Перекрестная ссылка на родственные заявки
Данное изобретение притязает на приоритет в соответствии с предварительными заявками на выдачу патента США № 60/483711, поданной 30 июня 2003 года, и 60/558616, поданной 1 апреля 2004 года.
Область техники
Данное изобретение относится к способам получения 2'-дезокси- или 2'-модифицированных нуклеозидов, в частности, β-L-2'-дезокситимидина. Настоящее изобретение представляет собой улучшенный способ, который легко масштабируется в целях промышленного производства. Соединения, полученные способом согласно настоящему изобретению, имеют важное значение в качестве противовирусных средств, антинеопластических средств и промежуточных продуктов синтеза фармацевтических соединений и композиций.
Уровень техники
HBV является второй причиной злокачественных опухолей человека, уступая только табаку. Механизм, посредством которого HBV индуцирует злокачественную опухоль, не известен, хотя предполагается, что он может непосредственно запускать развитие опухоли или косвенно запускать развитие опухоли посредством хронического воспаления, цирроза и регенерации клеток, связанных с инфекцией.
Вирус гепатита B достиг эпидемических уровней в мире. После инкубационного периода, составляющего от двух до шести месяцев, в течение которого хозяин не подозревает об инфекции, HBV-инфекция может приводить к острому гепатиту и повреждению печени, что вызывает боли в животе, желтуху и повышенные уровни в крови некоторых ферментов. HBV может вызывать скоротечный гепатит, быстро прогрессирующий, часто в форме смертельного заболевания, при котором разрушаются огромные участки печени.
Пациенты обычно излечиваются от острого гепатита. Однако у некоторых пациентов высокие уровни вирусного антигена сохраняются в крови в течение длительного или неограниченного периода времени, вызывая хроническую инфекцию. Хронические инфекции могут приводить к хроническому персистирующему гепатиту. Наиболее часто пациенты, инфицированные хроническим персистирующим HBV, встречаются в развивающихся странах. К середине 1991 года было примерно 225 миллионов хронических носителей HBV только в Азии, а во всем мире почти 300 миллионов носителей. Хронический персистирующий гепатит может вызывать утомление, цирроз печени и гепатоклеточную карциному, первичный рак печени.
В заявке WO 96/40164, поданной Emory University, UAB Research Foundation и национальным центром научных исследований (CNRS), описан ряд β-L-2',3'-дидезоксинуклеозидов для лечения гепатита B.
В заявке WO 95/07287, также поданной Emory University, UAB Research Foundation и национальным центром научных исследований (CNRS), описаны 2'- или 3'-дезокси- и 2',3'-дидезокси-β-L-пентофуранозилнуклеозиды для лечения ВИЧ-инфекции.
В заявке WO 96/13512, поданной Genencor International, Inc., и Lipitek, Inc., описано получение L-рибофуранозилнуклеозидов в качестве противоопухолевых средств и противовирусных средств.
Idenix Pharmaceuticals, Ltd. описывают 2'-дезокси-L-эритропентофуранонуклеозиды и их применение при лечении HBV в патентах США №№ 6395716; 6444652; 6566344 и 6539837. А также см. WO 00/09531. Описан способ лечения инфекции гепатита B у людей и других животных-хозяев, который заключается во введении эффективного количества биологически активного 2'-дезокси-β-L-эритропентофуранонуклеозида (альтернативно называемого β-L-dN или β-L-2'-dN) или его фармацевтически приемлемой соли, сложного эфира или пролекарства, включая β-L-дезоксириботимидин (β-L-dT), β-L-дезоксирибоцитидин (β-L-dC), β-L-дезоксирибоуридин (β-L-dU), β-L-дезоксирибогуанозин (β-L-dG), β-L-дезоксирибоаденозин (β-L-dA) и β-L-дезоксирибоинозин (β-L-dI), вводимых либо отдельно, либо в комбинации, необязательно в фармацевтически приемлемом носителе. Также заявлены 5'- и N4- (цитидин) или N6- (аденозин) ацилированные или алкилированные производные активного соединения или 5'-фосфолипиды или 5'-эфиры липидов.
Von Janta-Lipinski et al., в J. Med. Chem., 1998, 41 (12), 2040-2046, описывают применение L-энантиомеров 3'-фтор-модифицированных β-2'-дезоксирибонуклеозид-5'-трифосфатов для ингибирования полимераз гепатита B. В частности, 5'-трифосфаты 3'-дезокси-3'-фтор-β-L-тимидина (β-L-FTTP), 2',3'-дидезокси-3'-фтор-β-L-цитидина (β-L-FdCTP) и 2',3'-дидезокси-3'-фтор-β-L-5-метилцитидина (β-L-FMethCTP) заявлены в качестве эффективных ингибиторов ДНК-полимераз HBV. Кроме того, von Janta-Lipinski et al. описывают биологическую активность трифосфата β-L-тимидина (но не β-L-2'-dC) в качестве нуклеозидного ингибитора эндогенных ДНК-полимераз HBV и DHBV. Однако оценивали только трифосфорилированный β-L-тимидин, но не заявленную нефосфорилированную форму, и статья не содержит комментария по поводу того, фосфорилируются ли указанные β-L-нуклеозиды в клетках или in vivo, или, что более важно, не содержит комментария относительно эффективности фосфорилирования β-L-тимидина in vivo. Поэтому в статье не говориться о том, что β-L-тимидин может обладать какой-либо активностью по отношению к гепатиту B в клетке или in vivo. См. также WO 96/1204.
В Европейской заявке на выдачу патента № 0352248 A1 Johansson et al. описывают применение соединений L-рибофуранозила для лечения гепатита B.
Verri et al. описывает применение 2'-дезокси-β-L-эритропентофуранонуклеозидов в качестве антинеопластических средств и в качестве противогерпетических средств (Mol. Pharmacol. (1997), 51(1), 132-138 и Biochem. J. (1997), 328(1), 317-20). Saneyoshi et al. продемонстрировали применение 2'-дезокси-L-рибонуклеозидов в качестве ингибиторов обратной транскриптазы (I) для борьбы против ретровирусов и для лечения СПИДа, Jpn. Kokai Tokkyo Koho JP06293645 (1994).
Giovanni et al., в частности, тестировали 2'-дезокси-β-L-эритропентофуранонуклеозиды против вируса псевдобешенства (PRV), Biochem. J. (1993), 294 (2), 381-5.
Химиотерапевтические применения 2'-дезокси-β-L-эритропентофуранонуклеозидов исследовали Tyrsted et al. (Biochim. Biophys. Acta (1968), 155 (2), 619-22) и Bloch, et al. (J. Med. Chem. (1967), 10 (5), 908-12).
Morris S. Zedeck et al. впервые описали β-L-dA для ингибирования синтеза индуцируемых ферментов у Pseudomonas testosteroni, Mol. Phys. (1967), 3 (4), 386-95.
Кроме того, производные цитозина применимы в качестве промежуточных продуктов для получения таких лекарственных средств, как цитидиндифосфатхолин, родовое название которых цитиколин.
В публикации патента США № 20030083306, Idenix Pharmaceuticals, Ltd., описаны 3'-пролекарства 2'-дезокси-β-L-нуклеозидов для лечения HBV. См. также WO 01/96353.
В патенте США № 4957924 Beauchamp описывает различные терапевтические сложные эфиры ацикловира.
17-21 апреля 2002 года на конференции Европейской ассоциации по исследованию печени в Мадриде, Испания, Siihnel et al., Gilead Sciences, Inc., представили стендовое сообщение, свидетельствующее, что комбинации адефовира с β-L-2'-дезокситимидином дает аддитивный эффект против HBV in vitro.
Синтез нуклеозидов
Способы получения нуклеозидов и промежуточных фуранозильных соединений хорошо известны в предшествующем уровне техники. В 1952 Pratt и соавторы сообщили о синтезе L-дезокситимидина (LdT) из арабинозы (J. W. Pratt et al., J. Am. Chem. Soc., 1952, 74: 2200-2205). Путь синтеза, описанный Pratt, заключается в образовании метилгликозида из L-арабинозы с последующим превращением в метилтиотиокарбонат и восстановлением до дезоксисахара. Альтернативно 2-гидроксигруппу превращали в соответствующую ей группу мезилата, которую затем подвергали восстановительному расщеплению, чтобы получить конечный продукт LdT (J. W. Pratt et al., J. Am. Chem. Soc., 1952, 74:2200-2205; H. Urata et al., Nucleic Acids Res., 1992, 20:3325-3332).
Вариации синтеза LdT осуществляли Shull et al., Sznaidman et al., Wang et al. и Stick et al., каждый из которых превращал L-арабинозу в метил-2'-дезоксирибофуранозид через промежуточный гликаль (B.K. Shull et al., J. Carbohydr. Chem., 1996, 15:955-64; M. L. Sznaidman et al., Nucleosides, Nucleotides & Nucleic Acids, 2002, 21:155-63; Z.X. Wang et al., Nucleosides, Nucleotides & Nucleic Acids, 2001, 20:11-40; и R.V. Stick et al., Aust. J. Chem. 2002, 55:83-85).
В 1969 году Niedballa и Vorbruggen описали способ получения β-нуклеозидов посредством сочетания силилированного N-гетероциклического соединения, в частности пиримидина, с 1-O-алкил- или предпочтительно 1-ацил-защищенным сахаром, таким как 1-ацил-защищенная рибоза, дезоксирибоза, арабиноза или глюкоза. В реакции использовали реагент Фриделя-Крафтcа в качестве катализатора, и реакция протекала при температурах окружающей среды (DE 1919307, Schering Aktiengesellschaft). Авторы изобретения отмечали, что данный способ неожиданно давал почти исключительно β-аномерный продукт и мог работать в случае урацила и цитозина, но не в случае тимидина (DE 1919307, примеры 1-10 и 12-15).
В своих примерах Niedballa и Vorbruggen сообщили только об 1-O-ацетил-, 1-ацетил- и 1-O-метил-производных соединениях рибозы, дезоксирибозы и арабинофуранозы в качестве исходных реагентов (DE 1191307, примеры 1-16). Нигде не использовали 1-галогенсахар. К тому же авторы изобретений отмечали, что применение 1-галогенсахара в качестве реагента нежелательно из-за его нестабильности (DE 1191307; JP 63026183, Sato et al.). В одном примере, в котором цитозиновое основание подвергали взаимодействию с сахаром 2'-дезоксирибозой, исходным соединением была 1-O-метил-2-дезокси-3,5-дитолуилрибоза (DE 1919307, пример 7). Не является неожиданным то, что в данной реакции образовывался β-аномер почти при полном исключении α-аномера, так как известно, что 3'-сложноэфирные производные рибозы обычно предпочтительно образуют β-аномер по сравнению с α-аномерным продуктом.
В последующих патентах Vorbruggen et al. ссылались на свой более ранний способ синтеза (1969) как «очень невыгодный», так как отделение солей кислот Льюиса или катализаторов Фриделя-Крафтcа, образованных во время реакции, приводило к необходимости в многочисленных трудоемких стадиях при заключительной обработке, что давало более низкий процент выхода конечного продукта (DE 2508312, эквивалентная заявка Великобритании GB 1542442). В GB 1542442 сообщалось о замене в способе кислот Льюиса триметилсилиловыми сложными эфирами неорганических кислот и об исходных реагентах, которые представляли собой 1-галоген-, 1-O-алкил- или 1-O-ацил-сахар. Как указано ранее, во всех иллюстративных примерах использовали исходный реагент 1-O-ацетил-β-D-рибофуранозу, и следовательно, не был неожиданным полученный β-аномерный продукт при почти полном отсутствии α-аномера (GB 1542442, примеры 1-13).
Подобным образом в патенте США 4209613 Vorbruggen описал одностадийный синтез нуклеозидов, который заключался во взаимодействии силилированного основания нуклеозида с 1-O-ацил-, 1-O-алкил- или 1-галоген-производным защищенного сахара в присутствии катализатора Фриделя-Крафтcа, выбранного из любого катализатора из группы кислот Льюиса (US 4209613). Как указано ранее, во всех иллюстративных примерах использовали исходный реагент 1-O-ацетил-β-D-рибофуранозу, и опять-таки не был неожиданным полученный β-аномерный продукт при почти полном отсутствии α-аномера (US 4209613, примеры 1-16).
В патенте США 5750676, Vorbruggen et al. сообщили о способе, который заключался во взаимодействии свободного сахара с N-гетероциклическим основанием в присутствии силилирующего агента и инертного растворителя, содержащего кислоту Льюиса, при этом усовершенствование состояло в персилилировании свободного сахара. Не было сделано пояснений относительно аномерных соотношений продукта, и не говорится о преимуществе одной кислоты Льюиса. Однако примеры показали, что требовались многочисленные стадии обработки, чтобы получить конечные продукты, явный недостаток для производства в промышленном масштабе (US 5750676, примеры 1-3).
Еще один способ получения нуклеозидов, о котором сообщали Vorbruggen et al., заключался в синтезе в одном сосуде с использованием сложного триалкилсилилового эфира неорганической или сильной органической кислоты, главным образом катализатора Фриделя-Крафтcа, основания нуклеозида и 1-O-ацил-, 1-O-алкил- или 1-галогензамещенного производного защищенного производного сахара (US 4209613).
Промежуточный продукт в виде хлор-сахара
Хлор-сахар является важным промежуточным продуктом в образовании LdT и существуют многочисленные способы его синтеза. Неограничивающие примеры синтеза хлор-сахаров включают следующие способы.
Isbell, Bock et al. и Lundt et al. сообщали о синтезе LdT из D-ксилозы способом, в который включен промежуточный 1,4-лактон (H.S. Isbell, Methods in Carbohydrate Research, 1963, 2:13-14; K. Bock et al., Carbohydrate Research, 1981, 90:17-26; K. Bock et al., Carbohydrate Research, 1982, 104:79-85; и I. Lundt и R. Madsen, Topics in Current Chemistry, 2001, 215:177-191).
Bock et al. и Humphlett использовали D-галактозу в качестве исходного вещества, которую подвергали окислительному расщеплению и бромировали, получая D-ликсонолактон. Последующие стадии избирательного гидролиза и превращений давали промежуточный хлор-сахар, который затем можно было использовать для получения LdT (K. Bock et al., Carbohydrate Research, 1981, 90:17-26; K. Bock et al., Carbohydrate Research, 1979, 68:313-319; K. Bock et al., Acta Chem. Scand. B, 1984, 38:555-561; и W.J. Humphlett, Carbohydrate Research, 1967, 4:157-164).
Bock et al. также получали LdT из D-глюконолактона обработкой последнего водным раствором брома и гидразина и затем избытком водного раствора гидроксида калия с образованием первичного эпоксида. Затем они осуществляли перегруппировку Пейна первичного эпоксида во вторичный эпоксид на лактоне, и подвергали лактон окислительному расщеплению с образованием промежуточного хлор-сахара, который затем можно было использовать для получения LdT (K. Bock et al., Carbohydrate Research, 1979, 68:313-316; K. Bock et al., Acta Chem. Scand. B, 1984, 38:555-561). В некоторых упомянутых журнальных статьях Bock et al. описали образование хлор-сахара из D-галаконолактона и бромированием D-манноно-1,4-лактона.
Liotta и Hager сообщали о синтезе хлор-сахара из коммерчески доступного лактона способом синтеза, который включает стадию стереоселективной циклизации, а также способом синтеза, в котором используют промежуточный альдегид и модификацию Хорнера-Эммонса реакции Виттига (D. C. Liotta et al., Tetrahedron Letters, 1992, 33:7083-7086; и US 5414078).
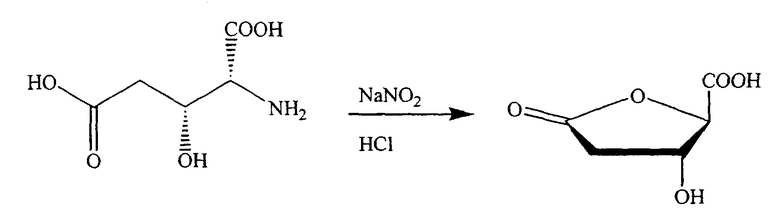
Schinazi et al., Ravid et al. и Taniguchi et al. описали способы получения промежуточных хлор-сахаров из гидроксиглутаминовой кислоты, которую подвергают циклизации до производного рибонолактона, который затем может быть превращен в хлор-сахар (US 6348587 B1, R. F. Schinazi et al.; U. Ravid et al., Tetrahedron, 1978, 34:1449-1452; и M. Taniguchi et al., Tetrahedron, 1974, 30:3547-3552).
Jung et al. сообщили об использовании эпоксидирования по Шарплессу на коммерчески доступном спирте, чтобы получить эпоксид, который затем обрабатывали спиртом, получая диол, который затем превращали в ацетонид. Ацетонид подкисляли, получая требуемую рибофуранозу, которую затем превращали в хлор-сахар. Альтернативно эпоксиспирт подвергали гидроборированию, используя окисление по Сверну, и хлор-сахар образовывали из дитолуоил-производного (M. E. Jung et al., Tetrahedron Letters, 1998, 39:4615-4618).
Yadav et al. и Harada et al. описали синтезы, при которых использовали аллилбромид и озонолиз или 2-бромметил[1,3]диоксолан без озонолиза, чтобы получить хлор-сахара (J. S. Yadav et al., Tetrahedron Letters, 2002, 43:3837-3839; T. Harada et al., Chem. Lett., 1981, 1109-1110), тогда как Ohuri et al., Cheng et al. и Abramski et al. сообщали об обработке гликаля кислым метанолом с получением 2-дезоксирибофуранозы, которую затем превращали в требуемый хлор-сахар.
В JP 09059292 Takeya Mori описал синтез в одном сосуде 4-аминопиримидинового нуклеозида из 4-гидроксипиримидинового нуклеозида посредством защиты гидроксильных групп реагента триметилсилильными группами, последующим взаимодействием с оксихлоридом фосфора или 4-хлорфенилфосфордихлоридатом и аминированием водным раствором аммиака.
Chu сообщил о способе получения 2'-дезоксинуклеозидов, который заключается во взаимодействии нуклеозида, имеющего 2'- и 3'-гидроксильные группы, со смесью ацилбромида или ацилхлорида и бромистоводородной или хлористоводородной кислоты при умеренных температурах с получением производного галогенацилнуклеозида, защиту которого удаляли с образованием требуемого нуклеозидного продукта (US 5200514).
В Nucleosides and Nucleotides, 1996, 15 (1-3):749-769 Kamaike et al. описали образование 2'-дезоксирибонуклеозидов посредством реакций нуклеофильного замещения 4-азолил-1-β-D-рибофуранозилпиримидин-2(1H)она, полученного превращением из уридина с использованием [15N]фталимида в присутствии триэтиламина или DBU, с получением N4-фталоил[4-15N]цитидина с высокими выходами.
В JP 71021872 Sankyo Co. Ltd. представили реакцию силилированного основания цитозина, урацила, тимина или азаурацила с галогенидом сахара, таким как галогенированная рибоза или глюкоза, в присутствии растворителя и галогенида ртути.
D-ксилоза
Используя D-ксилозу в качестве исходного вещества, можно синтезировать 2'-дезоксинуклеозиды согласно способам, описанным в предшествующем уровне техники.
Okabe et al. описали синтез 2-дезокси-3,5-ди-O-пара-толуоил-α-L-эритропентофуранозилхлорида, который затем может быть подвергнут взаимодействию с получением β-L-2'-дезокситимидина (LdT) (Okabe et al., J. Org Chem., 1991, 56(14): 4392; Bock et al., Carbohydr. Res., 1981, 90:17-26; Bock et al., Carbohydr. Res., 1982, 104:79-85).
Далее следует неограничивающий список способов, используемых для получения промежуточных продуктов синтеза 2'-дезоксинуклеозидов, и в частности, 2'-дезокситимидина, из D-ксилозы.
Takahata et al. и Graf et al. сообщили об образовании 2,5-дибром-2,5-дидезокси-D-ликсо-1,4-лактона в результате взаимодействия ликсо-1,4-лактона с йодидом калия в ацетоне (Takahata et al., J. Org. Chem., 1994, 59:7201-7208; Graf et al., Liebigs Ann. Chem., 1993, 1091-1098).
Lundt et al., Bock et al. и Choi et al. описали инверсию 5-бром-2,5-дидезокси-D-треопентоно-1,4-лактона с образованием 2-дезокси-L-рибоно-1,4-лактона (Lundt et al., Topics in Current Chemistry, 2001, 215:177-191; Bock et al., Carbohydr. Res., 1981, 90:17-26; WO 01/72698, Y-R. Choi et al.).
Urata et al. и Zhang et al. сообщили о превращении 2-дезокси-3,5-ди-O-толуоил-α,β-L-рибозы в 2-дезокси-3,5-ди-O-пара-толуоил-α-L-эритропентофуранозилхлорид либо прямо из лактола в результате взаимодействия с хлористоводородной и уксусной кислотами, либо опосредованно через промежуточный 2-дезокси-7-метокси-3,5-ди-O-толуоил-α,β-L-рибозу в результате взаимодействия с уксусной и хлористоводородной кислотами (H. Urata et al., Nucleic Acids Res., 1992, 20(13):3325-3332; Zhang et al., Nucleosides and Nucleotides, 1999, 18 (11-12):2357).
Urata et al. также описали получение 2'-дезокси-3',5'-ди-O-пара-толуоил-L-тимидина из 2-дезокси-3,5-ди-O-пара-толуоил-α-L-эритропентофуранозилхлорида и силилированного тимина в присутствии хлороформа с последующим удалением защиты с образованием 2'-L-дезокситимидина (H. Urata et al., Nucleic Acids Res., 1992, 20 (13):3325).
Промежуточный 2,2'-ангидро-1-фуранозилнуклеозид
Показано, что 2'-дезокси- и 2'-замещенные нуклеозиды, и в частности, 2'-дезокси- или 2'-замещенные нуклеозиды, которые имеют пиримидиновые основания, стабилизируют олигонуклеотиды от разрушения нуклеазами. Разрушение нуклеазами является проблемой в области олигонуклеотидных терапевтических средств (Huryn et al., (1992), Chem. Rev. 92:1745-88; English et al., (1991), Angew. Chem. 30:613-722). Однако до настоящего времени модификацию пиримидиновых нуклеозидов во 2'-положении осуществили только в жестких условиях и способами синтеза, которые являются неэффективными, как правило, с низкими выходами продукта (Verheyden et al., (1971), J. Org. Chem. 36:250-254).
Tronchet et al. описали восстановление оксимного производного 2'-кетоуридина с помощью BH3, которое преимущественно дает 2'-гидрокси- или 2'-аминонуклеозиды в арабино-конфигурации (Tronchet et al., (1990), Tetrahedron Lett. 31:351). Указанная работа Tronchet является одной из немногих попыток стереоселективного синтеза 2'-рибофуранозиламино- или 2'-рибофуранозилгидроксилпиримидинов.
Ранние подходы к синтезу 2'-дезокси- или 2'-замещенных пиримидиновых нуклеозидов были сфокусированы на подходящих защитных группах для рибозы, ксилозы и арабинозы, которые были исходными реагентами в синтезах. Например, были предприняты многочисленные подходы к синтезу перацилированной рибофуранозы в качестве промежуточного продукта в способах получения нуклеозидов. Подходы включают i) 7-стадийный стереоспецифичный способ, который начинался с D-рибозы и давал β-D-2'-дезоксирибофуранозилтимидин с выходом конечного продукта около 40% (M. Jung и Y. Xu, Tetrahedron Lett. (1997), 38:4199); ii) 3-стадийный способ, начинающийся с L-рибозы и приводящий к выходу продукта, составляющему 56% (E. F. Recondo и H. Rinderknecht, Helv. Chim. Acta, (1959) 42:1171); iii) 8-стадийный способ с использованием L-арабинозы в качестве исходного вещества и получением примерно 20% выхода продукта (J. Du et al., Nucleosides and Nucleotides, (1999), 18:187); iv) 6-стадийный способ, начиная с L-ксилозы (% выхода конечного продукта не известен) (E. Moyroud и P. Strazewski, Tetrahedron (1999) 55:1277); и v) многостадийный способ, начинающийся с D-рибозы, которую сначала превращали в три-O-ацетилтимидин (патент США № 4914233).
В 1959 году E. F. Recondo сообщил о 5-стадийном способе получения толуоил-, бензоил- и ацетил-защищенного рибофуранозила с выходом примерно 70-80% из D-рибозы (E. F. Recondo, Helv. Chim. Acta, (1959) 121:1171). Codington, Doerr и Fox описали синтез 2,2'-ангидро-1-(5-O-тритил-β-D-арабинофуранозил)тимина из β-D-тимидина в результате взаимодействия β-D-тимидина с тритилхлоридом и пиридином в течение 24 часов при комнатной температуре и затем примерно при 70°C в течение 3 часов, чтобы защитить 5'-OH на β-D-тимидине; затем посредством взаимодействия 5'-защищенного β-D-тимидина с тозилхлоридом (TsCl) и пиридином при 0°C, которое давало тозил-защищенную 2'-группу; и, наконец, посредством взаимодействия 5'-тритил-O-защищенного, 2'-тозил-O-защищенного β-D-тимидина с бензоатом натрия (NaOBz) и ацетамидом при 100°C в течение 1 часа с получением 2,2'-ангидро-1-(5'-O-тритил-β-D-арабинофуранозил)тимина с выходом 61% (Codington et al., J. Org. Chem., (1963) 29:558-64).
Сообщалось о ферментативном синтезе β-D-тимидина с использованием E. coli и гипоксантина на первой стадии и взаимодействием полученного в результате 2-монофосфорилированного соединения рибофуранозила с уридинфосфорилазой и извлечением требуемого продукта β-D-тимидина с выходом 45% с помощью хроматографии на колонке (А.И. Зинченко, Химия природных соединений, (1989), 4:587-88).
Другой способ синтеза нуклеозидов заключается в образовании промежуточного 5-метил-2,2'-ангидроуридина из «раскрытого нуклеозида». Раскрытый нуклеозид образуют посредством реакции внутримолекулярного нуклеофильного замещения, которая дает 2,2'-ангидро-1-(β-D-арабинофуранозил)нуклеозид в результате раскрытия цикла 2,2'-ангидронуклеозида. В заявке Японии Kokai № 8149398 (выложенная 2 мая 1981 года) описан синтез ангидронуклеозидов, который требует в качестве промежуточного продукта кислотно-аддитивной соли ацилированного иминоарабино[1',2':4,5]оксазолина. О применении доступного углеводного производного аминооксазолина в качестве предшественника ангидронуклеозида сообщали в 1971 году (J. Mol. Biol., (1970) 47:537).
Rao et al. сообщили о 6-стадийном синтезе, в котором использовали D-ксилозу в качестве исходного реагента, образуя 1-β-D-ксилофуранозилтимин, который затем обрабатывали PhOCOOPh (дифенилкарбонатом) и катализатором NaHCO3 в присутствии ДМФА при 140-150°C в течение примерно 4 часов, получая 2,2'-ангидро-1-(β-арабинофуранозил)тимин с выходом 55% (A. V. Rama Rao et al., J. Chem. Soc. Comm., (1994), p.1255; EP 0683171 B1). И Schinazi et al. и Manfredi et al. описали синтез, сходный с синтезом, описанным Rao et al., в котором использовали такие же реагенты, за исключением использования 1-β-D-арабинофуранозилтимина, а не 1-β-D-ксилофуранозилтимина (Schinazi et al., J. Med. Chem., (1979) 22:1273; Manfredi et al., Bioorg. Med. Chem. Letters, (2001) 11:1329-32).
Ранняя попытка образования 3',5'-дибензоил-защищенного 2,2'-ангидро-1-(β-рибонофуранозил)тимина описана Anton Holy et al. Holy et al. использовали β-D-рибонофуранозилтимин в качестве исходного соединения, его подвергали взаимодействию с 1,4 эквивалента PhOCOOPh и катализатором NaHCO3 в HMPA в течение примерно 20 минут при температуре около 150°C, чтобы образовать 2,2'-ангидро-1-(β-D-рибофуранозил)тимин (5-метилуридин), который подвергали взаимодействию с PhCOCN в ДМФА, чтобы защитить 3'- и 5'-OH-группы посредством образования 2,2'-ангидро-1-(β-3',5'-ди-O-бензоил)рибофуранозилтимин с входом примерно 87% (A. Holy et al., Collect. Czech. Commun., (1974), 39:3157-67). Holy et al. также сообщили о безуспешной попытке превратить 2-амино-β-D-арабинофурано[1',2':4,5]-2-оксазолин в O2,2'-ангидро-1-(β-D-арабинофуранозил)тимин (там же, 1377).
Fraser et al. усовершенствовали способ Holy, используя такой же исходный реагент и подвергая его взаимодействию с 1,2 эквивалента PhOCOOPh и катализатором NaHCO3 в присутствии HMPA примерно при 150°C в течение примерно 2 часов, получая 2,2'-ангидро-1-β-D-рибофуранозилтимин. Однако способ Fraser et al. давал сниженный процент выхода продукта, составляющий примерно 77%, по сравнению с выходом примерно 87%, полученным при синтезе согласно Holy et al. (Allister Fraser et al., J. Heterocycl. Chem., (1993) 30 (5):1277-88).
Yukio Aoyama et al. описали образование силил-защитного цикла, который охватывает как 3'-, так и 5'-положения β-1-D-(2-Br-рибофуранозил)тимина с выходом примерно 96% (Aoyama et al., Nucleosides and Nucleotides, (1996), 15 (1-3):733-8). 1-β-D-рибофуранозилтимин использовали в качестве исходного вещества и подвергали взаимодействию с TPDSCl2 и пиридином при комнатной температуре, получая 3'-, 5'-силил-защищенную циклическую структуру. Затем силил-защищенную структуру подвергали взаимодействию с TfCl и DMAP в CH2Cl2 при комнатной температуре с образованием промежуточного 2,2'-ангидропродукта и, наконец, промежуточный 2,2'-ангидропродукт подвергали взаимодействию с LiBr, BF3-OEt в 1,4-диоксане примерно при 60°C, получая конечный продукт, 1-β-D-2'-Br,3',5'-три-O-ди(диметил)силил)рибофуранозилтимин.
Mitsui Chemicals, Inc. сообщали о способах получения 2,2'-ангидро-1-(β-L-арабинофуранозил)тимина и 2,2'-ангидро-5,6-дигидроциклоуридина, который применим в качестве промежуточных продуктов при синтезе L-нуклеиновых кислот (публикация PCT № WO 02/044194; EP 1348712 A1). 7-стадийный способ согласно Mitsui включает: a) взаимодействие L-арабинозы с цианамидом с получением L-арабиноаминооксазолина; b) взаимодействие L-арабиноаминооксазолина с производным акриловой кислоты с образованием производного L-арабиноаминооксазолина, содержащего сложный эфир метилакриловой кислоты, связанный с N-атомом остатка оксазолина; c) взаимодействие продукта со стадии (b) с таким основанием, как, например, щелочной металл, алкоксид щелочного металла, карбонат щелочного металла, бикарбонат щелочного металла, гидроксид щелочного металла, гидрид щелочного металла, органическое основание, основная ионообменная смола и тому подобное, любое из которых образует при этом трициклическое кольцо, которое является производным L-2,2'-ангидронуклеиновой кислоты; d) изомеризацию производного L-2,2'-ангидронуклеиновой кислоты со стадии (c) с получением 2,2'-ангидро-1-(β-L-арабинофуранозил)тимина; e) 2,2'-ангидро-1-(β-L-арабинофуранозил)тимин со стадии (d) подвергают либо галогенированию и последующей защите, либо защите и последующему галогенированию, либо одновременному галогенированию и защите с образованием галогенированного по 2'-положению производного L-тимидина; f) дегалогенирование галогенированного производного L-тимидина со стадии (e); и g) деблокирование 3'- и 5'-положений продукта со стадии (f) с получением L-тимидина. Хотя Mitsui сообщал о хороших выходах продукта в результате данного синтеза, желательно иметь способ, который требует меньше стадий, чтобы его было легче адаптировать для крупномасштабного промышленного производства.
Вторым близким способом, найденным в предшествующем уровне техники, является способ, сообщенный Pfizer в EP 0351126 B1. Способ Pfizer заключается в новом пути образования O2,2'-ангидро-1-(β-D-арабинофуранозил)тиминовых нуклеозидов (ангидронуклеозидов), которые могут быть легко превращены в производные β-тимина. Способ включает реакцию конденсации между 2-амино-β-D-арабинофурано[1',2':4,5]-2-оксазолином или его 5'-тритил- или силил-защищенной формой предпочтительно с метил-2-формилпропионатом в H2O и NaOH при pH 8,1 в течение 48 часов при комнатной температуре с последующей обработкой водным раствором кислоты с получением O2,2'-ангидро-1-(β-D-арабинофуранозил)тимина с выходом примерно 42%. Альтернативы использования метил-2-формилпропионата включают применение метил-3-бромметилакрилата в присутствии DMAP и Et3N примерно при 80°C в течение 4 дней, что давало примерно 25% выход конечного продукта ангидротимидина; применение этил-2-формилпропионата в водном MeOH и Et3N при комнатной температуре в течение примерно 24 часов и затем примерно при 60°C еще в течение 24 часов для выхода ангидротимидинового продукта, составляющего примерно 8%; и применение метил-3-метоксиметакрилата в ДМСО примерно при 80°C в течение 4 дней с получением ангидротимидинового продукта с выходом примерно 32%.
Реакция конденсации по Pfizer включает использование в предпочтительном варианте основных катализаторов. Такими катализаторами являются третичные амины и неорганические соли, и предпочтительными среди них являются диметиламинопиридин, триэтиламин, N-метилморфолин и их комбинации. Pfizer сообщил, что предпочтительным способом превращения O2,2'-ангидро-1-(β-D-арабинофуранозил)тимина в β-тимидин была реакция ангидротимидина с HBr с последующим удалением Br в результате взаимодействия с BaSO4-отравленным Pd-катализатором. Желательно получить промышленно масштабируемый синтез, который мог бы исключить необходимость применения отравленного катализатора данного типа.
Boehringer-Ingelheim Pharma GMBH сообщили о 4-стадийном способе получения β-L-2'-дезокситимидина, в котором использовали L-арабинозу в качестве исходного вещества (публикация PCT № WO 03/087118). Способ включал: a) взаимодействие L-арабинозы с цианамидом в водном или водно-спиртовом растворе или в другом полярном растворителе, например, таком как ДМФА, пиридин или N-метилпирролидин, при температуре 80-100°C в присутствии основного катализатора, такого как NH3, Et3N или триэтилкарбонат, основный карбонат или двухосновный карбонат, с образованием производного L-арабинофуранозиламинооксазолина; b) взаимодействие производного L-арабинофуранозиламинооксазолина со стадии (a) с 2-метил-C-3-кислотой или ее активированным производным в инертном растворителе в условиях осаждения водой, например, таких как в присутствии ДМФА, ДМСО, NMP, ацетона, бензола, толуола или циклогексана и катализатора в виде основания третичного амина или неорганической соли, подобного DMAP, Et3N или N-метилморфолину, примерно при 20-80°C; c) взаимодействие β-L-2,2'-ангидротимидина со стадии (b) с нуклеофильным реагентом, таким как галогенводородная кислота, подобная HCl, HI или HBr, толуолсульфоновая кислота или тиоуксусная кислота, в растворителе ДМФА или трифторуксусной кислоте, чтобы разорвать связь C-O во 2'-положении; и d) взаимодействие β-L-2'-галогентимидина с катализатором, предпочтительно либо с Pd, либо с никелем Ренея, чтобы удалить группу галогена из 2'-положения и получить β-L-тимидин в качестве конечного продукта.
Предпочтительно перед выполнением стадий синтеза (a) или (b) любые свободные гидроксильные группы защищают, чтобы предотвратить их взаимодействие с производным аминооксазолина или с 2-метил-C-3-кислотой.
В указанном способе синтеза по Boehringer предпочтительные защитные группы включают бензил, дифенилметил, трифенилметил или силил, где три заместителя на силиле могут представлять собой C1-6-алкил или фенил, и фенильные группы необязательно могут быть дополнительно замещены. Любые защитные группы могут быть удалены на конечной стадии синтеза, и также могут быть добавлены стадии кристаллизации или очистки.
К сожалению, первая стадия способа, раскрытого Boehringer, требовала как минимум двух стадий экстракции, фильтрования и кристаллизации; вторая стадия способа требовала использования кипящего циклогексана и конечной очистки хроматографией; и четвертая стадия способа требовала использования катализатора Pd или никеля Ренея. Сообщенный выход промежуточного β-L-2,2'-ангидроарабинофуранозилтимина составлял примерно 49%. Таким образом, существует необходимость в способе синтеза, который избегает применения катализатора Pd или никеля Ренея и который обеспечивает более высокие проценты выхода промежуточного 2,2'-ангидротимидина.
Holy и Pragnacharyulu et al. описали применение L-арабинозы в качестве исходного вещества, которую подвергают взаимодействию с цианамидом, получая производное 1,2-оксазолина; производное оксазолина подвергают взаимодействию с этиловым эфиром пропионовой кислоты, получая промежуточный O2,2'-ангидро-L-тимидин, который подвергают бензоилированию и восстановительному расщеплению или обрабатывают хлористым водородом, получая требуемый хлор-сахар. (A. Holy, Coll. Czech. Chem. Commun. 1972, 37, 4072-4087).
Abushanab et al. сообщили о синтезе хлор-сахара, который заключается во взаимодействии сложного эфира метилоксиранкарбоновой кислоты с оксазолином с получением промежуточного O2,2'-ангидро-L-тимидина (E. Abushanab и P. V. P Pragnacharyula, патент США 5760208, 2 июня 1998 года), тогда как Asakura et al., Hirota et al. и A. Holy описали реакцию этилпропиолата с оксазолином с получением O2,2'-ангидро-L-уридина, который затем защищали по его 3'- и 5'-положениям и подвергали взаимодействию с хлористым водородом, получая 2'-дезокси-2'-хлор-сахар в качестве промежуточного продукта (J.-I. Asakura и M. J. Robins, J. Org. Chem. 1990, 55, 4928-4933; J.-I. Asakura и M. J. Robins, Tetrahedron Lett. 1988, 29, 2855-2858; K. Hirota, Y. Kitade, Y. Kanbe, Y. Isobe и Y. Maki, Synthesis, 1993, 210, 213-215; и A. Holy, Coll. Czech. Chem. Commun. 1972, 37, 4072-4087).
В 2003 году Abushanab и Pragnacharyulu сообщили о способе получения пиримидиновых нуклеозидов, который включает реакцию конденсации типа реакции Михаэля, между арабинорибофуранозиламинооксазолином и замещенным производным эпоксиметилата; последующее ацилирование конденсированного продукта обработкой пивалоилхлоридом, чтобы поместить группу хлора вo 2'-положении тимидина; и, наконец, дегалогенирование, чтобы удалить хлорный заместитель, если требуемым продуктом является 2'-дезокситимидин (патент США № 6596859).
Однако известно, что пивалоилхлорид вызывает раскрытие ангидроцикла и размещение группы хлора во 2'-положении тимидина, затем требует дополнительной стадии синтеза, чтобы удалить группу хлора. Также было бы полезно избежать применения дорогостоящего реагента метил-2-метилглицидата, который Abushanab и Pragnacharyulu используют в реакции конденсации в своем способе, а также применения ацетонитрила, используемого на второй стадии способа, и хроматографических разделений, требуемых на каждой стадии синтеза.
Pragnacharyulu et al. также сообщили об образовании 2,2'-ангидроаминооксазолина из L-арабинозы в результате взаимодействия L-арабинозы с H2NCN, которое обеспечивает возможность внутримолекулярного элиминирования одного концевого OH и одного H, чтобы получить промежуточный 2,2'-ангидроаминооксазолин (Pragnacharyulu et al., (1995), J. Org. Chem. 60:3096-99).
Sawai et al. описали стадию прямой циклизации при образовании 2,2'-ангидро(арабинофуранозил)тимина из D-арабинозы. Синтез этих авторов включал (1) получение D-арабинофуранозиламинооксазолина из D-арабинозы способами, известными в данной области; (2) взаимодействие D-арабинофуранозиламинооксазолина с этил-α-(бромметил)акрилатом в диметилацетамиде с получением оксазолино-N-разветвленного промежуточного продукта с выходом примерно 88%; и (3) взаимодействие промежуточного продукта, образованного на стадии (2), с KOtBu и трет-BuOH с получением 2,2'-ангидро(арабинофуранозил)тимина с выходом примерно 30%, или альтернативно, использование йодида водорода для раскрытия связи O2,2'-ангидро-L-тимидина и затем взаимодействие ациклического продукта с йодидом калия, чтобы получить ди-O-бензоил-2'-дезокситимидин (Sawai et al., (1994), Nucleosides and Nucleotides, 13 (6-7):1647-54; Sawai et al., Chem. Lett., 1994, 605-606). В данном способе выгодно избегают использования катализаторов, подобных отравленному Pd/BaSO4, но в результате получают более низкие выходы продуктов в %.
В патенте США № 4914233 Freskos et al. описали селективное выделение β-тимидина из смеси α- и β-аномеров посредством 5-стадийного способа, который заключается в образовании три-O-ацил-β-риботимидина и превращении 2,2'-ангидро-β-тимидина в 2'-галоген-2'-дезокси-5-метилуридин с последующим превращением последнего в β-тимидин.
В патенте США № 5212293 Green et al. сообщили о синтезе 2',3'-дидезоксинуклеозидов в результате взаимодействия защищенного ангидротимидина с галогенобразующим агентом, который содержал алюмоорганическое соединение для повышенной растворимости реагента.
В патенте США № 5596087 Alla et al. включили образование 2,2'-ангидротимидина, который подвергали бромированию и затем восстанавливали способами, известными специалистам в данной области, чтобы получить β-тимидин.
В патенте США № 6369040 Acevedo et al. описали 3',5'-защищенный 2,2'-ангидроуридин для синтеза соответствующих арабинозидов.
McGee и Murtiashaw, каждый, сообщили о получении промежуточного хлор-сахара из L-арабинозы в качестве исходного вещества, которое включает образование промежуточного O2,2'-ангидро-L-тимидина, полученного из других соединений реагентов, отличных от соединений, используемых Holy или Pragnacharyulu et al. (D. McGee, Boehringer Ingelheim Proposal to Novirio Pharmaceuticals, Inc., May 17, 2002; C. W. Murtiashaw, Eur. Patent, 0351126 B1, January 18, 1995).
McGee et al. описали способ получения 2'-модифицированных нуклеозидов в результате реакции внутримолекулярного замещения (патент США № 6090932). McGee et al. сообщили о введении заместителя во 2'-положении 2,2'-ангидроуридина при тщательном выборе 3'-заместителя, который может быть активирован, вызывая стереоспецифичное восстановление во 2'-положении. Синтез включал защиту 5'-OH уридина посредством взаимодействия с DMT с образованием 5'-O-(4,4'-диметокситритил)уридина и давал конечный продукт, 2'-дезокситимидин, с выходом примерно 24%.
Хотя McGee et al. сообщали, что их способ может быть масштабирован для промышленных целей, известно, что диоксан легко воспламеняется и подвержен образованию пероксида, и поэтому противопоказан для промышленных целей. Кроме того, McGee et al. умалчивают о том, дает ли их способ D- или L-энантиомер 2'-дезокситимидина, или требуется разделение энантиомеров.
Таким образом, существует необходимость в простом, рентабельном и безопасном способе получения 2'-дезоксинуклеозидов, их солей, аналогов и пролекарств, включая β-L-2'-дезоксинуклеозиды, такие как β-L-2'-дезокситимидин, который дает возможность избегать применения опасных, токсичных, рискованных и/или трудноподвергаемых обработке реагентов, которые сами по себе не приспособлены для промышленного производства.
Также существует необходимость в обеспечении синтеза для получения 2'-дезоксинуклеозидов, их солей, аналогов и пролекарств, включая β-L-2'-дезоксинуклеозиды, такие как β-L-2'-дезокситимидин, при котором используют безопасные материалы и реагенты.
Также существует необходимость в обеспечении синтеза для получения 2'-дезоксинуклеозидов, их солей, аналогов и пролекарств, включая β-L-2'-дезоксинуклеозиды, такие как β-L-2'-дезокситимидин, в умеренных условиях реакциях.
Также существует необходимость в обеспечении эффективного и рентабельного способа синтеза 2'-дезоксинуклеозидов, их солей, аналогов и пролекарств, включая β-L-2'-дезоксинуклеозиды, такие как β-L-2'-дезокситимидин, в умеренных условиях реакции.
Также существует необходимость в обеспечении синтеза, который является эффективным, требуя минимального количества стадий.
Также существует необходимость в способе, который требует немного или не требует стадий разделения продуктов.
Также существует необходимость в обеспечении промышленно масштабируемого способа синтеза 2'-дезоксинуклеозидов, их солей, аналогов и пролекарств, включая β-L-2'-дезоксинуклеозиды, такие как β-L-2'-дезокситимидин, который является рентабельным и дает конечный продукт с высоким выходом.
Также существует необходимость в обеспечении промышленно масштабируемого синтеза β-2'-дезоксинуклеозидов, их солей, аналогов и пролекарств, включая β-L-2'-дезоксинуклеозиды, такие как β-L-2'-дезокситимидин, который дает β-аномерную форму требуемого соединения в избытке по сравнению с α-аномерной формой с хорошими выходами.
Также существует необходимость в обеспечении синтеза аминокислотных пролекарств 2'-дезоксинуклеозидов, их солей и аналогов, включая β-L-2'-дезоксинуклеозиды, такие как β-L-2'-дезокситимидин.
Сущность изобретения
Настоящее изобретение раскрывает новые эффективные способы синтеза для получения 2'-, 3'- и/или 5'-замещенных нуклеозидов и 2'-, 3'- и/или 5'-дезоксинуклеозидов, таких как 2'-замещенные и 2'-дезоксинуклеозиды, полученные из природных и неприродных карбоциклических, гетероциклических и гетероароматических нуклеозидных оснований, и, в частности, β-L-2'-дезокситимидина (LdT) и его солей, пролекарств, стереоизомеров и энантиомеров. Также предлагаются способы получения стереоизомерных, диастереоизомерных и энантиомерных форм соединений согласно настоящему изобретению на основе соответствующих исходных веществ. Соединения, полученные согласно настоящему изобретению, могут быть использованы в качестве промежуточных продуктов при получении широкого ряда других нуклеозидных аналогов или могут быть использованы непосредственно в качестве противовирусных и/или антинеопластических средств.
В одном варианте 2'-дезоксинуклеозиды и 2'-замещенные нуклеозиды имеют встречающиеся в природе пиримидиновые нуклеозидные основания. В конкретном варианте способ направлен на синтез β-L-2'-дезокситимидина (LdT). В другом варианте 2'-дезоксинуклеозиды и 2'-замещенные нуклеозиды имеют не встречающиеся в природе подобные пиримидину нуклеозидные основания. В одном конкретном варианте не встречающееся в природе подобное пиримидину нуклеозидное основание может быть получено способом синтеза, раскрытым в настоящем изобретении.
В одном варианте способ согласно настоящему изобретению не требует разделения изомеров и поэтому является усовершенствованием по сравнению с предшествующим уровнем техники.
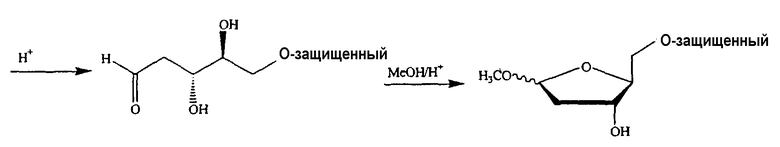
В одном варианте введение функциональных групп вo 2'-положении или удаление таких функциональных групп, чтобы получить 2'-дезоксинуклеозид, осуществляют посредством селективных реакций, в которых используют D-ксилозу, L-арабинозу, L-рибозу, D-галактозу, D-глюконолактон, D-галактонолактон, D-глюкозу, D-гидроксиглутаминовую кислоту (для рибонолактона), спирт или эпоксиспирт, изопропилиденглицеральдегиды или замещенный диоксолан в качестве исходного реагента.
В одном конкретном варианте изобретения синтезы протекают через промежуточный хлор-сахар. Таким образом, одним конкретным промежуточным продуктом в способах синтеза, приведенных в данном описании, который не содержит внутримолекулярных перегруппировок, является соединение хлор-сахара.
В другом конкретном варианте изобретения синтез протекает через внутримолекулярное нуклеофильное замещение. Таким образом, одним конкретным промежуточным продуктом способов синтеза, приведенных в данном описании, является 2,2'-ангидро-1-фуранозилнуклеозидное кольцо.
В одном варианте изобретения один из ключевых промежуточных продуктов получают посредством восстановления лактона таким восстановителем, как Red-Al, следующим образом:
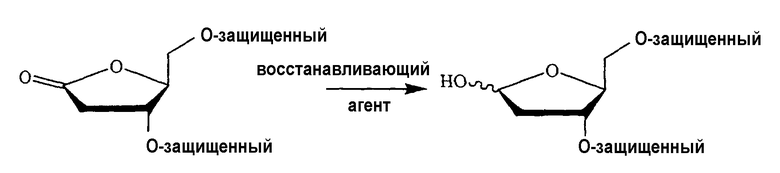
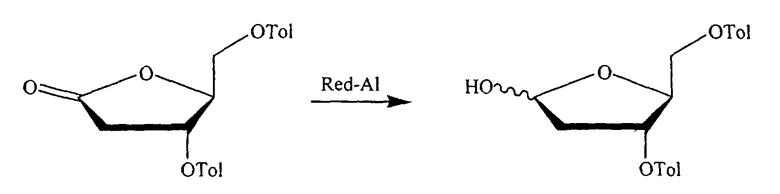
В одном конкретном варианте защитной группой кислорода является толуоил.
В другом конкретном варианте промежуточный продукт получают следующим образом:
Таким образом, в варианте настоящего изобретения способ синтеза включает стадии:
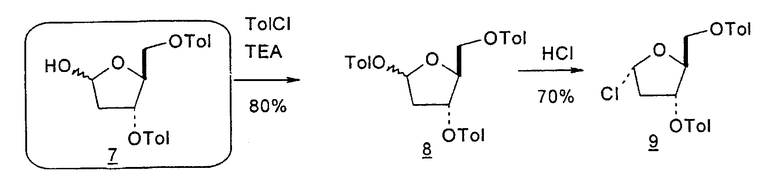
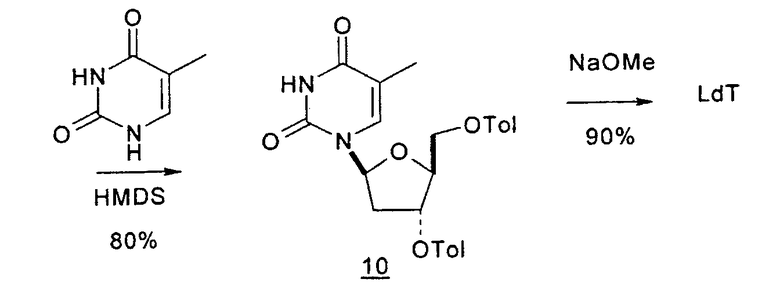
Альтернативный синтез согласно настоящему изобретению для получения 2'-дезокситимидина включает следующие стадии способа:
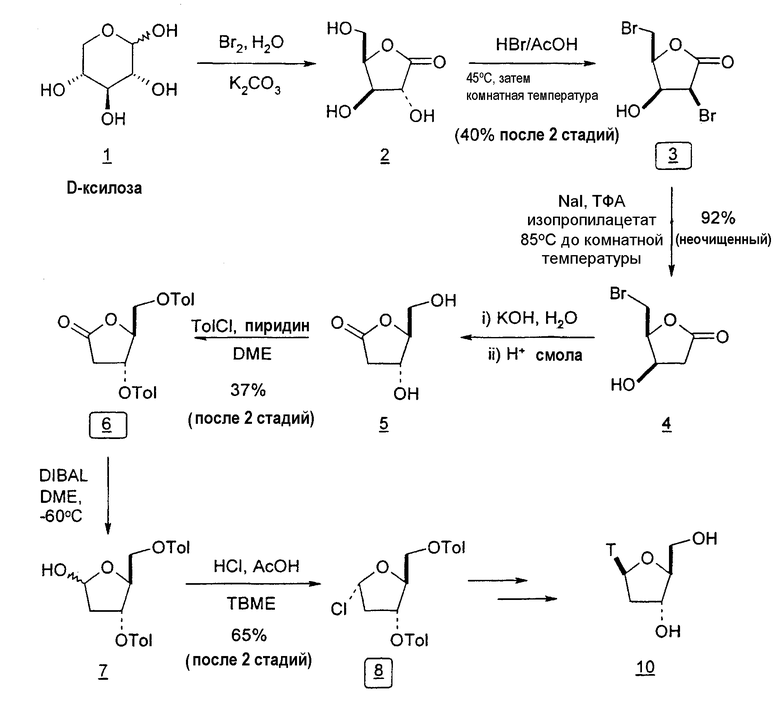
В еще одном варианте настоящего изобретения предлагается способ получения 2'-дезокситимидина из D-ксилозы, который включает 2-дезокси-3,5-ди-O-пара-толуоил-α-L-эритропентофуранозилхлорид в качестве ключевого промежуточного продукта.
В альтернативном варианте предлагается синтез с использованием мезилатного промежуточного продукта:
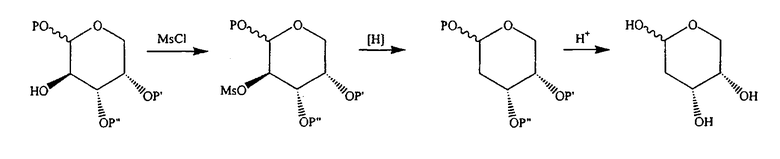
где P, P' и P” независимо означают H, алкил или подходящую защитную группу кислорода. В одном варианте P означает метил. В другом варианте P' и P” объединены с образованием изопропилидена.
Таким образом, в одном конкретном варианте предлагается синтез с использованием мезилатного промежуточного продукта:
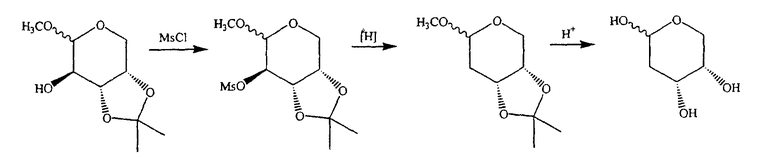
В альтернативном варианте один из ключевых промежуточных продуктов получают следующим способом:
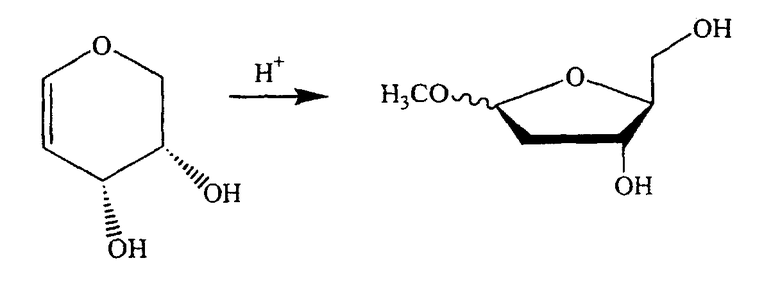
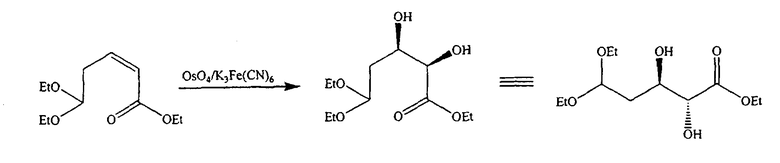
В альтернативном варианте один из ключевых промежуточных продуктов получают цис-окислением алкена с использованием подходящего окисляющего агента, способного к цис-окислению, такого как OsO4, следующим способом:
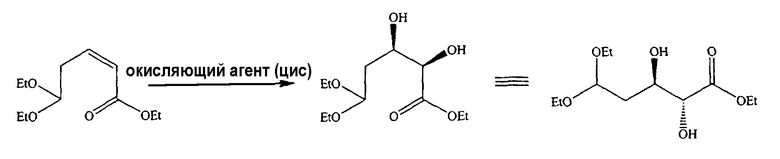
Таким образом, в одном конкретном варианте ключевой промежуточный продукт получают цис-окислением алкена с использованием OsO4 следующим способом:
В альтернативном варианте один из ключевых промежуточных продуктов получают следующим способом:
В альтернативном варианте один из ключевых промежуточных продуктов получают следующим способом:
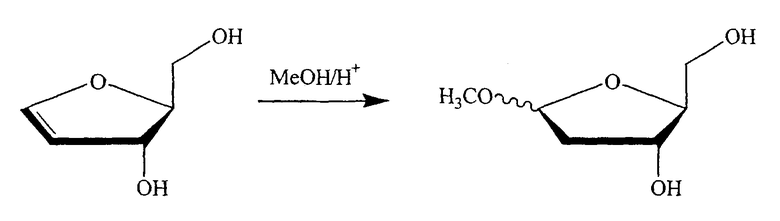
В альтернативном варианте один из ключевых промежуточных продуктов получают посредством реакции с раствором спирта/кислоты одним из следующих способов:
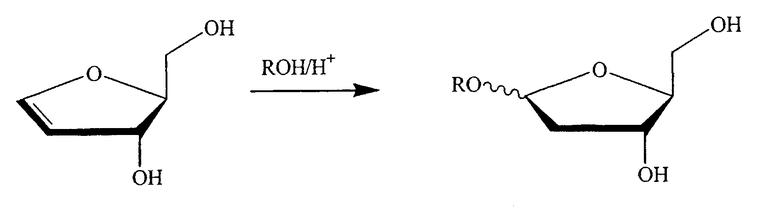
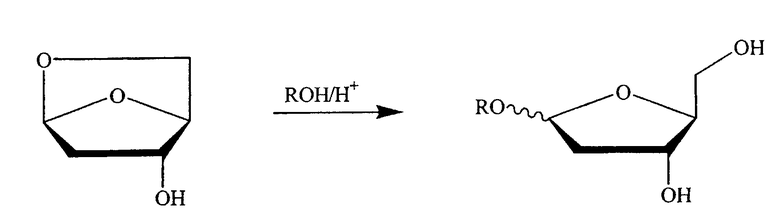
где R означает алкил, предпочтительно низший алкил, такой как метил или этил, и, в частности, метил.
В одном варианте изобретения спирт выбран из группы, состоящей из метанола, этанола, пропанола, изопропанола, бутанола, изобутанола, трет-бутанола, s-бутанола, пентанола, гексанола или их смеси. В конкретном варианте спирт является метанолом или этанолом. В другом конкретном варианте спирт является метанолом.
Таким образом, в конкретном варианте изобретения ключевой промежуточный продукт получают посредством взаимодействия с раствором спирта/кислоты одним из следующих способов:
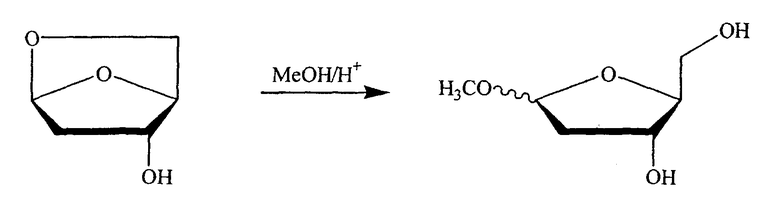
Другой типичный способ согласно настоящему изобретению заключается в использовании восстановителя, такого как Red-Al, в комбинации с комплексообразующим агентом, таким как 15-краун-5-эфир, чтобы расщепить циклический промежуточный 2,2'-ангидро-1-фуранозилнуклеозид и получить требуемый нуклеозидный продукт.
Неожиданно обнаружено, что применение комплексообразующего агента, такого как 15-краун-5-эфир, дает более высокий выход продукта в процентах в том случае, когда выбранной защитной группой является диметокситритил, но более низкий выход продукта в процентах в том случае, когда в качестве защитной группы используют только тритил. Поэтому в одном варианте изобретения предлагается способ, который включает стадию расщепления циклического промежуточного 2,2'-ангидро-1-фуранозилнуклеозида с образованием требуемого нуклеозидного продукта в отсутствие комплексообразующего агента. В конкретном варианте настоящего изобретения предлагается способ, который включает стадию расщепления циклического промежуточного 2,2'-ангидро-1-фуранозилнуклеозида с образованием требуемого нуклеозидного продукта в отсутствие комплексообразующего агента в том случае, когда защитной группой является тритил.
Предлагаются способы применения соответствующего нуклеофильного агента, например, металлоорганического соединения (например, реагента Гриньяра или алкиллитиевого реагента), если требуется алкильный заместитель, чтобы расщепить циклический промежуточный 2,2'-ангидро-1-фуранозилнуклеозид с получением требуемого 2'-замещенного нуклеозидного продукта.
В одном варианте настоящее изобретение относится к способу получения 2'-дезоксинуклеозида или 2'-модифицированного нуклеозида, который включает: (a) необязательно защиту одной или нескольких гидроксильных групп фуранозильного кольца, такого как рибо-, арабино- или ксилофуранозил, посредством взаимодействия с защитной группой; (b) конденсацию фуранозильного кольца со стадии (a) с необязательно замещенным природным или неприродным нуклеозидным основанием с образованием нуклеозида; (c) взаимодействие нуклеозида со стадии (b) с конденсирующим агентом при повышенной температуре с получением 2,2'-ангидро-1-фуранозилнуклеозида; (d) взаимодействие 2,2'-ангидро-1-фуранозилнуклеозида со стадии (c) с восстановителем, таким как Red-Al, и комплексообразующим агентом, таким как 15-краун-5-эфир, предпочтительно в полярном растворителе при низкой температуре, чтобы получить необязательно защищенный 2'-дезоксинуклеозид или 2'-замещенный нуклеозид; и (e) удаление защиты необязательно защищенных гидроксильных групп, если это необходимо или желательно, например, посредством добавления кислот или кислых смол при температуре примерно 50°C.
В другом варианте предлагается способ получения 2'-дезокситимидина, который включает: (a) необязательно защиту одной или нескольких гидроксильных групп фуранозильного кольца посредством взаимодействия с защитной группой; (b) взаимодействие необязательно защищенного фуранозильного кольца с цианамидом с образованием необязательно защищенного фуранозиламинооксазолина; (c) взаимодействие необязательно защищенного фуранозиламинооксазолина с циклизующим или конденсирующим агентом, чтобы получить необязательно защищенный 2,2'-ангидро-1-фуранозилтимидин; (d) взаимодействие необязательно защищенного 2,2'-ангидро-1-фуранозилтимидина с восстановителем, таким как Red-Al, и комплексообразующим агентом, таким как 15-краун-5-эфир, предпочтительно в полярном растворителе при низкой температуре с получением необязательно защищенного 2'-дезокситимидина; и (e) удаление защиты необязательно защищенного 2'-дезокситимидина, если это необходимо или желательно, например, посредством взаимодействия с кислотами или кислыми смолами примерно при 50°C с получением 2'-дезокситимидина.
В еще одном варианте настоящее изобретение относится к способу получения 2'-дезокситимидина, который включает стадии (a)-(e), приведенные выше, но не включает применение комплексообразующего агента, который указан для стадии (d).
В еще одном варианте настоящее изобретение относится к способу получения 2'-дезоксинуклеозида или 2'-модифицированного нуклеозида, который включает: (a) необязательно защиту одной или нескольких гидроксильных групп фуранозильного кольца, такого как рибо-, арабино- или ксилофуранозил, посредством взаимодействия с защитной группой; (b) конденсацию фуранозильного кольца со стадии (a) с необязательно замещенным природным или неприродным нуклеозидным основанием с образованием нуклеозида; (c) взаимодействие нуклеозида со стадии (b) с конденсирующим агентом при повышенной температуре, чтобы получить 2,2'-ангидро-1-фуранозилнуклеозид; (d) взаимодействие 2,2'-ангидро-1-фуранозилнуклеозида со стадии (c) с восстановителем, таким как Red-A1, в отсутствие комплексообразующего агента, такого как 15-краун-5-эфир, предпочтительно в полярном растворителе при низкой температуре с получением необязательно защищенного 2'-дезоксинуклеозида или 2'-замещенного нуклеозида; и (e) удаление защиты необязательно защищенных гидроксильных групп, если это необходимо или желательно, например, посредством добавления кислот или кислых смол при температуре примерно 50°C.
В другом варианте предлагается способ получения 2'-дезокситимидина, который включает: (a) необязательно защиту одной или нескольких гидроксильных групп фуранозильного кольца посредством взаимодействия с защитной группой; (b) взаимодействие необязательно защищенного фуранозильного кольца с цианамидом с образованием необязательно защищенного фуранозиламинооксазолина; (c) взаимодействие необязательно защищенного фуранозиламинооксазолина с циклизующим или конденсирующим агентом, чтобы получить необязательно защищенный 2,2'-ангидро-1-фуранозилтимидин; (d) взаимодействие необязательно защищенного 2,2'-ангидро-1-фуранозилтимидина с восстановителем, таким как Red-Al, в отсутствие комплексообразующего агента, такого как 15-краун-5-эфир, предпочтительно в полярном растворителе при низкой температуре, чтобы получить необязательно защищенный 2'-дезокситимидин; и (e) удаление защиты необязательно защищенного 2'-дезокситимидина, если это необходимо или желательно, например, посредством взаимодействия с кислотами или кислыми смолами примерно при 50°C с получением 2'-дезокситимидина.
В объем настоящего изобретения включены способы получения 2'-модифицированных нуклеозидов, фосфорамидитов 2'-модифицированных нуклеозидов, 3'- и 5'-моно-, ди- и трифосфатов 2'-модифицированных нуклеозидов и олигонуклеотидов, которые содержат по меньшей мере один нуклеозид, модифицированный согласно способу, предлагаемому в настоящем изобретении. Также включены способы получения внутримолекулярных функциональных групп, которые включают ангидронуклеозиды в других положениях, отличных от 2'-положения цикла фуранозы, например, в 3'- и/или 5'-положении. Способы согласно настоящему изобретение также включают модификацию функциональных групп, чтобы получить, например, соответствующие 5'-диацилглицерофосфатные или 5'-диалкилглицерофосфатные производные, которые могут быть использованы в качестве пролекарств.
В описании и примерах, приведенных в данном описании, предлагаются дополнительные варианты настоящего изобретения.
Краткое описание схем
Фиг.1 является схемой способа согласно настоящему изобретению для получения LdT из L-арабинозы через промежуточный мезилат.
Фиг.2 является схемой способа согласно настоящему изобретению для получения LdT из L-арабинозы через промежуточный гликаль.
Фиг.3 является схемой способа согласно настоящему изобретению получения LdT из L-арабинозы через промежуточный гликаль и стадию восстановительного элиминирования.
Фиг.4 является схемой способа согласно настоящему изобретению получения LdT из L-ксилозы через ди-O-толуоил-производное.
Фиг.5 является схемой способа согласно настоящему изобретению получения LdT из D-галактозы.
Фиг.6 является схемой способа согласно настоящему изобретению получения LdT из D-глюконолактона.
Фиг.7 является схемой способа согласно настоящему изобретению получения LdT из D-галактонолактона.
Фиг.8 является схемой способа согласно настоящему изобретению получения LdT из фуронолактона, неуглеводного ахирального исходного вещества.
Фиг.9 является схемой способа согласно настоящему изобретению получения LdT из этил-3,3-диэтоксипропаноата.
Фиг.10 является схемой способа согласно настоящему изобретению получения LdT из гидроксиглутаминовой кислоты.
Фиг.11 является схемой способа согласно настоящему изобретению получения LdT из коммерчески доступного спирта посредством эпоксидирования.
Фиг.12 является схемой способа согласно настоящему изобретению получения LdT из эпоксиспирта.
Фиг.13 является схемой способа согласно настоящему изобретению получения LdT из 1,2-O-изопропилиден-L-глицеральдегида.
Фиг.14 является схемой способа согласно настоящему изобретению получения LdT из 2-бромметил[1,3]диоксолана.
Фиг.15 является схемой способа согласно настоящему изобретению получения LdT из гликаля, обработанного кислым метанолом.
Фиг.16 является схемой способа согласно настоящему изобретению получения LdT из L-арабинозы и цианамида.
Фиг.17 является схемой способа согласно настоящему изобретению получения LdT из L-арабинозы посредством раскрытия хлористым водородом O2,2'-связи соединения.
Фиг.18 является схемой способа согласно настоящему изобретению получения LdT из L-арабинозы, как на фиг.17, с использованием альтернативных реагентов для раскрытия O2,2'-связи соединения.
Фиг.19 является схемой способа согласно настоящему изобретению получения LdT из L-арабинозы, как на фиг.17, с использованием йодистого водорода для раскрытия O2,2'-связи соединения.
Фиг.20 является схемой способа согласно настоящему изобретению получения LdT из L-арабинозы, который включает взаимодействие сложного эфира 2-метилоксиран-2-карбоновой кислоты с 1,2-оксазолином.
Фиг.21 является схемой способа согласно настоящему изобретению получения LdT из L-арабинозы через промежуточный O2,2'-ангидро-L-уридин.
Фиг.22 является схемой способа согласно настоящему изобретению получения LdT из L-арабинозы, как на фиг.21, проходящего через промежуточный 2'-дезокси-5-этоксиметил-L-уридин.
Фиг.23 является схемой способа согласно настоящему изобретению получения LdT из D-ксилозы через промежуточный 2-дезокси-3,5-ди-O-пара-толуоил-α-L-эритропентофуранозилхлорид.
Фиг.24 является схемой способа согласно настоящему изобретению получения β-L-дезокситимидина, при котором 5'-OH промежуточного арабинофуранозиламинооксазолина защищают группой тритила перед образованием промежуточного 2,2'-ангидро-1-(β-арабинофуранозил)тимидина и его восстановительным расщеплением с использованием Red-Al и 15-краун-5-эфира.
Фиг.25 является схемой способа согласно настоящему изобретению получения β-L-дезокситимидина, где защита 5'-OH остатка L-арабинофуранозила происходит после образования промежуточного 2,2'-ангидро-1-(β-арабинофуранозил)тимидина и его восстановительного расщепления Red-Al и 15-краун-5-эфиром.
Фиг.26 является схемой способа согласно настоящему изобретению получения β-D-дезокситимидина из D-рибозы, который включает защиту и удаление защиты OH-защитной группой во 2'-, 3'- и 5'-положениях рибофуранозила, и затем использование тритила в качестве защитной группы в 5'-положении отдельно перед восстановительным расщеплением Red-Al и 15-краун-5-эфиром.
Фиг.27 является схемой способа согласно настоящему изобретению, при котором промежуточный 2,2'-ангидро-1-(β-рибофуранозил)тимидин образуют непосредственно из тимидина, затем защищают его 5'-OH тритильной группой и, наконец, расщепляют, используя Red-Al и 15-краун-5-эфир.
Фиг.28 является схемой способа согласно настоящему изобретению, в котором используют L-рибозу в качестве исходного вещества и который осуществляют посредством защиты и удаления защиты ее гидроксильных групп любой подходящей защитной группой перед образованием промежуточного 2,2'-ангидро-1-(β-рибофуранозил)тимидина, который затем защищают по положению 5'-OH перед восстановительным расщеплением Red-Al и 15-краун-5-эфиром.
Фиг.29 является схемой способа согласно настоящему изобретению получения β-D-дезокситимидина из 2,2'-ангидро-1-β-D-арабинофуранозилтимина без применения комплексообразующего агента во время восстановления.
Подробное описание изобретения
Настоящее изобретение раскрывает новые эффективные способы синтеза для получения 2'-, 3'- и/или 5'-замещенных нуклеозидов и 2'-, 3'- и/или 5'-дезоксинуклеозидов, таких как 2'-замещенные и 2'-дезоксинуклеозиды, полученные из природных и неприродных карбоциклических, гетероциклических и гетероароматических нуклеозидных оснований, и, в частности, β-L-2'-дезокситимидина (LdT), и их солей, пролекарств, стереоизомеров и энантиомеров. В изобретение включены способы получения стереоизомерных, диастереоизомерных и энантиомерных форм соединений согласно настоящему изобретению на основе соответствующих исходных веществ. Соединения, полученные согласно настоящему изобретению, можно использовать в качестве промежуточных продуктов при получении широкого ряда других нуклеозидных аналогов или можно использовать непосредственно в качестве противовирусных и/или антинеопластических средств.
В одном варианте 2'-дезоксинуклеозиды и 2'-замещенные нуклеозиды имеют встречающиеся в природе пиримидиновые нуклеозидные основания. В конкретном варианте способ направлен на синтез β-L-2'-дезокситимидина (LdT). В другом варианте 2'-дезоксинуклеозиды и 2'-замещенные нуклеозиды имеют не встречающиеся в природе подобные пиримидину нуклеозидные основания. В одном конкретном варианте не встречающееся в природе подобное пиримидину нуклеозидное основание может быть получено способом синтеза, раскрытым в настоящем изобретении.
В одном варианте способ согласно настоящему изобретению не требует разделения изомеров и поэтому является усовершенствованием по сравнению с предшествующим уровнем техники.
В одном варианте введение функциональных групп во 2'-положении или удаление таких функциональных групп, чтобы получить 2'-дезоксинуклеозид, осуществляют посредством селективных реакций, в которых используют D-ксилозу, L-арабинозу, L-рибозу, D-галактозу, D-глюконолактон, D-галактонолактон, D-глюкозу, D-гидроксиглутаминовую кислоту (для рибонолактона), спирт или эпоксиспирт, изопропилиденглицеральдегиды или замещенный диоксолан в качестве исходного реагента.
В одном конкретном варианте изобретения синтезы протекают через промежуточный хлор-сахар. Поэтому одним конкретным промежуточным продуктом в способах синтеза, приведенных в данном описании, который не содержит внутримолекулярных перегруппировок, является соединение хлор-сахара.
В другом конкретном варианте изобретения синтез протекает через внутримолекулярное нуклеофильное замещение. Поэтому одним конкретным промежуточным продуктом способов синтеза, приведенных в данном описании, является 2,2'-ангидро-1-фуранозилнуклеозидный цикл.
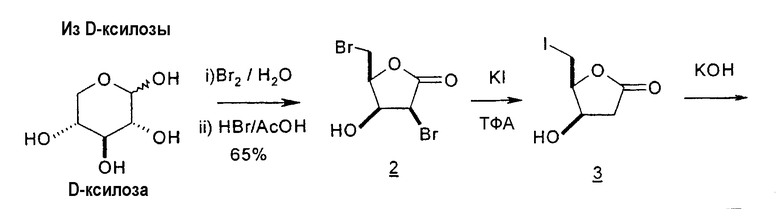
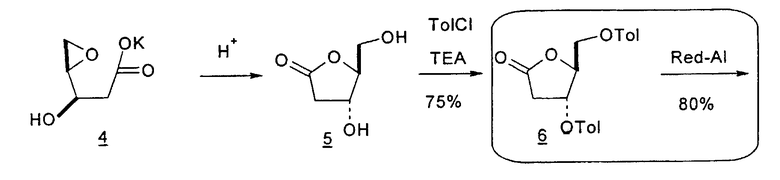
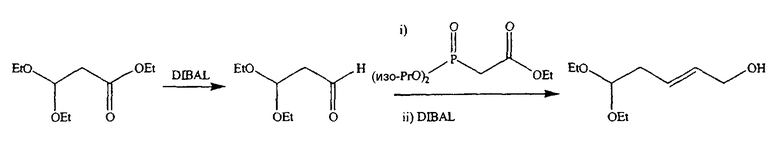
В первом варианте 2'-дезокситимидин получают из D-ксилозы в качестве исходного вещества (фиг.4). Указанный синтез включает: (a) окисление D-ксилозы сначала водным раствором брома, а затем уксусной и бромистоводородной кислотой с образованием 2,5-дибром-2,5-дидезокси-D-ликсоно-1,4-лактона (2); (b) взаимодействие лактонного продукта со стадии (a) с йодидом калия в трифторуксусной кислоте (ТФА), чтобы получить соответствующее 5-йод-соединение с избирательным удалением атома брома у C-2, чтобы получить 5-йод-2-дезоксилактон (3); (c) воздействие на 5-йод-2-дезоксилактон водного раствора гидроксида калия с получением 4,5-эпоксидного производного (4); (d) обработку 4,5-эпоксидного производного с водным раствором кислоты с получением соответствующего 2-дезокси-L-рибонолактона посредством стереоспецифичной инверсии по C-4 (5); (e) защиту положений C-3 и C-5 посредством взаимодействия с любой защитной группой, такой как толуоилхлорид в TEA (6); (f) избирательное восстановление защищенного 2-дезокси-L-рибонолактона восстановителем Red-Al, чтобы получить соответствующий лактол (7); и (g) превращение лактола со стадии (f) в требуемый промежуточный хлор-сахар (9).
Во втором варианте предлагается альтернативный синтез для получения 2'-дезокситимидина, в котором также используют D-ксилозу в качестве исходного вещества, используя альтернативные реагенты и предпочтительно исключают три хроматографические очистки, в которых используются высокополярные водорастворимые УФ-неактивные реагенты (фиг.23). Способ включает: (a) окисление D-ксилозы сначала бромом/водой и карбонатом калия с получением D-ликсоно-1,4-лактона (2); (b) взаимодействие лактона со стадии (a) с уксусной и бромистоводородной кислотой, например, при 45°C в течение 1 часа и затем при комнатной температуре с перемешиванием примерно в течение 1,5 часа, чтобы получить 2,5-дибром-2,5-дидезокси-D-ликсоно-1,4-лактон (3); (c) взаимодействие лактона со стадии (b) с изопропилацетатом и йодидом натрия в ТФА, и, например, нагревание реакционной смеси примерно до 85°C в течение примерно 1,5 часа с образованием 5-бром-2,5-дидезокси-D-треопентоно-1,4-лактона (4); (d) взаимодействие лактона со стадии (c) с гидроксидом калия и водой и, например, через 3 часа нагревание реакционной смеси примерно до 80°C в течение 30 минут, затем охлаждение смеси до комнатной температуры при перемешивании в течение ночи, чтобы получить 2-дезокси-L-рибоно-1,4-лактон (5); (e) добавление толуоильных защитных групп к C-3 и C-5 посредством взаимодействия лактона со стадии (d) с пара-толуоилхлоридом, например с пиридином в DME, (6); (f) взаимодействие 2-дезокси-3,5-ди-O-пара-толуоил-L-рибоно-1,4-лактона с DIBAL и, например, DME примерно при -60°C примерно в течение 1 часа с получением 2-дезокси-3,5-ди-O-пара-толуоил-L-рибозы (7); (g) взаимодействие продукта со стадии (f) с сухим газом HCl в уксусной кислоте с получением 2-дезокси-3,5-ди-O-пара-толуоил-α-L-эритропентофуранозилхлорида (8), который затем может быть подвергнут взаимодействию способами, известными специалистам в данной области, чтобы получить 2'-дезокситимидин в качестве конечного требуемого продукта.
В некоторых вариантах L-арабинозу используют в качестве исходного вещества для получения 2'-дезоксинуклеозидов, в частности 2'-дезокситимидина. Указанные способы включают стадии: (a) превращение L-арабинозы в соответствующий метилгликозид, при этом защищая гидроксильные группы у C-3 и C-4 в виде производных ацетонида (2); (b) дезоксигенирование гидроксильной группы C-2 посредством превращения ее в соответствующую мезилатную группу (3); и затем (c) подвергают промежуточный мезилат восстановительному расщеплению (5), используя две дополнительные стадии способа, чтобы получить ключевой промежуточный хлор-сахар (фиг.1).
Альтернативно L-арабиноза может быть превращена в соответствующее производное гликаля посредством стадии восстановительного элиминирования, см., например, фиг.2 и 3, стадии (1) и (2) соответственно, и полученный в результате промежуточный гликаль затем может быть превращен в метил-2-дезоксирибофуранозид, стадии (4) и (5), соответственно.
В других вариантах настоящего изобретения в качестве исходного вещества используют L-арабинозу. Такие способы включают стадии: (a) взаимодействие L-арабинозы с цианамидом с получением промежуточного 1,2-оксазолина (1), (b) взаимодействие промежуточного продукта со стадии (a) со сложноэфирным производным 3-оксопропионовой кислоты или этилпропиолатом с получением 2,2'-ангидро-1-фуранозилнуклеозидного цикла (2) и (c) раскрытие цикла со стадии (b) с использованием различных реагентов и в разных условиях реакции, чтобы получить LdT (фиг.16-22).
Альтернативно 2'-дезоксинуклеозиды также могут быть образованы из галактозы в качестве исходного вещества. В том случае, когда в качестве исходного вещества используют D-галактозу, ее подвергают окислительному расщеплению и бромированию, чтобы получить 2,5-дибром-2,5-дидезокси-D-ликсоно-1,4-лактон, и полученный лактон подвергают селективному гидрогенолизу, получая 5-бром-2-дезоксилактон, который подвергается ряду превращений с получением ключевого промежуточного хлор-сахара (фиг.5).
Подобным образом глюконолактоны могут служить в качестве исходных веществ для синтеза 2'-дезоксинуклеотидов. Глюконолактон превращают в 2,6-дибром-2,6-дидезокси-D-манноно-1,4-лактон (1), последовательно обрабатывают гидразином и водным раствором гидроксида калия, подкисляют, вызывая инверсию в положении C-4 и C-5, которая дает 2-дезоксилактон (6), подвергают перегруппировке эпоксида Пейна (5), подвергают окислительному расщеплению и восстановлению, получая лактон (7), который легко может быть превращен в требуемый хлор-сахар (11) (фиг.6).
Альтернативно 2'-дезоксинуклеозиды также могут быть образованы из галактонолактонов в качестве исходного вещества. В том случае, когда в качестве исходного вещества используют галактонолактон, его превращают в ацетилированный дибромлактон (2), обрабатывают гидразином и бромируют, получая 2-дезоксилактон (3), который затем деацетилируют, подвергают окислительному расщеплению и восстанавливают, используя NaBH4, получая 2-дезокси-L-рибоно-1,4-лактон (5), и полученный лактон защищают посредством взаимодействия с толуоилхлоридом, подвергают восстановлению Red-Al и хлорированию, получая конечный требуемый продукт хлор-сахар (9) (фиг.7).
Настоящее изобретение также относится к дополнительным способам получения 2'-дезоксинуклеозидов, и в частности, 2'-дезокситимидина, из исходных веществ, которые не являются углеводами (фиг.8), являются диоксаланил-производными (фиг.14), кислотами, сложными эфирами и альдегидами (фиг.9, 10, 13), гликалем (фиг.15) и спиртами (фиг.11 и 12). Подробности указанных синтезов можно найти в примерах, приведенных в данном описании, которые являются предпочтительными вариантами (см. фиг.1-23).
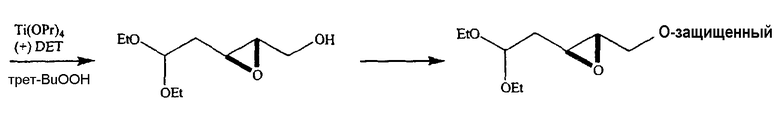
Также предлагаются способы применения восстановителя, такого как Red-Al, в комбинации с комплексообразующим агентом, таким как 15-краун-5-эфир, для того, чтобы расщепить циклический промежуточный 2,2'-ангидро-1-фуранозилнуклеозид и получить требуемый 2'-дезоксинуклеозидный продукт. Альтернативно предлагаются способы применения восстановителя, такого как Red-Al, в отсутствие комплексообразующего агента, чтобы расщепить циклический промежуточный 2,2'-ангидро-1-фуранозилнуклеозид и получить требуемый 2'-дезоксинуклеозидный продукт. Альтернативно циклический промежуточный 2,3'-ангидро-1-фуранозилнуклеозид можно использовать для образования соответствующего 3'-дезоксинуклеозида.
Можно использовать любые восстановители, известные в данной области, которые обеспечивают необходимое хемоселективное и региоселективное восстановление. Подходящие восстановители включают Red-Al, Red-Al (бис[2-метоксиэтокси]алюмогидрид натрия), NaHTe, SmI2, H2+Pd-фосфиновый катализатор и LiAl(OtBu)3H (три-трет-бутоксиалюмогидрид лития).
Реакцию раскрытия цикла можно осуществлять при любой температуре, при которой достигаются требуемые результаты, например, которая подходит для того, чтобы реакция протекала с приемлемой скоростью, не стимулируя распад или избыточное образование побочных продуктов, предпочтительно при пониженных температурах, таких как примерно 0-5°C.
Может быть выбран любой растворитель в реакции, который может достичь необходимой температуры и который может растворять компоненты реакции. Неограничивающими примерами любой полярный апротонный растворитель, включая, но, не ограничивая указанным, дихлорметан (ДХМ) или дихлорэтан, ацетон, этилацетат, дитианы, ТГФ, 1,2-диметоксиэтан (DME), диоксан, ацетонитрил, диэтиловый эфир, пиридин, диметилформамид (ДМФА), диметилсульфоксид (ДМСО), диметилацетамид или любую их комбинацию, хотя предпочтительно ТГФ и/или DME.
Альтернативно предлагаются способы применения подходящего нуклеофильного агента, например, металлоорганического агента (например, реагента Гриньяра или алкиллитиевого реагента), если требуется алкильный заместитель, чтобы раскрыть цикл промежуточного 2,2'-ангидро-1-фуранозилнуклеозида с получением требуемого 2'-замещенного нуклеозидного продукта. В другом варианте циклический промежуточный 2,3'-ангидро-1-фуранозилнуклеозид или циклический промежуточный 2,5'-ангидро-1-фуранозилнуклеозид можно использовать для образования требуемого 3'-замещенного или 5'-замещенного нуклеозидного продукта.
В частности, в одном варианте настоящее изобретение относится к способу получения 2'-дезоксинуклеозида или 2'-модифицированного нуклеозида, который включает: (a) необязательно защиту одной или нескольких гидроксильных групп фуранозильного кольца, такого как цикл рибо-, арабино- или ксилофуранозила, посредством взаимодействия с защитной группой (2); (b) конденсацию необязательно защищенного фуранозильного кольца со стадии (a) с необязательно замещенным природным или неприродным нуклеозидным основанием с образованием нуклеозида (3); (c) взаимодействие нуклеозида со стадии (b) с конденсирующим агентом при повышенной температуре с получением 2,2'-ангидро-1-фуранозилнуклеозида (5); (d) взаимодействие 2,2'-ангидро-1-фуранозилнуклеозида со стадии (c) с восстановителем, таким как Red-Al, и комплексообразующим агентом, таким как 15-краун-5-эфир, предпочтительно в полярном растворителе при низкой температуре, чтобы получить необязательно защищенный 2'-дезоксинуклеозид или 2'-замещенный нуклеозид (8); и (e) удаление защиты необязательно защищенных гидроксильных групп, если это необходимо или желательно, например, посредством добавления кислот или кислых смол при температуре примерно 50°C (9) (фиг.26).
В другом варианте предлагается способ получения 2'-дезокситимидина, который включает: (a) необязательно защиту одной или нескольких гидроксильных групп фуранозильного кольца посредством взаимодействия с защитной группой (2); (b) взаимодействие необязательно защищенного фуранозильного кольца с цианамидом с образованием необязательно защищенного фуранозиламинооксазолина (3); (c) взаимодействие необязательно защищенного фуранозиламинооксазолина с циклизующим или конденсирующим агентом, чтобы получить необязательно защищенный 2,2'-ангидро-1-фуранозилтимидин (5); (d) взаимодействие необязательно защищенного 2,2'-ангидро-1-фуранозилтимидина с восстановителем, таким как Red-Al, и комплексообразующим агентом, таким как 15-краун-5-эфир, предпочтительно в полярном растворителе при низкой температуре с получением необязательно защищенного 2'-дезокситимидина (8); и (e) удаление защиты необязательно защищенного 2'-дезокситимидина, если это необходимо или желательно, например, посредством взаимодействия с кислотами или кислыми смолами примерно при 50°C с получением 2'-дезокситимидина (9) (фиг.28).
В еще одном варианте настоящее изобретение относится к способу получения 2'-дезоксинуклеозида или 2'-модифицированного нуклеозида, который включает (a) конденсацию фуранозильного кольца с необязательно замещенным природным или неприродным нуклеозидным основанием с образованием нуклеозида; (b) взаимодействие нуклеозида со стадии (a) с конденсирующим агентом при повышенной температуре, получая 2,2'-ангидро-1-фуранозилнуклеозид (1); (c) взаимодействие 2,2'-ангидро-1-фуранозилнуклеозида со стадии (b) с защищающим агентом, таким как тритильная защитная группа, чтобы защитить 5'-положение нуклеозида (2); (d) добавление восстановителя, такого как Red-Al, предпочтительно в полярном растворителе при низкой температуре, чтобы получить необязательно защищенный 2'-дезоксинуклеозид или 2'-замещенный нуклеозид (3); и (e) удаление защиты необязательно защищенных гидроксильных групп, если это необходимо или желательно, например, посредством добавления кислот или кислых смол при температуре примерно 50°C (4) (фиг.29).
Предпочтительные варианты представлены на фиг.1-29.
Определения
В настоящем изобретении термин «выделенный» относится к композиции нуклеозида, которая содержит по меньшей мере 85 или 90 мас.%, предпочтительно 95-98 мас.% и еще более предпочтительно 99-100 мас.% нуклеозида, при этом остальная часть содержит другие химические виды молекул или энантиомеры.
Термин «защищенный» в используемом в данном описании смысле, и если не оговорено особо, относится к группе, которую добавляют к атому кислорода, азота или фосфора, чтобы предотвратить его дальнейшее взаимодействие или для других целей. Специалистам в области органического синтеза известно широкое множество защитных групп кислорода, азота и фосфора.
Примеры подходящих защитных групп включают, но не ограничены указанным, бензоил; замещенные или незамещенные алкильные группы, замещенные или незамещенные арильные группы, замещенные или незамещенные силильные группы; замещенные или незамещенные ароматические или алифатические сложные эфиры, например, такие как ароматические группы, подобные бензоилу, толуоилам (например, пара-толуоил), нитробензоилу, хлорбензоилу; эфирные группы, например, такие как -C-O-аралкил, -C-O-алкил или -C-O-арил; и алифатические группы, подобные ацильным или ацетильным группам, включая любые замещенные или незамещенные ароматические или алифатические ацил, -(C=O)-аралкил, -(C=O)-алкил или -(C=O)-арил; где ароматический или алифатический остаток ацильной группы может иметь прямую или разветвленную цепь; все группы, кроме того, могут быть необязательно замещены группами, на которые не влияют реакции, включающие улучшенный синтез (см. Greene et al., Protective Groups in Organic Synthesis, John Wiley and Sons, 2nd Edition (1991)). Например, в одном варианте изобретения защитные группы замещены группами, на которые не влияет выбранный восстановитель, такой как Red-Al. В случае применения в качестве защитных групп сложных эфиров внимание обратили на патент США 6229008 Saischek et al., включенный в данное описание в виде ссылки, в котором сообщается, что применение простого эфира в качестве защитной группы может давать значительные преимущества, особенно в 5'-положении пентофуранозида, в отношении стабильности к реагентам и условиям процесса. Это дает преимущество для разделения, выделения и очистки требуемого продукта и, следовательно, в отношении процентного выхода продукта.
Защитными группами гидроксила сахара в качестве неограничивающих примеров могут быть силил, бензоил, пара-толуоил, пара-нитробензоил, пара-хлорбензоил, ацил, ацетил, -(C=O)-алкил и -(C=O)-арил, все из которых могут быть незамещенными или замещенными одной или несколькими группами, на которые не влияет выбранный восстановитель. В одном варианте защитной группой гидроксила сахара является бензоил. Защитными группами аминокислот предпочтительно являются BOC (бутоксикарбонил), -(C=O)-аралкил, -(C=O)-алкил или -(C=O)-арил. В одном варианте изобретения защитной группой аминогруппы является BOC (бутоксикарбонил).
Термин «алкил» в используемом в данном описании смысле, и если не оговорено особо, включает насыщенный или ненасыщенный, с прямой цепью, разветвленный или циклический, первичный, вторичный или третичный углеводород, обычно C1-C10, и, в частности, включает метил, трифторметил, этил, пропил, изопропил, циклопропил, бутил, изобутил, трет-бутил, пентил, циклопентил, изопентил, неопентил, гексил, изогексил, циклогексил, циклогексилметил, метилпентил и диметилбутил. Термин включает как замещенные, так и незамещенные группы алкила, алкилена, алкенила, алкенилена, алкинила и алкинилена. Остатки, которыми алкильная группа может быть замещена в одном или нескольких положениях, выбраны из группы, состоящей из галогена (включая фтор, хлор, бром или йод), гидроксила (например, CH2OH), амино (например, CH2NH2, CH2NHCH3 или CH2N(CH3)2), алкиламино, ариламино, алкокси, арилокси, нитро, азидо (например, CH2N3), циано (CH2CN), сульфоновой кислоты, сульфата, фосфоновой кислоты, фосфата или фосфоната, любой из которых или все могут быть незащищены или дополнительно защищены при необходимости, как известно специалистам в данной области и как указано, например, в Greene et al., Protective Groups in Organic Synthesis, John Wiley and Sons, 2nd Edition (1991).
Термин «арил» в используемом в данном описании смысле, и если не оговорено особо, относится к фенилу, бифенилу или нафтилу. Термин включает как замещенные, так и незамещенные остатки. Арильная группа может быть замещена одним или несколькими остатками, включая без ограничения гидроксил, амино, алкиламино, ариламино, алкокси, арилокси, нитро, циано, сульфоновую кислоту, сульфат, фосфоновую кислоту, фосфат или фосфонат, любой из которых или все могут быть незащищены или при необходимости дополнительно защищены как известно специалистам в данной области и как указано, например, в Greene et al., Protective Groups in Organic Synthesis, John Wiley and Sons, 2nd Edition (1991).
Термин «ацил» включает группу -C(=O)-R, в которой некарбонильный остаток R представляет собой, например, имеющий прямую цепь, разветвленный или циклический алкил или низший алкил, алкоксиалкил, включая метоксиметил, аралкил, включая бензил, арилоксиалкил, такой как феноксиметил, арил, включая фенил, необязательно замещенный галогеном, C1-C4-алкил или C1-C4-алкокси, сульфонатные сложные эфиры, такие как алкил- или аралкилсульфонил, включая метансульфонил, моно-, ди- или трифосфатный сложный эфир, тритил или монометокситритил, замещенный бензил, триалкилсилил, такой как, например, диметил-трет-бутилсилил или дифенилметилсилил. Арильные группы в сложных эфирах оптимально содержат фенильную группу. Термин «низший ацил» относится к ацильной группе, в которой некарбонильным остатком является низший алкил.
Термин пиримидиновое нуклеозидное основание включает пиримидиновое основание или аналог пиримидинового основания. Примеры пиримидиновых оснований или аналогов пиримидиновых оснований включают, но не ограничены указанным, тимин, цитозин, 5-фторцитозин, 5-метилцитозин, 6-азапиримидин, включая 6-азацитозин, 2- и/или 4-меркаптопиримидин, урацил, 5-галогенурацил, включая 5-фторурацил, C5-алкилпиримидины, C5-бензилпиримидины, C5-галогенпиримидины, C5-винилпиримидин, C5-ацетиленовый пиримидин, C5-ацилпиримидин, C5-амидопиримидин, C5-цианопиримидин, C5-нитропиримидин, C5-аминопиримидин, 5-азацитидинил, 5-азаурацилил, триазолопиридинил, имидазолопиридинил, пирролопиримидинил и пиразолопиримидинил. Функциональные группы кислорода и азота основания могут быть защищены, если это необходимо или желательно. Подходящие защитные группы хорошо известны специалистам в данной области и включают триметилсилил, диметилгексилсилил, трет-бутилдиметилсилил и трет-бутилдифенилсилил, тритил, алкильные группы и ацильные группы, такие как ацетил и пропионил, метансульфонил и пара-толуолсульфонил. Альтернативно пиримидиновое основание или аналог пиримидинового основания необязательно могут быть замещены так, чтобы образовалось пригодное пролекарство, которое может расщепляться in vivo. Примеры подходящих заместителей включают остаток ацила, амин или циклопропил (например, 2-амино-, 2,6-диамино- или циклопропилгуанозин).
Другие реагенты, используемые в способе согласно настоящему изобретению или в предшествующем уровне техники, определяют следующим образом: AIBN означает азобис(изобутиронитрил); BSA (бис(триметилсилил)ацетамид); CAN означает нитрат аммония-церия; DIBAL означает гидрид диизобутилалюминия; TMSCl означает хлортриметилсилан; ТФА означает трифторуксусную кислоту; TEA означает триэтиламин; TFAA означает трифторуксусный ангидрид; TBDPSCl означает трет-бутилдифенилсилилхлорид; TBDMSCl означает трет-бутилдиметилсилилхлорид; TBTN означает гидрид три-н-бутилолова; DET означает диэтилтартрат; TBS означает трет-бутилдиметилсилил; DMTrCl означает диметокситритилхлорид; DME означает 1,2-диметоксиэтан; «Pyr» используют в качестве сокращения пиридина; DMAP означает 4-диметиламинопиридин; DIBAL означает гидрид диизобутилалюминия; PhOCO2Ph означает дифенилкарбонат; HMDS означает гексаметилдисилазид; и ДХМ означает дихлорметан.
Способ согласно настоящему изобретению не ограничен применением приведенных в качестве примеров нуклеозида и реагентов. Подходящие альтернативные реагенты могут быть использованы для настоящего изобретения вместо реагентов, приведенных выше. Например, DME (1,2-диметоксиэтан) может быть заменен любым подходящим полярным апротонным растворителем, таким как ТГФ (тетрагидрофуран) или любой простой эфир; и Red-Al (бис[2-метоксиэтокси]алюмогидрид натрия) в толуоле может быть заменен NaHTe, SmI2, H2+Pd-фосфиновым катализатором или LiAl(OtBu)3H (три-трет-бутоксиалюмогидрид лития), все из которых обеспечивают хемоселективное и региоселективное восстановление.
Подробное описание стадий способа
Циклический промежуточный ангидро-1-фуранозилнуклеозид
Одним из ключевых соединений для способов согласно настоящему изобретению является 2,2'-ангидро-1-фуранозилнуклеозид, например, α- или β-, D- или L-, 2,2'-ангидро-1-фуранозилнуклеозид общей формулы:
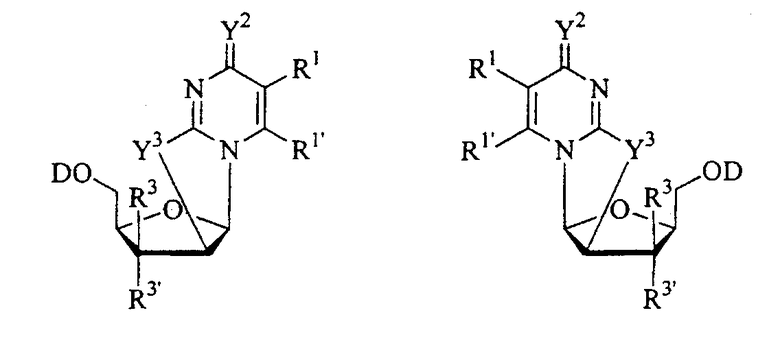
где:
каждый D означает водород или подходящую защитную группу гидроксила, такую как замещенный или незамещенный алкил, замещенный или незамещенный арил, замещенный или незамещенный ацил, силил или аминокислоту;
каждый R1 и R1' независимо означает водород, замещенный или незамещенный низший алкил, замещенный или незамещенный низший алкенил, замещенный или незамещенный низший алкинил, замещенный или незамещенный арил, алкиларил, галоген (F, Cl, Br или I), NH2, NHR5, NR5R5', NHOR5, NR5NHR5', NR5NR5'R5”, OH, OR5, SH, SR5, NO2, NO, CH2OH, CH2OR5, CO2H, CO2R5, CONH2, CONHR5, CONR5R5' или CN;
каждый R3 и R3' независимо означает водород или галоген (F, Cl, Br или I), OH, SH, OCH3, SCH3, NH2, NHCH3, CH3, C2H5, CH=CH2, CN, CH2NH2, CH2OH или CO2H;
каждый Y2 означает O, S, NH или NR6;
каждый Y3 означает O, S, NH или NR7; и
каждый R5, R5', R6 и R7 независимо означает водород, замещенный или незамещенный низший алкил C1-C6, арилалкил или замещенный или незамещенный арил.
2,2'-ангидро-1-фуранозилнуклеозид может быть приобретен или синтезирован любым способом, известным в данной области, включая синтез из фуранилсахаров с 2'-гидроксилом с использованием стандартного способа связывания сахара с последующей конденсацией с образованием 2,2'-ангидро-соединения, или альтернативно связыванием сахара с цианамидом, чтобы получить промежуточный оксазолин, затем построением основания с использованием необходимого циклизующего или конденсирующего агента.
В конкретных вариантах настоящего изобретения β- или α-, D- или L-, 2'-дезокси или 2'-замещенный нуклеозид получают через 2,2'-ангидро-1-фуранозилнуклеозид согласно следующим протоколам.
Из арабинофуранозы
В одном способе согласно настоящему изобретению можно использовать фуранозу, такую как L-фураноза, в частности L-арабинозу, в качестве исходного вещества для получения 2,2'-ангидро-1-фуранозилнуклеозида, который затем восстанавливают согласно настоящему изобретению, получая 2'-дезоксинуклеозид, такой как β-L-2'-дезоксинуклеозид, в частности β-L-2'-дезокситимидин. Альтернативно 2,2'-ангидро-1-фуранозилнуклеозид может быть подвергнут взаимодействию с нуклеофильным агентом, например металлоорганическим агентом (например, реагентом Гриньяра или алкиллитиевым реагентом), чтобы получить требуемый 2'-замещенный нуклеозид.
На фиг.24 в настоящем изобретении в качестве исходного вещества использована L-арабиноза (1), чтобы получить β-L-2'-дезокситимидин в ходе 5-стадийного синтеза. L-арабинозу (1) сначала подвергают взаимодействию с цианамидом в условиях, указанных в предшествующем уровне техники, с образованием промежуточного L-арабинофуранозиламинооксазолина (2) (см. WO 02/44194). Затем 5'-OH арабинозного остатка промежуточного L-арабинофуранозиламинооксазолина (2) защищают посредством взаимодействия с тритилхлоридом (TrCl) и пиридином при температуре примерно 45°C (3). Добавление защитной группы OH в указанных условиях также хорошо известно специалистам в данной области (Greene et al., Protective Groups in Organic Synthesis (1991), John Wiley and Sons, 2nd Edition).
Стадия 3 способа изображена на фиг.24, где показано взаимодействие в подходящих условиях, которые указаны в предшествующем уровне техники, 5'-тритил-защищенного L-арабинофуранозиламинооксазолина (3) с циклизующим или конденсирующим агентом, выбранным из следующей группы:
Например, если в качестве конденсирующего или циклизующего агента используют структуру (ii), показанную выше, то реакцию осуществляют в присутствии Na2CO3/H2O и последующую изомеризацию осуществляют при добавлении Pd/Al2O3/H2O (см. WO 02/44194). Однако если используют конденсирующий или циклизующий агент (i), то реакцию осуществляют в метил-2-формилпропионате при температуре образования флегмы в течение 1 часа (см. EP 0351126). Циклизация приводит к образованию 2,2'-ангидро-1-(L-арабинофуранозил)тимидина (4).
Следующая стадия согласно настоящему изобретению включает восстановление 2,2'-ангидро-1-(L-арабинофуранозил)тимидина (4) таким восстановителем, как Red-Al, и комплексообразующим агентом, таким как 15-краун-5-эфир, в присутствии полярного растворителя, такого как ТГФ и/или DME, предпочтительно при пониженных температурах, таких как примерно 0-5°C, с получением β-L-5'-тритил-2'-дезокситимидина (5).
Использование комплексообразующего агента, такого как 15-краун-5-эфир, на данной стадии полезно вследствие повышения растворимости 2,2'-ангидро-1-(L-арабинофуранозил)тимидина, что приводит к более высокому процентному выходу продукта и позволяет избегать использования таких реагентов, как палладиевые катализаторы, удаление которых требует трудоемких усилий. Кроме того, применение 15-краун-5-эфира позволяет обходиться без использования HBr, чтобы раскрыть ангидроциклическую структуру, требующего использования H2 с отравленным катализатором Pd-BaSO4, чтобы удалить бромид (см. EP 0351126), таким образом позволяя избегать применения опасных реагентов, обнаруженных в некоторых способах предшествующего уровня техники. Наконец, способ согласно настоящему изобретению позволяет избегать применения диоксана в качестве реагента. Это имеет преимущество, так как диоксан легко воспламеняется и не подходит для синтеза в промышленном масштабе.
Конечной стадией в способе, показанном на фиг.24, является удаление тритильной защитой группы из 5'-положения β-L-2'-дезокситимидина (5) обработкой 80% AcOH при температуре примерно 50°C, чтобы удалить L-2'-дезокситимидин (6).
Альтернативно избирательная защита L-арабинозы (1) в C-5-положении с использованием тритила возможна в результате взаимодействия L-арабинозы (1) с TrCl (тритилхлоридом) и пиридином при температуре примерно 45°C с образованием 5-TrO-L-арабинозы (структура не показана). Затем 5-TrO-L-арабинозу подвергают взаимодействию с цианамидом в условиях, указанных в предшествующем уровне техники, с образованием промежуточного 5-TrO-L-арабинофуранозиламинооксазолина (3) (см. WO 02/44194). Остальные стадии способа такие же, как стадии, приведенные на фиг.24 для образования структур (4), (5) и (6).
На фиг.25 показан синтез согласно настоящему изобретению, который сходен с синтезом, изображенным на фиг.24, но отличается стадиями, на которых защищают OH промежуточных продуктов. Как и на фиг.24, L-арабинозу (1) используют в качестве исходного вещества и подвергают взаимодействию с цианамидом, получая L-арабинофуранозиламинооксазолин в качестве промежуточного продукта (2). Затем L-арабинофуранозиламинооксазолин (2) подвергают взаимодействию с любым из циклизующих/конденсирующих реагентов, приведенных выше в структурах (i)-(ix), в подходящих условиях, которые указаны в предшествующем уровне техники, получая 2,2'-ангидро-1-(L-арабинофуранозил)тимидин (3). 2,2'-ангидро-1-(L-арабинофуранозил)тимидин (3) затем подвергают взаимодействию с TrCl и пиридином при температуре примерно 45°C, чтобы защитить 5'-OH остатка арабинозы 2,2'-ангидросоединения (4). Указанную стадию необходимо сравнить со стадией 2 на фиг.24, где группу тритила добавляют к 5'-положению арабиноостатка арабинофуранозиламинооксазолина до его взаимодействия с циклизующим или конденсирующим реагентом. Последние 2 стадии 5-стадийного способа, показанные на фиг.25, идентичны последним 2 стадиям, приведенным на фиг.24, и дают 5'-тритил-защищенный тимидин (5) и 2'-дезокситимидин с удаленной защитой (6).
Заявители пришли к выводу, что способ, описанный на фиг.24, является более эффективным, чем способ, изображенный на фиг.25, на основании того, что присоединение тритильной защитной группы в 5'-положении имеет место на стадии, которую осуществляют при синтезе раньше, чем осуществляли в предшествующем уровне техники.
Из рибофуранозы
В другом способе согласно настоящему изобретению можно использовать фуранозу, такую как L-фураноза, в частности L-рибоза, в качестве исходного вещества для получения 2,2'-ангидро-1-фуранозилнуклеозида, который затем восстанавливают согласно настоящему изобретению, получая 2'-дезоксинуклеозид, такой как β-L-2'-дезоксинуклеозид, в частности β-L-2'-дезокситимидин. Альтернативно 2,2'-ангидро-1-фуранозилнуклеозид можно подвергать взаимодействию с нуклеофильным агентом, например металлоорганическим агентом (например, реагентом Гриньяра или алкиллитиевым реагентом), получая требуемый 2'-замещенный нуклеозид.
На фиг.26 показан 7-стадийный синтез для получения 2'-дезокситимидина с использованием D-рибозы в качестве исходного вещества. На первой стадии данного способа все OH-группы D-рибозы защищают таким образом, как, например, группой ацетила или бензоила, как известно специалистам в данной области. Затем защищенную D-рибозу подвергают взаимодействию с тимином в присутствии, например, SnCl4, HMDS и TMSCl, как известно в предшествующем уровне техники, получая тимидин, который имеет защитные группы в 2'-, 3'- и 5'-положениях нуклеозида. Защитные группы удаляют на стадии 3 реагентами и в условиях, подходящих для удаления конкретной связанной защитной группы. Промежуточным продуктом, полученным на стадии 3, является тимидин.
На стадии 4, на фиг.26, вводят стадию циклизации/конденсации непосредственно из тимидина, а не из фуранозиламинооксазолина, как показано на фиг.24 и 25. В данном случае тимидин подвергают взаимодействию с PhOCOOPh и катализатором NaHCO3 в присутствии ДМФА при температуре примерно 150°C, получая 2,2'-ангидро-1-(рибофуранозил)тимидин.
Структуры 2,2'-ангидро-1-(рибофуранозил)тимидина 5 и 6 представляют собой два отдельных варианта согласно настоящему изобретению, так как структуру 5 получают из структуры тимидина 4 с защитными группами в 3'- и 5'-положениях рибо-остатка тимидина, а структуру 6 получают из структуры тимидина, в которой 3'- и 5'-положения рибо-остатка тимидина не защищены. В любом случае 2'-OH тимидина должен быть свободной OH-группой, чтобы он мог принимать участие во взаимодействии, давая структуру 2,2'-ангидро-1-(рибофуранозил)тимидина. Если синтез проходит через структуру 5, в одном варианте 5'-защитной группой является тритил; затем может быть осуществлена дополнительная стадия, чтобы удалить защитную группу из 3'-положения рибо-остатка перед восстановлением таким восстановителем, как Red-Al, и комплексообразующим агентом, таким как 15-краун-5-эфир, с образованием структуры (8).
В варианте согласно настоящему изобретению, изображенному на фиг.26, синтез проходит через структуру 6, где TrCl и пиридин подвергают взаимодействию с 2,2'-ангидро-1-(рибофуранозил)тимидином примерно при 45°C, получая 5'-тритилированный 2,2'-ангидро-1-(рибофуранозил)тимидин, который представляет собой структуру (7). Затем 5'-тритилированный 2,2'-ангидро-1-(рибофуранозил)тимидин восстанавливают посредством его взаимодействия с таким восстановителем, как Red-Al, и комплексообразующим агентом, таким как 15-краун-5-эфир, в полярном растворителе, таком как ТГФ и/или DME, предпочтительно при температуре примерно 0-5°C. Указанная стадия дает 5'-тритилированный 2'-дезокситимидин (8), защиту которого затем удаляют в результате его взаимодействия с 80% AcOH примерно при 50°C, получая D-2'-дезокситимидин (9). Способ, показанный на фиг.26, обеспечивает способ получения 2,2'-ангидрофуранозилтимидина непосредственно из тимидина или защищенного тимидина без участия промежуточного фуранозиламинооксазолидина и сопровождающей стадии конденсации или циклизации.
На фиг.27 показан 5-стадийный способ получения L-2'-дезокситимидина, начиная с L-рибозы. В данном синтезе L-рибозу подвергают взаимодействию с тимином и SnCl4 в TMSCl и HMDS с образованием тимидина (2). Затем тимидин подвергают взаимодействию с PhOCOOPh и катализатором NaHCO3 в ДМФА примерно при 150°C, получая L-2,2'-ангидрорибофуранозилтимидин (3). Полученный L-2,2'-ангидрорибофуранозилтимидин подвергают взаимодействию с TrCl и пиридином примерно при 45°C, получая 5'-тритил-защищенный L-2,2'-ангидрорибофуранозилтимидин (4) и его в свою очередь восстанавливают посредством взаимодействия с таким восстановителем, как Red-Al, и комплексообразующим агентом, таким как 15-краун-5-эфир, в полярном растворителе, таком как ТГФ и/или DME, предпочтительно при температурах примерно 0-5°C, получая 5'-тритил-L-2'-дезокситимидин (5). Наконец, удаляют защиту соединения (5) при взаимодействии с 80% AcOH примерно при 50°C с образованием L-2'-дезокситимидина (6). Указанный синтез является эффективным по количеству требуемых стадий, а также позволяет избегать образования рибофуранозиламинооксазолидина.
На фиг.28 изображен 8-стадийный способ получения 2'-дезокситимидина из L-рибозы. Сначала L-рибозу (1) защищают любой защитной группой в условиях, подходящих для использования такой защитной группой (2), которые известны специалисту в данной области. Защищенную L-рибозу (2) подвергают взаимодействию с тимином и SnCl4 в присутствии TMSCl и HMDS, на стадии, известной в предшествующем уровне техники, чтобы образовать тимидин, который имеет защитные группы в 2'-, 3'- и 5'-положениях (3). Защищенный тимидин (3) подвергают удалению защиты (4), используя реагенты и условия, подходящие для удаления конкретной используемой защитной группы, и незащищенный тимидин (4) подвергают взаимодействию с PhOCOOPh и катализатором NaHCO3 в присутствии ДМФА примерно при 140-150°C с образованием 2,2'-ангидро-1-рибофуранозилтимидина (5) или (6). Следует отметить, что если полученным промежуточным продуктом является 2,2'-ангидро-1-рибофуранозилтимидин (5), то требуется дополнительная стадия после образования промежуточного продукта (4), на которой 3'- и 5'-положения тимидина подвергают взаимодействию, чтобы поместить в этих положениях защитные группы. Тритильные группы являются предпочтительными защитными группами для данного промежуточного продукта. Если получают промежуточный продукт (6), то он может быть получен непосредственно из структуры тимидина (4).
Затем промежуточный продукт (5) в случае его использования должен быть подвергнут удалению защиты в его 3'-положении реагентами и в условиях, подходящих для удаления защитной группы из данного положении, чтобы получить 5'-тритил-защищенный 2,2'-ангидро-1-рибофуранозилтимидин (7). Однако, если используют промежуточный продукт (6), то он может быть подвергнут взаимодействию с TrCl и пиридином примерно при 45°C, чтобы получить 5'-тритил-защищенный 2,2'-ангидро-1-рибофуранозилтимидин (7).
Затем 5'-тритил-защищенный 2,2'-ангидро-1-рибофуранозилтимидин (7) восстанавливают таким восстановителем, как Red-Al, и комплексообразующим агентом, таким как 15-краун-5-эфир, в полярном растворителе, таком как ТГФ и/или DME, предпочтительно при температуре примерно 0-5°C, получая 5'-тритил-защищенный тимидин (8), защиту которого затем удаляют посредством взаимодействия с 80% AcOH при температуре примерно 50°C, получая L-рибо-2'-дезокситимидин (9).
Синтез, изображенный на фиг.28, позволяет избегать прохождения реакции через промежуточный фуранозиламинооксазолидин, который требует дополнительной стадии конденсации для образования соответствующего 2,2'-ангидросоединения. Это также обеспечивает выбор в отношении того, какие промежуточные продукты должны быть защищены на разных стадиях способа.
В другом способе согласно настоящему изобретению синтез требуемых соединений может быть осуществлен в отсутствие комплексообразующего агента (см. фиг.29). Применение комплексообразующего агента, например, такого как 15-краун-5-эфир, дает более высокий процентный выход продукта в том случае, когда защитной группой является диметокситритил, однако в том случае, когда в качестве защитной группы используют только тритил, применение комплексообразующего агента, такого как, например, 15-краун-5-эфир, дает более низкий процентный выход продукта. Таким образом, в некоторых вариантах изобретения синтез требуемых соединений может быть осуществлен в отсутствие комплексообразующего агента, когда в качестве защитной группы используют тритил, как на фиг.29.
На фиг.29 изображен 3-стадийный способ получения 2'-дезокситимидина. Способ включает:
(a) получение 2,2'-ангидро-1-(5-O-тритил-β-D-арабинофуранозил)тимина (2) из 2,2'-ангидро-1-(β-D-арабинофуранозил)тимина (1) суспендированием 2,2'-ангидро-1-(β-D-арабинофуранозил)тимина (1), например, в пиридине и DMAP, и добавлением порциями тритилхлорида, например, при комнатной температуре. Реакционную смесь можно поддерживать при комнатной температуре или при необходимости нагревать, например, реакционную смесь можно поддерживать при комнатной температуре в течение примерно 1 часа и затем нагревать до 45°C (внутренняя температура) в течение примерно 15 часов. Реакцию можно контролировать, например, ТСХ (Rf исходного вещества 0,15; Rf продукта 0,43). Затем реакционную смесь можно погасить и очистить требуемый продукт, например, охлаждением примерно до 0°C и медленным добавлением насыщенного водного раствора NaHCO3 в течение 15-минутного периода времени без изменения внутренней температуры. Сразу же можно осадить белое твердое вещество из раствора, и белую суспензию можно перемешивать в течение 30 минут при комнатной температуре. Твердое вещество можно выделить фильтрованием через воронку Бюхнера и затем промыть водой. Оставшееся твердое вещество можно поместить в дихлорметан и перемешивать в течение примерно 30 минут при комнатной температуре. Остаток может быть выделен фильтрованием через воронку Бюхнера, промыт дихлорметаном и высушен в вакууме в течение ночи, чтобы получить 2,2'-ангидро-1-(5-O-тритил-β-D-арабинофуранозил)тимин (2) с выходом примерно 73% в виде белого твердого вещества;
(b) получение 2'-дезокси-5'-O-тритил-β-D-тимидина (3) из 2,2'-ангидро-1-(5-O-тритил-β-D-арабинофуранозил)тимина (2) восстановлением 2,2'-ангидро-1-(5-O-тритил-β-D-арабинофуранозил)тимина (2), например, суспендированием (2) в безводном тетрагидрофуране и охлаждением суспензии примерно до 0-5°C на бане со льдом. В отдельной колбе, погруженной в баню со льдом, 65 мас.% раствор Red-Al в толуоле может быть разбавлен подходящим растворителем, например, добавлением к безводному тетрагидрофурану. Затем полученный разбавленный раствор Red-Al может быть охлажден примерно до 0-5°C и по каплям через шприц добавлен к суспензии 2,2'-ангидро-1-(5-O-тритил-β-D-арабинофуранозил)тимина (2). Скорость добавления капель раствора Red-Al является важной для реакции, и добавление может быть завершено примерно за 1 час. Полученный в результате прозрачный раствор поддерживали примерно при 0-5°C в течение 1 часа, после указанного периода времени анализ ТСХ показал наличие исходного вещества (Rf 0,34), требуемого продукта (Rf 0,47) и примесей (Rf 0,42 и 0,26). Анализ ВЭЖХ показал наличие исходного вещества (11,35 мин, 36,5% AUC), продукта (12,60 мин, 24%) и немного основной примеси (11,7 мин, 2,9%). Примерно после 2 часов суммарно при температуре около 0-5°C по каплям через шприц добавляли дополнительную порцию «неразбавленного» 65 мас.% раствора Red-Al в толуоле в течение периода времени, составляющего примерно 20 минут, к реакционной смеси, которую поддерживали примерно при 0-5°C. Еще через 1 час анализы ТСХ и ВЭЖХ показали наличие исходного вещества (11,35 мин, 3,2%). По каплям добавляли следующую порцию 65 мас.% раствора Red-Al в толуоле и реакционную смесь поддерживали примерно при 0-5°C еще в течение 45 минут. Через указанный период времени анализ ТСХ показал только следовое количество оставшегося исходного вещества. Реакцию гасили добавлением насыщенного раствора NH4Cl и слой тетрагидрофурана декантировали. Водный слой экстрагировали изопропилацетатом и полученную в результате эмульсию расслаивали медленным добавлением 5 н. раствора HCl. Органический слой отделяли, объединяли со слоем тетрагидрофурана и промывали насыщенным раствором NH4Cl и затем насыщенным раствором соли. В этой точке pH слоя насыщенного раствора соли составлял от 6,5 до 7, и органический слой сушили над Na2SO4, фильтровали и концентрировали в вакууме, получая пенообразное твердое вещество. Грубый остаток выпаривали совместно с толуолом, концентрировали в вакууме и полученный в результате остаток собирали в толуоле при нагревании примерно до 45°C. Смесь охлаждали до комнатной температуры и перемешивали при данной температуре вплоть до того, как белое твердое вещество начинало выпадать в осадок. По каплям добавляли воду и полученную в результате смесь перемешивали при комнатной температуре в течение примерно 3 часов. Твердое вещество выделяли фильтрованием и осадок на фильтре промывали водой и толуолом. Твердое вещество сушили примерно при 45°C в условиях высокого вакуума в течение примерно 1 часа и затем при комнатной температуре в вакууме в течение ночи, получая 2'-дезокси-5'-O-тритил-β-D-тимидин 3 с выходом примерно 41%;
(c) получение 2'-дезокси-D-тимидина (4) из 2'-дезокси-5'-O-тритил-β-D-тимидина (3) (1,215 г, 2,5 ммоль) суспендированием (3) в метаноле и нагреванием реакционной смеси примерно до 45°C на водяной бане вплоть до растворения (3). Затем колбу охлаждали до комнатной температуры и концентрировали. К смеси добавляли HCl и перемешивали при комнатной температуре. Примерно через 25 минут белое твердое вещество начинало выпадать в осадок из раствора. Через 1 час анализ ТСХ показал, что исходного вещества не оставалось (Rf 0,53) и образовывался основной продукт (Rf 0,21). К реакционной смеси добавляли порцию н-гептана и перемешивали при комнатной температуре в течение примерно 15 минут. Белое твердое вещество выделяли фильтрованием. Фильтрат разделяли на два слоя и слой в метаноле экстрагировали н-гептаном и затем концентрировали в вакууме до объема 2 мл. Остаток объединяли с 405 мг белого твердого вещества, суспендированного в TBME, и перемешивали при комнатной температуре в течение 1 часа. Белое твердое вещество выделяли фильтрованием, промывали TBME и сушили в вакууме в печи, получая 2'-дезокси-D-тимидин (4) с выходом примерно 78%.
Следует понимать, что все способы синтеза, описанные на фиг.1-29, и все примеры в равной мере применимы к любой стереохимической форме, α- или β-, D- или L-, любого исходного вещества, и что исходные соединения не ограничены рибозой, ксилозой и арабинозой, которые указаны в данном описании, а также включают 5- и 6-членные циклы, имеющие S, N или CH2 вместо O, указанного в неограничивающих примерах и на фиг.1-29.
Настоящее изобретение лучше описано в следующей неограничивающей серии примеров. Конкретные растворители, реагенты и/или условия реакции, приведенные в данном описании, могут быть заменены эквивалентными, сходными или подходящими растворителями, реагентами и/или условиями реакции, не отходя от сути и не выходя за рамки объема изобретения.
ПРИМЕРЫ
Пример 1
L-арабинозу превращают в соответствующий метилгликозид и 3- и 4-гидроксильные группы защищают в виде производного ацетонида. На схеме ниже показан простой способ дезоксигенирования 2-гидроксильной группы соединения 2 посредством ее превращения в соответствующую мезилатную группу, и подвергая указанный промежуточный мезилат воздействию условий восстановительного расщепления, чтобы получить 2-дезокси-промежуточный продукт 4. См. H. Urata, E. Ogura, K. Shinohara, Y. Ueda and M. Akagi, Nucleic Acids Res. 1992, 20, 3325-3332; и J. W. Pratt, N. K. Richtmyer and C. S. Hudson, J. Am. Chem. Soc. 1952, 74, 2200-2205.
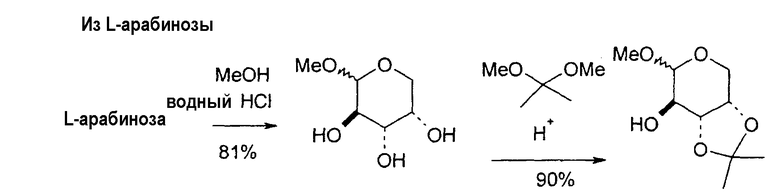
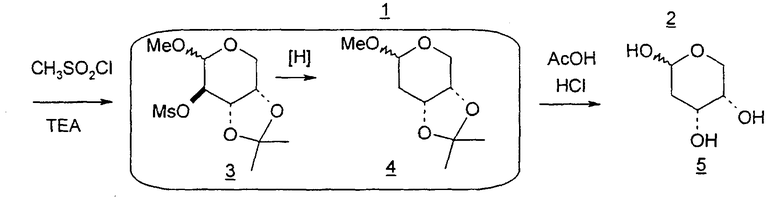
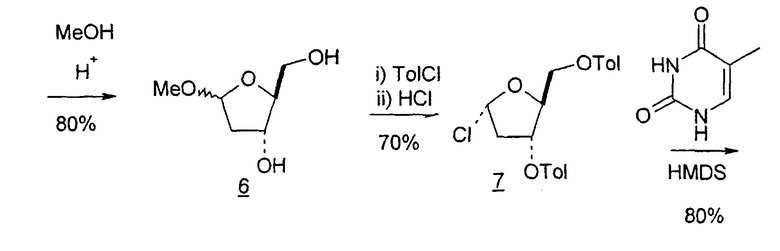
Пример 2
L-арабинозу превращают в соответствующее производное гликаля через ключевую стадию восстановительного элиминирования и превращением полученного в результате промежуточного гликаля в метил-2-дезоксирибофуранозид. См. B. K. Shull, Z. Wu и M. Koreeda, J. Carbohydr. Chem. 1996, 15, 955-964; M. L. Sznaidman, M. R. Almond и A. Pesyan., Nucleosides, Nucleotides & Nucleic Acids 2002, 21, 155-163; и Z.-X. Wang, W. Duan, L. I. Wiebe, J. Balzarini, E. D. Clercq и E. E. Knaus, Nucleosides, Nucleotides & Nucleic Acids 2001, 20, 11-40.
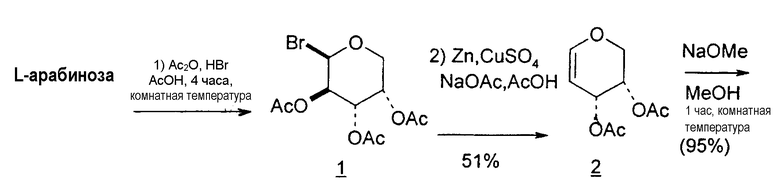
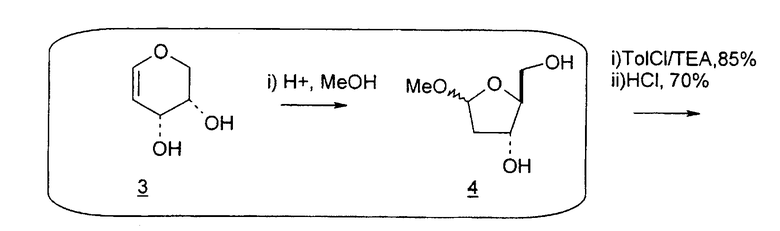
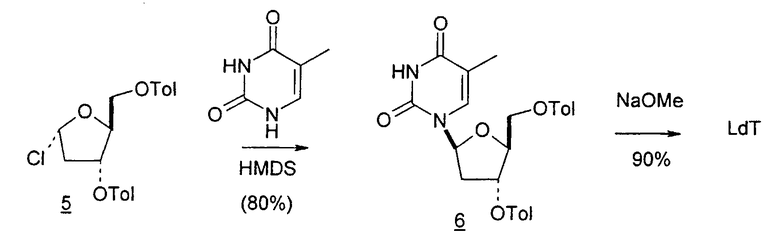
Пример 3
L-арабинозу превращают в соответствующее производное гликаля через ключевую стадию восстановительного элиминирования и превращением полученного в результате промежуточного гликаля в метил-2-дезоксирибофуранозид. См. M. L. Sznaidman, M. R. Almond и A. Pesyan., Nucleosides, Nucleotides & Nucleic Acids 2002, 21, 155-163; и Z.-X. Wang, W. Duan, L. I. Wiebe, J. Balzarini, E. D. Clercq и E. E. Knaus, Nucleosides, Nucleotides & Nucleic Acids 2001, 20, 11-40; и R. V. Stick, K. A. Stubbs, D. M. G. Tilbrook и A. G. Watts, Aust. J. Chem. 2002, 55, 83-85.
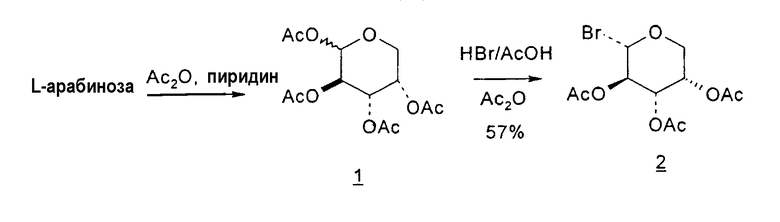
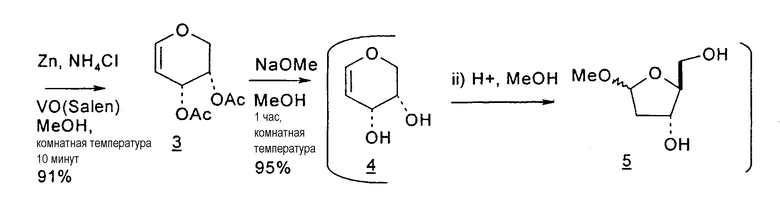
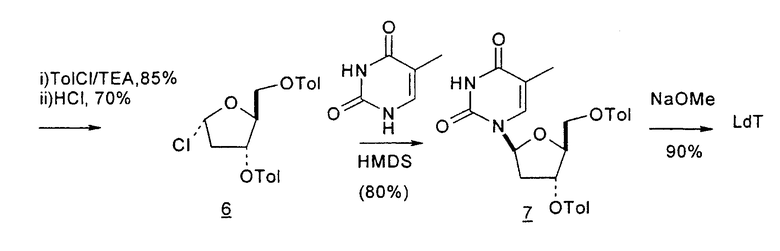
Пример 4
D-ксилозу окисляют бромом/водой и затем полученный в результате 1,4-лактон подвергают воздействию смеси HBr/уксусная кислота, чтобы получить 2,5-дибром-2,5-дидезокси-D-ликсоно-1,4-лактон 2. Обработка дибромлактона 2 йодидом калия в ТФА дает соответствующее 5-йод-соединение, а также приводит к селективному удалению атома брома у C-2, давая 5-йод-2-дезоксилактон 3. Подвергая указанный 5-йодлактон 3 воздействию водного раствора гидроксида калия, получают 4,5-эпоксидное производное, которое при обработке водным раствором кислоты дает соответствующий 2-дезокси-L-рибонолактон посредством стереоспецифичной инверсии по C-4. Защищенный 2-дезокси-L-рибонолактон 6 селективно восстанавливают до соответствующего лактола 7, используя Red-Al. Затем лактол 7 превращают в требуемый хлор-сахар 9. См. H.S. Isbell, Methods in Carbohydrate Research 1963, 2, 13-14; K. Bock, I. Lundt и C. Pedersen, Carbohydrate Research 1981, 90, 17-26; K. Bock, I. Lundt и C. Pedersen, Carbohydrate Research 1982, 104, 79-85; и I. Lundt и R. Madsen, Topics in Current Chemistry 2001, 215, 177-191.
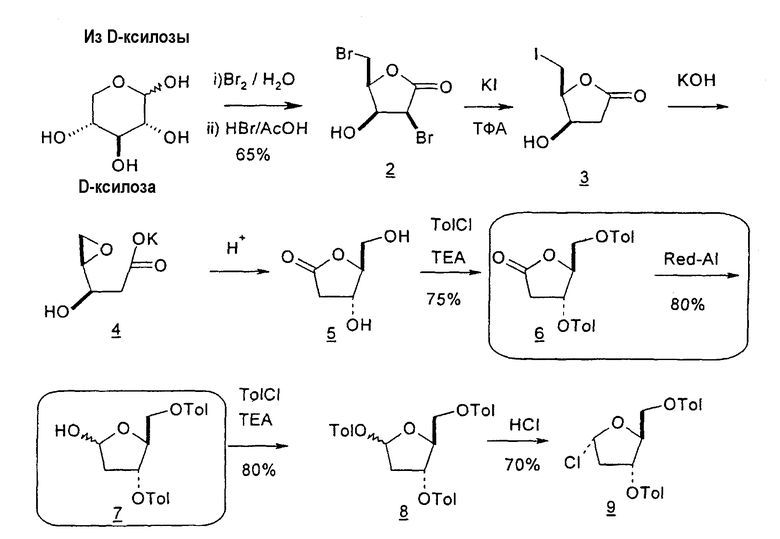
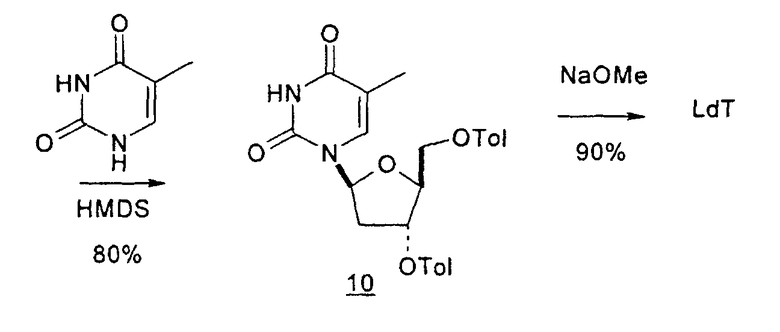
Пример 5
D-галактозу подвергают окислительному расщеплению, получая D-ликсонолактон, который затем подвергают бромированию, чтобы получить 2,5-дибром-2,5-дидезокси-D-ксилоно-1,4-лактон 2. Селективный гидрогенолиз 2 дает 5-бром-2-дезоксилактон 3, который затем подвергают последовательным превращениям, подобным превращениям, показанным в примере 7, получая ключевой промежуточный хлор-сахар 8. См. K. Bock, I. Lundt и C. Pedersen, Carbohydrate Research 1981, 90, 17-26; K. Bock, I. Lundt и C. Pedersen, Carbohydrate Research 1979, 68, 313-319; K. Bock, I. Lundt и C. Pedersen, Acta Chem. Scand. B 1984, 38, 555-561; и W. J. Humphlett, Carbohydrate Research 1967, 4, 157-164.
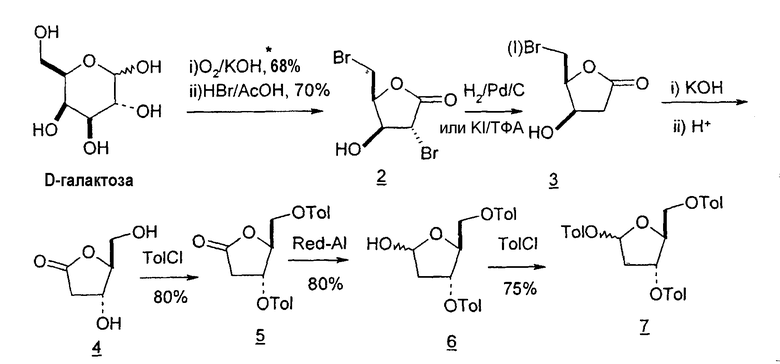
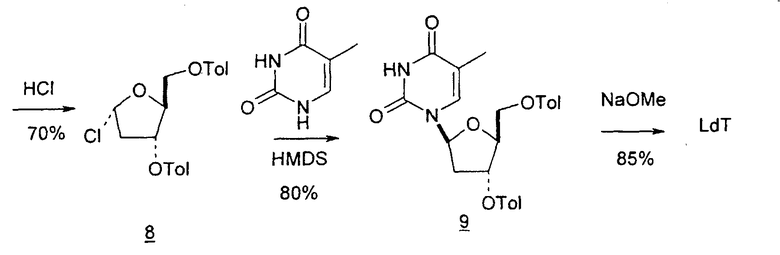
Пример 6
D-глюконо-1,4-лактон превращают в 2,6-дибром-2,6-дидезокси-D-манноно-1,4-лактон 1. Обработка лактона 1 гидразином и затем водным раствором брома дает 6-бром-2,6-дидезокси-D-арабиногексоно-1,4-лактон 3. Взаимодействие 3 с избытком водного раствора гидроксида калия с последующим подкислением приводит к инверсии по C-4 и C-5, давая 2-дезокси-L-рибогексоно-1,4-лактон 6. Указанное превращение включает раскрытие цикла лактона посредством перегруппировки Пейна первичного эпоксида 4 во вторичный эпоксид 5. 2-дезокси-L-рибогексоно-1,4-лактон 6 подвергают окислительному расщеплению с последующим восстановлением полученного в результате альдегида с получением лактона 7, который превращают в требуемый хлор-сахар, используя последовательность реакций, сходную с последовательностью реакций, показанной в примере 5. См. K. Bock, I. Lundt и C. Pedersen, Carbohydrate Research 1979, 68, 313-319; и K. Bock, I. Lundt и C. Pedersen, Acta Chem. Scand. B 1984, 38, 555-561.
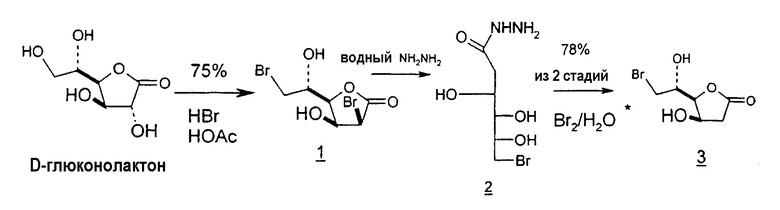
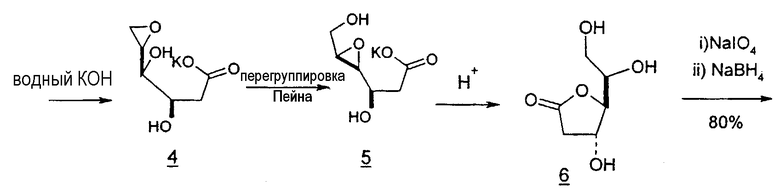
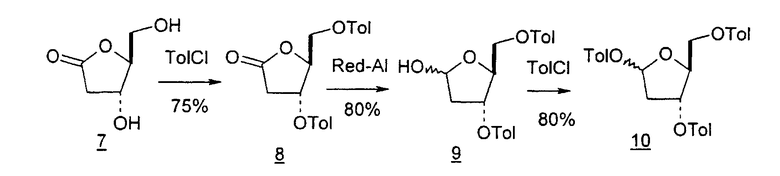
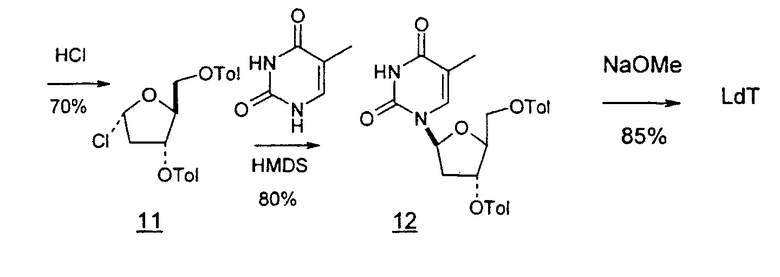
Пример 7
D-галактоно-1,4-лактон превращают в ацетилированный дибромлактон 2, который при обработке гидразином с последующим бромированием дает 2-дезоксилактон 3. Лактон 3 деацетилируют и подвергают окислительному расщеплению с последующим восстановлением полученного в результате альдегида с использованием NaBH4, чтобы получить 2-дезокси-L-рибоно-1,4-лактон 5. См. K. Bock, I. Lundt и C. Pedersen, Carbohydrate Research 1979, 68, 313-319; и K. Bock, I. Lundt и C. Pedersen, Acta Chem. Scand. B 1984, 38, 555-561.
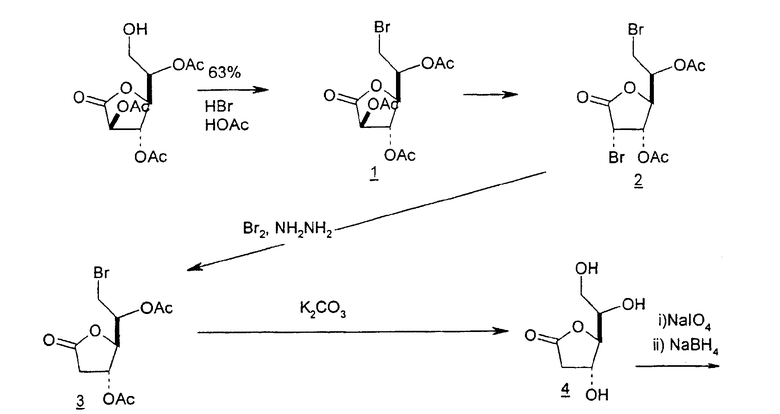
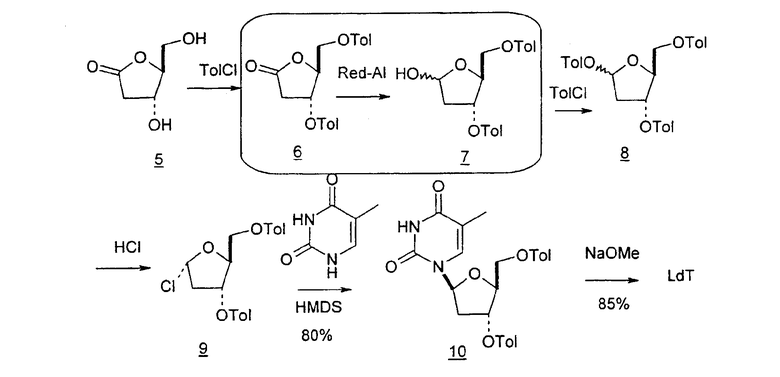
Пример 8
Фуранолактон, коммерчески доступное неуглеводное и ахиральное соединение, используют в качестве исходного вещества, чтобы получить хлор-сахар. Ключевой стадией в данном способе является асимметричное дигидроксилирование сложного эфира 2Z-пентаноата 2 с образованием производного 2(R),3(R)-пентаноата 3. Промежуточный продукт 3 подвергают стереоселективной циклизации, получая 2-дезокси-L-сахар 4. Соединение 4 превращают в требуемый защищенный хлор-сахар посредством трех превращений прямым синтезом. См. D. C. Liotta и M. W. Hager, патент США 5414078, 9 мая 1995 года.
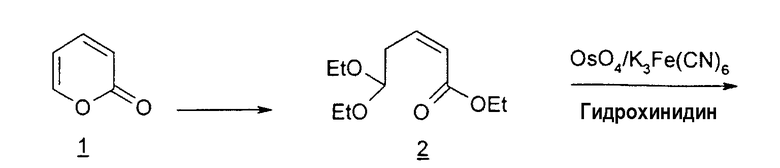
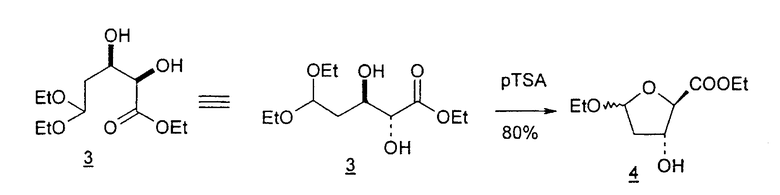
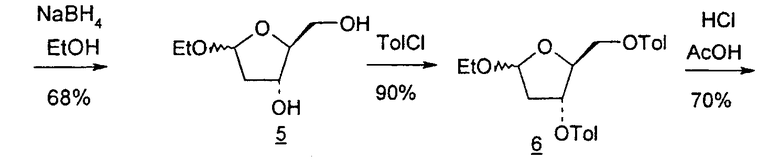
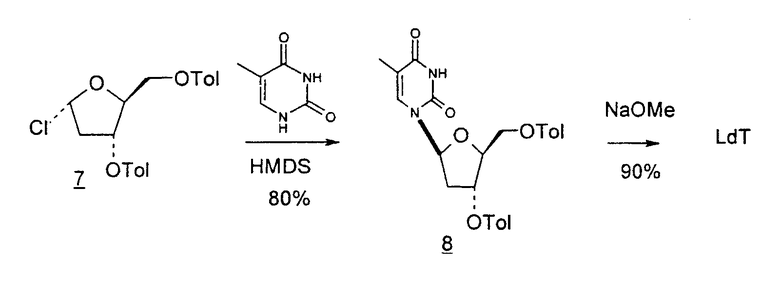
Пример 9
Этил-3,3-диэтоксипропаноат 1 является недорогим неуглеводным ациклическим и ахиральным исходным веществом. Соединение 1 восстанавливают до соответствующего альдегида 2, используя DIBAL. На следующей стадии альдегид 2 превращают в α,β-ненасыщенный сложный эфир, 5,5-диэтокси-2E-пентеноат, используя модификацию Хорнера-Эммонса реакции Виттига, с использованием диизопропил(этоксикарбонил)метилфосфоната, и полученный в результате сложный эфир восстанавливают до производного 2E-пентен-1-ола 3. Полученный прохиральный аллиловый спирт 3 превращают в соответствующий 2(S),3(S)-эпоксиспирт 4, используя условия ассиметричного эпоксидирования по Шарплессу. Полученный в результате эпоксиспирт 4 защищают и затем подвергают кислотному гидролизу, получая ключевой промежуточный продукт 6. Соединение 6 циклизуют до производного 2-дезокси-L-рибофуранозы 7, которую затем превращают в требуемый L-хлор-сахар 9. См. D. C. Liotta и M. W. Hager, патент США 5414078, 9 мая 1995 года; и M. W. Hager и D. C. Liotta, Tetrahedron Lett. 1992, 33, 7083-7086.
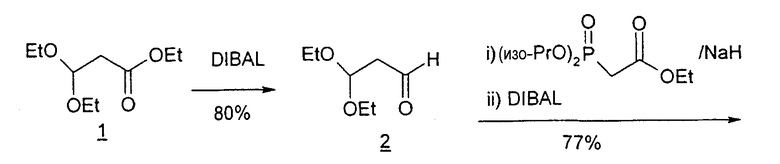
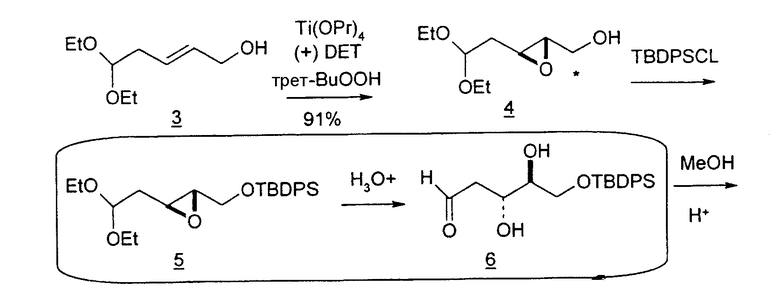
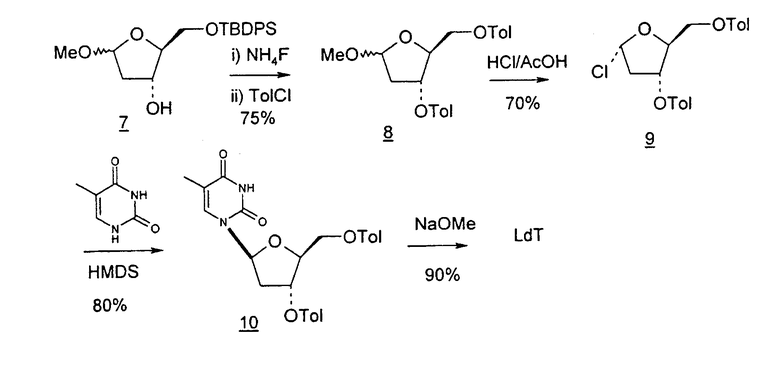
Пример 10
Гидроксиглутаминовую кислоту 1 подвергают циклизации, получая производное 2-дезокси-L-1,4-рибонолактон 2; который затем превращают в требуемый хлор-сахар 5 в четыре стадии. См. R. F. Schinazi, D. C. Liotta, C. K. Chu, J. J. McAtee, J. Shi, Y. Choi, K. Lee и J. H. Hong, патент США 6348587B1, 19 февраля 2002 года; U. Ravid, R. M. Silverstein и L. R. Smith, Tetrahedron 1978, 34, 1449-1452; и M. Taniguchi, K. Koga и S. Yamada, Tetrahedron 1974, 30, 3547-3552.
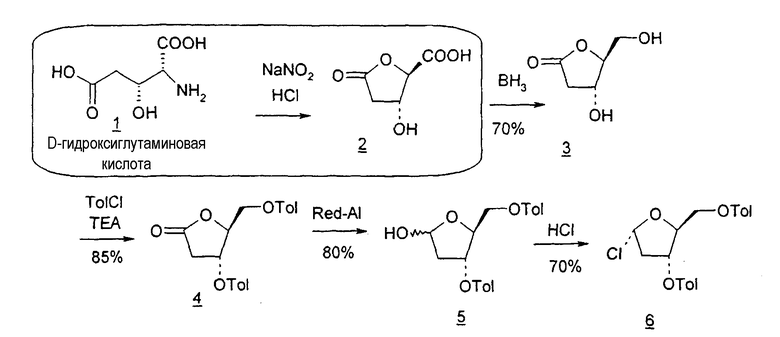
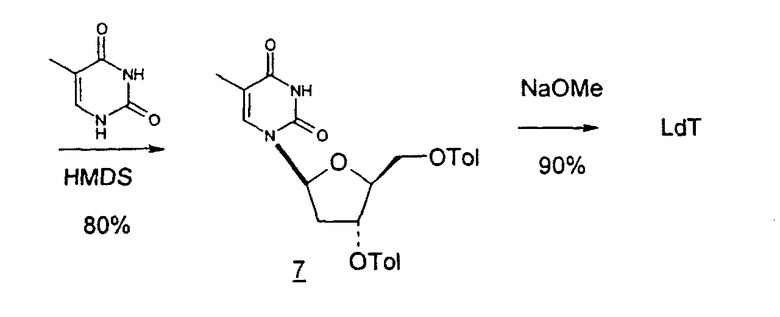
Пример 11
Коммерчески доступный спирт 1 подвергают воздействию условий эпоксидирования по Шарплессу, получая эпоксид 2. Эпоксиспирт 2 обрабатывают бензиловым спиртом в присутствии Ti(Oi-Pr)4, получая диол 3, который превращают в соответствующее ацетонидное производное 4. Соединение 4 окисляют, используя условия реакции Уокера, чтобы получить альдегид 5, который обрабатывают водным раствором хлористоводородной кислоты, получая 5-O-бензил-2-дезокси-L-рибофуранозу 6. Соединение 6 превращают в требуемый хлор-сахар 9 в ходе четырех простых стадий. См. M. E. Jung и C. J. Nichols, Tetrahedron Lett. 1998, 39, 4615-4618.
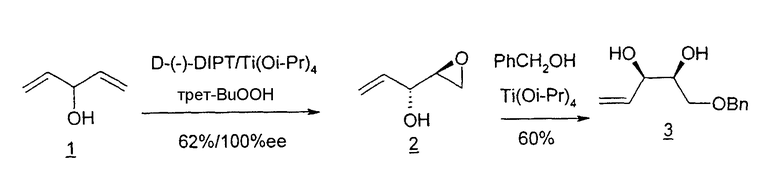
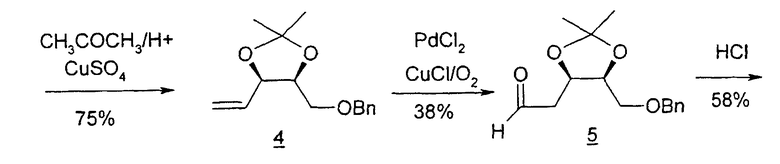
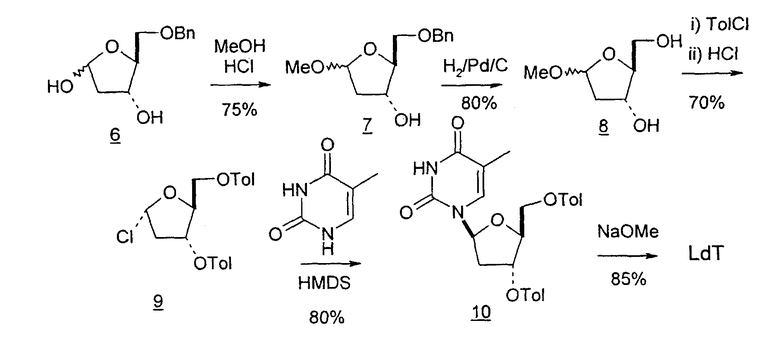
Пример 12
Эпоксиспирт 1 защищают в виде бензилового эфира 2 и раскрывают эпоксид, используя бензилат натрия в бензиловом спирте, затем защищают полученный в результате спирт 3, получая трис-бензиловый эфир 4. Превращение соединения 4 в альдегид 6 (который является защищенной 2-дезокси-L-рибозой) осуществляют, используя гидроборирование/окисление H2O2 с последующим окислением по Сверну полученного в результате спирта 5. Удаление защиты бензиловых эфиров 6 с использованием гидроксида палладия на угле дает смесь трех этил-2-дезокси-L-рибозидов 7a, 7b и 7c в соотношении 2:2:1. Защита 7a и 7b с использованием толуоилхлорида и обработка полученного в результате дитолуоил-производного хлористым водородом дает требуемый хлор-сахар 8. См. M. E. Jung и C. J. Nichols, Tetrahedron Lett. 1998, 39, 4615-4618.
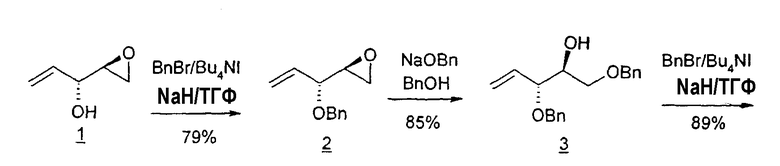
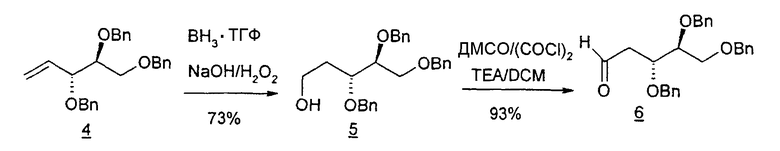
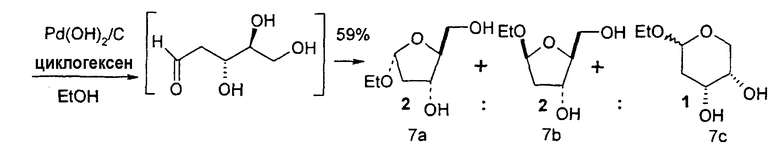
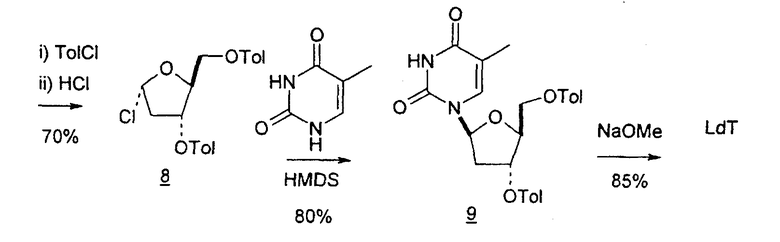
Пример 13
1,2-O-изопропилиден-L-глицеральдегид 1 обрабатывают аллилбромидом в присутствии цинка и водного раствора хлорида аммония, получая соответствующий гомоаллиловый спирт 2. Группу изопропилидена 2 удаляют, используя водный раствор уксусной кислоты, получая промежуточный продукт 3. Последующий озонолиз и восстановление диметилсульфидом соединения 3 дает 2-дезокси-L-рибофуранозу 4, которую превращают в защищенный хлор-сахар 7 в три стадии. См. J. S. Yadav и C. Srinivas, Tetrahedron Lett. 2002, 43, 3837-3839; и T. Harada и T. Mukaiyama, Chem. Lett. 1981, 1109-1110.
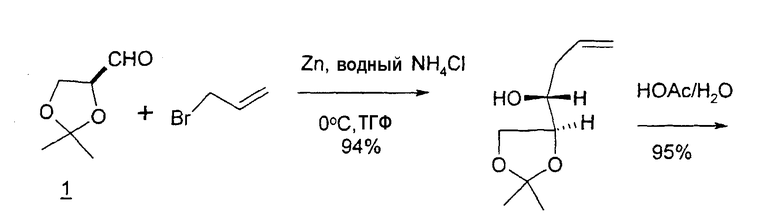
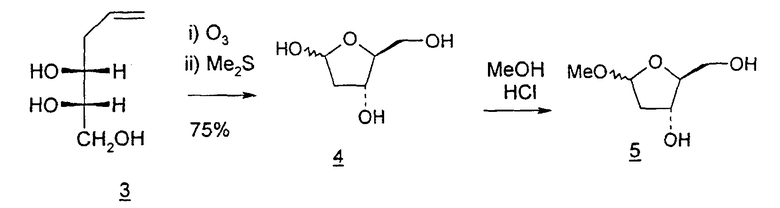
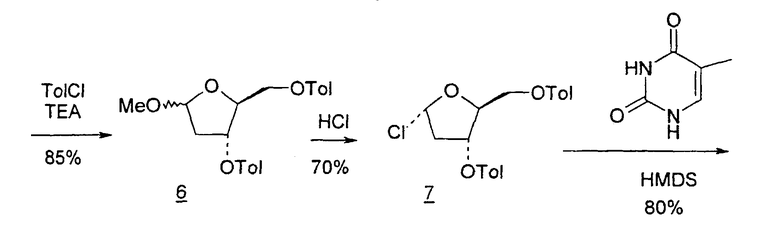
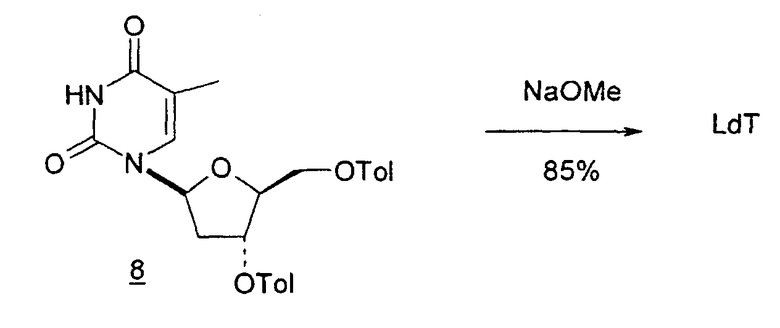
Пример 14
В данном примере используют 2-бромметил[1,3]диоксолан вместо аллилбромида, который использован в примере 13, что исключает необходимость в озонолизе и последующем восстановлении промежуточного продукта 3. См. J. S. Yadav и C. Srinivas, Tetrahedron Lett. 2002, 43, 3837-3839; и T. Harada и T. Mukaiyama, Chem. Lett. 1981, 1109-1110.
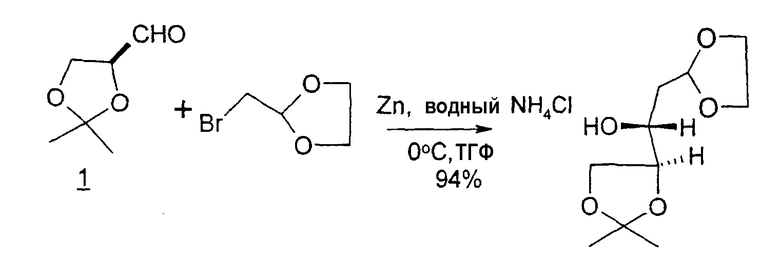
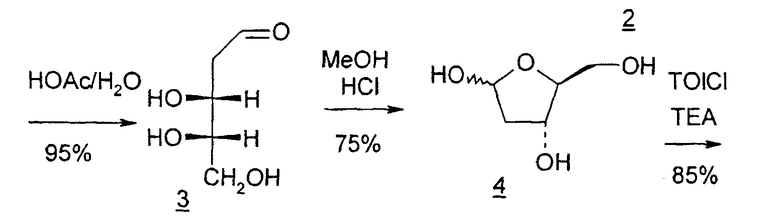
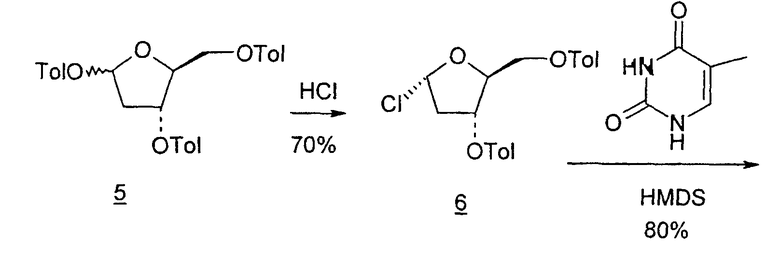
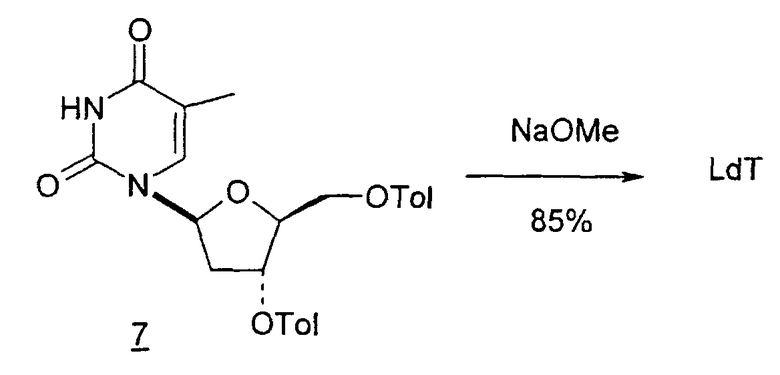
Пример 15
Гликаль 1 (который может быть получен из L-рибозы в четыре стадии) обрабатывают кислым метанолом, чтобы получить 2-дезокси-L-рибозу 3, которую превращают в защищенный хлор-сахар 5 в две стадии. См. H. Ohrui и J. J. Fox, Tetrahedron Lett. 1973, 1951-1954; W. Abramski и M. Chmielewski, J. Carbohydr. Chem. 1994, 13, 125-128; и J. C.-J. Cheng, U. Hacksell и G. D. Daves, Jr., J. Org. Chem. 1985, 50, 2778-2780.
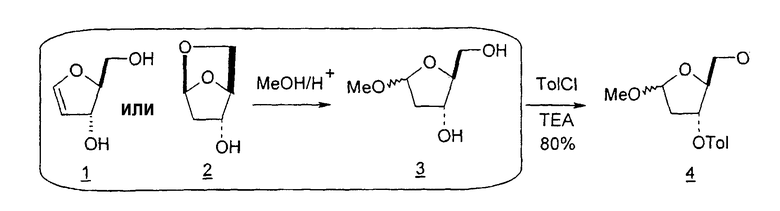
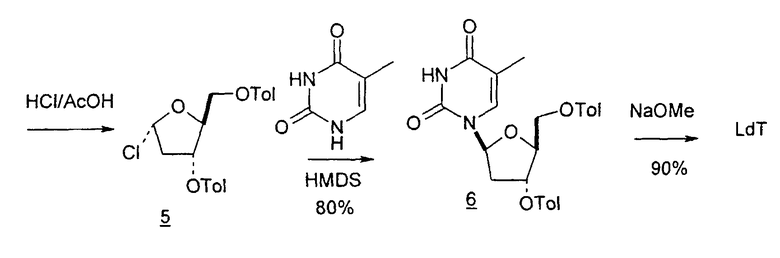
Исходное вещество 1 может быть получено из L-рибозы
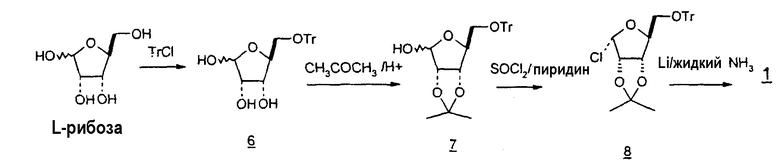
Пример 16
L-арабинозу подвергают взаимодействию с цианамидом, получая производное 1,2-оксазолина 1. В случае обеспечения взаимодействия с этиловым эфиром 2-метил-3-оксопропионовой кислоты соединение 1 дает O2,2'-ангидро-L-тимидин 2. Соединение 2 подвергают бензоилированию и полученное в результате ди-O-бензоил-производное 3 подвергают воздействию условий восстановительного расщепления, чтобы получить 3',5'-ди-O-бензоил-LdT 4. Соединение 4 обрабатывают метанольным раствором метоксида натрия, получая LdT. См. A. Holy, Coll. Czech. Chem. Commun. 1972, 37, 4072-4087; и P. V. P. Pragnacharyulu, C. Vargeese, M. McGregor и E. Abushanab, J. Org. Chem. 1995, 60, 3096-3099.
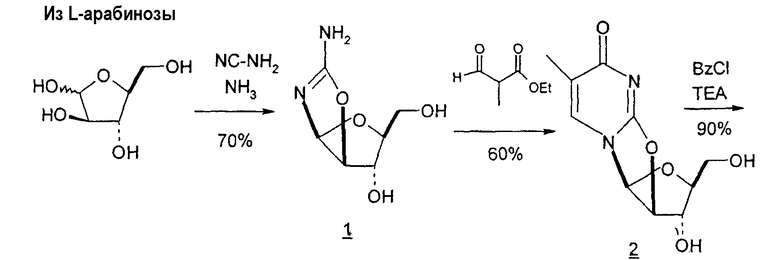
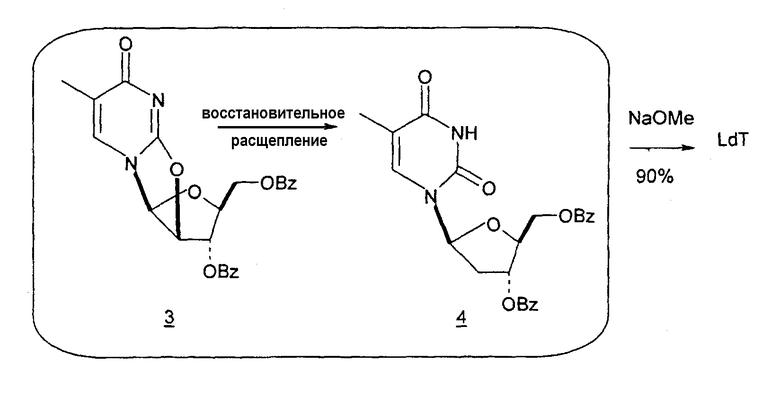
Пример 17
В данном примере используют другой способ раскрытия O2,2'-связи соединения 3, используя хлористый водород. Полученное в результате 2'-дезокси-2'-хлор-производное 4 обрабатывают TBTH/AIBN, получая 2'-дезокси-защищенный нуклеозид 5, который при дезацилировании дает LdT. См. P. V. P. Pragnacharyulu, C. Vargeese, M. McGregor и E. Abushanab, J. Org. Chem. 1995, 60, 3096-3099.
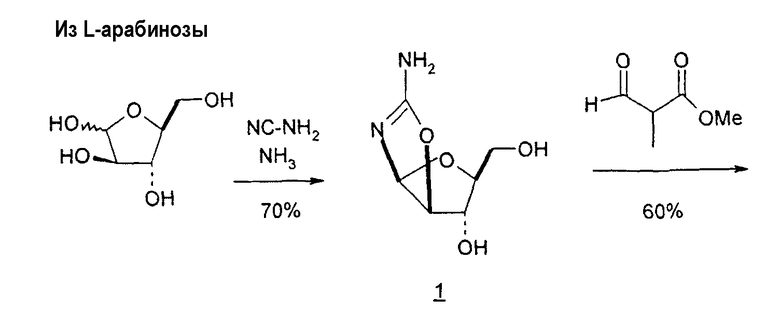
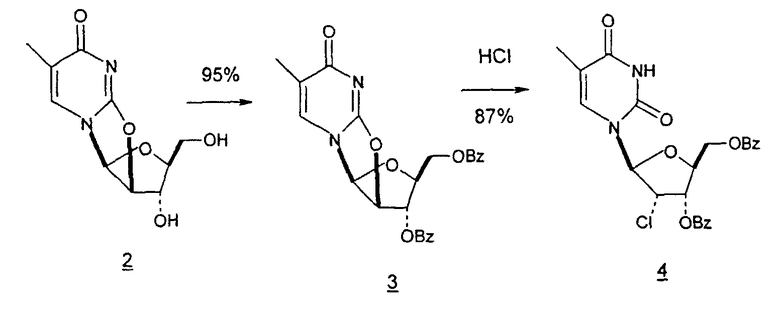
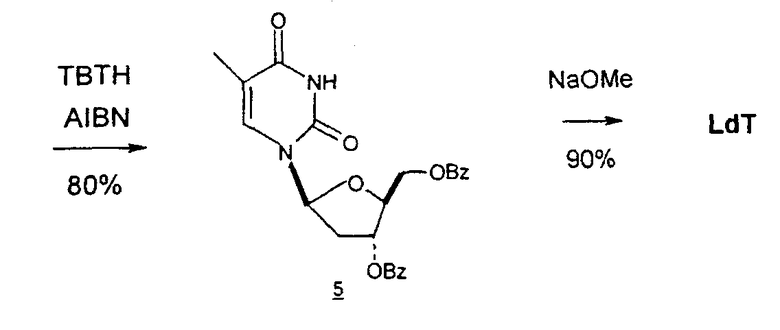
Пример 18
Данный пример отличается от примера 17 тем, что производное 1,2-оксазолина 1 подвергают взаимодействию с другими соединениями, чтобы получить O2,2'-ангидро-L-тимидин 2. См. D. McGee, Boehringer Ingelheim Proposal to Novirio Pharmaceuticals, Inc. May 17, 2002; и C.W. Murtiashaw, Европейский патент 0351126 B1, 18 января 1995 года.
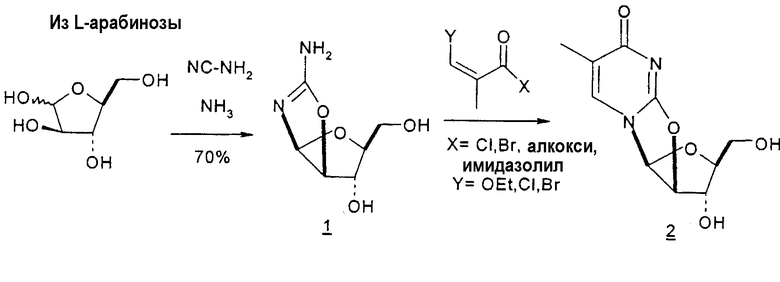
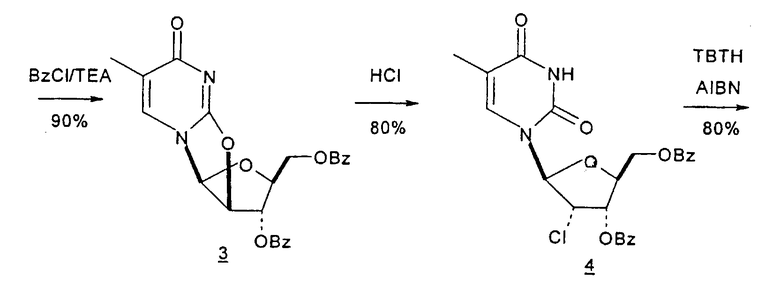
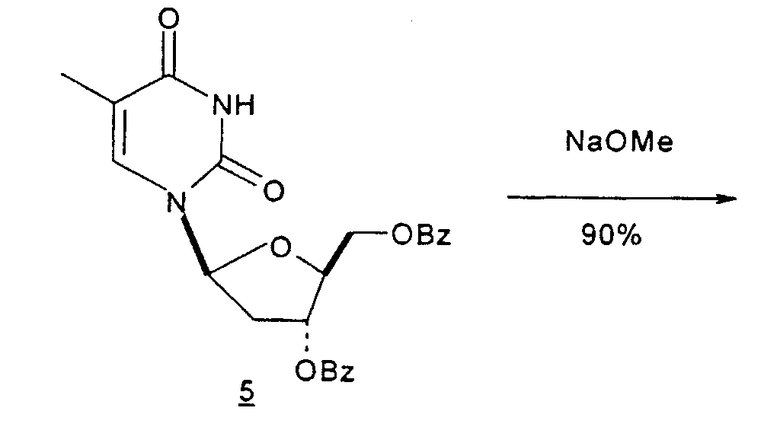
Пример 19
Йодид водорода используют для раскрытия O2,2'-связи соединения 3, чтобы получить 2'-дезокси-2'-йод-производное 4. Соединение 4 обрабатывают йодидом калия, получая 3',5'-ди-O-бензоил-2'-дезокси-L-тимидин 5. Соединение 5 подвергают воздействию метанольным раствором метоксида натрия, получая LdT. См. H. Sawai, A. Nakamura, H. Hayashi и K. Shinozuka, Nucleosides & Nucleotides 1994, 13, 1647-1654; и H. Sawai, H. Hayashi и S. Sekiguchi, Chemistry Lett. 1994, 605-606.
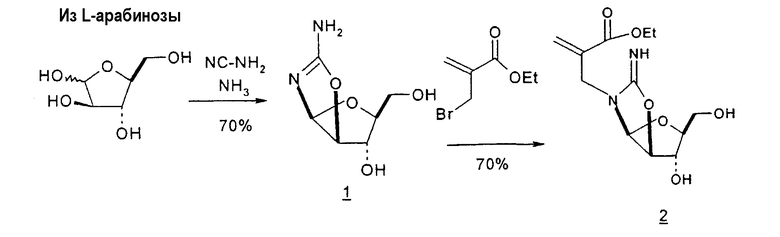
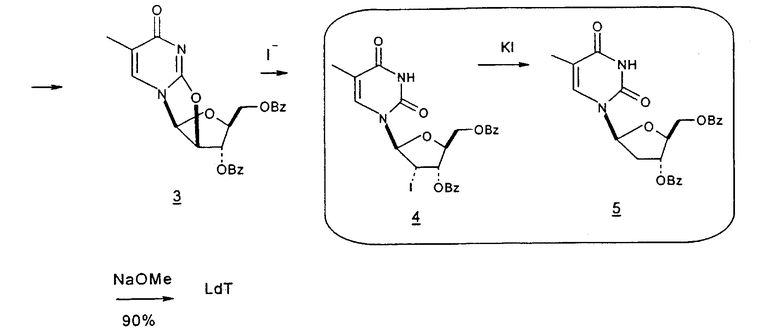
Пример 20
Сложный эфир 2-метилоксиран-2-карбоновой кислоты подвергают взаимодействию с 1,2-оксазолином 1, получая производное O2,2'-ангидро-L-тимидина 2. Соединение 2 обрабатывают пивалоилхлоридом, чтобы защитить 3'- и 5'-гидроксильные группы, а также расщепить O2,2'-связь и получить 2'-дезокси-2'-хлорнуклеозид 3. Соединение 3 обрабатывают тионилхлоридом, чтобы элиминировать гидроксильную группу остатка основания, и полученное в результате соединение восстанавливают, используя TBTH/AIBN, чтобы удалить 2'-хлор и получить защищенный LdT 5. Соединение 5 обрабатывают метоксидом натрия в метаноле, получая LdT. См. E. Abushanab и P. V. Pragnacharyula, патент США 5760208, 2 июня 1998 года.
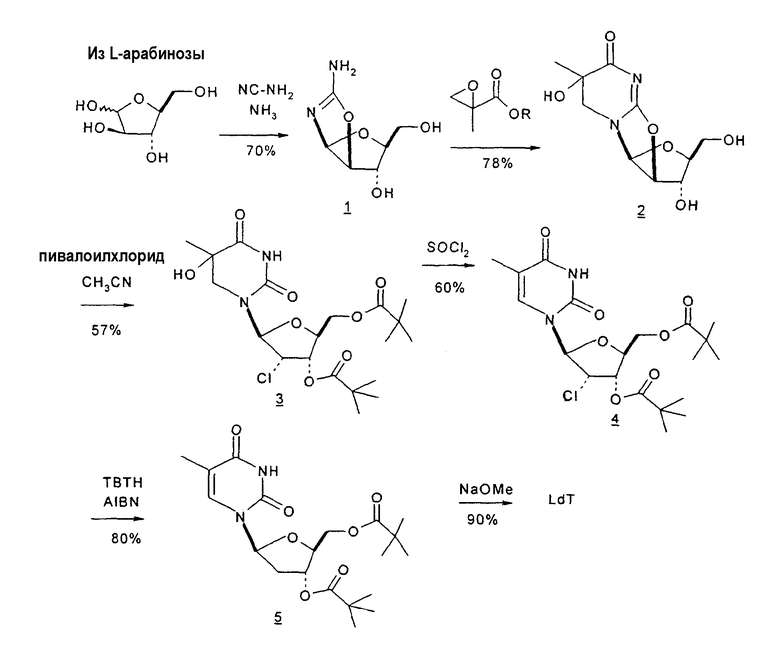
Пример 21
Этилпропиолат подвергают взаимодействию с 1,2-оксазолином 1, получая O2,2'-ангидро-L-уридин 2. Соединение 2 защищают и полученное в результате 3',5'-ди-O-бензоил-производное 3 обрабатывают хлористым водородом, получая 2'-дезокси-2'-хлорнуклеозид 4. Соединение 4 затем обрабатывают TBTH/AIBN, чтобы удалить 2'-хлор, и затем 2'-дезокси-производное подвергают воздействию условий йодирования, чтобы получить 5-йоднуклеозидное производное 6. 5-йод-группу соединения 6 заменяют метильной группой, используя AlMe3 и (Ph3P)4Pd, получая 3',5'-ди-O-бензоил-LdT 7, который при обработке метоксидом натрия в метаноле дает LdT. См. A. Holy, Coll. Czech. Chem. Commun. 1972, 37, 4072-4087; J.-I. Asakura и M. J. Robins, J. Org. Chem. 1990, 55, 4928-4933; J.-I. Asakura и M. J. Robens, Tetrahedron Lett. 1988, 29, 2855-2858; и K. Hirota, Y. Kitade, Y. Kanbe, Y. Isobe и Y. Maki, Synthesis, 1993, 210, 213-215.
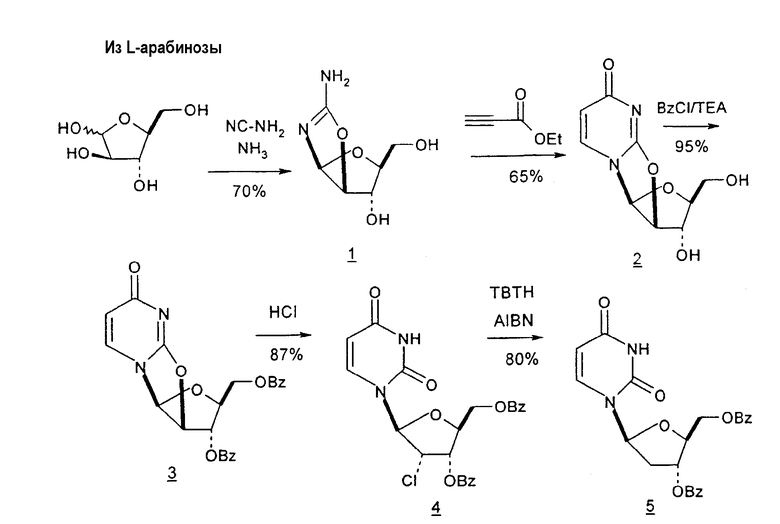
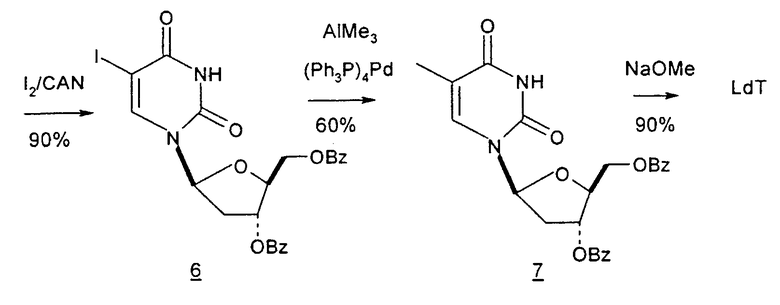
Пример 22
Данный пример отличается от примера 21 только способом введения метильной группы в положение 5 производного 2'-дезокси-L-уридина 5. Соединение 5 обрабатывают формальдегидом в щелочной среде, получая 5-гидроксиметил-производное 6, которое при воздействии кислого этанола дает 2'-дезокси-5-этоксиметил-L-уридин 7. Соединение 7 дает LdT в условиях каталитического гидрирования. См. A. Holy, Coll. Czech. Chem. Commun. 1972, 37, 4072-4087.
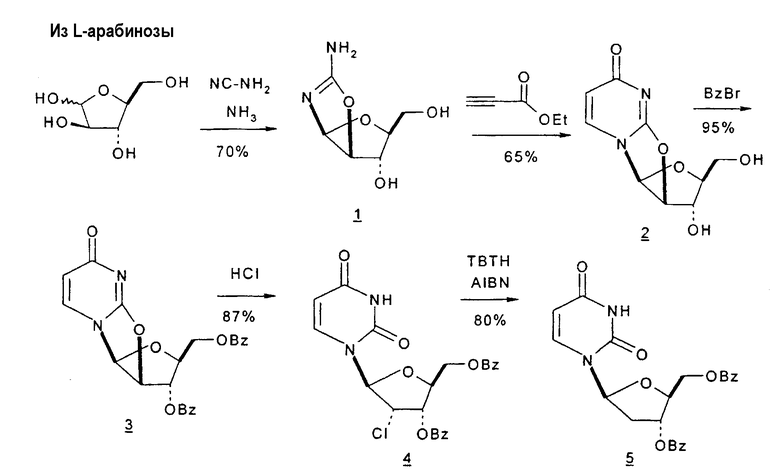
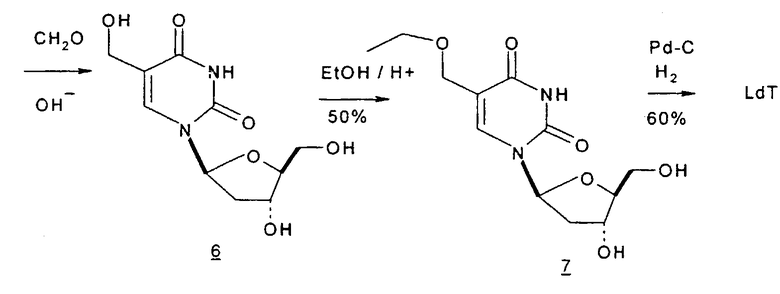
Пример 23
Синтез ключевого промежуточного 2-дезокси-3,5-ди-O-пара-толуоил-β-L-эритропентофуранозилхлорида из D-ксилозы
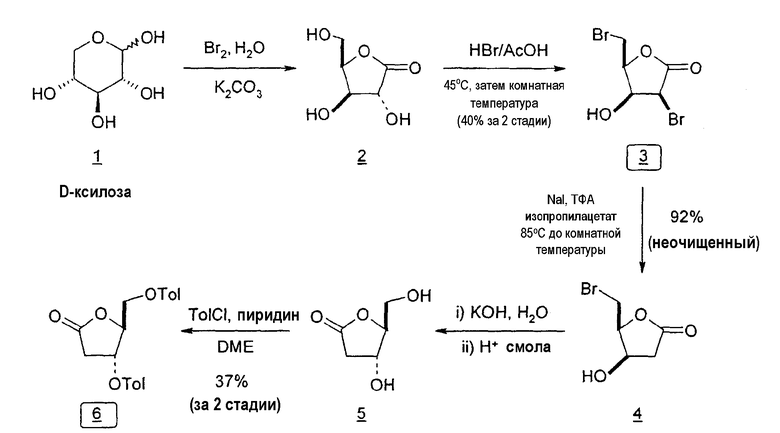
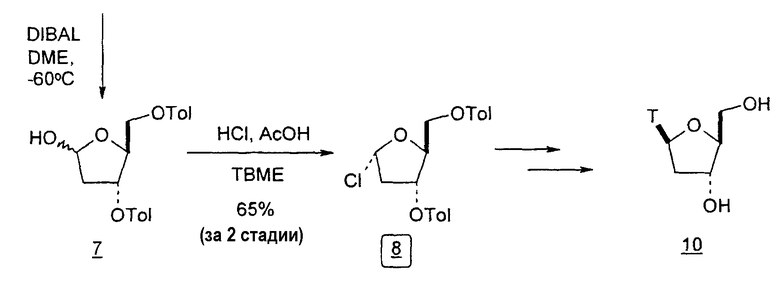
Пример 23(a)
D-ксилоно-1,4-лактон (2) из D-ксилозы (1) посредством окисления бромом
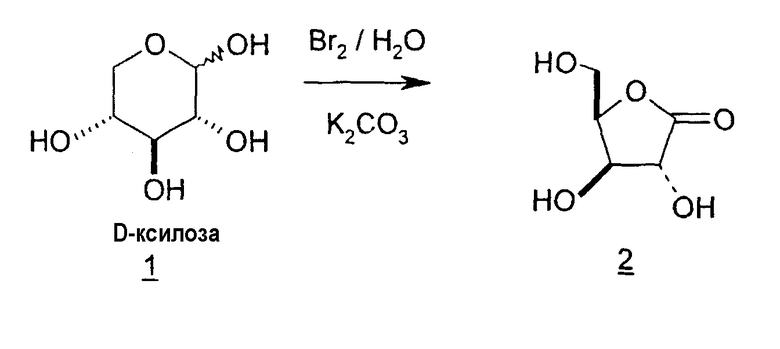
D-ксилозу 1 (100 г, 666,1 ммоль) растворяли в дистиллированной воде (270 мл) и охлаждали до 0°C при перемешивании сверху в трехгорлой круглодонной колбе объемом 1 л. Порциями добавляли карбонат калия (113,2 г, 819,3 ммоль), поддерживая температуру ниже 20°C. Затем по каплям добавляли бром (39,4 мл, 766,0 ммоль) при 0-5°C в течение периода времени, составляющего 2 часа, при этом поддерживая температуру ниже 10°C. Реакционную смесь поддерживали при температуре примерно 5-10°C еще в течение 30 минут, затем нагревали до комнатной температуры и перемешивали в течение ночи. Примерно через 8 часов анализ ТСХ (10% метанол в этилацетате, визуализация с использованием ванилина) показала отсутствие исходного вещества (Rf 0,0) и наличие нового продукта (Rf 0,3). Реакционную смесь перемешивали с муравьиной кислотой (6,6 мл) в течение примерно 15 минут и затем концентрировали в вакууме при 45°C до объема примерно 50 мл. Осуществляли совместное выпаривание с уксусной кислотой (200 мл) и концентрирование в вакууме примерно при 45°C до объема 60 мл и неочищенный D-ксилоно-1,4-лактон 2 переносили для использования в таком виде на следующей стадии.
Преимущества данной стадии синтеза заключаются в переключении с BaCO3, известного в предшествующем уровне техники, на K2CO3, который давал лучшее соотношение при загрузке (50 г D-ксилозы 1 в 135 мл воды по сравнению с 400 мл в случае BaCO3); лактон можно было использовать без дополнительной очистки/удаления соли KBr на следующей стадии; остаточный KBr можно было использовать для следующей реакции; и совместное выпаривание с уксусным ангидридом, чтобы удалить остаточную воду, приводит к образованию менее полярных продуктов при анализе ТСХ.
Пример 23(b)
2,5-дибром-2,5-дидезокси-D-ликсо-1,4-лактон (3) из D-ксилоно-1,4-лактона (2)
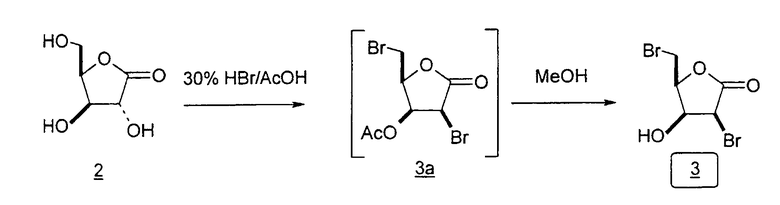
D-ксилоно-1,4-лактон (2) (неочищенный в уксусной кислоте, 666,08 ммоль) переносили к колбу объемом 3 л, используя теплую уксусную кислоту (200 мл) и к перемешиваемой суспензии медленно добавляли 30% HBr-AcOH (662 мл, 3330 ммоль). Реакционную смесь нагревали до 45°C в течение 1 часа и затем охлаждали и перемешивали в течение 1,5 часа при комнатной температуре. Через указанный период времени анализ ТСХ (1:1, этилацетат:гексан) показал наличие двух основных продуктов (Rf 0,63 [3-O-ацетил-2,5-дибром-2,5-дидезокси-D-ликсо-1,4-лактон 3a] и Rf 0,5 [2,5-дибром- 2,5-дидезокси-D-ликсо-1,4-лактон 3]) и некоторое количество оставшегося исходного вещества (анализ ТСХ, 10% метанол в этилацетате, Rf 0,0, визуализация с использованием ванилина). Реакционную смесь охлаждали при 0°C и добавляли метанол (850 мл) в течение 1 часа, поддерживая температуру ниже 20°C. Затем реакционной смеси давали возможность нагреться до комнатной температуры и перемешивали в течение ночи. После указанного периода времени анализ ТСХ (1:1, этилацетат:гексан) показал превращение одного продукта (Rf 0,63) в другой продукт (Rf 0,44). Реакционную смесь фильтровали через воронку Бюхнера, чтобы удалить остаточный KBr (173,89 г), и затем концентрировали в вакууме и выпаривали совместно с водой (2×250 мл). Добавляли этилацетат (800 мл) и воду (250 мл) и слои разделяли. Затем водный слой экстрагировали этилацетатом (2×300 мл) и объединенные органические экстракты промывали насыщенным водным раствором гидрокарбоната натрия (400 мл) и водой (100 мл). Слои разделяли, водный слой экстрагировали этилацетатом (2×300 мл) и объединенные органические экстракты сушили, используя сульфат натрия (125 г), фильтровали и концентрировали в вакууме при 50°C. Перед концентрированием досуха органический экстракт упаривали совместно с гептаном (200 мл), получая коричневое полутвердое вещество. Растирание полученного твердого вещества осуществляли, используя 20% гептан в изопропиловом эфире (100 мл гептана и 500 мл изопропилового эфира), и сушили в вакууме при 30-35°C в течение ночи, получая 2,5-дибром-2,5-дидезокси-D-ликсо-1,4-лактон 3 в виде чистого светло-коричневого твердого вещества (71,9 г, 40% за 2 стадии).
Т.пл. 92-94°С [Лит. 92-93°С]; δН (d6-ДМСО, 400 МГц): 3,65 (1Н, дд, J4,5'=8,1 Гц, J5,5'=10,7 Гц, Н-5'), 3,73 (1H, дд, J4,5=5,9 Гц, J5,5'=10,7 Гц), 4,4 (1Н, м, Н-3 или Н-4), 4,73 (1Н, м, Н-4 или Н-3), 5,31 (1Н, д, J2,3=4,4 Гц, Н-2), 6,38 (1Н, ушир.с, 3-ОН); δН (CDCl3, 400 МГц): 3,65 (1Н, дд, J5,5'=10,3 Гц, J4,5=5,9 Гц, Н-5'), 3,72 (1H, a-т, J5,5'=9,88 Гц, Н-5), 4,63 (1Н, м, Н-3), 4,71 (1Н, м, Н-4), 4,86 (1Н, д, J2,3=4,4 Гц, Н-2).
Преимущества данной стадии синтеза заключаются в возможности осуществлять реакцию при 45°C, значительно уменьшая время реакции по сравнению с 24 часами, как было известно в предшествующем уровне техники; удаление соли KBr фильтрованием было возможно после обработки метанолом, и это важно, так как обеспечивает простую экстракцию продукта; и контроль точной температуры реакции гашения очень важен для того, чтобы предотвратить образование побочных продуктов.
Пример 23(c)
5-бром-2,5-дидезокси-D-треопентоно-1,4-лактон (4)
Способ 1 - йодид натрия
2,5-дибром-2,5-дидезокси-D-ликсо-1,4-лактон 3 (35 г, 127,8 ммоль) растворяли в изопропилацетате (300 мл) и добавляли йодид натрия (76,6 г, 511,2 ммоль) и трифторуксусную кислоту (14,8 мл) при комнатной температуре. Реакционную смесь нагревали примерно до 85°C (внутренняя температура) в течение 1,5 часа. После указанного периода времени анализ ТСХ (1:1, этилацетат:гексан) показал небольшое количество оставшегося исходного вещества (Rf 0,44) и наличие нового продукта (Rf 0,19). Реакционную смесь охлаждали до комнатной температуры и перемешивали в течение 4 часов. Анализ ТСХ показал отсутствие исходного вещества, поэтому реакционную смесь концентрировали в вакууме до 20 мл, чтобы удалить трифторуксусную кислоту, и разбавляли изопропилацетатом (200 мл). Реакционную смесь промывали насыщенным водным раствором гидрокарбоната натрия (200 мл) и слои разделяли. Затем водный слой экстрагировали изопропилацетатом (3×200 мл). Объединенные органические экстракты обрабатывали водным раствором тиосульфата натрия (48 г в 160 мл воды). Водный слой экстрагировали изопропилацетатом (2×200 мл) и объединенные органические экстракты сушили, используя сульфат натрия (20 г), фильтровали и концентрировали в вакууме, получая 5-бром-2,5-дидезокси-D-треопентоно-1,4-лактон 4 (16,38 г, грубый выход 92%) в виде маслянистого коричневого остатка. Полученный продукт растворяли в воде и использовали как таковой для последующей реакции. В других случаях изопропилацетат заменяли водой и водный раствор использовали как таковой для реакции с KOH.
δH (D2O, 400 МГц): 2,64 (1H, д, J2,2'=18,3 Гц, H-2'), 3,12 (1H, дд, J2,2'=18,0 Гц, J2,3=5,49 Гц, H-2), 3,45 (0,125H, дд, H-5' и H-5 для йодида 4I); 3,70 (2H, a-д, J=6,71 Гц, H-5, H-5'), 4,74 (1H, a-т, H-3), 4,87 (1H, м, H-4). δС (D2O, 100 МГц): 27,1 (C-5), 39,0 (C-2), 67,9 (C-3), 84,6 (C-4), 178,8 (C-1); δН (d6-ДМСО, 400 МГц): 2,34 (1H, a-д, J2,2'=17,1 Гц, J2',3=6,3 Гц, H-2'), 2,95 (1H, дд, J2,2'=17,1 Гц, J2,3=5,4 Гц, H-2), 3,39 (0,125H, дд, J=7,3 Гц, J=6,8 Гц, J=11,2 Гц, H-5' и H-5 для йодида 4I), 3,60 (1H, дд, J5,5=10,7 Гц, J4,5'=8,3 Гц, H-5'), 3,70 (1H, дд, J5,5=10,7 Гц, J4,5=5,4 Гц, H-5), 4,39 (1H, м, H-3), 4,63 (1H, м, H-4), 5,61 (1H, д, J3,ОH=4,4 Гц, 3-OH); m/z (ES-ve): 253 (М+AcOH)-; Найдено: С, 30,69, Н, 3,55, Br, 41,22%; С5Н7ВrO3 требует С, 30,80, Н, 3,62, Вr, 40,97%.
Способ 2 - гидрирование
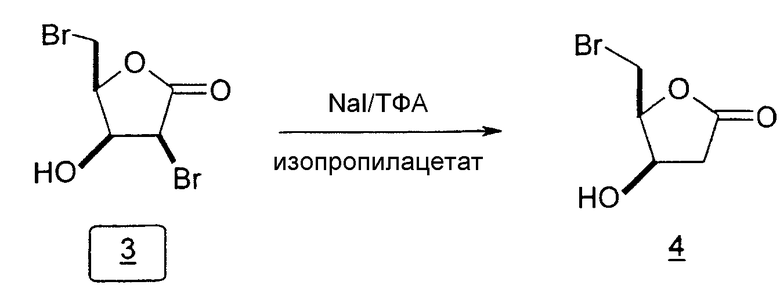
2,5-дибром-2,5-дидезокси-D-ликсо-1,4-лактон 3 (7,5 г, 27,6 ммоль) растворяли в этилацетате (120 мл) и к перемешиваемому раствору добавляли триэтиламин (4 мл, 28,7 ммоль). Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода (атмосферное давление) в присутствии 5% безводного палладия на угле (1 г) в течение примерно 1,5 часа. После указанного периода времени анализ ТСХ (1:1, этилацетат:гексан) показал наличие нового продукта (Rf 0,16), остаточного исходного вещества (Rf 0,44). Поэтому реакционную смесь продували аргоном (три раза) и затем перемешивали в атмосфере водорода еще в течение 2 часов. После указанного периода времени анализ ТСХ показал наличие небольшого количества исходного вещества, поэтому реакционную смесь фильтровали через целит (этилацетат в качестве элюента), промывали 4 М HCl (30 мл) и водный слой дополнительно экстрагировали этилацетатом (2×20 мл). Объединенные органические экстракты сушили, используя сульфат натрия (10 г), фильтровали и концентрировали в вакууме, получая 5-бром-2,5-дидезокси-D-треопентоно-1,4-лактон 4 (5,15 г, грубый выход 96%) в виде неочищенного бледно-желтого масла.
δH (D2O, 400 МГц): 2,65 (1H, д, J2,2'=18,3 Гц, H-2'), 3,09 (1H, дд, J2,2'=18,3 Гц, J2,3=5,8 Гц, H-2), 3,71 (2H, a-д, J=7,01 Гц, H-5, H-5'), 4,74 (1H, a-т, J=4,9 Гц, J=4,6 Гц, H-4), 4,87 (1H, м, J=3,7 Гц, H-3); δС (D2O, 100 МГц): 27,7 (C-5), 39,5 (C-2), 68,5 (C-3), 85,2 (C-4), 179,5 (C-1); δН (d6-ДМСО, 400 МГц): 2,34 (1H, д, J2,2'=17,1 Гц, H-2'), 2,93 (1H, дд, J2,2'=17,1 Гц, J2,3=5,4 Гц, H-2), 3,60 (1H, дд, J5,5=10,7 Гц, J4,5'=8,3 Гц, H-5'), 3,70 (1H, дд, J5,5=10,7 Гц, J4,5=5,4 Гц, H-5), 4,40 (1H, м, H-3), 4,65 (1H, м, H-4), 5,58-5,64 (1H, ушир.с, 3-OH); δС (d6-ДМСО, 100 МГц): 29,6 (C-5), 39,5 (C-2), 67,2 (C-3), 83,2 (C-4), 175,4 (C-1).
Указанные стадии в способе обеспечивали преимущества, связанные с реакцией гидрирования, при которой требовалось значительно меньше растворителя для экстракции, замена йодида калия на йодид натрия и нагревание реакционной смеси укорачивало время реакции с 6 часов до 2 часов, и замена ацетона на изопропилацетат улучшала экстракцию продукта из водного слоя.
Пример 23(d)
2-Дезокси-L-рибоно-1,4-лактон 5 из 5-бром-2,5-дидезокси-D-треопентоно-1,4-лактона 4
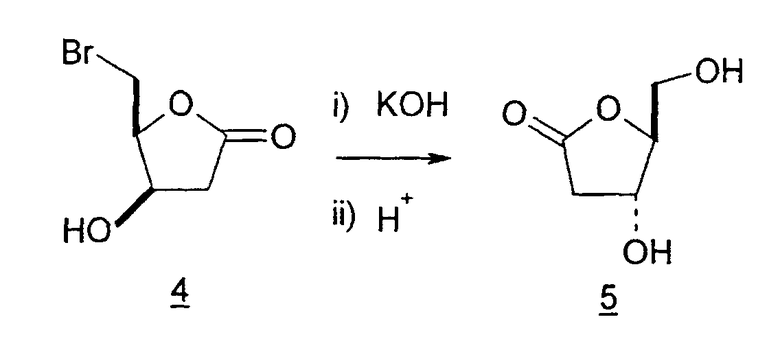
Гидроксид калия (14,9 г, 230,7 ммоль) растворяли в воде (124 мл) и охлаждали до 15°C. Полученный раствор добавляли к перемешиваемому раствору 5-бром-2,5-дидезокси-D-треопентоно-1,4-лактона 4 (15 г, 76,9 ммоль) в воде (62 мл) при комнатной температуре. Спустя примерно 3 часа анализ ТСХ (2% метанол в этилацетате) показал отсутствие исходного вещества (Rf 0,55) и наличие нового продукта (Rf 0,0). Реакционную смесь нагревали до 80°C (внутренняя температура) в течение 30 минут, охлаждали до комнатной температуры и не наблюдали изменения при анализе ТСХ. Добавляли амберлит IR-120 плюс кислую смолу (50 г) и реакционную смесь перемешивали при комнатной температуре в течение 30 минут, и измеренное в этой точке значение pH составляло 3. Добавляли дополнительное количество смолы (40 г) и реакционную смесь перемешивали при комнатной температуре еще в течение 30 минут, и измеренное в этой точке значение pH составляло 1. Реакционную смесь перемешивали при комнатной температуре в течение ночи, после указанного периода времени анализ ТСХ показал образование нового продукта (Rf 0,21). Смолу удаляли фильтрованием через пористую воронку (вода в качестве элюента, 200 мл) и концентрировали в вакууме. Не доводя досуха, осуществляли совместное выпаривание с 1,2-диметоксиэтаном (2×100 мл). Красный остаток растворяли в 1,2-диметоксиэтане (200 мл) и перемешивали с MgSO4 (10 г) в течение 40 минут при комнатной температуре. Фильтрование, промывка 1,2-диметоксиэтаном (75 мл) и концентрирование в вакууме при 45°C давали 2-дезокси-L-рибоно-1,4-лактон 5 (10,47 г, грубый выход 90%) в виде неочищенного красного твердого вещества. В других случаях продукт хранили в DME и использовали как таковой для последующей реакции.
δН (d6-ДМСО, 400 МГц): 2,22 (1H, дд, J2,2'=17,6 Гц, J2',3=2,0 Гц, H-2'), 2,80 (1Н, дд, J2,2'=18,1 Гц, J2,3=6,3 Гц, H-2), 3,50 (1H, дд, J5,5'=12,2 Гц, J4,5'=3,9 Гц, H-5'), 3,54 (1H, дд, J5,5'=12,2 Гц, J4,5=4,2 Гц, H-5), 4,26 (2H, м, H-3 и H-4), 4,7-5,0 (2Н, ушир.с, ОН).
Пример 23(e)
2-дезокси-3,5-ди-O-пара-толуоил-L-рибоно-1,4-лактон 6 из 2-дезокси-L-рибоно-1,4-лактона 5
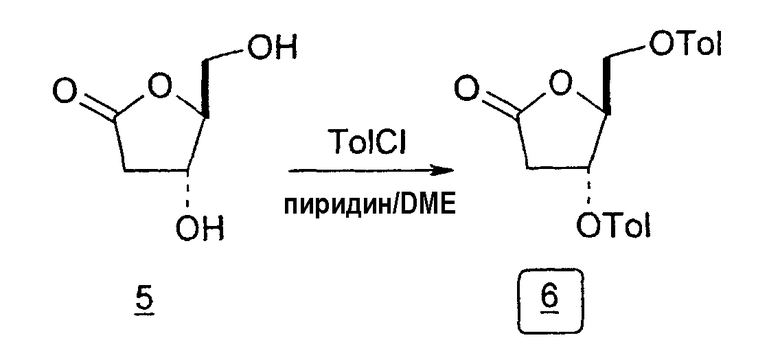
Раствор 2-дезокси-L-рибоно-1,4-лактона 5 (10,47 г, 76,9 ммоль) и пиридина (31,1 мл, 384,4 ммоль) в 1,2-диметоксиэтане (100 мл) охлаждали до температуры от 0 до -5°C в атмосфере аргона. Из капельной воронки добавляли пара-толуоилхлорид (21,4 г, 138 ммоль) в течение 20 минут, поддерживая температуру от 0 до -5°C. Через 3,5 часа поддерживания температуры от 0 до -5°C анализ ТСХ (30% этилацетат в гексане) показал наличие нового продукта (Rf 0,76) и отсутствие оставшегося исходного вещества (Rf 0,36) и анализ ВЭЖХ показал, что реакция завершена. Реакционную смесь охлаждали (0°C) и гасили раствором гидрокарбоната натрия (25 г в 300 мл). От реакционной смеси отделяли коричневое масло, которое постепенно затвердевало при перемешивании при комнатной температуре. Через 1,5 часа твердое вещество собирали фильтрованием, промывали водой (150 мл) и неочищенное твердое вещество (25,84 г) сушили в течение ночи. Неочищенный лактон растворяли в дихлорметане (150 мл) и перемешивали с MgSO4 (10 г) в течение 1 часа. Твердое вещество собирали фильтрованием, промывали дихлорметаном (50 мл) и фильтрат концентрировали при 40°C примерно до 50 мл. Добавляли TBME (100 мл) и смесь концентрировали при 40°C примерно до 50 мл. Затем оставшийся концентрированный раствор перемешивали при комнатной температуре, и он образовывал густую взвесь. Добавляли TBME (50 мл) и перемешивание продолжали при комнатной температуре в течение 2 часов. Твердое вещество собирали фильтрованием, промывали TBME (50 мл) и сушили в вакууме при 30-35°C в течение ночи, получая 2-дезокси-3,5-ди-O-пара-толуоил-L-рибоно-1,4-лактон 6 (10,46 г, 37% за 3 стадии) в виде бледно-коричневого твердого вещества.
δН (CDCl3, 400 МГц): 2,42, 2,43 (2×с, 2×СН 3Аr, 2×3Н), 2,82 (1Н, дд, J2,2'=18,7 Гц, J2',3=1,8 Гц, H-2'), 3,16 (1Н, дд, J2,2'=18,7 Гц, J2,3=7,3 Гц, H-2), 4,61 (1Н, дд, J5,5'=12,4 Гц, J4,5'=3,3 Гц, H-5'), 4,71 (1Н, дд, J5,5'=12,1 Гц, J4,5=3,7 Гц, H-5), 4,95 (1Н, м, H-4), 5,61 (1Н, a-д, J=7,69 Гц, H-3), 7,25-7,28 (4Н, м, 2×ArH), 7,86-7,93 (2×2Н, 2×д, J=8,4 Гц, 2×ArH); δC (CDCl3, 100 МГц): 21,9, 35,3, 63,9, 71,8, 82,8, 125,1, 126,5, 129,4, 129,5, 129,6, 130,0, 130,4, 144,6, 145,0, 166,0, 166,1 (2×ArCO2), 174,2 (C-1).
Преимущества данной стадии общего способа заключались в том, что толуоилангидрид удаляли из процесса посредством обработки TBME, и стадия хроматографии на колонке была исключена из способа.
Пример 23(f)
2-дезокси-3,5-ди-O-пара-толуоил-L-рибоза 7 из 2-дезокси-3,5-ди-O-пара-толуоил-L-рибоно-1,4-лактона 6
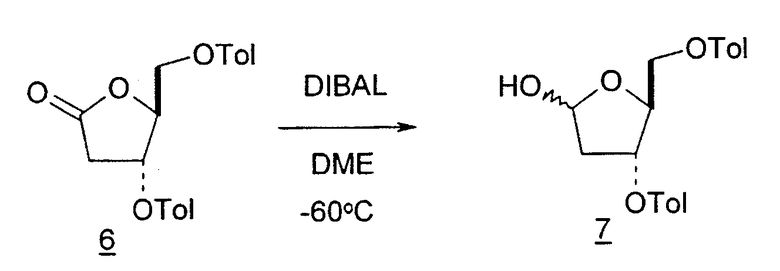
Раствор 2-дезокси-3,5-ди-O-пара-толуоил-L-рибоно-1,4-лактона 6 (9,0 г, 24,42 ммоль) в 1,2-диметоксиэтане (90 мл) охлаждали примерно до -60°C в аргоне с верхним перемешиванием. Пo каплям через капельную воронку добавляли 1 М раствор гидрида диизобутилалюминия в толуоле (32,4 мл, 32,4 ммоль) в течение 15 минут. Внутреннюю температуру поддерживали при -60°C в течение 1 часа, и анализ ВЭЖХ показал завершение реакции. Реакционную смесь гасили добавлением ацетона (10 мл) в течение 2 минут и затем добавлением 5 н. HCl (30 мл) в течение 5 минут. Смесь перемешивали при комнатной температуре в течение 30 минут и концентрировали в вакууме при 35°C примерно до 30 мл. Оставшееся масло объединяли с насыщенным раствором соли (24 г в 60 мл) и экстрагировали этилацетатом (3×100 мл). Объединенные органические экстракты сушили сульфатом натрия (10 г), концентрировали до объема 50 мл и выпаривали совместно с TBME, получая 2-дезокси-3,5-ди-O-пара-толуоил-α,β-L-рибозу 7. Часть концентрировали в вакууме досуха для 1H-ЯМР-анализа. Полученный продукт дополнительно не характеризовали, так как его использовали неочищенным на следующей стадии.
δН (CDCl3, 400 МГц, соотношение аномеров составляет 0,75:1): 2,2-2,6 (м, 2×СН
3Ar и Н-2/Н-2' для α- и β-аномеров), 4,4-4,75 (м, Н-4 и Н-5/Н-5' для α- и β-аномеров), 5,4-5,8 (4×м, Н-1 и Н-3 для А и  7,18-8,02 (м, 7Н, ароматические протоны для А и В).
7,18-8,02 (м, 7Н, ароматические протоны для А и В).
Пример 23(g)
2-дезокси-3,5-ди-O-пара-толуоил-β-L-эритропентофуранозилхлорид 8
Способ 1: Непосредственно из лактола 7
Раствор 2-дезокси-3,5-ди-O-пара-толуоил-α,β-L-рибозы 7 (примерно 7,0 г, 18,91 ммоль) в TBME (30 мл) разбавляли TBME (15 мл) и перемешивали в течение 20 минут при комнатной температуре. Добавляли уксусную кислоту тремя порциями по 1 мл с перемешиванием вплоть до получения прозрачного коричневого раствора. Раствор охлаждали до 0°C в аргоне и пропускали сухой газ HCl постоянным потоком в течение 25 минут. Через 10 минут при 0°C аликвоту гасили безводным этанолом (1,2 мл) и оставляли стоять при комнатной температуре с периодическим встряхиванием в течение 10 минут, получая при этом прозрачный раствор. Реакционную смесь выдерживали при 0-5°C и через 65 минут реакционную смесь фильтровали. Твердый продукт промывали TBME (30 мл) и сушили в вакууме в течение 5 часов, получая 2-дезокси-3,5-ди-O-пара-толуоил-β-L-эритропентофуранозилхлорид 8 (4,79 г, 65%) в виде белого кристаллического твердого вещества. Т.пл. 118-121°C.
δН (CDCl3, 400 МГц): 2,41, 2,43 (2×с, 2×СН 3Аr, 2×3Н), 2,74 (1Н, a-д, J2,2'=14,6 Гц, H-2'), 2,87 (1Н, ддд, J2,2'=12,7 Гц, J2,3=7,3 Гц, J1,2=5,3 Гц, H-2), 4,59 (1Н, дд, J5,5'=12,2 Гц, J4,5'=4,4 Гц, H-5'), 4,68 (1Н, дд, J5,5'=12,2 Гц, J4,5=3,4 Гц, H-5), 4,86 (1Н, м, J=3,4 Гц, H-3/H-4), 5,56 (1Н, a-дд, J=1,95 Гц, J=6,3 Гц, H-3/H-4), 6,47 (1Н, д, J1,2=5,4 Гц, H-1), 7,23-7,28 (4H, м, 2×АrН), 7,89, 7,99 (2×2Н, 2×д, J=8,3 Гц, 2×АrН). [а]D 25 = -117 (с, 1,0 в СНСl3) [СМS химические Ltd: [а]D 20 = -118,9 (с, 1 в ДХМ)].
Способ 2: Через метоксид 7-OMe
Раствор 1% HCl в метаноле получали добавлением ацетилхлорида (0,2 мл) к метанолу (10 мл), предварительно охлажденному до 5°C. 2-дезокси-3,5-ди-O-пара-толуоил-α,β-L-рибозу 7 (470 мг, 1,27 ммоль) растворяли в безводном метаноле (9 мл) и охлаждали примерно до 10°C. Добавляли порцию 1% раствора HCl в метаноле (1 мл) и реакционную смесь выдерживали при 10-15°C в течение 1,5 часа. После указанного периода времени анализ ВЭЖХ показал наличие непрореагировавшего исходного вещества, поэтому добавляли дополнительную порцию 1% HCl в метаноле (1 мл) и перемешивание продолжали при комнатной температуре еще в течение 1,5 часа. Анализ ВЭЖХ показал, что реакция была близка к завершению, и реакционную смесь концентрировали в вакууме при 30°C и выпаривали совместно с TBME (10 мл). Остаток растворяли в TBME (10 мл) и наблюдали суспендированное белое твердое вещество. Добавляли этилацетат (15 мл), чтобы растворить суспензию, и раствор сушили сульфатом натрия (2 г), фильтровали и концентрировали в вакууме, получая метил-2-дезокси-3,5-ди-O-пара-толуоил-α,β-L-рибозид 7-OMe (480 мг, грубый выход 98%) в виде коричневого масла.
δН (CDCl3, 400 МГц, соотношение аномеров составляет 1:1): 2,2-2,6 (м, 2×СН
3Аr и H-2/H-2' для α- и β-аномеров), 3,36 (с, 3Н, ОСН3 для 3,42 (с, 3Н, ОСН3 для 4,4-4,6 (м, H-4 и H-5/H-5' для α- и β-аномеров), 5,19 (д, 1H, H-1 для 5,21 (дд, 1H, H-1 для 5,41 (м, 1H, H-3 для 5,59 (м, 1H, H-3 для 7,18-8,02 (м, 8H, ароматический). δС (CDCl3, 100 МГц): 21,9, 39,5, 55,3, 55,4, 64,5, 65,3, 74,8, 75,6, 81,2, 82,1, 105,3, 105,8, 127,1, 127,2, 127,3, 127,4, 129,3, 129,3, 129,9, 130,0, 130,0, 143,9, 144,0, 144,1, 144,2, 166,3, 166,5, 166,6, 166,7.
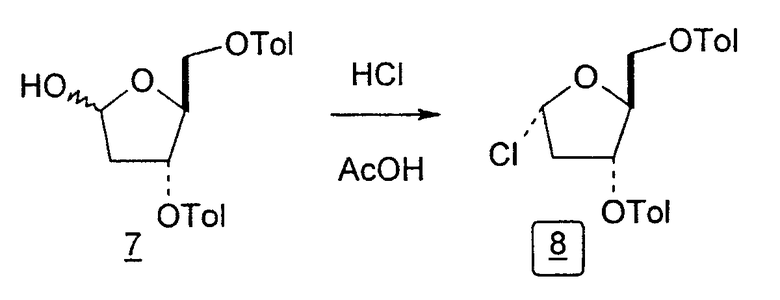
Метил-2-дезокси-3,5-ди-O-пара-толуоил-α,β-L-рибозид 7-OMe (480 мг, 1,25 ммоль) растворяли в TBME (3 мл) и уксусной кислоте (1 мл) и раствор охлаждали до 0°C в аргоне. Полученный раствор барботировали сухим газом HCl в течение 15 минут и реакционной смеси давали возможность перемешиваться при 0-5°C еще в течение 10 минут. Анализ ВЭЖХ показал наличие оставшегося исходного вещества даже после того, как реакция продолжалась еще в течение 1 часа и 10 минут. Белое твердое вещество, которые кристаллизовалось из реакционной смеси, собирали фильтрованием, промывали (TBME) и сушили в вакууме, получая 2-дезокси-3,5-ди-O-пара-толуоил-β-L-эритропентофуранозилхлорид 8 (228 мг, 47% за 3 стадии) в виде белого кристаллического твердого вещества. Выделенное соединение во всех отношениях было идентично соединению, указанному выше в случае способа 1.
Пример 23(h)
2'-дезокси-3',5'-ди-O-пара-толуоил-L-тимидин 9
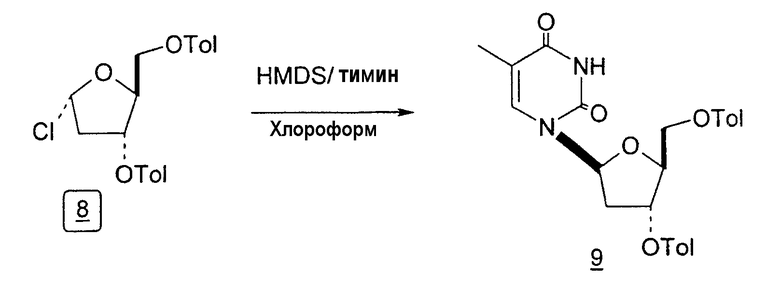
Смесь тимина (1,0 г, 7,92 ммоль), HMDS (1,66 г, 10,28 ммоль) и сульфата аммония (100 мг, 0,76 ммоль) нагревали примерно при 145°C в течение 2 часов, и в это время тимин растворялся. Еще через 4 часа при 145°C реакционную смесь концентрировали в вакууме при 60°C. Полученный таким образом силилированный тимин (7,92 моль) суспендировали в безводном хлороформе (15 мл) и охлаждали до 15°C. Порциями добавляли 2-дезокси-3,5-ди-O-пара-толуоил-β-L-эритропентофуранозилхлорид 8 (1,48 г, 3,8 ммоль) в течение 2-3 минут и реакционную смесь (желтый раствор) перемешивали при комнатной температуре в атмосфере аргона. Через 2 часа анализ ВЭЖХ показал, что исходное вещество отсутствовало. Реакционную смесь охлаждали примерно до 5°C и гасили 90% этанолом (0,25 мл). Происходило выпадение белого твердого вещества в осадок, и реакционную смесь перемешивали при комнатной температуре в течение 20 минут. Реакционную смесь фильтровали через целит (10 г) и промывали дихлорметаном (60 мл). Фильтрат промывали водой (2×25 мл) и оставляли стоять, чтобы эмульсия расслоилась. Затем органический слой промывали водным раствором бикарбоната натрия (2 г в 25 мл воды) и насыщенным раствором соли (10 г NaCl в 30 мл воды). Органический слой сушили сульфатом натрия (5 г) и фильтровали через целит (8 г). Фильтрат концентрировали в вакууме при 40°C и остаток суспендировали в гексане (20 мл), который перемешивали при комнатной температуре в течение 1,5 часа, чтобы получить однородную дисперсию. Смесь фильтровали, осадок на фильтре промывали гексаном (10 мл) и недолго сушили в вакууме, получая белое твердое вещество. Полученное твердое вещество перемешивали в этаноле (30 мл) при 60°C в течение 40 минут, концентрировали в вакууме (удаляли 15 мл) и оставшуюся взвесь охлаждали до комнатной температуры. Взвесь фильтровали, промывали холодным этанолом (10 мл) и TBME (5 мл) и сушили в вакууме при 55°C в течение ночи, получая 2'-дезокси-3',5'-ди-O-пара-толуоил-L-тимидин 9 (1,38 г, 76%) в виде белого твердого вещества. ВЭЖХ: отношение α:β=268:1.
δН (CDCl3, 400 МГц): 1,61 (3H, с, 5-Ме), 2,31 (1Н, м, H-2”), 2,42, 2,43 (2×3Н, 2×с, СН 3Аr), 2,71 (1Н, дд, J=4,8 Гц, J2',2"=14,3 Гц, H-2'), 4,52 (1Н, м, H-4'), 4,64 (1Н, дд, J4',5"=3,3 Гц, J5',5"=12,5 Гц, H-5”), 4,77 (1Н, дд, J4',5'=2,6 Гц, J5',5”=12,5 Гц, H-5'), 5,64 (1Н, a-д, J=6,6 Гц, H-3'), 6,47 (1Н, дд, J1,2=5,5 Гц, J1,2=8,8 Гц, H-1'), 7,2-7,3 (5H, м, H-6 и ArH), 7,91-7,96 (4H, м, Ar-H), 8,6 (1Н, ушир.с, NH); δC (CDCl3, 100 МГц): 12,3, 21,5, 21,9, 38,2, 64,4, 75,1, 83,0, 85,0, 111,9, 126,3, 126,7, 129,5, 129,7, 129,7, 130,0, 134,6, 144,8, 150,6, 163,8, 166,2, 166,3.
Пример 23(i)
2'-дезокси-L-тимидин 10
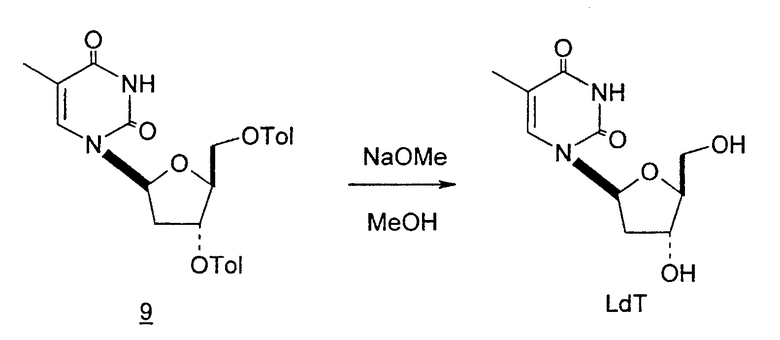
Перемешиваемую суспензию 2'-дезокси-3',5'-ди-O-пара-толуоил-L-тимидина 9 (500 мг, 1,05 ммоль) в безводном метаноле (6 мл) охлаждали до 5°C в аргоне. Одной порцией добавляли метоксид натрия (64 мг, 1,19 ммоль). Через 5 минут охлаждающую баню убирали и реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Реакционную смесь нагревали до 45-50°C в течение 1 часа, и она оставалась в виде почти нерастворимой суспензии. Добавляли безводный тетрагидрофуран (4 мл) и получали прозрачный раствор. Температуру поддерживали при 45-50°C еще в течение 30 минут, и в этой точке анализ ВЭЖХ показал наличие оставшегося исходного вещества. Поэтому еще через 30 минут (всего 2,5 часа) добавляли дополнительную порцию метоксида натрия (31 мг, 0,57 ммоль) и реакционную смесь выдерживали при 45-50°C. Через 2,5 часа (всего 5 часов) анализ ВЭЖХ показал, что реакция не завершилась, поэтому еще через 1 час (всего 6 часов) добавляли дополнительную порцию метоксида натрия (25 мг, 0,46 ммоль) и реакционную смесь перемешивали в течение ночи при 40-45°C. После указанного периода времени ВЭЖХ и анализ ТСХ (10% метанол в этилацетате) показали полное превращение исходного вещества (Rf 0,73) в продукт (Rf 0,15) [образец, приготовленный для ВЭЖХ и анализа ТСХ = аликвота со смолой Dowex-H+ смола, разбавляли метанол, фильтровали и анализировали]. Реакционную смесь охлаждали до комнатной температуры и добавляли ионообменную смолу DOWEX 50W × 2-200(H) (предварительно промытую метанолом (3×10 мл)). После перемешивания в течение 30 минут при комнатной температуре значение pH было равно 3, и реакционную смесь фильтровали, используя пористую стеклянную воронку, промывали метанолом (5 мл) и фильтрат концентрировали в вакууме при 45°C. Оставшийся метанол выпаривали совместно со смесью TBME и дихлорметана (1:1, 10 мл) и остаток растворяли в TBME (10 мл). Твердые вещества диспергировали при 40-45°C в течение 1 часа, охлаждали до комнатной температуры и собирали фильтрованием. Полученное в результате твердое вещество промывали TBME (5 мл) и сушили в вакууме, получая 2-дезокси-L-тимидин 10 (225 мг, 88%) в виде белого твердого вещества.
Обнаружено, что полученное соединение было идентично во всех отношениях аутентичному образцу 2-дезокси-L-тимидина 10.
Пример 24
2,2'-ангидро-1-(5-O-диметокситритил-β-D-арабинофуранозил)тимин (2) из 2,2'-ангидро-1-β-D-арабинофуранозил)тимина (1)
К предварительно охлажденной (0-5°C) смеси 2,2'-ангидро-1-β-D-арабинофуранозил)тимина 1 (2,40 г, 10,0 ммоль) и DMAP (111 мг, 0,9 ммоль) в безводном пиридине (15 мл) порциями добавляли 4,4'-диметокситритилхлорид (3,56 г, 10,5 ммоль) в течение периода времени, равного 3 минутам. Полученную в результате смесь перемешивали при 0-5°C в атмосфере аргона, и через 1,5 часа анализ ТСХ (пластинки диоксида кремния, метанол:дихлорметан 1:9) показал отсутствие оставшегося исходного вещества. Реакционную смесь концентрировали в вакууме при 45°C. Остаток собирали в дихлорметане (50 мл) и насыщенном растворе бикарбоната натрия (20 мл). После перемешивания при комнатной температуре в течение 10 минут слои разделяли, органический слой промывали дистиллированной водой (2×20 мл) и сушили безводным сульфатом натрия. Реакционную смесь концентрировали в вакууме при 50°C и остаток упаривали совместно с толуолом (2×10 мл). Полученный в результате грубый остаток растирали в дихлорметане (5 мл) и TBME (25 мл). После перемешивания при комнатной температуре в течение 1 часа твердое вещество собирали фильтрованием при пониженном давлении и промывали TBME (15 мл). Желтое твердое вещество сушили в вакууме, получая 2,2'-ангидро-1-(5-O-диметокситритил-β-D-арабинофуранозил)тимин 2 (5,3 г, выход 97,8%, 91% AUC (область под кривой) анализом ВЭЖХ).
δН (d6-ДМСO, 400 МГц): 1,78 (3H, с, Мe), 2,76 и 2,90 (2H, ABX, H-5' и H-5”), 3,72 (6Н, с, 2×OМe), 4,22-4,28 (2H, 2×м, H-3' и H-4'), 5,17 (1Н, д, J1',2'=5,9 Гц, H-2'), 5,93, (1Н, д, J3',OH=4,4 Гц, 3'-OH), 6,30 (1Н, д, J1',2'=5,9 Гц, Н-1'), 6,79-7,28 (13H, м, Ar-H), 7,84 (1Н, с, H-6).
Пример 25
2'-дезокси-5'-O-диметокситритил-β-D-тимидин (3) из 2,2'-ангидро-1-(5-O-диметокситритил-β-D-арабинофуранозил)тимин (2)
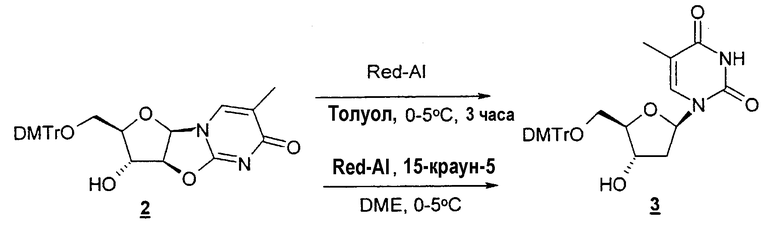
Пример 25 иллюстрирует сравнение между процентным выходом 5'-DMTrO-защищенного 2'-дезокситимидина (3), полученного способом 1, в котором используют Red-Al в толуоле, с выходом в % продукта (3), полученного способом 2, в котором используют Red-Al в комбинации с 15-краун-5-эфиром в DME. Способ 1 осуществляли, используя известные методики (например, патент США № 6090932), и получали продукт (3) с выходом 21%. Способ 2 осуществляли согласно способу, предлагаемому в настоящем изобретении, в котором используют Red-A1 в комбинации с 15-краун-5-эфиром в DME, который давал продукт (3) с выходом 35%.
Способ 1: Взаимодействие Red-Al с 2,2'-ангидро-1-(5-O-диметокситритил-β-D-арабинофуранозил)тимином 2 в толуоле
К предварительно охлажденному (0-5°C) раствору 2,2'-ангидро-1-(5-O-диметокситритил-β-D-арабинофуранозил)тимина 2 (1,08 г, 2,0 ммоль) в безводном толуоле (50 мл) по каплям добавляли Red-Al (65 мас.% в толуоле, 0,90 мл, 3,0 ммоль) в течение периода времени, составляющего 10 минут. Смесь выдерживали при перемешивании при 0-5°C в атмосфере аргона. Реакцию контролировали посредством ТСХ (диоксид кремния, 5:95 метанол в дихлорметане) и анализа ВЭЖХ. После перемешивания в течение 2 часов при 0-5°C к реакционной смеси добавляли дополнительное количество Red-Al (0,5 экв., 65 мас.% в толуоле, 0,30 мл, 1,0 ммоль). После перемешивания в течение 45 минут брали аликвоту из реакционной смеси в ТГФ, чистом для ВЭЖХ (примерно 1 мл), гасили, добавляя по каплям дистиллированную воду, и инъецировали в прибор для ВЭЖХ. Результат показал соотношение 1:1 продукта (37,4% AUC) к исходному веществу (36% AUC). Реакцию гасили добавлением насыщенного раствора соли (30 мл) при 5°C. После перемешивания еще в течение 30 минут смесь фильтровали через подушку целита и промывали этилацетатом (60 мл). Фильтрат распределяли в делительной воронке. Органический слой промывали насыщенным водным раствором NH4Cl (30 мл) и насыщенным раствором соли (2×25 мл) и сушили, используя безводный сульфат натрия. Реакционную смесь концентрировали в вакууме при 40°C. Грубый остаток (1,01 г, желтое пенообразное твердое вещество) очищали хроматографией на колонке (силикагель, 5% метанол в дихлорметане), получая 2'-дезокси-5'-O-диметокситритил-β-D-тимидин 3 (0,23 г, выход 21%) в виде светло-желтого твердого вещества.
δH (d6-ДМСO, 400 МГц): 1,43 (3H, с, Мe), 2,14 и 2,22 (2×1H, 2×м, H-2' и H-2”), 3,18 (2H, м, H-5” и H-5'), 3,72 (с, 6H, 2×OМe), 3,87 (1H, м, H-4'), 4,30 (1H, м, H-3'), 5,32 (1H, д, J3',OH=4,4 Гц, 3'-OH), 6,19 (1H, м, Н-1'), 6,85-7,39 (13H, м, DМTr), 7,50 (1H, с, H-6), 11,38 (1H, с, NH); МС (ESI+, М+1=545,3, М+Na+=567,3).
Способ 2: Взаимодействие Red-Al с 2,2'-ангидро-1-(5-O-диметокситритил-β-D-арабинофуранозил)тимином 2 в DME в присутствии 15-краун-5
К предварительно охлажденному (0-5°C) раствору 2,2'-ангидро-1-(5-O-диметокситритил-β-D-арабинофуранозил)тимина 2 (120 мг, 0,22 ммоль) и 15-краун-5 (65 мкл, 0,33 ммоль) в безводном DME (5 мл) по каплям добавляли Red-Al (65 мас.% в толуоле, 0,10 мл, 0,33 ммоль) в течение периода времени, составляющего 4 минутам. Реакционную смесь выдерживали при 0-5°C в атмосфере аргона. Реакцию контролировали анализом ВЭЖХ. После перемешивания в течение 3,5 часа к реакционной смеси добавляли дополнительное количество Red-Al (0,5 экв., 65 мас.% в толуоле, 0,030 мл, 0,10 ммоль). После перемешивания в течение 30 минут результаты анализа ВЭЖХ показали, что соотношение продукт:исходное вещество возрастало с 1,7:1 до 2,4:1. Реакционной смеси давали возможность нагреться до комнатной температуры и выдерживали при комнатной температуре в течение 16 часов. Анализ ВЭЖХ показал, что отношение продукта к исходному веществу немного возросло до 2,8:1. Затем реакционную смесь охлаждали до 0-5°C и к реакционной смеси добавляли еще 0,5 эквивалента Red-Al (65 мас.% в толуоле, 0,030 мл, 0,10 ммоль). После перемешивания при 0-5°C в течение 1 часа результаты ВЭЖХ показали, что отношение продукта к исходному веществу возрастало до 4,0:1 (62,7% AUC продукта по сравнению с 15,8% AUC исходного вещества). Дальнейшее добавление 15-краун-5 и Red-A1 не приводило к улучшению образования продукта. К реакционной смеси добавляли небольшое количество ацетона (примерно 0,1 мл). После перемешивания в течение 10 минут реакционную смесь концентрировали в вакууме при 40°C и остаток упаривали совместно с изопропилацетатом (10 мл). Остаток распределяли между изопропилацетатом (20 мл) и дистиллированной водой (5 мл). Органический слой промывали насыщенным водным раствором NH4Cl (5 мл) и насыщенным раствором соли (5 мл) и сушили безводным сульфатом натрия. После концентрирования в вакууме при 40°C грубый остаток (120 мг, желтое пенообразное твердое вещество) очищали хроматографией на колонке (силикагель, 5% метанол в дихлорметане), получая 2'-дезокси-5'-O-диметокситритил-β-D-тимидин 3 в виде светло-желтого твердого вещества (45 мг, выход 35%). Спектр 1H-ЯМР соответствует структуре, полученной согласно способу 1.
Пример 26
2,2'-ангидро-1-(5-O-тритил-β-D-арабинофуранозил)тимин (4) (из 2,2'-ангидро-1-β-D-арабинофуранозил)тимина (1)
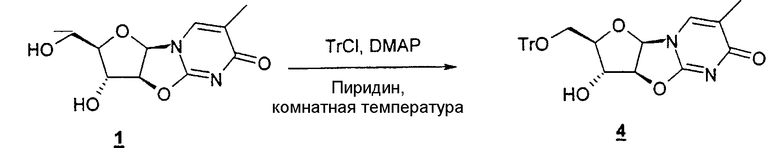
2,2'-ангидро-1-β-D-арабинофуранозил)тимин 1 (500 мг, 2,08 ммоль) растворяли в безводном пиридине (5 мл) и к перемешиваемому раствору добавляли DMAP (12,5 мг, 0,1 ммоль). Порциями добавляли тритилхлорид (1,28 г, 2,29 ммоль) в течение 3 минут при комнатной температуре. Полученную в результате реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем при 40°C в течение ночи в атмосфере аргона. После указанного периода времени анализ ТСХ (пластина диоксида кремния, метанол:дихлорметан 2:8) показал, что исходного вещества не оставалось (Rf 0,3) и образовался новый продукт (пластина диоксида кремния, метанол:дихлорметан 1:9, Rf 0,17). Реакционную смесь охлаждали до 0°C, используя баню со льдом, порциями медленно добавляли насыщенный раствор NaHCO3 (15 мл), и белое твердое вещество выпадало из раствора в осадок. Полученную в результате суспензию перемешивали при комнатной температуре в течение 30 минут, белое твердое вещество собирали фильтрованием и затем промывали дистиллированной водой (25 мл). Неочищенное твердое вещество (3 г) собирали в TBME (18 мл) и перемешивали при комнатной температуре в течение 30 минут. Белое твердое вещество собирали фильтрованием, промывали TBME (8 мл) и затем сушили в вакууме, получая 2,2'-ангидро-1-(5-O-тритил-β-D-арабинофуранозил)тимин 4 (844 мг, 84%) в виде белого твердого вещества.
δH (d6-ДМСO, 400 МГц): 1,77 (3H, с, Мe), 2,73 (1H, дд, J4',5”=7,4 Гц, J5',5”=10,2 Гц, H-5”), 2,92 (1H, дд, J4',5'=4,3 Гц, J5',5”=10,2 Гц, H-5'), 4,24-4,28 (2×1H, 2×м, H-3' и H-4'), 5,16 (1H, д, J1',2'=5,86 Гц, H-2'), 5,94 (1H, д, J3',ОH=4,23 Гц, 3'-OH), 6,29 (1H, д, J1',2'=5,45 Гц, Н-1'), 7,2-7,27 (15H, м, Tr), 7,83 (1H, ушир.с, H-6).
Пример 27
2'-дезокси-5'-O-тритил-β-D-тимидин (5) из 2,2'-ангидро-1-(5-O-тритил-β-D-арабинофуранозил)тимина (4)
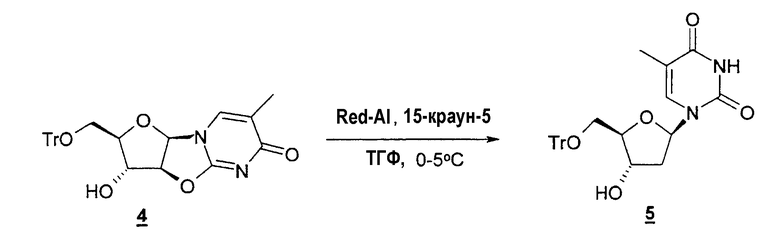
К предварительно охлажденной (0-5°C) смеси 2,2'-ангидро-1-(5-O-тритил-β-D-арабинофуранозил)тимина 4 (241 мг, 0,5 ммоль) и 15-краун-5 (0,15 мл, 0,75 ммоль) в безводном ТГФ (10 мл) по каплям добавляли Red-Al (65 мас.% в толуоле, 0,23 мл, 0,75 ммоль) в течение периода времени, составляющего 5 минут. Смесь поддерживали при 0-5°C в атмосфере аргона. Реакцию контролировали анализом ТСХ (диоксид кремния, 5:95 метанол в дихлорметане) и ВЭЖХ. После перемешивания при 0-5°C в течение 1 часа из реакционной смеси брали аликвоту в ТГФ, чистом для ВЭЖХ (примерно 1 мл), гасили добавлением по каплям дистиллированной воды и инъецировали в прибор для ВЭЖХ. Результаты показали, что оставалось только 8% AUC (площадь под кривой) исходного вещества и присутствовало 70,8% продукта. Реакцию гасили добавлением насыщенного водного раствора NH4Cl (5 мл) при 5°C и перемешивали в течение 15 минут. После указанного периода времени слои разделяли и водный слой затем экстрагировали изопропилацетатом (10 мл). Органические слои объединяли, промывали насыщенным раствором соли (5 мл) и сушили, используя безводный сульфат натрия. После концентрирования в вакууме при 40°C грубый остаток (287 мг, белое пенообразное твердое вещество) очищали хроматографией на колонке (силикагель, 5% метанол в дихлорметане), получая 2'-дезокси-5'-O-тритил-β-D-тимидин 5 (106 мг, выход 44%) в виде белого твердого вещества.
δН (d6-ДМСO, 400 МГц): 1,45 (3H, с, Мe), 2,15 (1H, м, H-2”), 2,22 (1H, м, H-2'), 3,15 (1H, дд, J4',5”=2,6 Гц, J5',5”=10,5 Гц, H-5”), 3,22 (1H, дд, J4',5'=4,8 Гц, J5',5”=10,5 Гц, H-5'), 3,87 (1H, м, H-4'), 4,31 (1H, м, H-3'), 5,33 (1H, д, J3,ОН=4,83 Гц, 3'-OH), 6,19 (1H, a-т, J=6,6 Гц, J=7,0 Гц, Н-1'), 7,25-7,39 (15H, м, Tr), 7,49 (1H, ушир.с, H-6), 11,35 (1H, с, NH).
δС (d6-ДМСO, 100 МГц): 11,7, 54,9, 70,4, 83,7, 85,4, 86,4, 109,6, 127,2, 128,0, 128,3, 135,7, 143,5, 150,4, 163,7.
Удаляли защиту полученного соединения (5), используя уксусную кислоту примерно при 50°C, получая 2'-дезокси-D-тимидин в качестве конечного продукта, который был идентичен во вех отношениях аутентичному образцу известного 2'-дезокси-D-тимидина.
Пример 28
Образование 2'-дезокси-D-тимидина (4) из 2,2'-ангидро-1-(β-D-арабинофуранозил)тимина (1)
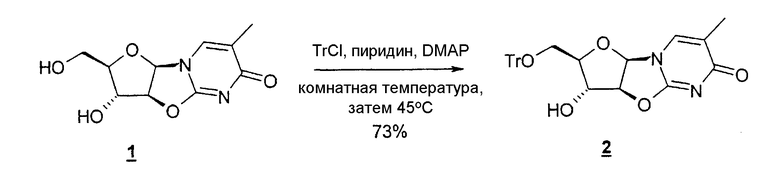
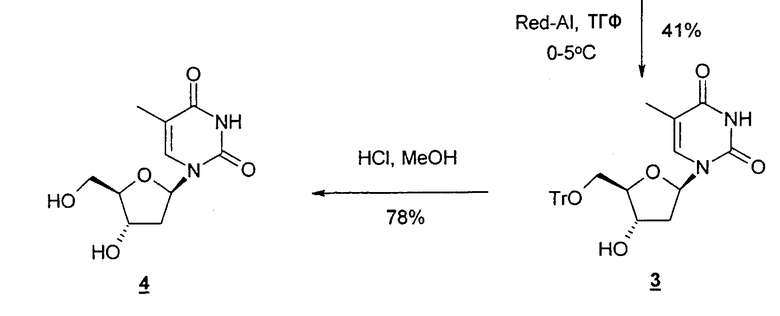
Пример 28(a)
2,2'-ангидро-1-(5-O-тритил-β-D-арабинофуранозил)тимин (2) из 2,2'-ангидро-1-(β-D-арабинофуранозил)тимина (1)

2,2'-ангидро-1-(β-D-арабинофуранозил)тимин (1) (10,0 г, 41,62 ммоль) суспендировали в пиридине (100 мл) и DMAP (254 мг, 2,08 ммоль) и порциями добавляли тритилхлорид (25,48 г, 91,56 ммоль) при комнатной температуре. Реакционную смесь выдерживали при комнатной температуре в течение примерно 1 часа и затем нагревали примерно до 45°C (внутренняя температура). Примерно через 5 часов анализ ТСХ (10% метанол в дихлорметане, визуализация с использованием 1% KMnO4 и УФ) показал наличие значительного количества исходного вещества (Rf 0,15) и образование продукта (Rf 0,43). Поэтому реакционную смесь выдерживали примерно при 45°C еще в течение примерно 15 часов (в течение ночи). После указанного периода времени анализ ТСХ показал, что исходного вещества не оставалось (Rf 0,15). Реакционную смесь охлаждали примерно до 0°C и медленно в течение 15 минут добавляли насыщенный водный раствор NaHCO3 (320 мл) (изменения внутренней температуры не происходило). Из раствора сразу же выпадало в осадок белое твердое вещество, и белую суспензию перемешивали в течение примерно 30 минут при комнатной температуре. Твердое вещество выделяли фильтрованием через воронку Бюхнера и промывали водой (3×100 мл). Оставшееся твердое вещество собирали в дихлорметане (150 мл) и перемешивали в течение примерно 30 минут при комнатной температуре. Остаток выделяли фильтрованием через воронку Бюхнера, промывали дихлорметаном (20 мл) и сушили в вакууме в течение ночи, получая 2,2'-ангидро-1-(5-O-тритил-β-D-арабинофуранозил)тимин (2) (14,66 г, 73%) в виде белого твердого вещества.
δН (d6-ДМСO, 400 МГц): 1,77 (3H, с, 5-Мe), 2,76 (1Н, дд, J5',5”=10,3 Гц, J4',5”=7,8 Гц, H-5”), 2,94 (1Н, дд, J5',5”=10,3 Гц, J4',5'=3,9 Гц, H-5'), 4,26 (1Н, м, H-4'), 4,29 (1Н, м, H-3'), 5,17 (1Н, a-д, J=5,9 Гц, H-2'), 5,98 (1Н, ушир.с, 3-OH), 6,30 (1Н, д, J1',2'=5,37 Гц, Н-1'), 7,2-7,27 (15H, м, Tr), 7,83 (1Н, с, H-6); δC (d6-ДМСO, 125 МГц): 13,5 (5-Мe), 63,2 (C-5'), 74,8 (C-3'), 85,9 (TrC), 86,7 (C-4'), 88,1 (C-2'), 89,9 (С-1'), 116,9 (С-6), 127,0, 127,7, 127,9, 128,0 (Tr), 132,1 (С-5), 143,3 (Tr), 158,8 (С-2), 171,3 (C-4).
Пример 28(b)
2'-дезокси-5'-O-тритил-β-D-тимидин 3 из 2,2'-ангидро-1-(5-O-тритил-β-D-арабинофуранозил)тимина 2
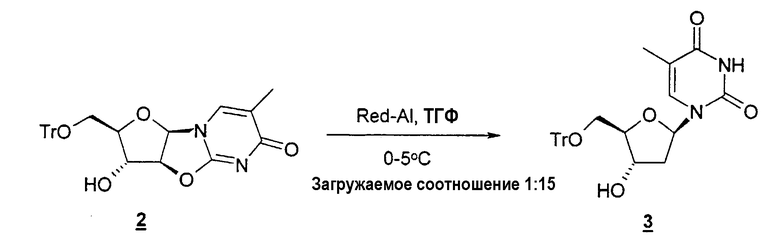
2,2'-ангидро-1-(5-O-тритил-β-D-арабинофуранозил)тимин (2) (4,30 г, 8,91 ммоль) суспендировали в безводном тетрагидрофуране (43 мл) и охлаждали примерно до 0-5°C, используя баню со льдом. В отдельной колбе, погруженной в баню со льдом, примерно при 0-5°C, 65 мас.% разбавляли раствор Red-Al в толуоле (3,26 мл, 10,69 ммоль) добавлением к безводному тетрагидрофурану (21,5 мл). Полученный разбавленный раствор Red-Al охлаждали примерно до 0-5°C и добавляли по каплям через шприц к суспензии 2,2'-ангидро-1-(5-O-тритил-β-D-арабинофуранозил)тимина (2) в тетрагидрофуране. Скорость добавления по каплям раствора Red-Al является важной для реакции, и добавление завершали примерно за 1 час. Полученный в результате прозрачный раствор выдерживали примерно при 0-5°C в течение 1 часа, после указанного периода времени анализ ТСХ (10% метанол в дихлорметане) показал наличие исходного вещества (Rf 0,34), требуемого продукта (Rf 0,47) и примесей (Rf 0,42 и 0,26). Анализ ВЭЖХ показал наличие исходного вещества (11,35 мин, 36,5% AUC), продукта (12,60 мин, 24%) и небольшое количество основной примеси (11,7 мин, 2,9%). Спустя всего примерно 2 часа при температуре около 0-5°C, по каплям через шприц в течение периода времени, составляющего примерно 20 минут, добавляли дополнительную порцию «неразбавленного» 65 мас.% раствора Red-Al в толуоле (1,63 мл, 5,35 ммоль) к реакционной смеси, которую выдерживали примерно при 0-5°C. Спустя примерно еще 1 час анализ ТСХ и ВЭЖХ показали наличие исходного вещества (11,35 мин, 3,2%). По каплям добавляли следующую порцию 65 мас.% раствора Red-Al в толуоле (0,26 мл, 0,85 ммоль) и реакционную смесь выдерживали примерно при 0-5°C еще в течение 45-минутного периода. После указанного периода времени анализ ТСХ показал наличие только следового количества оставшегося исходного вещества. Реакцию гасили добавлением насыщенного раствора NH4Cl (40 мл) и слой тетрагидрофурана декантировали. Водный слой экстрагировали изопропилацетатом (50 мл) и полученную в результате эмульсию подвергали расслоению медленным добавлением 5 н. раствора HCl (15 мл). Органический слой отделяли, объединяли со слоем тетрагидрофурана и промывали насыщенным раствором NH4Cl (30 мл) и затем насыщенным раствором соли (30 мл). В данной точке значение pH слоя насыщенного раствора соли составляло от 6,5 до 7, и органический слой сушили, используя Na2SO4, фильтровали и концентрировали в вакууме, получая пенообразное твердое вещество (4,4 г). Неочищенный остаток упаривали совместно с толуолом (30 мл), концентрировали в вакууме и полученный в результате остаток собирали в толуоле (25 мл), нагревая примерно до 45°C. Смесь охлаждали до комнатной температуры и перемешивали при указанной температуре вплоть до того, как начинало выпадать в осадок белое твердое вещество. По каплям добавляли воду (8,5 мл) и полученную в результате смесь перемешивали при комнатной температуре в течение примерно 3 часов. Твердое вещество выделяли фильтрованием и осадок на фильтре промывали водой (5 мл) и толуолом (3 мл). Твердое вещество сушили примерно при 45°C в условиях высокого вакуума в течение примерно 1 часа и затем при комнатной температуре в вакууме в течение ночи, получая 2'-дезокси-5'-O-тритил-β-D-тимидин (3) (1,77 г, 41%) в виде белого твердого вещества.
δН (d6-ДМСO, 400 МГц): 1,45 (3H, с, Мe), 2,15 (1H, м, H-2”), 2,22 (1H, м, H-2'), 3,15 (1H, дд, J4',5”=2,6 Гц, J5',5”=10,5 Гц, H-5”), 3,22 (1H, дд, J4',5'=4,8 Гц, J5',5”=10,5 Гц, H-5'), 3,87 (1H, м, H-4'), 4,31 (1H, м, H-3'), 5,33 (1H, д, J3',ОН=4,83 Гц, 3'-OH), 6,19 (1H, a-т, J=6,6 Гц, J=7,0 Гц, Н-1'), 7,25-7,39 (15H, м, Tr), 7,49 (1H, ушир.с, H-6), 11,35 (1H, с, NH). δС (d6-ДМСO, 100 МГц): 11,7, 54,9, 70,4, 83,7, 85,4, 86,4, 109,6, 127,2, 128,0, 128,3, 135,7, 143,5, 150,4, 163,7.
Пример 28(c)
2'-дезокси-D-тимидин (4) из 2'-дезокси-5'-O-тритил-β-D-тимидина (3)

2'-дезокси-5'-O-тритил-β-D-тимидин (3) (1,215 г, 2,5 ммоль) суспендировали в метаноле (9,6 мл) и реакционную смесь нагревали примерно до 45°C на водяной бане вплоть до растворения. Затем колбу охлаждали до комнатной температуры, к смеси добавляли концентрированную HCl (200 мкл, 2,5 ммоль) и перемешивали при комнатной температуре. Спустя примерно 25 минут белое твердое вещество начинало выпадать в осадок из раствора. Примерно через 1 час анализ ТСХ (10% метанол в дихлорметане, визуализация ванилином и УФ) показал, что исходное вещество не оставалось (Rf 0,53) и образовывался основной продукт (Rf 0,21). К реакционной смеси добавляли порцию н-гептана (10 мл) и перемешивали при комнатной температуре в течение примерно 15 минут. Белое твердое вещество выделяли фильтрованием (405 мг твердого вещества). Фильтрат делили на два слоя, метанольный слой экстрагировали н-гептаном (10 мл) и затем концентрировали в вакууме до объема 2 мл. Остаток объединяли с 405 мг белого твердого вещества, суспендировали в TBME (10 мл) и перемешивали при комнатной температуре в течение примерно 1 часа. Белое твердое вещество выделяли фильтрованием, промывали TBME (3 мл) и сушили в вакууме в печи, получая 2'-дезокси-D-тимидин (4) (471 мг, 78%). Полученный продукт был идентичен при анализе 1H-ЯМР и ВЭЖХ аутентичному образцу.
Изобретение описано со ссылкой на различные конкретные и предпочтительные варианты и методики. Однако следует понимать, что многие изменения и модификации будут очевидны для специалистов в данной области на основание приведенного выше подробного описания изобретения и могут быть осуществлены, не отходя от сути и не выходя за рамки объема изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| β-L-2'-ДЕЗОКСИНУКЛЕОЗИДЫ ДЛЯ ЛЕЧЕНИЯ ГЕПАТИТА В | 1999 |
|
RU2300381C2 |
| БЕТА-L-2' ДЕЗОКСИНУКЛЕОЗИДЫ ДЛЯ ЛЕЧЕНИЯ ГЕПАТИТА В | 1999 |
|
RU2424016C2 |
| СРЕДСТВО ДЛЯ ИНГИБИРОВАНИЯ ФЕРМЕНТА ПОЛИ(АДФ-РИБОЗО)ПОЛИМЕРАЗЫ-1 ЧЕЛОВЕКА | 2009 |
|
RU2411948C1 |
| СПОСОБ ПОЛУЧЕНИЯ 3'-АЗИДО-2',3'-ДИДЕЗОКСИТИМИДИНА | 1994 |
|
RU2102399C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ 1-О-АЦИЛ-2-ДЕЗОКСИ-2-ФТОР-4-ТИО-β-D-АРАБИНОФУРАНОЗ | 2010 |
|
RU2559364C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКИЛЗАМЕЩЕННЫХ 2-ДЕЗОКСИ-2-ФТОР-D-РИБОФУРАНОЗИЛ-ПИРИМИДИНОВ И ПУРИНОВ И ИХ ПРОИЗВОДНЫХ | 2005 |
|
RU2407747C2 |
| МОДИФИЦИРОВАННЫЕ НУКЛЕОЗИД-5'-ТРИФОСФАТЫ КАК АНТИВИРУСНЫЕ АГЕНТЫ | 1996 |
|
RU2183213C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2'-ДЕЗОКСИКСИЛОТИМИДИНА, ПРОИЗВОДНЫЕ D-КСИЛОФУРАНОЗЫ, ПРОИЗВОДНЫЕ КСИЛОТИМИДИНА | 1994 |
|
RU2108339C1 |
| 3' И/ИЛИ 2'-АМИНО- ИЛИ ТИОЛМОДИФИЦИРОВАННЫЕ НУКЛЕОЗИДЫ, НУКЛЕОТИДЫ ИЛИ ОЛИГОНУКЛЕОТИДЫ, СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ | 1991 |
|
RU2073682C1 |
| L-НУКЛЕОЗИДЫ, ОБЛАДАЮЩИЕ АНТИ-HBV ИЛИ АНТИ-EBV АКТИВНОСТЬЮ, СПОСОБ ИНГИБИРОВАНИЯ HBV ИЛИ EBV ИНФЕКЦИИ | 1995 |
|
RU2171809C2 |
Изобретение относится к способу получения 2'-дезокси-β-L-тимидина, включающему взаимодействие 5'-O-тритил- или 5'-O-диметокситритил-защищенного 2,2'-ангидро-1-β-L-арабинофуранозилтимина с восстановителем RedAl и комплексообразующим агентом 15-краун-5-эфиром в полярном растворителе 1,2-диметоксиэтане (DME) или тетрагидрофуране (ТГФ) с получением 5'-O-тритил- или 5'-O-диметокситритил-защищенного 2'-дезокси-(3-L-тимидина, подвергающегося при необходимости удалению защиты. Изобретение относится также к способу получения 2'-дезокси-(3-L-тимидина, включающему взаимодействие L-арабинозы с цианамидом с последующим взаимодействием промежуточного продукта - L-арабинофуранозиламинооксазолина - с циклизующим или конденсирующим агентом с образованием 2,2'-ангидро-1-(3-L-арабинофуранозилтимина; взаимодействие последнего с восстановителем RedAl и комплексообразующим агентом 15-краун-5-эфиром в полярном растворителе 1,2-диметоксиэтане (DME) или тетрагидрофуране (ТГФ) с получением 2'-дезокси-β-L-тимидина, причем L-арабинофуранозиламинооксазолин может быть защищен тритилом или диметокситритилом в положении 5' до или после взаимодействия с циклизующим или конденсирующим агентом; и удаление защиты необязательно защищенного 2'-дезокси-β-L-тимидина, если это необходимо или желательно. Применение в данных способах такого восстановителя, как Red-Al, и такого комплексообразующего агента, как 15-краун-5-эфир, вызывает реакцию внутримолекулярного замещения и образование требуемого нуклеозидного продукта с хорошими выходами. Соединение, полученное способами по настоящему изобретения, имеет важное значение в качестве противовирусных и/или антинеопластических средств. 2 н. и 11 з.п. ф-лы, 29 ил.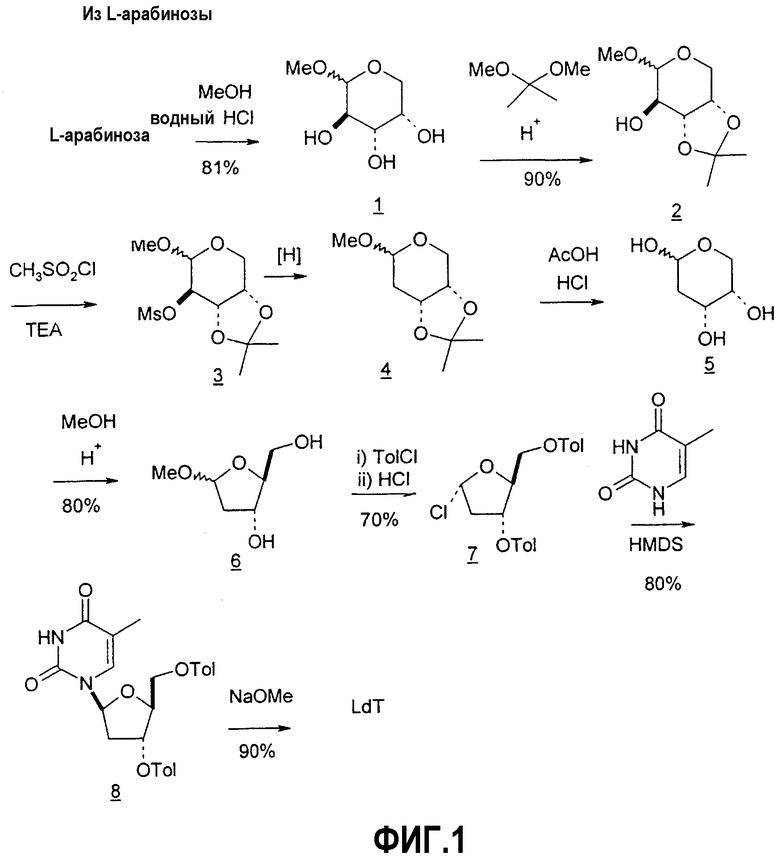
1. Способ получения 2'-дезокси-(β-L-тимидина, включающий
a) получение 5'-O-тритил- или 5'-O-диметокситритил - защищенного 2,2'-ангидро-1-β-L-арабинофуранозилтимина;
b) взаимодействие 5'-O-тритил- или 5'-O-диметокситритил - защищенного 2,2'-ангидро-1-β-L-арабинофуранозилтимина со стадии (а) с восстановителем RedAl и комплексообразующим агентом 15-краун-5-эфиром в полярном растворителе 1,2-диметоксиэтане (DME) или тетрагидрофуране (ТГФ) с получением 5'-O-тритил- или 5'-O-диметокситритил - защищенного 2'-дезокси-β-L-тимидина; и
c) удаление защиты продукта со стадии (b), если это необходимо или желательно.
2. Способ по п.1, в котором на стадии (с) удаление защиты осуществляют посредством добавления кислоты или кислой смолы при температуре примерно 50°С.
3. Способ по п.1, в котором на стадии (b) температура реакции составляет примерно 0-5°С.
4. Способ по п.1, в котором указанный 5'-O-тритил- или 5'-O-диметокситритил - защищенный 2,2'-ангидро-1-β-L-арабинофуранозилтимин получают с помощью следующих стадий:
a) защиты L-рибофуранозы посредством взаимодействия с диметокситритильной или тритильной защитной группой;
b) конденсации продукта со стадии (а) с тимином с образованием нуклеозида и
с) взаимодействия нуклеозида со стадии (b) с конденсирующим агентом с образованием 5'-O-тритил- или 5'-O-диметокситритил - защищенного 2,2'-ангидро-1-β-L-арабинофуранозилтимина.
5. Способ по п.4, в котором на стадии (b) конденсацию осуществляют в присутствии растворителя и необязательно катализатора.
6. Способ по п.4, в котором на стадии (с) конденсирующим агентом является диалкил- или диарилкарбонат в присутствии основания и органического растворителя.
7. Способ по п.6, в котором конденсирующим агентом является PhOCOOPh/NaHCO3 и органическим растворителем является ДМФА.
8. Способ по п.4, в котором на стадии (с) взаимодействие происходит при повышенных температурах.
9. Способ по п.8, в котором температура составляет примерно 140-150°С.
10. Способ получения 2'-дезокси-β-L-тимидина, включающий
a) взаимодействие L-арабинозы с цианамидом с образованием L-арабинофуранозиламинооксазолина;
b) взаимодействие продукта со стадии (а) с циклизующим или конденсирующим агентом с образованием 2,2'-ангидро-1-β-L-арабинофуранозилтимина;
c) взаимодействие 2,2'-ангидро-1-β-L-арабинофуранозилтимина с восстановителем RedAl и комплексообразующим агентом 15-краун-5-эфиром в полярном растворителе 1,2-диметоксиэтане (DME) или тетрагидрофуране (ТГФ) с получением 2'-дезокси-β-L-тимидина, причем L-арабинофуранозиламинооксазолин может быть защищен тритилом или диметокситритилом в положении 5' до или после взаимодействия с циклизующим или конденсирующим агентом; и
d) удаление защиты необязательно защищенного 2'-дезокси-β-L-тимидина, если это необходимо или желательно.
11. Способ по п.10, в котором на стадии (d) удаление защиты осуществляют посредством добавления кислоты или кислой смолы при температуре примерно 50°С.
12. Способ по п.10, в котором на стадии (b) циклизующий или конденсирующий агент выбран из группы, состоящей из:
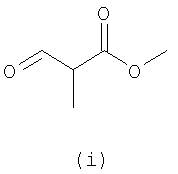 ,
,  ,
, 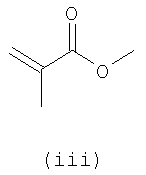
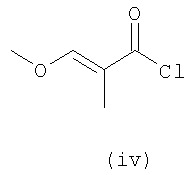 ,
, 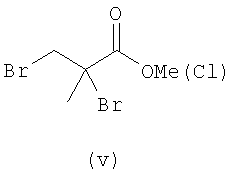 ,
, 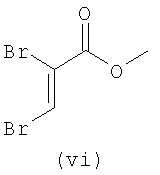 ,
,
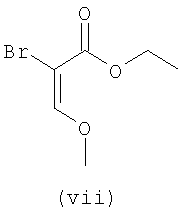 ,
, 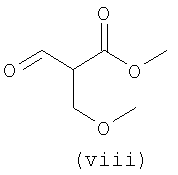 и
и 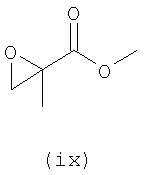
13. Способ по п.10, в котором на стадии (с) температура реакции составляет примерно 0-5°С.
| US 6090932 А, 18.07.2000 | |||
| Химическая энциклопедия | |||
| - М.: Советская энциклопедия, 1990, т.2, с.984-987 | |||
| WO 00/09531, 24.02.2000 | |||
| СПОСОБ ПОЛУЧЕНИЯ 2'-ДЕЗОКСИКСИЛОТИМИДИНА, ПРОИЗВОДНЫЕ D-КСИЛОФУРАНОЗЫ, ПРОИЗВОДНЫЕ КСИЛОТИМИДИНА | 1994 |
|
RU2108339C1 |

