Изобретение относится к стереоспецифичному способу получения флорфеникола, в частности к способу получения флорфеникола, способу региоизбирательного открытия хирального эпоксида и к способу получения его R,S-изомера.
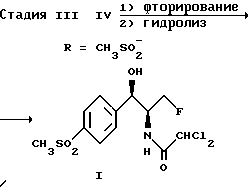
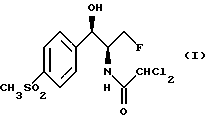
Известен способ получения флорфеникола [R-(R*, S*)]-2,2-дихлор-N-1-(фторметил)-2-гидрокси-2-[4-(метилсульфонил)-фенил] -этилацетамида формулы (I)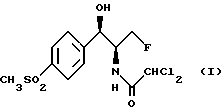
который заключается в том, что производное оксазолина формулы А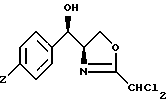
где Z означает метансульфонил, подвергают взаимодействию с реагентом, вызывающим равновесие с получением равновесной смеси, содержащей производное оксазолина вышеприведенной формулы А и производное оксазолина формулы Б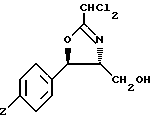
где Z имеет вышеуказанное значение, и полученное производное оксазолина формулы Б обрабатывают агентом фторирования с последующим гидролизом кислотой с получением флорфеникола (см WO 92,07824 А1, кл. C 07 C 315/04, 14.05.92).
Задачей изобретения является расширение технологической базы эффективного и экономического получения флорфеникола.
Данная задача решается предлагаемым способом получения флорфеникола формулы (I)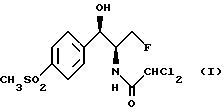
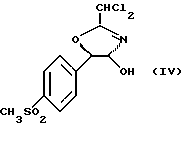
включающим получение оксазолина формулы (IV)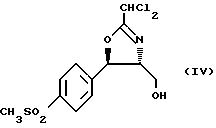
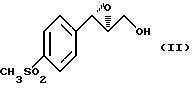
обработку его агентом фторирования с последующим гидролизом кислотой, который заключается в том, что получение оксазолина вышеприведенной формулы (IV) включает стадию региоизбирательного открытия хирального эпоксида формулы (II)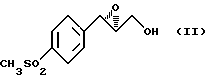
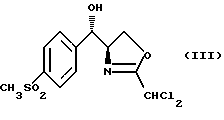
путем последовательной обработки сильным основанием, кислотой Льюиса и дихлорацетонитрилом с получением оксазолина формулы (III)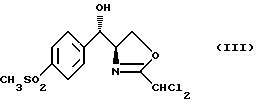
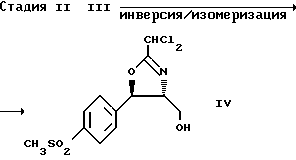
и стадию избирательной инверсии/изомеризации полученного оксазолина формулы (III) путем последовательной обработки третичным амином и низшим алкилсульфонилхлоридом, водной кислотой и гидроксидом щелочного металла.
Дополнительным объектом изобретения является способ региоизбирательного открытия хирального эпоксида вышеприведенной формулы (II), заключающийся в том, что он включает последовательную обработку сильным основанием, кислотой Льюиса и дихлорцетонитрилом с получением оксазолина формулы (III)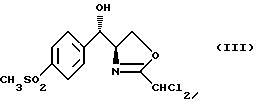
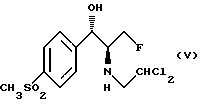
Дальнейшим объектом изобретения является способ получения R,S-изомера флорфеникола вышеприведенной формулы (I), заключающийся в том, что S,S-изомер формулы (V)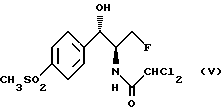
подвергают изомеризации путем последовательной обработки третичным амином и низшим алкилсульфонилхлоридом, водной кислотой и гидроксилом щелочного металла.
Предлагаемый способ асимметричного получения флорфеникола иллюстрируется следующей реакционной схемой A: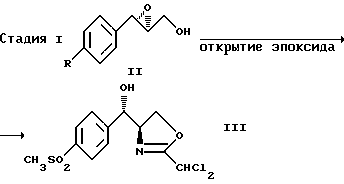
Хиральный эпоксид формулы (II) можно получать общеизвестными методами.
На стадии 1 эпоксид формулы (II) в виде раствора в пригодном растворителе, например, тетрагидрофуране медленно добавляют предпочтительно каплями к суспензии гидрида натрия в тетрагидрофуране при температуре от -10oC до +20oC, предпочтительно от 0oC до +10oC, наиболее предпочтительно примерно +5oC, и реакционную смесь оставляют стоять в течение около 30 минут. К получаемой таким образом холодной смеси добавляют раствор безводного хлористого цинка в тетрагидрофуране. Примерно через 30 минут при температуре от -10oC до 20oC, предпочтительно от 0oC до +10oC, наиболее предпочтительно примерно +5oC, к смеси добавляют раствор дихлорацетонитрила в тетрагидрофуране и молекулярное сито величиной пор 4А. Смесь перемешивают в в течение около 15 минут, нагревают до температуры 30o - 80oC, предпочтительно 50o - 60oC, наиболее предпочтительно 55oC, в течение 16 часов. Получаемую смесь охлаждают до комнатной температуры, добавляют водный раствор бикарбоната натрия и экстрагируют природным растворителем, например этилацетатом, получаемый сырой продукт суспендируют в изопропаноле, фильтруют и сушат с выделением хирального оксасолина формулы (III).
На стадии II оксазолин формулы (III) смешивают с триалкиламином, предпочтительно триэтиламином, и пиридином при комнатной температуре. Смесь охлаждают до комнатной температуры -20o - +20oC, предпочтительно 0o - +10oC, наиболее предпочтительно около +5oC, после чего к смеси медленно добавляют, предпочтительно каплями, алкилсульфонилхлорид, предпочтительно метансульфонилхлорид, и перемешивают в течение приблизительно 2 часов. Значение pH смеси доводят до 1,9 - 4,0, предпочтительно примерно 2,0, добавлением водной кислоты, предпочтительно серной кислоты, наиболее предпочтительно примерно 3,0-н. серной кислоты. Добавляют тетрагидрофуран для получения гомогенной смеси. После нагревания до комнатной температуры в течение 10 минут смесь обрабатывают гидроксидом щелочного металла, предпочтительно гидроксидом натрия, наиболее предпочтительно в виде 50%-ного водного гидроксида натрия для доведения до pH > 9,5, предпочтительно около 12,5. Затем смесь сгущают и экстагируют пригодным растворителем, например, этилацетатом, в результате чего получают изомеризованный оксазолиновый продукт формулы (IV).
Предлагаемый способ получения R,S-изомера флорфеникола формулы (I) иллюстрируется следующей реакционной схемой B: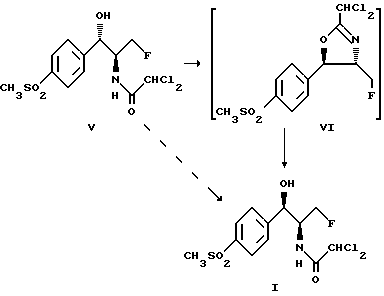
Изомеризацию осуществляют аналогично вышеописанной стадии II. S,S-изомер флорфеникола смешивают с триалкиламином, предпочтительно триэтиламином, и пиридином при комнатной температуре. Смесь охлаждают до температуры -20 до +20oC, предпочтительно 0 - 10oC, наиболее предпочтительно примерно +5oC, после чего к смеси медленно, предпочтительно каплями, добавляют алкилсульфонилхлорид, предпочтительно метансульфонилхлорид, и перемешивают в течение приблизительно 2 часов. Затем реакционную смесь обрабатывают гидроксидом щелочного металла, предпочтительно гидроксидом натрия, наиболее предпочтительно в качестве 50%-ного водного гидроксида натрия, для доведения значения pH до примерно 12,5. Получаемый при этом промежуточный оксазолин формулы (VI) обрабатывают водным раствором кислоты, например уксусной кислоты, при комнатной температуре с получением R,S-изомера флорфеникола формулы (I).
Следующие примеры поясняют настоящее изобретение.
Исходное соединение
Пример 1.
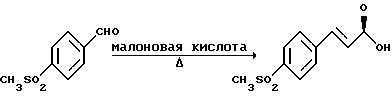
Смесь 312 г (2,99 моль) малоновой кислоты, 596 мл пиридина, 30 мл пиперидина и 300 г (1,49 моль) п-метилсульфонил-безальдегида нагревают при 95 - 100o в течение 4 часов. Смесь охлаждают до комнатной температуры и медленно подают в 3 л смеси соляной кислоты, воды и льда. Выпавший осадок фильтруют, твердое вещество сушат и получают 340 г (83%) природного Е-коричной кислоты. Температура плавления: 294 - 296oC. 1Н-ЯМР (ДМСО-d6, ч/мил.): 7,96 (c, 4H); 7,78 (д, J = 16 Гц, 1H); 6,71 (д, J = 16 Гц, 1H); 3,5 (шир. C, 3H).
Пример 2.
Стадия А: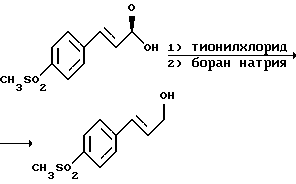
Смесь 96 мл (1,35 моль) тионилхлорида и 50 г (0,225 моль) продукта примера 1 нагревают с обратным холодильником в течение часа. Избыточный тионилхлорид отгоняют, после чего добавляют 100 мл дихлорметана. Получаемый раствор прикапывают к предварительно охлажденной (до -5oC) смеси 42 г (1,1 моль) борана натрия и 500 мл этанола. Реакционную смесь размешивают при 10oC в течение часа, после чего ее подают в смесь соляной кислоты, воды и льда, три раза экстрагируют дихлорметаном, взятым в количестве по 300 мл, экстракты объединяют, сгущают и фильтруют. Получают 28 г (74%) аллилового спирта.
Температура плавления: 126 - 127oC. H1-ЯМР (CDCl3, 4/милл.): 7,85 (д, J = 9 Гц, 2H); 6,52 (дт, J = 16 Гц, 5 Гц, 1H); 4,38 (дд, J = 5 Гц, 1Гц, 2H); 3,05 (c, 3H); 1,93 (шир. c, 1H);
Стадия Б:
К 8 г молекулярного сита величиной пор 4А добавляют 1,74 г (7,4 моль) L -диизопропилвинной кислоты и 2,16 г (7,4 ммоль) изопропанолята титана (IV) при -20oC в безводных условиях и получаемую смесь перемешивают в течение 30 минут. Прикапывают раствор 8,1 г (37 моль) аллилового спирта, получаемого на стадии А, в 500 мл дихлорметана и 13,4 мл 3,0-м. раствора трет.-бутилгидропероксида в 2,2,4-триметилпентане. Смесь перемешивают при -20oC в течение 4 часов, после чего добавляют 6,0 мл диметилсульфоксида, фильтруют, к фильтрату добавляют 250 мл насыщенного водного раствора фтористого натрия и перемешивают при 25oC в течение 16 часов. Фильтруют на кизельгуре марки Целите и фильтрат три раза экстрагируют дихлорметаном, взятым в количестве по 100 мл. Экстракты объединяют, дважды промывают водой, взятой в количестве по 100 мл, сгущают и фильтруют. Получают 7,15 г (82%) S,S-эпоксида, оптическая чистота которого составляет 97%.
Температура плавления: 104 - 106oC. H1-ЯМР (CDCl3): 7,92 (д, J = 8 Гц, 2H); 7,49 (д, J = 8 Гц, 2H); 4,10 (дд, J = 17 Гц, 3 Гц, 1H); 4,04 (с, 1H); 3,85 (дд, J = 17 Гц, 3 Гц, 1H); 3,20 - 3,17 (м, 1H); 3,05 (c, 3H); 1,88 (шир. c, 1H);
Оптическую чистку эпоксида определяют хиральной хроматографией на колонке марки Chracel OJ размером 25 см x 4,6 мм при температуре 25oC с применением в качестве элюента смеси гексана и изопропанола в соотношении 1:1, содержащей 1% ацетонитрила.
Стадия В: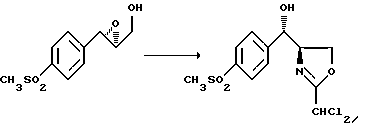
Смесь 6,1 г (153 ммоль) 60%-ной дисперсии гидрида натрия в масле и 90 мл тетрагидрофурана охлаждают до 5oC. К получаемой суспензии прикапывают раствор 30 г (128 ммоль) эпоксида стадии Б в 300 мл тетрагидрофурана, перемешивают при 5oC в течение 30 минут, после чего прикапывают 17,8 г (128 ммоль) безводного хлористого цинка в качестве раствора в 250 мл тетрагидрофурана и перемешивают при 5oC в течение 30 минут. Прикапывают раствор 17,0 г (153 ммоль) дихлорацетонитрила в 10 мл тетрагидрофурана, после чего добавляют 1 г молекулярного сита величиной пор 4А и продолжают перемешивать при температуре 5oC в течение 15 мин. Реакционную смесь нагревают до 55oC, перемешивают в течение 16 часов, охлаждают до комнатной температуры и добавляют водный раствор бикарбоната натрия. Экстрагируют четыре раза этилацетатом, взятым в количестве по 400 мл, экстракты объединяют и сгущают. Получаемый остаток суспендируют в изопропане, фильтруют и твердое вещество сушат. Получают 16,0 г оксазолина, который перекристаллизовывают из метилизобутилкетона и получают очищенный оксазолин, чистота которого составляет 97%.
Температура плавления: 156,5 - 157, 5oC. 1H-ЯМР (CDCl3): 7,93 (д, J = 8,4 Гц, 2H); 7,60 (д, J = 8,4 Гц, 2H); 6,26 (c, 1H); 5,14 (д, J = 4 Гц, 1H); 4,57 (ддд, J = 8,7 Гц 7,9 Гц, 4,0 Гц, 1H); 4,46 (дд, J = 9,7 Гц, 7,9 Гц, 1H); 4,28 (дд, J = 9,7 Гц, 8,7 Гц, 1H); 3,05 ((c, 3H); 2,60 (шир. c, 1H).
Стадия Г: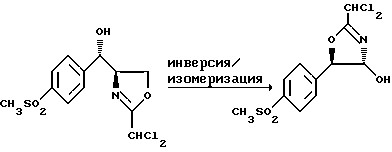
К 3,5 г (10 ммоль) оксазолина стадии В добавляют 5 мл пиридина и 2,8 мл триэтиламина при 25oC, получаемую смесь охлаждают до 5oC и прикапывают 0,95 мл (12 ммоль) метансульфонилхлорида. Реакционную смесь перемешивают при 5oC в течение 2 часов, после чего добавлением 3,00 н. водной серной кислоты pH доводят до 2. Затем к реакционной смеси добавляют 5 мл тетрагидрофурана, нагревают до комнатной температуры и перемешивают в течение 10 минут. Добавлением 50%-ной водной гидроокиси натрия pH доводят до 12,5. Смесь сгущают и три раза экстрагируют этилацетатом, взятым в количестве по 30 мл. Экстракты объединяют и сгущают, в результате чего получают продукт оптической чистоты > 99,9%. Продукт очищают хроматографией на силикагеле.
Температура плавления: 144 - 145oC. Н1-ЯМР (ДМСО-d6): 8,00 (д, J = 8,3 Гц, 2H); 7,26 (7,60 (д, J = 8,3 Гц, 2H); 5,75 (д, J = 6,4 Гц, 1H); 5,18 (т, J = 5,6 Гц, 1H); 4,10 - 4,05 (м, 1H); 3,75 - 3,65 (м, 1H); 3,60 - 3,55 (м, 1H); 3,23 (c, 3H).
Оптическую чистоту оксазолина определяют хиральной хроматографией на колонке марки Chiracel OJ размером 25 см x 4,6 мм при температуре 25oC с применением в качестве элюента смеси гексана, изопропанола и ацетонитрила в соотношении 69 : 30 : 1.
Стадия Д: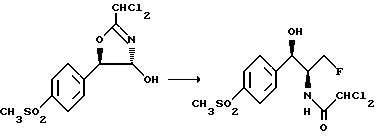
В автоклаве емкостью 30 мл подают 8,9 мл 23,3%-ного по весу раствора N-(1,1,2,3,3,3-гексафторпропил)диэтиламина в метиленхлориде, после чего добавляют 2,0 г водной суспензии дихлорметилоксазолина стадии Г. Автоклав закрывают и нагревают при температуре 100oC на масляной бане в течение 3 часов. Затем охлаждают до 0oC, автоклав охлаждают до 0oC, автоклав открывают и его содержимое перемещают с 0,35 г ацетата калия и 2,0 мл метанола. Органические растворители отгоняют и добавляют 20 мл смеси 2-пропанола и воды в соотношении 65 : 35. Остаток метиленхлорида отгоняют в вакууме и водную фазу нагревают с обратным холодильником в течение 3 часов. Половину растворителя отгоняют в вакууме и оставшуюся водную фазу охлаждают до - 5oC при перемешивании. Получаемые кристаллы фильтруют, промывают водой и сушат. Выход: 82% флорфеникола с чистотой 98,5%.
Пример 3.
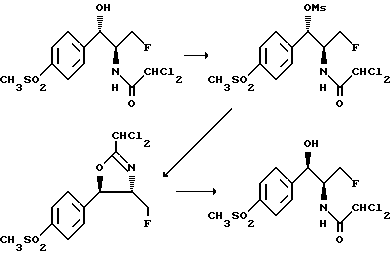
Смесь 3,0 г (8,4 ммоль) S,S-изомера флорфеникола, 3 мл тетрагидрофурана, 3 мл пиридина и 2,3 мл триэтиламина при перемешивании охлаждают до 10 - 15oC. Медленно прикапывают раствор 1,9 г (16.8 ммоль) метансульфонилхлорида в 2 мл тетрагидрофурана, поддерживая температуру при 10 - 15oC. Получаемую смесь перемешивают при 10oC в течение 30 минут, потом нагревают до комнатной температуры и перемешивают в течение часа. В этот момент образование промежуточного мезилата закончено. К реакционной смеси добавляют 10 мл воды, охлаждают до 10oC, после чего медленно прикапывают раствор 2,7 г 50%-ного водного гидроксида натрия, разбавленного в 5 мл воды. Перемешивают в течение 10 минут при 10oC, добавляют еще 0,5 г 50%-ного водного гидроксида натрия и продолжают перемешивать еще 10 минут до завершения образования промежуточного оксазолина. Добавлением 1,0-н. водной серной кислоты pH доводят до 6,5 - 6,0, после чего смесь дополнительно подкисляют добавлением уксусной кислоты до pH 5,5 - 5,0. Образуется осадок, который растворяют в ацетоне. Получаемый раствор перемешивают при комнатной температуре в течение 16 часов, сгущают в вакууме для удаления тетрагидрофурана, пиридина и ацетона и получаемую смесь перемешивают в течение 30 минут при комнатной температуре. Фильтруют, твердое вещество сушат и получают 2,5 г флорфеникола вышеприведенной формулы (I), имеющего чистоту 99%.
Изобретение относится к способу асимметричного получения флорфеникола формулы I, который состоит из следующих стадий: 1) стадии региоизбирательного открытия хирального эпоксида формулы II путем последовательной обработки сильным основанием, кислотой Льюиса и дихлорацетонитрилом с получением оксазолина формулы (III); II) стадию избирательной инверсии/изомеризации полученного оксазолина формулы (III) путем последовательной обработки третичным амином и низшим алкилсульфонилхлоридом, водной кислотой и гидроксидом щелочного металла с получением оксазолина формулы (IV); III) стадию обработки оксазолина формулы IV агентом фторирования с последующим гидролизом кислотой. Дополнительным объектом изобретения является способ региоизбирательного открытия хирального эпоксида вышеприведенной формулы II путем последовательной обработки сильным основанием, кислотой Льюиса и дихлорацетонитрилом с получением оксазолина формулы III. Дальнейшим объектом изобретения является способ получения RS -изомера флорфеникола вышеприведенной формулы I, заключающийся в том, что S,S-изомер флорфеникола формулы (V) подвергают изомеризации путем последовательной обработки третичным амином и низшим алкилсульфонилхлоридом, водной кислотой и гидроксидом щелочного металла. Изобретение расширяет технологическую базу эффективного и экономичного получения флорфеникола. 3 c.п. ф-лы.
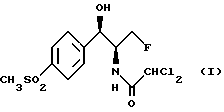
включающий получение оксазолина формулы IV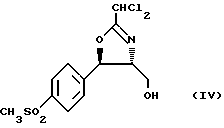
обработку его агентом фторирования с последующим гидролизом кислотой, отличающийся тем, что получение оксазолина вышеприведенной формулы IV включает стадию региоизбирательного открытия хирального эпоксида формулы II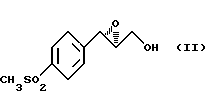
путем последовательной обработки сильным основанием, кислотой Льюиса и дихлорацетонитрилом с получением оксазолина формулы III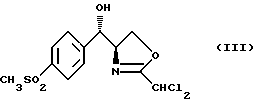
и стадию избирательной инверсии-изометрии полученного оксанозолина формулы III путем последовательной обработки третичным амином и низшим алкилсульфонилхлоридом, водной кислотой и гидроксидом щелочного металла.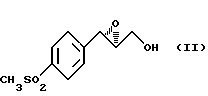
отличающийся тем, что включает последовательную обработку сильным основанием, кислотой Льюиса и дихлорацетонитрилом с получением оксазолина формулы III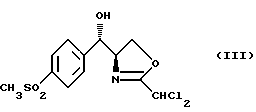
3. Способ получения R,S-изомера флорфеникола формулы I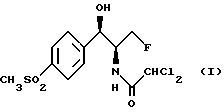
отличающийся тем, что S,S-изомер флорфеникола формулы V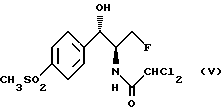
подвергают изомеризации путем последовательной обработки третичным амином и низшим алкилсульфонилхлоридом, водной кислотой и гидроксидом щелочного металла.
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| УСТРОЙСТВО ДЛЯ УПАКОВКИ ТОВАРОВ В СЕТЧАТЫЙ ЭЛАСТИЧНЫЙ МАТЕРИАЛ | 1972 |
|
SU423705A1 |
| US 4235892 A, 25.11.80 | |||
| US 4876352 A, 24.10.89. | |||

