Современная терапия для регуляции повышенного внутриглазного давления (ВГД) или повышенного глазного давления, которые, как полагают, являются фактором появления и прогрессирования глаукомы, обычно заключаются в применении различных средств местного применения, которые делятся на 4 категории: β -блокаторы, симпатомиметические средства, парасимпатомиметические средства и ингибиторы холинэстеразы. Практикуется вспомогательное пероральное введение ингибитора карбоангидразы (CA1), в том случае, когда побочные действия описанных выше средств местного применения ограничивают их применение и/или не удается достичь адекватной регуляции (ВГД). Активные при пероральном введении CA1 могут обнаруживать серьезные побочные эффекты, такие как анорексия, желудочно-кишечное расстройство и парастезиаз. Поэтому был предпринят интенсивный и продолжающийся поиск активных при местном введении CA1, не обнаруживающих таких побочных действий вследствие способа введения и специфичности органа-мишени. Этот поиск привел к обнаружению класса соединений, раскрытых в Baldwin et al. (US Patent 4797413), общей формулы ,
,
в которой R и R1 обозначают низший алкил, в частности, дорзоламида, в котором R обозначает метил и R1 обозначает метил. US патент 4797413 описывает новые ароматические сульфонамиды, применимые для лечения повышенного глазного давления, и их композиции.
Реакция Риттера хорошо известна в этой области и она состоит в обработке алифатического гидроксила нитрилом и сильной кислотой с образованием амида.
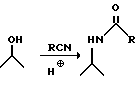
Реакция проходит через образование иона карбония в месте присоединения -OH так, что если в исходном материале имеется хиральный центр, то хиральность теряется во время реакции и образуется рацемический продукт.
В изобретении реакцию Риттера применяют для введения содержащей азот группы в 4-е положение молекулы, причем исходным материалом является чистый этантиомер, и хиральность неожиданно сохраняется в получаемом продукте.
Это изобретение касается способа синтеза соединения дорзоламидного типа с высоким выходом и высокой энантиомерной частотой. Ключевой стадией в этом новом способе является реакция Риттера с неожиданной и непредсказуемой тенденцией проходить с сохранением хиральности.
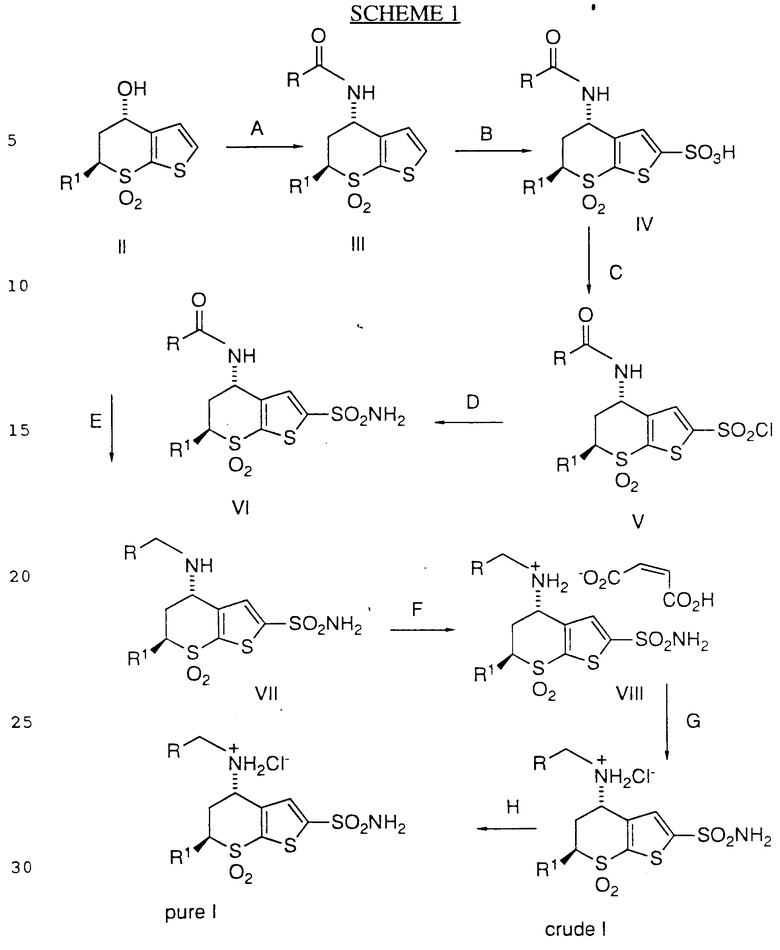
Новый способ изобретения может быть изображен, как показано в cхеме 1 (см. в конце текста).
где R и R1, одинаковые или различные, обозначают C1-3-алкил.
Стадия A схемы 1 является реакцией Риттера или ее модификацией, предусматривающей медленное добавление приблизительно 10-15-кратного молярного избытка сильной кислоты, такой как концентрированная серная кислота, или смеси концентрированной серной кислоты и дымящей серной кислоты к перемешиваемому холодному раствору 11 в нитриле структуры RCN, где R обозначает C1-3-алкил, такой как метил, этил, пропил или изопропил. Количество воды, присутствующей во время реакции Риттера является критическим для оптимального сохранения хиральности и оно варьируется (0,5-10%) в зависимости от применяемой кислоты. Промышленно доступная серная кислота может вводить слишком много воды в систему и ее количество уменьшают добавлением дымящей серной кислоты. Однако безводные кислоты, такие как метансульфоновая, трифторуксусная или эфират трехфтористого бора, требуют добавления воды к реакции. Удовлетворительными являются температуры приблизительно -20 - 0oC, особенно -5oC. После добавления кислоты смеси дают нагреться спонтанно при перемешивании до завершения реакции приблизительно в течение 12-18 ч. Реакцию останавливают добавлением смеси к воде, кислоту нейтрализуют добавлением основания, такого как гидроксид натрия, и продукт экстрагируют органическим растворителем, таким как этилацетат.
Стадия B включает сульфонилирование 111 путем добавления его к охлажденной хлоросульфоновой кислоте или дымящей серной кислоте приблизительно при 0oC при такой скорости, чтобы поддерживать температуру ниже приблизительно 20oC. Образующуюся смесь затем нагревают при приблизительно 40-60oC до завершения реакции в течение приблизительно 10-15 ч. Этот материал непосредственно используют в следующей стадии.
Стадия C, хлорирование, представляет собой медленное добавление тионилхлорида к охлажденному (15-25oC) раствору IV с последующим нагреванием при приблизительно 40-60oC в течение 4-8 ч.
Реакцию останавливают медленным добавлением смеси к перемешиваемой охлажденной воде с последующим сбором продукта фильтрованием.
Стадия D, или процесс амидирования, с образованием сульфонамида  предусматривает медленное добавление
предусматривает медленное добавление  к охлажденному (-15 - 0oC) раствору водного аммиака в THF при скорости, достаточной для поддержания температуры ниже приблизительно 0oC с последующим перемешиванием при приблизительно 0oC в течение приблизительно 0,5-2 ч. Продукт выделяют доведением pH до 3-5 концентрированной серной кислотой, отделением органических слоев, разбавлением водой и концентрированием, вызывающим кристаллизацию.
к охлажденному (-15 - 0oC) раствору водного аммиака в THF при скорости, достаточной для поддержания температуры ниже приблизительно 0oC с последующим перемешиванием при приблизительно 0oC в течение приблизительно 0,5-2 ч. Продукт выделяют доведением pH до 3-5 концентрированной серной кислотой, отделением органических слоев, разбавлением водой и концентрированием, вызывающим кристаллизацию.
Стадия E включает в себя восстановление карбониламида соединения VI путем медленного добавления кислоты Льюиса, такой как эфират трифтористого бора, или хлористого алюминия, или безводной сильной кислотой, такой как метансульфоновой кислоты или трифторуксусной кислоты, к перемешиваемой суспензии VI и борогидрида натрия в сухом THF при приблизительно (-5) - (+5)oC с последующим перемешиванием в течение приблизительно 4-6 ч при (-5) - (+5)oC, а затем в течение приблизительно 12-18 ч при приблизительно 25-40oC. По завершении реакции реакционную смесь медленно добавляли к охлажденной разбавленной кислоте с последующим выделением согласно стандартным способам, если желательно. Специалисту в данной области понятно, что описанная выше реакция с борогидридом натрия в сухом THF и кислотой, такой как эфират трифтористого бора или метансульфоновая кислота, приводит к получению борантетрагидрофурана в процессе образования соединения VII. Альтернативно, соединение VII может быть получено восстановлением соединения VI боран-тетрагидрофураном или борандиметилсульфидом, без применения кислоты.
Соль малеат VIII получают стандартными методами, превращают в неочищенную соль I соляной кислоты и перекристаллизуют с образованием чистого I.
Эти стадии реакции иллюстрируются примером, где R и R1 оба обозначают метил.
Продукт нового способа изобретения представляет собой эффективный при местном применении ингибитор карбоангидразы, применимый для лечения повышенного глазного давления. Его вводят непосредственно в глаз обычно в виде раствора, содержащего приблизительно 0,1-15 мас.% соединения, по 1 или 2 капли 1-4 раза в день.
Пример 1. Стадия A: Методика проведения реакции Риттера с серной кислотой
К механически перемешиваемому охлажденному (-5±5oC) раствору гидроксисульфона II (25,0 г, 0,114 моль: 98:2 транс/цис) в ацетонитриле (300 мл) медленно добавляют концентрированную серную кислоту (18 M, 86 мл, 1,52 моль) в течение 0,5 ч, поддерживая внутреннюю температуру при -5±5oC.
Смеси дают нагреться до 20±5oC и перемешивают при этой температуре в течение 12 - 18 ч или пока реакция не завершится (согласно ЖХВД).
Методика анализа: Аликвоту (0,1 мл) разбавляли до 50,0 мл водой и затем анализировали при помощи следующего способа ЖХВД.
Прибор: Spectra Physics 8800
Колонка: 4,1 х 250 мм Uetrasphere C-8 (Altex inc.)
Элюент А: H2O (0,1% (об./об.) H3PO4)
Элюент B: MeCN
Изократическая элюция: 87:13 A:B в течение 7 мин, затем
Градиентная элюция: 87:13 - 35:65 А:B в течение 14 минут
Скорость течения: 2,0 мл/мин.
Температура: 45oC
Инъекция: 10,0 мкл
Детектирование% УФ (230 нм)
Время удерживания: Гидроксисульфон II (цис-изомер) 6,0 мин. Гидроксисульфон II (транс-изомер) 6,6 мин. Ацетамидосульфон III (цис-изомер) 7,6 мин. Ацетамидосульфон III (транс-изомер) 8,5 мин.
Реакцию считали завершенной, когда оставалось менее 1% гидроксисульфона II (US. продукта ацетамидосульфона III). В конце реакции отношение транс/цис ацетамидосульфона III в продукте было 92,4:7,6.
После завершения реакции реакционную смесь медленно добавляли к перемешиваемой механически предварительно охлажденной (0 - 5oC) гасящей реакцию смеси этилацетата (1,7 л) и воды (800 мл). В то же самое время 50% (мас. /мас. ) водного гидроксида натрия (185 мл) добавляли к гасящей смеси при такой скорости, что pH поддерживался между 3 и 5 и внутренняя температура поддерживалась ниже 25oC. Затем pH доводили до 7,0 - 7,5 дополнительным добавлением гидроксида натрия и смесь перемешивали 1 ч при 30oC. Смесь фильтровали для удаления сульфата натрия и фильтровальный осадок промывали этилацетатом (300 мл). Фильтрат и промывки осадка объединяли и смесь разделяли на фазы. Органические (верхние) фазы объединяли и затем концентрировали в вакууме [10 мБар (1• 103Па), 50oC] до объема 100 мл. Гексан (300 мл) добавляли медленно и смесь перемешивали в течение 1 ч при 20 - 22oC. Смесь фильтровали и фильтровальный осадок промывали гексаном (1 объем слоя). Продукт сушили на воздухе, затем сушили в вакууме [100 мБар (1 • 104Па) ток азота, 30 - 35oC] до постоянного веса.
Выход: 31,0 г (95% чистоты согласно HPLC, мас.%) неочищенного ацетамидосульфона III в виде белого твердого вещества. Неочищенный продукт содержит также небольшое количество ацетамида и ацетата натрия.
1H-ЯМР: (DMSO - d6) δ 8,57 (шир. д, 1H, J = 8,5 Гц), 8,53 (шир. д, 1H, J = 11,7 Гц), 7,96 (д, 1H, J = 5,0 Гц), 7,94 (д, 1H, J = 5,0 Гц), 7,03 (д, 1H, J = 5,0 Гц), 6,95 (д, 1H, J = 5,0 Гц), 5,21 - 5,14 (м, 2H), 3,84 - 3,76 (м, 2H), 2,51 - 2,36 (м, 2H), 2,29 - 2,2 (м, 2H), 1,84 (c, 3H), 1,75 (c, 3H), 1,35 (д, 3H, J = 6,8 Гц), 1,32 (д, 3H, J = 6,2 Гц).
ЖХВД: 93:7 транс/цис (описанный выше способ)
Микроанализ: Аналитически рассчитано для C10H13NO3S2:
C 46,32, H 5,05, N 5,40, S 24,73.
Определено: C 46,41, H 4,94, N 5,34, S 24,55.
Стадия А (Альтернатива): Методика проведения реакции Риттера с серной кислотой/дымящей серной кислотой
К механически перемешиваемому охлажденному (-5±5oC) раствору гидроксисульфона 11 (10,0 г, 45,8 ммоль, 98:2 транс/цис) в ацетонитриле (50 мл) медленно добавляли концентрированную серную кислоту (18 М, 9,0 мл, 162 моль), поддерживая внутреннюю температуру при 10oC, с последующим добавлением 30% дымящей серной кислоты (1,2 мл). Смесь перемешивали в течение 2 ч при 15 - 20oC, затем 3 ч при 20 - 22oC, (при температуре выше 25oC образуются значительные количества ацетамида). Ход реакции наблюдали при помощи ЖХВД (способ описан в примере 1). Реакцию считали законченной, когда оставалось менее 1% гидроксисульфона II (US. продукта ацетамидосульфона III). В конце реакции отношение транс/цис продукта ацетамидосульфона III было 93,5:6,5. После завершения реакции смесь выливали в лед (100 г) и pH смеси доводили до 3,5 - 5,5 путем медленного добавления 50% водного гидроксида натрия (приблизительно 20 мл). Смесь экстрагировали этилацетатом (2 х 100 мл). Экстракты объединяли и промывали солевым раствором (1 х 50 мл). Затем раствор концентрировали в вакууме [100 мБар (1 • 104Па), 35 - 40oC] до объема 20 мл. Добавляли этилацетат (100 мл) и концентрирование повторяли (конечный объем 20 мл) для гарантии полного удаления ацетонитрила. Гексан (100 мл) добавляли медленно в смесь перемешивали 2 ч при 20 - 22oC (1 объем слоя). Продукт сушили на воздухе, затем сушили в вакууме [100 мБар (1 • 104Па), ток азота, 30 - 35oC] до постоянного веса.
Выход: 11,5 г (97%) неочищенного ацетамидосульфона III в виде белого твердого вещества. В этом случае неочищенный продукт не содержит ацетамида и ацетата натрия.
1H-ЯМР: совпадающий.
ЖХВД: 93,5 : 6,5 транс/цис (способ описан выше).
Стадия B: Методика сульфонилирования
К механически перемешиваемой, охлажденной (0oC) хлоросульфоновой кислоты (70 мл, 1,05 моль) добавляли неочищенный ацетамидосульфонамид III (29,7 г, 0,114 моль, 93: 7) по частям при скорости, пригодной для поддержания внутренней температуры < 20oC. Темную реакционную смесь для сульфонилирования нагревали до 50oC в течение 12 ч или до завершения реакции согласно ЖХВД. (Примечание: во время реакции выделялась соляная кислота (0,114 моль).
Методика анализа: Аликвоту (0,1 мл) разбавляли до 100,0 мл водой и затем анализировали согласно следующему способу ЖХВД.
Прибор: Spectra Physics 88800/
Колонка: 4,1 х 250 мм Ultrasphere C-8 (Altex Inc.)
Элюент А: H2O (0,1% об./об. H3PO4).
Элюент B: MeC/
Градиент: 97:3 - 35:65 A:B в течение 25 мин.
Скорость течения: 2,0 мл/мин.
Температура: 45oC.
Инъекция: 10,0 мкл.
Время удерживания: Сульфоновая кислота IV (цис/транс изомеры) 5,0 мин. Ацетамидосульфон III (цис-изомер) 9,0 мин. Ацетамидосульфон III (транс-изомер) 10,0 мин.
Реакцию сульфонилирования считали завершенной, когда оставалось менее 1% ацетамидосульфона III (US. сульфоновой кислоты IV).
Стадия C: Методика хлоросульфонилирования
После завершения реакции стадии B смесь охлаждали до 20oC. Затем медленно добавляли тионилхлорид (70 мл, 0,96 моль) при скорости, позволяющей контролировать выделение гидрогенхлорида (0,114 моль) и диоксида серы (0,114 моль). После добавления смесь нагревали до 50oC в течение 6 ч или до завершения реакции согласно ЖХВД.
Методика анализа: Аликвоту (0,1 мл) разбавляли до 50,0 мл ацетонитрилом и затем сразу анализировали согласно описанному выше способу ЖХВД (для уменьшения гидролиза сульфонилхлорида V).
Время удерживания: Сульфоновая кислота IV (цис/транс-изомеры) 5,0 мин. Сульфонилхлорид V (цис/транс-изомеры) 19 мин.
Реакцию считали завершенной, когда оставалось менее 1% сульфоновой кислоты IV (US. продукта сульфонилхлорида V). После завершения реакции смесь охлаждали до 15 - 20oC и затем дозировали медленно в интенсивно перемешиваемую воду (1,4 л), предварительно охлажденную до 0 - 5oC, при скорости, позволяющей поддерживать температуру < 5oC. (Примечание: внутренняя температура не должна превышать 5oC для уменьшения гидролиза продукта сульфонилхлорида V).После добавления приблизительно 10% реакционной смеси, погашенную смесь можно охлаждать далее до -5±5oC. Во время гашения реакции образуются значительные количества соляной и сернистой кислоты. Смесь перемешивали в течение 1 ч при 0 - 5oC, фильтровали и отфильтрованный осадок затем промывали холодной (5oC) водой ( 1 л). Отфильтрованный осадок хорошо отсасывали для удаления как можно большего количества воды.
Выход: 68 г неочищенного сульфонилхлорида Y в виде влажного твердого вещества (примерно 40 мас.% воды), который сразу использовали в следующей стадии.
1H-ЯМР: (CDCl3) δ 7,74 (c, 1H), 8,07 (шир.д, J=8,1 Гц), 5,45 - 5,35 (м, 1H), 3,63 - 3,56 (м, 1H), 2,64 - 2,56 (м, 2H), 2,09 (с, 3H), 1,57 (д, 1H, J = 6,9 Гц).
Стадия D: Методика амидирования
К механически перемешиваемому, охлажденному (-10±5oC) раствору концентрированного водного аммиака (15 M, 43 мл), 0,65 моль) в тетрагидрофуране (THF, 300 мл) добавляли по частям сульфонилхлорид Y (68 г влажного, примерно 40,9 г, 0,114 моль) при скорости, поддерживающей внутреннюю температуру ниже 0oC. После завершения добавления смесь перемешивали при 0 - 5oC в течение 1 ч или до полного завершения реакции согласно ЖХВД.
Методика анализа: Аликвоту (0,1 мл) разбавляли до 50,0 мл ацетонитрилом, и затем сразу анализировали по способу ЖХВД, описанному в примере 3 (для уменьшения гидролиза исходного материала сульфонилхлорида V).
Время удерживания: Ацетамидосульфонамид VI (цис-изомер) 9,0 мин. Ацетамидосульфонамид VI (транс-изомер) 10,0 мин. Сульфонилхлорид V (цис/транс-изомер) 19 мин.
Реакцию считали завершенной, когда оставалось менее 1% сульфонилхлорида V (US. продукта ацетамидосульфонамида VI). После завершения реакции pH смеси доводили до 3 - 5 добавлением по каплям концентрированной серной кислоты (18 M, приблизительно 12,2 мл, 0,218 моль), поддерживая внутреннюю температуре ниже 20oC. Смеси давали осесть и слои разделяли. Водную (нижнюю) фазу экстрагировали THF (70 мл). Два органических слоя объединяли и затем разбавляли водой (250 мл). Затем раствор концентрировали перегонкой до объема 125 мл. Во время концентрирования продукт спонтанно кристаллизовался. Суспензию разбавляли водой до объема 250 мл и смесь затем перемешивали в течение 12 - 18 ч при 20 - 25oC. Смесь фильтровали и фильтровальный осадок промывали водой (150 мл). Продукт сушили на воздухе, затем сушили в вакууме [100 мБар, 1•104 Па), ток азота, 55oC] до постоянного веса.
Выход: 29,5 г (выход 76% от гидроксисульфона 11) ацетамидосульфонамида VI в виде белого кристаллического твердого вещества.
ЖХВД: 95:5 транс/цис (способ описан выше).
1H-ЯМР: (DMSO-d6) δ 8,65 (шир. д, 1H, J = 9,5 Гц), 8,60 (шир. д, 1H, J = 9,5 Гц), 8,05 (шир. с. 4H), 7,42 (с, 1H), 7,31 (с, 1H), 5,32 - 5,15 (м, 2H), 4,10 - 3,80 (м, 2H), 2,53 - 2,41 (м, 2H), 2,34 - 2,18 (м, 2H), 1,91 (с, 3H), 1,87 (с, 3H), 1,37 (д, 3H, J = 7,0 Гц), 1,34 (д, 3H, J = 7,6 Гц).
Микроанализ: Аналитически рассчитано для C10H14O5N2S3:
C 35,49, H 4,17, N 8,28, S 28,42.
Определено: C 35,60, H 4,04, N 8,21, S 28,40.
Стадия E: Методика восстановления через образованный in situ боран
К механически перемешиваемой, охлажденной (0 - 5oC) суспензии ацетамидосульфонамида VI (29,5 г, 87,1 моль, 95:5 транс/цис) и борогидрида натрия (16,9 г, 447 ммоль) в сухом THF (290 мл) добавляли неразбавленный эфират трихлористого бора (8,13 M, 73 мл, 593 ммоль) в течение 0,5 ч, поддерживая внутреннюю температуру ниже 5oC. (Предостережение: во время реакции образуется водород, так как борогидрид натрия и/или диборан реагирует с протонами сульфонамида). После завершения добавления смесь перемешивают в течение 5 ч при 0 - 5oC и затем при 30 - 35oC в течение 12 - 18 ч или до завершения реакции согласно ЖХВД.
Методика анализа: Аликвоту (0,1 мл) разбавляли до 50,0 мл водой и затем анализировали способом ЖХВД, описанным в примере 3.
Время удерживания: Аминосульфонамид VII (цис-изомер) 4,5 мин.
Аминосульфонамид VII (транс-изомер) 5,0 мин, Ацетамидосульфонамид VI (цис-изомер) 9,0 мин. Ацетамидосульфонамид VI (транс-изомер) 10,0 мин. Комплекс амин-боран 14 - 20 мин.
Реакцию считали законченной, когда оставалось менее 1% ацетамидосульфонамида VI (US. аминосульфонамида VII). После завершения реакции реакционную смесь добавляли медленно к механически перемешиваемому, предварительно охлажденному (0 - 5oC) раствору 1 M водной серной кислоты (400 мл) при такой скорости, чтобы поддерживать внутреннюю температуру ниже 20oC (предостережение: во время гашения реакции выделяется водород). Смесь перемешивали в течение 2 ч при 20 - 25oC или до тех пор, пока не прекращалось выделение водорода. Затем смесь концентрировали перегонкой (1 атм.) до объема 400 мл. Полученный водный раствор охлаждали до 10oC и pH осторожно доводили до 4 - 5 добавлением по каплям 50% водного гидроксида натрия (например 37 мл, 0,7 моль), поддерживали внутреннюю температуру ниже 20oC. Добавляли этилацетат (600 мл) и pH доводили далее до 7,5 - 8,0 путем добавления насыщенного водного бикарбоната натрия (примерно 75 мл, 90 ммоль). Смесь фильтровали для удаления сульфата натрия, образовавшегося во время первоначального доведения pH, и осадок на фильтре промывали этилацетатом (100 мл). Фильтрат и промывки осадка объединяли и полученную смесь разделяли на фазы. Водную (нижнюю) фазу экстрагировали этилацетатом (100 мл). Органические слои объединяли и затем промывали солевым раствором (100 мл). Этот раствор, содержащий неочищенный продукт аминосульфонамид VII (примерно 27,9 г), использовали в таком виде в следующей стадии.
ЖХВД: 95:5 транс/цис (способ описан выше)
Стадия F: Методика образования соли малеиновой кислоты
Раствор в этилацетате аминосульфонамида VII (примерно 27,9 г, 86 ммоль, 95:5 транс/цис) из стадии 5 концентрировали дистилляцией (1 атм) (1,0133•105 Па) до объема 70 мл. Добавляли ацетоном (250 мл) и концентрирование повторяли до объема 70 мл. Эту операцию повторяли, концентрируя на этот раз до объема 160 мл. Добавляли малеиновую кислоту (9,98 г, 86 ммоль). Смесь перемешивали, пока соль не кристаллизовалась, и затем перемешивали в течение 12 - 18 ч при 20 - 22oC. Смесь фильтровали и полученный осадок промывали ацетоном (1 объем слоя). Продукт сушили на воздухе, затем сушили в вакууме [100 мБар, (1•104 Па), ток азота, 75oC] до постоянного веса.
Выход: 33,0 г (92%) соли малеата VIII в виде белого кристаллического твердого вещества.
ЖХВД: 99:1 транс/цис (способ описан выше)
1H-ЯМР: (DMSO - d5) δ 8,17 (шир. с, 2H), 7,81 (с, 1H), 6,05 (с, 2H), 4,61 (шир. с, 1H), 4,08 - 4,00 (м, 1H), 3,24 - 3,14 (м, 1H), 3,06 - 2,93 (м, 1H), 2,7 - 2,45 (м, 2H), 1,39 (д, 3H, J=6,7 Гц), 1,20 (т, 3H, J=7,1 Гц).
Микроанализ: Аналитически рассчитано для C14H20N2O4S3:
C 38,17 H 4,58, N 6,39, S 21,83.
Определено: C 38,19, H 4,58, N 6,29, S 21,60.
Стадия G: Методика получения неочищенной соли соляной кислоты
К механически перемешиваемой смеси этилацетата (250 мл) и насыщенного водного бикарбоната натрия (120 мл) добавляли соль малеиновой кислоты VIII (33,0 г, 75 ммоль 99: 1 транс/цис). Смесь перемешивали при 20 - 25oC до полного растворения твердого вещества, когда две фазы становились прозрачными. Смеси давали отстояться и затем разделяли слои. Водную (нижнюю) фазу экстрагировали этилацетатом (50 мл). Органические слои объединяли и затем промывали насыщенным водным хлоридом натрия (50 мл). К хорошо перемешанному раствору в этилацетате медленно добавляли концентрированную соляную кислоту (12 M, 6,25 мл, 75 ммоль). Во время добавления продукт кристаллизовался. Смесь концентрировали в вакууме (200 мБар (2•104Па), заменяя этилацетат по необходимости, до тех пор, пока содержание воды раствора не становилось менее 0,1 мг/мл при объеме 150 мл. Смесь охлаждали до 20 - 22oC и затем перемешивали в течение 12 - 18 ч при этой температуре. Смесь фильтровали и осадок на фильтре промывали этилацетатом (2х55 мл). Продукт сушили на воздухе, затем сушили в вакууме [100 мБар (1•104 Па) ток азота, 45 - 50oC] до постоянного веса.
Выход: 26,4 г (98% выход, 64% общий выход из гидроксисульфона 11) неочищенного гидрохлорида аминосульфонамида 1 в виде белого кристаллического твердого вещества.
ЖХВД: более 99% (способ описан выше).
Стадия H: Методика перекристаллизации
Механически перемешиваемую суспензию неочищенной соли гидрохлорида аминосульфонамида 1 (26,4 г, 73 ммоль) в воде (70 мл) нагревали при 90 - 95oC до полного растворения твердого вещества. К горячему раствору добавляли активированный уголь (Darco кв® , 0,26 г) и смесь перемешивали в течение 15 мин при 90 - 95oC. Смесь фильтровали в горячем виде (85 - 90oC) через хорошо промытый слой ускорителя фильтрования (Super Cel). Фильтрованный осадок промывали кипящей водой (9 мл). Фильтрат и промывку осадка объединяли и продукту давали кристаллизоваться путем охлаждения хорошо перемешиваемого раствора до 60oC. Смесь перемешивали в течение 1 ч при 60oC или до тех пор, пока продукт не превращался в термодинамически более стабильную полугидратную кристаллическую форму. Затем смесь медленно охлаждали до 3oC и затем перемешивали в течение 1 ч при этой температуре. Смесь фильтровали холодным фильтрованием, используя маточный раствор для ополаскивания осадка. Продукт сушили на воздухе, затем сушили в вакууме [100 мБар (1•104 Па), ток азота, 45 - 50oC] до постоянного веса.
Выход: 24,2 г (92% выход, 59% общий выход из гидроксисульфона 11) чистой соли гидрохлорида аминосульфонамида 1 в виде белого кристаллического твердого вещества.
ЖХВД: 99,9% (по площади) (254 нм); 99,6 мас.% против наружного стандарта; 99% (4,6) в виде производного N-TFA;
Удельное вращение: /(α)589/589 = -17,1oC (с = 1,00, H2O)
Т.пл.: 238oC (DS C, наклон 2oC /мин.)
1H-ЯМР (DMSO - d6) δ 9,91 (шир. с., 1H), 9,63 (шир. с, 1H), 8,21 (c, 2H), 8,02 (с, 1H), 4,68 (шир. с., 1H), 4,37 (м, 2H), 3,19 (шир. с., 1H), 3,04 (шир. с, 1H), 2,80 (д, 1H), 2,55 (м, 1H), 1,39 (д, 3H), 1,29 (д, 3H).
13C-ЯМР: (DMSO - d6) δ 149,7 (с), 141,9 (с), 137,4 (с), 130,7 (с), 51,6 (с), 49,2 (с), 40,8 (с), 30,7 (с), 11,1 (с), 10,0 (с).
Микроанализ: Аналитически рассчитано для C10H17N2O4S3Cl:
C 33,28, H 4,75, N 7,76, S 26,66, Cl 9.84.
Определено: C 33,33, H 4,70, N 7,67, S 26,60, Cl 9,77.
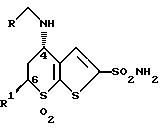
7,7-Диоксид 5,6-дигидро-4-(этиламино)-6-метил-5H-тиено (2,3-в) тиопиран-сульфонамида получают взаимодействием гидроксипроизводного 7,7-диоксида тиено (2,3-в) тиопирана с ацетонитрилом с последующим введением во второе положение гетероциклической системы сульфонамидной группы. Настоящий способ позволяет провести синтез с сохранением хиральности молекулы. Соединение используется в фармакологии для регуляции внутриглазного давления. 2 с. и 6 з.п.
где R и R1 одинаковые или различные и представляют собой C1 - C3-алкил,
в оптической форме по C-6 и в кристаллическом виде с сохранением трансстереохимического соотношения между заместителями C-4 и C-6, или его хлористоводородной соли, отличающийся тем, что а) соединение общей формулы II
обрабатывают нитрилом формулы
RCN
и сильной кислотой так, чтобы количество присутствующей воды, зависящее от используемой кислоты, было в пределах 0,5 - 10,0 мас.%, с образованием соединения общей формулы III
с последующей b обработкой соединения III хлоросульфоновой кислотой с образованием соединения формулы IV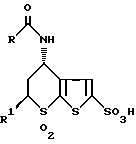
c обработкой соединения IV тионилхлоридом с образованием соединения формулы V
d обработкой соединения V аммиаком с образованием соединения формулы VI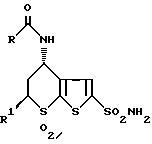
e обработкой соединения VI борогидридом натрия и синильной кислотой или комплексом боран-тетрагидрофуран или борандиметилсульфид с образованием соединения формулы I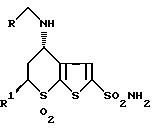
f выделением соединения I в свободном виде или в виде соли малеиновой кислоты VIII, g превращением соединения VIII в соль хлористоводородной кислоты I и h очисткой гидрохлорида соединения формулы I.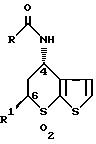
в котором фиксирована хиральность при C-6 и сохраняется трансстереохимическое соотношение между C-4 и C-6 заместителями;
R и R1 являются одинаковыми или различными и представляют собой C1-C3-алкил,
отличающийся тем, что соединение формулы II
обрабатывают нитрилом формулы
RCN
и сильной минеральной кислотой так, чтобы количество присутствующей воды, зависящее от используемой кислоты, было в пределах 0,5 - 10,0 мас.%.
| US 4797413, 1989, A 61 K 31/38 | |||
| EP, 296879, 1988, C 07 D 495/04. |

