Настоящее изобретение касается нового промежуточного соединения и способа синтеза соединений, которые ингибируют протеазу вируса иммунодефицитного состояния человека (HIV) и, в частности, соединений, раскрытых и упомянутых как "соединение J" в EPO 541168, который опубликован 12 мая 1993 г, или их фармацевтически приемлемых солей.
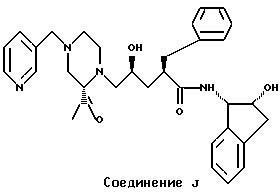
Эти соединения имеют значение при предотвращении инфекции, вызванной HIV, лечении инфекции, вызванной HIV, и лечении синдрома приобретенного иммунодефицита (СПИДа) (AIDS).
Более конкретно, настоящий способ включает получение эпоксидного промежуточного соединения для получения указанного выше соединения "J", являющегося ингибитором протеазы HIV. Способ относится к образованию иодогидрина из аллилацетонида, при этом сначала образуется промежуточное соединение иодоиминолактон. Циклизация иодогидрина, вызванная основанием, приводит затем к получению эпоксидного промежуточного соединения. Образование иодогидрина происходит при высокой диастереоселективности, при этом в этом процессе наблюдается реальное отсутствие гидролиза амидной связи.
Ретровирус, обозначенный как вирус иммунодефицитного состояния человека (HIV), представляет причинный фактор комплекса заболеваний, который включает прогрессирующую деструкцию иммунной системы (синдром приобретенного иммунодефицита: AIDS) и дегенерацию центральной и периферической нервной системы. Этот вирус ранее был известен как LAV, HTLV-III или ARV. Общей особенностью репликации ретровируса является экстенсивная посттрансляционная обработка предшественника полипротеинов посредством вирусно-кодированной протеазы для генерации зрелых вирусных белков, необходимых для скопления вирусов и их функционирования. Ингибирование такой обработки приводит к предотвращению получения обычного инфекционного вируса. Kohl, N.E. et. al., Proc. Nat'l Acad. Sci., 85, 4686(1988) показали, что генетическая инактивация протеазы HIV приводит к получению недоразвитых неинфекционных вирусных частиц. Эти результаты показывают, что ингибирование протеазы HIV представляет жизнеспособный способ лечения AIDS и предотвращения или лечения инфекции, вызванной HIV.
Нуклеотидная последовательность HIV показывает присутствие pol гена в одной открытой рамке для чтения [Ratner, L. et. al.. Nature, 313, 277, (1985)] . Гомология аминокислотной последовательности обеспечивает доказательство того, что pol последовательность кодирует ревертирующую транскриптазу, эндонуклеазу и протеазу HIV. [Toh, Н. et. al., EMBO J., 4, 1267 (1985): Power, M.D. et. al., Science, 231, 1567 (1986); Pearl, L.H. et. al.. Nature, 329, 351 (1987)]. Соединения конечного продукта, включая соединение J, которые могут быть получены из новых промежуточных соединений и способа этого изобретения, представляют ингибиторы протеазы HIV, и раскрыты в EPO 541168, который опубликован 12 мая 1993 г.
Ранее синтез Соединения J и родственных соединений осуществляли посредством 12-стадийного процесса, в котором использовали гидроксизащищенный дигидро-5(S)-гидроксиметил-3-(2H)фуранон, который алкилировали и который включал замену спиртовой отщепляемой группы на алкилированный фуранон с пиперидиновой составляющей. Затем связанный продукт гидролизовали для разрыва фуранонового кольца на оксикислотную составляющую и, в конечном счете, кислоту связывали в 2(R)-гидрокси- 1(S)-аминоиндан. Эта методика описана в EPO 541168. Чрезвычайная продолжительность этого процесса (12 стадий) приводит к большим трудозатратам, и он требует применения многих дорогостоящих реагентов и дорогостоящего исходного материала. Способ, для реализации которого требуется меньшее количество стадий реакции и/или более эффективных реагентов, обеспечит экономическую выгоду и снижение продолжительности процесса.
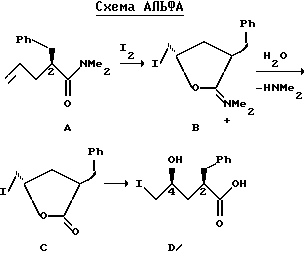
Иодлактамизация олефинового третичного амида А происходит, как известно, с последующим гидролизом заряженного промежуточного соединения иодоиминолактама B, при этом в качестве единственного выделенного продукта получают иодолактон C (Схема АЛbФА). Смотри Tamaru Y. et. al., J.Am.Chem.Soc., 106, 1079 - 1085 (1984): Trost, B.M. et. al., eds. Comprehensive Organic Synthesis: Selectivity, Strategy, & Efficiency in Modern Organic Chemistry, Volume 4, Pergamon Press, New York, 1991, p.398-421. Известно, что в этом процессе происходит очень эффективное перемещение хиральности из 2-положения в 4-положение, при этом получают 2,4-синтетические продукты (представленные соответствующей оксикислотой D) с высокой диастереоселективностью.
В другом существующем способе ацетонид взаимодействует с (S)-глицидилтозилатом в присутствии сильного основания LHMDS с образованием эпоксида (смотри схему БЭТА). Поскольку как исходный материал (S)-глицидилтозилат, так и продукт представляют эпоксиды, анион ацетонида взаимодействует также с полученным эпоксидом, поэтому, кроме полученного эпоксида с выходом 71%, образуется около 20% двойных побочных продуктов присоединения. После кристаллизации из MeOH для обеспечения эпоксида, не содержащего димера, необходима дополнительная перекристаллизация из МТВЕ; в результате общий выход из ацетонида может находиться в диапазоне 56-61%. Образование двойных нуклеофильных продуктов присоединения представляет проблему, свойственную электрофильному глицидилтозилату. (S)-глицидилтозилат является в настоящее время также наиболее дорогим исходным материалом при синтезе Соединения J.

Известно, что реакция энолата лития ацетонидного промежуточного соединения с электрофилами происходит с очень высокой селективностью и при высоком выходе желательных 2(R)-продуктов. Смотри Askin, D. et. al., J. Org. Chem. , 57, 2771-2773 (1992); Askin, D. et. al., Tetrahedron Lett., 35, 673-676 (1994). Известно также, что производные галогенгидрина беспримесно превращаются в желательное эпоксидное промежуточное соединение для Соединения J. Однако не ожидалось, что условия настоящего изобретения приведут к выделению иодогидрина из ацетонидных промежуточных соединений с превосходным выходом, превышающим 70%. Смотри также EPO 541168, EPO 521686 и Tamasa, Y., et. al., J.Am.Chem.Soc., 106, 1079 (1984).
Сущность изобретения
Представлен способ синтеза эпоксида формулы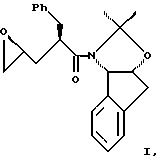
включающий первую стадию образования галогенгидрина из аллилацетонидного реагента, после которой следует стадия циклизации, вызванная основанием. Дополнительная первая стадия включает аллилирование ацетонидного реагента с образованием аллилацетонида. Продукты являются пригодными в качестве промежуточных соединений для синтеза ингибиторов ренина или протеазы HIV или других протеаз.
Защитная группа
BOC(Boc) - трет-бутилоксикарбонил
CBZ(Cbz) - бензилоксикарбонил(карбобензокси)
TBS(TBDMS) - трет-бутил-диметилсилил
Активирующая группа
Ts или тозил или тозилат - p-толуолсульфонил
Ns или нозил или нозилат - 3-нитробензолсулъфонил
Tf или трифлил или трифлат - трифторметансульфонил
Ms или мезил или мезилат - метансульфонил
Реагент сочетания
BOP реагент - бензотриазол-1-илокситрис-(диметиламино)- фосфоний гексафторфосфат
BOP-Cl - бис(2-оксо-3-оксазолидинилфосфинхлорид
EDC - 1-этил-3-(3-диметиламинопропил)карбодиимидгидрохлорид
Другие
BOC-ON - 2-(трет-бутилкарбонилоксиимино)-2-фенилацетонитрил
(BOC)2O(BOC2O или Boc2) - ди-трет-бутилбикарбонат
n-Bu4N+F- - тетрабутиламмонийфторид
n-BuLi(n-Buli) - n-бутиллитий
(S)-CSA - (1S)-(+)-10-камфорсульфоновая кислота
DABCO - диазабициклооктан
DBN - диазабициклононан
DBU - диазабициклоундекан
Dl - деионизованная
DIFA или DIPEA - диизопропилэтиламин
DMA - диметилацетамид
DME - диметоксиэтан
DMF - диметилформамид
DMPU - диметилтетрагидропиримидинон
DMSO - диметилсульфоксид
Et2N - триэтиламин
EtOAc - этилацетат
h - час(ы)
IPA - 2-пропанол
KF - титрование Карла Фишера для воды
LDA - диизопропиламид лития
LHMDS - гексаметилдисилазид лития
L-PGA - (L)-пироглутаминовая кислота
MeCH - ацетонитрил
MTBE - метил-трет-бутиловый эфир
NMP - N-метилпирролидинон
r.t - комнатная температура
TFA - трифторуксусная кислота
TG - термогравиметрия: потери при нагреве
THF - тетрагидрофуран
TLC - тонкослойная хроматография
TMEDA - тетраметилэтилендиамин
TMU - тетраметилмочевина
Подробное описание изобретения
Способ настоящего изобретения поясняется следующей схемой: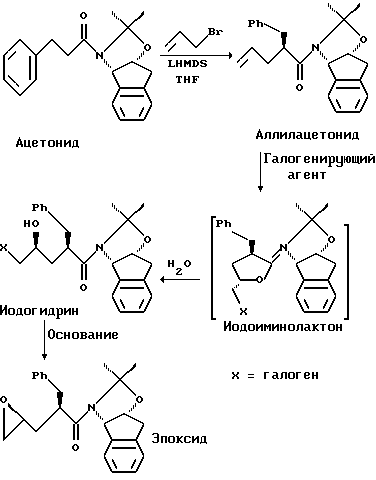
Вышеприведенная схема иллюстрирует три стадии. Сначала происходит аллилирование ацетонида, после которого следует образование галогенгидрина и затем циклизация, вызванная основанием, с образованием полученного эпоксида.
В настоящем изобретении способ синтеза эпоксида формулы I
включает стадии:
(a) контактирования одного эквивалента аллилацетонида формулы II
с галогенирующим агентом в количестве от около одного до двух эквивалентов в растворителе, смешанном с водным раствором слабого основания при температуре в диапазоне между около -40oC и около 100oC с образованием галогенгидрина формулы III, и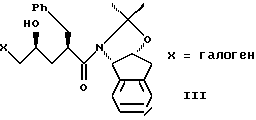
(b) добавления основания в растворителе или растворителях для установления образования эпоксида формулы I.
Настоящий способ синтеза эпоксида формулы I из ацетонидного реагента: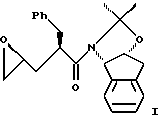
включает стадии:
(a) взаимодействия одного эквивалента ацетонида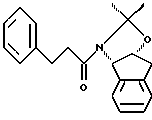
с приблизительно одним эквивалентом аллилгалогенида в сильном основании с получением аллилацетонида формулы II,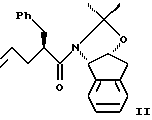
(b) смешивания с ним галогенирующего агента в количестве от около одного до двух эквивалентов в растворителе, смешанном с водным раствором слабого основания при температуре в диапазоне между от около -40oC и около 100oC с образованием галогенгидрина формулы III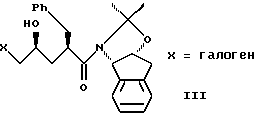
(с) добавления основания в растворителе или растворителях для установления образования эпоксида формулы I.
При получении аллилацетонида схемы ГАММА предпочтительные реагенты аллилирования включают аллилгалогениды, такие как аллилхлорид, аллилбромид или аллилиодид, а также другие аллилэлектрофилы, например, аллилметансульфонат или аллиловые эфиры, в присутствии катализатора переходного металла. Наиболее предпочтительные реагенты аллилирования включают аллилгалогениды, например, аллилхлорид, аллилбромид и аллилиодид.
Для этой реакции аллилирования предпочтительными основаниями являются сильные основания, которые включают амидные основания, например, литиевые, натриевые, калиевые или магниевые соли аминов, например, диэтиламина, диизопропиламина, дициклогексиламина, пиперидина, пирролидина или бистриметилсилиламина; металлалкилы, например C1-6-алкиллитий, такие как n-, изо-, втор, и трет-бутиллитий, метил-, этил- или ариллитий, например фениллитий: реагенты Гриньяра, например, метил-, этил-, пропил- или бутилмагнийгалогенид: алкоксиды, например, метоксид, этоксид, изопропоксид, трет-бутоксид, трет-амилоксидалкоксиды лития, натрия, калия или магния.
В реакции аллилирования наиболее предпочтительным основанием является литийгексаметилдисилазид (LHMDS).
В реакции аллилирования предпочтительные растворители включают эфирные растворители, например, THF, DME, MTBE, диэтиловый эфир, диглим или дибутиловый эфир; углеводородные растворители, например, пентан, гексан, гептан, бензол, толуол или этилбензол; или другие растворители, совместимые с основаниями и металлорганическими реагентами, например, DMSO, DMPU, NMP, TMU, TMEDA и краун-эфиры; и также смеси этих растворителей.
Наиболее предпочтительными растворителями для аллилирования являются эфирные растворители для аллилирования, например, THF, DME и MTBE.
Предпочтительный температурный диапазон для аллилирования составляет от -78oC до +30oC. Инкубационный период продолжается, по крайней мере, 15 минут и обычно до 3-х часов.
Для образования галогенгидрина предпочтительные галогенирующие агенты включают галогены, интергалогенные соединения, галогенидные соли или гипогалитные соли или эфиры, оксигалогеновые соли или кислоты, галогенамиды, галогенмочевины, галогенкарбаматы, галогенсульфонамиды, галогенамины или другие галогенированные азотные соединения или их сочетания или с галогенидными солями, или с межфазными катализаторами, или с теми и другими. Предпочтительные галогенирующие агенты представляют гипогалитные соли или сложные эфиры, галогенамиды, мочевины, карбаматы, сульфонамиды, амины или другие галогенированные азотные соединения, например, N-иодсукцинимид, N-бромсукцинимид с иодидной солью или N-хлорсукцинимид с иодидной солью. Наиболее предпочтительными галогенирующими агентами являются N-иодсукцинимид, N-бромсукцинимид в сочетании с иодидной солью или N-хлорсукцинимид в сочетании с иодидной солью.
Реакционными условиями для образования галогенгидринов являются растворы, суспензии или другие двухфазные системы, содержащие слабые основания, например, бикарбонат натрия, карбонат кальция, гидроксид магния, основный оксид алюминия, нейтральный оксид алюминия, ацетат натрия, вторичный кислый фосфат натрия, вторичный кислый фосфат калия, фторид калия, другие соли или вода в обычных органических растворителях. Предпочтительными реакционными условиями являются слабые основания, например, бикарбонат натрия, основный оксид алюминия, фторид калия или вода. Наиболее предпочтительными условиями реакции являются основный оксид алюминия или бикарбонат натрия. Растворители должны быть совместимы с условиями реакции и включают простые эфиры, ароматические хлорированные углеводороды, сложные эфиры, спирты, MeCN, DMF, DMPU или кетоны. Предпочтительными являются хлорированные углеводороды, простые эфиры и сложные эфиры. Наиболее предпочтительными являются дихлорметан, IPAC, EtOAc, DME и MTBE. Температурный диапазон находится между около - 40oC и около 10oC, но предпочтительно между 0 и около 35oC. Инкубация продолжается, по крайней мере, около 10 минут, и обычно ее прекращают до истечения около 48 часов.
Циклизацию, вызванную основанием, в результате которой образуется эпоксид, осуществляют путем обработки галогенгидрина основанием. Предпочтительные основания для такой циклизации включают гидроксиды и оксиды лития, натрия, калия, магния, кальция или тетраалкиламмония: алкоксиды, например, метоксид, этоксид, n- и изо-пропоксид, n-, изо-, втор- и трет-бутоксид лития, натрия, калия, магния и тетраалкиламмония. Другие подходящие основания включают третичные и затрудненные амины, например, триэтиламин, DIEA, DBU, DBN, DABCO, метилморфолин, диизопропиламин, дициклогексиламин, бис-триметилсилиламин или тетраметилпиперидин, а также их металламидные соли. Наиболее предпочтительными основаниями являются гидроксиды лития, натрия, калия или тетраалкиламмония: алкоксиды, например метоксид, этоксид, изо-пропоксид или трет-бутоксид лития, натрия и калия; или третичные амины, например, DIEA. Гидроксид щелочного металла означает LiOH, KOH или NaOH или их смеси.
Предпочтительными растворителями для циклизации, вызванной основанием, являются простые эфиры, сложные эфиры, углеводороды, ароматические растворители, хлорированные углеводороды, кетоны, вода, спирты, DMSO, MeCN, DMF или DMPU или другие полярные растворители или их смеси. Наиболее предпочтительными растворителями являются простые эфиры, сложные эфиры, спирты или полярные апротонные растворители.
Циклизацию, вызванную основанием, осуществляют в температурном диапазоне между около -40oC и около 100oC. Инкубация продолжается, по крайней мере, около 10 минут, и обычно ее прекращают до истечения около 48 часов.
В способе настоящего изобретения может быть использовано широкое множество растворителей, если не указано по-иному. Углеводородные растворители включают пентан, гексан, гептан, циклогексан, метилциклогексан, бензол, толуол и ксилол. Ароматические вещества в качестве растворителей включают бензол, толуол, ксилол и этилбензол. Хлорированные углеводороды в качестве растворителей включают метиленхлорид, хлороформ, углеродтетрахлорид, дихлорэтан, трихлорэтан, тетрахлорэтан, трихлорэтилен, тетрахлорэтилен, хлорбензол и дихлорбензол. Простые эфиры в качестве растворителей включают диэтиловый эфир, дибутиловый эфир, тетрагидрофуран, диметоксиэтан, диэтоксиэтан и MTBE. Сложные эфиры в качестве растворителей включают этилацетат, IPAC и этоксиэтилацетат. Кетоны в качестве растворителей включают ацетон, MEK и MIBK. Спирты в качестве растворителей включают метанол, этанол, пропанол, изопропанол, бутанол и метоксиэтанол. Полярные апротонные растворители в качестве растворителей включают DMF, DMA, DMSO, DMPU, TMU, NMP и ацетонитрил. Третичные амины в качестве растворителей включают триэтиламин, диизопропилэтиламин, пиридин, DABCO, DBU, DBN, пентаметилпиперидин и DMAP.
В одном варианте настоящего изобретения способ синтеза эпоксида формулы I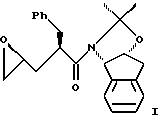
включает стадии:
(а) контактирования одного эквивалента аллилацетонида формулы II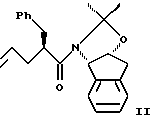
с галогенирующим агентом в количестве от около одного до двух эквивалентов в растворителе, смешанном с водным раствором слабого основания, при температуре в диапазоне между от около -40oC и около 100oC с образованием галогенгидрина формулы III,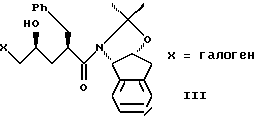
при этом галогенирующий агент выбирают из группы, состоящей из N-иодсукцинимида, N-бромсукцинимида или N-хлорсукцинимида, два последних из которых можно смешать с иодидной солью; растворитель выбирают из группы, состоящей из дихлорметана, IPAC, EtOAc, DME и MTBE, слабое основание выбирают из основного оксида алюминия или бикарбоната натрия, и
(b) добавления основания в воде для установления образования зпоксида формулы I, при этом основание выбирают из группы, состоящей из гидроксида лития, гидроксида натрия, гидроксида калия, тетраалкиламмонийгидроксида; C1-4-алкоксида лития, натрия или калия; и DIEA.
В другом варианте настоящего изобретения, включающем использование ацетонидного реагента, настоящий способ синтеза эпоксида формулы I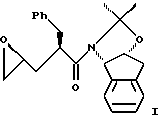
включает стадии:
(а) взаимодействия одного эквивалента ацетонида
с приблизительно одним эквивалентом аллилгалогенида в сильном основании, при этом аллилгалогенид выбирают из аллилхлорида, аллилбромида и аллилиодида, с получением аллилацетонида формулы II,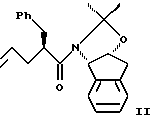
(b) смешивания с ним галогенирующего агента в количестве от около одного до двух эквивалентов в растворителе, смешанном с водным раствором слабого основания при температуре в диапазоне от около -40oC до около 100oC с образованием галогенгидрина формулы III,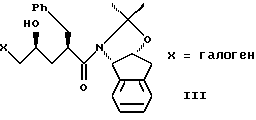
при этом галогенирующий агент выбирают из группы, состоящей из N-иодсукцинимида, N-бромсукцинимида или N-хлорсукцинимида, два последних из которых можно смешать с иодидной солью; растворитель выбирают из группы, состоящей из дихлорметана, IPAC, ETOAc, DME и MTBE; слабое основание выбирают из основного оксида алюминия или бикарбоната натрия, и
(с) добавления основания в воде для установления образования эпоксида формулы I, при этом основание выбирают из группы, состоящей из гидроксида лития, гидроксида натрия, гидроксида калия, тетраалкиламмонийгидроксида, C1-4-алкоксида лития, натрия или калия, и DIEA.
В другом варианте настоящего изобретения способ синтеза эпоксида формулы I,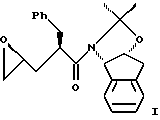
включает стадии:
(а) контактирования одного эквивалента аллилацетонида формулы II,
растворенного в изопропилацетате с N-иодосукцинимидом в количестве от около одного до двух эквивалентов в ~0.5М водном растворе бикарбоната натрия при комнатной температуре с образованием иодогидрина формулы III, и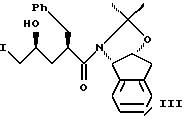
(b) добавления гидроксида щелочного металла в воде для установления образования эпоксида формулы I.
В другом варианте настоящего изобретения способ синтеза эпоксида формулы I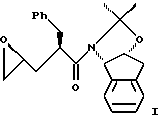
включает стадии:
(а) взаимодействия одного эквивалента ацетонида,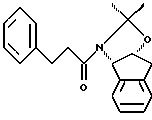
растворенного в эфирном растворителе, с приблизительно одним эквивалентом аллилбромида и приблизительно одним эквивалентом ~ 1.0-2.0М литийгексаметилдисилилазида (в эфирном растворителе) с получением аллилацетонида формулы II: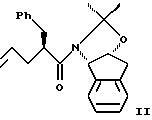
(b) смешивания с ним N- иодсукцинимида в количестве от одного до двух эквивалентов в ~0.5М водном растворе бикарбоната натрия при комнатной температуре с образованием иодгидрина формулы III, и
(с) добавления гидроксида щелочного металла в воде для установления образования эпоксида формулы I.
Способы и промежуточные соединения этого изобретения являются пригодными для получения соединений конечного продукта, которые пригодны при ингибировании протеазы HIV, предотвращении или лечении инфекции, вызванной вирусом иммунодефицитного состояния человека (HIV), и при лечении последующих паталогических состояний, например AIDS. Лечение AIDS или предотвращение или лечение инфекции, вызванной HIV, определяют как включающее, но не ограниченное, лечение широкого круга состояний, связанных с инфекцией, вызванной HIV: AIDS, ARC (комплекс, родственный с AIDS), как симптомокомплекс, так и асимптомокомплекс, и действительное или потенциальное подвержение HIV. Соединения конечного продукта, которые могут быть получены с помощью способов и промежуточных соединений этого изобретения, являются пригодными при лечении инфекции, вызванной HIV, после предполагаемого подвержения в прошлом HIV, например, при переливании крови, трансплантации органа, обмене жидкости в организме, острых болях, при случайном заражении во время иглоукалывания или при воздействии на кровь пациента во время хирургического вмешательства.
Конечный продукт - ингибиторы протеазы HIV - являются также пригодными при получении и выполнении ситового анализа антивирусных соединений. Соединения конечного продукта являются пригодными, например, для выделения ферментативных мутантов, которые являются превосходным средством для отбора более сильных антивирусных соединений. Кроме того, такие соединения являются пригодными при установлении или определении места связывания других антивирусов в протеазу HIV, например, посредством конкурентного ингибирования. Таким образом, соединения конечного продукта, которые получены с помощью способов и промежуточных соединений этого изобретения, представляют коммерческие продукты, продаваемые для этих целей.
Соединения, ингибирующие протеазу HIV, которые могут быть получены с помощью промежуточных соединений и способов настоящего изобретения, представлены в EPO 541164. Соединения, ингибирующие протеазу HIV, могут быть введены пациентам, в случае необходимости такого лечения, в виде лекарственных препаратов, включающих фармацевтический носитель и терапевтически эффективные количества соединения или его фармацевтически приемлемой соли. В EPO 541164 раскрыты подходящие лекарственные препараты, способы их введения, солевые формы и дозировки соединений.
Соединения настоящего изобретения могут иметь ассиметрические центры и встречаются в виде рацематов, рацемических смесей и в виде отдельных диастереомеров или энантиомеров, при этом в настоящее изобретение включены все изомерные формы.
Когда в составляющей какая-либо переменная (например, арил, гетероцикл, R, R1, R2, n, X и т.д.) встречается более одного раза, ее определение при каждом проявлении не зависит от ее определения при каждом другом проявлении. Являются также допустимыми комбинации заместителей и/или переменных, если только такие комбинации приводят к получению устойчивых соединений.
Как используется здесь, за исключением того, где отмечено по-иному, "алкил" включает насыщенные алифатические углеводородные группы как с разветвленной, так и с прямой цепью, имеющие конкретное количество углеродных атомов (Me представляет метил, Et является этилом, Pr представляет пропил, Bu является бутилом, t-Bu представляет трет-бутил): "Галоген", как используется здесь, означает фтор, хлор, бром и иод. Как используется здесь, "арил" означает фенил (Ph) или нафтил.
Характерные экспериментальные методики, в которых используют новый способ, описаны подробно ниже. Эти методики служат только примером и не являются ограничениями для нового способа этого изобретения.
Пример 1.
Превращение ацетонида в аллилацетонид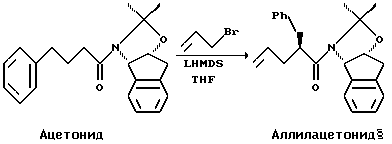
Ацетонид - 32.1 г
Аллилбромид - 12.70 г
Литийгексаметилдисилилазид (LHMDS) 1.0М в THF - 105 мл
Тетрагидрофуран (THF) - 200 мл
В 200 мл THF в 100 мл трехгорлой колбе, снабженной капельной воронкой, растворили ацетонид и дегазировали путем барботирования в азоте в течение 20 минут. Смесь охладили до -25oC и через шприц добавили аллилбромид. В капельную воронку под давлением азота через канюлю перенесли LHMDS. LHMDS медленно, в течение 20 минут, прокапали в реакционную смесь, перемешиваемую магнитной мешалкой. В то время, когда температура охлаждающей ванны была равна -30oC, внутренняя температура достигла -14oC. Смесь выдержали в течение 30 минут при температуре от -20oC до -15oC. Добавили воду (100 мл) и IPAC (100 мл) и температуру подняли до 5oC. Нижнюю водную фазу выбросили за ненадобностью, и органическую фазу промыли 100 мл 0.2HCl в 3% водном растворе NaCl, 30 мл рассола и 30 мл 0.5М бикарбоната натрия. Органическую фазу выпарили (55oC, 100 торр) до образования масла, добавили 40 мл IPAC, и смесь опять выпарили до образования масла. В этот момент сырой аллилацетонид можно было непосредственно подать на следующую стадию или очистить кристаллизацией из гексана-IPAC 30: 1 или метилциклогексана-IPAC 30:1 до получения аллилацетонида в виде белого кристаллического твердого вещества с выходом 87%.
Данные 13C ЯМР для основного ротамера аллилацетонида (62,5 MHz) указаны в табл.1.
Пример 2
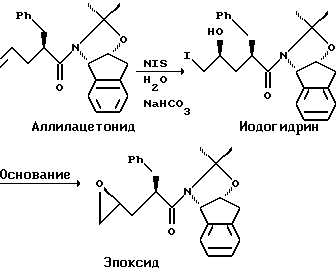
Превращение аллилацетонида в иодогидрин и циклизация до эпоксида с помощью NIS.
Аллилацетонид (сырой из вышеприведенного получения) ~0,1 моля
N-иодсукцинимид (NIS) - 29,24 г
Водный раствор бикарбоната натрия (0,5 M) - 350 мл
Изопропилацетат (IPAC) - 300 мл
Сырой аллилацетонид растворили в IPAC и перемешали с водным раствором бикарбоната натрия и NIS в течение 17 часов. Добавили водный раствор бисульфата натрия (38-40%) и отделили верхнюю органическую фазу. Органическую фазу промыли 300 мл воды и 2х100 мл рассола. В этот момент раствор сырого иодогидрина в IPAC можно было непосредственно перенести на следующую стадию или раствор можно было выпарить и кристаллизовать из метилциклогексана-IPAC до получения иодогидрина в виде бледно-желтого кристаллического твердого вещества, 13C ЯМР, вращение при точке плавления.
Иодогидрин (раствор сырого иодогидрина в
IPAC из вышеприведенного получения) - ~0,1 моль
Моногидрат гидроксида лития - 50 г
Вода - 200 мл
Данные 13C ЯМР для основного ротамера иодогидрина (62,5 MHz) указаны в табл.2.
Пример 3
Превращение аллилацетонида в иодогидрин и циклизация до эпоксида с помощью NCS/Nal.
Иодогидрин в IPAC смешали с гидроксидом лития в воде в течение 3-х часов при 25-30oC. Верхнюю органическую фазу промыли 200 мл воды и 200 мл рассола и сушили над ~ 2 г сульфата магния. Раствор IPAC отфильтровали и выпарили (50-60oC, 100 торр) до ~50 мл, когда начал кристаллизоваться эпоксид. Смесь охладили до 25oC в течение 30 минут и 10 мл порциями в течение 30 минут при перемешивании добавили 75 мл метилциклогексана. Смесь выдержали в течение 1 часа, и кристаллы отфильтровали и промыли 2х20 мл метилциклогексана и сушили до получения 24,10 г (64%) эпоксида в виде белого кристаллического твердого вещества со степенью чистоты 99,9А%, определенной HPLC. Маточный раствор и промывки выпарили до масла и растворили в 40 мл IPAC. Раствор обработали 10 г углерода Дарко G60 в течение 2-х часов при 25oC и фильтровали через прокладку из Solka Floc. Фильтрат выпарили до ~20 мл и добавили 40 мл метилциклогексана. Кристаллический эпоксид отфильтровали и промыли 2х10 мл метилциклогексана до получения еще 4,96 г (13%) эпоксида со степенью чистоты 96,2А%, определенной HPLC. Превращение иодогидрина в эпоксид можно также осуществлять путем добавления 1,7 М калий-трет-бутоксида в THF (0,70 мл, 1,2 ммоля) или 5 М гидроксида калия в метаноле (0,24 мл, 1,2 ммоля) или DIEA (155 мг, 1,2 ммоля) к раствору иодогидрина (505 мг, 1,0 ммоль) в IPAC (2-3 мл), последующей промывки 2х2 мл воды и кристаллизации из метилциклогексана-IPAC.
Аллилацетонид - 26,15 г
N-хлорсукцинамид (NCS) - 22,7 г
Иодид натрия - 25,5 г
Водный раствор бикарбоната натрия (0,5 М) - 350 мл
Изопропилацетат (IPAC) - 300 мл
NCS и Nal смешали вместе в 200 мл воды в течение 20 минут. Когда смесь стала темно-коричневой, тогда тотчас же отделили темное твердое вещество. Твердое вещество растворили и при дополнительной выдержке цвет поблекнул и стал светло-желтым. В IPAC растворили сырой аллилацетонид и перемешали с водным раствором бикарбоната натрия и полученным выше светло-желтым раствором в течение 17 часов. Добавили водный раствор бисульфита натрия (38-40%) и отделили верхнюю органическую фазу. Органическую фазу промыли 300 мл воды и 2х100 мл рассола. В этот момент раствор сырого иодогидрина в IPAC можно было непосредственно перенести на следующую стадию или раствор можно было выпарить и кристаллизовать из метилциклогексана-IPAC до получения иодогидрина в виде бледно-желтого кристаллического твердого вещества.
Пример 4.
Получение Амида 1.

Раствор (-)-цис-1-аминоиндан-2-ола (884 г, 5,93 моля) в 17.8 л сухого THF (тетрагидрофурана) (KF - 55 мг/мл) (KF означает титрование Карла Фишера для воды) и триэтиламин (868 мл, 6.22 моля) в 50 л круглодонной колбе, снабженной термопарой, механической мешалкой, приспособлением, имеющим входное отверстие для азота, и барботером, охладили до 15oC. Затем в течение 75 минут добавили 3-фенил-пропионилхлорид (1000 г, 5,93 моля) при установлении внутренней температуры между 14-24oC с помощью охлаждающей ванны из смеси льда и воды. После добавления смесь выдерживали при температуре от 18oC до 20oC в течение 30 минут и контролировали посредством анализа HPLC (высокоэффективной жидкостной хроматографией) на исчезновение (-)-цис-1-аминоиндан-2-ола.
Ход реакции контролировали посредством анализа высокоэффективной жидкостной хроматографией (HPLC): 25 см колонка C8-RX фирмы Дупонт, ацетонитрил/10 мМ (KH2PO4/K2HPO4) 60: 40, 1.0 мл/мин, объем впрыскивания = 20 мл, обнаружение = 200 нм, получение пробы при 500 х разбавлении. Приблизительные величины времени удерживания:
время удерживания (мин) - идентификация
6.3 - цис-аминоинданол
Реакционную смесь обработали пиридиний-р-толуолсульфонатом (241 г, 0.96 моля, 0.16 экв. ) и перемешивали в течение 10 минут. (pH смеси после разбавления 1 мл пробы равным объемом воды находился между 4.3-4.6). Затем добавили 2-метоксипропен (1.27 л, 13.24 моля, 2.2 экв) и реакционную смесь нагрели до 38-40oC в течение 2-х часов. Реакционную смесь охладили до 20oC и разделили этилацетатом (12 л) и 5% водным раствором NaHCO3 (10 л). Смесь перемешали и слои разделили. Этилацетатный экстракт промыли 5% водным раствором NaHCO3 (10 л) и водой (4 л). Этилацетатный экстракт сушили посредством атмосферной перегонки и растворитель изменили на циклогексан (общий объем ~ 30 л). В конце перегонки и концентрирования раствор горячего циклогексана медленно охладили до 25oC для кристаллизации продукта. Затем полученную суспензию охладили до 10oC и выдерживали в течение 1 часа. Продукт выделили фильтрацией и влажный фильтровальный осадок промыли холодным (10oC) циклогексаном (2х800 мл). Промытый осадок сушили под вакуумом (давление ртути 26 дюймов = 66.04 см) при 40oC до получения 1.65 кг ацетонида 1 (86.4%, площадь определенная HPLC, 98%), 1ЯМР (300. 13 MHz, CDCl3, более важный ротамер) δ 7.36-7.14 (m, 9H), 5.03 (d, J=4.4, 1H), 4.66 (m, 1H) 3.15 (m, 2H), 3.06 (широк., s, 2H), 2.97 (m, 2H), 1.62 (s, 3H), 1.37 (s, 3Н): 13ЯМР (75.5 MHz, CDCl3, более важный ротамер) δc 168.8, 140.9, 140.8, 140.6, 128.6, 128.5, 128.4, 127.1, 126.3, 125.8, 124.1, 96.5, 78.6, 65.9, 38.4, 36.2, 31.9, 26.5, 24.1.
Аналит. вычислено для C21H23NO2:
C, 78.47; H, 7.21; N, 4.36.
Найдено: C, 78.65: H, 7.24; N, 4.40.
Пример 5.
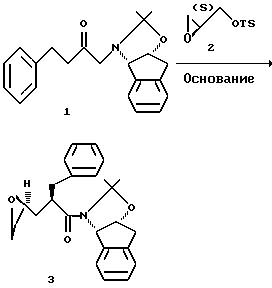
Получение эпоксида 3 с использованием тозилата.
Раствор ацетонида 1 (1000 г, 3.11 моля) и 2(S)-глицидилтозилата 2 (853 г, 3.74 моля, 1.2 экв.) в 15.6 л THF (KF = 22 мг/мл) в 50 л четырехгорлой круглодонной колбе, снабженной термопарой, механической мешалкой, капельной воронкой и приспособлением, имеющим входное отверстие для азота, 3 раза дегазировали посредством продувки азотом в вакууме и охладили до -56oC. Затем в течение 2-х часов добавили гексаметилдисилазид лития (LiN[(CH3)3Si] 2) (2.6 л, 1.38 М., 1.15 экв.) при поддержании внутренней температуры между -50oC и -45oC. Реакционную смесь перемешали при температуре от -45oC до -40oC в течение одного часа и затем нагрели в течение 1 часа до температуры -25oC. Смесь перемешивали при температуре от -25oC до -22oC в течение 4-х часов (или до тех пор, пока исходный ацетонид находился на 3% площади).
Ход реакции контролировали посредством анализа HPLC: 25 см х 4.6 нм колонка Зорбакса с диоксидом кремния, 20% этилацетат в гексане, 2.0 мл/мин, объем впрыскивания = 20 мл, обнаружение = 254 нм, получение пробы = 100 x разбавление.
Приблизительные значения времени удерживания:
время удерживания (мин) - идентификация
5.5 - амид 1
6.5 - глицидилтозилат 2
13.5 - эпоксид 3
Реакционную смесь резко охладили DI водой (6.7 л) при температуре -15oC и раздели с помощью этилацетата (10 л). Смесь перемешали и слои разделили. Этилацетатный экстракт промыли смесью 1% водного раствора NaHCO3 (5 л) и насыщенным NaCl (0.5 л). Этилацетатный экстракт (28.3 л) концентрировали вакуумной перегонкой (давление ртути 28 дюймов = 71.12 см) и добавили дополнительное количество этилацетата для завершения замены растворителя на этилацетат (конечный объем = 11.7 л). Затем растворитель этилацетатного концентрата заменили на MeOH для кристаллизации продукта и концентрирования до конечного объема 3.2 л. Остаточный растворитель этилацетат удалили путем загрузки 10 л метанола и сбора 10 л дистиллята. Полученную суспензию перемешивали при 22oC в течение 1 часа, затем охладили до 5oC и выдерживали в течение 0.5 часа. Продукт выделили фильтрацией и влажный фильтровальный осадок промыли холодным метанолом (2 х 250 мл). Промытый осадок сушили под вакуумом (давление 26 дюймов Hg = 66.04 см) при 25oC до получения 727 г эпоксида 3 (61.2%, как показала HPLC, площадь основного продукта эпоксида 98.7%):
13C ЯМР(300 MHz, CDCl3) δ 171.1, 140.6, 140.5, 139.6, 129.6, 128.8, 128.2, 127.2, 126.8, 125.6, 124.1, 96.8, 79.2, 65.8, 50.0, 48.0, 44.8, 39.2, 37.4, 36.2, 26.6, 24.1.
Пример 6.
Получение пиналтимата 6.

Суспензию 2(S)-трет-бутилкарбоксамид-4-N-Boc-пиперазина 4 (1950 г, 6.83 моля, ее > 99.5% (ее - энантиомерный избыток)) и эпоксид 3 (2456 г, смесь 4S/R эпоксидов при соотношении 97.5 : 2.5, 6.51 моля) в изопропаноле (2-пропанол, 18.6 л) в 72 л круглодонной колбе с 4-мя входными отверстиями для азота, снабженной механической мешалкой, парциальным конденсатором горячего орошения, паровой баней, термопарой, покрытой тефлоном, и входным отверстием для азота, нагревали до образования флегмы (внутренняя температура составила 84-85oC). Через 40 минут получили гомогенный раствор. Смесь нагревали в колбе с обратным холодильником в течение 28 часов.
Внутренняя температура во время нагревания в колбе с обратным холодильником составила 84-85oC. Ход реакции контролировали посредством анализа высокоэффективной жидкостной хроматографией;. 25 см колонка C8-RX фирмы Дупонт, ацетонитрил/10 мМ (KH2PO4/K2HPO4 60:40), 1.0 мл/мин, обнаружение - 220 нм, полученная проба = 2 μл, реакционную смесь разбавили в ацетонитриле до 1 мл. Приблизительные значения времени удерживания:
время удерживания (мин) - идентификация
4.8 - пиперазин 4
8.9 - эпоксид 3
15.2 - связанный продукт 5
Через 28 часов оставшийся эпоксид 3 и связанный продукт 5 (анализ HPLC) находились соответственно на 1.5% площади и 91-93% площади. Смесь охладили до температуры от 0oC до 5oC и во время поддержания температуры ниже 15oC добавили 20.9 л 6N HCl. После завершения добавления смесь нагрели до 22oC. В этот момент наблюдалось выделение газа (изобутилена). Смесь выдерживали в течение 6 часов при температуре от 20oC до 22oC.
Ход реакции контролировали анализом HPLC: те же самые условия, которые приведены выше. Приблизительные величины времени удерживания:
время удерживания (мин) - идентификация
7.0 - цис-аминоинданол
11.9 - пиналтимат 6
15.1 - связанный продукт 5
Смесь охладили до 0oC и медленно добавили 7.5 л 50% NaOH для того, чтобы установить pH смеси равным 11.6, при поддержании температуры во время добавления менее 25oC. Смесь разделили этилацетатом (40 л) и водой (3 л). Смесь перемешали и слои разделили. Органическую фазу (60 л) концентрировали под пониженным давлением (давление 29 дюймов Hg = 73.66 см), растворитель заменили на DMF (N,N-диметилформамид) и концентрировали до конечного объема 10.5 л (KF=1.8 мг/мл). Анализ HPLC показал, что выход 6 в этилацетате составил 86.5%. Пиналтиматное соединение 6 в DMF использовали непосредственно на следующей стадии без дополнительной очистки. Для выделенного соединения 6:
13C ЯМР (75.4 MHz, CDCl3) δ 175.2, 170.5, 140.8, 140.5, 139.9, 129.1, 128.5, 127.9, 126.5, 125.2, 124.2, 73.0, 66.0, 64.8, 62.2, 57.5, 49.5, 47.9, 46.4, 45.3, 39.6, 38.2, 28.9.
Пример 7.
Получение моногидрата Соединения J.
В раствор соединения 6 в DMF (10.5 л, KF = 10 мг/мл) с предыдущей стадии загрузили 8 л высушенного на сите DMF (KF < 30 мг/л) и смесь нагрели с помощью паровой бани под вакуумом 30 дюймов Hg (76.20 см) для того, чтобы отогнать, главным образом, воду и/или остаточный изопропанол или этилацетат. Конечный объем концентрата составил 13.5 л (KF=1.8 мг/мл), и затем к раствору при 25oC добавили триэтиламин (28.6 л, 20.5 молей), после которого добавили 3-пиколилхлоридгидрохлорид (96%, 1287 г, 7.84 моля). Полученную суспензию нагрели до 68oC. Ход реакции контролировали посредством анализа HPLC при использовании тех же самых условий, что и на предыдущей стадии. Приблизительные значения времени удерживания:
Время удерживания (мин) - Идентификация
2.7 - DMF
4.2 - 3-пиколилхлорид
4.8 - Соединение J.
9.1 - пиналтимат 6
Смесь выдерживали при 68oC до тех пор, пока остаток пиналтиматного соединения 6 находился на поверхности < 0.3%, как следовало из анализа HPLC.
Смесь перемешали в течение 4-х часов при 68oC, затем охладили до 25oC и разделили этилацетатом (80 л) и смесью, состоящей из 24 л насыщенного водного раствора NaHCO3 и дистиллированной воды (14 л). Смесь перемешали при 55oC и слои разделили. Этилацетатный слой три раза промыли водой (20 л) при 55oC. Промытый этилацетатный слой концентрировали при атмосферном давлении до конечного объема котла 30 л. В конце атмосферного концентрирования к горячему раствору добавили воду (560 мл), смесь охладили до 55oC и внесли затравку моногидрата Соединения J. Смесь охладили до 4oC и фильтровали для сбора продукта. Продукт промыли холодным этилацетатом (2х3 л) и сушили при 25oC и атмосферном давлении до получения 2905 г (70.7%) моногидрата соединения J в виде белого твердого вещества.
Пример 8.
Пиразин-2-трет-бутилкарбоксамид 9.
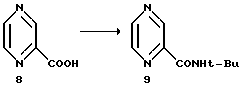
2-Пиразинкарбоновая кислота - 3.35 кг (27 молей)
Оксалилхлорид - 3.46 кг (27.2 моля)
трет-бутиламин (KF=460 μг г/мл) - 9.36 л (89 молей)
EtOAc (KF=56 μг г/мл) - 27 л
DMF - 120 мл
1-Пропанол - 30 л
Карбоновую кислоту 8 суспендировали в 27 л EtOAc и 120 мл DMF в 72 л трехгорлой колбе при механическом перемешивании под азотом и суспензию охладили до 2oC. Добавили оксалилхлорид, поддерживая температуру между 5 и 8oC.
Добавление завершили через 5 часов. Во время экзотермического добавления выделились CO и CO2. Образованный HCl остался большей частью в растворе. Присутствующий осадок представлял, по всей вероятности, хлорид пиразиновой кислоты. Анализ образования хлорида кислоты осуществляли путем резкого охлаждения безводной пробы реакционного продукта трет-бутил-амином. После завершения резкого охлаждения осталось < 0.7% кислоты 8.
Анализ завершения образования хлорида кислоты является важным, потому что незавершенная реакция приводит к образованию примеси бис-трет-бутилоксамида.
Реакцию можно контролировать посредством HPLC: 25 см колонка Зорбакса RXC8 фирмы Дупонт при скорости потока 1 мл/мин и обнаружении при 250 нм; линейный градиент от 98% 0.1% водного раствора H3PO4 и 2% CH3CN до 50% водного раствора H3PO4 и 50% CH3CN при времени, равном 30 мин. Время удерживания: кислоты 8 = 10.7 мин, амида 9 = 28.1 мин.
Реакционную смесь выдерживали при 5oC в течение 1 часа. Полученную суспензию охладили до 0oC и добавили трет-бутиламин при такой скорости, чтобы сохранить внутреннюю температуру ниже 20oC.
Добавление осуществляли в течение 6 часов, так как реакция была сильно экзотермической. Из реакционной смеси устранили небольшую часть образованного трет-бутиламмонийгидрохлорида в виде пушистого белого твердого вещества.
Смесь выдерживали при 18oC в течение еще 30 минут. Осажденные аммониевые соли удалили фильтрацией. Фильтровальный осадок промыли 12 л EtOAc. Смешанные органические фазы промыли 6 л 3% NaHCO3 и 2х2 л насыщенного водного раствора NaCl. Органическую фазу обработали 200 г углерода Дарко G60, фильтровали через Solka Flok и осадок промыли 4 л EtOAc.
Обработка углеродом позволила эффективно удалить пурпурный цвет продукта.
Раствор EtOAc 9 концентрировали при давлении 10 миллибар до 25% от первоначального объема. Добавили 30 л 1-пропанола и перегонку продолжили до тех пор, пока достигли конечного объема, равного 20 л.
В этот момент концентрация EtOAc была ниже предела обнаружения в 1ЯМР (<1%). Внутренняя температура в этом растворителе составила < 30oC. При выпаривании аликвоты получили рыжевато-коричневое твердое вещество, точка плавления 87-88oC. 13C ЯМР (75 MHz, CDCl3, част. на миллион) 161,8; 146,8; 145,0; 143,8; 142,1; 51,0; 28,5.
Пример 9.
Рац-2-трет-бутилкарбоксамидпиперазин 10.
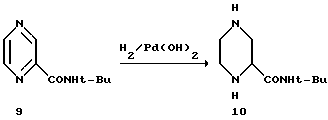
Материалы.
Пиразин-2-трет-бутилкарбоксамид 9 2,4 кг (13,4 моля) в растворе 1-пропанола, 12 л 20% Pd(OH)2/C, 16 вес.% воды 144 г.
Пиразин-2-трет-бутилкарбоксамид 9/раствор 1-пропанола поместили в автоклав вместимостью 5 галлонов (18,927 л). Добавили катализатор и смесь гидрировали при 65oC и давлении H2, равном 40 фунтов/дюйм2 (3 атм).
Через 24 часа в реакции израсходовалось теоретическое количество водорода, и GC (газовая хроматография) показала, что содержание 9 составляет < 1%. Смесь охладили, продули азотом и фильтрацией через Solka Flok удалили катализатор. Катализатор промыли 2 л подогретого 1-пропанола.
Обнаружили, что применение во время промывки фильтровального осадка подогретого 1-пропанола улучшает фильтрацию и снижает потери продукта с фильтровальным осадком.
Реакцию контролировали GC (газовой хроматографией): 30 м колонка Мегабора, от 100oC до 160oC при скорости потока 10oC/мин, выдержка 5 мин, затем при 10oC/мин, до 250oC, время удерживания: 9=7,0 мин, 10=9,4 мин. Реакцию можно также контролировать посредством TLC (тонкослойной хроматографии) с помощью EtOAc/MeOH (50:50) в качестве растворителя и нингидрина в качестве проявляющего вещества.
Выпаривание аликвоты показало, что выход во время амидирования и гидрирования составлял 88% и что концентрация 10 равна 133 г/л.
При выпаривании аликвоты получили 10 в виде белого твердого вещества, точка плавления 150-151oC; 13C ЯМР (75 MHz, D2O, част. на миллион) 173,5; 59,8; 52,0; 48,7; 45,0; 44,8; 28,7.
Пример 10
Соль(S) (S)-2-трет-бутилкарбоксамидпиперазин-бис(S)- камфорсульфоновой кислоты-11.
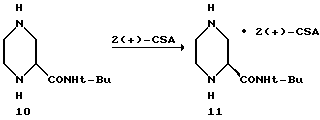
Материалы
Рац-2-трет-бутилкарбоксамидпиперазин 10 в растворе 1-пропанола - 4.10 кг (22.12 моля) в 22.5 кг растворителя
(S)-(+)-10-камфорсульфоновая кислота - 10.0 кг (43.2 моля)
1-пропанол - 12 л
ацетонитрил - 39 л
Вода - 2.4 л
Раствор амина 10 в 1-пропаноле загрузили в 100 л колбу, снабженную прикрепленным загрузочным концентратором. Раствор концентрировали при 10 миллибарах и температуре < 25oC до объема приблизительно 12 л.
В этот момент продукт высадился из раствора, но когда смесь нагрели до 50oC, он опять перешел в раствор.
Анализ гомогенной аликвоты показал, что концентрация 10 составила 34 г/л. Концентрацию определили HPLC: 25 см колонка Зорбакса RXC8 фирмы Дупонт при скорости потока 1.5 мл/мин и обнаружении при 210 нм; изократно CH3CN/0.1% водный раствор H3PO4 (98/2). Время удерживания 10: 2.5 минуты.
Для получения прозрачного слегка коричневатого раствора добавили ацетонитрил (39 л) и воду (2.4 л).
Определение содержания воды путем титрования KF и отношения CH3CN/1-пропанол с помощью интегрирования 1H ЯМР показало, что отношение CH3CN/1-пропанол/H2O составляет 26/8/1.6. Концентрация в растворе составила 72.2 г/л.
В течение 30 минут четырьмя порциями при 20oC загрузили (S)-10-камфорсульфоновую кислоту. После добавления CSA температура поднялась до 40oC. Через несколько минут образовался плотный белый осадок. Для растворения всех твердых частиц белую суспензию нагрели до 76oC, слегка коричневый раствор затем охладили в течение 8 часов до 21oC.
Продукт осадили при 62oC. Продукт фильтровали без выдержки при 21oC, и фильтровальный осадок промыли 5 л растворяющей смеси, состоящей из CH3CN/1-пропанола/H2O при соотношении компонентов в смеси 26/8/1.6. Его сушили в вакуумной печи при 35oC и продувке азотом до получения 5.6 кг (39%) соединения 11 в виде белого кристаллического твердого вещества, точка плавления: 288-290oC (с разлож.). α D25 = 18,9o (с=0,37 H2O). 13C ЯМР (75 MHz, D2O, част. на миллион) 222,0; 164,0; 59,3; 54,9; 53,3; 49,0; 48,1; 43,6; 43,5; 43,1; 40,6; 40,4; 28,5; 27,2; 25,4; 19,9; 19,8.
В соответствии с последующим анализом хиральной HPLC ее материала составил 95%: аликвоту соединения 11 (33 мг) суспендировали в 4 мл EtOH и 1 мл Et3N. Добавили Boc2O (11 мг) и реакционную смесь выдержали в течение 1 часа. Полностью в вакууме удалили растворитель, остаток растворили в приблизительно 1 мл EtOAc и фильтровали через пипетку Пастера с SiO2 при использовании в качестве злюента EtOAc. Фракции подвергнутого выпариванию продукта повторно растворили в гексане при концентрации ~1 мг/мл.
Энантиомеры разделили в колонке AS Дайцеля при использовании в качестве растворяющей системы гексана/IPA (97: 3) при скорости потока 1 мл/мин, и обнаружении при 228 нм. Время удерживания: S-антипода=7,4 мин, R=9,7 мин.
Пример 11.
(S)-2-трет-бутилкарбоксамид-4-трет-бутоксикарбонил-пиперазин 4 из соли 11.
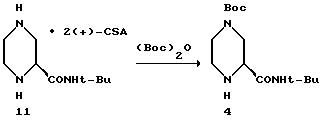
Материалы
(S)-2-трет-бутилкарбоксамидпиперазин бис(S)-(+)-CSA соль 11, ее 95% - 5,54 кг (8,53 моля)
Ди-трет-бутилбикарбонат - 1,86 кг (8,53 моля)
Et3N - 5,95 л (42,6 моля)
EtOH, испытанный 200 раз - 55 л
EtOAc - 2 л
К (S)-CSA соли 11 в 100 литровой трехгорлой колбе, снабженной капельной воронкой, под азотом добавили EtOH, затем при 25oC добавили триэтиламин. Твердое легко растворили при добавлении Et3N. В EtOAc растворили Boc2O и загрузили в капельную воронку. Раствор Boc2O в EtOAc добавили при такой скорости, чтобы сохранить температуру ниже 25oC. Добавление осуществляли в течение 3-х часов. После завершения добавления раствора Boc2O реакционную смесь выдерживали в течение одного часа.
Реакцию можно контролировать посредством HPLC: 25 см колонка Зорбакса RXC8 фирмы Дупонт, при скорости потока 1 мл/мин и обнаружении при 228 нм; изократно CH3CN/0.1M KH2PO4 (50/50), pH равный 6,8 установили с помощью NaOH. Время удерживания соединения 4=7,2 мин. Хиральный анализ осуществляли с использованием такой же системы, как и на предыдущей стадии. Реакцию можно также контролировать посредством TLC при использовании в качестве растворителя 100% EtOAc (Rf=0,7).
Затем раствор концентрировали до ~10 л при внутренней температуре <20oC в концентраторе загрузочного типа под вакуумом 10 миллибар. Изменение растворителя завершили путем медленного растворения в 20 л EtOAc и повторного концентрирования до ~ 10 л. Реакционную смесь промыли в экстракторе 60 л EtOAc. Органическую фазу промыли 16 л 5% водного раствора Na2CO3, 2х10 л дистиллированной воды и 2х6 л насыщенного водного раствора хлорида натрия. Смешанные водные промывки опять экстрагировали 20 л EtOAc, и органическую фазу промыли 2х3 л воды и 2х4 л насыщенного водного раствора хлорида натрия. Смешанные EtOAc экстракты концентрировали под вакуумом 10 миллибар при внутренней температуре < 20oC в 100 л концентраторе загрузочного типа до ~8 л. Изменения растворителя на циклогексан достигли путем медленного растворения в ~ 20 л циклогексана и повторного концентрирования до ~8 л. К суспензии добавили 5 л циклогексана и 280 мл EtOAc и смесь нагрели до образования флегмы, когда все перешло в раствор. Раствор охладили и при 58oC добавили затравку (10 г). Суспензию охладили до 22oC в течение 4-х часов и после 1 часа выдержки при 22oC продукт выделили фильтрацией. Фильтровальный осадок промыли 1,8 л циклогексана и сушили в вакуумной печи при 35oC при продувке азотом до получения 1,87 кг (77%, как показала HPLC, поверхность > 99,9%, R-изомер ниже уровня обнаружения) соединения 4 в виде слегка рыжевато-коричневого порошка, [ α]D 25=22,0o (с=0,20 MeOH). Точка плавления 107oC: 13C ЯМР (75 MHz, CDCl3, част. на миллион) 170,1; 154,5; 79,8; 58,7; 50,6; 46,6; 43,6; 43,4; 28,6; 28,3.
Хотя в предшествующем описании приведены принципы настоящего изобретения, а с целью его иллюстрации обеспечены примеры, следует понимать, что практика изобретения охватывает все возможные варианты, переделки и модификации, устранения или добавления методик и протоколов, описанных здесь, как входящие в область далее приведенной формулы изобретения и ее эквивалентов.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ HIV ПРОТЕАЗЫ | 1994 |
|
RU2137768C1 |
| ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ СИНТЕЗА ИНГИБИТОРОВ ВИЧ-ПРОТЕАЗ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1994 |
|
RU2125561C1 |
| СПОСОБ ОКИСЛЕНИЯ ΔСТЕРОИДНОГО АЛКЕНА, СОЕДИНЕНИЕ, СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЯ | 1995 |
|
RU2149875C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ, ИСПОЛЬЗУЕМЫХ ДЛЯ СИНТЕЗА ИНГИБИТОРОВ ВИЧ-ПРОТЕАЗЫ | 1994 |
|
RU2134263C1 |
| СПОСОБ РАЦЕМИЗАЦИИ ОПТИЧЕСКИ ЧИСТОГО ИЛИ ОБОГАЩЕННОГО ПИПЕРАЗИН-2- ТРЕТ.БУТИЛКАРБОКСАМИДНОГО СУБСТРАТА И РАЦЕМИЧЕСКИЙ 2-ТРЕТ-БУТИЛКАРБОКСАМИД-4-(3- ПИКОЛИЛ)ПИПЕРАЗИН | 1995 |
|
RU2135482C1 |
| СПОСОБ ПОЛУЧЕНИЯ АНТАГОНИСТОВ ЛЕЙКОТРИЕНА, ДИЦИКЛОГЕКСИЛАМИНОВАЯ СОЛЬ, СПОСОБЫ ПОЛУЧЕНИЯ КРИСТАЛЛИЧЕСКОЙ НАТРИЕВОЙ СОЛИ И 1-/МЕРКАПТОМЕТИЛ/- ЦИКЛОПРОПАНУКСУСНОЙ КИСЛОТЫ, КРИСТАЛЛИЧЕСКОЕ СОЕДИНЕНИЕ | 1994 |
|
RU2140909C1 |
| СПОСОБ ПОЛУЧЕНИЯ 5-(1,2,4-ТРИАЗОЛ-1-ИЛ)ТРИПТАМИНОВЫХ СОЕДИНЕНИЙ И 2-[5-(1,2,4-ТРИАЗОЛ-1-ИЛ- МЕТИЛ)-1Н-ИНДОЛ-3-ИЛ/ЭТИЛОВЫЙ СПИРТ | 1995 |
|
RU2138496C1 |
| ПРОИЗВОДНЫЕ ФЕНОКСИФЕНИЛУКСУСНОЙ КИСЛОТЫ | 1994 |
|
RU2139273C1 |
| СОЛЬ АНТАГОНИСТА CCR-2 | 2004 |
|
RU2317295C1 |
| СПОСОБ ПОЛУЧЕНИЯ ФЕНИЛЬНЫХ ГЕТЕРОЦИКЛОВ И ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ | 1995 |
|
RU2160257C2 |


Способ получения эпоксида формулы I включает следующие стадии (а) взаимодействие 1 эквивалента соединения формулы II с 1-2 эквивалентами галогентрующего агентапри (-40)-(100)oС в растворителе, смешанном с водным раствором слабого основания и (б) циклизация полученного на стадии (а) галогенгидрина формулы III добавлением основания в растворителе. Соединение формулы I - новое промежуточное соединение, которое используется в синтезе ингибиторов протеазы вируса иммунодефицитного состояния человека. 4 с. и 2 з. п. ф-лы, 2 табл.
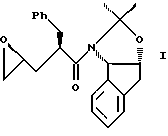
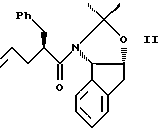
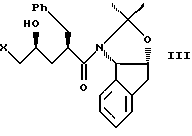
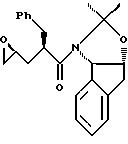
отличающийся тем, что проводят следующие стадии: (а) вводят в контакт один эквивалент аллилацетонида формулы II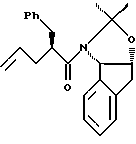
с галогенирующим агентом в количестве от около одного до двух эквивалентов в растворителе, смешанном с водным раствором слабого основания, при температуре в диапазоне между около -40°С и около 100°С с образованием галогенгидрина формулы III, и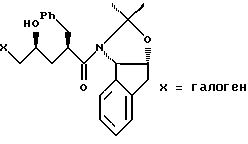
(b) добавляют основание в растворителе или растворителях для установления образования эпоксида формулы I.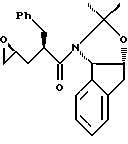
отличающийся тем, что проводят следующие стадии: (а) вводят в контакт один эквивалент ацетонида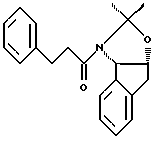
с приблизительно одним эквивалентом аллилгалогенида в сильном основании с получением аллилацетонида формулы II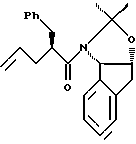
(b) смешивают с галогенирующим агентом в количестве от около одного до двух эквивалентов в растворителе, смешанном с водным раствором слабого основания, при температуре в диапазоне между около -40°С и около 100°С с образованием галогенгидрина формулы III, и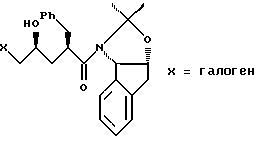
(с) добавляют основание в растворителе или растворителях для установления образования эпоксида формулы I.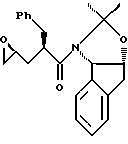
отличающийся тем, что проводят следующие стадии: (а) вводят в контакт один эквивалент аллилацетонида формулы II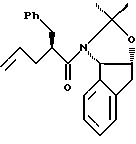
с галогенирующим агентом в количестве от около одного до двух эквивалентов в растворителе, смешанном с водным раствором слабого основания, при температуре в диапазоне между около -40°С и около 100°С с образованием галогенгидрина формулы III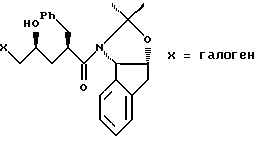
при этом галогенирующий агент выбирают из группы, состоящей из N-иодсукцинимида, N-бромсукцинимида или N-хлорсукцинимида, два последних из которых можно смешать с иодидной солью; растворитель выбирают из группы, состоящей из дихлорметана, изопропилацетата (IРАС), этилацетата (EtOAc), диметоксиэтана (DМЕ) и метил-трет-бутилового эфира (МIВЕ); слабое основание выбирают из основного оксида алюминия или бикарбоната натрия, и (b) добавляют основание в воде для установления образования эпоксида формулы I, при этом основание выбирают из группы, состоящей из гидроксида лития, гидроксида натрия. гидроксида калия, тетраалкиламмонийгидроксида; С1-С4-алкоксида лития, натрия ИЛИ калия; и диизопропилэтиламин.
отличающийся тем, что проводят следующие стадии: (а) вводят в контакт один эквивалент ацетонида
с приблизительно одним эквивалентом аллилгалогенида в сильном основании, при этом аллилгалогенид выбирают из аллилхлорида, аллилбромида и аллилиодида, с получением аллилацетонида формулы II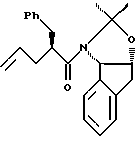
(b) смешивают с галегенирующим агентом в количестве от около одного до двух эквивалентов в растворителе, смешанном с водным раствором слабого основания, при температуре в диапазоне между около -40°С и около 100°С с образованием галогенгидрина формулы III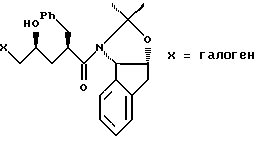
при этом галогенирующий агент выбирают из группы, состоящей из N-иодсукцинимида, N-бромсукцинимида, или N-хлорсукцинимида, два последних из которых можно смешать с иодидной солью; растворитель выбирают из группы, состоящей из дихлорметана, изопропилацетата (IРАС), этилацетата (EtOAc), диметоксиэтана (DME) и метил-трет-бутилового эфира (МТВЕ); слабое основание выбирают из основного оксида алюминия или бикарбоната натрия, и (с) добавляют основание в воде для установления образования эпоксида формулы I, при этом основание выбирают из группы, состоящей из гидроксида лития, гидроксида натрия, гидроксида калия, тетраалкиламмонийгидроксида; C1-4-алкоксида лития, натрия или калия; и диизопропилэтиламин.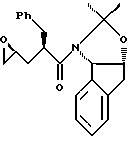
отличающийся тем, что проводят следующие стадии: (а) вводят в контакт один эквивалент аллилацетонида формулы II,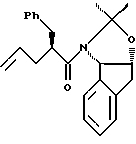
растворенного в изопропилацетате, с N-иодсукцинимидом в количестве от около одного до двух эквивалентов в ~0.5 М водном растворе бикарбоната натрия при комнатной температуре с образованием иодогидрина формулы III, и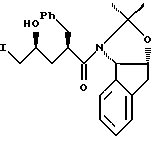
(b) добавляют гидроксид щелочного металла в воде для установления образования эпоксида формулы I.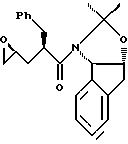
отличающийся тем, что проводят следующие стадии: (а) вводят в контакт один эквивалент ацетонида,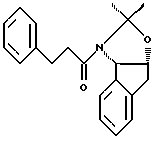
растворенный в эфирном растворителе, с приблизительно одним эквивалентом аллилбромида и приблизительно одним эквивалентом ~1.0 - 2.0 М литийгексаметилдисилазида (в эфирном растворителе) с получением аллилацетонида формулы II,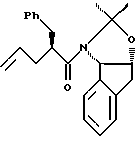
(b) смешивают с ним N-иодсукцинимид в количестве от одного до двух эквивалентов в ~ 0.5 М водном растворе бикарбоната натрия при комнатной температуре с образованием иодогидрина формулы III,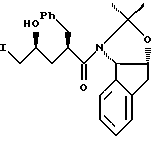
и (с) добавляют гидроксид щелочного металла в воде для установления образования эпоксида формулы I.
| 0 |
|
SU158577A1 | |
| Устройство для возведения двоичных чисел в степень | 1974 |
|
SU541168A1 |
| EP 0521686 A1, 1993 | |||
| David Askin et al | |||
| Highly diastereoselective reaction of a chiral non-racemic amide enolate with (s)-glicidyl tosilate | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Tetrahedron Letters, 1994, v.35, № 5, р.673 - 676. | |||

