Данная заявка является частичным продолжением заявки США N 07/562180, поданной 3 августа 1990 г.; заявки США N 07/582287, поданной 13 сентября 1990 г.; заявки США N 07/582456, поданной 13 сентября 1990 г.; и заявки США N 07/582457, поданной 13 сентября 1990 г.
Настоящее изобретение относится к соединениям, композициям и методам ингибирования генной экспрессии. Предлагаемые соединения содержат
(1) олигонуклеозидные последовательности, включающие примерно от 6 до примерно 200 оснований, с трехатомным мостиком между нуклеозидами или
(2) олигонуклеозидные последовательности, включающие примерно от 9 до 200 оснований, с диолом у любого или обоих их концов.
Антисмысловое соединение относится к любому соединению, которое связывается или гибридизируется с нуклеотидной последовательностью в любой нуклеиновой кислоте, то есть ДНК или РНК, и тем самым ингибирует функцию или синтез указанной нуклеиновой кислоты. Вследствие способности антисмысловых соединений к гибридизации как с РНК, так и с ДНК, такие соединения могут препятствовать экспрессии генов на уровне транскрипции, процессинга РНК или трансляции.
Антисмысловые молекулы можно построить и синтезировать для предотвращения транскрипции специфических генов на мРНК путем гибридизации с геномной ДНК и ингибирования прямо или косвенно действия РНК полимеразы. Преимущество доставки ДНК к мишени заключается в том, что для обеспечения лечебного эффекта необходимо только незначительные количества антисмысловых соединений. В альтернативном варианте антисмысловые соединения можно сконструировать и синтезировать гибридизацией с РНК для ингибирования механизмов пост-транскрипционной модификации (РНК процессинга) или синтеза белка (трансляции). В качестве примеров РНКаз-носителей можно указать матричную РНК (мРНК), транспортную РНК (тРНК), рибосомную РНК (рРНК) и тому подобное. К примерам механизмов процессинга и трансляции относится сплайсинг пре-мРНК с удалением нитронов, кэппинг 5' - конца мРНК, остановка гибридизации и нуклеаза-опосредованный мРНК-гидролиз.
В настоящее время разработка практических применений антисмысловых технологий в научных и терапевтических целях, однако, затруднена рядом технических задач, Klausner, A, Biotechnology, 8: 303 - 304, 1990 г. Синтетические антисмысловые молекулы подвержены быстрой деградации нуклеазой, которая присутствует в клетках-мишенях. Олигонуклеозидные последовательности антисмысловых ДНК или ЗНК, например разрушаются экзонуклеазами, действующими как у 5'-, так и 3'- конца нуклеиновой кислоты. Кроме того, эндонуклеазы могут расщеплять ДНК или РНК по внутренним фосфодиэфирным связям между отдельными нуклеозидами. В результате такого расщепления эффективный полупериод жизни вводимых антисмысловых соединений очень короткий, что неизбежно приводит к использованию больших, часто принимаемых лекарственных доз.
Другая проблема связана с чрезвычайно высокой себестоимостью ДНК или РНК при использовании для их получения имеющихся в настоящее время полуавтоматических синтезаторов ДНК. Недавно получены расчетные данные о том, что производство одного грамма антисмысловой ДНК составляет примерно 100 000 ам. долларов (Armstrong, L., Business Week, 5 марта 1990, стр. 89).
Следующая проблема относится к доставке антисмысловых веществ к органу - или клетке-мишени. Антисмысловые вещества должны иметь доступ в клеточное ядро (то есть такие вещества должны проникать в плазменную или ядерную мембрану). Необходимость увеличения мембранной проницаемости (повышенной гидрофобности) должна быть, однако, сбалансирована относительно потребности водной растворимости (повышенной гидрофильности) в пространствах организма с жидким содержимым, например, как плазменный и клеточный цитозоль.
Еще одна проблема связана с устойчивостью антисмысловых веществ независимо от того, действуют ли они самостоятельно в организме, или гибридизованы с нуклеиновой кислотой-мишенью. Олигонуклеотидные последовательности, например как антисмысловая ДНК, подвержены стерической рекомбинации вокруг хиральных фосфорных центров.
Доставка генных продуктов с помощью антисмысловых агентов - неизбежный следующий этап в производстве создания лекарственных препаратов для человека (см. выше Armstrong, стр. 88). Успешное применение антисмысловых технологий в терапевтических целях требует, однако, нахождения решений вышеуказанных задач.
Один подход решения задачи получения антисмысловых соединений, обладающих стабильностью, резистентностью к действию, нуклеазы, низкой себестоимостью, и способностью к доступу в и гибридизации с мишенями нуклеиновых кислот по всему организму, заключается в синтезировании олигонуклеозидных последовательностей с модификациями структуры нормальной фосфат-углеводной цепи.
В настоящее время, известно, по-существу, два типа олигонуклеозидов с модифицированными цепями. Первый тип включает в себя модификации нормальной межолигонуклеозидной фосфодиэфирной связи. Второй тип включает модификацию с заменой фосфодиэфирной связи на нефосфатные межолигонуклеозидные связи (Uhlmann, E. и Peyman, A. Chemical Reviews, 9 (4), стр. 544-584, 1990.
В качестве фосфодиэфирных связей, как известно, в настоящее время используют фосфотиоаты, алкилфосфотриэфиры, метилфосфонаты и алкилфосфорамидаты.
Фосфортиоат-модифицированные фосфодиэфирные связи означают фосфодиэфирные связи, где один или несколько атомов мостикового кислорода заменен серой. Такие связи, однако, не пригодны для использования в антисмысловых соединениях. Сохранение хирального фосфорного центра приводит к стерической модификации монотиоатов. Более того, как у моно- так и у дитиоатов отсутствует участок специфичной гибридизации, и оба быстро элиминируют из плазмы. Высокая аффинность фосфотиоатов к стеклу и пластику также делает синтез указанных соединений трудновыполнимым и неэффективным.
Метил- и этилфосфотриэфиры получены путем взаимодействия фосфодиэфир-связанных олигонуклеозидов с безводным метанолом или этанолом. (Miller, P.S. и др., J. Am. Chem. Soc., 93, стр. 6657-6665, 1971).
Триэфирная связь в олигодезоксирибонуклеотид-этилфосфотриэфирах обладает устойчивостью в обычных физиологических условиях pH, хотя оно его можно гидролизовать сильной кислотой или основанием. Метилфосфотриэфиры менее устойчивы, чем этил- и другие алкилфосфотриэфиры при нейтральной pH благодаря возможности нуклеофильного замещения метильной группы триэфира при введении растворителя. Олигодезоксирибонуклеотидэтилфосфотриэфиры, как оказывается, совершенно не восприимчивы к гидролизу нуклеазами, и не подвержены гидролизу нуклеазами или эстеразами, обнаруженными в фетальной телячьей сыворотке или сыворотке крови человека, (см. выше Uhlmann).
Метилфосфонаты имеют несколько существенных недостатков с точки зрения их лечебного потенциала, включая их слабую растворимость в воде, хиральные фосфорные центры, неспособность регулирования стереоизбирательного синтеза с высоким выходом продукта, быстрый плазменный клиренс и выделение их с мочой.
Олигодезоксирибонуклеозид-фосфорамидаты имеют межнуклеозидные связи, содержащие азот-фосфорные мостики. Такие аналоги нуклеиновых кислот можно получить из фосфорамидатных промежуточных форм или реакцией окисления промежуточных Н-фосфонатов в присутствии первичных или вторичных аминов. Получение Н-фосфонатных аналогов и реакцию окисления можно легко проводить в промышленном ДНК-синтезаторе.
Известно и синтезировано множество неионогенных олигонуклеотидных последовательностей, содержащих нефосфатные межнуклеозидные связи, например, как карбонатная, ацетатная, карбаматная или диалкил- или диарилсилилпроизводные.
Хотя карбонатная связь устойчива к кислотному гидролизу, ее довольно легко расщепить основанием, и поэтому при удалении защитных групп в конце синтеза необходимо соблюдать особые меры предосторожности. Хотя устойчивые дуплексы обнаружены между поли(dA)-аналогами, содержащими карбоксиметильные межнуклеотидные связи, и поли(U)аналогами, другие основания еще не изучены. Так, например, не ясно, мешает ли карбонатная связь, или нет, точному воспроизведению дуплекса.
Известно, что карбаматные межнуклеозидные связи обладают большей водорастворимостью по сравнению с другими межнуклеозидными мостиками. Применение карбаматных мостиков однако ограничено из-за того, что тиминкарбаматы не образуют гибридов с комплементарной ДНК, в то время как цитозинкарбаматы не гибридизуются с гуаниновыми олигомерами.
Карбаматная связь, аналогично карбонатной, обладает стабильностью в физилогических условиях. В отличие от карбонатных связей, карбаматный мостик устойчив в отношении гидролиза основаниями, и эта способность упрощает синтез олигомеров, содержащих такую связь. Карбаматный мостик обладает устойчивостью к нуклеазному гидролизу.
Так же как карбонатные и ацетатные связи, карбаматный мостик не имеет сходства с формой фосфодиэфирной межнуклеотидной связи. Молекулярные модели, однако, предлагают, чтобы указанный мостик обеспечивал бы принятие олигомером конформаций, в результате которых он мог бы образовывать водород-связанные комплексы с комплементарными нуклеиновыми кислотами.
Карбамат-связанный олигомер, содержащий шесть тимидиновых звеньев не образует комплексов ни с A(pA)5, ни с dA(pA)5. C другой стороны, карбамат-связанный олигомер, содержащий шесть дезоксицитозиновых звеньев образует стабильные комплексы с d-(pG)6 и поли(dG). Межнуклеозидная связь диалкил- или дифенилсилилпроизводных олигомерных аналогов почти сходна с формой тетраэдра нормальной фосфодиэфирной межнуклеотидной связи. Указанные олигомеры получают в растворе путем взаимодействия надлежаще защищенного нуклеозид-3'-O-диалкил- или дифенилсилилхлорида или трифторметансульфонильного производного с 3'-защищенным нуклеозидом в безводном пиридине. Последний можно получить при взаимодействии 5'-O-тритилнуклеозида с диалкил- или дифенилдихлорсиланом, либо с бис(трифторметансульфонил)диизопропилсиланом.
Ввиду того, что диалкил- и дифенилсилильные группы чувствительны к кислотному гидролизу, необходимо быть особенно внимательным при подборе защитных групп для синтеза. Нуклеозидные димеры и гексамеры с силоксановыми межнуклеозидными связями и метод синтеза таких полимеров описывают Ogilvie и Cormier, см. , например, Ogilvie K.K. и Cormier J.F., Tetrahedron Letters, 26 (35), стр. 4159-4162 (1985); Ogilvie K.K и Cormier J.F., Nucleic Acid Research, 16 (10), стр. 4583-4594, 1988 г.
Хотя олигонуклеозидные последовательности, связанные карбонатным, карбаматным и силильным мостиками, обладают необходимой устойчивостью к нуклеазе, что делает заманчивым использовать их в качестве потенциальных антисмысловых реагентов, об этой способности пока еще не сообщалось. Более того, нигде не сообщалось о способности указанных олигомеров к включению клетками в культуре. Потенциальный недостаток этих олигомеров заключается, как сообщают, в низкой растворимости их в водной среде. До сих пор не выяснена возможность получения необходимых их титров для эффективного использования их в биологических экспериментах, хотя растворимость можно по-существу повысить введением гидрофильных групп в молекулы.
Настоящее изобретение предлагает олигонуклеотидные аналоги, композиции на их основе, промежуточные соединения для их получения и методы синтеза указанных новых олигонуклеотидных аналогов, обладающих стабильностью, стойкостью к нуклеазам, специфичностью к клеткам-мишеням и липидной растворимостью.
Краткое изложение сущности изобретения
Настоящее изобретение предлагает нуклеотидные аналоги, содержащие олигонуклеозидные последовательности, включающие от примерно 6 до 200 оснований, и имеющие трехатомный межнуклеозидный мостик. Указанный трехатомный межнуклеозидный мостик таких олигонуклеозидных последовательностей представлен формулой
-D-D-D-
в которой каждый D означает независимо CHR, кислород или NR6, где R означает независимо водород, OH, SH, или NH2, R6, водород или C1-C2-алкил при условии, что только один из D означает кислород или группу NR6.
В предпочтительном варианте осуществления настоящего изобретения, олигонуклеозидные последовательности включают основания, выбранные из группы, состоящей из аденина, цитозина, гуанина, урацила, тимина и их производных.
Более конкретно, предлагаемые соединения содержат олигонуклеозидные последовательности формулы I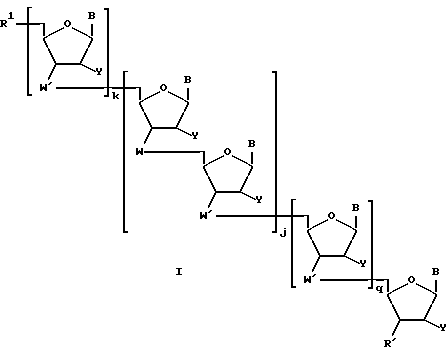
где W означает -D-D-D-, в которой каждый радикал D означает независимо CHR, кислород или NR6, где R означает независимо водород, OH, SH, или NH2, R6 водород или C1-C2-алкил при условии, что только один из D означает кислород или группу NR6;
каждый W' независимо означает W или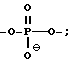
где каждый R1 означает OH, SH, группу NR2R3, в которой R2 и R3 каждый независимо означает водород или C1-C6-алкил, или группу NHR4, где R4 означает C1-C12-алкил;
R4 означает C1-C12-ацил;
каждый Y независимо означает H или OH;
каждый B независимо означает аденин, цитозин, гуанин, урацил, тимин и их модификации;
j означает целое число от 1 до примерно 200;
k означает 0 или целое число от 1 до примерно 197; и
q означает 0 или целое число от 1 до примерно 197, при условии, что сумма j + k + q равна примерно 4 - 200.
Предлагаемые соединения содержат олигонуклеотидные или олигонуклеозидные последовательности, которые могут содержать диол на любом или обоих их концах.
В качестве диолов предпочтительно использовать 1,2-диолы (гликоли).
В качестве примеров гликолей можно указать полиалкиленгликоли, предпочтительно полиэтиленгликоли или полипропиленгликоли. К предпочтительным гликолям относятся тетраэтиленгликоль и гексаэтиленгликоль. В качестве диолов можно также использовать полиолы, у которых все гидроксилы, кроме двух, защищены.
В случае, когда соединения предлагаемого изобретения представляют собой олигонуклеозидные последовательности с диолом у любого или обоих их концах, то указанные соединения выражаются формулой II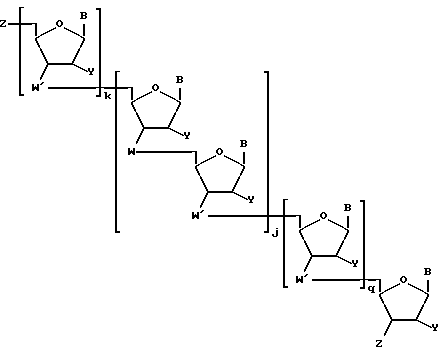
где каждый Z независимо означает R1 или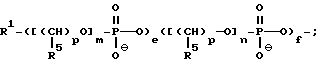
где каждый R1 означает OH, SH, группу MR2R3, в которой R2 и R3 каждый независимо означает водород или C1-C6-алкил, или группу NHR4, где R4 означает C1-C12-ацил;
каждый R5 независимо означает водород или C1-C12-алкил;
каждый из W, W', Y, B, j, k и q имеет вышеуказанные значения;
каждое e и f независимо друг от друга принимает значения от 0 до 50 при условии, что по крайней мере одно из значений e и f равно по крайней мере 1;
каждое m и n независимо друг от друга принимает значения от 1 до 200; каждое p независимо принимает значение от 2 до 4.
В предпочтительном варианте осуществления изобретения, сумма j+k+q составляет примерно от 9 до 50 оснований, более предпочтительно от примерно 12 до 50 оснований и наиболее предпочтительно от примерно 15 до 18 оснований. В этом варианте осуществления изобретения предлагаемые соединения содержат олигонуклеозиды формулы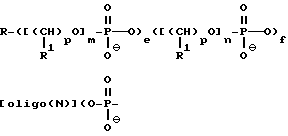
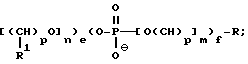
где R означает OH, SH, группу NR2R3, в которой R2 и R3 каждый независимо означает водород или C1-C6-алкил, или группу NHR4, где R4 означает C1-C12-ацил;
R1 означает водород или C1-C12-алкил;
олиго (N) означает нативную или модифицированную олигонуклеотидную последовательность, содержащую от примерно 9 до 200 оснований;
каждое e и f независимо друг от друга принимает значения от 0 до 50 при условии, что по крайней мере одно из значений e и f равно по крайней мере 1;
каждое m и n независимо друг от друга принимает значение от 1 до 200;
каждое p независимо принимает значение от 2 до 4.
В предпочтительном варианте осуществления изобретения, указанный олигонуклеотид, содержит, например в гомополимерной или гетерополимерной форме любую из комбинаций dA, dC, dG, T.
В случае, когда в качестве гликоля используют полиэтиленгликоль, в указанном варианте осуществления изобретения соединения содержат олигонуклеозиды формулы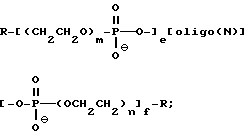
где R означают OH, SH, группу NR2R3, в которой R2 и R3 каждый независимо означает водород или C1-C6-алкил, или группу NHR4, где R4 означает C1-C12-ацил;
R1 означает водород или C1-C12-алкил;
олиго(N) означает олигонуклеотидную последовательность, содержащую от примерно 9 до 50 оснований;
e и f независимо друг от друга принимает значения от 0 до 50 при условии, что по крайней мере одно из значений e и f равно по крайней мере 1;
m и n независимо друг от друга принимает значение от 0 до 200 при условии, что по крайней мере одно из значений m и n составляет от 1 до 200.
Олигонуклеотиды настоящего изобретения могут содержать известные межнуклеозидные линкерные группы, например как фосфодиэфирные, силильные и другие хорошо известные специалистам группы связи, при условии, что они содержат эффективное количество предлагаемых линкерных и/или диольных групп, завершающих синтез - D-D-D-последовательности настоящего изобретения.
Настоящее изобретение также относится к нуклеозидным димерам формулы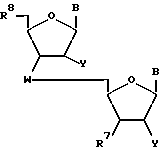
где W означает D-D-D-,
в которой каждый радикал D означает независимо CHR, кислород или NR6, где R означает независимо водород, OH, SH, или NH2, R6 - водород или C1-C2-алкил при условии, что только один из D означает кислород или группу NR6;
каждый B независимо означает аденин, цитозин, гуанин, урацил, тимин и их модификации;
R7 означает OH, трет-бутилдиметилсилилокси или фосфорамидитную группу, и R8 означает OH, защитную группу или трет-бутилдиметилсилилоксигруппу.
Настоящее изобретение также предлагает метод ингибирования нуклеазного гидролиза соединений, содержащих олигонуклеозидные последовательности. Указанный метод включает прикрепление диола к любому 5' - или 3' - концу указанного соединения, либо каждому из них. Такие диолы прикрепляют к 5' - и/или 3' - концу в результате взаимодействия указанных олигонуклеозидов с алкокситритилдиолцианофосфином, предпочтительно диметокситритилгликольцианофосфином или монометокситритилгликольцианофосфином.
Настоящее изобретение также предлагает метод ингибирования нуклеазного гидролиза нативных или модифицированных нуклеотидных соединений, заключающийся в том, что получают последовательности олигонуклеозидов из примерно 6 до примерно 200 оснований, имеющие трехатомную межнуклеозидную связь, представленную вышеуказанной формулой
-D-D-D-.
Настоящее изобретение также предлагает композиции, предназначенные для ингибирования экспрессии генов, включающие соединения, содержащие последовательности олигонуклеозидов из примерно 6 - 200 оснований, имеющие вышеуказанную трехатомную межнуклеозидную связь, и физиологически приемлемый носитель. Указанное соединение может содержать диол y любого из или обоих концов. В качестве диолов предпочтительно используют полиэтиленгликоли.
Настоящее изобретение предлагает также метод ингибирования экспрессии генов, заключающийся в том, что любому млекопитающему при необходимости лечения вводят эффективную дозу соединения, содержащего последовательность олигонуклеозидов из примерно 6 - 200 оснований, имеющие вышеуказанную трехатомную межнуклеозидную связь. Указанные соединения могут содержать диол у любого из или обоих их концов. В качестве диолов предпочтительно используют полиэтиленгликоли.
Краткое описание чертежей.
На фиг. 1a показан синтетический путь получения альдегиднуклеозида (Соединение 1).
На фиг. 1b показан синтетический путь получения нуклеозидного комплекса с йодистым фосфонием (Соединение II).
На фиг. 2 показан синтетический путь получения нуклеозидных димеров, соединенных 3 атомным углеродным мостиком между нуклеозидами с использованием альдегид-нуклеозида и фосфонийодид-нуклеозида, полученных согласно схеме фиг. 1a и 1b, соответственно.
На фиг. 3 показан синтетический путь получения тимидинового димера при использовании альдегид-тимидина и фосфоной-иодид-тимидина (Соединение I и II, соответственно).
На фиг. 4 показан синтетический путь получения нуклеозидного димера, соединенного мостиком с 2 атомами углерода и 1 атомом азота между нуклеозидами в форме 3'-C-C-N-5'. Указанные димеры получают при взаимодействии нуклеозидов, которые содержат альдегид (CHO) с нуклеозидами, содержащими функциональную аминогруппу (NH2) в условиях восстановления.
На фиг. 5 показан синтетический путь получения нуклеозидного димера, соединенного мостиком с 2 атомами углерода и 1 атомом азота между нуклеозидами в форме 3'-C-C-N-5'. Указанные димеры получают при взаимодействии нуклеозидов, которые содержат альдегид (CHO) и функциональную аминогруппу (NH2) в условиях восстановления.
На фиг. 6 показан синтетический путь получения тимидинового димера, соединенного мостиком с 2 атомами углерода и 1 атомом азота между нуклеозидами в форме 3'-C-C-N-5'. Указанные димеры получают при взаимодействии тимидинов, которые содержат альдегид (CHO) с тимидинами, содержащими функциональную аминогруппу (NH2) в условиях восстановления.
На фиг. 7 показан синтетический путь получения тимидинового димера, соединенного мостиком с 2 атомами углерода и 1 атомом азота между нуклеозидами в форме 3'-C-C-N-5'. Указанные димеры получают при взаимодействии тимидинов, которые содержат альдегид (CHO) функциональную аминогруппы (NH2) в условиях восстановления.
Подробное описание сущности изобретения.
Предлагаемые соединения по существу относятся к олигонуклеотидным или олигонуклеозидным последовательностям, которые устойчивы к нуклеазному гидролизу.
Используемый в данном описании термин "нуклеозид" означает соединение, состоящее из пуринового или пиримидинового основания с пятиуголеродным сахаром (пентозой).
Используемый в данном описании термин "нуклеотид" означает сложный эфир фосфорной кислоты любого нуклеозида.
Используемый в данном описании термин "нуклеотид" означает полинуклеотид, содержащий только фосфодиэфирные межнуклеозидные связи, например, "нативную" ДНК или РНК.
В качестве нуклеозидов можно использовать аденозин (А), гуанозин (G), цитидин (C), уридин (U), дезоксиаденозин (dA), дезоксигуанозин (dG), дезоксицитидин (dC), и тимидин.
Соединения настоящего изобретения содержат олигонуклеозидные последовательности, состоящие из примерно 6 - 200 оснований, с фосфодиэфирной или трехатомной межнуклеозидной связью. Трехатомная межнуклеозидная связь (-D-D-D-) содержит
1) три атома углерода,
2) два атома углерода и один атом кислорода или
3) два атома углерода и один атом азота.
К олигонуклеозидным последовательностям относятся последовательности из нативных или модифицированных нуклеозидов.
Используемый в данном описании термин "межнуклеозидная связь" означает атомы и молекулы, образующие мостик между атомом углерода в положении 3 у сахарного остатка одного основания нативного или модифицированного нуклеозида и атомом углерода в положении 5 у сахарного остатка соседнего основания такого нуклеозида. Таким образом, предлагаемые нуклеозиды включают в свой состав A, C, G, U,dA, dC, T или их производные, например, 5-бром или 5-йодурацил, 5-метилцитозин, изоцитозин-(2-амино-4-оксопиримидин), изогуанин (2-оксо-6-аминопурин), инозин(6-оксопурин), 5-винилурацил и 5-винилцитозин.
Трехатомная межнуклеозидная связь представлена формулой
-D-D-D-
в которой каждый D означает независимо CHR, кислород или NR6, где R означает независимо водород, OH, SH, или NH2 , R6 водород или C1-C2 алкил при условии, что только один из D означает кислород или группу NR6.
Предлагаемые соединения содержат олигонуклеозидные последовательности формулы I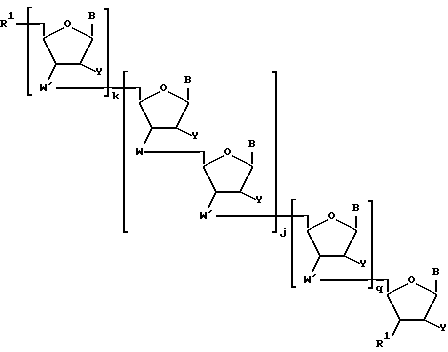
где W означает -D-D-D, в которой каждый радикал D означает независимо CHR, кислород или NR6, где R означает независимо водород, OH, SH, или NH2, R6 - водород или C1-C2-алкил, при условии, что только один из D означает кислород или группу NR6;
каждый W1 означает, независимо означает W или
где каждый R1 означает OH, SH, группу NR2R3, в которой R2 и R3 каждый независимо означает водород или C1-C6-алкил, или группу NHR4, где R4 означает C1-C12-алкил;
R4 означает C1-C12-ацил;
каждый Y независимо означает H или OH;
каждый B независимо означает аденин, цитозин, гуанин, урацил, тимин и их производные;
j означает целое число от 1 до примерно 200;
k означает 0 или целое число от 1 до примерно 197; и
q означает 0 или целое число от 1 до примерно 197 при условии, что сумма j+k+q равна примерно 4 - 200.
В предпочтительном варианте осуществления изобретения, сумма j+k+q составляет примерно от 9 до 50 оснований, более предпочтительно от примерно 12 до 25 оснований и наиболее предпочтительно от примерно 15 до 18 оснований.
Предлагаемые соединения могут содержать диол у любого или каждого двух концов. К предпочтительным диолам относятся гликоли, известные также как 1,2-диолы, содержащие две гидроксильные группы у соседних атомов углерода. В качестве предпочтительных гликолей используют полиалкиленгликоли. Термин "алкилен", его как используют в данном описании, означает радикалы с открытой и разветвленной цепью, содержащие от 2 до 4 атомов углерода, возможно замещенные, как указано в данном описании. К примерам таких радикалов относятся этилен, пропилен, изобутилен и тому подобное. В качестве предпочтительных полиалкиленгликолей используют полиэтиленгликоли, например, как гексаэтиленгликоль и тетраэтиленгликоль. В качестве диолов можно также использовать полиолы, каждый из гидроксилов которых, кроме двух, имеет защитные группы.
Указанные диолы прикреплены к любому из 5' - или 3'-конца таких олигонуклеозидов, либо каждому из них через фосфодиэфирные связи. В одном варианте осуществления настоящего изобретения указанные диолы прикреплены только к одному концу олигонуклеозидной цепи.
Концевой диол связан с остатком, выбранным из группы, состоящей из гидроксила (OH), сульфгидрила (SH), аминогруппы (NH2), алкиламиногруппы (NH-алкилзамещенной группы), диалкиламино (NH[алкил]2) и амидогруппы NH[ацил]).
В случае, когда гликоли присутствуют у любого или обоих концах, то соединения предлагаемого изобретения содержат олигонуклеозидные последовательности формулы II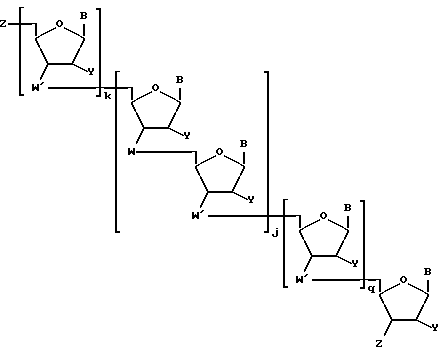
где каждый Z независимо означает R1 или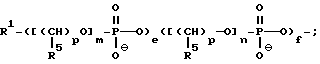
где каждый R1 означает OH, SH, группу NR2R3, в которой R2 и R3 каждый независимо означает водород или C1-C6- алкил, или группу NHR4, где R4 означает C1-C12-ацил;
каждый R5 независимо означает водород или C1-C12 алкил;
каждый из W, W', Y, B, j, k и q имеет вышеуказанные значения; каждое e и f независимо друг от друга принимает значения от 0 до 50 при условии, что по крайней мере одно из значений e и f равно по крайней мере 1;
каждое m и n независимо друг от друг принимает значения от 1 до 200;
каждое p независимо принимает значение от 2 до 4.
В предпочтительном варианте осуществления изобретения, сумма j+k+q составляет примерно от 9 до 50 оснований, более предпочтительно от примерно 12 до 25 оснований и наиболее предпочтительно от примерно 15 до 18 оснований.
В другом варианте осуществления настоящего изобретения, предлагаемые соединения содержат олигонуклеотидные последовательности из примерно 9 до 200 оснований с диолом у любого или каждого из двух их концов. Еще в одном варианте осуществления настоящего изобретения, соединения настоящего изобретения содержат олигонуклеотидные последовательности из примерно 2 - 200 оснований, имеющий предлагаемую (-D-D-D-)-связь. К предпочтительным диолам относятся гликоли, известные также как 1,2- диолы, содержащие две гидроксильные группы у соседних атомов углерода. В качестве предпочтительных гликолей используют полиалкиленгликоли. Термин "алкилен", его как используют в данном описании, означает радикалы с открытой и разветвленной цепью, содержащие от 2 до 4 атомов углерода, возможно замещенные, как указано в данном описании. К примерам таких радикалов относятся этилен, пропилен бутилен и тому подобное. В качестве предпочтительных полиалкиленгликолей используют полиэтиленгликоли. К предпочтительным полиалкиленгликолям относятся полиэтиленгликоли. Наиболее предпочтительны гексаэтиленгликоль и тетраэтиленгликоль.
Указанные диолы прикреплены к любому из 5'- или 3'- конца таких олигонуклеозидов, либо каждому из них через фосфодиэфирные связи. В одном варианте осуществления настоящего изобретения указанные диолы прикреплены только к одному концу олигонуклеозидной цепи.
Концевой диол связан с остатком, выбранным из группы, состоящей из гидроксила (OH), сульфгидрила (SH), аминогруппы (NH2), алкиламиногруппы (NH-алкилзамещенной группы), диалкиламино (NH[алкил]2) и амидогруппы (NH[ацил]). Как используют в данном описании термин "алкилен" означает радикалы с неразветвленной или разветвленной цепью, содержащие от 1 до 12 атомов углерода, возможно замещенные как указано в данном описании. В качестве примеров алкильных и диалкиламиногрупп можно указать метил-, этил-, пропил-, бутил-, пентил-, гексил-, диметил-, диэтил-, дипропил-, дибутил-, дипентил-, и дигексиламины и тому подобное. Как используют в данном описании, "NH-(ацил)" или "амино" радикалы неразветвленной или разветвленной цепью, содержащие от 1 до 12 атомов углерода с концевой O=CHNH2 группой. В качестве примеров амидных групп можно указать метанамид, этанамид, пропанамид, бутанамид, пентанамид, гексанамид, гептанамид, октанамид, нонанамид, деканамид, ундеканамид и додеканамид.
В одном варианте осуществления изобретения предлагаемые соединения содержат олигонуклеозиды формулы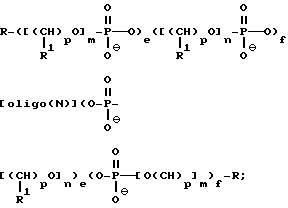
где R означает OH, SH, группу NR2R3, в которой R2 и R3 каждый независимо означает водород или C1-C6 -алкил, или группу NHR4, где R4 означает C1-C12 -ацил;
R1 означает водород или C1-C12-алкил;
олиго (N) означает нативную или модифицированную олигонуклеотидную последовательность, содержащую от примерно 9 до 200 оснований;
каждое e и f независимо друг от друга принимает значения от 0 до 50 при условии, что по крайней мере одно из значений e и f равно по крайней мере 1;
каждое m и n независимо друг от друга принимает значение от 1 до 200;
каждое p независимо принимает значение от 2 до 4.
Указанная последовательность олигонуклеотидов предпочтительно представляет собой гомополимерную или гетерополимерную последовательность, содержащую любую из комбинаций dA, dC, dG, T или их аналогов.
В предпочтительном варианте осуществления изобретения, m и n независимо друг от друга принимают значения от 1 до 8, более предпочтительно, если m, так и n равны 4. В предпочтительном варианте олигонуклеотидные последовательноcти содержат примерно от 9 до 50 оснований, более предпочтительно от примерно 12 до 25 оснований и наиболее предпочтительно от примерно 15 до 18 оснований.
В предпочтительном варианте, предлагаемые антисмысловые соединения содержат полиэтиленгликоль как у 5'-, так и 3' - конца и представлены формулой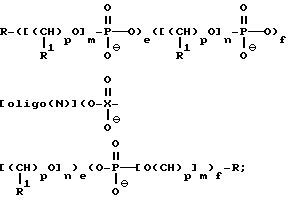
где R означает OH, SH, группу NR2R3, в которой R2 и R3 каждый независимо означает водород или C1-C6 алкил, или группу NHR4, где R4 означает C1-C12 ацил;
R1 означает водород или C1-C12 алкил;
олиго (N) означает нативную или модифицированную олигонуклеотидную последовательность, содержащую от примерно 9 до 200 оснований;
каждое e и f независимо друг от друга принимает значения от 0 до 50 при условии, что по крайней мере одно из значений e и f равно по крайней мере 1;
каждое m и n независимо друг от друга принимает значение от 1 до 200;
каждое p независимо принимает значение от 2 до 4.
В случае, когда в качестве гликоля используют полиэтиленгликоль, в указанном варианте осуществления изобретения соединения содержат олигонуклеозиды формулы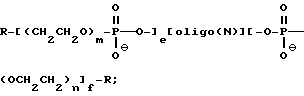
где R означает OH, SH, группу NR2R3, в которой R2 и R3 каждый независимо означает водород или C1-C6 алкил, или группу NHR4, где R4 означает C1-C12 ацил;
олиго (N) означает олигонуклеотидную последовательность, содержащую от примерно 9 до 50 оснований;
e, f, m и n каждый независимо друг от друга принимает значение от 1 до 50.
В предпочтительном варианте осуществления изобретения, указанный олигонуклеотид, содержит, например, в гомополимерной или гетерополимерной форме любую из комбинаций dA, dC, dG, T.
В других предпочтительных вариантах осуществления настоящего изобретения, в качестве полиэтиленгликоля используют тетраэтиленгликоль (ТЭГ), при условии, что как m, так и n равны 4, или гексаэтиленгликоль, если как m, так и n равны 6.
Соединения предлагаемого изобретения пригодны для использования в качестве антисмысловых агентов. Антисмысловые агенты гибридизуются с комплементарной нуклеозидной последовательностью в нуклеиновой кислоте-мишени, и тем самым ингибируют трансляционную и транскрипционную функцию указанной нуклеиновой кислоты-мишени. В качестве нуклеиновой кислоты мишени можно использовать как PHK, так и ДНК.
Антисмысловые соединения предлагаемого изобретения содержат олигонуклеозидные последовательности из примерно 6 до 200 оснований, имеющие гомополимерные или гетерополимерные последовательности, которые включают основания, выбранные из группы: аденина (А), цитозина (С), гуанина (G), урацила (U), тимина (T) и их производные. Специфичные последовательности выбирают на основе их заданных мишеней. Выбранную последовательность гибридизуют с указанной нуклеиновой кислотой-мишенью. В качестве мишеней можно указать MYC-онкоген, RAC-онкоген и вирусные нуклеиновые кислоты.
Соединения предлагаемого изобретения можно получить по следующим методикам:
А. Соединения с межнуклеозидным мостиком из трех атомов углерода.
Олигонуклеозиды, соединенные трехуглеродным межнуклеозидной мостиком синтезируют путем взаимодействия нуклеозидов с функциональными альдегидными и илидными группами в положениях 3' и 6', соответственно по методу Виттига.
Получение нуклеозид-альдегида и фосфонийиодид-нуклеозида из имеющихся в продаже реагентов, проиллюстрировано на фиг. 1a и 1b , соответственно. Указанный альдегид (Соединение 1, полученное согласно схеме фиг. 1a) синтезируют из известного 3'-аллил-3'-дезокси-5'-0-трет-бутилдиметилсилил-3'-тимидина (Соединение A, фиг.1). Полученное аллильное производное подвергают окислению региоизбирательным методом при использовании каталитически эффективного количества четырехокиси осмия и N-метилморфолиноксида в качестве сооксиданта. Полученный диол (Соединение B, фиг. 1a) гидролизуют перйодатом натрия с выходом альдегида с почти количественным выходом.
Синтез фосфониййодид-нуклеозида (Соединение II, фиг.1b) осуществляют из имеющегося в продаже 5'-трифенилметилнуклеозида (Соединение C, фиг. 1b). В этот тритилированный нуклеозид вводят силильную группу в положении 3'-конца при использовании трет-бутилдиметилсилилхлорида с последующим удалением тритильной группы в кислых условиях. Полученный первичный гидроксил (Соединение E, фиг. 1b) окисляют в условиях Сверна с выходом альдегида (Соединение F, фиг. 1b). Неочищенный альдегид сразу же подвергают взаимодействию с илидом, полученным из метилтрифенилфосфонийбромида, в результате чего получают 4'-винилдезокси-3'- трет-бутилдиметилсилилнуклеозид с высоким выходом. Полученное винильное соединение подвергают региоизбирательному взаимодействию с гидроборатом с получением первичного спирта (Соединение G, фиг. 1b). Первичный спирт в свою очередь превращают в соответствующий йодид (Соединение H, фиг. 1b) при использовании трифенилфосфин-йодида в присутствии имидазола с превосходным выходом йодида. И наконец, полученный йодид превращают в целевой нуклеозид-фосфониййодид при использовании трифенилфосфина в ацетонитриле.
Из нуклеозид-фосфониййодида получают илид при использовании трет-бутилата в качестве основания, который сразу же подвергают взаимодействию с альдегидом с высоким выходом соединения Виттига. Полученное соединение Виттига гидрируют водородом региоизбирательным методом на 10% палладиевой черни (10% Pd-C) при атмосферном давлении с количественным выходом для насыщения двойной связи указанного мостика. С полученного насыщенного соединения (Соединение 2, фиг. 2) снимает силильную группу фторидом тетрабутиламмония, в результате чего получают диол (Соединение 3, фиг. 2). 5'-Концевой первичный гидроксил диола затем защищают региоизбирательным методом при использовании диметилокситритилхлорида, и полученный 3'-гидроксил (Соединение 4, фиг.2) превращают в фосфорамидит (Соединение 5, фиг. 2) при взаимодействии его с 2-цианоэтил-N,N-диизопропилхлорфосфорамидитом.
Нуклеозидные димеры или высшие олигомеры с триалкилсилилоксизащитными группами коньюгируют с образованием олигонуклеотидов любой заданной длины цепи. При завершении реакции роста цепи у полученных олигомеров снимают защиту традиционными методами. Для дальнейшего наращивания цепи в твердофазном синтезаторе, где олигомеры связаны фосфатными мостиками, концевые гидроксильные группы указанных олигомеров у 5'- и 3'-конца надлежащим образом функционализируют, при использовании тритилирующих реагентов, например, как диметокситритилхлорид и фосфорамидит, соответственно.
В. Соединения с междунуклеозидным мостиком с 2 атомами углерода и 1 кислорода.
Олигонуклеозидные последовательности с межнуклеозидным мостиком, содержащим 2 атома углерода и 1 атом азота, синтезируют путем взаимодействия 3'-силилированного, 5'-толуолсульфонильного нуклеозида с 5'-защищенным нуклеозидом.
3'-Ацетил-5'-альдегид-нуклеозид получают из имеющегося на рынке 3'-ацетил-нуклеозида по традиционным методом, хорошо знакомым специалистам. Полученный 3'-ацетил-5'-альдегид-нуклеозид затем превращают в 3'-ацетил-5'-карбоксиметоксиметиленнуклеозид при использовании модифицированной реакции Виттига.
5'-Метиленовый отросток восстанавливают борогидридом натрия в спирте, предпочтительно изопропаноле с последующим снятием защиты с 3'- ацетильной группы метилатом натрия в спирте, предпочтительно метаноле. После этого 3'-гидроксигруппу защищают силильной группой. В предпочтительном варианте осуществления изобретения в качестве силильной группы используют третбутилдиметилсилил.
Затем 3'-Ацетил-5'-карбоксиметоксиметиленнуклеозид дополнительно восстанавливают диизобутилалюминийгидридом (DJBAL) в тетрагидрофуране до 3'-0-силил-5'-дезокси-3'-(2''-этанол) производного нуклеозида. 5'-Этанольную группу превращают в пара-толуолсульфонильную группу при введении пара-толуолсульфонилхлорида в пиридине. Экзоциклическую аминогруппу основного остатка 5'-пара-толуолсульфонилнуклеозида при желании можно защитить по общеизвестным методам. В качестве защитной группы экзоциклической аминогруппы аденина и цитозина используют бензоильный остаток. Предпочтительной защитной группой экзоциклической аминогруппой гуанина является изобутиловый остаток. Гуанин можно также защитить в положении 06.
3'-O-силил-5'-пара-толуолсульфонилнуклеозид затем подвергают взаимодействию с 5'-защищенным нуклеозидом, образуя в результате 3'-O-силил-5'-защищенный нуклеозидный димер с двууглеродным-однокислородным мостиком между нуклеозидами. В качестве 5'-O-защитной группой предпочтительно используют тритил 6 и более предпочтительно диметокситритил. При желании 3'-O-силил-5'-O-защитный нуклеозид может иметь защитную группу в экзоциклических аминогрупп основного остатка нуклеозида.
После этого снимают защиты с указанных нуклеозидных димеров и модифицируют их в положении атома углерода у 3'-конца цианофосфиновым реагентом, предпочтительно 2-цианоэтоксидиизопропиламинофосфоном для последующего использования в методе твердофазного синтеза по фосфорамидитному механизму наращивания цепи. (см. выше Gait).
Нуклеозидные димеры или высшие олигомеры с триалкилсилилоксизащитными группами конъюгируют с образованием олигонуклеозидов любой заданной длины цепи. При завершении реакции роста цепи у полученных олигомеров снимают защиту традиционными методами. Для дальнейшего наращивания цепи в твердофазном синтезаторе, где олигомеры связаны фосфатными мостиками, концевые гидроксильные группы указанных олигомеров у 5'- и 3'-конца надлежащим образом функционализируют, при использовании тритилирующих реагентов, например, как диметокситритилхлорид и фосфорамидит, соответственно.
C. Соединения с межнуклеозидным мостиком из 2 атомов углерода и 1 азота.
Олигонуклеозидные последовательности, соединенные межнуклеозидным мостиком, содержащим 2 атома углерода и 1 азота, представленного в виде C-C-N, получают при взаимодействии нуклеозидов, содержащих альдегиды, с нуклеозидами, включающими аминогруппы в условиях восстановления, как показано на рис. 4.
Как альдегидные производные, так и аминопроизводные получают из 3'-аллил-3'-дезокси-5'-O-трет-бутилдиметилсилил-3'-тимидина. Полученное аллильное производное подвергают окислению региоизбирательным методом при использовании каталитически эффективного количества четырехокиси осмия и N-метилморфолина, N- оксида в качестве сооксиданта с выходом диола. Полученный диол гидролизуют перйодатом натрия с выходом альдегида с почти количественным выходом.
Аминопроизводные получают из имеющихся в продаже нуклеозидов. Согласно стандартной методике, первичную гидроксильную группу нуклеозида превращают региоизбирательными методами в тозилатную группу пара-толуолсульфонилхлоридом, с последующим превращением полупродукта в йодид. 3'-Гидроксилированный промежуточный йодид защищают трет-бутилдиметилсилилхлоридом и азидной группой, введенной при взаимодействии с азидом натрия. Функциональную азидную группу можно эффективно превратить в целевой амин путем восстановления с использованием 10% палладиевой черни в атмосфере водорода или при восстановлении катализатором Ренея.
Полученный амин и альдегид связывают друг с другом (реакцией восстановительного аминирования) в присутствии цианоборгидрида натрия в забуференной среде. Образуемый олигонуклеозидный димер с C-C-N-мостиком между нуклеозидами получают с высоким выходом. Полученный олигонуклеозид подвергают взаимодействию со смесью трифторуксусного ангидрида и триэтиламина для защиты вторичного алифатического азота. С защищенного олигонуклеозида снимают силильную группу третрабутиламмонийфторидом, а первичную гидроксильную группу полученного диола избирательно защищают диметокситритилхлоридом. Оставшуюся вторичную гидроксильную группу превращают в целевой фосфорамидит путем взаимодействия ее с 2-цианоэтил-N,N-диизопропилхлорфосфорамидитом.
Олигонуклеозиды, соединенные межнуклеозидным мостиком, содержащим 2 атома углерода и 1 азота, представленного в виде N-C-C, получают при взаимодействии нуклеозидов, содержащих функциональные альдегидные и аминогруппы в положениях 3'- и 5'-конца, соответственно, в условиях восстановления, как показано на фиг. 5.
Аминные и альдегидные составляющие синтезируют из имеющихся в продаже соединений. Амин получают из 3-азидо-3-дезокситимидина (AZT). Первичную гидроксильную группу AZT защищают диметокситритилхлоридом, и полученный азид превращают в требуемый амин региоизбирательным методом при использовании 10% черни в атмосфере водорода или катализатора Ренея.
Альдегид синтезируют из имеющегося в продаже 5'-диметокситритилтимидина. В тритилированный тимидин вводят силильную группу при использовании трет-бутилдиметилсилилхлорид, после чего тритильную группу удаляют в кислой среде. Полученную первичную гидроксильную группу окисляют в среде Сверна с выходом альдегида. Полученный альдегид не выделяют, а используют немедленно для его реакции с (карбэтоксиметилен)трифенилфосфораном, что дает ненасыщенный сложный эфир. Указанный ненасыщенный сложный эфир подвергают региоизбирательному гидрированию на 10% палладиевой черни с образованием насыщенного сложного эфира с количественным выходом. Полученный насыщенный сложный эфир в свою очередь превращают в целевой альдегид при использовании диизобутилалюминийгидрида (DIBAL-H) высокоизбирательным методом.
Амин и альдегид связывают друг с другом в присутствии цианборгидрида в условиях восстановительного аминирования в забуференной среде. Образованный N-C-C мостик между нуклеозидами получают с хорошим выходом. Вторичный алифатический азот олигонуклеозида защищают смесью трифторуксусной кислоты и триэтиламина. С защищенного димера или высшего олигонуклеозидного производного снимают силильную группу, и полученный гидроксил превращают в фосфорамидит при использовании 2-цианоэтил-N,N-диизопропилхлорфосфорамидита.
Нуклеозидные димеры или высшие олигомеры с триалкилсилилоксизащитными группами конъюгируют с образованием олигонуклеозидов любой заданной длины цепи. При завершении реакции роста цепи у полученных олигомеров снимают защиту традиционными методами. Для дальнейшего наращивания цепи в твердофазном синтезаторе, где олигомеры связаны фосфатными мостиками, концевые гидроксильные группы указанных олигомеров -5'- и 3'-конца надлежащим образом функционализируют, при использовании тритилирующих реагентов, например, как диметокситритилхлорид и фосфорамидит, соответственно.
D. Соединения, содержащие диол у любого и каждого из их концов
В случае необходимости, диолы прикрепляют к любому или каждому из концов нуклеиновых кислот путем модификации метода твердофазного синтеза по фосфорамидитному механизму (Oligonucleotide Synthesis: A Practical Approach, публик. M.J. Gait, стр. 35-81, IRL Press, Вашингтон D.C., 1984).
В соответствии с предлагаемой модификацией метода твердофазного синтеза любой диол прикрепляют к одному или каждому из концов олигонуклеотида по методике, где диол взаимодействует с алкокситритильным реагентом с образованием тритилированного диола. В качестве диола предпочтительно используют гликоль, более предпочтительно полиалкиленгликоль, в качестве алкокситритильного предпочтительно используют монометокситритилхлорид или диметокситритилхлорид, и наиболее предпочтительно диметокситритилхлорид. Полученные тритилированные диолы затем подвергают взаимодействию с цианофосфиновым реагентом с образованием тритилдиолцианофосфина, который используют в качестве фосфорамидитного реагента (упоминаемого далее в описании как диоловый фосфорамидитный реагент) в твердофазном синтезе соединений настоящего изобретения.
На начальной стадии твердофазного синтеза нуклеозид прикрепляют к твердофазному носителю, предпочтительно к носителю на основе стекла с регулируемой пористостью (CGP). Указанный нуклеозид предпочтительно прикрепляют к такому стеклянному носителю через сукцинатный мостик у 3'-гидроксильной группы. Известные другие средства иммобилизации нуклеозидов на твердофазных носителях и любой специалист в области олигонуклеозидного синтеза может легко их реализовать. В альтернативном варианте для введения диола в 3'-конец диол-фосфорамидитный субстрат можно иммобилизовать на твердофазном носителе до введения первого нуклеозида. Любой специалист легко поймет средства модификации таких методов синтеза с использованием диол-фосфорамидитных реагентов. К твердофазному носителю можно прикрепить любое количество диолов перед введением первого нуклеозида. В предпочтительном варианте используют от 1 до примерно 50 диолов. Если диолы прикрепляют только к 5'-концу, то нельзя иммобилизовать на твердофазном носителе ни одного диола.
После прикрепления первого нуклеозида или диола (диолов) к твердофазному носителю, рост олигонуклеозидной цепи происходит последовательными стадиями, заключающимися в том, что снимают защитную группу 5'-гидроксила (функциональную тритильную группу), затем активируют 5'-гидроксил в присутствии фосфорамидитного реагента, то есть 5'-тритилнуклеозид, 3'-фосфорамидит, осуществляют кэппинг непрореагировавших нуклеозидов и окисляют фосфорную связующую группу.
Защитную группу у 5'-гидроксила снимают кислотой, предпочтительно трихлоруксусной кислотой.
Специалисты хорошо знают активирующие агенты, которые можно использовать в соответствии с предлагаемым методом. В качестве предпочтительных активаторов используют тетразол и активирующее золото (Beckman Instr. Inc., Palo Alto, Ca).
Активирование происходит в присутствии введенного нуклеозид-фосфоримидитного или диол-фосфорамидитного реагента, причем последний заменяют на нуклеозид-фосфорамидитный субстрат, используемый в традиционных методах твердофазного синтеза, если диол вводят в конец или оба конца полинуклеотидной цепи. Непрореагировавшие полинуклеотидные цепи отщепляют или осуществляют их кэппинг при использовании кэппирующих агентов, например, как уксусный ангидрид и N-метилимидазол.
Неустойчивую трехвалентную фосфорную связь окисляют, например, в предпочтительном варианте йодом для образования стабильной пятивалентной фосфодиэфирной связи олигонуклеотида.
После завершения сборки требуемой олигонуклеотидной цепи фосфатные защитные группы удаляют, полученные олигонуклеотидные звенья снимают с твердофазного носителя и основные защитные группы удаляют традиционными методами (см. выше Gaits, стр. 67 - 70).
Специалистам ясно, что другие методы синтеза олигонуклеотидов можно модифицировать аналогичным образом, чтобы получить антисмысловые олигонуклеотиды с концевыми диольными группами.
Предлагаемые изобретения пригодны для использования в лечении млекопитающих, страдающих наследственными болезнями, или заболеваниями, вызванными нарушением механизмов экспрессии генетической информации. В настоящее время проводятся исследования по созданию терапевтических методов с использованием антисмысловых соединений в лечении вирусных инфекций, вызванных, например, ВИЧ, вирусом цитомегалии, вирусом герпеса обыкновенного, гепатита B, папилломы, пикорнавирусом; раковых болезней легких, кишечника, шейки матки, груди и яичника; аллергических болезней иммунной системы, например, как синдром приобретенного иммунодефицита (СПИД), гематологическая неоплазия и гиперпролиферативные нарушения (см. выше Armstrong и др., стр. 89: Klausher и др., стр. 303 - 304, см. выше).
Композиции предлагаемого изобретения, пригодные для ингибирования генной экспрессии, включают физиологически приемлемые носители и
1) соединения, содержащие олигонуклеотидные последовательности, состоящие примерно из 9 - 200 оснований, с межнуклеозидной связью формулы -D-D-D; как оговорено в данном описании, возможно, содержащие диол у любого или обеих их концах или
2) соединения, включающие олигонуклеотидные последовательности, состоящие из примерно 9 - 200 оснований с диолом у любого или обоих их концов.
Предлагаемые композиции пригодные для ингибирования генной экспрессии включают в свой состав одно или несколько предлагаемых соединений вместе с одним или несколькими нетоксичными физиологически приемлемыми носителями, адъювантами или переносчиками, обобщенно упоминаемые в данном описании как носители для парентерального введения, перорального введения в твердой или жидкой форме, ректального или местного введения и тому подобное.
Указанные композиции можно вводить человеку и животным как перорально, ректально, парентерально (внутривенно, внутримышечно или подкожно), интрацистернально, интравагинально, внутрибрюшинно, местно (порошки, эмульсии или капли) или трансбуккально или назально в виде аэрозоли.
Композиции предлагаемого изобретения пригодные для парентерального введения могут включать физиологически приемлемые стерильные водные или неводные растворы, эмульсии, суспензии или дисперсии и стерильные порошки для получения перед использованием инъекционных растворов или дисперсий. В качестве примеров пригодных для использования водных или неводных носителей, разбавителей, растворителей или переносчиков можно указать воду, этанол, полиолы (пропиленгликоль, полиэтиленгликоль, глицерин и тому подобное), соответствующие их смеси, растительные масла (например, как оливковое масло) и инъекционные сложные эфиры, например, как этилолеат. Необходимую сыпучесть можно достигнуть путем использования покровной оболочки, например, лецитина, обеспечением требуемого гранулометрического состава в случае дисперсий и использованием ПАВ.
Указанные композиции могут также содержать адъюванты, например, как консерванты, смачивающие, эмульгирующие и разъединяющие агенты. Защиту от действия микроорганизмов можно обеспечить различными противобактериальными и противогрибковыми агентами, например, как парабены, хлорбутанол, фенол, сорбиновая кислота и тому подобное. Кроме того, при желании, они могут включать в свой состав изотоники, например сахара, хлористый натрий и тому подобное. Пролонгированную абсорбцию инъекционной формы можно обеспечить при использовании агентов, задерживающих всасывание, например, моностеарата алюминия и геля.
При желании, и для более эффективного распределения, предлагаемые соединения можно включать в субстраты с пролонгированным их высвобождением или целенаправленной их доставкой к органу-мишени, например, полимерные матрицы, липосомы и микросферы. Предлагаемые композиции можно стерилизовать, например, фильтрованием через задерживающий бактерии фильтр или введением асептических средств в виде стерильных твердых составов, которые можно растворять как в стерильной воде, так и некоторых других стерильных инъекционных растворах непосредственно перед употреблением.
В качестве твердых лекарственных форм для орального введения можно использовать капсулы, таблетки, пилюли, порошки и гранулы. В таких твердых лекарственных формах активное соединение смешивают вместе с по крайней мере одним инертным традиционным наполнителем (или носителем), например, как цитрат натрия или дикальцийфосфат, или как с (a) заполнителями, так и сухими разбавителями, например, как крахмалы, лактоза, сахароза, глюкоза, маннит и кремневая кислота, (b) связующими, например, как карбоксиметилцеллюлоза, желатин, поливинилпирродлидон, сахароза и акация, (c) увлажнителями, например, глицерином, (d) дезинтеграторами, например, как агар-агар, карбонат кальция, картофельный или маниоковый крахмал, альгиновая кислота, некоторые комбинированные силикаты и карбонат натрия, (e) агентами, задерживающими всасываемость, например, как парафин, (f) ускорителями всасывания, например, как четвертичные соли аммония, (g) смачивающими агентами, например, как цетиловый спирт и моностеарат глицерина, (h) адсорбентами, например, каолином и бентонитом и (i) замасливателями, например, тальком, стеаратом кальция, магния, твердыми полиэтиленгликолями, лаурилсульфатом натрия или их смесями. В случае использования в виде капсул, таблеток и пилюль, такие лекарственные формы могут также содержать буферные вещества.
Твердые лекарственные формы аналогичного типа можно также использовать в качестве наполнителей в твердых и мягких желатиновых капсулах при использовании, например, лактозы или молочного сахара, а также высокомолекулярных порлиэтиленгликолей и тому подобное.
Твердые лекарственные формы, например, таблетки, драже, капсулы, пилюли и гранулы можно получить с покрытием или в оболочке, например, как энтеросолюбильные покрытия и с другими общеизвестными в данной области покрытиями. Эти формы могут включать инкапсулирующие вещества, и быть такого состава, который обеспечивает пролонгированное высвобождение активного ингредиента или ингредиентов в определенной части желудочно-кишечного тракта в пролонгированной манере. К примерам таких инкапсулированных композиций, пригодных для использования относятся полимерные вещества и воски.
Жидкие лекарственные формы для орального введения могут быть выполнены в виде физиологически приемлемых эмульсий, растворов, суспензий, сиропов и эликсиров. Кроме активных соединений жидкие лекарственные формы могут содержать инертные разбавители, традиционно используемые в данной области, например, как вода, или другие растворители, солюбилизирующие агенты и эмульгаторы, например, как этиловый, изопропиловый спирты, этилацетат и этилкарбонат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, растительные масла, в частности, хлопковое, арахисовое, кукурузное, оливковое и кунжутное масла, глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот сорбита или смеси указанных веществ, и тому подобное.
Кроме инертных разбавителей, предлагаемые композиции могут также включать адъюванты, например, смачивающие средства, эмульгаторы и суспендирующие средства, подслащивающие средства, отдушки и ароматические вещества.
Суспензии, кроме активных соединений, могут содержать суспендирующие средства, например, этоксилированные изостеариловые спирты, этерифицированный полиоксиэтиленовый сорбит и сорбитан, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, или смеси указанных веществ и тому подобное.
Композиции для ректального применения используют предпочтительно в виде суппозиториев, полученных при смешивании соединений предлагаемого изобретения соответствующими инертными наполнителями или носителями, например, как шоколадное масло, полиэтиленгликоль или воск для суппозиториев, который при нормальных температурах находится в твердом состоянии, но при температуре тела становится жидким, и таким образом расплавляясь в ректальном или вагинальном отверстии высвобождает активный ингредиент.
К лекарственным формам для местного применения соединения предлагаемого изобретения относятся мази, порошки, аэрозоли и ингаляторы. Активный ингредиент смешивают в стерильных условиях с физиологически приемлемым носителем и любыми необходимыми консервантами, буферами или газами-вытеснителями, если это необходимо. Офтальмологические лекарственные формы, в частности, глазные мази, присыпки и растворы также включены в объем настоящего изобретения.
Соединения предлагаемого изобретения можно кроме того вводить в липосомальной форме. Известно, что липосомы обычно получают из фосфолипидов или других липидных веществ. Липосомы образуются под действием моно- или многослоистых гидратированных жидких кристаллов, которые диспергированы в водной среде. Можно использовать любой нетоксичный, физиологически приемлемый и метаболизируемый липид, обладающий способностью образовывать липосомы. Предлагаемые композиции в липосомальной форме и кроме липоксигеназу-ингибирующих соединений настоящего изобретения могут содержать стабилизаторы, консерванты, наполнители и тому подобное. В качестве предпочтительных липидов используют фосфолипиды и фосфатидилхолины (лецитины), как природные, так и синтетические.
Известны способы получения липосом, см., например, Methods in Cell Biology, публикация Prescott, том. XI, Academic Press, Нью-Йорк штат Нью-Йорк, стр. 33 и последующие, 1976.
Фактическую концентрацию активного ингредиента в композициях предлагаемого изобретения для одноразового приема можно изменять так, чтобы получить количество активного ингредиента, которое эффективно для создания требуемого терапевтического эффекта от конкретной композиции и методе введения. Выбор одноразовой дозы, следовательно зависит от требуемого терапевтического эффекта, метода приема, требуемого курса лечения и других факторов.
Общая суточная доза соединений предлагаемого изобретения, вводимая в организм-хозяин однократно или дробно может составлять, например, от примерно 1 ммоль до 5 мкл на кг массы тела. Разовая лекарственная форма препарата настоящего изобретения может содержать однократную или разделенную его дозу, которые можно использовать для введения суточной дозировки. Следует иметь ввиду, однако, что подбор индивидуальной дозировки препарата для любого конкретного больного зависит от множества факторов, в том числе массы тела, общего состояния здоровья, пола, диеты, времени и способа приема, скорости всасывания и выделения, комбинирования с другими лекарственными средствами и тяжести болезни для назначения курса лечения.
Следующие примеры иллюстрируют наилучший вариант осуществления настоящего изобретения и ни в коем случае их не следует рассматривать в качестве ограничения объема данного описания и формулы изобретения.
Пример 1.
Получение 5'-O-диметокситритил-3'-O-трет-бутил-диметилсилилтимидина.
Диметокситритилтимидин (5.0 г, 9.2 ммоль) и имидазол (1.2 г, 18.4 моль) растворяют в 15 мл безводного диметилформамида (ДМФ) и полученный раствор прибавляют к трет-бутилдиметилилсилилхлориду (1.7 г, 11.5 ммоль).
Реакционную смесь перемешивают в течение 4 часов при комнатной температуре, разводят этилацетатом, промывают водой, насыщенным раствором хлористого натрия и сушат сульфатом натрия. Названное соединение получают при количественном выходе.
Пример 2.
Получение 3'-O'-трет-бутилдиметилсилилтимидина.
5-O'-диметокситритил-3-O'-трет-бутилиметилсилилтимидин, (0.7 г, 1.1 ммоль), полученный по методике Примера 1, обрабатывают в течение 1 часа при комнатной температуре 13 мл 3% трифторуксусной кислоты в растворе хлористого метилена. Полученную смесь затем нейтрализуют 5% (масс/об) раствором бикарбоната натрия. Органический слой сушат сульфатом натрия. Полученное названное соединение очищают флэш-хроматографией при использовании 0 - 30% градиента этилацетата в метиленхлориде. Выход продукта составляет 85%.
Пример 3.
Получение 3'-O'-трет-бутилдиметилсилилтимидин-4'-альдегида.
К тщательно перемешанному раствору сухого хлористого метилена при температуре - 78oC прибавляют оксалилхлорид (2.88 мл, 33.0 ммоль) с последующим прибавлением по каплям диметилсульфоксида (ДМСО) (3.12 мл, 4.4 ммоль). Через 10 минут прибавляют по каплям в течение 2 мин. спирта (5.6 г, 15.7 ммоль), полученного по методике Примера 2, в 20 мл CH2Cl2 и полученную смесь перемешивают в течение 45 минут. После этого к реакционному раствору прибавляют Et3N (58.1 мл, 8.1 ммоль) и полученную смесь перемешивают в течение еще 45 минут. Затем реакционную смесь доводят до комнатной температуры, промывают вначале водой (2х10 мл), затем солевым раствором (10 мл) и сушат сульфатом натрия. Полученный неочищенный альдегид используют на следующей стадии синтеза.
Пример 4.
Получение 5'-винил-5'-дезокси-3'-O'-трет-бутилдиметилсилилдезокситимидина.
К раствору метилтифенилфосфонийбромида (0.7 ммоль) в сухом тетрагидрофуране при температуре 0oC прибавляют по каплям раствор, содержащий бис-триметилсилиламид натрия (0.6 ммоль). Через 30 мин к полученному раствору прибавляют по каплям в атмосфере азота раствор, содержащий соответствующий 4'-альдегид в ТГФ. Реакционную смесь перемешивают в течение 2 часов, разбавляют этилацетатом, промывают водой, затем солевым раствором и сушат сульфатом натрия. Полученное вышеназванное соединение очищают флэш-хроматографией при использовании 20% этилацетата в гексане. Выход составляет 55 - 60%.
Пример 5.
Получение 3'-O'-трет-бутилдиметилсилил-5'-дезокси-5' -гидроксиметилтимидина.
К раствору, содержащему 2M 2-метил-2-бутена (1.6 эк., 1.5 мл, 3 ммоль) в 3 мл безводного ТГФ при температуре 0oC, медленно вводят 1.6 экв. 1M боран-тетрагидрофуранового комплекса (3 мл, 2 ммоль) в атмосфере азота.
полученный реакционный раствор перемешивают в течение 10 минут с последующим прибавлением винилтимидина, полученного по методике Примера 4 (0.7 г, 1.9 ммоль) в 5 мл безводного ТГФ. Реакционную смесь перемешивают в течение 45 минут и помещают в холодильник на 4 дня.
Субстрат приготавливают при использовании водного раствора, содержащего 3.1 экв. 2М едкого натра и 3.1 экв. 30% перекиси водорода (предпочтительно прибавляя по каплям перекись водорода к водному раствору гидроокиси натрия при температуре 0oC при перемешивании в течение 10 минут). Затем полученный раствор медленно прибавляют через капельную воронку к реакционной смеси при температуре 0oC, перемешивают в течение 1 часа, снимают с ледяной бани, разбавляют этилацетатом, промывают водой, затем насыщенным раствором хлористого натрия и сушат сульфатом натрия. Названное соединение очищают флэш-хроматографией при использовании 20 - 80% градиента этилацетата в гексане. Выход продукта составляет 62%.
Пример 6.
Получение 5'-Йодметил-5'-дезокси-3'-O'- трет-бутилдиметилсилилтимидина.
К раствору, содержащему 3'-O'-трет-бутилдиметилсилил-5'-дезокси-5'-гидроксиметилтимидин, полученному по методике Примера 5 (0.3 г, 0.9 ммоль), в сухом ацетонитриле (5 мл) и простом эфире (3.4 мл) прибавляют 3 эквивалента трифенилфосфина (0.7 г, 2.8 ммоль), 4 эквивалента имидазола (0.3 г, 3.7 ммоль) и 2.2 эквивалента йода (0.5 г, 2.8 ммоль). Полученную реакционную смесь перемешивают в течение 45 мин, и растворитель упаривают. К остатку прибавляют этилацетат и выпавший осадок промывают водой, насыщенным раствором хлористого натрия и сушат сульфатом натрия. Названное соединение очищают флэш-хроматографией при использовании 30 - 50% градиента этилацетата в гексане. Выход продукта составляет 90%.
Пример 7.
Получение 3'-O'-трет-бутилдиметилсилил-5'-дезокси-5'-тимидилметилфосфониййодида.
К перемешанному раствору, содержащему 5'-йодметил-5'-дезокси-3'-O'-трет-бутилдиметилсилилтимидин (480 мг, 1 ммоль), полученному по методике Примера 6, прибавляют трифенилфосфин (1.57 г, 6 ммоль), и полученную смесь нагревают с обратным холодильником в течение 12 часов при температуре 90oC. Реакционную смесь охлаждают, после чего растворитель отгоняют. Названное соединение очищают флэш-хроматографией при использовании 5% MeOH в CH2Cl2. Полученный продукт получают с 95 - 96% выходом.
Пример 8.
Получение 5'-трет-бутилдиметилсилил-3'-дезокси-3'-(1'', 2''- дигидрокси-3''-пропил)тимидина.
Четырехокись осмия (OSO4) (4 капли, 2.5% (мас./об.) в бутаноле прибавляют к перемешанной смеси, состоящей из 3'-дезокси-(2''-пропенил)-3'-дезокси-5'-O'-трет-бутилдиметилсилил- тимидина, полученного по методике, описанной в журнале J.Org.Chem. 1989, 54, стр. 2 767 - 2 769 (C.K.Chu и др.) (183 мг, 0.5 ммоль) и 4- метилморфолин-N-оксида (53 мг, 0.45 ммоль) в безводном тетрагидрофуране при температуре 0oC. Полученную реакционную смесь затем гасят 10% водным раствором метансульфита натрия (2.0), перемешивают в течение 20 минут, фильтруют через слой двуокиси кремния и разводят этилацетатом (25.0 мл). Органическую фазу промывают водой (5.0) и солевым раствором, а затем сушат сульфатом натрия. Упаривают растворитель, и полученное вышеназванное соединение очищают флэш-хроматографией.
Пример 9.
Получение 5'-трет-бутилдиметилсилил-3'-дезокситимид-3'-мл-ацетальдегида.
Перйодат натрия (214 мг, 1 ммоль) прибавляют к перемешанному раствору тимидин-диола, полученному по методике Примера 2, (200 мг, 0.5 ммоль) в ТГФ-H2O (4:1;5,0 мл). Через 1 час реакционную смесь разбавляют этилацетатом (25 мл), промывают водой (2х5 мл), а затем солевым раствором и сушат. Названное соединение очищают флэш-хроматографией при использовании 70% этилацетата в гексане.
Четырехокись осмия (OSO4) (4 капли, 2.5 % (мас./об.) в бутаноле прибавляют к перемешенной смеси, состоящей из 3'-дезокси-(2''-пропенил)-3'-дезокси-5'-O'-трет-бутилдиметилсилил -тимидина, полученного по методике, описанной в журнале J.Org.Chem., 1989, 54, стр. 2767 - 2769 (C.K.Chu и др.) (183 мг, 0.5 ммоль) и 4-метилморфолин-N-оксида (53 мг, 0.45 ммоль) в безводном тетрагидрофуране при температуре 0oC.
Полученную реакционную смесь затем гасят 10% водным раствором метасульфита натрия (2.0), перемешивают в течение 20 минут, фильтруют через слой двуокиси кремния и разводят этилацетатом (25.0 мл). Органическую фазу промывают водой (5.0) и солевым раствором, а затем сушат сульфатом натрия. Упаривают растворитель, и полученное вышеназванное соединение очищают флэш-хроматографией.
Пример 10.
Получение тимидиновых димеров с трехуглеродной межнуклеозидной связью.
Стадии синтеза Примера 10a-10e проиллюстрированы на фиг. 3.
10a. К перемешанной суспензии соединения на основе йодистого фосфония, полученного по методике Примера 7, (241 мг, 0.326 ммоль) в сухом ТГФ (2.0 мл) прибавляют трет-бутилат натрия (0.62 мл, 1M раствор в ТГФ, 0.62 ммоль) при температуре -78oC в атмосфере азота. Через 20 минут к полученной реакционной смеси прибавляют 3'-ацетальдегидное производное, получение по методике Примера 9, (80 мг, 0.22 ммоль). Спустя 60 минут реакционную смесь разводят этилацетатом (30 мл), промывают водой (2 x 5 мл) и сушат сульфатом натрия. Растворитель упаривают и олефиновый продукт очищают флэш-хроматографией с использованием 70% этилацетата в гексане. Выход продукта колеблется в диапазоне 55 - 60%.
10b. 10% палладиевую чернь (20 мг) прибавляют к перемешанному раствору соединения 1 (109 мг) в метаноле при температуре 25oC при давлении водорода 1 атм. Через 4 часа катализатор фильтруют через слой целита, и растворитель отгоняют. После этого полученное соединение 2 отделяют и очищают флэш-хроматографией в 80% растворе этилацетата, растворенном в гексане.
10c. Пример 2.8 эквивалентов тетрабутиламмонийфторида при 0oC прибавляют к перемешанному раствору, содержащему Соединение 2 (350 мг) в 5.0 мл ТГФ. Через 3 часа растворитель отгоняют и полученное Соединение 3 очищают флэш-хроматографией при использовании 10% метанола в CH2Cl2.
10d. Примерно 0,05 эквивалентов 4-диметиламинопиридина, 1.4 эквивалента триэтиламина и 1.2 эквивалента 4,4-диметокситритилхлорида прибавляют к перемешанному раствору Соединения 3 (0,6 ммоль) в сухом пиридине (4.0 мл). Через 2 часа реакционную смесь гасят 2 мл воды, а затем разводят 2.0 мл этилацетата. Органический слой отделяют, промывают солевым раствором и сушат. Полученное Соединение 4 очищают флэш-хроматографией при использовании 5% метанола в метиленхлориде.
10e. Примерно 2.0 эквивалента диизопропилэтиламина и 1.0 мл сухого дихлорметана (CH2Cl) прибавляют к перемешанному раствору Соединения 4 (0.5 ммоль). Через 30 минут в реакционную смесь прибавляют по каплям 0.75 эквивалента 2-цианоэтил-N,N- диизопропилхлорфосфорамидита в течение 20 минут и перемешивание продолжают еще в течение 1 часа. Растворитель упаривают и полученное Соединение 5 очищают флэш-хроматографией при использовании этилацетата (содержащего 1% триэтиламин) в атмосфере азота.
Вышеуказанные стадии синтеза a-d используют для получения димеров, содержащих трехуглеродные межнуклеозидные связи, где каждый из трех атомов углерода представлен формулой -CH2-.
Любой или каждый из углеродных атомов может быть гидроксилирован путем модификации метода, показанного на фиг. 3, исходя из нижеследующего. Каплю 2.5% (мас. /об) раствора четырехокиси осмия в трет-бутаноле при температуре 0oC прибавляют к перемешанному раствору, содержащему Соединение 1 и 4-морфолин-N-оксид (9.1 мг) в 0.8 мл ТГФ. Полученную реакционную смесь выдерживают при температуре 0oC в течение 24 часов, затем гасят ее водным раствором метабисульфита натрия, разводят этилацетатом и промывают сначала водой, а затем солевым раствором. Растворитель упаривают, и полученный гидроксилированный димер очищают тонкослойной хроматографией при использовании этилацетата в качестве элюента. Указанный гидроксилированный димер затем защищают, и 5'- и 3'-концы модифицируют согласно вышеуказанных стадий b-d.
Пример 11.
Получение гидроксилированных трехуглеродных межнуклеозидных связей.
Тимин-димер-фосфорамидиты, полученные на стадиях a-d, используют в методе твердофазного синтеза с фосфорамидитной модификацией с образованием олигонуклеозидных последовательностей, приведенных в таблице 1.
Олигодезоксинуклеотиды синтезируют от 3' - в направлении 5'-конца.
Таблица 1
Последовательность - Ссылочн. код
5' Tp Tp Tp Tp Tp [TcT] p Tp Tp Tp Tp yp yp T3' - 1
5' Tp Tp Tp Tp Tp Tp Tp [TcT] p Tp Tp pyp уT3' - 2
5' Tp Tp Tp Tp Tp Tp Tp Tp [TcT] p yp T3' - 3
T = тимидин
C = -CH2-CH2-CH2-
Y = тетраэтиленгликоль
Синтез затем протекает в соответствии с модифицированным фосфорамидитным методом. 5' - Концевую гидроксильную группу прикрепленного тимидина подвергают взаимодействию с трихлоруксусной кислотой со снятием защиты 5'-гидроксила. После снятия защиты, прикрепленный тимидин подвергают взаимодействию с активатором, в частности, с тетразолом и фосфорамидитным реагентом, содержащим диметокситритилтетраэтиленгликольцианофосфин. После активации осуществляют кэппинг непрореагировавших 5' гидроксилов при использовании уксусного ангидрида и N-метилмидазола. Полученную фосфорную связь затем окисляют йодом в соответствии со стандартными методиками. В последовательностях 1 и 2, содержащих два тетраэтиленгликолевых остатка (ТЭГ) повторяют вышеописанные стадии снятия защиты, активирования, кэппинга и окисления.
Рост цепи затем протекает проведением обычных последовательных операций снятия защиты, активирования, кэппинга и окисления при использовании модификации, заключающейся в том, что связанный трехуглеродным мостиком тимидиновый димер, полученный по методикам примеров 1 - 9, вводят в цепь, при необходимости, во время стадии активирования.
В конце сборки цепи тимидиновые олигомеры снимают с носителя из микропористого стекла при использовании концентрированного раствора гидроксида аммония. Полученный раствор затем обрабатывают при температуре 55oC в течение примерно 8 - 15 часов для удаления всех защитных групп Y экзоциклическиаминогрупп оснований.
Пример 12.
Получение 3'-O-ацетил-5'-карбометоксиметил-5'-дезокситимидина.
Примерно 0.39 г борогидрида натрия прибавляют к охлажденной (на ледяной бане) перемешанной смеси из 3.17 г 3'-O-ацетил-5'-карбометоксиметилен-5'-дезокситимидина в 95 мл изопропанола. Полученную смесь перемешивают при температуре 0oC в атмосфере азота в течение 30 минут, затем при комнатной температуре еще 4.5 часа.
Закаленную смесь гасят 20 мл метанола, затем в течение 30 минут 200 мл дистиллированной воды, после чего экстрагируют несколькими порциями этилацетата. Объединенные органические экстракты обрабатывают солевым раствором и сушат безводным сульфатом магния. Указанный осушитель отфильтровывают, и растворитель упаривают. Получают названное соединение в виде остатка на стеклянном носителе в количестве 2.7 г.
Пример 13.
Получение 5'-карбоксиметоксиметил-5'-дезокситимидина
Примерно 10 капель (25% (масс/об) метанольного раствора метилата натрия прибавляют к охлажденному перемешанному раствору, содержащему 2.22 г 3'-O-ацетил-6'-карбоксиметил-5'-дезокситимидина, полученного по методике Примера 12, в примерно 300 мг сухого (пропущенного через слой нейтральной двуокиси алюминия) метанола. Полученную смесь перемешивают в атмосфере азота без охлаждения на ледяной бане в течение 20 минут.
Небольшое количество катионообменной смолы (Biorad AB 50WX8) прибавляют к реакционной смеси с последующим ее перемешиванием в течение 30 минут. Растворитель упаривают при пониженном давлении с выходом остатка на стекле в количестве 2.1 г, который затем обрабатывают теплым толуолом и после охлаждения, фильтрования и промывания циклогексаном получают неочищенный продукт в виде белого твердого вещества, 1.64 г.
Названное соединение далее очищают от следового количества исходного материала колоночной хроматографией на силикагеле, элюируя этилацетатом. После перекристаллизации из смеси этилацетат/гексан получают белые кристаллы.
Пример 14.
Получение 3'-O-трет-бутилдиметилсилил-5'-(2''-гидроксиэтил)тимидина.
Около 19 мл 1M раствора, содержащего диизобутилалюминия гидрид в ТГФ прибавляют в охлажденный (-40o - 30oC) перемешанный раствор, содержащий 1.88 г 3'-O-третбутилдиметилсилил-5'- карбоэтоксиметил-5'-дезокситимидина в 40 мл безводного ТГФ в атмосфере азота. Реакционную температуру затем постепенно повышают до -20oC.
Охлажденную смесь затем гасят путем введения примерно 3.5 мл метанола, и реакционную температуру затем опять повышают до -10oC. Около 18 мл воды в 36 мл ТГФ прибавляют к подогретой реакционной смеси, и температуру опять повышают до 10oC. Большую часть ТГФ упаривают при пониженном давлении, и остаток разбавляют примерно двумя объемами воды. Водную фазу экстрагируют несколько раз смесью этилацетата и хлороформа. Объединенные экстракты промывают охлажденной 2N соляной кислотой, а затем солевым раствором, сушат безводным сульфатом натрия и фильтруют. Растворитель удаляют из фильтра при пониженном давлении с выходом названного соединения (примерно 1.6 г).
Пример 15.
Получение 3'-O-трет-бутилдиметилсилил-5'-(2''-йодэтил)-5'- дезокситимидина.
Примерно 1 г пара-толуолсульфонилхлорида прибавляют к раствору, содержащему 3'-O-трет-бутилдиметилсилил-5'-(2''-гидроксиэтил)тимидин, полученный по методике Примера 14, в 25 - 30 мл безводного пиридина, и полученную смесь выдерживают при температуре примерно 5oC в течение примерно 19 часов.
Затем реакционную смесь прибавляют к примерно 200 мл воды со льдом и экстрагируют несколько раз простым диэтиловым эфиром. Объединенные органические экстракты промывают охлажденной 2Н соляной кислотой, затем водой и солевым раствором. Промытые экстракты сушат безводным сульфатом натрия и отфильтровывают. Растворитель удаляют из фильтрата при пониженном давлении с выходом остатка на стеклянном носителе в количестве 1.24 г в виде пара-толуолсульфонилпроизводного (деривата).
Примерно 0.54 г пара-толуолсульфонильного производного и 0.38 г иодистого натрия растворяют в 55 мл сухого (молекулярные сита 4A) ацетона в течение трех дней с последующим введением еще 0.19 г иодистого натрия при перемешивании в последний день.
Реакционную смесь очищают хроматографией на силикагеле, 85 г при элюировании в 25% растворе этилацетата в гексане. Растворитель упаривают. Получают 0.4 г названного 3'-O-трет-бутилдиметилсилил-5'-(2''-йодэтил)-5'-дезокситимидина. Растворитель упаривают. Получают 0.4 г названного 3'-O-трет-бутилдиметилсилил-5'-(2''-йодэтил)-5'-дезокситимидина.
Пример 16.
Получение 5'-карбометоксиметилен-5'-дезокситимидина.
Примерно 10 капель 25% (мас./об.) метанольного раствора метилата натрия прибавляют к перемешанному раствору, содержащему 1.55 г 3'-O-ацетил-5'-карбометоксиметилен-5'-дезокситимидина в 150 мл сухого метанола (пропущенного через слой нейтральной двуокиси алюминия). Полученную смесь перемешивают при комнатной температуре в атмосфере азота в течение 6 часов.
Небольшое количество катионнообменной смолы (Biorad AB 50WX8) прибавляют к реакционной смеси с последующим ее перемешиванием в течение 10 минут. Растворитель упаривают при пониженном давлении с выходом белого твердого остатка в количестве 1.3 г, который затем порошкуют дважды теплым толуолом, обрабатывают в горячем этаноле, фильтруют, охлаждают и после сушки получают названное соединение в виде белого кристаллического вещества, 0.85 г.
Пример 17.
Получение 3'-O-трет-бутилдиметилсилил-5'-(2''-гидроксиэтилен)-5'- дезокситимидина
Раствор, содержащий 296 мг 5'-карбэтоксиметилен-5'-дезокситимидина прибавляют по каплям в атмосфере азота к охлажденному (водяная баня со льдом) перемешанному раствору, содержащему 205 мг имидазола и 227 мг трет-бутилдиметилсилихлорида в 1 мл безводного дитметилформамида. После завершения введения, полученную смесь снимают с ледяной бани, и перемешивают ее при температуре окружающей среды в течение двух часов, затем при температуре 35oC еще два часа и наконец, при температуре 40oC в течение 30 минут.
Охлажденную смесь затем гасят путем введения примерно 2 мл метанола, с последующим прибавлением 2 - 3 объемов воды. Водную фазу экстрагируют несколько раз этилацетатом. Объединенные экстракты промывают водой, насыщенным раствором бикарбоната натрия, а затем солевым раствором, сушат безводным сульфатом натрия и фильтруют. Растворитель удаляют из фильтрата при пониженном давлении с выходом 0.40 г 3'-O-трет-бутилдиметилсилил-5'-(2''-гидроксиэтилен)-5'-дезокситимидина.
Примерно 4 мл 1М раствора, содержащего диизобутилалюминийгидрида в тетрагидрофуране прибавляют по каплям в охлажденный до от -30 до -35oC раствор, содержащий 0.37 г.
3'-O-третбутилдиметилсилил-5'-карбометоксиметилен-5'- дезокситимидина растворяют в 10 мл безводного тетрагидрофурана при температуре ниже 30oC. После полного введения реагента, полученную реакционную смесь перемешивают в атмосфере азота в течение еще двух часов одновременно поддерживая внутреннюю температуру в интервале от примерно -30o до примерно -20oC.
Примерно 0.8 мл метанола прибавляют к реакционной смеси с последующим добавлением раствора, содержащего 4 мл воды в 8 мл тетрагидрофурана. Большую часть более летучих компонентов ТГФ упаривают при пониженном давлении. Водный остаток разбавляют примерно двумя объемами воды и экстрагируют несколько раз этилацетатом. Объединенные экстракты промывают охлажденной 2N соляной кислотой, а затем солевым раствором, сушат безводным сульфатом натрия и фильтруют. После упаривания фильтрата получают в остатке названное соединение, 0.267 г. Часть указанного продукта очищают до аналитической чистоты методом хроматографии на силикагеле при использовании 50% раствора этилацетата в гексане в качестве элюента.
Пример 18.
Получение тимидиновых димеров, содержащих межнуклеозидную связь с 2 атомами углерода и 1 атомом кислорода (3'-O-C-C-5').
18a. К перемешанному раствору, содержащему 5'-O-тритилтимидин, прибавляют эквимолярное количество каждого основания и 3'-O-трет-бутилдиметилсилил-5'-(2''-йодэтил)-5'-дезокситимидина. Ходом реакции управляют с помощью тонкослойной хроматографии (ТСХ). После завершения реакции, целевой димер выделяют и очищают флэш-хроматографией.
18b. К перемешанному раствору, содержащему 3'-O-третбутилдиметилсилил-5'-(2''-йодэтил)-5'-дезокситимидин, поддерживая температуру примерно при -5oC, прибавляют 2 эквивалента основания, после чего растворитель упаривают досуха. Полученный остаток перерастворяют в диметилформамиде, и прибавляют эквивалент 5'-диметокситритил-2', 3'-циклотимидина. Полученную реакционную смесь нагревают до температуры примерно 40oC, и синтез димера контролируют ТСХ. После завершения синтеза целевой димер выделяют и очищают флэш-хроматографией.
Пример 19.
Снятие защиты с 3'-конца димера, полученного в примере 18.
3'-Трет-бутилдиметилсилильную защитную группу защищенных дисеров снимают путем обработки ТГФ раствора, содержащего димер по Примеру 18 при использовании 2.8 эквивалентов тетрабутиламмонийфторида при температуре 0oC. После завершения гидролиза (примерно 3 часа), растворитель упаривают, и целевой димер выделяют и очищают флэш-хроматографией.
Пример 20.
Получение функционального димерного звена для автоматизированного синтеза.
Димерный продукт, полученный в примере 19, растворяют в дихлорметане, и прибавляют 2 эквивалента диизопропилэтиламина. Смесь перемешивают в течение 30 минут с последующим прибавлением по каплям 0.75 эквивалентов 2-цианоэтил-N, N-диизопропилхлорфосфорамидита в течение примерно 20 минут. Перемешивание продолжают еще в течение 1 часа, и после упаривания растворителя полученный функционализированный димер выделяют и очищают флэш-хроматографией при использовании этилацетата в инертной среде.
Пример 21.
Получение 5'-трет-бутилдиметилсилил-3'-дезокси-3'-(1'',2''-дигидрокси-3''-пропил)тимидина.
Четырехокись осмия (OsO4) (4 капли, 2.5 % (мас./об.) в бутаноле прибавляют к перемешанной смеси, состоящей из 3'-(2''-пропенил)-3'-дезокси-5'-O'-трет-бутилдиметилсилилтимидина (183 мг, 0.5 ммоль), полученного по методике, описанной в литературе, и 4-метилморфолин-N-оксида (53 мг, 0.45 ммоль) в 5.0 мл безводного тетрагидрофурана при температуре 0oC. Полученную реакционную смесь затем гасят 10% водным раствором метабисульфита натрия (2.0), перемешивают в течение 20 минут, фильтруют через слой двуокиси кремния и разводят этилацетатом (25.0 мл). Органическую фазу промывают водой (5.0) и солевым раствором, а затем сушат сульфатом натрия. Упаривают растворитель, и полученное вышеназванное соединение очищают флэш-хроматографией.
Пример 22.
Получение 3'-дезокситимид-3'ил-ацетальдегид-5'-O-трет- бутилдиметилсилилтимидина.
Перйодат натрия (214 мг, 1 ммоль) прибавляют к перемешанному раствору тимидин-диола, полученного по методике Примера 21 (200 мг, 0.5 ммоль) в ТГФ-H2O (4 : 1, 5, 0 мл). Через 1 час реакционную смесь разбавляют этилацетатом (25 мл), промывают водой (2х5 мл), а затем солевым раствором и сушат. Названное соединение очищают флэш-хроматографией при использовании 70% этилацетата в гексане.
Пример 23.
Получение 5'-O-(пара-толуолсульфонил)-тимидина.
К перемешанному раствору тимидина (20 г, 82.6 ммоль) в сухом пиридине (200 мл) прибавляют пара-толуолсульфонилхлорид (47.2 г, 247.6 ммоль) при 0oC в атмосфере азота. Через три часа реакционную смесь выливают на лед и затем экстрагируют этилацетатом. После упаривания растворителя, полученный продукт перекристаллизовывают из смеси этилацетата и метанола. Названное соединение получают в виде белого кристаллического вещества с выходом 70 - 75%.
Пример 24.
Получение 5'-иод-5'-дезокситимидина.
К перемешанному раствору тимидин-тозилата (10.65 г, 26.9 ммоль), полученного по методике Примера 23, в сухом ацетоне (75 мл) прибавляют иодистый натрий (10 г, 66.7 ммоль) и полученную смесь нагревают с обратным холодильником в течение 16 часов. Растворитель упаривают и остаток разводят этилацетатом. Органическую фазу промывают водой (2х20 мл) и солевым раствором (10 мл), а затем сушат сульфатом натрия. Названное соединение кристаллизуют из метанола в виде белого кристаллического продукта с выходом его 90 - 95%.
Пример 25.
Получение 3'-O-трет-бутилдиметилсилил-5'-йод-5'-дезокситимидина.
К перемешанному раствору тимидинйодида (8.0 г, 24.7 ммоль), полученного по методике Примера 24 в сухом ДМФ, прибавляют имидазол (4.2 г, 61.7 ммоль). Через пять минут трет-бутилдиметилсилилхлорид (4.47 г, 29.64 ммоль) прибавляют к реакционной смеси с последующим перемешиванием в течение 4 часов. Реакционную смесь затем разводят этилацетатом (250 мл), промывают водой (2х100 мл) и солевым раствором. После сушки сульфатом натрия названное соединение очищают флеш-хроматографией при использовании 60% этилацетата в гексане при выходе его 92%.
Пример 26.
Получение 3'-O-трет-бутилдиметилсилил-5'-азидо-5'- дезокситимидина.
К перемешанному раствору 3'-O-трет-бутилдиметилсилил-5'-йод-5'-дезокситимидина (10.8 г, 20 ммоль), полученному по методике Примера 25 в сухом ДМФ, прибавляют азид натрия (3.9 г, 60 ммоль), и полученную смесь нагревают при 0oC в течение 12 часов. Затем реакционную смесь разводят этилацетатом (200), промывают водой (2х50 мл) и солевым раствором (50 мл), а затем сушат сульфатом натрия. Названное соединение очищают флэш-хроматографией при использовании 50% этилацетата в гексане. Выход продукта составляет 92%.
Пример 27.
Получение 3'-O-трет-бутилдиметилсилил-5'-амино-5'- дезокситимидина.
К перемешанному раствору тимидин-азида (5.0 г, 13.1 ммоль), полученного по методике Примера 26, в MeOH, прибавляют 200 мг палладиевой черни в атмосфере азота. Азот затем выпускают и заменяют на водород. Операции выпуска и замены газов повторяют дважды, при этом перемешивание продолжают при давлении водорода 1 атм. в течение 12 часов. Водород удаляют, а катализатор фильтруют через слой целита. Растворитель упаривают в вакууме. После очистки неочищенного продукта флэш-хроматографией при использовании 5 - 10% раствора MeOH в дихлорметане получают 85 - 87% названного соединения.
Пример 28.
Получение 3'-азидо-3'-дезокси-5'-O-диметокситритилтимидина.
К перемешанному раствору, содержащему 3'-азидо-3'-дезокситимидин (2.67 г, 10 ммоль) в сухом пиридине (50 мл) прибавляют 4-диметиламинопиридин (61 мг, 0.5 ммоль), триэтиламин (1.9 мл, 14 ммоль) и 4,4'-диметокситритилхлорида (4.1 г, 12 ммоль) в последовательном порядке. Через 3 часа прибавляют воду, и полученный раствор экстрагируют этилацетатом (250 мл). Органическую фазу отделяют, промывают солевым раствором (50 мл) и сушат сульфатом натрия. Названное соединение очищают флэш-хроматографией при использовании 5% метанола в метиленхлориде. Выход продукта составляет 80 - 85%.
Пример 29.
Получение 3'-амино-3'-дезокси-5'-O-диметокситритилтимидина.
К перемешанному раствору тимидин-азида (3.99 г, 40 ммоль), полученного по методике Примера 2, в MeOH, прибавляют 200 мг 10% палладиевой черни в атмосфере аргона. Газообразный аргон затем выпускают и вводят водород. Эту операцию повторяют дважды, при этом перемешивание продолжают при давлении водорода 1 атм. в течение 12 часов. Водород затем удаляют, и катализатор фильтруют через слой целита. Растворитель упаривают в вакууме. После очистки неочищенного продукта флэш-хроматографией при использовании 5% раствора MeOH в метиленхлориде получают 90 - 93% названного соединения.
1H ЯМР (300 МГц, CDCl3) δ 7.61 (с, 1H), 7.60 - 7.21 (м, 1H), 6.83 - 6.87 (m, 3H), 6.85 (т, J = 5 Гц, 1H), 3.80 (с, 6H), 3.81 - 3.73 (m, 2H), 3.53 - 3.49 (m, 1H), 3.38 - 3.33 (m, 1H), 2.36 - 2.33 (m, 1H), 2.25 - 2.20 (m, 1H), 1.5 (с, 3H), ИК (чистый) νмакс 3 020, 1 697, 1 605, 1 512, 1 246, 1 030 см.
Пример 30.
Получение 5'-O-диметокситритил-3'-O-трет-бутил-диметилсилилтимидина.
Диметокситритилтимидин (5.0 г, 9.2 ммоль) и имидазол (1.2 г, 18.4 моль) растворяют в 15 мл безводного диметилформамида (ДМФ) и полученный раствор прибавляют к трет-бутилдиметилсилилхлориду (1.7 г, 11.5 ммоль).
Реакционную смесь перемешивают в течение 4 часов при комнатной температуре, разводят этилацетатом, промывают водой, насыщенным раствором хлористого натрия и сушат сульфатом натрия. Названное соединение получают при количественном выходе.
Пример 31.
Получение 3'-O'-трет-бутилдиметилсилилтимидина.
5-O'-диметокситритил-3-O'-трет-бутилдиметилсилилтимидин, (0.7 г, 1.1 ммоль), полученный по методике Примера 30, обрабатывают в течение 1 часа при комнатной температуре 13 мл 3% трифторуксусной кислоты в растворе хлористого метилена. Полученную смесь затем нейтрализуют 5% (мас./об) раствором бикарбоната натрия. Органический слой сушат сульфатом натрия. Полученное названное соединение очищают флэш-хроматографией при использовании 0 - 30% градиента этилацетата в метиленхлориде. Выход продукта составляет 85%.
Пример 32.
Получение 3'-O'-трет-бутилдиметилсилилтимидина.
К тщательно перемешанному раствору сухого хлористого метилена при температуре -78oC прибавляют оксалилхлорид (2.88 мл, 33.0 ммоль) с последующим прибавлением по каплям диметилсульфоксида (ДМСО) (3.12 мл, 4.4 ммоль). Через 10 минут прибавляют по каплям в течение 2 мин. спирта (5.6 г, 15.7 ммоль), полученного по методике Примера 31 в 20.0 мл CH2Cl2 и полученную смесь перемешивают в течение 45 минут. После этого к реакционному раствору прибавляют Et3N (58.1 мл, 8.1 ммоль) и полученную смесь перемешивают в течение еще 30 минут. Затем реакционную смесь доводят до температуры -23oC в течение 30 минут. После этого прибавляют карбэтоксиметилентрифенилфосфоран (10.94 г, 31.4 ммоль), и реакционную смесь перемешивают в течение 12 часов при комнатной температуре. Реакционную смесь затем разводят водой (2 х 125 мл) и солевым раствором (50 мл) и сушат (Na2SO4). Неочищенный продукт очищают флэш-хроматографией при использовании вначале 20% этилацетата в гексане, а затем 40% этилацетата в гексане. В результате получают как транс- так и цис-изомеры названного соединения в соотношении 3 : 1, соответственно. Суммарный выход продукта составляет примерно 72 - 76%.
Аналитические данные по транс-изомеру
ИК (чистый) νмакс 3 125, 3 180, 2 982, 2 960, 1 698, 1 490, 1 274, см-1;
1H ЯМР (300 МГц, CDCl3 ) δ 7.04 (с, 1H), 6.87 (дд, J = 15.6 и 5.4 Нц, 1H), 6.23 (т, J = 6.7 Гц, 1H), 6.03 (дд, J = 15.6 и 1.6 Гц, 1H), 4.33 - 4.28 (m, 1H), 4.14 (к, J = 71 Гц, 2H), 4.16 - 4.12 (m, 1H), 2.28 - 2.19 (m, 1H), 2.09 - 1.98 (m, 1H), 1.87 (с, 3H), 1.23 (т, J = 7.1 Гц, 3H), 0.81 (с, 9H), 0.01 (с, 6H); Рассчитано для C20H32O6N2Si;
C 56.58; H 7.60; N 6,60; Найдено
C 56.36; H 7.30; N 6.60.
Пример 33.
Получение 3'-O-трет-бутилдиметилсилил-5'- карбэтоксиметил-5'-дезокситимидина.
К перемешанному раствору ненасыщенного тимидинэфира (4.24 г, 10 ммоль), полученному по методике Примера 32, в EtOAc, прибавляют 200 мг 10% палладиевой черни в атмосфере азота. Газообразный азот удаляют под вакуумом и вводят водород. Эту операцию повторяют дважды, при этом перемешивание продолжают при давлении водорода 1 атм. в течение 12 часов. Водород затем удаляют, и катализатор фильтруют через слой целита. Растворитель упаривают в вакууме. Полученный продукт кристаллизуют из смеси гексана и этилацетата. Названное соединение получают с 95% его выходом.
ИК (чистый) νмакс 3 180, 2 925, 2 950, 1 696, 1 486, 1 260, 1 240 см-1;
1H ЯМР (300 МГц, CDCl3) δ 7.20 (с, 1H), 6.11 (т, J = 6.6 Гц, 1H), 4.07 (к, J = 7.1 Гц, 2H), 4.03 - 3.98 (m, 1H), 2.51 - 2.32 (m, 2H), 24 - 2.15 (m, 1H), 1.18 (т, J = 7.1 Гц, 3H), 0.81 (с, 9H), 0.01 (с, 6H);
Для C20H34O6N2Si;
Рассчитано C 56.31; H 8.03; N 6.57;
Найдено C 55.91; H 7.74; N 6.50.
Пример 34.
Получение 3'-трет-бутилдиметилсилил-5'-дезокси-тимид-5'ил-ацетальдегида.
К перемешанному раствору тимидинэфира (3.41 г, 8 ммоль), полученному по методике Примера 33, в 60 мл сухого CH2Cl2 при температуре -78oC прибавляют по каплям Di-BAL-H (16.4 мл, 1.0 М раствор в гексане, 16.4 ммоль) в течение 3 минут. Через 20 минут реакционную смесь разводят 300 мл EtOAc и промывают дважды 50 мл насыщенного раствора натрий-калиевого тартрата. Органическую фазу промывают солевым раствором и сушат (сульфатом натрия). Названное соединение очищают флэш-хроматографией 50 - 70% этилацетата. Выход продукта составляет 85 - 87%.
Пример 35.
Получение дезокситимидинового димера, содержащего межнуклеозидную связь 3'-C-C-N-5' (фиг. 3)
35a. К перемешанному раствору, содержащему тимидин-амин из Примера 27 (1.07 г, 3 ммоль) и тимидин-альдегида из Примера 22 (1.38 г, 3.6 ммоль) в 50 мл этанола и 10 мл водного буферного раствора (pH 5.5. NaH2PO4 - NaOH) прибавляют по каплям раствор NaCNBH3 в ТГФ (12 мл, 1.0M раствор в ТГФ, 12 ммоль) при температуре 5oC в течение 1 часа. Реакционную смесь перемешивают еще в течение 4 часов, а затем разводят при использовании 2.50 мл этилацетата. Реакционную смесь промывают водой (2 х 40 мл) и солевым раствором (25 мл), и сушат сульфатом натрия. Соединение 1 (фиг. 6) очищают флэш-хроматографией при элюировании сначала этилацетатом, а затем 5 - 8% MeOH в CH2Cl2 с 62 - 64% выходом.
1H ЯМР (300 МГц, CDCL3) δ 7.60 (с, 1H), 7.19 (с, 1H), 6.18 (т, J = 6.6 Гц, 1H), 6.087 (т, J = 3.9 Гц, 1H), 4.29 - 4.23 (м, 1H), 4.15 - 3.98 (м, 1H), 3.91 - 1.85 (м, 1H), 3.70 - 3.78 (м, 2H), 2.95 - 2.87 (м, 1H), 2.84 - 2.66 (м, 3H), 2.35 - 2.05 (м, 5H), 1.94 (с, 3H), 1.93 (с, 3H), 1.80 - 1.63 (м, 1H), 1.55 - 1.45 (м, 1H), 0.93 (с, 9H), 0.69 (с, 9H), 0.11 (c, 6H), 0.07 (с, 6H).
35b. Соединение 1 (фиг. 6) (1.66 мг, 0.23 ммоль) прибавляют к перемешанному раствору, содержащему трифторуксусный ангидрид (0.32 мл, 2.3 ммоль) и триэтиламин (0.64 мл, 4.6 ммоль) в CH2Cl2 (5.0 мл). Через 2 часа реакционную смесь гасят водным раствором NaHCO3 (5.0 мл) и разводят EtOAc (25 мл). Органическую фазу промывают водой (2 х 10 мл), солевым раствором (5 мл) и сушат (Na2SO4). Соединение 2 (фиг. 6) очищают флэш-хроматографией при использовании 7% MeOH в CH2Cl2 с 91 - 93% его выходом.
35c. К перемешанному раствору, содержащему Соединение 2 (164 мг, 0.2 ммоль) в ТГФ (4.0 мл) прибавляют тетрабутиламмонийфторид (68 ммоль) при 0oC. Через 2 часа растворитель упаривают, и полученное Соединение 3 (фиг. 6) очищают флэш-хроматографией при использовании возрастающей 5 - 8% полярности раствора MeOH в CH2Cl2. Выход продукта составляет 90%.
35d. К перемешанному раствору, содержащему Соединение 3 (151 мг, 0.26 ммоль) в сухом пиридине (3.0 мл) прибавляют 4.4-диметиламинопиридин (1.6 мг, 0.0128 ммоль) и триэтиламин (0.057, 0.42 ммоль). Через 5 минут к реакционной смеси прибавляют диметокситритилхлорид (121 мг, 0.358 ммоль) и перемешивание продолжают. Через два часа реакционную смесь разводят этилацетатом (25 мл) и промывают водой (2 х 10 мл), солевым раствором (5 мл) и сушат (Na2 SO4). Полученный неочищенный продукт очищают флэш-хроматографией при использовании 7% MeOH в CH2Cl2 с 91 - 93% выходом Соединения 4 (фиг. 3).
35е. Сухой диизопропиловый этиламин (0.15 мл, 0.67 ммоль) прибавляют к Соединению 4 (150 мг, 0.168 ммоль), а затем прибавляют CH2Cl2 (0.5 мл). Затем колбу встряхивают для растворения спирта и прибавляют 2-цианоэтил-N,N-диизопропилхлорфосфорамидита (0.056 мл, 0.25 ммоль) в течение 20 сек. Через 45 минут реакционную смесь гасят CH3OH (1.0 мл), разводят EtOAc (50 мл) и Et3N (1.0 мл), промывают 10% водным раствором K2CO3 (2 x 5.0 мл), а затем солевым раствором (5 мл) и сушат (Na2SO4). Полученный неочищенный продукт очищают флэш-хроматографией при использовании EtOAc с 91 - 93% выходом Соединения 5 (фиг. 6).
Пример 36.
Получение дезокситимидинового димера, содержащего межнуклеозидную связь 3'-N-C-C-5' (фиг. 4).
36a. К перемешанному раствору, содержащему амин из Примера 29 (2.72 г, 5 ммоль) и альдегид из Примера 34 (2.29 г, 6 ммоль) в 50 мл этанола и 10 мл водного буферного раствора (pH 5.5, Na2H2PO4 - NaOH) прибавляют по каплям раствор NaCNBH3 в ТГФ (12 мл, 1.0 М раствор в ТГФ, 12 ммоль) при температуре 5oC в течение 1 часа. Реакционную смесь перемешивают еще в течение 4 часов, а затем разводят при использовании 250 мл этилацетата. Реакционную смесь промывают водой (2 x 60 мл) и солевым раствором, и сушат сульфатом натрия. Соединения 1 (фиг. 7) очищают флэш-хроматографией при элюировании сначала этилацетатом, а затем 5 - 8% MeOH в CH2Cl2. Соединение 1 (фиг. 7) получают с 72 - 74% выходом.
1H ЯМР (300 МГц, CDCl3) δ 7.56 (m, 1H), 7.36 - 7.34 (m, 2H), 7.29 - 7.15 (m, 8H), 7.03 (c, 1H), 6.77 (m, 3H), 6.20 (т, J = 6.0 Гц, 1H), 6.08 (т, J = 6.7 Гц, 1H), 4.01 - 3.97 (m, 2H), 3.84 - 3.72 (m, 1H), 3.72 (c, 6H), 3.71 - 3.63 (m, 1H), 3.48 - 3.22 (m, 2H), 3.30 - 3.22 (m, 1H), 3.48 - 3.32 (m, 2H), 3.30 - 3.32 (m, 1H), 7.52 - (m, 2H), 2.27 (m, 3H), 2.08 - 1.97 (m, 1H), 1.83 (c, 1H), 1.67 - 1.48 (m, 3H), 1.43 (c, 3H), 1.22 - 1.15 (m, 1H), 0.01 (c, 6H).
36b. К перемешанному раствору, содержащему трифторуксусный ангидрид (0.32 мл, 2.3 ммоль) и триэтиламин (0.64 мл, 4.6 ммоль) в CH2Cl2 (5.0 мл) прибавляют Соединение 1 (фиг. 7) (210 мг, 0.23 ммоль). Через 2 часа реакционную смесь гасят водным раствором NaHCO3 (5.0 мл) и разводят EtOAc (25 мл). Органическую фазу промывают водой (2 x 10 мл), солевым раствором (5 мл) и сушат (Na2SO4). Соединения 2 (фиг. 7) очищают флэш-хроматографией при использовании 7% MeOH в CH2Cl2. Выход Соединения 2 составляет 89 - 91%.
36c. К перемешанному раствору, содержащему Соединение 2 (180 мг, 0.2 ммоль) в ТГФ (4.0 мл) прибавляют тетрабутиламмонийфторид (0.4 ммоль, 1.0 М раствора ТГФ, 0.4 ммоль) при 0oC. Через 2 часа растворитель упаривают, и полученный продукт очищают флэш-хроматографией при использовании возрастающей полярности, то есть раствора MeOH с 5 до 8% в CH2Cl2. Выход Соединения 3 (фиг. 7) составляет 89%.
36d. Сухой диизопропиловый этиламин (0.15 мл, 0.67 ммоль) прибавляют к Соединению 3 (150 мг, 0.168 ммоль), а затем прибавляют CH2Cl2 (0.5 мл). Затем колбу встряхивают для растворения спирта и прибавляют 2-цианоэтил-N,N-диизопропилхлорфосфорамидита (0.056 мл, 0.25 ммоль) в течение 20 сек. Через 45 минут реакционную смесь гасят CH3OH (1.0 мл), разводят EtOAc (50 мл) и Et3N (1.0 мл), промывают 10% водным раствором K2CO3 (2 x 5.0 мл), а затем солевым раствором ( 5 мл) и сушат (Na2SO4). Полученный неочищенный продукт очищают флэш-хроматографией при использовании EtOAc с 70 - 75% выходом Соединения 4.
Пример 37.
Синтез дезокситимидиновых олигомеров с межнуклеозидной связью 3'-N-C-C-5'.
Тимидин-димер-фосфорамидиты, полученные на стадиях a - d используют в модифицированном методе твердофазного синтеза по фосфорамидитному механизму с получением олигонуклеозидных последовательностей, приведенных в Таблице 2.
Таблица 2
Последовательность - Ссылочн. код
5' TpTpTpTpTpTpTpTp[TnT]pT 3' - 4
5' TpTpTpTpTp[TnT]pTpTpTpT 3' - 5
5' [TnT]pTpTpTpTpTpTpTpTpT 3' - 6
T = тимидин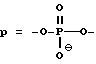
m = 3'-N-C-C-5'
Олигодезоксинуклеотиды синтезируют от 3' - в направлении 5'-конца.
Начальная стадия синтеза заключается в прикреплении, то есть через 3'-сукцинатную связь, 5'-диметокситритилдезокситимидина к СРА-носителю (стеклянная подложка с регулируемой пористостью). 5'-O-диметокситритил-защитную группу прикрепленного тимидина подвергают взаимодействию с трихлоруксусной кислотой для снятия защиты 5'-гидроксила.
Рост цепи затем протекает проведением обычных последовательных операций снятия защиты, активирования, кэппинга и окисления при использовании модификации, приводящей к тому, что (-N-C-C-)-связанный тимидиновый димер, полученный по методикам Примеров 30 - 37, вводят в цепь, при необходимости во время стадии активирования.
В конце сборки цепи, тимидиновые олигомеры снимают с носителя из микропористого стекла при использовании концентрированного раствора гидроксида аммония. Полученный раствор затем обрабатывают при температуре 55oC в течение примерно 8 - 15 часов для удаления всех защитных групп у экзоциклическиаминогрупп оснований.
Пример 38.
Получение тетраэтиленгликоль-концевой последовательности ДНК анти-RAS онкогена.
38a. Получение диметокситритилтетраэтиленгликоля (ДМТТЭГ). Избыток тетраэтиленгликоля (ТЭГ) (примерно 100 мл) смешивают с примерно 7 мл (5.1 г; 40 ммоль) основания Hunig в круглодонной колбе. Примерно 3.08 г (10 ммоль) диметокситритилхлорида (ДМТХ) прибавляют тетраэтиленгликолевой смеси, и полученную ДМТХ-ТЭГ-смесь поддерживают при постоянном перемешивании при комнатной температуре (примерно 25oC) в течение примерно от 8 до 12 часов с образованием ДМТТЭГ.
38b. Получение диметокситритилтетраэтилен-гликольцианофосфина (ДМТТЭГЦФ).
6 г ДМТТЭГ, полученного на стадии (a) смешивают с 20 мл сухого дихлорметана. Примерно 6.2 мл основания Hunig прибавляют к полученной смеси с последующим прибавлением по каплям хлорфосфиновой смеси в результате чего ДМТТЭГЦФ. Хлорфосфиновую смесь приготавливают путем растворения 1.67 г 2-цианоэтил-N,N-диизопропилхлорфосфорамидита в 5 мл сухого дихлорметана.
38c. Получение ТЭГ-концевой ДНК-последовательности анти-RAS онкогена.
Олигодезоксинуклеотиды таблицы 3 получают модифицированным фосфоримидитным методом твердофазного синтеза (см. выше GAIT). Указанные олигодезоксинуклеотиды получают от 3'- в направлении к 5'-концу.
Таблица 3
Последовательность - Ссылочн.код.
5' X GGA GCT GGT GGC GTA X (A)3' - 7
5' XX GGA GCT GGT GGC GTA XX (A)3' - 8
5' X CCT CGA CCA CCG CAT X (A)3' - 9
5' XX CCT CGA CCA CCG CAT XX (A)3' - 10
5' CCT CGA CCA CCG CAT 3' - 11
X означает TEG;
A, C, G и T означает дезокси-аденозин, цитидин, гуанозин и тимидин, соответственно.
К указанной стеклянной подложке прикрепляют как аденозин (7, 8, 9, 10), так и тимидин (II) при использовании сукцинатной связи (см. выше GAIT). Синтез тимидина (II) протекает согласно традиционным методам твердофазного синтеза при использовании форсфорамидитного реагента. В последовательностях 7, 8, 9 и 10 синтез протекает по методам модификации фосфорамидитного реагента. 5'-гидроксил прикрепленного аденозина подвергают взаимодействию с трихлоруксусной кислотой для снятия защиты с 5'-гидроксила. После снятия защиты, прикрепленный аденозин подвергают взаимодействию с активатором, в частности, с тетразолом и фосфорамидитным реагентом, содержащим диметокситритилтетраэтиленгликольцианофосфин (ДМТТЭГЦФ), полученный в вышеуказанных стадиях a и b. Стадия активации сопровождается кэппингом непрореагировавших 5'гидроксилов при использовании уксусного ангидрида и N-метилимидазола. Полученную фосфорную связь затем окисляют йодом в соответствии со стандартными методиками. В последовательностях 8 и 10, содержащих два тетраэтиленгликольостатка (ТЭГ) повторяют вышеописанные стадии снятия защиты, активирования, кэппинга и окисления. Рост цепи затем протекает проведением обычных последовательных операций снятия защиты, активирования, кэппинга и окисления по методике, описанной выше, но с модификацией, заключающейся в том, что фосфорамидитный реагент в целевом нуклеозиде заменяют на ДМТТЭГЦФ во время стадии активирования. После прикрепления концевого заданного нуклеозида любой или каждый из двух ТЭГ-остатков прикрепляют к 5'-концу аналогично связыванию ТЭГ с 3'-концом.
В конце сборки цепи, нить ДНК снимают с носителя на основе стекла с регулируемой пористостью при использовании концентрированного раствора гидроксида аммония. Полученный раствор затем обрабатывают при температуре 55oC в течение примерно 8 - 15 часов для удаления всех защитных групп у экзоциклическиаминогрупп оснований.
Пример 39.
Получение гексаэтиленгликоль-концевой (ГЭГ) ДНК-последовательности анти-RAS-онкогена.
Гексаэтиленгликоль-концевую ДНК-последовательность анти-RAS-онкогена получают по методикам Примера 38. ГЭГ подвергают взаимодействию с ДМТХ (DMTC1) с образованием ДМТГЭГ (DMTHEG). ДМТГЭГ затем подвергают взаимодействию с цианофосфиновым соединением в результате чего образуется ДМТГЭГЦФ (DMTHEGCP), который используют в модифицированном методе твердофазного синтеза по фосфорамидатному механизму согласно Примеру 38 (с) с получением ГЭГ-концевой ДНК-последовательности анти-RAS- онкогена. Последовательности указанных олигонуклеотидов показаны в нижеследующей таблице 4.
Таблица 4
Последовательность
5' X GGA GCT GGT GGC GTA X (A) 3'
5' XX GGA GCT GGT GGC GTA XX (A) 3'
5' X CCT CGA CCA CCG CAT X (A) 3'
5' XX CCT CGA CCA CCG CAT XX (A) 3'
5' CCT CGA CCA CCG CAT 3'
где X означает ГЭГ;
A, C, G и T означают дезокси-аденозин, цитидин, гуанозин и тимидидин, соответственно.
Пример 40.
Устойчивость к нуклеазе ТЭГ-концевой ДНК-последовательности анти-RAS-онкогена.
Полученные олигонуклеотиды, приведенные в таблице 4 растворяют в воде. После этого определяют концентрации ДНК путем измерения полосы поглощения образцов на длине волны 260 нм (на Lambda 4C-спектрофотомере фирмы Perkin Elmer при комнатной температуре) с использованием расчетных коэффициентов экстинкции (метод Cantor-Warsaw, CRC Handbook of Biochemistry and Molecular Biology, 3-е издание, том 1, CRC Press, стр. 589, 1975 г).
Олигонуклеотиды инкубируют в течение 2 часов при температуре 37oC при суммарной концентрации всей ДНК-цепи примерно 6 или 7 мкмоль в среде для культивирования клеток, содержащей RPMI 1 640 ; 20 ммоль N-(2-гидроксиэтил)пиперазин-N'-(2-этансульфокислоты), pH 7.4; и 10% фетальную телячью сыворотку (ФТС) (GIBCO Laboratories. Grand Islands, штат Нью-Йорк). ФТС инактивируют путем нагревания при температуре 56oC в течение 0.5 часа до ее использования. Образцы затем помещают на лед и снимают защиту при пятикратном экстрагировании смесью растворителей хлороформ:изопропиловый спирт, 24: 1. Обработанные образцы затем либо хранят в замороженном состоянии при температуре -20oC, либо немедленно помещают в охлаждаемый (4oC) автоинжектор ВЭЖ хроматографа WISP (Waters).
Степень гидролиза олигонуклеотидов оценивают по выделевшемуся количеству исходного соединения. Олигонуклеотиды (выделенные из реакционной смеси) разделяют на двухинжекционной хроматографической системы LKB Ultrachrome, оснащенной детектором с фиксированной длиной волны (260 нм) и самописцем-интегратором, при использовании колонки с анионообменной смолой фирмы GenPak FAX (Waters), сбалансированной в буфере A (1 мМ EDTA; 15 мМ фосфат натрия, pH 8.5). Температуру в колонке удерживают на уровне 60oC при использовании термостата фирмы Waters. Используют 50 мкл дозаторы для ввода образцов. Олигонуклеотиды элюируют при использовании линейного градиента 0% относительно 100% буфера A (буфер A содержит 0.5 М раствор NaCl) в промежутке 60 минут. Расход буферного раствора составляет 1 мл/мин.
После инкубации (2 часа) в присутствии ФТС-связанной экзонуклеазой не отмечено никакой деградации соединений 7 или 10 (таблица 5, в конце описания, см.% разложения главного пика). В течение такого же инкубационного периода не повергается гидролизу 87.0% и 82% образца 9 и 8, соответственно. В противоположность этому, только 24.7% олигомера II остается на носителе после аналогичного инкубационного периода.
Все 4 ТЭГ-прикрепленные олигомеры устойчивы к гидролизу ФТС-связанных экзонуклеаз. Бис-диТЭГ-прикрепленные олигомеры (7 и 10), как отмечено, полностью устойчивы к нуклеазному гидролизу. ТЭГ-производные олигодезоксинуклеотиды обнаруживают значительное увеличение устойчивости к экзонуклеазному гидролизу по сравнению с немодифицированными соединениями.
Пример 41.
Способность ТЭГ-антисмысловых олигомеров к ингибированию белковой экспрессии и роста в линии опухолевых клеток человека и к стимуляции ФГА периферических лимфоцитов.
Было продемонстрировано другими (Heikkila, R., и др., Nature, вып. 328, стр. 445 - 449), что немодифицированные антисенсовые олигонуклеотиды, связанные с областью инициации кодона c-myc онкогена могут ингибировать экспрессию c-myc-белка с ФГА-стимулированных лимофцитах периферической крови (PBL), приводя таким образом к блокированию прогрессии клеток в направлении S-фазы клеточного цикла. C-myc-направляемая антисмысловая ДНК, как продемонстрировано, также ингибирует клетки эритролейкемии человека HL-60 in vitro (Wickstrom T.L., и др., Proc. Natl. Acad. Sci. USA, вып. 85, стр. 1 028 - 1 032, 1988). Последовательности, показанные в таблице 6, получены и исследованы по методикам Примера 41a и 41b.
Таблица 6
5' AAC GNN GAG GGG CFT 3'
5' XX FFC GNN GAG GGG CAT XX A 3'
(X = TEG)
41a. Сравнительные исследования влияния модифицированной (ТЭГ) и немодифицированной C-myc антисмысловой ДНК на прогрессию ФГА-стимулированных периферических лимфоцитов в S-фазу клеточного цикла.
Периферические лимфоциты человека стимулировали ФГА в течение 48 часов в присутствии или без антисмысловых олигонуклеотидных последовательностей, приведенных в таблице 6. Процентное содержание популяции клеток в каждой обработанной группе в S-фазе клеточного цикла в сравнении с контролем определена при использовании стандартных методов проточной цитометрии. Полученные результаты приведены в таблице 7 (см. в конце описания).
Указанные данные свидетельствуют о том, что присутствие ТЭГ у обоих 3'- и 5'-концах не влияет на ингибирующее действие антисмысловой ДНК.
41b. Сравнительное исследование влияния модифицированной (в присутствии ТЭГ) немодифицированной C-myc антисмысловой ДНК на белковую экспрессию в T-клеточно-опосредованных человеческих лейкозных клетках MOLT-4.
Асинхронные экспоненциально растущие Molt 4-клетки инкубируют в течение 8 часов при введении или без введения 60 мкмоль c-myc-опосредованной антисмысловой ДНК. Клетки затем инкубируют в течение 45 минут при добавлении 35 S-метионина, и содержание белка определяют методом иммунной преципитации с использованием c-myc-антитела. Полученные результаты приведены в таблице 8. (см. в конце описания).
ТЭГ-содержащая антисмысловая ДНК незначительно более действенна по сравнению с немодифицированной антисмысловой ДНК.
41c. Сравнительное исследование влияния модифицированной (в присутствии ТЭГ) и немодифицированной c-myc антисмысловой ДНК на ингибирование фактора роста CCRF-CEM T-клеточно-опосредованной лейкемии in vitro.
Асинхронные экспоненциально растущие CCRF-CEM клетки инкубируют в течение 48 часов в присутствии или без антисмысловой ДНК, после чего определяют цитолитический индекс в каждой обработанной группе. После этого определяют концентрацию антисмысловой ДНК, необходимую для 50% ингибирования клеточного роста (ИК 50). Как модифицированная, так и немодифицированная антисмысловая ДНК, приведенные в таблице 5, демонстрируют примерно одинаковую ИК 50 40 мкмоль.
Эти данные свидетельствуют о том, что наличие ТЭГ у 3'-и 5'-конец антисмысловой ДНК не влияет на способность такой антисмысловой ДНК к гибридизации с нуклеиновыми кислотами-мишенями и ингибированию их функции.
Пример 42.
Экзонуклеазо-устойчивые олигонуклеотидные димеры.
Экзонуклеаза-устойчивые олигонуклеотидные димеры, приведенные в таблице 9, получают по методике Примера 38.
Специалистам ясно, что различные варианты и модификации могут иметь место в пределах объема и сущности предлагаемого изобретения. (01) (19) (11) (21) (51) 6 (54) (57) (08) (09) (00)о
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ СОЕДИНЕНИЯ НУКЛЕОЗИДОВ 3'-5'-МЕЖНУКЛЕОТИДНЫМ СИЛИЛЬНЫМ ЗВЕНОМ | 1991 |
|
RU2079508C1 |
| СИНТЕЗ β-L-2'-ДЕЗОКСИНУКЛЕОЗИДОВ | 2004 |
|
RU2361875C2 |
| МОДИФИЦИРОВАННЫЕ ОЛИГОДЕЗОКСИРИБОНУКЛЕОТИДЫ, КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1994 |
|
RU2111971C1 |
| СПОСОБЫ ЛЕЧЕНИЯ СИНДРОМА АЛЬПОРТА | 2018 |
|
RU2791694C2 |
| НУКЛЕОЗИДЫ С МОДИФИЦИРОВАННЫМИ САХАРАМИ И ОЛИГОНУКЛЕОТИДЫ | 1995 |
|
RU2145964C1 |
| СПОСОБ ПОЛУЧЕНИЯ 3'-АЗИДО-2',3'-ДИДЕЗОКСИТИМИДИНА | 1994 |
|
RU2102399C1 |
| КОМПОЗИЦИИ И СПОСОБЫ МОДУЛИРОВАНИЯ ЭКСПРЕССИИ HBV И TTR | 2014 |
|
RU2782034C2 |
| КОМПОЗИЦИИ И СПОСОБЫ МОДУЛИРОВАНИЯ ЭКСПРЕССИИ HBV И TTR | 2014 |
|
RU2670614C9 |
| КОМПОЗИЦИИ И СПОСОБЫ | 2014 |
|
RU2686080C2 |
| НУКЛЕИНОВЫЕ КИСЛОТЫ, МОДИФИЦИРОВАННЫЕ АМИНОКИСЛОТАМИ | 1995 |
|
RU2154638C2 |


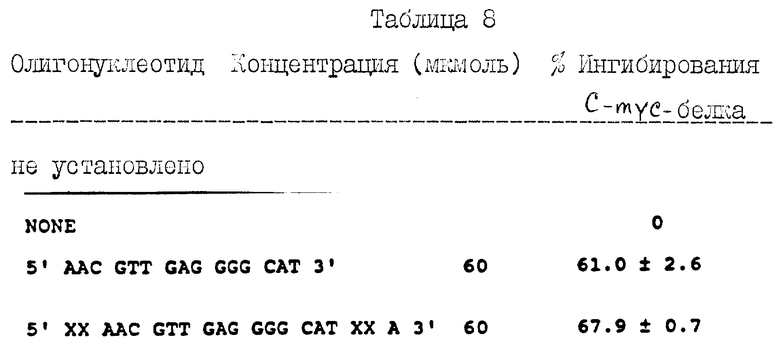


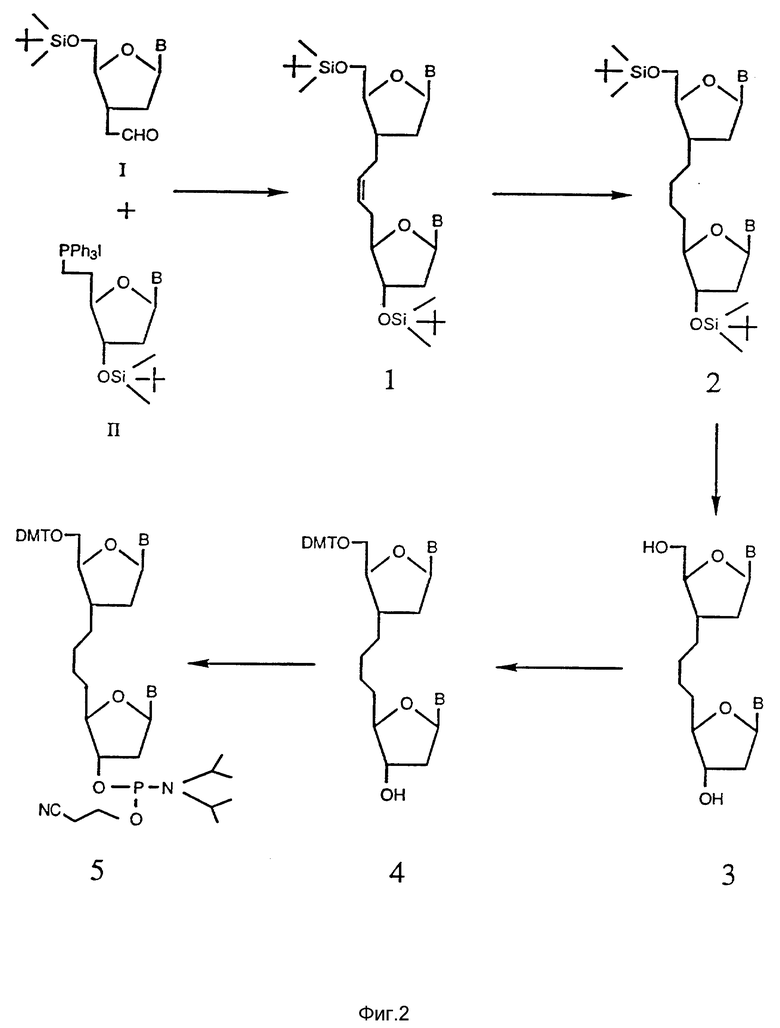
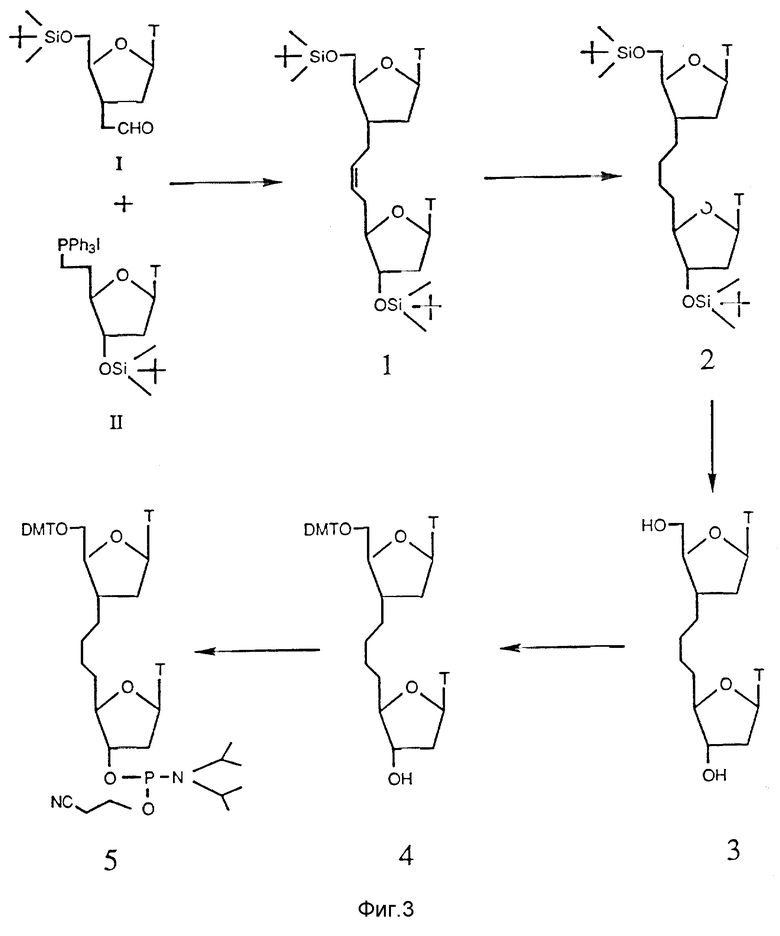
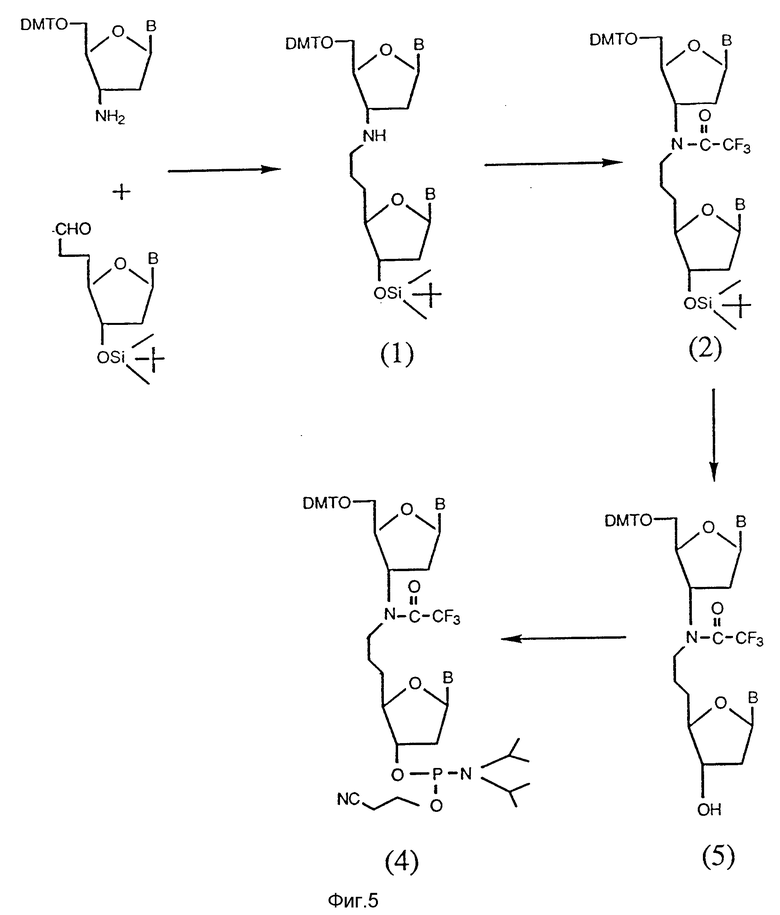
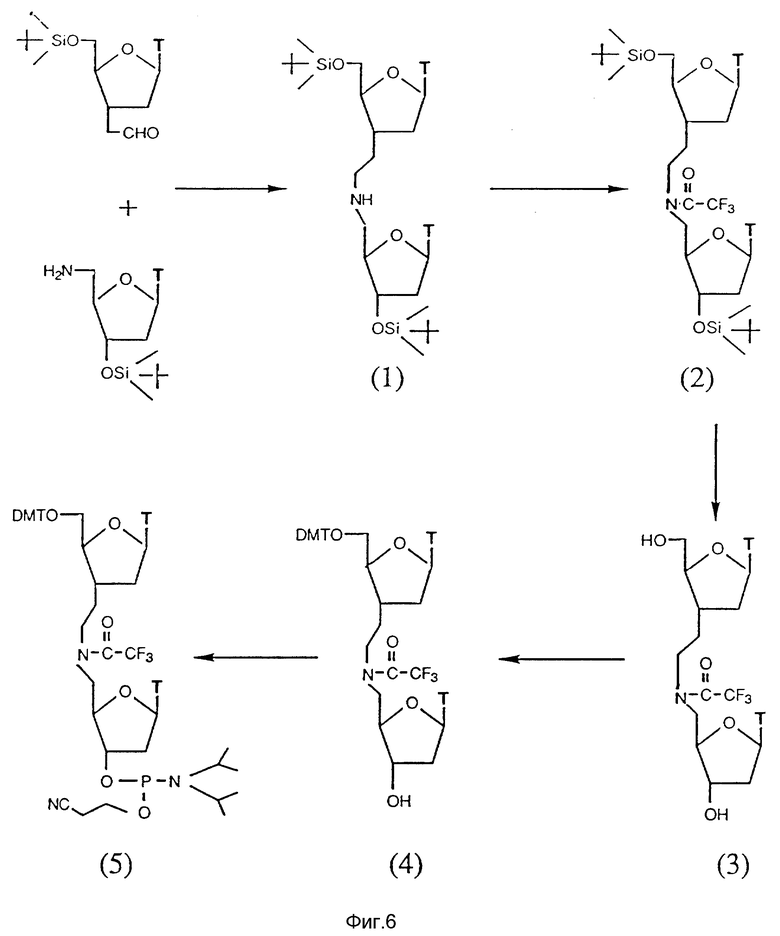
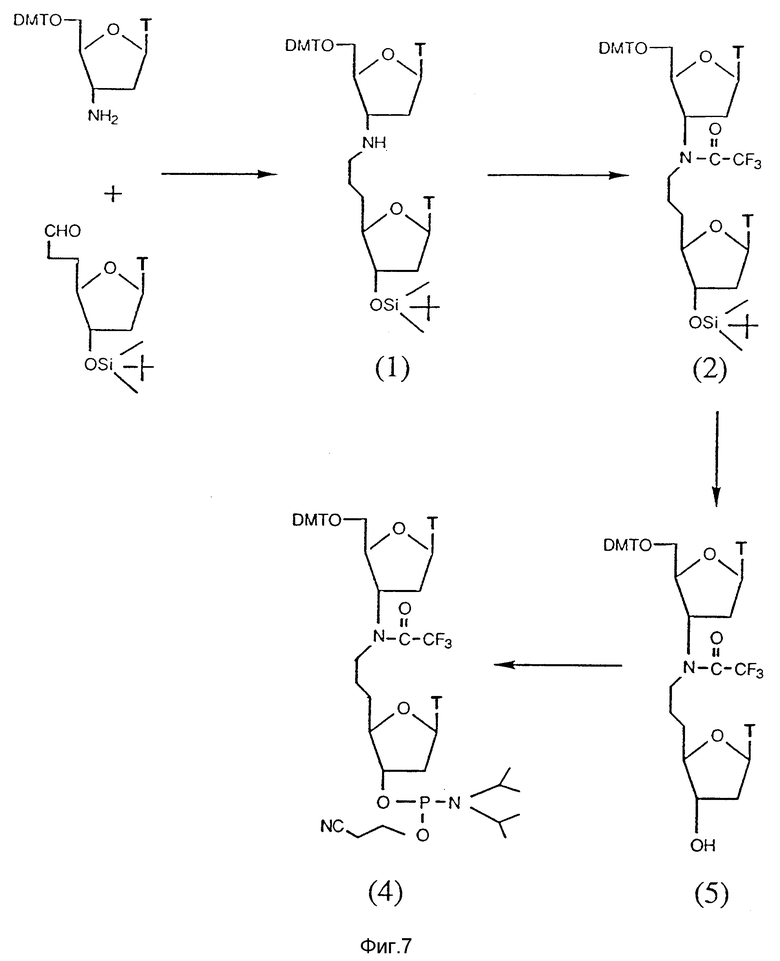
Описывается новый резистентный к нуклеазе олигонуклеозид, содержащий последовательность общей формулы I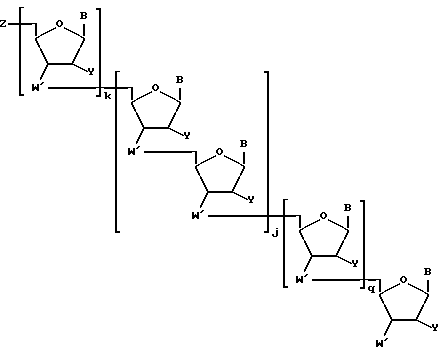
где каждый Z означает независимо R1 или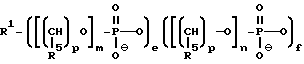
где W означает -D-D-D-, где каждый D независимо является CHR, NR6, где R означает независимо водород, ОН, R6 означает водороду при условии, что только одно из D означает группу NR6,
W1 означает независимо W или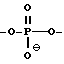
R1 независимо является OH, SH, NR2R3, где R2 и R3 независимо Н или С1-6-алкил, каждый R5 означает независимо Н или С1-12-алкил;
каждый Y означает независимо Н или ОН;
каждый B означает независимо аденин, цитозин, гуанин, тимин, урацил или их модификации;
каждое e и f независимо целое число от 0 до 50 при условии, что по крайней мере одно из e и f принимает по крайней мере значение 1;
j - целое число от 1 до примерно 200;
K - 0 или целое число от 1 до примерно 197;
каждое m и n независимо целое число от 1 до 200;
каждое p независимо число от 2 до 4;
q или целое число от 1 до примерно 197 при условии, что сумма j + k + q составляет примерно от 5 до 200. 4 c. и 4 з.п. ф-лы, 9 табл., 7 ил.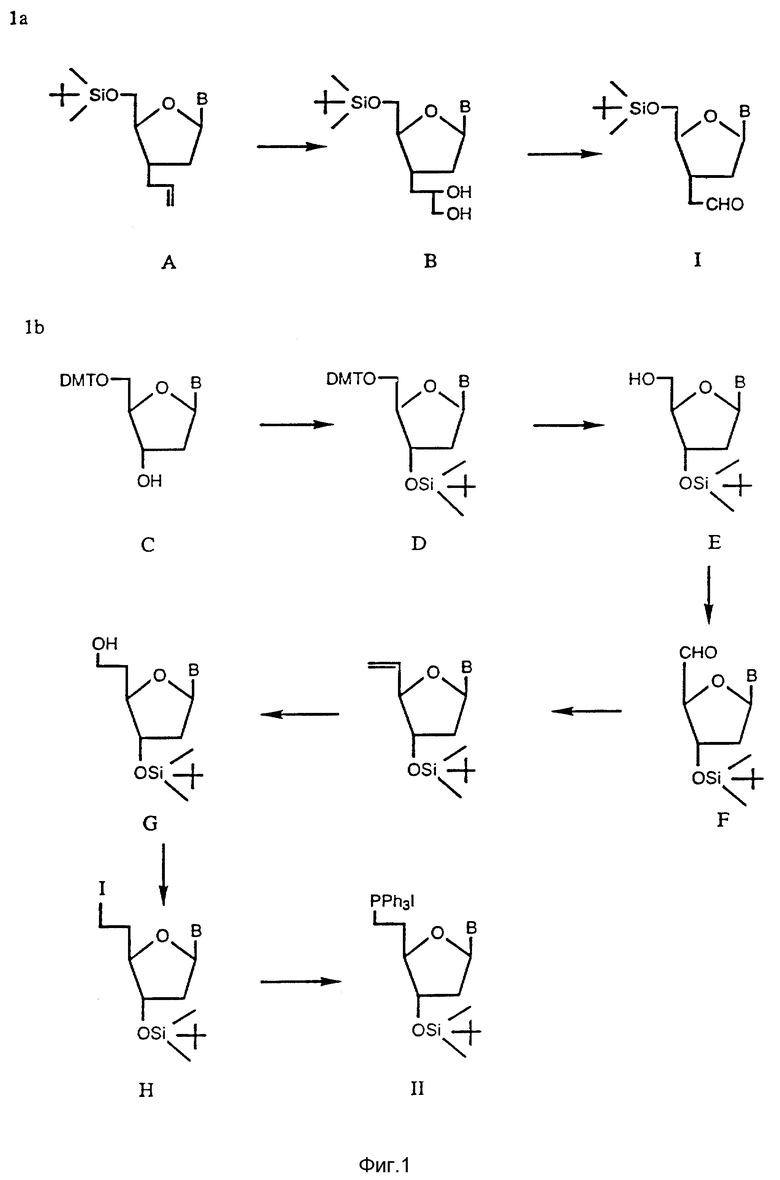
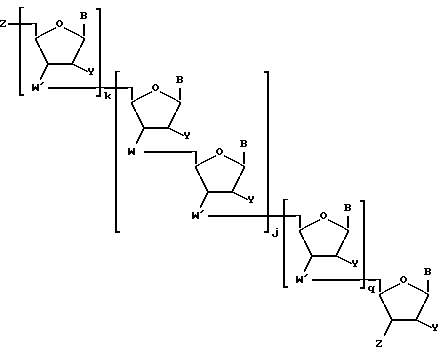
где каждый Z означает независимо R1 или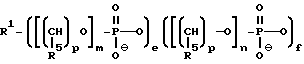
где W означает -D-D-D, где каждый D независимо является CHR, NR6, где R означает независимо водород, ОН, R6 - водород, при условии, что только одно из D означает группу NR6;
W' означает независимо W или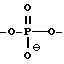
R1 независимо является ОН, SH, Nr2R3, где R2 и R3 независимо Н или С1 - С6-алкил;
каждый R5 - означает независимо Н или С1 - С12 алкил;
каждый У означает независимо Н или ОН, каждый В - означает независимо аденин, цитозин, гуанин, тимин, урацил или их модификации;
каждое е и f - независимо целое число от 0 до 50 при условии, что по крайней мере одно из е и f принимает по крайней мере значение 1,
j - целое число от 1 до примерно 200;
к - 0 или целое число от 1 до примерно 197;
каждое m и n независимо целое число от 1 до 200;
каждое Р независимо число от 2 до 4;
q - 0 или целое число от 1 до примерно 197 при условии, что сумма j + k + q составляет примерно от 5 до 200.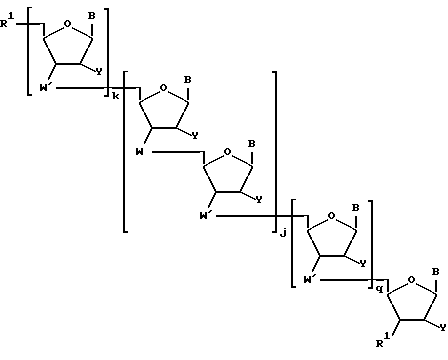
где W - означает D D D, где каждый D независимо является CHR, NR6, где R означает независимо водород, ОН, R6 означает водород, при условии, что только одно из D означает группу NR6, каждый W' означает W или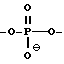
каждый R1 независимо является ОН, SH, NR2R3, где R2 и R3 независимо Н или С1 - С6-алкил;
каждый У является независимо Н или ОН;
каждый В является независимо аденином, цитозином, урацилом или их модификациями;
J - среднее число от 1 до 200;
к - 0 или среднее число от 1 до 197;
q - 0 или среднее число от 1 до 197 при условии j + k + q составляет от 4 до 200.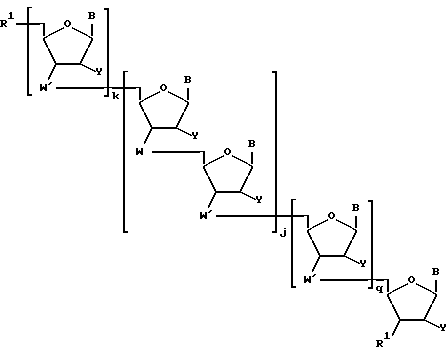
где W означает -D-D-D-, в которой каждый радикал D означает независимо CHR, кислород или группу NR6, где R - независимо водород, ОН, SH или NH2, R6 водород или С1 - С2-алкил при условии, что только одно из D означает кислород или группу NR6;
каждый W1 означает независимо W или
R1 независимо OH, SH, NR2R3, где R2 и R3 независимо водород или С1 - С6-алкил, или NHR4, где R4 - С1 - С12-ацил;
каждый У независимо Н или ОН;
каждый В независимо аденин, цитозин, урацил или их модификации;
j - целое число от 1 до примерно 200;
к - 0 или целое число от 1 до примерно 197;
q - 0 или целое число от 1 до примерно 197, при условии, что сумма j + k + q составляет 4 - 200.
где B, W, Y имеют значения, указанные в п.1;
R7 -ОН или фосфорамидатная группа;
R8 - ОН или трет-бутилдиметилсилилоксигруппа.
| Tetrahedron Letters | |||
| Способ очистки нефти и нефтяных продуктов и уничтожения их флюоресценции | 1921 |
|
SU31A1 |
| Способ размножения копий рисунков, текста и т.п. | 1921 |
|
SU89A1 |
| Способ минералогического анализа порошков | 1955 |
|
SU101985A1 |
