Настоящее изобретение относится к способу получения артроподицидных оксадиазинов и промежуточных соединений их синтеза.
В международных заявках WO 92/11249 и 93/19045 раскрываются оксадиазины, обладающие артроподицидной активностью. Однако, существует необходимость улучшения способов получения таких соединений с целью повышения эффективности производства. В соответствии с этим, настоящее изобретение предлагает совершенную технологическую схему получения предпочтительных артроподицидных оксадиазинов.
Краткое изложение сущности изобретения
Данное изобретение относится к способу получения соединения формулы I, которое является рацемическим или энантиомерно обогащенным у хирального центра. соединением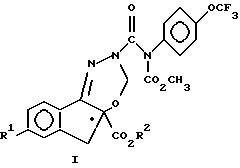
где R1 означает F, Cl или C1-C3-фторалкоксигруппу, а R2 означает C1-C3-алкил, включающий:
(a) взаимодействие соединения формулы II, возможно энантиомерно обогащенного у ассиметрического атома углерода.,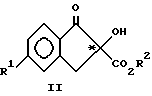
с соединением формулы III, в присутствии кислотного катализатора
H2N-NHR3
с образованием соединения формулы IV: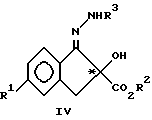
где R3 означает защитную группу, например CO2CH2(C6H5) и тому подобную;
(b) взаимодействие соединения формулы IV с ди(C1-C3-алкокси)-метаном в присутствии кислоты Льюиса с образованием соединения формулы V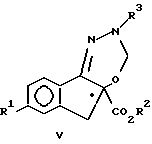
(c) гидрирование указанного соединения формулы V с образованием соединения формулы VI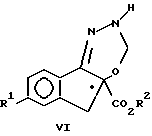
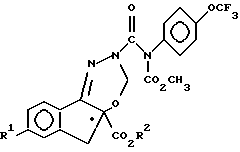
(d) взаимодействие соединения формулы VI с соединением формулы VII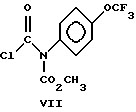
с образованием соединения формулы I, имеющего абсолютную идентичную конфигурацию, что и соединение формулы II.
Настоящее изобретение также включает в свой объем способ получения соединения формулы I, энантиомерно обогащенного у хирального центра . , включающий стадии a - d, отличающийся тем, что соединение формулы II, образованное из стадии a, является энантиомерно обогащенным у ассиметрического атома углерода* соединением, имеющим идентичную конфигурацию, что и целевое соединение формулы I.
Настоящее изобретение также относится к способу получения формулы I, энантиомерно обогащенного у хирального центра ., содержащему стадии a - d, и включающему дополнительные стадии:
(i) взаимодействия пара-замещенного галоидангидрида фениуксусной кислоты с этиленом в присутствии кислоты Льюиса с образованием соединений формулы VIII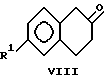
(ii) взаимодействия соединения формулы VIII с любой надкислотой с образованием соединений формулы IX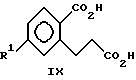
(iii) взаимодействия соединения формулы IX с C1-C3-спиртом в присутствии кислотного катализатора с образованием соединений формулы X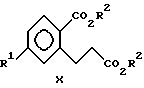
(iv) взаимодействия соединения формулы X с основанием с образованием соединений формулы XI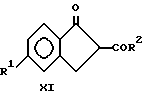
(v) взаимодействия соединения формулы XI с гидроперекисью в присутствии хирального основания с образованием энантиомерно обогащенного производного формулы II, отличающийся тем, что энантиомерно обогащенное соединений формулы II, полученное на стадии v, используют в стадии a, и в котором R1 и R2 имеют вышеуказанные значения.
Настоящее изобретение включает также в свой объем индивидуальные стадии a, b, c и d и многостадийные способы a, b; a, b, c; b, c; b, c, d и c, d синтеза предлагаемых соединений.
Настоящее изобретение включает также в свой объем одностадийный способ получения энантиомеров формулы II из соединений формулы XI; 5-стадийный (i-v) способ получения соединений формулы II; 4-стадийный (i-iv) способ получения соединений формулы XI из пара-замещенного галоидного фенилацетила; 2-стадийный (i-ii) способ получения соединений формулы IX; одностадийный (ii) способ получения соединений формулы IX; и двухстадийный (ii-iii) способ получения соединений формулы X.
Настоящее изобретение также включает в свой объем (+) энантиомеры соединений формулы II: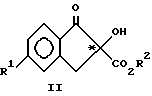
где R1 выбирают из группы, состоящей из F, Cl и C1-C3-фторалкоксигруппы, а R2 означает C1-C3-алкил, являющиеся чистыми (+) энантиомерами.
Настоящее изобретение также включает в свой объем рацемические и энантиомерно обогащенные соединения формул IV, V и VI: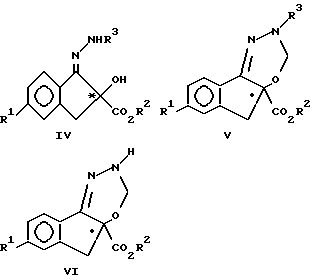
где R1 означает F, Cl и C1-C3-фторалкоксигруппу, R2 означает C1-C3-алкил, а R3 означает группу CO2CH2(C6H5).
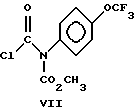
Настоящее изобретение также включает в свой объем соединение формулы VII.
Настоящее изобретение также включает в свой объем соединения формул IX и X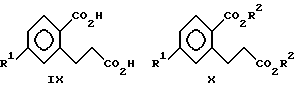
где R1 выбирают из группы, состоящей из F, Cl и C1-C3-фторалкоксигруппы, а R2 означает C1-C3-алкил.
В вышеуказанных определениях термин "галогенид" означает фторид, хлорид, бромид или иодид. Термин "C1-C3-алкил" означает алкил с нормальной или разветвленной углеродной цепью, например метил, этил, н-пропил или изопропил. Термин "C1-C3-алкоксигруппа" означает метокси, этокси, н-пропокси или изопропокси. Термин "C1-C3-фторалкоксигруппа" означает метокси, этокси, н-пропокси или изопропокси, частично или полностью замещенные атомами фтора и в качестве которых можно привести, например, CF3O или CF3CH2O. Термин "C1-C3-спирт" означает метиловый, этиловый, н-пропиловый или изопропиловый спирт.
К предпочтительным соединениям формулы IV, V VI относятся соединения, в которых R2 означает метил, а R1 означает хлор, CF3O или CF3CH2O. Наибольшее предпочтение отдают фенилметил[5-хлор-2,3-дигидро-2-гидрокси-2-(метоксикарбонил)- 1H-инден-1-илиден]гидразинкарбоксилату (обозначенному как соединение IVa);
4a-метил-2-(фенилметил)-7-хлориндено[1,2-e] [1,3,4]-оксадиазин- 2,4a(3H, 5H)дикарбоксилату (обозначенному как соединение Va) и
метил-7-хлор-2,5-дигидроиндено[1,2-e][1,3,4]-оксадиазин- 4a(3H)карбоксилату (обозначенному как соединение VIa).
К предпочтительным соединениям формул II, IX и X относятся соединения, где R2 означает метил, а R1 означает хлор, бром, CF3O или CF3CH2O. Наибольшее предпочтение отдают (+)-форме метил-5-хлор-1,3-дигидро-2-гидрокси-2- оксо-2H-инден-2-карбоксилата (обозначенному как (+)-изомер IIa);
2-карбокси-5-хлорбензолпропановой кислоте (обозначенной как соединение IXa); и
метил-5-хлор-2-(метоксикарбонил)бензолпропаноату (обозначенному как соединение Xa).
Подробное описание сущности изобретения
Один вариант осуществления настоящего изобретения относится к способу получения соединений формулы I, содержащей стадии a - d, который обычно проводят следующим образом.
На стадии a) образуется соединение формулы IV при взаимодействии соединения формулы II (полученного из замещенного инданона, например, 5-хлор-1-инданона по методике, описанной в заявке WO 9211249), с примерно одним моль-эквивалентом соединения формулы III в присутствии кислотного катализатора, например, пара-толуолсульфокислоты, серной или уксусной кислоты возможно в инертном растворителе, например, метаноле, изопропаноле, тетрагидрофуране, дихлорметане и 1,2-дихлорэтане. Указанные реакции обычно проводят при температурах в диапазоне от примерно 40oC до 120oC, предпочтительно от 65o до 85oC в течение примерно от 0.5 часа до 25 часов. Соединение IV можно отделить общепринятыми методами, например фильтрованием, возможно после разбавления реакционной смеси водой. В альтернативном варианте соединение IV можно экстрагировать растворителем и экстракт использовать непосредственно на следующей стадии синтеза без выделения указанного соединения.
На стадии b) получают соединение формулы V путем взаимодействия IV с ди(C1-C3-алкокси)метаном, например диметоксиметаном или диэтоксиметаном в присутствии кислоты Льюиса, при желании в инертном растворителе, например, как дихлорметан, 1,2-дихлорэтан, хлорбензол, α,α,α -трифтортолуол и тому подобные. Ди(C1-C3-алкокси)метан можно использовать в молярном избытке. Кислоты Льюиса включают в свой состав P2O5, BF3, SO3, которые для достижения наилучшего эффекта обычно целесообразно использовать в количестве от 0.9 до 4.0 моль-эквивалентов (относительно соединения V); причем указанные кислоты также включают в свой состав трифторметансульфонаты металлов (предпочтительно скандия, иттербия, иттрия и цинка), которые можно использовать примерно от 0.1 до 0.5 моль-эквивалентов относительно соединения V. К наиболее предпочтительным льюисовским кислотам, которые используют на этой стадии синтеза, относятся P2O5 и SO3; SO3 может быть в форме комплексного соединения, например DMF • SO3 (DMF - диметилформамид). Указанные реакции обычно проводят при температурах в диапазоне от примерно 20oC до 150oC, предпочтительно от 50o до 60oC и давлении от примерно 100 до 700 кПа, предпочтительно от 100 до 300 кПа в течение примерно от 0.5 часа до 48 часов. Побочный продукт C1-C3-спирта целесообразно непрерывно удалять в течение проведения реакции путем перегонки в тех случаях, когда используют "не-жертвенную" льюисовскую кислоту, например, трифторметансульфонат щелочноземельного металла. Соединение V можно отделить общепринятыми методами, например фильтрацией, и использовать его на следующей стадии без дополнительной очистки. В альтернативном варианте, в случае, когда трифторметансульфонаты металла используют в качестве льюисовской кислоты, соединение V можно выделить путем упаривания реакционной массы с последующим необязательным разведением полученного остатка инертным несмешивающимся с водой растворителем, например этилацетатом, промыванием водой для удаления трифторметансульфонатов металла, упариванием органической фазы и выкристаллизовыванием соединения V из остатка, прибавляя при желании к нему подходящий растворитель, например водный раствор метанола, гексана и тому подобное.
На стадии c) образуется соединение формулы VI путем реакции соединения V с водородом, поступаемого из H2-источника водорода, или предпочтительно с самим молекулярным водородом, в присутствии катализатора гидрогенолиза на основе металла, например палладия, предпочтительно на носителе, например, на угле, в любом инертном растворителе, например, метилацетате, этилацетате, толуоле, диэтоксиметане или C1-C3-спирте. Указанные реакции обычно проводят при температурах в диапазоне от примерно 0oC до 30oC, предпочтительно при температуре около 20oC и давлении от примерно 105 до 140 кПа, предпочтительно при давлении примерно 35 кПа в течение примерно 3 часов. Соединение VI можно выделить общепринятыми методами, например путем промывания реакционной смеси разведенной в воде кислотой или водным раствором хлористого натрия с последующим сгущением органической фазы с отгонкой растворителя и выкристаллизовыванием из остатка соединения VI, прибавляя при желании к нему подходящий растворитель, например водный C1-C3-спирт, ацетонитрил или алифатический углеводород, например, гексан. В предпочтительном варианте соединение VI используют на следующей стадии синтеза без его выделения из раствора в органической фазе.
На стадии d) образуется соединение формулы IV путем реакции соединения формулы VI с примерно одним моль-эквивалентом соединения формулы VII возможно в присутствии примерно 1.0-1.5 моль-эквивалентов акцептора кислоты, например, триалкиламина, пиридина или, предпочтительно водного раствора карбоната или бикарбоната натрия в инертном растворителе, например, толуоле, ксилоле, метилацетате, этилацетате, дихлорметане, 1,2-дихлорэтане, диэтоксиэтане и тому подобного. Указанные реакции обычно проводят при температурах в диапазоне от примерно 0oC до 30oC в течение примере от 0.2 часа до 2 часов. Соединение I можно выделить общепринятыми методами, например путем промывания реакционной смеси разведенной в воде кислотой или водным раствором хлористого натрия с последующим сгущением органической фазы и выкристаллизовыванием из остатка соединения I, можно с прибавлением, например C1-C3-спирта, спирто-водных смесей или алифатического углеводорода, например, гексана. Стадии c и d можно объединить в одноемкостный реакционный процесс путем прибавления соединения VII и возможно акцептора кислоты во время гидрогенолиза соединения V. При этом, соединение VII ацилируется сразу же после его образования с выходом соединения I. В качестве растворителей для объединенных стадий синтеза a и d можно использовать метилацетат, этилацетат, толуол, ксилол, дихлорметан, 1,2-дихлорэтан и тому подобные. В качестве акцепторов кислоты можно использовать триалкиламин, например, трипропиламин, трибутиламин, триизопропилэтиламин и тому подобные или неорганическое соединение в виде твердого вещества, например, бикарбонат натрия, оксид кальция, пирофосфат натрия, тринатриевую соль лимонной кислоты и тому подобные.
Стадии a - d проводимых реакций протекают, главным образом, с сохранением устойчивой конфигурации у хирального центра.. В предпочтительном варианте, соединение формулы II, используемое на стадии a, образуется в энантиоморфно обогащенной форме, что позволяет получить энантиоморфно обогащенное соединение формулы I, которое имеет такую же абсолютную конфигурацию, что и соединение II. Под определением "энантиоморфно обогащенный" подразумевается то, что образец указанного соединения в сыпучем состоянии имеет избыток либо (+)-, либо (-)-энантиомера и содержит в несколько большей пропорции, чем 1 : 1 (рацемическую) смесь указанных оптических антиподов, составляющих вплоть до 100%, и, в том числе, 100% чистого энантиомера. Так, например, обогащенное соединение, имеющее 25% (-)-энантиомера изомера и 75% (+)-энантиомера, можно рассматривать как смесь, состоящую из 50% рацемата и 50% чистого (+)-энантиомера и обозначить как соединение, содержащее 50% энантиоморфный избыток (+)-энантиомера. В особенно предпочтительном варианте осуществления настоящего изобретения, соединение формулы II обогащено (+)-энантиомером, что приводит к образованию соединения формулы I, обогащенного (+)-энантиомером, причем установлено, что указанная (+)-форма является более артропоцидно активным соединением. Предпочтительно, чтобы обогащение соединение формулы II (+)-энантиомером составляло по меньшей мере 10% и особенно предпочтительно по меньшей мере 20%.
Энантиоморфно обогащенные соединения формулы II можно получить, например, путем физического разделения оптических изомеров из рацемической смеси в соответствии с общепринятыми методами. Однако, такие методы разделения труднореализуемы в широком масштабе и часто приводят к непроизвольным затратам времени из-за необходимости удаления нежелательного энантиомера. В предпочтительном варианте осуществления настоящего изобретения, энантиоморфно обогащенное соединение формулы II получают энантиоселективным реакционным процессом, включающим 5 стадий, i - v. Под термином "энантиоселективный" подразумевается то, что в ходе реакции образуется преимущественно, хотя вовсе не обязательно, требуемый энантиомер из хирального продукта. Стадии синтеза i-v обычно проводят по следующей методике.
На стадии i) образуется соединение формулы VIII путем взаимодействия соответствующим образом замещенным галоген-ангидридом фенилуксусной кислоты, который можно приобрести в продаже (например у фирмы Spectrum Chemical Manufacturing Co.) или получить из указанных кислот общеизвестными способами, и при желании, образовать на месте, с примерно 1 - 4 моль-эквивалентами, предпочтительно 2 моль-эквивалентами этилена и примерно 0.9 - 1.5 моль-эквивалентами кислоты Льюиса, например хлориде алюминия в примерно 3 - 10 мас. частях инертного растворителя, например, дихлорметане, дихлорэтане, сероуглероде или орто-дихлорбензоле. Указанные реакции обычно проводят при температурах в диапазоне от примерно -20oC до +30oC, предпочтительно от -5o до 0oC и давлении в диапазоне от примерно 60 до 400 кПа в течение примерно от 0.5 до 8 часов. Соединение VIII можно выделить общепринятыми методами, или в случае использования растворителя, например, дихлорметана или дихлорэтана, реакционную смесь можно использовать в следующей стадии синтеза без выделения соединения формулы VIII. В предпочтительном варианте, реакционную смесь из стадии i используют на стадии ii без выделения соединения VIII.
На стадии ii) образуется соединение формулы IX путем взаимодействия соединения VIII с примерно 2.5 - 3.5 эквивалентами надкарбоновой кислоты, предпочтительно надуксусной кислоты, в инертном растворителе, например, уксусной кислоте, дихлорметане, о-дихлорбензоле или 1,2-дихлорэтане. Типичные условия включают проведение реакции при температурах в диапазоне от примерно 15oC до 55oC, предпочтительно от 25 до 45oC в течение промежутка времени примерно от 5 - 35 часов. Температуру поддерживают при низких значениях из-за соображений безопасности. Предпочтительно, однако вовсе не обязательно проведение реакции в присутствии 0.5 - 2.5 моль-эквивалентов реагента, обладающего буферным действием, например ацетата натрия. Скорость подачи надкарбоновой кислоты в раствор соединения VIII находится под контролем для предотвращения накопления избытка надкарбоновой. Целевой продукт можно выделить, например, путем гашения реакции водой с необязательным прибавлением к реакционной массе восстановителя диоксида серы для удаления избытка оксиданта и последующим фильтрованием. При необходимости, pH реакционной массы можно довести до значения > 3 перед отфильтровыванием полученного продукта.
На стадии iii) образуется соединение X этерификацией соединение IX в соответствии с общепринятыми методами. В предпочтительном варианте осуществления изобретения соединение IX реагирует со спиртовым растворителем (взятом в количестве примерно 2 до 20 мас. частей) в присутствии от 1 до 20 моль-эквивалентов существующего карбонатного производного спирта в качестве осушителя и примерно от 0.001 до 0.2 моль-эквивалентов кислотного катализатора, например, серной или пара-толуолсульфокислоты. Типичные условия проведения реакции включают температуры в диапазоне примерно от 75oC до 105oC, и давление в интервале примерно от 100 до 500 кПа в течение промежутка времени примерно от 10 до 30 часов. Соединение X можно выделить общепринятыми методами. В альтернативном варианте, реакционную смесь можно использовать на следующей стадии синтеза без выделения соединения X. В предпочтительном варианте, соединение X, перед проведением стадии iv, не выделяют из реакционной массы.
На стадии iv) образуется соединение XI путем взаимодействия соединения X с сильным основанием, например окисью или гидридом щелочных металлов в подходящем растворителе, например, соответствующем спирте, в частности бензоле, толуоле или смеси ксилола. Типичные условия проведения реакции включают температуры в диапазоне примерно от 60oC до 90oC, и давление в интервале примерно от 100 до 500 кПа в течение промежутка времени примерно от 0.5 часа до 10 часов. Полученный продукт можно извлечь в виде соли щелочного металла и отделить, например, фильтрованием. В альтернативном варианте, полученный продукт можно вначале нейтрализовать добавлением кислоты, например ледяной уксусной кислоты или разбавить водным раствором минеральной кислоты; после этого продукт выделяют, например, фильтрованием или экстракцией.
На стадии v) образуется энантиоморфно обогащенное соединение II при взаимодействии соединения XI с примерно 0.9 - 1.5 эквивалентами гидропероксида, например, перекиси водорода и простого моноэфира перекиси водорода в присутствии примерно от 0.001 до 1.5 эквивалентов оптически-активного аминового основания и возможно в присутствии инертного растворителя. К предпочтительным моноэфирам перекиси водорода относятся трет-бутилгидропероксид, гидроперекись кумола и различные их комбинации. В качестве растворителей можно использовать алифатические углеводороды, например, циклогексан, ароматические углеводороды, например, толуол, ксилолы, этилбензолы, мезитилол и кумол, галогенуглеводороды, например, дихлорметан, дихлорэтан, и ортодихлорбензол, кетоны, например, метилэтилкетон, метилизобутилкетон и метилизопропилкетон, сложные эфиры, например, метилацетат, сложные эфиры, этилацетат, изопропилацетат и простые эфиры, например диэтиловый эфир и тетрагидрофуран. Предпочтительно использование растворителей на основе ароматических углеводородов. Типичные условия включают проведение реакции при температурах в диапазоне от примерно -5oC до 50oC в течение промежутка времени примерно от 2 часов до 8 дней. В качестве аминооснования предпочтительно использование цинхонового алкалоида или его производного. В предпочтительном варианте для получения соединения II, обогащенного (+)-энантиомером (обозначенного как (+)-изомер II), в качестве цинхонового алкалоида используют цинхонин, хинидин, соответствующие гидропроизводные цинхонина или хинидина, или любая комбинация вышеуказанных соединений. Обогащенные (-)-энантиомером соединения формулы II получают при использовании оснований, например, цинхонидина, хинина и их производных, которые имеют 8-(S),9-(R)-конфигурацию. Требуемый продукт можно разделить общепринятыми методами, например фильтрацией, с последующим разбавлением, при желании, либо достаточным количеством водного раствора кислоты для удаления катализатора, либо неполярным растворителем, например как гексаны. В альтернативном варианте смесь полученных продуктов можно разбавить полярным, несмешивающимся с водой растворителем, например, этилацетатом, которую затем промывают водным раствором кислоты для удаления катализатора, упаривают и кристаллизуют. Соединение II можно, при желании, растереть в порошок и перекристаллизовать в подходящем растворителе, например, изопропилацетате для выделения чистого оптически-активного изомера из обогащенной энантиоморфной смеси.
В предпочтительном варианте осуществления изобретения, в качестве растворителя на стадии v используют растворитель, в котором соединение XI обладает значительно большей растворимостью, чем соответствующее соединение формулы II. При использовании таких растворителей, соединение II выпадает в осадок и его можно отделить фильтрованием, а фильтрат, содержащий любой остаток нерастворенного II, непрореагировавшего соединения XI и катализатор, может быть легко рециркулирован в последующей загрузке исходного сырья. В предпочтительном варианте используют также несмешивающийся с водой растворитель для того, чтобы фильтрат перед его использованием в последующей загрузке сырья можно было бы промыть водным раствором основания и/или водой для снижения содержания загрязняющих кислотных примесей и водо-растворимых побочных продуктов. Рециркуляция фильтрата снижает до минимума потерю продукта и обеспечивает более эффективное использование катализатора. Ароматические углеводороды, например ксилолы, особенно предпочтительны для использования в качестве растворителя по этому методу, особенно для получения любого соединения, например как Iia.
Пример 1
Иллюстрация стадий a - d с образованием соединения формулы I.
Стадия A: Образование фенилметил-[5-хлоро-2,3-дигидро-2- гидрокси-2-(метоксикарбонил)-1H-инден-1-илиден]гидразинкарбоксилата (Соединение Iva)
В 1-литровую трехгорловую колбу, оснащенную навесной мешалкой, термометром, парциальным холодильником горячего орошения и отверстием для подачи азота, загружают смесь, содержащую 87 г (0.363 моль) метил-5-хлор-2,3-дигидро-2-гидрокси-1-оксо-1H-инден-2-карбоксилата, 63.5 г (0.380 моль) фенилметилгидразин-карбоксилата (полученного синтезом Ланкастера), 1.8 г (1,01 моль) моногидрата паратолуолсульфокислоты и 300 мл метанола. Полученную суспензию нагревают в колбе с обратным холодильником (67oC) с получением оранжевого раствора, из которого целевой продукт постепенно выпадает в осадок. Через 14 - 16 часов реакционную смесь охлаждают до 5oC и фильтруют. Фильтровальную лепешку промывают 100 мл холодного метанола и сушат при температуре 60oC в вакууме с продувкой азотом в течение 2 часов. В результате получают 135 г (96% выход в расчете на инден-карбоксилат) соединения IV в виде белого кристаллического вещества. Аналитический образец получают перекристаллизацией из ацетонитрила, т. пл. 187 - 188oC; 1H ЯМР (CDCl3): δ 3.23 (д, 1H, J = 18 Гц), 3.48 (д, 1H, J = 18 Гц), 3.7 (с, 3H), 4.58 (ш.с, 1H), 5.19 (ш AB кв., 2H), 7.18 (д, 1H), 7.25 (д.д, 1H), 7.45 (м, 5H), 7.75 (ш. д, 1H), 9.55 (ш. с, 1H). Продукт кажется исключительно близким к Z-(син)-изомеру.
Стадия B: Образование 4a-метил-2-(фенилметил)-7- хлориндено[1,2-e] [1,3,4] -оксадиазин-2,4a(3H,5H)-дикарбоксилату (обозначенному как соединение Va)
В сухую 1-литровую трехгорловую колбу, оснащенную навесной мешалкой, термометром, парциальным холодильником горячего орошения и отверстием для подачи азота загружают смесь, содержащую 42 г диатомовой земли, 500 мл 1,2-дихлорметоксиметана. Затем прибавляют фосфорный ангидрид (42 г, 0.31 моль) в атмосфере азота при охлаждении (баня при 20oC). Полученную смесь перемешивают в течение 15 мин при температуре от 20o до 25oC перед прибавлением к ней порциями 97 г (0.25 моль) соединения IVa. Полученную смесь нагревают до температуры 55o - 60oC в течение 2 часов и затем фильтруют. Фильтровальную лепешку промывают двумя 100 мл порциями 1,2-дихлорэтана и объединенный фильтрат сгущают перегонкой до объема 150 мл. Затем pH реакционной смеси повышают примерно с 1.5 до 4 путем прибавления около 5 г NaOAc в 300 мл метанола, и остаток дихлорэтана удаляют отгонкой примено 150 мл растворителя. После этого к остатку прибавляют окло 30 мл воды, и смесь охлаждают до 5oC и фильтруют. Отфильтрованный продукт промывают 100 мл холодного метанола и сушат на фильтре путем отсоса в течение ночи, получают 89 г (89% выход в расчете на IVa) соединения Va. Аналитический образец получают перекристаллизацией из изопропанола, т. пл. 122 - 124oC; 1H ЯМР (CDCl3): δ 3.16 (д, 1H, J = 16 Гц), 3.42 (д, 1H, J = 16 Гц), 3.64 (с, 3H), 5.12 (д, 1H, J = 10 Гц), 5.26 (AB кв., 2H, J = 12 Гц), 5.53 (шир. д, 1H, J = 10 Гц), 7.2 - 7.45 (м, 7H), 7.65 (д, 1H, J = 9 Гц).
Стадия C: Образование метил-7-хлор-2,5-дигидроиндено[1,2-e][1,3,4] -оксадиазин-4a(3H)-дикарбоксилата (Соединение Via).
1-литровую трехгорловую колбу, оснащенную магнитной мешалкой, впускным клапаном для ввода газа с 3-ходовым запорным краном продувают азотом и загружают смесь, содержащую 27,3 г (0.13 моль) моногидрата лимонной кислоты, 100 мл воды, 10.4 (0.13 моль) 50% водного NaOH, 0.6 г 5% палладиевой черни, 500 мл метилацетата и 52.0 г (0.13 моль) соединения Va. Реакционный сосуд продувают азотом, и смесь интенсивно перемешивают примерно в течение 3 часов при температуре 5o - 10oC и 300 мл с одновременным прохождением в подповерхностном слое потока H2. Протекание реакции контролируют ВЖЭХ на отсутствие соединения Va; при завершении реакции (примерно через 4 часа), реакционный сосуд продувают азотом и палладиевую чернь отфильтровывают на подушку из диатомовой земли и промывают 50 мл метилацетата и 20 мл воды. Фильтрат разделяют и органический слой, содержащий соединение VIa, используют непосредственно на следующей стадии синтеза. При раздельной нагрузке исходных материалов вышеуказанную методику стадии c повторяют, и соединение VIa выделяют отгонкой примерно 400 мл растворителя, прибавлением 100 мл смеси гексана с последующим фильтрованием и сушкой путем отсоса перекристаллизованного продукта, т. пл. 124 - 127oC; 1H ЯМР (CDCl3): δ 3.18 (д, 1H, J = 17 Гц), 3.40 (д, 1H, J = 17 Гц), 3.65 (д, 3H), 4.43 (д, 1H, J = 7 Гц), 4.79 (д, 1H, J = 7 Гц), 6.10 (шир. с, 1H), 7.25 (м, 2H), 7.54 (д, 1H, J = 8 Гц).
Стадия D: Образование метил-7-хлор-2,5-дигидро-2- [[(метоксикарбонил)-4-(трифторметокси)фенил] амино] карбонил]- индено[1,2-e][1,3,4]-оксадиазин-4a(3H)-дикарбоксилата (Соединение Ia).
К органической фазе из стадии c, содержащей соединение VIa, прибавляют водный насыщенный раствор NaHCO3 (140 г, 0.15 моль), а затем 41 г (0.14 моль) метил-(хлоркарбонил)[4-(трифторметокси)- фенил]карбамата (Соединение VII), и полученную смесь перемешивают примерно в течение 1 часа при температуре 10o - 15oC. Органическую фазу отделяют, сушат (MgSO4), упаривают в вакууме для удаления примерно 400 мл метилацетата, и остаток растворителя подвергают реакции обмена путем перегонки с 300 мл метанола до тех пор, пока температура головной фракции не достигнет 64oC. Реакционную смесь охлаждают до 5oC, и полученный продукт отфильтровывают, промывают 70 мл холодного метанола и после сушки путем отсоса получают 58 г соединения Ia (85% суммарный выход в расчете на соединение Va из Стадии c, т. пл. 139 - 141oC; 1H ЯМР (CDCl3): δ 3.25 (д, 1H, J = 16 Гц), 3.48 (д, 1H, J = 16 Гц), 3.70 (с, 3H), 3.71 (с, 3H), 5.29 (д, 1H, J = 10 Гц), 5.69 (д, 1H, J = 10 Гц), 7.2 - 7,4 (м, 6H), 7.50 (д, 1H, J = 8 Гц).
Пример 2
Иллюстрация стадий i - v с образованием соединения формулы II.
Стадия i: Образование 6-хлор-3,4-дигидро-2(1H)-нафталина (Соединение VIII)
В колбу загружают смесь, содержащую 34 г (0.20 моль) 4-хлорфенилуксусной кислоты (PCPA) и 150 мл 1,2-дихлорметана. Полученную суспензию перемешивают, прибавляют затем 25 г (0.21 моль) тионилхлорида, и полученный раствор нагревают при температуре 80o - 90oC в течение 2 - 3 часов. Затем прикрепляют дистилляционную насадку, и отгоняют 25 мл растворителя для удаления остаточного SO2 и HCl. Бледно-оранжевый раствор хлорангидрида охлаждают до 5oC, прибавляют алюминия хлорид (30 г, 0.22 моль) при температуре от -5oC до 0oC, и дистиллятор заменяют баллоном. Этилен (12 г, 0.43 моль) порциями загружают в баллон, при этом поддерживая температуру от -5o до 0oC. Полученный красный раствор переносят постепенно с помощью канюли в 200 мл 5oC воды с такой скоростью, чтобы температура закалки удерживалась в диапазоне 20o - 30oC. Затем смесь перемешивают в течение 1 часа при 25oC, нижний органический слой, содержащий соединение VIIIa, отделяют и промывают 100 мл 5% водного раствора HCl.
Стадия ii: Образование 2-карбокси-5-хлорбензолпропановой кислоты (Соединение Ixa)
Раствор, содержащий соединение VIIIa, образованное на предыдущей стадии, выливают в сосуд, оснащенный навесной мешалкой. Затем загружают ацетат натрия (? г, 0.20 моль), и полученную смесь перемешивают при температуре 25o - 30oC при охлаждении с одновременным прибавлением к указанной смеси непрерывно 114 г (0.60 моль) 32% перуксусной кислоты через капельную воронку в течение 3 - 4 часов. Затем реакционную смесь перемешивают еще 20 минут при температуре 25oC, после чего прибавляют 300 мл 0.8 н HCl, и полученную суспензию охлаждают до 5oC. Реакционную смесь фильтруют, прибавляют последовательно холодным 5% водным раствором NaHSO3, водой, остаток сушат путем отсоса, а затем в течение ночи в вакуумной сушилке при температуре 50oC и пониженном давлении с выходом 35 - 36 г (76 - 78% выход в расчете на PCPA) соединения IXa 99% чистоты в виде белого кристаллического вещества, т. пл. 156 - 158oC.
Стадия iii: Образование метил-5-хлор-2-(метоксикарбонил) бензол-пропаноата (Соединение Xa).
В колбу, оснащенную терморегулятором и навесной мешалкой, загружают смесь, содержащую 45.7 г (0.200 моль) соединения IXa, 5 мл метанола и 100 мл диметилкарбоната. Затем прибавляют 1 г серной кислоты, и полученную смесь перемешивают в атмосфере азота при температуре 85oC в течение 20 часов. Кислоту нейтрализуют путем добавления 3 г 25% раствора метилата натрия и из реакционной колбы перегоняют весь объем диметилкарбоната (DMC). Во время перегонки добавляют метанол (100 - 200 мл) с образованием изотропной смеси метанола и диметилкарбоната (62oC) для облегчения удаления DMC, который в противном случае образует дистиллят при 90oC. Полученный на этой стадии продукт используют в следующей стадии без выделения.
Стадия iv: Образование метил-5-хлор-1-оксо-2,3-дигидроинден- 2-карбоксилата (Соединение Xia).
После удаления большей части диметилкарбоксилата, к метанольному раствору, содержащему Xa, образованному на предыдущей стадии, прибавляют 150 мл метанола, а затем 47.5 г (0.22 моль) 25% NaOMe в метаноле. Реакционный раствор поддерживают при температуре 70oC, и метанол отгоняют до минимального уровня, необходимого для эффективного перемешивания смеси. После завершения реакции, реакционную массу охлаждают до температуры окружающей среды. Затем прибавляют уксусную кислоту (3 г, 0.05 моль), и к полученной смеси добавляют достаточное количество 1 н HCl для доведения pH до 5 - 6. Смесь охлаждают до 5oC, фильтруют и неочищенный твердый продукт промывают водой, затем смесью холодного гексана с выходом 40 - 42 г (89 - 93% выход) соединения XIa в виде твердого продукта, т. пл. 80 - 82oC.
Стадия v: Образование (+)-изомера метил-5-хлор-1,3-дигидро-2- гидрокси-1-оксо-2H-инден-2-карбоксилата (Соединение (+)IIa)
Смесь, содержащую 10.0 г соединения XIa, 17 мл (51 ммоль) 3.0М трет-бутилгидроперекиси в изооктане, 70 мл изопропилацетата и 0.2 г цинхонина (продукт фирмы Aldrich® Chemical Co.), перемешивают при температуре окружающей среды в течение 6 дней. К смеси затем прибавляют около 100 мл этилацетата, 30 мл разбавленного водой бисульфита натрия и 20 мл 2н соляной кислоты. Смесь перемешивают встряхиванием и разделяют. Органический экстракт промывают последовательно водой и солевым раствором. Растворитель отгоняют в вакууме, и полученный неочищенный твердый продукт промывают гексаном. Получают 7.31 г соединения IIa (68% выход) с соотношением энантиомеров (+)-формы 72% и (-)-формы 28%, определенного хиральной ВЖЭХ. (+)-формы соединения IIa перекристаллизовывают из изопропилацетата с выходом 5 г чистого (+)-энантиомера IIa, т. пл. 163 - 165oC; [α]
Пример 3
Иллюстрация альтернативного варианта осуществления стадий a - d, начиная с энантиоморфно обогащенного соединения IIa и кончая образованием энантиоморфно обогащенного соединения Ia.
Стадия a: Образование (+)-изомера IVa
В 1-литровую одногорловую колбу, оснащенную прибором Дина-Старка и отверстием для подачи азота, загружают смесь, содержащую 75 г (0.312 моль) (+)-изомера IIa (50% энантиоморфный избыток), 54.6 г (0.358 моль) фенилметилгидразинкарбоксилата, 1.78 г (0.0094 моль) моногидрата пара-толуолсульфокислоты ( Aldrich® Chemical Company) и 275 мл 1,2-дихлорэтана. Полученную суспензию нагревают с обратным холодильником с получением оранжевого раствора, из которого постепенно выпадает в осадок целевой продукт. Водную фазу собирают в лоток прибора Дина-Старка. Через 2 часа реакционную смесь охлаждают до комнатной температуры. Реакционную массу используют непосредственно в следующей стадии b.
Стадия b: Образование (+)-изомера Va
В 2-литровую трехгорловую колбу, оснащенную навесной мешалкой, термометром, парциальным холодильником горячего орошения и отверстием для подачи азота, загружают 88.5 г диатомовой земли ( Celite® ) и 300 мл 1,2-дихлорэтана. Затем прибавляют к содержимому фосфорный ангидрид (88.5 г 0.623 моль) и 120 мл диметоксиметана. Прибавляют суспензию (+)-изомера IV в 1,2-дихлорэтане из стадии a. Полученную смесь нагревают до температуры 35o - 40oC в течение 5 часов, и затем охлаждают до 30oC и фильтруют. Фильтровальную лепешку промывают 135 мл 1,2-дихлорэтана, и объединенный фильтрат перегоняют до минимального объема. К остатку прибавляют метанол и отгонку продолжают. Когда отогнан весь объем 1,2-дихлорэтана, но в сосуде остается примерно 500 мл метанола, процесс перегонки прекращают, и сосуд охлаждают до 45oC. Когда продукт начинает выпадать в осадок, прибавляют 120 мл воды и смесь охлаждают до 20oC. Смесь фильтруют и фильтровальную лепешку промывают 370 мл смеси метанола и воды, 3 : 1. Полученное твердое вещество сушат в течение ночи при температуре 80oC в вакууме с выходом 100.5 г (80.5% выход за две стадии) (+)-изомера Va. Данные 1H ЯМР-спектра сопоставимы с полученными для соединения Va в примере 1. Чистота составляет 99.3% по данным ВЭЖХ. Анализ образца хиральной ВЭЖХ продукта свидетельствует о 43% энантиоморфном избытке (+)-энантиомера.
Стадия c: Образование (+)-изомера Via
500- мл трехгорловую колбу, оснащенную магнитной мешалкой, термометром, впускным клапаном для отвода газа с 3-ходовым запорным краном, продувают азотом и загружают 50 мл метилацетата, 50 мл 0.5 М натрия бифосфатного буфера (pH 3.5) и 0.2 г 5% палладиевой черни, увлажненного 50% воды. Двухфазную суспензию перемешивают при температуре окружающей среды в течение 0.5 часа. В отдельную колбу к 50 мл метилацетата прибавляют 10 г (0.025 моль) (+)-изомера Va в атмосфере азота, полученную смесь нагревают до температуры 35oC и перемешивают до полного ее растворения. Полученный раствор, содержащий (+)-форму Va, прибавляют к суспензии Pd-катализатора, и полученную смесь охлаждают до 10oC. Из реакционного сосуда сбрасывают давление и смесь интенсивно перемешивают при температуре 10oC с одновременным прохождением в подповерхностном слое потока H2. Протекание реакции контролируют тонкослойной и газовой хроматографией на отсутствие в анализируемой пробе (+)-изомера Va; при завершении реакции (примерно через 1.5 часа), из реакционного сбрасывают давление и продувают азотом; реакционную смесь фильтруют через слой диатомовой земли и остаток на фильтре промывают 20 мл метилацетата. Жидкие фазы отделяют и метилацетатный слой, содержащий (+)-энантиомер VIa, используют непосредственно на следующей стадии синтеза d.
Стадия d: Образование (+)-изомера Ia
Метилацетатный раствор из стадии c, содержащий (+)-энантиомер VI, прибавляют к раствору, содержащему 3 г NaHCO3 в 38 мл воды. Полученную смесь охлаждают до 10oC в атмосфере азота и затем прибавляют одной порцией 7.43 г (0.025 моль) соединения VII. Реакционную смесь перемешивают при температуре 10oC в течение 1 часа. Метилацетатную фазу отделяют и упаривают при пониженном давлении с удалением примерно 100 мл растворителя. К остатку прибавляют 50 мл метанола, и полученную суспензию упаривают для удаления оставшегося метанола в виде азеотропной смеси ацетата и метанола. Прибавляют последнюю 50 мл порцию метанола, и суспензию кипятят с обратным холодильником. Затем к суспензии прибавляют диатомовую землю (0.4 г) в ходе ее кипячения, а затем по каплям добавляют воду. Полученную суспензию охлаждают, фильтруют, осадок промывают 33 мл смеси метанола и воды, 2 : 1 и после вакуумной сушки получают 11.16 г энантиоморфно обогащенного (+)-энантиомера Ia (78% суммарный выход за стадии c и d в расчете на соединение Va). Анализ образца ВЭЖХ свидетельствует о 42% избытке (+)-энантиомера.
Пример 4
Иллюстрация альтернативного варианта осуществления стадий c и d.
Стадия c: Образование соединения Via
1-л трехгорловую колбу, оснащенную магнитной мешалкой, термометром, впускным клапаном для отвода газа с 3-ходовым запорным краном, продувают азотом и загружают 580 мл метилацетата, 0.164 г натрия ацетата (2 мол.%) и 0.8 г 5% палладиевой черни в качестве катализатора. Из реакционной смеси удаляют отгонкой примерно 200 мл растворителя, и полученную суспензию сухого растворителя/катализатора охлаждают до температуры 50oC, и затем прибавляют к ней 40.0 г (0.1 моль) соединения Va в одной порции. Реакционный сосуд продувают азотом, смесь интенсивно перемешивают при температуре окружающей среды пока поток H2 проходит через подповерхностный слой. Реакцию контролируют на отсутствие в анализируемой пробе Va; при завершении реакции (примерно через 3 часа) из реакционного сосуда откачивают газ и продувают азотом; катализатор на основе палладиевой черни отфильтровывают на подушку из диатомовой земли и фильтрат промывают 50 мл метилацетата. Фильтрат используют непосредственно на следующей стадии синтеза d.
Стадия d: Образование соединения Ia
Метилацетатный раствор из стадии c, содержащий соединение VI, прибавляют к раствору, содержащему 12 г NaHCO3 в 150 мл воды. Полученную смесь охлаждают до 10oC в атмосфере азота и затем прибавляют порциями 29.7 г (0.01 ммоль) соединения VII в течение 0.5 часа; смесь затем перемешивают при температуре от 10 до 15oC в течение 1 часа. Метилацетатную фазу отделяют и упаривают в вакууме с удалением примерно 400 мл растворителя. К остатку прибавляют 50 мл метанола, и растворитель опять отгоняют в вакууме. Затем к остатку прибавляют 70% водный раствор метанола (100 г), и смесь перемешивают в течение 45 минут при охлаждении на ледяной бане. Полученный продукт фильтруют, промывают 25 мл охлажденного 70% водного раствора метанола и после вакуумной сушки получают 51 г названного продукта (86% суммарный выход из соединения Va в расчете на 88.9% выход по данным анализа ВЭЖХ), т. пл. 135 - 138oC.
Пример 5
Иллюстрация альтернативного варианта осуществления стадии c.
Стадия v: Образование (+)-изомера Iia)
Суспензию, приготовленную из 11.25 г соединения Va, 70 мл смеси ксилола и 1.4 г (4.8 ммоль) цинхонина ( Aldrich® Chemical Co.), перемешивают в атмосфере азота, и прибавляют 7.0 г (70 ммоль) 90% водного раствора третбутилгидропероксида ( Aldrich® Chemical Co.). Полученный раствор перемешивают при комнатной температуре в течение 24 часов. Реакционную смесь затем разбавляют 100 мл этилацетата, и затем промывают последовательно двумя 50 мл порциями насыщенного водного раствора бикарбоната натрия, 50 мл 1 н соляной кислоты и 50 мл насыщенного водного бисульфита натрия. Органическую фазу сушат (MgSO4), и после отгонки растворителя в вакууме получают 10.6 г обогащенного (+)-энантиомером соединения IIa (86% чистота, 76% выход в расчете на Va). Данные анализа образца методом ВЖЭХ свидетельствуют о 45% энантиоморфном избытке (+)-энантиомера.
Пример 6
Иллюстрация альтернативного варианта осуществления стадии b.
Стадия b: Образование соединения Va
В сухую 500-мл трехгорловую колбу, оснащенную магнитной мешалкой, термометром и двумя впускными отверстиями для подачи газа, загружают 49.9 г (0.128 моль) соединения Va и 250 мл диэтоксиметана. Смесь затем охлаждают до -10oC, и из реакционного сосуда сбрасывают давление (до примерно 24 см рт. столба). Затем в охлажденный реакционный сосуд подают газообразную трехокись серы со скоростью, например, при которой температура реакционной смеси поддерживается в интервале от -10o до 0oC. Сразу же после введения SO3, пропускают азот для ликвидации вакуума. Смесь доводят до комнатной температуры, перемешивают в течение 4.75 часа, прибавляют 50 мл воды при комнатной температуре при быстром перемешивании и перемешивают реакционную массу еще 2 часа. Реакционную смесь фильтруют и органическую фазу отделяют от фильтрата и упаривают. Остаток растворяют в 125 мл метанола и объединяют с осадком, полученным при фильтрации. К полученной суспензии прибавляют по каплям 125 мл воды, и затем смесь перемешивают 1.5 часа и фильтруют. Фильтровальную лепешку сушат в вакууме при комнатной температуре с получением 46.3 г (90% выход в расчете на IVa) соединения Va. Небольшую порцию продукта перекристаллизовывают из метанола с получением аналитического образца, т.пл. и данные 1H ЯМР-спектра сопоставимы с результатами, полученными для соединения Va в примере 1, стадия b.
Пример 7
Получение метил-(хлоркарбонил)[4-(трифторметокси)фенил] карбамата (Соединение VII)
В первом реакционном сосуде растворяют 70.5 г (0.30 моль) метил-4-(трифторметокси)фенил] карбамата в 700 мл дихлорметана. Затем к полученному раствору прибавляют 14.0 г 60% эмульсии гидрида натрия (0.35 моль) в минеральном масле, а затем добавляют 60 мл диглима (диметилового эфира этиленгликоля) в течение 15 мин. Происходит экзотермическая реакция, и температура реакционной смеси становится немного выше температуры окружающей среды (комнатной температуры). Реакционную смесь перемешивают примерно в течение ночи (около 16 часов) без внешнего нагрева. Во втором реакционном сосуде, оснащенном дистилляционной колонкой, растворяют 120 г (1.2 моль) фосгена в 300 мл дихлорметана, который охлаждают до температуры 5o - 10oC. Реакционную массу из первого сосуда, в виде густой суспензии, медленно вводят во второй сосуд с раствором фосгена при температуре 5o - 10o. После завершения введения суспензии, избыток фосгена удаляют путем дистилляции до тех пор, пока по температуре головного погона не будет видно, что только дихлорметан выходит сверху колонны. Перегонку прекращают, и реакционную смесь охлаждают до примерно 0oC. Для растворения побочного продукта, натрия хлорида, к реакционной массе прибавляют 200 мл ледяной воды. Дихлорметановый слой отделяют от водного слоя, фильтруют и сушат (MgSO4). Высушенный дихлорметановый раствор, стадии c, содержащий соединение VII, затем перегоняют для удаления дихлорметана, и при реакции обмена прибавляют гексан 400 мл в общем объеме (методика обмена растворителем). Сразу же после отгонки дихлорметана и начала выхода гексанового дистиллята перегонку прекращают. Гексановый раствор затем охлаждают до 5oC, после чего осаждают соединение VII (возможно будет необходимо использование затравки), и полученный продукт отделяют фильтрованием, промывают дополнительной порцией холодного гексана и сушат. Выход продукта обычно составляет 94% в расчете на 97 - 98% выход чистого соединения VII, т. пл. 97 - 99oC; 1H ЯМР (CDCl3): δ 3.80 (S, 3), 7.29 (S, 4).
Описывается способ получения производных оксадиазинов формулы I, которые являются рацемическими или энантиоморфно обогащенными у хирального центра соединениями, где R1 означает F, Сl или C1-С3-фторалкоксигруппу, R2 означает С1-С3-алкил, соединение формулы VI, в которой R1 и R2 имеют вышеуказанные значения, подвергают взаимодействию с соединением формулы VII. Предложенный способ позволяет получить соединения, обладающие артропозицидной активностью, значительно проще, чем известные способы. 5 с. и 13 з.п. ф-лы.
где R1 означает F, Cl или C1 - C3-фторалкоксигруппу;
R2 означает C1 - C3-алкил,
отличающийся тем, что соединение формулы VI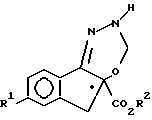
в которой R1 и R2 имеют указанные значения,
подвергают взаимодействию с соединением формулы VII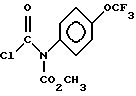
2. Способ по п.1, отличающийся тем, что соединение формулы 1 представляет собой (+) метил-5-хлор-1,3-дигидро-2-гидрокси-1-оксо-2H-инден-2-карбоксилат.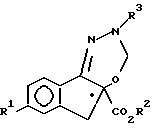
где R3 - защитная группа CO2CH2(C6H5);
R1 и R2 имеют значения, указанные в п.1.
•
где R1, R2 и R3 имеют указанные значения,
с ди(C1 - C3-алкокси)метаном в присутствии кислоты Льюиса.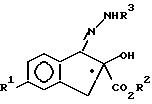
в которой R1 и R2 имеют значения, приведенные в п.1,
с соединением формулы III в присутствии кислотного катализатора формулы
H2N - NHR3
с образованием соединения формулы IV, определенного выше.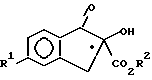
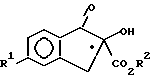
в которой R1 и R2 имеют значения, указанные в п.1,
с соединением формулы III, в присутстчии кислотного катализатора формулы
H2N - NHR3
с образованием соединения формулы IV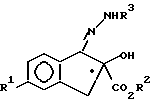
где R3 означает защитную группу CO2CH2(C6H5);
R1 и R2 имеют указанные в п.1 значения;
b) взаимодействие формулы IV с ди(C1 - C3-алкокси)метаном в присутствии кислоты Льюиса с образованием соединения формулы V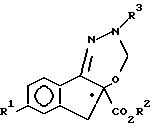
в котором R1, R2 и R3 имеют значения, указанные в п.3
c) гидрирование соединения форпмулы V с образованием соединения формулы VI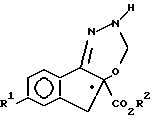
в которой R1 и R2 имеют значения, указанные выше,
d) взаимодействие соединения формулы VI с соединением формулы VII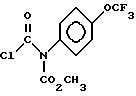
с образованием соединения формулы I, имеющего конфигурацию, абсолютно идентичную конфигурации соединения формулы II.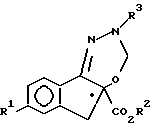
в которой значения R1, R2 и R3 приведены в п.1,
получают путем a) взаимодействия соединения формулы II, возможно энантиомерно обогащенного у асимметрического атома углерода •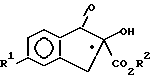
в которой R1 и R2 имеют значения, приведенные в п.1,
с соединением формулы III в присутствии кислотного катализатора
H2N - NHR3
с образованием соединения формулы IV
где R1 означает F, Cl и C1 - C3-фторалкоксигруппу;
R2 означает C1 - C3-алкил;
R3 означает защитную группу CO2CH2(C6H5),
b) взаимодействия соединения формулы IV с ди(C1 - C3-алкокси) метаном в присутствии кислоты Льюиса.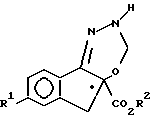
получают гидрированием соединения формулы V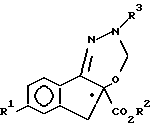
где значения R1, R2 и R3 приведены в п.1.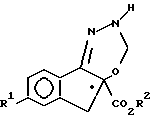
в которой значения R1 и R2 приведены в п.1,
получают взаимодействием соединения формулы IV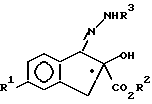
где R1 означает F, Cl и C1 - C3-фторалкоксигруппу;
R2 означает C1 - C3-алкил;
R3 означает защитную группу CO2CH2(C6H5),
с ди(C1 - C3-алкокси) метаном в присутствии кислоты Льюиса с образованием соединения V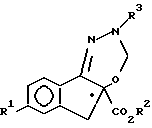
в которой R1, R2 и R3 имеют указанные значения,
с последующим гидрированием соединения формулы V.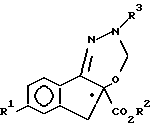
где R3 означает защитную группу CO2CH2(C6H5),
с образованием соединения формулы VI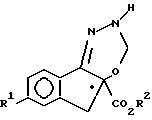
где R1 и R2 имеют указанные значения,
и взаимодействие соединения формулы VI с соединением формулы VII
12. Рацемическое или энантиомерно-обогащенное соединение общей формулы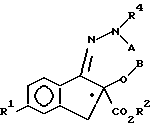
где A - R3 и B - водород, либо A и B, взятые вместе, представляют группу -CH2-, таким образом, образуя оксадиазиновое кольцо;
R1 - фтор, хлор или C1 - C3-фторалкокси;
R2 - C1 - C3-алкил;
R3 - CO2CH2(C6H5);
R4 выбран из H и CO2CH2(C6H5), при условии, что R4 всегда - водород, когда A - R3.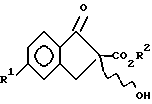
в котором R1 выбирают из группы, состоящей из F, Cl и C1 - C3-фторалкоксигруппы;
R2 означает C1 - C3-алкил,
получают путем i) взаимодействия паразамещенного галогенангидрида фенилуксусной кислоты с этиленом в присутствии кислоты Льюиса с образованием соединений формулы VIII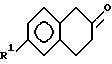
ii) взаимодействия соединения VIII с надкислотой с образованием соединений формулы IX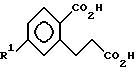
iii) взаимодействия соединения IX с C1 - C3-спиртом в присутствии кислотного катализатора с образованием соединений формулы X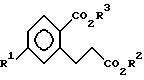
iv) взаимодействия соединения X с основанием с образованием соединений формулы XI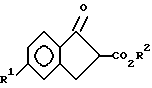
v) взаимодействия соединения IX с гидроперекисью в присутствии хирального основания с образованием энантиомерно обогащенного соединения II.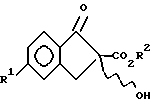
в котором R1 выбирают из группы, состоящей из F, Cl и C1 - C3-фторалкоксигруппы;
R2 означает C1 - C3-алкил.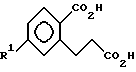
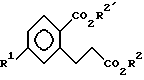
где R1 выбирают из группы, состоящей из F, Cl и C1 - C3-фторалкоксигруппы;
R2 означает H или C1 - C3-алкил.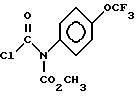
Приоритет по пунктам:
20.04.94 по пп.1 - 18;
31.08.94 уточнение признаков по пп.1 - 18.
| WO 9211249 A1, 1992 | |||
| Способ получения фторсодержащих 1,3,5-4н-оксадиазинов | 1978 |
|
SU743997A1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ДАВЛЕНИЯ МЕТАЛЛА НА ОПРАВКУ ПРИ ПРОДОЛЬНОЙ ПРОКАТКЕ ТРУБ В КАЛИБРЕ МЕСДОЗАМИ | 0 |
|
SU192150A1 |
| WO 9220682 A1, 1992. | |||
