Изобретение относится к ряду новых модифицированных олигодезоксирибонуклеотидов, которые обладают превосходной противовирусной активностью, а также представляет способы и композиции, основанные на использовании этих олигодезоксирибонуклеотидов, для лечения и профилактики вирусных инфекций и для ингибирования пролиферации неопластических (опухолевых) клеток. Изобретение представляет также способы получения этих соединений. Соединения настоящего изобретения особенно эффективны против вируса иммунодефицита человека (ВИЧ), который в настоящее время считается обычно причиной синдрома приобретенного иммунодефицита (AIDS).
Известно, что олигодезоксирибонуклеотиды (антисмысловые олигодезоксирибонуклеотиды), имеющие последовательность, комплементарную к гену, ингибируют функциональную экспрессию данного гена. Сообщалось также, что антисмысловые олигодезоксирибонуклеотиды, комплементарные к вирусному гену или онкогену, могут ингибировать репликацию вируса или размножение клетки путем ингибирования функции соответствующих генов (P.C. Zamecnik, M. L. Stephenson, Proc. Natl. Acad. Sci. США, 75 (I), 280 (1978) и P.C. Zamecnik. J. Goodchild, Y. Taguchi, Serin Proc. Natl Acad Sci. США, 83, (6) 4143 (1986).
Ранее считалось, что для того, чтобы антисмысловой олигодезоксирибонуклеотид проявлял желаемую активность, этот олигодезоксирибонуклеотид должен быть способен образовывать стабильный гибрид с целевой РНК или ДНК in vivo, и что соответственно он должен иметь длину цепи из 15 или более нуклеотидов. Обычно, однако, трудно синтезировать такие олигодезоксирибонуклеотиды с выходами и чистотой, которые дают возможность использовать их на практике. Кроме того, антисмысловые олигодезоксирибонуклеотиды не имеют достаточной активности для ингибирования репликации вируса или для ингибирования размножения клеток, чтобы дать возможность использовать их для лечения; кроме того, токсичность этих соединений по отношению к обычным или нормальным клеткам хозяина является относительно высокой.
Хотя известны олигодезоксирибонуклеотиды, имеющие короткую длину цепи, раньше считалось, что они обладают плохой ингибирующей активностью. В результате большинство исследователей сконцентрировали свои усилия на исследовании олигодезоксирибонуклеотидов, имеющих более длинные цепи, чем соединения настоящего изобретения. Так, например, хотя заявка PCT N WO 88/07544 (которая считается ближайшим аналогом настоящего изобретения) включает в своем объеме олигонуклеотиды, имеющие всего 4 основных звена, на практике ясно, что единственными испытанными материалами являются те, которые имеют значительно большее число звеньев, более длинные, чем олигонуклеотиды настоящего изобретения. В настоящее время авторами обнаружено, что некоторые модифицированные олигодезоксирибонуклеотиды, состоящие из различных последовательностей оснований и полученные путем введения различных заместителей в 5' - и/или 3' - концевые положения, проявляют превосходную активность против вируса AIDS и что токсичность этого нуклеотида по отношению к нормальным клеткам животного-хозяина является низкой. Более того, важным практическим фактором является то, что модифицированные олигодезоксирибонуклеотиды настоящего изобретения могут легко синтезироваться с использованием простых известных приемов.
Таким образом, одной из целей настоящего изобретения является представление ряда новых модифицированных олигодезоксирибонуклеотидов.
Дальнейшей и более конкретной целью изобретения является представление таких модифицированных олигодезоксирибонуклеотидов, которые обладают способностью ингибировать репликацию чужеродных нуклеиновых кислот в нормальных клетках и могут поэтому использоваться для лечения и профилактики вирусных инфекций, включая AIDS, и опухоли.
Связанным с указанной целью изобретения является представление процессов получения новых олигодезоксирибонуклеотидных соединений и промежуточных продуктов для использования в процессах получения.
Другие цели и преимущества изобретения станут очевидными по мере описания изобретения.
Соединениями настоящего изобретения являются модифицированные олигодезоксирибонуклеотиды формулы (1):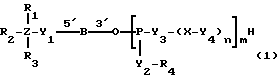
в которой
R1, R2 и R3 независимо выбраны из группы, состоящей из атомов водорода, алкильных групп с 1 - 4 атомами водорода, арильных групп, определенных ниже, и антрахинонильных групп, которые являются незамещенными или замещенными по крайней мере одним заместителем, предпочтительно выбранным из группы, состоящей из заместителей 1, определенных ниже;
Z представляет атом углерода или кремния;
или
R2, R3 и Z вместе представляют флуоренильную или ксантенильную группу;
R4 представляет атом водорода, незамещенную алкильную группу, имеющую от 1 до 4 атомов углерода, замещенную алкильную группу, которая имеет от 1 до 4 атомов углерода и замещена по крайней мере одним заместителем, предпочтительно выбранным из группы, состоящей из заместителей 2, определенных ниже, арильную группу, определенную ниже, или арилкильную группу, определенную ниже;
Y1, Y3 и Y4 независимо выбраны из группы, состоящей из атомов кислорода, атомов серы и групп формулы 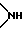 ;
;
Y2 представляет атом кислорода, атом серы, группу формулы 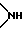 , алкиленовую группу, имеющую от 1 до 4 атомов углерода, или фениленовую группу;
, алкиленовую группу, имеющую от 1 до 4 атомов углерода, или фениленовую группу;
X представляет незамещенную алкиленовую группу, имеющую от 1 до 10 атомов углерода, или алкиленовую группу, которая имеет от 1 до 10 атомов углерода и которая замещена по крайней мере одной гидрокси группой;
m и n каждый независимо представляет 0 или целое число от 1 до 10;
B представляет олигодезоксирибонуклеотид, имеющий цепь длиной от 3 до 9;
указанная арильная группа представляет ароматическую карбоциклическую группу, которая имеет от 6 до 20 кольцевых атомов углерода и которая является незамещенной или замещена по крайней мере одним заместителем, выбранным из группы, состоящей из заместителей 1, определенных ниже;
указанная аралкильная группа представляет алкильную группу, которая имеет от 1 до 4 атомов углерода и которая замещена по крайней мере одной арильной группой, определенной выше;
указанные заместителя 1 выбраны из группы, состоящей из алкильных групп, имеющих от 1 до 4 атомов углерода, галоидалкильных групп, имеющих от 1 до 4 атомов углерода, атомов галогена, нитрогрупп, цианогрупп, аминогрупп, алкоксигрупп, имеющих от 1 до 4 атомов углерода, алкилтиогрупп, имеющих от 1 до 4 атомов углерода, арильных групп, определенных выше, арилоксигрупп, в которых арильная часть имеет значения, определенные выше, и аралкилоксигрупп, в которых аралкильная часть является такой, как определена выше, при условии, что, когда указанный заместитель 1 представляет арильную группу или группу, содержащую арильную группу, которая замещена дополнительной арильной группой или группой, содержащей арильную группу, эта дополнительная группа сама не замещена арильной группой или группой, содержащей арильную группу; и
указанные заместители 2 выбраны из группы, состоящей из аминогрупп, алкоксигрупп, имеющих от 1 до 4 атомов углерода, и атомов галогена.
Изобретение представляет также композицию для лечения или профилактики вирусных инфекций, которая включает эффективное количество по крайне мере одного олигодезоксирибонуклеотида, в которой указанным олигодезоксирибонуклеотидом является соединение формулы (1), определенной выше.
Изобретение также представляет способ лечения или профилактики вирусной инфекции млекопитающего, которым может быть человек, который предусматривает назначение указанному млекопитающему эффективного количества по крайней мере одного олигодезоксирибонуклеотида, в котором указанным олигодезоксирибонуклеотидом является соединение формулы (1), определенной выше.
Изобретение представляет также способы и промежуточные соединения для получения соединений настоящего изобретения, при этом способы описаны более подробно далее.
Новые модифицированные олигодезоксирибонуклеотиды настоящего изобретения обычно представляются в свободной от побочных продуктов реакции форме.
В соединениях настоящего изобретения, в которых R1, R2 или R3 представляет алкильную группу, последняя может быть алкильной группой с прямой или разветвленной цепью, имеющей от 1 до 4 атомов углерода, и примеры их включают метильную, этильную, пропильную, изопропильную, бутильную, втор-бутильную и трет-бутильную группы. Из них мы предпочитаем трет-бутильную группу.
Когда R1, R2 или R3 представляет арильную группу, ею может быть ароматическая карбоциклическая группа, которая имеет от 6 до 20 кольцевых атомов углерода, более предпочтительно от 6 до 16 кольцевых атомов углерода, еще более предпочтительно от 6 до 10 кольцевых атомов углерода, и наиболее предпочтительно 6 или 10 атомов углерода, и которая является незамещенной или замещенной по крайней мере одним заместителем, выбранным из группы, состоящей из заместителей 1, определенных и проиллюстрированных примерами ниже. Примеры незамещенных групп включают фенильную, 1-нафтильную, 2-нафтильную, фенантрен-4-ильную, антрацен-9-ильную, антрацен-2-ильную и пиренильную группы. Из них более предпочтительными являются нафтильная и фенильная группы, причем фенильная группа является наиболее предпочтительной. Замещенными группами могут быть любые из этих групп, и они могут быть замещены одним или более из заместителей 1, определенных выше и проиллюстрированных примерами ниже. Нет никаких особых ограничений в отношении числа заместителей, за исключением тех, которые могут налагаться числом замещаемых положений (например, 5 в случае фенильной группы или 7 в случае нафтильной группы) и, возможно, стерическими сдерживающими факторами. Наиболее обычно, однако, предпочтительно от 1 до 5 таких заместителей, более предпочтительно от 1 до 3, и наиболее предпочтительно 1 или 2 заместителя. Конкретные примеры заместителей 1 включают:
алкильные группы, имеющие от 1 до 4 атомов углерода, такие как метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, и трет-бутильная группа, из которых предпочтительны метильная и трет-бутильная группы;
галоидалкильные группы, имеющие от 1 до 4 атомов углерода, такие как фторметил, трифторметил, трихлорметил, 2,2,2,-трифторэтил, 2,2,2,-трихлорэтил, 2 - фторэтил, 2 - хлорэтил, 2 - йодэтил, 3 - хлорпропил и 4 - фторбутильная и 6 - йодгексильная группы, из которых предпочтительны 2,2,2, - трихлорэтильная и трифторметильная группы;
атомы галогена, такие как атомы фтора, хлора, брома и йода, из которых предпочитаются атомы хлора и фтора;
нитрогруппы, цианогруппы, аминогруппы;
алкоксигруппы, имеющие от 1 до 4 атомов углерода, также как метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси и трет-бутоксигруппы, из которых предпочтительны метокси и трет-бутоксигруппы;
алкилтиогруппы, имеющие от 1 до 4 атомов углерода, такие как метилтио, этилтио, пропилтио, изопропилтио, бутилтио, изобутилтио, втор-бутилтио и трет-бутилтио группы, из которых предпочтительны метилтио и трет-бутилтиогруппы;
арильные группы, определенные и проиллюстрированные примерами выше, и наиболее предпочтительно фенильную группу;
арилоксигруппы, из которых арильная часть является такой, как определена и показана примерами выше, и наиболее предпочтительно феноксигруппу; и
аралкилоксигруппы, в которых аралкильная часть является такой, как определена выше, такие как бензилокси и дибензилоксибензилокси [особенно 3,5 - дибензилоксибензилоксигруппу].
Следует заметить, что, когда арильная группа (или группа, содержащая арильную группу) может быть сама замещена дополнительной такой группой, на такое дополнительное замещение накладывается ограничение, касающееся того, что когда указанный заместитель 1 представляет арильную группу или группу, содержащую арильную группу, которая замещена дополнительной арильной группой или группой, содержащей арильную группу, эта дополнительная группа сама не замещена арильной группой или группой, содержащей арильную группу.
Конкретные примеры таких замещенных арильных групп включают 4-метилфенил, 4-трет-бутилфенил, 2-фенилфенил, 4-фенил-фенил, 4-фторфенил, 2-хлорфенил, 4-хлорфенил, 4-бромфенил, 4-йодфенил, 2,4-дифторфенил, 4-нитрофенил, 4-трет-бутоксифенил, 4-метоксифенил, 4-этоксифенил, 3-феноксифенил, 4-феноксифенил, 2-бензилоксифенил, 4-бензилоксифенил, 3,4-дибензилоксифенил, 3,5-дибензилоксифенил и 3,5-бис(3,5-дибензилоксибензилокси)фенильную группу. Из замещенных и незамещенных арильных групп мы предпочитаем фенильную, 4-метоксифенильную, 3,4-дибензилоксифенильную, 3,5-дибензилоксифенильную и 3,5-бис(3,5-дибензилоксибензилокси)фенильную группы.
Когда R1, R2 или R3 представляет антрахинонильную группу, она может быть незамещенной или замещенной одним или большим числом заместителей 1, определенных и показанных на примерах выше. Когда эта группа является замещенной, она может быть замещена по крайней мере одним заместителем, выбранным из группы, состоящей из заместителей 1, которые определены и представлены примерами выше. Нет каких-либо особых ограничений в отношении числа заместителей, за исключением таких, которые могут налагаться числом замещаемых положений и, возможно, пространственными сдерживающими факторами. Предпочтительны от 1 до 5 таких заместителей, более предпочтительно от 1 до 3, и наиболее предпочтительно 1 заместитель. Примеры таких групп включают 9, 10-антрахинон-1-ил, 9,10-антрахинон-2-ил, 4-метил-9,10-антрахинон-1-ил, 5-метокси-9,10-антрахинон-1-ил, 7-хлор-9,10-антрахинон-1-ил, 8-фтор-9,10-антрахинон-2-ил, 6-этил-9,10-антрахинон-2-ил, 8-этокси-9,10-антрахинон-2-ил и 6-гидрокси-9,10-антрахинон-1-ил. Из них предпочтительны незамещенные антрахинонильные группы.
Альтернативно, R2, R3 и Z могут вместе представлять флуоренил или ксантенильную группу, в этом случае ею предпочтительно является флуорен-9-ильная или ксантен-9-ильная группа.
Когда R4 представляет алкильную группу, ею может быть любая из групп, примеры которых приведены выше в отношении R1 и др., и она может быть незамещенной или замещенной. Если она замещена, она является замещенной по крайней мере одним из заместителей 2, определенных выше. Примеры таких заместителей 2 включают аминогруппы, алкоксигруппы, имеющие от 1 до 4 атомов углерода, и атомы галогена, в качестве примеров которых могут быть названы примеры групп, приведенных выше в отношении заместителей 1. Конкретные примеры таких замещенных и незамещенных алкильных групп включают метильную, этильную, 2-аминоэтильную, 2-метоксиэтильную, пропильную, изопропильную, бутильную, изобутильную, втор-бутильную и трет-бутильную группы. Из них предпочтительны метильная, этильная, 2-аминоэтильная, 2-метоксиэтильная и пропильная группы.
Когда R4 представляет арильную группу, ею может быть любая из арильных групп, примеры которых приведены выше в отношении R1 и др., и она может быть замещенной или незамещенной группой. Примеры таких групп включают: фенильную группу, алкилфенильные группы, такие как 2-метилфенильная или 3-этилфенильная группа, галоидированные фенильные группы, такие как 2-фторфенильная, 2-хлорфенильная, 4-хлорфенильная, 2-бромфенильная или 2-йодфенильная группа; нитрофенильные группы, такие как 2-нитрофенильная или 4-нитрофенильная группа; алкоксифенильные группы, такие как 4-метоксифенильная или 4-этоксифенильная группа; алкилтиофенильные группы, такие как 4-метилтиофенильная или 4-этилтиофенильная группа; и нафтильная, фенантренильная, антраценильная и пиренильная группы. Из них предпочтительны незамещенная фенильная, галоидированная фенильная и нитрофенильная группы.
Когда R4 представляет аралкильную группу, ею может быть алкильная группа, которая имеет от 1 до 4 атомов углерода и которая замещена по крайней мере одной (и предпочтительно 1, 2 или 3, более предпочтительно 1) из арильных групп, которые могут быть такими, как определены и показаны примерами выше. Примеры таких групп включают бензильную, метилбензильную, этилбензильную, метоксибензильную, этоксибензильную, фторбензильную, хлорбензильную, бромбензильную, хлорнафтилметильную, инденилметильную, фенантренилметильную, антраценилметильную, дифенилметильную, трифенилметильную, 1-фенэтильную, 2-фенэтильную, 2,2-дифенилэтильную, 2,2,2-трифенилэтильную, 3,3,3-трифенилпропильную, 1-нафтилэтильную, 2-нафтилэтильную, 1-фенилпропильную, 2-фенилпропильную, 3-фенилпропильную, 1-нафтилпропильную, 2-нафтилпропильную, 3-нафтилпропильную, 1-фенилбутильную, 2-фенилбутильную, 3-фенилбутильную, 4-фенилбутильную, 1-нафтилбутильную, 2-нафтилбутильную, 3-нафтилбутильную, 4-нафтилбутильную, 1-фенилпентильную, 2-фенилпентильную, 3-фенилпентильную, 4-фенилпентильную, 5-фенилпентильную, 1-нафтилпентильную, 2-нафтилпентильную, 3-нафтилпентильную, 4-нафтилпентильную, 5-нафтилпентильную, 1-фенилгексильную, 2-фенилгексильную, 3-фенилгексильную, 4-фенилгексильную, 5-фенилгексильную, 6-фенилгексильную, 1-нафтилгексильную, 2-нафтилгексильную, 3-нафтилгексильную, 4-нафтилгексильную, 5-нафтилгексильную и 6-нафтилгексильную группы. Из них предпочтительна незамещенная бензильная или 2-фенэтильная группа.
Когда Y2 представляет алкиленовую группу, ею может быть метиленовая, этиленовая, пропиленовая, тетраметиленовая или пентаметиленовая группа. Из них предпочтительна метиленовая группа.
Y1, Y3 и Y4 каждый предпочтительно представляет атом кислорода.
Y2 представляет предпочтительно атом кислорода или серы.
Z представляет предпочтительно атом углерода.
Когда X представляет прямую или разветвленную цепь, необязательно замещенную гидроксигруппой, примеры включают метиленовую, метилметиленовую, этиленовую, пропиленовую, тетраметиленовую, метилэтиленовую, 1-метилтриметиленовую, 2-метилтриметиленовую, 2-метилтетраметиленовую, 3-метилтриметиленовую, пентаметиленовую, гексаметиленовую, гептаметиленовую, октаметиленовую, нонаметиленовую, декаметиленовую, 2-гидрокситриметиленовую или 2-гидрокситетраметиленовую группу. Из них предпочтительны метиленовая, метилметиленовая, этиленовая или метилэтиленовая группа.
Предпочтительными значениями для каждого из символом m и n являются целые числа от 0 до 6. Особенно предпочтительно, чтобы m представляло целое число от 0 до 4.
Олигодезоксирибонуклеотиды, представленные символом B, предпочтительно имеют длину цепи 4 - 8; более предпочтительно длину цепи 5 или 6. Кроме того, предпочитается, чтобы четвертый дезоксирибонуклеотид от 5'-терминального конца представлял гуаниндезоксирибонуклеотид.
Типичными предпочтительными иллюстративными олигодезоксирибонуклеотидами являются соединения в следующей " α- группе" и более предпочтительными соединениями являются соединения в следующей " β- группе", при этом сокращения, используемые в следующих ниже α- и β- группах, имеют следующие значения:
A : адениндезоксирибонуклеотид,
G : гуаниндезоксирибонуклеотид,
C : цитозиндезоксирибонуклеотид,
T : тиминдезоксирибонуклеотид,
mС : 5-метилцитозиндезоксирибонуклеотид, и
mG : O6 - метилгуаниндезоксирибонуклеотид.
Термин "левый конец" означает 5'-терминальный конец, и термин "правый конец" означает 3'-терминальный конец при условии, что нет гидроксигрупп ни на 5'-, ни на 3'-терминальном концах каждого олигодезоксирибонуклеотида.
" α- группа" :
TGGGAG, TGGGA, TGGGG, TGGG, TGGGAGG, CGGGAGG, TTGGAGG, TTGGGAGG, TGCGAGG, GGGGAGG, mCGGGAGG, mCGmCGAGG, CTGGGAGG, GGGCGGGGC, TAGGAGG, TGGGAGGT, TGGGCGCAG, CCG, TCGGAGG, TGmCGAGG, CTGGGAGG, TGG, TGGGAmGG, TGGGAGA, AATGGGAGG, TTGGGG, TGGGGG, CGGGG, CGCGG, CGGGT, TGGGC, TGGGT.
" β- группа"
TGGGAG, TGGGA, TGGGG, TGGG, TGGGAGG, CGGGAGG, TTGGAGG, TTGGGAGG, TGCGAGG, GGGGAGG, mCGGGAGG, mCGmCGAGG, CTGGGAGG, TTGGGG, TGGGGG, CGGGG, CGCGG.
Примеры предпочтительной группы формулы:
R1, R2, R3,Z - Y1 на 5' - терминальном конце являются трифенилметилокси, 3,4-(дибензилокси)бензилокси, 3,5-(дибензилокси)бензилокси, 3,5-бис[3,5-(дибензилокси)бензилокси] бензилокси, трет-бутилдифенилсилолокси, фенилфтулоренилокси или фенилксантенилоксигруппы; более предпочтительно трифенилметилокси, 3,4-(дибензилокси)бензилокси или 3,5-(дибензилокси)бензилоксигруппа.
Примеры предпочтительной группы формулы: [P (O) (Y2R4)-Y3-(X-Y4)n]mH - на 3'-терминальном конце включают атом водорода, метилфосфорильную, 2-хлорфенилфосфорильную, - O - метилтиофосфорильную, метилфосфонильную, метилтиофосфонильную, фенилфосфонильную, 2-гидроксиэтилфосфорильную, -O-(2-гидроксиэтил)тиофосфорильную, фенилфосфорильную, 4-хлорфенилфосфорильную, 2-нитрофенилфосфорильную, 4-нитрофенилфосфорильную, этилфосфорильную или O-этилтиофосфорильную группу; более предпочтительно атом водорода, метилфосфорильную, 2-хлорфенилфосфорильную, -O-метилтиофосфорильную, метилфосфонильную, метилтиофосфонильную, фенилфосфонильную, 2-гидроксиэтилфосфорильную или -O-(2-гидроксиэтил)тиофосфорильную группу.
В общем, предпочтительными соединениями настоящего изобретения являются соединения, в которых:
/1/ длина цепи B составляет от 4 до 8;
/2/ длина цепи B составляет 5 или 6;
/3/ длина цепи B составляет от 4 до 8, и четвертый дезоксирибонуклеотид от 5'-терминала B представляет гуаниндезоксирибонуклеотид;
/4/ группа формулы: R1R2R3Z - Y1 на 5'-терминальном конце представляет трифенилметилокси, 3,4-(дибензилокси)бензилокси, 3,5-(дибензилокси)бензилокси, 3,5-бис[3,5-(дибензилокси)бензилокси] бензилокси, трет-бутилдифенилсилилокси, фенилфлуоренилокси или фенилксантенилоксигруппу;
группа формулы: [P(O) (Y2R4)-Y3-(X-Y4)n] mH на 3'-терминальном конце представляет атом водорода, метилфосфорильную, 2-хлорфенилфосфорильную, -O-метилтиофосфорильную, метилфосфонильную, метилтиофосфонильную, фенилфосфонильную, 2-гидроксиэтилфосфорильную, -O-(2-гидроксиэтил)тиофосфорильную, фенилфосфорильную, 4-хлорфенилфосфорильную, 2-нитрофенилфосфорильную, 4-нитрофенилфосфорильную, этилфосфорильную или -O-этилтиофосфорильную группу;
и B представляет собой TGGGG, TGGG, TGGGAGG, CGGGAGG, TTGGAGG, TTGGGAGG, TGCGAGG, GGGGAGG, mCGGGAGG, mCGmCGAGG, CTGGGAGG, GGGCGGGGC, TAGGAGG, TGGGAGGT, TGGGCGCAG, CCG, TCGGAGG, TGmCGAGG, CTGGGAGG, TGG, TGGGAmGG, TGGGAGA, AATGGGAGG, TTGGGG, TGGGGG, CGGGG, CGCGG, CGGGT, TGGGC или TGGGT;
/5/ группа формулы: R1R2R3Z - Y1 на 5'-терминальном конце представляет трифенилметилокси, 3,4-(дибензилокси)бензилокси, 3,5-(дибензилокси)бензилокси, 3,5-бис-[3,5-(дибензилокси)бензилокси]бензилокси, трет-бутилдифенилсилилокси, фенилфлуорентилокси или фенилксантенилокси группу;
группа формулы: [P(O) (Y2R4)-Y3-(X-Y4)n] mH на 3'-терминальном конце представляет атом водорода, метилфосфорильную, 2-хлорфенилфосфорильную, -O-метилтиофосфорильную, метилфосфонильную, метилтиофосфонильную, фенилфосфонильную, 2-гидроксиэтилфосфорильную, -O-(2-гидроксиэтил)тиофосфорильную, фенилфосфорильную, 4-хлорфенилфосфорильную, 2-нитрофенилфосфорильную, 4-нитрофенилфосфорильную, этилфосфорильную или -O-этилтиофосфорильную группу;
и B представляет TGGGAG, TGGGA, TGGGG, TGGG, TGGGAGG, GGGGAGG, TTGGAGG, TTGGGAGG, TGCGAGG, GGGGAGG, mCGGGAGG, mCGmCGAGG, CTGGGAGG, TTGGGG, TGGGGG, CGGGG или CGCGG;
/6/ группа формулы: R1R2R3Z-Y1 на 5'-терминальном конце представляет трифенилметилокси, 3,4-(дибензилокси)бензилокси, 3,5-(дибензилокси)бензилокси, 3,5-бис[3,5-(дибензилокси)бензилокси] бензилокси, трет-бутилдифенилсилилокси, фенилфлуоренилокси или фенилксантенилоксигруппу;
группа формулы: [P (O) (Y2R4)-Y3-(X-Y4)n]mH на 3'-терминальном конце представляет атом водорода, метилфосфорильную, 2-хлорфенилфосфорильную, -O-метилтиофосфорильную, метилфосфонильную, метилтиофосфонильную, фенилфосфонильную, 2-гидроксиэтилфосфорильную, или -О-(2-гидроксиэтил)тиофосфорную группу;
и B представляет TGGGAG, TGGGA, TGGGG, TGGG, TGGGAGG, CGGGAGG, TTGGAGG, TTGGGAGG, TGCGAGG, GGGGAGG, mCGGGAGG, mCGmCGAGG, CTGGGAGG, TTGGGG, TGGGGG, CGGGG или CGCGG;
/7/ группа формулы: R1R2R3Z-Y1 на 5'-терминальном конце представляет трифенилметилокси, 3,4-(дибензилокси)бензилокси или 3,5-(дибензилокси)бензилоксигруппу:
группа формулы: [(P (O) (Y2R4)-Y3-(X-Y4)n]mH на 3'-терминальном конце представляет атом водорода, метилфосфорильную, 2-хлорфенилфосфорильную, -O-метилтиофосфорильную, метилфосфонильную, метилтиофосфонильную, фенилфосфонильную, 2-гидроксиэтилфосфорильную, -O-(2-гидроксиэтил)тиофосфорильную, фенилфосфорильную, 4-хлорфенилфосфорильную, 2-нитрофенилфосфорильную, 4-нитрофенилфосфорильную, этилфосфорильную или -O-этилтиофосфорильную группу;
и B представляет TGGGAG, TGGGA, TGGGG, TGGG, TGGGAGG, CGGGAGG, TTGGAGG, TTGGGAGG, TGCGAGG, GGGGAGG, mCGGGAGG, mCGmCGAGG, CTGGGAGG, TTGGGG, TGGGGG, CGGGG или CGCGG;
/8/ группа формулы : R1R2R3Z-Y1 на 5'-терминальном конце представляет трифенилметилокси, 3,4-( дибензилокси)бензилокси или 3,5-(дибензилокси)бензилоксигруппу;
группа формулы : [(P (O) (Y2P4)-Y3-(X-Y4)n]mH на 3'-терминальном конце представляет атом водорода, метилфосфорильную, 2-хлорфенилфосфорильную, -O-метилтиофосфорильную, метилфосфонильную, метилтиофосфонильную, фенилфосфонильную, 2-гидроксиэтилфосфорильную или -O-(2-гидроксиэтил)тиофосфорильную группу;
и B представляет TGGGAG, TGGGA, TGGGG, TGGG, TGGGAGG, CGGGAGG, TTGGAGG, TTGGGAGG, TGCGAGG, GGGGAGG, mCGGGAGG, mCGmCGAGG, CTGGGAGG, TTGGGG, TGGGGG, CGGGG или CGCGG.
Примеры соединений настоящего изобретения перечислены в табл. 1. Эти примеры не следует рассматривать как ограничивающие данное изобретение.
В табл. 1 для обозначения некоторых групп используются следующие сокращения:
2-Anq - антрахинон-2-ил
2-Ant - антрацен-2-ил
9-Ant - антрацен-9-ил
Bdbbp - 3,5-бис[3,5-(дибензилокси)бензилокси]фенил
Bu - бутил
tBu - трет-бутил
Bz - бензил
3,4-Dbp - 3,4-(дибензилокси)фенил
3,5-Dbp - 3,5-(дибензилокси)фенил
Decm - декаметилен[-(CH2)10-]
Et - этил
Ete - этилен [-CH2CH2-]
Hepm - гептаметилен [-(CH2)7-]
Hexm - гексаметилен [-(CH2)6-]
Hpr - 2-гидроксипропилен[-CH2C(OH)(CH3)-]
Me - метил
Mee - метилен [-CH2-]
Nonm - нонаметилен[-(CH2)9-]
Npe - нафталенил[например, 2-Npe представляет нафтален-2ил или 1-Npe представляет нафтален-1-ил]
Octm - октаметилен[-(CH2)8-]
Penm - пентаметилен[-(CH2)5-]
Ph - фенил
Pha - фенантренил[например, 4-Pha представляет фенантрен-4-ил]
1,4-фенилен
Pr - пропил
Pre - пропилен [-CH2CH(CH3)-]
1-Pyr - пирен-1-ил
Tetm - тетраметилен[-(CH2)4-]
Trim - триметилен[-(CH2)3-]
В дополнение к сказанному, последовательность, представленная символом "В" в формуле /1/, идентифицируется следующими кодовыми номерами:
1 TGGGAG
2 TGGGA
3 TGGGG
4 TGGG
5 TGGGAGG
6 CGGGAGG
7 TTGGAGG
8 TTGGGAGG
9 TGCGAGG
10 GGGGAGG
11 mCGGGAGG
12 mCGmCGAGG
13 CTGGGAGG
14 GGGCGGGC
15 TAGGAGG
16 TGGGAGGT
17 TGGGCGCAG
18 CCG
19 TCGGAGG
20 TGmCGAGG
21 GTGGGAGG
22 TGG
23 TGGGAmGG
24 TGGGAGA
25 AATGGGAGG
26 TTGGGG
27 TGGGGG
28 CGGGG
29 CGCGG
30 CGGGT
31 TGGGC
32 TGGGT.
Кроме того, когда в табл. 1 показана флуоренильная или ксантенильная группа, она представлена символами R2, R3 и Z вместе.
Из перечисленных выше соединений предпочтительными являются соединения NN с 1 по 440, 454, 476, 586, 696, 806, 916, 1026, 1136, 1246, 1356, 1466, 1576, 1686, 1763, 1773, 1793, 1979, 1980, с 1990 по 1994, 2250 и с 2326 по 2906.
Более предпочтительными являются соединения NN 1-110, 113, 221-330, 333, 454, 1763, 1773, 1793, 1979, 1980, 1990-1994, 2250 и 2334-2906.
Наиболее предпочтительными являются соединения NN 1, 2, 3, 4, 12, 13, 14, 15, 2555, 2556, 2557, 2558, 2566, 2567, 2568, 2569, 2665, 2666, 2667, 2668, 2676, 2677, 2678 и 2679.
Соединения настоящего изобретения могут быть представлены в форме солей, особенно фармацевтически приемлемых солей с катионами. Примерами подходящих солей являются неорганические или органические соли, например соли с щелочными металлами, такими как натрий или калий, с щелочно-земельными металлами, такими как кальций; соли с аммиаком; основными аминокислотами, такими как лизин или аргинин; и соли с алкиламинами, такими как триэтиламин. Предпочтительными солями являются соли щелочных металлов, таких как натрий или калий.
Некоторые способы получения соединений настоящего изобретения иллюстрируются с помощью следующих реакционных схем.
Обычно соединения общей формулы (I) могут быть получены с помощью конденсации подходящего дериватизированного нуклеотида, являющегося нуклеотидом в 5'-конце желаемого соединения, с защищенным олигонуклеотидом, в котором не достает 5'-концевого нуклеотида, причем нуклеотиды олигонуклеотида с указанным недостатком дают с указанным 5'-концевым нуклеотидом соответствующую нуклеотидную последовательность желаемого олигодезоксирибонуклеотидного соединения, причем защищенный нуклеотид с недостатком связан с полимерной подложкой (носителем). В типичном случае данный процесс включает реакцию соединения R1R2R3Z - Y'-[5'-концевой нуклеотид] с соединением [протектор]-[олигонуклеотид с недостатком] - линкер - полимер.
Более конкретно, настоящее изобретение представляет процесс, который включает конденсацию соединения следующей ниже формулы (2) с соединением вида [протектор] -O-F-W, где F представляет олигонуклеотид с недостатком и W представляет линкер и полимерную подложку. При использовании ДМТ в качестве протектора примеры соединений для реакции с соединением (2) включает соединения формул: ДМТ-O-F-W1 (3), ДМТ-O-F-W2a (4a), ДМТ-O-F-W2b (4b), ДМТ-O-F-W3 (5), ДМТ-O-F-W4a (6a), ДМТ-O-F-W4b (6b), ДМТ-O-F-W4c (6c), ДМТ-O-F-W4d (6d), ДМТ-O-F-W5a (7a) и ДМТ-O-F-W5b (7b), согласно методу C-1, C-2 или C-3. Реагент (2), используемый в данной реакции, может подходящим образом получаться по способу A-1 или A-2, а реагенты (3, 4a, 4b, 5, 6a - 6b, 7a и 7b) могут подходящим образом получаться по способу B-1, B-2, B-3, B-4 или B-5. Способы A-1, A-2, B-1, B-2, B-3, B-4 и B-5 представлены в конце описания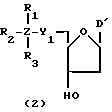
В различных соединениях общей формулы (2) символы R1, R2, R3, Y1 и Z имеют значения, определенные выше для указанной общей формулы (1).
D' представляет основание, выбранное из следующей "группы основания" или соответствующее защищенное основание, причем указанным основанием является основание в 5'-концевом фрагменте указанной эффективной последовательности оснований (обозначаемое здесь далее как 5'-концевой фрагмент указанной эффективной последовательности оснований или просто 5'-концевое основание), где "группа основания 5'" представляет аденин, гаунин, цитозин, тимин или 5-метилцитозин.
Способ B-1 включает получение соединения (3) с одним недостающим нуклеотидным фрагментом с 5' конца указанной эффективной последовательности, синтезируемого с использованием стекла с регулируемыми порами (называемого далее CPG), включающего линкер, связанный с защищенным нуклеозидом, используемым для синтеза ДНК в 3'-концевом фрагменте указанной эффективной последовательности оснований (называемым далее 3'-концевой нуклеозид) и нуклеотидным звеном, промышленно выпускаемым для ДНК-синтезатора (называемым здесь далее нуклеотидным звеном).
Способ B-2 включает получение соединения (4a), имеющего один нуклеотидный короткий фрагмент с 5' конца указанной эффективной последовательности, синтезируемого с помощью защиты одной концевой гидроксильной группы C2-C10 - алкилендиола, имеющего гидроксильные группы в концевых положениях, или алкилендиола, имеющего защищенные гидроксильную и аминогруппы диметокситритильной (ДМТ) группой, взаимодействия другой гидроксильной группы с янтарным ангидридом с получением моноэфира янтарной кислоты, связывания карбоксильной группы моноэфира с CPG, удаления концевой ДМТ-группы и, наконец, взаимодействия нуклеотидного звена со связанным с CPG алкиленовым спиртом (4 - 6) в регулярной последовательности на ДНК-синтезаторе; и получение соединения (4b) путем превращения связанного с CPG алкиленового спирта (4 - 6) в 3'-концевой нуклеозидный фосфортиоат, нанесенный на CPG, (4 - 7) в соответствии с общепринятыми приемами, используемыми при получении тиоатных нуклеотидов на ДНК-синтезаторе, и затем взаимодействия с нуклеотидным звеном таким же образом, как выше.
Способ B-3 включает получение соединения (5), имеющего один нуклеотидный короткий фрагмент с 5' конца указанной эффективной последовательности, синтезируемого с помощью конденсации 3'-концевого нуклеозид-фосфоната, который получается с помощью реакции гидроксильной группы в 3'-положении 3'-концевого нуклеозида с фосфоновой кислотой, с получением сложноэфирной связи, со связанными с CPG этиленгликолем (5 - 1), получаемым с использованием янтарного ангидрида в способе B-2, реакции с алкиламином с получением 3'-концевого нуклеозид-фосфорамидита, нанесенного на CPG (5 - 4), и, наконец, взаимодействия с нуклеотидным звеном в регулярной последовательности на ДНК-синтезаторе.
Способ B-4 включает получение известного соединения (6 - 3) [M. Durand и др. Nucleuc Acids Res., 18, 6353/1990/] с помощью защиты гидроксильной группы тексаэтиленгликоля ДМТ-группой и взаимодействия другой гидроксильной группы с реагентом для получения фосфорамитной группы.
Впоследствии по аналогии с процедурой, описанной в способе B-3, CPG - связанный защищенный ДМТ-гликоль (6 - 6) может быть получен с помощью конденсации указанного защищенного ДМТ-группой гексаметиленгликоля (6 - 2) с CPG с использованием янтарной кислоты.
После удаления ДМТ-группы из связанного с CPG защищенного ДМТ-гликоля (6 - 6) на ДНК-синтезаторе гликоль подвергается реакции с нуклеотидным звеном, давая желаемое соединение (6a), имеющее один нуклеотидный короткий фрагмент с 5' конца указанной синтезированной эффективной последовательности. Гликоль, нанесенный на CPG, который получается при удалении ДМТ-группы из связанного с CPG защищенного ДМТ-гликоля (6 - 6) на ДНК-синтезаторе, подвергается реакции один, два или три раза с указанным фосфорамидитом с последующим взаимодействием с нуклеотидным звеном, включающим один нуклеотидный короткий фрагмент с 5 - конца указанной эффективной последовательности оснований, с получением соединения формулы (6b), (6c) или (6d) соответственно.
Способ B-5 включает получение соединения (7 - 3) с помощью снятия защиты ДМТ-группой из промышленно доступного защищенного 2'-дезоксинуклеозида, нанесенного на CPG через линкер (называемого здесь далее D'''- CPG) и взаимодействия с (2-цианоэтокси) - 2 - (2' - 0-4,4' - диметокситритилоксиэтилсульфонил)этокси-N, N-диизопропиламинофосфином (7 - 1), описанное авторами Horn и др. в Tetrahedron Letters, 27, 4705 /1986/; получение соединения (7 - 5) с помощью снятия защиты ДМТ-группой из соединения (7 - 3), полученного выше, и конденсации деблокированного соединения с соединением (7 - 4), имеющим арил - или алкил-фосфат гидроксильной группы в 3'-положении 3'-концевого нуклеотида с использованием конденсирующего агента; и получение соединения (7a) с помощью реакции соединения (7 - 5), полученного выше, с нуклеотидным звеном, включающим один нуклеотидный короткий фрагмент с 5' конца указанной эффективной синтезированной последовательности.
Аналогичным образом, как описано выше, соединение (7 - 7) может получаться с помощью реакции соединения (7 - 3), освобожденного от ДМТ-группы, с соединением (7 - 6), имеющим алкил- или арил-фосфорамидитную группу в 3'-положении 3'-концевого нуклеозида, и обработки по аналогии с синтезом триэфира фосфорной кислоты или фосфортиоатного триэфира на ДНК-синтезаторе. Продукт, полученный таким образом, вводится в реакцию с нуклеотидным звеном (единицей) в регулярной последовательности с получением соединения (7a), имеющего один нуклеотидный короткий фрагмент с 5' конца указанной эффективной последовательности оснований.
Способ C-1 (приведен в конце описания) включает получение желаемого соединения общей формулы (1) с помощью взаимодействия соединения (2) с фосфорилирующим агентом с получением 3'-фосфорного производного (8), взаимодействия полученного таким образом продукта с каждым олигомером формул (3, 4a, 4b, 5, 6a, 6d, 7a и 7b), нанесенным (или осажденным) на CPG, который получается по способам B-1 - B-5, после деблокирования ДМТ-группы, окисления конденсированного продукта окисляющим агентом, отщепления нуклеотидной цепи от CPG и, наконец, удаления защитной группы за исключением замещающего фрагмента, связанного с атомом углерода в 5'-положении 5'-концевого нуклеозида.
Способ C-2 (приведен в конце описания) включает получение требуемого соединения соединения (1) с помощью реакции соединения (2) с фосфорилирующим агентом с получением 3'-фосфорно-кислотного производного (9), конденсации продукта с указанным соединением каждой из формул (3, 4a, 4b, 5, 6a - 6d, 7a и 7b) после деблокирования ДМТ-группы, одной с образованием фосфорно-кислотной триэфирной связи, отщепления нуклеотидной цепи от CPG, удаления защитной группы за исключением заместителя-фрагмента, связанного с атомом углерода в 5'-положении 5'-концевого нуклеозида, и, наконец, очистки с помощью обычных приемов.
Способ C-3 (приведен в конце описания) включает получение желаемого соединения (1) введением фосфоновокислотной группы в 3'-положении соединения (2) с получением соединения (10), конденсацией продукта с указанным соединением каждой из формул (3, 4a, 4b, 5, 6a - 6d, 7a и 7b) с использованием галоидангидрида кислоты в присутствии основания, окислением конденсированного продукта окисляющим агентом с образованием фосфорно-кислотной диэфирной связи, отщеплением нуклеотида от CPG, удалением защитной группы за исключением заместителя фрагмента, связанного с атомом углерода в 5'-положении 5'-концевого нуклеозида, и, наконец, очистки с помощью обычных приемов.
Способы от A до C поясняются более подробно следующим образом.
В реакционных схемах, приведенных в способах A - C, символы R1, R2, R3, R4, Z, Y1, Y2, Y3, Y4, X, n, m и B имеют значения, определенные выше;
A1 представляет тритильную (Tr) группу, монометокситритильную (ММТ) группу или диметокситритильную (DМТ) группу, которая обычно используется для защиты, в частности, первичной гидроксильной группы нуклеозида;
A2 представляет три-замещенную силильную группу, такую как трет-бутилдиметилсилильная (TBDMS) или триизопропилсилильную (TIPS) группу; тригалоидэтоксикарбонильную группу, такую как трихлорэтоксикарбонильная (Tr ос) группа; или аралкилоксикарбонильную группу, такую как бензилоксикарбонильная (Z) группа.
D' представляет основание 5'-концевого нуклеотида, аминогруппа которого защищена ацильной группой для синтезирования ДНК указанной эффективной последовательности оснований;
D'' представляет основной фрагмент нуклеотида в 3'-конце; и D'' представляет фрагмент основания необязательного нуклеотидного звена, используемого в синтезе ДНК, т.е. основание, выбранное из группы 5'-основания или соответствующего защитного основания.
F представляет собой олигонуклеотидную часть, имеющую один короткий нуклеотидный фрагмент с 5' конца указанной эффективной последовательности и представляет часть основания (основная часть) или соответствующий олигонуклеотид, защищенный защитной группой фосфорно-кислотной части, которая обычно используется для синтеза ДНК, но не содержит какой-либо гидроксильной группы в 5'-положении 5'-концевого нуклеозида и в 3'-положении 3'-концевого нуклеозида.
V представляет собой защитную группу фосфорно-кислотной части в случае синтеза ДНК.
U представляет аминогруппу амидитной части.
W1 - W5 представляет фрагмент от CPG до атома кислорода гидроксильной группы в 3'-положении 3'-концевого нуклеотида олигонуклеотида (F) конечного требуемого соединения согласно процедуре, описанной в способах B-1 - B-5. представляет целое число от 1 до 9;
E представляет атом водорода или необязательно защищенную гидроксильную или аминогруппу;
K представляет атом кислорода или серы.
R6 представляет метильную, этильную, пропильную, бутильную, фенильную, метокси, этокси, пропокси, бутокси, цианоэтилокси или необязательно замещенную фенилоксигруппу.
Каждая из стадий поясняется более подробно следующим образом. В том случае, если стадия способна к проведению ее по аналогии с процедурой, описанной в предыдущей стадии, первая поясняется как характерная.
Стадии 1, 8 и 18
На этих стадиях соединение (2 - 2), в котором гидроксильная группа только в 5'-положении селективно защищена, может получаться с помощью взаимодействия соединения (2 - 1) с гидроксилзащищающим реагентом в инертном растворителе. Когда основная часть представляет A, G или C, аминогруппы, содержащиеся в основании, защищаются путем ацилирования на предыдущей стадии, и реакция защиты может осуществляться по способу, который сам по себе известен, например, по аналогии с процедурой, описанной в [J. Am. Chem. Soc. 104, 1316 (1982)]. В качестве аминозащищающей группы в реакцию конденсации обычно вступает алифатическая низшая ацильная или ароматическая ацильная группа. Примеры ацильных групп включают: алифатические низшие ацильные группы, такие как формильная, ацетильная, пропионильная, бутирильная, изобутирильная, пентаноильная, пивалоильная, валерильная или изовалерильная группа; и ароматические ацильные группы, такие как бензоильная, 4 - ацетоксибензоильная, 4 - метоксибензоильная, 4-метилбензоильная или 1-нафтоильная группа; предпочтительно, если основная часть представлена A или C, бензоильная группа, и, в случае, когда основная часть представляет G, - изобутирильная группа.
Примеры предпочтительных растворителей включают: ароматические углеводороды, такие как бензол, толуол или ксилол; галоидированные углеводороды, такие как дихлорметан, хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол или дихлорбензол; сложные эфиры, такие как этилформиат, этилацетат, пропилацетат, бутилацетат или диэтилкарбонат; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан или диэтиленгликольдиметиловый эфир; кетоны, такие как ацетон, метилэтилкетон, метилизобутилкетон, изофорон или циклогексанон; нитросоединения, такие как нитроэтан или нитробензол; нитрилы, такие как ацетонитрил или изобутиронитрил; амиды, такие как формамид, диметилформамид (ДМФ), диметилацетамид или гексаметилфосфорный триамид; сульфоксиды, такие как диметилсульфоксид или сульфолан; алифатические третичные амины, такие как триметиламин, триэтиламин или N-метилморфолин; ароматические амины, такие как пиридин или пиколин; более предпочтительно галоидированные углеводороды (особенно дихлорметан) и амиды (особенно ДМФ).
Нет каких-либо особых ограничений в отношении природы реагента, используемого для защиты, при условии, что он может использоваться для конкретной защиты гидроксильной группы только в 5'-положении и способен к удалению в кислых или нейтральных условиях. Примеры предпочтительных защищающих реагентов включают триарилметилгалогениды, такие как тритилхлорид, монометокситритилхлорид или диметокситритилхлорид.
Когда защитным реагентом является триарилметилгалогенид, реакция обычно осуществляется в присутствии основания.
Примеры подходящих оснований включают: гетероциклические амины, такие как пиридин, диметиламинопиридин или пирролидинопиридин; и алифатические третичные амины, такие как триметиламин или триэтиламин; предпочтительно органические основания (особенно пиридин, диметиламинопиридин и пирролидинопиридин).
Когда в качестве растворителя используются органические амины, не обязательно использовать другой устраняющий кислотность агент, потому что органические амины сами действуют как агенты, устраняющие кислотность.
Температура реакции изменяется в зависимости от характера исходного материала и используемого растворителя, а также других реакционных условий, но обычно реакция осуществляется при температуре от 0 до 150oC, предпочтительно от 20 до 100oC.
Время, требуемое для реакции, варьирует в зависимости от используемого исходного материала и растворителя, а также от реакционной температуры, и обычно реакция завершается за перйод 1-100 ч, предпочтительно в течение 2-24 ч.
После завершения реакции требуемое соединение может выделяться из реакционной смеси, например, выливанием реакционной смеси в воду, экстрагированием не смешиваемым с водой растворителем, таким как бензол, простой эфир или этилацетат, и, наконец, отгонкой растворителя из экстракта. Продукт, полученный таким образом, может использоваться в последующей реакции без дополнительной очистки, но, при необходимости, он может очищаться с помощью общепринятых приемов, например с помощью различных методов хроматографии или перекристаллизации.
Стадия 2.
На данной стадии соединение (2-3) может получаться с помощью взаимодействия соединения (2-2) с гидроксилзащищающим реагентом в присутствии инертного растворителя.
Примеры предпочтительных растворителей включают: ароматические углеводороды, такие как бензол, толуол или ксилол; галоидированные углеводороды, такие как дихлорметан, хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол или дихлорбензол; сложные эфиры, такие как этилформиат, этилацетат, пропилацетат, бутилацетат, диэтилкарбонат; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан или диэтиленгликольдиметиловый эфир; кетоны, такие как ацетон, метилэтилкетон, метилизобутилкетон, изофорон или циклогексанон; нитросоединения, такие как нитроэтан или нитробензол; нитрилы, такие как ацетонитрил или изобутиронитрил; амиды, такие как формамид, диметилформамид, диметилацетамид или гексаметилфосфорный триамид; сульфоксиды, такие как диметилсульфоксид или сульфолан; более предпочтительно простые эфиры (особенно тетрагидрофуран), галоидированные углеводороды (особенно дихлорметан), ароматические углеводороды (особенно толуол) и амиды (особенно ДМФ).
Нет никаких особых ограничений в отношении характера используемого гидроксилзащищающего реагента при условии, что может деблокироваться данная защитная группа в отличие от защитной группы в 5'-положении. Примеры таких защищающих реагентов включают: силилгалогениды, такие как трет-бутилдиметилсилилхлорид или триизопропилсилилхлорид; галоидалкоксикарбонилгалогениды, такие как трихлорэтоксикарбонилхлорид; и аралкилоксикарбонилгалогениды, такие как бензилоксикарбонилхлорид.
Когда защищающим реагентом являются силилгалогениды, галоидалкоксикарбонилгалогениды или аралкилоксикарбонилгалогениды, реакция защиты обычно осуществляется в присутствии основания.
Примеры предпочтительных оснований включают органические основания (особенно триэтиламин, пиридин, N-метилморфолин, ДВИ и имидазол).
Температура реакции меняется в зависимости от характера реагентов, исходного соединения и растворителя и от других условий реакции, но обычно реакция осуществляется при температуре от -20 до 150oC, предпочтительно от -10 до 50oC.
Время, требуемое для реакции, изменяется в зависимости от характера используемого исходного соединения и растворителя, а также от реакционной температуры, и обычно реакция завершается за период от 1 до 100 ч, предпочтительно за 1 - 24 ч.
После завершения реакции желаемое соединение может выделяться из реакционной смеси. Пример одного из таких приемов включает: выливание реакционной смеси в воду, экстрагирование не смешиваемым с водой растворителем, таким как бензол, диэтиловый эфир или этилацетат; и, наконец, отгонку растворителя из экстракта. Полученный таким образом продукт может обычно использоваться в последующей реакции без дополнительной очистки, но, если необходимо, может очищаться с помощью разнообразных приемов хроматографии, перекристаллизации или аналогичных способов.
Стадии 3, 11, 22 и 25.
На этих стадиях соединения (2-4), (4-6), (6-7) и (6-9) могут получаться по реакции соединений (2-3), (4-5), (6-6) и (6-8) с деблокирующим реагентом в присутствии инертного растворителя для селективного удаления гидроксилзащищающей группы в 5'-положении.
Примеры предпочтительных растворителей включают: ароматические углеводороды, такие как бензол, толуол или ксилол; галоидированные углеводороды, такие как дихлорметан, хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол или дихлорбензол; сложные эфиры, такие как этилформиат, этилацетат, пропилацетат, бутилацетат или диэтилкарбонат; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан или диэтиленгликольдиметиловый эфир; спирты, такие как метанол, этанол, н-пропанол, изопропанол, н-бутанол, изобутанол, трет-бутанол, изоамиловый спирт, диэтиленгликоль, глицерин, октанол, циклогексанол или метилцеллозольв; кетоны, такие как ацетон, метилэтилкетон, метилизобутилкетон, изофорон или циклогексанон, нитросоединения, такие как нитроэтан или нитробензол; нитрилы, такие как ацетонитрил или изобутиронитрил; амиды, такие как формамид, диметилформамид, диметилацетамид или гексаметилфосфорный триамид; и сульфоксиды, такие как диметилсульфоксид или сульфолан; более предпочтительно спирты (особенно метанол и этанол) и дихлорметан, и в случае когда деблокирующим реагентом является уксусная кислота - смесь уксусной кислоты с водой.
Нет каких-либо особых ограничений в отношении природы используемого деблокирующего реагента при условии, что он может обычно использоваться для обычного деблокирования. Когда защитной группой является триарилметильная группа, она может удаляться с использованием кислот, таких как уксусная кислота, дихлоруксусная кислота, трифторуксусная кислота, соляная кислота или кислота Льюиса, такая как бромид цинка, предпочтительно уксусная кислота, дихлоруксусная кислота или трифторуксусная кислота.
Температура реакции варьирует в зависимости от характера реагента, исходного соединения, используемого растворителя и других реакционных условий, но обычно реакция осуществляется при температуре от -10 до 100oC, предпочтительно 0-50oC.
Время, требуемое для реакции, варьирует в зависимости от природы используемого исходного соединения и растворителя, а также от температуры реакции, и обычно реакция завершается за период от 1 мин до 50 ч, предпочтительно в пределах 1 мин - 24 ч.
После завершения реакции требуемое соединение может выделяться из реакционной смеси. На стадии 3 одним из примеров таких приемов является нейтрализация реакционной смеси, выливание ее в воду; экстракция не смешиваемым с водой растворителем, таким как бензол, эфир или этилацетат; и отгонка растворителя из экстракта. Полученный таким образом продукт может обычно использоваться в последующей реакции без дополнительной очистки, но при желании может очищаться с помощью разнообразных приемов хроматографии, перекристаллизации или аналогичных способов. На стадиях 11, 22 и 25 пример таких приемов включает: отделение желаемого соединения с помощью фильтрования и промывку его метиленхлоридом.
Стадии 4 и 6
На этих стадиях соединение (2-6) или (2) может получаться с помощью реакции соединения (2-4) или (2-1) с соединением (2-5) (в указанной форме Ha1 обозначает атом галогена) в присутствии инертного растворителя и основания.
Примером используемого галогенидного фрагмента соединения (2-5) является хлор, бром или йод, предпочтительно хлор или бром.
Примеры предпочтительных растворителей включают: ароматические углеводороды, такие как бензол, толуол или ксилол; галоидированные углеводороды, такие как дихлорметан, хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол или дихлорбензол; сложные эфиры, такие как этилформиат, этилацетат, пропилацетат, бутилацетат или диэтилкарбонат; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан или диэтиленгликольдиметиловый эфир; кетоны, такие как ацетон, метилэтилкетон, метилизобутилкетон, изофорон или циклогексанон; нитросоединения, такие как нитроэтан или нитробензол; нитрилы, такие как ацетонитрил или изобутиронитрил; амиды, такие как формамид, диметилформамид, диметилацетамид или гексаметилфосфорный триамид; и сульфоксиды, такие как диметилсульфоксид или сульфолан; более предпочтительно простые эфиры (особенно тетрагидрофуран), кетоны (особенно ацетон), галоидированные углеводороды (особенно дихлорметан), амиды (особенно ДМФ) и ароматические амины (особенно пиридин).
Примеры предпочтительных оснований включают: органические основания (особенно триэтиламин, пиридин, N-метилморфолин, DBU и аналогичные основания), гидриды щелочных металлов (особенно гидрид натрия) и карбонаты щелочных металлов (особенно карбонат натрия и карбонат лития), и, если Z соединения (2-5) представляет атом кремния, наиболее предпочтительным органическим основанием является имидазол.
Реакционная температура не является особенно существенной, и обычно реакция осуществляется при температуре от 0 до 100oC, предпочтительно в интервале 20 - 60oC.
Время, требуемое для реакции, обычно варьирует от 5 мин до 30 ч. Когда реакция осуществляется при 50oC, она завершается за 10 ч.
После завершения реакции желаемое соединение может выделяться из реакционной смеси, например, с помощью нейтрализации реакционной смеси, отфильтровывания нерастворимых веществ, если они есть, добавления воды и не смешиваемого с водой органического растворителя, такого как этилацетат, отделения органического слоя, содержащего требуемое соединение, промывки экстракта водой, сушки экстракта над безводным сульфатом магния и др., и, наконец, отгонки растворителя. Полученное таким образом требуемое соединение, если необходимо, может очищаться с помощью общепринятых приемов, например перекристаллизации, переосаждения, хроматографии или аналогичных средств.
Стадия 5
На данной стадии соединение (2) может получаться с помощью взаимодействия соединения (2-5) с деблокирующим реагентом в присутствии инертного растворителя.
/1/ Когда гидроксилзащищающей группой в 3'-положении является силильная группа, она может обычно удаляться с помощью обработки соединением, способным давать фторидный ион, таким как тетрабутиламмоний - фторид.
Нет никаких особых ограничений в отношении природы используемого растворителя при условии, что он не оказывает вредного воздействия на реакцию. Предпочтительные примеры включают простые эфиры, такие как тетрагидрофуран или диоксан.
Температура реакции не является особенно критической, и обычно реакция осуществляется при температуре от -30 до 100oC, предпочтительно при 0 - 30oC.
Время, необходимое для реакции, составляет обычно от 5 мин до 30 ч. Когда реакция осуществляется при 20oC, она завершается за 10 ч.
После завершения реакции требуемое соединение может выделяться из реакционной смеси, например, с помощью надлежащей нейтрализации реакционной смеси, отфильтровывания нерастворимых веществ, если они имеются, добавления воды и не смешиваемого с водой органического растворителя, такого как этилацетат, отделения органического слоя, содержащего требуемое соединение, промывки экстракта водой, сушки экстракта над безводным сульфатом магния и др. , и, наконец, отгонки растворителя. Полученный таким образом желаемый продукт, если необходимо, может очищаться с помощью обычных средств, например перекристаллизации, переосаждения, хроматографии или аналогичных приемов.
Полученный таким образом желаемый продукт, если необходимо, может дополнительно очищаться с помощью обычных приемов, например перекристаллизации, переосаждения, хроматографии или аналогичных средств.
/2/ Когда гидроксилзащищающей группой в 3'-положении является галоидалкоксикарбонильная группа, она может обычно удаляться с помощью обработки цинковым порошком.
Нет никаких особых ограничений в отношении природы используемого растворителя при условии, что он не оказывает отрицательного воздействия на реакцию. Примеры подходящих растворителей включают уксусную кислоту, спирты или смеси воды и одного или более из этих растворителей.
Температура реакции не является особенно существенной, и реакция обычно осуществляется при температуре 0 - 100oC, предпочтительно при комнатной температуре.
Время, необходимое для реакции, обычно составляет от 5 мин до 30 ч. Когда реакция осуществляется при комнатной температуре, она завершается за 10 ч.
После завершения реакции требуемое соединение может выделяться из реакционной смеси, например, с помощью нейтрализации реакционной смеси или отфильтровывания нерастворимых веществ, если они есть, добавления воды и не смешиваемого с водой органического растворителя, такого как этилацетат, отделения органического слоя, содержащего требуемое соединение, промывки экстракта водой, сушки над безводным сульфатом магния и др., и, наконец, отгонки растворителя. Полученный таким образом реакционный продукт может дополнительно очищаться с помощью обычных средств, например, перекристаллизации, переосаждения, хроматографии или аналогичных средств.
/3/ Когда гидроксилзащищающей группой в 3'-положении является арилкилоксикарбонильная группа, она может обычно удаляться с помощью каталитического восстановления или окисления.
Нет никаких особых ограничений в отношении природы катализатора, используемого для восстановления, при условии, что он обычно может использоваться при каталитическом восстановлении. Примеры предпочтительных катализаторов включают палладий на активированном угле, никель Ренея, окись платины, платиновую чернь, родий на окиси алюминия, сочетание трифенилфосфина и хлорида родия и палладия на сульфате бария.
Давление водорода, при котором осуществляется реакция, не особенно существенно, но реакция обычно осуществляется при давлении в интервале от 1 до 10 атм.
Температура реакции и время, требуемое для реакции, варьируют в зависимости от характера исходного соединения и используемого растворителя, а также от типа катализатора и других условий реакции, и реакция осуществляется при температуре от 0 до 100oC и завершается за период времени от 5 мин до 24 ч.
Нет каких-либо особых ограничений в отношении природы используемого растворителя при деблокировании с помощью окисления при условии, что он не оказывает вредного воздействия на реакцию. Примерами предпочтительных растворителей являются смеси воды и одного или более органических растворителей.
Примеры предпочтительных органических растворителей включают: кетоны, такие как ацетон; галоидированные углеводороды, такие как дихлорметан, хлороформ или четыреххлористый углерод; нитрилы, такие как ацетонитрил; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран или диоксан; амиды, такие как диметилформамид, диметилацетамид или гексаметилфосфорный триамид; и сульфоксиды, такие как диметилсульфоксид.
Нет никаких особых ограничений в отношении природы используемого окисляющего агента при условии, что он может использоваться при обычном окислении. Предпочтительными примерами являются персульфат калия, персульфат натрия, цериевый нитрат аммония (CAN) и 2,3-дихлор-5,6-дициано-n-бензохинон (DDQ).
Температура реакции и время, требуемое для реакции, изменяются в зависимости от природы исходного соединения и растворителя, а также от типа используемого катализатора и других условий, но обычно реакция осуществляется при температуре 0 - 150oC, и завершается она за период от 10 мин до 24 ч.
Гидроксилзащищающая группа может также удаляться с помощью обработки щелочными металлами, такими как литий или натрий в жидком аммиаке или спиртах, таких как метанол или этанол, при температуре от -78 до -20oC.
Далее защитная группа может удаляться с использованием сочетания хлористого алюминия и йодистого натрия или алкилсилилгалогенидов, таких как триметилсилилиодид, в растворителе.
Нет никаких особых ограничений в отношении природы используемого растворителя при условии, что он не оказывает отрицательного влияния на реакцию. Предпочтительными примерами растворителей являются нитрилы, такие как ацетонитрил, галоидированные углеводороды, такие как дихлорметан или хлороформ, и смеси двух или более этих растворителей.
Реакционная температура и время, требуемое для реакции, варьируют в зависимости от природы исходного соединения и растворителя и от других условий реакции, но обычно реакция осуществляется при температуре 0 - 50oC и завершается за период от 5 мин до 3 дн.
Когда субстрат содержит атом серы, деблокирование может предпочтительно достигаться при использовании сочетания хлорида алюминия и йодистого натрия.
После завершения реакции требуемое соединение выделяется из реакционной смеси, например, с помощью соответствующей нейтрализации реакционной смеси или отфильтровывания нерастворимых материалов, если они есть, добавления воды и не смешиваемого с водой органического растворителя, такого как этилацетат, отделения органического слоя, содержащего желаемое соединение, промывки экстракта водой, сушки над безводным сульфатом магния и др., и, наконец, отгонки растворителя. Полученное таким образом требуемое соединение, если необходимо, может далее очищаться с помощью общепринятых средств, например с помощью перекристаллизации, переосаждения, хроматографии или аналогичных приемов (стадии 7, 12 ,14, 17, 23, 26, 28, 30, 33 и 35).
На этих стадиях связанные с CPG олигодезоксирибонуклеотиды (3), (4a), (4b), (5), (6a) - (6d), (7a) и (7b) могут получаться с помощью использования CPG-связанных нуклеозидов, включающих 3'-концевые нуклеозиды указанной эффективной последовательности оснований, повторения стадии удлинения ДНК-цепи на ДНК-синтезаторе, и, наконец, получения одного нуклеотидного короткого фрагмента с 5' конца указанной эффективности последовательности.
Удлинение ДНК-цепи на ДНК-синтезаторе поясняется в соответствии с "фосфорамидитным" подходом, но следует понимать, что данный метод представлен здесь с целью описания, но не для ограничения им.
На стадии 7 промышленно доступный CPG-связанный D" (3-1) обрабатывается реагентом деблокирования ДМТ-группы на ДНК-синтезаторе для удаления 5'-терминальной ДМТ-группы и затем конденсируется с нуклеотидным звеном, выпускаемым для ДНК-синтезатора, с последующим образованием фосфористокислотной триэфирной связи, которая впоследствии окисляется в триэфир фосфористой кислоты с использованием соответствующего окисляющего агента. После синтезирования одного нуклеотидного короткого фрагмента с 5' конца указанной эффективной последовательности путем повторения этих стадий получается CPG-связь ODN (3), имеющая 5'-терминальную ДМТ-группу. CPG-связанный ODN требуемой нуклеотидной последовательности оснований, 5'-терминал которой защищен ДМТ-группой, может синтезироваться в соответствии с процедурой, описанной авторами H. Koster и др. в Nucleic Acid Res., 12, 4539 /1984/, или модифицированной процедурой с использованием синтезатора, основанного на фосфорамидитном методе, например Модель 380В (продукт фирмы Эпплайд Биосистемз Инк) или Циклон Плюс (продукт Милиген /Биосерч).
Фрагмент основания (основной фрагмент) нуклеотидного звена, используемого для синтезирования ODN, является фрагментом, который защищается ацильной группой. Примерами предпочтительных ацильных групп являются бензоильная группа в случае, когда основной фрагмент представляет A или C, и изобутирильная группа, когда им является G.
Примеры кислотных веществ, используемых в качестве катализатора в реакции конденсации данной стадии, включают кислые вещества, включающие тетразолы, предпочтительно тетразол.
Нет каких-либо особых ограничений в отношении природы используемого растворителя при условии, что он не оказывает отрицательного воздействия на реакцию. Примерами предпочтительных растворителей являются ацетонитрил и тетрагидрофуран.
Реакция осуществляется при температуре от -30 до 50oC, обычно при комнатной температуре.
Время, необходимое для реакции, варьирует в зависимости от температуры реакции, и реакция завершается за период от 1 мин до 20 ч. Когда реакция осуществляется при комнатной температуре, она заканчивается в пределах 10 мин.
Нет каких-либо особых ограничений в отношении природы используемого на данных стадиях окисляющего агента при условии, что он может использоваться в качестве окисляющего агента для обычных реакций окисления. Примеры предпочтительных окисляющих агентов включают: окисляющие агенты на основе неорганических металлических агентов, включающие окислы марганца, такие как пенманганат калия или двуокись калия; окислы рутения, такие как тетраокись рутения; селеновые соединения, такие как двуокись селена; соединения железа, такие как хлорид железа (3); соединения осмия, такие как тетраокись осмия; соединения серебра, такие как окись серебра; ртутные соединения, такие как ацетат ртути; окисносвинцовые соединения, такие как окись свинца или тетраокись свинца; хромовокислотные соединения, такие как хромат калия, комплексы сульфата хрома и серной кислоты или комплексы хромовой кислоты и пиридина; соединения церия, такие как аммониевый нитрат церия (CAN); неорганические окисляющие агенты, включающие молекулы галогена, такие как молекулярный хлор, бром или йод; периодаты, такие как периодат натрия; озон; перекись водорода; азотистые соединения, такие как азотистая кислота; соединения на основе хлористой кислоты, такие как хлорит калия или хлорит натрия; соединения надсерной кислоты, такие как персульфат калия или персульфат натрия; органические окисляющие агенты, включающие реагенты, используемые при окислении с использованием DMCO (комплекс диметилсульфоксида и дициклогексилкарбодиимида, оксалилхлорида, уксусного ангидрида или пятиокиси фосфора, или комплексы пиридина и трехокиси серы); перекиси, такие как трет-бутилгидроперекись; устойчивые катионы, такие как трифенилметильный катион; сукцинимиды, такие как N-бромсукцинимид; соединения гипохлористой кислоты, такие как трет-бутилгипохлорит; соединения азодикарбоновой кислоты, такие как азодикарбоксилат; сочетания трифенилфосфина и дисульфидов, такие как диметилдисульфид или дифенилдисульфид; азотистые сложные эфиры, такие как метилнитрит; тетрагалоидуглероды; такие как тетрабромметан; хиноны, такие как 2,3-дихлор-5,6-дициано-n-бензохинон (DDQ); предпочтительно йод.
Нет каких-либо особых ограничений в отношении характера используемого растворителя при условии, что он не оказывает отрицательного воздействия на реакцию. Примеры предпочтительных растворителей включают: ароматические углеводороды, такие как бензол, толуол или ксилол; галоидированные углеводороды, такие как дихлорметан или хлороформ; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан или диметоксиэтан; амиды, такие как диметилформамид, диметилацетамид или гексаметилфосфорный триамид; сульфоксиды, такие как диметилсульфоксид; спирты, такие как метанол, этанол, н-пропанол, изопропанол, н-бутанол, изобутанол или изоамиловый спирт; разбавленные кислоты, такие как водная серная кислота; разбавленные основания, такие как водный раствор гидроокиси натрия; воду; кетоны, такие как ацетон или метилэтилкетон; гетероциклические амины, такие как пиридин; и нитрилы, такие как ацетонитрил; предпочтительно гетероциклические амины (особенно пиридин); нитрилы (особенно ацетонитрил), простые эфиры (особенно тетрагидрофуран) и галоидированные углеводороды (особенно дихлорметан).
Реакция осуществляется при температуре от -50 до 100oC. Время, требуемое для реакции, варьируется в зависимости от температуры реакции, а также от характера исходного соединения и растворителя, и реакция осуществляется при температуре от -50 до -100oC и завершается обычно за период от 30 мин до 15 ч. Реакция окисления, описанная выше, ускоряется добавлением катализатора фазового переноса, такого как триэтилбензиламмонийхлорид или трибутилбензиламмонийбромид.
Стадии 12, 14, 17, 23, 26, 28, 30, 33 и 35 являются аналогичными.
Стадии 9 и 20
На этих стадиях полуэфир дикарбоновой кислоты может приготавливаться с помощью реакции свободной гидроксильной группы соединения (4-3) или (6-2) с ангидридом дикарбоновой кислоты, таким как янтарный ангидрид в присутствии катализатора-основания (основного катализатора).
Нет каких-либо особых ограничений в отношении характера используемой дикарбоновой кислоты. Предпочтительными дикарбоновыми кислотами являются кислоты, которые содержат 2-10 атомов углерода, и наиболее предпочтительной дикарбоновой кислотой является янтарная или глутаровая кислота. Примеры подходящих основных катализаторов включают: аминопиридины, такие как диметиламинопиридин или пирролидинопиридин; третичные амины, такие как триметиламин или триэтиламин; бикарбонат натрия; и карбонаты щелочных металлов, такие как карбонат калия; наиболее предпочтительно диметиламинопиридин.
Нет каких-либо особых ограничений в отношении природы используемого растворителя при условии, что он не оказывает отрицательного воздействия на реакцию и способен растворять исходный материал в какой-то степени. Примеры предпочтительных растворителей включают: ароматические углеводороды, такие как бензол, толуол или ксилол; галоидированные углеводороды, такие как дихлорметан или хлороформ; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан или диметоксиэтан; амиды, такие как диметилформамид, диметилацетамид или гексаметилфосфорный триамид; сульфоксиды, такие как диметилсульфоксид; спирты, такие как метанол, этанол, н-пропанол, изопропанол, н-бутанол, изобутанол или изоамиловый спирт; разбавленные кислоты, такие как водная серная кислота; разбавленные основания, такие как водный раствор гидроокиси натрия; воду; кетоны, такие как ацетон или метилэтилкетон, гетероциклические амины, такие как пиридин; и нитрилы, такие как ацетонитрил, более предпочтительно нитрилы (особенно ацетонитрил), простые эфиры (особенно тетрагидрофуран) и галоидированные углеводороды (особенно дихлорметан).
Реакция осуществляется при температуре от -50 до 100oC. Время, требуемое для реакции, варьируется в зависимости главным образом от температуры реакции, а также от характера исходного соединения и используемого растворителя, но обычно реакция завершается в течение периода от 30 мин до 15 ч.
Стадии 10 и 21
На этих стадиях требуемые соединения (4-5) и (6-6) могут получаться с помощью взаимодействия полуэфира янтарной кислоты (4-3) и (6-4), полученного на стадиях 9 и 20, с фенолами, такими как пентахлорфенол, в присутствии конденсирующего агента с получением активированного сложного эфира, и впоследствии реакции продукта с CPG-аминами (4-4) и (6-5) в присутствии основания.
Нет каких-либо особых ограничений в отношении природы используемых в данной реакции фенолов. Примерами предпочтительных фенолов являются пентахлорфенол и 4-нитрофенол.
Нет каких-либо особых ограничений в отношении природы используемого растворителя при условии, что он не оказывает отрицательного воздействия на реакцию и способен в некоторой степени растворять исходный материал. Примеры подходящих растворителей включают: амиды, такие как диметилформамид, диметилацетамид или гексаметилфосфорный триамид; сульфоксиды, такие как диметилсульфоксид; кетоны, такие как ацетон или метилэтилкетон; гетероциклические амины, такие как пиридин; и нитрилы, такие как ацетонитрил, предпочтительно амиды, такие как диметилформамид.
Нет никаких особых ограничений в отношении используемого основания при условии, что оно может использоваться в качестве основания в обычной реакции. Примеры предпочтительных оснований включают: органические основания, такие как триэтиламин, трибутиламин, диизопропилэтиламин, N - метилморфолин, пиридин, 4-(N,N-диметиламино)пиридин, N,N-диметиланилин, N,N - диэтиланилин, 1,5 - диазабицикло(4, 3, 0)нон-5-ен, 1,4-диазабицикло(2,2,2)октан (DАВСО) или 1,8-диазабицикло(5,4,0)ундец-7-ен(DВU); более предпочтительно органические основания, особенно триэтиламин, пиридин, П-метилморфолин и DВU.
Реакция осуществляется при температуре от -50 до 100oC.
Время, требуемое для реакции, варьирует главным образом в зависимости от температуры реакции, а также от природы исходного соединения или используемого растворителя, но, когда реакция осуществляется при комнатной температуре, она обычно завершается в течение периода 30 - 50 ч.
Стадии 13 и 32
На стадии 13 связанный с CPG нуклеозид (4-7), содержащий тиоатную группу, может быть получен с помощью реакции соединения (4-6), полученного на стадии П, с промышленного доступным 5' - О-ДМТ - нуклеозид-3'-фосфорамидитным реагентом на ДНК - синтезаторе и последующей реакции с реагентом, дающим тиоат (тиоирующий агент).
Нет каких-либо особых ограничений в отношении природы используемого агента образования тиоата при условии, что он способен образовывать тиоатную группу при реакции с трехвалентным фосфором. Примеры предпочтительных тиоатобразующих реагентов включают: в дополнение к сере тетраэтилтиурамдисульфид (TETD) (продукт фирмы Эпплайд Биосистемз Инк) и реагент Веаисаде (продукт Миллиген/Биосерч). Требуемое соединение (4-7), в котором 3' - концевой нуклеозид указанной эффективной последовательности оснований осажден на CPG через тиоатную группу, может получаться с помощью обработки тетраэтилтиурамдисульфидом (TETD) согласно процедуре, описанной в Tetrahedron Letters, 32, 3005 /1991/, или реагентом Beaucage согласно процедуре, описанной в J. Am. Chem. Soc. , 112, 1253 /1990/ или в соответствии с ее модифицированной процедурой.
На стадии 32 CPG - связанный нуклеозид (7-7), имеющий фосфоротриэфирную или тиоатную группу, может получаться с помощью реакции соединения (7-3) с фосфорамидитным реагентом с последующей обработкой с помощью обычных средств или тиоатобразующим агентом.
Стадия 15
На данной стадии CPG-связанное соединение (5-3), имеющее фосфоновокислотную диэфирную группу, может получаться с помощью реакции CPG-связанного соединения (5-1), освобожденного от ДМТ-группы, которое получается по аналогии с получением CPG-связанного соединения (4-6), с промышленно доступным фосфоновокислотным моноэфирным соединением (5-2) в присутствии конденсирующего агента и агента, устраняющего кислотность. Нет каких-либо особых ограничений в отношении природы используемого конденсирующего агента при условии, что он может образовывать ангидрид кислоты с фосфоновокислотным моноэфиром. Примеры предпочтительных конденсирующих агентов включают адамантан-1-карбонилхлорид и пивалоилхлорид. Нет каких-либо особых ограничений в отношении характера используемого агента, устраняющего кислотность, при условии, что он может использоваться в качестве устраняющего кислотность агента в случае, если ацилирование осуществляется с использованием хлорангидрида кислоты. Примеры предпочтительных устраняющих кислотность агентов обычно включают ароматические амины, такие как пиридин. Нет каких-либо особых ограничений в отношении характера используемого растворителя при условии, что он не оказывает отрицательного воздействия на реакцию. Примеры предпочтительных растворителей включают нитрилы, такие как безводный ацетонитрил. Когда реакция осуществляется при комнатной температуре, она завершается в течение 5 - 60 мин.
Стадия 16
На данной стадии фосфоновокислотная диэфирная группа CPG-связанного соединения (5-3) трансформируется в фосфорамидатную группу с помощью реакции с алкиламином и четыреххлористым углеродом. В качестве алкиламина следует иметь в виду требуемые алкиламины. Нет каких-либо особых ограничений в отношении характера используемого растворителя при условии, что он не оказывает отрицательного воздействия на реакцию. Примеры предпочтительных растворителей включают обычно неполярные растворители, такие как четыреххлористый углерод. Температура реакции не является особенно существенной, и обычно реакция осуществляется при температуре от -50 до 100oC. Когда реакция проводится при комнатной температуре, она завершается в течение 1 - 10 ч.
Стадии 19 и 36
На этих стадиях 3' - фосфористокислотные производные (6-3) и (8) могут получаться по реакции соединений (6-2) и (2) с хлорфосфорамидитом (6-3'), который используется в качестве фосфотилирующего агента, в присутствии инертного растворителя и агента, устраняющего кислотность. Символ U, используемый при определении соединения (6-3'), означает диалкиламиногруппу, такую как диметиламино или диизопропиламиногруппа, или гетероциклическую группу, имеющую 1 или 2 атома кислорода и/или азота в кольце. Символ V, используемый при определении соединения (6-3'), может быть любой группой при условии, что она может удаляться после образования фосфатной связи. Примеры таких групп включают предпочтительно низкие алкилокси группы, такие как метоксигруппа, и цианоалкилоксигруппы, такие как цианоэтилоксигруппа. В качестве соединения (6-3) могут быть упомянуты особенно фосфины, такие как хлорморфолинометоксифосфин, хлорморфолиноцианоэтоксифосфин, хлордиметиламинометоксифосфин, хлордиметиламиноцианоэтоксифосфин, хлордиизопропиламинометоксифосфин и хлордиизопропиламиноцианоэтоксифосфин; предпочтительно хлорморфолинометоксифосфин, хлорморфолиноцианоэтоксифосфин, хлордиизопропиламинометоксифосфин и хлордиизопропиламиноцианоэтоксифосфин.
Нет каких-либо особых ограничений в отношении природы используемого растворителя при условии, что он не оказывает отрицательного воздействия на реакцию. Предпочтительными растворителями являются простые эфиры, такие как тетрагидрофуран, диэтиловый эфир или диоксан. Примеры устраняющих кислотность агентов включают: гетероциклические амины, такие как пиридин или диметиламинопиридин, и алифатические амины, такие как триметиламин, триэтиламин или диизопропилэтиламин, особенно алифатические амины (особенно диизопропилэтиламин).
Температура реакции не является особенно существенной, и обычно реакция осуществляется при температуре от -50 до 50oC, предпочтительно при комнатной температуре.
Время, требуемое для реакции, варьирует в зависимости от характера исходного соединения и реагента, а также от температуры реакции, и обычно реакция заканчивается за период от 5 мин до 30 ч. Когда реакция предпочтительно осуществляется при комнатной температуре, она завершается в течение 30 мин.
Требуемое соединение может выделяться из реакционной смеси, например, с помощью надлежащей нейтрализации реакционной смеси или отфильтровывания нерастворимых веществ, если они есть, добавления воды и не смешиваемого с водой растворителя, такого как этилацетат, отделения органического слоя, содержащего желаемое соединение, промывки экстракта водой, сушки над безводным сульфатом магния и проч., и, наконец, отгонки растворителя.
Полученное таким образом требуемое соединение, если необходимо, может далее очищаться с помощью общепринятых приемов, например, перекристаллизации, переосаждения, хроматографии или аналогичных средств.
Стадия 24.
На данной стадии CPG-связанное соединение (6-8), имеющее фосфористотриэфирную группу, может быть получено по аналогии с процедурой, описанной для стадии 7, но с использованием соединения (6-3) вместо нуклеозид-фосфорамидитного соединения, используемого в качестве нуклеотидного звена, на ДНК-синтезаторе.
Стадии 27 и 29
На данных стадиях CPG-связанные соединения (6-10 и 6-11), имеющие 2 или 3 фосфористо-триэфирные группы, могут получаться из CPG-связанного соединения (6-9), полученного на стадии 25, с помощью обработки по аналогии с процедурой, описанной для стадии 24.
Стадия 31
На данной стадии может получаться CPG-связанное соединение (7-3), имеющее 4,4'-диметокситритилоксиэтилсульфонилэтоксигруппу, с помощью обработки и промышленно доступного CPG - связанного соединения (7-2), освобожденного от 5'-ДМТ-группы, (2-цианоэтокси)-2-[2'-0-(4,4'-диметокситритилоксиэтилсульфонил)] этокси-N,N -диизопропиламинофосфином (7-1) /T. Horn и др., Tetrahedron Letters, 27, 4705 /1986// по аналогии с процедурой, описанной на стадии 24.
Стадия 34
На данной стадии CPG-связанное соединение (7-5) может получаться с помощью делокирования ДМТ-группы у CPG-связанного соединения (7-3), полученного на стадии 31, а затем конденсации с нуклеотидом (7-4), имеющим ДМТ-группу в 5'-положении и группу алкилфосфата, фенилфосфата, алкилфосфоната или фенилфосфоната в 3'-положении, с использованием конденсирующего агента.
Нет каких-либо особых ограничений в отношении характера используемого растворителя при условии, что он не оказывает отрицательного воздействия на реакцию. Предпочтительными растворителями являются ароматические амины, такие как пиридин. В качестве конденсирующих агентов могут использоваться дициклогексилкарбодиимид (DCC), мезитиленсульфонилхлорид (M-CI), триизопропилбензолсульфонилхлорид, мезитиленсульфонилтриазол (MST), мезитиленсульфонил-3-нитротриазол (MSNT), триизопропилбензолсульфонилтетразол (TPS-Te), триизопропилбензолсульфонилнитроимидазол (TPS-NI) и триизопропилбензолсульфонилпиридилтетразол, предпочтительно MSN, TPS - Te и TRS - NI.
Температура реакции не является особенно критической, и реакция осуществляется при температуре от -10 до 100oC. Время, требуемое для реакции, варьирует в зависимости от характера используемого растворителя и температуры реакции. Когда реакция осуществляется при комнатной температуре с использованием пиридина в качестве растворителя, она завершается в течение 30 мин.
Стадия 37
На данной стадии конечный продукт (1) может получаться с помощью конденсации соединения (8), полученного на стадии 36, с CPG-связанным ODN (3, 4a, 4b, 5, 6a - 6d, 7a или 7b), который синтезируется на ДНК-синтезатора, и деблокируется только 5'-терминальная ДМТ-группа, с использованием кислотного катализатора, с образованием фосфитного триэфира, окисления с использованием соответствующего окисляющего агента с получением фосфористокислотного триэфира, отщепления от CPG, удаления защитных групп и, наконец, очистки.
В качестве кислотного катализатора, используемого в реакции конденсации, можно использовать такие кислотные вещества, как тетразолы; предпочтительно тетразол.
Нет каких-либо особых ограничений в отношении природы окисляющего агента, который можно использовать, при условии, что он может использоваться в качестве окисляющего агента в реакциях окисления. Примеры предпочтительных окисляющих агентов включают: неорганические окиси металлов, включающие окиси марганца, такие как перманганат калия или двуокись марганца, окиси рутения, такие как тетраокись рутения; окиси селена, такие как двуокись селена; соединения железа, такие как хлорное железо; соединения осмия, такие как тетраокись осмия; соединения серебра, такие как окись серебра, ртутные соединения, такие как ацетат ртути, свинцовоокисные соединения, такие как окись свинца или тетраокись свинца; хромовокислотные соединения, такие как хромат калия, комплекс хромовой кислоты и серной кислоты или комплекс хромовой кислоты и пиридина, и соединения церия, такие как цериевый нитрат аммония (CAN); неорганические окисляющие агенты, включающие молекулярные галогены, такие как молекулярный хлор, бром или йод; периодаты, такие как периодат натрия, озон, перекись водорода; азотистые соединения, такие как азотистая кислота; соединения хлористой кислоты, такие как хлорид натрия; персульфатные соединения, такие как персульфат калия или персульфат натрия; и органические окисляющие агенты, включающие реагенты, используемые при окислении с ДМСО (сочетание диметилсульфоксида и дициклогексилкарбодиимида, оксалилхлорида, уксусного ангидрида или пятихлористого фосфора, или комплекс пиридина и серного ангидрида); перекиси, такие как трет-бутилгидроперекись; устойчивые катионы, такие как трифенилметильный катион; сукцинимиды, такие как П - бромсукцинимид; соединения гипохлористой кислоты, такие как трет-бутилгипохлорит; соединения азодикарбоновой кислоты, такие как азодикарбоксилат; сочетание дисульфидов, такие как диметилдисульфид, дифенилдисульфид или дипиридилдисульфид, и трифенилфосфина; нитриты, такие как метилнитрит; тетрагалоидированные соединения, такие как тетрабромметан; и хиноновые соединения, такие как 2,3 - дихлор - 5,6 - дициано-n-бензохинон (DDQ); более предпочтительно йод.
Нет каких-либо особых ограничений в отношении природы используемого растворителя при условии, что он не оказывает отрицательного воздействия на реакцию и способен в некоторой степени растворять исходные материалы. Примеры предпочтительных растворителей включают: ароматические углеводороды, такие как бензол, толуол или ксилол; галоидированные углеводороды, такие как дихлорметан или хлороформ; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан или диметоксиэтан; амиды, такие как диметилформамид, диметилацетамид или гексаметилфосфорный триамид; сульфоксиды, такие как диметилсульфоксид; спирты, такие как метанол, этанол, н-пропанол, изопропанол, н-бутанол, изобутанол или изоамиловый спирт; разбавленные кислоты, такие как водная серная кислота; разбавленные основания, такие как водный раствор гидроокиси натрия; воду; кетоны, такие как ацетон или метилэтиленкетон; гетероциклические амины, такие как пиридин; нитрилы, такие как ацетонитрил, предпочтительно гетероциклические амины (особенно пиридин), нитрилы (особенно ацетонитрил), простые эфиры (особенно тетрагидрофуран) и галоидированные углеводороды (особенно дихлорметан).
Реакция проводится при температуре от -50 до 100oC. Время, требуемое для реакции, варьируется главным образом в зависимости от температуры реакции и характера исходного соединения и составляет от 30 мин до 15 ч. Реакция окисления, описанная выше, ускоряется добавлением катализатора фазового переноса, такого как триэтилбензиламмонийхлорид или трибутилбензиламмонийбромид.
Стадия 38
На данной стадии может получаться промежуточное соединение мононуклеотид (19) с помощью реакции соединения (2) с фосфорилирующим агентом, пригодным для введения YP(=O) (OH) - например, с использованием бистриазолид, в присутствии инертного растворителя, и добавления воды с последующей обработкой.
Нет каких-либо особых ограничений в отношении природы используемого растворителя при условии, что он не оказывает отрицательного воздействия на реакцию. Растворитель обычно выбирается из ароматических аминов, таких как пиридин. Нет никаких особых ограничений в отношении характера радикала Y, используемого при определении фосфорилирующего агента, при условии, что он может удаляться в условиях удаления защитной группы основного фрагмента после завершения реакции конденсации стадии 39. Символ Y обозначает обычно о-хлорфеноксигруппу.
Хотя температура реакции не является особенно критической, реакция осуществляется при температуре от -20 до 100oC, обычно при комнатной температуре. Время, необходимое для реакции, варьирует в зависимости от природы используемого растворителя и температуры реакции. Когда реакция осуществляется при комнатной температуре с использованием пиридина в качестве растворителя, она завершается в течение 1 ч.
Стадия 39
На данной стадии конечный продукт (1) может получаться с помощью конденсации мононуклеотида (9) с CPG-связанным ODN (3, 4a, 4b, 5, 6a - 6d, 7a или 7b), который синтезируется на ДНК-синтезаторе, и деблокируется 5'-терминальная ДМТ-группа, имеющая защитные группы во фрагментах основания и фосфорной кислоты, с использованием конденсирующего агента с образованием фосфорно-кислотной триэфирной группы, отщепления от CPG с помощью обычных средств, деблокирования защитной группы и, наконец, очистки. Нет никаких особых ограничений в отношении характера используемого растворителя при условии, что он не оказывает отрицательного воздействия на реакцию.
Примеры конденсирующих агентов включают: дициклокарбодиимид (ДОС), мезитиленсульфонилхлорид (Ms - Cl), триизопропилбензолсульфонилхлорид, мезитиленсульфонилтриазол (MST), мезитиленсульфонил-3-нитротриазол (MSNT), триизопропилбензолсульфонилтетразол (TPS - Te), триизопропиленбензолсульфонилнитроимидазол (TPS-NI) и триизопропилбензолсульфонилпиридилтетразол, предпочтительно MSNT, TPS-Te и TPS-NI.
Температура реакции не имеет особого значения, и реакция осуществляется при температуре от -10 до 100oC, обычно при комнатной температуре. Время, требуемое для реакции, варьирует в зависимости от природы используемого растворителя и температуры реакции. Когда реакция осуществляется при комнатной температуре с использованием пиридина в качестве растворителя, она завершается за 30 мин.
Отщепление ODN от CPG-связанного ODN и удаление защитных групп за исключением 5' - терминального заместителя осуществляются в соответствии с известными методами (J. Am. Chem. Soc., 103, 3185 /1981/).
Реакционная смесь, содержащая соединения общей формулы (1), очищается с помощью общепринятых приемов очистки, например с помощью различных методов хроматографии, включая обращенно-фазовую и ионно-обменную хроматографию (включающую высокоскоростную жидкостную хроматографию), с получением соединений, имеющих указанную общую формулу (1).
Стадия 40
На данной стадии трис(1,2,4)-триазолилфосфит, который ранее получен из треххлористого фосфора и 1,2,4-триазола согласно процедуре, описанной авторами B. C.Freohler, P.G.Ng и M.D. Matteucci в Nucleic Acids Res, 14, 5399 /1986/, подвергается реакции с соединением (2) в инертном растворителе, и реакция прекращается добавлением воды с последующей обработкой с получением нуклеозид 3'-H- фосфоната (10). Нет каких-либо особых ограничений в отношении природы используемого растворителя при условии, что он не оказывает отрицательного воздействия на реакцию. Предпочтительным растворителем является галоидированный углеводород, такой как дихлорметан Реакционная температура не является особенно критической, и обычно реакция осуществляется при температуре от -20 до 100oC. Обычно при комнатной температуре.
Время, требуемое для реакции, меняется в зависимости от природы используемого растворителя и реакционной температуры. Когда реакция осуществляется при комнатной температуре с использованием дихлорметана в качестве растворителя, она завершается в течение 10 мин.
Стадия 41
На данной стадии конечный продукт (1) может получаться с помощью взаимодействия нуклеозид 3'-H-фосфоната (10), полученного на стадии 40, с CPG-связанным ODN(3, 4a, 4b, 5, 6a - 6d, 7a или 7b), который синтезируется на синтезаторе, и деблокируется только 5'-терминальная ДМТ-группа, имеющая защитные группы во фрагментах основания и фосфорной кислоты, с использованием конденсирующего агента, такого как пивалоилхлорид, в присутствии агентов, устраняющих кислотность, с получением H-фосфоновокислотной диэфирной связи, преобразования H-фосфоновокислотной группы в фосфорно-кислотную диэфирную группу с использованием окисляющего агента, отщепления ODN от CPG, связанного ODN, и параллельно удаления защитной группы основного фрагмента в основных условиях, и, наконец, очистки. Нет каких-либо особых ограничений в отношении природы используемого растворителя при условии, что он не оказывает отрицательного воздействия на реакцию. Предпочтительным растворителем является безводный ацетонитрил. Примеры конденсирующих агентов включают хлорангидриды кислот и хлористый фосфор, предпочтительно пивалоилхлорид.
Нет никаких особых ограничений в отношении характера окисляющего агента, используемого для окисления H-фосфоновокислотной группы ODN в фосфорно-кислотную диэфирную группу, при условии, что он может использоваться в качестве окисляющего агента в реакциях окисления. Примеры подходящих окисляющих агентов включают: неорганические окиси металлов, включающие окиси марганца, такие как перманганат калия или двуокись марганца, окиси рутения, такие как четырехокись рутения, окиси селена, такие как двуокись селена; соединения железа, такие как хлорное железо, соединения осмия, такие как тетраокись осмия, соединения серебра, такие как окись серебра, ртутные соединения, такие как ацетат ртути; окисно-свинцовые соединения, такие как окись свинца или тетраокись свинца; соединения хромовой кислоты, такие как хромат калия, комплексы хромовой кислоты и серной кислоты или комплекс хромовой кислоты и пиридина; и соединения церия, такие как аммонийнитрат церия (CAN); неорганические окисляющие агенты, включающие молекулы галогенов, такие как молекулярный хлор, бром или йод; периодиды, такие как периодид натрия, озон; перекись водорода; азотистые соединения, такие как азотистая кислота; соединения хлористой кислоты, такие как хлорит натрия; и персульфатные соединения, такие как персульфат калия или персульфат натрия; и органические окисляющие агенты, включающие реагенты, используемые при окислении с применением ДМСО (сочетание диметилсульфоксида и дициклогексилкарбодиимида, оксалилхлорида, уксусного ангидрида или пятихлористого фосфора или комплекс пиридина и серного ангидрида); перекиси, такие как трет-бутилгидроперекись; устойчивые катионы, такие как трифенилметильный катион; сукцинимиды, такие как N-бромсукцинимид; соединения гипохлористой кислоты, такие как трет-бутилгипохлорит; соединения азодикарбоновой кислоты, такие как азодикарбоксилат; сочетание дисульфидов, таких как диметилдисульфид, дифенилдисульфид или дипиридилдисульфид, и трифенилфосфина, нитриты, такие как метилнитрит, тетрагалоидированные соединения, такие как тетрабромметан, и хиноновые соединения, такие как 2,3-дихлор-5,6-дициано-n-бензохинон (DDQ); более предпочтительно йод.
В качестве агентов, устраняющих кислотность, пригодны гетероциклические амины, такие как пиридин или диметиламинопиридин, и алифатические амины, такие как триметиламин, триэтиламин или диизопропилэтиламин, предпочтительно алифатические амины (особенно диизопропилэтиламин). Температура реакции не является особенно критической, и реакция обычно осуществляется при температуре от -50 до 50oC, предпочтительно при комнатной температуре.
Время, требуемое для реакции, варьирует в зависимости от характера исходного соединения и используемого растворителя, а также от реакционной температуры, и обычно реакция длится от 5 мин до 30 ч. Когда реакция осуществляется при комнатной температуре, она предпочтительно завершается в течение 30 мин.
Отщепление ODN от CPG-связанного ODN и удаление защитных групп, за исключением 5'-терминального заместителя осуществляются согласно известным методам (J. Am. Chem. Soc., 103, 3185 /1981/).
Реакционная смесь, содержащая соединения общей формулы (1), очищается с помощью обычных приемов очистки, например различных приемов хроматографии, включая хроматографию с обращенной фазой и ионно-обменную хроматографию (включающую высокоскоростную жидкостную хроматографию) с получением соединений, имеющих указанную общую формулу (1).
Настоящее изобретение далее представляет новый способ синтеза олигодезоксирибонуклеотидов и олигодезоксирибонуклеотидных производных. Новые олигодезоксирибонуклеотиды формулы (11), полученные с помощью нового процесса, в типичном случае включают новые соединения общей формулы (1). Кроме того, для использования в новом процессе настоящее изобретение представляет новые промежуточные соединения, как это будет определено более подробно.
В следующем описании для замещающих групп применяются различные определения, но между соответствующими определениями можно легко увидеть соответствие. Так, например, можно увидеть сходство между замещающей группой R1R2R3Z - соединений общей формулы (1) и замещающей группой R4R5R6Z-, показанной для соединений, применяемых и получаемых по новому способу. Кроме того, необходимо заметить, что группы, показанные с подстрочными указателями, такие как R1, R2, Y1 и др., не всегда идентичны с группами, показанными с надстрочными указателями, такими как R1, R2, Y1 и др.
В данном аспекте изобретения процесс, представляемый изобретением, включает получение олигодезоксирибонуклеотида, имеющего замещенный фосфат на 3'-конце. Наиболее подробно настоящее изобретение представляет новые линкерные соединения для использования с полимерными носителями (или подложками) при получении твердофазных материалов для синтеза олигодезоксирибонуклеотидов. Твердофазные материалы могут иметь защищенную гидроксигруппу на конце линкера с полимерным носителем, которая может затем деблокироваться и вводиться в реакцию для присоединения нуклеотидов.
Так, например, настоящее изобретение представляет линкерные соединения, такие как следующие соединения (12), которые с полимерными носителями могут давать защищенные твердофазные материалы (14) для деблокирования в соединения (22) и последующей реакции с получением соединений (24), (25) и (27) с присоединенными нуклеотидами.
В одном из аспектов настоящий процесс предназначен для получения соединения, представленного формулой (11):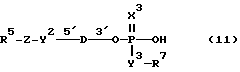
где
R4, R5 и R6 могут быть одинаковыми или отличными друг от друга и каждый независимо представляет атом водорода, акильную группу, имеющую 1-4 атома углерода, необязательно замещенную арильную группу или необязательно замещенную антрахинонильную группу; Z представляет атом углерода или кремния, R5, R6 и Z могут вместе представлять флуоренильную группу или ксантенильную группу; или R4, R5, R6 и Z могут вместе представлять атом водорода;
R7 представляет атом водорода, необязательно замещенную алкильную группу, имеющую 1-4 атома углерода, необязательно замещенную арильную группу или необязательно замещенную аралкильную группу, Y2 представляет атом кислорода, атом серы или NH-группу, Y3 представляет атом кислорода, атом серы, NH-группу, алкиленовую группу, имеющую 1 - 4 атома углерода; или фениленовую группу, X3 представляет атом кислорода или серы; и D представляет олигодезоксирибонуклеотид, имеющий длину цепи 2 - 30, при условии, что гидроксильные группы на 5' - и 3' - терминалах не включены в D;
указанный процесс включает:
реакцию соединения, представленного формулой (12):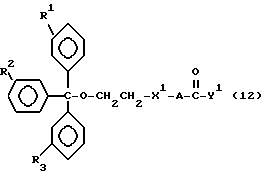
где
R1, R2 и R3 могут быть одинаковыми или различными и каждый независимо представляет атом водорода или алкоксигруппу, имеющую 1-4 атома углерода; X1 представляет группу -S-, -SO- или -SO2; A представляет группу - (CH2)h - (где h представляет целое число от 1 до 16), группу - (CH2)hO - (где h представляет целое число от 1 до 16), группу -(CH2)j-O-CO-(CH2)k - (где j представляет положительное целое число, k представляет 0 или положительное целое число и j + k равно от 2 до 16), или группу - (CH2)j; NH - CO - (CH2)k - (где j представляет положительное целое число, k представляет 0 или положительное целое число и j + k составляет от 2 до 16), группу - (CH2)j- S - CO - (CH2)k - (где j представляет положительное целое число, k представляет 0 или положительное целое число и j + k составляет 2 - 16) или группу -(CH2)n-O-CO-CH2(OCH2CH2)p- OCH2- (где n - положительное целое число, p -0 или положительное целое число) и Y1 представляет гидроксильную группу, необязательно замещенную фенилоксигруппу или этилоксигруппу, которая может быть замещена галогеном (с полимерным материалом, представленным общей формулой (13)):
(где
X2 представляет атом кислорода, атом серы или NH-группу, P представляет полимерный материал (при условии, что, когда Y1 в общей формуле (12) представляет гидроксильную группу, соединение общей формулы (12) подвергается взаимодействию с карбоновокислотным активирующим агентом перед реакцией с полимерным материалом (13) с образованием полимерного материала, представленного общей формулой (14):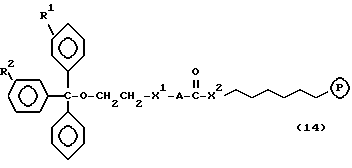
(где
R1, R2, R3, X1, A, X2 и P имеют значения, определенные выше; и подтверждение получающегося полимерного материала формулы (14) обработке с удлинением ДНК-цепи с использованием ДНК-синтезатора.
Данный процесс настоящего изобретения показан на схеме, приведенной в конце описания.
На приведенных схемах R1, R2, R3, R4, R5, R6, R7, X1, X2, X3, P, Y1, Y2, Y3, A и Z имеют значения, определенные выше (однако в соединениях иных, чем соединения (11), R4, R5, R6 и Z вместе не являются атомом водорода). R8a представляет метильную группу или цианоэтильную группу; R8b представляет фенильную группу, необязательно замещенную хлором; R8 представляет метильную группу, цианоэтильную группу или фенильную группу, необязательно замещенную хлором; R9a представляет атом водорода или водородную группу, имеющую защитную группу; X представляет атом галогена (предпочтительно хлор, бром и йод); A1 представляет алкиленовую группу, имеющую от 1 до 20 атомов углерода; m представляет целое число от 1 до 14; n - целое число от 1 до 28; B, B' и B'' каждый представляет основной фрагмент аденин-нуклеотида, гуанин-нуклеотида, тимин-нуклеотида, урацил-нуклеотида и цитозин-нуклеотида, защищенного в основном и фосфатном фрагментах (они должны выбираться для получения желаемой последовательности оснований); при условии, что B' представляет основной фрагмент 3'-терминального нуклеотида D, имеющего желаемую последовательность основания, B'' представляет основной фрагмент 5'-терминального нуклеотида D, имеющего желаемую последовательность оснований, D представляет желаемый олигодезоксирибонуклеотид (при условии, что 5'-терминальная гидроксильная группа и 3'-терминальная гидроксильная группа не включены); O представляет галоидальную группу, галоидфенильную группу или нитрофенильную группу, такую как 2,2,2 - трихлорэтильная группа, 2,2 - дихлорэтильная, 2 - хлорэтильная, 2,2,2 - трибромэтильная, 2,2 - дибромэтильная, 2 - бромэтильная, 2 - нитрофенильная, 4 - нитрофенильная, 2,4 - динитрофенильная, 2,4,5 - трихлорфенильная и 2,3,4,5,6 - пентахлорфенильная группа (предпочтительно трихлорэтильную и ортонитрофенильную группу);
V представляет низшую алкильную группу (предпочтительно метильную, этильную и изопропильную группы, особенно изопропильную группу).
Далее каждая стадия будет описана подробно.
Стадия 1
Данная стадия является стадией получения соединения (17) с помощью взаимодействия 2 - меркаптоэтанола (16) с ω -гилоидкарбоновой кислотой (15) в инертном растворителе в присутствии связывающего кислоту агента.
Применяемый растворитель особенно не ограничивается, если он не оказывает влияния на реакцию, и он включает ароматические углеводороды, такие как бензол, толуол и ксилол; галоидированные углеводороды, такие как метиленхлорид и хлороформ; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан и диметоксиэтан; амиды, такие как диметилформамид, диметилацетамид и гексаметилфосфортриамид; сульфоксиды, такие как диметилсульфоксид; спирты, такие как метанол, этанол, н-пропанол, изопропанол, н-бутанол, изобутанол и изоамиловый спирт; разбавленные кислоты, такие как водная серная кислота; водные основания, такие как водная гидроокись натрия; воду; кетоны, такие как ацетон, метилэтилкетон; гетероциклические амины, такие как пиридин; и нитрилы, такие как ацетонитрил, предпочтительно кетоны, такие как ацетон; и спирты, такие как этанол и пропанол.
Применяемый связывающий кислоту агент включает карбонаты щелочных металлов, такие как карбонат калия и карбонат натрия; бикарбонаты щелочных металлов, такие как бикарбонат натрия; гидроокиси щелочных металлов, такие как гидроокись натрия и гидроокись калия; и органические амины, такие как триэтиламин, предпочтительно карбонаты щелочных металлов (особенно карбонат калия).
Температуру и время реакции варьируют в зависимости от растворителя, связывающего кислоту агента и др., применяемых в реакции, когда используется карбонат калия и при проведении реакции при нагревании в условиях дефлегмации, реакция осуществляется в течение 8 ч.
После завершения реакции требуемое соединение выделяется из реакционной смеси с помощью обычных приемов.
Например, после того, как реакционная смесь нейтрализуется соответствующим образом и нерастворимые вещества, если они присутствуют, удаляются с помощью фильтрования, к реакционной смеси добавляется не смешиваемый с водой органический растворитель, такой как этилацетат. После того, как получающаяся в результате смесь промывается водой, органический слой, содержащий требуемое соединение, отделяется и сушится над безводным сульфатом магния с последующим выпариванием растворителя, давая желаемое соединение.
Требуемое соединение, полученное таким образом, может далее очищаться с помощью общепринятых процедур, таких как перекристаллизация, переосаждение, хроматография и др., если необходимо.
Стадия 2
Эта стадия является стадией получения соединения (18), которое соответствует соединению (17), гидроксильная группа которого защищается с помощью реакции соединения (17) с защитным реагентом, который может удаляться (предпочтительно диметокситритилхлоридом) в кислых условиях в инертном растворителе, в присутствии связывающего кислоту агента.
Применяемый растворитель особенно не ограничивается, если он не оказывает влияния на реакцию и может растворять до некоторой степени исходные материалы, и он включает ароматические углеводороды, такие как бензол, толуол и ксилол; галоидированные углеводороды, такие как метиленхлорид и хлороформ; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан и диметоксиэтан; амиды, такие как диметилформамид, диметилацетамид и гексаметилфосфоротриамид; сульфоксиды, такие как диметилсульфоксид; кетоны, такие как ацетон, метилэтилкетон; гетероциклические амины, такие как пиридин; и нитрилы, такие как ацетонитрил, предпочтительно гетероциклические амины (особенно пиридин).
Применяемый защитный реагент включает тритилгалогениды, такие как тритилхлорид, монометокситритилхлорид, диметокситритилхлорид и триметокситритилхлорид, предпочтительно диметокситритилхлорид.
Применяемый связывающий кислоту агент особо не ограничивается, если он не оказывает вредного влияния на реакцию и не разлагает продукт и исходные материалы, и он предпочтительно включает ароматические амины, такие как пиридин и диметиламинопиридин.
Хотя температура реакции и время реакции изменяются в зависимости от вида защитного реагента и применяемого связующего кислоту реагента, реакция осуществляется при комнатной температуре в течение 2 ч, когда в качестве защитного реагента используется диметокситритилхлорид, а в качестве растворителя, а также агента, связывающего кислоту, используется пиридин.
После завершения реакции требуемое соединение выделяется из реакционной смеси с помощью обычных приемов.
Например, после того, как реакционная смесь нейтрализуется соответствующим образом и нерастворимые вещества, если они присутствуют, удаляются фильтрованием, к реакционной смеси добавляется не смешиваемый с водой органический растворитель, такой как этилацетат. После промывки получающейся в результате смеси водой, органический слой, содержащий требуемое соединение, отделяется и сушится над безводным сульфатом магния с последующим выпариванием растворителя, давая требуемое соединение.
Желаемое соединение, полученное таким образом, может далее, если необходимо, очищаться с помощью общепринятых приемов, таких как перекристаллизация, переосаждение, хроматография и др.
Стадия 3
Данная стадия предназначена для получения соединения (18) с помощью реакции соединения (19) с дикарбоновым ангидридом (20) в инертном растворителе.
Применяемый растворитель особо не ограничивается, постольку поскольку он не оказывает отрицательного воздействия на реакцию и может растворять до некоторой степени исходные материалы, и он включает ароматические углеводороды, такие как бензол, толуол и ксилол; галоидированные углеводороды, такие как метиленхлорид и хлороформ; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан и диметоксиэтан; амиды, такие как диметилформамид, диметилацетамид и гексаметилфосфоротриамид; сульфоксиды, такие как диметилсульфоксид; кетоны, такие как ацетон, метилэтилкетон; гетероциклические амины, такие как пиридин; и нитрилы, такие как ацетонитрил, предпочтительно галоидированные углеводороды, такие как метиленхлорид.
Применяемый агент, связывающий кислоту, включает пиридины, такие как пиридин, диметиламинопиридин и 4-пирролидинопиридин, предпочтительно диметиламинопиридин.
Хотя применяемый дикарбоновый ангидрид особенно не ограничивается, коль скоро он является ангидридом α,ω -алкилдикарбоновой кислоты, имеющей 3 - 16 атомов углерода, им предпочтительно является янтарный ангидрид.
Хотя температура и время реакции изменяются в зависимости от вида применяемых ангидридов кислоты, агента, связывающего кислоту, и др., реакция осуществляется при комнатной температуре в течение 30 мин, когда в качестве агента, связующего кислоту, применяется янтарный ангидрид и диметиламинопиридин.
После завершения реакции желаемое соединение выделяется из реакционной смеси с помощью общепринятых процедур.
Например, после того, как реакционная смесь соответствующим образом нейтрализуется и нерастворимые вещества, если они присутствуют, отфильтровываются, к реакционной смеси добавляется не смешиваемый с водой органический растворитель, такой как этилацетат. После промывки получающейся смеси водой органический слой, содержащий требуемое соединение, отделяется и сушится над безводным сульфатом магния, с последующим выпариванием растворителя, давая желаемое соединение.
Полученное таким образом требуемое соединение может далее, если необходимо, очищаться с помощью обычных процедур, таких как перекристаллизация, переосаждение, хроматография и др.
Стадия 4
Эта стадия - стадия образования активного сложного эфира (12) с помощью реакции карбоксильной группы соединения (18), имеющего свободную карбоксильную группу, с реагентом, образующим сложный эфир, с последующим взаимодействием с необязательно замещенным фенолом.
Применяемый растворитель особо не ограничивается, если он не оказывает отрицательного воздействия на реакцию, и он включает ароматические углеводороды, такие как бензол, толуол и ксилол; галоидированные углеводороды, такие как метиленхлорид, хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол и дихлорбензол; сложные эфиры, такие как этилацетат, этилформиат, пропилацетат, бутилацетат и диэтилкарбонат; кетоны, такие как ацетон, метилэтиленкетон, метилизобутилкетон, изофорон и циклогексанон; нитросоединения, такие нитроэтан и нитробензол; нитрилы, такие как ацетонитрил и изобутиронитрил; амиды, такие как формамид, диметилформамид (ДМФ), диметилацетамид и гексаметилфосфатотриамид, и сульфоксиды, такие как диметилсульфоксид и сульфоран, предпочтительно галоидированные углеводороды (особенно метиленхлорид) и амиды (особенно диметилформамид).
Применяемый фенол особенно не ограничивается, если он может использоваться как активный сложный эфир, и он включает 4-нитрофенол, 2,4-динитрофенол, 2,4,5-трихлорфенол, пентахлорфенол и 2,3,5,6-тетрафторфенол, предпочтительно пентахлорфенол.
Применяемый реагент, образующий сложный эфир, включает, например, N - гидроксисоединения, такие как N - гидроксисукцинимид, 1 - гидроксибензотриазол и N-гидрокси-5-норборнен-2,3-дикарбоксиимид; диимидазольные соединения, такие как 1,1'-оксалилдиимидазол и N,N'-карбонилдиимидазол; дисульфидные соединения, такие как 2,2'-дипиридилдисульфид; соединения янтарной кислоты, такие как N,N'-дисукцинимидилкарбонат; фосфиновохлоридные соединения, такие как N, N'-бис(2-оксо-3-оксазолидинил)фосфиновый хлорид; оксалатные соединения, такие как N, N'-дисукцинимидилоксалат (DSO), N,N-дифталимидилоксалат (DPO), N,N'-бис(норборненилсукцинимид)оксалат (BNO), 1,1'-бис(бензотриазолил)оксалат (BBTO), 1,1'-бис(6- хлорбензотриазолил)оксалат (BCTO) и 1,1'-бис(6- трифторметилбензотриазолил)оксалат (BTBO); и карбодиимиды, такие как дициклогексилкарбодиимид (DCC), предпочтительно диимидазольные соединения и карбодиимиды (особенно DCC).
Хотя реакционная температура и время реакции варьируют в зависимости от вида реагента, образующего сложный эфир, и применяемого растворителя, реакция осуществляется при 0 - 100oC в течение 5 - 50 ч, и особенно, когда используются пентахлорфенол и ДСС в ДМФ, реакция осуществляется при комнатной температуре в течение 18 ч.
После завершения реакции желаемое соединение выделяется из реакционной смеси с помощью общепринятых приемов.
Например, когда реакционная смесь соответствующим образом нейтрализуется, и нерастворимые вещества, если они есть, удаляются фильтрованием, к реакционной смеси добавляется не смешиваемый с водой органический растворитель, такой как этилацетат. После того как получающаяся смесь промывается водой, органический слой, содержащий желаемое соединение, отделяется и сушится над безводным сульфатом магния с последующим выпариванием растворителя, давая желаемое соединение.
Требуемое соединение, полученное таким образом, если необходимо, может далее очищаться с помощью общепринятых процедур, таких как перекристаллизация, переосаждение, хроматография и др.
Стадия 5
Данная стадия является стадией получения соединения (12) с помощью реакции соединения (19) с соединением (21) в инертном растворителе в присутствии связывающего кислоту агента.
Применяемый растворитель особо не ограничивается, если он не оказывает отрицательного воздействия на реакцию, и растворитель включает: ароматические углеводороды, такие как бензол, толуол и ксилол; галоидированные углеводороды, такие как метиленхлорид, хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол и дихлорбензол; сложные эфиры, такие как этилформиат, этилацетат, пропилацетат, бутилацетат и диэтилкарбонат; кетоны, такие как ацетон, метилэтилкетон, метилизобутилкетон, изофорон и циклогексанон; нитросоединения, такие как нитроэтан и нитробензол; нитрилы, такие как ацетонитрил и изобутиронитрил; амиды, такие как формамид, диметилформамид (ДМФ), диметилацетамид и гексаметилфосфоротриамид; сульфоксиды, такие как диметилсульфоксид и сульфолан, предпочтительно галоидированные углеводороды, особенно метиленхлорид.
Применяемый связывающий кислоту агент включает органические основания, такие как триэтиламин, трибутиламин, диизопропилэтиламин, N-метилморфолин, пиридин, 4-(N, N-диметиламино)пиридин, N,N-диметиламилин, N,N-диэтиланилин, 1,5-диазабицикло(4,3,0)нон-5-ен, 1,4-диазабицикло(2,2,2)октан (ДАВСО) и 1,8-диазабицикло (5,4,0)унден- 7-ен (DBU), предпочтительно органические основания, особенно пиридин и N-метилморфолин.
Хотя температура и время реакции меняются в зависимости от вида применяемого связывающего кислоту агента, реакция обычно осуществляется при 10 - 40oC в течение 1 - 5 ч.
После завершения реакции желаемое соединение выделяется из реакционной смеси с помощью обычных процедур.
Например, после того, как реакционная смесь соответствующим образом нейтрализуется и нерастворимые вещества, если они присутствуют, удаляются с помощью фильтрования, к реакционной смеси добавляется не смешиваемый с водой органический растворитель, такой как этилацетат. После того, как получающаяся смесь промывается водой, органический слой, содержащий желаемое соединение, отделяется и сушится над безводным сульфатом магния, с последующим выпариванием растворителя, давая требуемое соединение.
Полученное таким образом целевое соединение, если необходимо, может далее очищаться с помощью общепринятых приемов, таких как перекристаллизация, переосаждение, хроматография и др.
Стадия 6
Данная стадия - стадия получения полимерного производного (14), применяемого в качестве носителя для синтезирования олигонуклеотида с помощью реакции соединения (12), имеющего активированную карбоксильную группу, полученного на стадии 5, с полимерным материалом (13), таким как стекло с регулируемыми порами (OPG), с которым аминогруппа, гидроксильная группа, сульфгидрильная группа и др. связаны через алкиленовую группу, в инертном растворителе.
Хотя полимерный материал (13), применяемый на данной стадии, особо не ограничивается, пока он может обычно использоваться в качестве носителя, следует проверять размер частиц, площадь поверхности трехмерной сетчатой структуры, количество сайтов с гидрофильными группами, химический состав, стойкость к давлению и другие показатели носителя.
Применяемый носитель включает: полисахаридные производные, такие как целлюлоза, декстран и агароза; синтетические полимеры, такие как полиакриламидный гель, полистирольные смолы и полиэтиленгликоль; и неорганические материалы, такие как силикагель, пористое стекло и окиси металлов, типичными представителями которых, но не ограниченных ими, являются промышленно доступные носители, такие как Аминопропил - CPG и длинноцепочечный аминоалкил - CPG (производимые фирмой CPG Инк); Космосил NH2 и Коскосил Диол (производимые Накараи Тесуку); CPG - кремнеземный носитель, покрытый силаном, Аминопропил - CPG - 550 A, Аминопропил - CPG - 1400A и полиэтиленгликоль 5000 монометиловый эфир (производимый фирмой Фурука Ко.), n-алкоксибензиловоспиртовая смола, аминометильная смола и гидроксиметильная смола (производимая фирмой Кокусан Кагаку К.К.); и полиэтиленгликоль 14000 монометиловый эфир (производимый фирмой Юнион Карбайд).
Далее функциональная группа, связанная с носителем, предпочтительно включает аминогруппу, сульфогидрильную группу и гидроксильную группу.
Применяемый растворитель особо не ограничивается до тех пор, пока он не оказывает отрицательного влияния на реакцию и может растворять исходные материалы в некоторой степени, и он предпочтительно включает: ароматические углеводороды, такие как бензол, толуол и ксилол; галоидированные углеводороды, такие как метиленхлорид, хлороформ, четыреххлористый углерод, дихлорэтан, хлоробензол и дихлорбензол; сложные эфиры, такие как этилформиат, этилацетат, пропилацетат, бутилацетат и диэтилкарбонат; кетоны, такие как ацетон, метилэтилкетон, метилизобутилкетон, изофорон и циклогексанон; нитросоединения, такие как нитроэтан и нитробензол; нитрилы, такие как аценитрил и изобутиронитрил; амиды, такие как формамид, диметилформамид, диметилацетамид и гексаметилфосфорный триамид; сульфоксиды, такие как диметилсульфоксид и сульфолан, предпочтительно галоидированные углеводороды (особенно метиленхлорид) и амиды (особенно диметилформамид).
Температура реакции обычно составляет от -20 до 150oC, предпочтительно от 0 до 50oC.
Хотя время реакции изменяется в зависимости от исходных материалов, применяемого растворителя, температуры реакции, обычно оно составляет 1 - 200 ч, предпочтительно 24 - 100 ч.
После завершения реакции желаемое соединение выделяется из реакционной смеси с помощью общепринятых процедур.
Например, желаемый полимерный носитель выделяется из реакционной смеси с помощью фильтрования, промывается органическим растворителем, таким как метиленхлорид, и сушится при пониженном давлении, давая желаемое соединение.
Стадия 7
Данная стадия является стадией получения соединения (22) с помощью реакции соединения (14) с деблокирующим реагентом в инертном растворителе для селективного устранения защитной группы - гидроксильной группы.
Стадии с 7 по 11 обычно проводят в ДНК - синтезаторе.
Применяемый растворитель предпочтительно включает: ароматические углеводороды, такие как бензол, толуол и ксилол; галоидированные углеводороды, такие как метиленхлорид, хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол и дихлорбензол; сложные эфиры, такие как этилформиат, этилацетат, пропилацетат, бутилацетат и диэтилкарбонат; простые эфиры, такие как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан и дитиленгликольдиметиловый эфир; спирты, такие как метанол, этанол, н-пропанол, изопропанол, н-бутанол, изобутанол, трет-бутанол, изоамиловый спирт, диэтиленгликоль, глицерин, октанол, циклогексанол и метилцеллозольв; кетоны, такие как ацетон, метилэтилкетон, метилизобутилкетон, изофорон и циклогексанон; нитросоединения, такие как нитроэтан и нитробензол; нитрилы, такие как ацетонитрил и изобутиронитрил; амиды, такие как формамид, диметилформамид, диметилацетамид и гексаметилфосфоротриамид; сульфоксиды, такие как диметилсульфоксид и сульфолан, более предпочтительно спирты (особенно метанол и этанол) и метиленхлорид, и, когда используется в качестве деблокирующего реагента уксусная кислота, включается смесь уксусной кислоты и воды.
Хотя применяемый деблокирующий реагент особенно не ограничивается, коль скоро он используется обычно, и, если защитной группой является триарилметильная группа, в качестве примеров могут быть приведены уксусная кислота, дихлоруксусная, трифторуксусная, соляная кислота и кислота Льюиса, такая как бромистый цинк, предпочтительно могут использоваться уксусная кислота, дихлоруксусная и трифторуксусная кислота.
Хотя температура реакции меняется в зависимости от применяемых реагентов, исходных материалов и растворителей, обычно она составляет от -10 до 100oC, предпочтительно от 0 до 50oC.
Хотя время реакции варьирует в зависимости от исходных материалов, растворителя и применяемой температуры реакции, обычно оно составляет от 1 мин до 50 ч, предпочтительно от 1 мин до 24 ч.
После завершения реакции желаемое соединение выделяется из реакционной смеси с помощью общепринятых процедур.
Например, полимерный носитель выделяется с помощью фильтрования из реакционной смеси, промывается органическим растворителем, таким как метиленхлорид, и сушится при пониженном давлении, давая требуемое соединение.
Стадия 8
Данная стадия является стадией получения соединения (24) с помощью реакции соединения (23), (23') или (23"), имеющего диметокситритильную группу при 5'-гидроксильной группе, в котором основной фрагмент является фрагментом 3' конца желаемой последовательности оснований, и 3'-гидроксильная группа имеет через связанный с ней фосфор, желаемую алкилокси, фенилокси, аралкилокси, алкильную, аралкильную или фенильную группу, с полимерным материалом (22) с последующим тиоированием или алкиламинированием, если необходимо, давая соединение (24), имеющее желаемое 3'-терминальное нуклеотидное звено, связанное с полимерным материалом, таким как CPG.
Применяемые соединения (23), (23') и (23") будут соответственно описаны ниже.
/a/ Если на данной стадии вводится в реакцию соединение (23), применяется кислотный материал.
Применяемый кислотный материал включает такие соединения, как тетразол и др. , предпочтительно тетразол. Хотя окисляющий агент, применяемый в реакции окисления на данной стадии, особо не ограничивается, если только он обычно используется в реакциях окисления, он предпочтительно включает окисляющие агенты на основе неорганических металлических агентов, таких как окислы марганца, например перманганат калия и двуокись марганца; окислы рутения, например тетраокись рутения, соединения селена, например двуокись селена, соединения железа, например хлорное железо (III); соединения осмия, например тетраокись осмия; соединения серебра, например окись серебра, соединения ртути, например ацетат ртути; окисно-свинцовые соединения, например окись свинца и тетраокись свинца, хромовокислотные соединения, например хромат калия, комплекс хромовой кислоты и серной кислоты и комплекс хромовая кислота-пиридин; и соединения церия, например аммонийнитрат церия (CAN); неорганические окисляющие агенты, такие как молекулы галогенов, например молекулярный хлор, бром и йод; периодные кислоты, например периодат натрия, озон, водную перекись водорода; соединения азотистой кислоты, например азотистую кислоту; соединения на основе хлористой кислоты, например хлорит калия и хлорит натрия, и надсернокислотные соединения, например персульфат калия и персульфат натрия; и органические окисляющие агенты, такие как реагенты, применяемые для окисления с применением ДМСО (комплекс диметилсульфоксида и дициклогексилкарбодиимида, оксалилхлорида, уксусного ангидрида или пятиокиси фосфора, или комплекс пиридин - серный ангидрид); перекиси, например трет-бутилгидроперекись; устойчивые катионы, например трифенилметильный катион; сукцинимиды, например N-бромсукциинимид, соединения гипохлористой кислоты, например трет-бутилгипохлорит; соединения азодикарбоновой кислоты, например эфир азодикарбоновой кислоты; дисульфиды, например диметилдисульфид, дифенилдисульфид и дипиридилсульфид, и трифенилфосфин, эфиры азотистой кислоты, например метилнитрит; тетрагалогениды углерода, например тетрабромистый метан; и хиноновые соединения, например 2,3-дихлор-5,6-дициано-n-бензохинон (DDQ), предпочтительно йод. Применяемый растворитель особо не ограничивается, если он не оказывает отрицательного влияния на реакцию и может до некоторой степени растворять исходные материалы, и он включает: ароматические углеводороды, такие как бензол, толуол и ксилол; галоидированные углеводороды, такие как метиленхлорид и хлороформ; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан и диметоксиэтан; амиды, такие как диметилформамид, диметилацетамид и гексаметилфосфоротриамид; сульфоксиды, такие как диметилсульфоксид; кетоны, такие как ацетон, метилэтилкетон; гетероциклические амины, такие как пиридин; и нитрилы, такие как ацетонитрил, предпочтительно гетероциклические амины, (особенно пиридин), нитрилы (особенно ацетонитрил), простые эфиры (особенно тетрагидрофуран) и галоидированные углеводороды (особенно метиленхлорид).
Реакция осуществляется при температуре от -50 до 100oC, и хотя время реакции варьирует главным образом в зависимости от температуры реакции, типов исходных соединений и применяемого растворителя, оно обычно составляет от 5 мин до 15 ч.
/b/ Когда соединение (23') подвергается реакции, применяемый растворитель особо не ограничивается, если он не влияет пагубно на реакцию, и предпочтительно используется ароматический амин, такой как пиридин.
В случае, когда вводится в реакцию соединение (23'), обычно используется агент конденсации.
Применяемый агент конденсации включает дициклокарбодиимид (DCC), хлорангидрид мезитиленсульфоновой кислоты (Ms - Cl), хлорангидрид триизопропилбензолсульфоновой кислоты, триазолид мезитиленсульфоновой кислоты (MST), 3-нитротриазолид мезитиленсульфоновой кислоты (MSNT), тетразолид триизопропилбензолсульфоновой кислоты (TPS - Te), нитроимидазолид триизопропилбензолсульфоновой кислоты (TPS - NI)и пиридилтетразолид триизопропилбензолсульфоновой кислоты, предпочтительно MS NT, TPS - Te и TPS - NI. Хотя температура реакции особо не ограничивается и находится в интервале от -10 до 100oC, реакция обычно осуществляется при комнатной температуре. Хотя время реакции варьирует в зависимости от применяемого растворителя и температуры реакции, оно составляет 30 мин в том случае, когда реакция осуществляется при комнатной температуре с использованием в качестве растворителя для реакции пиридина.
/c/ Хотя применяемый растворитель, когда подвергается реакции соединение (23'), особенно не ограничивается, если он не оказывает отрицательного воздействия на реакцию, предпочтительно используется безводный ацетонитрил. В качестве реагента, используемого как агент конденсации, используются хлорангидриды кислот, таких как карбоновая кислота и фосфорная кислота, и предпочтительно используется пивалоилхлорид.
Хотя окисляющий агент для окисления олигонуклеотида, имеющего H-фосфонатную связь с олигонуклеотидом фосфодиэфирного типа, особо не ограничивается, если он обычно используется для реакции окисления, он предпочтительно включает неорганические металлические окисляющие агенты, такие как окиси марганца, например перманганат калия и двуокись марганца; окислы рутения, например тетраокись рутения; селеновые соединения, например двуокись селена; соединения железа, например хлорид железа (III); соединения осмия, например тетраокись осмия, соединения серебра, например окись серебра, ртутные соединения, например ацетат ртути; свинцовоокисные соединения, например окись свинца и тетраокись свинца; хромовокислотные соединения, например хромат калия, комплекс хромовая кислота - серная кислота и комплекс хромовая кислота - пиридин; и соединения церия, например аммонийнитрат церия (CAN); неорганические окисляющие агенты, такие как галогеновые молекулы, например молекулярный хлор, молекулярный бром и молекулярный йод; периодные кислоты, например периодат натрия, озон, водную перекись водорода; соединения азотистой кислоты, такие как азотистая кислота; соединения хлористой кислоты, например хлорит калия и хлорит натрия; и надсернокислотные соединения, например персульфат калия и персульфат натрия; и органические окисляющие агенты, такие как реагенты, применяемые для окисления с применением DMCO (комплекс диметилсульфоксида и дициклогексилкарбодиимида, оксалилхлорида, уксусного ангидрида или пятиокиси фосфора, или комплекс пиридина и серного ангидрида); перекиси, например трет-бутилгидроперекись, устойчивые катионы, например трифенилметильный катион; сукцинимиды, например N-бромсукцинимид, соединения гипохлористой кислоты, например трет-бутилгипохлорит; соединения азодикарбоновой кислоты, например метилазодикарбоксилат; дисульфиды, например диметилдисульфид, дифенилдисульфид и дипиридилдисульфид, и трифенилфосфин, эфиры азотистой кислоты, например метилнитрит; тетрагалогениды углерода, например тетрабромид метана; и хиноновые соединения, например 2,3-дихлор-5,6-дициано-n-бензохинон (DDQ), предпочтительно молекулярный йод.
Применяемый связывающий кислоту агент включает гетероциклические амины, такие как пиридин и диметиламинопиридин, и алифатические амины, такие как триметиламин, триэтиламин и диизопропилэтиламин, предпочтительно гетероциклические амины (особенно пиридин). Хотя температура реакции особо не ограничивается, она обычно составляет от -50 до 50oC, предпочитается комнатная температура.
Хотя время реакции меняется в зависимости от исходных материалов, применяемого реагента, температуры и др., оно обычно составляет от 5 мин до 30 ч, предпочтительно 30 мин, когда реакция осуществляется при комнатной температуре.
Тиоирование, которое осуществляется, если это необходимо, на данной стадии, проводится следующим образом.
После того, как соединение (23) связывается с соединением (22), вводится в реакцию агент тиоирования для осуществления тиоирования вместо окисления молекулярным йодом и др., давая соединение (24), которое представляет полимерный носитель, имеющий тиоатную связь, такой как CPG.
Реагент тиоирования особо не ограничивается, если только он может образовывать тиоат с помощью реакции с трехвалентным фосфором, и он предпочтительно включает серу, а также тетраэтилтиурамдисульфид (ТЕТД), поставляемый фирмой Эпплайд Биосистемз, и реагент Beaucage, поставляемый Миллиген/Биосерч. В случае использования первого, может использоваться без ограничений процесс, описанный в известной литературе (Tetrahedron Letters, 32, 3005 (1991)) или его разновидности, и в случае последнего - процесс, описанный в литературе (J. Am. Chem. Soc. 112, 1253 (1990)) или его разновидности.
Алкиламинирование, осуществляемое, если необходимо, на данной стадии, может проводиться следующим образом.
После того, как соединение (23'') связывается с соединением (22), как описано выше, с ними вводится в реакцию желаемый алкиламин при комнатной температуре вместо окисляющего агента, такого как молекулярный йод. Согласно данному процессу может получаться соединение (24), в котором 3' - концевой нуклеотид, имеющий желаемую последовательность, оснований, связан с полимерным материалом, таким как CPG, через фосфорамидатную связь.
Стадия 9.
Данная стадия осуществляется с помощью
1) обработки полимерного материала (24), полученного на стадии 8, кислотой для удаления ДМТ-группы,
2) взаимодействия обработанного таким образом полимерного материала с амидитным реагентом и кислотным катализатором,
3) подвержения получающего материала реакции окисления с использованием окисляющего агента,
4) подвержения непрореагировавшего фрагмента реакции кэппинг с использованием уксусного ангидрида.
На данной стадии указанные выше процедуры 1) - 4) повторяются с получением соединения (25), с которым еще не связан только 5'-терминальный нуклеотид, имеющий желаемую последовательность оснований. Реакция наращивания ДНК-цепи на ДНК-синтезаторе, применяемая на данной стадии, проводится, например, с помощью видоизменения метода Stec (J.Am.Chem.Soc., 106, 6077 - 6089 (1984)) с использованием фосфорамидитного метода, использующего Эпилайд Биосистемз Модель 380B, или фосфорамидитного метода (метод H. Koester и др. в Nucleic Acids. Res., 12, 4539 (1984)) с использованием Циклон Плюс ДНК-синтезатора Миллиген / Биосерч, но способ не ограничивается указанными.
Стадия 10.
Данная стадия является стадией получения полимерного материала (27), имеющего желаемую последовательность оснований и заместители, а также сохраняющего защитную группу, путем удаления 5' - терминальной ДМТ-группы полимерного материала (25), полученного на стадии 9, согласно процедурам стадии 7 с последующей той же обработкой, как на стадии 8, с использованием соединений (26), (26') и (26''), как описано ниже, вместо соединений (23), (23') и (23'').
Соединения (26), (26') и (26''), каждое, представляют 2' - дезоксирибонуклеотид, имеющий основной фрагмент, в котором 5' - терминальный нуклеотид, желаемой последовательности оснований защищен, или рибонуклеозид, имеющий защищенную гидроксильную группу в 2' - положении, с желаемой модифицирующей группой в 5' - положении. Далее соединение (26) имеет в 3' - положении его фосфорамитную связь, включающую защитную группу фосфатного фрагмента, используемого для обычного ДНК-синтеза.
Аналогичным образом, соединение (26') имеет фосфодиэфирную связь, а соединение (26'') имеет H-фосфонатную связь.
Стадия 11.
Данная стадия является стадией получения желаемого олигонуклеотида (11) путем отщепления олигонуклеотидной части от полимерного материала (27), полученного на стадии 10, который имеет желаемую последовательность оснований и заместители на 5' - 3' - терминалах, и удаления защитных групп, иных, чем желаемые заместители у 5' - терминала и/или 3' - терминала. Удаление защитных групп может осуществляться с помощью известных процедур (J. Am. Chem. Soc., 103, 3185 (1981)).
Полученная таким образом реакционная смесь, содержащая соединение общей формулы (11), очищается с помощью обычных приемов обработки по очистке, применяемых для очистки нуклеиновой кислоты, например различных методов хроматографии, таких как с обращенной фазой и/или ионно-обменная хроматография /включая жидкостную хроматографию высокой разрешающей способности/ и др., давая соединение, имеющее общую формулу (11).
Стадия 12.
Данная стадия - стадия получения полуэфира дикарбоновой кислоты, т.е. соединения (15), которое является исходным материалом в настоящем изобретении, с помощью реакции свободной гидроксильной группы соединения (29) или свободной сульфгидрильной группы соединения (30) с дикарбоновым ангидридом, таким как соединение (28), в инертном растворителе, в присутствии основного катализатора.
Применяемый дикарбоновый ангидрид особенно не ограничивается и предпочтительно включает дикарбоновые ангидриды, имеющие 1-16 атомов углерода, и наиболее предпочтительно янтарный ангидрид или глутаровый ангидрид. Применяемый основной катализатор включает предпочтительно аминопиридины, такие как диметиламинопиридин и пирролидинопиридин; третичные амины, такие как триметиламин и триэтиламин, и карбонаты щелочных металлов, такие как бикарбонат натрия и карбонат калия, наиболее предпочтительно диметиламинопиридин и пирролидинопиридин.
Применяемый растворитель особо не ограничивается, если он не оказывает отрицательного воздействия на реакцию и может растворять до некоторой степени исходные материалы, и растворитель предпочтительно включает: ароматические углеводороды, такие как бензол, толуол и ксилол; галоидированные углеводороды, такие как метиленхлорид и хлороформ; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, диоксан и диметоксиэтан; амиды, такие как диметилформамид, диметилацетамид и гексаметилфосфоротриамид; сульфоксиды, такие как диметилсульфоксид; спирты, такие как метанол, этанол, н-пропанол, изопропанол, н-бутанол, изобутанол и изоамиловый спирт; разбавленные кислоты, такие как водная серная кислота; разбавленные основания, такие как водная гидроокись натрия; воду; кетоны, такие как ацетон, метилэтилкетон; гетероциклические амины, такие как пиридин; и нитрилы, такие как ацетонитрил, предпочтительно нитрилы (особенно ацетонитрил), простые эфиры (особенно тетрагидрофуран) и галоидированные углеводороды (особенно метиленхлорид).
Реакция осуществляется при температуре от -50 до 100oC, и время всей реакции варьирует в зависимости от температуры реакции, вида исходных материалов и применяемого растворителя, обычно оно составляет от 30 мин до 15 ч.
После завершения реакции желаемое соединение выделяется из реакционной смеси с помощью общепринятых процедур.
Например, после того, как реакционная смесь соответствующим образом нейтрализуется и нерастворимые вещества, если они присутствуют, удаляются фильтрованием, к реакционной смеси добавляется не смешиваемый с водой органический растворитель, такой как этилацетат. После того как реакционная смесь промывается водой, органический слой, содержащий требуемое соединение, отделяется и сушится над безводным сульфатом магния, с последующим выпариванием растворителя, давая желаемое соединение.
Требуемое соединение, полученное таким образом, может, если необходимо, далее очищаться с помощью общепринятых процедур, таких как перекристаллизация, переосаждение, хроматография и др.
Стадия 13.
Данная стадия является стадией получения соединения (18) с помощью реакции соединения (19) с дикарбоновой кислотой (31) в инертном растворителе в присутствии реагента, образующего сложный эфир.
Применяемый растворитель особо не ограничивается, если он не оказывает отрицательного влияния на реакцию, и он включает: ароматические углеводороды, такие как бензол, толуол и ксилол; галоидированные углеводороды, такие как метиленхлорид, хлороформ, четыреххлористый углерод, дихлорэтан, хлорбензол и дихлорбензол; сложные эфиры, такие как этилформиат, этилацетат, пропилацетат, бутилацетат и диэтилкарбонат; кетоны, такие как ацетон, метилэтилкетон, метилизобутилкетон, изофорон и циклогексанон; нитросоединения, такие как нитроэтан и нитробензол; нитрилы, такие как ацетонитрил и изобутиронитрил; амиды, такие как формамид, диметилформамид (ДМФ), диметилацетамид и гексаметилфосфоротриамид; и сульфоксиды, такие как диметилсульфоксид и сульфолан; предпочтительно галоидированные углеводороды (особенно метиленхлорид) и амиды (особенно диметилформамид).
Применяемый образующий сложный эфир реагент включает, например, N - гидроксисоединения, такие как N - гидроксисукцинимид, 1 - гидроксибензотриазол и N - гидрокси - 5-норборнен - 2,3 - дикарбоксимид, диимидазольные соединения, такие как 1,1' - оксалилдиимидазол и N,N' - карбонилдиимидазол, дисульфидные соединения, такие как 2,2' - дипиридилдисульфид, соединения янтарной кислоты, такие как N,N' - дисукцинимидилкарбонат, фосфиновохлоридные соединения, такие как N, N' - бис(2 - оксо-3 - оксазолидинил)фосфиновый хлорид, оксалатные соединения, такие как N,N'- дисукцинимидилоксалат (DSO), N,N - диифталимидилоксалат (DPO), N,N' - бис (норборненилсукцинимидил)оксалат (BNO), 1,1' - бис (бензотриазолил)оксалат (BBTO), 1,1' - бис(6-хлорбензотриазолил)оксалат (ВСТО) и 1,1'-бис(6-трифторметилбензотриазолил)оксалат (BTBO); и карбодиимиды, такие как дициклогексилкарбодиимид (DCC), предпочтительно диимидазольные соединения и карбодиимиды (особенно DCC).
Хотя температура и время реакции варьируют в зависимости от вида реагента, образующего сложный эфир, и применяемого растворителя, реакция осуществляется при 0 - 100oC в течение 5 - 50 ч.
После завершения реакции желаемое соединение выделяется из реакционной смеси с помощью обычных процедур.
Например, после того, как реакционная смесь нейтрализуется соответствующим образом и нерастворимые вещества, если они присутствуют, удаляются фильтрованием, к реакционной смеси добавляется не смешиваемый с водой органический растворитель, такой как этилацетат. После промывания получающейся в результате смеси водой, органический слой, содержащий желаемое соединение, отделяется и сушится над безводным сульфатом магния с последующим выпариванием растворителя, давая желаемое соединение.
Требуемое соединение, полученное таким образом, может далее очищаться, если необходимо, с помощью общепринятых процедур, таких как перекристаллизация, переосаждение, хроматография и др.
Согласно новому процессу настоящего изобретения модифицированный олигонуклеотид, имеющий желаемый заместитель на 3' конце олигонуклеотида, присоединенный через фосфат, может легко синтезироваться на ДНК-синтезаторе, обеспечивая таким образом получение модифицированного олигонуклеотида с высокой чистотой в больших количествах, с использованием простой процедуры очистки и при низкой стоимости. Полезный олигонуклеотид, имеющий заместитель на 3' конце, который обладает анти-СПИД активностью, описывается в японской патентной заявке N Hei - 301744.
Соединения настоящего изобретения проявляют специфическую цитопатическую активность против вируса СПИД (ВИЧ-1) и могут, в частности, ингибировать пролиферацию вируса в инфицированных клетках. Соответственно соединения изобретения могут использоваться для лечения и профилактики или предотвращения СПИДа.
Настоящее изобретение поясняется конкретно с помощью иллюстрации следующими примерами.
/Пример испытания 1/ Измерение анти-ВИЧ-1-активности модифицированных олигодезоксирибонуклеотидов
Анти-ВИЧ-1-активность определялась по методу Pauel и др. (R. Pauel и др. , J. Virological Methods, 20, 309 - 321 (1988)). Вкратце, клетки MT-4 в экспоненциальной фазе роста центрифугировались в течение 5 мин при 150 x g. Полученная лепешка клеток суспендировалась в культуральной среде и инфицировалась ВИЧ-1 (Тип III B) в течение 1 ч при 37oC при концентрации 10 CCLD50. Инфицированные ВИЧ-1 МТ-4 клетки получались с помощью центрифугирования в культурной среде RPMI-1640, содержащей 10%-ную сыворотку плодного теленка (называемой здесь ниже "сывороточная культуральная среда").
Инфицированные ВИЧ-1 клетки MT-4 и неинфицированные ВИЧ-1 клетки MT-4 суспендировались в сывороточной культуральной среде так, чтобы достигалась концентрация каждой среды 4•105 клеток /мл. 100 мкл раствора ступенчато разбавленного образца соединения (растворенного в сывороточной культуральной среде), полученного заранее, помещалось в каждую лунку 98 - луночных титрационных микропланшетов. Затем в каждую лунку титрационных микропланшетов добавлялось 100 мкл каждой из упомянутых выше суспензий клеток, и клетки культивировались в течение 6 дн в присутствии 5% углекислого газа.
В качестве контроля таким же самым образом культивировались инфицированные ВИЧ-1 клетки MT-4 и неинфицированные ВИЧ-1 клетки MT-4, в которые не добавлялся образец соединения.
После культивирования определялось число живых клеток на основе MTT (3-(4,5-диметилтиазол-2-ил)-2,5-дифенил- тетразолийбромид) метода (L.M. Green и др. J. Immunol, Methods, 70, 257 - 268 (1984)). Затем определялась цитопатическая активность ВИЧ-1. По цитопатической активности инфицированных ВИЧ-1 клеток МТ-4, к которым не добавлялись образцы соединения, которая принималась за 100%, и цитопатической активности не инфицированных ВИЧ-2 (тип ШВ) клеток MT-4, к которым не добавлялся образец соединения, и принятой за 0%, измерялась концентрация образца, которая ингибировала цитопатическую активность инфицированных ВИЧ-1 клеток МТ-4 на 50% (IC50). Цитотоксичность образца соединения определялась в виде концентрации, которая ингибировала размножение неинфицированных ВИЧ-1 клеток MT-4 на 50% (CC50). Результаты этих определений показаны в табл. 2.
Согласно приведенным данным, результаты испытаний отчетливо показали, что все модифицированные олигодезоксирибонуклеотиды, перечисленные в табл. 2, обнаруживают особенно интенсивную анти-ВИЧ-1-активность при концентрации менее 10 мкг/мл.
Если не указано иное, последовательность оснований (например, TGGGAGG), использованная в следующих химических структурах, означает триэтиламиновую соль соответствующего олигодезоксирибонуклеотида, не имеющего гидроксигрупп и в 5'- и в 3'-положениях.
Изобретение далее иллюстрируется следующими ниже примерами. В примерах все размеры в мешах применяют стандарт Тилера, а спектры ядерно-магнитного резонанса получались с использованием триметилсилана в качестве внутреннего стандарта, где имеется сокращенное указание "TMC".
Количество олигодезоксирибонуклеотидного производного, полученного в каждом из этих примеров, измерялось с помощью оптической плотности при 260 нм (OD (260 нм)).
Пример 1
1/a/ 5'-O-Тритилтимидин-2-цианоэтил, N,N-диизопропилфосфорамидит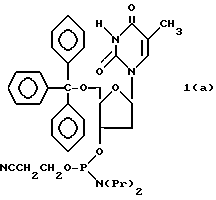
969 мг (2 ммоль) 5'-O-тритилтимидина [J. Am. Chem. Soc., 80, 6112 (1958)] сушились три раза с помощью азеотропной перегонки с пиридином, после чего он растворялся в 10 мл тетрагидрофурана. Затем к раствору добавлялось 1,39 мл (8 ммоль) N,N-диизопропил-N-этиламина и 0,822 мл (4 ммоль) 2-цианоэтил N,N-диизопропилхлорфосфорамидита, и получающаяся смесь перемешивалась при комнатной температуре в течение 30 мин в атмосфере аргона. В конце данного периода получающийся в результате осадок отфильтровывался из реакционной смеси, и фильтрат освобождался от растворителя с помощью перегонки при пониженном давлении. Получающийся остаток растворялся в 100 мл этилацетата, и раствор промывался дважды, каждый раз 50 мл охлажденного льдом 10% вес/об. водного раствора карбоната натрия. Раствор затем сушился над безводным сульфатом натрия, после чего растворитель удалялся с помощью перегонки при пониженном давлении. Получающийся остаток очищался с помощью хроматографии на колонке при пропускании через 40 г силикагеля (70 - 230 меш) с использованием 45:45:10 по объему смеси метиленхлорида, этилацетата и триэтиламина в качестве элюента, давая 1,35 г (выход 98%) названного в заголовке соединения.
1H Спектр ядерно-магнитного резонанса (ЯМР) (CDCl3, 270 МГц, TMC), δ , млн. дол.:
7,62, 7,57 (вместе 1H, два синглета), 7,46 - 7,20 (15H, мультиплет), 6,46 - 6,37 (1H, мультиплет), 4,68 (1H, широкий синглет), 4,19. 4,15 (вместе 1H, два широких синглета), 3,90 - 3,30 (6H, мультиплет), 2,63 - 2,28 (4H, мультиплет), 1,47 (3H, синглет), 1,23 - 1,00 (12H, мультиплет).
1 (b) Соединение формулы 1(b):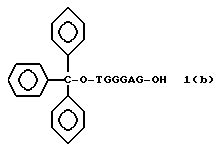
Синтез проводился с использованием ДНК/РНК - синтезатора Циклон Плюс (торговая марка), производимого МиллиГен /Биосерч (подразделение Миллипор Лтд, Япония). В него загружались химические реагенты, соответствующие нуклеотидным остаткам в приведенной выше формуле 1(b), для синтеза указанного выше олигонуклеотида. В устройство вставлялся программный патрон для амидитного метода. Синтез осуществлялся в масштабе 15 мкмоль. В данном случае 35 мМ ацетонитрильный раствор 5' - O -тритилтимидин-2-цианоэтил, N, N - диизопропилфосфорамидита [полученного, как описано в стадии (a) выше] использовался вместо раствора 2 - цианоэтилфосфорамидита, соответствующего тимидину. Реакционная колонка была колонкой для реакции масштаба 15,0 мкмоль, наполненной 5 мкмоль G - CPG [т.е. гуаниндезоксирибонуклеотида (G) в сочетании со стеклом с регулируемыми порами (CPG)]. Концентрация нуклеотида на стеклянном фильтре была 35-44 мкмоль/г, а CPG-наполнитель имел средний размер пор 50 нм. Вставлялась требуемая последовательность оснований TGGGAG (как это обычно, последовательность оснований, указанная здесь, а также последовательности, упоминаемые здесь ниже, включают основание, которое связано с CPG), и осуществлялась программа без обработки кислотой после образования связи с терминальным T, давая производное, в котором желаемый защищенный олигонуклеотид сочетался со стеклом с регулируемыми порами (CPG). Данное вещество сушилось в вакууме, удалялось с колонки и погружалась примерно в 10 мл 29%-ного водного аммиака. Затем ему давали возможность реагировать при комнатной температуре в течение примерно 2 дн в запаянном сосуде. В конце данного периода CPG удалялся фильтрованием и промывался дважды, каждый раз 10 мл воды; фильтрат и промывные воды затем объединялись. Объединенная смесь затем промывалась три раза 30 мл диэтилового эфира каждый раз, после чего аммиак и диэтиловый эфир удалялись выпариванием в вакууме. Получающийся водный раствор концентрировался до примерно 3 мл с помощью выпаривания при пониженном давлении, и концентрированный раствор фильтровался с помощью миллипористого фильтра (0,45 мкм). Фильтрат разделялся на 3 части, и каждая порция очищалась с помощью жидкостной хроматографии высокой разрешающей способности или высокоэффективной жидкостной хроматографии (ВЭЖХ) с обращенной фазой при пропускании через 20,0 х 250 мм колонку Инертсил PREP - ODS (товарный знак), которая предварительно уравновешивалась 0,1 М водным триэтиламмоний - ацетатным буфером (TEAA) (pH 7,3), содержащим 20% по объему ацетонитрила, и контролировалась ультрафиолетовым светом при 254 нм. Желаемый продукт в течение 30 мин элюировался с использованием градиентного метода элюирования 0,1 М TEАA, содержащим ацетонитрил с концентрацией, варьирующей от 20 до 50% по объему, с линейным градиентом 8 мл/мин. Элюированные после 17,2 мин фракции собирались, раствор освобождался от ацетонитрила упариванием при пониженном давлении, после чего полученный в результате водный раствор лиофилизировался. Полученный таким образом продукт растворялся в 50 мл воды и снова лиофилизировался, давая 94,8 OD (260 нм) соединения формулы 1(b) в виде аморфного твердого вещества.
УФ - спектр поглощения (H2O), λ макс., нм: 254
Время удерживания: 23,3 мин.
Высокоэффективная жидкостная хроматография (УМС - A-312, S - 5, 120A, ODS элюент A: 0,1 М TEAA, pH 7,3; элюент B: ацетонитрил, 20-->50% по объему, 30 мин, линейный градиент, 1 мл/мин; 254 нм).
Пример 2
(2a) 5'-Тритиламино - 5' -дезокситимидин
557 мг Тритилхлорида добавлялись к раствору 560 мг (2 ммоль) 3'- O -ацетил- 5' -амино- 5'-дезокситимидина [который получался с помощью процедуры, описанной J.P.Horwits и др. в J. Org. Chem., 27, 3045 (1962)] в 20 мл сухого пиридина, получающаяся смесь нагревалась с обратным холодильником в течение 1 ч при исключении влаги. После исчезновения исходного соединения, о чем свидетельствовали данные тонкослойной хроматографии (с использованием метиленхлорида, содержащего 5% по объему метанола в качестве проявляющего растворителя), растворитель удалялся из реакционной смеси с помощью перегонки при пониженном давлении. Получающийся остаток смешивался с 10 мл каждого из метанола и воды, а затем смесь концентрировалась с помощью перегонки при пониженном давлении. Данная операция повторялась три раза, после чего остаток растворялся в 30 мл этилацетата, и получающийся раствор промывался 20 мл каждого из насыщенного водного раствора хлористого натрия, 0,2 н. водной соляной кислоты и 5% водного раствора бикарбоната натрия. Органический раствор затем сушился над безводным сульфатом магния, и раствор затем упаривался досуха при пониженном давлении. Остаток растворялся в 30 мл метанола, насыщенного аммиачным газом, и колба с раствором плотно закрывалась. Затем она оставалась стоять на протяжении ночи при комнатной температуре. В конце данного периода времени реакционная смесь освобождалась от растворителя с помощью перегонки при пониженном давлении, и остаток растворялся в 10 мл метиленхлорида. Раствор очищался с помощью хроматографии на колонке при пропускании через силикагель, с использованием метиленхлорида, содержащего 5% по объему метанола, в качестве элюента. Фракции, содержащие требуемое соединение, объединялись и выпаривались досуха при пониженном давлении. С помощью лиофилизации остатка из бензола получалось 338 мг указанного в заголовке соединения в виде белого порошка.
1H Спектр ЯМР (CDCl3, 270 МГц, ТМС), δ, млн.дол. :
8,38 (1H, широкий синглет), 7,48-7,18 (15H, мультиплет), 7,04 (1H, мультиплет), 6,67 (1H, триплет, J = 6,60 Гц), 4,35 - 4,29 (1H, мультиплет), 4,01 - 3,95 (1H, мультиплет), 2,62 - 2,06 (5H, мультиплет), 1,84 (3H, синглет).
2 (b) 5'-Тритиламино-5'-дезокситимидин-2-цианоэтил N, N -диизопропилфосфорамидит
0,15 г (0,31 ммоль) 5'-тритиламино-5'-дезокситимидина [полученного, как описано в стадии (a) выше] сушилось с помощью азеотропной перегонки с пиридином и затем растворялось в 2 мл метиленхлорида. Затем к получающемуся раствору добавлялось 0,23 мл (1,2 ммоль) N,N-диизопропил - N -этиламина, после чего добавлялось 0,08 мл (0,36 ммоль) 2-цианоэтил N, N -диизопропилхлорфосфорамидита на протяжении 2 мин в токе азота. Получающаяся смесь затем перемешивалась при комнатной температуре в течение 60 мин. В конце данного периода по данным тонкослойной хроматографии подтверждалось, что исходное соединение исчезло, и реакционная смесь разбавлялась 30 мл этилацетата. Получающийся органический раствор промывался насыщенным водным раствором бикарбоната натрия и насыщенным водным раствором хлористого натрия в указанном порядке. Органический слой фильтровался через IPS (торговая марка) фильтровальную бумагу (Ватман), и фильтрат освобождался от растворителя с помощью перегонки при пониженном давлении. Получающийся остаток очищался с помощью хроматографии на колонке с пропусканием через 15 г силикагеля (70-230 меш), с использованием этилацетата в качестве элюента, давая 0,19 г (выход 89%) указанного в заготовке соединения в виде пенистого вещества.
1H Спектр ЯМР (CDCl3, 270 МГц, ТМС), δ , млн. дол:
7,50 - 7,19 (15H, мультиплет), 7,15, 7,06 (вместе 1H, два синглета), 6,30 - 6,26 (1H, мультиплет), 4,55 - 4,35 (1H, мультиплет), 4,12 - 4,07 (1H, мультиплет), 3,84 - 3,53 (4H, мультиплет), 2,64 - 2,09 (6H, мультиплет), 1,86, 1,84 (вместе 3H, два синглета), 1,19 - 1,10 (12H, мультиплет).
2 (с) Соединение формулы 2 (с):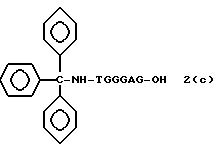
С помощью процедуры, аналогичной процедуре, описанной в примере 1(b), но при использовании 5'-тритиламино-5'-дезокситимидин-2-цианоэтил N,N - диизопропилфосфорамидита [полученного, как описано в получении 2 (b)] вместо 5'-  -тритилтимидин-2-цианоэтил N,N -диизопропилфосфорамидита получалось целевое соединение. Реакционный продукт очищался с помощью хроматографии через силикагельную колонку с обращенной фазой (препаративная C18, Уотерс, 1,5 х 15 см, 50 мМ водный триметиламмоний - бикарбонатный буфер (TEAB),pH 7,5, 20-->50% ацетонитрил, линейный градиент, 254 нм). Элюат обрабатывался аналогично описанному в примере 1 b, давая 168 OD (260 нм) целевого соединения в виде аморфного твердого вещества. Анализ с помощью высокоэффективной хроматографии с обращенной фазой (Инертсил ODS 6 х 150 нм, элюент A: 0,1 М TEAA, pH 7,5, элюент B: ацетонитрил, 10 --> 60% по объему, 20 мин, линейный градиент, 1 мл/мин, 260 нм) показал, что образец имел время удерживания 19-20 мин.
-тритилтимидин-2-цианоэтил N,N -диизопропилфосфорамидита получалось целевое соединение. Реакционный продукт очищался с помощью хроматографии через силикагельную колонку с обращенной фазой (препаративная C18, Уотерс, 1,5 х 15 см, 50 мМ водный триметиламмоний - бикарбонатный буфер (TEAB),pH 7,5, 20-->50% ацетонитрил, линейный градиент, 254 нм). Элюат обрабатывался аналогично описанному в примере 1 b, давая 168 OD (260 нм) целевого соединения в виде аморфного твердого вещества. Анализ с помощью высокоэффективной хроматографии с обращенной фазой (Инертсил ODS 6 х 150 нм, элюент A: 0,1 М TEAA, pH 7,5, элюент B: ацетонитрил, 10 --> 60% по объему, 20 мин, линейный градиент, 1 мл/мин, 260 нм) показал, что образец имел время удерживания 19-20 мин.
Спектр УФ-поглощения (H2O), λ, нм: 254.
Пример 3
3(а) 5'-O-Бензгидрилтимидин
350 мг (8 ммоль) гидрида натрия (в виде 55% вес/вес дисперсии в минеральном масле) добавлялось к раствору 1,426 г (4 ммоль) 3'-O-[(1,1-диметилэтил)диметилсилил] тимидана [Can. J. Chem., 56, 2768 (1978)] в 8 мл тетрагидрофурана в атмосфере аргона, и получающаяся смесь перемешивалась при 60oC в течение 2 ч, а затем оставлялась охлаждаться до комнатной температуры. Затем к смеси по каплям добавлялся раствор 988 мг (4 ммоль) бензилбромида в 2 мл тетрагидрофурана, а затем 300 мг (2 ммоль) йодида натрия. Реакционная смесь затем перемешивалась при комнатной температуре в течение 17 ч, после чего она освобождалась от растворителя с помощью перегонки при пониженном давлении. Концентрат растворялся в 50 мл этилацетата, и получающийся раствор промывался дважды, каждый раз 50 мл насыщенного водного раствора хлорида натрия. Затем он сушился над безводным сульфатом магния, и растворитель отгонялся при пониженном давлении. Остаток растворялся в 4 мл тетрагидрофурана, и к раствору добавлялось 4 мл 1 M тетрагидрофуранового раствора тетрабутиламмонийфторида. Получающаяся смесь затем перемешивалась при комнатной температуре в течение 2 ч. В конце данного времени растворитель удалялся с помощью перегонки при пониженном давлении, и остаток растворялся в 50 мл этилацетата. Получающийся раствор промывался дважды, каждый раз 50 мл насыщенного водного раствора хлористого натрия. Смесь сушилась над безводным сульфатом магния, а затем растворитель удалялся перегонкой при пониженном давлении. Остаток очищался с помощью хроматографии на колонке через 60 г силикагеля (230-400 меш) с использованием метода градиентного элюирования метиленхлоридом, содержащим от 1 до 2,5% по объему метанола в качестве элюента, давая 377,7 мг (выход 23%) указанного в заголовке соединения.
1Н Спектр ЯМР (CDCl3, 270 МГц, TMC), δ, млн. дол.:
9,85 (1H, широкий синглет), 7,56 (1H, синглет), 7,38-7,20 (10H, мультиплет), 6,45 (1H, триплет, J=6,92 Гц), 6,40 (1H, синглет), 4,62 - 4,58 (1H, мультиплет), 4,17-4,15 (1H, мультиплет), 3,75 - 3,58 (2H, мультиплет), 2,47 - 2,22 (2H, мультиплет), 2,47 - 2,22 (2H, мультиплет), 1,36 (3H, синглет).
3 (b) 5'-O-Бензгидрилтимидин-2-цианоэтил N,N-диизопропилфосфорамидит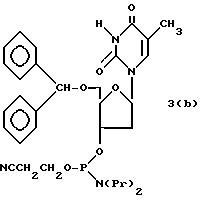
204,2 мг (0,5 ммоль) 5'-О-бензгидрилтимидина [полученного, как описано в стадии (а) выше] сушилось три раза с помощью азеотропной перегонки с пиридином, а затем растворялось в 2,5 мл тетрагидрофурана. К раствору в атмосфере аргона добавлялось 0,348 мл (2 ммоль) N,N-диизопропиламина и 0,223 мл (1 ммоль) 2-цианоэтил N,N-диизопропилхлорфосфорамидита, и получающаяся смесь перемешивалась при комнатной температуре в течение 30 мин. В конце данного времени реакционная смесь освобождалась от осадков путем фильтрования, и фильтрат концентрировался выпариванием при пониженном давлении. Концентрат растворялся в 50 мл этилацетата, и получающийся раствор промывался два раза, каждый раз 50 мл охлажденного льдом 10% вес./об. водным раствором карбоната натрия, и сушился над безводным сульфатом натрия. Растворитель затем удалялся перегонкой при пониженном давлении. Остаток очищался с помощью колоночной хроматографии через 30 г силикагеля (70-230 меш) с использованием 45: 45: 10 по объему смеси метиленхлорида, этилацетата и триэтиламина в качестве элюента. Фракции, содержащие целевое соединение, объединялись и растворитель отгонялся. Повторялась та же процедура хроматографии, и получалось 213,4 мг (выход 70%) целевого соединения.
1H Спектр ЯМР (CDCl3, 270 МГц, TMC) δ, млн. дол.:
7,55, 7,51 (вместе 1H, два синглета), 7,43 - 7,22 (10H, мультиплет), 6,48 - 6,42 (1H, мультиплет), 5,44 - 5,42 (вместе 1H, два синглета), 4,73 - 4,64 (1H, мультиплет), 4,25, 4,19 (вместе 1H, два широких синглета), 3,90 - 3,55 (6H, мультиплет), 2,68 - 2,24 (4H, мультиплет), 1,39 (3H, синглет), 1,30 - 1,10 (12H, мультиплет).
3 (с) Соединение формулы 3 (с);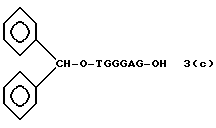
С помощью процедуры, аналогичной процедуре, описанной в примере 1 (b), но с использованием 5'- -бензгидрилтимидин-2-цианоэтил N,N-диизопропилфосфорамидита [полученного, как описано в стадии (b) выше] вместо 5'-
-бензгидрилтимидин-2-цианоэтил N,N-диизопропилфосфорамидита [полученного, как описано в стадии (b) выше] вместо 5'-  -тритилтимидин-2-цианоэтил N, N-диизопропилфосфорамидита, получалось указанное в заголовке соединение. Реакционный продукт разделялся на три порции, и каждая порция очищалась с помощью хроматографии через колонку для высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил PREP - ODS 20,0х250 нм, элюент A: 0,1 M TEAA, pH 7,3; элюент B: ацетонитрил, 15 --> 45% B по объему, 30 мин, линейный градиент, 7 мл/мин, 254 нм). Фракции, элюируемые через 20,2 мин, объединялись и обрабатывались аналогично тому, как описано в примере 1 (b), давая [76 OD/ 260 нм] целевое соединение в виде аморфного твердого вещества.
-тритилтимидин-2-цианоэтил N, N-диизопропилфосфорамидита, получалось указанное в заголовке соединение. Реакционный продукт разделялся на три порции, и каждая порция очищалась с помощью хроматографии через колонку для высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил PREP - ODS 20,0х250 нм, элюент A: 0,1 M TEAA, pH 7,3; элюент B: ацетонитрил, 15 --> 45% B по объему, 30 мин, линейный градиент, 7 мл/мин, 254 нм). Фракции, элюируемые через 20,2 мин, объединялись и обрабатывались аналогично тому, как описано в примере 1 (b), давая [76 OD/ 260 нм] целевое соединение в виде аморфного твердого вещества.
УФ-спектр поглощения (H2O), λмакс, нм: 255.
Время удерживания: 17,3 мин.
Высокоэффективная жидкостная хроматография (УMC-Pack A-312, S-5 120A, ODS, элюент A: 0,1 M TEAA, pH 7,3; элюент B: ацетонитрил, 10 ---> 40% B по объему, 30 мин, линейный градиент, 2 мл/мин, 254 нм).
Пример 4.
Соединение формулы 4(а):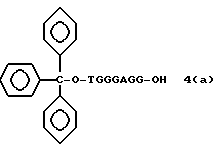
С использованием процедуры, аналогичной процедуре, описанной в примере 1(b), но помещая основную последовательность TGGGAGG в ДНК/РНК-синтезатор, описанный в примере 1(b), получалось целевое соединение. Реакционный продукт очищался с помощью хроматографии через силикагельную колонку с обращенной фазой (Препаратив C18, Уотерс, 1,5 х 15 см, 50 мМ ТЕАВ, pH 7,5, 20 ---> 50% ацетонитрила, линейный градиент, 254 нм). Обработка, аналогичная описанной в примере 1(b), давала 180 ОД (260 нм) целевого соединения в виде аморфного твердого вещества. Анализ с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсид ODS, 6 x 150 мм, элюент A: 0,1 М TEAA, pH 7,5, элюент B: ацетонитрил, 10 ---> 60% B по объему, 20 мин, линейный градиент, 1 мл/мин, 260 нм) показал, что образец имеет время удерживания 18,22 мин.
УФ-спектр поглощения (вода), λмакс, нм : 255.
Пример 5.
5(a) 5'-O-[(1,1-Диметилэтил)дифенилсилил]тимидин
1,43 мл (5,5 ммоль) трет-бутилдифенилсилилхлорида добавлялось к раствору 1,21 г (5 ммоль) тимидина и 0,749 г (1 ммоль) имидазола в 10 мл диметилформамида в атмосфере аргона, получающаяся смесь перемешивалась при комнатной температуре в течение 140 мин. В конце данного периода времени растворитель удалялся перегонкой при пониженном давлении, и получающийся остаток растворялся в 100 мл метиленхлорида. Раствор затем промывался 5 раз, каждый раз 100 мл воды. Раствор затем сушился над безводным сульфатом магния, после чего растворитель отгонялся при пониженном давлении. Остаток очищался с помощью колоночной хроматографии через 100 г силикагеля (70 - 230 меш) с использованием метода градиентного элюирования метиленхлоридом, содержащим от 0,5 до 3% по объему метанола, в качестве элюента, давая 1,5 г (выход 62%) целевого соединения.
1H Спектр ЯМР (CDCl3, 270 МГц, ТМС), δ, млн. дол.:
10,52 (1H, широкий синглет), 7,73 - 7,32 (10H, мультиплет), 7,57 (1H, синглет), 6,53 - 6,47 (1H, мультиплет), 4,63 (1H, широкий синглет), 4,48 (1H, широкий синглет), 4,11 (1H, широкий синглет), 4,04 - 3,85 (2H, мультиплет), 2,58 - 2,16 (2H, мультиплет), 1,59 (3H, синглет), 1,10 (9H, синглет).
5 (b) 5'-O-[(1,1-Диметилэтил)дифенилсилил]тимидин 2-цианоэтил N,N-диизопропилфосфорамидит.
С помощью процедуры, аналогичной процедуре, описанной в примере 3 (b), но с использованием 240 мг (0,5 ммоль) 5'-O-[(1,1-диметилэтил)дифенилсилил] тимидина [полученного, как описано в стадии (a) выше], получалось 254,4 мг (выход 74,7%) целевого соединения.
1H Спектр ЯМР (CDCl3, 270 МГц, TMC), δ, млн. дол.:
7,69 - 7,36 (11H, мультиплет), 6,43 - 6,38 (1H, мультиплет), 4,70 - 4,62 (1H, мультиплет), 4,16 - 4,09 (1H, мультиплет), 4,04 - 3,55 (6H, мультиплет), 2,67 - 2,15 (4H, мультиплет), 1,61 (3H, синглет), 1,22 - 1,05 (12H, мультиплет).
5 (c) Соединение формулы 5 (c):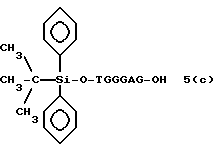
С помощью процедуры, аналогичной описанной в примере 1 (b), но с использованием 5'-  -[(1,1-диметилэтил)дифенилсилил]тимидин-2-цианоэтил N, N-диизопропилфосфорамидита [полученного, как описано в стадии (b) выше] вместо 5'-O-тритилтимидин-2-цианоэтил N,N-диизопропилфосфорамидита получалось целевое соединение. Реакционный продукт разделялся на три порции, и каждая порция очищалась с помощью хроматографии при пропускании через колонку для жидкостной хроматографии высокой разрешающей способности с обращенной фазой (Инертсил PREP - ODS, 220,0 x 250 мм, элюент A: 0,1 М TEAA, pH 7,3 ; элюент B: ацетонитрил 20 ---> 50% B по объему, 30 мин, линейный градиент, 7 мл/мин, 254 нм). Фракции, элюируемые через 22,4 мин, объединялись и обрабатывались по способу, аналогичному описанного в примере 1(b), давая 54 OD (260 нм) целевого соединения в виде аморфного твердого вещества.
-[(1,1-диметилэтил)дифенилсилил]тимидин-2-цианоэтил N, N-диизопропилфосфорамидита [полученного, как описано в стадии (b) выше] вместо 5'-O-тритилтимидин-2-цианоэтил N,N-диизопропилфосфорамидита получалось целевое соединение. Реакционный продукт разделялся на три порции, и каждая порция очищалась с помощью хроматографии при пропускании через колонку для жидкостной хроматографии высокой разрешающей способности с обращенной фазой (Инертсил PREP - ODS, 220,0 x 250 мм, элюент A: 0,1 М TEAA, pH 7,3 ; элюент B: ацетонитрил 20 ---> 50% B по объему, 30 мин, линейный градиент, 7 мл/мин, 254 нм). Фракции, элюируемые через 22,4 мин, объединялись и обрабатывались по способу, аналогичному описанного в примере 1(b), давая 54 OD (260 нм) целевого соединения в виде аморфного твердого вещества.
УФ-спектр поглощения (вода), λмакс , нм: 255.
Время удерживания : 22,0 мин.
Высокоэффективная жидкостная хроматография (УМС - Pack A-312, S-5, 120A, ODS, элюент A: 0,1 М TEAA, pH 7,3, элюент B: ацетонитрил, 20 _→ 50% B по объему, 30 мин, линейный градиент, 1 мл/мин, 254 нм).
Пример 6
6 (a) 5'-O-(3,5-Дибензилоксибензил)тимидин
175 мг (4 ммоль) гидрида натрия (в виде 55% вес./вес. дисперсии в минеральном масле) добавлялось к раствору 713 мг (2 ммоль) 3'-O-[(1,1-диметилэтил)диметилсилил] тимидина [Can. J. Chem., 56, 2768 (1978)] в 4 мл тетрагидрофурана в атмосфере аргона, и получающаяся смесь перемешивалась при 60oC в течение 2 ч. В конце данного периода смесь оставлялась охлаждаться до комнатной температуры, а затем по каплям добавлялся раствор 767 мг (2 ммоль) 3,5-дибензилоксибензилбромида [Chem. Ber. , 102, 2887 (1969)] в 1 л тетрагидрофурана. Затем к получающейся смеси добавлялось 149,9 мг (1 ммоль) йодида натрия, после чего смесь перемешивалась при комнатной температуре в течение 16 ч. В конце данного периода растворитель удалялся перегонкой при пониженном давлении, остаток растворялся в 50 мл этилацетата, и получающийся раствор промывался дважды, каждый раз 50 мл 0,01 н. водной соляной кислоты. Раствор затем сушился над безводным сульфатом магния, после чего растворитель отгонялся при пониженном давлении. Остаток очищался с помощью колоночной хроматографии через 100 г силикагеля (230 - 400 меш), с использованием метода градиентного элюирования, с использованием метиленхлорида, содержащего от 0,5 до 3% по объему метанола, в качестве элюента, давая 637,3 мг смеси, содержащей 3'-O-[(1,1-диметилэтил) диметилсилил]-5'-  -(3,5-дибензилоксибензил)тимидин.
-(3,5-дибензилоксибензил)тимидин.
Вся смесь растворялась в 1,93 мл тетрагидрофурана. К раствору добавлялось 1,93 мл 1 М тетрагидрофуранового раствора тетрабутиламмонийфторида, и получающаяся смесь перемешивалась при комнатной температуре в течение 30 мин. В конце данного времени растворитель удалялся перегонкой при пониженном давлении, и получающийся остаток растворялся в 50 мл этилацетата. Данный раствор промывался дважды, каждый раз 50 мл насыщенного водного раствора хлорида натрия. Раствор затем сушился над безводным сульфатом магния, растворитель отгонялся при пониженном давлении, и остаток очищался с помощью хроматографии через 30 г силикагеля (230 - 400 меш), с использованием метода градиентного элюирования, метиленхлорид, содержащий от 1 до 3% по объему метанола в качестве элюента, давая 258, 5 мг (выход 23,7%) целевого соединения.
1H Спектр ЯМР (CDCl3, 270 МГц, TMC), δ, млн. дол. :
7,87 (1H, синглет), 7,52 (1H, синглет), 7,43 - 6,52 (13H, мультиплет), 6,37 (1H, триплет, J = 6,75 Гц), 5,03 (4H, синглет), 4,51 (2H, дублет, J = 3,30 Гц), 4,50 - 4,44 (1H, мультиплет), 4,06-4,03 (1H, мультиплет), 3,77-3,63 (2H, мультиплет), 2,32-2,12 (2H, мультиплет), 1,67 (3H, синглет).
6 (b) 5'-O-(3,5-Дибензилоксибензил)тимидин-2-цианоэтил N, N-диизопропилфосфорамидит.
С помощью процедуры, аналогичной процедуре, описанной в примере 3 (b), но с использованием 258,5 мг (0,475 ммоль) 5'-O-(3,5-дибензилоксибензил)тимидина [полученного, как описано в стадии (а) выше], получалось 307,7 мг (87%) указанного в заголовке соединения.
1H Спектр ЯМР (CDCl3, 270 МГц, TMC), δ , млн.дол.:
7,56, 7,53 (вместе 1H, два синглета), 7,42-6,56 (13H, мультиплет), 6,40 (1H, триплет, J = 6,60 Гц), 5,02 (4H, синглет), 4,67-4,58 (1H, мультиплет), 4,53, 5,51 (вместе 2H, два синглета), 4,23, 4,17 (вместе 1H, два широких синглета), 3,90-3,52 (6H, мультиплет), 2,68-2,53 (2H, мультиплет), 2,49-2,12 (2H, мультиплет), 1,65 (3H, синглет), 1,18 (12H, дублет, J = 5,94 Гц).
6 (c) Соединение формулы 6 (c)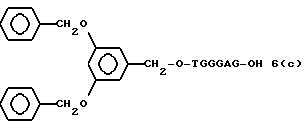
С помощью процедуры, аналогичной процедуре, описанной в примере 1 (b), но с использованием 5'-  -(3,5-дибензилоксибензил)тимидин-2-цианоэтил N,N, -диизопропилфосфорамидита, полученного, как описано в стадии (b) выше, вместо 5'-
-(3,5-дибензилоксибензил)тимидин-2-цианоэтил N,N, -диизопропилфосфорамидита, полученного, как описано в стадии (b) выше, вместо 5'-  -тритилтимидин-2-цианоэтил N,N-диизопропилфосфорамидита получалось целевое соединение. Продукт реакции разделялся на три порции, и каждая порция очищалась с помощью хроматографии через колонку для высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил PREP-ODS, 20,0 x 250 мм, элюент A: 0,1 M TEAA, pH 7,3; элюент B: ацетонитрил, 20 ---> 50% B по объему, 30 мин, линейный градиент, 7 мл/мин, 254 нм). Фракции, элюированные через 19,8 мин, объединялись и обрабатывались по способу, аналогичному описанному в примере 1 (b), давая 64,6 OD (260 нм) целевого соединения в виде аморфного твердого вещества.
-тритилтимидин-2-цианоэтил N,N-диизопропилфосфорамидита получалось целевое соединение. Продукт реакции разделялся на три порции, и каждая порция очищалась с помощью хроматографии через колонку для высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил PREP-ODS, 20,0 x 250 мм, элюент A: 0,1 M TEAA, pH 7,3; элюент B: ацетонитрил, 20 ---> 50% B по объему, 30 мин, линейный градиент, 7 мл/мин, 254 нм). Фракции, элюированные через 19,8 мин, объединялись и обрабатывались по способу, аналогичному описанному в примере 1 (b), давая 64,6 OD (260 нм) целевого соединения в виде аморфного твердого вещества.
УФ-спектр поглощения (вода), λмакс, нм : 255.
Время удерживания: 22,8 мин.
Высокоэффективная жидкостная хроматография (УМС-Pack A-312, S-5, 120A, ODS; элюент A: 0,1 M TEAA, pH 7,3; элюент B: ацетонитрил 20 ---> 50% B по объему, 30 мин; линейный градиент; 1 мл/мин, 254 нм).
Пример 7
7 (а) 5'-О-(3,5-Дибензилоксибензил)тимидин
713 мг (2 ммоль) 3'-0-[(1,1-диметилэтил)]диметилсилил тимидина [Can. J. Chem., 56, 2768 (1978)] растворялось в 5 мл тетрагидрофурана, и к получающемуся раствору в атмосфере аргона добавлялось 175 мг (4 ммоль) гидрида натрия (в виде 55% вес./вес. дисперсии в минеральном масле). Смесь затем перемешивалась при 60oC в течение 2 ч. В конце данного периода времени температура смеси понижалась до комнатной температуры, и затем добавлялось 678 мг (2 ммоль) 3,4-дибензилоксибензилхлорида, а затем 149,9 мг (1 ммоль) йодистого натрия, и полученная в результате смесь перемешивалась при комнатной температуре в течение 19 ч и затем при 60oC в течение 5 ч. В конце этого времени растворитель отгонялся при пониженном давлении, и остаток растворялся в 50 мл этилацетата. Полученный раствор дважды промывался порциями по 50 мл 0,01 N водным раствором соляной кислоты, затем он сушился над безводным сульфатом магния. Растворитель удалялся перегонкой при пониженном давлении. Остаток растворялся в 4 мл тетрагидрофурана, и к раствору добавлялось 4 мл 1 M раствора тетрабутиламмонийфторида в тетрагидрофуране. Смесь затем перемешивалась при комнатной температуре в течение 2 ч. В конце этого времени растворитель удалялся перегонкой при пониженном давлении, и остаток растворялся в 50 мл этилацетата. Полученный в результате раствор дважды промывался порциями по 50 мл насыщенного водного раствора хлористого натрия и сушился над безводным сульфатом магния. Растворитель затем удалялся перегонкой при пониженном давлении, и остаток подвергался хроматографированию на колонке (Lichroprep /торговая марка/ (Si 60, 40-63 μм Grosse C. Merk) и элюировался, с использованием метода градиентного элюирования, метиленхлоридом, содержащим от 1 до 4% по объему метанола, при этом получалось 345,4 мг (выход 31,7%) целевого соединения.
1H Спектр ЯМР (CDCl3, 270 МГц, TMS), δ, млн.дол. :
8,26 (1H, синглет), 7,50 (1H, синглет); 7,47-7,28 (10H, мультиплет); 6,91-6,79 (3H, мультиплет); 6,36 (1H, триплет, J = 6,75 Гц); 5,17 (4H, синглет); 4,47, 4,45 (вместе 2H, два синглета); 4,41-4,36 (1H, мультиплет); 4,04-4,01 (1H, мультиплет); 3,72-3,56 (2H, мультиплет); 2,31-2,05 (2H, мультиплет); 1,62 (3H, синглет).
7 (b) 5'-O-(3,4-дибензилоксибензил)тимидин-2-дианоэтил N,N- диизопропилфосфорамидит
271,7 мг (0,499 ммоль) 5'-O-(3,4-дибензилоксибензил)тимидина, приготовленного, как описано выше в стадии (а), три раза подвергалось сушке путем азеотропной перегонки с пиридином, после чего он растворялся в 2,5 мл тетрагидрофурана. К получающемуся раствору добавлялось 0,223 мл (1 ммоль) 2-дианоэтил N, N-диизопропилхлорфосфорамида и 0,348 мл (2 ммоль) N,N-диизопропил-N-этиламина, и смесь перемешивалась при комнатной температуре в атмосфере аргона в течение 2 ч. В конце данного периода времени растворитель удалялся с помощью перегонки при пониженном давлении. Остаток растворялся в 50 мл этилацетата, и получающийся раствор промывался дважды, каждый раз 50 мл охлажденного льдом 10% в/о (вес/объем) водного раствора карбоната натрия, а затем сушился над безводным сульфатом натрия. Растворитель удалялся с помощью перегонки при пониженном давлении, и получающийся остаток наносился на колонку, содержащую 30 г (70-230 меш) силикагеля. Затем он элюировался смесью 45:45:10 по объему метиленхлорида, этилацетата и триэтиламина. Фракции, содержащие желаемый продукт, объединялись, растворитель удалялся перегонкой при пониженном давлении, и получающийся остаток наносился на колонку, содержащую 30 г (70 - 230 меш) силикагеля. Остаток затем элюировался 6:3:1 по объему смесью метиленхлорида, этилацетата и триэтиламина, давая 286,9 мг (выход 77%) целевого соединения.
1H Спектр ЯМР (CDCl3, 270 МГц, ТМС), δ, млн. дол.:
8,07 (1H, широкий синглет), 7,53, 7,51 (вместе 1H, два синглета), 7,45 - 7,25 (10H, мультиплет), 6,92 - 6,81 (3H, мультиплет), 6,37 (1H, триплет, J = 6,60 Гц), 5,15 (4H, синглет), 4,62 - 4,53 (1H, мультиплет), 4,47 (2H, синглет), 4,22, 4,14 (вместе 1H, два широких синглета), 3,90 - 3,53 (6H, мультиплет), 2,68 - 2,53 (2H, мультиплет), 2,47 - 2,05 (2H, мультиплет), 1,58 (3H, синглет), 1,17 (12H, дублет, J = 5,94 Гц).
7 (c) Соединение формулы 7 (c)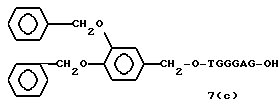
В соответствии с процедурой, аналогичной процедуре, описанной в примере 1 (b), но при использовании 5'-  -(3,4-дибензилоксибензил)тимидин-2-цианоэтил N,N-диизопропилфосфорамидоита [полученного, как описано в стадии (b) выше] вместо 5'-
-(3,4-дибензилоксибензил)тимидин-2-цианоэтил N,N-диизопропилфосфорамидоита [полученного, как описано в стадии (b) выше] вместо 5'-  - тритилтимидин-2-цианоэтил N,N,-диизопропилфосфоримидита, получалось целевое соединение. Реакционный продукт затем разделялся на три порции, и каждая порция очищалась с помощью хроматографии при пропускании через колонку для высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил PREP-ODS, 20,0 x 250 нм, элюент A: 0,1 M TEAA, pH 7,3; элюент B: ацетонитрил, 20 _→ 50% B по объему, 30 мин, линейный градиент, 7 мл/мин; 254 нм). Фракции, элюируемые через 22,5 мин, объединялись и обрабатывались по способу, аналогичному описанному в примере 1 (b), давая 55,7 OD (260 нм) целевого соединения в виде аморфного твердого вещества.
- тритилтимидин-2-цианоэтил N,N,-диизопропилфосфоримидита, получалось целевое соединение. Реакционный продукт затем разделялся на три порции, и каждая порция очищалась с помощью хроматографии при пропускании через колонку для высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил PREP-ODS, 20,0 x 250 нм, элюент A: 0,1 M TEAA, pH 7,3; элюент B: ацетонитрил, 20 _→ 50% B по объему, 30 мин, линейный градиент, 7 мл/мин; 254 нм). Фракции, элюируемые через 22,5 мин, объединялись и обрабатывались по способу, аналогичному описанному в примере 1 (b), давая 55,7 OD (260 нм) целевого соединения в виде аморфного твердого вещества.
УФ-спектр поглощения (вода), λмакс, нм : 254.
Время удерживания: 13,6 мин.
Высокоэффективная жидкостная хроматография (УМС- Pack A-312, S-5, 120A, ODS; элюент A: 0,1 M TEAA, pH 7,3; элюент B: ацетонитрил; 20 ---> 50% по объему, 30 мин, линейный градиент; 2 мл/мин; 254 нм).
Пример 8
Соединение формулы 8 (a)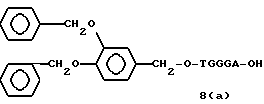
В соответствии с процедурой, аналогичной процедуре, описанной в примере 1 (b), но при использовании 5'-  -(3,4-дибензилоксибензил)тимидин 2-дианоэтил N, N-диизопропилфосфорамидита [полученного, как описано в примере 7 (b)] вместо 5'-O-тритилтимидин-2-цианоэтил N,N-диизопропилфосфорамидита, с использованием колонки, наполненной 5 мкмоль A-CPG (т.е. адениндезоксирибонуклеотид (A), присоединенный к стеклу с регулируемыми порами) и при помещении основной последовательности TGGGA в синтезатор, получалось целевое соединение. Продукт реакции разделялся на три порции, и каждая порция очищалась с помощью хроматографии при пропускании через колонку высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил-PREP-ODS, 20,0 x 250 нм; элюент A: 0,1 M TEAA, pH 7,3; элюент B: ацетонитрил; 20 ---> 50% по объему, 30 мин.; линейный градиент, 7 мл/мин, 254 нм). Фракции, элюированные через 23,3 мин, объединялись и обрабатывались по способу, аналогичному описанному в примере 1 (b), давая 86,2 OD (260 нм) целевого соединения в виде аморфного твердого вещества.
-(3,4-дибензилоксибензил)тимидин 2-дианоэтил N, N-диизопропилфосфорамидита [полученного, как описано в примере 7 (b)] вместо 5'-O-тритилтимидин-2-цианоэтил N,N-диизопропилфосфорамидита, с использованием колонки, наполненной 5 мкмоль A-CPG (т.е. адениндезоксирибонуклеотид (A), присоединенный к стеклу с регулируемыми порами) и при помещении основной последовательности TGGGA в синтезатор, получалось целевое соединение. Продукт реакции разделялся на три порции, и каждая порция очищалась с помощью хроматографии при пропускании через колонку высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил-PREP-ODS, 20,0 x 250 нм; элюент A: 0,1 M TEAA, pH 7,3; элюент B: ацетонитрил; 20 ---> 50% по объему, 30 мин.; линейный градиент, 7 мл/мин, 254 нм). Фракции, элюированные через 23,3 мин, объединялись и обрабатывались по способу, аналогичному описанному в примере 1 (b), давая 86,2 OD (260 нм) целевого соединения в виде аморфного твердого вещества.
УФ-спектр поглощения (вода), λмакс, нм : 256.
Время удерживания : 14,2 мин.
Высокоэффективная жидкостная хроматография (УМС-Pack A-312, S-5, 120A, ODS; элюент A: 0,1 M TEAA, pH 7,3; элюент B: ацетонитрил; 20 ---> 50% B по объему, 30 мин, линейный градиент, 2 мл/мин, 254 нм).
Пример 9
Соединение формулы 9 (а)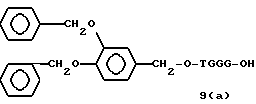
В соответствии с процедурой, аналогичной процедуре, описанной в примере 1 (b), но при использовании 5'-  - (3,4 - дибензилоксибензил)тимидин 2 - цианоэтил, N, N - диизопропилфосфорамидита [полученного, как описано в примере 7 (b) выше ] вместо 5'-
- (3,4 - дибензилоксибензил)тимидин 2 - цианоэтил, N, N - диизопропилфосфорамидита [полученного, как описано в примере 7 (b) выше ] вместо 5'-  - тритилтимидин 2 - цианоэтил N,N -диизопропилфосфорамидита и при помещении основной последовательности ТGGG в синтезатор получалось целевое соединение. Реакционный продукт разделялся на три порции, и каждая порция очищалась с помощью хроматографии при пропускании через колонку высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил PREP - ODS, 20,0 х 250 нм; элюент A: 0,1 M TEAA, pH 7,3; элюент B: ацетонитрил, 25 ---> 55% B по объему, 30 мин; линейный градиент; 7 мл/мин; 254 нм). Фракции, элюированные через 19,5 мин, объединялись и обрабатывались по способу, аналогичному описанному в примере 1 (b), давая 77,5 OD (260 нм) целевого соединения в виде аморфного твердого вещества.
- тритилтимидин 2 - цианоэтил N,N -диизопропилфосфорамидита и при помещении основной последовательности ТGGG в синтезатор получалось целевое соединение. Реакционный продукт разделялся на три порции, и каждая порция очищалась с помощью хроматографии при пропускании через колонку высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил PREP - ODS, 20,0 х 250 нм; элюент A: 0,1 M TEAA, pH 7,3; элюент B: ацетонитрил, 25 ---> 55% B по объему, 30 мин; линейный градиент; 7 мл/мин; 254 нм). Фракции, элюированные через 19,5 мин, объединялись и обрабатывались по способу, аналогичному описанному в примере 1 (b), давая 77,5 OD (260 нм) целевого соединения в виде аморфного твердого вещества.
УФ-спектр поглощения (вода), λмакс, нм: 255.
Время удерживания: 14,8 мин.
Высокоэффективная жидкостная хроматография (УМС-Ресk A-312, S - 5, 120A, ODS, элюент A: 0,1 M TEAA, pH 7,3; элюент B: ацетонитрил; 20 ---> 50% B по объему, 30 мин, линейный градиент; 2 мл/мин, 254 нм).
Пример 10
Соединение формулы 10 (а)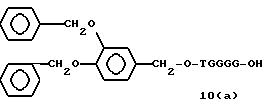
В соответствии с процедурой, аналогичной процедуре, описанной в примере 1 (b), но с использованием 5'-  -(3,4 - дибензилоксибензил)тимидин - 2 - цианоэтил N,N - диизопропилфосфорамидита [полученного, как описано в примере 7 (b) выше] вместо 5' -
-(3,4 - дибензилоксибензил)тимидин - 2 - цианоэтил N,N - диизопропилфосфорамидита [полученного, как описано в примере 7 (b) выше] вместо 5' -  - тритилтимидин 2 - цианоэтил N,N - диизопропилфосфорамидита и помещая основную последовательность TGGGG в синтезатор, получали целевое соединение. Реакционный продукт разделялся на три порции, и каждая порция очищалась с помощью хроматографии при пропускании через колонку высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил PREP-ODS, 20,0 х 250 нм; элюент A: 0,1 M TEAA, pH 7,3; элюент B: ацетонитрил; 20 _→ 50% B по объему, 30 мин; линейный градиент; 7 мл/мин, 254 нм). Фракции, элюированные через 22,9 мин, объединялись и обрабатывались по способу, аналогичному описанному в примере 1 (b), давая 19,8 OD (260 нм) целевого соединения в виде аморфного твердого вещества.
- тритилтимидин 2 - цианоэтил N,N - диизопропилфосфорамидита и помещая основную последовательность TGGGG в синтезатор, получали целевое соединение. Реакционный продукт разделялся на три порции, и каждая порция очищалась с помощью хроматографии при пропускании через колонку высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил PREP-ODS, 20,0 х 250 нм; элюент A: 0,1 M TEAA, pH 7,3; элюент B: ацетонитрил; 20 _→ 50% B по объему, 30 мин; линейный градиент; 7 мл/мин, 254 нм). Фракции, элюированные через 22,9 мин, объединялись и обрабатывались по способу, аналогичному описанному в примере 1 (b), давая 19,8 OD (260 нм) целевого соединения в виде аморфного твердого вещества.
УФ - спектр поглощения (вода), λмакс , нм: 254
Время удерживания: 14,1 мин.
Высокоэффективная жидкостная хроматография (УМС - Pack A-312, S - 5, 120A, ODS; элюент A: 0,1 M TEAA, pH 7,3; элюент B: ацетонитрил; 20 ---> 50% B по объему, 30 мин, линейный градиент; 2 мл/мин; 254 нм).
Пример 11
11 (a) 5'-O-[(Пирен-1-ил)метил]тимидин
Смесь 875 мг гидрида натрия (в виде 55% в/в дисперсии в минеральном масле) в 5 мл диметилсульфоксида перемешивалась при комнатной температуре в течение 30 мин в атмосфере азота, а затем к получающейся смеси добавлялся по каплям раствор 2,42 г (10 ммоль) тимидина в 5 мл диметилсульфоксида. Смесь перемешивалась при комнатной температуре в течение 30 мин, после чего добавлялась суспензия 2,51 г (10 ммоль) 1 - (хлорметил)пирена [Acta Chem. Scand., 10, 1362 (1956)] в 15 мл диметилсульфоксида. Смесь затем перемешивалась при комнатной температуре в течение 90 мин. В конце данного периода времени реакционная смесь выливалась в 100 мл смеси лед-вода, и водная смесь экстрагировалась 100 мл этилацетата, а затем 100 мл метиленхлорида. Экстракт сушился над безводным сульфатом магния, а затем растворитель удалялся перегонкой при пониженном давлении. Остаток очищался с помощью колоночной хроматографии при пропускании через 150 г силикагеля (230-400 меш), с использованием градиентного метода элюирования, смесями метиленхлорида, содержащего от 0 до 4% по объему метанола, в качестве элюента, давая 292,4 мг (выход 6,4%) целевого соединения.
1H Спектр ЯМР (гексадейтерированный диметилсульфоксид, 270 МГц, ТМС),
δ, млн. дол.:
8,44 - 8,07 (9H, мультиплет), 7,44 (1H, синглет), 6,19 (1H, триплет, J = 6,75 Гц); 5,33 (1H, дублет, J = 5,4 Гц), 5,29 (2H, синглет), 4,37 - 4,30 (1H, мультиплет), 4,00 - 3,96 (1H, мультиплет), 3,92 - 3,79 (2H, мультиплет), 2,10 - 2,04 (2H, мультиплет), 1,20 (3H, синглет).
11 (b) 5'-O-[(Пирен-1-ил)метил] тимидин 2-цианоэтил N,N - диизопропилфосфорамидит.
91,3 мг (0,2 ммоль) 5' -  - [(пирен-1-ил)метил]тимидина [полученного, как описано в стадии (a) выше] сушилось три раза с помощью азеотропной перегонки с пиридином, после чего реагент суспендировался в 2 мл тетрагидрофурана. К суспензии в атмосфере аргона добавлялось 139 мкл (0,8 ммоль) N,N - диизопропил - N - этиламина и 789 мкм (0,4 ммоль) 2-цианоэтил N,N - диизопропилхлорфосфорамидита, и получающаяся смесь перемешивалась при комнатной температуре в течение 30 мин. В конце данного периода времени осадки удалялись фильтрованием, и фильтрат освобождался от растворителя с помощью перегонки при пониженном давлении. Остаток растворялся в 20 мл этилацетата, и получающийся раствор промывался дважды, каждый раз 20 мл охлажденного льдом 10% в/о водного раствора карбоната натрия. Раствор сушился над безводным сульфатом натрия, а затем растворитель удалялся с помощью перегонки при пониженном давлении. Получающийся в результате остаток очищался с помощью колоночной хроматографии при пропускании через 30 г силикагеля (70 - 230 меш), с использованием 6:3:1 по объему смеси метиленхлорида, этилацетата и триэтиламина в качестве элюента, давая 100,2 мг (выход 76%) целевого соединения.
- [(пирен-1-ил)метил]тимидина [полученного, как описано в стадии (a) выше] сушилось три раза с помощью азеотропной перегонки с пиридином, после чего реагент суспендировался в 2 мл тетрагидрофурана. К суспензии в атмосфере аргона добавлялось 139 мкл (0,8 ммоль) N,N - диизопропил - N - этиламина и 789 мкм (0,4 ммоль) 2-цианоэтил N,N - диизопропилхлорфосфорамидита, и получающаяся смесь перемешивалась при комнатной температуре в течение 30 мин. В конце данного периода времени осадки удалялись фильтрованием, и фильтрат освобождался от растворителя с помощью перегонки при пониженном давлении. Остаток растворялся в 20 мл этилацетата, и получающийся раствор промывался дважды, каждый раз 20 мл охлажденного льдом 10% в/о водного раствора карбоната натрия. Раствор сушился над безводным сульфатом натрия, а затем растворитель удалялся с помощью перегонки при пониженном давлении. Получающийся в результате остаток очищался с помощью колоночной хроматографии при пропускании через 30 г силикагеля (70 - 230 меш), с использованием 6:3:1 по объему смеси метиленхлорида, этилацетата и триэтиламина в качестве элюента, давая 100,2 мг (выход 76%) целевого соединения.
1H Спектр ЯМР (CDCl3, 270 МГц, ТМС), δ, млн. дол.:
8,36 - 7,99 (9H, мультиплет), 7,50, 7,46 (вместе 1H, два синглета), 6,35 (1H, триплет, J = 6,60 Гц), 5,38 - 5,22 (2H, мультиплет), 4,60 - 4,50 (1H, мультиплет), 4,25 - 4,17 (1H, мультиплет), 4,01 - 3,77 (2H, мультиплет), 3,73 - 3,44 (4H, мультиплет), 2,78 - 2,09 (4H, мультиплет), 1,31, 1,29 (вместе 3H, два синглета), 1,10 - 1,05 (12H, мультиплет).
11 (c) Соединение формулы 11 (c):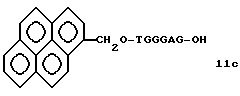
Целевое соединение получали, следуя процедуре, описанной в примере 1 (b), но используя 5'-  -[(пирен-1-ил)метил]тимидин 2 - цианоэтил N,N - диизопропилфосфорамидит [приготовленный, как описано выше в стадии (b)] вместе 5'-
-[(пирен-1-ил)метил]тимидин 2 - цианоэтил N,N - диизопропилфосфорамидит [приготовленный, как описано выше в стадии (b)] вместе 5'-  - тритилтимидин 2 - цианоэтил N,N - диизопропилфосфорамидита. Продукт реакции разделялся на три порции, и каждая из них очищалась с помощью хроматографии через колонку для высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил PREP - ODS, 20,0 х 250 мм; элюент A: 0,1 M TEAA, pH 7,3; элюент B: ацетонитрил, 20 ---> 50% B по объему, 30 мин, линейный градиент, 7 мл/мин; 254 нм). Фракции, элюированные спустя 15,4 мин, объединялись и обрабатывались по способу, аналогичному описанному в примере 1 (b), давая 80 OD (260 нм) целевого соединения в виде аморфного твердого вещества.
- тритилтимидин 2 - цианоэтил N,N - диизопропилфосфорамидита. Продукт реакции разделялся на три порции, и каждая из них очищалась с помощью хроматографии через колонку для высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил PREP - ODS, 20,0 х 250 мм; элюент A: 0,1 M TEAA, pH 7,3; элюент B: ацетонитрил, 20 ---> 50% B по объему, 30 мин, линейный градиент, 7 мл/мин; 254 нм). Фракции, элюированные спустя 15,4 мин, объединялись и обрабатывались по способу, аналогичному описанному в примере 1 (b), давая 80 OD (260 нм) целевого соединения в виде аморфного твердого вещества.
УФ - спектр поглощения (вода), λмакс, нм: 243.
Время удерживания: 16,4 мин.
Высокоэффективная жидкостная хроматография (УМС-Pack A-312, S - 5, 120A, ODS; элюент A: 0,1 M TEAA, pH 7,3; элюент B: ацетонитрил, 20 ---> 50% B по объему, 30 мин, линейный градиент, 1 мл/мин; 254 нм).
Пример 12
12 (a) O - Диметокситритилэтиленгликоль
3,1 г (50 ммоль) этиленгликоля сушилось с помощью азеотропной перегонки с пиридином и затем растворялось в 40 мл пиридина. К раствору добавлялось 3,38 г (10 ммоль) 4,4' - диметокситритилхлорида, и получающаяся смесь перемешивалась при комнатной температуре в течение 2 ч. После исчезновения исходного соединения, которое подтверждалось данными тонкослойной хроматографии, к реакционной смеси добавлялось 5 мл метанола, и смесь затем концентрировалась до примерно половины ее объема с помощью перегонки при пониженном давлении. Концентрат растворялся в 100 мл метиленхлорида, и получающийся раствор промывался насыщенным водным раствором бикарбоната натрия. Органический раствор фильтровался через IPS-фильтровальную бумагу (Ватман), и фильтрат освобождался от растворителя перегонкой при пониженном давлении. Остаток очищался с помощью хроматографии на колонке, заполненной 100 г силикагеля (70 - 230 меш) с использованием метиленхлорида, содержащего 1% по объему метанола в качестве элюента, давая 1,97 г целевого соединения в виде камедеообразного вещества.
1H Спектр ЯМР (CDCl3, 270 МГц, ТМС), δ, млн. дол.:
7,45 - 6,82 (13H, мультиплет), 3,79 (6H, синглет), 3,75 (2H, триплет, J = 4,95 Гц), 3,25 (2H, триплет, J = 4,62 Гц), 1,95 (1H, триплет).
12 (b) Соединение формулы 12 (b):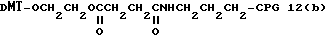
0,18 г (0,5 ммоль) O-диметокситритилэтиленгликоля [полученного, как описано в стадии (a) выше] подвергалось сушке с помощью азеотропной перегонки с пиридином, а затем растворялось в 2 мл метиленхлорида. Затем к раствору добавлялось 75 мг (0,75 ммоль) янтарного ангидрида и 92 мг (0,75 ммоль) 4-(диметиламино)пиридина, и получающаяся смесь перемешивалась в течение 1 ч. После исчезновения исходного материала, которое определялось с помощью тонкослойной хроматографии, реакционная смесь разбавлялась метиленхлоридом, и разбавленный раствор промывался 0,5 М водным раствором первичного кислого фосфата калия (pH 5,0) и водой в указанном порядке. Органический слой сушился над безводным сульфатом натрия, и растворитель удалялся перегонкой при пониженном давлении, давая моносукцинат O-диметокситритилэтиленгликоля.
Все указанное соединение подвергалось сушке азеотропной перегонкой с пиридином и затем растворялось в 3 мл диметилформамида. К раствору добавлялось 0,16 г (0,75 ммоль) пентахлорфенола и 0,16 г (0,75 ммоль) 1,3 - дициклогексилкарбодиимида, и получающаяся смесь перемешивалась при комнатной температуре в течение 42 ч. В конце данного периода времени нерастворимые вещества отфильтровывались, и фильтрат концентрировался перегонкой при пониженном давлении. Остаток смешивался с бензолом, и получающиеся нерастворимые вещества снова отфильтровывались, после чего растворитель удалялся перегонкой при пониженном давлении.
0,25 г (0,2 ммоль) остатка, полученного, как описано выше, растворялось в 4 мл диметилформамида, и к раствору добавлялось 60 мкл (0,44 ммоль) триэтиламина и 2,0 г (3 - аминопропил) - CPG (содержащего 0,129 ммоль/г аминогрупп). Получающаяся смесь затем оставлялась стоять при комнатной температуре в течение 36 ч. В конце данного времени CPG-носитель собирался фильтрованием, промывался метиленхлоридом, а затем сушился в вакууме. CPG-носитель смешивался с 10 мл каждого из A Вкэп растворов (Миллипор Лтд., Япония), и смесь оставлялась стоять в течение 10 мин для того, чтобы ацетилировать любые аминогруппы, остающиеся для реакции. Продукт реакции затем промывался пиридином и метиленхлоридом в указанном порядке, после чего он сушился в вакууме, давая названное в заголовке соединение формулы 12 (b). Кэп A раствор представляет 1: 9 по объему смесь уксусного ангидрида и тетрагидрофурана, а кэп B раствор представляет 1:4 по объему смесь N - метилимидазола и тетрагидрофурана.
Количество остатков, получающихся от O-диметокситритилэтиленгликоля, вводимых в соединение примера 12 (b), количественно анализировалось следующим образом. Смесь 9,6 мг соединения примера 12 (b) и деблокирующий раствор (раствор дихлоруксусной кислоты в метиленхлориде; Миллипор Лтд., Япония) встряхивалась в течение 3 мин, и затем к раствору добавлялось достаточное количество метиленхлорида для доведения общего объема до 20 мл. 0,4 мл данного раствора отбиралось в опытную пробирку (трубку) и использовалось для измерения. Образец раствора освобождался от растворителя перегонной при пониженном давлении, и остаток растворялся в 3:2 по объему смеси перхлорной кислоты и этанола. Поглощение диметокситритильного катиона в растворе определялось при 500 нм.
В результате было найдено, что вводимое количество диметокситритильных групп составляло 40,0 мкмоль/г.
12 (c) Соединение формулы 12 (c):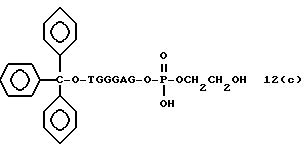 .
.
Согласно процедуре, аналогичной описанной в примере 1 (b), но при использовании колонки, наполненной 125 мг (5 мкмоль) соединения примера 12 (b), и помещении основной последовательности TGGGAGZ (в которой Z представляет модельный генетический код, как поясняется в примере 17) в синтезатор, получалось целевое соединение с использованием ДНК/РНК-синтезатора, как описано в примере 1 (b). Реакционный продукт очищался с помощью хроматографии на силикагельной колонке с обращенной фазой (Preparative C18, Уотерс, 1,5 x 15 см; 50 мМ ТЕАВ, pH 7,5; 20 ---> 50% ацетонитрила; линейный градиент, 254 нм) и обрабатывался по способу, аналогичному способу, описанному в примере 1 (b), давая 165 OD (250 нм) целевого соединения в виде аморфного твердого вещества. Анализ с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил ODS, 6 x 150 мм; элюент A: 0,1 M TEAA, pH 7,5; элюент B: ацетонитрил; 10 ---> 60% по объему, 20 мин, линейный градиент, 1 мл/мин, 260 нм) показал, что образец имеет время удерживания 18,76 мин.
УФ - спектр поглощения (вода), λмакс, нм:254.
Пример 13
Соединение формулы 13 (a):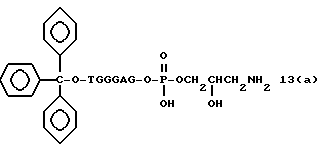
С использованием процедуры, аналогичной процедуре, описанной в примере 12 (c), но при применении 151 мг (5 мкмоль) 3' - амино- ON CPG (торговая марка продукта Клонтек), получалось целевое соединение. Продукт очищался с помощью хроматографии при пропускании через силикагельную колонку с обращенной фазой (Preparative C18, Уотерс, 1,5 x 15 см; 50 мМ TEAB, pH 7,5; 20 ---> 50% ацетонитрил, линейный градиент, 254 нм) и обрабатывался по способу, аналогичному описанному в примере 1 (b), давая 165 OD (260 нм) целевого соединения в виде аморфного твердого вещества. Анализ высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил ODS, 6 x 150 мм: элюент A: 0,1 M TEAA, pH 7,5; элюент B: ацетонитрил; 10 ---> 60% B по объему, 20 мин, линейный градиент; 1 мл/мин; 260 нм) показал, что образец имеет время удерживания 18,76 мин.
УФ - спектр поглощения (вода), λмакс, нм: 255.
Пример 14
Соединение формулы 14 (a):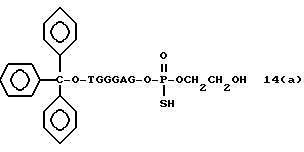
Повторялась процедура, аналогичная процедуре, описанной в примере 12 (c), но с использованием следующей модификации. Соединение примера 12 (b) сначала сочеталось с гуанидиндезоксирибонуклеотид-β- амидитом при 3' - терминальном положении, и колонка затем удалялась из синтезатора без окисления окисляющим раствором. 5 мл ацетонитрильного раствора тетраэтилтиурамидисульфида (TETD) (Эпплайд Биосистемз) затем добавлялось к колонке, которая затем оставлялась стоять при комнатной температуре в течение 15 мин. Колонка затем промывалась ацетонитрилом и устанавливалась в синтезатор.
На стадии очистки хроматография осуществлялась через колонку из силикагеля с обращенной фазой (Preparative C18, Уотерс, 1,5 x 15 см; 50 мМ TEAB, pH 7,5; 20 ---> 50% ацетонитрил, линейный градиент, 254 нм).
С помощью обработки по способу, аналогичному описанному в примере 1 (b), получалось 33 OD (260 нм) названного в заголовке соединения в виде аморфного твердого вещества. Анализ с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил ODS, 6 x 150 мм; элюент A: 0,1 M TEAA, pH 7,5; элюент B: ацетонитрил, 10 ---> 60% по объему, 20 мин, линейный градиент, 1 мл/мин; 260 нм) показал, что образец имел время удерживания 18,92 мин.
УФ - спектр поглощения (вода), λмакс, нм: 254.
Пример 15
15 (a) 5'-O-Тритил-N-бензоил-2'-дезоксицитидин
3,31 г (10 ммоль) N - бензоил-2'-дезоксицитидина добавлялось к 25 мл пиридина, а затем к суспензии добавлялось 3,07 г (11 ммоль) тритилхлорида. Получающаяся суспензия перемешивалась при 100oC в течение 1 ч, после чего оставлялась охлаждаться до комнатной температуры. Пиридин затем удалялся перегонкой при пониженном давлении. Получающийся остаток растворялся в смеси этилацетата и насыщенного водного раствора хлористого натрия. Органический слой отделялся и промывался дважды, каждый раз насыщенным водным раствором хлорида натрия. Раствор затем сушился над безводным сульфатом магния, после чего растворитель удалялся перегонкой при пониженном давлении. Получающийся остаток очищался с помощью хроматографии на колонке из силикагеля (Лобер колонка Si-60, Размер C) с использованием метода градиентного элюирования смесями метанола и метиленхлорида с изменением соотношения от 2:98 до 5:95 по объему в качестве элюента, давая 1,47 г (выход 26%) целевого соединения.
1H Спектр ЯМР (CDCl3, 270 МГц, ТМС), δ, млн.дол.:
8,72 (1H, широкий синглет), 8,26 (1H, дублет, Q = 7,3 Гц), 7,89 (1H, дублет, J = 7,3 Гц), 7,26 - 7,64 (20H, мультиплет), 6,29 (1H, триплет, J = 5,9 Гц), 4,48 - 4,53 (1H, мультиплет), 4,12 - 4,16 (1H, мультиплет), 3,42 - 3,57 (2H, мультиплет), 2,23 - 2,77 (2H, мультиплет), 2,51 (1H, широкий синглет).
15 (b) 5'-O-Тритил-N-бензоил-2'-дезоксицитидин-2-цианоэтил N,N-диизопропилфосфорамидит
402 мг (0,7 ммоль) 5'-O-тритил-N-бензоил-2'-дезоксицитидина [полученного, как описано в стадии (a) выше] подвергались сушке с помощью азеотропной перегонки с пиридином и затем растворялись в 3,5 мл метиленхлорида. К раствору добавлялось 60 мг (0,35 ммоль) диизопропиламмонийтетразолида. Затем по каплям в атмосфере аргона добавлялось 245 мкл (0,77 ммоль) 2-цианоэтил N,N,N1,N1-тетраизопропилфосфордиамидита. Получающаяся смесь перемешивалась при комнатной температуре в течение 3,5 ч в той же атмосфере. К концу данного времени метиленхлорид удалялся с помощью перегонки при пониженном давлении. Получающийся остаток растворялся в этилацетате, и раствор промывался 10% в/о водным раствором карбоната натрия и насыщенным водным раствором хлорида натрия в данном порядке. Раствор затем сушился над безводным сульфатом магния, после чего растворитель удалялся перегонкой при пониженном давлении, и остаток очищался с помощью хроматографии на колонке, наполненной 23 г силикагеля (70 - 230 меш), с использованием этилацетата в качестве элюента, давая 337 мг (выход 62%) целевого соединения в виде пенообразного вещества.
1H Спектр ЯМР (CDCl3, 270 МГц, ТМС), δ, млн. дол.:
8,87 (1H, широкий синглет), 8,52 и 8,43 (1H, дублет, J = 7,3 Гц), 8,12 и 8,11 (1H, дублет, J = 7,3 Гц), 7,23 - 7,86 (20H, мультиплет), 6,49 - 6,55 (1H, мультиплет), 4,79 - 4,91 (1H, мультиплет), 4,46 - 4,47 (1H, мультиплет), 3,60 - 4,10 (6H, мультиплет), 2,46 - 3,08 (4H, мультиплет), 1,29 - 1,40 (12H, мультиплет).
15 (c) Соединение формулы 15 (c)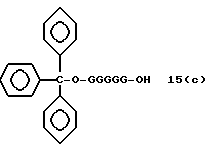
Повторялась процедура, аналогичная процедуре, описанной в примере 1(b), за исключением того, что в сосуд для амидита (называемый "X") помещалось 35 мМ ацетонитрильного раствора соединения примера 15(b), и в синтезатор вводилась основная последовательность XGGGG. 90%-ный водный раствор формамида, содержащий целевое соединение, полученное таким образом, нагревался при 95oC в течение 5 мин и затем сразу же охлаждался льдом. Реакционная смесь затем разделялась на четыре порции, и каждая порция очищалась с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил PREP-ODS 20,0 х 250 мм; элюент A: 0,1 М TEAA, pH 7,0; элюент B: ацетонитрил; 10 ---> 60% B по объему, 30 мин, линейный градиент; 7 мл/мин, 260 нм, температура колонки 60oC). Фракции, элюированные через 20,9 мин, собирались и обрабатывались по способу, аналогичному описанному в примере 1(b), давая 68 OD (260 нм) целевого соединения в виде аморфного твердого вещества.
УФ-спектр поглощения (вода), λмакс , нм: 254.
Время удерживания: 13,25 мин.
Высокоэффективная жидкостная хроматография (Инертсил ODS -2; 6 х 150 мм; элюент A: 0,1 М TEAA, pH 7,5; элюент B: ацетонитрил; 10 ---> 60% B по объему, 20 мин, линейный градиент, 1 мл/мин; 260 нм).
Пример 16
16 (a) Соединение формулы 16 (a):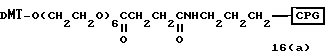
0,26 г (0,5 ммоль) O-диметокситригексаэтиленгликоля [Nucleic Acids Res., 18, 6353, (1990)] подвергалось сушке с помощью азеотропной перегонки с пиридином, а затем растворялось в 2 мл метиленхлорида. Затем к раствору добавлялось 75 мг (0,75 ммоль) янтарного ангидрида и 92 мг (0,75 ммоль) 4-(диметиламино)пиридина, и получающаяся смесь перемешивалась в течение 2 ч. После завершения реакции, о котором судили по данным тонкослойной хроматографии, реакционная смесь разбавлялась метиленхлоридом, и разбавленная смесь промывалась 0,5 М водным раствором первичного кислого фосфора калия (pH 5,0) и водой в указанном порядке. Органический слой затем сушился над безводным сульфатом натрия, и растворитель удалялся перегонкой при пониженном давлении, давая моносукцинат O-диметокситритилгексаэтиленгликоля.
Весь данный продукт сушился с помощью азеотропной перегонки с пиридином, а затем растворялся в 3 мл диметилформамида. К раствору добавлялось 0,23 г (0,37 ммоль) пентахлорфенола и 0,12 г (0,56 ммоль) 1,3-дициклогексилкарбодиимида, и получающаяся смесь перемешивалась при комнатной температуре в течение 16 ч. В конце данного периода нерастворимые вещества, которые выпадали в осадок, отфильтровывались, и фильтрат освобождался от растворителя с помощью выпаривания при пониженном давлении. Остаток растворялся в бензоле, и получающиеся нерастворимые вещества снова отфильтровывались, после чего растворитель удалялся перегонкой при пониженном давлении.
30 мкл (0,22 ммоль) триэтиламина и 1,0 г (3-аминопропил)-CPG (содержащего 0,129 ммоль/г аминогрупп) добавлялось к раствору 0,23 г (0,18 ммоль) остатка в 5 мл диметилформамида, и получающаяся смесь оставлялась стоять при комнатной температуре в течение 24 ч. В конце данного периода времени CPG-носитель собирался фильтрованием и промывался метиленхлоридом, после чего он сушился в вакууме. Для ацетилирования аминогрупп, оставшихся непрореагированными, CPG- носитель смешивался с 3 мл каждого из кэп растворов A и B (Миллипор Лтд., Япония), и смесь оставлялась стоять в течение 10 мин. Реакционный продукт затем промывался пиридином и метиленхлоридом, в указанном порядке, после чего он сушился в вакууме, давая целевое соединение.
Количество O-диметокситритилгексаэтиленгликольных остатков, введенных в соединение примера 16(a), определялось следующим образом. 9,7 мг Соединения примера 16(a) точно взвешивалось и к нему добавлялся деблокирующий раствор (метиленхлоридный раствор дихлоруксусной кислоты, Миллипор Лтд, Япония). Смесь встряхивалась в течение 3 мин, и затем добавлялось достаточное количество метиленхлорида до объема 20 мл. В опытную пробирку бралось 0,4 мл данного вещества, и растворитель удалялся перегонкой при пониженном давлении. К остатку добавлялось 3 мл 3:2 по объему смеси перхлорной кислоты и этанола, и измерялось поглощение (ε = 71700) диметокситритильного катиона при 500 нм.
Количество введенных диметокситритильных групп составляло 59,1 мкмоль/г.
16 (b) Соединение формулы 16 (b):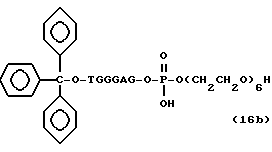
В соответствии с процедурой, аналогичной процедуре, описанной в примере 12 (c), но с использованием колонки, наполненной 85 мг (5 мкмоль) соединения примера 16 (a), получалось указанное в заголовке соединение.
Продукт очищался с помощью хроматографии через силикагельную колонку с обращенной фазой (Preparative C18, Уотерс, 1.5 x 15 см; 50 мМ TEAB, pH 7,5; 5 _→ 50% по объему ацетонитрила, линейный градиент; 254 нм).
С помощью обработки по способу, аналогичному описанному в примере 1 (b), получалось целевое соединение формулы 16 (b), имеющее 151 OD (260 нм) в виде аморфного твердого вещества. Анализ с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил ODS, 6 x 150 мм; элюент A: 0,1 М TEAA, pH 7,5, элюент B: ацетонитрил, 10 _→ 60% B по объему, 20 мин, линейный градиент, 1 мл/мин, 260 нм) показал, что образец имел время удержания 18,50 мин.
УФ-спектр поглощения (вода), λмакс, нм: 254.
Пример 17
Соединение формулы 17 (a)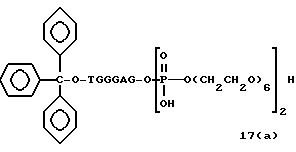
В соответствии с процедурой, аналогичной описанной в примере 1 (b), но с использованием колонки, наполненной 85 мг (5 мкмоль) соединения примера 16 (a), и с использованием 35 мМ ацетонитрильного раствора O-диметокситритилгексаэтиленгликоль - O- 2 -цианоэтил N,N - диизопропилфосфорамидита [Nucleic Acids Res. , 18, 6353 (1990)], помещаемого в амидитный сосуд (упоминаемый здесь ниже как "X"), и при внесении последовательности оснований TGGGAGXZ в синтезатор, получалось целевое соединение. В данном примере и далее X имеет значения, определенные выше, и Z представляет модельный код, т.е. генетический код, поступавший в синтезатор, который не вносит действительное основание, при внесении модельного генетического кода в синтезатор может вноситься любой из кодов A, G, C, T или X, но это не приводит в результате к добавлению основания к последовательности. Продукт очищался с помощью хроматографии при пропускании через силикагельную колонку с обращенной фазой (Preparative C18, Уотерс, 1,5 x 15 см, 50 мМ TEAB, pH 7,5; 5 ---> 50% ацетонитрила, линейный градиент, 254 нм). С помощью обработки по способу, аналогичному описанному в примере 1 (b), получалось целевое соединение, имеющее 136 OD (260 нм) в виде аморфного твердого вещества. Анализ с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил ODS 6 x 160 мм; элюент A: 0,1 М TEAA, pH 7,5, элюент B: ацетонитрил; 10 ---> 60% B по объему, 20 мин, линейный градиент, 1 мл/мин, 260 нм) показал, что образец имел время удерживания 18,32 мин.
УФ-спектр поглощения (вода), λмакс , нм: 254.
Пример 18
Соединение формулы 18 (a):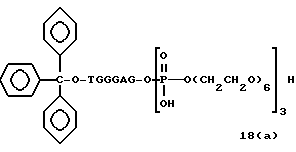
В соответствии с процедурой, аналогичной процедуре, описанной в примере 17, но при внесении последовательности оснований TGGGAGXXZ (в которой X и Z имеют значения, определенные в примере 17) в синтезатор получалось целевое соединение. Продукт очищался с помощью хроматографии при пропускании через силикагельную колонку с обращенной фазой (Preparative C18, Уотерс, 1,5 x 15 см; 50 мМ TEAB, pH 7,5; 5 _→ 50% ацетонитрила, линейный градиент, 254 нм). Затем он обрабатывался по способу, аналогичному описанному в примере 1 (b), давая 136 OD (260 нм) целевого соединения формулы 18 (a) в виде аморфного твердого вещества. Анализ высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил ODS, 6 x 150 нм; элюент A: 0,1 М TEAA, pH 7,5; элюент B: ацетонитрил; 10 ---> 60% B по объему, 20 мин, линейный градиент, 1 мл/мин; 260 нм) показал, что образец имеет время удерживания 18,20 мин.
УФ-спектр поглощения (вода), λмакс , нм: 255
Пример 19
Соединение формулы 19 (a):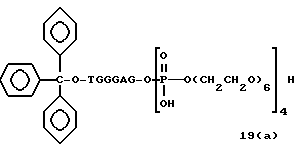
В соответствии с процедурой, аналогичной процедуре, описанной в примере 17, но при внесении в синтезатор последовательности оснований TGGGAGXXX (в которой X и Z имеют значения, определенные в примере 17), получалось целевое соединение. Реакционный продукт очищался с помощью хроматографии на силикагельной колонке с обращенной фазой (Preparative C18, Уотерс, 1,5 x 15 см; 50 мМ TEAB, pH 7,5, 5 _→ 50% ацетонитрила, линейный градиент, 254 нм). Продукт обрабатывался по способу, аналогичному описанному в примере 1 (b), давая целевое соединение, имеющее 110 OD (260 нм) в виде аморфного твердого вещества,
Анализ высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил ODS, 6 x 150 мм; элюент A: 0,1 М TEAA, pH 7,5, элюент B: ацетонитрил, 10 ---> 60% B по объему, 20 мин; линейный градиент, 1 мл/мин, 260 нм) показал, что образец имеет время удерживания 17,99 мин.
УФ-спектр поглощения (вода), λмакс , нм: 254.
Пример 20
Соединение формулы 20 (a):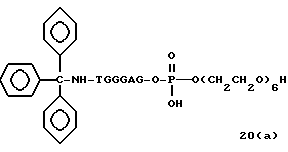
В соответствии с процедурой, аналогичной процедуре, описанной в примере 2 (c), но при использовании колонки, наполненной 85 мг (5 мкмоль) соединения примера 16 (a), и при внесении последовательности оснований TGGGAGZ (в которой Z имеет значения, определенные в примере 17) в ДНК /РНК-синтезатор, описанной в примере 1 (b), получалось целевое соединение. Продукт очищался с помощью хроматографии при пропускании через силикагельную колонку с обращенной фазой (Preparative C18, Уотерс, 1,5 x 15 см; 50 мМ TEAB, pH 7,5; 20 _→ 50% ацетонитрила, линейный градиент, 254 нм). Продукт обрабатывался по способу, аналогичному описанному в примере 1 (b), давая целевое соединение, имеющее 177 OD (260 нм) в виде аморфного твердого вещества. Анализ с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил ODS, 6 x 150 мм; элюент A: 0,1 М TEAA, pH 7,5; элюент B: ацетонитрил; 10 ---> 60% B по объему, 20 мин, линейный градиент, 1 мл/мин; 260 нм) показал, что образец имеет время удерживания 19,10 мин.
УФ-спектр поглощения (вода), λмакс , нм : 254.
Пример 21
21 (a) Соединение формулы 21 (a):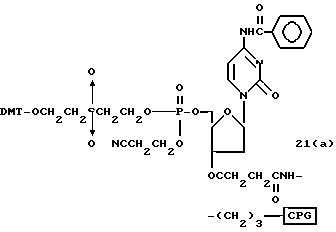
Раствор 250 мг 5'-фосфата ON (торговая марка продукта фирмы Клонтек) в 5,2 мл ацетонитрила и колонка, наполненная 15 мкмоль C-CPG (Миллипор Лтд., Япония), использовались в ДНК/РНК-синтезаторе, описанном в примере 1 (b), давая количественно целевое соединение.
21 (b) Соединение формулы 21 (b):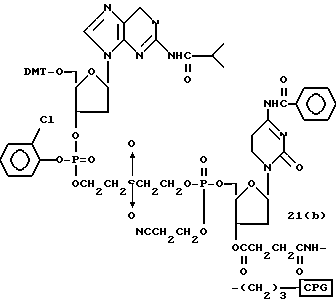
5 мл деблокирующего раствора (Миллипор Лтд., Япония) добавлялось на снабженную фильтром колонку, наполненную 115 мг (4 мкмоль) соединения примера 21 (a). Затем все оставлялось стоять в течение 1 мин, после чего колонка промывалась 5 мл метиленхлорида, а затем обрабатывалась деблокирующим раствором в течение 1 мин. Смесь затем промывалась 5 мл метиленхлорида и 5 мл пиридина, в указанном порядке, после чего она азеотропно перегонялась с пиридином. Затем к дистилляту добавлялся раствор 20 мг триэтиламмоний 5'-O-диметокситрили-2-N - изобутирил- 2'-дезоксигуанозин - 3'-0/2- хлорфенил/фосфата (Nucleic Acids Res., 8, 5461 /1980/) в 0,5 мл пиридина, после чего растворитель удалялся перегонкой при пониженном давлении. К остатку добавлялся раствор 40 мг 2,4,6 - триметилбензолсульфонил-3-нитротриазола в 0,5 мл пиридина, и получающаяся смесь нагревалась до 40oC в течение 30 мин. Реакционная смесь затем промывалась три раза, каждый раз 5 мл пиридина, а затем к смеси добавлялось 2,5 мл кэп раствора A (Миллипор Лтд., Япония) и 2,5 мл кэп раствора B (Миллипор Лтд., Япония). Смесь оставлялась стоять в течение 3 мин, после чего она промывалась три раза, каждый раз 5 мл метиленхлорида, давая целевое соединение.
21 (c) Соединение формулы 21 (c):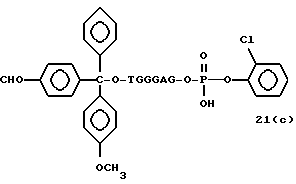
В соответствии с процедурой, аналогичной процедуре, описанной в примере 1 (b), но с использованием колонки, наполненной 115 мг (4 мкмоль) соединения примера 21 (b) и тиминдезоксирибонуклеотид- β -амидита (Миллипор Лтд., Япония) вместо 5' - O - тритилтимидин 2- цианоэтил N,N - диизопропилфосфорамидита, получалось целевое соединение. Реакционный продукт очищался хроматографией при пропускании через силикагельную колонку с обращенной фазой (Preparative C18, Уотерс, 1,5 x 15 см; 50 мМ TEAB, pH 7,5; 5 ---> 50% ацетонитрила, линейный градиент, 254 нм). Затем он обрабатывался по способу, аналогичному способу, описанному в примере 1 (b), давая целевое соединение, имеющее 85 OD (260 нм) в виде аморфного твердого вещества. Анализ с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил ODS, 6 x 150 мм; элюент A: 0,1 М TEAA, pH 7,5; элюент B: ацетонитрил; 10 ---> 50% B по объему, 20 мин, линейный градиент, 1 мл/мин, 260 нм) показал, что образец имел время удерживания 21,5 мин.
УФ-спектр поглощения (вода), λмакс , нм: 254.
Пример 22
22 (a) Триэтиламмоний 5' - O - диметокситритил - 2 - N - изобутирил-2'- дезоксигуанозин-3'- O - фенилфосфат
0,8 г (11,55 ммоль) 1,2,4 - триазола подвергалось сушке азеотропной перегонкой с пиридином и затем суспендировалось в 10 мл диоксана. К суспензии добавлялось 0,52 мл (3,5 ммоль) фенилдихлорфосфата, а затем 1,0 мл (7,35 ммоль) триэтиламина при охлаждении льдом и перемешивании. Температура затем поднималась до комнатной. Смесь перемешивалась в течение 2 ч., после чего выпавший в осадок хлоргидрат отфильтровывался, давая диоксановый раствор, содержащий 0,35 ммоль/мл фенил фосфоробис(триазолидата).
1 мл (0,35 ммоль) диоксанового раствора фенил бис/триазолид/ фосфата, полученного, как описано выше, добавлялся к 0,15 г (0,23 ммоль) 5'-O-диметокситритил-2 -N - изобутирил-2'-дезоксигуанозина (Methods Enzymol I., 65, 610 /1980/), и получающаяся смесь перемешивалась при комнатной температуре в течение 3 ч. В конце данного периода добавлялось около 10 мл 30% по объему водного пиридина. Реакционная смесь экстрагировалась затем 30 мл метиленхлорида, и экстракт промывался 0,1 М TEAB. Растворитель удалялся с помощью перегонки при пониженном давлении, и получающийся остаток растворялся в 1 мл метиленхлорида и растирался с 30 мл 1:1 по объему смеси гексана и диэтилового эфира, содержащей 1% по объему триэтиламина, давая 81 мг (выход 40%) целевого соединения в виде порошка.
1H Спектр ЯМР (CDC13, 270 Мгц, TMC), δ, млн.дол.:
12,07 (1H, широкий синглет), 9,67 (1H, широкий синглет), 7,80 - 7,64 (1H, мультиплет), 7,47 - 6,91 (18H, мультиплет), 6,12 - 6,10 (1H, мультиплет), 5,48 - 5,45 (1H, мультиплет), 4,64 - 4,58 (1H, мультиплет), 4,28 (1H, мультиплет), 3,74 (6H, синглет), 3,38 - 3,18 (2H, мультиплет), 2,98 - 2,89 (6H, мультиплет), 2,73 - 2,51 (2H, мультиплет), 2,34 - 2,29 (1H, мультиплет), 1,25 - 1,19 (9H, мультиплет), 1,12 - 0,99 (6H, мультиплет).
22 (b) Соединение формулы 22 (b):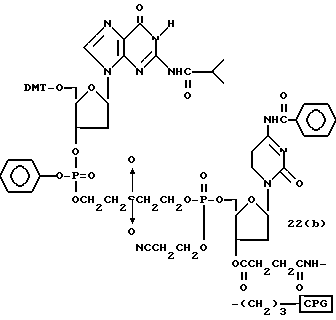
В соответствии с процедурой, аналогичной описанной в примере 21 (b), но с использованием 143 мг (5 ммоль) соединения примера 21 (a) и соединения примера 22 (a) получалось целевое соединение.
22 (c) Соединение формулы 22 (c):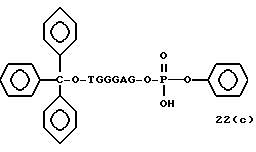
В соответствии с процедурой, аналогичной процедуре, описанной в примере 1 (b), но при использовании колонки, наполненной 143 мг (5 мкмоль) соединения примера 22 (b), получалось целевое соединение. Реакционный продукт затем очищался с помощью хроматографии при пропускании через силикагельную колонку с обращенной фазой (Preparative C18, Уотерс, 1,5 x 15 см; 50 мМ TEAB, pH 7,5; 20 _→ 50% ацетонитрила, линейный градиент, 254 нм). Затем он обрабатывался по способу, аналогичному описанному в примере 1 (b), давая целевое соединение, имеющее 66 OD (260 нм) в виде аморфного твердого вещества. Анализ с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил ODS, 6 x 150 мм; элюент A: 0,1 М TEAA, pH 7,5, элюент B: ацетонитрил; 10 _→ 60% B по объему, 20 мин, линейный градиент, 1 мл/мин, 260 нм) показал, что образец имеет время удерживания 18,22 мин.
Уф-спектр поглощения (вода), λмакс , нм: 255.
Пример 23
Продукт примера 23 (a):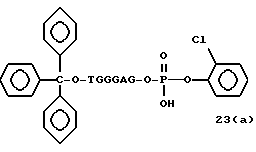
В соответствии с процедурой, аналогичной процедуре, описанной в примере 1 (b), но при использовании колонки, наполненной 115 мг (4 мкмоль) соединения примера 21 (b), получалось целевое соединение. Реакционный продукт очищался с помощью хроматографии на силикагельной колонке с обращенной фазой (Preparative Уотерс, 1,5 х 15 см; 50 мМ ТЕАВ, pH 7,5; 20 ---> 50% ацетонитрила; линейный градиент; 254 нм). Продукт обрабатывался по способу, аналогичному описанному в примере 1 (b), давая целевое соединение, имеющее 56 OD (260 нм) в виде аморфного твердого вещества. Анализ с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил ODS 6 х 150 мм; элюент A: 0,1 M TEAA, pH 7,5; элюент B: ацетонитрил; 10 ---> 60% B по объему, 20 мин, линейный градиент, 1 мл/мин; 260 нм) показал, что образец имел время удержания 18,38 мин.
УФ-спектр поглощения (вода), λмакс , нм: 255.
Пример 24
24 (a) Соединение формулы 24(a):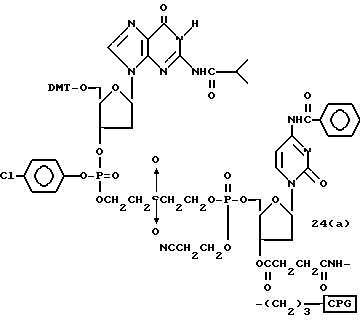
В соответствии с процедурой, аналогичной процедуре, описанной в примере 21 (b), но при использовании 143 мг (5 мкмоль) соединения примера 21 (a) и триэтиламмоний 5'-O - диметокситритил - 2 N - изобутирил- 2'- дезоксигуанозин - 3'- (4-хлорфенил)фосфата [Methods Enzymol, 65, 610 (1980)], получалось целевое соединение.
24 (b) Соединение формулы 24 (b):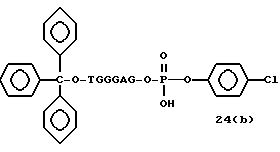
Целевое соединение получали, следуя процедуре, сходной с описанной в примере 1 (b), за исключением того, что колонка заполнялась 143 мг (4 мкмоль) соединения примера 24 (a). Реакционный продукт очищался хроматографией при пропускании через силикагельную колонку с обратной фазой (Preparative C 18, Waters 1,5 х 15 см; 50 мМ TAEB, pH 7,5; 20 _→ 50% ацетонитрила; линейный градиент, 254 нм). Он затем обрабатывался тем же способом, что и в примере 1 (b), давая целевое соединение, имеющее 55 OD (260 нм) в качестве аморфного твердого вещества. Анализ с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Unteptcus ODS 6 х 150 мм, элюент A: 0,1 M TEAA, pH 7,5; элюент B: ацетонитрил, 10 ---> 10% B по объему, 20 мин, линейный градиент; 1 мл/мин, 260 нм) показал, что образец имел время удерживания 18,55 мин.
Спектр УФ-поглощения (вода), λмакс , нм: 255.
Пример 25
DMT - O - TGGGAG - OH
Требуемая последовательность оснований (5'-TGGGAG-3') помещалась в синтезатор 380B (продукт фирмы Эпплайд Биосистемз Инк), и к нему присоединялась колонка со стеклом с регулируемыми порами (масштаб 1 мкмоль), связанная с соответствующим нуклеозидом (2'-дезоксигуанозин) в 3'-терминале. Синтез проводился в масштабе 1 мкмоль. Аппарат устанавливался так, чтобы не деблокировать ДМТ-группу после завершения реакции, и со смолы автоматически высвобождался продукт, имеющий ДМТ-группу, давая олигодезоксирибонуклеотид в виде аммиачного раствора. Данный раствор герметизировался в сосуде и нагревался при 55oC в течение 8 ч, и аммиак выпаривался в токе азота. Получающийся остаток растворялся в небольшом количестве 0,1 M TEAA (pH 7,0). Данный раствор фильтровался через миллипор-фильтр (0,2 мкм), и фильтрат очищался с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Космосил 5С18-АР, 20 х 250 мм), в условиях, перечисленных ниже. Фракции, элюированные спустя 13,5 мин, собирались и освобождались от растворителя (ацетонитрила) перегонкой при пониженном давлении, с последующей лиофилизацией, давая 800 мкг целевого соединения,
Время элюирования: 17,0 мин.
Условия высокоэффективной препаративной хроматографии приведены в табл. 3.
Буферный раствор A : 0,1 M триэтиламмонийацетатный буфер (pH 7,0).
Буферный раствор B : 100% ацетонитрил.
Скорость потока : 9 мл/мин.
Примеры 26 - 38
В соответствии с процедурой, аналогичной процедуре, описанной в примере 25, синтезировались соединения примеров 26 - 38. Наименования соединений и их время элюирования указаны в табл. 4.
Пример 82
82 (a) Соединение формулы 82 (a) :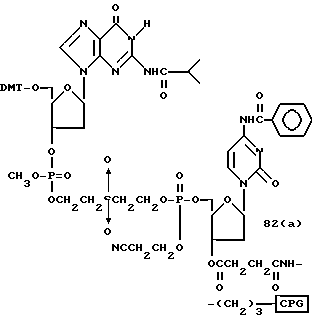
Требуемые реагенты для синтеза, подробно описанного ниже, подавались в ДНК/РНК-синтезатор Циклон /торговая марка/ Плюс Миллиген /биосерч/ Миллипор Лтд. , Япония/. Устанавливалась также программа для амидитного метода (15 мкмоль). Использовался приблизительно 35 мМ ацетонитрильный раствор 5'-O - диметокситритил- 2 - N - изобутрил-2'-дезоксигуанозин-3'-О- (метил N,N - диизопропилфосфорамидита) (Сигма) вместо β -цианоэтиламидита, соответствующего гуанозину. С помощью использования пустой колонки (15,0 мкмоль), заполненной 143 мг/мм (5 мкмоль) соединения примера 21 (a), введения в синтезатор последовательности оснований GX и программы обработки без кислотной обработки после связывания с основанием G получалось целевое соединение.
82 (b) Соединение формулы 82 (b) :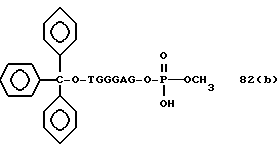
В соответствии с процедурой, аналогичной процедуре, описанной в примере 1 (b), но при использовании колонки, наполненной 143 мг (5 мкмоль) соединения примера 82 (a), получалось названное в заголовке соединение. Продукт очищался с помощью хроматографии при пропускании через силикагельную колонку с обращенной фазой (Preparative C18, Уотерс, 1,5 х 15 см; 50 мМ TEAB, pH 7,5; 20 _→ 50% ацетонитрила, линейный градиент; 254 нм). Затем он обрабатывался по способу, аналогичному описанному в примере 1 (b), давая названное в заголовке соединение, имеющее 66 OD (260 нм) в виде аморфного твердого вещества. Анализ высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил ODS, 6 х 250 мм; элюент A: 0,1 M TEAA, pH 7,5; элюент B: ацетонитрил; 10 _→ 60% В по объему, 20 мин; линейный градиент; 1 мл/мин; 260 нм) показал, что образец имеет время удерживания 17,54 мин.
УФ-спектр поглощения (вода), λмакс, нм: 254.
Пример 83
83 (a) Соединение формулы 83 (a)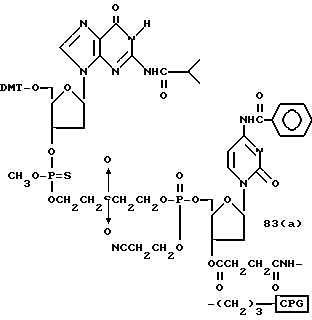
В соответствии с процедурой, аналогичной процедуре, описанной в примере 82 (a), но при использовании 143 мг (5 мкмоль) соединения примера 21 (a), получалось названное в заголовке соединение. Подробнее, после соединения с 5'-O-диметокситритил-2-N-изобутирил-2'-дезоксигуанозин-3'-O- (метил-N, N-диизопропил)фосфорамидитом колонка убиралась из ДНК/РНК-синтезатора без окисления окисляющим раствором. К колонке добавлялось 5 мл ацетонитрильного раствора тетраэтилтиурамдисульфида (ТЕТД) (Эпплайд Биосистемз), и колонка оставлялась стоять при комнатной температуре в течение 15 мин. Колонка затем промывалась ацетонитрилом и устанавливалась в синтезатор.
83 (b) Соединение формулы 83 (b):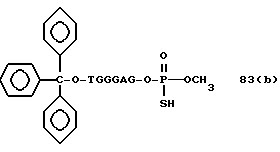
В соответствии с процедурой, аналогичной процедуре, описанной в примере 1 (b), но при использовании колонки, наполненной 143 мг (5 мкмоль) соединения примера 83 (a), получалось указанное в заголовке соединение. Реакционный продукт очищался с помощью хроматографии на силикагельной колонке с обращенной фазой (Preparative C18. Уотерс, 1,5 х 15 см; 50 мМ ТЕАВ, pH 7,5; 20 _→ 50% ацетонитрила, линейный градиент; 1 мл/мин; 260 нм). Затем продукт обрабатывался по способу, аналогичному описанному в примере 1 (b), давая целевое соединение, имеющее III OD (260 нм) в виде аморфного твердого вещества. Анализ с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил ODS, 6 х 150 мм; элюент А: 0,1 М ТЕАА, pH 7,5; алюент B: ацетонитрил; 10 _→ 60% по объему, 20 мин, линейный градиент, 1 мл/мин; 260 нм) показал, что образец имеет время удерживания 17,72 мин.
УФ-спектр поглощения (вода), λмакс, нм: 254.
Пример 84
84 (a) Соединение формулы 84 (a):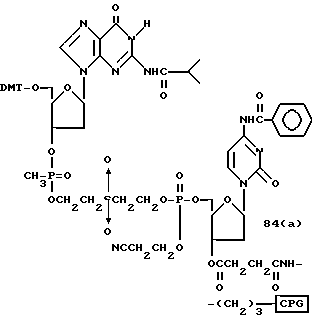
Требуемые реагенты для синтеза, как подробно описано ниже, подавались в Циклон (торговая марка) Плюс ДНК/РНК-синтезатор Миллиген/биосеры /Миллипор Лтд. , Япония/. Устанавливалась также программа для амидитного метода (15 мкмоль). Использовался приблизительно 35 мМ тетрагидрофурановый раствор дезоксигуанозин (N-изобутил)метилфосфонамидита (Амирокан Бионетик Инк. ) вместо β -цианоэтиламидитного раствора, соответствующего гуанозину. При использовании пустой колонки (15,0 мкмоль), заполненной 143 мг (4 мкмоль) соединения примера 21 (a) в качестве твердого носителя, введения последовательности оснований GZ (где Z имеет значения, определенные в примере 17) и программы обработки без обработки кислотой после связывания с основанием G получалось указанное в заголовке соединение.
84 (b) Соединение формулы 84 (b):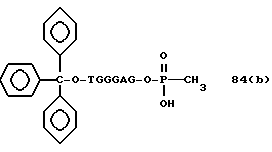
В соответствии с процедурой, аналогичной процедуре, описанной в примере 1 (b), но при использовании колонки, наполненной 143 мг (5 мкмоль) соединения примера 84 (a), получалось названное в заголовке соединение. Продукт реакции очищался с помощью хроматографии при пропускании через силикагельную колонку с обращенной фазой (Preparative C18, Уотерс 1,5 х 15 см; 50 мМ ТАЕВ, pH 7,5; 20 _→ 50% ацетонитрила, линейный градиент; 254 нм). Затем он обрабатывался по способу, аналогичному описанному в примере 1 (b), давая указанное в заголовке соединение, имеющее 159 OD (260 нм) в виде аморфного твердого вещества. Анализ с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил ODS, 6 х 150 мм; элюент A: 0,1 M TEAA, pH 7,5; элюент B: ацетонитрил; 10 _→ 60% B по объему, 20 мин; линейный градиент; 1 мл/мин; 260 нм) показал, что образец имеет время удерживания 18,03 мин.
УФ-спектр поглощения (вода), λмакс, нм: 254.
Пример 85
85 (a) Соединение формулы 85 (a):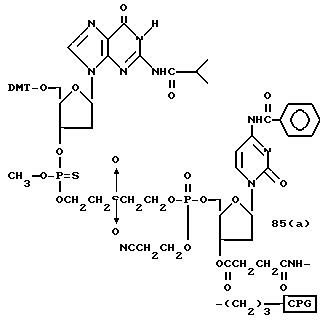
В соответствии с процедурой, аналогичной процедуре, описанной в примере 84 (a), но с использованием колонки, наполненной 143 мг (5 мкмоль) соединения примера 21 (a), получалось указанное в заголовке соединение. Более подробно, после присоединения к дезоксигуанозин (N-изобутил)метилфосфонамидиту, колонка удалялась из синтезатора без окисления окисляющим раствором, а затем к колонке добавлялось 5 мл ацетонитрильного раствора тетраэтилтиурамдисульфида (ТЕТД) (Эпплайд Биосистемз). Колонка оставлялась стоять при комнатной температуре в течение 15 мин. После чего она промывалась ацетонитрилом и снова устанавливалась в синтезатор.
85 (b) Соединение формулы 85 (b):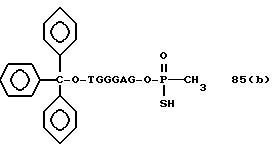
В соответствии с процедурой, аналогичной процедуре, описанной в примере 1 (b), но с использованием колонки, наполненной 143 мг (5 мкмоль) соединения примера 85 (a), получалось указанное в заголовке соединение. Продукт реакции очищался с помощью хроматографии через силикагельную колонку с обращенной фазой (Preparative C18, Уотерс, 1,5 х 15 см; 50 мМ ТЕАВ, pH 7,5; 20 _→ 50% ацетонитрила, линейный градиент; 254 нм). Затем он обрабатывался по способу, аналогичному способу, описанному в примере 1 (b), давая указанное в заголовке соединение, имеющее 124 OD (260 нм) в виде аморфного твердого вещества. Анализ с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил ODS, 6 x 160 мм; элюент A: 0, 1 M TEAA, pH 7,5; элюент B: ацетонитрил; 10 _→ 60% B по объему, 20 мин; линейный градиент; 1 мл/мин; 260 нм) показал, что образец имеет время удерживания 18,07 мин.
УФ-спектр поглощения (вода), λмакс , нм: 254.
Пример 86
Соединение формулы 86 (a):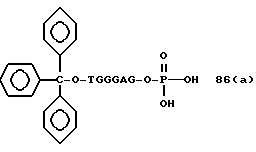
В соответствии с процедурой, аналогичной процедуре, описанной в примере 1 (b), но при использовании колонки, заполненной 143 мг (5 мкмоль) соединения примера 21(a), и помещении последовательности оснований TGGGAGZ (в которой Z имеет значения, определенные в примере 17) в ДНК/РНК-синтезатор, описанный в примере 1(b), приготавливалось целевое соединение. Реакционный продукт очищался с помощью хроматографии через силикагельную колонку с обращенной фазой (Preparative C18, Уотерс, 1,5 х 15 см; 50 мМ TEAB, pH 7,5; 20 _→ 50% ацетонитрил; линейный градиент; 254 нм). Затем продукт обрабатывался по способу, аналогичному описанному в примере 1(b), давая целевое соединение, имеющее 54 OD (260 нм) в виде аморфного твердого вещества. Анализ с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил ODS, 6 х 150 мм; элюент A: 0,1 M TEAA, pH 7,5; элюент B: ацетонитрил, по объему, 15 _→ 65%, 20 мин; линейный градиент; 1 мл/мин, 260 нм), показал, что образец имеет время удерживания 15,20 мин.
УФ-спектр поглощения (вода), λмакс , нм: 255.
Пример 87
Соединение формулы 87 (a):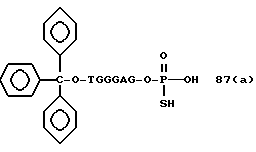
В соответствии с процедурой, аналогичной процедуре, описанной в примере 86, но при использовании колонки, наполненной 143 мг (5 мкмоль) соединения примера 21 (a), получалось целевое соединение. Более подробно, после первого присоединения к G - β- амидиту в 3' - концевом положении колонка удалялась из ДНК/РНК-синтезатора без окисления окисляющим раствором. К колонке добавлялись 5 мл ацетонитрильного раствора тетраэтилтиурамидсульфида (ТЕТД) (Эпплайд Биосистемз), и колонка оставлялась стоять при комнатной температуре в течение 15 мин, после чего она промывалась ацетонитрилом и устанавливалась в синтезатор.
Реакционный продукт очищался с помощью хроматографии через силикагельную колонку с обращенной фазой (Preparative C 18, Уотерс, 1,5 х 15 см; 50 мМ TEB, pH 7,5; 20 _→ 50% ацетонитрил; линейный градиент; 254 нм). Продукт затем обрабатывался по способу, аналогичному описанному в примере 1(b), давая указанное в заголовке соединение, имеющее 136 OD (260 нм) в виде аморфного твердого вещества. Анализ с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил ODS, 6 х 150 мм; элюент A: 0,1 M TEAA, pH 7,5; элюент B: ацетонитрил; 10 ---> 60% B по объему, 20 мин; линейный градиент; 1 мл/мин; 260 нм) показал, что образец имеет время удерживания 18,02 мин.
УФ-спектр поглощения (вода), λмакс , нм: 254.
Пример 88
Соединение формулы 88(a):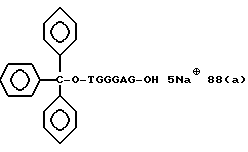
Колонка заполнялась ионно-обменной смолой Дауэкс 50W - X2 (торговая марка продукции Дау Кемикал Co. ; H-форма, около 1 мл), и через колонку пропускалось 3 мл 20% о/о водного пиридина, колонка затем промывалась водой, давая пиридиниевую форму смолы в колонке. В соответствии с аналогичной процедурой, но при использовании 3 мл 1 н. водного раствора гидроокиси натрия, на еще одной колонке приготавливалась натриевая форма смолы. Соединение примера 1(b), имеющее 27 OD, наносилось последовательно на сочетание колонки из смолы в пиридиниевой форме и на колонку со смолой в натриевой форме, в указанном порядке, а затем колонки элюировались водой, давая целевое соединение (натриевую соль), имеющее 27 OD (260 нм) в виде аморфного твердого вещества. Анализ с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил ODS, 6 х 150 мм; элюент A: 0,1 M TEAA, pH 7,5; элюент B: ацетонитрил; 10 _→ 60% B по объему, 20 мин; линейный градиент, 1 мл/мин; 260 нм) показал, что образец имеет время удерживания 18,22 мин.
УФ-спектр поглощения (вода), λмакс , нм: 256.
Пример 89
Соединение формулы 89(a):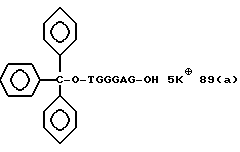
Колонка заполнялась ионно-обменной смолой Дауэкс 50W - X2 (торговая марка продукта фирмы Дау Кеминал Ко., Н-форма, около 1 мл), и через колонку, которая затем промывалась водой, наносилось 3 мл 20% о/о водного пиридина, это давало пиридиниевую форму смолы в колонке. В соответствии с аналогичной процедурой, но при использовании 3 мл 1 н. водного раствора гидроокиси калия, в другой колонке получалась калиевая форма смолы. Соединение примера 1(b), имеющее 27 OD, наносилось последовательно на колонку со смолой в пиридиниевой форме и колонку со смолой в калиевой форме, и колонки элюировались водой, давая целевое соединение (калиевую соль), имеющее 27 OD (260 нм) в виде аморфного твердого вещества. Анализ с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил ODS 6 х 150 мм; элюент A: 0,1 М TEAA, pH 7,5; элюент B: ацетонитрил; 10 _→ 60% B по объему, 20 мин; линейный градиент 1 мл/мин, 260 нм) показал, что образец имеет время удерживания 18,18 мин.
УФ-спектр поглощения (вода), λмакс , нм: 254.
Пример 90
Соединение формулы 90(a)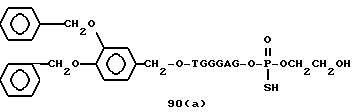
В соответствии с процедурой, аналогичной процедуре, описанной в примере 14(a), но при использовании 5'-O-(3,4-дибензилоксибензил)тимидин 2 - дианоэтил N, N - диизопропилфосфорамидита [полученного, как описано в примере 7(b)] вместо 5'-O-тритилтимидин 2 - цианоэтил N, N - диизопропилфосфорамидита, получалось целевое соединение. Реакционный продукт очищался с помощью хроматографии через силикагельную колонку с обращенной фазой (Preparative C18, Уотерс, 1,5 х 15 см, 50 мМ TEAB, pH 7,5; 10 _→ 50% ацетонитрил, линейный градиент, 254 нм). Затем продукт обрабатывался по способу, аналогичному способу, описанному в примере 1(b), давая целевое соединение, имеющее 119 OD (260 нм) в виде аморфного твердого вещества. Анализ с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил ODS 6 х 150 мм; элюент A: 0,1 М TEAA, pH 7,5; элюент B: ацетонитрил, 10 _→ 60% B по объему, 20 мин, линейный градиент, 1 мл/мин, 250 нм) показал, что образец имеет время удерживания 19,20 мин.
УФ-спектр поглощения (вода), λмакс , нм: 254.
Пример 91
Соединение формулы 91(a):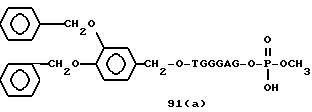
Согласно процедуре, аналогичной описанной в примере 82 (a), но с использованием 5'-О-(3,4-дибензилоксибензил)тимидин 2-цианоэтил N,N-диизопропилфосфорамидита [полученного, как описано в примере 7(b)] вместо 5'-O-тритилтимидин 2-цианоэтил N,N-диизопропилфосфорамидита, получалось целевое соединение. Реакционный продукт очищался с помощью хроматографии через силикагельную колонку с обращенной фазой (Preparative C18, Уотерс, 1,5 х 15 см; 50 мМ ТЕАВ; pH 7,5; 20 _→ 50% ацетонитрила; линейный градиент; 254 нм). Продукт затем обрабатывался по способу, аналогичному описанному в примере 1 (b), давая целевое соединение, имеющее 50 OD (260 нм) в виде аморфного твердого вещества. Анализ с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил ODS, 6 х 150 мм; элюент А: 0,1 M TEAA, pH 7,5 : элюент B: ацетонитрил, 10 _→ 60% В по объему, 20 мин, линейный градиент; 1 мл/мин; 260 нм) показал, что данный образец имеет время удерживания 19,15 мин.
УФ-спектр поглощения (вода), λмакс, 254 нм.
Пример 92
Соединение формулы 92 (a)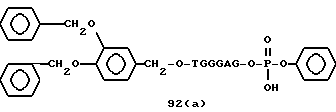
Согласно процедуре, аналогичной описанной в примере 22 (c), но с использованием 5'-O-(3,4-дибензилоксибензил)тимидин 2-цианоэтил N,N-диизопропилфосфорамидита [полученного, как описано в примере 7(b)] вместо 5'-O-тритилтимидин 2-цианоэтил N,N-диизопропилфосфорамидита, получалось целевое соединение. Продукт реакции очищался хроматографией через силикагельную колонку с обращенной фазой (Preparative C18, Уотерс 1,5 х 15 см; 50 мМ TEAB, pH 7,5; 15 _→ 50% ацетонитрил, линейный градиент, 254 нм). Затем он обрабатывался по способу, аналогичному описанному в примере 1 (b), давая целевое соединение, имеющее 67 OD (260 нм) в виде аморфного твердого вещества. Анализ высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил ODS 6 x 150 мм; элюент A: 0,1 М TEAA, pH 7,5; элюент B: ацетонитрил; 10 _→ 60% B по объему, 20 мин; линейный градиент; 1 мл/мин, 260 нм) показал, что данный образец имеет время удерживания 19,51 мин.
УФ-спектр поглощения (вода), λмакс, нм: 255.
Пример 93
Соединение формулы 93(a)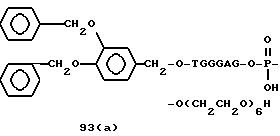
Согласно процедуре, аналогичной описанной в примере 16 (b), но с использованием 5'-O-(3,4-дибензилоксибензил) тимидин 2-цианоэтил N,N-диизопропилфосфорамидита [полученного, как описано в примере 7 (b)] вместо 5'-O-тритилтимидин 2-цианоэтил N,N-диизопропилфосфорамидита, получалось целевое соединение. Продукт реакции очищался с помощью хроматографии через колонку из силикагеля с обращенной фазой (Preparative C18, Уотерс, 1,5 х 15 см; 50 мМ TEAB, pH 7,5; 15 _→ 50% ацетонитрил; линейный градиент; 254 нм). Продукт затем обрабатывался по способу, аналогичному описанному в примере 1 (b), давая целевое соединение, имеющее 98 OD (260 нм) в виде аморфного твердого вещества. Анализ путем высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил ODS 6 x 150 мм; элюент A: 0,1 M TEAA, pH 7,5; элюент B: ацетонитрил; 10 _→ 60% B по объему, 20 мин, линейный градиент; 1 мл/мин; 260 нм) показал, что данный образец имеет время удерживания 19,26 мин.
УФ-спектр поглощения (вода), λмакс, нм: 254.
Пример 94
Соединение формулы 94 (a):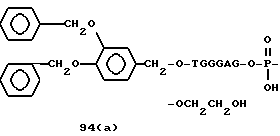
В соответствии с процедурой, аналогичной описанной в примере 12 (c), но с использованием 5'-O-(3,4-дибензилоксибензил) тимидин 2-цианоэтил N,N-диизопропилфосфорамидита [полученного, как описано в примере 7 (b)] вместо 5'-O-тритилтимидин 2-цианоэтил N,N-диизопропилфосфорамидита, получалось целевое соединение. Продукт реакции очищался с помощью хроматографии через силикагельную колонку с обращенной фазой (Preparative C18, Уотерс, 1,5 х 15 см; 50 мМ TEAB, pH 7,5; 15 _→ 50% ацетонитрила; линейный градиент; 254 нм). Продукт затем обрабатывался по способу, аналогичному описанному в примере 1 (b), давая целевое соединение, имеющее 97 OD (260 нм) в виде аморфного твердого вещества. Анализ с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил ODS 6 x 150 мм; элюент A: 0,1 M TEAA, pH 7,5; элюент B: ацетонитрил; 10 _→ 60% B по объему, 20 мин; линейный градиент; 1 мл/мин; 260 нм) показал, что данный образец имеет время удерживания 19,16 мин.
УФ-спектр поглощения (вода), λмакс, нм: 254.
Пример 95
Соединение формулы 95 (a):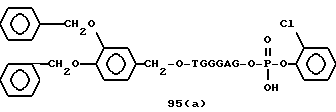
В соответствии с процедурой, аналогичной описанной в примере 23 (a), но при использовании 5'-O-(3,4-дибензилоксибензил) тимидин 2-цианоэтил N,N-диизопропилфосфорамидита [полученного, как описано в примере 7 (b)] вместо 5'-O-тритилтимидин 2-цианоэтил N,N-диизопропилфосфорамидита, получалось целевое соединение. Продукт реакции очищался с помощью хроматографии через силикагельную колонку с обращенной фазой (Preparative C18, Уотерс, 1,5 х 15 см; 50 мм TEAB, pH 7,5; 15 _→ 50% ацетонитрил; линейный градиент; 254 нм). Продукт затем обрабатывался по способу, аналогичному описанному в примере 1 (b), давая целевое соединение, имеющее 83 OD (260 нм) в виде аморфного твердого вещества. Анализ с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил ODS 6 x 150 мм; элюент A: 0,1 M TEAA, pH 7,5; элюент B: ацетонитрил, 10 _→ 60% B по объему, 20 мин; линейный градиент; 1 мл/мин; 260 нм) показал, что данный образец имеет время удерживания 19,80 мин.
УФ-спектр поглощения (вода), λмакс, нм: 255.
Пример 96
Соединение формулы 96 (a):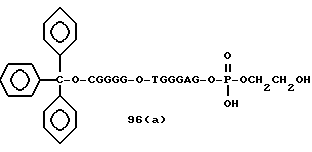
Повторялась процедура, аналогичная процедуре, описанной в примере 15 (c), но с использованием колонки, наполненной 125 мг (5 мкмоль) соединения примера 12 (b), и помещением последовательности оснований XGGGGZ (в которой Z имеет значения, определенные в примере 17) в синтезатор. Затем 90% о/о водный формамидный раствор, содержащий полученное таким образом целевое соединение, нагревался при 95oC в течение 5 мин. После данного нагревания раствор быстро охлаждался. Реакционная смесь затем разделялась на четыре части, и каждая часть очищалась с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил PPEP-O 20,0 х 250 нм % 250 мм; элюент A: 0,1 М ТЕАА, pH 7,0; элюент B: ацетонитрил; 10 _→ 60% B по объему, 30 мин, линейный градиент; 7 мл/мин, 260 нм; температура колонки 60oC). Фракции, элюированные через 20,7 мин, собирались и обрабатывались по способу, аналогичному описанному в примере 1 (b), давая 79 OD (260 нм) целевого соединения в виде аморфного твердого вещества.
УФ-спектр поглощения (вода), λмакс, нм : 253.
Время удерживания: 13,22 мин.
Высокоэффективная жидкостная хроматография (Инертсил ODS-2; 6 х 150 нм; элюент A: 0,1 М ТЕАА, pH 7,5; элюент B: ацетонитрил; 10 _→ 60% B по объему, 20 мин; линейный градиент, 1 мл/мин, 260 нм).
Пример 97
Соединение формулы 97 (a)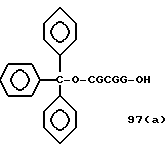
Повторялась процедура, аналогичная процедуре, описанной в примере 1 (b), за исключением того, что в амидитном сосуде X использовался 35 мМ ацетонитрильный раствор соединения примера 15 (b), и последовательность оснований XGCGG помещалась в синтезатор в качестве программы. 90%-ный водно-формамидный раствор, содержащий целевое соединение, полученное таким образом, нагревался при 95oC в течение 5 мин. После данного нагревания раствор быстро охлаждался. Реакционная смесь затем разделялась на четыре части, и каждая часть очищалась с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил PREP - ODS 20,0 х 250 мм; элюент A: 0,1 М ТЕАА, pH 7,0; элюент B: ацетонитрил, 10 _→ 60% B по объему, 30 мин, линейный градиент, 7 мл/мин, 260 нм, температура колонки 60oC). Фракции, элюированные спустя 21,7 мин, собирались и обрабатывались по способу, аналогичному описанному в примере 1 (b), давая 123 OD (260 нм) целевого соединения в виде аморфного твердого вещества.
УФ-спектр поглощения (вода), λмакс , нм : 255.
Время удерживания: 13,58 мин.
Высокоэффективная жидкостная хроматография (Инертсил ODS-2; 6 х 150 нм; элюент A: 0,1 М ТЕАА, pH 7,6; элюент B: ацетонитрил; 10 _→ 60% B по объему, 20 мин; линейный градиент; 1 мл/мин; 260 нм).
Пример 98
Соединение формулы 98 (a)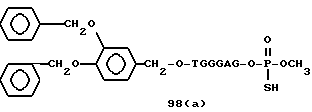
В соответствии с процедурой, аналогичной описанной в примере 83 (b), но с использованием 5'-O-(3,4-дибензилоксибензил)тимидин 2-цианоэтил N, N -диизопропилфосфорамидита [полученного, как описано в примере 7 (b)] вместо 5'-O-тритилтимидин 2- цианоэтил N, N - диизопропилфосфорамидита, получалось целевое соединение. Реакционной продукт очищался с помощью хроматографии через силикагельную колонку с обращенной фазой (Preparative C18, Уотерс, 1,5 х 15 см; 50 мМ ТЕАВ, pH 7,5; 15 _→ 50% ацетонитрил; линейный градиент; 254 нм). Продукт затем обрабатывался по способу, аналогичному описанному в примере 1 (b), давая целевое соединение, имеющее 92 OD (260 нм) в виде аморфного твердого вещества. Анализ высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил ODS 6 х 150 мм; элюент A: 0,1 М ТЕАА, pH 7,5; элюент B: ацетонитрил; 10 _→ 60% B по объему, 20 мин, линейный градиент; 1 мл/мин; 260 нм) показал, что данный образец имеет время удерживания 19,46 мин.
УФ-спектр поглощения (вода), λмакс , нм : 254,
Пример 99
Соединение примера 99 (a):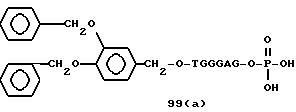
Согласно процедуре, аналогичной описанной в примере 86, но при использовании 5'-O-(3,4 - дибензилоксибензил)тимидин 2-цианоэтил N, N - диизопропилфосфорамидита [полученного, как описано в примере 7 (b)] вместо 5'-O-тритилтимидин 2 - цианоэтил N,N - диизопропилфосфорамидита, получалось целевое соединение. Продукт реакции очищался с помощью хроматографии через силикагельную колонку с обращенной фазой (Preparative C18, Уотерс 1,5 х 15 см; 50 мМ ТЕАВ, pH 7,5; 15 _→ 50% ацетонитрила; линейный градиент; 254 нм). Продукт затем обрабатывался по способу, аналогичному описанному в примере 1 (b), давая целевое соединение, имеющее 43 OD (260 нм) в виде аморфного твердого вещества. Анализ высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил ODS 6 х 150 мм; элюент A: 0,1 М ТЕАА, pH 7,5; элюент B: ацетонитрил; 10 _→ 60% B по объему, 20 мин, линейный градиент; 250 н/м) показал, что данный образец имеет время удерживания 19,12 мин.
УФ-спектр поглощения (вода), λмакс , нм : 254.
Пример 100
Соединение формулы 100 (a)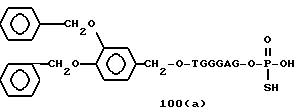
В соответствии с процедурой, аналогичной процедуре, описанной в примере 87, но при использовании 5'-O- (3,4 - дибензилоксибензил)тимидин 2 - цианоэтил N, N -диизопропилфосфорамидита [полученного, как описано в примере 7 (b)] вместо 5'-тритилтимидин 2-цианоэтил N, N - диизопропилфосфорамидита, получалось целевое соединение. Продукт реакции очищался с помощью хроматографии через силикагельную колонку с обращенной фазой (Preparative 18, Уотерс, 15 х 15 см: 50 мМ ТЕАВ, pH 7,5 ; 15 _→ 50% ацетонитрила; линейный градиент; 254 нм). Продукт затем обрабатывался по способу, аналогичному описанному в примере 1 (b), давая целевое соединение, имеющее 36 OD (260 нм) в виде аморфного твердого вещества. Анализ с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил ODS, 6 х 150 мм; элюент A: 0,1 М ТЕАА, pH 7,5; элюент B: ацетонитрил; 10 _→ 60% B по объему, 20 мин; линейный градиент; 260 нм) показал, что образец имеет время удерживания 19,11 мин.
УФ-спектр поглощения (вода), λмакс , нм: 255.
Пример 101
Соединение формулы 101 (a):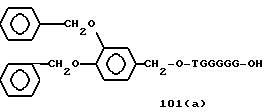
Согласно процедуре, аналогичной описанной в примере 1 (b), но при использовании 35 мМ ацетонитрильного раствора 5'-O-(3,4-дибензилоксибензил)тимидин 2-цианоэтил N, N-диизопропилфосфорамидита [полученного, как описано в примере 7(b)] в амидитном сосуде X и помещении в синтезатор в качестве программы последовательности оснований XTGGGG, получалось целевое соединение. Продукт реакции разделялся на три части, и каждая часть очищалась с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил PREP-ODS, 20,0 х 250 мм; элюент A: 0,1 М TEAA, pH 7,3; элюент B: ацетонитрил; 25 _→ 55% B объему; 30 мин; линейный градиент, 7 мл/мин; 254 нм). Фракции, элюированные спустя 19,8 мин, собирались и обрабатывались по способу, аналогичному способу, описанному в примере 1(b), давая 173,1 OD (260 нм) целевого соединения в виде аморфного твердого вещества.
УФ-спектр поглощения (вода), λмакс , нм: 255.
Время удерживания: 15,1 мин.
Высокоэффективная жидкостная хроматография (УМС-Pack A-312, S-5, 120A, ODS элюент A: 0,1 М TEAA, pH 7,3; элюент B: ацетонитрил; 20 _→ 60% B по объему, 30 мин; линейный градиент; 2 мл/мин; 254 нм).
Пример 102
Соединение формулы 102(a):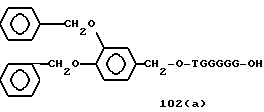
В соответствии с процедурой, аналогичной описанной в примере 1(b), но при использовании 5'-O-(3,4-дибензилоксибензил)тимидин 2-цианоэтил N,N-диизопропилфосфорамидита [полученного, как описано в примере 7(b)] вместо 5'-O-тритилтимидин 2-цианоэтил N, N-диизопропилфосфорамидита и помещении в синтезатор в виде программы последовательности оснований TGGGGG, получалось целевое соединение. Продукт реакции разделялся на три порции, и каждая порция очищалась с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил PREP-ODS 20,0 х 250 мм; элюент A: 0,1 М TEAA, pH 7,3; элюент B: ацетонитрил; 25 _→ 55% B по объему, 30 мин, линейный градиент; 7 мл/мин; 254 нм). Фракции, элюированные через 17,4 мин, собирались и обрабатывались по способу, аналогичному описанному в примере 1(b), давая 120,2 OD (260 нм) целевого соединения в виде аморфного твердого вещества.
УФ-спектр поглощения (вода), λмакс , нм: 254.
Время удерживания: 13,1 мин.
Высокоэффективная жидкостная хроматография (УМС - Pack A-312, S - 5, 120A, ODS элюент A: 0,1 М TEAA, pH 7,3; элюент B: ацетонитрил; 20 _→ 50% B по объему, 30 мин, линейный градиент; 2 мл/мин; 254 нм).
Пример 103
103(a) 5'-O-[(Нафтален-2-ил)метил]тимидин
В соответствии с процедурой, аналогичной описанной в примере 6(a), но при использовании 442 мг (2 ммоль) 2-бромметилнафталина, получалось 253,1 мг (выход 33%) целевого соединения.
1H ЯМР-спектр (CDCl3, 270 МГц, ТМС), δ, млн. дол.:
8,12 (1H, широкий синглет), 7,88-7,43 (8H, мультиплет), 6,40 (1H, триплет, J=6,75 Гц), 4,76 (2H, синглет), 4,61-4,54 (1H, мультиплет), 4,12-4,10 (1H, мультиплет), 3,88-3,72 (2H, мультиплет), 2,40-2,20 (2H, мультиплет), 2,05 (1H, широкий синглет), 1,58 (3H, синглет).
103(b) 5'-O-[(Нафтален-2-ил)метил] тимидин 2-цианоэтил N,N-диизопропилфосфорамидит
Согласно процедуре, аналогичной описанной в примере 11(b), но при использовании 77 мг (0,2 ммоль) 5'-O-[(Нафтален-2-ил)метил]тимидина [полученного, как описано в стадии (a) выше], получалось 62,5 мг (выход 54%) указанного в заголовке соединения.
1H Спектр ЯМР (CDCl3, 270 МГц, ТМС), δ, млн. дол.:
7,88-7,43 (8H, мультиплет), 6,39 (1H, триплет, J=7,26 Гц), 4,75 (2H, широкий синглет), 4,68-4,60 (1H, мультиплет), 4,25, 4,20 (вместе 1H, два широких синглета), 3,90-3,53 (6H, мультиплет), 2,68-2,18 (4H, мультиплет), 1,57, 1,55 (вместе 3H, два синглета), 1,17 (12H, дублет, J=6,60 Гц).
103(c) Соединение формулы 103(c):
В соответствии с процедурой, аналогичной описанной в примере 1(b), но при использовании 5'-O-[(нафтален-2-ил)метил]тимидин 2-цианоэтил N,N-диизопропилфосфорамидита [полученного, как описано в стадии (b) выше] вместо 5'-O-тритилтимидин 2-цианоэтил N,N-диизопропилфосфорамидита, получалось целевое соединение. Продукт реакции разделялся на три части, и каждая часть очищалась с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил PREP - ODS 20,0 x 250 мм; элюент A: 0,1 М TEAA, pH 7,3; элюент B: ацетонитрил; 20 _→ 50% B по объему, 30 мин, линейный градиент; 7 мл/мин; 254 нм). Фракции, элюированные спустя 12,8 мин, собирались и обрабатывались по способу, аналогичному описанному в примере 1(b), давая 257,5 OD (260 нм) целевого соединения в виде аморфного твердого вещества.
УФ-спектр поглощения (вода), λмакс , нм: 255:
Время удерживания: 13,9 мин.
Высокоэффективная жидкостная хроматография (УМС-Pack A-312, S-5, 120A, ODS элюент A: 0,1 М TEAA, pH 7,3; элюент B: ацетонитрил; 10 _→ 40% B по объему, 30 мин; линейный градиент; 2 мл/мин; 254 нм).
Пример 104
104(a) 5'-O-(4-Фенилбензил)тимидин
Согласно процедуре, аналогичной описанной в примере 6(a), но при использовании 405 мг (2 ммоль) 4-фенилбензилхлорида, получалось 387,8 мг (выход 47%) целевого соединения.
1H Спектр ЯМР (CDCl3+CD3OD, 4:1 о/о, ТМС, 270 МГц), δ, млн. дол.:
7,65-7,33 (10H, мультиплет), 6,37 (1H, триплет, J=7,26 Гц), 4,64 (2H, синглет), 4,55-4,48 (1H, мультиплет), 4,13-4,09 (1H, мультиплет), 3,87-3,70 (2H, мультиплет), 2,38-2,15 (2H, мультиплет), 1,63 (3H, синглет).
104(b) 5'-O-(4-фенилбензил)тимидин 2-цианоэтил-N,N-диизопропилфосфорамидит
В соответствии с процедурой, аналогичной описанной в примере 11(b), но при использовании 82 мг (0,2 ммоль) 5'-O-(4-фенилбензил)тимидина [полученного, как описано в стадии (а) выше], получалось 71,8 мг (выход 59%) указанного в заголовке соединения.
1H Спектр ЯМР (CDCl3, 270 МГц, ТМС), δ, млн. дол.:
7,62-7,34 (10H, мультиплет), 6,40 (1H, триплет, J=6,60 Гц), 4,70-4,60 (3H, мультиплет), 4,27, 4,20 (вместе 1H, два широких синглета); 3,90-3,55 (6H, мультиплет), 2,67-2,57 (2H, мультиплет), 2,53-2,16 (2H, мультиплет), 1,63 (3H, синглет), 1,19 (12H, дублет, J= 6,60 Гц).
104(c) Соединение формулы 104(c):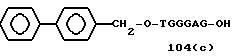
Следуя процедуре, аналогичной описанной в примере 1(b), но используя 5'-O-(4-фенилбензил)тимидин 2-цианоэтил N, N-диизопропилфосфорамидит [приготовленный, как описано выше в стадии (b)] вместо 5'-O-тритилтимидин 2-цианоэтил N,N-диизопропилфосфорамидита, получали целевое соединение. Продукт реакции разделялся на три порции и каждая из них очищалась с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Inertsil PReP-ODS, 20,0 x 250 мм, элюент A: 0,1 М TEAA, pH 7,3; элюент B: ацетонитрил; 20 _→ 50% B по объему, 30 мин; линейный градиент; 7 мл/мин; температура 60oC; 254 нм).
Элюированные фракции спустя 16,5 мин собирались и обрабатывались по способу, описанному в примере 1(b), давая 281,0 OD (260 нм) целевого соединения в виде аморфного твердого вещества.
Спектр УФ-поглощения (вода), λмакс , нм: 255.
Время удерживания: 7,1 мин.
Высокоэффективная жидкостная хроматография (УМС- Pack A-312, S-5, 120 A, ODS; элюент A: 0,1 М TEAA, pH 7,3; элюент B: ацетонитрил; 20 _→ 50% B по объему, 30 мин, линейный градиент; 2 мл/мин; 60oC; 254 нм).
Пример 105
105(a) 5'-O-(2-Фенилбензил)тимидин
Следуя процедуре, аналогичной описанной в примере 6(a), но используя 365 мкм (2 ммоль) 2-фенилбензилбромида, получали 133,2 мг (выход 16%) целевого соединения.
1H спектр ЯМР (CDCl3, 270 МГц, ТМС), δ, млн. дол.:
8,13 (1H, широкий синглет); 7,50-7,28 (10H, мультиплет); 6,34 (1H, триплет, J=6,75 Гц); 4,57; 4,50 (1H, два дублета, J=13,0 Гц); 4,30 (1H, широкий синглет); 3,98 (1H, широкий синглет); 3,73-3,52 (2H, мультиплет); 2,32-2,05 (2H, мультиплет); 1,90 (1H, широкий синглет), 1,51 (3H, синглет).
105(b) 5'-O-(2-Фенилбензил)тимидин 2-цианоэтил N,N-диизопропилфосфорамидит
В соответствии с процедурой, аналогичной процедуре, описанной в примере 11(b), но при использовании 82 мг (0,2 ммоль) 5'-O-(2-фенилбензил)тимидина [полученного, как описано в стадии (a) выше], получалось указанное в заголовке соединение в количестве 89,9 мг (выход 73%).
1H Спектр ЯМР (CDCl3, 270 МГц, ТМС), δ, млн. дол.:
7,51-7,29 (10H, мультиплет), 6,36 (1H, триплет, J=7,92 Гц), 4,55-4,45 (3H, мультиплет), 4,19, 4,11 (вместе 1H, два синглета), 3,90-3,52 (6H, мультиплет), 2,66-2,54 (2H, мультиплет), 2,45-2,03 (2H, мультиплет), 1,51 (3H, синглет), 1,19 (12H, дублет, J=6,60 Гц).
105(c) Соединение формулы 105(c):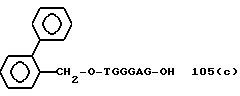
В соответствии с процедурой, аналогичной описанной в примере 1(b), но при использовании 5'-O-(2-фенилбензил)тимидин 2-цианоэтил N,N-диизопропилфосфорамидита [полученного, как описано выше в стадии (b)] вместо 5'-O-тритилтимидин 2-цианоэтил N, N-диизопропилфосфорамидита, получалось целевое соединение. Продукт реакции разделялся на три порции, и каждая порция очищалась с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил PREP - ODS, 20,0 x 250 мм; элюент A: 0,1 М TEAA, pH 7,3; элюент B: ацетонитрил; 20 _→ 50% B по объему, 30 мин; линейный градиент; 7 мл/мин; 60oC, 254 нм). Фракции, элюированные спустя 14,8 мин, собирались и обрабатывались по способу, аналогичному способу, описанному в примере 1(b), давая 172,2 OD (260 нм) целевого соединения в виде аморфного твердого вещества.
УФ-спектр поглощения (вода), λмакс , нм: 252.
Время удерживания: 6,1 мин.
Высокоэффективная жидкостная хроматография (УМС - Pack A-312, S-5, 120A, ODS; элюент A: 0,1 М TEAA, pH 7,3; элюент B: ацетонитрил; 20 ---> 50% B по объему, 30 мин, линейный градиент; 2 мл/мин; 60oC; 254 нм).
Пример 106
106(a) 3'-O-Триизопропилсилил-5'-O-(4,4'-диметокситритил)тимидин
1,485 г (2,73 ммоль) 5'-O-(4,4'-диметокситритил)тимидина и 0,37 г (5,45 ммоль) имидазола подвергались сначала сушке азеотропной перегонкой при пониженном давлении с пиридином, а затем растворялись в 6 мл диметилформамида. 875 мкл (4,09 ммоль) триизопропилсилилхлорида добавлялись к данному раствору, и получающаяся смесь перемешивалась на протяжении ночи при комнатной температуре. В конце данного периода времени к смеси добавлялось 875 мкл триизопропилсилилхлорида и 0,37 г имидазола, и смесь затем перемешивалась в течение суток. Реакционная смесь затем разбавлялась 100 мл этилацетата, и разбавленная смесь промывалась дважды, каждый раз 100 мл 5% в/о водного раствора бикарбоната натрия. Органический слой сушился над безводным сульфатом натрия, и растворитель удалялся перегонкой при пониженном давлении. Получающийся остаток очищался с помощью хроматографии на колонке при пропускании через 100 г силикагеля (70-230 меш) с использованием метиленхлорида, содержащего от 0 до 3% по объему метанола, в качестве элюента, давая 1,861 г (выход 97%) указанного в заголовке соединения.
1H Спектр ЯМР (CDCl3, 270 МГц, ТМС), δ млн. дол.:
8,08 (1H, синглет), 7,67 (1H, синглет), 7,42-6,80 (13H, мультиплет), 6,39 (1H, триплет, J=5,94 Гц), 4,60-4,55 (1H, мультиплет), 4,06-4,03 (1H, мультиплет), 3,79 (6H, синглет), 3,53-3,26 (2H, мультиплет), 2,41-2,18 (2H, мультиплет), 1,50 (3H, синглет), 1,03-0,90 (21H, мультиплет).
106(b) 3'-O-(Триизопропилсилил)тимидин
1,6 мл Трифторуксусной кислоты добавлялось по каплям к раствору 1,861 г (2,655 ммоль) 3'-O-триизопропилсилил-5'-O-(4,4'- диметокситритил)тимидина [полученного, как описано на стадии (a) выше] в 80 мл хлороформа при охлаждении на ледяной бане и перемешивании. Получающаяся смесь перемешивалась в течение 10 мин, а затем добавлялось 2 мл пиридина. Реакционная смесь промывалась дважды, каждый раз 100 мл 5% вес/объем водного раствора бикарбоната натрия и сушилась над безводным сульфатом натрия. Растворитель удалялся перегонкой при пониженном давлении, и получающийся остаток очищался с помощью хроматографии на колонке с пропусканием через 50 г силикагеля (70-230 меш) с использованием метиленхлорида, содержащего 4% по объему метанола, в качестве элюента, давая 0,9965 г (выход 94%) указанного в заголовке соединения.
1H Спектр ЯМР (CDCl3, 270 МГц, ТМС), δ, млн. дол.:
8,17 (1H, широкий синглет), 7,36 (1H, синглет), 6,12 (1H, триплет, J= 5,94 Гц), 4,63-4,59 (1H, мультиплет), 4,03-4,00 (1H, мультиплет), 3,96-3,74 (2H, мультиплет), 2,48 (1H, широкий синглет), 2,45-2,21 (2H, мультиплет), 1,91 (3H, синглет), 1,15-1,00 (21H, мультиплет).
106(c) 3'-O-Триизопропилсилил-5'-O-[(4-бензилокси)бензил]тимидин
88 мг (2 ммоль) гидрида натрия (в виде 55% вес/вес дисперсии в минеральном масле) добавлялось к раствору 398 мг (1 ммоль) 3'-O-(триизопропилсилил) тимидина [полученного, как описано в стадии (b) выше] в 2,5 мл тетрагидрофурана, и получающаяся смесь оставлялась стоять при 60oC в течение 2 ч, а затем охлаждалась до комнатной температуры. К смеси добавлялось 232 мг (1 ммоль) 4-бензилоксибензилхлорида и 75 мг (0,5 ммоль) йодида натрия, и реакционная смесь затем перемешивалась на протяжении ночи при комнатной температуре, а затем при 55oC в течение 8 ч. В конце данного периода времени растворитель удалялся с помощью перегонки при пониженном давлении, и остаток растворялся в 100 мл этилацетата. Получающийся раствор промывался дважды, каждый раз 100 мл 0,01 н. водной соляной кислоты. Органический слой сушился над безводным сульфатом магния, и растворитель удалялся перегонкой при пониженном давлении. Получающийся остаток очищался с помощью хроматографии на колонке с 30 г силикагеля (230-400 меш) с использованием метиленхлорида, содержащего от 1 до 2% по объему метанола, в качестве элюента, давая 237 мг (выход 40%) указанного в заголовке соединения.
1H Спектр ЯМР (CDCl3, 270 МГц, ТМС), δ , млн. дол.:
8,00 (1H, широкий синглет), 7,59 (1H, синглет), 7,48-6,90 (9H, мультиплет), 6,36 (1H, триплет, J=6,60 Гц), 5,07 (2H, синглет), 4,56-4,46 (1H, мультиплет), 4,50 (2H, синглет), 4,07-4,03 (1H, мультиплет), 3,80-3,60 (2H, мультиплет), 2,32-2,08 (2H, мультиплет), 1,56 (3H, синглет), 1,10-0,97 (21H, мультиплет).
106(d) 5'-O-[(4-Бензилокси)бензил]тимидин
1,5 мл 1 М тетрагидрофуранового раствора тетрабутиламмонийфторида добавлялось к раствору 237 мг (0,4 ммоль) 3'-O-триизопропилсилил-5'-O-[(4-бензилокси)бензил] тимидина [полученного, как описано в стадии (c) выше] в 1,5 мл тетрагидрофурана, и получающаяся смесь перемешивалась при комнатной температуре в течение 3 ч. В конце данного периода реакционная смесь освобождалась от растворителя перегонкой при пониженном давлении, и получающийся остаток растворялся в 100 мл этилацетата. Данный раствор промывался дважды, каждый раз 100 мл насыщенного водного раствора хлористого натрия, и затем сушился над безводным сульфатом магния. Растворитель удалялся с помощью перегонки при пониженном давлении, и получающийся остаток очищался с помощью хроматографии на колонке, заполненной 30 г силикагеля (230-400 меш), с использованием метиленхлорида, содержащего от 1 до 4% по объему метанола, в качестве элюента, давая 108,5 мг (выход 61%) указанного в заголовке соединения.
1H Спектр ЯМР (CDCl3, 270 МГц, ТМС), δ, млн. дол.:
8,11 (1H, широкий синглет), 7,56 (1H, синглет), 7,47-6,93 (9H, мультиплет), 6,38 (1H, триплет, J= 7,26 Гц), 5,06 (2H, синглет), 4,57-4,47 (1H, мультиплет), 4,52 (2H, синглет), 4,10-4,06 (1H, мультиплет), 3,80-3,64 (2H, мультиплет), 2,38-2,15 (2H, мультиплет), 1,67 (3H, синглет).
106(e) 5'-O-[(4-Бензилокси)бензил]тимидин 2-цианоэтил N,N-диизопропилфосфорамидит
В соответствии с процедурой, аналогичной процедуре, описанной в примере 11(b), но при использовании 88 мг (0,2 ммоль) 5'-O-[(4-бензилокси)бензил]тимидита [полученного, как описано в стадии (d) выше], получалось 104,8 мг (выход 82%) указанного в заголовке соединения.
1H Спектр ЯМР (CDCl3, 270 МГц, ТМС), δ, млн. дол.:
7,58, 7,56 (вместе 1H, два синглета), 7,46-6,93 (9H, мультиплет), 6,37 (1H, триплет, J= 6,60 Гц), 5,06 (2H, синглет), 4,64-4,54 (1H, мультиплет), 4,60, 4,59 (2H, 2 синглета), 4,25-4,15 (1H, мультиплет), 3,90-3,50 (6H, мультиплет), 2,66-2,54 (2H, мультиплет), 2,50-2,10 (2H, мультиплет), 1,65 (3H, синглет), 1,18 (12H, дублет, J=7,26 Гц).
106 (f) Соединение формулы 106 (f):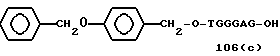
В соответствии с процедурой, аналогичной процедуре, описанной в примере 1 (b), но с использованием 5'-O-[4 - бензилокси)бензил]тимидин 2 - цианоэтил N, N - диизопропилфосфорамидита [полученного, как описано в стадии (e) выше] вместо 5'-O-тритилтимидин 2-цианоэтил N, N -диизопропилфосфорамидита, синтезировалось целевое соединение. Реакционный продукт разделялся на три порции, и каждая порция очищалась с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил PREP - ODS, 20,0 х 250 мм; элюент A: 0,1 М ТЕАА, pH 7,3; элюент B: ацетонитрил; 20 _→ 50% B по объему, 30 мин; линейный градиент; 7 мл/мин; 60oC; 254 нм). Фракции, элюируемые через 17,0 мин, собирались и обрабатывались по способу, аналогичному описанному в примере 1 (b), давая 208,8 OD (260 нм) целевого соединения в виде аморфного твердого вещества.
УФ-спектр поглощения (вода), λмакс, нм: 254.
Время удерживания : 8,1 мин.
Высокоэффективная жидкостная хроматография (УМС- Pack A-312, S- 5, 120A, ODS; водный раствор 0,1 М ТЕАА, pH 7,3; элюент B: ацетонитрил, 20 _→ 50% B по объему, 30 мин, линейный градиент, 2 мл/мин; 60oC; 254 нм).
Пример 107
107 (a) 5'-O-(9- Фенилксантен- 9- ил)тимидин
585 мг (2 ммоль) 9- хлор-9-фенилксантена добавлялось к раствору 484 мг (2 ммоль) тимидина в 20 мл пиридина, и получающаяся смесь перемешивалась при комнатной температуре в течение 1 ч при затенении ее от света. Реакционная смесь затем разбавлялась 200 мл метиленхлорида, и разбавленная смесь промывалась дважды, каждый раз 200 мл 5% вес/объем водного раствора бикарбоната натрия. Органический слой сушился над безводным сульфатом натрия, и растворитель удалялся перегонкой при пониженном давлении. Остаток очищался с помощью хроматографии на колонке через 35 г силикагеля (70-230 меш), с использованием метиленхлорида, содержащего от 0 до 2,5% по объему метанола в качестве элюента, давал 647,8 мг (выход 65%) указанного в заготовке соединения.
1H Спектр ЯМР (CDCl3, 270 МГц, ТМС), δ, млн.дол.:
8,33 (1H, широкий синглет), 7,62 (1H, синглет), 7,45 - 7,02 (13H, мультиплет), 6,36 (1H, триплет, J=5,94 Гц), 4,49 - 4,41 (1H, мультиплет); 4,02 - 3,96 (1H, мультиплет), 3,29 - 3,12 (2H, мультиплет), 2,52 - 2,24 (2H, мультиплет); 1,99 (1H, дублет, J=3,96 Гц), 1,67 (3H, синглет).
107 (b) 5'-O-(9-Фенилксантен-9- ил)тимидин-2-цианоэтил N, N - диизопропилфосфорамидит.
В соответствии с процедурой, аналогичной процедуре, описанной в примере 11 (b), но с использованием 100 мг (0,2 ммоль) 5'-O-(9- фенилксантен-9-ил) тимидина [полученного, как описано в стадии (a) выше], получалось 134,9 мг (выход 99%) указанного в заголовке соединения.
1H Спектр ЯМР (CDCl3, 270 МГц, ТМС), δ, млн. дол :
8,03 (1H, широкий синглет), 7,69, 7,64 (вместе 1H, два синглета), 7,40 - 7,03 (13H, мультиплет), 6,40 - 6,32 (1H, мультиплет), 4,58 - 4,47 (1H, мультиплет), 4,15 - 4,05 (1H, мультиплет), 3,90 - 3,10 (6H, мультиплет), 2,82 - 2,28 (4H, мультиплет), 1,63, 1,62 (вместе 3H, два синглета), 1,16 (12H,дублет, J = 6,6 Гц).
107 (c) Соединение формулы 107 (c):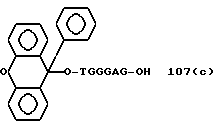
В соответствии с процедурой, аналогичной процедуре, описанной в примере 1 (b), но при использовании 5'-O- (фенилксантен-9-ил)тимидин 2-цианоэтил N, N - диизопропилфосфорамидита [полученного, как описано в стадии (b) выше] вместо 5'-O- тритилтимидин 2-цианоэтил N, N - диизопропилфосфорамидита, синтезировалось указанное в заготовке соединение. Продукт реакции разделялся на три части, и каждая часть очищалась с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил PREP - ODS, 20,0 х 250 мм; элюент A: 0,1 М ТЕАА, pH 7,3; элюент B: ацетонитрил, 20 _→ 50% B по объему, 30 мин, линейный градиент, 7 мл/мин; 60oC; 254 нм). Фракции, элюируемые спустя 19,7 мин, собирались и обрабатывались по способу, аналогично описанному в примере 1 (b), давая 145,6 OD (260 нм) целевого соединения в виде аморфного твердого вещества.
УФ-спектр поглощения (вода), λмакс, нм: 249.
Время удерживания: 10,9 мин.
Высокоэффективная жидкостная хроматография (УМС -Pack A-312, S-5, 120A, ODS; элюент A: 0,1 М ТЕАА, pH 7,3; элюент B: ацетонитрил, 2 _→ 50% В по объему, 30 мин; линейный градиент; 2 мл/мин, 60oC 254 нм).
Пример 108
108 (a) 5'-O-(9-Фенилфлуорен-9- ил) тимидин
770 мг (2,4 ммоль) 9-бром-9-фенилфлуорена добавлялось к раствору 242 мг (1 ммоль) тимидина в 10 мл пиридина, и получающаяся смесь перемешивалась при 100oC в течение 8 ч. В конце данного периода времени реакционная смесь концентрировалась выпариванием при пониженном давлении, и концентрат растворялся в 100 мл метиленхлорида. Раствор промывался 100 мл 5% вес/объем водного раствора бикарбоната натрия и сушился над безводным сульфатом натрия. Растворитель удалялся с помощью перегонки при пониженном давлении, и получающийся остаток очищался с помощью хроматографии на колонке с пропусканием через 30 г силикагеля (230 - 400 меш) с использованием метиленхлорида, содержащего от 1 до 3% по объему метанола, в качестве элюента, давая 194,2 мг (выход 37%) указанного в заголовке соединения.
1H Спектр ЯМР (CDCl3, 270 МГц, ТМС), δ, млн. дол. :
8,06 (1H, широкий синглет), 7,76 (1H, синглет), 7,72 - 7,20 (13H, мультиплет), 6,41 (1H, триплет, J=6,60 Гц), 4,64 - 4,58 (1H, мультиплет), 3,95 - 3,90 (1H, мультиплет), 3,49 - 3,13 (2H, мультиплет), 2,45 - 2,38 (2H, мультиплет), 1,88 (1H, дублет, J = 3,96 Гц), 1,48 (3H, синглет).
108 (b) 5'-O-(9-Фенилфлуорен-9-ил) тимидин 2- цианоэтил N, N -диизопропилфосфорамидит
В соответствии с процедурой, аналогичной описанной в примере 11 (b), но с использованием 105 мг (0,2 ммоль) 5'-O- (9 - фенилфлуорен-9-ил)тимидина [полученного, как описано в стадии (a) выше], получалось 11,8 мг (выход 76%) указанного в заголовке соединения.
1H Спектр ЯМР (CDCl3, 270 МГц, ТМС), δ, млн. дол.:
8,00 (1H, широкий синглет), 7,80 (1H, синглет), 7,78 - 7,20 (13H, мультиплет), 6,44 - 6,37 (1H, мультиплет), 4,72 - 4,62 (1H, мультиплет), 4,10 - 4,00 (1H, мультиплет), 3,90 - 3,10 (6H, мультиплет), 2,75 - 2,34 (4H, мультиплет), 1,46, 1,45 (вместе 3H, два синглета), 1,18 (12H, дублет, J = 6,60 Гц).
108 (c) Соединение формулы 108 (c):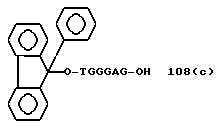
В соответствии с процедурой, аналогичной описанной в примере 1(b), но с использованием 5'-O-(9-фенилфлуорен-9-ил)тимидин 2-цианоэтил N,N-диизопропилфосфорамидита [полученного, как описано в стадии (b) выше] вместо 5'-O-тритилтимидин 2-цианоэтил N, N-диизопропилфосфорамидита, синтезировалось указанное в заголовке соединение. Продукт реакции разделялся на три порции, и каждая порция очищалась с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил PREP - ODS 20,0 x 250 мм, элюент A: 0,1 M TEAA, pH 7,3; элюент B: ацетонитрил, 20 _→ 50% B по объему, 30 мин; линейный градиент; 7 мл/мин, 60oC; 254 нм). Фракции, элюированные через 19,1 мин, собирались и обрабатывались по способу, аналогичному приведенному в примере 1 (b), давая 291,2 OD (260 нм) целевого соединения в виде аморфного твердого вещества.
УФ-спектр поглощения (вода), λмакс, нм: 255.
Время удерживания: 10,4 мин.
Высокоэффективная жидкостная хроматография (УМС - Pack A - 312, S - 5, 120A, ODS; элюент A: 0,1 M TEAA, pH 7,3; элюент B: ацетонитрил; 20 _→ 50% B по объему; 30 мин; линейный градиент; 2 мл/мин, 60oC, 254 нм).
Пример 109
109 (a) 5'-(S-Трифенилметил) тио-5'-дезокситимидин 2-цианоэтил N,N-диизопропилфосфорамидит
В соответствии с процедурой, аналогичной описанной в примере 11 (b), но с использованием 100 мг (0,2 ммоль) 5'- /S-трифенилметил/ тио-5'-дезокситимидина (B. S.Sproate, B.Beijer, P.Rider, P.Neuner, Nucleic Acids Res, 15, /12/ 4837 /1987/), получалось 88 мг (выход 62%) указанного в заголовке соединения.
1H Спектр ЯМР (CDCl3, 270 МГц), δ, млн. дол.:
7,45 - 7,18 (16H, мультиплет), 6,23 - 6,17 (1H, мультиплет), 4,31 - 4,20 (1H, мультиплет), 4,11 - 4,00 (1H,мультиплет), 3,85 - 3,43 (4H, мультиплет), 1,56 (3H, синглет), 1,20 - 1,05 (12H, мультиплет).
109 (b) Соединение формулы 109 (b):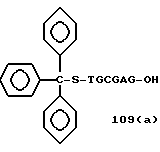
В соответствии с процедурой, аналогичной описанной в примере 1 (b), но с использованием 5'-(S-трифенилметил) тио 5'-дезокситимидин 2-цианоэтил N,N-диизопропилфосфорамидита [приготовленного, как описано выше в стадии (a)] вместо 5'-O-тритилтимидин 2-цианоэтил N,N-диизопропилфосфорамидита, синтезировалось целевое соединение. Продукт реакции очищался с помощью хроматографии на колонке из силикагеля с обращенной фазой (Preparative C18, Уостерс, 1,5 х 15 см; элюент A: 50 мМ TEAB, pH 7,5; элюент B: ацетонитрил; 20 _→ 50% B по объему, линейный градиент; 254 нм). Затем он обрабатывался тем же способом, что и в примере 1 (b), давая 122,3 OD (260 нм) целевого соединения в виде аморфного твердого вещества.
УФ-спектр поглощения (вода), λмакс, нм : 254.
Время удерживания : 19,0 мин.
Высокоэффективная жидкостная хроматография (Интерсил 6 х 1500 мм, элюент A: 0,1 M TEAA, pH 7,5; элюент B: ацетонитрил; 10 _→ 60% B по объему, 20 мин, линейный градиент, 1 мл/мин; 60oC; 260 нм).
Пример 110
Соединение формулы 110 (a):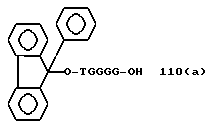
В соответствии с процедурой, аналогичной описанной в примере 1 (b), но с использованием 5'-O-(9-фенилфлуорен-9-ил)тимидин 2-цианоэтил N,N-диизопропилфосфорамидита [приготовленного, как описано в примере 108 (b)] вместо 5'-O-тритилтимидин 2-цианоэтил N,N-диизопропилфосфорамидита и внесением основной последовательности TGGGG в синтезатор, получалось целевое соединение. Продукт реакции разделялся на три порции и каждая из них очищалась с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил PRED-ODS 20,0 х 250 мм, элюент A: 0,1 M TEAA, pH 7,3; элюент B: ацетонитрил; 25 _→ 55% B по объему, 30 мин; линейный градиент; 7 мл/мин, 60oC; 254 нм). Спустя 14,6 мин элюированные фракции собирались и обрабатывались тем же способом, что и в примере 1 (b), давая 28,3 OD (260 нм) целевого соединения в виде аморфного твердого вещества.
УФ-спектр поглощения (вода), λмакс, нм: 256.
Высокоэффективная жидкостная хроматография (УMC - Pack A - 312, S - 5, 120A. ODS; элюент A: 0,1 M TEAA, pH 7,3; элюент B: ацетонитрил; 20 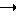 50% B по объему, 30 мин, линейный градиент, 2 мл/мин; 60oC; 254 нм).
50% B по объему, 30 мин, линейный градиент, 2 мл/мин; 60oC; 254 нм).
Пример 111
Соединение формулы 111 (a):
Повторялась процедура, аналогичная описанной в примере 15 (c), но с использованием колонки, заполненной 125 мг (ммоль) соединения примера 12 (b), и внесением последовательности оснований XGGGGZ ( в которой Z имеет значения, указанные в примере 17 (b)) в ДНК/РНК-синтезатор, описанный в примере 1 (b). Полученный таким образом 90%-ный водный раствор формамида, содержащий целевое соединение, нагревался при 95oC в течение 5 мин. После этого нагревания раствор быстро охлаждался. Реакционная смесь разделялась на четыре порции и каждая из них очищалась с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Интертсил PREP - ODS 20,0 x 250 мм, элюент A: 0,1 M TEAA; pH 7,0; элюент B: ацетонитрил; 10 _→ 60% B по объему, 30 мин; линейный градиент; 7 мл/мин; 260 нм; температура колонки 60oC). Фракции, элюированные спустя 20,8 мин, собирались и обрабатывались тем же способом, что в примере 1 (b), давая 82 OD (260 нм) целевого соединения в виде аморфного твердого вещества.
УФ-спектр поглощения (вода), λмакс, нм: 254.
Время удерживания : 13,38 мин.
Высокоэффективная жидкостная хроматография (Интертсил ODS-2; 6 х 150 нм; элюент A: 0,1 M TЕAA, pH 7,5; элюент B: ацетонитрил, 10 _→ 60% B по объему, 20 мин, линейный градиент; 1 мг/мин; 260 нм).
112 (a) 5'-[(3,4-Дибензилокси)бензилтио]-5'-дезокситимидин
258 мг (1 ммоль) 5'-дезокси-5'-меркаптотимидина добавлялось к 20 мл ацетона. Затем к раствору в атмосфере азота добавлялись 406 мг (1,2 ммоль) 3,4-дибензилоксибензилхлорида и 1 г безводного карбоната калия. Полученная смесь перемешивалась при одновременном нагревании с обратным холодильником в течение 1 ч. В конце этого времени осадки отфильтровывались из реакционной смеси, и фильтрат освобождался от растворителя перегонкой при пониженном давлении. Остаток растворялся в небольшом количестве метиленхлорида и очищался хроматографией на колонке через 10 г силикагеля с использованием в качестве элюента 5 : 95 по объему смеси метанола и метиленхлорида, давая 452 мг целевого соединения в виде бесцветного карамелеподобного остатка. Этот остаток лиофилизировался из бензола, давая 180 мг (выход 32%) целевого соединения в виде белых кристаллов.
1H Спектр ЯМР (CDCl3, 270 МГц, ТМС), δ, млн. дол.:
8,26 (1H, широкий синглет), 6,78 - 7,46 (14H, мультиплет), 6,20 (1H, триплет, J = 6,6 Гц), 5,15 и 5,17 (вместе 4H, два синглета), 4,20 - 4,27 (1H, мультиплет), 3,87 - 3,91 (1H, мультиплет), 3,67 - 3,68 (2H, мультиплет), 2,61 - 2,70 (2H, мультиплет), 2,09 - 2,40 (2H, мультиплет), 1,91, 1,92 (3H, мультиплет).
112 (b) 5'-[(3,4-Дибензилокси)бензилтио]-5'-тезокситимидин 2-цианоэтил N,N-диизопропилфосфорамидит
112 мг (0,2 ммоль) 5'-[3,4-дибензилокси)бензилтио]-5'-дезокситимидина [приготовленного, как описано выше в стадии (a)] сушились с помощью азеотропной перегонки с пиридином и затем растворялись в 1 мл метиленхлорида. Затем к раствору добавлялось 17 мг (0,1 ммоль) диизопропиламмонийтетразолида. Затем к этой смеси по каплям добавлялось 70 мкл (0,22 ммоль) 2-цианоэтил N,N, N', N'-тетраизопропилфосфордиамидита в атмосфере аргона. Получающаяся смесь перемешивалась при комнатной температуре в течение 3 ч в той же атмосфере. Метиленхлорид затем удалялся с помощью перегонки при пониженном давлении. Получающийся остаток растворялся в 10 мл этилацетата, и раствор промывался 10% в/о водным раствором карбоната натрия и насыщенным водным раствором хлористого натрия, в указанном порядке. Раствор затем сушился над безводным сульфатом натрия, после чего растворитель удалялся с помощью перегонки при пониженном давлении, и получающийся в результате остаток очищался с помощью хроматографии на колонке с пропусканием через 10 г силикагеля (70-230 меш) с использованием этилацетата в качестве элюента, давая 115 мг (выход 76%) указанного в заголовке соединения в виде карамелеобразного вещества.
1H Спектр ЯМР (CDCl3, 270 МГц, ТМС), δ, млн. дол.:
8,16 (1H, широкий синглет), 6,76 - 7,45 (14H, мультиплет), 6,23 - 6,29 (1H, мультиплет), 5,14 - 5,16 (4H, мультиплет), 4,33 - 4,46 (1H, мультиплет), 4,08 - 4,16 (1H, мультиплет), 3,50 - 3,90 (6H, мультиплет), 2,09 - 2,80 (6H, мультиплет), 1,91 - 1,93 (3H, мультиплет), 1,08 - 1,58 (12H, мультиплет).
112 (c) Соединение формулы 112 (c) .
.
В соответствии с процедурой, аналогичной описанной в примере 1 (b), но с использованием 5'-[(3,4-дибензилокси)бензилтио] -5'-дезокситимидин 2-цианоэтил N, N-диизопропилфосфордиамидита [полученного, как описано в стадии (b) выше] вместо 5'-O-тритилтимидин 2-цианоэтил N,N-диизопропилфосфорамидита, получалось указанное в заголовке соединение. Продукт реакции очищался с помощью хроматографии при пропускании через силикагельную колонку с обращенной фазой (Preparative C18, Уотерс, 1,5 х 150 мм; элюент A: 50 мМ водный триэтиламмонийбикарбонатный буфер (TEAB); pH 7,5; элюент B: ацетонитрил; 15 _→ 50% B по объему; линейный градиент; 260 нм). Элюированные фракции, содержащие указанное в заголовке соединение, собирались и обрабатывались по способу, аналогичному описанному в примере 1 (b), давая 57 OD (260 нм) целевого соединения в виде аморфного твердого вещества.
УФ-спектр поглощения (вода), λмакс, нм: 253.
Время удерживания: 16,85 мин.
Высокоэффективная жидкостная хроматография (Инертсил - 2, 6 х 150 нм; элюент A: 0,1 M TEAA, pH 7,5; элюент B: ацетонитрил; 10 _→ 60% B по объему, 20 мин; линейный градиент, 1 мл/мин; 260 нм).
Пример 113
113 (a) 5'-[(Антрацен-9-ил)метилтио]-5'-дезокситимидин
258 мг (1 ммоль) 5'-дезокси-5'-меркаптотимидина добавлялось к 20 мл ацетона. Затем к раствору в атмосфере азота добавлялось 272 мг (2,2 ммоль) 9-хлорметилантрацена и 1 г безводного карбоната калия. Получающаяся смесь затем перемешивалась при нагревании с обратным холодильником в течение 2 ч. В конце данного периода осадки отфильтровывались от реакционной смеси, и фильтрат освобождался от растворителя перегонкой при пониженном давлении. Остаток кристаллизовался из 10 мл этанола, давая 156 мг (выход 35%) указанного в заголовке соединения в виде бледно-желтых кристаллов.
1H Спектр ЯМР (гексадейтерированный диметилсульфоксид, 270 МГц, ТМС), δ, млн. дол.:
8,56 (1H, синглет), 8,40 (2H, дублет, J = 8,7 Гц), 8,09 (2H, дублет, J = 8,2 Гц), 7,51 - 7,61 (5H, мультиплет), 6,24 - 6,28 (1H, мультиплет), 5,43 (1H, широкий синглет), 4,87 - 4,89 (2H, мультиплет), 4,24 - 4,25 (1H, триплет, J = 2,7 Гц), 4,03 - 4,07 (1H, мультиплет), 3,05 (2H, дублет, J = 6,5 Гц), 2,07 - 2,29 (2H, мультиплет), 1,72 - 1,73 (3H, мультиплет).
113 (b) 5'-[(Антрацен-9-ил)метилтио] -5'-дезокситимидин 2-цианоэтил N, N-диизопропилфосфорамидит
74 мг (0,165 ммоль) 5'-[(антрацен-9-ил)метилтио]-5'- дезокситимидина [полученного, как описано в стадии (a) выше] подвергались сушке азеотропной перегонкой с пиридином, а затем растворялись в 1 мл тетрагидрофурана. К раствору добавлялось 0,115 мл (0,659 ммоль) N,N-диизопропил N-этиламина. Затем по каплям к смеси добавлялось 49 мкл (0,22 ммоль) 2-цианоэтил N,N-диизопропилхлорфосфорамидита в атмосфере аргона. Получающаяся смесь перемешивалась при комнатной температуре в течение 1,75 ч в той же атмосфере. Тетрагидрофуран затем удалялся перегонкой при пониженном давлении. Получающийся остаток растворялся в 10 мл этилацетата, и раствор промывался 10% вес/объем водным раствором карбоната натрия и насыщенным водным раствором хлористого натрия, в указанном порядке. Раствор сушился над безводным сульфатом натрия, а затем растворитель удалялся перегонкой при пониженном давлении, и остаток очищался с помощью хроматографии на колонке при пропускании через 10 г силикагеля (70 - 230 меш) с использованием этилацетата в качестве элюента, давая 85 мг (выход 79%) указанного в заголовке соединения в виде пенообразного вещества.
1H Спектр ЯМР (CDCl3, 270 МГц, ТМС), δ, млн. дол.:
7,26 - 8,43 (10H, мультиплет), 6,26 - 6,33 (1H, мультиплет), 4,74 - 4,93 (2H, мультиплет), 4,32 - 4,42 (1H, мультиплет), 4,20 - 4,28 (1H, мультиплет), 3,49 - 3,80 (4H, мультиплет), 2,89 - 3,11 (2H, мультиплет), 1,93 - 2,68 (4H, мультиплет), 1,71 - 1,76 (3H, мультиплет), 1,08 - 1,22 (12H, мультиплет).
113 (с) Соединение формулы 113 (c):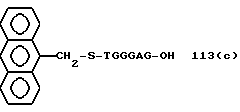
В соответствии с процедурой, аналогичной описанной в примере 1 (b), но при использовании 5'-[(антрацен-9-ил)метилтио]-5'-дезокситимидин 2-цианоэтил N, N-диизопропилфосфорамидита [полученного, как описано в стадии (b) выше] вместо 5'-O-тритилтимин 2-цеаноэтил N,N-диизопропилфосфорамидита, получалось указанное в заголовке соединение. Реакционный продукт очищался с помощью хроматографии при пропускании через силикагельную колонку с обращенной фазой (Preparative C18, Уотерс, 1,5 х 150 мм; элюент A: 50 мм TEAB; pH 7,5, элюент B: ацетонитрил; 15 _→ 50% B по объему; линейный градиент; 260 нм). Элюированные фракции, содержащие целевое соединение, собирались и обрабатывались по способу, аналогичному описанному в примере 1 (b), давая 30 OD (260 нм) целевого соединения в виде аморфного твердого вещества.
УФ-спектр поглощения (вода), λмакс , нм: 255.
Время удерживания: 13,52 мин.
Высокоэффективная жидкостная хроматография (Инертсил 0 - 2; 6 х 150 нм; элюент A: 0,1 М TEAA, pH 7,5; элюент B: ацетонитрил; 10 _→ 60% B по объему, 20 мин, линейный градиент, 1 мл/мин, 260 нм).
Пример 114
114 (a) 5'[2-Нафтил)метилтио]-5'-дезокситимидин
258 мг ( 1 ммоль) 5'-дезокси-5'-меркаптотимидина добавлялось к 20 мл ацетона. Затем к раствору в атмосфере аргона добавлялось 265 мг (1,2 ммоль) 2-бромметилнафталина и 1 г безводного карбоната калия. Получающаяся смесь перемешивалась при нагревании с обратным холодильником в течение 2 ч. В конце данного периода времени осадки отфильтровывались из реакционной смеси, и фильтрат освобождался от растворителя с помощью перегонки при пониженном давлении. Остаток кристаллизовался из 10 мл этанола, давая 100 мг (выход 25%) целевого соединения в виде белых кристаллов.
1H Спектр ЯМР (гексадейтерированный диметилсульфоксид, 270 МГц, ТМС), δ, млн. дол.:
7,84 - 7,90 (1H, широкий синглет), 7,78 (1H, синглет), 7,46 - 7,53 (4H, мультиплет), 6,16 - 6,19 (1H, мультиплет), 5,34 (1H, дублет, J = 4,4 Гц), 4,14 - 4,19 (1H, мультиплет), 3,92 - 3,99 (2H, мультиплет), 3,83 - 3,87 (1H, мультиплет), 2,62 - 2,78 (2H, мультиплет), 2,02 - 2,23 (2H, мультиплет), 1,75 - 1,79 (3H, мультиплет).
114 (b) 5'-[(2-Нафтил)метилтио]-5'-дезокситимидин 2-цианоэтил N,N-диизопропилфосфоримидит
79,7 мг (0,2 ммоль) 5'-[(2-нафтил)метилтио]-5'-дезокситимидина [полученного, как описано в стадии (a) выше] подвергались сушке с помощью азеотропной перегонки с пиридином, а затем растворялись в 2,5 мл тетрагидрофурана. К раствору добавлялось затем 0,084 мл (0,48 ммоль) диизопропиламина. К смеси в атмосфере аргона по каплям добавлялось 54 мкл (0,24 ммоль) 2-цианоэтил N,N-диизопропилхлорфосфорамидита. Получающаяся смесь перемешивалась при комнатной температуре в течение 1,75 ч в той же атмосфере. Тетрагидрофуран удалялся затем с помощью перегонки при пониженном давлении. Получающийся остаток растворялся в 10 мл этилацетата, и раствор промывался 10% вес/объем водным раствором карбоната натрия и насыщенным водным раствором хлористого натрия в указанном порядке. Раствор затем сушился над безводным сульфатом магния, после чего растворитель отгонялся при пониженном давлении, а остаток очищался с помощью хроматографии на колонке с пропусканием через 10 г силикагеля (70 - 230 меш) с использованием этилацетата в качестве элюента, давая 67 мг (выход 56%) указанного в заголовке соединения в виде пенообразного вещества.
1H Спектр ЯМР (CDCl3, 270 МГц, ТМС), δ, млн. дол.:
8,32 (1H, широкий синглет), 7,26 - 7,85 (8H, мультиплет), 6,27 (1H, мультиплет), 4,38 - 4,46 (1H, мультиплет), 4,11 - 4,19 (1H, мультиплет), 3,93 - 3,95 (2H, мультиплет), 3,47 - 3,90 (4H, мультиплет), 2,10 - 2,88 (6H, мультиплет), 1,62 - 1,90 (3H, мультиплет), 1,10 - 1,21 (12H, мультиплет).
114 (c) Соединение формулы 114 (c);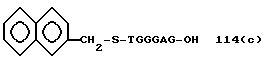
В соответствии с процедурой, аналогичной процедуре, описанной в примере 1 (b), но с использованием 5'-[(2-нафтил)метилтио]-5'-дезокситимидин 2-цианоэтил N,N-диизопропилфосфорамидита [полученного, как описано в стадии (b) выше] вместо 5'-O-тритилтимидин 2-цианоэтил N,N-диизопропилфосфорамидита, получалось указанное в заголовке соединение. Реакционный продукт очищался с помощью хроматографии при пропускании через силикагельную колонку с обращенной фазой (Preparative C 18, Уотерс, 1,5 х 150 мм; элюент A: 50 мМ TEAB; pH 7,5; элюент B: ацетонитрил; 15 _→ 50% B по объему, линейный градиент; 260 нм). Элюированные фракции, содержащие целевое соединение, собирались и обрабатывались по способу, аналогичному описанному в примере 1(b), давая 165 OD (260 нм) целевого соединения в виде аморфного твердого вещества.
УФ-спектр поглощения (вода), λмакс, нм: 254.
Время удерживания: 12,07 мин.
Высокоэффективная жидкостная хроматография (Инертсил ODS-2; 6 х 150 нм; элюент A: 0,1 М TEAA, pH 7,5; элюент B: ацетонитрил; 10 _→ 60% B по объему, 20 мин, линейный градиент; 1 мл/мин; 260 нм).
Пример 115
115 (a) 5'-{3,5- Бис [3,5 - (дибензилокси)бензилокси]бензилтио}- 5' -дезокситимидин
258 мг (1 ммоль) 5' -дезокси- 5'-меркаптотимидина добавлялось к 20 мл ацетона. Затем к раствору в атмосфере аргона добавлялось 969 мг (1,2 ммоль) 3,5 - бис [3,5 -(дибензилокси) бензилокси] бензилбромида и 1 г безводного карбоната калия. Получающаяся смесь перемешивалась при нагревании с обратным холодильником в течение 2 ч. В конце данного периода времени осадки отфильтровывались от реакционной смеси, и фильтрат освобождался от растворителя с помощью перегонки при пониженном давлении. Остаток лиофилизовался из бензола, давая 365 мг (выход 37%) указанного в заголовке соединения в виде белого порошка.
1H ЯМР- спектр (CDCl3, 270 МГц, ТМС), δ, млн. дол.:
7,99 (1H, широкий синглет), 7,23 - 7,42 (21H, мультиплет), 6,49 - 6,68 (9H, мультиплет), 4,98 - 5,03 (12H, мультиплет), 4,18 - 4,26 (1H, мультиплет), 3,87 - 3,91 (1H, мультиплет), 3,64 - 3,71 (2H, мультиплет), 2,67 (2H, дублет, J = 5,7 Гц), 2,00 - 2,64 (2H, мультиплет), 1,86 - 1,92 (3H, мультиплет).
115 (b) 5' -{3,5 - Бис [3,5-(дибензилокси)бензилокси]бензилтио}- 5' - дезокситимидин 2 -цианоэтил N,N - диизопропилфосфорамидит
197 мг (0,2 ммоль) 5'-{3,5 - бис [3,5 - (дибензилокси)бензилокси]бензилтио} - 5' - дезокситимидина [полученного, как описано в стадии (a) выше] сушились с помощью азеотропной перегонки с пиридином, а затем растворялись в 1 мл метиленхлорида. К раствору затем добавлялось 0,017 г (0,1 ммоль) диизопропиламмонийтетразолида. К смеси затем по каплям в атмосфере аргона добавлялось 70 мкл (0,22 ммоль) 2 - цианоэтил N,N,N'N' - тетраизопропилфосфордиамидита. Получающаяся смесь перемешивалась при комнатной температуре в течение 3 ч в той же атмосфере. Тетрагидрофуран затем удалялся перегонкой при пониженном давлении. Получающийся остаток растворялся в 10 мл этилацетата, и раствор промывался 10% вес/ объем водным раствором карбоната натрия и насыщенным водным раствором хлористого натрия в указанном порядке. Раствор затем сушился над безводным сульфатом магния, после чего растворитель удалялся с помощью перегонки при пониженном давлении, и остаток очищался с помощью хроматографии на колонке с пропусканием через 10 г силикагеля (70 - 230 меш) с использованием этилацетата в качестве элюента, давая 103 мг (выход 43%) целевого соединения данной стадии в виде карамелеобразного вещества.
1H Спектр ЯМР (CDCl3, 270 МГц, ТМС), δ, млн. дол. :
7,27 - 7,44 (20H, мультиплет), 6,51 - 6,69 (10H, мультиплет), 4,98 - 5,05 (12H, мультиплет), 4,44 - 4,51 (1H, мультиплет), 4,13 - 4,18 (1H, мультиплет), 3,47 - 3,87 (6H, мультиплет), 2,17 - 2,84 (6H, мультиплет), 1,92 (3H, синглет), 1,08 - 1,34 (12H, мультиплет).
115 (c) Соединение формулы 115 (c):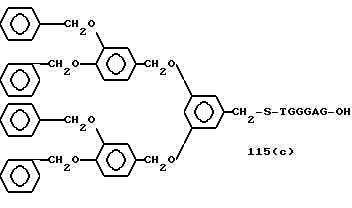
В соответствии с процедурой, аналогичной описанной в примере 1 (b), но с использованием 5'-{ 3,5 - бис [3,5-(дибензилокси)бензилокси]бензилтио} - 5'-дезокситимидин 2 - цианоэтил N,N - диизопропилфосфорамидита [полученного, как описано в стадии (b) выше] вместо 5'-O - тритилтимидин 2 - цианоэтил N,N - диизопропилфосфорамидита, получалось указанное в заголовке соединение. Продукт реакции очищался с помощью высокоэффективной жидкостной хроматографии с обращенной фазой (Инертсил PREP- ODS 20,0 х 250 мм ; элюент A: 0,1 М TEAA, pH 7,0; элюент B: ацетонитрил ; 30 _→ 100% B по объему, 30 мин, линейный градиент; 7 мл/мин; 260 нм; температура колонки 60oC). Фракции, элюированные через 26,5 мин, собирались и обрабатывались по способу, аналогичному описанному в примере 1 (b), давая 86 OD (260 нм) указанного в заголовке соединения в виде аморфного твердого вещества.
УФ - спектр поглощения (вода), λмакс, нм : 257.
Время удерживания : 18,44 мин.
Высокоэффективная жидкостная хроматография (Инертсил ODS - 2; 6 х 150 нм; элюент A: 0,1 M TEAA, pH 7,5; элюент B: ацетонитрил; 40 _→ 100% B по объему, 25 мин, линейный градиент; 1 мл/ мин; 260 нм).
Получение 1
2 -[2 - O - (4,4 - Диметокситритилокси)этилсульфонил] этилмоносукцината
1,37 г (3,0 ммоль) 2- [2 -O (4,4 - диметокситритилокси) этилсульфонил] этанола [Tetrahedron Lett. 27, 4705 (1986)] подвергалось сушке путем азотропной перегонки при пониженном давлении с пиридином, а затем растворялось в 12 мл метиленхлорида. К получающемуся раствору добавлялось 315 мг (3,15 ммоль) янтарного ангидрида и 384 мг (3,15 ммоль) 4-диметиламино / пиридина, и раствор затем перемешивался в течение 30 мин. После завершения реакции, что подтверждалось анализом тонкослойной хроматографии, к реакционной смеси добавлялось 80 мл метиленхлорида. Получающаяся смесь затем промывалась 0,5 M водным раствором первичного кислого фосфата калия (pH 5,0) и водой в указанном порядке, и органический слой сушился над безводным сульфатом натрия. Растворитель затем удалялся перегонкой при пониженном давлении, давая 1,60 г (выход 96%) целевого соединения.
1H Спектр ЯМР (CDCl3, 270 МГц, TMC), δ, млн.дол.:
7,40 - 6,83 (13H, мультиплет), 4,58 - 4,53 (2H, триплет, J = 5,94 Гц), 3,79 (6H, синглет), 3,68 - 3,64 (2H, триплет, J = 5,61 Гц), 3,50 - 3,45 (2H, триплет, J = 5,94 Гц), 3,18 - 3,14 (2H, триплет, J = 5,61 Гц), 2,71 - 2,61 (4H, мультиплет).
Получение 2
Соединение формулы 2 (p) :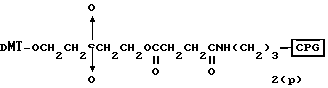
1,60 г (2,87 ммоль) 2-[2-O-(4,4' -диметокситритилокси)этилсульфонил] этилмоносукцината (полученного, как описано в получении 1) растворялось в 18 мл диметилформамида, и к получающемуся раствору добавлялось 0,96 г (3,6 ммоль) пентахлорфенола и 0,96 г (4,5 ммоль) 1,3 - дициклогексилкарбодиимида, и раствор затем перемешивался в течение 18 ч. Нерастворимые вещества, которые осаждались, отделялись фильтрованием, и растворитель удалялся из фильтрата выпариванием при пониженном давлении. К остатку добавлялся бензол, и дополнительные нерастворимые вещества, которые осаждались, отделялись фильтрованием. Растворитель снова удалялся из фильтрата выпариванием при пониженном давлении.
Остаток (0,16 г, 0,2 ммоль) растворялся в 4 мл диметилформамида, и к раствору добавлялось 15 мкл (0,11 ммоль) триэтиламина и 1,0 г аминопропил-CPG (85,7 мкмоль/г аминогрупп, введенных в него), и раствор затем оставлялся стоять при комнатной температуре в течение 24 ч.
В конце данного периода CPG-носитель собирался фильтрованием, промывался метиленхлоридом, а затем сушился выпариванием при пониженном давлении. К CPG-носителю добавлялось по 5 мл каждого из кэп A и B растворов (Ниппон Миллипор Лимитед), и смесь оставлялась стоять в течение 5 мин. Смесь затем промывалась пиридином и метиленхлоридом, в указанном порядке, и сушилась выпариванием при пониженном давлении, давая указанное соединение.
Количество соединения получения 1, введенного в целевое соединение, определялось в соответствии со следующим методом. Деблокирующий раствор (3% по объему раствора дихлоруксусной кислоты в метиленхлориде, Ниппон Миллипор Лимитед) добавлялся к 11,4 мг целевого соединения, и смесь встряхивалась в течение 3 мин, после чего добавлялось достаточное количество метиленхлорида для доведения общего объема до 20 мл. 0,4 мл растворителя отгонялось, а затем к остатку добавлялось 3 мл 3,2 по объему раствора перхлорной кислоты в этаноле, и измерялась абсорбция диметокситритильного катиона при 500 нм (ε = 71700).
Количество введенных диметокситритильных групп составило 53,1 мкмоль/г.
Получение 3
2 - [2 - O - (4,4' -Диметокситритилокси) этилсульфонил] этил 2,2,2, - трихлорэтоксикарбонат
1,1 г (2,4 ммоль) 2-[2-O - (4,4' -диметокситритилокси) этилсульфонил - этанола [Tetrahedron Lett 27, 4705 (1986)] сушилось с помощью азеотропной перегонки при пониженном давлении с пиридином, а затем растворялось в 12 мл метиленхлорида. К раствору добавлялось 0,35 мл (2,6 ммоль) 2, 2, 2 - трихлорэтоксикарбонилхлорида, и раствор затем перемешивался в течение 2 ч. После завершения реакции, что подтверждалось результатом тонкослойной хроматографии, реакционная смесь выливалась в смесь 100 мл метиленхлорида и 100 мл 5% в/о водного раствора бикарбоната натрия для разделения фаз. Органические слои промывались 5% в/о водным раствором бикарбоната натрия, а затем они собирались и сушились над безводным сульфатом натрия. Растворитель затем удалялся перегонкой при пониженном давлении, и остаток очищался с помощью хроматографии на колонке с обращенной фазой (Preparative C18, Уотерс, ⊘ 2,2 х 70 см, растворитель: 60% объем/объем водный ацетонитрил/, давая 1,35 г (выход 89%) целевого соединения в виде аморфного порошка.
1H Спектр ЯМР (CDCl3, 270 МГц, ТМС), δ, млн. дол.:
7,41 - 6,89 (13H, мультиплет), 4,75 (2H, синглет), 4,70 - 4,66 (2H, триплет, J = 5,94 Гц), 3,80 (6H, синглет), 3,68 - 3,64 (2H, триплет, J = 5,28 Гц), 3,61 - 3,56 (2H, триплет, J = 6,27 Гц), 3,23 - 3,19 (2H, триплет, J = 5,28 Гц).
Получение 4
Соединение формулы 4 (p):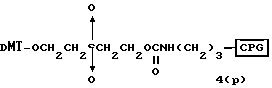
0,126 г (0,2 ммоль) 2 - [2 - О-(4,4' - диметокситритилокси)этилсульфонил]этил 2,2,2 - трихлорэтоксикарбоната (полученного, как описано в получении 3) растворялось в 5 мл диметилформамида, и к получающемуся раствору добавлялось 15 мкмл (0,11 ммоль) триэтиламина и 1,0 г аминопропил-CPG (85,7 мкмоль/г аминогруппы введены в него) и раствор промывался 1% о/о раствором триэтиламина в диметилформамиде. Смесь затем оставлялась стоять при комнатной температуре в течение 4 дн. В конце данного периода CPG-носитель собирался фильтрованием и промывался метиленхлоридом, после чего он сушился выпариванием при пониженном давлении.
К CPG-носителю добавлялось по 5 мл каждого из кэп A и B растворов (Ниппон Миллипор Лимитед), и смесь оставлялась стоять в течение 5 мин. Смесь затем промывалась пиридином и метиленхлоридом в, указанном порядке, и сушилась выпариванием при пониженном давлении, давая указанное в заготовке соединение.
Количество соединения получения 3, введенного в конечное соединение, определялось согласно следующему способу. Деблокирующей раствор (3% о/о раствор дихлоруксусной кислоты в метиленхлориде, Ниппон Миллипор Лимитед) добавлялся к 11,4 мг целевого соединения, и смесь встряхивалась в течение 3 мин, после чего добавлялось достаточное количество метиленхлорида для доведения общего объема до 20 мл. 0,4 мл растворителя отгонялось, и затем к остатку добавлялось 3 мл 3:2 по объему раствора перхлорной кислоты в этаноле и измерялась абсорбция диметокситритильных катионов при 500 нм ( ε = 71700).
Количество диметокситритильных введенных групп составило 24,0 мкмоль/г.
Получение 5
Соединение формулы 5 (p):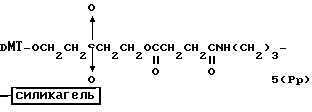
1,60 г (2,87 ммоль) 2-[2 - O-(4,4' - диметокситритилокси)этилсульфонил] этил моносукцината (полученного, как описано в получении 1) растворялось в 18 мл диметилформамида, и к раствору добавлялось 0,96 г (3,6 ммоль) пентахлорфенола и 0,96 г (4,5 ммоль) 1,3 - дициклогексилкарбодиимида. Получающаяся смесь затем перемешивалась при комнатной температуре в течение 18 ч. В конце данного времени осадки отфильтровывались из реакционной смеси, и фильтрат освобождался от растворителя перегонкой при пониженном давлении. К остатку добавлялся бензол. Получающиеся осадки отфильтровывались, и фильтрат снова освобождался от растворителя перегонкой при пониженном давлении. 1,0 г (0,92 ммоль) остатка использовался на следующей стадии.
1,0 г (0,92 ммоль) данного остатка, 75 мкл (0,55 ммоль) триэтиламина и Космосила 150 NH2 - 300 (имеющего 450 мкмоль аминогрупп на 1 г силикагеля, поставляемого фирмой Теске Ко. Лтд) добавлялись к 20 мл диметилформамида. Получающаяся смесь затем перемешивалась при комнатной температуре в течение 20 ч. Требуемый носитель, а именно замещенный Космосил 150 NH2 - 300, собирался, промывался метиленхлоридом и сушился выпариванием при пониженном давлении. К носителю добавлялось по 10 мл каждого из кэп A и B растворов (Ниппон Миллипор Лимитед), и смесь оставлялась стоять в течение 10 мин. Смесь затем промывалась пиридином и метиленхлоридом в указанном порядке и сушилась выпариванием при пониженном давлении, давая целевое соединение.
Количество соединения получения 1, введенного в целевое соединение, определялось согласно следующему методу. Деблокирующий раствор (3% о/о раствор дихлоруксусной кислоты в метиленхлориде, Ниппон Миллипор Лимитед) добавлялся к 5,4 мг целевого соединения, и смесь встряхивалась в течение 3 мин, после чего добавлялось достаточное количество метиленхлорида для доведения общего объема до 20 мл. 0,4 мл растворителя отгонялось при пониженном давлении, и к остатку добавлялось 3 мл 3:2 по объему раствора перхлорной кислоты в этаноле, и измерялась абсорбция диметокситритильного катиона при 500 нм (ε = 1700).
Введенное количество диметокситритильных групп было найдено составляющим 51,8 мкмоль/г.
Получение 6
Соединение формулы 6 (p):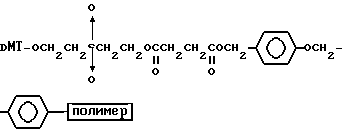
1,60 г (2,87 ммоль) 2-[2-O-(4,4'-диметокситритилокси)этилсульфонил] этилмоносукцината (полученного, как описано в получении 1) растворялось в 18 мл диметилформамида, и к раствору добавлялось 0,96 г (3,6 ммоль) пентахлорфенола и 0,96 г (4,5 ммоль) 1,3 - дициклогексилкарбодиимида. Получающаяся смесь затем перемешивалась при комнатной температуре в течение 18 ч. В конце данного периода осадки отфильтровывались из реакционной смеси, и фильтрат освобождался от растворителя перегонкой при пониженном давлении. К остатку добавлялся бензол. Получающиеся осадки отфильтровывались, и фильтрат снова освобождался от растворителя перегонкой при пониженном давлении. 0,48 г (0,6 ммоль) остатка использовались на следующей стадии.
0,48 г (0,6 ммоль) данного остатка, 110 мг (0,9 ммоль) 4-(диметиламино)пиридина и 1,0 г n-алкоксибензиловоспиртовой смолы (имеющей 0,9 ммоль аминогрупп на 1 г силикагельной поверхности, продуцируемой фирмой Кокусан Кагаку Ко. Лтд) добавлялись к 15 мл диметилформамида, и получающаяся смесь перемешивалась при комнатной температуре в течение 25 ч. В конце данного периода требуемый носитель, а именно замещенная n-алкоксибензиловоспиртовая смола, собирался, промывался метиленхлоридом, метанолом и диэтиловым эфиром, в указанном порядке, и сушился выпариванием при пониженном давлении. К остатку добавлялось по 10 мл каждого из кэп A и B растворов (Ниппон Миллипор Лимитед), и смесь оставлялась стоять в течение 15 мин. Смесь затем промывалась метиленхлоридом, метанолом и диэтиловым эфиром, в указанном порядке, и сушилась выпариванием при пониженном давлении, давая конечное соединение.
Количество соединения получения 1, введенное в конечное соединение, определялось в соответствии со следующим методом. Деблокирующий раствор (3% о/о раствор дихлоруксусной кислоты в метиленхлориде, Ниппон Миллипор Лимитед) добавлялся к 10,6 мг целевого соединения, и смесь встряхивалась в течение 3 мин, после чего добавлялось достаточное количество метиленхлорида для получения общего объема 20 мл. 0,4 мл растворителя отгонялось, а затем к остатку добавлялось 6 мл 3:2 по объему раствора перхлорной кислоты в этаноле и измерялась абсорбция диметокситритильного катиона при 500 нм (ε = 71700).
Количество введенных диметокситритильных групп было найдено равным 302 мкмоль/г.
Получение 7
Соединение формулы 7 (p):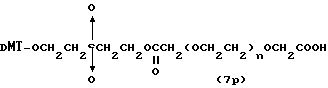
9,9 г (3,3 ммоль) поли(оксиэтилен) дигликоля сушилось с помощью азеотропной перегонки с пиридином и затем растворялось в 10 мл метиленхлорида. Затем к раствору при охлаждении льдом добавлялось 226 мг (1,1 ммоль) 1,3 - дициклогексилкарбодиимида. Получающаяся смесь затем перемешивалась при охлаждении льдом в течение 15 мин. В конце данного периода к раствору добавлялось 2 мл метиленхлоридного раствора, содержащего 0,5 г (1,1 ммоль) 2-[2-O-(4,4' - диметокситритилокси)этилсульфонил] - этанола (Tetrahedron Lett. , 27, 4705 /1986/) и 0,13 г (1,1 ммоль) 4-(диметиламино)пиридина. Получающаяся смесь перемешивалась при комнатной температуре в течение 18 ч. Осадки отфильтровывались. Реакционная смесь затем концентрировалась до 50 мл выпариванием при пониженном давлении. Затем к концентрированному раствору добавлялось 500 мл холодного диэтилового эфира, и полученные осадки собирались и сушились выпариванием при пониженном давлении. Получающиеся осадки растворялись в метиленхлориде и очищались с помощью хроматографии на колонке при пропускании через 200 г силикагеля (70 - 230 меш) с использованием 2:8 по объему смеси метанола и метиленхлорида в качестве элюента, давая 2,77 г (выход 72%) целевого соединения.
1H Спектр ЯМР (CDCl3, 270 МГц, ТМС), δ, млн. дол.:
7,09 - 7,43 (мультиплет), 6,81 - 6,88 (мультиплет), 4,58 - 4,64 (мультиплет), 3,65 (синглет), 3,50 - 3,53 (мультиплет), 3,46 - 3,49 (мультиплет), 3,31 - 3,34 (мультиплет).
Получение 8
Соединение формулы 8 (p):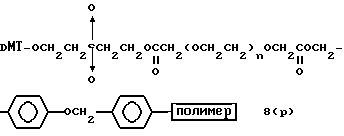
0,7 г (0,2 ммоль) соединения получения 7 растворялось в 6 мл метиленхлорида, и к раствору при охлаждении льдом добавлялось 2 мл диметилформамидного раствора, содержащего 63 мг (0,24 ммоль) пентахлорфенола и 64 мг (0,3 ммоль) 1,3-дициклогексилкарбодиимида. Получающийся раствор затем перемешивался при комнатной температуре в течение 7 дн. В конце данного периода осадки отфильтровывались из реакционной смеси, и фильтрат освобождался от растворителя перегонкой при пониженном давлении. Получающийся остаток растворялся в 10 мл метиленхлорида. К раствору добавлялось при охлаждении льдом 100 мл диэтилового эфира, и получающиеся осадки отфильтровывались. Фильтр освобождался от растворителя перегонкой при пониженном давлении. Фильтрат, 37 мг (0,3 ммоль) 4-(диметиламино)пиридина и 0,25 г n-алкоксибензилового спирта (смолы) (имеющей 0,9 ммоль аминогрупп на 1 г силикагельной поверхности, поставляемой фирмой Кокусан Кагаку Ко. Лтд) добавлялись к 6 мл диметилформамида. Получающаяся смесь оставлялась при комнатной температуре на 24 ч. Требуемый носитель, а именно замещенная n-алкоксибензиловоспиртовая смола, собирался, промывался пиридином, метиленхлоридом и диэтиловым эфиром, в указанном порядке, и сушился выпариванием при пониженном давлении. 10 мл каждого из кэп A и B растворов (Ниппон Миллипор Лимитед) добавлялись к носителю, и смесь оставлялась стоять в течение 15 мин. Смесь затем промывалась метиленхлоридом, метанолом и диэтиловым эфиром в указанном порядке и сушилась выпариванием при пониженном давлении, давая целевое соединение.
Количество соединения получения 1, введенное в целевое соединение, определялось согласно следующему способу. Деблокирующий раствор (3% о/о раствор дихлоруксусной кислоты в метиленхлориде) добавлялся к 6,7 мг целевого соединения, и смесь встряхивалась в течение 3 мин, после чего добавлялось достаточное количество метиленхлорида для достижения общего количества 20 мл. 0,4 мл растворителя удалялось перегонкой при пониженном давлении, и затем к остатку добавлялось 20 мл 3:2 по объему раствора перхлорной кислоты в этаноле, и измерялась абсорбция диметокситритильного катиона при 500 нм (ε = 71700).
Было найдено, что введенное количество диметокситритильных групп составляло 16 мкмоль/г.
Пример 1 препаративной формы
Инъекционная готовая форма препарата
Инъекционная препаративная форма приготавливалась с помощью перемешивания 1,5 мас. % соединения примера 25 в 10 об.% пропиленгликоля, добавления достаточного количества воды для инъекций для получения требуемого конечного объема и стерилизации.
Пример 2 препаративных форм
Таблетки, мг:
1/ Соединение примера 25 - 200
2/ Пирофосфат натрия - 5
3/ Аэросил 200 - 5
4/ Стеарат магния - 5
5/ Лактоза - 495
6/ Кукурузный крахмал - 154
7/ Апицел - 123
8/ HPL (L) - 10 - Всего 997
Таблетки, содержащие каждая 100 мг ингредиентов, приготавливались добавлением измельченной смеси указанных выше компонентов 1 - 4 к предварительно гранулированной смеси компонентов 5 - 8 и затем прессованием полученной смеси на подходящем таблетирующем устройстве.
Пример 3 препаративных форм
Капсула, мг:
1/ Соединение примера 25 - 200
2/ Фосфат калия - 200
3/ Силикат алюминия - 345
4/ Кристаллическая целлюлоза - 250
5/ Стеарат магния - 2 - Всего 997
В соответствии с общепринятыми средствами капсулы, включающие 200 мг указанных выше ингредиентов, приготавливались путем распыления или пульверизации смеси ингредиентов в подходящем смесителе, пропускания смеси через сито и, наконец, тщательного смешения.
Пример 4 препаративных форм
Твердая капсула
Единичные капсулы приготавливались смешением 100 мг порошкообразного соединения примера 25, 150 мг лактозы, 50 мг целлюлозы и 6 мг стеарата магния, заполнением указанной выше смеси в стандартные твердые желатиновые капсулы, состоящие из 2 ч., промывкой и, наконец, сушкой.
Пример 5 препаративных форм
Мягкая капсула
Мягкие капсулы, включающие 100 мг активного ингредиента, приготавливались смешением соединения примера 25 в перевариваемом масле (таком как соевое масло, хлопковое масло или оливковое масло), заполнением данной смеси в подходящие желатиновые капсулы с помощью насоса, промывкой и, наконец, сушкой.
Пример 6 препаративных форм
Таблетки
В соответствии с общепринятыми приемами таблетки приготавливались гранулированием смеси 100 мг соединения примера 25, 0,2 мг коллоидной двуокиси кремния, 5 мг стеарата магния, 275 мг тонкокристаллической целлюлозы, 11 мг крахмала и 98,8 мг лактозы.
Таблица последовательностей
Последовательность ID N: 1
Длина последовательности: 4
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TGGG
Последовательность ID N: 2
Длина последовательности: 5
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TGGGG
Последовательность ID N: 3
Длина последовательности: 5
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TGGGA
Последовательность ID N: 4
Длина последовательности: 6
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TGGGAG
Последовательность ID N: 5
Длина последовательности: 7
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TGGGAGG
Последовательность ID N: 6
Длина последовательности: 7
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: CGGGAGG
Последовательность ID N: 7
Длина последовательности: 7
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TTGGAGG
Последовательность ID N: 8
Длина последовательности: 7
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TGCGAGG
Последовательность ID N: 9
Длина последовательности: 7
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: GGGGAGG
Последовательность ID N: 10
Длина последовательности: 7
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: mCGGGAGG
Последовательность ID N: 11
Длина последовательности: 7
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: mCGmCGAGG
Последовательность ID N: 12
Длина последовательности: 8
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TTGCGAGG
Последовательность ID N: 13
Длина последовательности: 8
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: CTGGGAGG
Последовательность ID N: 14
Длина последовательности: 5
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: CTCGT
Последовательность ID N: 15
Длина последовательности: 5
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: CTCGC
Последовательность ID N: 16
Длина последовательности: 5
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: CTCGG
Последовательность ID N: 17
Длина последовательности: 5
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: CGGGT
Последовательность ID N: 18
Длина последовательности: 5
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: CGGGC
Последовательность ID N: 19
Длина последовательности: 5
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: CGGGG
Последовательность ID N: 20
Длина последовательности: 5
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: CGCGT
Последовательность ID N: 21
Длина последовательности: 5
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: CGCGC
Последовательность ID N: 22
Длина последовательности: 5
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: CGCGG
Последовательность ID N: 23
Длина последовательности: 5
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TTGGA
Последовательность ID N: 24
Длина последовательности: 5
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TTGGT
Последовательность ID N: 25
Длина последовательности: 5
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TTGGC
Последовательность ID N: 26
Длина последовательности: 5
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TTGGG
Последовательность ID N: 27
Длина последовательности: 5
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TTCGA
Последовательность ID N: 28
Длина последовательности: 5
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TTCGT
Последовательность ID N: 29
Длина последовательности: 5
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TTCGC
Последовательность ID N: 30
Длина последовательности: 5
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TTCGG
Последовательность ID N: 31
Длина последовательности: 5
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TGGGT
Последовательность ID N: 32
Длина последовательности: 5
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TGGGC
Последовательность ID N: 33
Длина последовательности: 5
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TGCGA
Последовательность ID N: 34
Длина последовательности: 5
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TGCGT
Последовательность ID N: 35
Длина последовательности: 5
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TGCGC
Последовательность ID N: 36
Длина последовательности: 5
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TGCGG
Последовательность ID N: 37
Длина последовательности: 5
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: CTGGT
Последовательность ID N: 38
Длина последовательности: 5
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: CTGGC
Последовательность ID N: 39
Длина последовательности: 4
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: CTCG
Последовательность ID N: 40
Длина последовательности: 4
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TTCG
Последовательность ID N: 41
Длина последовательности: 4
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: CTGG
Последовательность ID N: 42
Длина последовательности: 4
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: CGCG
Последовательность ID N: 43
Длина последовательности: 4
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TGCG
Последовательность ID N: 44
Длина последовательности: 4
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: CGGG
Последовательность ID N: 45
Длина последовательности: 4
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TTGG
Последовательность ID N: 46
Длина последовательности: 9
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: GGGCGGGGC
Последовательность ID N: 47
Длина последовательности: 7
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TAGGAGG
Последовательность ID N: 48
Длина последовательности: 8
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TGGGAGGT
Последовательность ID N: 49
Длина последовательности: 9
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TGGGCGCAG
Последовательность ID N: 50
Длина последовательности: 3
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: GCG
Последовательность ID N: 51
Длина последовательности: 7
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TCGGAGG
Последовательность ID N: 52
Длина последовательности: 7
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TGmCGAGG
Последовательность ID N: 53
Длина последовательности: 8
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: GIGGG AGG
Последовательность ID N: 54
Длина последовательности: 3
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TGG
Последовательность ID N: 55
Длина последовательности: 7
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TGGGAmGG
Последовательность ID N: 56
Длина последовательности: 7
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: TGGGAGA
Последовательность ID N: 57
Длина последовательности: 9
Тип последовательности: нуклеиновая кислота
Число нитей: однонитевая
Топология: линейная
Гипотетическая последовательность: нет
Антисмысловая: нет
Последовательность: AATGGGAGG
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ НУКЛЕОЗИДОВ ПИРИМИДИНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2085557C1 |
| ГЕКСАГИДРОНАФТАЛИНОВЫЕ СЛОЖНОЭФИРНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2104997C1 |
| ПЕПТИДНОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1993 |
|
RU2106357C1 |
| ПРОИЗВОДНЫЕ БИФЕНИЛА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2109736C1 |
| ПРОИЗВОДНЫЕ НЕЙРАМИНОВОЙ КИСЛОТЫ, ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ИЛИ ИХ СЛОЖНЫЕ ЭФИРЫ, А ТАКЖЕ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ СИАЛИДАЗУ-ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ | 1997 |
|
RU2124509C1 |
| АНАЛОГИ ЛИПИДА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1992 |
|
RU2076107C1 |
| α,ω ДИАРИЛАЛКАНЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2105752C1 |
| 13-ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ МИЛБЕМИЦИНА, АКАРИЦИДНАЯ И ИНСЕКТИЦИДНАЯ КОМПОЗИЦИЯ И СПОСОБ ЗАЩИТЫ РАСТЕНИЙ ОТ ПОВРЕЖДЕНИЯ ПАРАЗИТАМИ | 1995 |
|
RU2109744C1 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СЛОЖНЫЕ ЭФИРЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2086541C1 |
| ПРОИЗВОДНЫЕ 13-(ЗАМЕЩЕННОГО ТИО)АЦЕТОКСИМИЛБЕМИЦИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ИНСЕКТИЦИДНАЯ КОМПОЗИЦИЯ И СПОСОБ ЗАЩИТЫ РАСТЕНИЙ | 1992 |
|
RU2086552C1 |
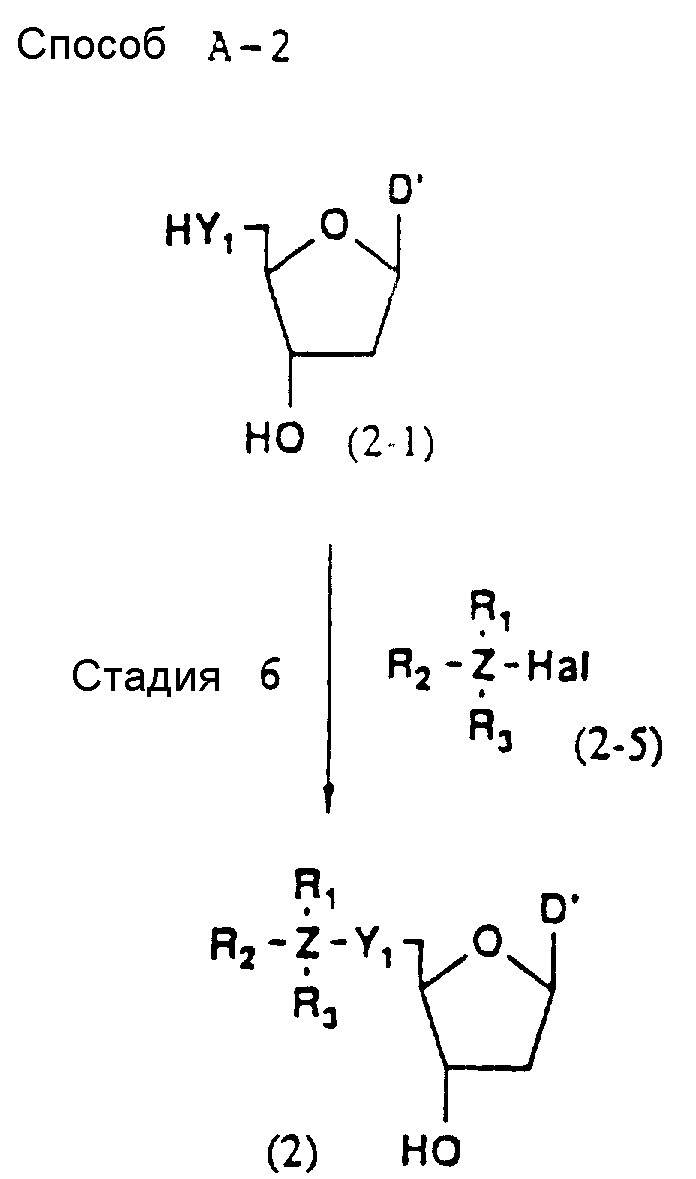
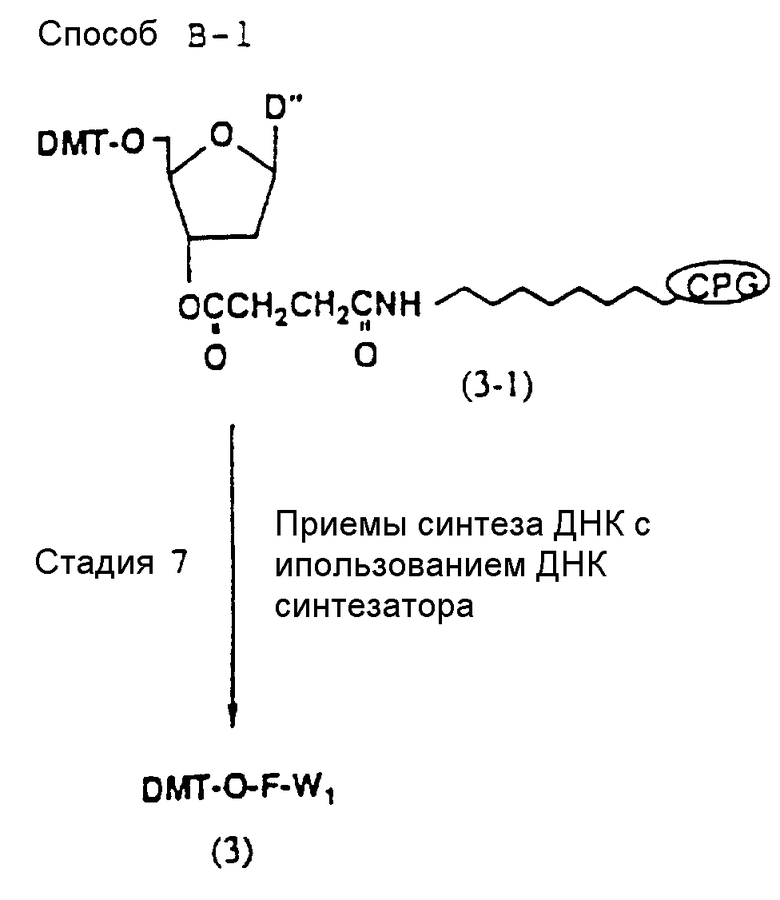
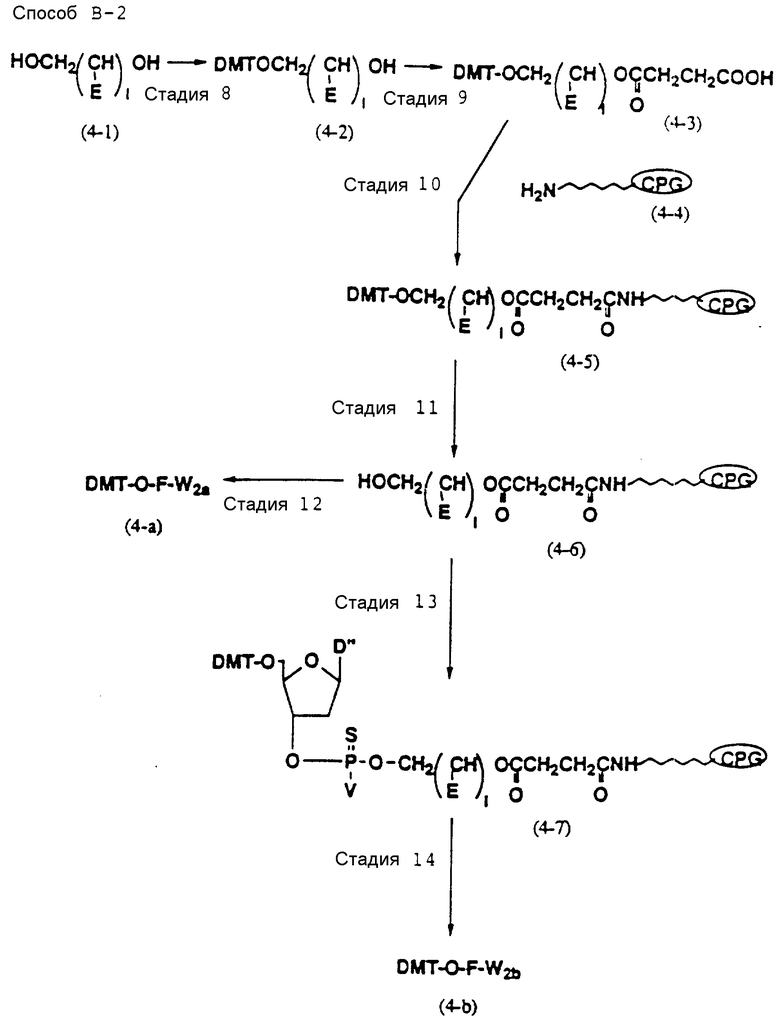
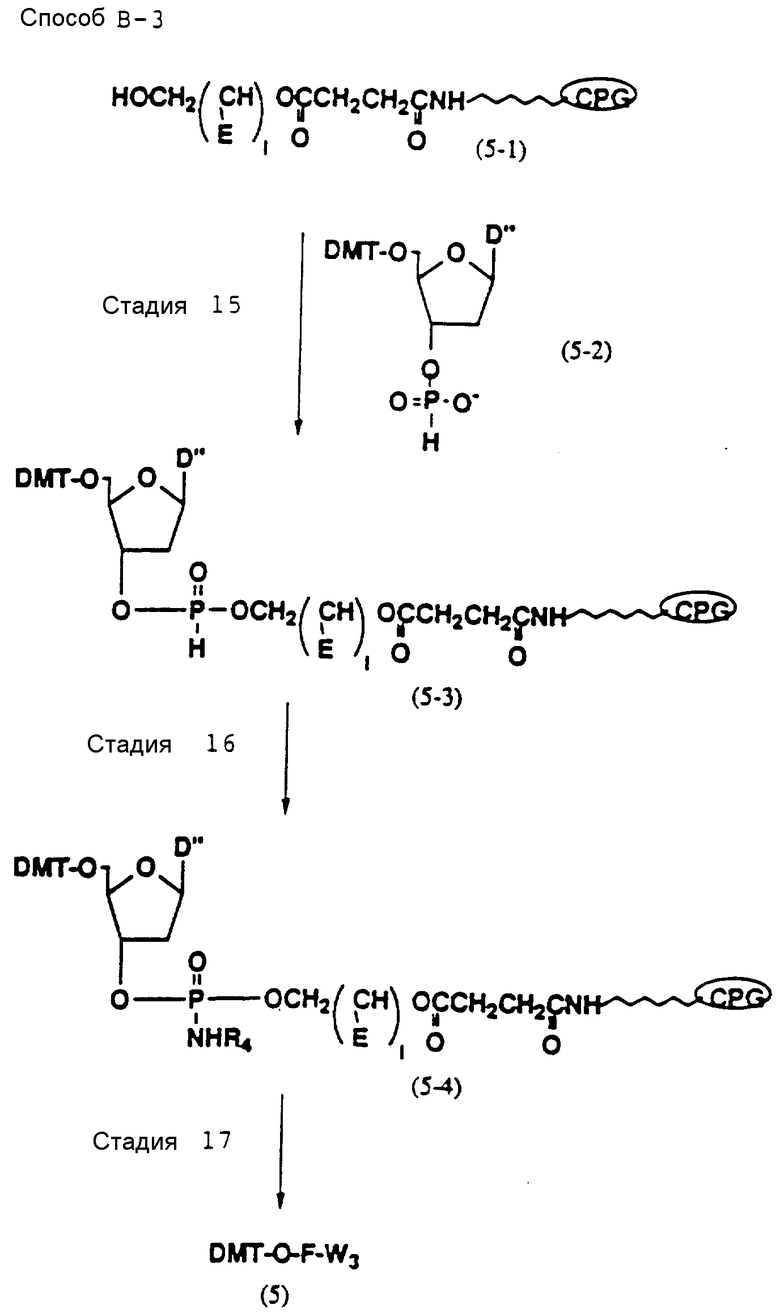
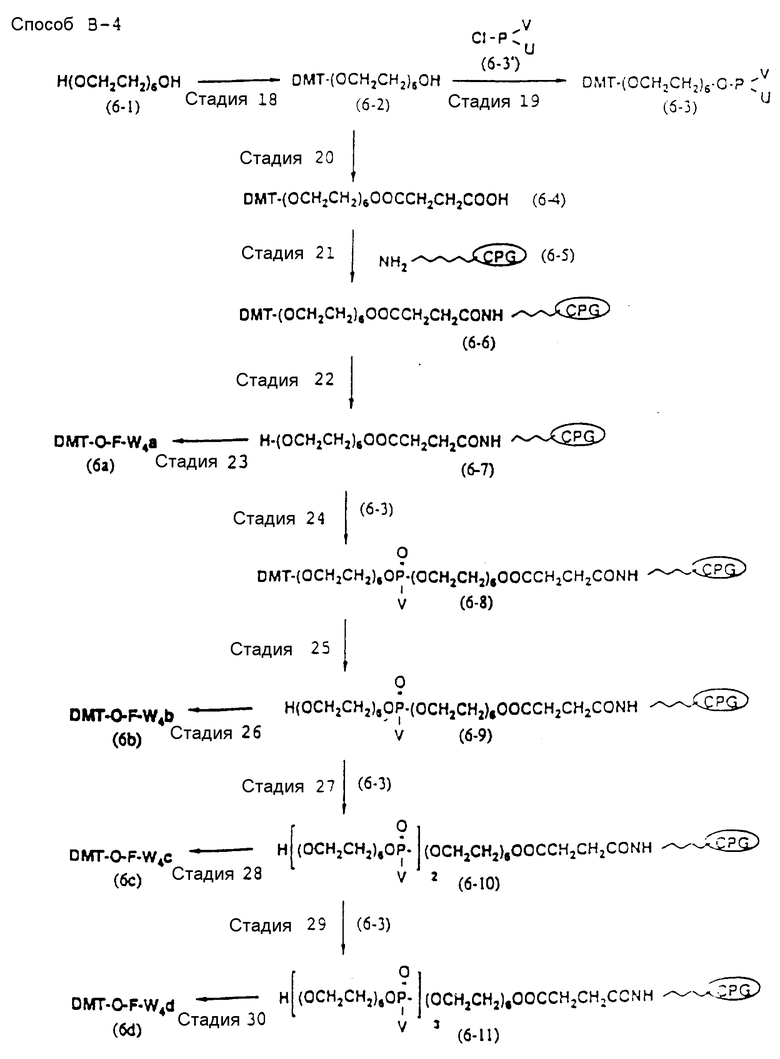
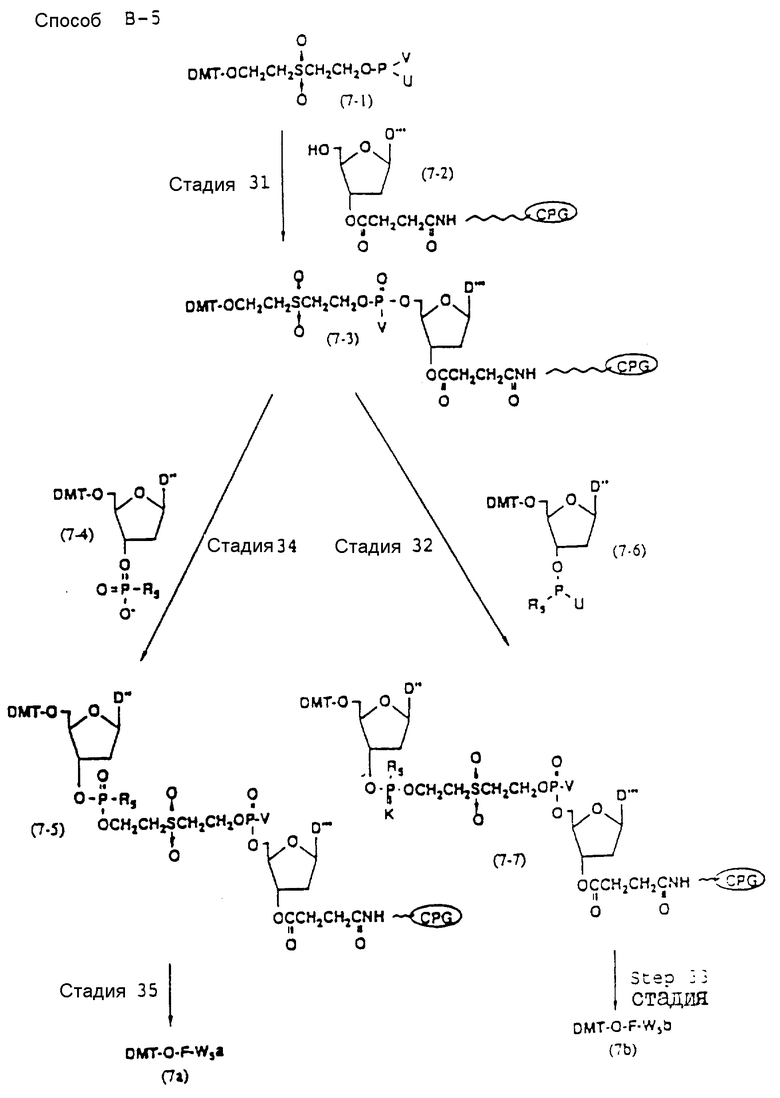
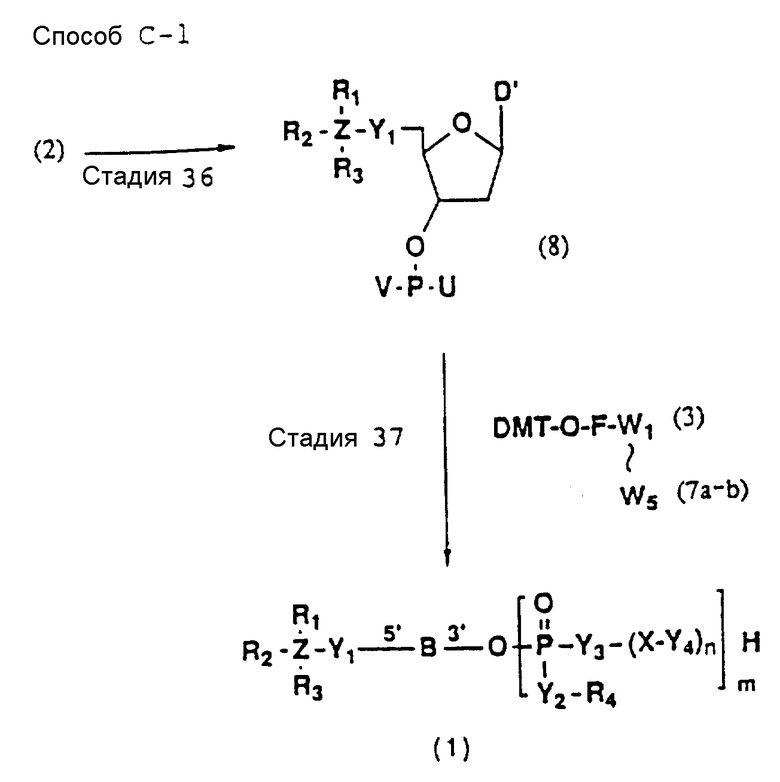
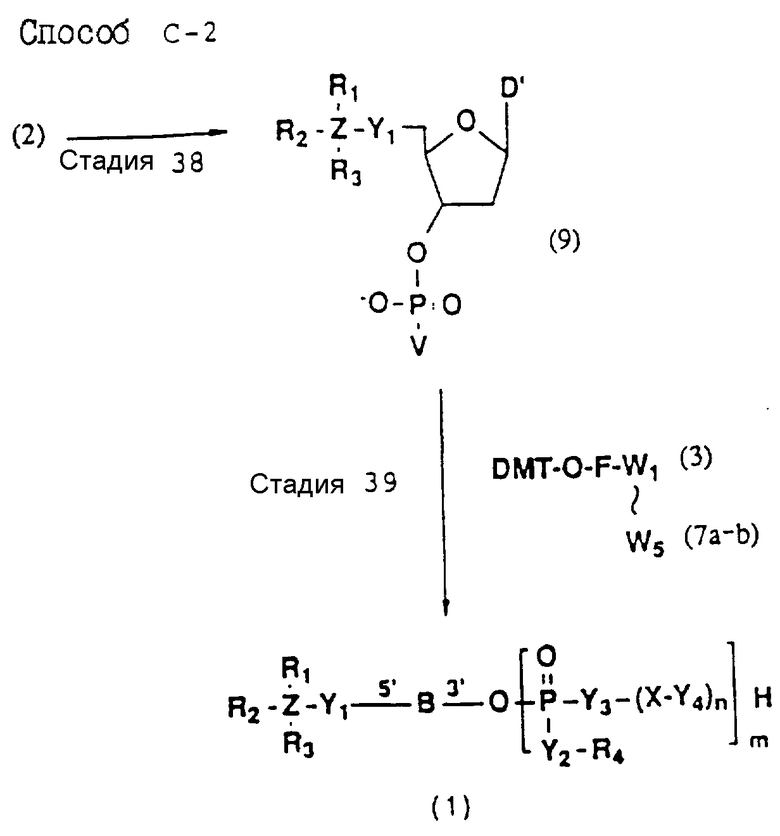
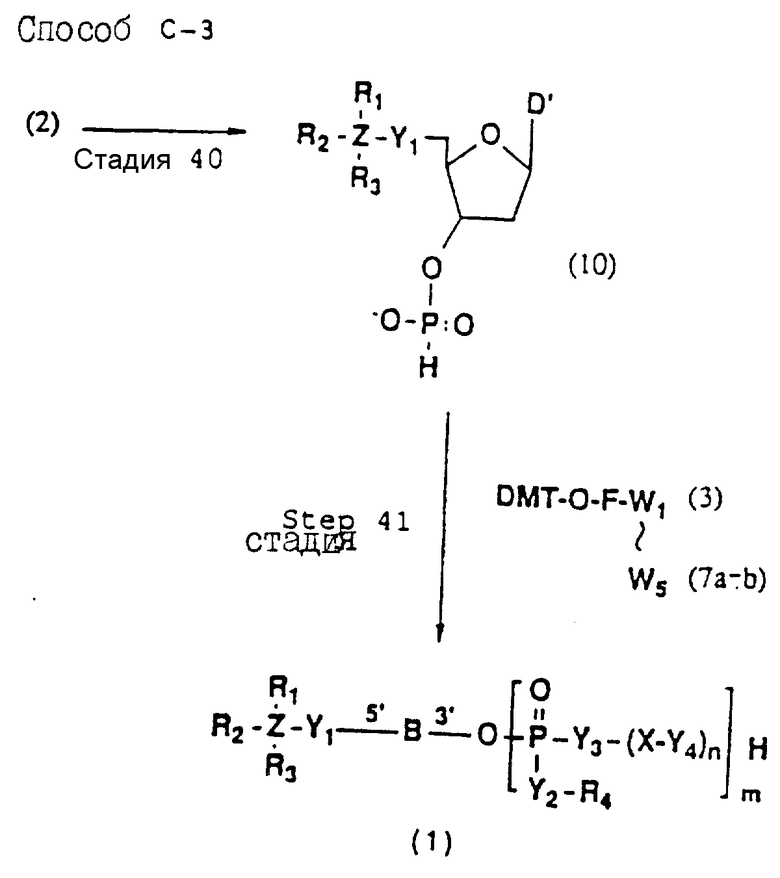
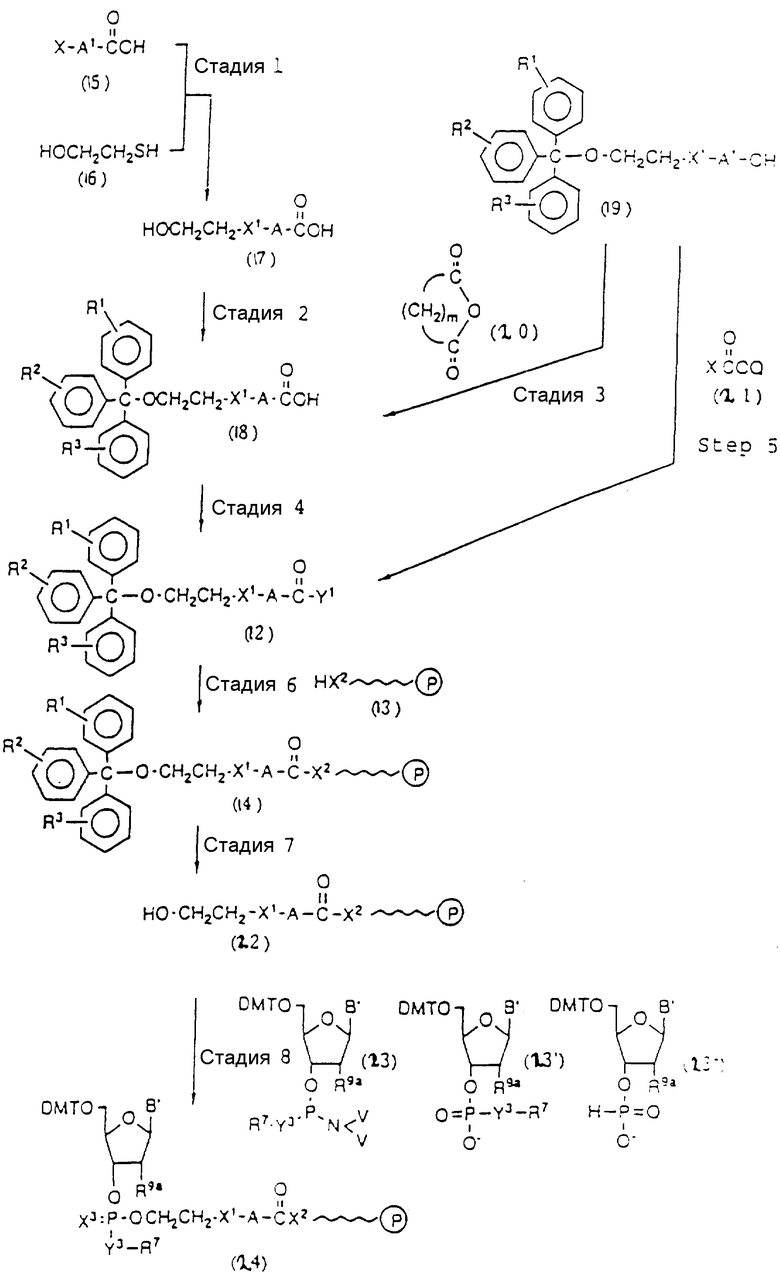
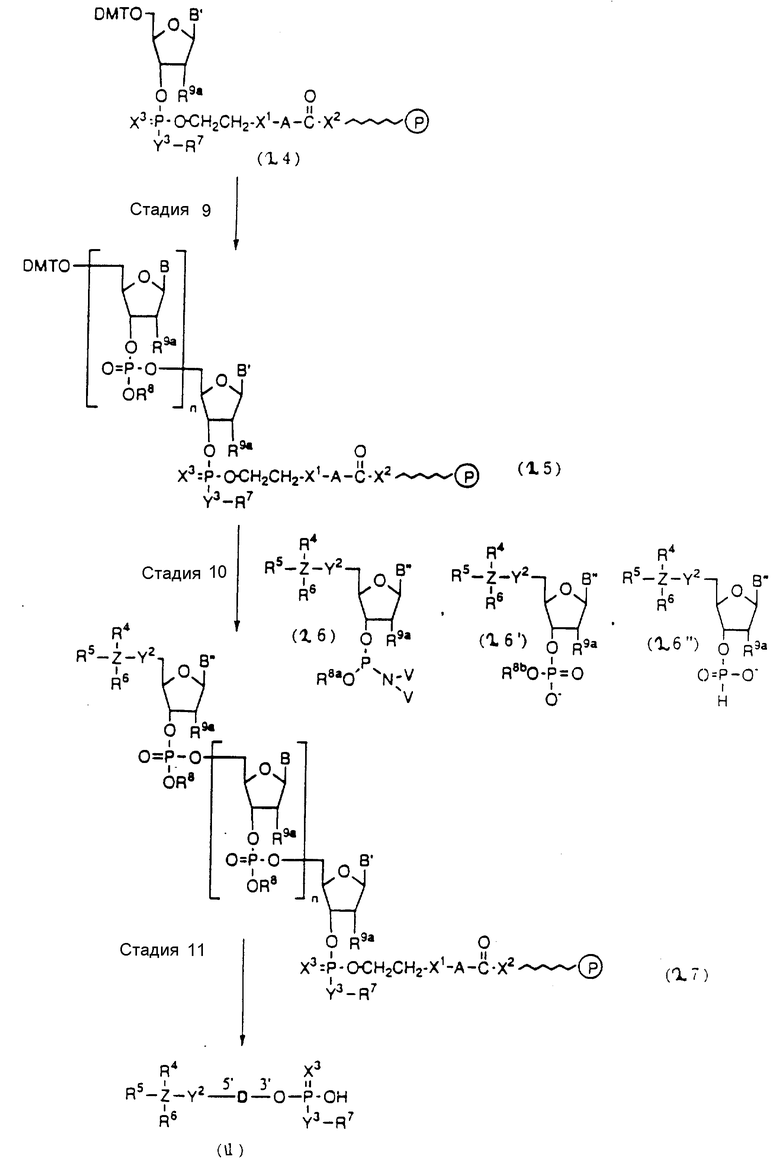
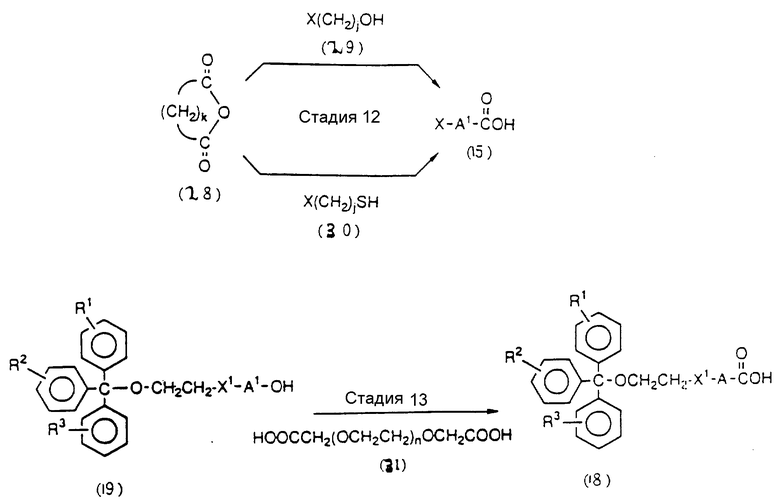
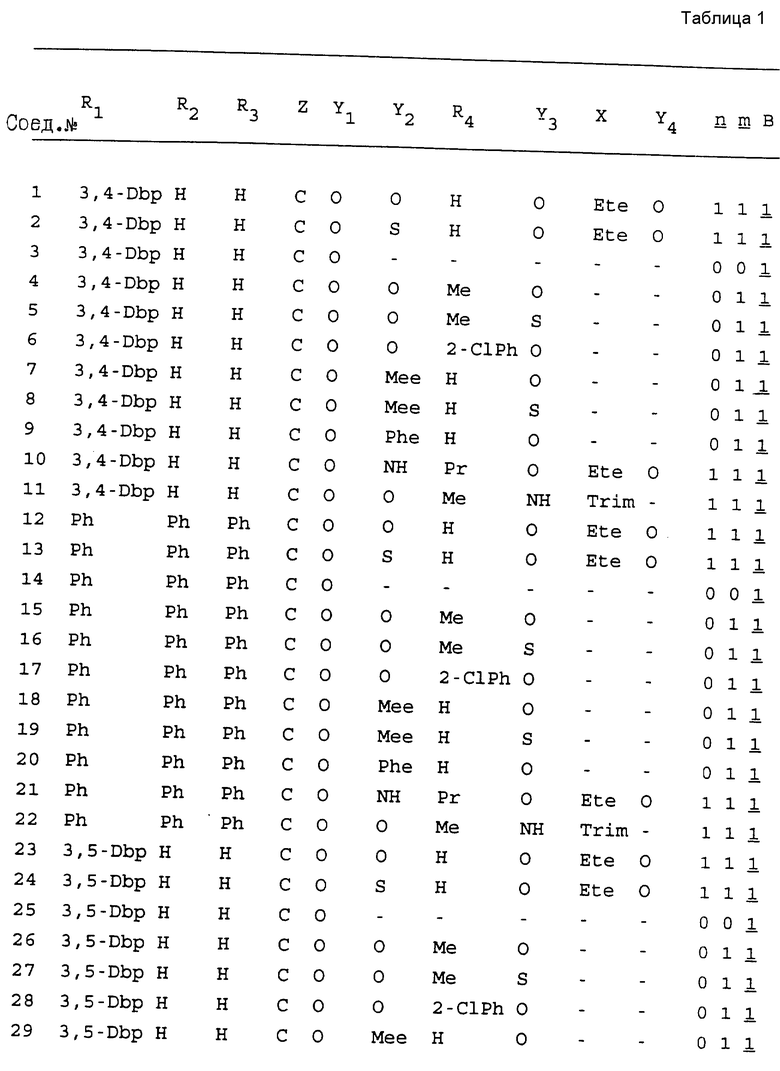
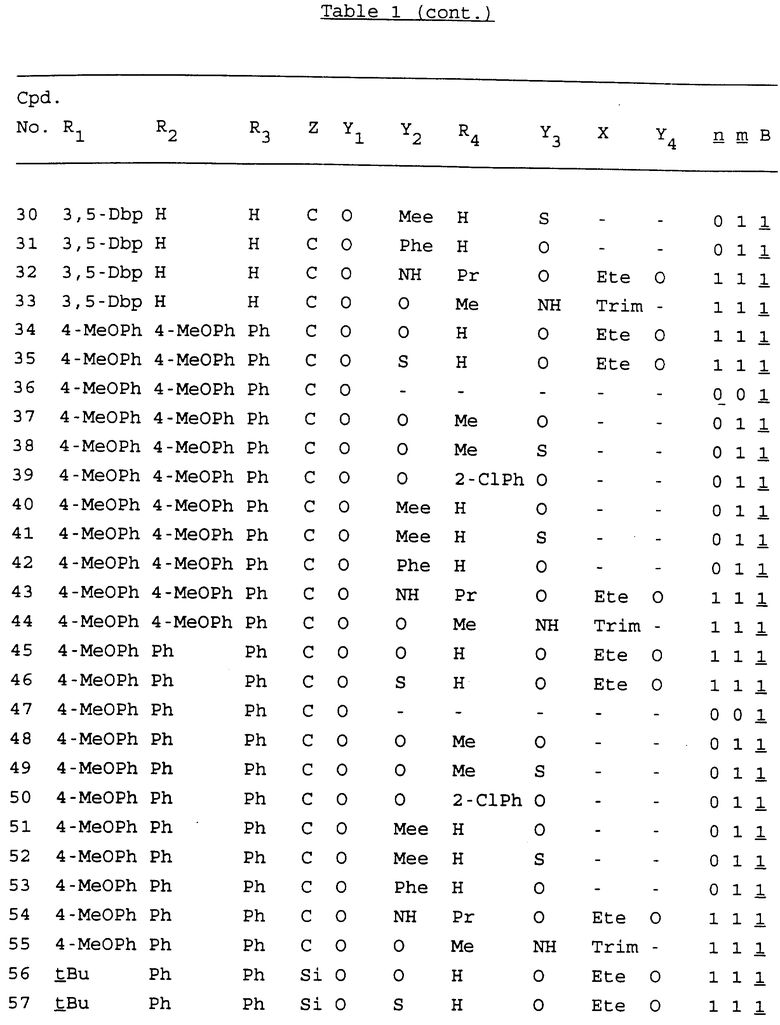
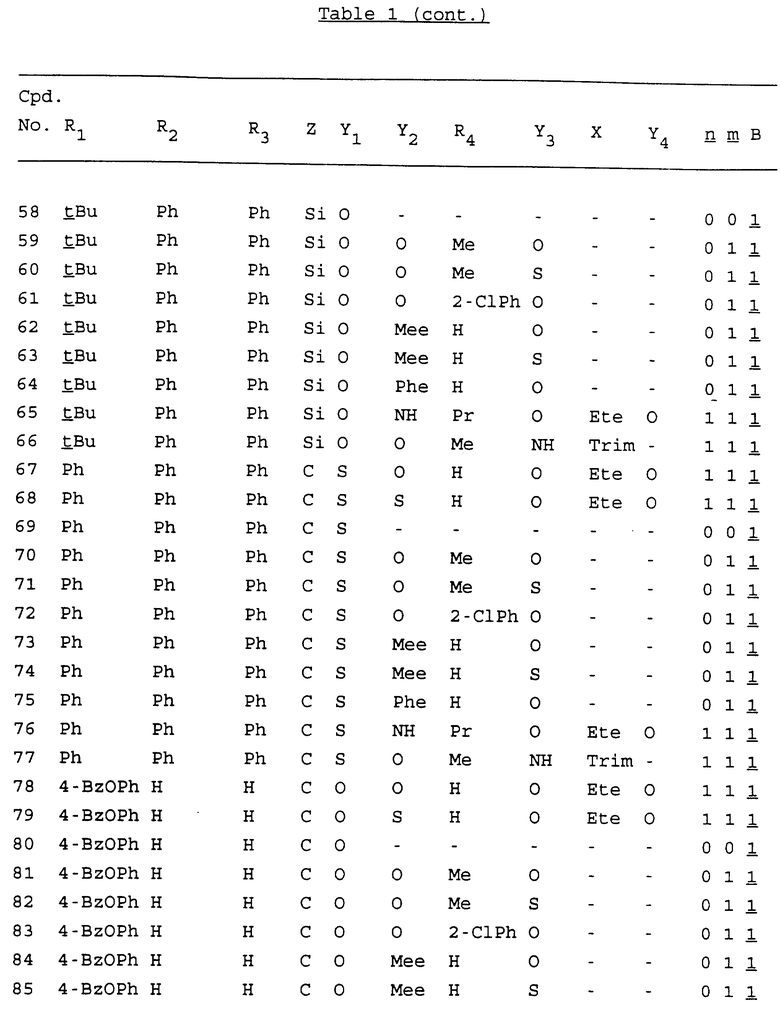
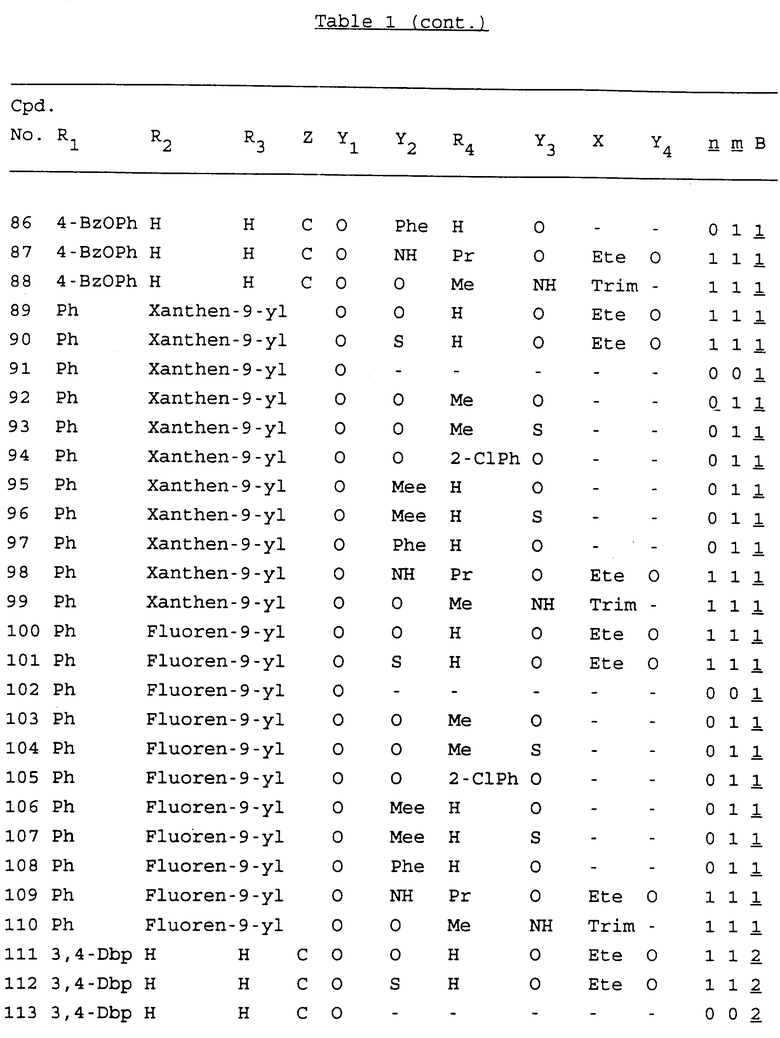
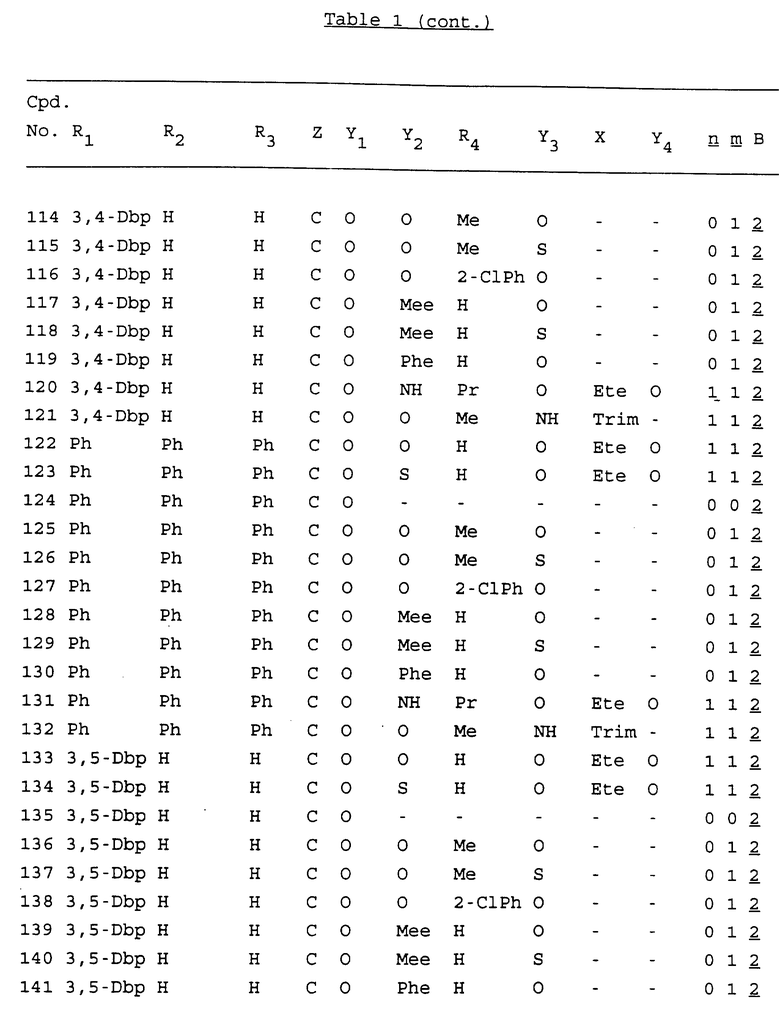
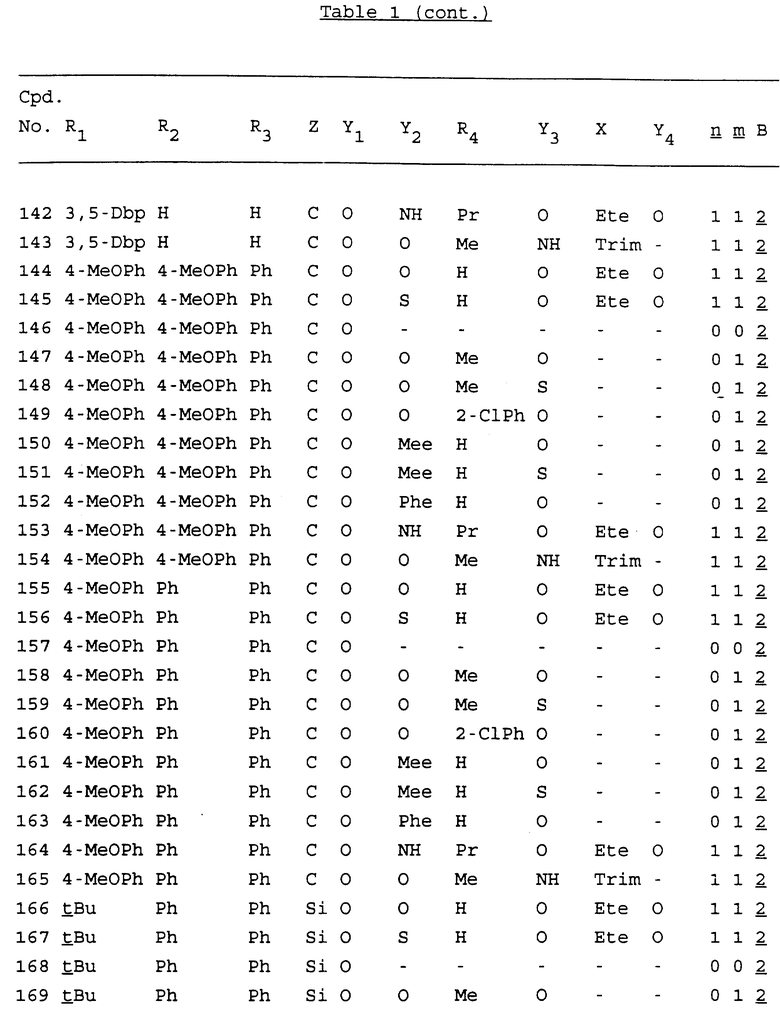
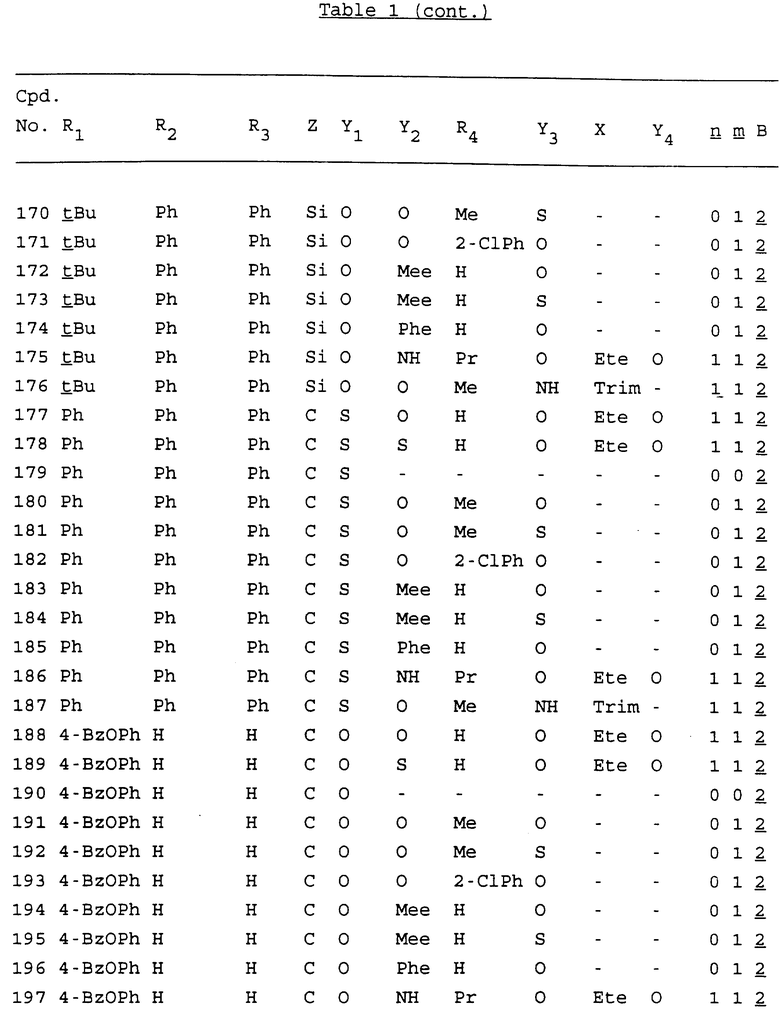
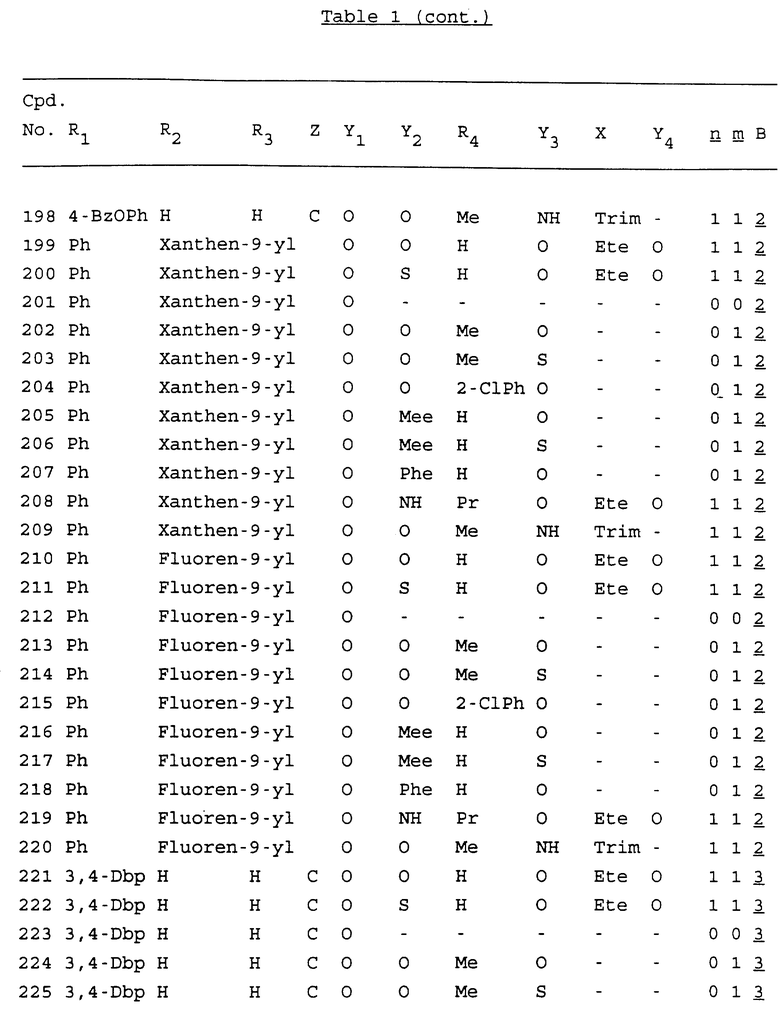
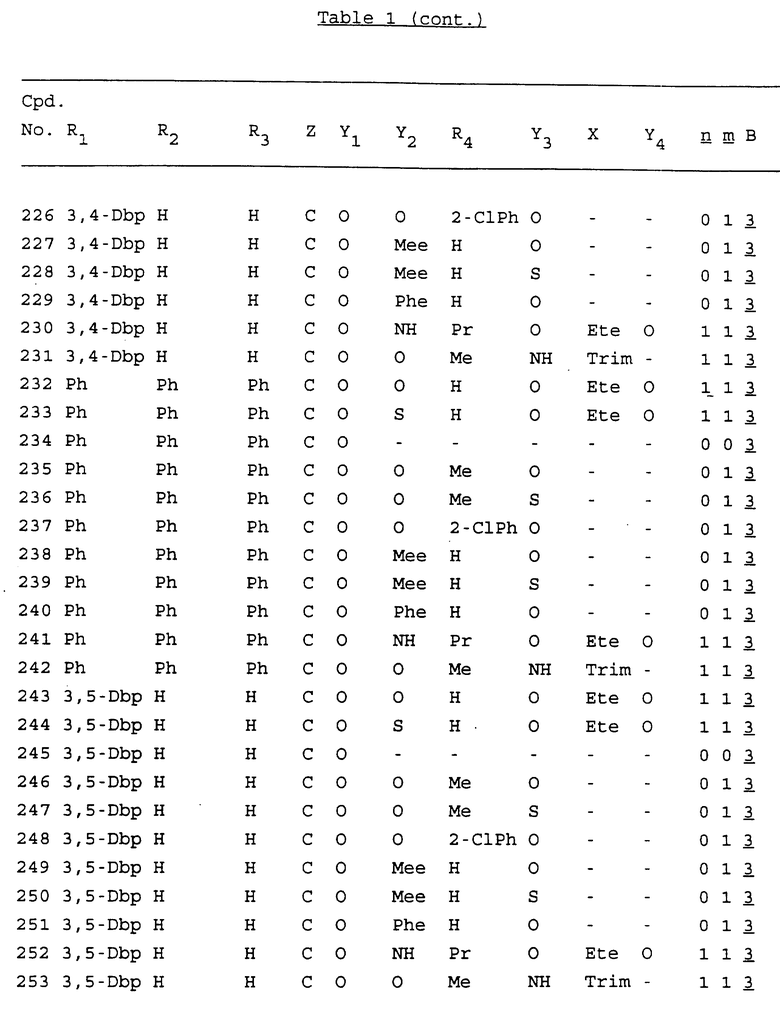
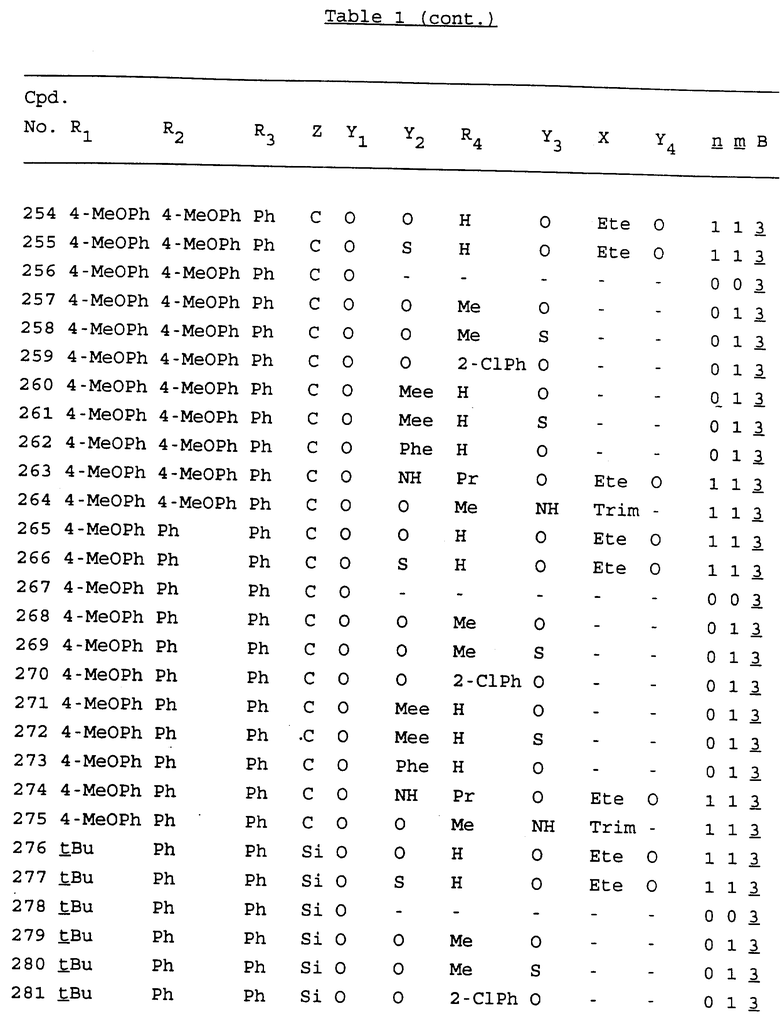
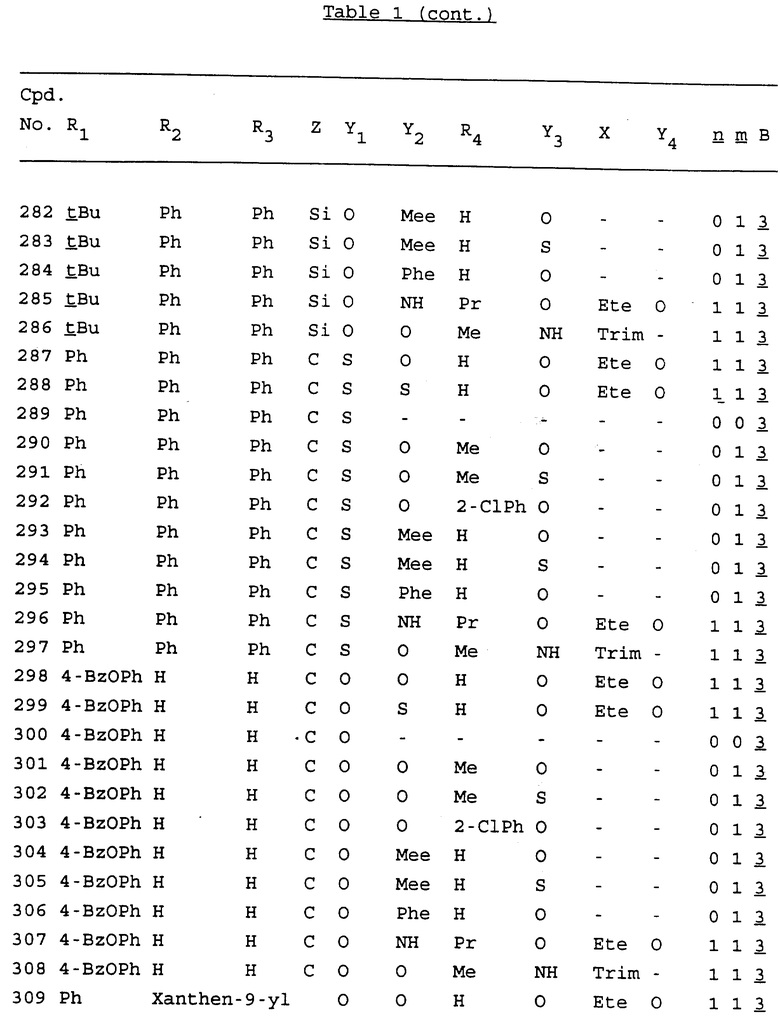
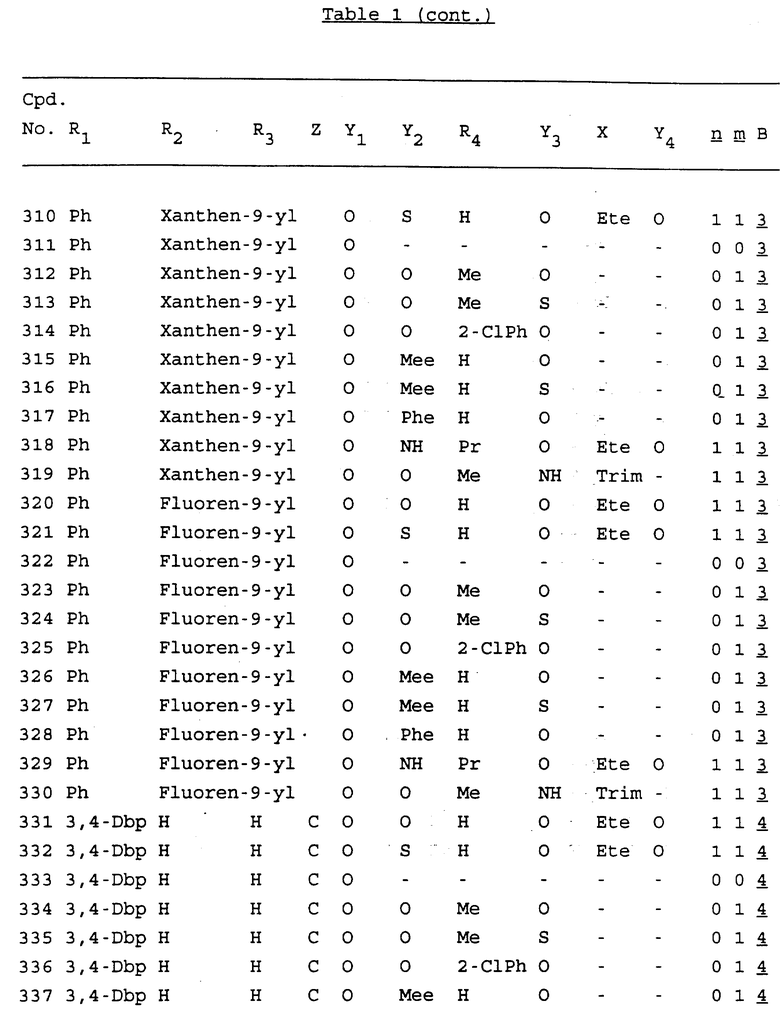
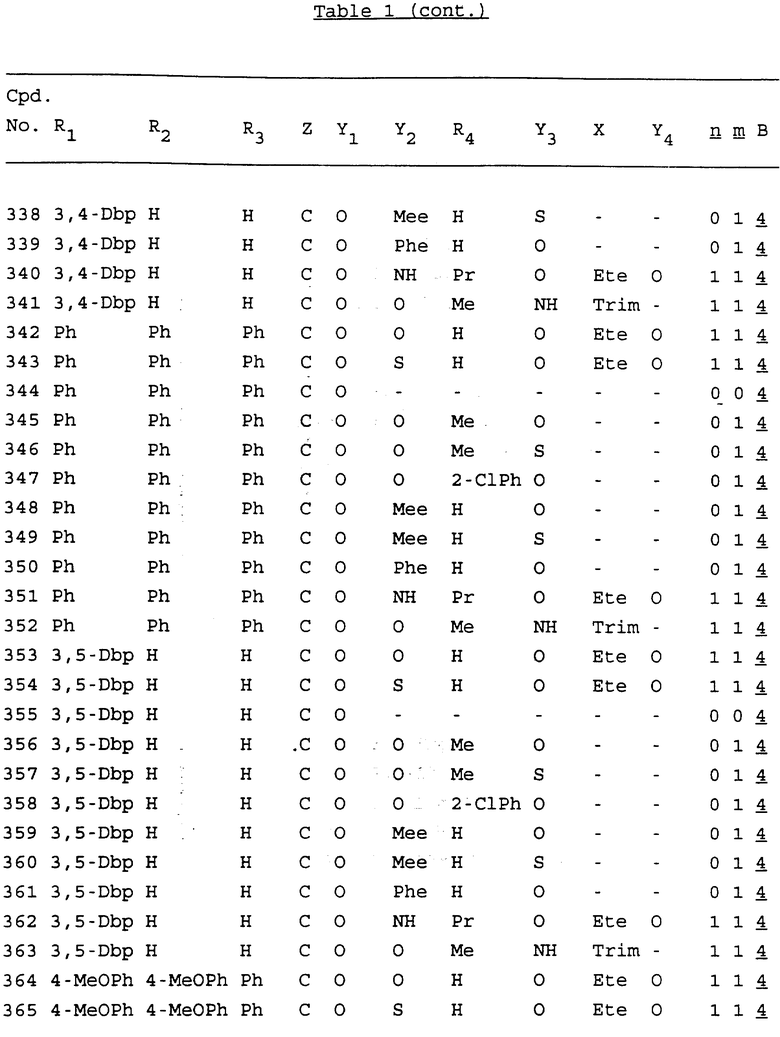
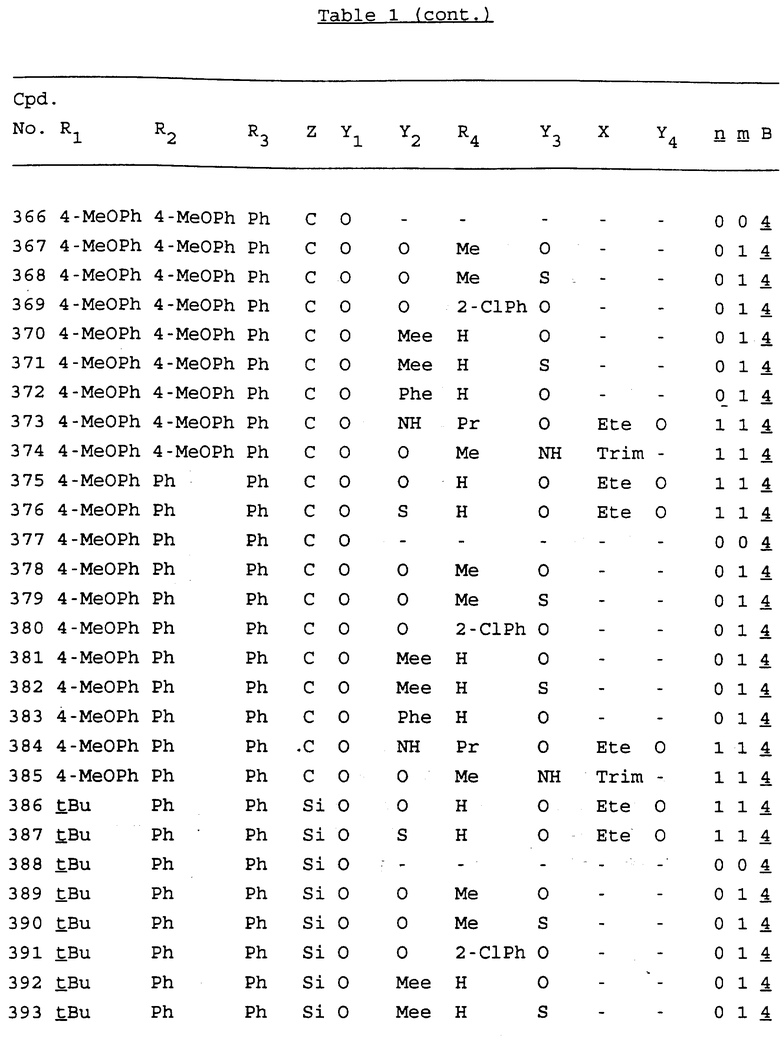
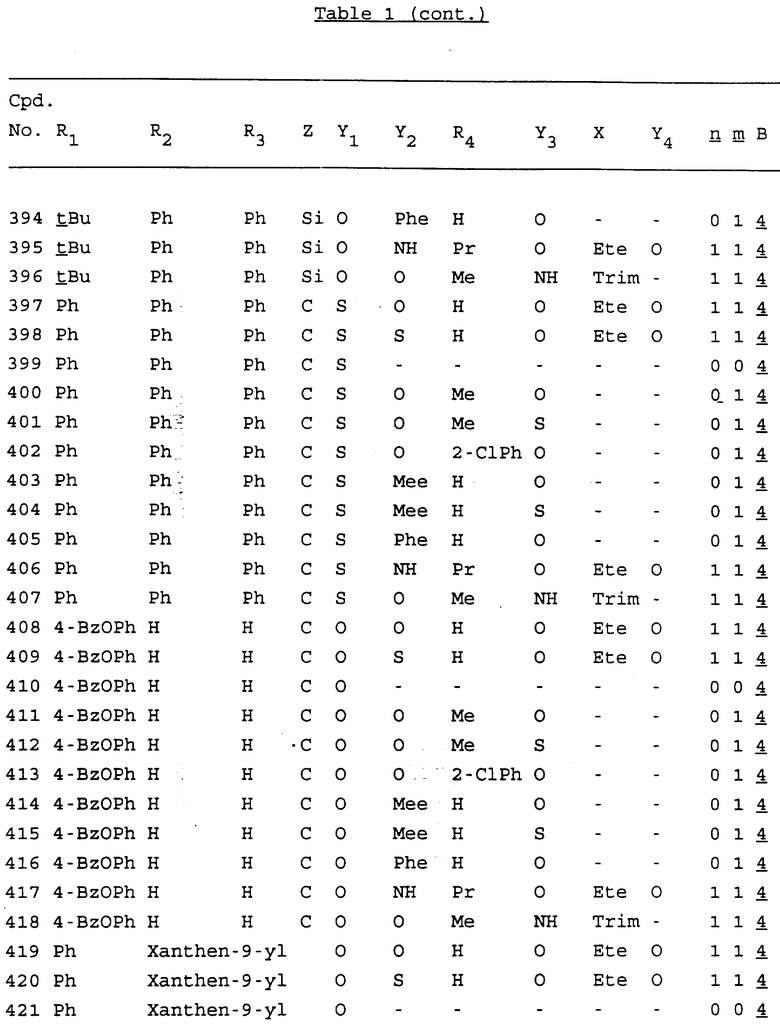
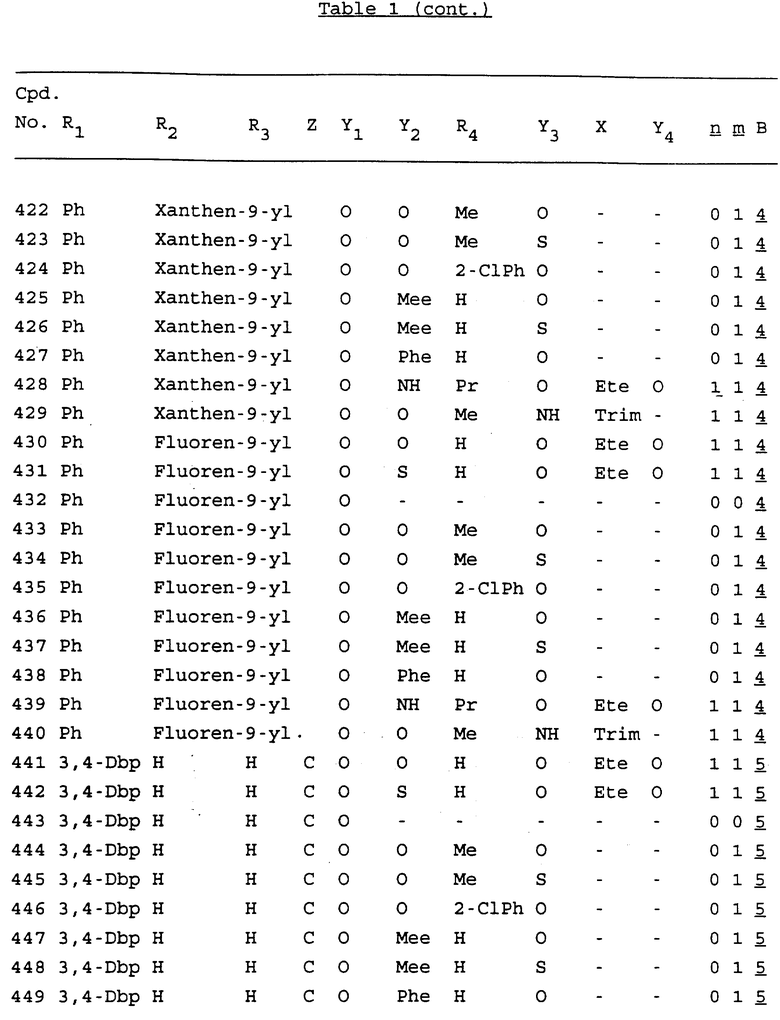
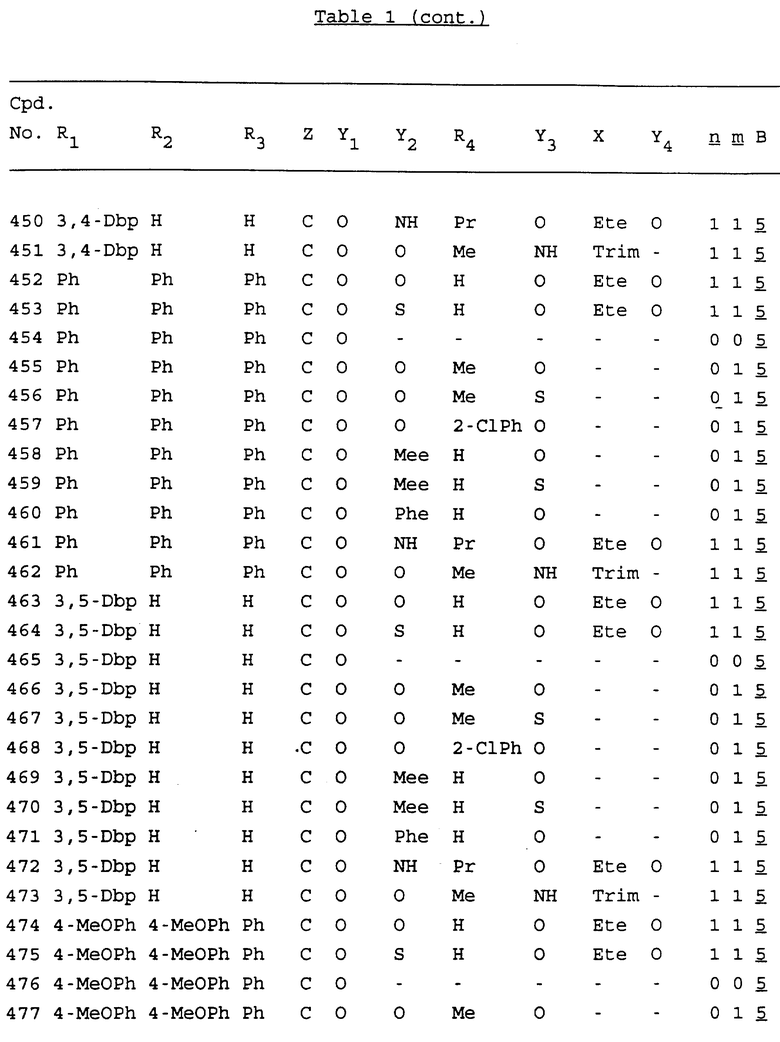
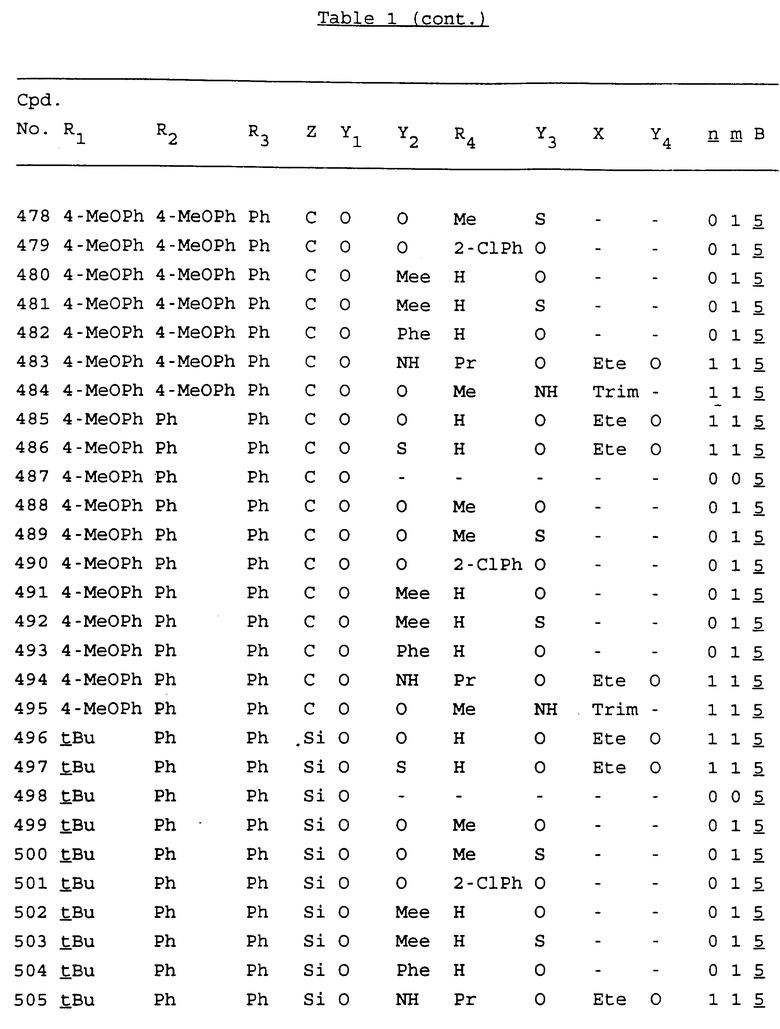
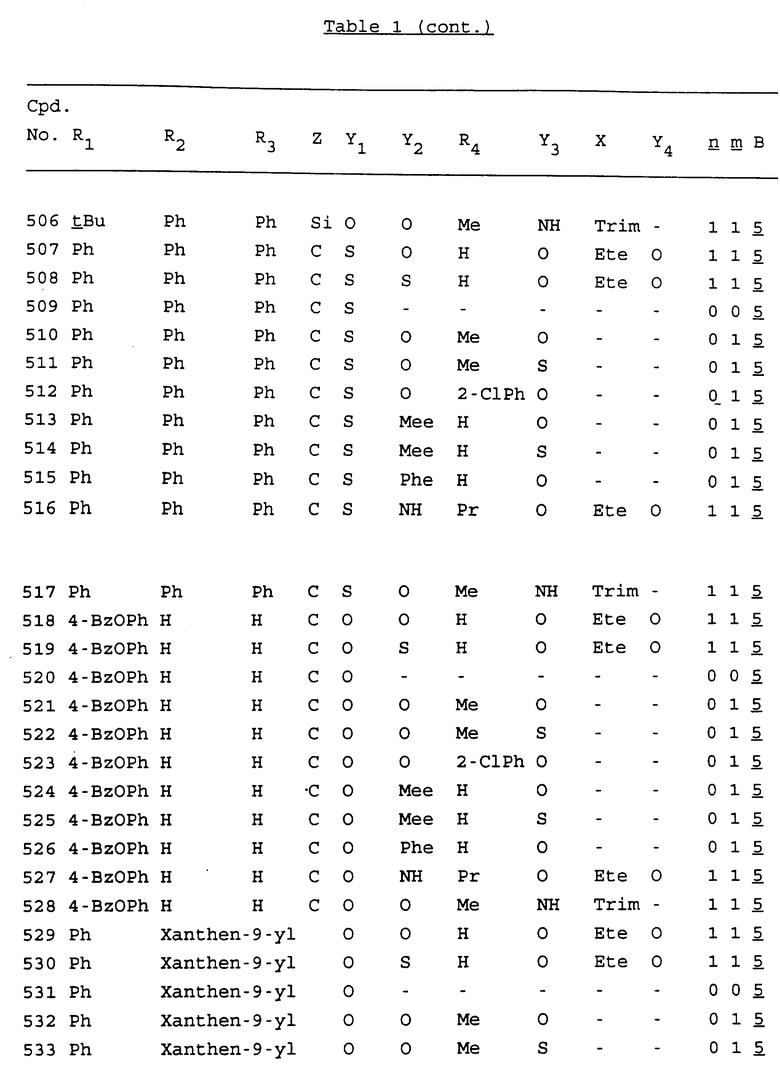
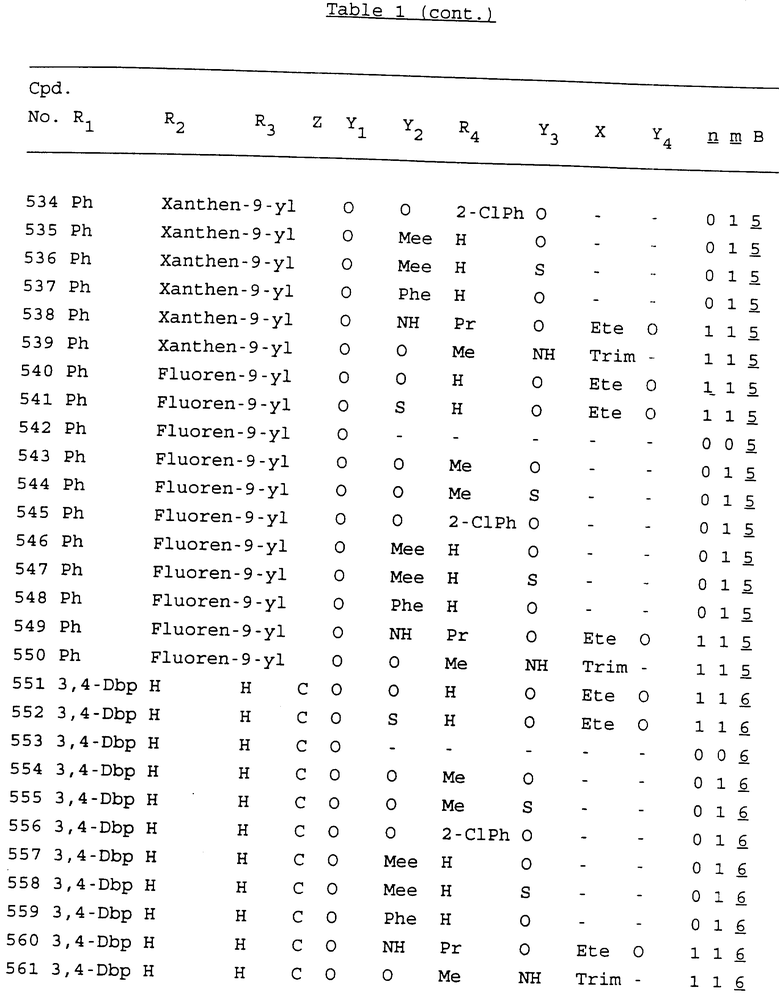
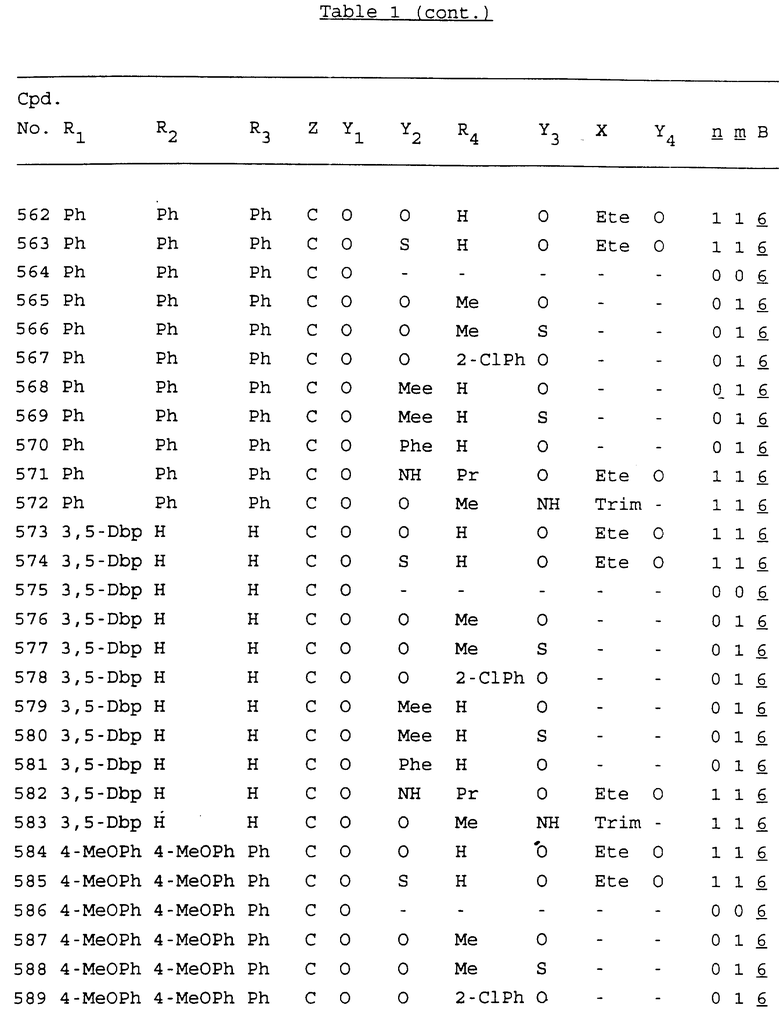
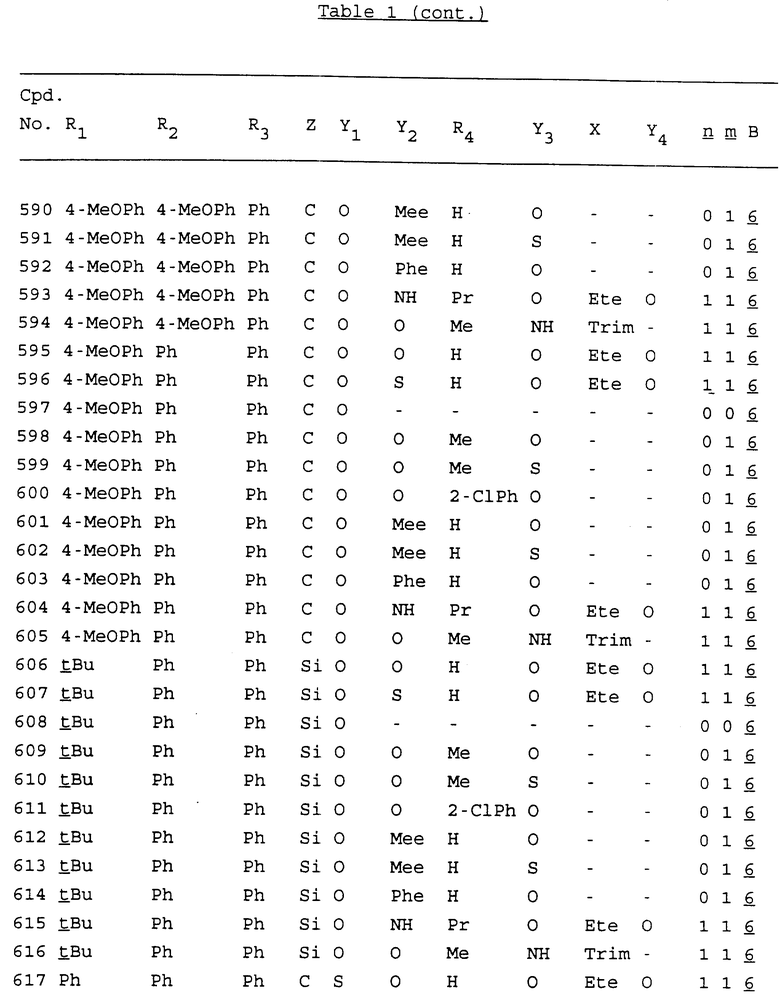
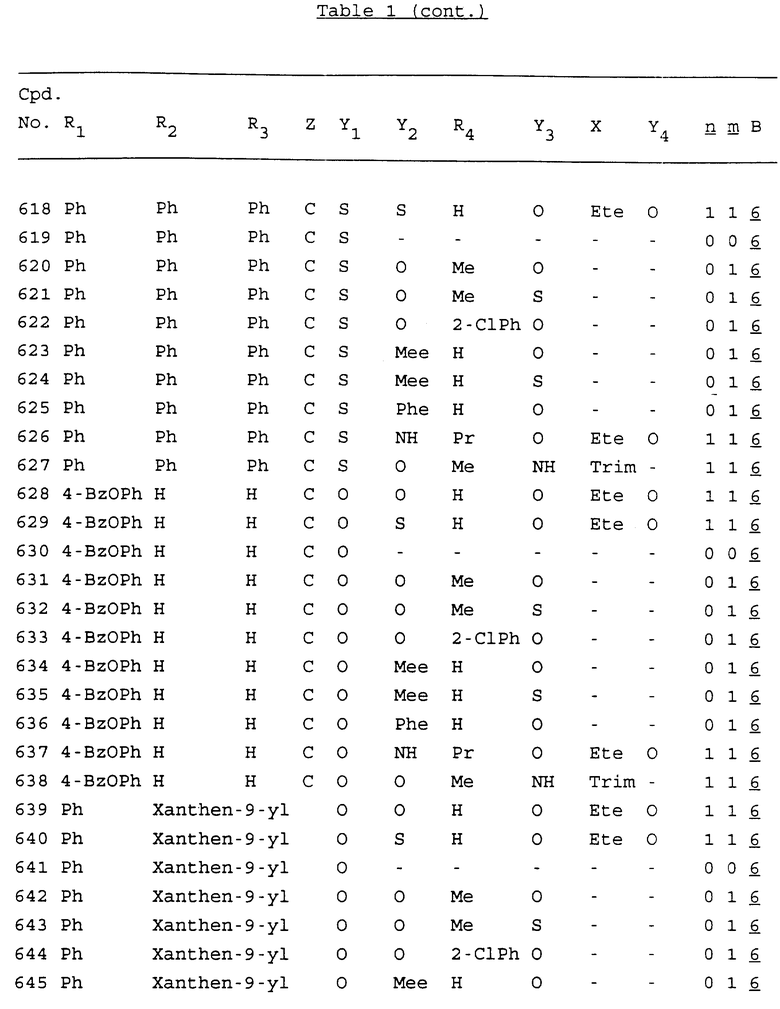
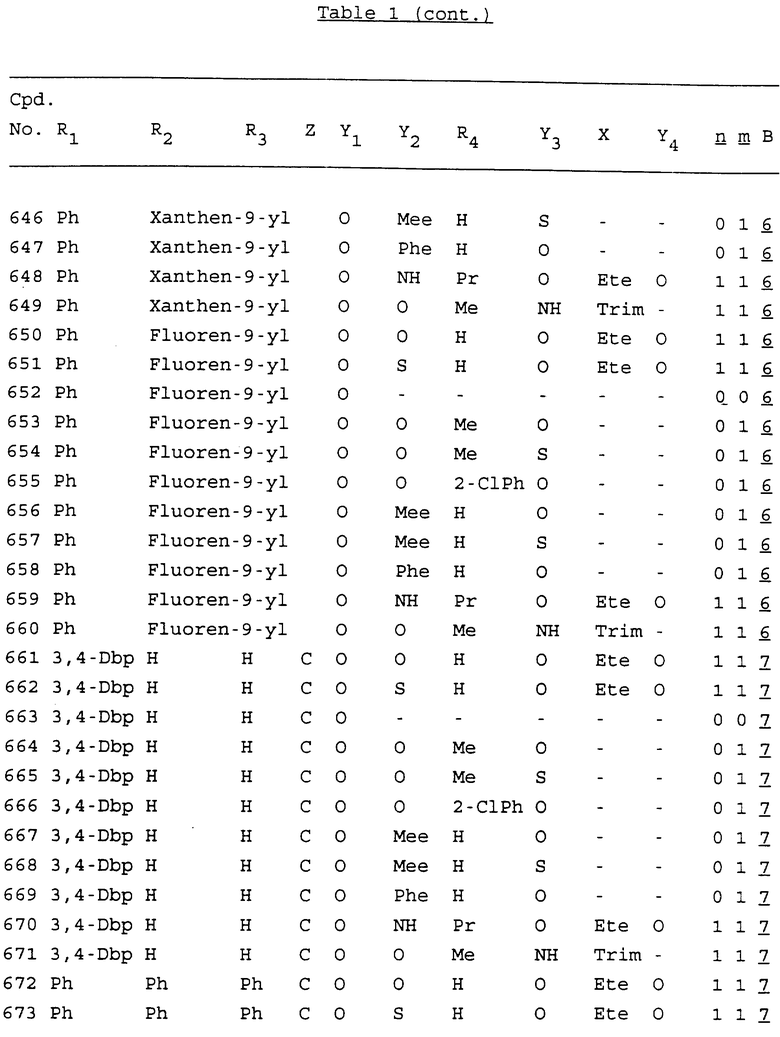
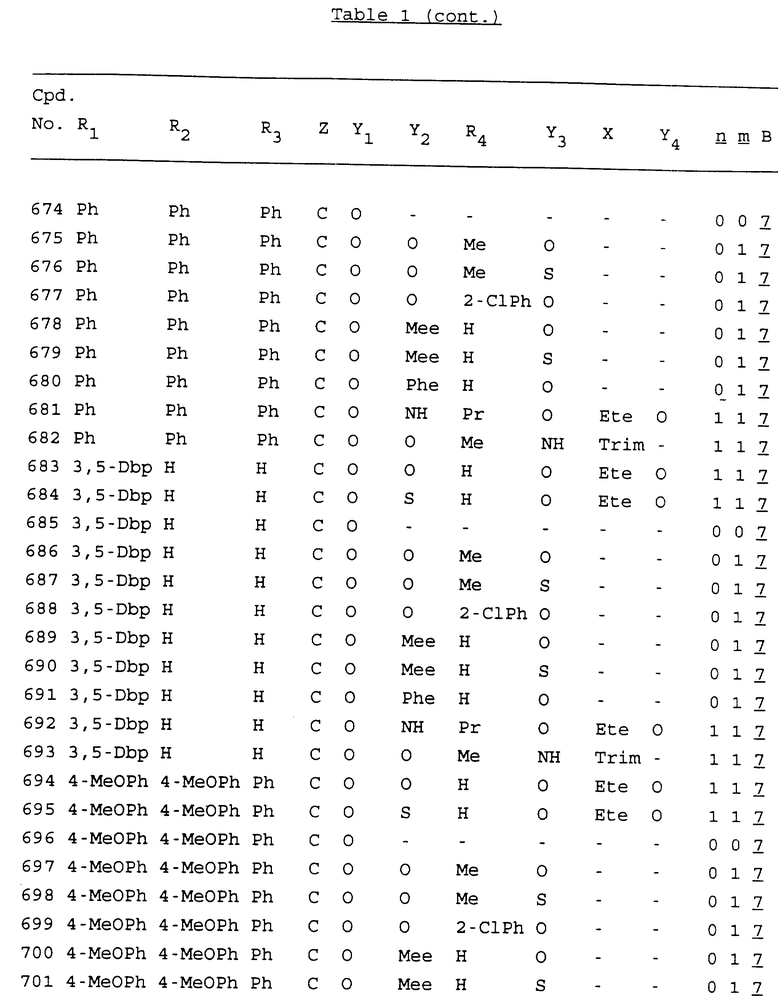
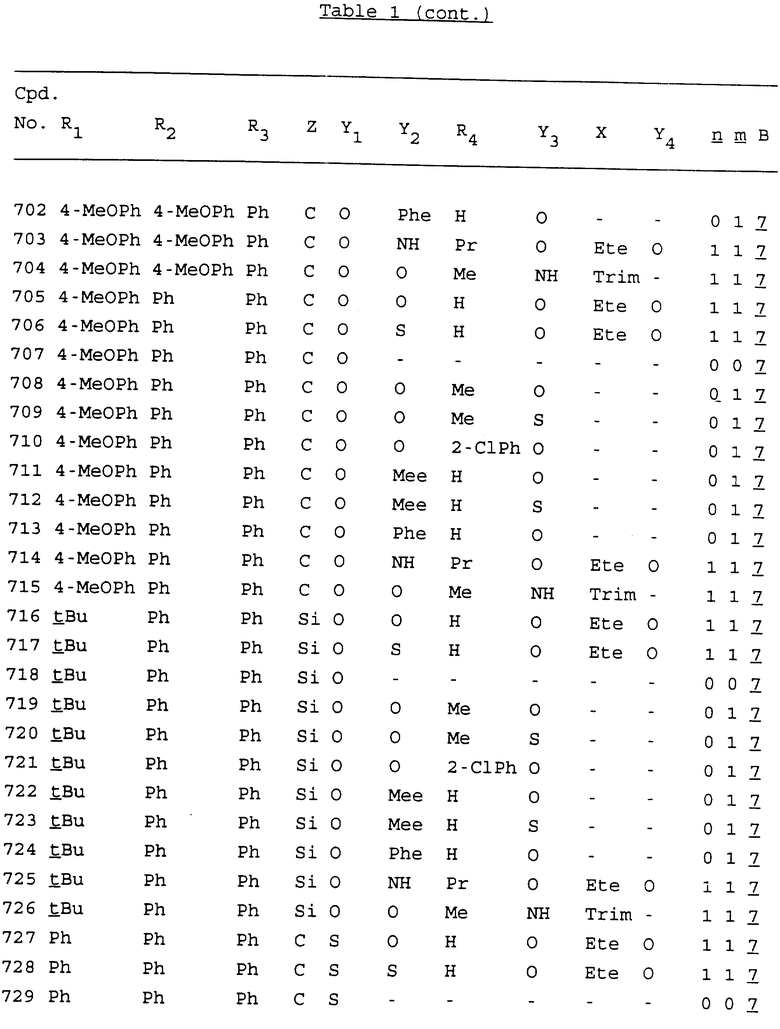
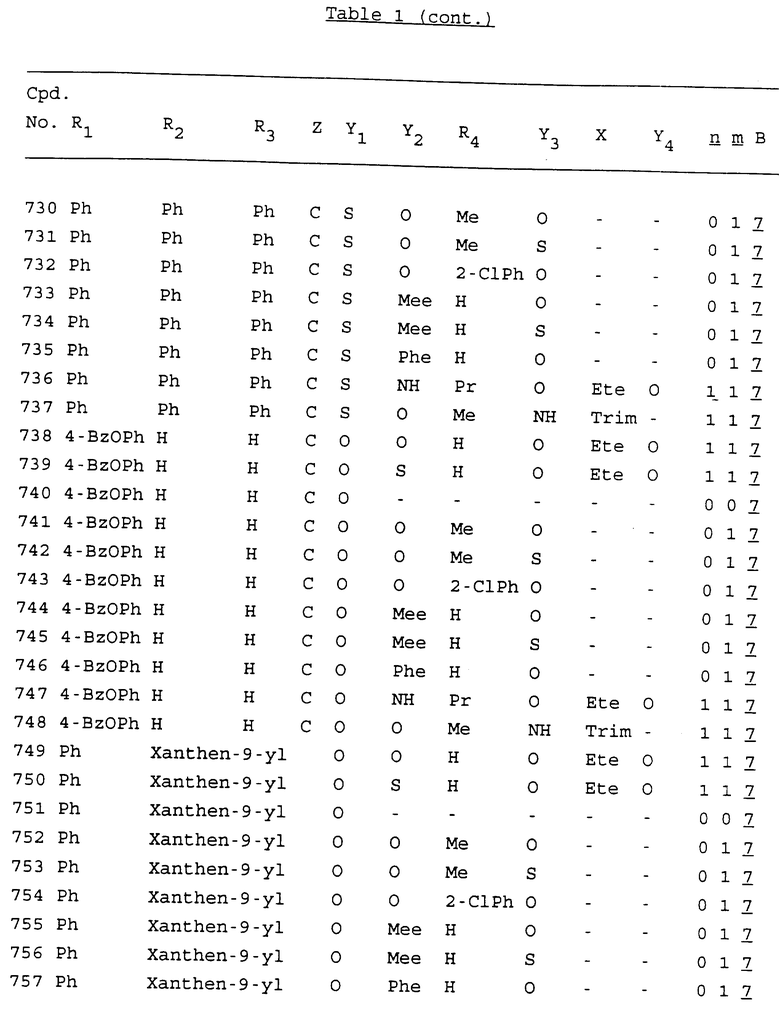
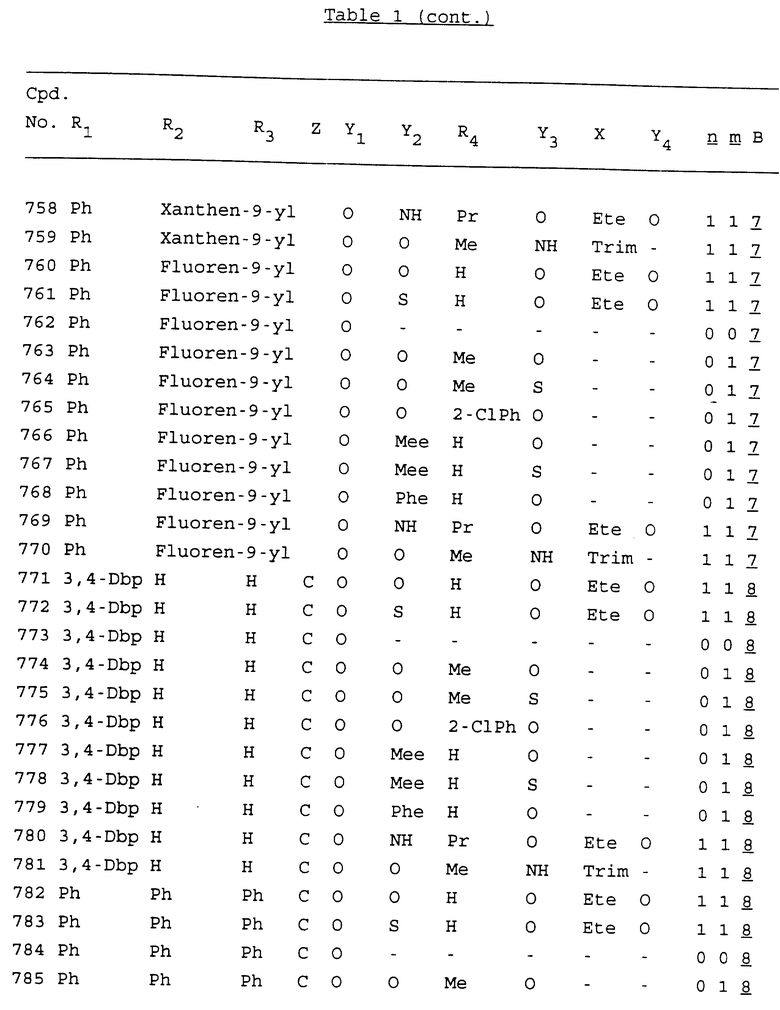
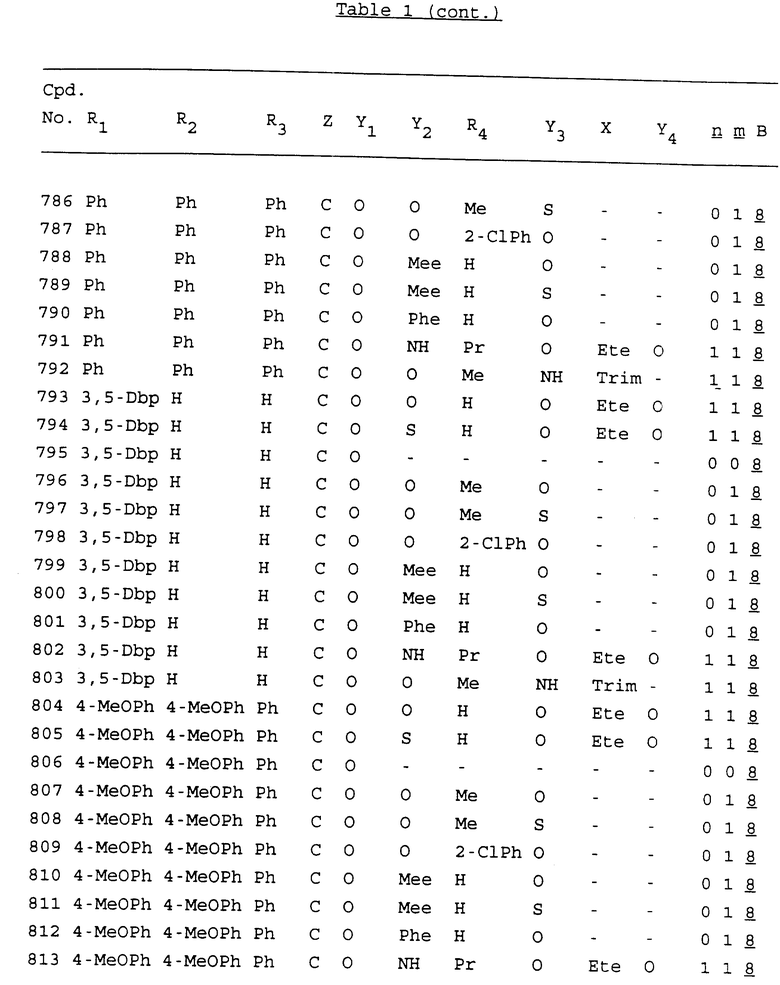
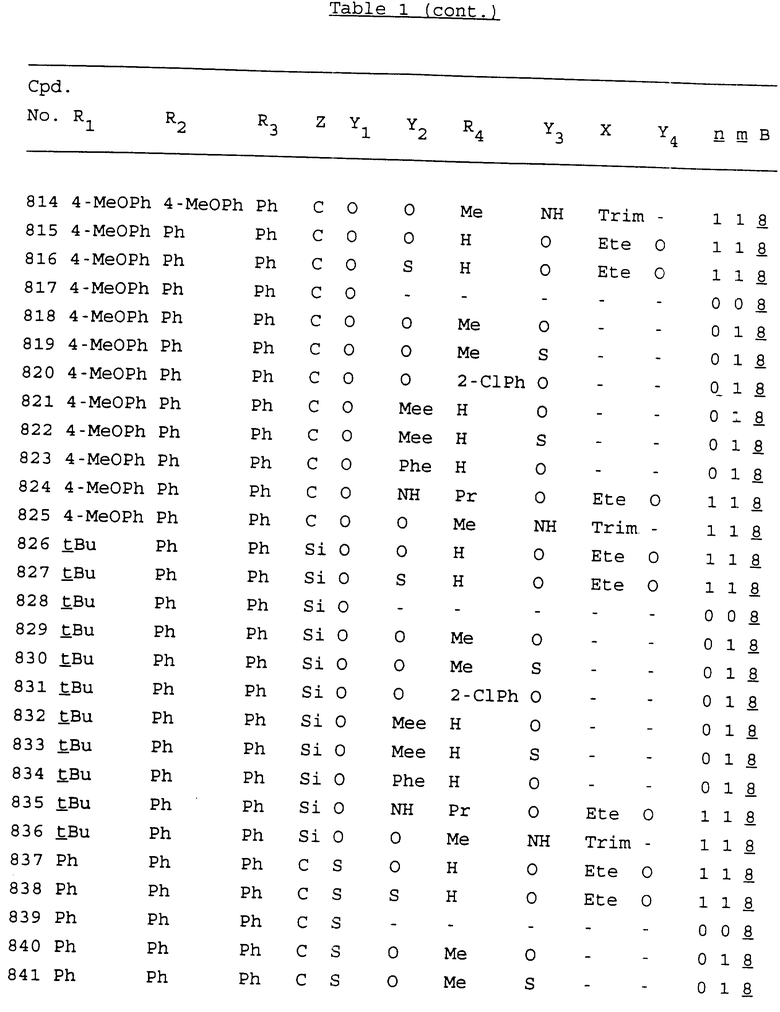
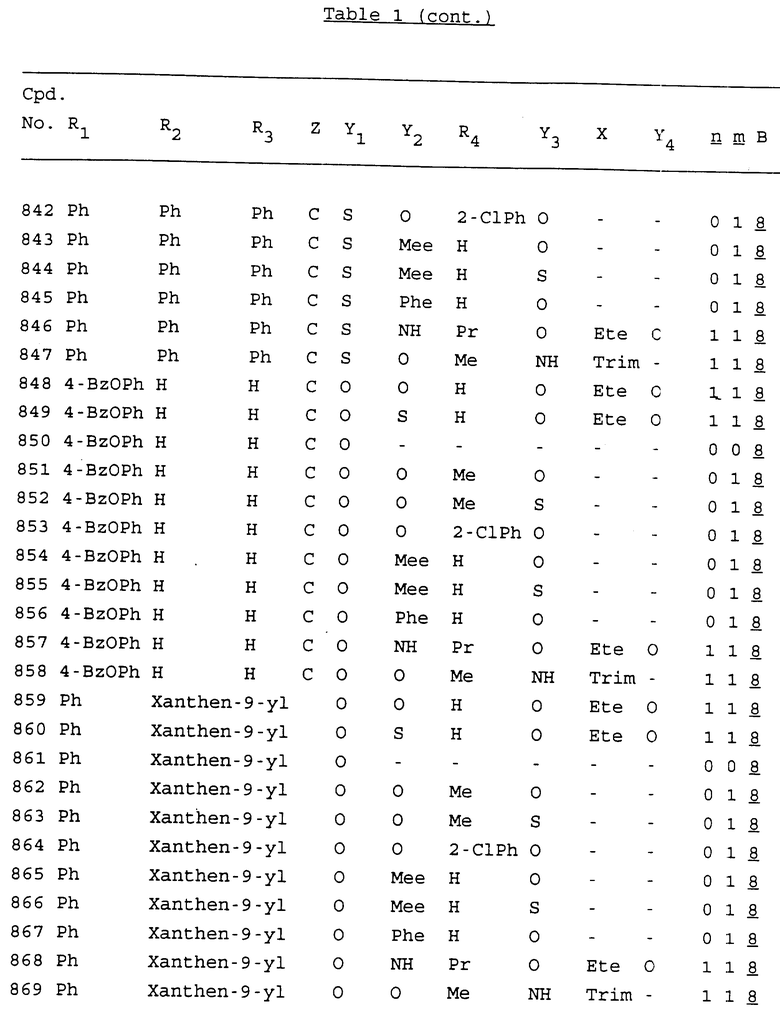
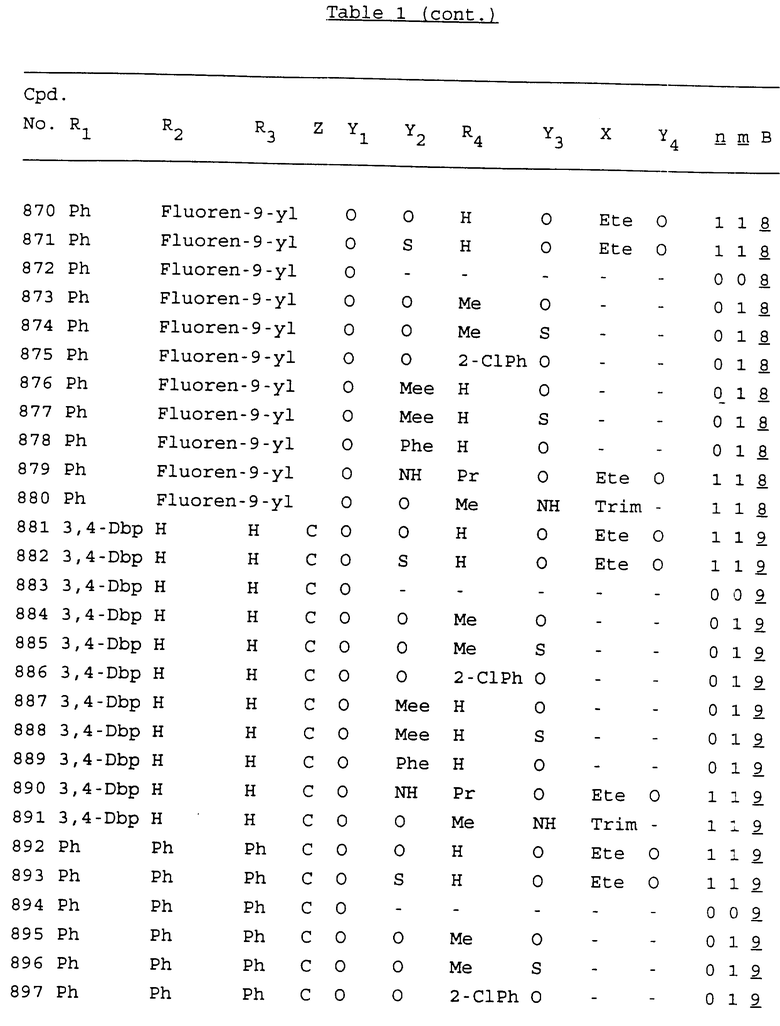
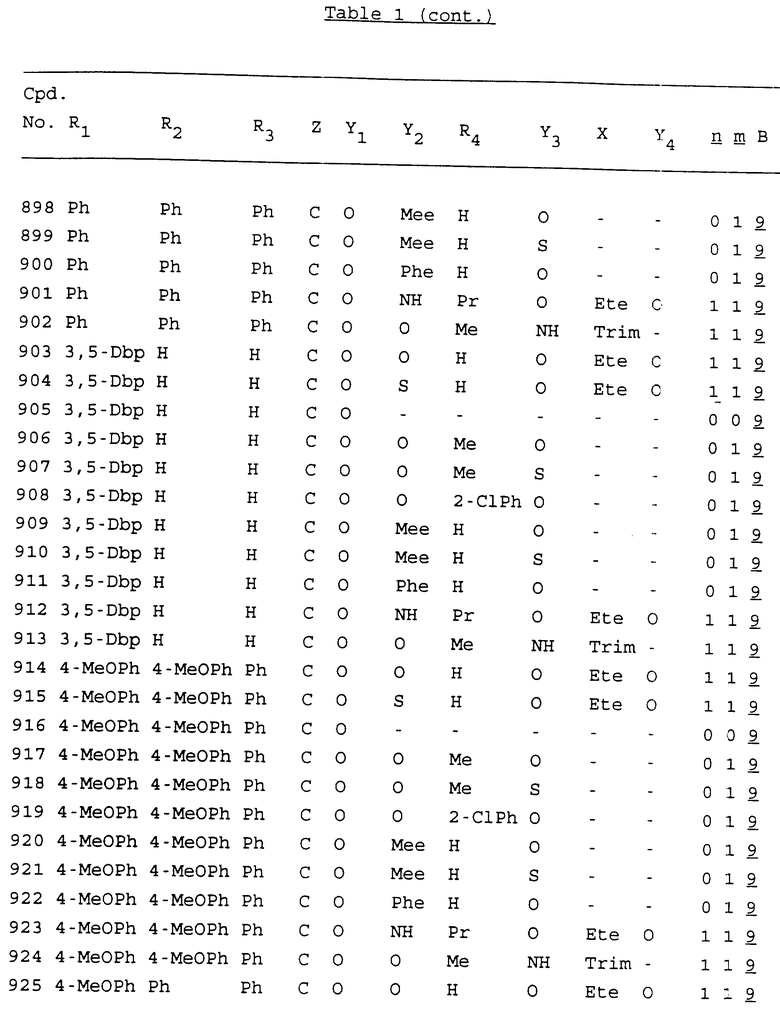
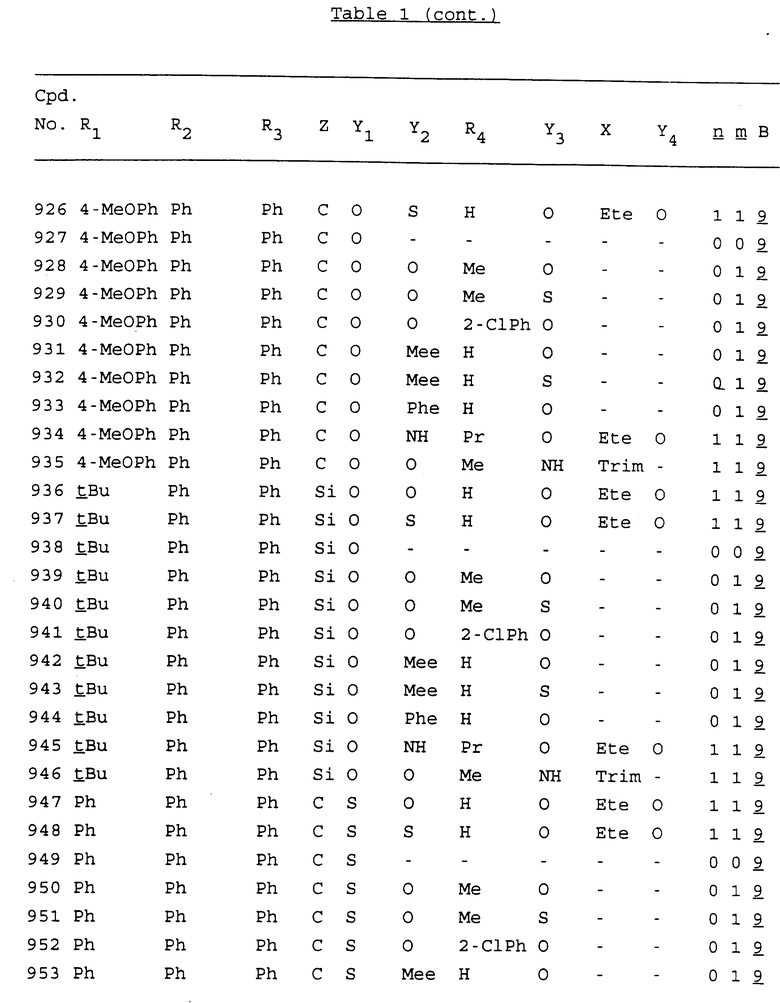
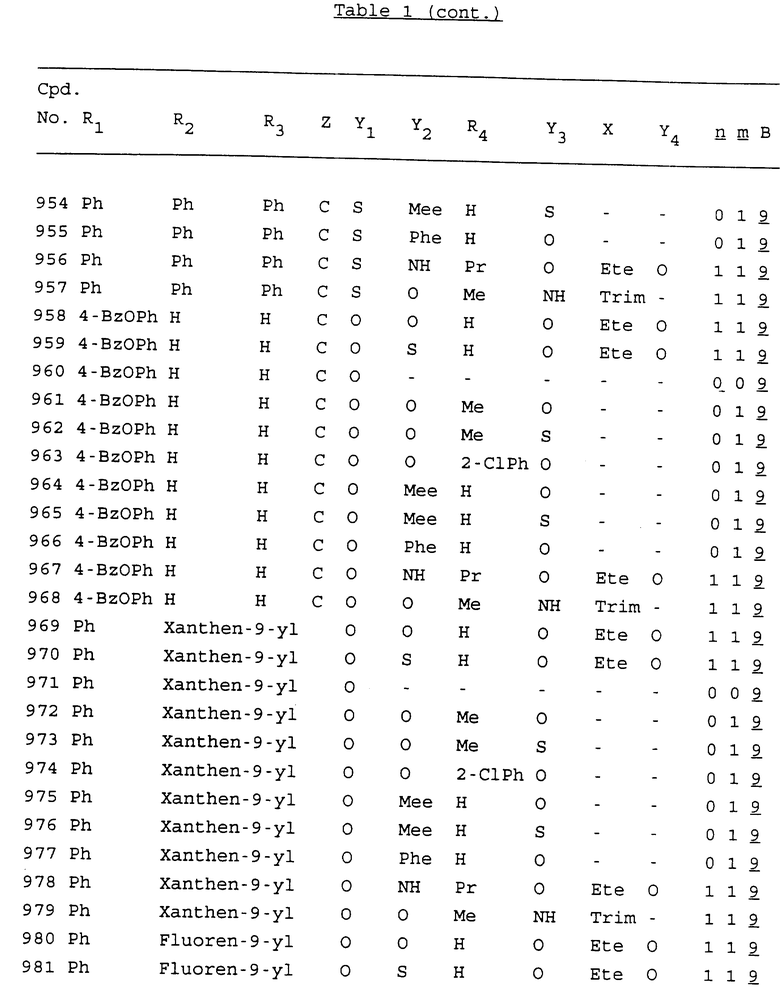
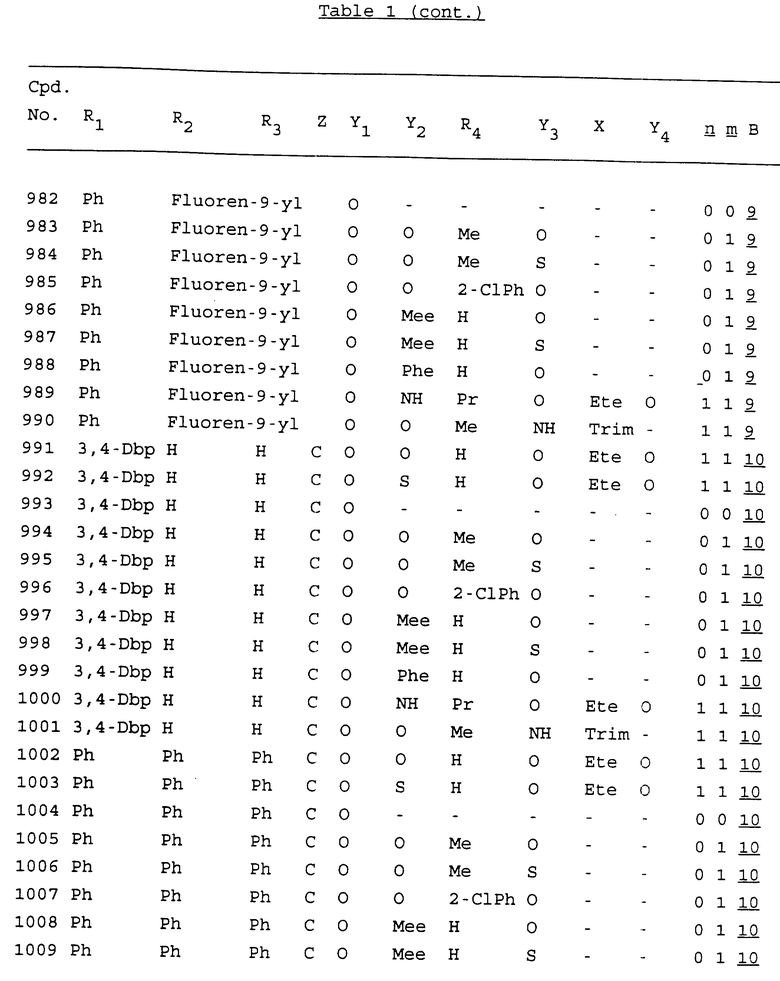
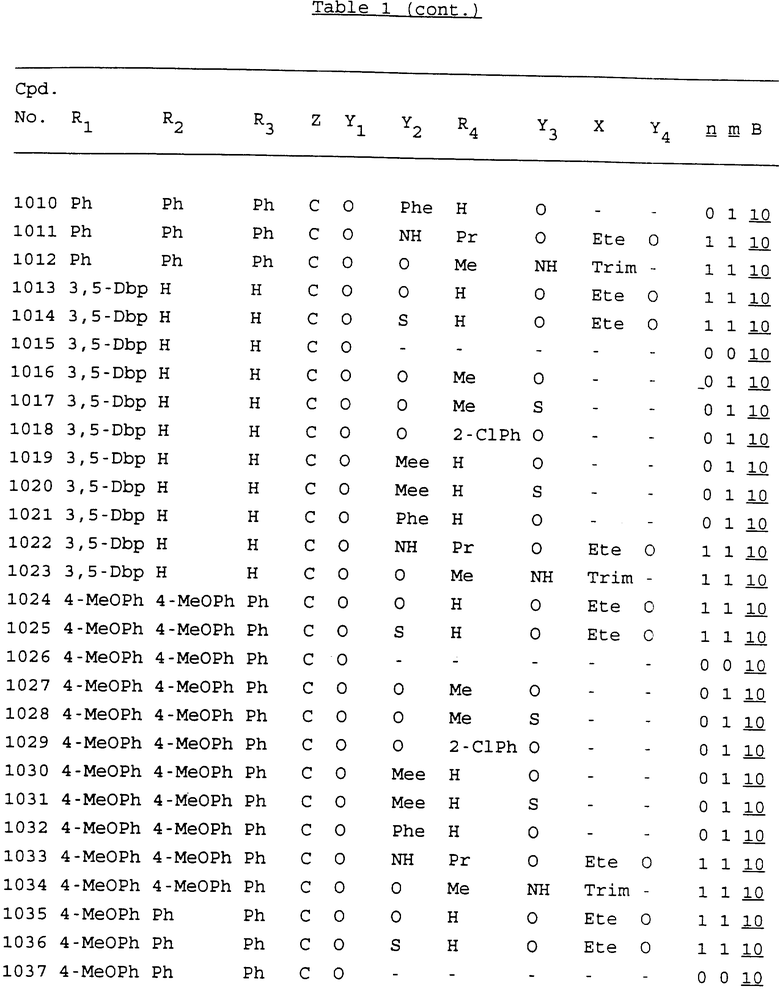
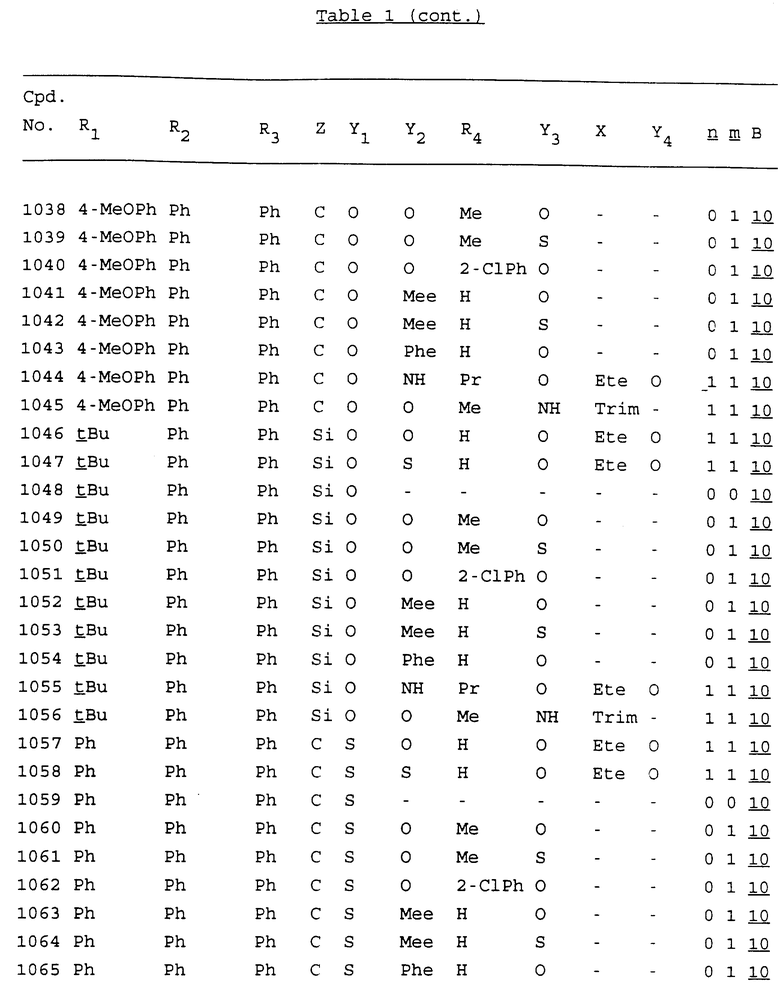
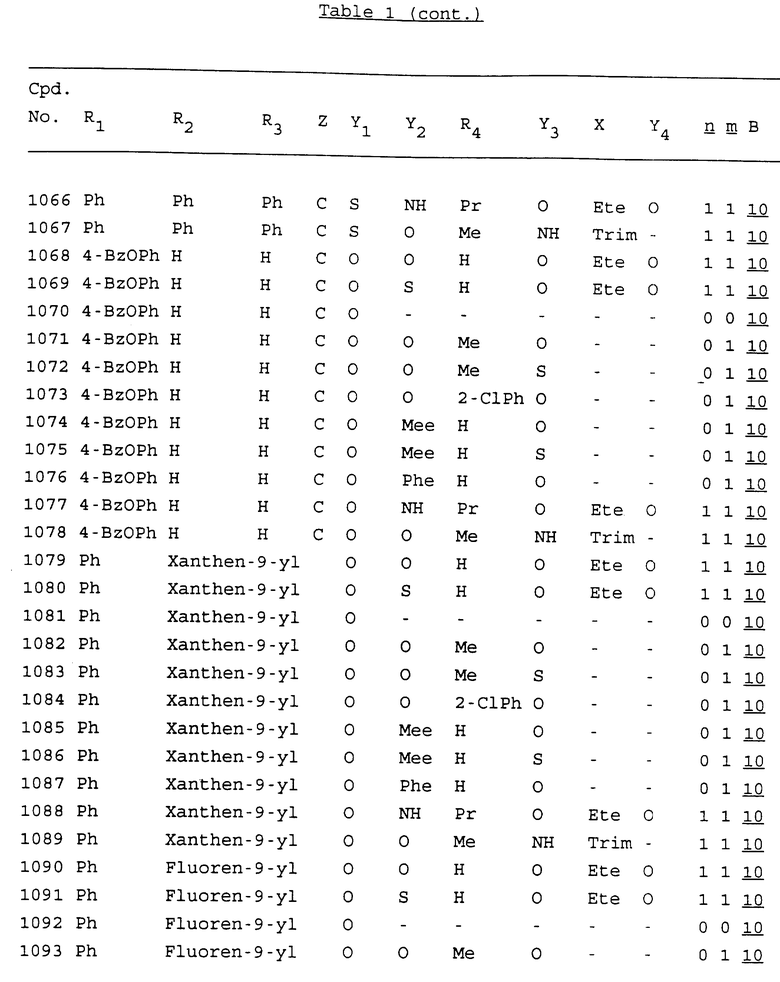
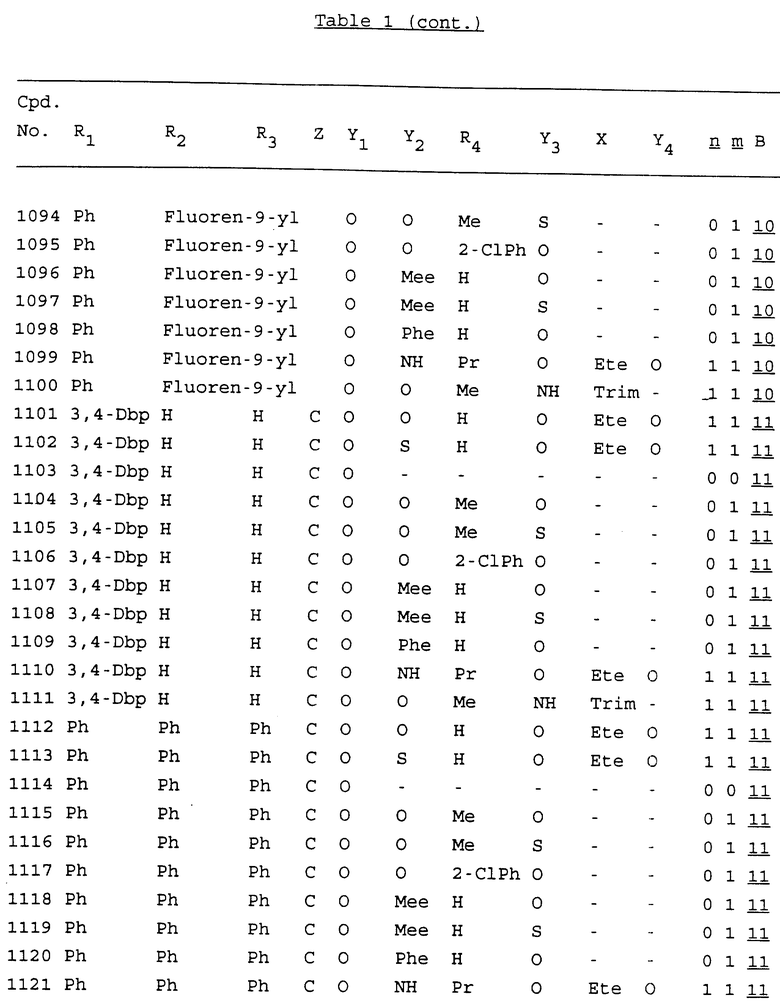
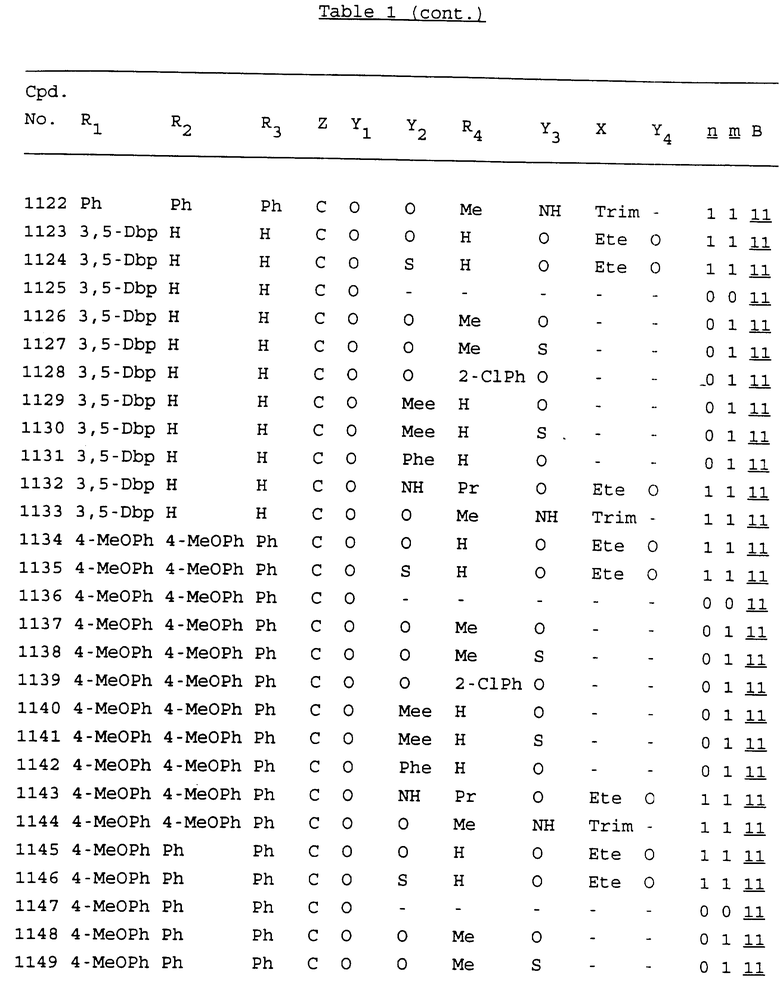
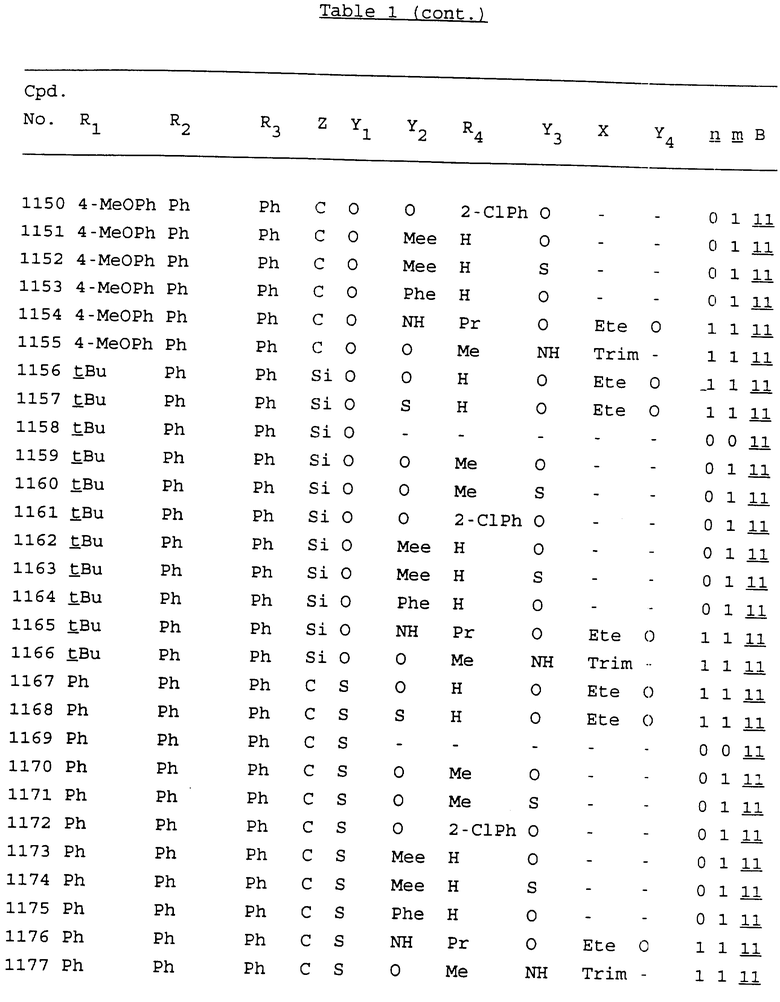
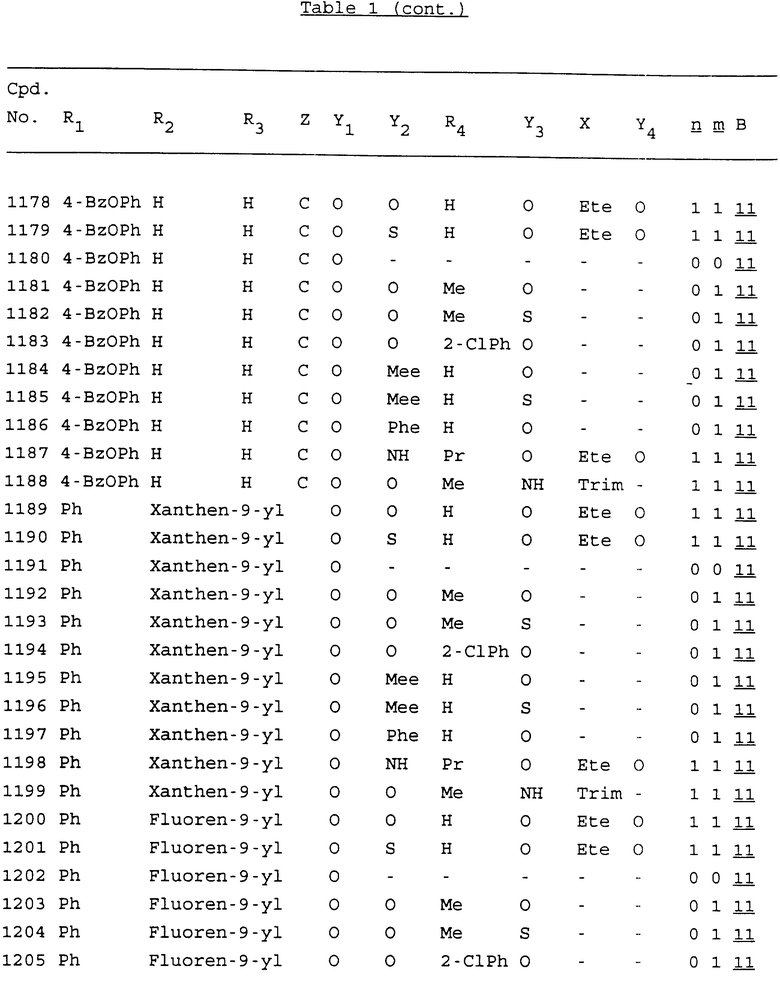
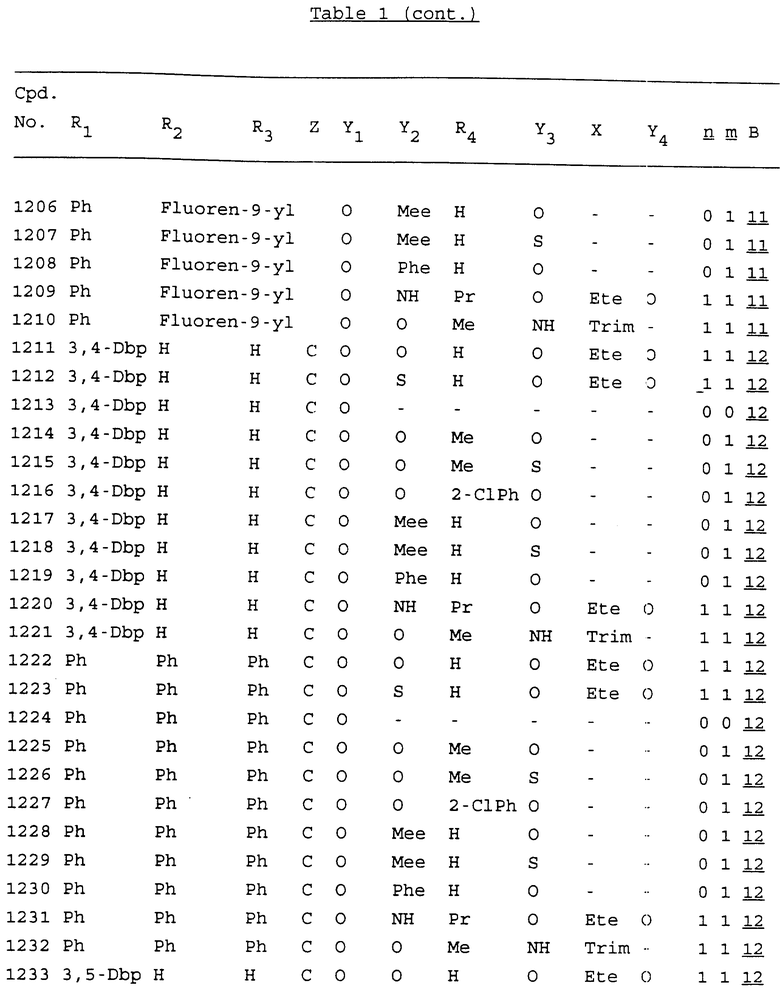
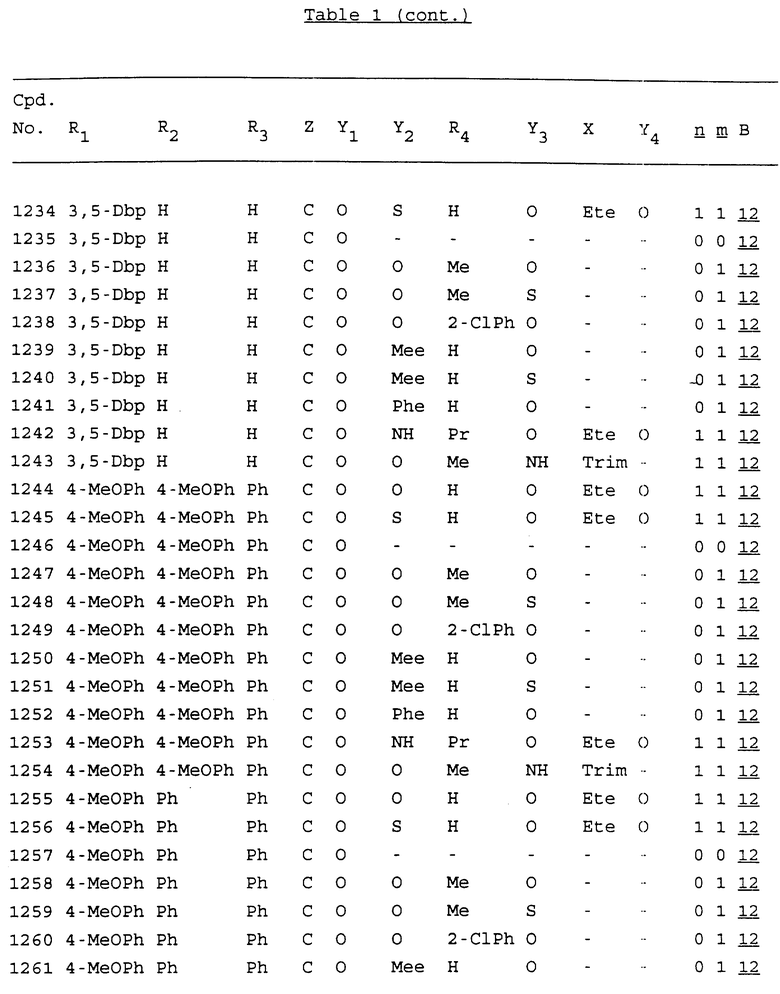
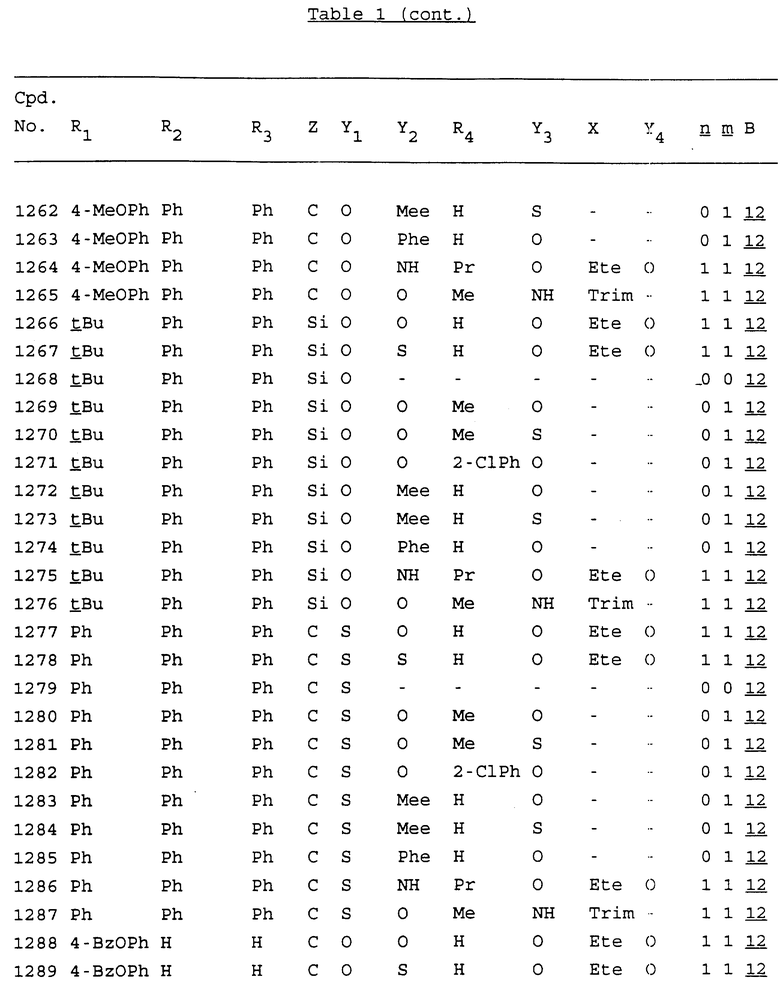
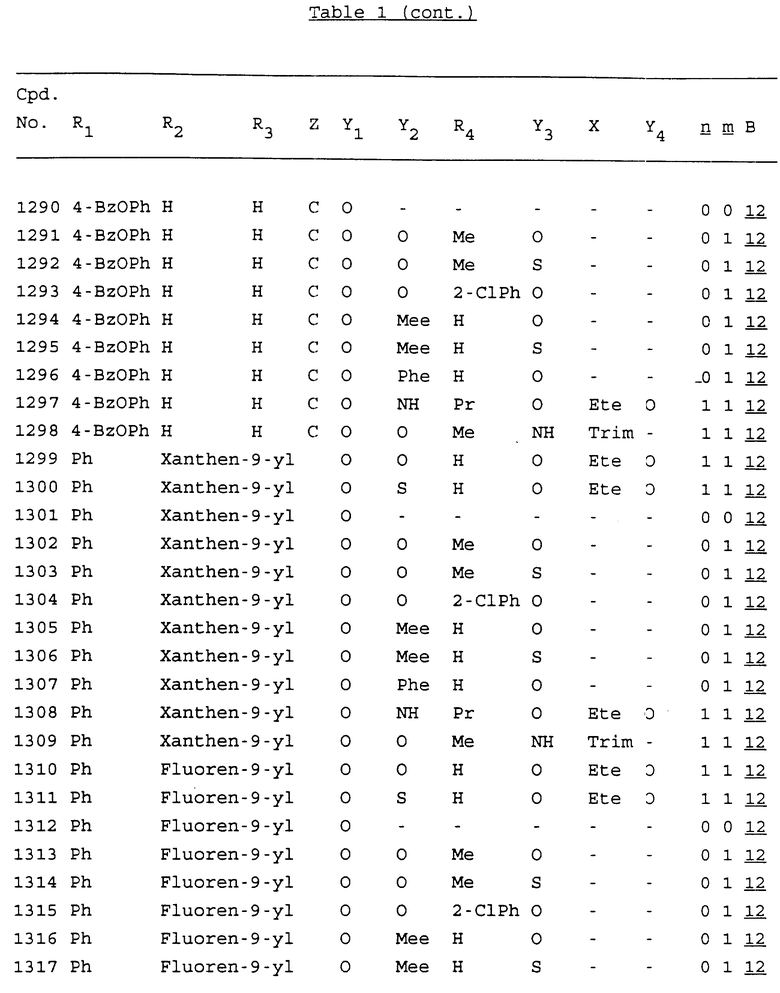
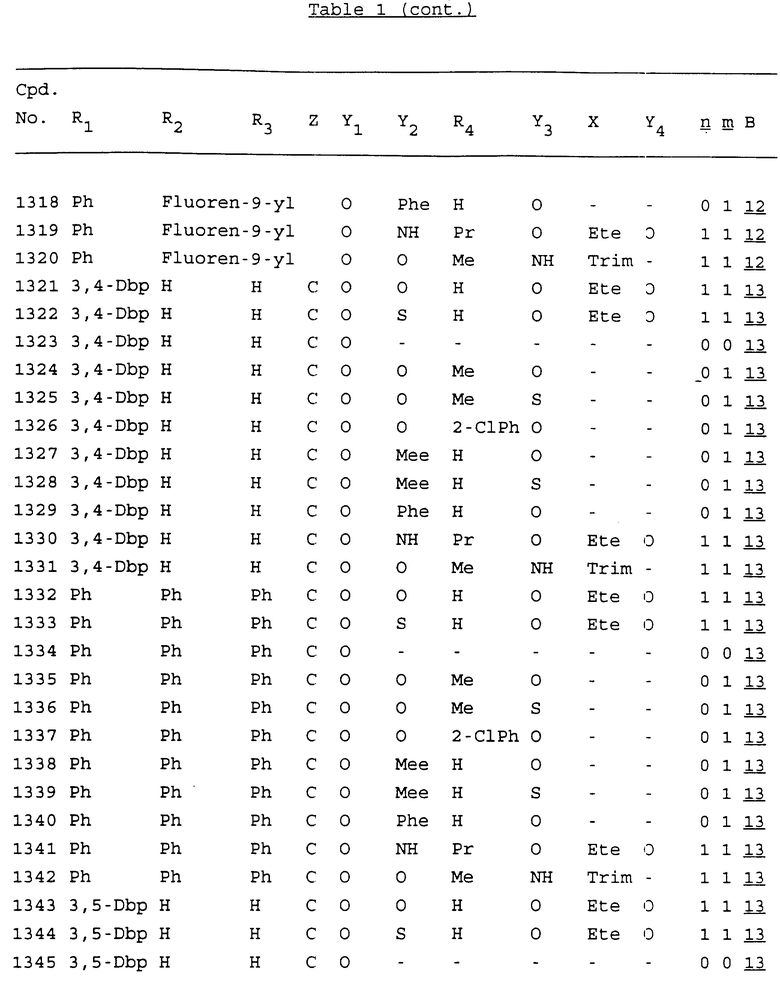
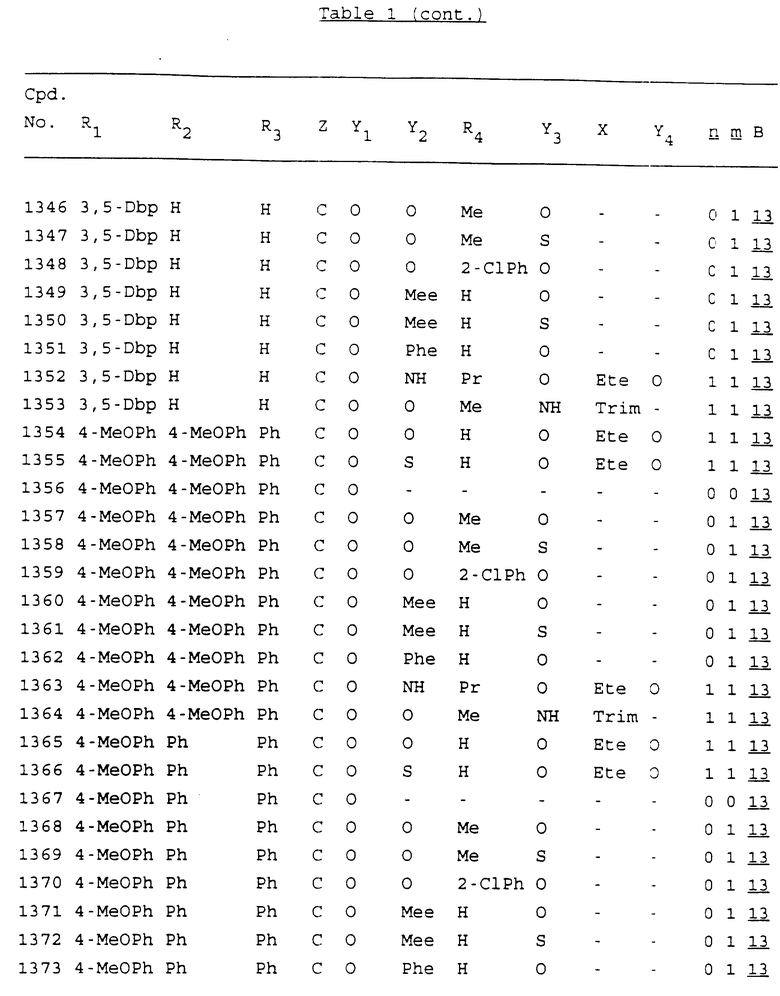
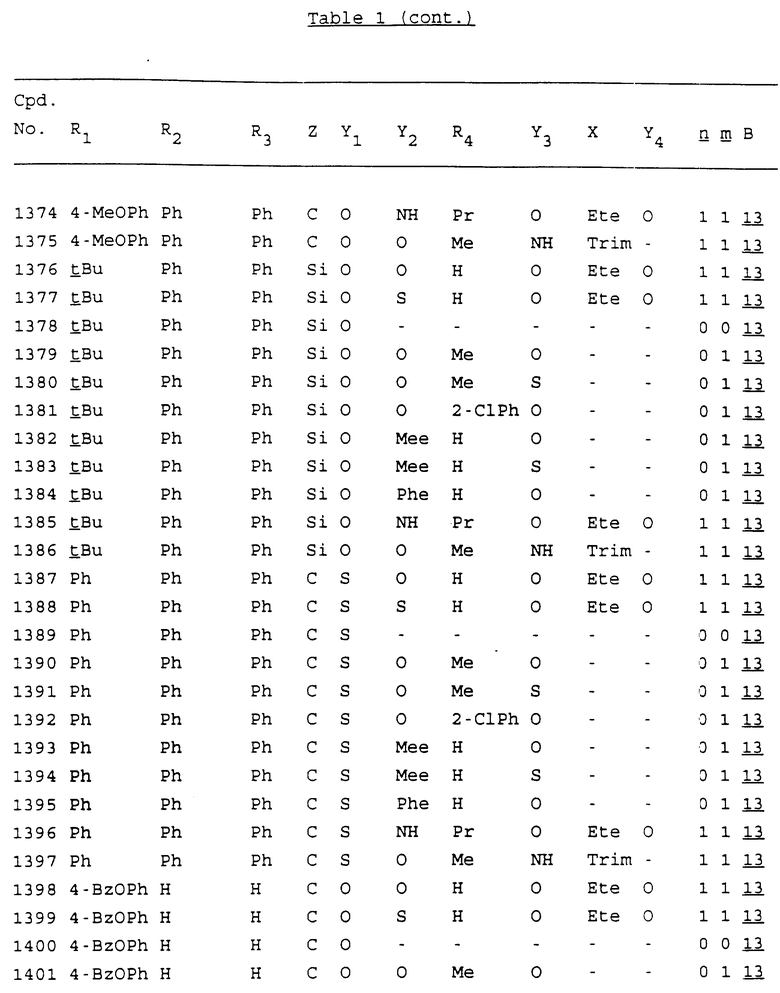
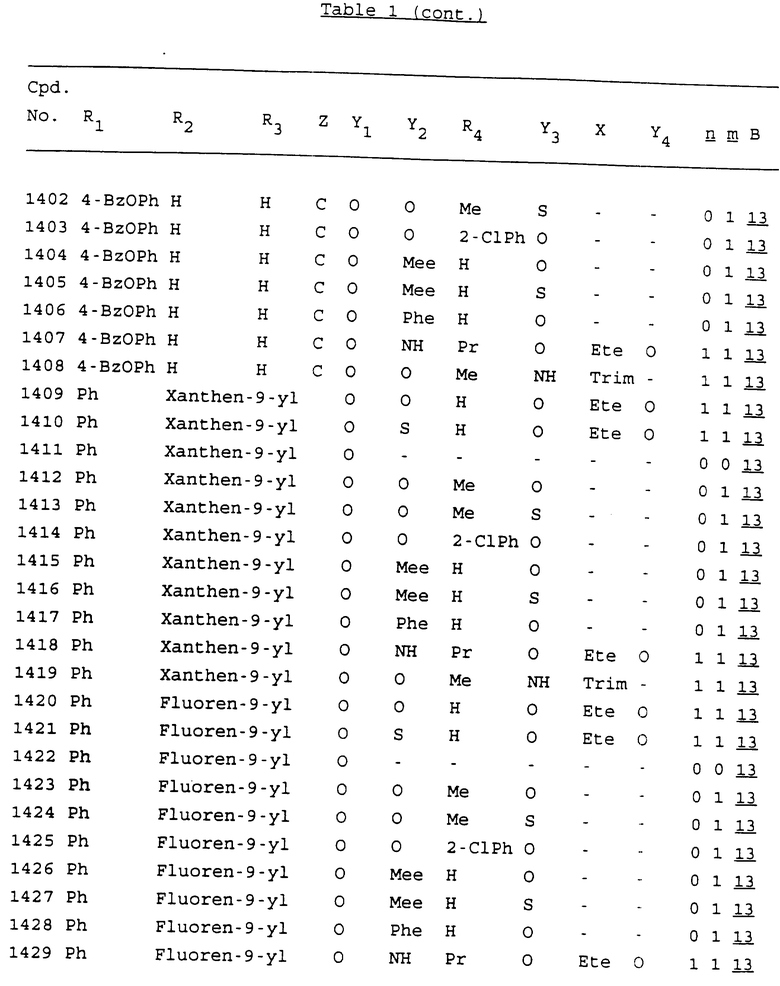
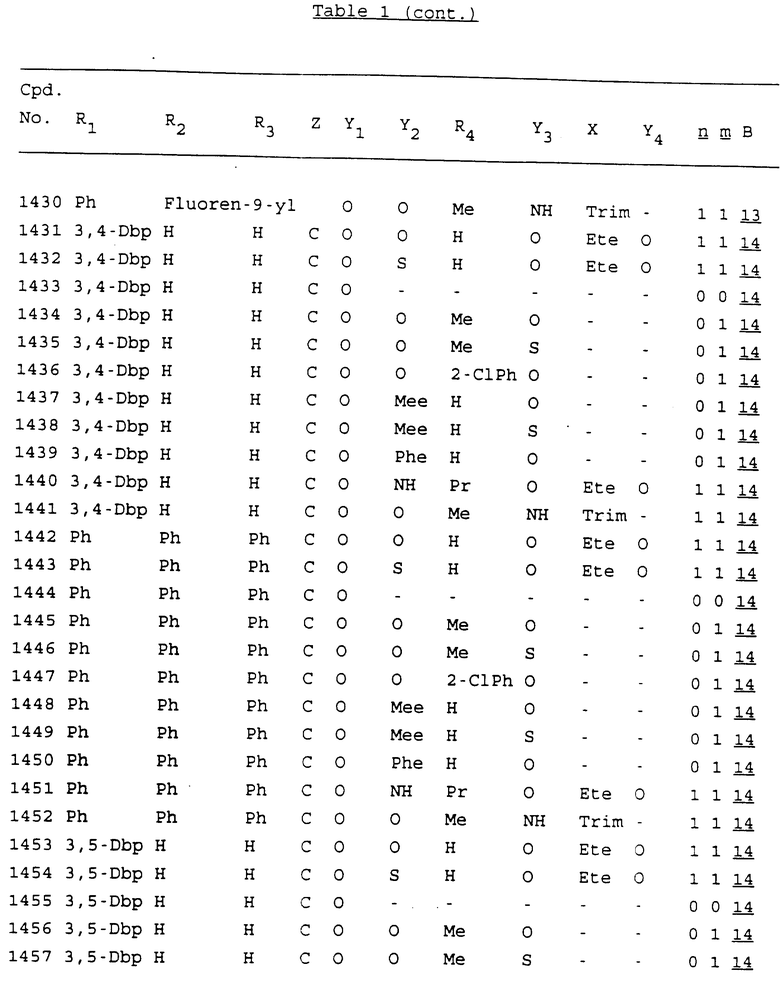
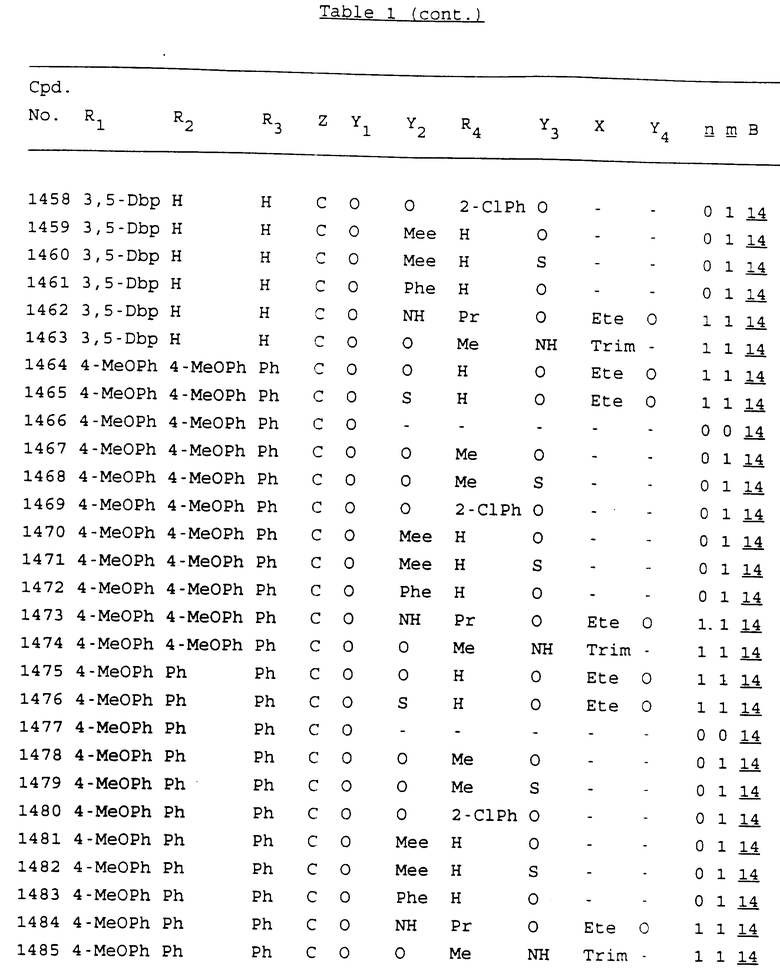
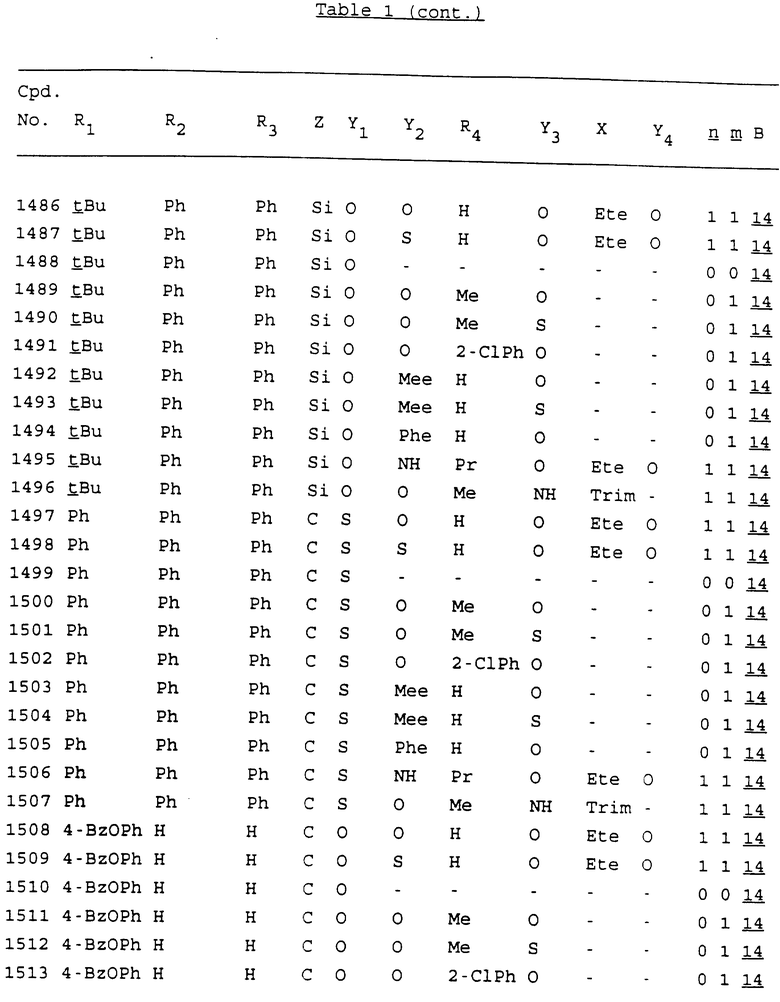
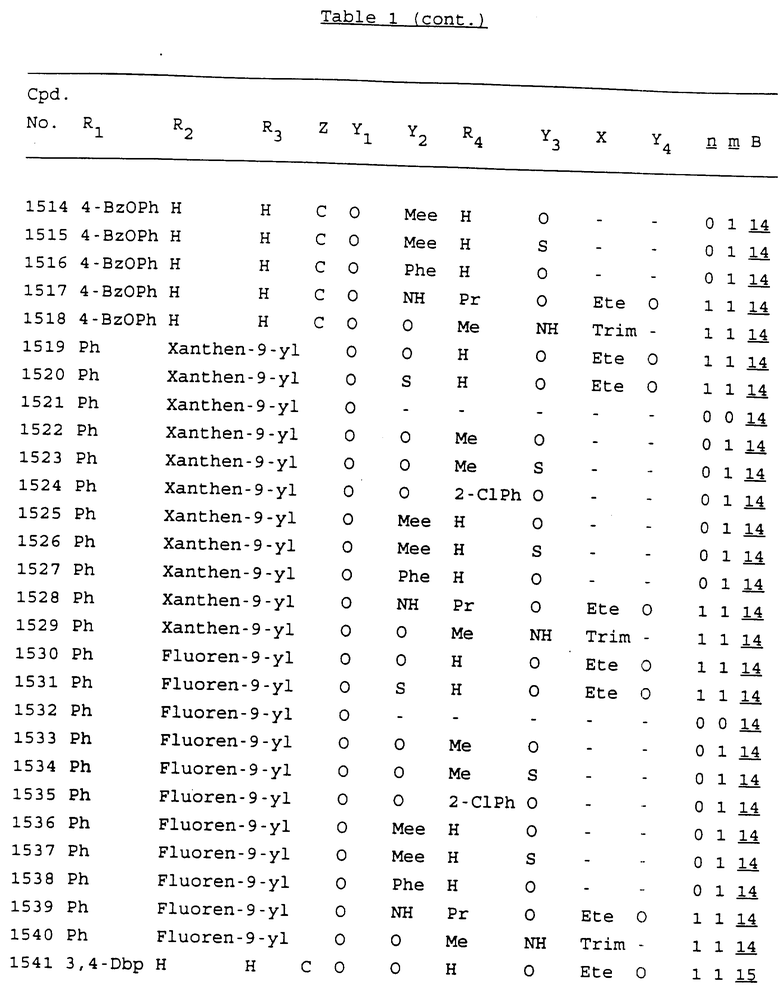
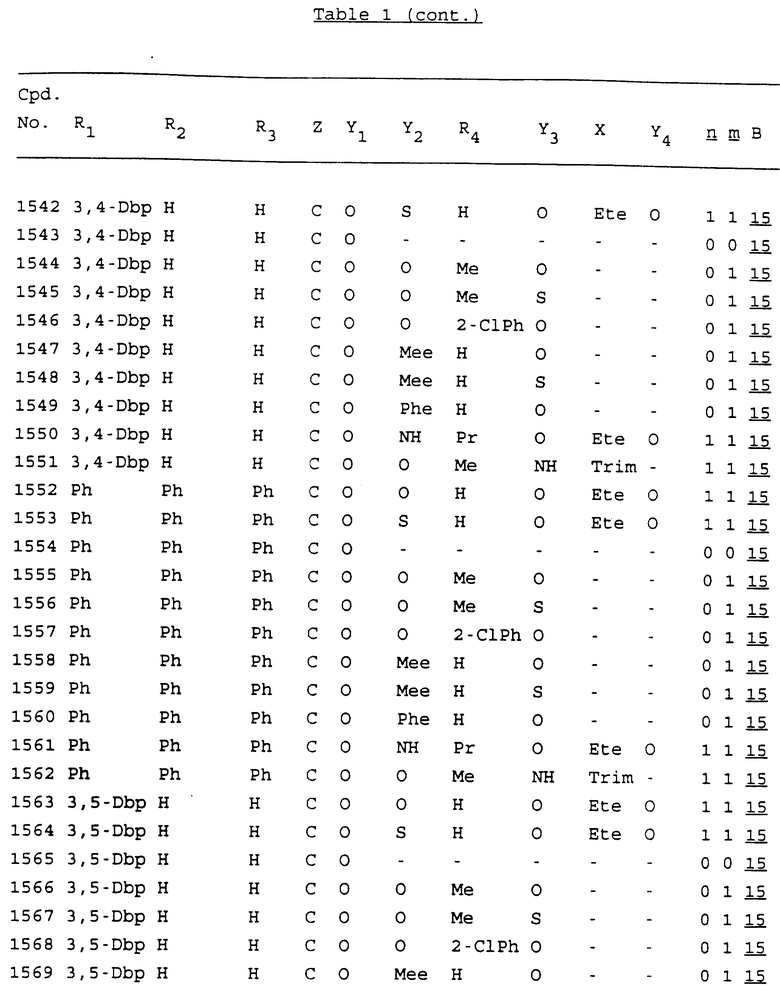
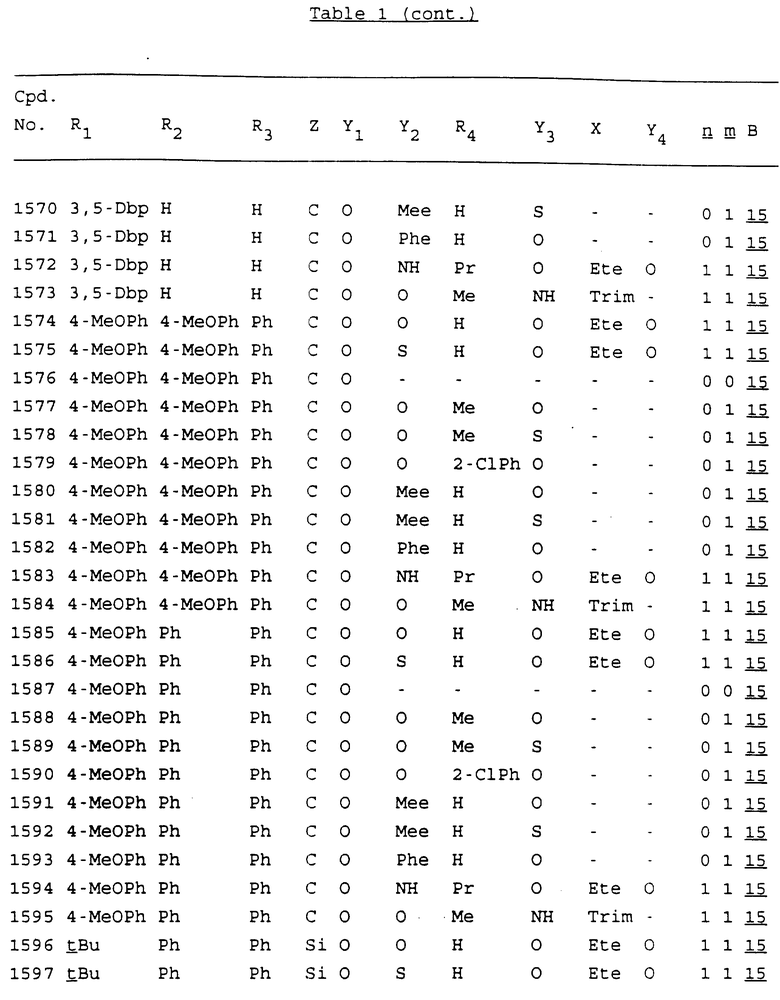
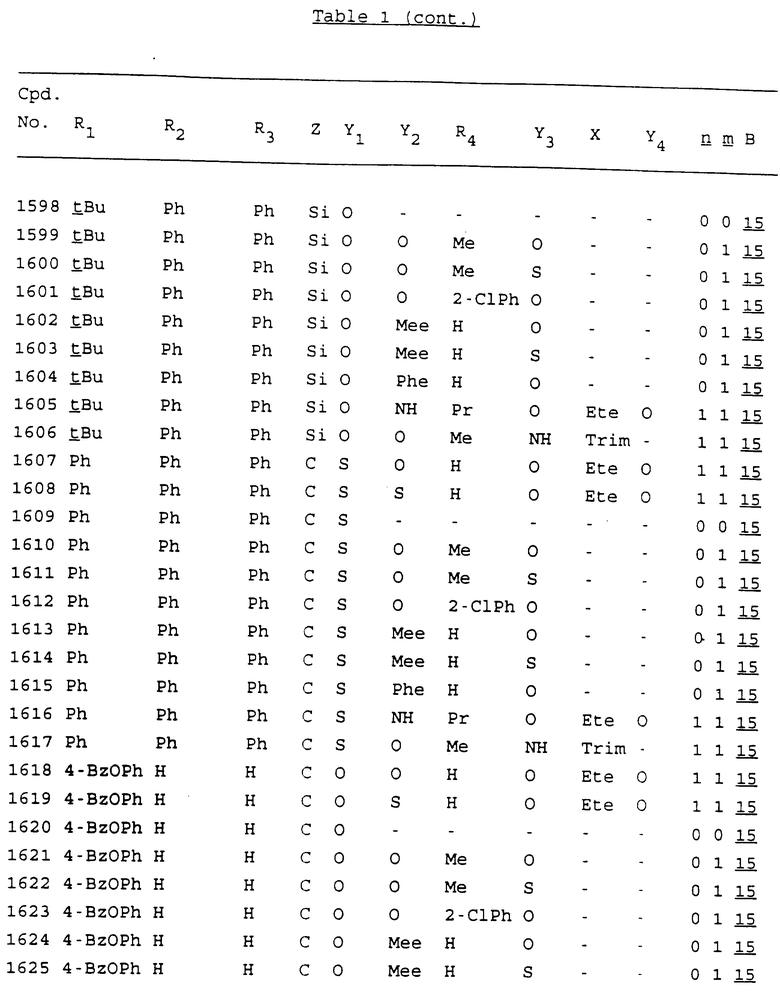
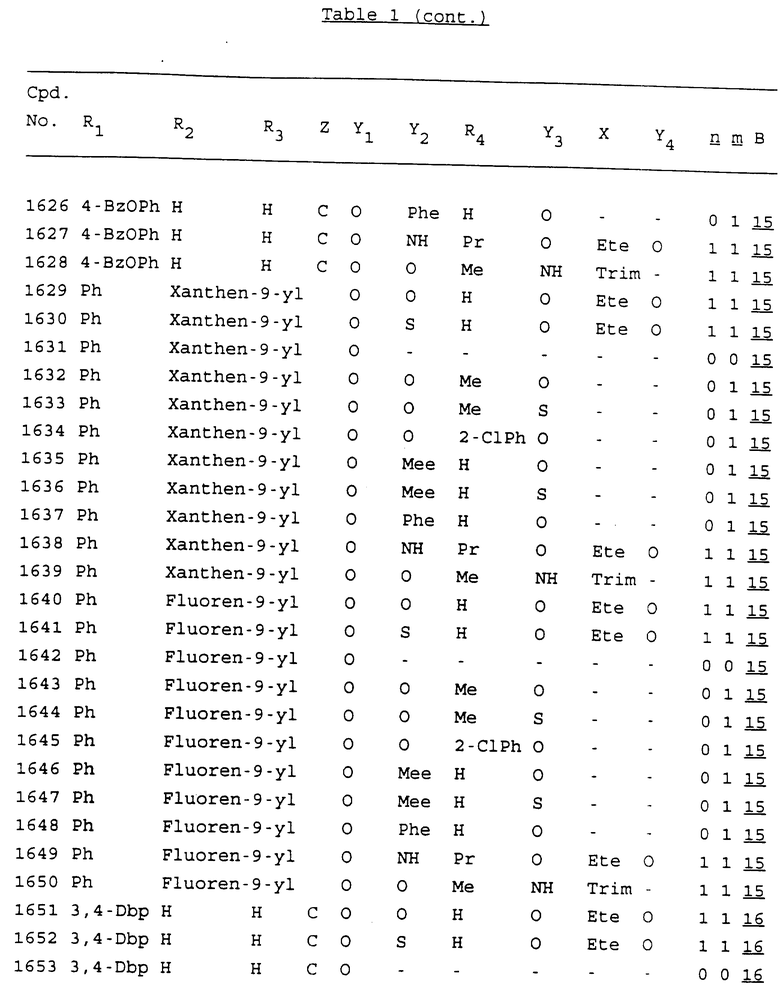
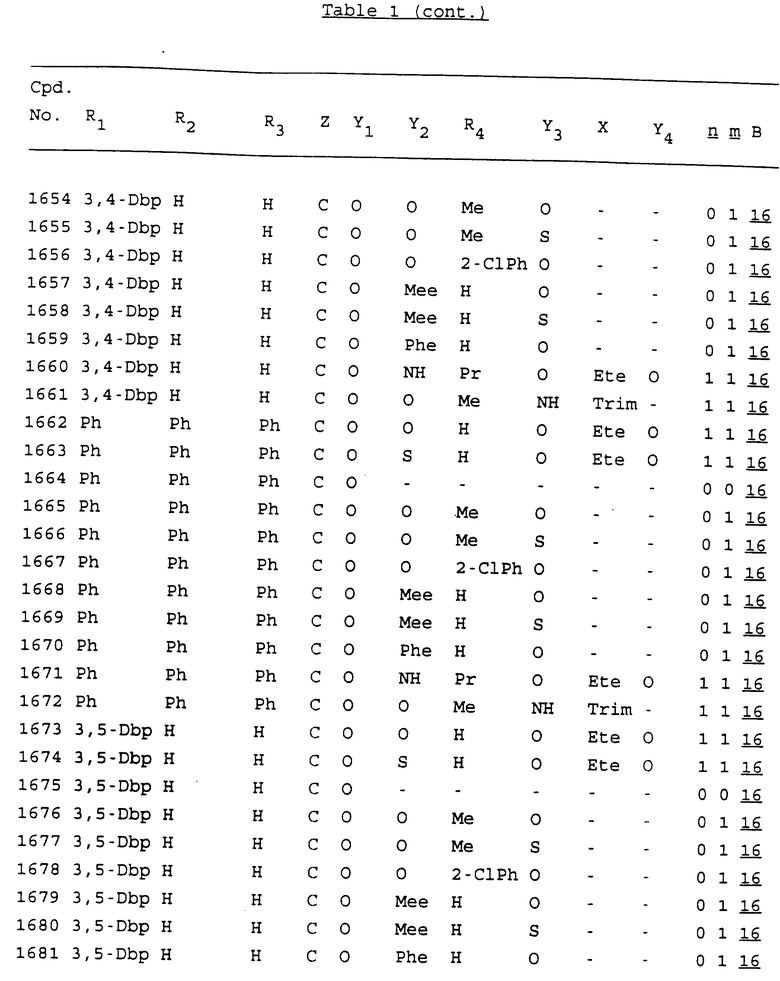
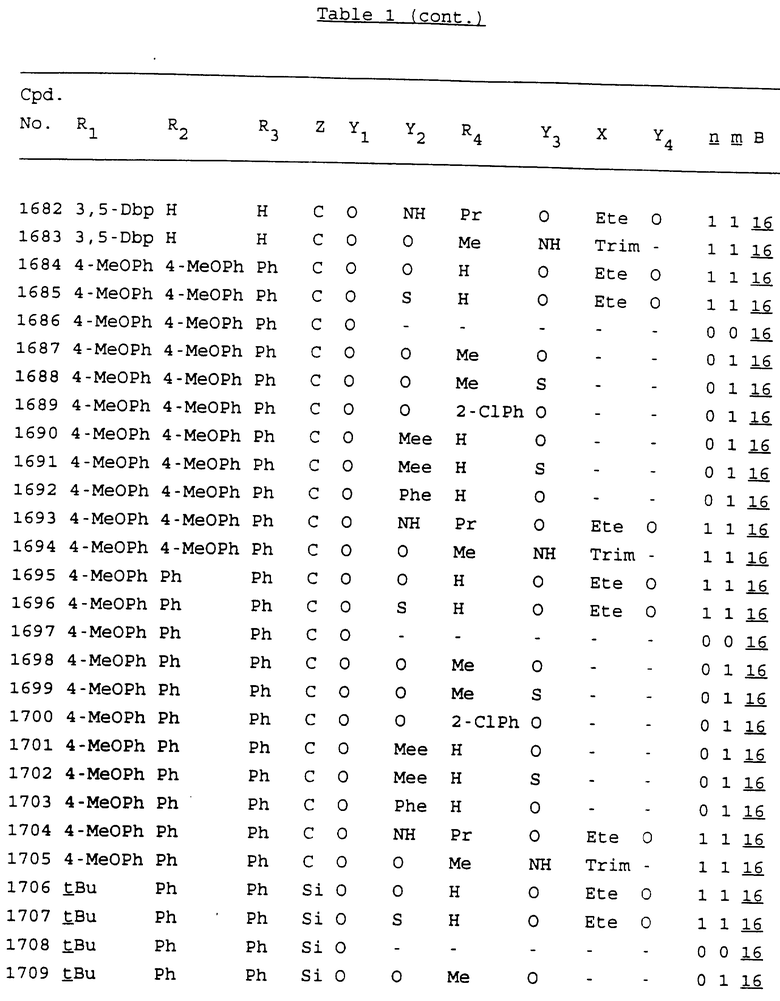
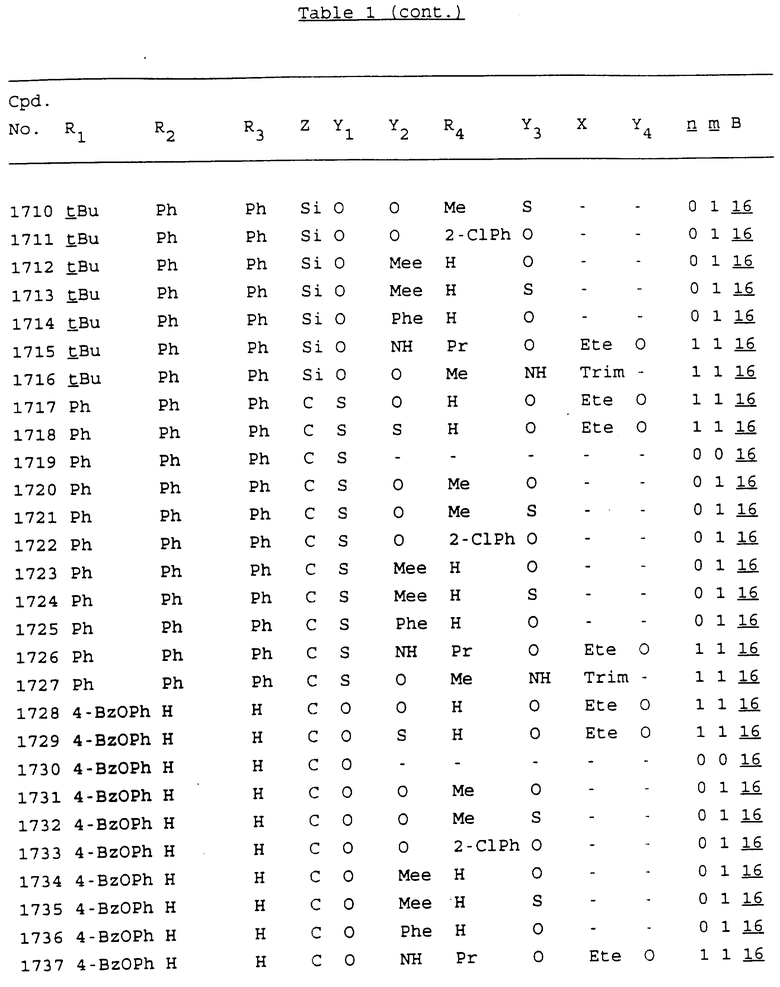
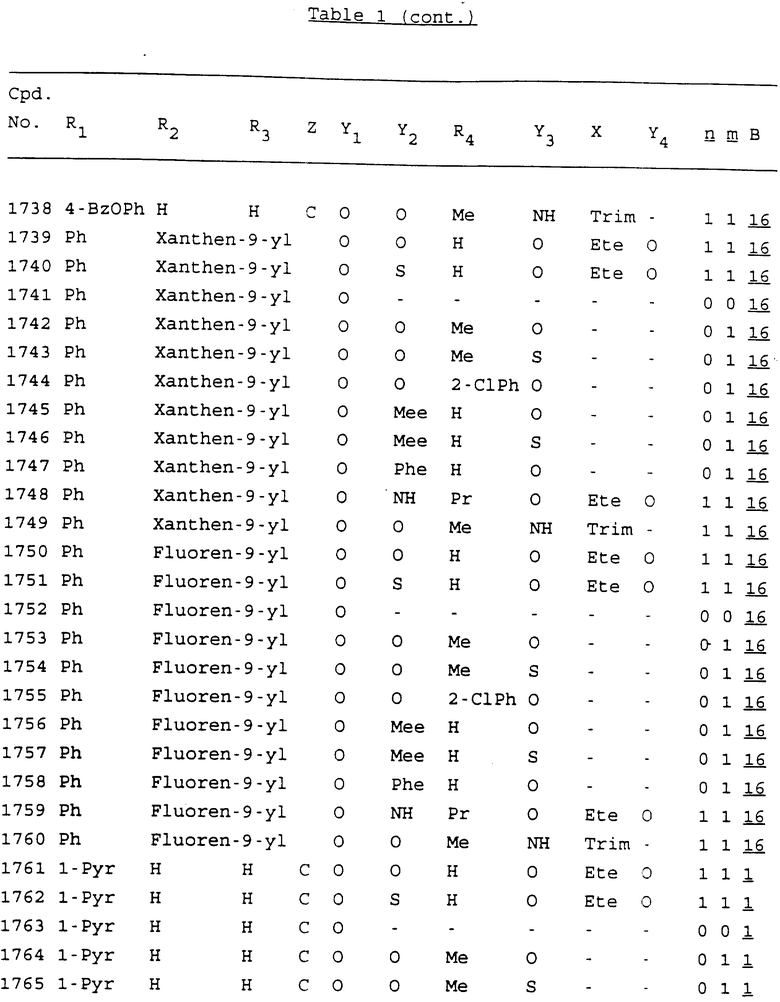
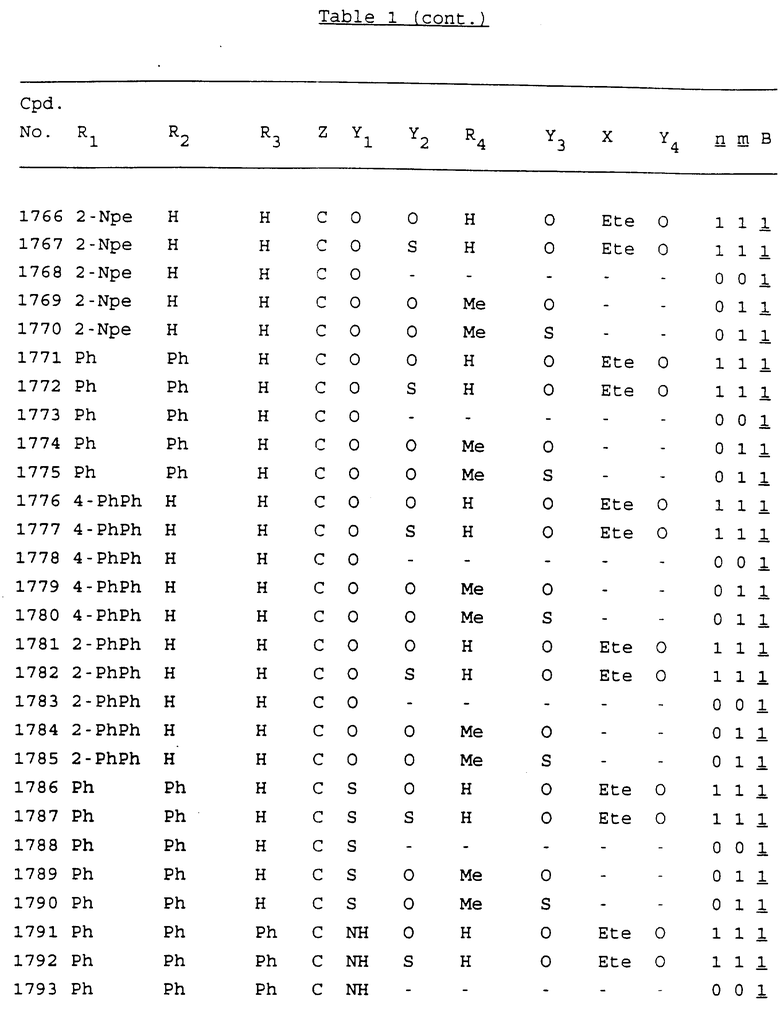
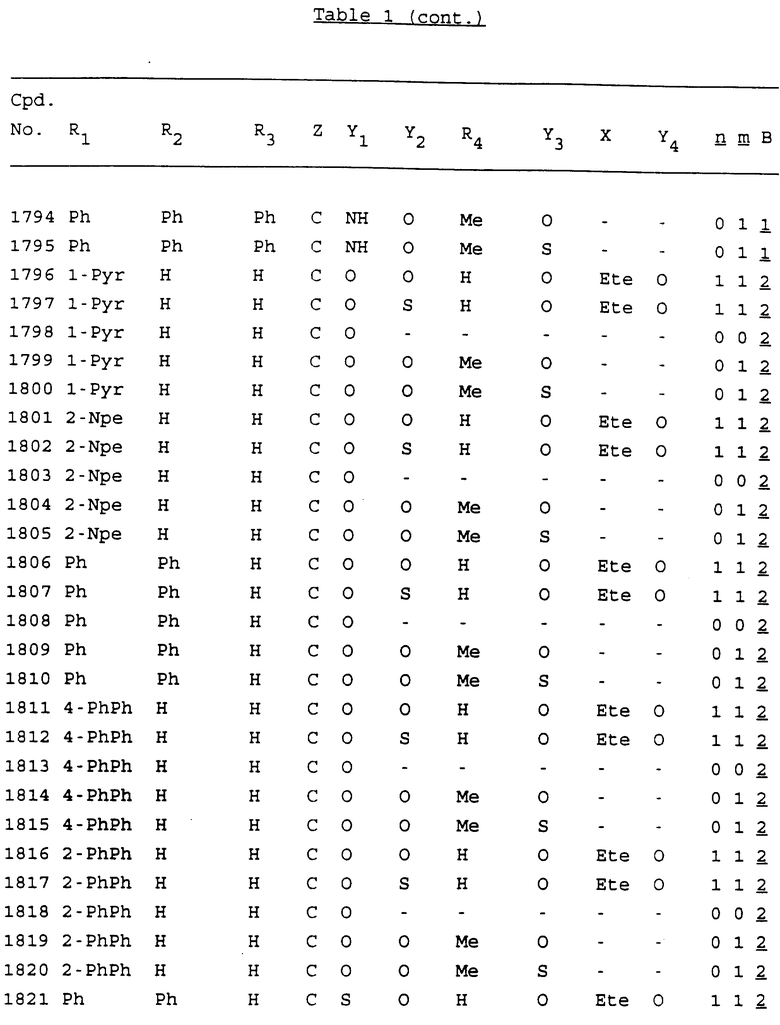
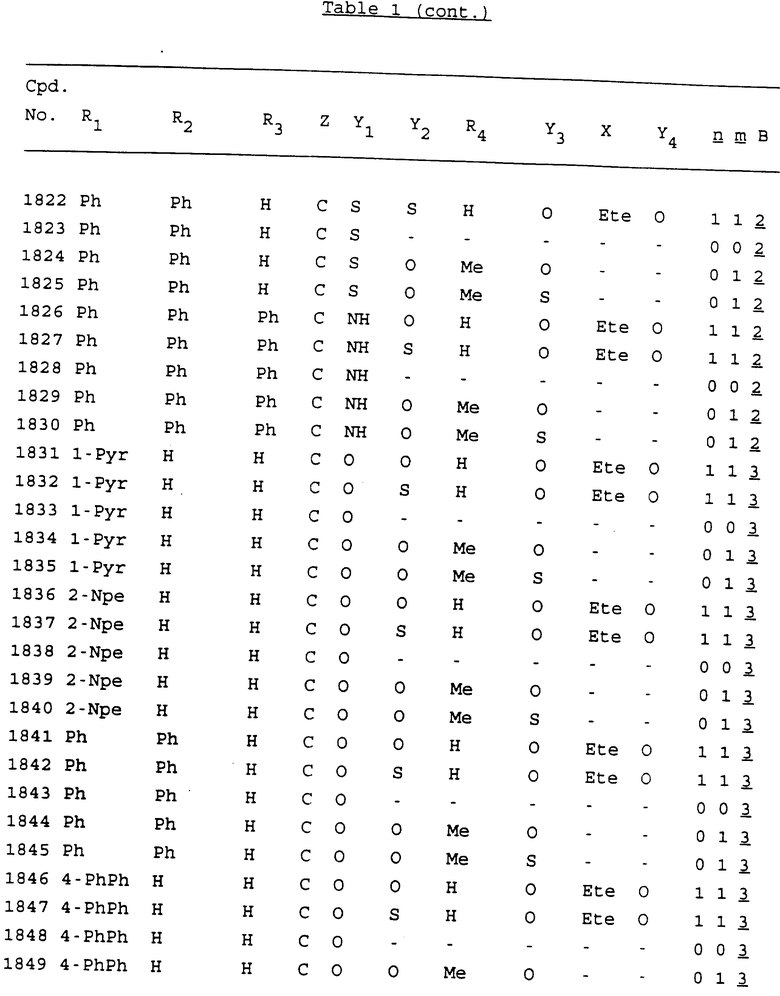
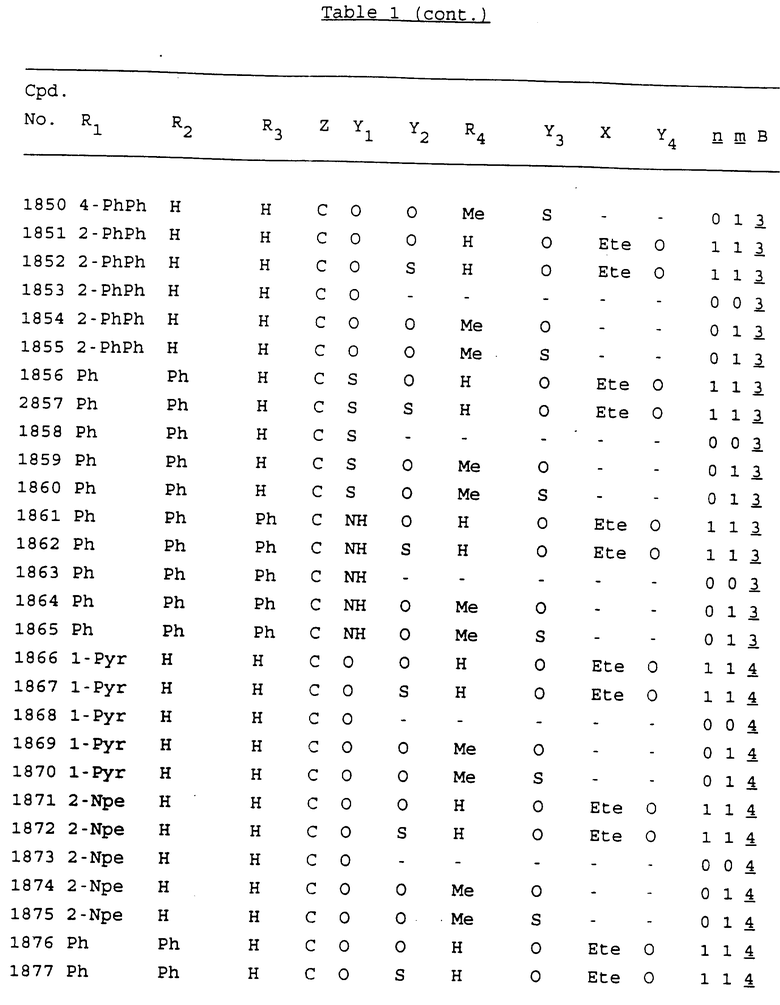
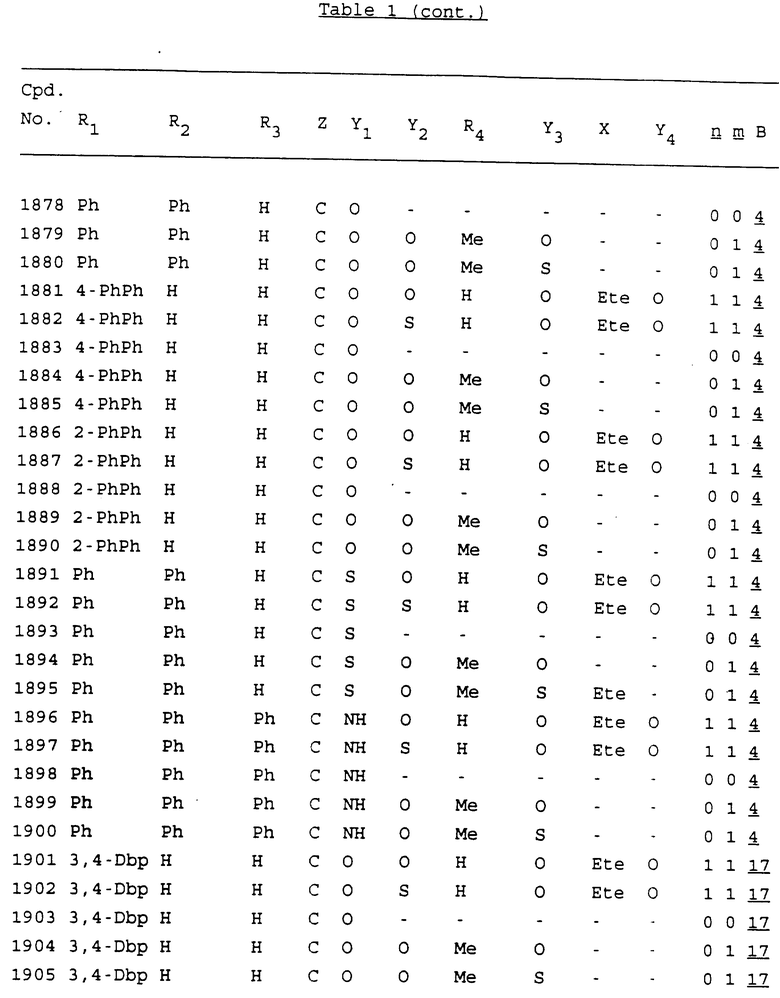
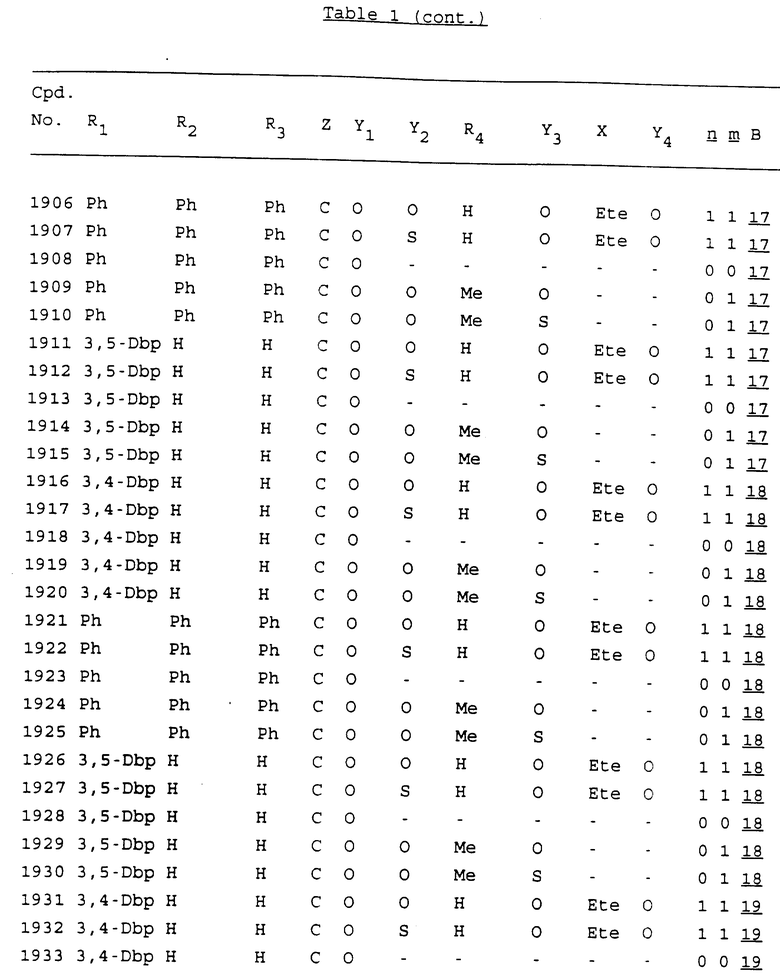
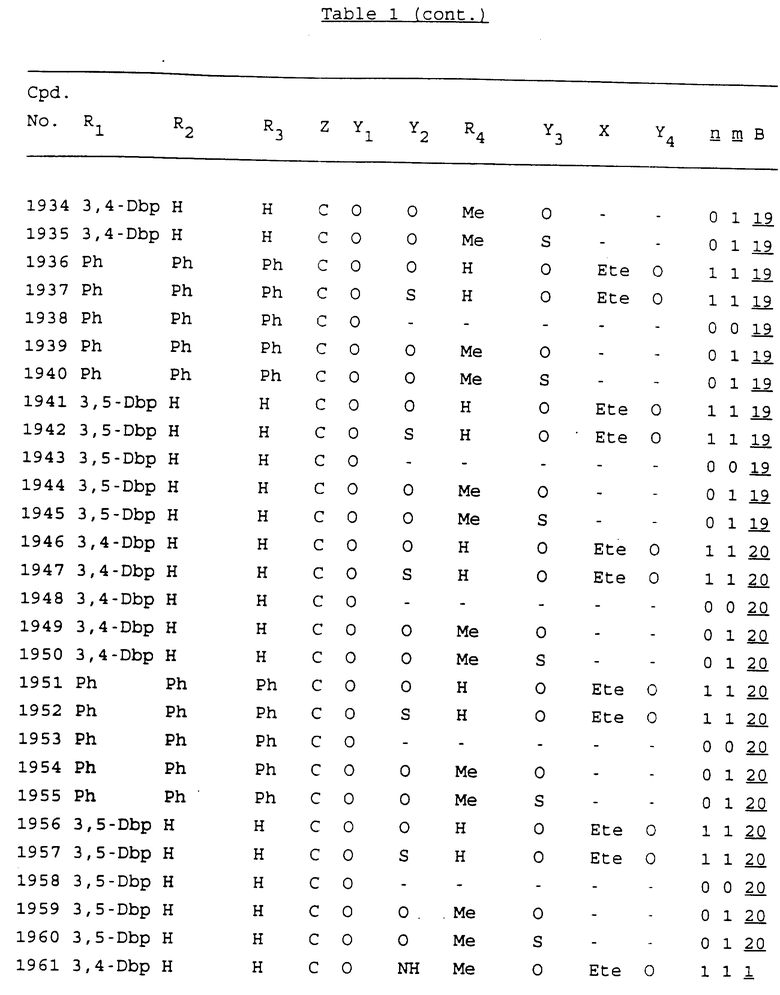
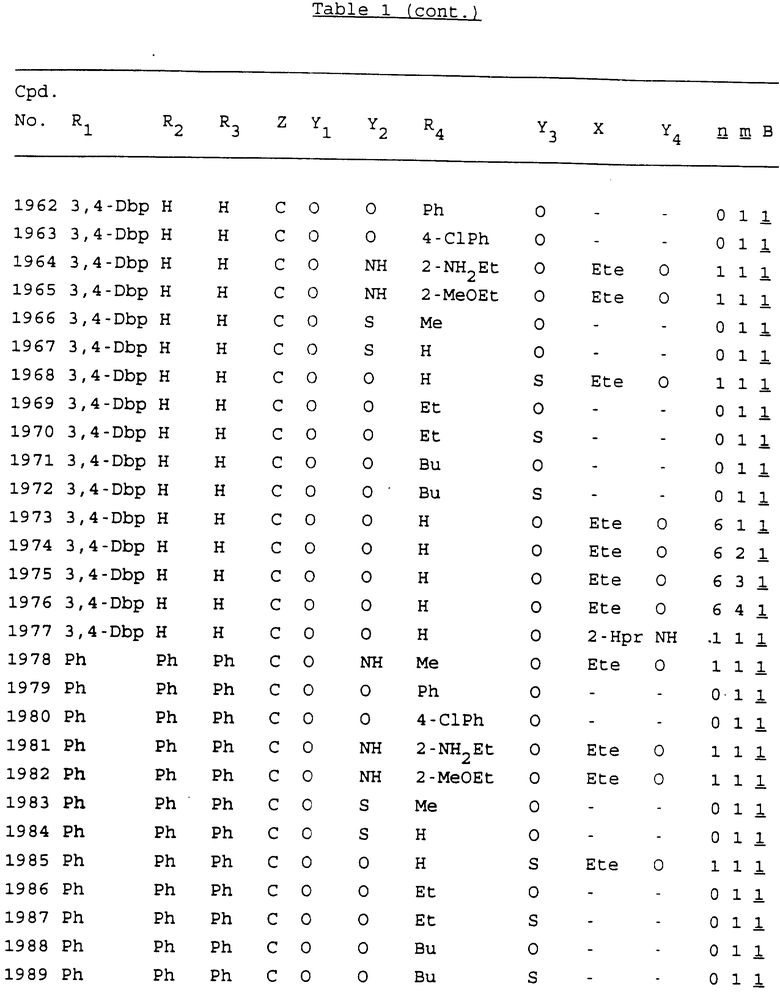
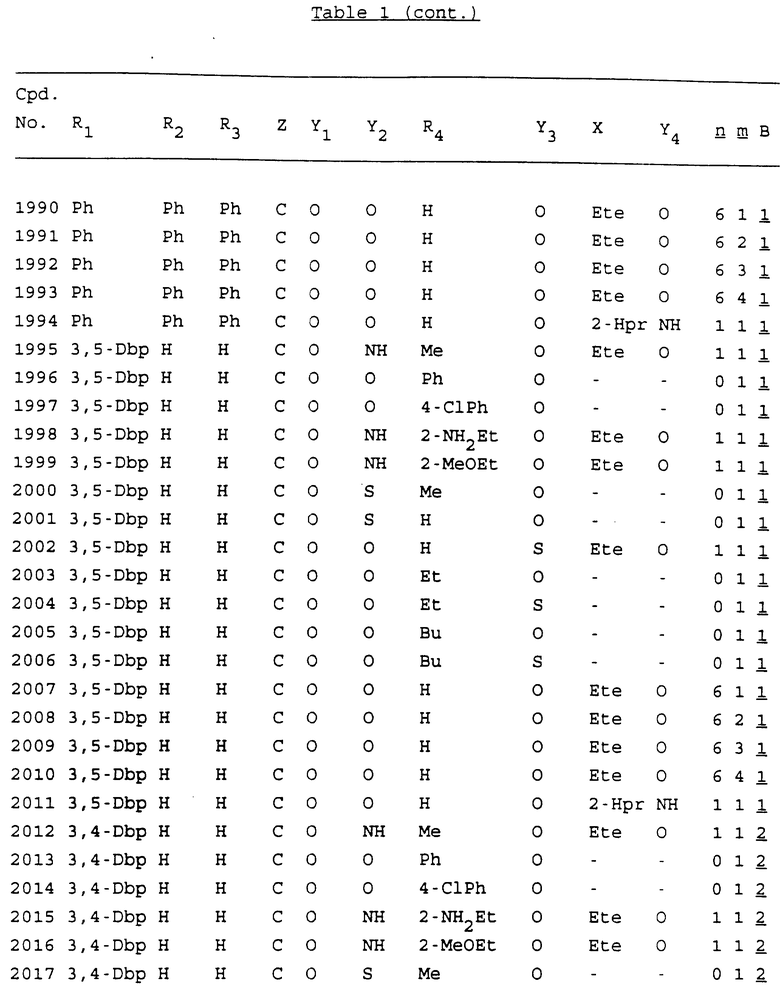
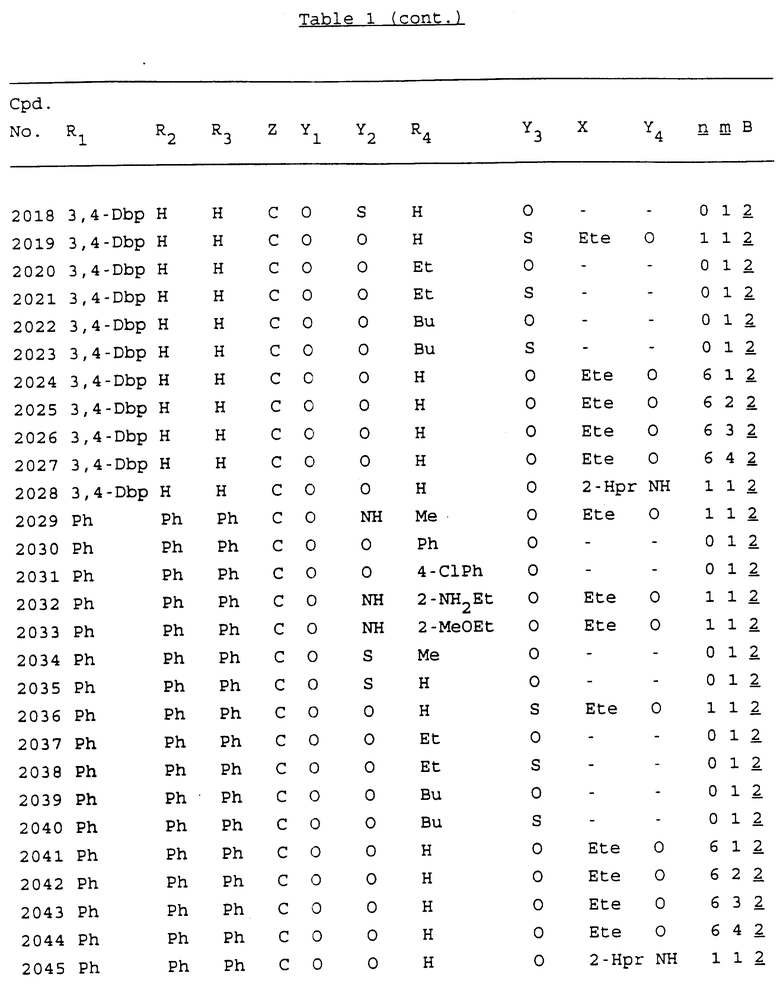
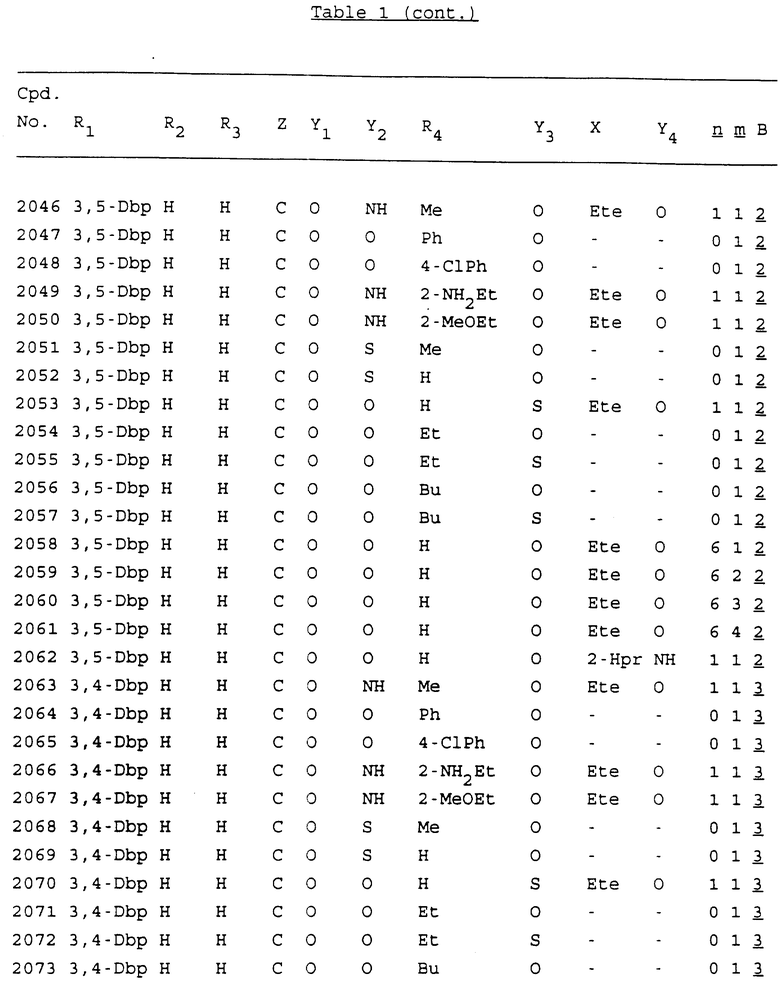
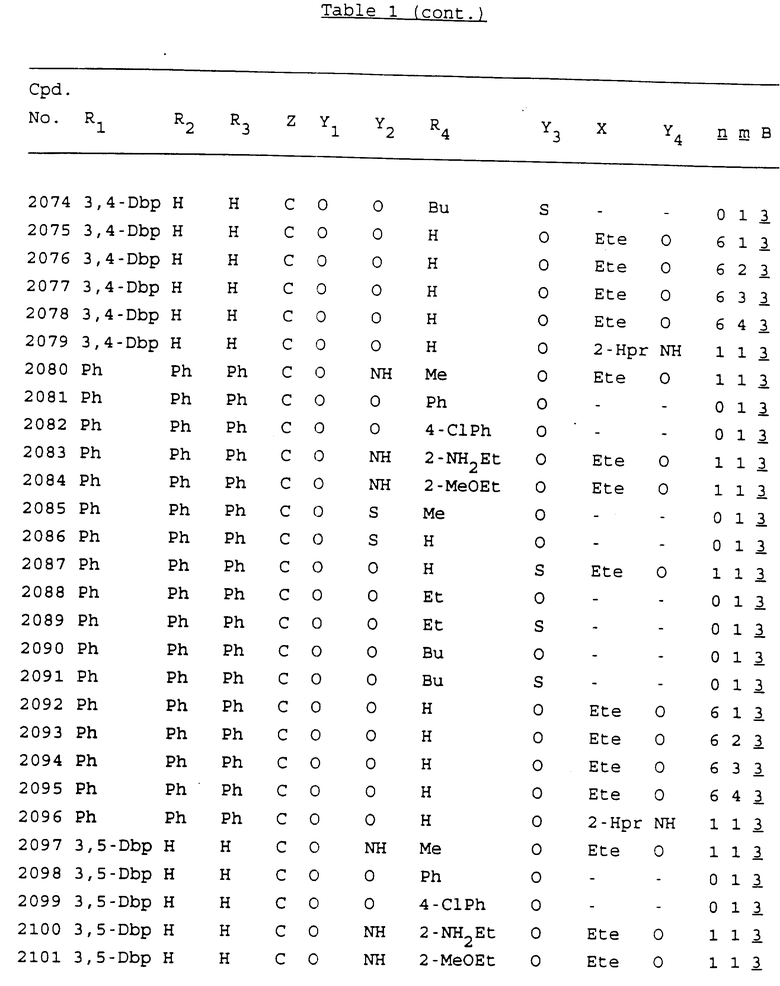
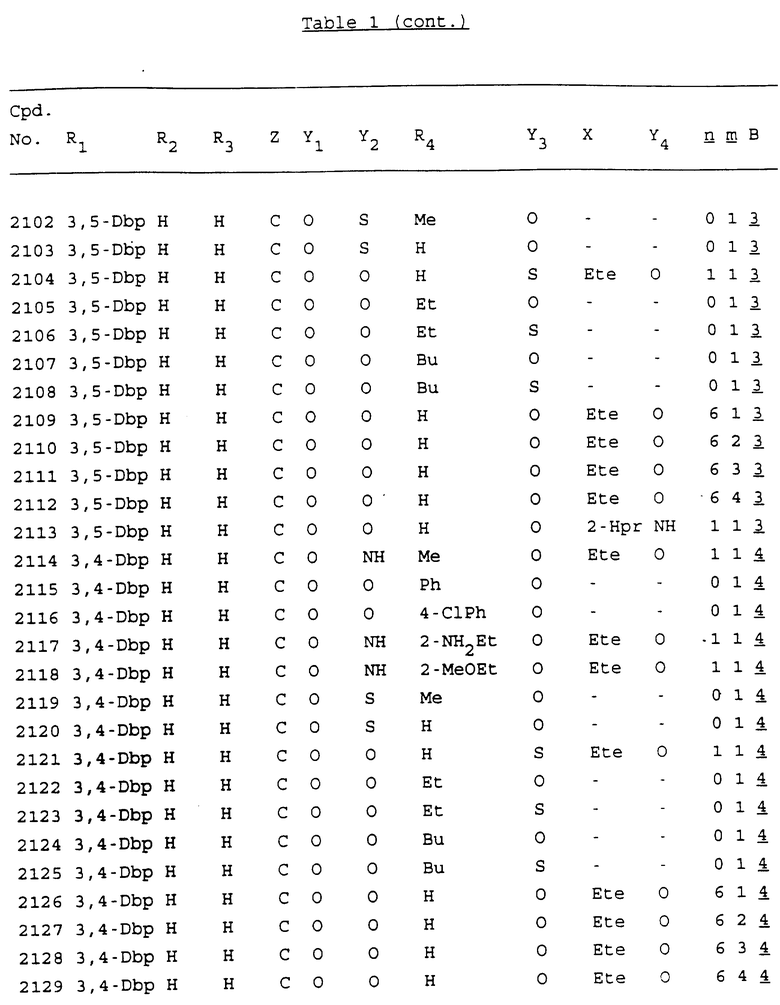
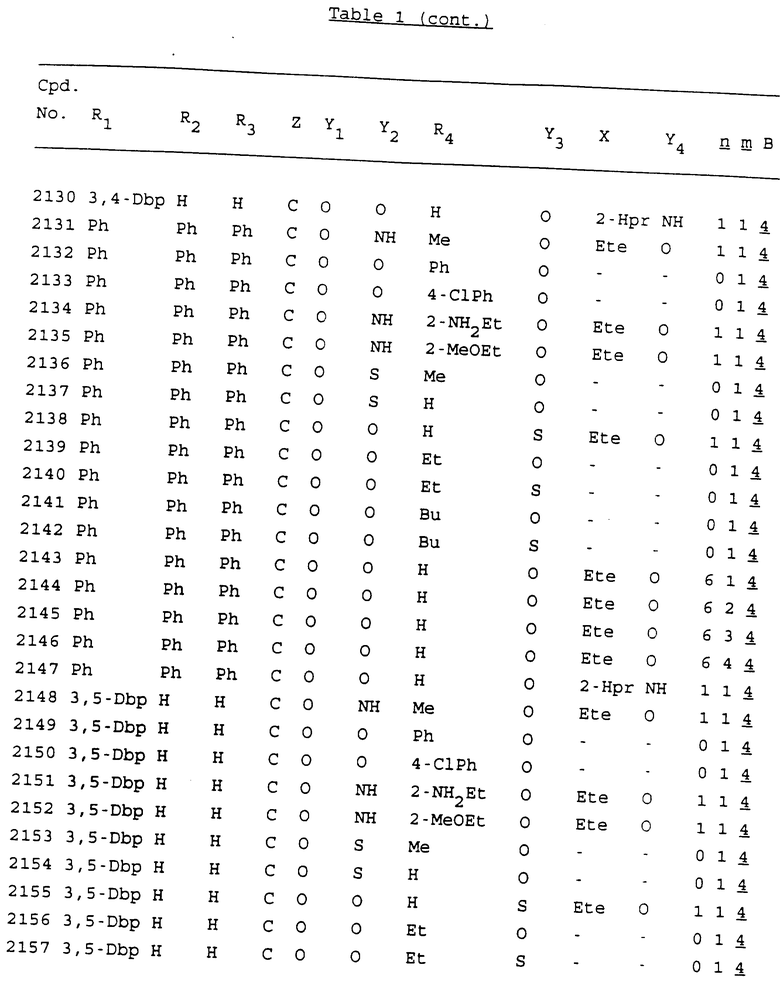
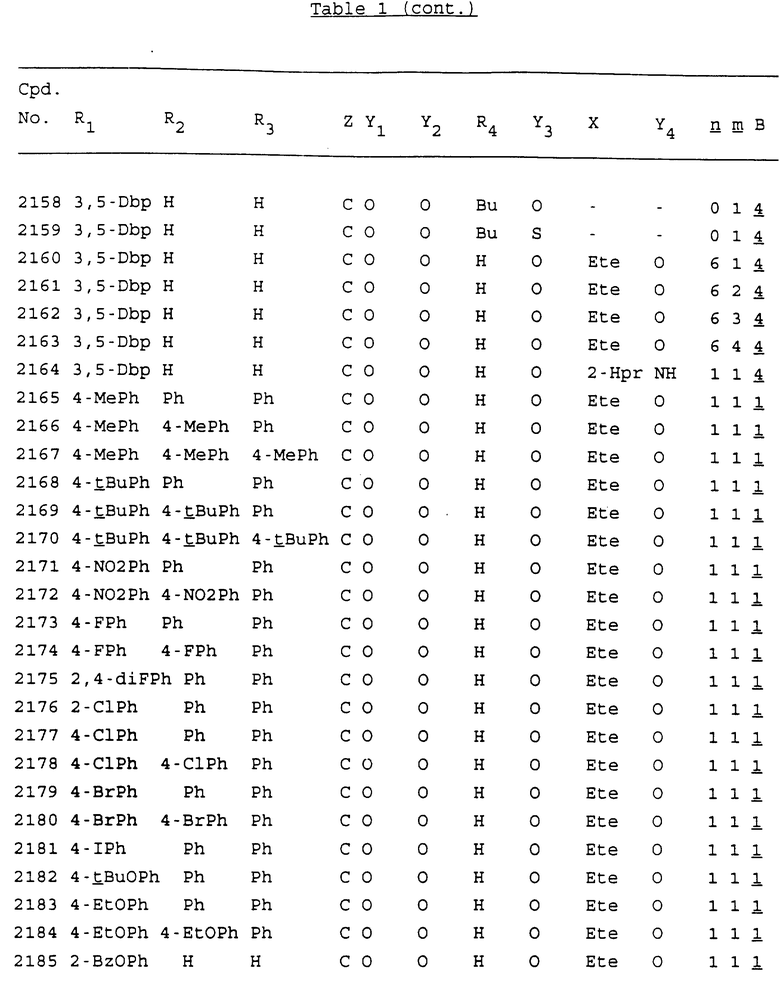
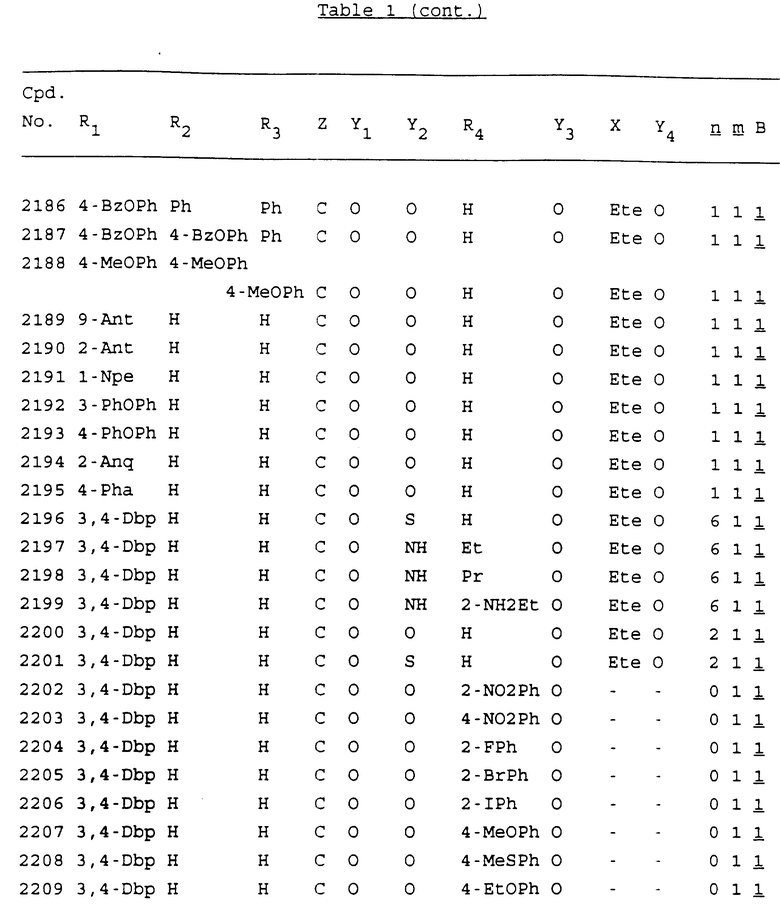
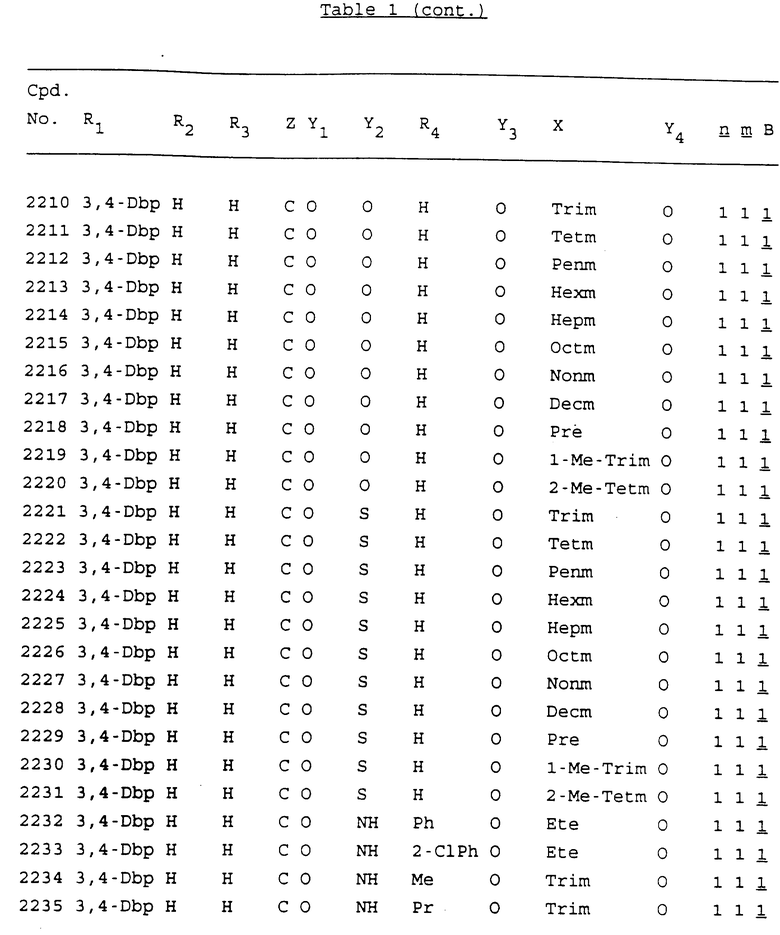
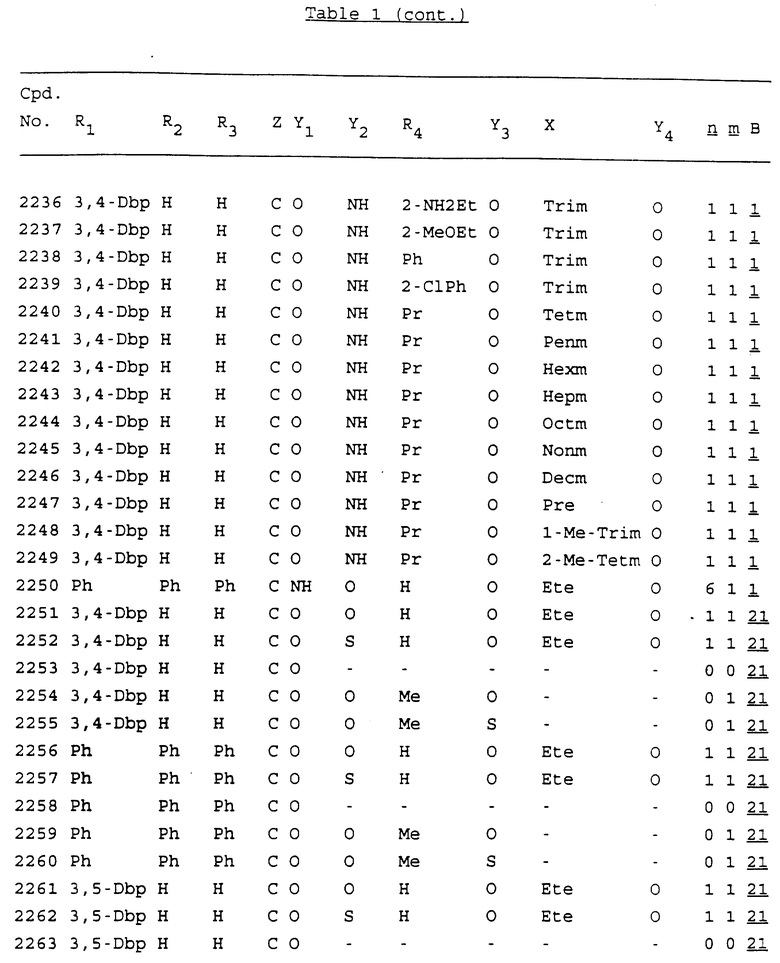
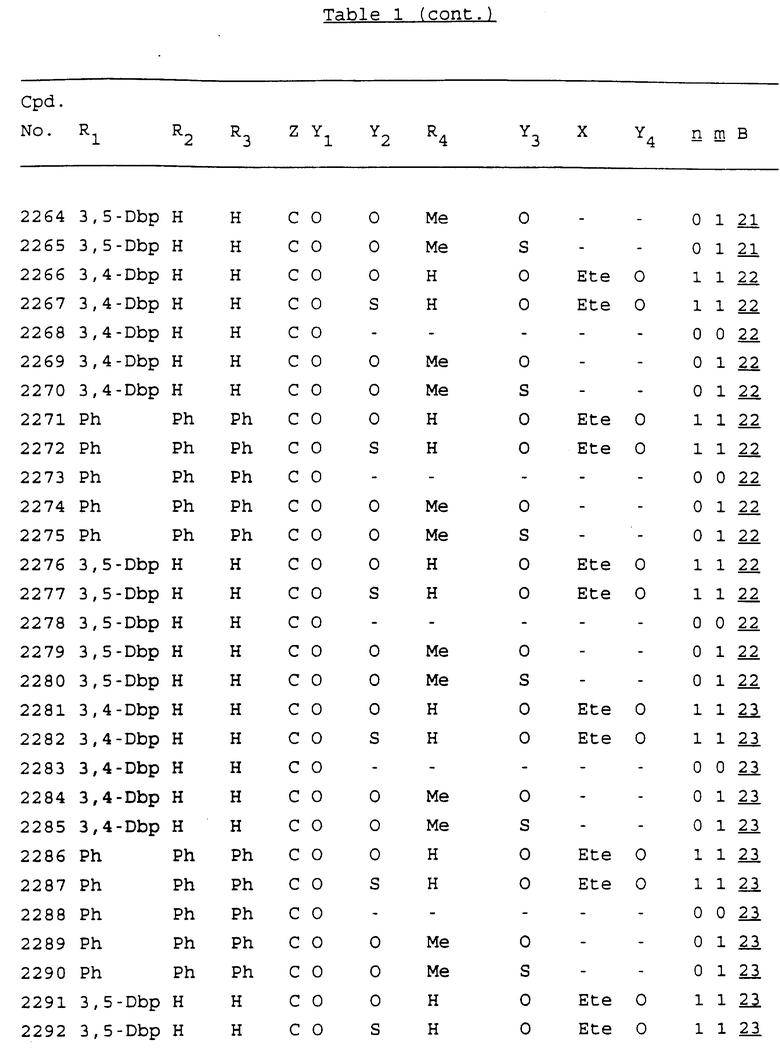
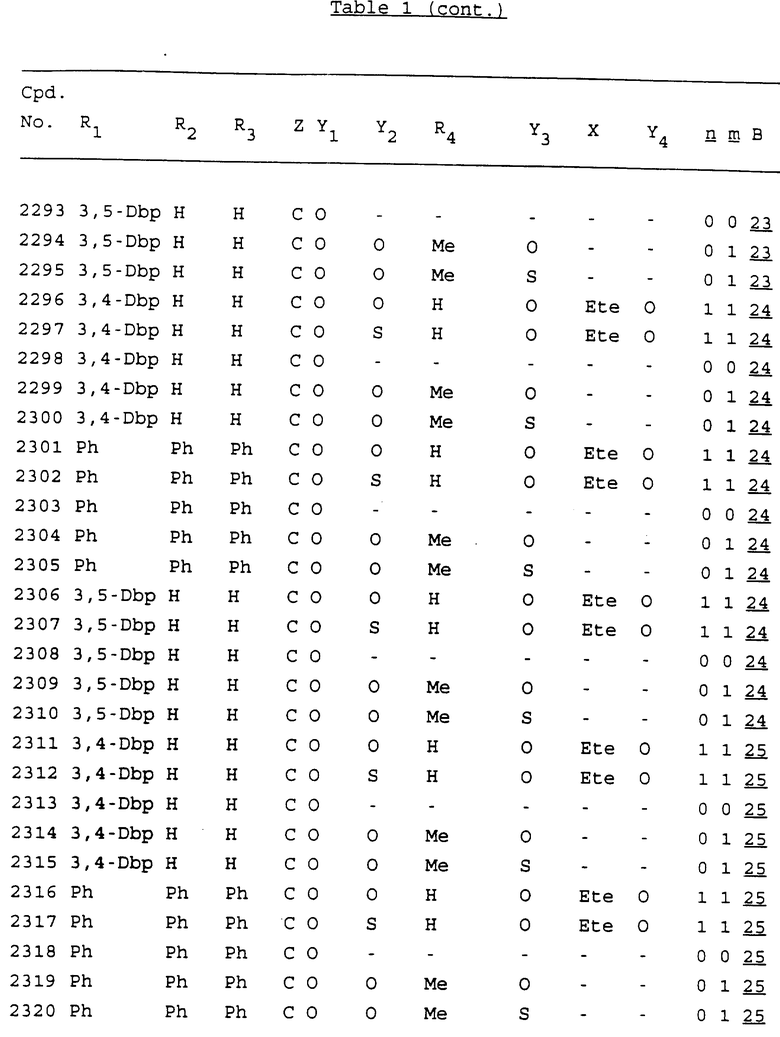
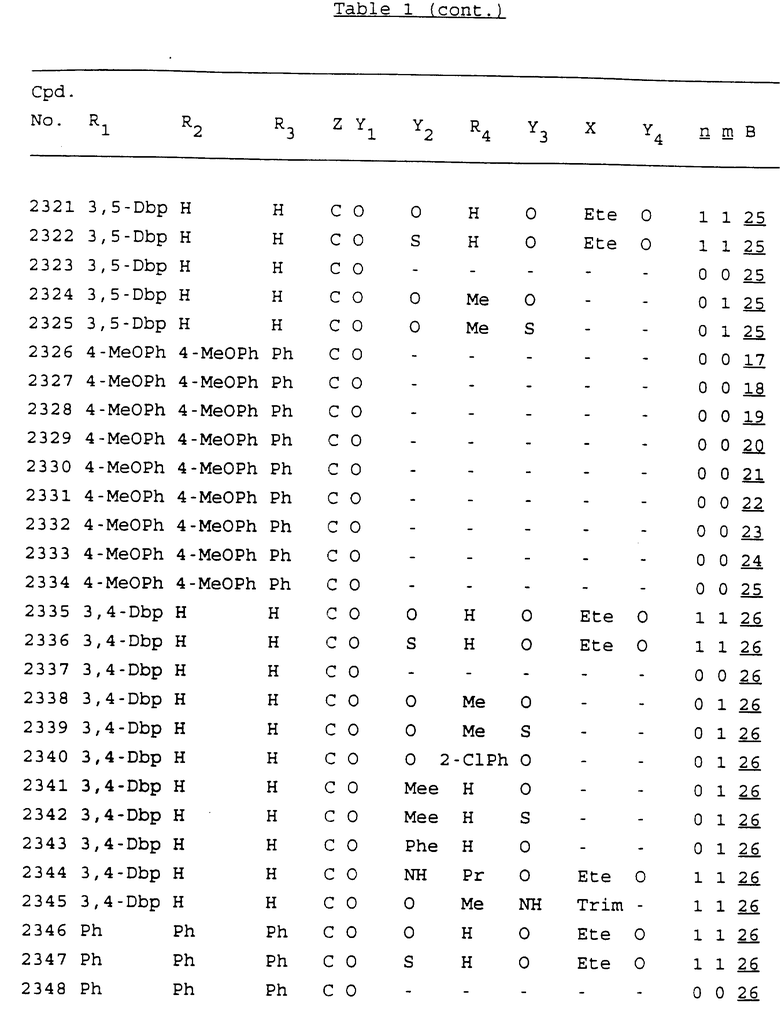
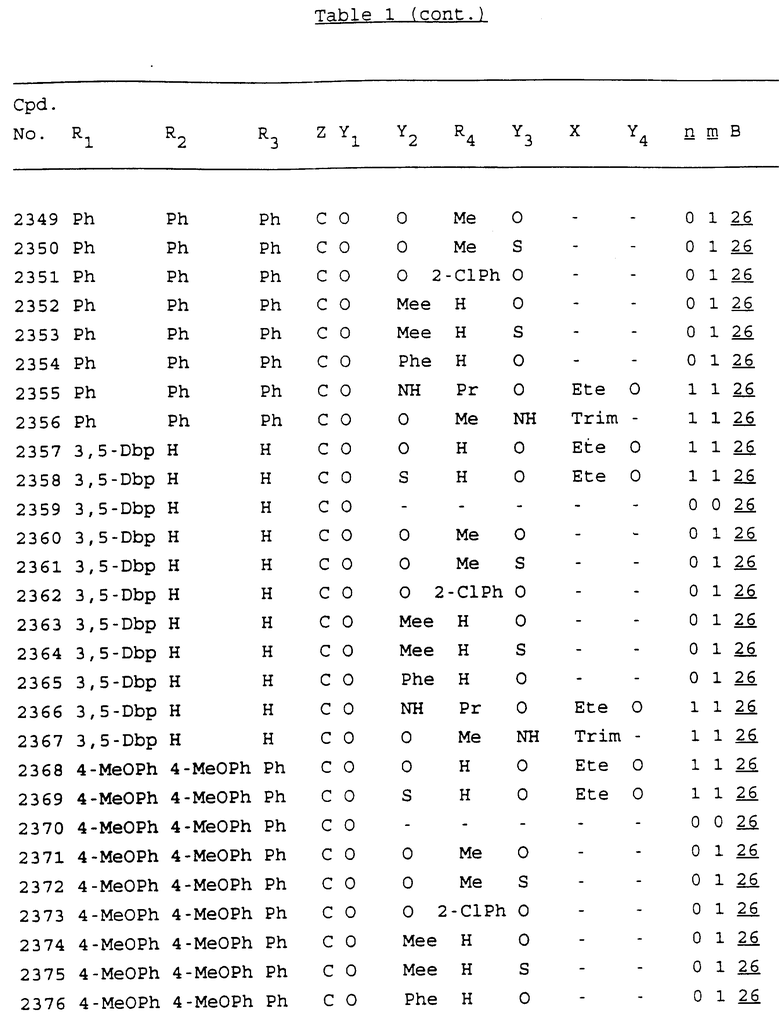
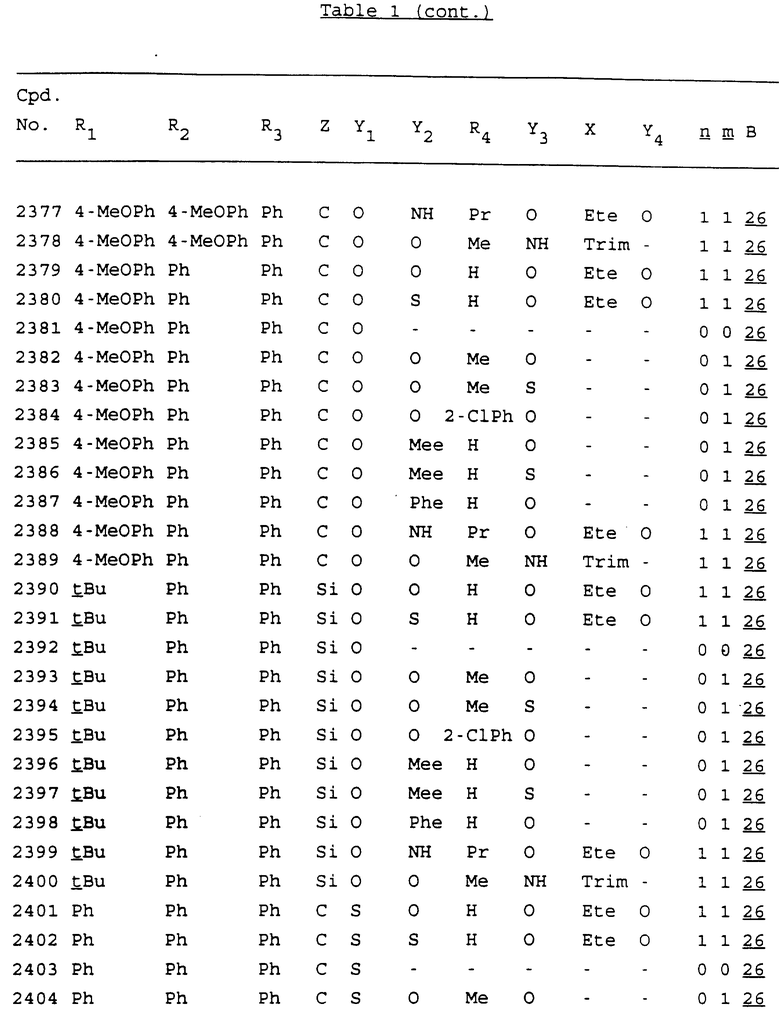
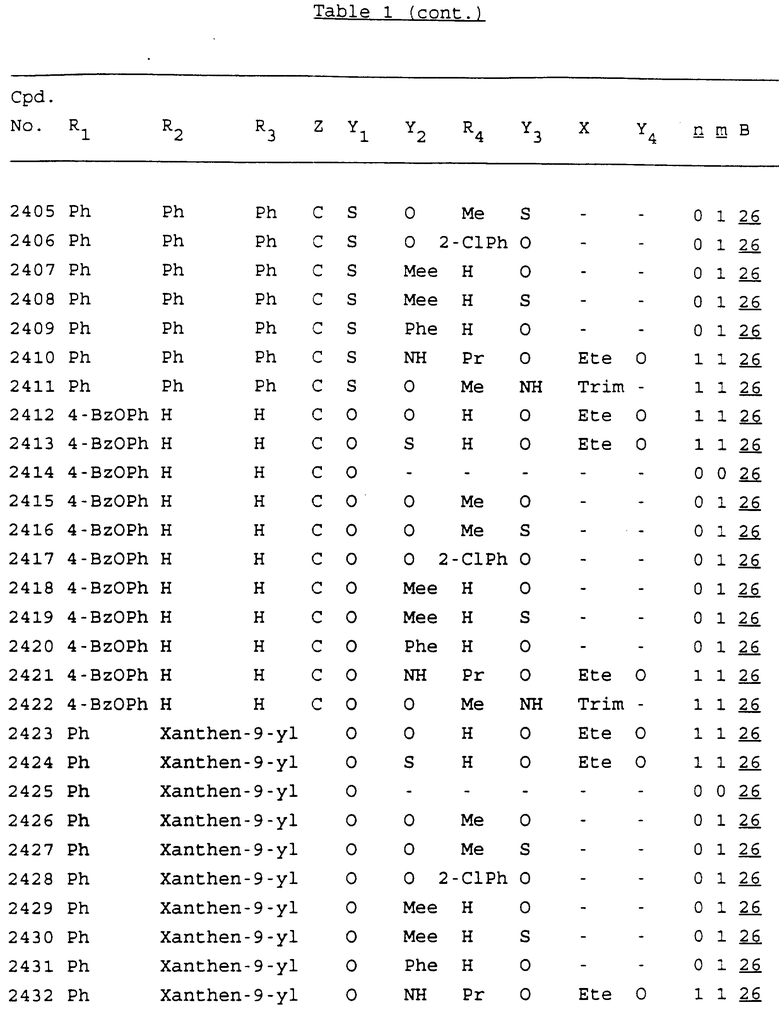
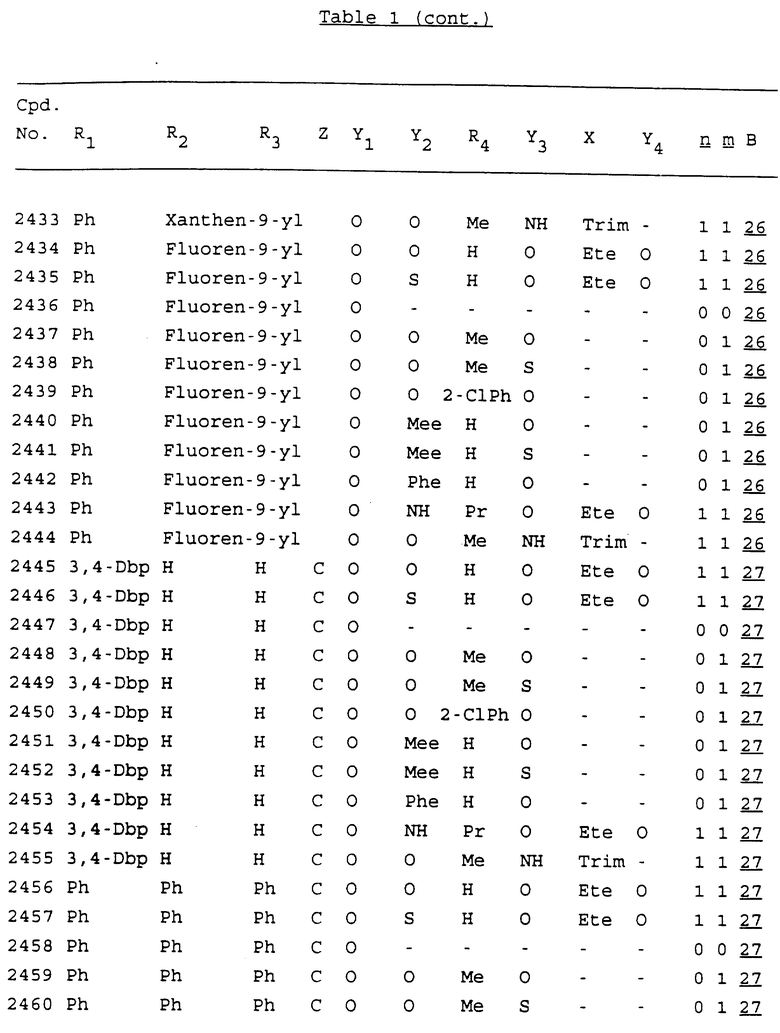
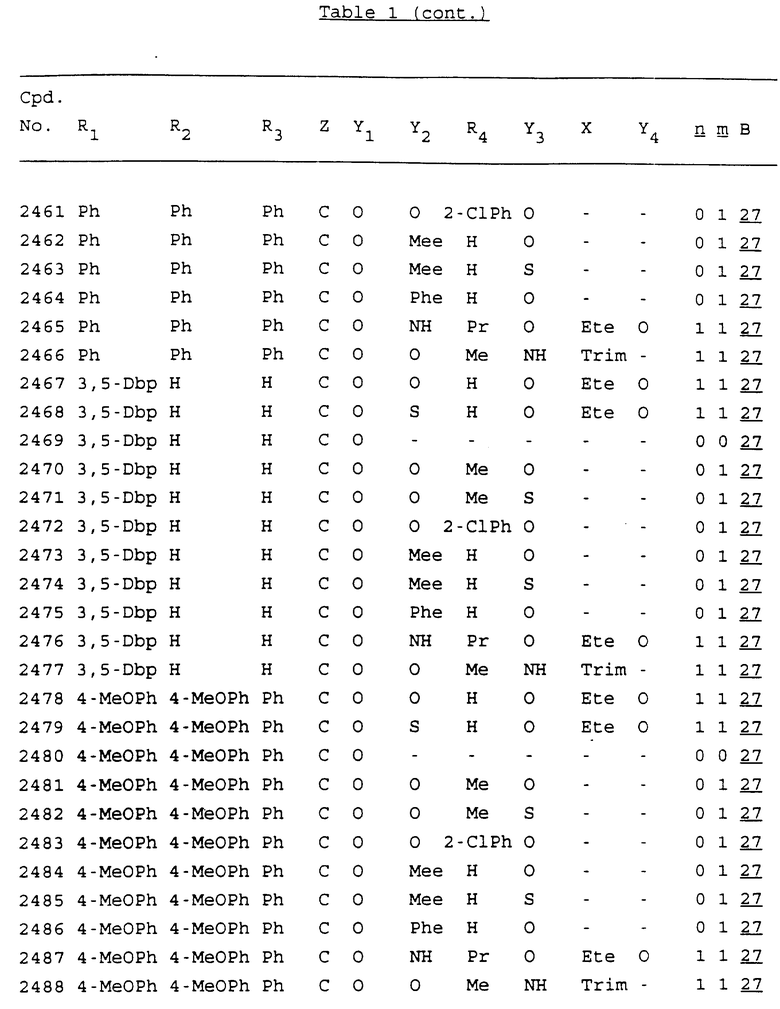
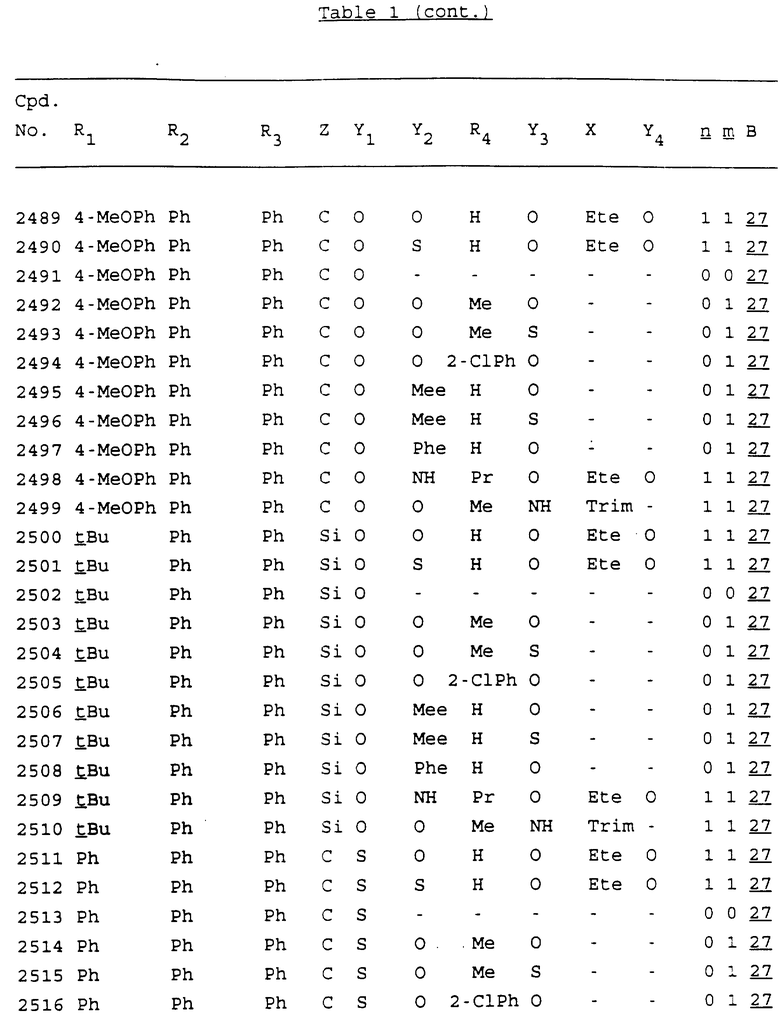
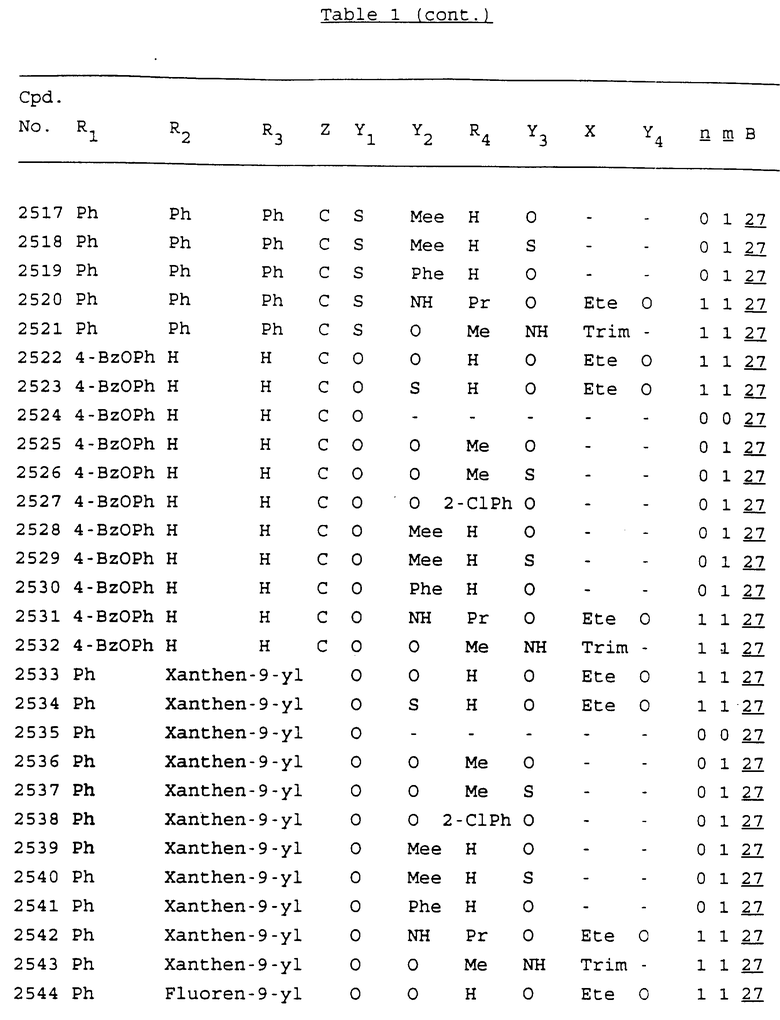
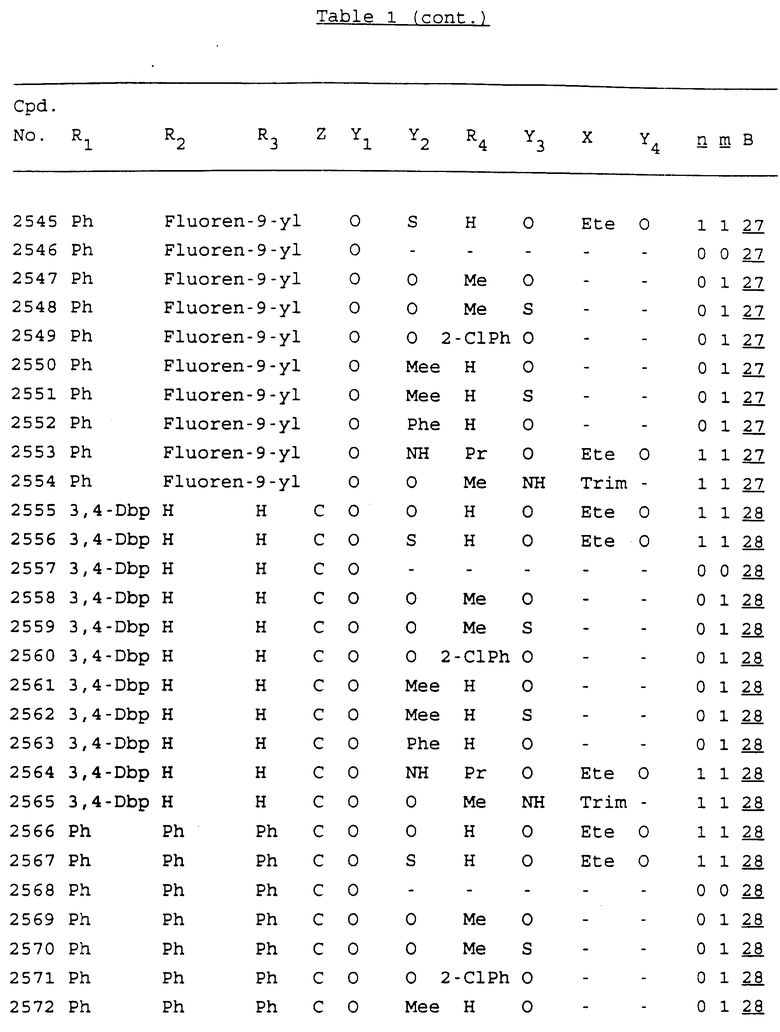
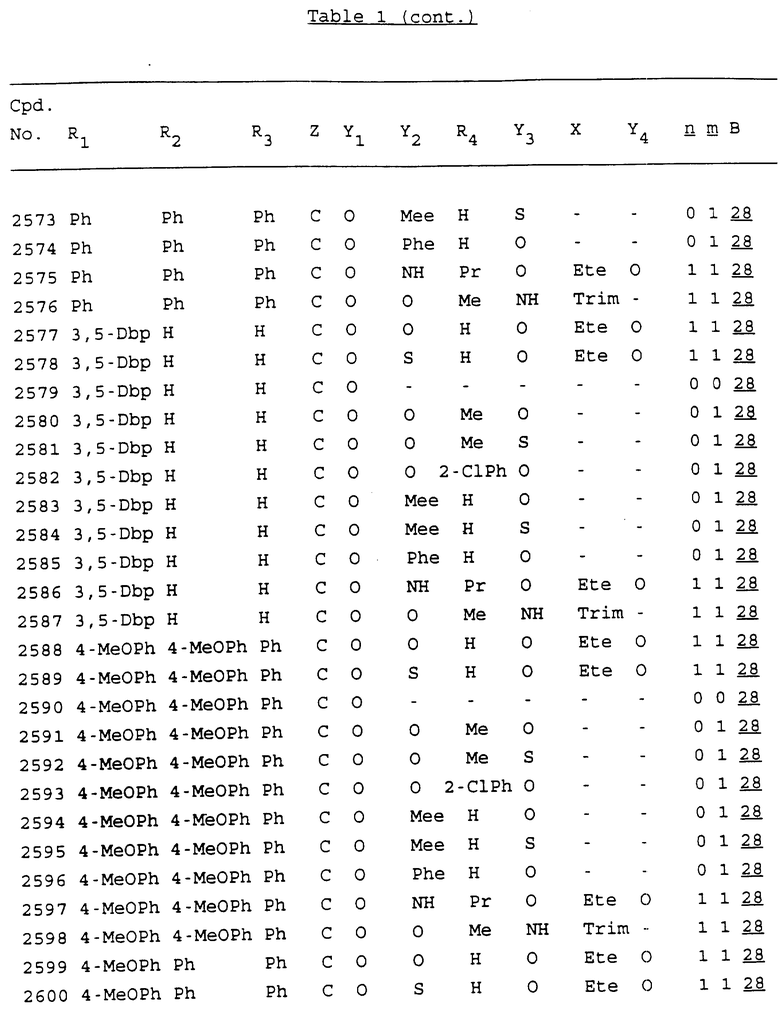
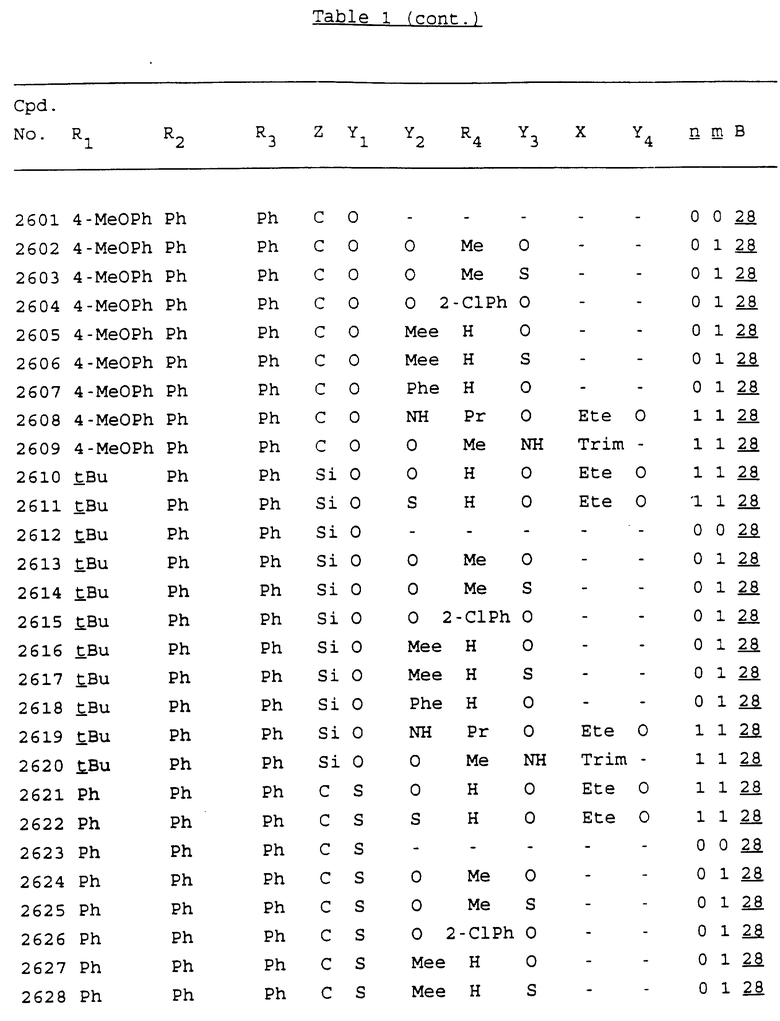
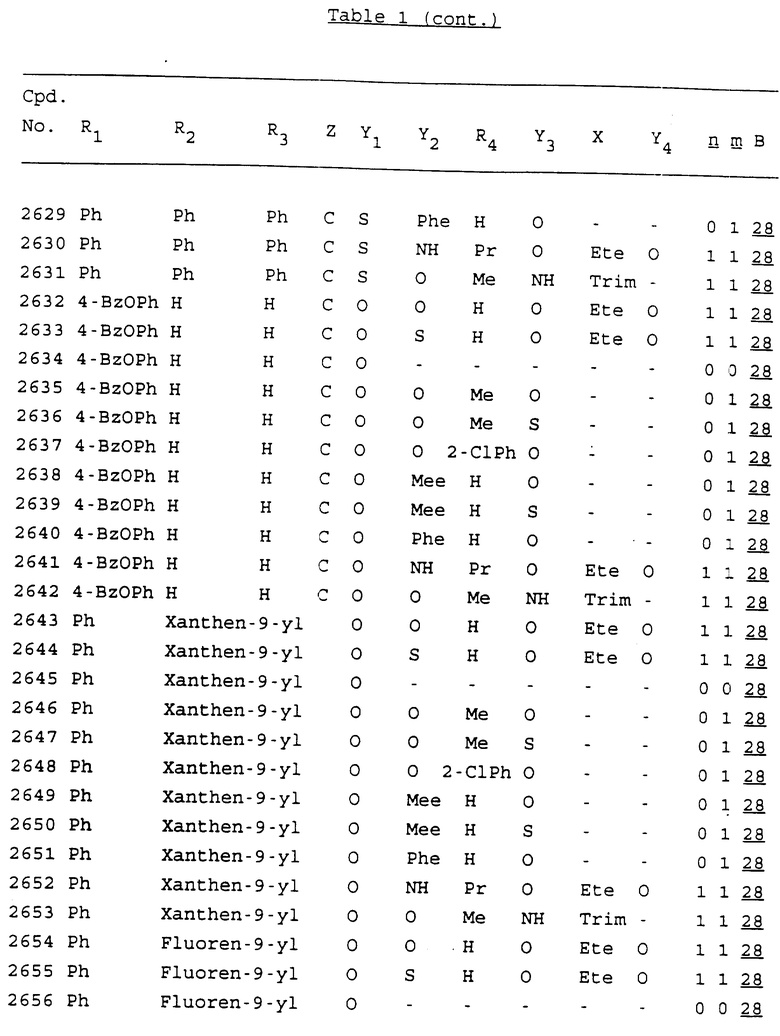
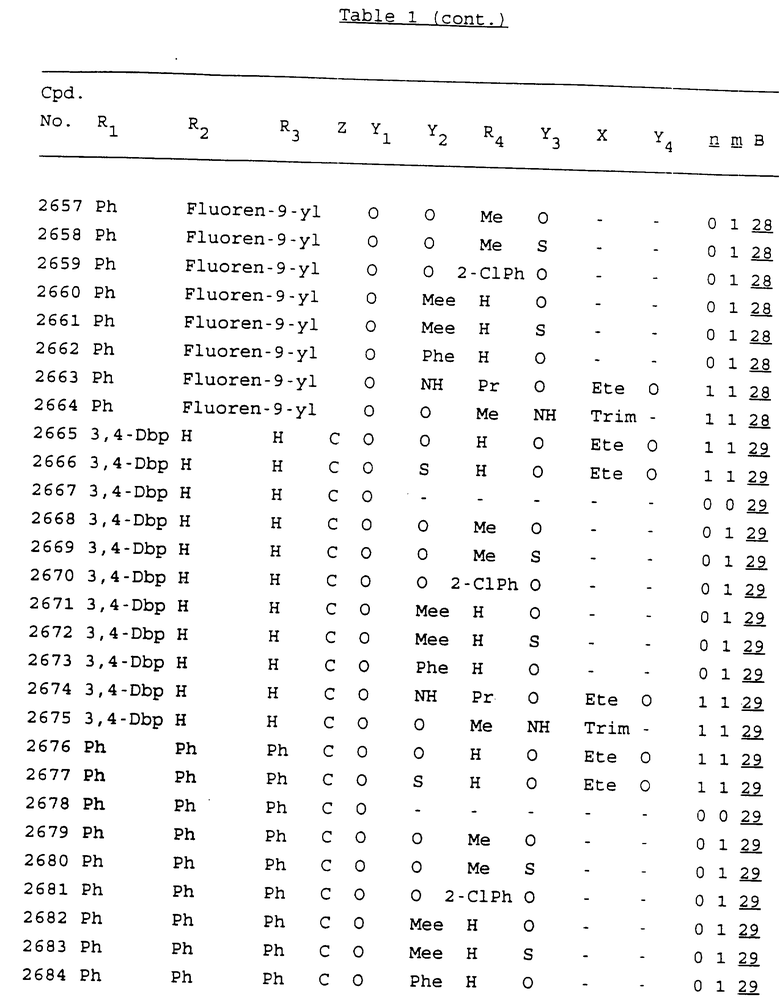
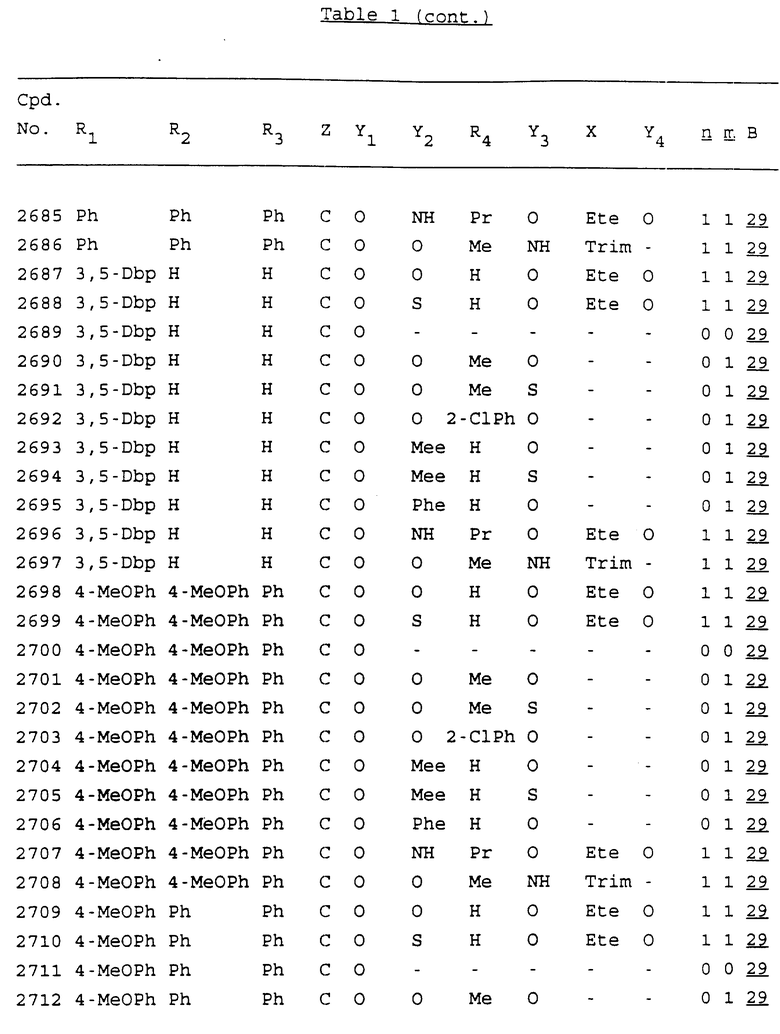
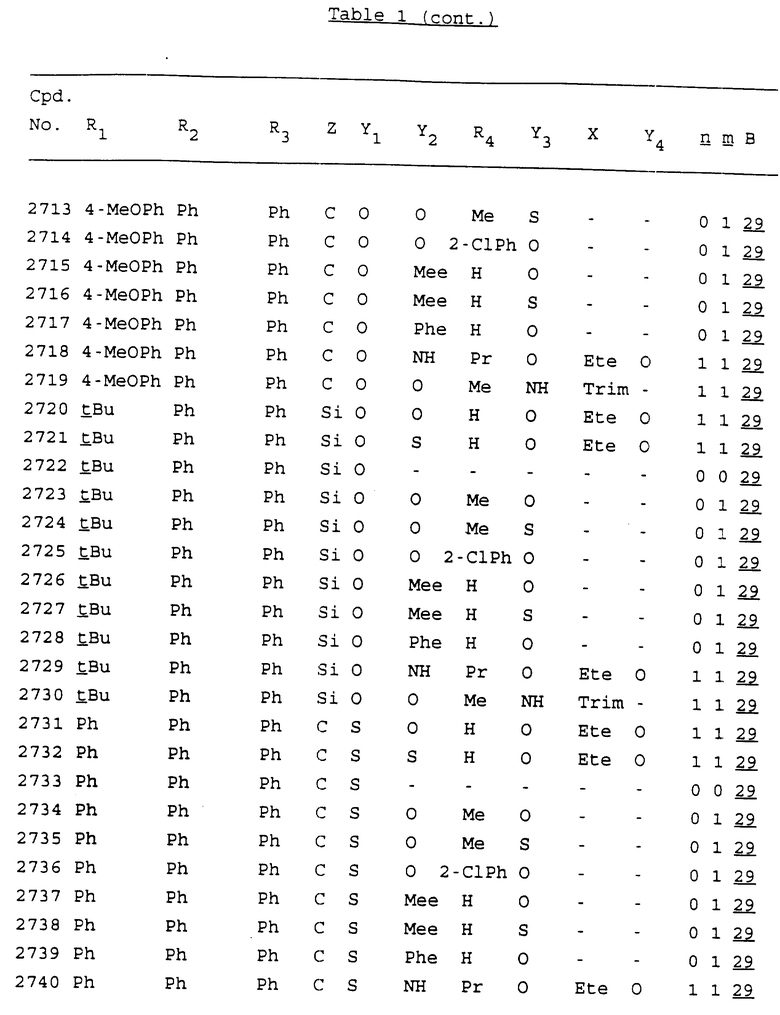
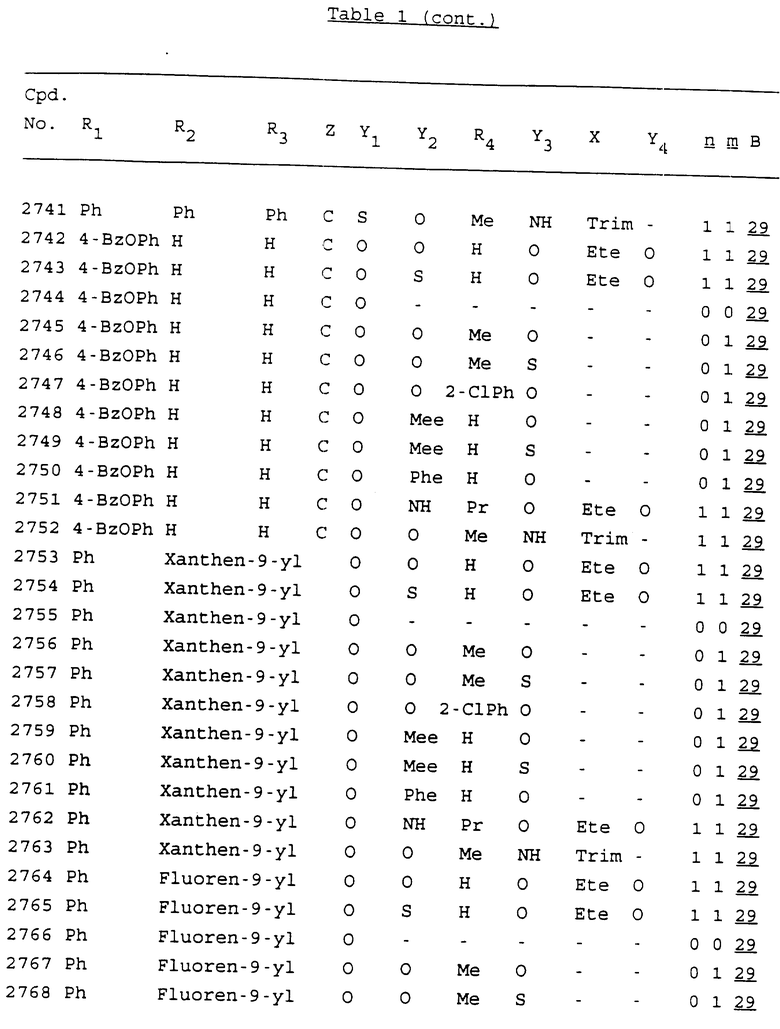
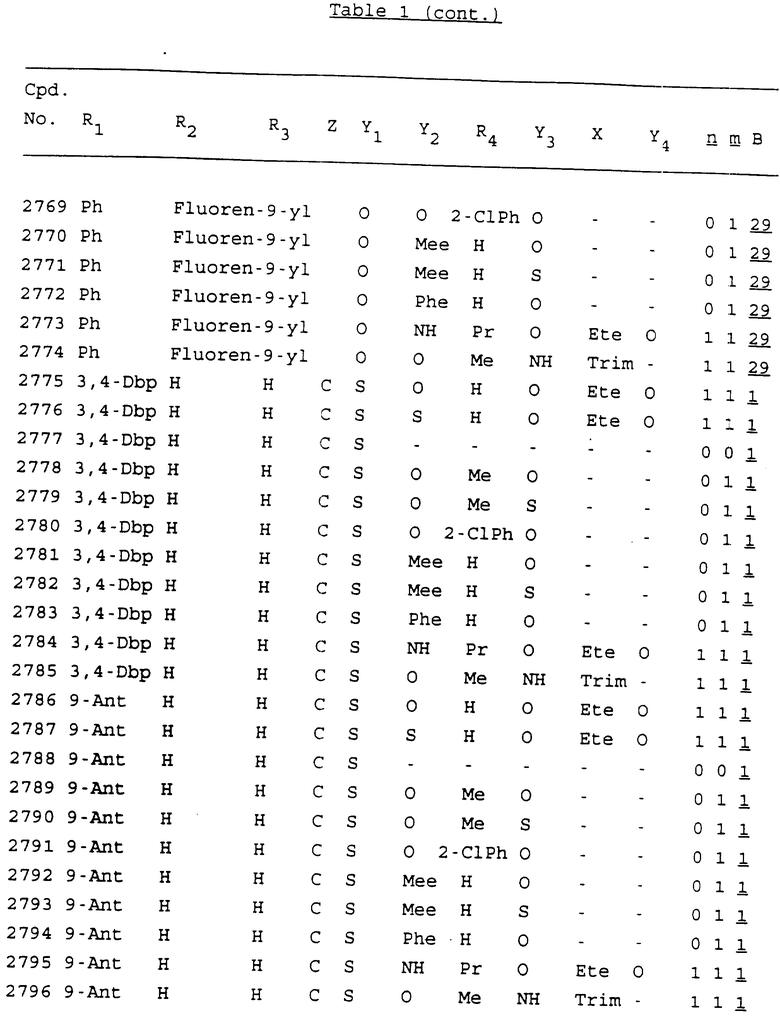
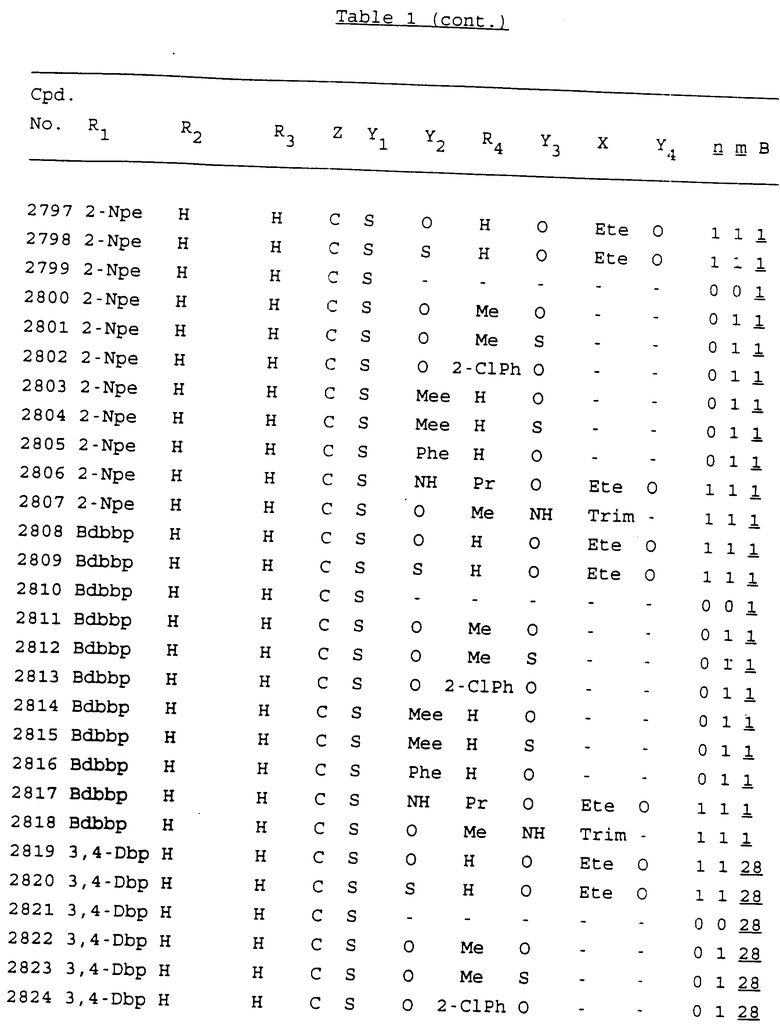
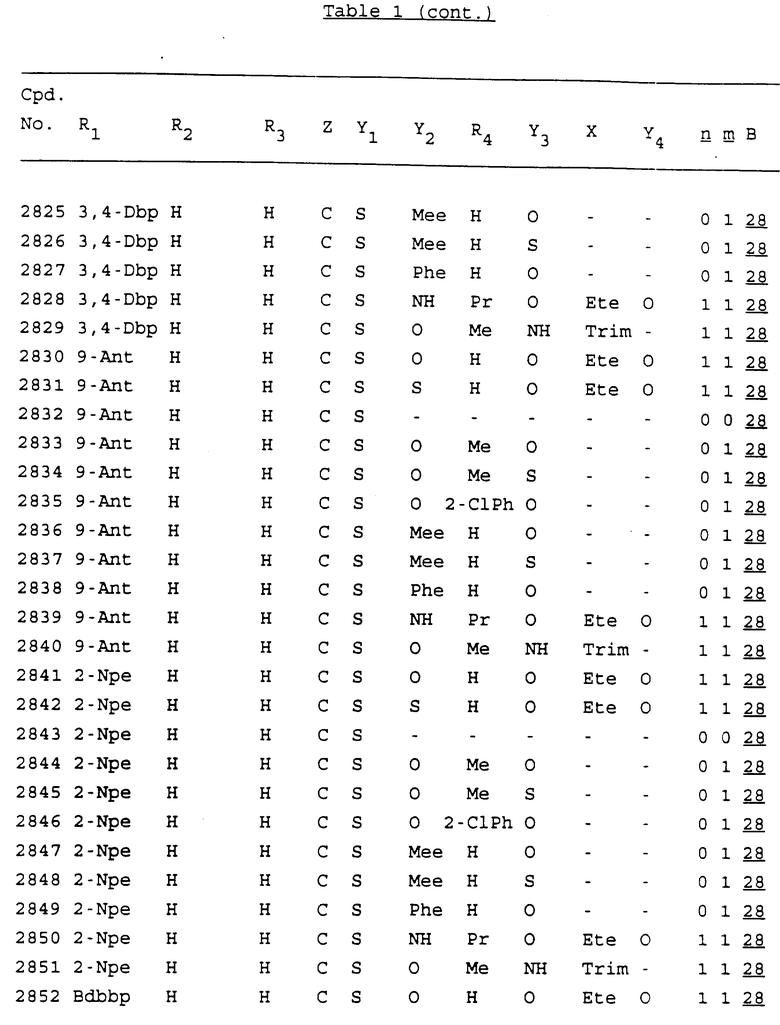
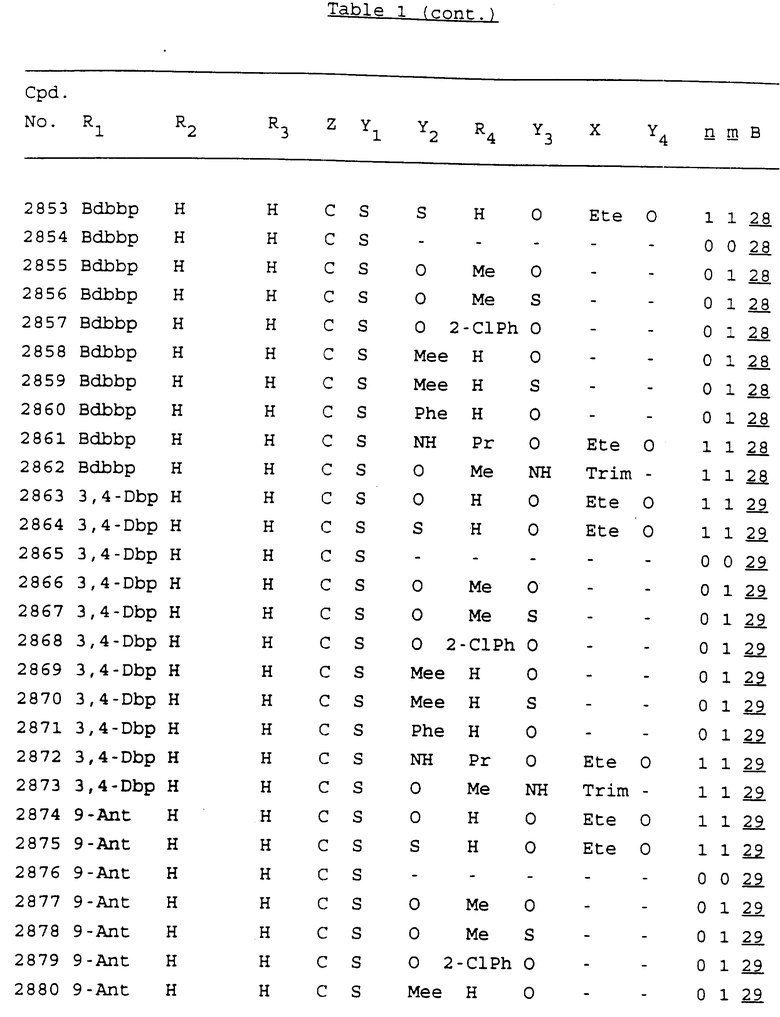
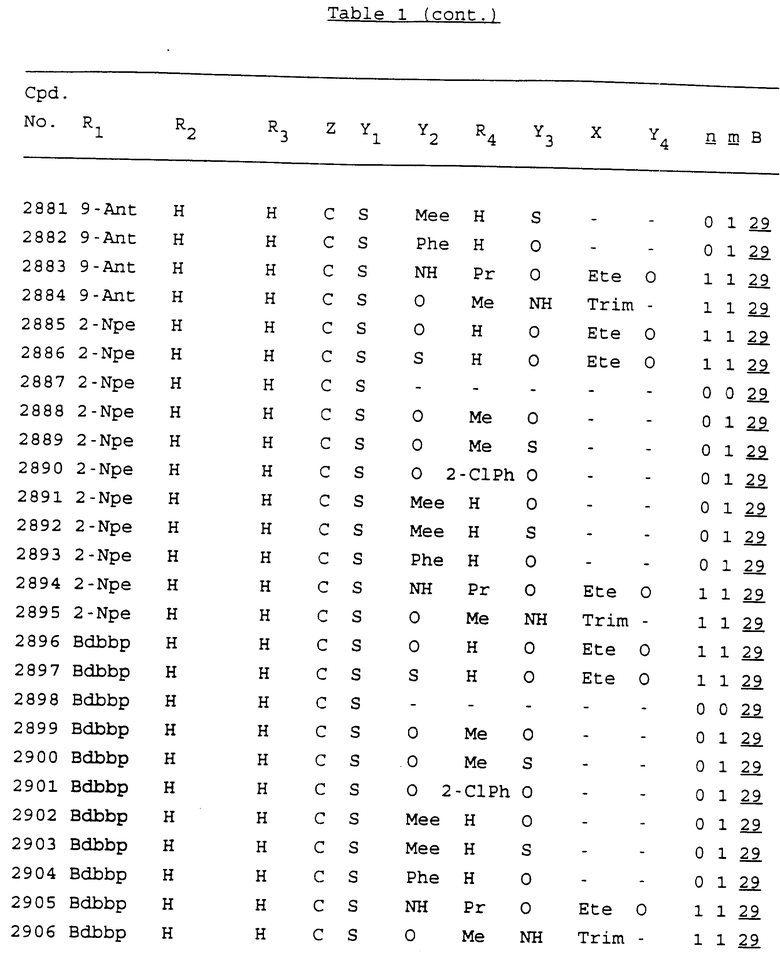
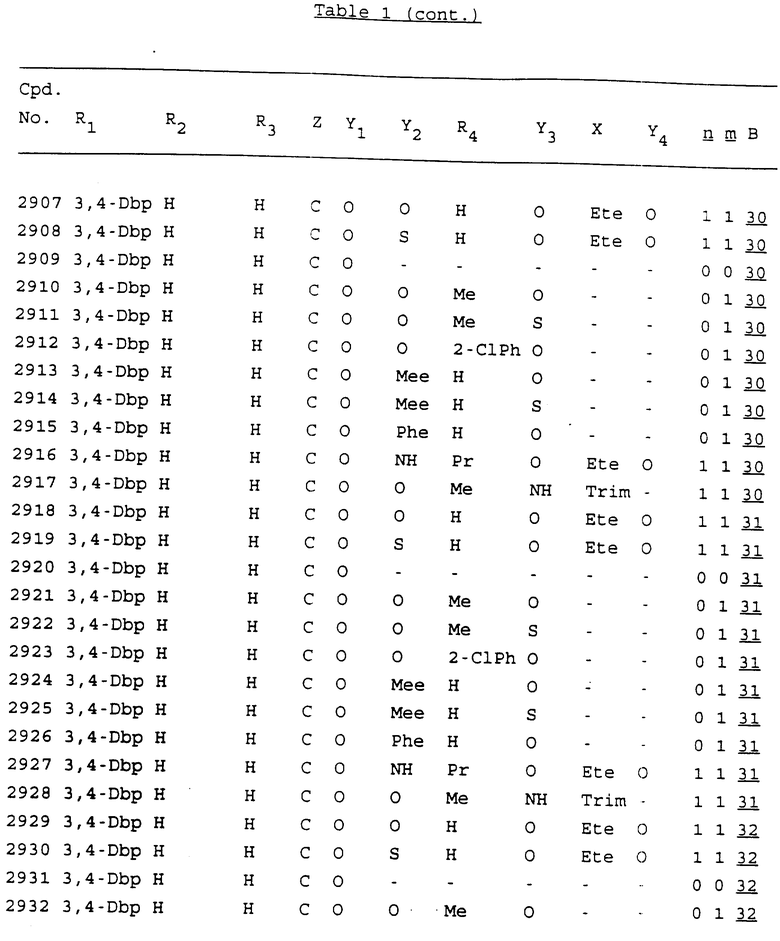
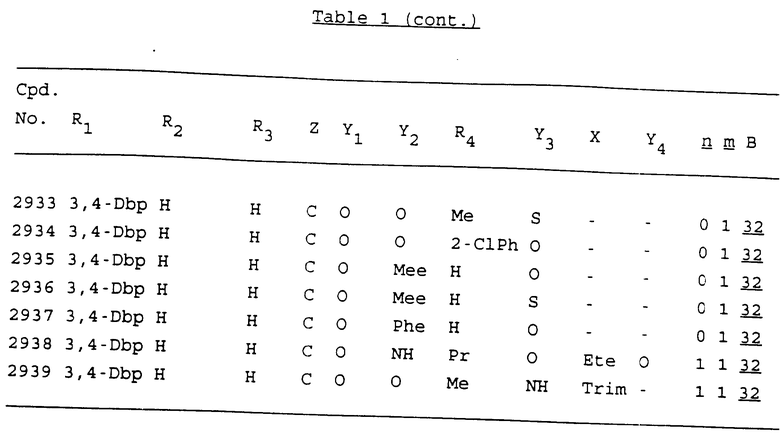
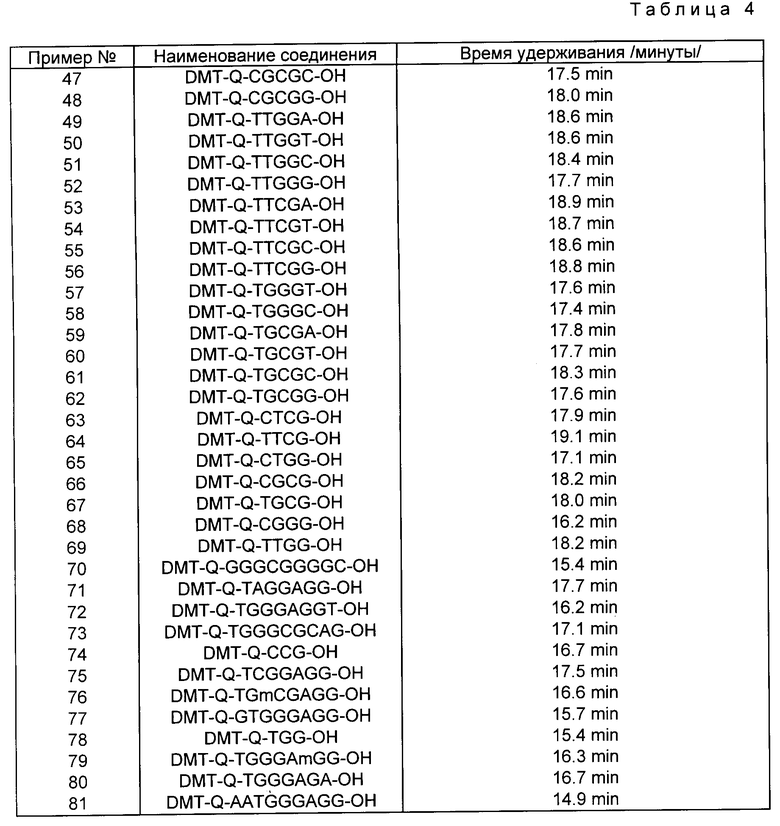
Предлагаются соединения для лечения или профилактики вирусных инфекций у млекопитающих, к которым относятся и люди. Соединения представляют собой производные олигодезоксирибонуклеиновой кислоты. Кроме того, предлагаются также новый способ получения таких соединений, а также промежуточные соединения, которые могут найти более общую область использования. Активные соединения являются соединениями общей формулы /1/ 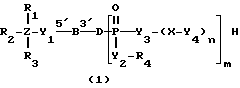 в которой R1, R2 и R3 представляют атомы водорода, алкильные группы, арильные группы, определенные в описании, и антрахинонильные группы, определенные в описании; Z представляет углерод или кремний; или R2, R3 и Z вместе представляют флуоренил или ксантенил; R4 представляет атом водорода, алкильную группу, как она определена в описании, арильную группу, определенную в описании, Y1, Y3 и Y4 представляют кислород, серу или группу
в которой R1, R2 и R3 представляют атомы водорода, алкильные группы, арильные группы, определенные в описании, и антрахинонильные группы, определенные в описании; Z представляет углерод или кремний; или R2, R3 и Z вместе представляют флуоренил или ксантенил; R4 представляет атом водорода, алкильную группу, как она определена в описании, арильную группу, определенную в описании, Y1, Y3 и Y4 представляют кислород, серу или группу  ; Y2 представляет кислород, серу,
; Y2 представляет кислород, серу,  , алкилен или фенилен, X - алкиленовая группа, как она определена в описании; m и n представляют 0 - 10; и B - олигодезоксирибонуклеотид цепи длиной 3 - 9, или их солями. 3 с. и 17 з.п. ф-лы. 4 табл.
, алкилен или фенилен, X - алкиленовая группа, как она определена в описании; m и n представляют 0 - 10; и B - олигодезоксирибонуклеотид цепи длиной 3 - 9, или их солями. 3 с. и 17 з.п. ф-лы. 4 табл.
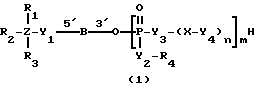
где R1, R2 и R3 независимо выбраны из группы, состоящей из атомов водорода, C1 - C4-алкильных групп, и арильных групп, указанных ниже;
Z - атом углерода или кремния;
или R2, R3 и Z вместе представляют флуоренильную или ксантенильную группу;
R4 - атом водорода, назамещенная C1 - C4-алкильная группа или фенильная группа, которая является незамещенной или замещена атомом галогена;
Y1, Y3 и Y4 независимо выбраны из группы, состоящей из атомов кислорода, атомов серы и группы формулы  ;
;
Y2 - атом кислорода, атом серы или C1 - C4-алкиленовая группа;
X - незамещенная C1 - C4-алкиленовая группа или C1 - C10-алкиленовая группа, и замещена по крайней мере одной гидроксигруппой;
m и n каждый независимо 0 или целое число от 1 до 10;
B - олигодезоксирибонуклеотид, имеющий длину цепи от 3 до 9;
указанная арильная группа представляет ароматическую карбоциклическую группу, которая имеет от 6 до 20 кольцевых углерода и является незамещенной или замещенной по крайней мере одним заместителем, выбранным из группы, состоящей из заместителей I, указанных ниже;
указанные заместители I выбраны из группы, состоящей из C1 - C4-алкоксигрупп, фенильных групп, бензилоксигрупп и 3,5-(дибензилокси)бензилокси групп,
или их соли.
R1R2R3Z - Y1
выбрана из группы, состоящей из трифенилметилокси, 3,4-(дибензилокси)бензилокси, 3,5-(дибензилокси)бензилокси, трет-бутилдифенилсилилокси, фенилфлуоренилокси и фенилксантенилоксигруппы, группа формулы
[P (O) (Y2R4) - Y3 - (X - Y4)n]m H
выбрана из группы, состоящей из атома водорода, метилфосфорила, 2-хлорфенилфосфорила, О-метилтиофосфорила, метилфосфонила, метилтиофосфонила; фенилфосфонила, 2-гидроксиэтилфосфорила, -О-(2-гидроксиэтил)тиофосфорила, фенилфосфорила, 4-хлорфенилфосфорила и этилфосфорила, B выбран из группы, состоящей из TGGGAG, TGGGA, TGGGG, TGGG, TGGGAGG, CGGGAGG, TTGGAGG, TTGGGAGG, TGCGAGG, GGGGAGG, mCGGGAGG, mCGmCGAGG, CTGGGAGG, GGGCGGGGC, TAGGAGG, TGGGAGGT, TGGGCGCAG, CCG, TCGGAGG, TGmCGAGG, CTGGGAGG, TGG, TGGGAmGG, TGGGAGA и AATGGGAGG.
R1R2R3Z - Y1
выбрана из группы, состоящей из трифенилметилокси, 3,4-(дибензилокси)бензилокси, 3,5-(дибензилокси)бензилокси, трет-бутилдифенил-силилокси, фенилфлуоренилокси и фенилксантенилоксигруппы, группа формулы
[P (O) (Y2R4) - Y3 - (X - Y4)n]mH
выбрана из группы, состоящей из атома водорода, метилфосфорила, 2-хлорфенилфосфорила, -О-метилтиофосфорила, метилфосфонила, метилтиофосфонила, фенилфосфонила, 2-гидроксиэтилфосфорила, -О-(2-гидроксиэтил)тиофосфорила, фенилфосфорила, 4-хлорфенилфосфорила и этилфосфорила; и B выбран из группы, состоящей из TGGGAG, TGGGA, TGGGG, TGGG, TGGGAGG, CGGGAGG, TTGGAGG, TTGGGAGG, TGCGAGG, GGGGAGG, mCGGGAGG, mCGmCGAGG и CTGGGAGG.
R1R2R3Z - Y1
выбрана из группы, состоящей из трифенилметилокси, 3,4-(дибензилокси)бензилокси, 3,5-(дибензилокси)бензилокси, трет-бутилдифенилсилилокси, фенилфлуоренилокси и фенилксантенилокси группы, группа формулы
[P (O) (Y2R4) - Y3 - (X - Y4)n]mH
выбрана из группы, состоящей из атома водорода, метилфосфорила, 2-хлорфенилфосфорила, -О-метилтиофосфорила, метилфосфонила, метилтиофосфонила, фенилфосфонила, 2-гидроксиэтилфосфорила, и -О-(2-гидроксиэтил)тиофосфорильной группы, и B выбран из группы, состоящей из TGGGAG, TGGGA, TGGGG, TGGG, TGGGAGG, CGGGAGG, TTGGAGG, TTGGGAGG, TGCGAGG, GGGGAGG, mCGGGAGG, mCGmCGAGG и CTGGGAGG.
R1R2R3Z - Y1
выбрана из группы, состоящей из трифенилметилокси, 3,4-(дибензилокси)бензилокси и 3,5-(дибензилокси)бензилоксигруппы, группа формулы
[P (O) (Y2R4) - Y3 - (X - Y4)n]mH
выбрана из группы, состоящей из атома водорода, метилфосфорила, 2-хлорфенилфосфорила, -О-метилфиофосфорила, метилфосфонила, метилтиофосфонила, фенилфосфонила, 2-гидроксиэтилфосфорила, -О-(2-гидроксиэтил)тиофосфорила, фенилфосфорила, 4-хлорфенил фосфорильной и этилфосфорильной группы, и B выбран из группы, состоящей из TGGGAG, TGGGA, TGGGG, TGGG, TGGGAGG, CGGGAGG, TTGGAGG, TTGGGAGG, TGCGAGG, GGGGAGG, mCGGGAGG, mCGmCGAGG и CTGGGAGG.
R1R2R3Z - Y1
выбрана из группы, состоящей из трифенилметилокси, 3,4-(дибензилокси)бензилокси и 3,5-(дибензилокси)бензилоксигруппы, группа формулы
[P (O) (Y2R4) - Y3 - (X - Y4)n]mH
выбрана из группы, состоящей из атома водорода, метилфосфорила, 2-хлорфенилфосфорила, -О-метилтиофосфорила, метилфосфонила, метилтиофосфонила, фенилфосфонила, 2-гидроксиэтилфосфорила, и -О-(2-гидроксиэтил)тиофосфорильной группы, и B выбран из группы, состоящей из TGGGAG, TGGGA, TGGGG, TGGG, TGGGAGG, CGGGAGG, TTGGAGG, TTGGGAGG, TGCGAGG, GGGGAGG, mCGGGAGG, mCGmCGAGG и CTGGGAGG.
R1R2R3Z - Y1
представляет 3,4-(дибензилокси)бензилоксигруппу, Y2 - атом кислорода, Y3 - атом кислорода, Y4 - атом кислорода, R4 - атом водорода, X - этиленовую группу, m = 1, n = 1 и B - TGGGAG.
R1R2R3Z - Y1
представляет 3,4-(дибензилокси)бензилоксигруппу, Y2 - атом серы, Y3 - атом кислорода, Y4 - атом кислорода, R4 - атом водорода, X - этиленовую группу, m = 1, n = 1 и B - TGGGAG.
R1R2R3Z - Y1
представляет 3,4-(дибензилокси)бензилоксигруппу, m = 0, n = 0 и B - TGGGAG.
R1R2R3Z - Y1
представляет 3,4-(дибензилокси)бензилоксигруппу, Y2 - атом кислорода, Y3 - атом кислорода, R4 - метильную группу, m = 1, n = 0 и B - TGGGAG.
R1R2R3Z - Y1
представляет трифенилметилоксигруппу, Y2 - атом кислорода, Y3 - атом кислорода, Y4 - атом кислорода, R4 - атом водорода, X - этиленовую группу, m = 1, n = 1 и B - TGGGAG.
R1R2R3Z - Y1
представляет триметилфенилоксигруппу, Y2 - атом серы, Y3 - атом кислорода, Y4 - атом кислорода, R4 - атом водорода, X - этиленовую группу, m = 1, n = 1 и B - TGGGAG.
R1R2R3Z - Y1
представляет триметилфенилоксигруппу, m = 0, n = 0 и B - TGGGAG.
R1R2R3Z - Y1
представляет триметилфенилоксигруппу, Y2 - атом кислорода, Y3 - атом кислорода, R4 - метильную группу, m = 1, n = 0 и B - TGGGAG.
R1R2R3Z - Y1
представляет 3,5-бис[3,5-(дибензилокси)бензилокси]бензилтио, группа формулы
[P (O) (Y2R4) - Y3 - (X - Y4)n]mH
представляет атом водорода и B - TGGGAG.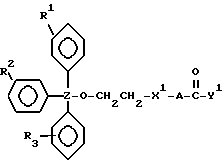 (12)
(12)
или общей формулы (14)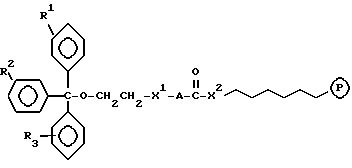 (14)
(14)
где R1, R2 и R3 - одинаковые или различные, каждый независимо атом водорода или C1 - C4-алкоксигруппа;
Z - атом углерода;
A - группа - (CH2)h - (где h - целое число от 1 до 16), группа - (CH2)hO - (где h - целое число от 1 до 16), группа -(CH2)j - O - CO - (CH2)k- (где j - положительное целое число, k - положительное целое число и j + k = 2 - 16) или группа -(CH2)n - O - CO - CH2(OCH2CH2)p - OCH2- (где n - положительное целое число, р = 0 или положительное целое число и j + k = 2 - 100);
P - полимерный материал;
X1 - группа -SO2-;
X2 - атом кислорода или NH-группа;
Y1 - гидроксильная группа или этилоксигруппа, которая замещена атомом галогена.
| WO, заявка 8807544, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

