 (SiO2 60; н-гексан-этилацетат, 3:2).
Пример 8. Синтез 2,6-ди-трет.-бутил-4-[9-(3-пиридил)-4-оксанонил]фенола 2,6-Ди-трет.-бутил-4-[9-(3-пиридил)-4-оксанонил]фенол.
Исходя из 5-(3-пиридил)пентанона (получение см. Стадию 2 примера 1 из 4-пентин-1-ола (E.R.H.Jones et al., Org. Synthesis Coll. Vol. 4,755 (1963)) по реакции образования простого эфира (методика описана выше в примере 7) получают 2,6-ди-трет.-бутил-4-[9-(3-пиридил)-4-оксанонил]фенол с выходом 57% в виде желтоватого вязкого масла.
(C27H41NO2; M.в. 411,6)
Rf: 0,52 (SiO2 60; н-гексан-этилацетат, 3:2).
Пример 9. Синтез 2,6-ди-трет.-бутил-4-[8-(3-пиридил)-2-оксаокт-7- инил] фенола.
По методикам, описанным в примере 1, стадия 2 (без стадии гидрирования) и стадия 3, получают 2,6-ди-трет.-бутил-4-[8-(3-пиридил)-2-оксаокт-7-инил] фенол в виде желтого вязкого масла с выходом 62% на последней стадии.
(C26H35NO2; M.в. 393,6)
Rf: 0,48 (SiO2 60; н-гексан-этилацетат, 2:1).
Кислотно-аддитивные соли
В качестве примеров кислот, приемлемых для образования кислотно-аддитивных солей в соответствии с обычной методикой, можно указать следующие минеральные кислоты: соляную, бромистоводородную, метансульфокислоту, изотионовую, серную, фосфорную и сульфаминовую; и органические кислоты: уксусную, пропионовую, малеиновую, фумаровую, винную, лимонную, щавелевую и бензойную. Предпочтительными кислотами являются соляная, лимонная и малеиновая. Если требуется, могут быть получены другие фармацевтически приемлемые кислотно-аддитивные соли, и одна кислотно-аддитивная соль может быть превращена в другую путем нейтрализации соли, например, хлороводородом, и обработкой другой выбранной минеральной или органической кислотой с получением другой фармацевтически приемлемой кислотно-аддитивной соли, как это уже объяснялось выше и как это принято в данной области.
Фармацевтические композиции
На основе заявленного соединения могут быть приготовлены фармацевтические композиции, содержащие фармацевтически приемлемый носитель или разбавитель в смеси с активным соединением. Такие композиции могут быть назначены животным, а также человеку, орально или парентерально. Например, твердые рецептуры или фармацевтические композиции настоящего изобретения могут иметь форму капсул, таблеток, пеллет, порошка или гранулятов, В таких твердых фармацевтических композициях активное соединение или пролекарство этого соединения смешивается с одним фармацевтическим приемлемым разбавителем или носителем, таким как тростниковый сахар, лактоза, крахмал, тальк или синтетические или природные смолы, связующим веществом, таким как желатин, смазочное вещество, такое как стеарат натрия, и/или диспергирующая добавка, такая как бикарбонат натрия. Для получения эффекта постоянного выделения в фармацевтические композиции может быть введено такое вещество, как гидроколлоид или другой полимер. Вспомогательные добавки, такие как смазочные вещества или буферы, обычное для данной области, также могут быть добавлены в рецептуры. Таблетки, пеллеты или грануляты могут быть покрыты энтеросолюбильным покрытием, если это необходимо. Жидкие препараты для орального применения могут иметь форму эмульсий, растворов или суспензий, содержащих обычные инертные разбавители, такие как вода. Кроме того, такие жидкие фармацевтические композиции могут также содержать смачиватели, эмульгаторы, дисперсанты или поверхностно-активные агенты, а также подслащивающие добавки, корригенты или ароматизирующие вещества.
Подходящими препаратами для парентерального применения могут быть наряду с другими стерильные водные или неводные растворы, суспензии, липосомы или эмульсии. Вспомогательные вещества, которые хорошо известны в данной области, могут быть использованы в качестве фармацевтически приемлемого разбавителя или носителя.
В зависимости от предполагаемого способа применения и продолжительности лечения точная доза активного соединения в препарате настоящего изобретения может изменяться в соответствии с представлениями врача или ветеринара. Активное соединение настоящего изобретения может применяться совместно с другими фармацевтически-активными агентами.
В композициях настоящего изобретения содержание активного агента или агентов может изменяться в широких пределах, при этом необходимо только, чтобы активный ингредиент изобретения или его пролекарство составляли или обеспечивали эффективное количество, то есть так, чтобы обеспечивалась приемлемая эффективная доза. Очевидно, что несколько дозирующих форм, а также несколько индивидуальных активных соединения могут применяться одновременно или почти одновременно или даже в одной и той же композиции или в одном и том же препарате.
Способ лечения
Как указывалось ранее, соединения настоящего изобретения предназначены особенно в форме фармацевтической композиции для орального или парентерального применения, причем точная индивидуальная дозировка, а также дневная дозировка для конкретного случая определяется в соответствии с хорошо известными медицинскими или ветеринарными принципами.
Кроме орального и парентерального применения можно использовать ректальное и/или внутривенное введение, причем дозировка обычно значительно ниже в случае парентерального введения, хотя предпочтительным является оральное.
Приемлемым является количество приблизительно от одного до трех грамм в день в виде нескольких доз. В зависимости от конкретного случая дозировка может осуществляться и в более широких пределах, от 0,5 до 10 г в день. Несмотря на то, что приемлемой дозировкой для использования в таблетках, как было установлено, является 500 мг активного ингредиента, индивидуальные дозировки могут изменяться от приблизительно 200 до 1000 мг, и предлагаемые для использования в таблетках 500 мг могут быть, конечно, введены орально, например, порциями от 1 до 3 раз в день. Само собой разумеется, что может быть введена более, чем одна таблетка для того, чтобы получить указанное выше предлагаемое дневное количество при оральном применении от 1 до 3 г в день.
Как уже говорилось, соединение настоящего изобретения или его пролекарство может вводиться животному и человеку любым из возможных путей, например, орально в виде капсул или таблеток, парентерально в форме стерильных растворов или суспензий, или путем имплантации пеллет, а в некоторых случаях внутривенно в виде стерильных растворов. Другими очевидными моделями введения являются накожный, подкожный, внутриротовой, внутримышечный и внутрибрюшинный, причем конкретный способ введения обычно выбирается врачом или ветеринаром.
Изучение токсичности
Цель данного эксперимента заключалась в том, чтобы получить информацию по токсичности соединений Mrz 3/124 и Mrz 3/161 после единичного внутрибрюшинного введения каждого соединения крысам в дозах 120 мг, 240 мг, 480 мг, 960 мг и 1920 мг/кг веса тела (в.т.)(см.табл.6).
Дополнительные испытания на токсичность
Действие Mrz 3/161 на ЦНС изучали на мышах через 40 минут после внутрибрюшинного введения лекарства. Дозировка находилась в интервале от 4 (наименьшая) до 324 (наибольшая) мг/кг.
Введение
Цель исследования заключалась в том, чтобы испытать активность Mrz 3/161 на центральную нервную систему после внутрибрюшинного введения мышам. Для изучения действия лекарства на токсичности, поведение и двигательную активность использовали серию из 19 испытаний.
Методы
Mrz 3/161 суспендируют в физиологическом солевом растворе с добавлением 1% метилцеллюлозы и 1% Tween 80. Испытание проводят на 25 мышах по 5 животных в каждой дозируемой группе для доз 4, 12, 36, 108 и 324 мг/кг.
Параметры испытаний
1. Смертность через 1 ч
2. Смертность через 24 ч
3. Треморы
4. Тонические судороги
5. Клонические судороги
6. Птоз
7. Седативный эффект
8. Возбуждение (заметное и сильное)
9. Потеря исследовательской активности
10. Потеря рефлекса наружного уха
11. Потеря установочного рефлекса
12. Мидриаз
13. Каталепсия
14. Потеря захвата стержня
15. Rotarod тест (1 мин)
16. Rotarod тест (2 мин)
17. Аналгезия
18. Защита от электрошока
19. Смертность после электрошока
Для каждого из параметра определяли общее количество реагирующих на воздействие мышей.
Результаты
Смертности нет при любой дозе как через 1 час (испытание 1), так и через 24 ч (испытание 2). Наблюдались значительные эффекты в отношении возбуждения (испытание 8) (13 баллов), rotarod активности при 1 мин (испытание 15) (8 баллов) и rotarod активности при 2 мин (испытание 16) (15 баллов).
Выводы
Mrz 3/161 умеренно воздействует как на возбуждение, так и на rotarod - активность.
Результаты (баллы) тех же испытаний для других соединений Mrz приведены в табл.7.
Вывод
Соединения проявляют различные величины LD50, причем другими заметными реакциями являются мидриаз, степень rotarod неустойчивости, анальгезия, защита от электрошока и смертность после электрошока.
Фармацевтически приемлемые кислотно-аддитивные соли
Получение. Для получения различных солей соединений Mrz растворы в эфире обрабатывают соответствующей кислотой, промывают эфиром и перекристаллизовывают. Получают соли от взаимодействия с соляной кислотой, перхлорной кислотой, серной кислотой, щавелевой кислотой и трифторуксусной кислотой. Следует отметить, что трифторацетат кристаллизуется с одной дополнительной связанной молекулой трифторуксусной кислоты. Когда для удаления кислоты используют вакуумную сушку, трифторацетат превращается в сиропообразное масло.
1) Mrz 3/124 • 2CF3COOH
3-[5-(3,5-дитретбутил-4-гидроксифенил)-2- оксапентил] пиридинтрифторацетат
Молекулярная формула: C23H33NO2 • 2CF2COOH
Молекулярный вес: 583,59
Температура плавления: 65 - 67oC
2) Mrz 3/124 • H2C2O4
3-[5-(3,5-дитретбутил-4-гидроксифенил)-2- оксапентил]пиридиноксалат
Молекулярная формула: C23H33NO2 • H2C2O4
Молекулярный вес: 445,59
Температура плавления: 110 - 112oC
3) Mrz 3/124 • HClO4
3-[5-(3,5-дитретбутил-4-гидроксифенил)-2- оксапентил]пиридинперхлорат
Молекулярная формула: C23H33NO2 • H2ClO4
Молекулярный вес: 456.01
Температура плавления: 145 - 147oC
4) Mrz 3/124 • H2SO4
3-[5-(3,5-дитретбутил-4-гидроксифенил)-2- оксапентил]пиридинсульфат
Молекулярная формула: C23H33NO2 • H2SO4
Молекулярный вес: 453.63
Температура плавления: 110 - 112oC
5) Mrz 3/124 • HCl
3-[5-(3,5-дитретбутил-4-гидроксифенил)-2- оксапентил]пиридингидрохлорид
Молекулярная формула: C23H33NO2 • HCl
Молекулярный вес: 391.98
Температура плавления: 148 - 150oC.
Приложение C
Примеры фармацевтических композиций
Таблетки, 8 мм ⊘, двухплоскостные, общая масса 150 мг
Mrz 3/124 (активный ингредиент) - 60 частей
ПВП (поливинилпирролидон; Povidone) 25 - 25 частей
Macrogol 10.000 (ПЭГ МВ пр. 10,000) - 8 частей
100 частей, сплавлены вместе, образуя 111 мг (74%) всей таблетки
добавление
Avicel PH 101 (коллоидная целлюлоза) - 30 мг
Ac-Di-Sol (сшитая натриевая соль карбоксиметилцеллюлозы) - 9 мг
Используется обычная методика таблетирования (на стандартном оборудовании).
Капсулы
Mrz 3/124 (активный ингредиент) - 100 мг
ПВП 25 - 300 мг
Карбоксиметилцеллюлоза (сшитая) - 16 мг
Оболочка капсулы - 116 мг
Процесс получения описан ниже.
Фармацевтические композиции других соединений аналогичны друг другу и могут быть получены по той же методике.
Способ получения фармацевтических композиций см. ниже и в табл.8.
Б. Состав порошкообразного наполнителя
Продукт соосаждения MRZ 3/124 (25%) - 4,192 г
Ac-Di-Sol тип SD-711 - 167,7 г
В. Способ получения продукта соосаждения MRZ 3/124
*Состав:
MRZ 3/124 (25%) - 1,125 г
ПВП 25 - 3,375 г
*смешение порошка
Переносят соединение 3.124 и ПВП 25 в барабанный смеситель и перемешивают в течение 10 мин, после чего смесь помещают на тефлоновую фольгу и равномерно распределяют таким образом, чтобы слой имел толщину 1 см.
*процесс сплавления
Сушильную камеру нагревают до 120oC и устанавливают в HCl подносы с порошком, предварительно продув камеру азотом в течение 7 мин. Выдерживают при 120oC в течение 1 ч, поддерживая при этом поток азота через камеру. Визуально проверяют, чтобы получить полностью гомогенный расплав. После охлаждения в течение ночи продукт удаляют с фольги и измельчают грубо вручную шпателем.
* Размалывание продукта соосаждения
Полученный выше грубоизмельченный продукт помещают в мельницу с последующим пропусканием через сито с отверстиями 0.7 и 1.9 мм. Повторяют этот процесс с остатком, просеивая его через сито 355 мкм. В конце пропускают остаток через малое круглое сито размеров 1.1 мм. Объединяют эту фракцию с фракцией менее 355 мкм и помещают в миксер на 5 мин.
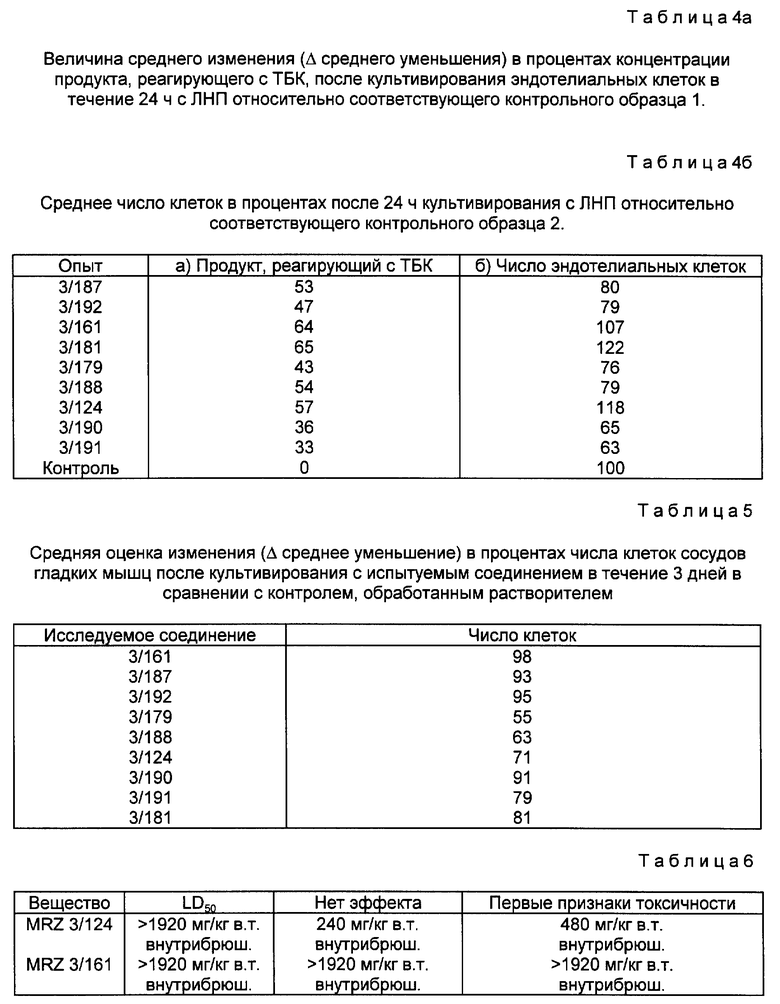
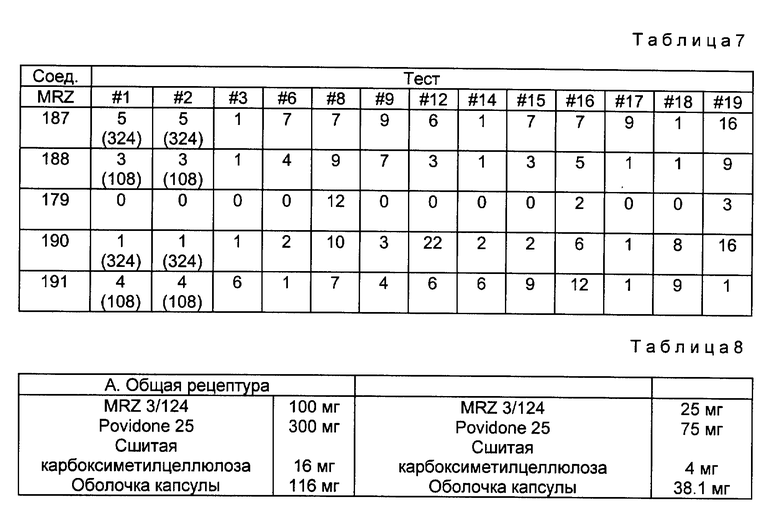
ω- Пиридиловые эфиры бутилированного гидрокситолуола формулы I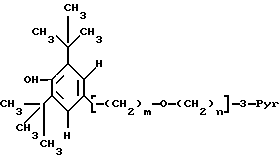
используют в фармацевтической композиции с антилипидемической и антиатеросклеротической активностью и в способе лечения гиперлипидемии и атеросклероза в дозе 200-1000 мг до 1-3 граммов в день. Соединение получают из ω- пиридилалкил-, алкенил- или алкинил-спиртов и 3,5-дитрет.-бутил-4-гидроксибензиловым спиртом в виде его ацетата. В соединении формулы I 3-Pyr представляет 3-пиридил; m = 1, n - 4-7, или m = 3, a n - 1-7 и где связь между двумя атомами углерода фрагмента (CH2)n, соседнего с пиридиновым кольцом является простой, двойной или тройной, и их фармацевтически приемлемые кислотно-аддитивные соли. 4 с. и 3 з.п.ф-лы, 8 табл.
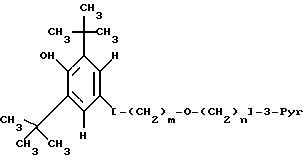
где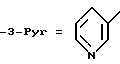
m = 1 и n = 4 - 7 или m = 3, а n = 1 - 7 и где связь между двумя атомами углерода фрагмента (CH2), соседнего с пиридиновым кольцом, является простой, двойной или тройной, и их фармацевтически приемлемый кислотно-аддитивные соли.
2,6-ди-трет.-бутил-4-[8-(3-пиридил)-2-оксаоктил]фенол (Mrz 3/161),
2,6-ди-трет.-бутил-4-[6-(3-пиридил)-2-оксагексил]фенол (Mrz 3/187),
2,6-ди-трет.-бутил-4-[7-(3-пиридил)-2-оксагептил]фенол (Mrz 3/192),
(Z)-2,6-ди-трет. -бутил-4-[8-(3-пиридил)-2-оксаокт-7-енил] фенол (Mrz 3/181).
2,6-ди-трет.-бутил-4-[9-(3-пиридил)-2-оксанонил]фенол (Mrz 3/188),
2,6-ди-трет.-бутил-4-[5-(3-пиридил)-4-оксапентил]фенол (Mrz 3/124),
2,6-ди-трет.-бутил-4-[7-(3-пиридил)-4-оксагептил]фенол (Mrz 3/190),
2,6-ди-трет.-бутил-4-[9-(3-пиридил)-4-оксанонил]фенол (Mrz 3/191),
2,6-ди-трет.-бутил-4-[8-(3-пиридил)-2-оксаокт-7-инил]фенол (Mrz 3/179).
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| DE, 2716125, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Corew et al | |||
| Proc | |||
| Nat - Acad Sci., USA, 84, 7725 - 7729, 1987. | |||
